
Submitted:
15 December 2023
Posted:
18 December 2023
You are already at the latest version
Abstract
Hepatic carboxylesterase 1(CES1) metabolizes many prodrugs into active ingredients or direct-acting drugs into inactive metabolites. We aim to develop a semi-physiologically based pharmacokinetic model(Semi-PBPK model) to simultaneously predict pharmacokinetics of CES1 substrates and their active metabolites in liver cirrhosis(LC) patients. Six prodrugs(enalapril, benazepril, cilazapril, temocapril, perindopril and oseltamivir) and three direct-acting drugs (flumazenil, pethidine and remimazolam) were selected. The parameters including organ blood flows, plasma binding protein concentrations, functional liver volume, hepatic enzymatic activity, glomerular filtration rate(GFR) and gastrointestinal transit rate were introduced into the simulation. Pharmacokinetic profiles of these drugs and their active metabolites were simulated in 100 virtual subjects. The developed semi-PBPK model, following validation in healthy subjects, was extrapolated to LC patients. Most of the observations are within the 5% and 95% quantile of simulations from 100 virtual patients. The estimated AUC and Cmax are within 0.5-2-fold of observation. The sensitivity analysis showed that the decreased plasma exposure of active metabolite due to the decreased CES1 was partly attenuated by the decreased GFR. Conclusion: The developed PBPK model has successfully predicted pharmacokinetics of CES1 substrates and their metabolites in healthy subjects and LC patients, which assists in tailoring dosages of CES1 substrates in LC patients.
Keywords:
carboxylesterase 1
; liver cirrhosis
; physiologically based pharmacokinetic model
; prodrugs
; pharmacokinetics.
1. Introduction
Liver cirrhosis (LC) is widely prevalent worldwide and results from different causes, such as obesity, non-alcoholic fatty liver disease, high alcohol consumption, hepatitis B or C infection, autoimmune diseases, cholestatic diseases, and iron or copper overload[1,2]. The Child-Pugh score is often used to classify liver cirrhosis into Child-Pugh A(CP-A), Child-Pugh B(CP-B) and Child-Pugh C(CP-C) according to the severity of LC[3,4]. In addition to impairment of hepatic functions, LC also leads to remarkable alterations in a series of other physiological parameters such as functional liver volume, hepatic artery blood flow, portal venous blood flow, glomerular filtration rate (GFR), α-acid glycoprotein, albumin content, drug-metabolizing enzymes and transporters. The alterations may directly affect the pharmacokinetics of drugs[5]. For example, Duthaler et al investigated effects of LC on pharmacokinetics of CYP450 cocktail probes caffeine(CYP1A2), efavirenz(CYP2B6), flurbiprofen(CYP2C9), omeprazole(CYP2C19), metoprolol(CYP2D6) and midazolam(CYP3A). They found that liver cirrhosis increased plasma exposure of tested probes, the extent of which is dependent on the type of probes and LC severity. The calculated ratio of AUC in patients to that in control subjects (AUCR)values of caffeine, efavirenz, flurbiprofen, omeprazole, metoprolol and midazolam in CP-C patients were 6.2, 0.8, 1.4, 10.5, 4.5 and 6.3, respectively. The calculated AUCR values of omeprazole in CP-A, CP-B and CP-C patients were separately 4.8, 6.5 and 10.5. The AUCR values of probes in LC patients were in line with those in the contents of hepatic CYP450s[6]. LC also affects renal excretion and intestinal absorption of drugs. Furosemide is mainly eliminated via kidney. It was reported[7] that clearance(CL) of furosemide was significantly decreased from 154 mL/min in control subjects to 91 mL/min in CP-B or CP-C patients, which mainly results from decreases in renal clearance(CLK). These results indicate that adjustment of drug dosage for LC patients is required according to the LC severity. Thus, regulatory agencies have recommended pharmacokinetic studies of drugs in LC patients[8]. However, pharmacokinetic study in LC patients is usually costly and time-consuming. More importantly, it is difficult to recruit into patients, especially patients with CP-C. Physiologically based pharmacokinetic (PBPK) modeling is considered an ideal technique for predicting pharmacokinetics of drugs in patients with altered physiology. The alterations in physiological parameters, expressions of hepatic drug-metabolizing enzymes and transporters under various degree severity of LC have been demonstrated. The possibilities for predicting pharmacokinetics of drugs in LC patients using the PBPK model have been demonstrated[9].
Carboxylesterase1 (CES1) is one of the most abundant drug-metabolizing enzymes in human livers, comprising approximately 1% of the entire liver proteome. CES1 is responsible for 80%-95% of total hydrolytic activity in the liver, which mediates the metabolism of a wide range of drugs, pesticides, environmental pollutants, and endogenous compounds[10]. CES1-mediated metabolism leads to the biotransformation of a pharmacologically active drug into its inactive metabolite, as exemplified by methylphenidate hydrolysis. CES1 also mediates the activation of some prodrugs. The typical examples are some angiotensin-converting enzyme inhibitors (such as enalapril, cilazapril and temocapril) and neuraminidase inhibitors(oseltamivir). CES1 also hydrolyzes cholesteryl ester in lipid metabolism in human macrophages and hepatocytes, inferring that CES1 is a potential drug target for the treatment of metabolic diseases, such as diabetes and atherosclerosis[10,11,12,13]. LC has been demonstrated to significantly downregulate expressions of hepatic CES1 protein[9] and alter plasma exposure of its substrate drugs such as enalapril and oseltamivir[14,15]. Moreover, metabolites of most CES1 substrates (such as enalapril and oseltamivir) are mainly eliminated via renal excretion. LC also injures renal functions, leading to decreases in renal clearance of the metabolites, indicating that alterations in plasma exposure of metabolites by LC are attributed to integrated effects of the decreases in hepatic CES1 activity and renal clearance.
The study aimed to develop a semi-PBPK model incorporating alterations in hepatic CES1 activity, liver/renal functions, gastrointestinal transit rate and relevant organ blood to simultaneously predict pharmacokinetics of nine CES1 drugs(enalapril, benazepril, cilazapril, perindopril, temocapril, oseltamivir, flumazenil, pethidine and remimazolam) and their metabolites in LC patients. The predicted results were compared with clinical studies in patients with different statuses of LC. The results will assist in tailoring dosages of CES1 substrates in LC patients.
2. Materials and Methods
2.1. General Workflow
The workflow for developing a PBPK model (Figure 1) for LC patients. Initially, a PBPK model was developed for a virtual healthy subject population validated using clinical pharmacokinetic studies in healthy subjects. Then, the developed PBPK model was translated to LC patients by replacing the values of system-specific model parameters. Finally, pharmacokinetic predictions were conducted in 100 virtual patient populations (CLint, CLint,K, fu,b, Vsystem, Peff, ka, KL:P, KG:P and KK:P vary from 80%-120% of the parameter values) and compared with clinic pharmacokinetic data from the literature.
2.2. Model Development
A semi-PBPK model was developed to simultaneously predict pharmacokinetics of CES1 substrate drugs and their metabolites in LC patients. The semi-PBPK model consists of stomach, intestinal wall, intestinal lumen, portal vein, liver, kidney and systemic compartment.
It was assumed that no absorption and metabolism of drugs occurs in stomach. The amount(A0) in stomach is controlled by the constant of gastric emptying rate (Kt,0), i,e
Small intestine is divided into duodenum, jejunum and ileum, which is further divided into the gut lumen and the gut wall. Drug amount (Ai) in the ith gut lumen is illustrated by
Where Kt,i represents the constant of intestinal transit rate. ka,i represents the absorption rate constant from the gut lumen to the gut wall, which may be calculated using equation,
Where ri is the intestinal radius. Peff A-B is effective permeability coefficient (Peff) from gut lumen to gut wall. The Peff (×10-4) values were estimated using the in vitro apparent permeability coefficient of drugs (Papp×10-6) in Caco-2 cells based the equation[16]:
The drug concentration in the ith gut wall (CGWi) is expressed as follows:
Where QGWi and KG:P represent the blood flow rate in ith gut wall and ratio of drug concentration in intestinal wall to plasma, respectively. CGWi and VGWi represent separately drug concentration in the ith intestinal and wall volume of the ith gut wall. Csys represent drug concentrations in the systemic compartment. Rb is the ratio of drug concentrations in blood to plasma.
The drug enters the liver through the portal vein and the concentration in the portal vein (CPV) is:
QPV and CPV represent portal vein blood flow rate and volume of portal vein, respectively.
It was assumed that metabolism of CES1-mediated drugs mainly occurs in liver. Drug concentration (CL) in liver is illustrated by
Where QLA and QL represent the hepatic artery blood flow rate to the liver and hepatic blood flow to the systemic compartment, respectively. VL and KL:P represent the volume of liver and ratio of drug concentration in liver to plasma, respectively. CLint and fu,b represent intrinsic clearance in the liver and free fraction of drug in blood, respectively. fu,b is generated from the fraction unbound in plasma (fu,p), i.e
CLint can be estimated using in vitro enzyme kinetics from human hepatic microsomes.
Where Vmax,i and Km,i represent the maximum velocity and Michaelis-Menten constant in vitro enzyme kinetic experiments, respectively.
Hepatic clearance (CLL) of a drug may be deprived from total CL(CLT) and renal CL(CLK), i.e
Thus, The CLint is also recalculated by hepatic blood clearance (CLL,b) using equation
The CL values by clinic are often plasma clearance of drug (CLp), which may be transferred to blood clearance (CLb) using equation 12.
Where Hct is hematocrit, 0.43 in healthy subjects[17].
Some metabolites of some drugs are also eliminated via bile. Amount of metabolites (AL,m) in liver is illustrated by equation 13.
Where CLint,b,m and CLint, m are intrinsic bile clearance and intrinsic metabolic clearance, respectively. If metabolism of the metabolite did not occur in the body, the CLint,b,m may be recalculated from CLK using equations 10 and 11.
Kidney is involved in elimination of drugs, especially their metabolites. Amount of drugs in kidney is illustrated by equation
Where QK and VK represent kidney blood flow and volume of the kidney, respectively. CLint,K and KK,P represent intrinsic clearance in kidney and tissue-to-plasma concentration ratio in the kidney, respectively. CLint,K was also estimated from CLK using equation 11.
Disposition of drugs in the systemic compartment is illustrated using one-compartment, two-compartment model or three-compartment model.
For one-compartment model
Drug concentration (Csys) in systemic compartment
For two-compartment model
Drug concentration (Csys) in systemic compartment.
For three-compartment model
Drug concentration (Csys) in systemic compartment.
Where Vsys represents the apparent distribution volume in systemic compartment. AP and AP1 are the amount of drug in two peripheral compartments. k12, k21, k13 and k31 represent the transfer rates between the systemic compartment and peripheral compartment, respectively.
All available information on anatomical physiological and ADME parameters of the tested drugs was collected for the initial model construction (Table 1 and Table 2). Coding and solving of the PBPK model were conducted on WinNonlin 8.1 (Pharsight, St. Louis, MO, USA). After the initial model was developed, part of plasma concentrations-curves of drugs from healthy subjects were used to estimate and optimize some parameters. Then, the developed PBPK model was validated using plasma concentration-time curves from the rest of clinical studies.
2.3. PBPK model development in LC patients
The anatomical and physiological parameters in healthy subjects were replaced with those (Table 1) in LC patients. The LC-induced alterations in parameters related to ADME were estimated according to their values in healthy(HT) subjects and the altered physiological parameters.
For CES1-mediated hepatic metabolism
Where CLint,CI,CES1 and CLint,HT,CES1 represent the values of CES1-mediated intrinsic clearance in liver of patients and healthy subjects, respectively. fCES1 and fliver represent the ratio of CES1 content in patients to healthy subjects and liver volume in patients to healthy subjects, respectively.
For hepatic elimination of drug mediated by other routes
Where CLint,cirr,other and CLint,heal,other represent the values of intrinsic clearance by other routes in liver of patients and healthy subjects, respectively. fother is ratio of other target content in patients to healthy subjects.
Among the tested drugs, pethidine binds mainly to α1-acid glycoprotein and the rest bind mainly to albumin[49,91,92,93,94,95,96,97] (no data on binding protein for temocapril, so binding to albumin was assumed based on pka < 7.4, acidic). Free fraction of drug in patient plasma was estimated using equation 23[21]:
Where fu,p,CI, fu,p,HT, Pprot,CI and Pprot,HT represent unbound fraction of the drug in plasma of patients and healthy subjects, concentration of drug-bound proteins in plasma of patient and healthy subjects, respectively.
It was assumed that the free apparent volume of distribution of the drug is unaltered, the apparent volume of distribution in cirrhosis patients (Vsys,CI) was derived from the apparent volume of distribution in healthy subjects, i,e.
Liver cirrhosis also impairs renal function and is characterized by decreases in glomerular filtration rate(GFR). The renal intrinsic clearance(CLint, K,CI ) in patients may be estimated using equation[17]:
Where CLint,k,HT, GFRHT and GFRCI represent renal intrinsic clearance in healthy subjects, GFR in healthy subjects and patients, respectively.
Lactulose/Rhamnose ratio is used to assess intestinal permeability[24]. The ratio of cirrhosis patients to healthy subjects was used to correct the absorption rate constant in LC patients.
One hundred virtual populations in healthy subjects, CP-A, CP-B and CP-C patients based on parameters (such as fu,b, CLint,K, CLint, L and ka,i) related to ADME process of drugs were randomly generated for population simulation. Effects of cirrhosis on plasma exposure of the tested drugs were indexed as AUCR or CmaxR
Or
Where AUCCI, AUCHT, CLCI, CLHT, Cmax,CI and Cmax,HT are respectively AUC, CL and Cmax of the tested drugs in cirrhosis patients and healthy subjects.
2.4. Criterion of the developed PBPK model.
The PBPK model was considered to be successful if the simulated AUC or Cmax fell within 0.5- to 2-fold of the observed data or the observed data were within the 5th and 95th percentiles of the simulation derived from 100 virtual subjects.
3. Results
3.1. Collection of data and selection of the tested drugs
Liver cirrhosis obviously alters CES1 enzyme content. The CES1 contents in CP-B patients and CP-C patients were respectively reported to decrease to 70% and 30% of healthy subjects, the CES1 enzyme content in CP-A patients was comparable to that of healthy subjects[9]. LC also leads to alterations in other physiological parameters such as liver volume, hepatic blood flow, renal blood flow, GFR and intestinal transit, which are listed in Table 1.
Clinical pharmacokinetic studies of the CES1 drugs were collected from data published on PubMed based on the following criteria. (1) the tested drug must be metabolized primarily by CES1. (2) pharmacokinetic parameters (such as AUC or plasma drug concentrations) following intravenous (i.v.) and/or oral (p.o.) administration to liver cirrhosis populations must be available. (3) the clinical pharmacokinetic data might come from different reports. Based on these criteria, nine CES1 substrates were included in the simulations. The nine drugs are primarily metabolized by CES1, which included six prodrugs (enalapril, benazepril, cilazapril, perindopril, temocapril and oseltamivir) and three direct-acting drugs (flumazenil, pethidine and remimazolam). Flumazenil and remimazolam are mainly administered by intravenous injection. Pethidine is administrated via intravenous or oral routes. The remaining drugs are administered as oral immediate-release formulations. The parameters related to drugs in the PBPK simulation are listed in Table 2. The detailed information on clinic reports of the tested drugs is illustrated in Table 3.
Enalapril and enalaprilat
Enalapril, an angiotensin-converting enzyme inhibitor (ACEI), is the prodrug, which is mainly metabolized to active product enalaprilat via hepatic CES1[12,98]. Enalaprilat is eliminated primarily through the kidneys[99]. In plasma, enalapril and enalaprilat are mainly bound to albumin, whose free factions in plasma are 0.55 and 0.5[27]. Five clinic reports including two reports involving liver cirrhosis were selected in the simulations.
Benazepril and benazeprilat
Benazepril, a prodrug, is metabolized by hepatic CES1 to the active product benazeprilat[12,98], showing inhibition of angiotensin-converting enzyme. Benazeprilat is eliminated via renal excretion. Benazepril and benazeprilat are mainly bound to albumin, belonging to drugs with high plasma binding, whose free factions in plasma are 0.03 and 0.05[63], respectively. Six clinic reports including one report involving liver cirrhosis were selected in the simulations.
Cilazapril and cilazaprilat
Cilazapril is also metabolized by hepatic CES1 into cilazaprilat[12,98]. Cilazaprilat is mainly eliminated via kidney[66]. Cilazapril and cilazaprilat are mainly bound to albumin, belonging to medium plasma binding, whose free factions in plasma are 0.70 and 0.76[33], respectively. Six clinic reports including one report involving liver cirrhosis were selected in the simulations.
Perindopril and perindoprilat
Prodrug perindopril is mainly metabolized by hepatic CES1 to perindoprilat, showing its inhibition of ACE. The bioavailability of perindopril is 66%[73]. Perindopril is primarily converted to perindoprilat in the liver and other major metabolites of perindopril are perindopril glucuronide and perindopril lactam[100]. Since it is not clear which isoenzyme of UGT metabolizes perindopril to perindopril glucuronide, the change rate of AUC0-inf (0.62) for metoprolol in cirrhosis was used as a variation coefficient of intrinsic clearance for UGT[101]. Perindoprilat is eliminated via renal excretion. Perindopril and perindoprilat are predominantly bound to albumin. Perindopril shows higher plasma binding(percent binding 60%) than that of perindoprilat(mean percent binding 15%)[77]. Four clinic reports including two reports involving liver cirrhosis were selected in the simulations. Cirrhosis in perindopril and perindoprilat have only pharmacokinetic parameters and no specific drug concentration-time profile, so only a comparison of parameters was made.

Table 3.
Clinic information about CES1 substrates in the simulations.
| No | Authors | Drug | Dose (mg) | Analytes | Subjects(n) | Ref |
| 1 | Ohnishi A et al. 1989 | enalapril maleate | 10, p.o | enalapril, enalaprilat | Healthy (7) | [14] |
| enalapril maleate | 10, p.o | enalapril, enalaprilat | CP-C (7) | |||
| 2 | Todd PA et al. 1986 | enalapril maleate | 10, p.o | enalapril, enalaprilat | Healthy(12) | [102] |
| 3 | Weisser K et al.1991 | enalapril maleate | 10, p.o | enalapril, enalaprilat | Healthy (8) | [103] |
| 4 | Dickstein K et al. 1987 | enalapril maleate | 10, p.o | enalapril, enalaprilat | Healthy(10) | [104] |
| 5 | Baba T et al. 1990 | enalapril maleate | 10 , p.o | enalapril, enalaprilat | CP-B (7) | [105] |
| 6 | Kaiser G et al. 1989 | benazepril.HC1 | 10, p.o | benazepril, benazeprilat | Healthy(59) | [106] |
| 7 | Schweizer C et al. 1993 | benazepril.HC1 | 10,p.o | benazepril, benazeprilat | Healthy(11) | [107] |
| 8 | Sioufi A et al.1994 | benazepril.HC1 | 20 , p.o | benazepril, benazeprilat | Healthy(24) | [108] |
| 9 | Waldmeier F et al. 1991 | benazepril.HC1 | 20, p.o | benazepril, benazeprilat | Healthy (4) | [109] |
| 10 | Kaiser G et al. 1990 | benazepril.HC1 | 20, p.o | benazepril, benazeprilat | CP-B(12) | [110] |
| 11 | Macdonald NJ et al.1993 | benazepril HCl | 10 , p.o | benazeprilat | Healthy(18) | [111] |
| 12 | Massarella J et al 1989 | cilazapril | 1.0,2.5,5, p.o | cilazapril, cilazaprilat | Healthy(24) | [65] |
| 13 | Williams PEO et al. 1990 | cilazapril | 2.5,p.o | cilazapril, cilazaprilat | Healthy(13) | [112] |
| 14 | Gross V et al.1993 | cilazapril | 1,p.o | cilazapril, cilazaprilat | Healthy(10) | [113] |
| cilazapril | 1,p.o | cilazapril, cilazaprilat | CP-B(9) | |||
| 15 | Williams PEO et al. 1989 | cilazapril | 1,p.o | cilazapril, cilazaprilat | Healthy(12) | [114] |
| 16 | Massarella JW et al. 1989 | cilazapril | 5.p.o | cilazapril, cilazaprilat | Healthy(16) | [115] |
| 17 | Francis RJ et al. 1987 | cilazapril | 1.25,2.5,5,10,p.o | cilazaprilat | Healthy(12) | [116] |
| 18 | Lecocq B et al. 1990 | perindoprila | 4,p.o | perindopril, perindoprilat | Healthy(12) | [117] |
| 19 | Tsai HH et al. 1989 | perindoprila | 8,p.o | perindopril, perindoprilat | CP-A(8) | [118] |
| 20 | Thiollet M et al. 1992 | perindoprila | 8,p.o | perindopril, perindoprilat | CP-B(10) | [119] |
| 21 | Lees KR et al. 1988 | perindoprila | 8,p.o | perindoprilat | Healthy(8) | [120] |
| 22 | Furuta S et al. 1993 | temocapril HCL | 1.p.o | temocapril, temocaprilat | Healthy(6) | [121] |
| temocapril HCL | 1,p.o | temocapril, temocaprilat | CP-C(7) | |||
| 23 | Abe M et al. 2006 | oseltamivirb | 75,p.o | oseltamivir,OC | Healthy(7) | [122] |
| 24 | Brewster M et al. 2006 | oseltamivirb | 75,p.o | oseltamivir,OC | Healthy(18) | [123] |
| 25 | Jittamala P et al. 2014 | oseltamivirb | 75,p.o | oseltamivir,OC | Healthy(12) | [124] |
| oseltamivirb | 150,p.o | oseltamivir,OC | Healthy(12) | |||
| 26 | Snell P et al. 2005 | oseltamivirb | 75, p.o | oseltamivir,OC | CP-B(11) | [15] |
| 27 | Amrei R et al. 1990 | flumazenil | 10mg,i.v. | flumazenil | Healthy(NA) | [125] |
| 28 | Breimer LTM et al. 1991 | flumazenil | 10/10 min,iv | flumazenil | Healthy(7) | [126] |
| 29 | Pomier-Layrargues G et al. 1989 | flumazenil | 2/5min,iv | flumazenil | CP-B(8) | [127] |
| flumazenil | 2/5min,iv | flumazenil | CP-C(8) | |||
| 30 | Klotz U, et al.1984 | flumazenil | 2.5,i.v | flumazenil | Healthy(6) | [82] |
| 31 | Janssen U,et al.1989 | flumazenil | 30 p.o | flumazenil | Healthy(8) | [128] |
| flumazenil | 2,i.v; 30 p.o | flumazenil | CP-C(8) | |||
| 32 | Verbeeck RK et al.1981 | pethidine HCL | 25,i.v | pethidine | Healthy(6) | [129] |
| pethidine HCL | 25,p.o | pethidine | Healthy(6) | |||
| 33 | Mather LE et al. 1975 | pethidine HCL | 50,i.v | pethidine | Healthy(4) | [130] |
| 34 | Kuhnert BR et al. 1980 | pethidine HCL | 50,i.v | pethidine | Healthy(7) | [131] |
| 35 | Guay DR et al. 1984 | pethidine HCL | 70,i.v | pethidine | Healthy(8) | [132] |
| 36 | Guay DR et al. 1985 | pethidine HCL | 70,i.v | pethidine | Healthy(8) | [133] |
| 37 | Pond SM et al. 1981 | pethidine HCL | 60, iv;112, po | pethidine | CP-A (5) | [134] |
| 38 | Pond SM et al. 1980 | pethidine HCL | 54.4, iv;108.8, po | pethidine | CP-B (4) | [135] |
| 39 | Mather LE et al. 1976 | pethidine HCL | 50, iv;100, po | pethidine | Healthy(4) | [136] |
| 40 | Klotz U et al. 1974 | pethidine HCL | 63.9,i.v | pethidine | Healthy(8) | [137] |
| pethidine HCL | 53.1,i.v | pethidine | CP-A(10) | |||
| 41 | Neal EA et al. 1979 | pethidine HCL | 56, iv; 56, po | pethidine | Healthy(4) | [138] |
| pethidine HCL | 56, iv; 56, po | pethidine | CP-A(8) | |||
| 42 | Sheng XY et al. 2020 | remimazolam besylate | 1.5425,3.315,i.v | remimazolam | Healthy(3) | [80] |
| remimazolam besylate | 4.8675,6.18,i.v | remimazolam | Healthy(7) | |||
| remimazolam besylate | 13.26,24.6,i.v | remimazolam | Healthy(8) | |||
| remimazolam besylate | 18.3,i.v | remimazolam | Healthy(10) | |||
| 43 | Stohr T et al. 2021 | remimazolam besylate | 10.4,i.v | remimazolam | CP-B(8) | [139] |
| remimazolam besylate | 8.2,i.v | remimazolam | CP-C(3) |
a, perindopril tert-butylamine; b, oseltamivir phosphate.
Temocapril and temocaprilat
Temocapril is also a prodrug and metabolized by hepatic CES1 to temocaprilat. Temocaprilat is eliminated via both bile and kidney. The biliary clearance of temocaprilat was about 2-fold of renal clearance[74]. The CLint,K of temocaprilat was calculated to be 949.84 mL/min[73]. Thus, CLbile,m of temocaprilat was estimated to be 1899.68 mL/min, assuming that the ratio of CLbile,m to CLint,K was 2.0. Biliary excretion of temocaprilat is considered to be mediated by multidrug resistance-associated protein2(MRP2)[140]. One clinic report involving both liver cirrhosis patients and healthy subjects was selected in the simulations.
Oseltamivir and oseltamivir carboxylate
Oseltamivir, a prodrug, is metabolized via hepatic CES1[12,98] to its active metabolite oseltamivir carboxylate (OC) which has an antiviral effect. About 80% of an orally administered dose of oseltamivir reaches the systemic circulation as the active metabolite. The absolute bioavailability of the active metabolite from orally administered oseltamivir is 75%[141]. About 60 to 70% of an oral oseltamivir dose appears in urine as the active metabolite, and less than 5% as oseltamivir. Oseltamivir carboxylate is primarily eliminated via renal excretion, accounting for 93% of intravenous dose[57]. CLint,K values of both oseltamivir and oseltamivir carboxylate exceed GFR, indicating that renal elimination occurs via a combination of glomerular filtration and renal tubular secretion. Both oseltamivir and oseltamivir carboxylate are primarily bound to albumin; their bound fractions in plasma were approximately 42% and less than 3%[28]. Four clinic reports including one report involving liver cirrhosis were selected in the simulations.
Flumazenil
Flumazenil, a benzodiazepine receptor antagonist, is usually administered by intravenous injection[84]. Flumazenil was inactivated by hepatic CES1 to flumazenil acid and probably by CYP450 catalyzed N-dealkylation to N-demethylated flumazenil[142]. Flumazenil was predominantly bound to serum albumin, whose plasma protein binding is about 40%[86]. Five clinic reports including two reports involving liver cirrhosis were selected in the simulations.
Pethidine
Pethidine(meperidine) is a synthetic opioid commonly used for analgesia in humans. Pethidine is metabolized in the body by two different pathways[49,98]. The predominant pathway is hepatic CES1 metabolism to pethidinic acid, an inactive metabolite. Another pathway is N-demethylation by CYP2B6 to normeperidine, a nonopioid active metabolite. Oral bioavailability of pethidine varies from 48% -56%[143]. Pethidine was predominantly bound to α1-acid glycoprotein. In the simulation for healthy subjects, free fraction of pethidine in plasma was 0.418[49]. Ten clinic reports including four reports involving liver cirrhosis were selected in the simulations.
Remimazolam
Remimazolam, as an ultrashort-acting sedative agent, is metabolized by hepatic CES1 to inactive carboxy acid metabolite. The plasma protein binding of remimazolam is approximately 92%, predominantly serum albumin[81]. In the clinic, remimazolam is normally administered intravenously. Two clinic reports including one report involving liver cirrhosis were selected in the simulations.
3.2. Development of PBPK model and validation using pharmacokinetic parameters from healthy subjects following i.v. or oral administrations
Plasma concentration-time profiles of the tested CES1 substrates and their active metabolites following i.v. or oral administration to healthy subjects were simulated using the developed PBPK model and compared with clinic observations. The results showed that most of the observed data of the tested agents fell within the 5th and 95th percentiles of the simulated data (Figure 2 and Figure S1). The corresponding pharmacokinetic parameters AUC, CL and Cmax were estimated using the mean of the simulated profiles derived from 100 virtual individuals and compared with clinic observations (Table S1-S9). Most of the simulated pharmacokinetic parameters (AUC, CL and Cmax) values for all drugs were also within two-fold of observations (Table S1-S9 and Figure 3). All the results demonstrated that the PBPK model was successfully developed.
3.3. Prediction of pharmacokinetic profiles for CES1 substrates and their active metabolites following i.v. or oral administration to LC patients using the developed PBPK model
The developed PBPK model, following validation in healthy subjects, was used to predict pharmacokinetic profiles of the selected CES1 substrates and their active metabolites following intravenous or oral administration to 100 virtual LC patients (Figure 3) and their pharmacokinetic parameters were estimated using the mean pharmacokinetic profile derived from 100 simulations (Table S1-S9). The results showed that except oral pethidine, the majority of the drug concentrations in LC patients were well within the 5% and 95% percentiles of pharmacokinetic profiles derived from 100 virtual LC patients. Most of the estimated pharmacokinetic parameters were also within 0.5-2.0-fold of observations (Figure 3), indicating that alterations in pharmacokinetic behaviors of CES1 substrates and their metabolites in LC patients may be predicted using the developed PBPK model.
Extents of pharmacokinetic parameters under liver cirrhosis, AUCR and CmaxR were also predicted using the estimated pharmacokinetic parameters (Figure 4 and Figure 5). AUC or Cmax values may come from different clinic reports or different doses, thus, the AUC or Cmax values were normalized by dose and their mean values were used for estimating AUCR or CmaxR. The results showed that the vast majority of the ratios of predicted AUCR and CmaxR are close to observed values, with only a few individual values differing significantly, indicating a good prediction. All these show that the PBPK model successfully predicted the pharmacokinetics of the drug in cirrhosis.
3.4. Sensitivity analysis of model parameters
Plasma concentration-time curve of enalapril and enalaprilat following oral dose(10mg) was exampled for pharmacokinetic sensitivity. Some parameters such as gastrointestinal motility rate (Kt), intestinal absorption (Peff), hepatic arterial blood flow rates (QLA), portal vein blood flow rates (QPV), hepatic CES1 activity (CLint,L), kidney blood flow rates (QK), GFR, fu,b and fu,b,m (free fraction of metabolites in blood) may affects pharmacokinetics of drugs, which were selected for sensitivity analysis. Variations of Kt, Peff, QLA, QPV, CLint,L and QK were 1/3, 1 and 3-fold. Variation of GFR was 0.5, 1 and 1.5-fold. Variation of fu,b was 0.74, 1 and 1.35-fold for enalapril and fu,b,m was 0.68, 1 and 1.47-fold for enalaprilat. The results (Figure 6) show that these tested parameters affect the pharmacokinetics profile of drugs in varying degrees, their contributions to AUC of enalapril were Peff>Kt>CLint,L>QPV>fu,b>GFR>QK>QLA, and to enalaprilat were Peff>Kt>GFR> CLint,L>QPV>fu,b,m>QK>QLA. In addition to impairment of liver failure, LC patients were associated with increases in intestinal transit rates, intestinal permeability of drugs, QLA and fu,b (due to decreases in plasma binding protein levels), decreases in GFR, QK, CES1 activity and QPV, although increases in QL were reported in CP-C patients. The contributions of LC-induced alterations in Kt, QPV, CLint, L, Peff, GFR, QK and fu,b to plasma concentrations of enalapril and enalaprilat following an oral dose of enalapril maleate(10 mg) to CP-C patients and their integrated effects were also simulated. The results showed that decreases in CLint,L and increases Peff of enalapril increased plasma concentrations of enalapril while the increases in fu,b, Kt and decreases in QPV obviously decreased plasma concentrations of enalapril following an oral dose of enalapril maleate, the net effects were to increase plasma concentrations of enalapril. For enalaprilat, increases in Peff and decreases in GFR, QK and QPV significantly increased plasma concentration profiles of enalaprilat, while decreases in CES1 activity and increases in Kt and fu,b,m of enalaprilat significantly decreased plasma concentrations following oral enalapril maleate. Their net effects were to decrease plasma concentrations of enalaprilat (Figure 6Q and Figure 6R).
4. Discussion
Hepatic CES1 mediates the inactivation of direct-acting drugs or activation of some prodrugs, most of whose active metabolites are mainly eliminated via kidney. In addition to impairment of liver function, LC patients are also associated with alterations in organ blood flow, decreases in plasma protein levels, increases in intestinal permeability of drugs and impairment of renal functions, commonly affecting pharmacokinetics of CES1 substrate drugs and their metabolites. The main contributions of the study were to successfully develop a semi-PBPK model involving intestinal absorption, hepatic metabolism and renal excretions to simultaneously predict pharmacokinetic profiles of nine CES1 substrates (six prodrugs and three direct-acting drugs) in both healthy subjects and LC patients. Most clinic observations were within 5%-95% quantiles of simulations derived from 100 virtual subjects. Most of the estimated AUC and Cmax were also within 0.5-2.0-fold of observations.
The extent of LC-induced alterations in plasma exposure of CES1 substrates and their metabolites were also assessed using AUCR and CmaxR. It was found that although most of the clinically observed plasma concentrations for the tested agents were within 5%-95% quantiles of simulations, poor predicted AUCR or CmaxR were found in benazepril, temocaprilat, perindopril and perindoprilat. The predicted AUCR values of flumazenil and pethidine were less than the clinic observations. Benazepril and temocaprilat belong to highly bound compounds, whose fu,b values were 0.03 and 0.025, respectively. In general, obtaining an accurate plasma binding measurement for highly bound compounds is difficult[144]. In addition to CES1, UGTs also mediate perindopril metabolism[100]. Which isoenzyme of UGT was involved in metabolism of perindopril was not identified. In the simulation, it was assumed that LC-induced alterations in CLint, UGT of perindopril were similar to that of metoprolol[101]. LC patients by different etiologies show different amounts of hepatic CES1. In addition to CES1, other enzymes also mediate metabolism of flumazenil[142]. Pethidine is co-metabolized by CES1 and CYP2B6[49,98]. Several reports have demonstrated extensive interindividual variability in the expression of CYP2B6[145] and CES1[98]. All may be reasons leading to these differences between the predicted and the observed AUCR values, which need further investigation.
In general, LC-induced impairments of hepatic CES1 activity increase plasma exposure of CES1 substrates, but sensitivity analysis showed that the increases in plasma concentrations of CES1 substrates under LC patients were only partly attributed to impairment of hepatic CES1. The increases in intestinal permeability of drugs were also observed in LC patients, contributing to increased plasma exposures of CES1 substrates. In contrast, LC-induced increases in intestinal transit rate and decreases in plasma binding protein and QPV obviously decreased plasma exposure of CES1 substrates, which partly attenuated the increases in plasma exposures of CES1 substrates by liver cirrhosis. Metabolites of the tested CES1 substrates are eliminated via kidney. The decreases in plasma exposure of metabolites induced by impairment of hepatic CES1 activity were also partly attenuated by LC-induced alterations in GFR and QK. Even under some conditions, levels of the metabolites are increased rather than decreased due to impairment of renal function. For example, AUC values of perindoprilat in CP-A and CP-B patients were obviously higher than those in healthy humans, the observed AUCR values were 2.89 and 1.2, respectively, which were near to predictions (1.97 in CP-A patients and 2.03 in CP-B patients). These findings may partly explain clinical findings that although liver cirrhosis obviously increases plasma levels of enalapril and perindopril, but the magnitude of serum ACE lowering effects by the two drugs was fairly comparable between LC patients and healthy humans[14,118,119].
Plasma levels of the direct-acting drugs flumazenil, pethidine and remimazolam following administration to LC patients were also successfully simulated. Although the observed AUCR values of remimazolam in LC patients could not be calculated due to lack of the observed pharmacokinetic parameters in LC patients, it was contrasted to our expectation that the predicted AUCR values in CP-B patients and CP-C patients were 0.77 and 0.62, which may be explained by the fact that the increased plasma concentration by impairment of hepatic CES1 may be attenuated by increases in hepatic arterial blood flow and increases in fu,b (Figure S2).
However, the study also has some limitations. The predictions for healthy subjects were based on “ideal” healthy subjects (body weight assumed to be 70 kg) without considering gender, body weight, race and gene variance of CES1. Genetic variation in CES1 also affects pharmacokinetics of the CES1 substrates[98]. During the simulation in LC patients, LC patients were considered “ileal” CP-A, CP-B or CP-C patients without considering LC etiology, gender and race. It was reported that the amount of CES1 protein in hepatitis C cirrhotic patients was about 1.47-fold of the alcoholic cirrhosis[146]. Similarly, it was reported that flumazenil might improve memory in alcoholic cirrhotic patients but not in nonalcoholic cirrhotic patients[147].
5. Conclusions
The developed PBPK model may successfully be applied simultaneously to predict the pharmacokinetics of CES1 substrate drugs and their active metabolites in healthy subjects and LC patients.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Observed and predicted values of AUC0-t and Cmax of enalapril and enalaprilat following oral enalapril maleate to healthy (HT) subjects and liver cirrhosis patients. Table S2: Observed and predicted values of AUC0-t and Cmax of benazepril and benazepril following benazepril hydrochloric to healthy(HT) subjects and cirrhosis. Table S3: Observed and predicted values of AUC0-t and Cmax of cilazapril following oral cilazapril to healthy(HT) subjects and LC patients. Table S4: Observed and predicted values of AUC0-t and Cmax of perindopril following oral perindopril tert-butylamine to healthy(HT) subjects and LC patients. Table S5: Observed and predicted values of AUC0-t and Cmax of temocapril and temocaprilat following oral temocapril hydrochloride to healthy (HT) subjects and LC patients. Table S6: Observed and predicted values of AUC0-t and Cmax of oseltamivir and oseltamivir carboxylate (OC) following oral oseltamivir phosphate to healthy (HT) subjects and cirrhosis. Table S7: Observed and predicted values of AUC0-t (μg×h/mL) or CL(L/min) and Cmax(ng/mL) of flumazenil to healthy(HT) subjects and LC patients. Table S8: Observed and predicted values of AUC0-t(μg×h/mL) or CL(L/min) and Cmax(ng/mL) of pethidine following oral and intravenous pethidine HCL to healthy (HT) subjects and LC patients. Table S9: Observed and predicted values of AUC0-t of remimazolam following intravenous remimazolam besylate to healthy (HT) subjects and LC patients. Figure S1: The observed(points) and predicted(lines) plasma concentrations of the tested CES1 substrates and their active metabolites following intravenous or oral administration to healthy subjects. Benazepril(A) and benazeprilat(B) following oral 10 mg benazepril hydrochloride; cilazapril(D,F) and cilazaprilat(C,E,G,H) following oral 1.25, 2.5, 5, 10 mg cilazapril; oseltamivir(I) and oseltamivir carboxylate(J) following oral 150 mg oseltamivir phosphate; flumazenil following intravenous 10 mg/1 min(K) and 10mg/10min(L); pethidine following intravenous 50 mg/1min(M), 25mg/1min(N), 0.8mg/kg,1min(O) and 0.8mg/kg,5min(P) pethidine hydrochloride and oral 25 mg(Q), 0.8mg/kg(R) pethidine hydrochloride; remimazolam following intravenous 0.05(S), 0.075(T), 0.2(U), 0.3(V), 0.4(W)mg/kg remimazolam besylate. Shaded areas indicate the 5% and 95% quantile of simulations derived from 100 virtual individuals. The dashed lines indicate the mean of the simulated profiles. Figure S2: Contributions of alterations in fu,b, CES1 activity, QLA and QPV by LC to plasma concentrations of remimazolam following 10.4mg(CP-B, A) and 8.2mg(CP-C, B) to healthy human and LC patients.
Author Contributions
Conceptualization, X.L. and Z.Z.; methodology, X.L. and Z.Z.; validation, X.L., R.M., and G.H.; formal analysis, X.L. and Z.Z.; investigation, X.L. and G.H.; resources, X.L., R.M. and G.H.; data curation, X.L. and R.M.; writing—original draft preparation, X.L.; writing—review and editing, L.L. and XD.L.; supervision, L.L. and XD.L.; project administration, L.L. and XD.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Natural Science Foundation of China (No. 82073922 and 82173844) and the “Double First-Class” university project (No. CPU2022QZ21).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 82073922 and 82173844) and the “Double First-Class” university project (No. CPU2022QZ21). The authors wish to thank Xiaodong Liu and Li Liu for their helpful advice in writing the English manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gines, P.; Krag, A.; Abraldes, J.G.; Sola, E.; Fabrellas, N.; Kamath, P.S. Liver cirrhosis. Lancet 2021, 398, 1359–1376. [Google Scholar] [CrossRef]
- Lee, N.Y.; Suk, K.T. The Role of the Gut Microbiome in Liver Cirrhosis Treatment. Int J Mol Sci 2020, 22. [Google Scholar] [CrossRef]
- El-Khateeb, E.; Darwich, A.S.; Achour, B.; Athwal, V.; Rostami-Hodjegan, A. Review article: time to revisit Child-Pugh score as the basis for predicting drug clearance in hepatic impairment. Aliment Pharmacol Ther 2021, 54, 388–401. [Google Scholar] [CrossRef]
- Pugh, R.N.; Murray-Lyon, I.M.; Dawson, J.L.; Pietroni, M.C.; Williams, R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg 1973, 60, 646–649. [Google Scholar] [CrossRef]
- Weersink, R.A.; Burger, D.M.; Hayward, K.L.; Taxis, K.; Drenth, J.P.H.; Borgsteede, S.D. Safe use of medication in patients with cirrhosis: pharmacokinetic and pharmacodynamic considerations. Expert Opin Drug Metab Toxicol 2020, 16, 45–57. [Google Scholar] [CrossRef]
- Duthaler, U.; Bachmann, F.; Suenderhauf, C.; Grandinetti, T.; Pfefferkorn, F.; Haschke, M.; Hruz, P.; Bouitbir, J.; Krahenbuhl, S. Liver Cirrhosis Affects the Pharmacokinetics of the Six Substrates of the Basel Phenotyping Cocktail Differently. Clin Pharmacokinet 2022, 61, 1039–1055. [Google Scholar] [CrossRef]
- Villeneuve, J.P.; Verbeeck, R.K.; Wilkinson, G.R.; Branch, R.A. Furosemide kinetics and dynamics in patients with cirrhosis. Clin Pharmacol Ther 1986, 40, 14–20. [Google Scholar] [CrossRef]
- Thakkar, N.; Slizgi, J.R.; Brouwer, K.L.R. Effect of Liver Disease on Hepatic Transporter Expression and Function. J Pharm Sci 2017, 106, 2282–2294. [Google Scholar] [CrossRef]
- Chen, Y.; Ke, M.; Xu, J.; Lin, C. Simulation of the Pharmacokinetics of Oseltamivir and Its Active Metabolite in Normal Populations and Patients with Hepatic Cirrhosis Using Physiologically Based Pharmacokinetic Modeling. AAPS PharmSciTech 2020, 21, 98. [Google Scholar] [CrossRef]
- Her, L.; Zhu, H.J. Carboxylesterase 1 and Precision Pharmacotherapy: Pharmacogenetics and Nongenetic Regulators. Drug Metab Dispos 2020, 48, 230–244. [Google Scholar] [CrossRef]
- Hosokawa, M. Structure and catalytic properties of carboxylesterase isozymes involved in metabolic activation of prodrugs. Molecules 2008, 13, 412–431. [Google Scholar] [CrossRef]
- Laizure, S.C.; Herring, V.; Hu, Z.; Witbrodt, K.; Parker, R.B. The role of human carboxylesterases in drug metabolism: have we overlooked their importance? Pharmacotherapy 2013, 33, 210–222. [Google Scholar] [CrossRef]
- Ross, M.K.; Streit, T.M.; Herring, K.L. Carboxylesterases: Dual roles in lipid and pesticide metabolism. J Pestic Sci 2010, 35, 257–264. [Google Scholar] [CrossRef]
- Ohnishi, A.; Tsuboi, Y.; Ishizaki, T.; Kubota, K.; Ohno, T.; Yoshida, H.; Kanezaki, A.; Tanaka, T. Kinetics and dynamics of enalapril in patients with liver cirrhosis. Clin Pharmacol Ther 1989, 45, 657–665. [Google Scholar] [CrossRef]
- Snell, P.; Dave, N.; Wilson, K.; Rowell, L.; Weil, A.; Galitz, L.; Robson, R. Lack of effect of moderate hepatic impairment on the pharmacokinetics of oral oseltamivir and its metabolite oseltamivir carboxylate. Br J Clin Pharmacol 2005, 59, 598–601. [Google Scholar] [CrossRef]
- Qian, C.Q.; Zhao, K.J.; Chen, Y.; Liu, L.; Liu, X.D. Simultaneously predict pharmacokinetic interaction of rifampicin with oral versus intravenous substrates of cytochrome P450 3A/P-glycoprotein to healthy human using a semi-physiologically based pharmacokinetic model involving both enzyme and transporter turnover. Eur J Pharm Sci 2019, 134, 194–204. [Google Scholar] [CrossRef]
- Edginton, A.N.; Willmann, S. Physiology-based simulations of a pathological condition: prediction of pharmacokinetics in patients with liver cirrhosis. Clin Pharmacokinet 2008, 47, 743–752. [Google Scholar] [CrossRef]
- Guo, H.; Liu, C.; Li, J.; Zhang, M.; Hu, M.; Xu, P.; Liu, L.; Liu, X. A mechanistic physiologically based pharmacokinetic-enzyme turnover model involving both intestine and liver to predict CYP3A induction-mediated drug-drug interactions. J Pharm Sci 2013, 102, 2819–2836. [Google Scholar] [CrossRef]
- Davies, B.; Morris, T. Physiological parameters in laboratory animals and humans. Pharm Res 1993, 10, 1093–1095. [Google Scholar] [CrossRef]
- Li, R.; Barton, H.A.; Maurer, T.S. A Mechanistic Pharmacokinetic Model for Liver Transporter Substrates Under Liver Cirrhosis Conditions. CPT Pharmacometrics Syst Pharmacol 2015, 4, 338–349. [Google Scholar] [CrossRef]
- Johnson, T.N.; Boussery, K.; Rowland-Yeo, K.; Tucker, G.T.; Rostami-Hodjegan, A. A semi-mechanistic model to predict the effects of liver cirrhosis on drug clearance. Clin Pharmacokinet 2010, 49, 189–206. [Google Scholar] [CrossRef]
- Karlsen, S.; Fynne, L.; Gronbaek, H.; Krogh, K. Small intestinal transit in patients with liver cirrhosis and portal hypertension: a descriptive study. BMC Gastroenterol 2012, 12, 176. [Google Scholar] [CrossRef]
- Rodriquez, A.; Martin, A.; Oterino, J.A.; Blanco, I.; Jimenez, M.; Perez, A.; Novoa, J.M. Renal function in compensated hepatic cirrhosis: effects of an amino acid infusion and relationship with nitric acid. Dig Dis 1999, 17, 235–240. [Google Scholar] [CrossRef]
- Zuckerman, M.J.; Menzies, I.S.; Ho, H.; Gregory, G.G.; Casner, N.A.; Crane, R.S.; Hernandez, J.A. Assessment of intestinal permeability and absorption in cirrhotic patients with ascites using combined sugar probes. Dig Dis Sci 2004, 49, 621–626. [Google Scholar] [CrossRef]
- Jacobsen, A.C.; Nielsen, S.; Brandl, M.; Bauer-Brandl, A. Drug Permeability Profiling Using the Novel Permeapad(R) 96-Well Plate. Pharm Res 2020, 37, 93. [Google Scholar] [CrossRef]
- Shin, B.S.; Yoon, C.H.; Balthasar, J.P.; Choi, B.Y.; Hong, S.H.; Kim, H.J.; Lee, J.B.; Hwang, S.W.; Yoo, S.D. Prediction of drug bioavailability in humans using immobilized artificial membrane phosphatidylcholine column chromatography and in vitro hepatic metabolic clearance. Biomed Chromatogr 2009, 23, 764–769. [Google Scholar] [CrossRef]
- Claassen, K.; Willmann, S.; Eissing, T.; Preusser, T.; Block, M. A detailed physiologically based model to simulate the pharmacokinetics and hormonal pharmacodynamics of enalapril on the circulating endocrine Renin-Angiotensin-aldosterone system. Front Physiol 2013, 4, 4. [Google Scholar] [CrossRef]
- Jogiraju, V.K.; Avvari, S.; Gollen, R.; Taft, D.R. Application of physiologically based pharmacokinetic modeling to predict drug disposition in pregnant populations. Biopharm Drug Dispos 2017, 38, 426–438. [Google Scholar] [CrossRef]
- Remko, M. Acidity, lipophilicity, solubility, absorption, and polar surface area of some ACE inhibitors. Chemical Papers 2007, 61. [Google Scholar] [CrossRef]
- Nishimuta, H.; Houston, J.B.; Galetin, A. Hepatic, intestinal, renal, and plasma hydrolysis of prodrugs in human, cynomolgus monkey, dog, and rat: implications for in vitro-in vivo extrapolation of clearance of prodrugs. Drug Metab Dispos 2014, 42, 1522–1531. [Google Scholar] [CrossRef]
- Sun, J.X.; Cipriano, A.; Chan, K.; John, V.A. Pharmacokinetic interaction study between benazepril and amlodipine in healthy subjects. Eur J Clin Pharmacol 1994, 47, 285–289. [Google Scholar] [CrossRef]
- Navia, M.; Chaturvedi, P. Design principles for orally bioavailable drugs. Drug Discovery Today 1996, 1, 179–189. [Google Scholar] [CrossRef]
- Wu, L.P.; Cui, Y.; Xiong, M.J.; Wang, S.R.; Chen, C.; Ye, L.M. Mixed micellar liquid chromatography methods: modelling quantitative retention-activity relationships of angiotensin converting enzyme inhibitors. Biomed Chromatogr 2008, 22, 1243–1251. [Google Scholar] [CrossRef]
- Sugihara, M.; Takeuchi, S.; Sugita, M.; Higaki, K.; Kataoka, M.; Yamashita, S. Analysis of Intra- and Intersubject Variability in Oral Drug Absorption in Human Bioequivalence Studies of 113 Generic Products. Mol Pharm 2015, 12, 4405–4413. [Google Scholar] [CrossRef]
- Kitagawa, S.; Takeda, J.; Sato, S. pH-dependent inhibitory effects of angiotensin-converting enzyme inhibitors on cefroxadine uptake by rabbit small intestinal brush-border membrane vesicles and their relationship with hydrophobicity and the ratio of zwitterionic species. Biol Pharm Bull 1999, 22, 721–724. [Google Scholar] [CrossRef]
- Ohura, K. [Evaluation of the Oral Absorption of Ester-type Prodrugs]. Yakugaku Zasshi 2020, 140, 369–376. [Google Scholar] [CrossRef]
- Vistoli, G.; Pedretti, A.; Testa, B. Chemodiversity and molecular plasticity: recognition processes as explored by property spaces. Future Med Chem 2011, 3, 995–1010. [Google Scholar] [CrossRef]
- Shitara, Y.; Maeda, K.; Ikejiri, K.; Yoshida, K.; Horie, T.; Sugiyama, Y. Clinical significance of organic anion transporting polypeptides (OATPs) in drug disposition: their roles in hepatic clearance and intestinal absorption. Biopharm Drug Dispos 2013, 34, 45–78. [Google Scholar] [CrossRef]
- Helal, F.; Lane, M.E. Transdermal delivery of Angiotensin Converting Enzyme inhibitors. Eur J Pharm Biopharm 2014, 88, 1–7. [Google Scholar] [CrossRef]
- Ono, A.; Tomono, T.; Ogihara, T.; Terada, K.; Sugano, K. Investigation of biopharmaceutical drug properties suitable for orally disintegrating tablets. ADMET and DMPK 2016, 4. [Google Scholar] [CrossRef]
- Sun, H. Capture hydrolysis signals in the microsomal stability assay: molecular mechanisms of the alkyl ester drug and prodrug metabolism. Bioorg Med Chem Lett 2012, 22, 989–995. [Google Scholar] [CrossRef]
- Hurst, M.; Jarvis, B. Perindopril: an updated review of its use in hypertension. Drugs 2001, 61, 867–896. [Google Scholar] [CrossRef]
- Zhou, J.; Curd, L.; Lohmer, L.L.; Ossig, J.; Schippers, F.; Stoehr, T.; Schmith, V. Population Pharmacokinetics of Remimazolam in Procedural Sedation With Nonhomogeneously Mixed Arterial and Venous Concentrations. Clin Transl Sci 2021, 14, 326–334. [Google Scholar] [CrossRef]
- Zhu, C.; Jiang, L.; Chen, T.M.; Hwang, K.K. A comparative study of artificial membrane permeability assay for high throughput profiling of drug absorption potential. Eur J Med Chem 2002, 37, 399–407. [Google Scholar] [CrossRef]
- Gottipati, G. Prediction of human systemic, biologically relevant pharmacokinetic (PK) properties using quantitative structure pharmacokinetic relationships (QSPKR) and interspecies pharmacokinetic allometric scaling (PK-AS) approaches for four different pharmacological classes of compounds. Doctor, Virginia Commonwealth University, Virginia, 2014.
- Ellison, C.A. Structural and functional pharmacokinetic analogs for physiologically based pharmacokinetic (PBPK) model evaluation. Regul Toxicol Pharmacol 2018, 99, 61–77. [Google Scholar] [CrossRef]
- Ghafourian, T.; Barzegar-Jalali, M.; Hakimiha, N.; Cronin, M.T. Quantitative structure-pharmacokinetic relationship modelling: apparent volume of distribution. J Pharm Pharmacol 2004, 56, 339–350. [Google Scholar] [CrossRef]
- Luttrell, W.E.; Castle, M.C. Species differences in the hydrolysis of meperidine and its inhibition by organophosphate compounds. Fundam Appl Toxicol 1988, 11, 323–332. [Google Scholar] [CrossRef]
- Alsmadi, M.M.; Idkaidek, N. The Analysis of Pethidine Pharmacokinetics in Newborn Saliva, Plasma, and Brain Extracellular Fluid After Prenatal Intrauterine Exposure from Pregnant Mothers Receiving Intramuscular Dose Using PBPK Modeling. Eur J Drug Metab Pharmacokinet 2023, 48, 281–300. [Google Scholar] [CrossRef]
- Holford, N.H.G. Basic Principles. In Basic & Clinical Pharmacology, Twelfth ed.; Katzung, B.G., Masters, S.B., Trevor, A.J., Eds.; McGraw·Hill: 2012; p. 39.
- Dahlgren, D.; Roos, C.; Sjogren, E.; Lennernas, H. Direct In Vivo Human Intestinal Permeability (Peff ) Determined with Different Clinical Perfusion and Intubation Methods. J Pharm Sci 2015, 104, 2702–2726. [Google Scholar] [CrossRef]
- Tarkiainen, E.K.; Tornio, A.; Holmberg, M.T.; Launiainen, T.; Neuvonen, P.J.; Backman, J.T.; Niemi, M. Effect of carboxylesterase 1 c.428G > A single nucleotide variation on the pharmacokinetics of quinapril and enalapril. Br J Clin Pharmacol 2015, 80, 1131–1138. [Google Scholar] [CrossRef]
- Gangnus, T.; Burckhardt, B.B.; consortium, C. Low-volume LC-MS/MS method for the pharmacokinetic investigation of carvedilol, enalapril and their metabolites in whole blood and plasma: Application to a paediatric clinical trial. Drug Test Anal 2021, 13, 694–708. [Google Scholar] [CrossRef]
- Faisal, M.; Cawello, W.; Burckhardt, B.B.; de Hoon, J.; Laer, S.; Consortium, L. Simultaneous Semi-Mechanistic Population Pharmacokinetic Modeling Analysis of Enalapril and Enalaprilat Serum and Urine Concentrations From Child Appropriate Orodispersible Minitablets. Front Pediatr 2019, 7, 281. [Google Scholar] [CrossRef]
- Hockings, N.; Ajayi, A.A.; Reid, J.L. Age and the pharmacokinetics of angiotensin converting enzyme inhibitors enalapril and enalaprilat. Br J Clin Pharmacol 1986, 21, 341–348. [Google Scholar] [CrossRef]
- Jhee, S.S.; Yen, M.; Ereshefsky, L.; Leibowitz, M.; Schulte, M.; Kaeser, B.; Boak, L.; Patel, A.; Hoffmann, G.; Prinssen, E.P.; et al. Low penetration of oseltamivir and its carboxylate into cerebrospinal fluid in healthy Japanese and Caucasian volunteers. Antimicrob Agents Chemother 2008, 52, 3687–3693. [Google Scholar] [CrossRef]
- He, G.; Massarella, J.; Ward, P. Clinical pharmacokinetics of the prodrug oseltamivir and its active metabolite Ro 64-0802. Clin Pharmacokinet 1999, 37, 471–484. [Google Scholar] [CrossRef]
- Snell, P.; Oo, C.; Dorr, A.; Barrett, J. Lack of pharmacokinetic interaction between the oral anti-influenza neuraminidase inhibitor prodrug oseltamivir and antacids. Br J Clin Pharmacol 2002, 54, 372–377. [Google Scholar] [CrossRef]
- Oh, J.; Lee, S.; Lee, H.; Cho, J.Y.; Yoon, S.H.; Jang, I.J.; Yu, K.S.; Lim, K.S. The novel carboxylesterase 1 variant c.662A>G may decrease the bioactivation of oseltamivir in humans. PLoS One 2017, 12, e0176320. [Google Scholar] [CrossRef]
- Hsueh, C.H.; Hsu, V.; Zhao, P.; Zhang, L.; Giacomini, K.M.; Huang, S.M. PBPK Modeling of the Effect of Reduced Kidney Function on the Pharmacokinetics of Drugs Excreted Renally by Organic Anion Transporters. Clin Pharmacol Ther 2018, 103, 485–492. [Google Scholar] [CrossRef]
- Wang, X.D.; Chan, E.; Chen, X.; Liao, X.X.; Tang, C.; Zhou, Z.W.; Huang, M.; Zhou, S.F. Simultaneous and rapid quantitation of benazepril and benazeprilat in human plasma by high performance liquid chromatography with ultraviolet detection. J Pharm Biomed Anal 2007, 44, 224–230. [Google Scholar] [CrossRef]
- Gatarić, B.B. Primena tehnika za naprednu analizu podataka u biofarmaceutskoj karakterizaciji lekova: identifikacija, klasifikacija i predviđanje faktora koji utiču na intestinalnu apsorpciju lekovitih supstanci. Doctor, Belgrade Faculty, Belgrade, 2021.
- Gengo, F.M.; Brady, E. The pharmacokinetics of benazepril relative to other ACE inhibitors. Clin Cardiol 1991, 14, IV44–50. [Google Scholar] [CrossRef]
- Chan, K.K.; Buch, A.; Glazer, R.D.; John, V.A.; Barr, W.H. Site-differential gastrointestinal absorption of benazepril hydrochloride in healthy volunteers. Pharm Res 1994, 11, 432–437. [Google Scholar] [CrossRef]
- Massarella, J.; DeFeo, T.; Lin, A.; Limjuco, R.; Brown, A. The pharmacokinetics and dose proportionality of cilazapril. Br J Clin Pharmacol 1989, 27 Suppl 2, 199S–204S. [Google Scholar] [CrossRef]
- Fillastre, J.P.; Moulin, B.; Godin, M.; Williams, P.E.; Brown, A.N.; Francis, R.J.; Pinta, P.; Manfredi, R. Pharmacokinetics of cilazapril in patients with renal failure. Br J Clin Pharmacol 1989, 27 Suppl 2, 275S–282S. [Google Scholar] [CrossRef]
- Kleinbloesem, C.H.; van Brummelen, P.; Francis, R.J.; Wiegand, U.W. Clinical pharmacology of cilazapril. Drugs 1991, 41 Suppl 1, 3–10. [Google Scholar] [CrossRef]
- Williams, P.E.; Brown, A.N.; Rajaguru, S.; Francis, R.J.; Walters, G.E.; McEwen, J.; Durnin, C. The pharmacokinetics and bioavailability of cilazapril in normal man. Br J Clin Pharmacol 1989, 27 Suppl 2, 181S–188S. [Google Scholar] [CrossRef]
- Maeda, K.; Ieiri, I.; Yasuda, K.; Fujino, A.; Fujiwara, H.; Otsubo, K.; Hirano, M.; Watanabe, T.; Kitamura, Y.; Kusuhara, H.; et al. Effects of organic anion transporting polypeptide 1B1 haplotype on pharmacokinetics of pravastatin, valsartan, and temocapril. Clin Pharmacol Ther 2006, 79, 427–439. [Google Scholar] [CrossRef]
- Puchler, K.; Eckl, K.M.; Fritsche, L.; Renneisen, K.; Neumayer, H.H.; Sierakowski, B.; Lavrijssen, A.T.; Thomsen, T.; Roots, I. Pharmacokinetics of temocapril and temocaprilat after 14 once daily oral doses of temocapril in hypertensive patients with varying degrees of renal impairment. Br J Clin Pharmacol 1997, 44, 531–536. [Google Scholar] [CrossRef]
- Ohura, K.; Nozawa, T.; Murakami, K.; Imai, T. Evaluation of transport mechanism of prodrugs and parent drugs formed by intracellular metabolism in Caco-2 cells with modified carboxylesterase activity: temocapril as a model case. J Pharm Sci 2011, 100, 3985–3994. [Google Scholar] [CrossRef]
- Oguchi, H.; Miyasaka, M.; Koiwai, T.; Tokunaga, S.; Hora, K.; Sato, K.; Yoshie, T.; Shioya, H.; Furuta, S. Pharmacokinetics of temocapril and enalapril in patients with various degrees of renal insufficiency. Clin Pharmacokinet 1993, 24, 421–427. [Google Scholar] [CrossRef]
- Song, J.C.; White, C.M. Clinical pharmacokinetics and selective pharmacodynamics of new angiotensin converting enzyme inhibitors: an update. Clin Pharmacokinet 2002, 41, 207–224. [Google Scholar] [CrossRef]
- Suzuki, H.; Kawaratani, T.; Shioya, H.; Uji, Y.; Saruta, T. Study on pharmacokinetics of a new biliary excreted oral angiotensin converting enzyme inhibitor, temocapril (CS-622) in humans. Biopharm Drug Dispos 1993, 14, 41–50. [Google Scholar] [CrossRef]
- Devissaguet, J.P.; Ammoury, N.; Devissaguet, M.; Perret, L. Pharmacokinetics of perindopril and its metabolites in healthy volunteers. Fundam Clin Pharmacol 1990, 4, 175–189. [Google Scholar] [CrossRef]
- Vrhovac, B.; Sarapa, N.; Bakran, I.; Huic, M.; Macolic-Sarinic, V.; Francetic, I.; Wolf-Coporda, A.; Plavsic, F. Pharmacokinetic changes in patients with oedema. Clin Pharmacokinet 1995, 28, 405–418. [Google Scholar] [CrossRef]
- Ghiadoni, L. Perindopril for the treatment of hypertension. Expert Opin Pharmacother 2011, 12, 1633–1642. [Google Scholar] [CrossRef]
- Li, Q.; Hao, Z.; Yu, Y.; Tang, Y. Bioequivalence study of two perindopril tert-butylamine tablet formulations in healthy Chinese subjects under fasting and fed conditions: A randomized, open-label, single-dose, crossover trial. Biomed Pharmacother 2021, 135, 111221. [Google Scholar] [CrossRef]
- Ogawa, R.; Stachnik, J.M.; Echizen, H. Clinical pharmacokinetics of drugs in patients with heart failure: an update (part 2, drugs administered orally). Clin Pharmacokinet 2014, 53, 1083–1114. [Google Scholar] [CrossRef]
- Sheng, X.Y.; Liang, Y.; Yang, X.Y.; Li, L.E.; Ye, X.; Zhao, X.; Cui, Y.M. Safety, pharmacokinetic and pharmacodynamic properties of single ascending dose and continuous infusion of remimazolam besylate in healthy Chinese volunteers. Eur J Clin Pharmacol 2020, 76, 383–391. [Google Scholar] [CrossRef]
- Kim, K.M. Remimazolam: pharmacological characteristics and clinical applications in anesthesiology. Anesth Pain Med (Seoul) 2022, 17, 1–11. [Google Scholar] [CrossRef]
- Klotz, U.; Ziegler, G.; Reimann, I.W. Pharmacokinetics of the selective benzodiazepine antagonist Ro 15-1788 in man. European Journal of Clinical Pharmacology 1984, 27, 115–117. [Google Scholar] [CrossRef]
- Patel, R.D.; Kumar, S.P.; Patel, C.N.; Shankar, S.S.; Pandya, H.A.; Solanki, H.A. Parallel screening of drug-like natural compounds using Caco-2 cell permeability QSAR model with applicability domain, lipophilic ligand efficiency index and shape property: A case study of HIV-1 reverse transcriptase inhibitors. Journal of Molecular Structure 2017, 1146, 80–95. [Google Scholar] [CrossRef]
- Karavokiros, K.A.; Tsipis, G.B. Flumazenil: a benzodiazepine antagonist. DICP 1990, 24, 976–981. [Google Scholar] [CrossRef]
- Paixao, P.; Gouveia, L.F.; Morais, J.A. Prediction of the in vitro intrinsic clearance determined in suspensions of human hepatocytes by using artificial neural networks. Eur J Pharm Sci 2010, 39, 310–321. [Google Scholar] [CrossRef]
- Klotz, U.; Kanto, J. Pharmacokinetics and clinical use of flumazenil (Ro 15-1788). Clin Pharmacokinet 1988, 14, 1–12. [Google Scholar] [CrossRef]
- Pond, S.M.; Kretschzmar, K.M. Effect of phenytoin on meperidine clearance and normeperidine formation. Clin Pharmacol Ther 1981, 30, 680–686. [Google Scholar] [CrossRef]
- Chan, K.; Tse, J.; Jennings, F.; Orme, M.L. Pharmacokinetics of low-dose intravenous pethidine in patients with renal dysfunction. J Clin Pharmacol 1987, 27, 516–522. [Google Scholar] [CrossRef]
- Paixao, P.; Gouveia, L.F.; Morais, J.A. Prediction of the human oral bioavailability by using in vitro and in silico drug related parameters in a physiologically based absorption model. Int J Pharm 2012, 429, 84–98. [Google Scholar] [CrossRef]
- Piscitelli, S.C.; Kress, D.R.; Bertz, R.J.; Pau, A.; Davey, R. The effect of ritonavir on the pharmacokinetics of meperidine and normeperidine. Pharmacotherapy 2000, 20, 549–553. [Google Scholar] [CrossRef]
- Toutain, P.L.; Lefebvre, H.P.; King, J.N. Benazeprilat disposition and effect in dogs revisited with a pharmacokinetic/pharmacodynamic modeling approach. J Pharmacol Exp Ther 2000, 280, 1087–1093. [Google Scholar]
- Pan, D.Q.; Jiang, M.; Liu, T.T.; Wang, Q.; Shi, J.H. Combined spectroscopies and molecular docking approach to characterizing the binding interaction of enalapril with bovine serum albumin. Luminescence 2017, 32, 481–490. [Google Scholar] [CrossRef]
- Lee, A.; Shirley, M. Remimazolam: A Review in Procedural Sedation. Drugs 2021, 81, 1193–1201. [Google Scholar] [CrossRef]
- Blei, A.T. Albumin dialysis for the treatment of hepatic encephalopathy. Journal of Gastroenterology and Hepatology 2004, 19, S224–S228. [Google Scholar] [CrossRef]
- Nafisi, S.; Vishkaee, T.S. Study on the interaction of tamiflu and oseltamivir carboxylate with human serum albumin. J Photochem Photobiol B 2011, 105, 34–39. [Google Scholar] [CrossRef]
- Obradovic, D.; Radan, M.; Dikic, T.; Nikolic, M.P.; Oljacic, S.; Nikolic, K. The evaluation of drug-plasma protein binding interaction on immobilized human serum albumin stationary phase, aided by different computational approaches. J Pharm Biomed Anal 2022, 211, 114593. [Google Scholar] [CrossRef]
- Anderson, P.J.; Critchley, J.A.; Tomlinson, B.; Resplandy, G. Comparison of the pharmacokinetics and pharmacodynamics of oral doses of perindopril in normotensive Chinese and Caucasian volunteers. Br J Clin Pharmacol 1995, 39, 361–368. [Google Scholar] [CrossRef]
- Chen, F.; Zhang, B.; Parker, R.B.; Laizure, S.C. Clinical implications of genetic variation in carboxylesterase drug metabolism. Expert Opin Drug Metab Toxicol 2018, 14, 131–142. [Google Scholar] [CrossRef]
- Gomez, H.J.; Cirillo, V.J.; Irvin, J.D. Enalapril: a review of human pharmacology. Drugs 1985, 30 Suppl 1, 13–24. [Google Scholar] [CrossRef]
- Grislain, L.; Mocquard, M.T.; Dabe, J.F.; Bertrand, M.; Luijten, W.; Marchand, B.; Resplandy, G.; Devissaguet, M. Interspecies comparison of the metabolic pathways of perindopril, a new angiotensin-converting enzyme (ACE) inhibitor. Xenobiotica 1990, 20, 787–800. [Google Scholar] [CrossRef]
- Duthaler, U.; Bachmann, F.; Ozbey, A.C.; Umehara, K.; Parrott, N.; Fowler, S.; Krahenbuhl, S. The Activity of Members of the UDP-Glucuronosyltransferase Subfamilies UGT1A and UGT2B is Impaired in Patients with Liver Cirrhosis. Clin Pharmacokinet 2023, 62, 1141–1155. [Google Scholar] [CrossRef]
- Todd, P.A.; Heel, R.C. Enalapril. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in hypertension and congestive heart failure. Drugs 1986, 31, 198–248. [Google Scholar] [CrossRef]
- Weisser, K.; Schloos, J.; Lehmann, K.; Dusing, R.; Vetter, H.; Mutschler, E. Pharmacokinetics and converting enzyme inhibition after morning and evening administration of oral enalapril to healthy subjects. Eur J Clin Pharmacol 1991, 40, 95–99. [Google Scholar] [CrossRef]
- Dickstein, K.; Till, A.E.; Aarsland, T.; Tjelta, K.; Abrahamsen, A.M.; Kristianson, K.; Gomez, H.J.; Gregg, H.; Hichens, M. The pharmacokinetics of enalapril in hospitalized patients with congestive heart failure. Br J Clin Pharmacol 1987, 23, 403–410. [Google Scholar] [CrossRef]
- Baba, T.; Murabayashi, S.; Tomiyama, T.; Takebe, K. The pharmacokinetics of enalapril in patients with compensated liver cirrhosis. Br J Clin Pharmacol 1990, 29, 766–769. [Google Scholar] [CrossRef]
- Kaiser, G.; Ackermann, R.; Brechbuhler, S.; Dieterle, W. Pharmacokinetics of the angiotensin converting enzyme inhibitor benazepril.HCl (CGS 14 824 A) in healthy volunteers after single and repeated administration. Biopharm Drug Dispos 1989, 10, 365–376. [Google Scholar] [CrossRef]
- Schweizer, C.; Kaiser, G.; Dieterle, W.; Mann, J. Pharmacokinetics and pharmacodynamics of benazepril hydrochloride in patients with major proteinuria. Eur J Clin Pharmacol 1993, 44, 463–466. [Google Scholar] [CrossRef]
- Sioufi, A.; Pommier, F.; Gauducheau, N.; Godbillon, J.; Choi, L.; John, V. The absence of a pharmacokinetic interaction between aspirin and the angiotensin-converting enzyme inhibitor benazepril in healthy volunteers. Biopharm Drug Dispos 1994, 15, 451–461. [Google Scholar] [CrossRef]
- Waldmeier, F.; Kaiser, G.; Ackermann, R.; Faigle, J.W.; Wagner, J.; Barner, A.; Lasseter, K.C. The disposition of [14C]-labelled benazepril HCl in normal adult volunteers after single and repeated oral dose. Xenobiotica 1991, 21, 251–261. [Google Scholar] [CrossRef]
- Kaiser, G.; Ackermann, R.; Gschwind, H.P.; James, I.M.; Sprengers, D.; McIntyre, N.; Defalco, A.; Holmes, I.B. The influence of hepatic cirrhosis on the pharmacokinetics of benazepril hydrochloride. Biopharm Drug Dispos 1990, 11, 753–764. [Google Scholar] [CrossRef]
- Macdonald, N.J.; Sioufi, A.; Howie, C.A.; Wade, J.R.; Elliott, H.L. The effects of age on the pharmacokinetics and pharmacodynamics of single oral doses of benazepril and enalapril. Br J Clin Pharmacol 1993, 36, 205–209. [Google Scholar] [CrossRef]
- Williams, P.E.; Brown, A.N.; Rajaguru, S.; Francis, R.J.; Bell, A.J.; Dewland, P.M. Pharmacokinetics of cilazapril during repeated oral dosing in healthy young volunteers. Eur J Drug Metab Pharmacokinet 1990, 15, 63–67. [Google Scholar] [CrossRef]
- Gross, V.; Treher, E.; Haag, K.; Neis, W.; Wiegand, U.; Scholmerich, J. Angiotensin-converting enzyme (ACE)-inhibition in cirrhosis. Pharmacokinetics and dynamics of the ACE-inhibitor cilazapril (Ro 31-2848). J Hepatol 1993, 17, 40–47. [Google Scholar] [CrossRef]
- Williams, P.E.; Brown, A.N.; Rajaguru, S.; Walters, G.E.; McEwen, J.; Durnin, C. A pharmacokinetic study of cilazapril in elderly and young volunteers. Br J Clin Pharmacol 1989, 27 Suppl 2, 211S–215S. [Google Scholar] [CrossRef]
- Massarella, J.W.; DeFeo, T.M.; Brown, A.N.; Lin, A.; Wills, R.J. The influence of food on the pharmacokinetics and ACE inhibition of cilazapril. Br J Clin Pharmacol 1989, 27 Suppl 2, 205S–209S. [Google Scholar] [CrossRef]
- Francis, R.J.; Brown, A.N.; Kler, L.; Fasanella d'Amore, T.; Nussberger, J.; Waeber, B.; Brunner, H.R. Pharmacokinetics of the converting enzyme inhibitor cilazapril in normal volunteers and the relationship to enzyme inhibition: development of a mathematical model. J Cardiovasc Pharmacol 1987, 9, 32–38. [Google Scholar]
- Lecocq, B.; Funck-Brentano, C.; Lecocq, V.; Ferry, A.; Gardin, M.E.; Devissaguet, M.; Jaillon, P. Influence of food on the pharmacokinetics of perindopril and the time course of angiotensin-converting enzyme inhibition in serum. Clin Pharmacol Ther 1990, 47, 397–402. [Google Scholar] [CrossRef]
- Tsai, H.H.; Lees, K.R.; Howden, C.W.; Reid, J.L. The pharmacokinetics and pharmacodynamics of perindopril in patients with hepatic cirrhosis. Br J Clin Pharmacol 1989, 28, 53–59. [Google Scholar] [CrossRef]
- Thiollet, M.; Funck-Brentano, C.; Grange, J.D.; Midavaine, M.; Resplandy, G.; Jaillon, P. The pharmacokinetics of perindopril in patients with liver cirrhosis. Br J Clin Pharmacol 1992, 33, 326–328. [Google Scholar] [CrossRef]
- Lees, K.R.; Green, S.T.; Reid, J.L. Influence of age on the pharmacokinetics and pharmacodynamics of perindopril. Clin Pharmacol Ther 1988, 44, 418–425. [Google Scholar] [CrossRef]
- Furuta, S.; Kiyosawa, K.; Higuchi, M.; Kasahara, H.; Saito, H.; Shioya, H.; Oguchi, H. Pharmacokinetics of temocapril, an ACE inhibitor with preferential biliary excretion, in patients with impaired liver function. Eur J Clin Pharmacol 1993, 44, 383–385. [Google Scholar] [CrossRef]
- Abe, M.; Smith, J.; Urae, A.; Barrett, J.; Kinoshita, H.; Rayner, C.R. Pharmacokinetics of oseltamivir in young and very elderly subjects. Ann Pharmacother 2006, 40, 1724–1730. [Google Scholar] [CrossRef]
- Brewster, M.; Smith, J.R.; Dutkowski, R.; Robson, R. Active metabolite from Tamiflu solution is bioequivalent to that from capsule delivery in healthy volunteers: a cross-over, randomised, open-label study. Vaccine 2006, 24, 6660–6663. [Google Scholar] [CrossRef]
- Jittamala, P.; Pukrittayakamee, S.; Tarning, J.; Lindegardh, N.; Hanpithakpong, W.; Taylor, W.R.; Lawpoolsri, S.; Charunwattana, P.; Panapipat, S.; White, N.J.; et al. Pharmacokinetics of orally administered oseltamivir in healthy obese and nonobese Thai subjects. Antimicrob Agents Chemother 2014, 58, 1615–1621. [Google Scholar] [CrossRef]
- Amrein, R.; Hetzel, W. Pharmacology of Dormicum (Midazolam) and Anexate (Flumazenil). Acta Anaesth Scand 1990, 34, 6–15. [Google Scholar] [CrossRef]
- Breimer, L.T.; Hennis, P.J.; Burm, A.G.; Danhof, M.; Bovill, J.G.; Spierdijk, J.; Vletter, A.A. Pharmacokinetics and EEG effects of flumazenil in volunteers. Clin Pharmacokinet 1991, 20, 491–496. [Google Scholar] [CrossRef]
- Pomier-Layrargues, G.; Giguere, J.F.; Lavoie, J.; Willems, B.; Butterworth, R.F. Pharmacokinetics of benzodiazepine antagonist Ro 15-1788 in cirrhotic patients with moderate or severe liver dysfunction. Hepatology 1989, 10, 969–972. [Google Scholar] [CrossRef]
- Janssen, U.; Walker, S.; Maier, K.; von Gaisberg, U.; Klotz, U. Flumazenil disposition and elimination in cirrhosis. Clin Pharmacol Ther 1989, 46, 317–323. [Google Scholar] [CrossRef]
- Verbeeck, R.K.; Branch, R.A.; Wilkinson, G.R. Meperidine disposition in man: influence of urinary pH and route of administration. Clin Pharmacol Ther 1981, 30, 619–628. [Google Scholar] [CrossRef]
- Mather, L.E.; Tucker, G.T.; Pflug, A.E.; Lindop, M.J.; Wilkerson, C. Meperidine kinetics in man. Intravenous injection in surgical patients and volunteers. Clin Pharmacol Ther 1975, 17, 21–30. [Google Scholar] [CrossRef]
- Kuhnert, B.R.; Kuhnert, P.M.; Prochaska, A.L.; Sokol, R.J. Meperidine disposition in mother, neonate, and nonpregnant females. Clin Pharmacol Ther 1980, 27, 486–491. [Google Scholar] [CrossRef]
- Guay, D.R.; Meatherall, R.C.; Chalmers, J.L.; Grahame, G.R. Cimetidine alters pethidine disposition in man. Br J Clin Pharmacol 1984, 18, 907–914. [Google Scholar] [CrossRef]
- Guay, D.R.; Meatherall, R.C.; Chalmers, J.L.; Grahame, G.R.; Hudson, R.J. Ranitidine does not alter pethidine disposition in man. Br J Clin Pharmacol 1985, 20, 55–59. [Google Scholar] [CrossRef]
- Pond, S.M.; Tong, T.; Benowitz, N.L.; Jacob, P.; Rigod, J. Presystemic metabolism of meperidine to normeperidine in normal and cirrhotic subjects. Clin Pharmacol Ther 1981, 30, 183–188. [Google Scholar] [CrossRef]
- Pond, S.M.; Tong, T.; Benowitz, N.L.; Jacob, P. Enhanced bioavailability of pethidine and pentazocine in patients with cirrhosis of the liver. Aust N Z J Med 1980, 10, 515–519. [Google Scholar] [CrossRef]
- Mather, L.E.; Tucker, G.T. Systemic availability of orally administered meperidine. Clin Pharmacol Ther 1976, 20, 535–540. [Google Scholar] [CrossRef]
- Klotz, U.; McHorse, T.S.; Wilkinson, G.R.; Schenker, S. The effect of cirrhosis on the disposition and elimination of meperidine in man. Clin Pharmacol Ther 1974, 16, 667–675. [Google Scholar] [CrossRef]
- Neal, E.A.; Meffin, P.J.; Gregory, P.B.; Blaschke, T.F. Enhanced Bioavailability and Decreased Clearance of Analgesics in Patients with Cirrhosis. Gastroenterology 1979, 77, 96–102. [Google Scholar] [CrossRef]
- Stohr, T.; Colin, P.J.; Ossig, J.; Pesic, M.; Borkett, K.; Winkle, P.; Struys, M.; Schippers, F. Pharmacokinetic properties of remimazolam in subjects with hepatic or renal impairment. Br J Anaesth 2021, 127, 415–423. [Google Scholar] [CrossRef]
- Ishizuka, H.; Konno, K.; Naganuma, H.; Sasahara, K.; Kawahara, Y.; Niinuma, K.; Suzuki, H.; Sugiyama, Y. Temocaprilat, a novel angiotensin-converting enzyme inhibitor, is excreted in bile via an ATP-dependent active transporter (cMOAT) that is deficient in Eisai hyperbilirubinemic mutant rats (EHBR). J Pharmacol Exp Ther 1997, 280, 1304–1311. [Google Scholar]
- Gao, G.; Law, F.; Wong, R.N.S.; Mak, N.K.; Yang, M.S.M. A physiologically-based pharmacokinetic model of oseltamivir phosphate and its carboxylate metabolite for rats and humans. ADMET DMPK 2019, 7, 22–43. [Google Scholar] [CrossRef]
- Kleingeist, B.; Bocker, R.; Geisslinger, G.; Brugger, R. Isolation and pharmacological characterization of microsomal human liver flumazenil carboxylesterase. Journal of pharmacy & pharmaceutical sciences : a publication of the Canadian Society for Pharmaceutical Sciences, Societe canadienne des sciences pharmaceutiques 1998, 1, 38–46. [Google Scholar]
- Tegeder, I.; L??tsch, J.r.; Geisslinger, G. Pharmacokinetics of Opioids in Liver Disease. Clinical Pharmacokinetics 1999, 37, 17–40. [Google Scholar] [CrossRef]
- Riccardi, K.; Cawley, S.; Yates, P.D.; Chang, C.; Funk, C.; Niosi, M.; Lin, J.; Di, L. Plasma Protein Binding of Challenging Compounds. J Pharm Sci 2015, 104, 2627–2636. [Google Scholar] [CrossRef]
- Turpeinen, M.; Zanger, U.M. Cytochrome P450 2B6: function, genetics, and clinical relevance. Drug Metabol Drug Interact 2012, 27, 185–197. [Google Scholar] [CrossRef]
- Prasad, B.; Bhatt, D.K.; Johnson, K.; Chapa, R.; Chu, X.; Salphati, L.; Xiao, G.; Lee, C.; Hop, C.; Mathias, A.; et al. Abundance of Phase 1 and 2 Drug-Metabolizing Enzymes in Alcoholic and Hepatitis C Cirrhotic Livers: A Quantitative Targeted Proteomics Study. Drug Metab Dispos 2018, 46, 943–952. [Google Scholar] [CrossRef]
- Kapczinski, F.; Sherman, D.; Williams, R.; Lader, M.; Curran, V. Differential effects of flumazenil in alcoholic and nonalcoholic cirrhotic patients. Psychopharmacology 1995, 120, 220–226. [Google Scholar] [CrossRef]
Figure 1.
Schematic structure of the semi-PBPK model. Kti represents the gastric emptying rate and intestinal transit rate, respectively. kai represents the rate of drug absorption into the gut wall. QGWi represents the blood flow rate in gut wall. QLA, QL and QPV represent the hepatic artery blood flow rate, hepatic blood flow rate and portal vein blood flow rate, respectively. CLint, CLbile and CLint,K represent the intrinsic hepatic clearance, biliary intrinsic clearance and renal intrinsic clearance, respectively.
Figure 1.
Schematic structure of the semi-PBPK model. Kti represents the gastric emptying rate and intestinal transit rate, respectively. kai represents the rate of drug absorption into the gut wall. QGWi represents the blood flow rate in gut wall. QLA, QL and QPV represent the hepatic artery blood flow rate, hepatic blood flow rate and portal vein blood flow rate, respectively. CLint, CLbile and CLint,K represent the intrinsic hepatic clearance, biliary intrinsic clearance and renal intrinsic clearance, respectively.
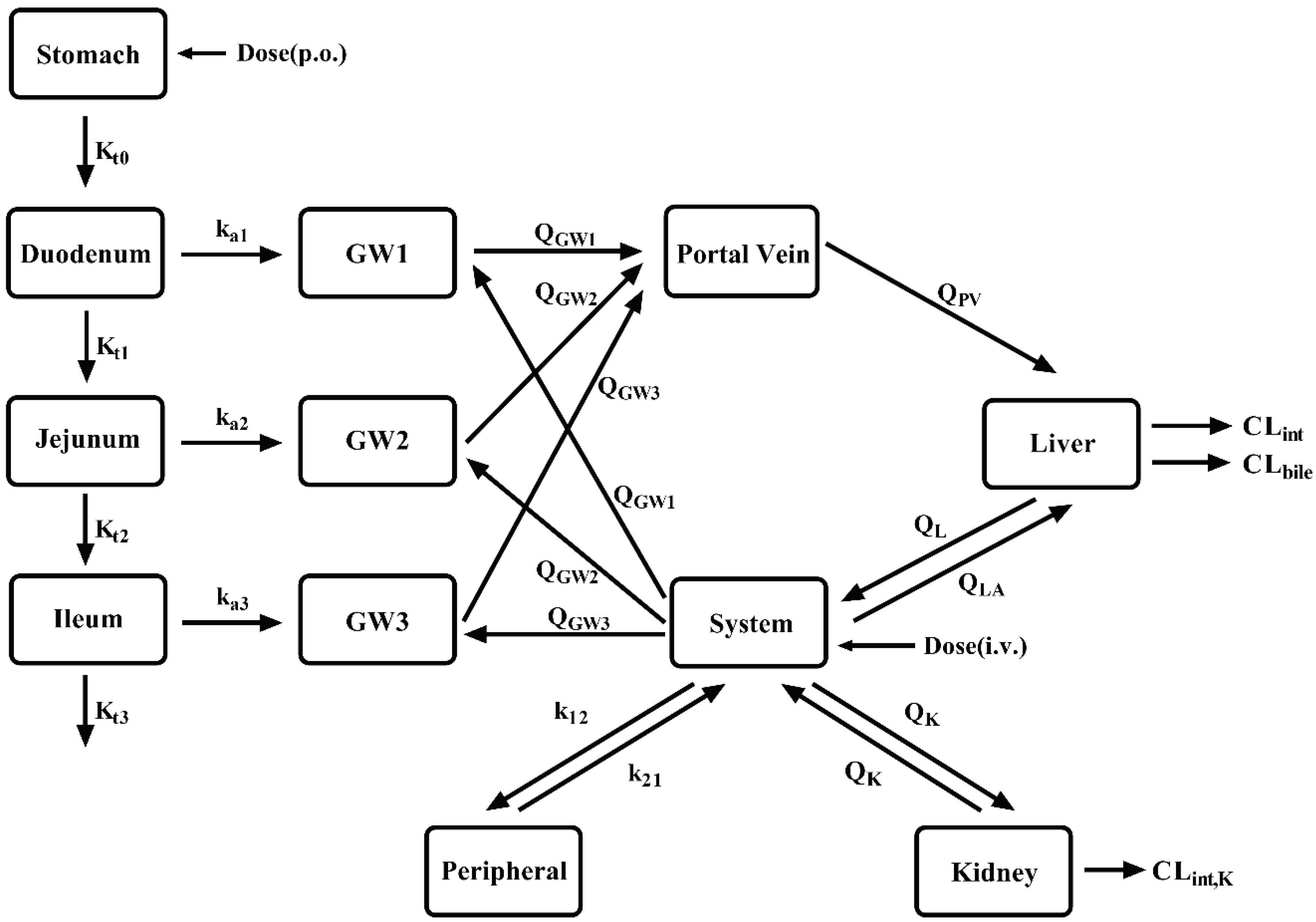
Figure 2.
The observed(points) and predicted(lines) plasma concentrations of the tested CES1 substrates and their active metabolites following intravenous or oral administration to healthy subjects. Enalapril(A) and enalaprilat(B) following oral 10 mg enalapril maleate; benazepril(C) and benazeprilat(D) following oral 20 mg benazepril hydrochloride; cilazapril(E) and cilazaprilat(F) following oral 1 mg cilazapril; perindopril(G) and perindoprilat(H) following oral 4 mg perindopril tert-butylamine; perindoprilat(I) following oral 8 mg perindopril tert-butylamine; temocapril(J) and temocaprilat(K) following 1 mg temocapril hydrochloride; oseltamivir(L) and oseltamivir carboxylate(M) following oral 75 mg oseltamivir phosphate; flumazenil following intravenous 2.5 mg/0.5 min(N) and oral 30 mg(O); pethidine following intravenous 70 mg/2 min pethidine hydrochloride(P) and oral 100 mg pethidine hydrochloride(Q); remimazolam(R) following intravenous 0.1 mg/kg remimazolam besylate. Shaded areas indicate the 5% and 95% quantile of simulations derived from 100 virtual individuals. The dashed lines indicate the mean of the simulated profiles.
Figure 2.
The observed(points) and predicted(lines) plasma concentrations of the tested CES1 substrates and their active metabolites following intravenous or oral administration to healthy subjects. Enalapril(A) and enalaprilat(B) following oral 10 mg enalapril maleate; benazepril(C) and benazeprilat(D) following oral 20 mg benazepril hydrochloride; cilazapril(E) and cilazaprilat(F) following oral 1 mg cilazapril; perindopril(G) and perindoprilat(H) following oral 4 mg perindopril tert-butylamine; perindoprilat(I) following oral 8 mg perindopril tert-butylamine; temocapril(J) and temocaprilat(K) following 1 mg temocapril hydrochloride; oseltamivir(L) and oseltamivir carboxylate(M) following oral 75 mg oseltamivir phosphate; flumazenil following intravenous 2.5 mg/0.5 min(N) and oral 30 mg(O); pethidine following intravenous 70 mg/2 min pethidine hydrochloride(P) and oral 100 mg pethidine hydrochloride(Q); remimazolam(R) following intravenous 0.1 mg/kg remimazolam besylate. Shaded areas indicate the 5% and 95% quantile of simulations derived from 100 virtual individuals. The dashed lines indicate the mean of the simulated profiles.

Figure 3.
The observed(points) and predicted(lines) plasma concentrations of the tested CES1 substrates and their active metabolites following intravenous or oral administration to LC patients. Enalapril(A,C) and enalaprilat(B,D) following oral 10 mg enalapril maleate to CP-B(A, B) and CP-C(C, D); benazepril(E) and benazeprilat(F) following oral 20 mg benazepril hydrochloride to CP-B; cilazapril(G) and cilazaprilat(H) following oral 1 mg cilazapril to CP-B; temocapril(I) and temocaprilat(J) following oral 1 mg temocapril hydrochloride to CP-B; oseltamivir(K) and oseltamivir carboxylate(L) following oral 75 mg oseltamivir phosphate to CP-B; flumazenil following intravenous 2 mg/1 min to CP-C(M), 2 mg/5 min to CP-C(N) and CP-B(O); flumazenil(P) following oral 30 mg to CP-C; pethidine following intravenous 0.8 mg/kg,1min(Q), 0.8mg/kg,5min(R), 0.8mg/kg(S,T) pethidine hydrochloride to CP-A(Q,R,S) and CP-B(T), pethidine following oral 0.8 mg/kg pethidine hydrochloride to CP-A(U), 1.6 mg/kg pethidine hydrochloride to CP-A(V) and CP-B(W); remimazolam following intravenous 0.1 mg/kg remimazolam besylate to CP-B(X) and CP-C(Y). Shaded areas indicate the 5% and 95% quantile of simulations derived from 100 virtual individuals. The dashed lines indicate the mean of the simulated profiles. Comparison of the predicted AUC(Z) and Cmax(Z-1)with observations in healthy subjects and LC patients. Solid, dashed and dotted lines respectively represent unity, 0.8-1.25-fold and 0.5-2-fold errors between observed and predicted data.
Figure 3.
The observed(points) and predicted(lines) plasma concentrations of the tested CES1 substrates and their active metabolites following intravenous or oral administration to LC patients. Enalapril(A,C) and enalaprilat(B,D) following oral 10 mg enalapril maleate to CP-B(A, B) and CP-C(C, D); benazepril(E) and benazeprilat(F) following oral 20 mg benazepril hydrochloride to CP-B; cilazapril(G) and cilazaprilat(H) following oral 1 mg cilazapril to CP-B; temocapril(I) and temocaprilat(J) following oral 1 mg temocapril hydrochloride to CP-B; oseltamivir(K) and oseltamivir carboxylate(L) following oral 75 mg oseltamivir phosphate to CP-B; flumazenil following intravenous 2 mg/1 min to CP-C(M), 2 mg/5 min to CP-C(N) and CP-B(O); flumazenil(P) following oral 30 mg to CP-C; pethidine following intravenous 0.8 mg/kg,1min(Q), 0.8mg/kg,5min(R), 0.8mg/kg(S,T) pethidine hydrochloride to CP-A(Q,R,S) and CP-B(T), pethidine following oral 0.8 mg/kg pethidine hydrochloride to CP-A(U), 1.6 mg/kg pethidine hydrochloride to CP-A(V) and CP-B(W); remimazolam following intravenous 0.1 mg/kg remimazolam besylate to CP-B(X) and CP-C(Y). Shaded areas indicate the 5% and 95% quantile of simulations derived from 100 virtual individuals. The dashed lines indicate the mean of the simulated profiles. Comparison of the predicted AUC(Z) and Cmax(Z-1)with observations in healthy subjects and LC patients. Solid, dashed and dotted lines respectively represent unity, 0.8-1.25-fold and 0.5-2-fold errors between observed and predicted data.
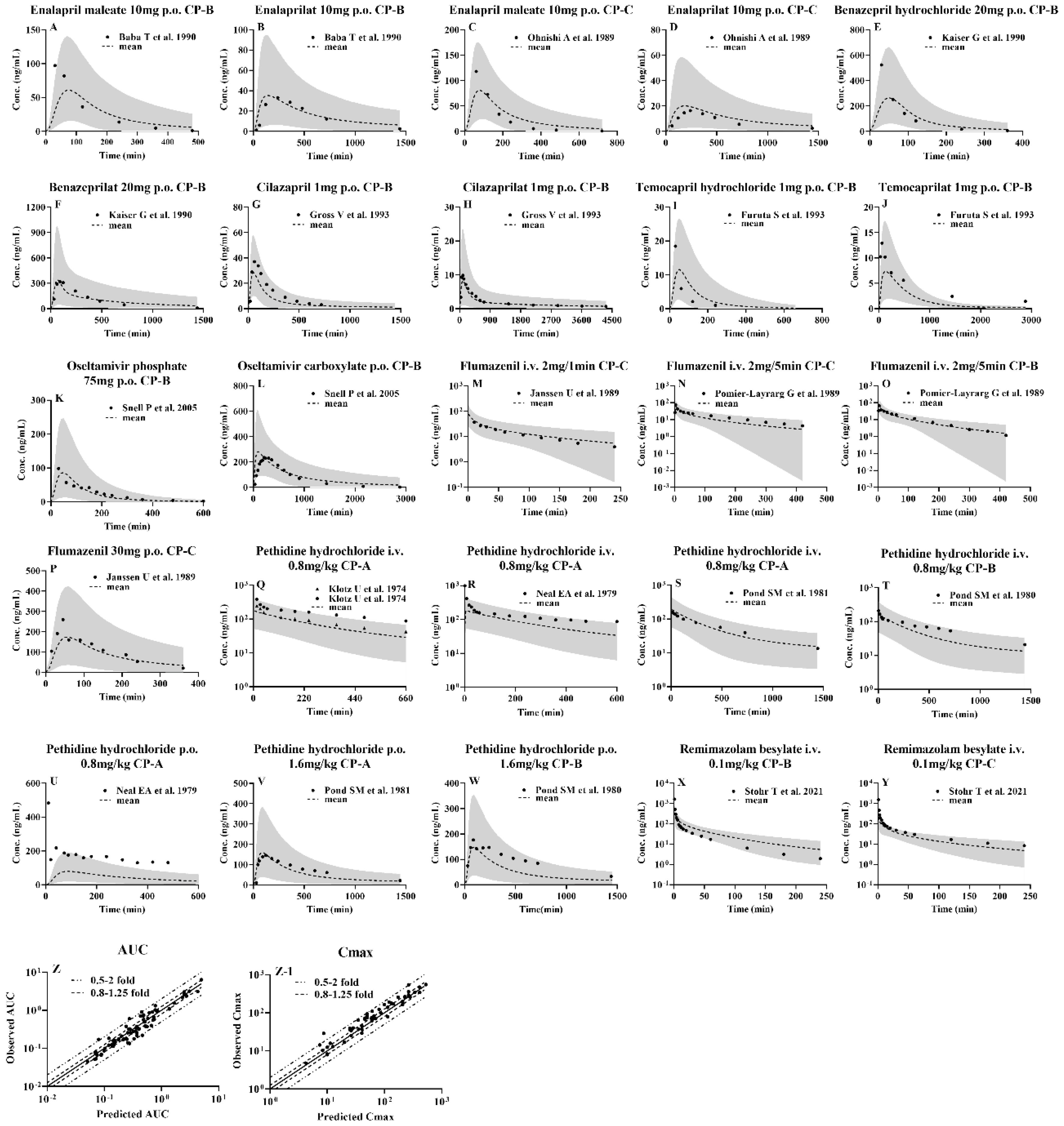
Figure 4.
AUCR was calculated from AUC (cirrhotic/healthy) or CL (healthy/cirrhotic) for cirrhotic status and healthy individuals, with the vast majority of parameters in the 0.5-2-fold range. (A) enalapril; (B) enalaprilat; (C) benazepril; (D) benazeprilat; (E) cilazapril; (F) cilazaprilat; (G) perindopril; (H) perindoprilat; (I) temocaprilat; (J) oseltamivir; (K) oseltamivir carboxylate; (L) flumazenil; (M) pethidine. Parameters not reported in the literature were excluded from the calculations; multiple doses were dose-normalized.
Figure 4.
AUCR was calculated from AUC (cirrhotic/healthy) or CL (healthy/cirrhotic) for cirrhotic status and healthy individuals, with the vast majority of parameters in the 0.5-2-fold range. (A) enalapril; (B) enalaprilat; (C) benazepril; (D) benazeprilat; (E) cilazapril; (F) cilazaprilat; (G) perindopril; (H) perindoprilat; (I) temocaprilat; (J) oseltamivir; (K) oseltamivir carboxylate; (L) flumazenil; (M) pethidine. Parameters not reported in the literature were excluded from the calculations; multiple doses were dose-normalized.
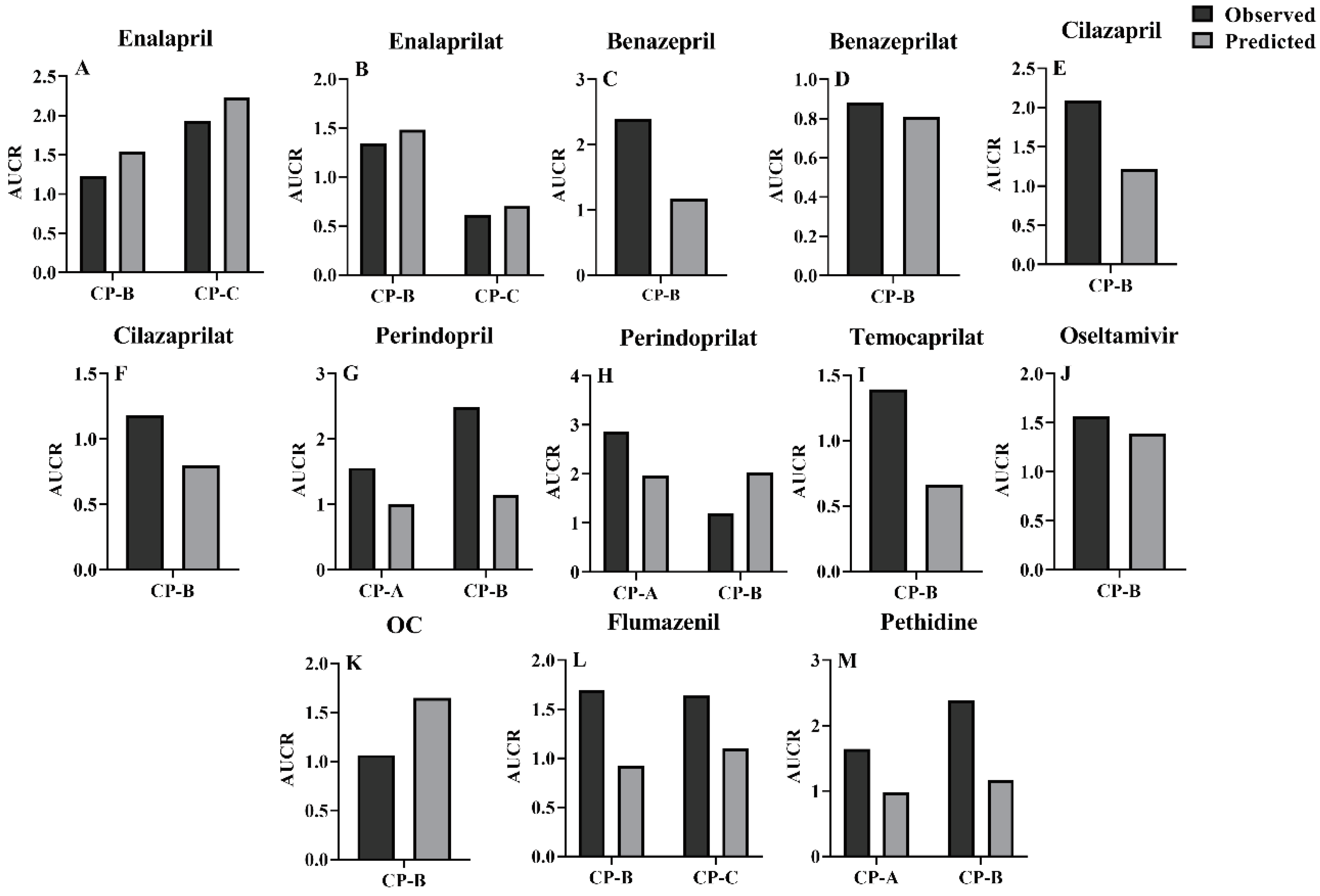
Figure 5.
CmaxR was calculated from Cmax for cirrhotic status and healthy individuals (cirrhotic/healthy), with the vast majority of parameters in the 0.5-2-fold range. (A) enalapril; (B) enalaprilat; (C) benazepril; (D) benazeprilat; (E) cilazapril; (F) cilazaprilat; (G) perindoprilat; (H) temocaprilat; (I) oseltamivir; (J) oseltamivir carboxylate; (K) flumazenil. Parameters not reported in the literature were excluded from the calculations; multiple doses were dose-normalized.
Figure 5.
CmaxR was calculated from Cmax for cirrhotic status and healthy individuals (cirrhotic/healthy), with the vast majority of parameters in the 0.5-2-fold range. (A) enalapril; (B) enalaprilat; (C) benazepril; (D) benazeprilat; (E) cilazapril; (F) cilazaprilat; (G) perindoprilat; (H) temocaprilat; (I) oseltamivir; (J) oseltamivir carboxylate; (K) flumazenil. Parameters not reported in the literature were excluded from the calculations; multiple doses were dose-normalized.
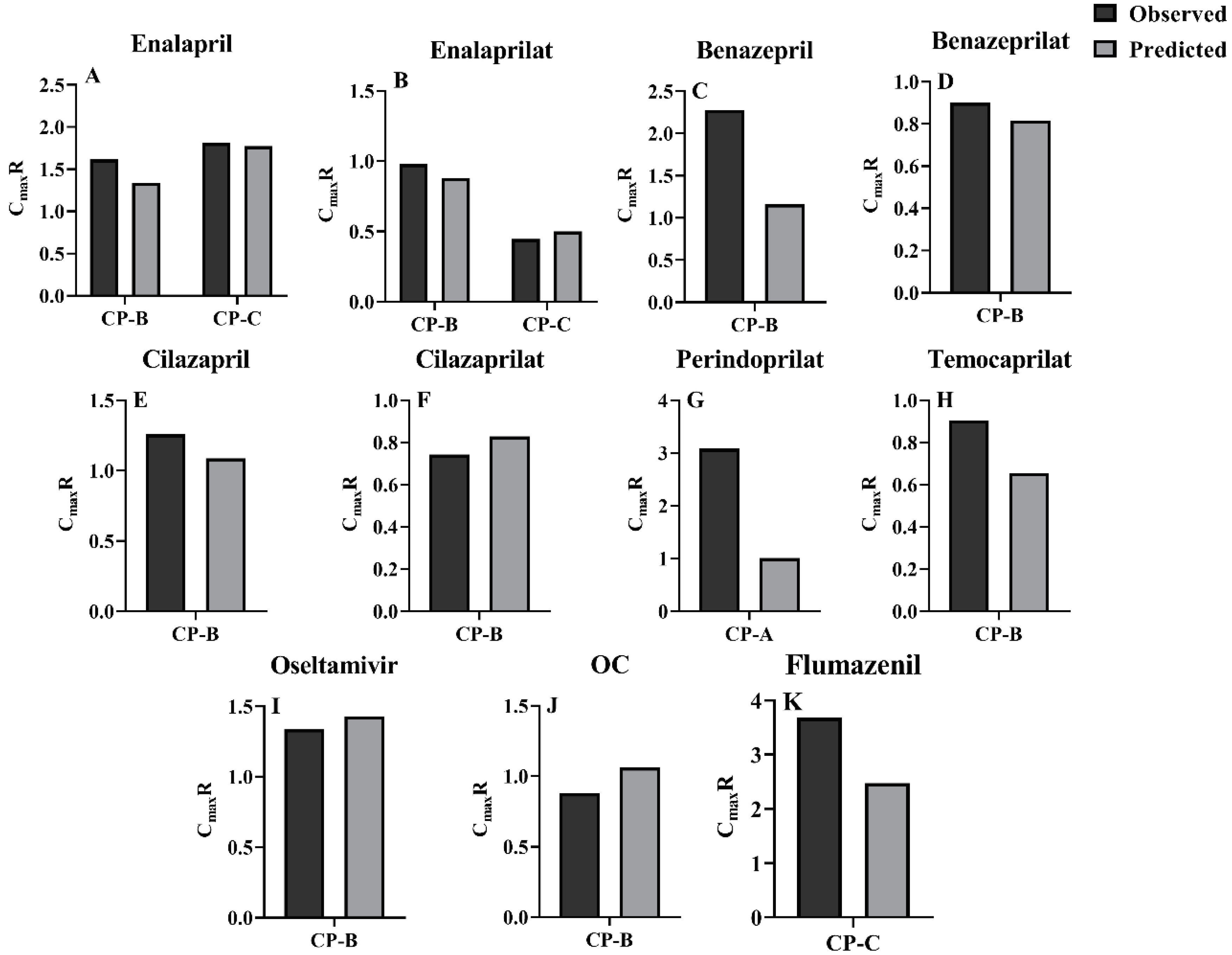
Figure 6.
Sensitivity analysis of enalapril and enalaprilat following oral 10 mg enalapril maleate. Enalapril: (A) Kt; (B) CLint,L; (C) GFR; (D) QLA; (E) QPV; (F) QK; (G) fu,b; (H) Peff; fu,b varies by 0.74-fold and 1.35-fold and GFR varies by 0.5-fold and 1.5-fold; the rest are varied by 1/3-fold and 3-fold. Enalaprilat: (I) Kt; (J) CLint,L; (K) GFR; (L) QLA; (M) QPV; (N) QK; (O) fu,b,m; (P) Peff. Where fu,b,m varies by 0.68-fold and 1.47-fold. Individual contributions of LC-induced alterations in Kt, CES1 activity, GFR, fu,b, Peff, QK and QPV to plasma concentrations of enalapril(Q) and enalaprilat(R) following oral 10 mg enalapril maleate to LC patients and their integrated effects.
Figure 6.
Sensitivity analysis of enalapril and enalaprilat following oral 10 mg enalapril maleate. Enalapril: (A) Kt; (B) CLint,L; (C) GFR; (D) QLA; (E) QPV; (F) QK; (G) fu,b; (H) Peff; fu,b varies by 0.74-fold and 1.35-fold and GFR varies by 0.5-fold and 1.5-fold; the rest are varied by 1/3-fold and 3-fold. Enalaprilat: (I) Kt; (J) CLint,L; (K) GFR; (L) QLA; (M) QPV; (N) QK; (O) fu,b,m; (P) Peff. Where fu,b,m varies by 0.68-fold and 1.47-fold. Individual contributions of LC-induced alterations in Kt, CES1 activity, GFR, fu,b, Peff, QK and QPV to plasma concentrations of enalapril(Q) and enalaprilat(R) following oral 10 mg enalapril maleate to LC patients and their integrated effects.
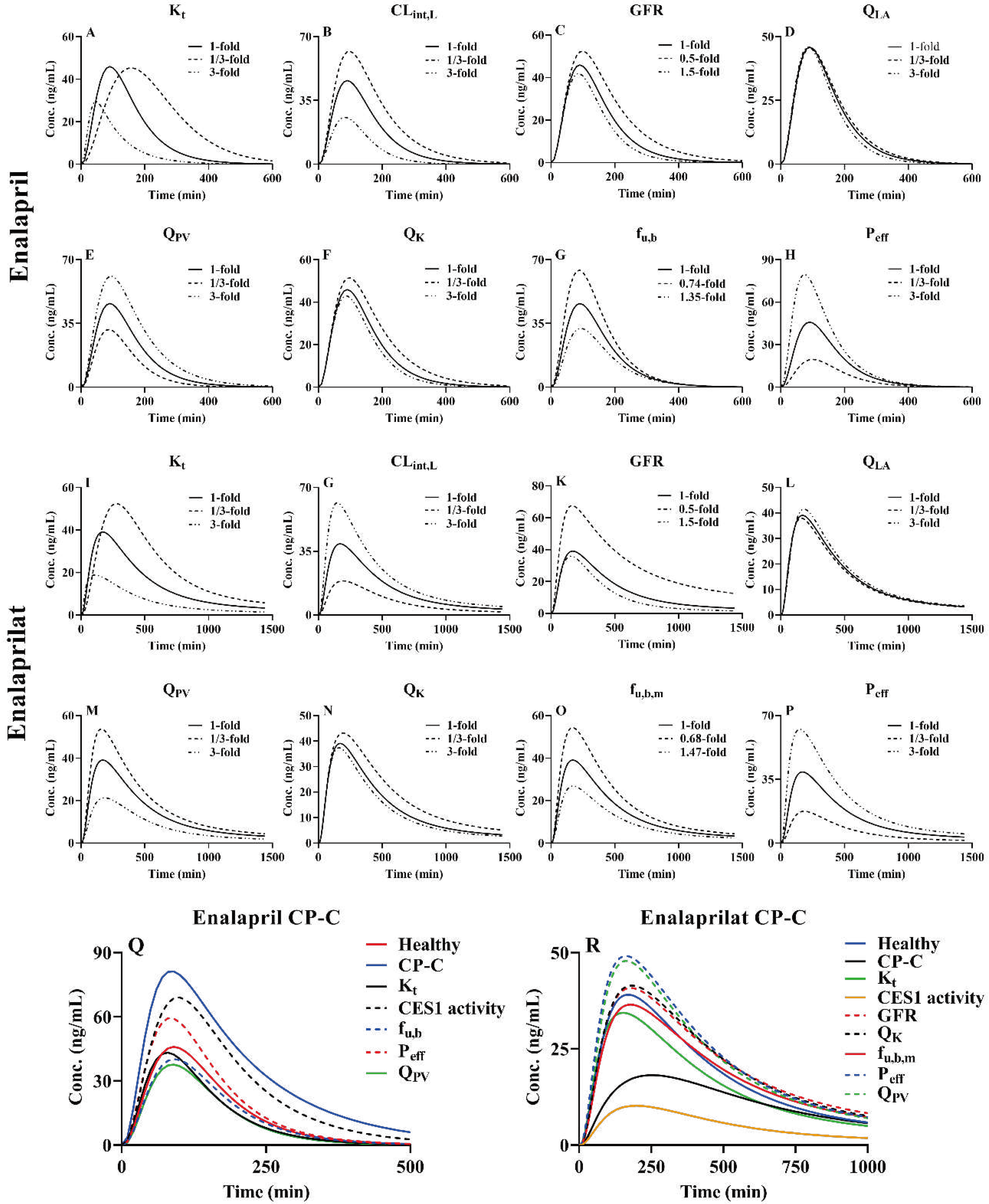
Table 1.
Physiological parameters used in the physiologically based pharmacokinetic model in normal adults[18,19] and cirrhosis.
| Normal | Child-Pugh class | Units | |||
| A | B | C | |||
| Blood flow rates | |||||
| Liver | 1450 | 1436.5 | 1176.9 | 1656.3 | mL/min |
| Hepatic arterial | 300 | 390[17] | 486.9[9] | 1020[17] | mL/min |
| Portal vein | 1150 | 1046.5[9] | 690[20] | 636.3[9] | mL/min |
| Kidney | 1240 | 1091.2[17] | 806[17] | 595.2[17] | mL/min |
| Duodenum | 45 | 45 | 45 | 45 | mL/min |
| Jejunum | 173 | 173 | 173 | 173 | mL/min |
| Ileum | 102 | 102 | 102 | 102 | mL/min |
| Volume | |||||
| Liver | 1690 | 1368.9[21] | 1098.5[21] | 895.7[21] | mL |
| Portal vein | 70 | 70 | 70 | 70 | mL |
| Kidney | 280 | 280 | 280 | 280 | mL |
| Duodenum | 21 | 21 | 21 | 21 | mL |
| Jejunum | 63 | 63 | 63 | 63 | mL |
| Ileum | 42 | 42 | 42 | 42 | mL |
| Transit rates | |||||
| Stomach | 0.04 | 0.0504[22] | 0.0504[22] | 0.0504[22] | min-1 |
| Duodenum | 0.07 | 0.0889[22] | 0.0889[22] | 0.0889[22] | min-1 |
| Jejunum | 0.03 | 0.0381[22] | 0.0381[22] | 0.0381[22] | min-1 |
| Ileum | 0.04 | 0.0508[22] | 0.0508[22] | 0.0508[22] | min-1 |
| Gut radius | |||||
| r1 | 2 | 2 | 2 | 2 | cm |
| r2 | 1.63 | 1.63 | 1.63 | 1.63 | cm |
| r3 | 1.45 | 1.45 | 1.45 | 1.45 | cm |
| Glomerular filtration rate | 105 | 82[23] | 82[23] | 82[23] | mL/min |
| Albumin | 44.7 | 36.2[17] | 30.4[17] | 26.3[9] | g/L |
| α1-acid glycoprotein | 0.8 | 0.57[21] | 0.52[21] | 0.46[21] | g/L |
| CES1 | 2.45 | 2.45[9] | 1.715[9] | 0.735[9] | mg/g Liver |
| CYP2B6 | 17 | 17[21] | 15.3[21] | 13.6[21] | pmol/mg |
| Lactulose/Rhamnose ratio | 0.037 | 0.046[24] | 0.052[24] | 0.057[24] | / |
| MRP2 ratio | 1 | 0.54[20] | 0.54[20] | 0.54[20] | / |
Table 2.
Simultaneously predicting the pharmacokinetics of CES1-metabolized drugs and their metabolites using the physiologically based pharmacokinetic model.
Table 2.
Simultaneously predicting the pharmacokinetics of CES1-metabolized drugs and their metabolites using the physiologically based pharmacokinetic model.
| Drug | logP | pka | CLint | Vmax | Km | KL;Pd | KG;Pd | KK;Pd | CLb |
| mL/min | nmoL/min/mg protein | μmol/L | mL/min | ||||||
| Enalapril | 0.59[25] | 5.20[25] | 784[26] | / | / | 1.66 | 2.29 | 1.79 | / |
| Enalaprilat | -0.74[27] | 2.03[27] | / | / | / | 1.12 | 1.04 | 1.25 | / |
| Oseltamivir | 0.36[28] | 7.7[28] | 20255.4[28] | / | / | 1.19 | 1.12 | 1.29 | / |
| OC | -1.3a | 4.19a | / | / | / | 1.71 | 1.89 | 1.91 | / |
| Benazepril | 1.11[29] | 4.74[29] | 6696[30] | / | / | 0.087 | 0.122 | 0.088 | 385.8[31] g |
| Benazeprilat | 0.56[29] | 1.97[29] | / | / | / | 0.093 | 0.088 | 0.101 | / |
| Cilazapril | 0.55[32] | 3.3[33] | 199.7 c | / | / | 1.32 | 1.31 | 1.43 | 205a |
| Cilazaprilat | -0.48a | 3.17 a | / | / | / | 1.28 | 1.22 | 1.42 | / |
| Temocapril | 2.102[34] | 2.8[35] | 5359.7[36] | / | / | 2.82 | 3.17 | 2.47 | / |
| Temocaprilat | 2.215[37] | 2.09[38] | / | / | / | 0.289 | 0.322 | 0.251 | / |
| Perindopril | -1.31[39] | 3.2[40] | 1011.15[41]156.47[42]c, j | / | / | 0.665 | 0.633 | 0.742 | / |
| Perindoprilat | -0.08a | 3.08a | / | / | / | 1.45 | 1.38 | 1.61 | / |
| Remimazolam | 3.68a | 5.99a | 79212.96c | / | / | 36.34 | 63.19 | 31.2 | 1180[43] |
| Flumazenil | 1.64[44] | 0.86[45] | 8169.9c | / | / | 2.57 | 2.71 | 2.41 | 1120[46] |
| Pethidine | 2.35[47] | 8.7[47] | / | 1.56[48] h5.382[49] i | 261[48] h356[49] i | 14.82 | 4.18 | 12.02 | / |
| Drug | Vsys | K12 | K21 | Peff,A-B | CLint,K | Rb | fu,b | F | ka |
| L | min-1 | min-1 | 10-4cm/s | mL/min | 1/min | ||||
| Enalapril | 40[50] | / | / | 1.60[51] | 624.6[52] | 0.74[53] | 0.74[27] | / | |
| Enalaprilat | 46.1[54] | 0.001[54] | 0.0009[54] | / | 186.4[55] | 0.73[53] | 0.68[27] | / | |
| Oseltamivir | 61.289[56] f | / | / | / | 1357.95[57] | 1e | 0.58[28] | / | 0.061[58] g |
| OC | 160.729[59] f | / | / | / | 438.5[60] | 1e | 0.97[28] | / | |
| Benazepril | 4.8[61] g | 0.0215[61] g | 0.0238[61] g | 1.21[62] | 8391.6c | 1e | 0.03[63] | 0.35[50] | |
| Benazeprilat | 1.204[64] f | 0.0438[64] f | 0.00837[64] f | / | 447.9[63] | 1e | 0.05[63] | / | |
| Cilazapril | 18.23[65] f | 0.00325[65] f | 0.00155[65] f | / | 118.095[66] | 1e | 0.7[32] | / | 0.099[67] g |
| Cilazaprilat | 10.3517[65] f | 0.00084[65] f | 0.008[65] f | / | 75.48[66] | 1e | 0.76[68] | / | / |
| Temocapril | 15.398[69] g | / | / | / | 110.2[70] | 1e | 0.3[38] | 0.65[71] | 0.065[69] g |
| Temocaprilat | 58.535[72] f | 0.00184[72] f | 0.000078[72] f | / | 949.84[73]1899.68[74] b | 1e | 0.025[38] | / | / |
| Perindopril | 13.119[75] g | 0.0028[75] g | 0.0024[75] g | 1.34[30] | 130.2[76] | 1e | 0.4[77] | 0.66[73] | / |
| Perindoprilat | 53.44[78] f | 0.271[78] f | 0.0996[78] f | / | 231.78[79] | 1e | 0.85[77] | / | / |
| Remimazolam | 15.0768[80] | 0.01638[80]0.3117[80](K13) | 0.000476[80]0.5057[80](K31) | / | / | 1e | 0.08[81] | / | |
| Flumazenil | 24.054[82] g | 0.0376[82] g | 0.0427[82] g | 3.78[83] | 1.67[84] | 1[85] | 0.6[86] | / | |
| Pethidine | 328.676[87] f | 0.002224[87] f | 0.0003697[87] f | / | 58.78[88] | 0.87[89] | 0.48[49] | / | 0.117[90] g |
a: Data from www.drugbank.com; b: bile intrinsic clearance of temocaprilat; c: recalculated from CLL,b; d: Calculations using Rodgers-Rowland method; e: Assumed values; f: Simulation by WinNonlin, cilazapril and cilazaprilat using 0.5 mg dose pharmacokinetic, remimazolam using 0.025mg/kg dose pharmacokinetic and flumazenil using T.F. pharmacokinetic to simulation; g: Calculated by WinNonlin; h: CES1-mediated CLint; i: CYP2B6-mediated CLint; j: UGT intrinsic clearance of perindopril.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



