
Submitted:
14 December 2023
Posted:
15 December 2023
You are already at the latest version
Abstract
This study employs molecular docking experiments to investigate the binding interactions of Hypericin with key proteins implicated in Alzheimer's disease (AD). Hypericin demonstrated a significant binding energy of -10.1 kcal/mol with ABAD (3-hydroxyacyl-CoA dehydrogenase type II). The analysis revealed multiple binding interactions, including hydrogen bonds with arginine 6, serine 7, and glutamic acid 140, as well as hydrophobic bonds involving leucine 11, aspartic acid 142, and aspartic acid 86. These findings illuminate the intricate molecular interactions between Hypericin and ABAD, suggesting its potential as a lead compound for further therapeutic development in the context of Alzheimer's disease. Additionally, a parallel docking investigation was conducted for Beta-secretase 1 (BACE1), a protein pivotal in the generation of beta-amyloid peptides—a hallmark of Alzheimer's disease. Inhibiting BACE1 activity is considered a promising therapeutic strategy for AD. Computational analyses suggest that Hypericin may serve as a potential inhibitor of BACE1. Molecular Docking results reveal a robust binding affinity between Hypericin and BACE1, supported by a significant binding energy score of -10.3 kcal/mol. Notably, both ABAD and BACE1 exhibit high binding affinity with Hypericin. This theoretical exploration provides an initial understanding of the intricate molecular pathways involved in Alzheimer's disease and proposes Hypericin as a potential therapeutic compound for mitigating its progression.
Keywords:
Alzheimer's disease
; Molecular Docking
; Hypericin
; BACE1
; ABAD (3-hydroxyacyl-CoA dehydrogenase type II
1. Introduction
Mitochondrial dysfunction is a pivotal feature in the pathology of Alzheimer’s disease (AD), primarily associated with neuronal toxicity induced by beta-amyloid (Aβ). Several studies sheds light on the molecular mechanisms linking Aβ and mitochondrial impairment [1]. Lustbader et al. (2004) demonstrate a direct binding between Aβ and alcohol dehydrogenase (ABAD) in mitochondria, establishing a molecular connection to mitochondrial toxicity [1]. Leuner et al. (2012) provide novel insights connecting mitochondrial dysfunction to Aβ formation [2]. Mao and Reddy (2011) highlight the contribution of aging and Aβ-induced oxidative DNA damage to mitochondrial dysfunction, proposing potential implications for early interventions [3]. Eckert et al. (2011) explore mitochondrial dysfunction as a crucial aspect in AD pathogenesis, examining separate and synergistic modes involving tau and Aβ [4]. Chen and Yan (2007) focus on Aβ-induced mitochondrial dysfunction as a key element in the etiology of Alzheimer’s. These studies collectively deepen our understanding of the interplay between mitochondrial function and Aβ in Alzheimer’s disease, offering potential avenues for further research and therapeutic development [5]. Mitochondrial dysfunction is implicated in various diseases and conditions, including neurodegenerative disorders, metabolic diseases, cardiovascular diseases, and aging [6,7]. In the context of neurodegenerative diseases like Alzheimer’s, as mentioned in a previous discussion, mitochondrial dysfunction is considered a hallmark and may play a role in the progression of the disease [1,2,3,4,5]. The consequences of mitochondrial dysfunction can include a decrease in ATP production, an increase in the generation of reactive oxygen species, and alterations in cellular metabolism [8,9,10]. These disruptions can lead to cell damage, impaired cellular function, and, ultimately, contribute to the development or progression of various diseases. The research conduced by Lustbader et al. study in 2004 [1] focused on the intricate relationship between beta-amyloid (Aβ) and mitochondrial dysfunction. The key finding is that Aβ directly contributes to mitochondrial toxicity through ABAD (3-hydroxyacyl-CoA dehydrogenase type II or Type II HADH). Recognizing this molecular link is crucial, given the acknowledged role of mitochondrial dysfunction in AD progression. The study suggests disrupting the ABAD-Aβ interaction as a potential therapeutic target to alleviate the adverse effects of Aβ on mitochondria [1].
This short communication centers on the molecular docking exploration of ABAD (3-hydroxyacyl-CoA dehydrogenase type II) with various natural compounds. The objective is to identify lead compounds with the potential to bind effectively to this target. Molecular docking [11] serves as a computational tool to predict and analyze the binding interactions between ABAD and selected natural compounds. The study aims to contribute to the discovery of promising candidates for further investigation and development as potential therapeutics targeting ABAD in the context of relevant biological processes.
2. Material and Methods
3-hydroxyacyl-CoA dehydrogenase type II (chain A) by Autodock Vina [12] by Pyrx program [13]. Several proteins were investigated in the Ligand Binding Site of these proteins:
-3-hydroxyacyl-CoA dehydrogenase type II was taken by Protein Data Bank and was accurately prepared before to perform docking investigation (PDB Code: 1SO8). Grid box Coordinates of binding Center X ( 84.0815), Y( 14.9621), Z(41.2509) ; size_X = 51.0352949142; size_Y = 45.7857791138; size_Z= 47.2655132866.
3. Results and Discussion
This research amalgamates findings from two pivotal studies focused on elucidating the molecular investigaitojn underlying Alzheimer’s disease (AD) pathogenesis. Lustbader et al. (2004) underscores the intricate relationship between beta-amyloid (Aβ) and mitochondrial dysfunction, highlighting Aβ’s direct contribution to mitochondrial toxicity through 3-hydroxyacyl-CoA dehydrogenase type II (ABAD). The recognition of this molecular link is pivotal, given the acknowledged role of mitochondrial dysfunction in AD progression. Lustbader et al. proposes disrupting the ABAD-Aβ interaction as a potential therapeutic avenue to mitigate Aβ-induced adverse effects on mitochondria.
In this work, molecular docking tests [11,12,13] were conducted with Hypericin, revealing a notable binding energy of -10.1 kcal/mol with ABAD (3-hydroxyacyl-CoA dehydrogenase type II). These results signify a substantial potential for Hypericin to bind effectively with ABAD. Docking analysis delineates multiple binding interactions, including hydrogen bonds with arginine 6, serine 7, and glutamic acid 140, as well as hydrophobic bonds with leucine 11, aspartic acid 142, and aspartic acid 86. These insights shed light on the molecular interactions between Hypericin and ABAD, accentuating its potential as a lead compound for further therapeutic development in the context of Alzheimer’s disease.
Furthermore, this docking investigation highlights the critical role of Beta-secretase 1 (BACE1) in the generation of beta-amyloid peptides, presenting the inhibition of its activity as a promising therapeutic approach for Alzheimer’s disease, characterized by the excessive accumulation of beta-amyloid. Computational analyses suggest that hypericin could serve as a potential inhibitor of BACE1. The results of Molecular Docking reveal a robust binding affinity between hypericin and BACE1, supported by a significant binding energy score of -10.3 kcal/mol. The formation of diverse chemical bonds, including hydrogen and hydrophobic bonds, indicates that hypericin may effectively engage with the active site of BACE1.
While the in silico findings are encouraging, further in vitro and in vivo studies are imperative to validate and deepen our understanding of hypericin’s inhibitory potential against BACE1. Additionally, the practical application of hypericin as a BACE1 inhibitor in therapeutics requires meticulous consideration of factors such as bioavailability, safety, and efficacy. This theoretical study provides an initial insight into the complex molecular pathways involved in Alzheimer’s disease and proposes a potential therapeutic strategy for mitigating its progression.
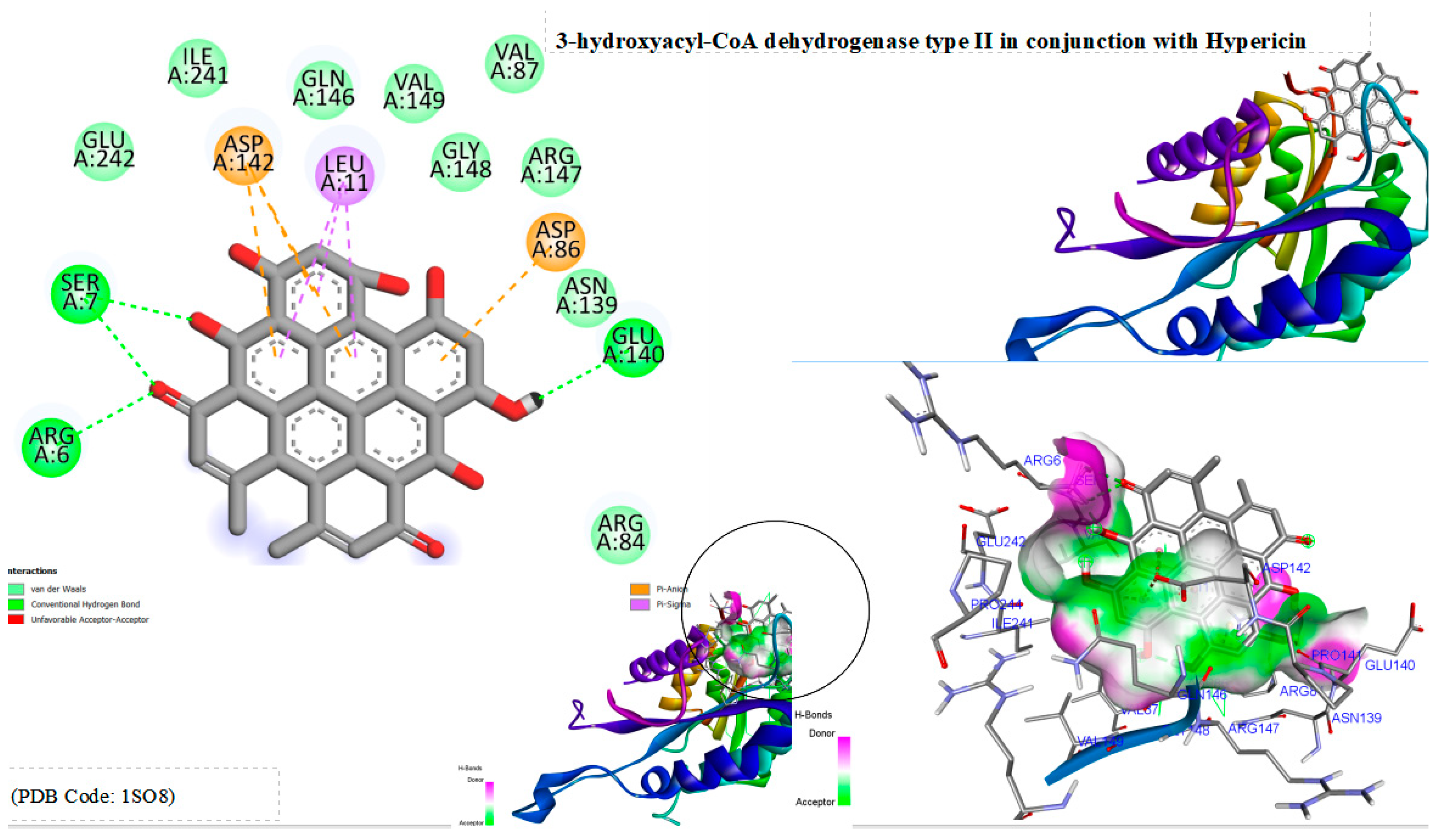
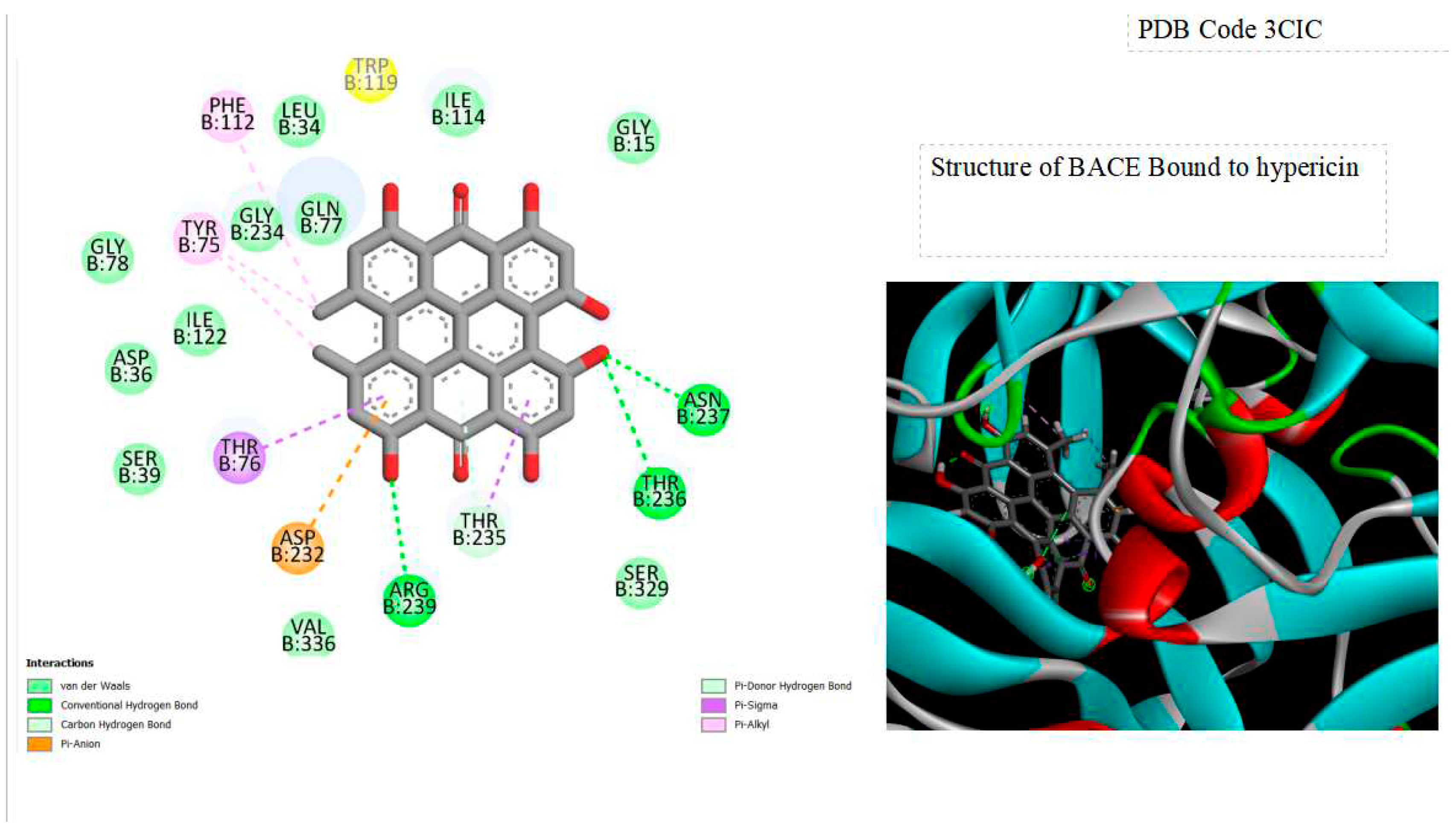
Figure 1.
Displays the docking outcomes of 3-hydroxyacyl-CoA dehydrogenase type II in conjunction with Hypericin within the potential Ligand Binding Site, as analyzed by Autodock Vina through the Pyrx program. On the left side, 2D diagrams illustrate the residue interactions between the protein and Hypericin. Meanwhile, the right side exhibits the potential Ligand Binding Site of the protein, highlighting the specific location of Hypericin.
Figure 1.
Displays the docking outcomes of 3-hydroxyacyl-CoA dehydrogenase type II in conjunction with Hypericin within the potential Ligand Binding Site, as analyzed by Autodock Vina through the Pyrx program. On the left side, 2D diagrams illustrate the residue interactions between the protein and Hypericin. Meanwhile, the right side exhibits the potential Ligand Binding Site of the protein, highlighting the specific location of Hypericin.
Figure 2.
Displays the docking outcomes of BACE-1 in conjunction with Hypericin within the potential Ligand Binding Site, as analyzed by Autodock Vina through the Pyrx program. On the left side, 2D diagrams illustrate the residue interactions between the protein and Hypericin. Meanwhile, the right side exhibits the potential Ligand Binding Site of the protein, highlighting the specific location of Hypericin.
Figure 2.
Displays the docking outcomes of BACE-1 in conjunction with Hypericin within the potential Ligand Binding Site, as analyzed by Autodock Vina through the Pyrx program. On the left side, 2D diagrams illustrate the residue interactions between the protein and Hypericin. Meanwhile, the right side exhibits the potential Ligand Binding Site of the protein, highlighting the specific location of Hypericin.
4. Conclusion
This investigation utilizes molecular docking experiments to explore the binding interactions between Hypericin and crucial proteins associated with Alzheimer’s disease (AD). Hypericin demonstrates a substantial binding energy of -10.1 kcal/mol with ABAD (3-hydroxyacyl-CoA dehydrogenase type II), revealing multiple binding interactions such as hydrogen bonds with arginine 6, serine 7, and glutamic acid 140, as well as hydrophobic bonds with leucine 11, aspartic acid 142, and aspartic acid 86. These findings highlight the complex molecular interactions between Hypericin and ABAD, indicating its potential as a lead compound for further therapeutic development in Alzheimer’s disease.Moreover, a parallel docking investigation examines Beta-secretase 1 (BACE1), a crucial player in the generation of beta-amyloid peptides—a hallmark of Alzheimer’s disease. Inhibiting BACE1 is a promising therapeutic strategy for AD. Computational analyses suggest that Hypericin may function as a potential BACE1 inhibitor. Molecular Docking results demonstrate a robust binding affinity between Hypericin and BACE1, supported by a significant binding energy score of -10.3 kcal/mol. Importantly, both ABAD and BACE1 exhibit high binding affinity with Hypericin. This theoretical exploration provides initial insights into the intricate molecular pathways of Alzheimer’s disease and proposes Hypericin as a potential therapeutic compound for mitigating its progression.
References
- Lustbader, J. W., Cirilli, M., Lin, C., Xu, H. W., Takuma, K., Wang, N., … & Wu, H. (2004). ABAD directly links Aß to mitochondrial toxicity in Alzheimer's disease. Science, 304(5669), 448-452.
- Leuner, K., Müller, W. E., & Reicher S. (2012). From mitochondrial dysfunction to amyloid beta formation: novel insights into the pathogenesis of Alzheimer’s disease. Molecular neurobiology, 46, 186-193.
- Mao, P., & Reddy, P. H. (2011). Aging and amyloid beta-induced oxidative DNA damage and mitochondrial dysfunction in Alzheimer's disease: implications for early intervention and therapeutics. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease, 1812(11), 1359-1370. 1359.
- Eckert, A., Schmitt, K., & Götz, J. (2011). Mitochondrial dysfunction-the beginning of the end in Alzheimer's disease? Separate and synergistic modes of tau and amyloid-β toxicity. Alzheimer's research & therapy, 3, 1-11.
- Chen, J. X., & Yan, S. D. (2007). Amyloid-β-induced mitochondrial dysfunction. Journal of Alzheimer's Disease, 12(2), 177-184.
- Elfawy, H. A., & Das, B. (2019). Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: Etiologies and therapeutic strategies. Life sciences, 218, 165-184.
- Bhat, A. H., Dar, K. B., Anees, S., Zargar, M. A., Masood, A., Sofi, M. A., & Ganie, S. A. (2015). Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomedicine & Pharmacotherapy, 74, 101-110.
- Trifunovic, A., & Larsson, N. G. (2008). Mitochondrial dysfunction as a cause of ageing. Journal of internal medicine, 263(2), 167-178.
- Brand, M. D., & Nicholls, D. G. (2011). Assessing mitochondrial dysfunction in cells. Biochemical Journal, 435(2), 297-312.
- Muravchick, S., Levy, R. J., & Warltier, D. C. (2006). Clinical implications of mitochondrial dysfunction. The Journal of the American Society of Anesthesiologists, 105(4), 819-837.
- Pagadala, N. S., Syed, K., & Tuszynski, J. (2017). Software for molecular docking: a review. Biophysical reviews, 9, 91-10.
- Trott, O., & Olson, A. J. (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of computational chemistry, 31(2), 455-461.
- Dallakyan, S., & Olson, A. J. (2015). Small-molecule library screening by docking with PyRx. Chemical biology: methods and protocols, 243-250.
- Ferrari, I.V. Molecular Insights into Hypericin: Unraveling its Theoretical Potential in Alzheimer's Disease Intervention. Preprints 2023, 2023111768. [Google Scholar] [CrossRef]
- Ferrari, I. V. (2023). Hypericin As A Possible Natural Molecule For The Treatment Of Alzheimer's Disease: Computationa Studies Focused On Beta-Secretase-1 (or BACE1). Int. J. Sci. Res. in Biological Sciences Vol, 10(5).
- Odhar, H. A., Rayshan, A. M., Ahjel, S. W., Hashim, A. A., & Albeer, A. A. M. A. (2019). Molecular docking enabled updated screening of the matrix protein VP40 from Ebola virus with millions of compounds in the MCULE database for potential inhibitors. Bioinformation, 15(9), 627.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



