
Submitted:
16 December 2023
Posted:
18 December 2023
You are already at the latest version
Abstract
We report the case of a four-year-old male patient with a complex medical history born prematurely as the result of intrauterine growth restriction due to placental insufficiency. His clinical manifestations include severe neurodevelopmental deficits, global developmental delay, Pierre-Robin sequence, and intractable epilepsy with both generalized and focal features. The proband’s low levels of citrulline and lactic acidosis provoked by administration of depakoke are evocative of a mitochondrial etiology. However, the genotype-phenotype correlation remains undefined in the absence of nuclear and mitochondrial pathogenic variants detected by deep sequencing of both genomes. However, live-cell mitochondrial metabolic investigations provide evidence of a deficient oxidative phosphorylation pathway responsible for ATP synthesis, leading to chronic energy crisis in the proband. In addition, our metabolic analysis reveals metabolic plasticity in favor of glycolysis for ATP synthesis. Our mitochondrial morphometric analysis by transmission electron microscopy confirms the suspected mitochondrial etiology, as the proband’s mitochondria exhibit an immature morphology with poorly developed and rare cristae. Thus, our results support the concept that sub-optimal levels of intrauterine oxygen and nutrients alter fetal mitochondrial metabolic reprogramming toward OXPHOS leading to a deficient postnatal mitochondrial energy metabolism. In conclusion, our collective studies shed light on the long-term postnatal mitochondrial pathophysiology caused by intrauterine growth restriction due to idiopathic placental insufficiency and its negative impact on the energy-demanding development of fetal and postnatal brain.
Keywords:
placental insufficiency
; fetal growth restriction
; neurodevelopmental deficits
; OXPHOS deficit
; metabolic reprogramming
; mitochondrial dysfunction
1. Introduction
The American College of Obstetricians and Gynecologists defines intrauterine growth restriction (IUGR) as an estimated fetal weight of less than 10th percentile, which is a problem in about 10-15% of pregnant women (Gordin et al., 2016; McCowan et al., 2018). This complex obstetric problem is commonly diagnosed antenatally. Its causes are multifactorial with consequences in fetal, neonatal, and adult life (Von Beckerath et al., 2013; Angelica et al., 2012; Barker, 2006). Fetal growth is essentially regulated by the interplay between maternal physiology and placental functions. The timing of inhibited fetal growth and its etiology dictate the type of growth abnormalities (Suhag and Berghella, 2013). Symmetric IUGR constitutes 20-30% of the IUGR cases and results in an undersized fetus based on its timing during the first 24 gestational weeks when fetal growth occurs via hyperplasia to rapidly increase the number of cells without affecting the cellular size. It is mainly caused by chromosomic abnormalities including (but not limited to) trisomy 21, 13 and 18, or fetal infections with toxoplasmosis and cytomegalovirus being the most frequent causes (Hendrix and Berghella, 2008). In contrast, asymmetric IUGR, which represents 70-80% of the IUGR cases, has a late onset during gestation during which cellular hypertrophy occurs. Thus, asymmetric IUGR leads to decreased fetal weight without affecting the total number of cells, but rather by reducing the overall cell size (Villar et al., 1982). The reduced fetal growth rate results from a curtailed transfer of oxygen and nutrients between maternal and fetal blood due to an unfavorable intrauterine environment caused by placental insufficiency, maternal malnutrition, maternal smoking, maternal hypertension, uterine malformations or chronic pulmonary disease (Hendrix and Berghella, 2008; Cosmi et al., 2011).
IUGR impairs postnatal neurodevelopment affecting motor, cognitive, and behavioral functions (Geva et al., 2006; Samuelsen et al., 2007; De Bie et al., 2010; Chernausek,, 2012). IUGR infants display reduced brain volumes accompanied by delayed and disrupted myelination (Dubois et al., 2008; Padilla et al., 2011; Ramenghi et al., 2011). The placenta is a nutrient sensor and supplier that plays a pivotal role in providing metabolites required for mitochondrial metabolism, to generate high levels of energy, which are essential for normal fetal growth and neural development (Cetin and Alvino, 2009). Two recent studies have provided the first evidence of mitochondrial abnormalities in IUGR pregnancies, more specifically in terms of placental mitochondrial DNA copy numbers and mitochondrial variants (Naha et al., 2020; Pandey et al., 2021). Thus, placental insufficiency due to IUGR has the potential to provoke dysfunctional mitochondrial metabolism in the fetus and subsequently the newborn.
In this study, we report the case of a 4-year-old male with a history of IUGR without a unifying genetic diagnosis for his complex medical history characterized by severe neurodevelopmental deficits, global developmental delay, Pierre-Robin sequence, intractable epilepsy with both generalized and focal features. His low levels of citrulline and lactic acidosis provoked by administration of Depakote are suggestive of a mitochondrial etiology. The proband’s genotype-phenotype correlation remains undefined since no nuclear and mitochondrial pathogenic variants were detected by deep sequencing of the proband’s nuclear and mitochondrial genomes. Our live-cell functional metabolic investigations provide evidence of dysregulation of the mitochondrial oxidative phosphorylation (OXPHOS) pathway responsible for ATP synthesis, congruent with the proband’s suspected mitochondrial etiology and phenotypic manifestations.
2. Methods
2.1. Editorial Policies and ethical considerations
This study was approved by the Institutional Review Board of the George Washington University and Children’s National Medical Center and was conducted in accordance with the ethical principles of the Declaration of Helsinki of 1975 (revised 1983). Patient skin biopsy was performed after receiving written informed consent with permission to study the derived dermal fibroblasts.
2.2. Skin biopsy and fibroblast culture
A 3mm skin biopsy was performed on a 4-year-old male proband from which dermal fibroblasts were derived in Dulbecco’s Modified Eagle Medium (DMEM; Gibco) supplemented with 2 mM glutamine, 2.5 mM pyruvate, 0.2 mM uridine, FGF-2 (10 ng/ml) and 20% fetal bovine serum, as described (Uittenbogaard et al., 2019). Derived dermal fibroblasts were frozen at passage 2 and never used beyond passage 10. Human primary dermal fibroblasts from a healthy 10-year old male (Cat# GM03377E) were obtained from the Coriell Cell Repositories (Camden, NJ).
2.3. Clinical genetic diagnosis
Whole exome sequencing (WES) and Long-Range PCR followed by massively parallel sequencing (LR-PCR-MPS) of the mitochondrial genome were performed by GeneDx using dermal fibroblasts derived from the proband’s skin biopsy.
2.4. Transmission electron microscopy
Dermal fibroblasts of the proband and a healthy subject (control) were fixed in 2.5% glutaraldehyde (Electron Microscopy Sciences), 1% paraformaldehyde in 0.12 M sodium cacodylate buffer (Electron Microscopy Sciences) for 20 min at room temperature followed by 40 min on ice, as described.18 Samples were imaged with a FEI Talos F200X-transmission electron microscope (Thermo Fisher).
2.5. Live-cell measurements of mitochondrial respiratory and glycolytic activity
The bioenergetic status of dermal fibroblasts derived from the patient and the healthy control subject was measured using the Seahorse Extracellular Flux XFp Analyzer (Agilent Technologies; Santa Clara, CA), as described (Uittenbogaard et al., 2019). Optimal cell density (5,000/well) and the uncoupler FCCP (fluoro 3-carbonyl cyanide-methoxyphenyl hydrazine; 2 µM) were determined using the Cell Energy Phenotype Test kit. Dermal fibroblasts were seeded in triplicate on poly-D lysine-coated plates and incubated for 24 hours at 37°C in 5% CO2 atmosphere. Prior to the assay, the supplemented DMEM medium was changed to unbuffered Base Medium supplemented with 2 mM glutamine (Invitrogen), 2 mM pyruvate (Sigma; St Louis, MO), and 7.1 mM glucose (Sigma) and adjusted to pH 7.4 with NaOH. Using the XFp Mito Stress Test kit, oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured under basal conditions and after sequential injections of oligomycin (1µM), FCCP (2 µM) and a mix of rotenone and antimycin A (1 µM) following the manufacturer’s recommendations. Quantification analyses of key functional parameters for mitochondrial respiration via OXPHOS were done using the Agilent XF Mito Stress Test Report Generator. OCR was normalized to the number of cells quantified after the conclusion of the assay using the Cytation 1 equipment (BioTek) and expressed in pmo/min/cell X 1000.
Using the Seahorse XFp real-time ATP Rate assay, we simultaneously quantified the rate of ATP produced by the two main bioenergetic pathways, OXPHOS and glycolysis, to calculate the total cellular rate of ATP production according to the manufacturer’s recommendations. Prior to the assay, the culture medium was switched to the phenol-red and bicarbonate-free medium assay containing 5 mM HEPES, 2cmM glutamine (Invitrogen), 2 mM pyruvate (Sigma), and 7.1 mM glucose (Sigma), and adjusted to pH 7.4 with NaOH. OCR and ECAR were measured under basal conditions and after sequential injections of oligomycin (1.5 µM) and a mix of rotenone and antimycin A (0.5 µM).
Using the XFp Glycolytic Rate Assay, we analyzed the glycolytic rate by quantifying the total proton efflux and the glycolytic proton efflux, as described (Uittenbogaard et al. 2018). Prior to the assay, the supplemented DMEM medium was changed to the XF base medium without phenol red supplemented with 2 mM glutamine, 10 mM glucose, 1 mM pyruvate, and 5.0 mM HEPES. OCR and ECAR were measured under basal conditions and after sequential injections of rotenone/antimycin A (0.5 µM) and 2-DG (50 mM).
All the data from three independent experiments, each including three technical replicates, were normalized to cell numbers after the assay and plotted as OCR (pmol/min/cell ± S.E.M.) and ECAR (mpH/min/cell ± S.E.M.), as a function of time using the Seahorse MultiReport Generator software. Statistical analyses were performed using the unpaired student t-test with a p-value of less than 0.05 considered statistically significant.
3. Results
3.1. Clinical history
The 4-year-old male proband was born at 36 week-gestational age with a history of IUGR and a complex medical history of Pierre Robin sequence (PRS) with the classical triad of micrognathia, glossoptosis with severe obstructive sleep apnea, and cleft palate, as well as of microcephaly, chronic static encephalopathy, global developmental delay, congenital hypotonia, intractable epilepsy with both generalized and focal features, suggestive of a mitochondrial etiology without a unifying genetic diagnosis (Figure 1).
The proband’s parents, a 30-year-old mother with a congenital heart defect and a healthy 29-year-old father, were of northern European descent and nonconsangineous. The mother had a healthy 5-year-old daughter with the proband’s father and a 10-year-old son diagnosed with attention-deficit/hyperactivity disorder with a separate partner. Worth noting was the mother’s two pregnancy losses before and after her daughter’s birth, one of which required a dilation and curettage procedure (Figure 2). The mother was a light smoker, who took prenatal vitamins and did not experience common maternal health conditions, such as diabetes, pre-eclampsia, thyroid problems, infection, rash, fever, hypertension, or bleeding during pregnancy. Moreover, the family history was noncontributory for the proband’s complex phenotypic manifestations (Figure 2).
During pregnancy, the mother did not gain much gestational weight and reported decreased fetal movements. Subsequently, she was monitored by a maternal fetal medicine specialist. The Panorama serum screen was normal. At 15 weeks, a fetal echocardiogram, which was prompted by the detection of ventricular enlargement by level II ultrasound, revealed a thickened mitral valve, bicuspid aortic valve, and small muscular ventricular septal defect. Amniocentesis and chorionic villus sampling were declined. At 26 weeks, a fetal ultrasound revealed ventriculomegaly. At 35 weeks, a postnatal head ultrasound confirmed supratentorial ventriculomegaly along with decreased cerebral white matter volume in keeping with white mattervolume loss and/or hypoplasia. A same-day computed tomography (CT) showed mandibular hypoplasia and a U-shaped cleft palate, both components of PRS (Figure 3A,B). At 36-5/7 weeks, vaginal delivery was induced due to IUGR and fetal growth arrest.
At birth, the proband weighed 3 lbs and 2 oz and was admitted to the newborn intensive care unit for IUGR and feeding difficulties. The proband’s dysmorphic facial features led to the diagnosis of PRS with the clinical triad of airway obstruction, glossoptosis with cleft palate, and micrognathia. He required phototherapy for jaundice and passed the newborn hearing screen before being discharged 21 days later. Between the age of two and five months, the proband was hospitalized due to poor weight gain and required placing a gastrotomy tube, and a mandibular osteotomy with fixation. Mandibular morphology was more normally proportioned on CT following surgical distraction (Figure 3C). Brain magnetic resonance imaging (MRI) at four months revealed corpus callosum dysgenesis, deficient myelination-related signal, basal ganglia hyperfusion, optic pathway volume loss and/or hypoplasia, and brain volume loss and/or hypoplasia (Figure 3D-G). He was then referred to our opthalmology clinic to evaluate the reduced optic pathway volume; he could blink to light, but could not fix or follow it.
At six-month-old, the proband underwent an epiglottoplasty to alleviate severe apnea. He also showed hypopigmentation of the skin, hair and fovea. However, the diagnosis of the Hermansky-pudlak syndrome was ruled out by a normal analysis of the multigene panel that included the AP3B1, AP3D1, BLOC1S3, BLOC1S5, BLOCS1S6, DTNBP1, HPS1, HPS3, HPS4, HPS5, and HPS6 genes. The proband’s karyotype was also normal. Thus, the proband was diagnosed with unspecified oculocutaneous albinism. At the age of seven months, the proband had an onset of seizures with abnormal electroencephalogram. He exhibited failure to thrive and developmental delays with a developmental level estimated to be that of a two-to-three month-old infant. The thyroid screen and sterol panel were normal.
At the age of 15 months, the proband’s brain MRI showed no evidence of an acute intracranial disease process, but revealed diffuse moderate cerebral gray and white matter with volume loss, mild brainstem hypoplasia, deficient myelination, diffuse thinning of the corpus callosum, and a lack of visibility of the rostrum, consistent with corpus callosum dysgenesis. These findings could reflect loss of white matter volume and superimposed mild corpus callosum dysgenesis, all suggestive of delayed myelination or hypomyelination.
At the age of 20 months, a brain CT showed further thining of the corpus callosum and stable diffuse decrease of the cerebral volume. At the age of 27 months, the proband’s brain MRI indicated no significant changes in his moderate volume loss of cerebral white matter and brainstem, but showed a slight increase of the associated ex-vacuo ventriculomegaly in proportion to proband’s head growth. At four years of age, an MRI revealed a relatively similar brain volume reduction when taking into account interval patient growth and development (Figure 3H). Although myelination progressed, it remained deficient for the proband’s age, and therefore consistent with hypomyelination.
Given the proband’s poor postnatal growth, microcephaly, static encephalopathy, focal and generalized epilepsy, and brain abnormalities without an unifying genetic diagnosis, pituitary abnormalities and growth hormone deficiency were explored. His IGF-1, IGFBP-3, and morning cortisol levels were normal. At the age of three, he was prescribed depakote for seizures, which triggered hyperammonemia and lactic acidosis. He was switched to keppra, clonazepam, and clonidine. When seizures lasted more than five minutes or with a frequency of three in one hour, the patient was administered diastat and immediately hospitalized. At the age of four, the proband had worsening refractory seizures, gastrointestinal dysmotility, acquired unequal leg length, and global developmental delay with absence of speech, movements limited to rolling from front to back, and regression of assisted walking with no weight bearing on the left.
Collectively, the proband’s symptoms of hypotonia, global developmental delays, intractable epilepsy with both generalized and focal features, static encephalopathy, reaction to depakote, and low levels of citrulline, led to a suspected mitochondrial etiology. However, the dual genome panel was uninformative in the absence of any mitochondrial pathogenic variants and large deletion of the mitochondrial genome. His dual genome panel only revealed a single heterozygous variant of uncertain significance, c.712C>T (p.R238C), mapping in the ADCK3 gene involved in the coenzyme Q synthesis pathway. Whole exome sequencing of the nuclear genome and LR-PCR-MPS of the mitochondrial genome failed to reveal any nuclear and mitochondrial pathogenic variants.
3.2. Functional studies of the ATP metabolism
To investigate whether an underlying mitochondrial etiology could contribute to the patient’s symptoms for a neurodevelopmental disorder of an unspecified genetic diagnosis, we performed a skin biopsy on the four-year-old proband. As a control subject, we used commercially available dermal fibroblasts from a healthy 10-year-old subject whose metabolic profile was already characterized and comparable to two other healthy subjects (Uittenbogaard et al., 2018). By means of OCR as a functional indicator of the mitochondrial energy metabolism, we assessed OXPHOS parameters using the Mitochondrial Stress test assay (Figure 4). We found that only the basal respiration and its related parameter ATP-linked respiration remained unchanged, when compared to those of a healthy subject (Figure 4C). In contrast, the maximal respiration capacity evoked by exposure to the protonophore FCCP decreased by 28% (Figure 4C). More critically was the 69% decline of the spare respiratory capacity, essential for the proband’s cells to avert an ATP crisis upon energy demand (Figure 4C). Thus, the proband’s fibroblasts exhibited a dysregulated OXPHOS pathway congruent with his severe neurological symptoms.
We next examined the glycolytic metabolism of the proband’s fibroblasts to determine whether they could switch to glycolysis for increased ATP synthesis. To this end, we used the Glycolysis Rate assay to accurately assess the glycolytic activity by correlating one-to-one with lactate accumulation. The total Proton Efflux Rate (PER) and the Glycolytic Proton Efflux Rate (GlycoPER) were measured using both OCR and ECAR values to account for mitochondrial (CO2) acidification from the mitochondrial TCA cycle (Figure 5A) (Mookerjee et al., 2015). We found that the proband’s fibroblasts showed a 180% increase in basal glycolysis, which was confirmed by an increased PER to 81%, compared to 66% in dermal fibroblasts from the healthy subject (Figure 5B). We next measured the compensatory glycolysis response as an indicator of metabolic reprogramming toward glycolysis subsequent to an OXPHOS crisis provoked by exposure to rotenone and antimycin A. We noticed that the proband’s fibroblasts were endowed with a strong compensatory glycolysis response that was 40% superior to that of healthy fibroblasts (Figure 5C).
We confirmed our OXPHOS and glycolytic results by measuring the basal rate of ATP production from glycolysis and the OXPHOS pathway using the XFp Real-Time ATP rate assay. Both OCR and ECAR were simultaneously measured upon injection of oligomycin followed by injection of rotenone and antimycin A to fully inhibit mitochondrial ATP production (Figure 6A). We detected similar basal rates of ATP production in the proband’s fibroblasts and healthy fibroblasts, corroborating our finding of comparable basal respiration from the OXPHOS pathway between the diseased and healthy fibroblasts (Figure 6B). Our results revealed a 233% increase in the rate of ATP production from basal glycolysis, validating our previous findings of metabolic switch from OXPHOS to glycolysis in the proband’s fibroblasts in order to increase ATP synthesis (Figure 6B).
3.3. Mitochondrial morphometric examination of the proband’s fibroblasts
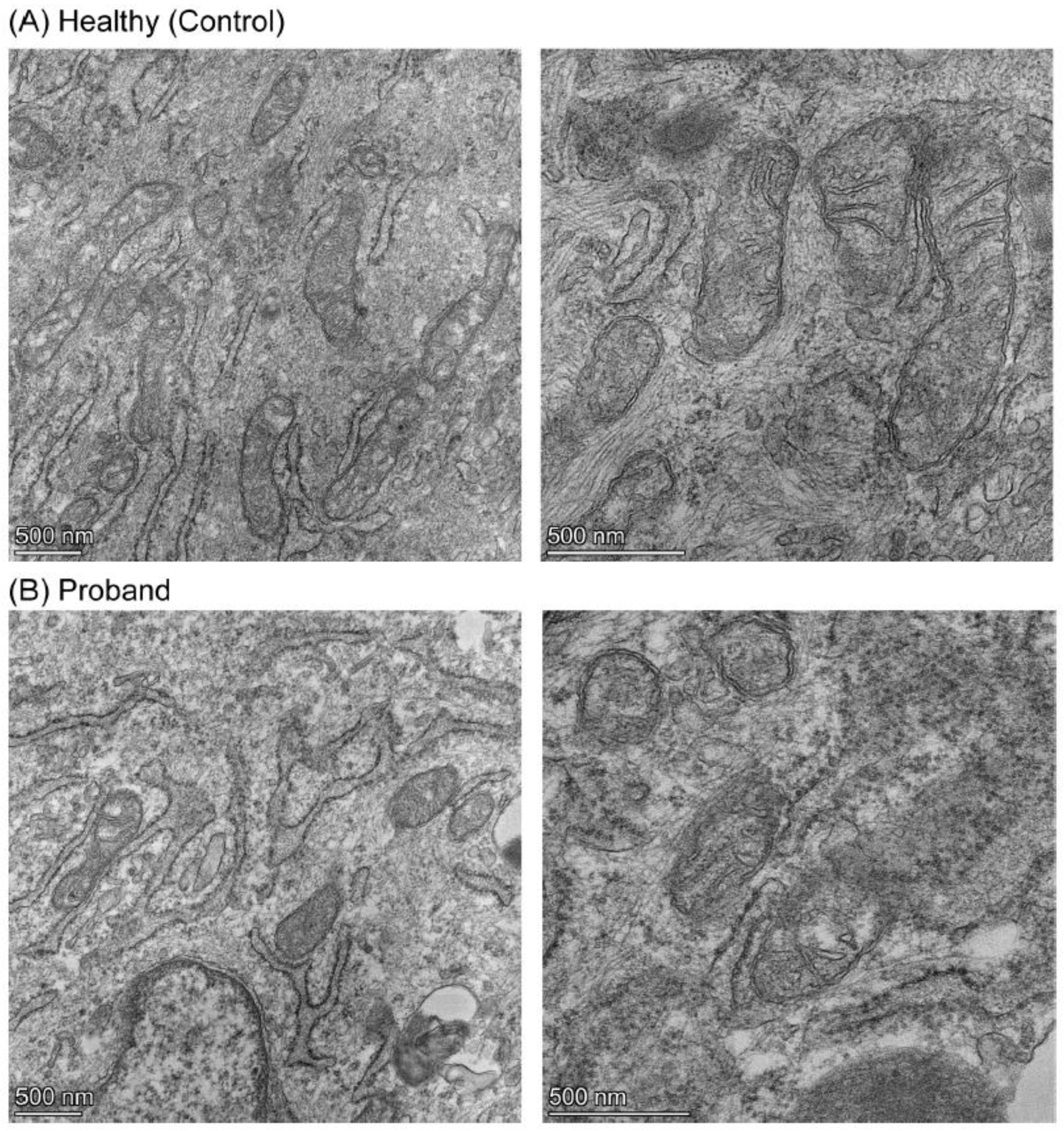
We then performed a mitochondrial morphometric analysis using transmission electron microscopy to investigate the proband’s mitochondrial ultrastructure. The proband’s fibroblasts harbored fewer mitochondria and of smaller size than those in healthy fibroblasts (Figure 7). More specifically, the proband’s mitochondria exhibited small and rare cristae, similar to those observed in immature mitochondria (Cogliati et al., 216), while mitochondria from the healthy subject have an elongated mitochondria with normal cristae (Figure 7). These mitochondrial morphometric results confirmed a deficiency in mitochondrial energy metabolism and reliance on glycolysis for ATP synthesis (Figure 4, Figure 5 and Figure 6).
4. Discussion
Our study reports a four-year-old male born prematurely due to IUGR. He has a complex medical history with profound neurodevelopmental and global defects, microcephaly, congenital hypotonia, intractable epilepsy with both generalized and focal features, and the dysmorphic syndrome Pierre-Robin sequence (PRS) with the classical clinical triad of micrognathia, glossoptosis with severe obstructive sleep apnea, and cleft palate. The absence of chromosomal abnormalities and genetic factors known to cause PRS, such as the critical chondrogenic regulator SOX9, supports the extrinsic abnormalities or neurological/neuromuscular abnormalities as potential embryonic pathogenic events of extrinsic abnormalities or neurological/neuromuscular abnormalities (Côté et al., 2015). Both are consistent with the proband’s congenital hypotonia and IUGR, evocative of an in utero compression of his mandible. Our live-cell mitochondrial metabolic analyses validate the patient’s suspected mitochondrial etiology based on his low levels of citrulline and lactic acidosis elicited by depakote. Moreover, our mitochondrial morphometric analysis reveals an immature mitochondrial morphology consistent with the patient’s OXPHOS deficiency and neurodevelopmental deficits.
The genetic causes of the proband’s complex phenotypic manifestations remain enigmatic in the absence of nuclear and/or mitochondrial pathogenic variants by whole exome sequencing of the nuclear genome and deep sequencing of the mitochondrial genome. Thus, the most probable cause is uteroplacental insufficiency and intrauterine growth restriction (IUGR). Placental insufficiency triggers inadequate supply of substrates such as glucose, fatty acids, amino acids and oxygen, which impairs the fetal energy metabolism and consequently fetal growth and development (Gagnon, 2003; Miller et al., 2016; Pendleton et al., 2021). IUGR is commonly associated with reduced blood flow through the placenta combined with limited invasion of the decidua and maternal blood vessels (Pardi et al., 2002). This leads to curtailed levels of oxygen and fuel substrates resulting in fetal mitochondrial dysfunction and the proband’s grey matter and white matter pathologies revealed by brain MRI, such as hypomyelination, corpus callosum dysgenesis, and decreased cerebral volume. Since mitochondrial ATP synthesis is coupled with oxygen consumption by Complex IV, low fetal levels of oxygen most likely decrease the overall OXPHOS activities to generate antenatal chronic energy deficit. It is well documented that the developing brain requires high levels of energy (Erecinska et al., 2004; Uittenbogaard and Chiaramello, 2014). This infers that deficits in brain structure and functions are the consequence of antenatal and postnatal alteration of the mitochondrial energy metabolism. Congruent with the patient’s brain MRI is the severe deficit in the spare respiratory capacity revealed by our live-cell functional mitochondrial OXPHOS investigations. Since firing neurons require 80% of the spare respiratory capacity for assuming synaptic activities (Nicholls, 2009), the proband’s developing brain does not have a sufficient reserve of energy to avert an ATP crisis upon such high energy demand. Congruent with our mitochondrial functional investigations is our mitochondrial morphometric analysis showing the proband’s immature mitochondrial morphology with poorly developed cristae, a morphological indicator of limited OXPHOS capacity and mitochondrial ATP synthesis (Uittenbogaard et al., 2018; Cogliati et al., 2016; Gropman et al., 2020). In keeping with the proband’s abnormal mitochondrial morphorlogy and deficit of the OXPHOS pathway is the proband’s metabolic adaptation to favor glycolysis for ATP synthesis demonstrated by an increased basal glycolytic response and compensatory glycolytic response as an attempt to overcome the OXPHOS-based energy deficit.
Interestingly, the immature mitochondrial morphology, the mitochondrial energy deficit, and the metabolic adaptive response toward glycolysis are hallmarks of mitochondria from undifferentiated human embryonic stem cells during the early stages of mammalian embryonic development (Facucho-Oliveira and St. John, 2009). This mitochondrial metabolic profile is programmed to support proliferation during the pre-implantation embryonic development and early fetal developmental stages (Anh and Metallo, 2015). More specifically, early fetal developmental stages are characterized by low levels of OXPHOS and oxygen consumption, combined with high glycolysis via passive diffusion of glucose through the placenta (Garden and Wale, 2013). At later fetal developmental stages, mitochondria undergo functional metabolic adaptation toward the OXPHOS metabolism to meet the high energy requirement for cell specification and differentiation (Chung et al., 2007). Such mitochondrial metabolic flexibility is necessary for proper fetal growth and development throughout the different in utero stages (Harvey, 2019; Rodríguez-Cano et al., 2020). Various animal models for IUGR support such metabolic plasticity toward glycolysis with an early activation of hepatic glucose production as a substrate for ATP synthesis, a reactive process that is absent in normal fetuses (Hay et al., 2013; Thorn et al., 2013).
In conclusion, our results support the concept that sub-optimal levels of intrauterine oxygen and nutrients alter fetal mitochondrial metabolic reprogramming toward OXPHOS leading to a deficient postnatal mitochondrial energy metabolism. Moroever, our collective studies shed light on the long-term postnatal mitochondrial pathophysiology caused by intrauterine growth restriction due to placental insufficiency and its negative impact on the energy-demanding development of fetal and postnatal brain.
Funding
This work was funded by the NIH National Center for Advanced Translational Studies [UG3TR003897] to AC and AG, and the NIH National Institute of Child Health and Development [1U54HD090257].
Abbreviations
COXPD = combined oxidative phosphorylation deficiency; CT = computed tomography ; DMEM = Dulbecco’s modified eagle medium; ECAR = extracellular acidification rate; EEG = electroencephalogram; FCCP = fluoro 3-carbonyl cyanide-methoxyphenyl hydrazine; FGF-2 = fibroblast growth factor-2; FIESTA = fast imaging employing steady-state acquisition; GlycoPER = glycolytic proton efflux rate; HEPES = 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; IUGR = Intrauterine growth restriction; LR-PCR-MPS = long range PCR followed by massively parallel sequencing; MRI = magnetic resonance imaging; NGS = next-generation sequencing; PER = proton exchange rate; PRS = Pierre Robin sequence; OCR = oxygen consumption rate; OXPHOS = oxidative phosphorylation; T2WI = T2-weighted image; WES = whole exome sequencing
References
- Angelica D, Giovanni O, Vassilios F. Physiopathology of intrauterine growth retardation: from classic data to metabolomics. J. Maternal-Fetal Neonatal. Med. 2012;25(S5):13-18.
- Anh CS, Metallo CM. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 2015;3:1. [CrossRef]
- Barker, JP. Barker JP. Adult consequences of fetal growth restriction. Clin. Obstet. Gynecol. 2006;49:270-283.
- Cetin A, Alvino G. Intrauterine growth restriction: implications for placental metabolism and transport. A review. Placenta 2009;30:S77-S82.
- Chernausek SD. Update: Consequences of abnormal fetal growth. J. Clin. Endocrinol. Metab. 2012;97:689-695. [CrossRef]
- Chung S, Dzeja PP, Faustino RS, Perz-Terzic C, Behfar A, Terzic A. Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nat. Clin. Pract. Cardovasc. Med. 2007;4:S60-S67. [CrossRef]
- Cogliati S, Enriquez JA, Scorrano L. Mitochondrial cristae: where beauty meets functionality. Trends Biochem. Sci. 2016;41:261-273. [CrossRef]
- Cosmi E, Fanelli T, Visentin S, Trevisanuto D, Zanardo V. Consequences in infants that were intrauterine growth restricted. J. Pregnancy 2011:364381. [CrossRef]
- Côté A, Fanous A, Almajed A, Lacroix Y. Pierre-Robin sequence: review of diagnostic and treatment challenges. Int. J. Pediatr. Otorhinolaryngol. 2015;79:451-464. [CrossRef]
- De Bie HMA, Oostrom KJ, Delemarre-van de Waal HA. Brain development, intelligence and cognitive outcome in children born small for gestational age. Horm. Res. Paediatr. 2010;73:6-14.
- Dubois J, Benders M, Borradori-Tolsa C, Cachia A, Lazeyras F, Ha-Vinh Leuchter R, Sizonenko SV, Warfield SK, Mangin JF, Hüppi PS. Primary cortical folding in the human newborn: an early marker of later functional development. Brain 2008;131:2028-2041. [CrossRef]
- Erecinska M, Cherian S, Silver IA. Energy metabolism in mammalian brain during development. Prog.Neurobiol. 2004; 73:397-445. [CrossRef]
- Facucho-Oliveira JM, St. John JC. The relationship between pluripotency and mitochondrial DNA proliferation during early embryo development and embryonic stem cell differentiation. Stem Cell Rev. and Rep. 2009; 5:140158. [CrossRef]
- Geva R, Eshel R, Leitner Y, Valevski AF, Harel S. Neuropsychological outcome of children with intrauterine growth restriction: a 9-year prospective study. Pediatrics 2006;118:91-101. [CrossRef]
- Gagnon R. Placental insufficiency and its consequences. Eur. J. Obstet. Gynecol. 2003;110:S99-S107. [CrossRef]
- Gardner DK, Wale PL. Analysis of metabolism to select viable human embryos for transfer. Fertil. Steril. 2013;99:1062-1072. [CrossRef]
- Gordijn SJ, Beune IM, Thilaganathan B, Papageorghiou A, Baschat AA, Baker PN, Silver RM, Wynia K, Ganzevoort W.. Consensus definition of fetal growth restriction: a Delphi procedure. Ultrasound Obstet. Gynecol. 2016;48:333-339. [CrossRef]
- Gropman A, Uittenbogaard M, Brantner CA, Wang Y, Wong LJ, Chiaramello A. Molecular genetic and mitochondrial metabolic analyses confirm the suspected mitochondrial etiology in a pediatric patient with an atypical form of alternating hemiplegia of childhood. Mol. Genet. Metab. Rep. 2020;24:100609. [CrossRef]
- Harvey AJ. Mitochondria in early developmemt: linking the microenvironment, metabolism and the epigenome. Reprod. 2019;157:R159-R179.
- Hay WW, Myers SA, Sparks JW, Wilkening RB, Meschia G, Battaglia FC. Glucose and lactate oxidation rates in the fetal lamb. Proc. Soc.Exp. Biol. Med. 1983;173:553-563. [CrossRef]
- Hendrix N, Berghella V. Non-placental causes of intrauterine growth restriction. Semin. Perinatol. 2008;32:161-165. [CrossRef]
- McCowan LM, Figueras F, Anderson NH. Evidence-based national guidelines for the management of suspected fetal growth restriction: comparison, consensus, and controversy. Am. J. Obstet. Gynecol. 2018;218:S855-S868. [CrossRef]
- Miller SL, Huppi PS, Mallard C. The consequences of fetal growth restriction on brain structure and neurodevelopmental outcome. J. Physiol. 2016;594:807-823. [CrossRef]
- Mookerjee SA, Goncalves RLS, Gerencser AA, Nicholls DG, Brand MD. The contributions of respiratory and glycolysis to extracellular acid production. Biochem. Biophys. Acta 2015;1847:171-181.
- Naha R, Anees A, Chakrabarty S, Naik PS, Pandove M, Pandey D, Satyamoorthy K. Placental mitochondrial DNA mutations and copy numbers in intrauterine growth restricted (IUGR) pregnancy. Mitochondrion 2020;55:85-94. [CrossRef]
- Nicholls DG. Spare respiratory capacity oxidative stress excitotoxicity. Biochem. Soc. Trans. 2009;37:1385-1388. [CrossRef]
- Padilla N, Falcón C, Sanz-Cortés M, Figueras F, Bargallo N, Crispi F, Eixarch E, Arranz A, Botet F, Gratacós E. Differential effects of intrauterine growth restriction on brain structure and development in preterm infants: a magnetic resonance imaging study. Brain Res. 2011;1382:98-108. [CrossRef]
- Pandey D, Yevale A, Naha R, Kuthethur R, Chakrabarty S, Satyamoorthy K. Mitochondrial DNA copy number variation – A potential biomarker for early onset preeclampsia. Pregnancy Hypertens. 2021;23:1-4. [CrossRef]
- Pardi G, Marconi AM, Cetin I. Placental-fetal interrelationships in IUGR fetuses – a review. Placenta 2002;23:S136-S141.
- Pendleton AL, Wesolowski SR, Regnault TRH, Lynch RM, Limesand SW. Dimming the powerhouse: mitochondrial dysfunction in the liver and skeletal muscle of intrauterine growth restricted fetuses. Front. Endocrinol. 2021;12:612888. [CrossRef]
- Ramenghi LA, Martinelli A, De Carli A, Brusati V, Mandia L, Fumagalli M, Triulzi F, Mosca F, Cetin I. Cerebral maturation in IUGR and appropriate for gestational age preterm babies. Reprod. Sci. 2011;18:469-475. [CrossRef]
- Rodríguez-Cano A, Calzada-Mendoza CC, Estrada-Gutierrez G, Mendoza-Ortega JA, Perichart-Perera O. Nutrients, mitochondrial function, and perinatal health. Nutrients 2020;12:2166. [CrossRef]
- Samuelsen GB, Packenberg B, Bogdanović N, Gundersen HJG, Larsen JF, Graem N, Laursen H. Severe cell reduction in the future brain cortex in human growth-restricted fetuses and infants. Am. J. Obstet. Gynecol. 2007;197:e1-56e7. [CrossRef]
- Suhag A, Berghella V. Intrauterine growth restriction (IUGR): etiology and diagnosis. Curr. Obstet. Gynecol. Rep. 2013;2:102-111. [CrossRef]
- Thorn SR, Brown LD, Rozance PJ, Hay WW, Friedman JE. Hepatic glucose production in fetal sheep with intrauterine growth restriction is not suppressed by insulin. Diabetis 2013;62:65-73.
- Uittenbogaard M, Chiaramello A. Mitochondrial biogenesis: a therapeutic target for neurodevelopmental disorders and neurodegenerative diseases. Curr. Pharm. Des. 2014;20:5574-5593. [CrossRef]
- Uittenbogaard M, Wang H, Zhang VW, Wong LJ, Brantner CA, Gropman A, Chiaramello A. The nuclear background influences the penetrance of the near-homoplasmic m.1630A>G MELAS variant in a symptomatic proband and asymptomatic mother. Mol. Genet. Metab. 2019;126:429-438. [CrossRef]
- Uittenbogaard M, Gropman A, Brantner CA, Chiaramello A. Novel metabolic signatures of compound heterozygous Szt2 variants in a case of early-onset of epileptic encephalopathy. Clin. Case Rep. 2018;9. [CrossRef]
- Uittenbogaard M, Brantner CA, Fang Z, Wong LJ, Gropman A, Chiaramello A. Novel insights into the functional metabolic impact of an apparent de novo m.8993T>G variant in the MT-ATP6 gene associated with maternally inherited form of Leigh syndrome. Mol. Genet. Metab. 2018;124:71-81.
- Villar J, Belizán JM, Spalding J, Klein RE. Postnatal growth of intrauterine growth retarded infants. Early Hum. Dev. 1982;6:265-271. [CrossRef]
- Von Beckerath AK, Kollman M, Rotky-Fast C, Karpf E, Lang U, Klaritsch P. Perinatal complications and long-term neurodevelopmental outcome of infants with intrauterine growth restriction. Am. J. Obstet. Gynecol. 2013;208:130.e1-6. [CrossRef]
Figure 1.
Highlights of the proband’s clinical history.
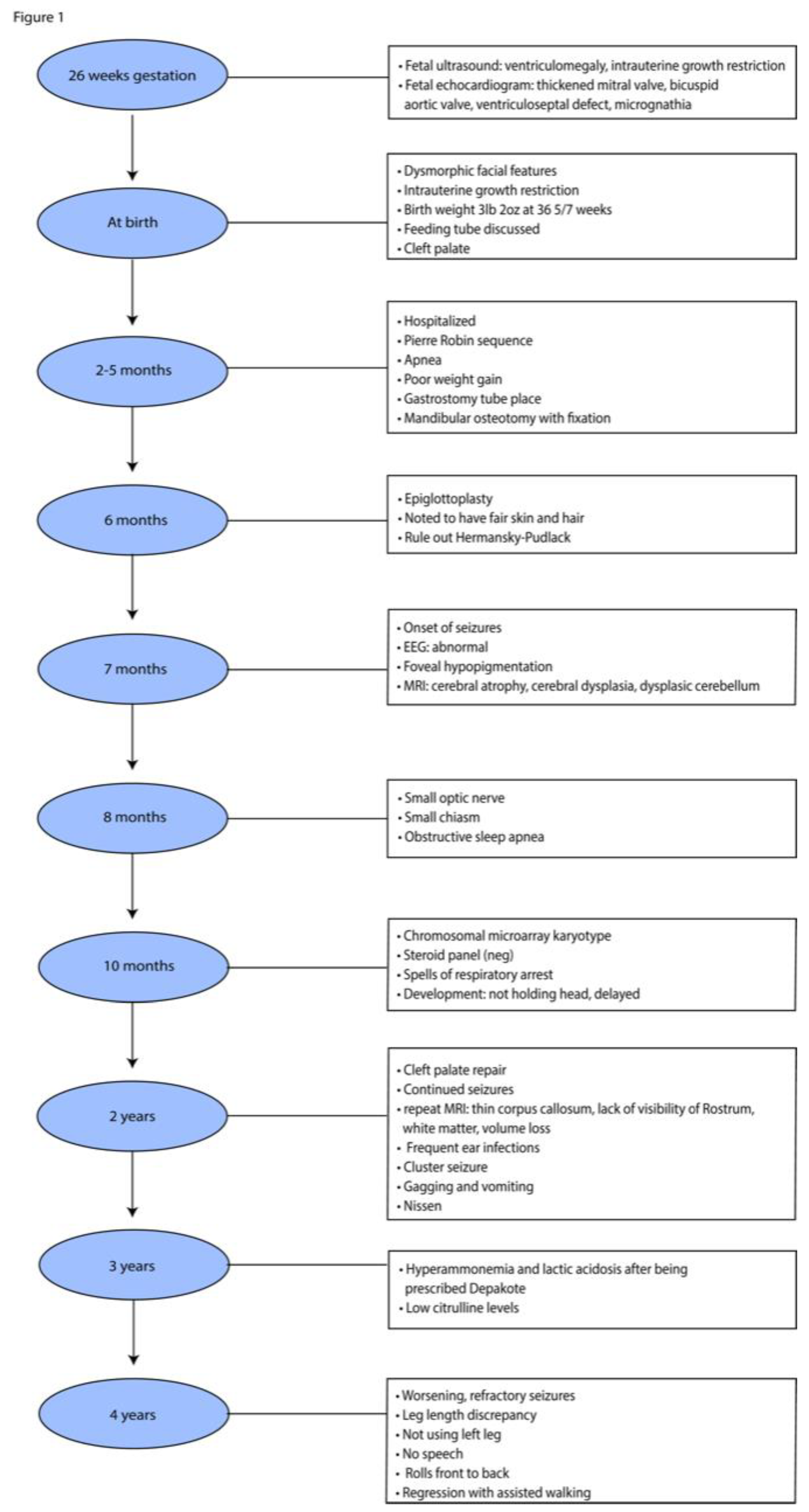
Figure 2.
Pedigree tree with the proband identified by an arrow and a blue square. Abbreviations: ADHD: attention deficit and hyperactivity disorder; SAB: spontaneous abortion; yo: years old.
Figure 2.
Pedigree tree with the proband identified by an arrow and a blue square. Abbreviations: ADHD: attention deficit and hyperactivity disorder; SAB: spontaneous abortion; yo: years old.
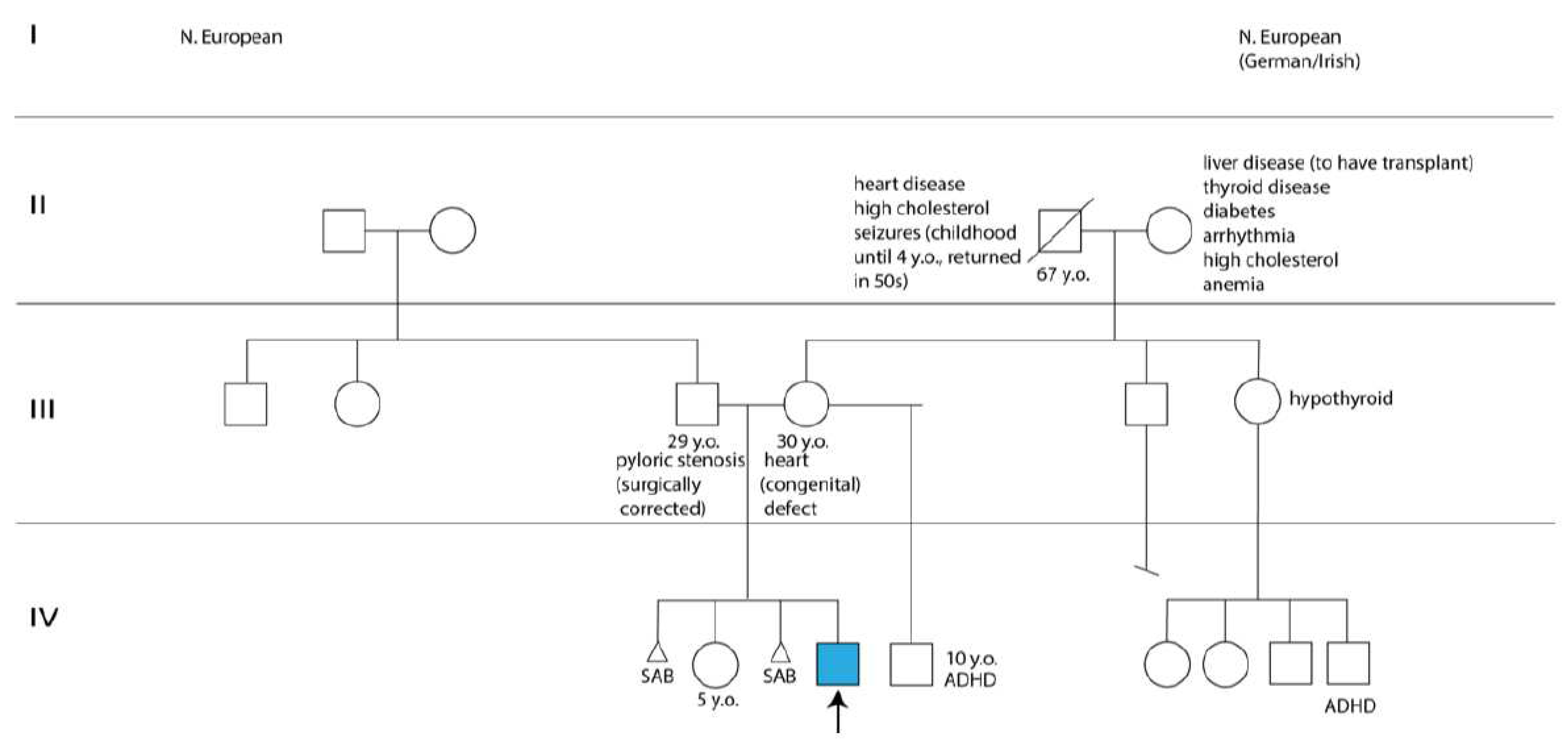
Figure 3.
Computed tomography and brain magnetic resonance imaging (MRI) of the proband. (A) Maxillofacial computed tomography (CT) in sagittal planes performed at 35 days shows mandibular hypoplasia. (B) Maxillofacial CT in coronal planes performed at 35 days shows a u-shaped cleft palate with the white arrow indicating components of Pierre-Robin sequence. (C) Sagittal image from a postoperative face CT at 3 months shows improved mandibular and maxillary proportions. (D) Brain MRI Axial T2WI image performed at 4 months illustrating myelination related signal hypointensity being mildly deficient consistent with hypomyelination or delayed myelination. (E) Brain MRI axial ASL perfusion image showing the basal ganglia being hyperperfused (orange and red hues). (F) Brain MRI coronal T2WI indicating the optic nerves are thin, consistent with volume loss and/or hypoplasia (arrows). (G) Brain MRI sagittal midline FIESTA image showing moderate diffuse cerebral gray and white matter and mild brainstem volume loss and/or hypoplasia with commissural thinning, decreased white matter depth, and ex-vaco ventriculomegaly. The corpus callosum is hypogenetic/dysgenetic and hypoplastic, being diffusely thin with absence of the rostrum. (H) Brain MRI at 4 year showing relatively similar cerebral and brainstem volume loss and/or hypoplasia.
Figure 3.
Computed tomography and brain magnetic resonance imaging (MRI) of the proband. (A) Maxillofacial computed tomography (CT) in sagittal planes performed at 35 days shows mandibular hypoplasia. (B) Maxillofacial CT in coronal planes performed at 35 days shows a u-shaped cleft palate with the white arrow indicating components of Pierre-Robin sequence. (C) Sagittal image from a postoperative face CT at 3 months shows improved mandibular and maxillary proportions. (D) Brain MRI Axial T2WI image performed at 4 months illustrating myelination related signal hypointensity being mildly deficient consistent with hypomyelination or delayed myelination. (E) Brain MRI axial ASL perfusion image showing the basal ganglia being hyperperfused (orange and red hues). (F) Brain MRI coronal T2WI indicating the optic nerves are thin, consistent with volume loss and/or hypoplasia (arrows). (G) Brain MRI sagittal midline FIESTA image showing moderate diffuse cerebral gray and white matter and mild brainstem volume loss and/or hypoplasia with commissural thinning, decreased white matter depth, and ex-vaco ventriculomegaly. The corpus callosum is hypogenetic/dysgenetic and hypoplastic, being diffusely thin with absence of the rostrum. (H) Brain MRI at 4 year showing relatively similar cerebral and brainstem volume loss and/or hypoplasia.
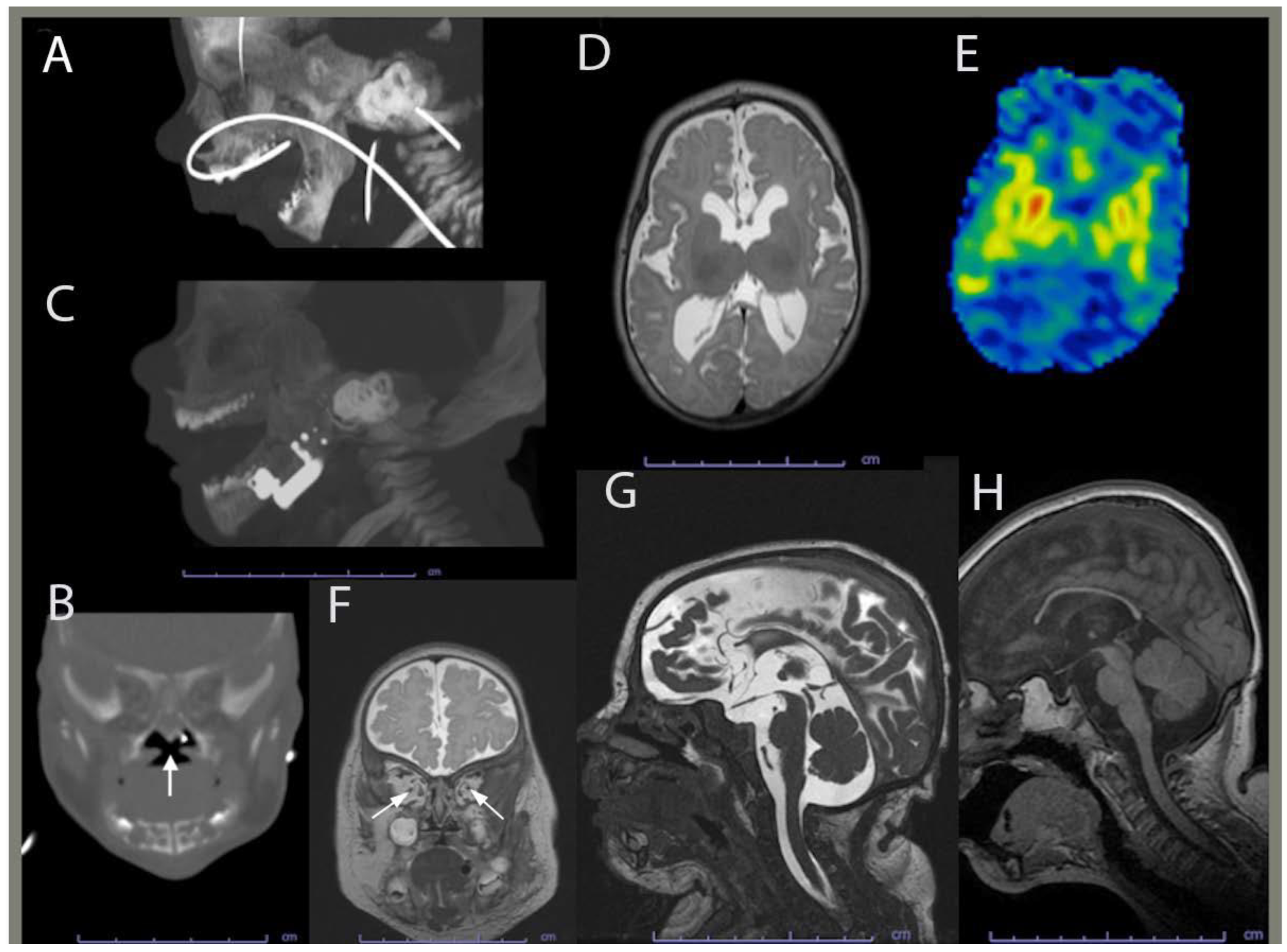
Figure 4.
The proband’s fibroblasts exhibit an impaired bioenergetic capacity. (A) Profile of the oxygen consumption rate (OCR) adapted from Agilent Technologies brochure of the Mitochondrial Stress Test. (B) Compared OCR responses between the proband and a healthy subject. (C) Quantitative data between the proband and the healthy subject of basal respiration, ATP-linked respiration, maximal respiration, and spare respiratory capacity. Data are represented as means ± S.E.M., n = 3 of three independent experiments. * indicates statistically significant differences with a p value of ≤0.005 between the proband and the healthy subject.
Figure 4.
The proband’s fibroblasts exhibit an impaired bioenergetic capacity. (A) Profile of the oxygen consumption rate (OCR) adapted from Agilent Technologies brochure of the Mitochondrial Stress Test. (B) Compared OCR responses between the proband and a healthy subject. (C) Quantitative data between the proband and the healthy subject of basal respiration, ATP-linked respiration, maximal respiration, and spare respiratory capacity. Data are represented as means ± S.E.M., n = 3 of three independent experiments. * indicates statistically significant differences with a p value of ≤0.005 between the proband and the healthy subject.
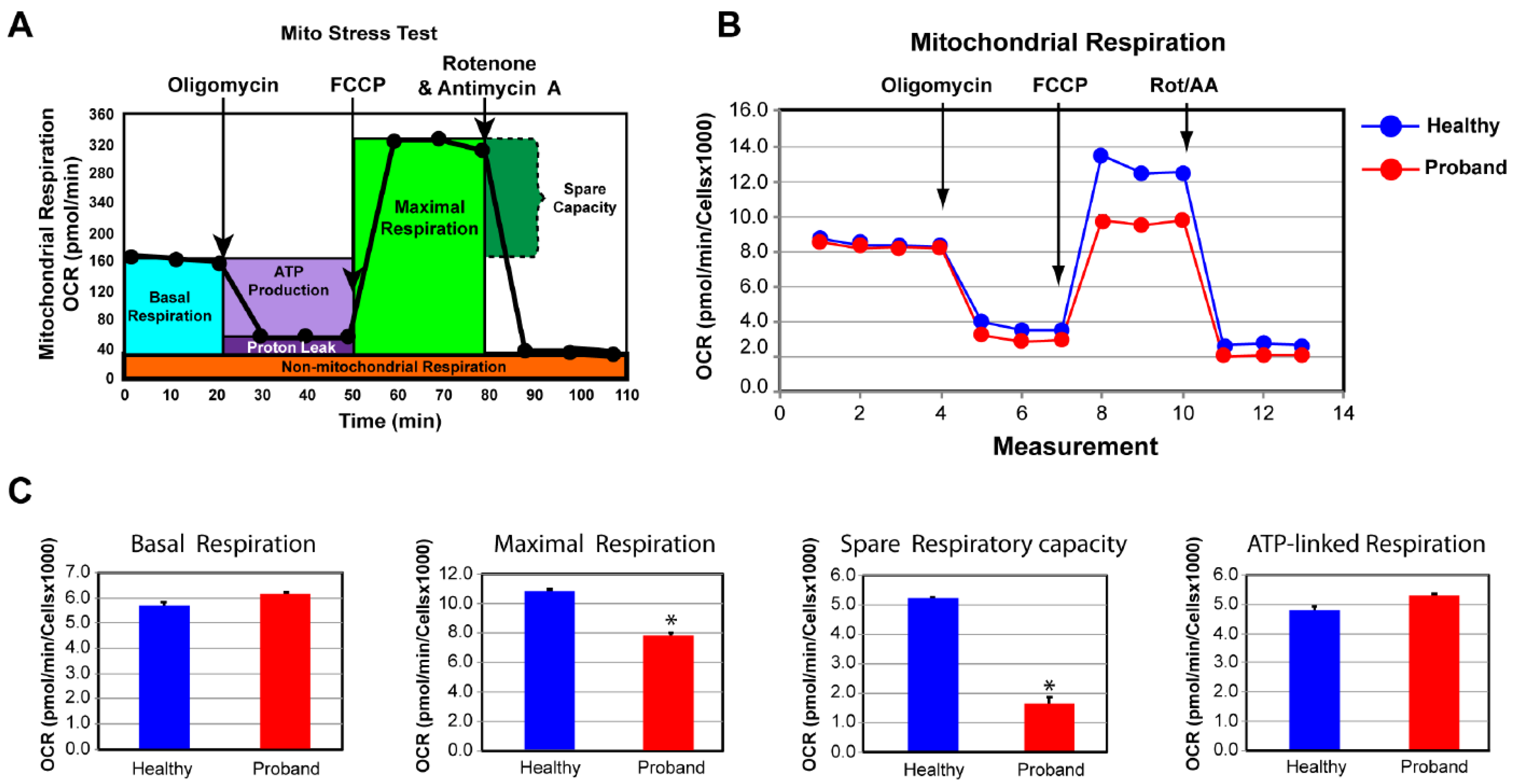
Figure 5.
The proband’s fibroblasts display increased proton efflux rate indicative of a stimulated glycolysis pathway. (A) Schematic representation of proton efflux adapted from the Agilent Technologies brochure of the Glycolytic Rate Test. (B) Quantitative data of basal glycolysis, compensatory glycolysis, and mitochondrial acidification. Data are represented as means ± S.E.M., n = 3 of three independent experiments. * and ** indicate statistically significant differences with a p value of 0.0001 and 0.0037 between the proband and the healthy subject.
Figure 5.
The proband’s fibroblasts display increased proton efflux rate indicative of a stimulated glycolysis pathway. (A) Schematic representation of proton efflux adapted from the Agilent Technologies brochure of the Glycolytic Rate Test. (B) Quantitative data of basal glycolysis, compensatory glycolysis, and mitochondrial acidification. Data are represented as means ± S.E.M., n = 3 of three independent experiments. * and ** indicate statistically significant differences with a p value of 0.0001 and 0.0037 between the proband and the healthy subject.
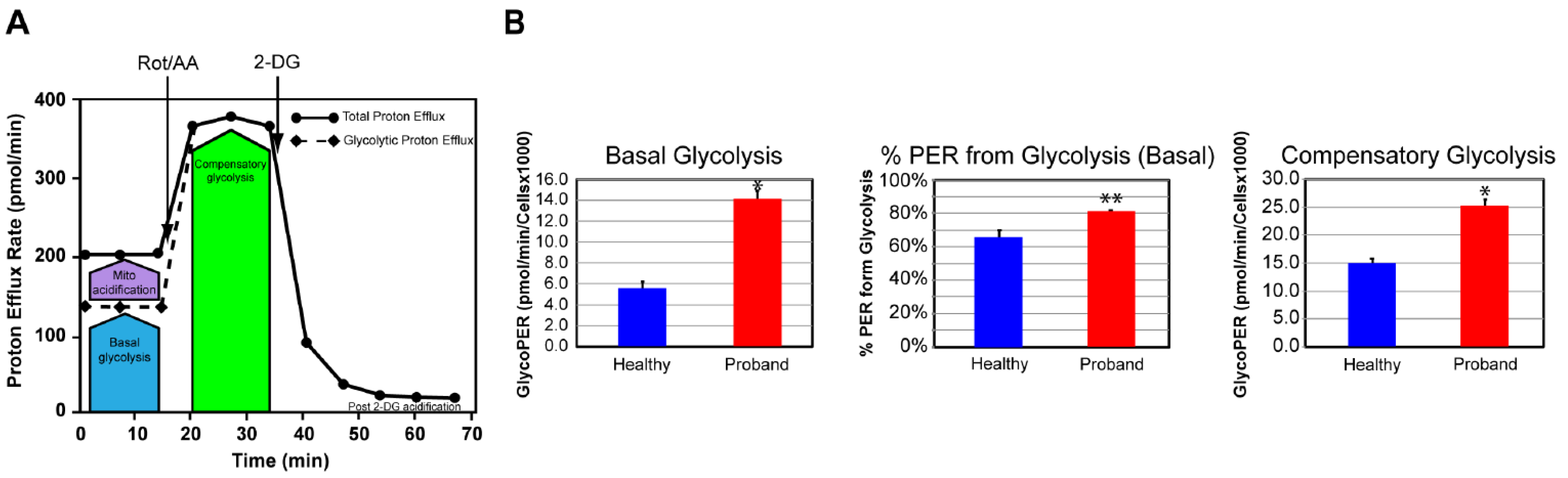
Figure 6.
The proband’s fibroblasts show a disturbed energy metabolic phenotype. (A) Schematic representation of the Agilent Seahorse XFp Real-Time ATP rate assay. Both OCR and ECAR of live fibroblasts from the proband or a healthy subject are simultaneously measured upon injection of the mitochondrial inhibitor oligomycin followed by a mixture of rotenone and antimycin A. (B) Quantification of the basal rate of ATP production from glycolysis (blue for the healthy subject and red for the proband) and mitochondrial OXPHOS (blue hatched column for the healthy subject and red hatched column for the proband). Data are represented as means ± S.E.M., n = 3 of three independent experiments. * indicates statistically significant differences with a p value of 0.0002 between the proband and the healthy subject.
Figure 6.
The proband’s fibroblasts show a disturbed energy metabolic phenotype. (A) Schematic representation of the Agilent Seahorse XFp Real-Time ATP rate assay. Both OCR and ECAR of live fibroblasts from the proband or a healthy subject are simultaneously measured upon injection of the mitochondrial inhibitor oligomycin followed by a mixture of rotenone and antimycin A. (B) Quantification of the basal rate of ATP production from glycolysis (blue for the healthy subject and red for the proband) and mitochondrial OXPHOS (blue hatched column for the healthy subject and red hatched column for the proband). Data are represented as means ± S.E.M., n = 3 of three independent experiments. * indicates statistically significant differences with a p value of 0.0002 between the proband and the healthy subject.
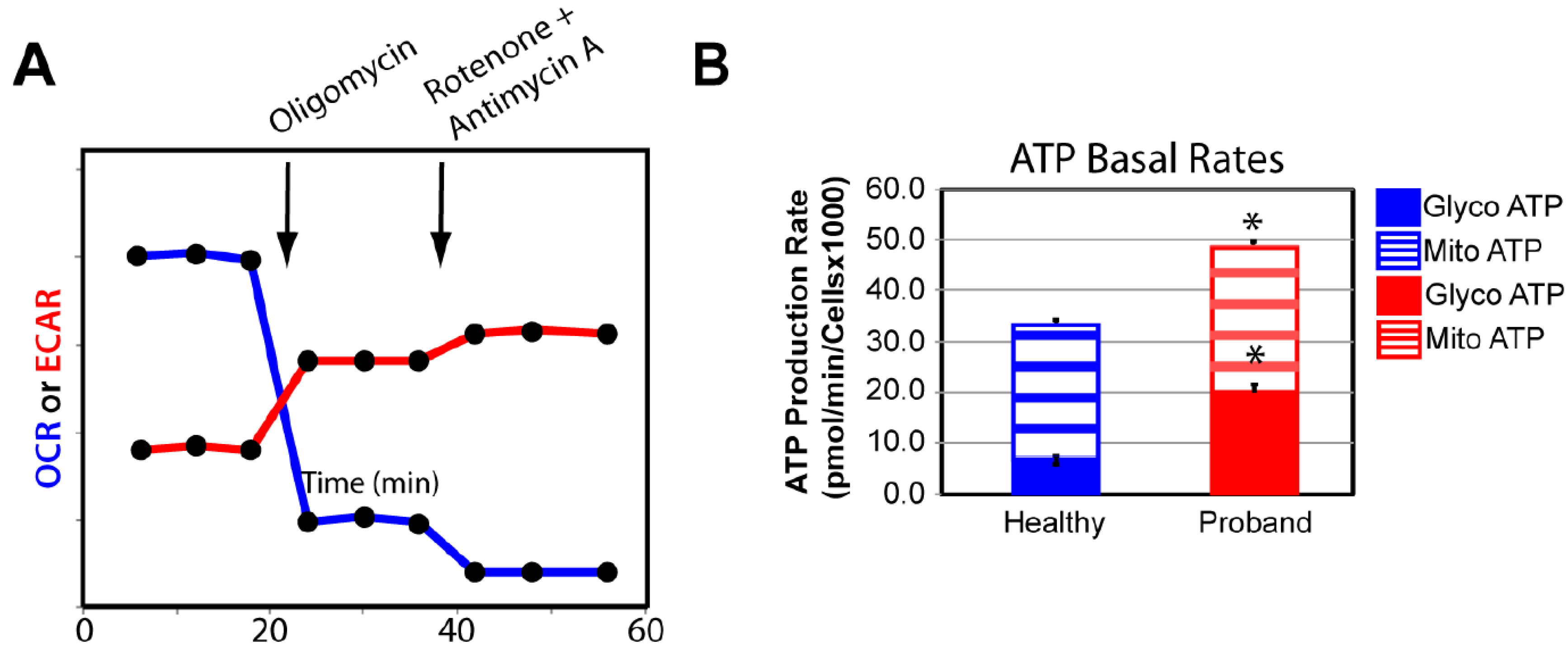
Figure 7.
Mitochondrial morphometric analysis illustrates the immature morphology and poorly developed cristae of the proband’s mitochondria. (A) Mitochondria from a healthy subject at low and high magnifications on the left and right panels, respectively . (B) Mitochondria from the proband at low and high magnifications on the left and right panels, respectively. The scale bar (500 nm) is indicated at the bottom left corner of each micrograph.
Figure 7.
Mitochondrial morphometric analysis illustrates the immature morphology and poorly developed cristae of the proband’s mitochondria. (A) Mitochondria from a healthy subject at low and high magnifications on the left and right panels, respectively . (B) Mitochondria from the proband at low and high magnifications on the left and right panels, respectively. The scale bar (500 nm) is indicated at the bottom left corner of each micrograph.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



