
Submitted:
14 December 2023
Posted:
15 December 2023
You are already at the latest version
Abstract
Plant cells are omnipotent and breeding of new varieties can be achieved by protoplast fusion. Such fusions can be achieved by treatment with poly(ethylene glycol) or by applying an electric field. Microfluidic devices allow for controlled conditions and targeted manipulation of small batches of cells down to single cell analysis. To provide controlled conditions for protoplast fusions and achieve high reproducibility, we developed and characterized a microfluidic device to reliably trap few of Arabidopsis thaliana protoplasts and induced a cell fusion by controlled addition of poly(ethylene glycol) (PEG, with a molecular weight of 6000). Experiments were conducted to determine the survival rate of isolated protoplasts in our microfluidic system. Afterwards, PEG induced fusion was studied. Our results indicate that the following fusion parameters had significant impact on the fusion efficiency and duration: PEG concentration, osmolarity of solution, and flow velocity. PEG concentration below 10% led to only partial fusion. Osmolarity of the PEG fusion solution was found to strongly impact the fusion process; complete fusion of two source cells sufficiently took part in slightly hyper osmotic solutions, whereas iso-osmotic solutions led to only partial fusion at 20% PEG concentration. We observed accelerated fusion for higher fluid velocities. Up to this study, it was common sense that fusion is one directional, i.e. once two cells were fused into one cell they stay fused. Here, we present for the first time reversible fusion of protoplasts. Our microfluidic device paves the way to a deeper understanding on the kinetics and processes of cell fusion.
Keywords:
Protoplast
; Arabidopsis thaliana
; induced cell fusion
; microfluidics
; Poly(ethylene glycol)
; single cell analysis
1. Introduction
Protoplasts are a versatile tool for plant cell biology [1]. For instance, they allow for transient transfection by osmotic shock with PEG-mediated plasmid import [2]. This method has been applied frequently for fluorescence microscopy: Multiple proteins have been localized this way, for instance V-ATPase subunits, malate dehydrogenases and peroxiredoxins [3,4,5,6]. Transient expression enabled the quantitative in vivo visualization of protein-protein interactions by bimolecular fluorescence complementation or Förster resonance energy transfer [7,8]. Transactivation of promoters by transcription factors has been addressed by using protoplasts, too [9]. Protoplasts are omnipotent cells which can be cultivated for callus formation or even plant growth, so that protoplasting became a breeding tool in combination with cell-cell fusions and the formation of hybrid cells [10,11]. Cell fusions are regular processes in flowering plants, such as the egg cell fertilization and synergid-endosperm fusion [12,13]. In the lab, fusion of protoplasts can be achieved by addition of PEG or by applying an electric field [14]. The resulting hybrid cell carries organelles, transcriptomes, proteomes and genomes of two source cells, so that analyzing the fate of all these turns cell fusion to a promising tool for the investigation of cellular organization and dynamics. Whereas the cellular organization subsequent fertilization and synergid-endosperm fusion is highly organized and regulated, the re-organization following protoplast fusion has to be either decisive or by chance. To investigate this process in detail, microfluidics is an appropriate and promising tool.
Standard devices like well-plates are insufficient for long-term observation of cells and single-cell observations. Microfluidic devices are versatile tool-boxes for single-cell studies because of the small dimensions and the well-controlled environment [15]. For instance, the channel dimensions of the devices can be adapted according to the respective needs; in this work, the channel height has to be slightly larger than the protoplasts to provide long-term observations, without the cell diffusing out of the focus, this cannot be achieved with conventional lab dishes. Microfluidic devices provide access to single-cell investigations with time-lapse microscopy and so yielding inside views in dynamics of processes [16]. Often, microfluidic devises are made monolithically from silicones that provide easy fabrication and tailoring of the devices` layout according to the respective studies [15]. So far, microfluidics has been used in numerous applications with plant cells. For instance, microfluidic devices were used to conduct cultivation studies providing deeper insight in cell mechanisms [15,17,18,19,20]. Microfluidics also was used to design small channels for investigation of pollen and root growth in confined structures [21,22,23,24]. Sakai et al used microfluidics to trap protoplasts over long time periods to observe cell wall reconstitution [25]. Ko et al compared cell division rates of tobacco protoplasts in PDMS microfluidic devices and Petri-dishes. They found that the microenvironment in the microfluidic devices did not harm the cells when cultured over several days and even provide better conditions for cell division than the Petri-dish [26]. Wu et al conducted tobacco protoplast fusion in a microfluidic device [27]. Their device consisted of a micro-column array that span the whole channel width preventing the protoplasts to pass by. They added PEG of high concentration and observed fusion of cells that were randomly distributed [28]. To the authors knowledge there has not been a microfluidic device that provides fusion of a controllable number of protoplasts, ideally two, so far. Here, we present for the first time a microfluidic device consisting tailored features to trap two A. thaliana protoplasts in close contact providing controlled PEG induced fusion. Those devices were used to study the impact of various parameter impacting the fusion efficiency, duration and reversibility.
2. Materials and Methods
2.1. Buffers and solutions
W5-buffer was used as resuspension medium after isolation of protoplasts. It contained 125 mM CaCl2, 5 mM KCl, 154 mM NaCl, 2 mM MES-KOH (pH 5.7), 5 mM glucose. The cell wall was lysed enzymatically by cellulose R-10 and macerozyme R-10. Lysis solution contained 1.5% cellulase R-10, 0.4% macerozyme R-10 (both SERVA Electrophoresis, Germany), 0.4 M mannitol, 20 mM KCl, 20 mM MES-KOH (pH 5.7), 10 mM CaCl2, 0.1% BSA fraction V. Murashige & Skoog-media (MS-media) [29] was purchased from Duchefa without vitamins (M0221) and supplemented with Gamborg B5 vitamins [30].
The PEG-fusion solution contained 0.147% CaCl2 and PEG6000 of respective (w/v)% in W5-buffer and green fluorescent beads (0.5 µm FluoSpheres (505/515), Thermo Fisher Scientific, Germany) for staining the flow. After adjusting the pH-value to 5.7 with KOH, the solution was autoclaved. Additionally 500 µM F108 (BASF, Germany) was added to all buffers used in the microfluidic devices serving as dynamic surface coating agent to suppress unspecific adsorption of the protoplasts to the channel surfaces and to increase the wettability of the hydrophobic PDMS. The surfaces of the PDMS became hydrophilic due to the surface coating [31], which also avoids trapping of air bubbles.
2.2. Fabrication of microfluidic devices
The microfluidic devices were fabricated by PDMS (poly(dimethylsiloxane)) soft lithography as described in [32]. First, a silicon wafer was covered with SU-8(50) photoresist by spin coating at 3,000 rpm for 30 s, leading to structure height of 56 µm. Beforehand, the wafer was cleaned in caroic acid as described elsewhere [33]. After spin coating the photoresist was prebaked at 95 °C for 20 min and illuminated through a chromium mask (DeltaMask, The Netherlands) for 29 s. The photoresist was hard baked at 95 °C for 8 min before developing in a mr-dev developer (Microresist, Germany) bath, submersion for 15 min and gentle rinsing every 2 min with acetone. After developing the negative relief structures were visible on the master wafer, which was hard baked before silanization with (APTES, Sigma Aldrich, USA).
PDMS elastomer (Sylgard 184, Dow Corning, USA) was poured over the master wafer and thermally cured at 85 °C for 4 h. After curing, the PDMS was peeled-off and reservoir holes were punched at the channel ends for fluidic access. The microfluidic channel was closed with a PDMS covered glass slide after oxygen plasma treatment and filled with buffer [33].
2.3. Measurement setup
A PMMA holder with 3 mm reservoir holes, to enlarge the reservoir volumes, was placed on the PDMS chip. Working buffer, PEG-solution, and protoplasts were pipetted into the respective reservoirs. The microfluidic chip was then placed either on an inverted microscope, using an additional filter (U-25LBD, Olympus, Japan) to gain true-colored images, or a confocal laser scanning microscope (CLSM). The fluids and cells were driven through the microfluidic device by applying pressure (MFCS, Fluigent, Germany).
2.4. Imaging
Cells in microfluidic devices were imaged with a bright field microscope (Axiovert 100 TV, Zeiss, Germany) with illumination filter (U-25LBD, Olympus, Japan) and a CCD-camera (HCD-905, Day&Night digital color camera). Images were processed with Fiji. Additionally, microfluidic experiments were conducted on a CLSM (Zeiss LSM780 with 10-fold magnification. Images were obtained upon 488 nm excitation; the emission range was 500 – 600 nm for fluorescent beads and vacuolar staining and 650 – 700 nm for chlorophyll autofluorescence, the beam splitter was MBS488. In addition, bright field images were obtained in the transmitted light mode. Images were processed with Zeiss Zen 3.7.
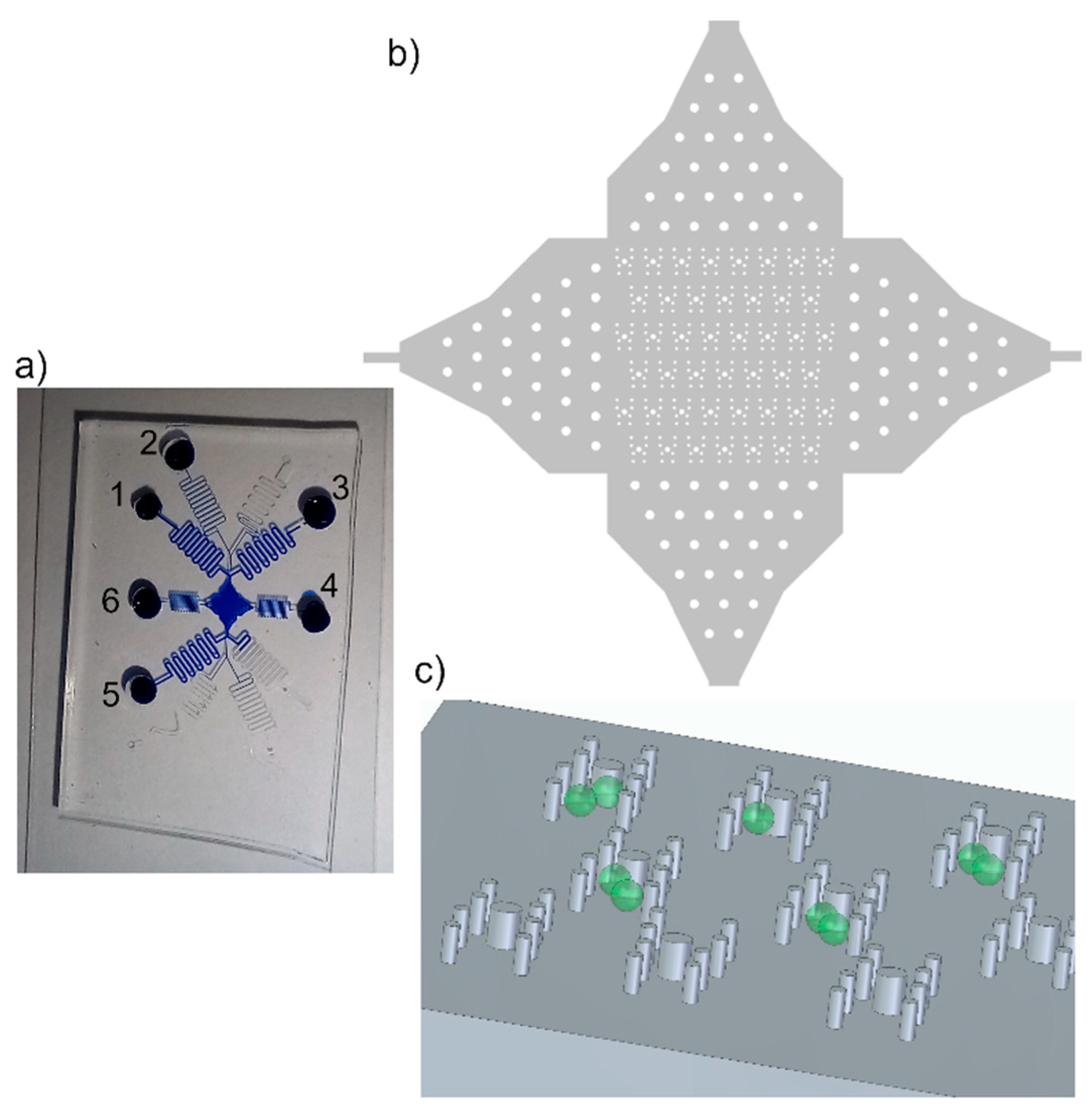
Figure 1.
Trapping features. a) Photograph of device filled with blue ink for better visualisation. The protoplasts were in reservoir 1, W5-buffer was in reservoirs 2,4-6. PEG fusion buffer was is reservoir 3. The channel lengths were about 20 cm to gain flow control. b) Overview of array of trapping region with posts. The posts in the channel entrances served for pre-distribution of cells. c) Sketch of posts, aligned in H-shape to form the trapping features, and cells (green). The posts reach from channel bottom to channel ceiling, 56 µm.
Figure 1.
Trapping features. a) Photograph of device filled with blue ink for better visualisation. The protoplasts were in reservoir 1, W5-buffer was in reservoirs 2,4-6. PEG fusion buffer was is reservoir 3. The channel lengths were about 20 cm to gain flow control. b) Overview of array of trapping region with posts. The posts in the channel entrances served for pre-distribution of cells. c) Sketch of posts, aligned in H-shape to form the trapping features, and cells (green). The posts reach from channel bottom to channel ceiling, 56 µm.
2.5. Protoplasts isolation, purification and fusion
Protoplasts were isolated as described before [34]. Briefly, approximately 2 g of four weeks old A. thaliana leaves were applied, this yields roughly 500,000 cells per ml or six million cells in total. The leaves were cut into leave stripes and vacuum-infiltrated with lysis solution and incubated at room temperature for 2 hours. Iso-osmotic solutions were achieved by adding 154 mM NaCl or 0.4 M mannitol to W5-solution, lysis solution, or fusion solution, respectively. Cells were separated from debris by a nylon mesh and harvested by centrifugation at 100 x g for three minutes. The viability tests were conducted either by detecting the chlorophyll absorption at 650 – 700 nm as a marker for viability or by vital staining with neutral red.
The fusion was induced by flushing the trapped cells with PEG-fusion solution of respective concentration.
2.6. Statistical analysis
For statistical analyses mean and standard deviations were calculated and student’s T-test was performed as indicated in the figure legends.
3. Results
3.1. Determination of cell vitality
The protoplast fusion is followed by a (re-)organization of the hybrid cell, which is expected to take at least hours. Therefore, the vitality of cells was monitored in the microfluidic device over time to test the vitality in the microfluidic devices. Cells were stained with neutral red as vital stain and the time point of the loss of the red staining was monitored. Second, the shape of the cells was analyzed. Neutral red-destaining occurred after 212 ± 68 minutes (mean ± SE, n=7) and the cells collapsed after 663 ± 141 minutes (mean ± SE, n=7). Cooling of the device resulted in no significant alteration of the cellular vitality (T-test, p=0.83).
Additionally, vitality of cells in microfluidic devices was analyzed by visual inspection, i.e. without staining to not interfere with the fusion process, during cell fusion. The analysis revealed that the cells kept their shape and size with small deviations, and thus, the fused protoplasts are assumed to be alive during the whole process, i.e. trapping, fusion and observation afterwards.
3.2. PEG-concentrations for fusions – batch experiments
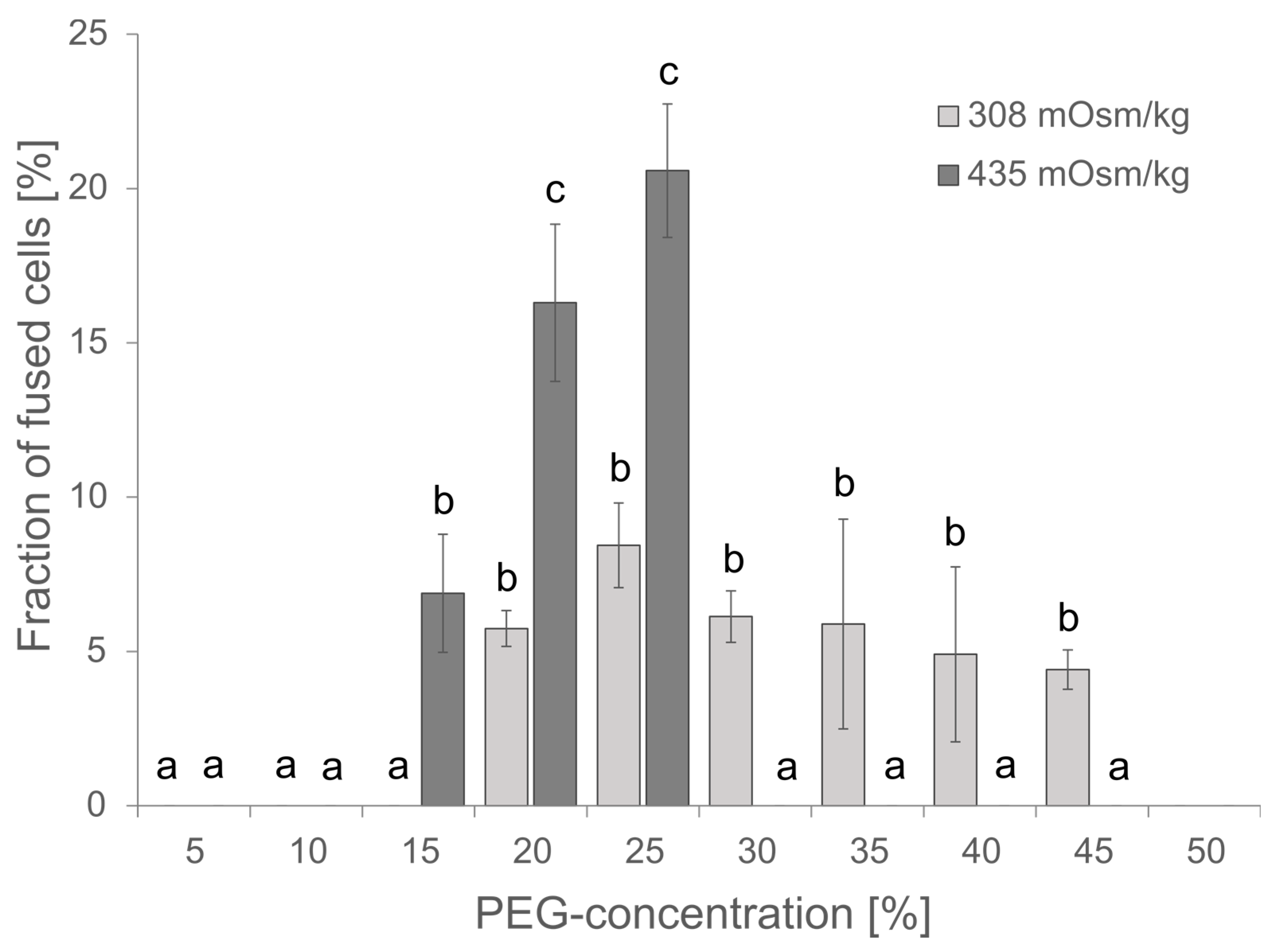
The PEG-concentration is critical for the fusion efficiency, but PEG might also be cytotoxic at higher concentrations of more than 25% as observed for the transfection of protoplasts from Elaeis guineensis [35]. Although the PEG-concentration has been estimated for protoplast fusions before by Xiao and co-workers and within their studies 20% PEG gave the best results [36], the conditions might be different within a microfluidic device. Vitality of cells and fusion efficiency have been investigated for PEG-concentrations in the range of 0 to 45 %. The highest fusion efficiency was observed at 20-25% PEG, the survival of cells was unaffected at PEG-concentration ≤ 20 % as indicated by insignificant changes in chlorophyll absorption (Suppl. Figure S1). Therefore, subsequent experiments were started with 20 % PEG. Usually, 0.4 M mannitol or 154 mM NaCl have been used as osmotically active component in Arabidopsis media, though both differ in their osmolarity of 435 and 308 mOsm kg-1, respectively. Comparing the effect of osmolarity on cell fusions revealed increased toxicity for higher osmolarity and a resulting lethality at PEG-concentrations of 30% and higher, but also an improved fusion rate and thus, more efficient fusion at 20-25 % PEG and 435 mOsm kg-1.
Figure 2.
Effect of PEG-concentration and osmolarity on the fusion efficiency. For each PEG-concentration 80-300 protoplasts were analysed for apparent cell fusions either in W5-media of 308 mOsm kg-1 or in 0.4 M Mannitol 2mM MES pH 5.7 of 435 mOsm kg-1. Mean ± SE is given, N=3 independent counts. Significance was proven by ANOVA followed by Scheffe posthoc-test and given letters (p<0.05).
Figure 2.
Effect of PEG-concentration and osmolarity on the fusion efficiency. For each PEG-concentration 80-300 protoplasts were analysed for apparent cell fusions either in W5-media of 308 mOsm kg-1 or in 0.4 M Mannitol 2mM MES pH 5.7 of 435 mOsm kg-1. Mean ± SE is given, N=3 independent counts. Significance was proven by ANOVA followed by Scheffe posthoc-test and given letters (p<0.05).
3.3. Microfluidic device fabrication and characterization
When the microfluidic devices were designed we had to consider the size of the protoplasts, before and after fusion, while the latter was estimated, because this directly set the bottom limit of the channel height and the dimensions of the trapping features. Protoplasts of Arabidopsis thaliana are about 30 µm in diameter. Taking also into account that some additional space has to be around the protoplast to enable sufficient flushing of both medium and protoplasts we set the channel height to a minimum of 55 µm. The minimum width of the trapping features was set to 90 µm to enable easy trapping of protoplasts. We observed that trapping of protoplasts in shallower features took longer because of directing the cells into the shallow trap entrance.
The first trapping features of the protoplasts were made of posts, reaching from channel bottom to ceiling, arranged in double U-shape, Figure 1. We tested two diameters, 20 µm and 40 µm, of the posts and evaluated them with respect to ease of fabrication and the fluid behavior in the final device. The microfluidic device was fabricated by PDMS soft lithography; see Section 2.2, where the channel structure was cast from a master wafer. Along fabrication of the master wafer, the recesses in the photoresist from which the posts were made were the largest challenge; this was because the removal of the uncured photoresist from the recess was difficult since the developer hardly penetrated that region and removed the resist and residuals remained in that area. This was intensified for photoresist layers of larger thickness.
We improved the photoresist development process by gentle shaking of the beaker with the wafer in the developer every minute starting after 8 min of developing. Therefore, the photoresist was removed from the post recesses and intact post structures could be molded from the master wafer with PDMS, Figure 1.
After fabrication of the microfluidic device we evaluated the fluid stream with special focus to the region of the trapping features. For that purpose we ran two fluid streams, one with pure water and one with the fluorescent dye fluorescein isothiocyanate (FITC) and monitored the streams by fluorescent microscopy. Though, the typical laminar flow could be observed in the channel, the posts of smaller diameter led to less disturbance of the fluid stream than the 40 µm posts. Therefore, we used posts with 20 µm diameter for all subsequent protoplast experiments.
Appropriate flow control via pressure driven flow was significantly improved by using channel length of 20 cm. This was necessary because of the large channel cross-section, channel width of 300 µm and the height of 56 µm, and thus low flow resistance.
Protoplasts were directed through the microfluidic device by applying sufficient pressures to the respective reservoirs, see Figure 1. The flow of the cells could be easily controlled due to the laminar flow; thus, the stream of cells in the trapping array was adapted by applying additional flow from perpendicular to the main flow direction, i.e. pressure was applied either to reservoir 4 or 6 to ensure most of the traps were filled with two protoplasts. Thus, the filling efficiency was not solely dependent on the cell distribution in the flow but could be controlled. Once, protoplasts entered the trapping feature they were reliably withhold by the posts, see Figure 3, i.e. trapped, until the fluid stream was inverted. Excess cells, i.e. if more than two cells were trapped, were removed by a flow perpendicular to the trapping features.
3.4. Characterization of PEG induced fusion in microfluidic devices
After fabrication and characterization of the microfluidic device for trapping protoplasts, we tested different PEG concentrations for induction of fusion of protoplasts. We flushed the microfluidic device with protoplasts until two to three protoplasts were trapped. The applied protoplast suspension had a concentration of roughly 500,000 cells per ml or six million cells in total.
Protoplast fusion was induced by introducing PEG-buffer into the trapping array after sufficient loading of the trapping features with protoplasts. We studied PEG-induced fusion at varying concentrations to determine the minimum concentration at which minimum PEG concentration reliable induced fusion was possible. This was done because PEG is known to be cytotoxic [37]. We tested concentrations from 30% down to 7.5%.
Reliable fusions were observed for all PEG concentrations of 10% and higher. The fused cells exhibited a perfectly spherical shape; see Figure 4a). At PEG concentrations of 8.75% and 7.5% only partial fusions were observed. For instance, two cells could be distinguished by eye as the partially fused cells did not reach a state of spherical shape, see Figure Figure 4b).
To gain further insight into the fusion process and its relevant parameter we determined the sizes of the source cells and fused cells and calculated the size differences, see Suppl. Table S1. Those measurements revealed that the fused cells were smaller than expected, based on the volume of the source cells. This was in agreement with observations during the fusion; cells that came in contact with the PEG solution immediately shrunk. Interestingly, we found that fusion always started about 35 s after the PEG fusion solution reached the source cells. That is, the two source cells started to move into each other, see also Suppl. video.
The osmolarity of the PEG solution was regulated by additional mannitol. However, we also tested PEG solution with 154 mM NaCl to regulate the osmolarity as that was used for the resuspension solutions as well. Fusion experiments with that solutions and PEG concentration of 20% only led to partially fused cells, similar to those in Figure Figure 4b). Furthermore, the cells did not change their volume when the PEG solution reached them, but remained constant.
Having close looks at the fully fused cells, we found structures that looked like the cells remained separated by a membrane like structure, see Suppl. Figure S2. First, we assumed that the new intracellular structure was a relic of previous cell membranes, which are highly permeable or fragmented, and thus provided the perfect spherical shape. But this was proven wrong after flushing the fused cells with resuspension buffer. As soon as the PEG fusion solution started to being replaced the fused cells started to separate again into the source cells (Figure 5). In all cases, we first observed a swelling of the fused cell following by an excrescence of one source cell. This process was slower than the fusion, but happened in all cases. Fusion and separation could be repeated as long as both source cells kept intact see Suppl. video. This was absolutely unexpected and not documented in literature so far. Analysis of the protoplast radii before fusion and after separation revealed that the size was the same, within expected measurement error margin.
Though, we focused the fusion on analysis of two fused cells even three cells fused. In any case both, 2-cell-fused and 3-cell-fused protoplasts, a rotation due to the fluid flow was observed, though by far slower for the 3-cell-fused protoplasts. Determination of the diameter of the fused cells revealed that it was almost as large as the channel height for the 3-cell-fused ones. Therefore, rotation was hindered.
4. Discussion
The microfluidic devices used in this work were fabricated monolithically with PDMS soft lithography. Fabrication of the negative relief structure, of the trapping features, on the masterwafer was challenging due to the small diameter of the recesses, which had a diameter of 20 µm and a depth of 56 µm. Adaptation of the development process with gentle shaking of the beaker with the wafer led to good penetration of the developer into the recesses and thus, removed the photoresist. Though this high aspect ratio of 2.8 was achieved due to optimized fabrication, this states a current limitation towards fabricating higher microfluidic devices. Therefore, other photoresists might be tested for the masterwafer in future studies.
The vitality of the cells was maintained in the microfluidic devices for at least three hours as observed by vital staining, analyzing the shape pointed to even ten hours without much adjustment of the conditions and media supply. This data demonstrates that the setup is suitable for long-term-observations subsequent fusion and allows for investigations of the cellular re-organization. The addition of up to 25 % of PEG did not significantly reduce the vitality of cells in the microfluidic device. This is in very good accordance to previous literature where microfluidics were used to study pollen or root growth [21,22,23,24].
Successful protoplast fusion was conducted in the microfluidic devices after trapping two protoplasts. This was the first time demonstration of a controlled fusion of two protoplasts. Previously, Wu et al conducted PEG induced fusion experiments of randomly distributed tobacco protoplast fusion in a microfluidic device [27,28]. Their device consists of a micro-column array that span the whole channel width preventing the protoplasts to pass by. We observed that fusion of the protoplasts depended on: 1) the PEG concentration of the fusion buffer, 2) the flow velocity, 3) the osmolarity.
1) PEG concentration had impact on both, if the fusion process was complete or partial and the duration of the fusion process. PEG in vicinity of membrane removes the hydration layer that impedes the close apposition of converging phospholipid bilayers [38]. We assume that this process was independent of the PEG concentration since the first cell approaching and merging was observed after about 35 s for all PEG concentrations tested. PEG is assumed to induce fusion by small damages in the cell membrane [39]. Those regions, when in contact with a second cell, might serve as starting point for fusion. PEG damage of membranes is assumed to be concentration dependent as diffusion and integration of PEG into the cell membrane is driven by the PEG concentration gradient, theoretically described by Fick´s law [40]. This is in very good agreement with our experimental findings that the total duration of fusion was longer for lower PEG concentrations. Additionally, a minimum concentration of 10% PEG was found in the microfluidic experiments. Only partial fusion was observed if the concentration was below 10% PEG. Hence, we could demonstrate successful fusion with reduced PEG concentration compared to previous experiments and batch experiments off-chip.
2) We assume the fusion velocity depend on the flow velocity due to both, a better contact of the two source protoplasts being pressed against each other and a shearing of the two membranes in close contact improving the membrane opening for fusion.
3) Analysis of the radii of source cells and fused cell revealed that the fused cells` volume was smaller than expected. This was in accordance with the osmolarity of the PEG fusion solution; in which we set the osmolarity with mannitol, the resuspension buffer contained NaCl to set the osmolarity. The latter was of lower osmolarity (308 and 435 mOsm/kg for resuspension buffer and fusion solution) and thus, the turgor decreased when the cells were flushed with PEG fusion buffer. We assume that osmolarity impacts the fusion efficiency; since fusion experiments with NaCl in the PEG fusion solution, i.e. being iso-osmotic, revealed both that the radii remained constant and no complete fusion took part. Higher osmolarity of the fusion buffer led to reduced turgor in the source cells, and thus enabled better nestling of the cells against each other providing improved cell contact. Additionally, reduced turgor and cell radii led to increased membrane curvature. Malinin et al. reported that vesicle radius strongly impacts the mixing of the vesicles` lipids [41]. Though protoplasts are not vesicles, their membranes can be compared with each other and so can their fusions or lipid mixing behaviors. For this reason we propose to use slightly hyperosmotic fusion solutions to improve fusion efficiency. Additionally, the separation of fused cells after removal of the hyperosmotic buffer also indicates that the fusion strongly depends on the osmotic strength, and thus the turgor. The separation of the fused cells took part in all experiments after removal of the mannitol PEG buffer. The separation of the fused cells also indicate that the fusion process was not completed within minutes but likely need more time to fully merge the two membranes into one. We assume that separation will take place as long as two vacuoles remain in the cell, i.e. don’t fuse into one. This needs to be proven in future studies.
A significant difference in fusion efficiency was found in our study comparing the fusion efficiency in microtiter wells, i.e. batch, and microfluidics. The batch experiments reached a maximum fusion efficiency of about 20% whereas the experiments in our microfluidic device had a fusion efficiency of more than 95%.
In rare events, we observed fusion of three source cells, though in most cases we flushed away additional cells excessing two trapped cells. Fusion of two protoplasts was faster than fusion of three protoplasts. We assume that this was due to the fact that the PEG more easily reaches larger parts of the cell membrane for two protoplasts trapped. Additionally, the channel height of 56 µm limited the cell rotation of fused cells if those were too large in diameter also perturbing successful fusion.
5. Conclusions and Outlook
In this work we designed and fabricated a monolithic microfluidic device consisting of trapping features to reliably trap two A. thaliana protoplasts at a time. The trapping features were designed such that the protoplasts were in close contact but could, at the same time, be flushed with media. This enabled us to demonstrate (for the first time) a reliable PEG induced fusion of two A. thaliana protoplasts in a microfluidic device. The experiments revealed that the fusion and its duration strongly depend on the PEG concentration, the osmolarity of the fusion solution and the applied flow velocity, i.e. shear forces. Additionally, our microfluidic device provided new insight in the reversibility of the fusion process. We observed separation of a fused cell in source cells after washing away the PEG fusion solution; this has not been reported in literature before.
In future, we will conduct further experiments to gain deeper understanding of the PEG-induced fusion. For instance, different fluorescent labelling of the vacuole and cell nucleus can be used to study if those also fuse into one vacuole and one nucleus. Additionally, future studies need to focus on the minimum concentration of PEG and exposure times that provides complete fusion and formation of two permanently fused cells.
Supplementary Materials
The following are available online at Preprints.org., SVideo1: Induced fusion (with 15% PEG) and separation. First, two source cells are trapped and fused. After removal of PEG the cells separated. After trapping a third cell again fusion was induced by PEG flushing and all three cells fused into one cell. Removal of PEG led to separation into three source cells. ESI document.
Author Contributions
PJA, IG and MV conducted the microfluidic fusion experiments; FI and ALB performed the cell viability experiments; ALB conducted batch fusion experiments; TSe isolated protoplasts; MV interpreted the data and wrote the manuscript; MV and TSe edited the manuscript; MV and TSe supervised the project. All authors read and approved the final manuscript.
Funding
This work has been funded by Bielefeld University.
Data Availability Statement
All data generated or analyzed during this study are included in this published article [and its supplementary information files]. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
Dario Anselmetti is gratefully acknowledged for supporting the studies. Lara Vogelsang is gratefully acknowledged for isolating and providing protoplasts. The authors acknowledge Tim Stricker and Carolin Buß for additional experimental work. We acknowledge support for the publication costs by the Open Access Publication Fund of Bielefeld University.
Conflicts of Interest
The authors declare that they have no competing interests.
References
- Yoo, S.-D.; Cho, Y.-H.; Sheen, J. Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat Protoc 2007, 2, 1565–1572. [Google Scholar] [CrossRef]
- Jen Sheen. Signal Transduction in Maize and Arabidopsis Mesophyll Protoplasts. PLANT PHYSIOLOGY 2001, 127, 1466–1475. [Google Scholar] [CrossRef]
- Kluge, C.; Seidel, T.; Bolte, S.; Sharma, S.S.; Hanitzsch, M.; Satiat-Jeunemaitre, B.; Ross, J.; Sauer, M.; Golldack, D.; Dietz, K.-J. Subcellular distribution of the V-ATPase complex in plant cells, and in vivo localisation of the 100 kDa subunit VHA-a within the complex. BMC Cell Biol. 2004, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Neubert, C.; Graham, L.A.; Black-Maier, E.W.; Coonrod, E.M.; Liu, T.-Y.; Stierhof, Y.-D.; Seidel, T.; Stevens, T.H.; Schumacher, K. Arabidopsis has two functional orthologs of the yeast V-ATPase assembly factor Vma21p. Traffic 2008, 9, 1618–1628. [Google Scholar] [CrossRef]
- Liszka, A.; Schimpf, R.; Cartuche Zaruma, K.I.; Buhr, A.; Seidel, T.; Walter, S.; Knuesting, J.; Dreyer, A.; Dietz, K.-J.; Scheibe, R.; et al. Three cytosolic NAD-malate dehydrogenase isoforms of Arabidopsis thaliana: on the crossroad between energy fluxes and redox signaling. Biochem J 2020, 477, 3673–3693. [Google Scholar] [CrossRef]
- Muthuramalingam, M.; Seidel, T.; Laxa, M.; Nunes de Miranda, S.M.; Gärtner, F.; Ströher, E.; Kandlbinder, A.; Dietz, K.-J. Multiple redox and non-redox interactions define 2-Cys peroxiredoxin as a regulatory hub in the chloroplast. Mol. Plant 2009, 2, 1273–1288. [Google Scholar] [CrossRef]
- Seidel, T.; Golldack, D.; Dietz, K.-J. Mapping of C-termini of V-ATPase subunits by in vivo-FRET measurements. FEBS LETTERS 2005, 579, 4374–4382. [Google Scholar] [CrossRef]
- Katia Schütze; Klaus Harter; Christina Chaban. Bimolecular Fluorescence Complementation (BiFC) to Study Protein-protein Interactions in Living Plant Cells. Plant Signal Transduction; Humana Press, Totowa, NJ, 2009; pp 189–202.
- Wolf, A.; Akrap, N.; Marg, B.; Galliardt, H.; Heiligentag, M.; Humpert, F.; Sauer, M.; Kaltschmidt, B.; Kaltschmidt, C.; Seidel, T. Elements of transcriptional machinery are compatible among plants and mammals. PLoS ONE 2013, 8, e53737. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhuang, X.; Wang, J.; Wang, H.; Lam, S.K.; Gao, C.; Wang, X.; Jiang, L. Vacuolar degradation of two integral plasma membrane proteins, AtLRR84A and OsSCAMP1, is cargo ubiquitination-independent and prevacuolar compartment-mediated in plant cells. Traffic 2012, 13, 1023–1040. [Google Scholar] [CrossRef]
- KAO, K.N.; CONSTABEL, F.; Michayluk, M.R.; GAMBORG, O.L. Plant protoplast fusion and growth of intergeneric hybrid cells. PLANTA 1974, 120, 215–227. [Google Scholar] [CrossRef]
- Maruyama, D.; Völz, R.; Takeuchi, H.; Mori, T.; Igawa, T.; Kurihara, D.; Kawashima, T.; Ueda, M.; ITO, M.; Umeda, M.; et al. Rapid Elimination of the Persistent Synergid through a Cell Fusion Mechanism. Cell 2015, 161. [Google Scholar] [CrossRef]
- Dresselhaus, T.; Sprunck, S.; Wessel, G.M. Fertilization Mechanisms in Flowering Plants. Curr. Biol. 2016, 26, R125–39. [Google Scholar] [CrossRef] [PubMed]
- Bruznican, S.; Eeckhaut, T.; van Huylenbroeck, J.; Keyser, E. de; Clercq, H. de; Geelen, D. An asymmetric protoplast fusion and screening method for generating celeriac cybrids. Sci. Rep. 2021, 11, 4553. [Google Scholar] [CrossRef] [PubMed]
- Ortseifen, V.; Viefhues, M.; Wobbe, L.; Grünberger, A. Microfluidics for Biotechnology: Bridging Gaps to Foster Microfluidic Applications. Front. Bioeng. Biotechnol. 2020, 8, 589074. [Google Scholar] [CrossRef]
- Greif, D.; Pobigaylo, N.; Frage, B.; Becker, A.; Regtmeier, J.; Anselmetti, D. Space- and time-resolved protein dynamics in single bacterial cells observed on a chip. Journal of biotechnology 2010, 149, 280–288. [Google Scholar] [CrossRef]
- Yu, F.B.; Willis, L.; Chau, R.M.W.; Zambon, A.; Horowitz, M.; Bhaya, D.; Huang, K.C.; Quake, S.R. Long-term microfluidic tracking of coccoid cyanobacterial cells reveals robust control of division timing. BMC Biol. 2017, 15, 11. [Google Scholar] [CrossRef]
- Li, J.; Wei, J.; Liu, Y.; Liu, B.; Liu, T.; Jiang, Y.; Ding, L.; Liu, C. A microfluidic design to provide a stable and uniform in vitro microenvironment for cell culture inspired by the redundancy characteristic of leaf areoles. Lab Chip 2017, 17, 3921–3933. [Google Scholar] [CrossRef] [PubMed]
- Bérut, A.; Chauvet, H.; Legué, V.; Moulia, B.; Pouliquen, O.; Forterre, Y. Gravisensors in plant cells behave like an active granular liquid. PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES OF THE UNITED STATES OF AMERICA 2018, 115, 5123–5128. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, Y.; Tanaka, H.; Kobayashi, Y.; Shikanai, T.; Nishimura, Y. Chloroplast nucleoids as a transformable network revealed by live imaging with a microfluidic device. Commun. Biol. 2018, 1, 47. [Google Scholar] [CrossRef]
- Shamsudhin, N.; Atakan, H.B.; Laubli, N.; Vogler, H.; Chengzhi, H.; Sebastian, A.; Grossniklaus, U.; Nelson, B.J. Probing the micromechanics of the fastest growing plant cell - the pollen tube. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. 2016, 2016, 461–464. [Google Scholar] [CrossRef]
- Yanagisawa, N.; Sugimoto, N.; Arata, H.; Higashiyama, T.; Sato, Y. Capability of tip-growing plant cells to penetrate into extremely narrow gaps. Sci. Rep. 2017, 7, 1403. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, N.; Sugimoto, N.; Higashiyama, T.; Sato, Y. Development of Microfluidic Devices to Study the Elongation Capability of Tip-growing Plant Cells in Extremely Small Spaces. JOVE-JOURNAL OF VISUALIZED EXPERIMENTS. [CrossRef]
- Grossmann, G.; Krebs, M.; Maizel, A.; Stahl, Y.; Vermeer, J.E.M.; Ott, T. Green light for quantitative live-cell imaging in plants. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Charlot, F.; Le Saux, T.; Bonhomme, S.; Nogué, F.; Palauqui, J.-C.; Fattaccioli, J. Design of a comprehensive microfluidic and microscopic toolbox for the ultra-wide spatio-temporal study of plant protoplasts development and physiology. Plant Methods 2019, 15, 79. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.-M.; Ju, J.; Lee, S.; Cha, H.-C. Tobacco protoplast culture in a polydimethylsiloxane-based microfluidic channel. PROTOPLASMA 2006, 227, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Isshiki, M.; Tsumoto, A.; Shimamoto, K. The serine/arginine-rich protein family in rice plays important roles in constitutive and alternative splicing of pre-mRNA. Plant Cell 2006, 18, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Heng Wu; Wenming Liu; Qin Tu; Na Song; Li Li; Jianchun Wang; Jinyi Wang. Culture and chemical-induced fusion of tobacco mesophyll protoplasts in a microfluidic device. Microfluidics and Nanofluidics 2010, 10, 867–876. [CrossRef]
- Murashige, T.; Skoog, F. A Revised Medium for Rapid Growth and Bio Assays with Tobacco Tissue Cultures. Physiol Plant 1962, 15, 473–497. [Google Scholar] [CrossRef]
- GAMBORG, O.L.; MILLER, R.A.; OJIMA, K. Nutrient requirements of suspension cultures of soybean root cells. Experimental Cell Research 1968, 50, 151–158. [Google Scholar] [CrossRef]
- Viefhues, M.; Manchanda, S.; Chao, T.-C.; Anselmetti, D.; Regtmeier, J.; Ros, A. Physisorbed surface coatings for poly(dimethylsiloxane) and quartz microfluidic devices. Anal Bioanal Chem 2011, 401, 2113–2122. [Google Scholar] [CrossRef]
- Sia, S.K.; Whitesides, G.M. Microfluidic devices fabricated in poly(dimethylsiloxane) for biological studies. Electrophoresis 2003, 24, 3563–3576. [Google Scholar] [CrossRef]
- Viefhues, M.; Regtmeier, J.; Anselmetti, D. Fast and continuous-flow detection and separation of DNA complexes and DNA in nanofluidic chip format. Methods Mol. Biol. 2015, 1274, 99–110. [Google Scholar] [CrossRef]
- Seidel, T.; Kluge, C.; Hanitzsch, M.; Ross, J.; Sauer, M.; Dietz, K.J.; Golldack, D. Colocalization and FRET-analysis of subunits c and a of the vacuolar H+-ATPase in living plant cells. Journal of biotechnology 2004, 112, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Masani, M.Y.A.; Noll, G.A.; Parveez, G.K.A.; Sambanthamurthi, R.; Prüfer, D. Efficient transformation of oil palm protoplasts by PEG-mediated transfection and DNA microinjection. PLoS ONE 2014, 9, e96831. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Huang, X.; Gong, Q.; Dai, X.-M.; Zhao, J.-T.; Wei, Y.-R.; Huang, X.-L. Somatic hybrids obtained by asymmetric protoplast fusion between Musa Silk cv. Guoshanxiang (AAB) and Musa acuminata cv. Mas (AA). PLANT CELL TISSUE AND ORGAN CULTURE 2009, 97, 313–321. [Google Scholar] [CrossRef]
- Guoqiang Liu; Yongsan Li; Lei Yang; Yen Wei; Xing Wang; Zhiming Wang; Lei Tao. Cytotoxicity study of polyethylene glycol derivatives, 7, 18252–18259. [CrossRef]
- Richard H., Guy; Francis, C. Szoka Jr. Perturbation of solute transport at a liquidPEG): implications for PEG-induced biomembrane fusion, 13, 5346. [CrossRef]
- L.F. De Filippis; R. Hampp; H. Ziegler. Membrane Permeability Changes and Ultrastructural Abnormalities Observed During Protoplast Fusion, 156, 628–634. [CrossRef]
- Philibert, J. One and a half century of diffusion: Fick, Einstein before and beyond.
- Vladimir, S. Malinin; Peter Frederik; Barry R. Lentz. Osmotic and Curvature Stress Affect PEG-Induced Fusion of Lipid Vesicles but Not Mixing of Their Lipids, 82, 2090–2100. [CrossRef]
Figure 3.
Bright field microscopy image of trapped protoplasts. The cells were flushed through the device from left to right.
Figure 3.
Bright field microscopy image of trapped protoplasts. The cells were flushed through the device from left to right.

Figure 4.
CLSM images of cells after induction of fusion. a) Cells after fusion with 13.75% PEG. Green fluorescent beads were used as dye to stain the PEG fusion solution. The shape is perfectly spherical. b) Fusion induction with PEG concentration less than 10% led to partial fusion, i.e. the cells stick together but did not exhibit a spherical shape.
Figure 4.
CLSM images of cells after induction of fusion. a) Cells after fusion with 13.75% PEG. Green fluorescent beads were used as dye to stain the PEG fusion solution. The shape is perfectly spherical. b) Fusion induction with PEG concentration less than 10% led to partial fusion, i.e. the cells stick together but did not exhibit a spherical shape.
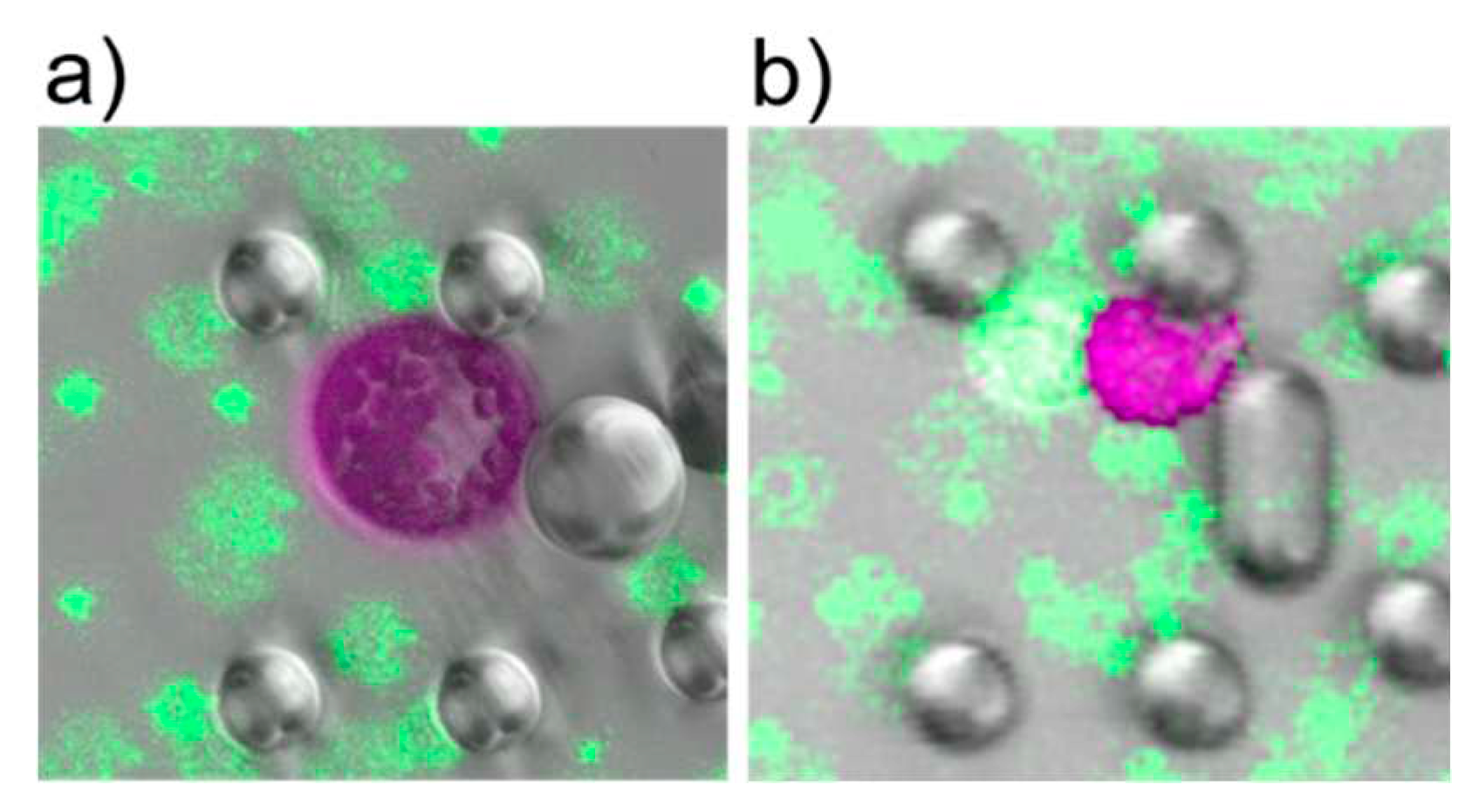
Figure 5.
Time-lapse study of PEG induced protoplast fusion (a1 – b5) and separation (c1 – e5). 15 s time intervals were between consecutive images. Impact of PEG on the source cells was observed about 35 s after first contact. After successful fusion image c5) the PEG fusion solution was washed away (c1-e5). The two source cells fully separated after removal of the PEG fusion solution.
Figure 5.
Time-lapse study of PEG induced protoplast fusion (a1 – b5) and separation (c1 – e5). 15 s time intervals were between consecutive images. Impact of PEG on the source cells was observed about 35 s after first contact. After successful fusion image c5) the PEG fusion solution was washed away (c1-e5). The two source cells fully separated after removal of the PEG fusion solution.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



