
Submitted:
18 December 2023
Posted:
19 December 2023
You are already at the latest version
Abstract
Serelaxin (RLX), the recombinant form of human Relaxin-2 hormone, protects the heart from the ischemia/reperfusion (I/R)-induced damage due to its anti-inflammatory, anti-apoptotic and antioxidant properties. RLX exerts its actions by binding to its specific RXFP1 receptor whereby it regulates multiple cellular transduction pathways. In this in vitro study we offer first evidence for the involvement of AMP kinase/Sirtuin1 (AMPK/SIRT1) pathway in the protection afforded by RLX against H/R-induced damage in H9c2 cells. Treatment of the H/R-exposed cells with RLX (17 nmol L−1) enhanced SIRT1 expression and activity. Inhibition of SIRT1 signaling with EX527 (10 µmol L-1) reduced the beneficial effect of the hormone on mitochondrial efficiency and cell apoptosis. Moreover, RLX upregulated AMPK pathway, as shown by the increase in the expression of phospho-AMPK active protein. Finally, AMPK pathway inhibition by Compound C (10 and 20 μmol L−1) abrogated the increase in SIRT1 expression induced by RLX, thus suggesting the involvement of AMPK pathway in this effect of RLX. The results of this study strengthen the concept that RLX exerts its cardioprotective effects against H/R-induced injury by multiple pathways which also include AMPK/SIRT1.
Keywords:
Serelaxin
; SIRT1
; AMPK
; hypoxia-reoxygenation
; apoptosis
; oxidative stress
; H9c2 cells
1. Introduction
Serelaxin (RLX), the recombinant form of the human Relaxin-2, is a natural peptide hormone originally known for its role on the female reproductive system [1]. RLX has long been recognized as a pleiotropic hormone, being able to mediate a broad range of actions across a variety of tissue and organs in both sexes, including the cardiovascular system [2,3]. Experimental evidence in vivo and in vitro, from independent research groups worldwide, attests that RLX is able to protect the heart from the damage induced by ischemia and reperfusion (I/R) [4,5,6,7,8,9]. The protective action of RLX on myocardial injury is attributable not only to its anti-inflammatory, anti-apoptotic, and antifibrotic properties, but also to its ability to reduce oxidative stress [9,10]. In fact, in H9c2 cardiac muscle cells exposed to hypoxia and reoxigenation (H/R), RLX reduces the production of reactive oxygen species (ROS), improves mitochondrial activity impaired by H/R and prevents the related cell apoptosis [9]. RLX, although does not possess intrinsic antioxidant properties, is capable to potentiate cardiomyocyte resistance to oxygen deprivation and nitroxidative stress by a mechanism that allows to increase the cellular availability of reduced glutathione (GSH), a ubiquitous endogenous antioxidant metabolite [9].
RLX exerts its multiple actions on the heart by binding to a specific G protein-coupled receptor, referred to as RLX family peptide receptor 1 (RXFP1), bioactive in rodent animal models and cells [11,12]. When RLX binds to RXFP1 receptor on cardiac cells activates multiple signal transduction pathways, including the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/Akt, Notch1, Extracellular signal-regulated kinase-1/2 (ERK1/2) [13,14] and AMP-activated protein kinase (AMPK) [15]. The ability of RLX/RXFP1 to up-regulate multiple cellular transduction signaling could explain the pleiotropic action of RLX on cardioprotection.
Silent information regulator 1 (SIRT1) is a nicotinamide adenine dinucleotide (NAD)-dependent histone deacetylase belonging to the highly conserved Sirtuin family which, in mammals, consists of seven members (SIRT1–SIRT7), with different subcellular localization, enzymatic activity, and binding targets [16,17,18]. SIRT1 is ubiquitously expressed and, by deacetylation of a variety of histones and non-histone proteins, regulates multiple biological processes, such as cellular senescence, metabolism, energy, redox balance and apoptosis [17,18]. SIRT1 is also an essential regulator of myocardial I/R-induced injury [19]. Numerous studies demonstrate that SIRT1 protects the heart from I/R-induced damage via multiple cellular mechanisms, that include the reduction of inflammation and apoptosis, preservation of mitochondrial function and attenuation of oxidative stress. SIRT1 exerts its antioxidant action by stimulating the upregulation of antioxidant enzymes and decreasing ROS production [19,20,21,22,23]. SIRT1 activity is enhanced by AMPK, a serine/threonine-protein kinase complex, which plays a crucial role in cell energy, metabolism and survival [17,24]. Stimulation of AMPK pathway by different stimuli, results in an increase of cellular NAD+ levels which in turn sustain the SIRT1 mediated deacetylation of downstream protein targets including antioxidant enzymes [23].
Aim of this in vitro study is to investigate the possible involvement of the AMPK/SIRT1 pathway in the noted protective action of RLX on H/R-induced damage.
2. Materials and Methods
2.1. Cell Cultures and Treatments
H9c2 embryonic rat cardiac muscle cells, a well-characterized and widely used cell line to study myocardial cell ischemia [25], were obtained from European Collection of Cell Cultures (ECACC, Salisbury, United Kingdom). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (Sigma-Aldrich, Milan, Italy), 2 mmol L−1 glutamine, 250 U mL−1 penicillin G and 250 μg mL−1 streptomycin (Sigma-Aldrich), in a humidified atmosphere with 5% CO2 at 37 °C.
H9c2 cells were subjected to H/R, simulated in vitro by substrate starvation plus hypoxia followed by reoxygenation, as previously described [13]. H9c2 cells were incubated in DMEM with no serum or glucose and placed in a hypoxic modular incubator chamber (Billups-Rothenberg, Inc., San Diego, CA, USA) gassed with 95% N2 and 5% CO2, humidified, and warmed at 37°C for 5 h. A flow meter was used to measure the quantity of gas mixture introduced into the chamber (25 L min−1). At end hypoxia, the medium was replaced with fresh complete culture medium and the cells were reoxygenated for 2h by incubation in normoxic conditions. Control normoxic cultures were also prepared.
Cells were treated or not with human recombinant RLX, kindly provided by the RRCA Relaxin Foundation (Florence, Italy), at concentrations of 17 nmol L−1. RLX was added at the onset of hypoxia and maintained throughout the experimental period, until the end of reoxygenation. This RLX concentration was similar to that used in the previous experiments to demonstrate its ability to protect H9c2 cells from H/R-induced injury [8]. The experiments were performed at least in triplicate.
To assess the involvement of SIRT1 pathway in the protective action of RLX against H/R-induced cell damage, H9c2 cells were treated with the specific SIRT1 inhibitor EX527 (1 and 10 μmol L−1) (Sigma Aldrich) 2h before the onset of hypoxia [26,27].
To investigate the involvement of the AMPK signaling in the possible upregulation of SIRT1 by RLX, H9c2 cells were treated for 24 h before the onset of hypoxia with 10 and 20 μmol L−1 of Compound C (Dorsomorphin) (MedChemExpress, NJ; USA), a selective and ATP-competitive AMPK inhibitor. The concentrations and the exposure time of Compound C were chosen, as reported [28].
2.2. Western Blotting
H9c2 cells from the different experimental groups were lysed in cold buffer (Cell Signaling Massachusetts, USA) composed of (mmol L−1): 20 Tris/HCl pH 7.5, 150 NaCl, 1 Na2EDTA, 1 EGTA, 1% Triton X-100, 2.5 sodium pyrophospate, 1 β-glycerophosphate, 1 Na3VO4, 1mg mL−1 leupeptin, and added with 1 mmol L−1 Phenylmethylsulfonyl fluoride (PMSF) protease inhibitor. Total protein content was measured spectrophotometrically using micro-BCATM Protein Assay Kit (Pierce, IL, USA). Forty µg of total proteins were electrophoresed by SDS–PAGE and blotted onto PVDF membranes (Millipore, Bedford, MA, USA). The membranes were incubated overnight at 4°C with the following primary antibodies: rabbit polyclonal anti-SIRT1 (1:800; Santa Cruz Biotechnology, Texas, USA), rabbit polyclonal anti-AMPKα (1:1000; Cell Signaling), rabbit polyclonal anti-phospho-AMPKα (1:1000; Cell Signaling), rabbit polyclonal anti-caspase 3 (1:1000; Cell Signaling) which detects both the full length and the cleaved form of the protein and mouse monoclonal anti-β-actin (1: 4500; Sigma Aldrich), assuming β-actin as control invariant protein. Specific bands were detected using rabbit or mouse peroxidase-labeled secondary antibodies (1:15.000; Vector Laboratories, Burlingame,CA, USA) and enhanced chemiluminescent substrate. Densitometric analysis of the bands was performed using Scion Image Beta 4.0.2 image analysis software (Scion Corp., Frederick, MD, USA) and the values were normalized to β-actin.
2.3. SIRT1 Activity
SIRT1 activity was measured using the SIRT1 Activity Fluorometric Assay Kit (Abcam, Cambridge, UK), according to the manufacturer's protocol. Briefly, H9c2 cells from each experimental group were lysed with an appropriate buffer not containing protease inhibitors to avoid their interference with the development of the assay. The lysis buffer was composed of (mmol L−1): 20 Tris-HCl (pH 7.5), 250 NaCl, 1 EDTA, 1 EGTA, 1% Triton X-100, 1 dithiothreitol (DTT). Four hundred µg of total protein extracts were immunoprecipitated for SIRT1. To preclean and minimize extra bands resulting from nonspecific precipitation, cell lysates were incubated with Protein G PLUS/Protein A-Agarose suspension (Calbiochem, Darmstadt, Germany) for 1h and 30 min, at 4°C, under stirring. After centrifugation, the samples were incubated with rabbit polyclonal anti-SIRT1 (1:100; Santa Cruz Biotechnology) overnight, at 4°C, under stirring. Then, Protein G PLUS/Protein A-Agarose suspension was added in each sample and incubated for 2h, at 4°C under stirring. After centrifugation, the obtained precipitates were used to measure SIRT1 activity. Fluorescence intensity was read for 30 to 60 minutes at 1-to-2-minute intervals using microtiter plate fluorometer with excitation at 340–360 nm and emission at 440–460 nm wavelength. The rate of reaction was measured and calculated while the reaction velocity remained constant.
2.4. Mitochondrial Activity
Cell mitochondrial activity was assayed by the resazurin reduction method (CellTiter-Blue, Promega Corp., Madison, WI, USA) [9], based on the ability of metabolically active cells to reduce resazurin to resorufin and dihydroresorufin proportionally to their number. H9c2 cells (5×104/well) were seeded in 24-well plates and subjected to H/R in the presence or absence of RLX and of the SIRT1 inhibitor EX527 (1 and 10 μmol L−1). At the end of the treatments, the culture medium was removed from each well and replaced with 300 μl of MTT (0.5 mg mL−1), followed by incubation for 4h at 37°C. Then, 350 μl of dimethyl sulfoxide (DMSO) (Sigma Aldrich) were added to each well to dissolve the formazan crystals. The plate was gently shaken for 5 min and read at 550 nm wavelength on a plate reader. Optical density was assumed as indicator of mitochondrial activity.
2.5. Statistical Analysis
The reported data are expressed as the mean ± SEM of at least 3 independent experiments. Statistical comparison of differences between groups was carried out using one-way ANOVA followed by Student-Newman-Keuls multiple comparison test. Student’s unpaired t-test was performed to compare the average values of two datasets. A p value <0.05 was considered significant. Calculations were done using GraphPad Prism 5.0 statistical program (GraphPad Software, San Diego, CA, USA).
3. Results
3.1. RLX Activates SIRT1 Signaling in H9c2 Cells Subjected to H/R
We investigate the involvement of the SIRT1 signaling in the mechanism of action by which RLX protects the cells from H/R-induced damage. Treatment of the control cells not subjected to H/R with RLX (17 nmol L−1) had no effect on SIRT1 expression (Figure 1A,B) or activity (Figure 2A). In H9c2 cells subjected to H/R, SIRT1 expression and activity were significantly reduced as compared with the control cells (Figures 1C,D and 2B). Treatment with RLX (17 nmol L−1) of the H/R-exposed cells significantly increased both SIRT1 expression (Figure 1C,D) and activity (Figure 2B).
3.2. RLX Affords Cell Protection by Up-Regulation of SIRT1 Signaling
In H9c2 cells subjected to H/R, the efficiency of the mitochondrial respiratory chain, measured by the fluorescent signal of reduced resazurin (MTT assay), was significantly reduced as compared with the control cells (Figure 3). Treatment with RLX (17 nmol L−1) significantly improved mitochondrial activity impaired by H/R. This beneficial effect of RLX was significantly reduced, albeit not abolished, in the presence of the SIRT1 inhibitor EX527 at the higher concentration (10 µmol L-1) (Figure 3). Treatment of the control H9c2 cells with RLX (17 nmol L−1) did not influence basal mitochondrial efficiency (Figure 3).
H/R challenge induced the expression of the cleaved, active caspase 3, leaving the expression of the full-length caspase 3 unaltered (Figure 4 A and B). As expected, treatment with RLX (17 nmol L−1) significantly reduced the expression of cleaved caspase 3 as compared with the H/R-induced cells (Figure 4A,B). The protective effect of RLX on apoptosis was significantly reduced, but not completely abolished, by co-treatment with the SIRT1 inhibitor EX527 at the higher concentration (10 µmol L-1) as judged by the observed increase in cleaved caspase 3 expression as compared with the RLX-treated cells (Figure 4A,B). Full-length caspase 3 expression was not affected by the different treatments (Figure 4A,B). In H9c2 cells not subjected to H/R, treated or not with RLX (17 nmol L−1), the active, cleaved form of caspase 3 was not detected (Figure 4A,B).
3.3. RLX Affects SIRT1 Signaling by Activation of AMPK Pathway
The possible molecular mechanism linking the activation of the RXFP1 receptor by RLX and the up-regulation of SIRT1 signaling was also investigated, focusing on the involvement of the AMPK pathway. In H9c2 cells exposed to H/R, phospho-AMPKα expression, the active form of the protein, is significantly decreased as compared with the control cells (Figure 5A,B). Treatment with RLX (17 nmol L−1) induced a significant increase in phospho-AMPKα expression as compared with HR-exposed cells, leaving total AMPKα expression unchanged (Figure 5A,B).
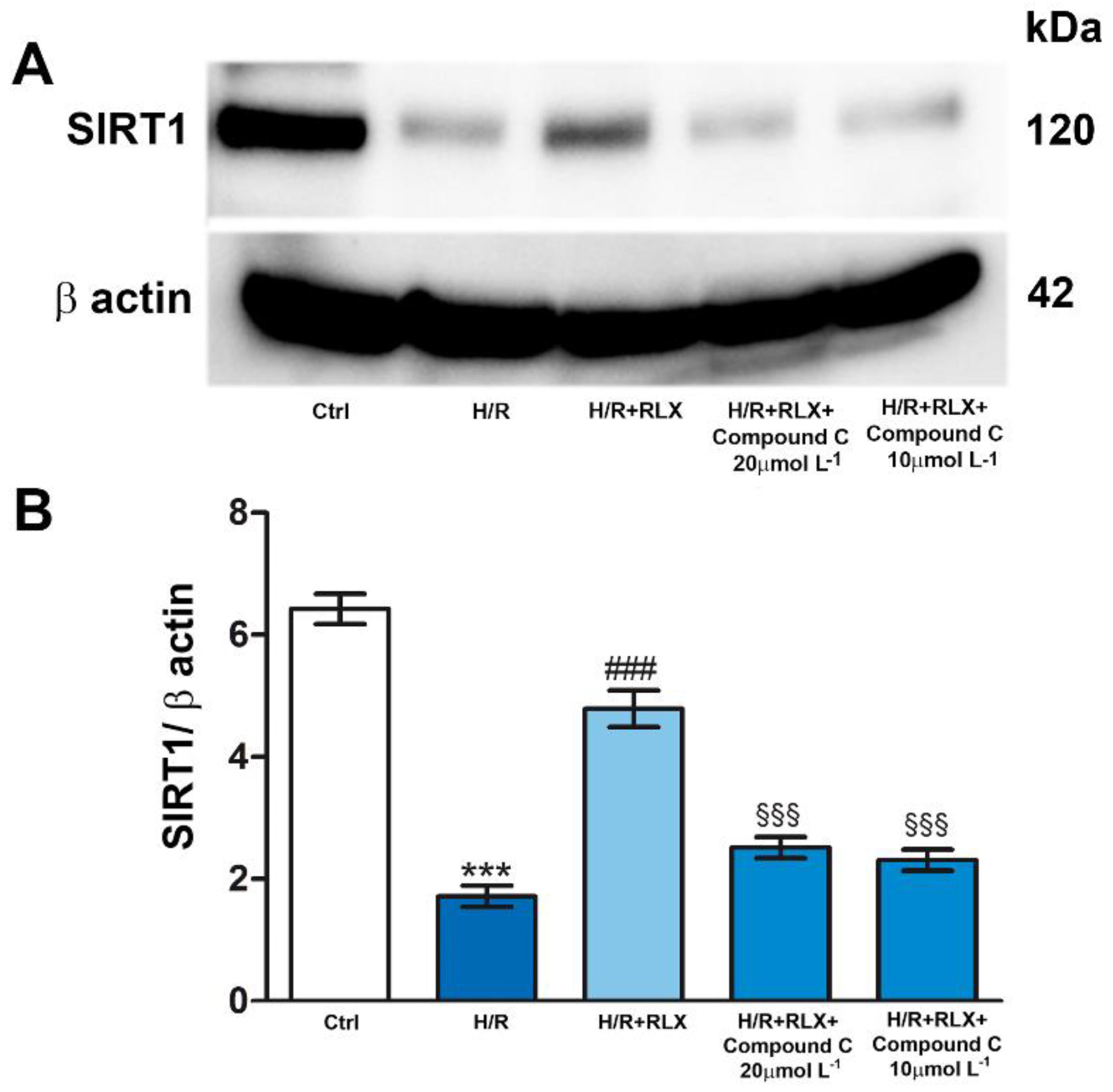
Then, to assess if the activation of AMPK pathway by RLX underlies the ability of the hormone to activate SIRT1 signaling, we evaluated SIRT1 signaling in the presence of the selective AMPK inhibitor Compound C. Co-incubation of H/R-exposed H9c2 cells with RLX and Compound C (10 and 20 µmol L-1) significantly abated the activation of SIRT1 signaling induced by RLX (Figure 6A,B).
4. Discussion
In recent years, robust evidence has been accumulating that the peptide hormone RLX can markedly reduce tissue and cell damage in pathological events that involve oxidative stress, such as vascular and lung injury caused by exposure to cigarette smoke [29,30] and myocardial damage induced by I/R [3,5,8]. RLX exerts its protective effects on oxidative stress by binding to its cognate G protein-coupled receptor RXFP1 and activating multiple intracellular transduction pathway by which it can regulate the expression and/or the activity of different cell protein kinases, including PI3K/AKT, ERK1/2 and AMPK [11,12,15]. This in vitro study provides first evidence that RLX is able to activate SIRT1 signaling in H/R-exposed H9c2 cells by the up-regulation of AMPK pathway. Treatment of H9c2 cells with RLX, from the onset of hypoxia to the end of reoxygenation, increases SIRT1 expression and activity which had been reduced by H/R. By this mechanism RLX protects these cells against the H/R induced oxidative damage. In fact, SIRT1 signaling inhibition by EX527 reduces, albeit not abolishes, the beneficial effects of the hormone on mitochondrial efficiency and cell apoptosis. The finding that SIRT1 inhibition does not completely abolish the effects of RLX confirms that multiple cytoprotective signaling pathways are operated by this hormone in cardiac muscle cells [8,9,13]. Of note, RLX is able to increase SIRT1 signaling only under pathological conditions, such as H/R, as it has no effects on SIRT1 expression and activity in the control cells under basal conditions. Within the limitation of this cellular model, SIRT1 seems to be only activated by RLX under stress conditions as a defensive mechanism. This agrees with the well-known cardioprotective properties of SIRT1. Numerous studies have shown that SIRT1 signaling plays a fundamental role in protecting the heart from I/R-induced injury [19,31] since its up-regulation inhibits inflammation, apoptosis, fibrosis, and oxidative stress [32,33], whereas its down-regulation or inactivation predisposes the myocardium to oxidative damage, leading to exacerbation of heart injury [17,34]. However, further studies need to be conducted to better understand the exact role of SIRT1 pathway in the cardioprotective action of RLX.
In addition, this study provides evidence that RLX stimulates SIRT1 signaling via the activation of AMPK, a serine/threonine-protein kinase complex whose up-regulation reduces inflammatory response and oxidative stress [35,36,37]. SIRT1 and AMPK pathways have a close interaction in the regulation of cellular energy, metabolism, aging and response to oxidative stress, since they can reciprocally enhance each other’s activity [38,39,40]. In fact, AMPK enhances SIRT1 activity by increasing the cellular levels of the SIRT1 cofactor NAD+ [24] and, in turn, SIRT1 stimulates AMPK pathway by deacetylation and activation of specific upstream regulators of AMPK signaling [41]. Moreover, activation of AMPK/SIRT1 signaling results in downregulation of pro-inflammatory cytokines, reduction of ROS production and upregulation of antioxidant enzymes [42]. In this study, we have demonstrated that RLX reverts the reduction of phospho-AMPK expression induced by H/R while leaving total AMPK unchanged. This suggests a direct regulatory action of the hormone on AMPK protein without affecting its gene expression. It is conceivable that SIRT1 activation may quickly occur after RLX binding to RXFP1, thus contributing to explain why the previously reported protective effects of RLX on myocardial H/R occurred in the short term, soon after RLX administration at reperfusion [5,8]. Moreover, treatment of H/R-induced H9c2 cells with RLX in the presence of the selective AMPK inhibitor Compound C abolished the ability of the hormone to activate SIRT1 pathway, thus confirming the involvement of AMPK on SIRT1 activation by RLX. The activation by RLX of AMPK/SIRT1 pathway could explain a possible mechanism by which RLX increases GSH bioavailability in H9c2 cells challenged with H/R [9]. It is well-known that SIRT1 up-regulates the expression and activity of nuclear factor erythroid 2-related factor 2 (Nrf2) [43], a transciption factor involved in the expression of glutathione reductase and de novo synthesis of GSH [44]. This hypothesis is worthy to be explored in future investigations.
5. Conclusions
The results of the present study expand the current knowledge on the molecular signal transduction mechanisms operated by RLX and its cognate receptor RXFP1 in cardiac H9c2 cells. The data obtained offer evidence for the involvement of the SIRT1 pathway in the protective action of this hormone against cell oxidative damage induced by H/R and strengthen the concept that RLX exerts its protective effects by multiple signaling pathways. In a clinical perspective, these results corroborate the current experimental background supporting RLX as a promising new therapeutic for heart ischemic diseases.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, S.N.; methodology, S.N., V.Z. and M.B.; formal analysis, S.N. V.Z. and D.B.; data curation, S.N., V.Z.; writing—original draft preparation, S.N. and D.B.; writing—review and editing, S.N and D.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by research funds from the University of Florence, Italy, issued to S.N.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sherwood, O.D. Relaxin’s physiological roles and other diverse actions. Endocrinol. Rev. 2004, 25, 205–234. [Google Scholar] [CrossRef] [PubMed]
- Jelinic, M.; Marshall, S.A.; Leo, C.H.; Parry, L.J.; Tare, M. From pregnancy to cardiovascular disease: Lessons from relaxin-deficient animals to understand relaxin actions in the vascular system. Microcirculation 2019, 26, e12464. [Google Scholar] [CrossRef] [PubMed]
- Bani, D. Recombinant human H2 relaxin (serelaxin) as a cardiovascular drug: aiming at the right target. Drug Discov. Today 2020, 25, 1239–1244. [Google Scholar] [CrossRef] [PubMed]
- Bani, D.; Masini, E.; Bello, M.G.; Bigazzi, M.; Bani Sacchi, T. Relaxin protects against myocardial injury caused by ischemia and reperfusion in rat heart. Am. J. Pathol. 1998, 152, 1367–1376. [Google Scholar] [PubMed]
- Perna, A.M.; Masini, E.; Nistri, S.; Briganti, V.; Chiappini, L.; Stefano, P.; Bigazzi, M.; Pieroni, C.; Sacchi, T.B.; Bani, D. Novel drug development opportunity for relaxin in acute myocardial infarction: evidence from a swine model. Faseb J. 2005, 19, 1525–1527. [Google Scholar] [CrossRef] [PubMed]
- Bonacchi, M.; Nistri, S.; Nanni, C.; Gelsomino, S.; Pini, A.; Cinci, L.; Maiani, M.; Zecchi-Orlandini, S.; Lorusso, R.; Fanti, S.; Silvertown, J.; Bani, D. Functional and histopathological improvement of the post-infarcted rat heart upon myoblast cell grafting and relaxin therapy. J. Cell. Mol. Med. 2009, 13, 3437–3448. [Google Scholar] [CrossRef] [PubMed]
- Raleigh, J.V.; Mauro, A.G.; Devarakonda, T.; Marchetti, C.; He, J.; Kim, E.; Filippone, S.; Das, A.; Toldo, S.; Abbate, A.; Salloum, F.N. Reperfusion therapy with recombinant human relaxin-2 (Serelaxin) attenuates myocardial infarct size and NLRP3 inflammasome following ischemia/reperfusion injury via eNOS-dependent mechanism. Cardiovasc. Res. 2017, 113, 609–619. [Google Scholar]
- Boccalini, G.; Sassoli, C.; Formigli, L.; Bani, D.; Nistri, S. Relaxin protects cardiac muscle cells from hypoxia/reoxygenation injury: involvement of the Notch-1 pathway. FASEB J. 2015, 29, 39–249. [Google Scholar] [CrossRef]
- Nistri, S.; Fiorillo, C.; Becatti, M.; Bani, D. Human Relaxin-2 (Serelaxin) Attenuates Oxidative Stress in Cardiac Muscle Cells Exposed In Vitro to Hypoxia-Reoxygenation. Evidence for the Involvement of Reduced Glutathione Up-Regulation. Antioxidants 2020, 9, 774. [Google Scholar] [CrossRef]
- Waza, A.A.; Hamid, Z.; Bhat, S.A.; Shah, N.U.D.; Bhat, M.; Ganai, B. Relaxin protects cardiomyocytes against hypoxia-induced damage in in-vitro conditions: Involvement of Nrf2/HO-1 signaling pathway. Life Sci. 2018, 213, 225–231. [Google Scholar] [CrossRef]
- Bathgate, R.A.; Halls, M.L.; van der Westhuizen, E.T.; Callander, G.E.; Kocan, M.; Summers, R.J. Relaxin family peptides and their receptors. Physiol. Rev. 2013, 93, 405–480. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.Y.; Li, X.; Hung, C.H.; Bahudhanapati, H.; Tan, J.; Kass, D.J.; Zhang, Y. The relaxin family peptide receptor 1 (RXFP1): An emerging player in human health and disease. Mol. Genet. Genomic. Med. 2020, 8, e1194. [Google Scholar] [CrossRef] [PubMed]
- Boccalini, G.; Sassoli, C.; Bani, D.; Nistri, S. Relaxin induces up-regulation of ADAM10 metalloprotease in RXFP1-expressing cells by PI3K/AKT signaling. Mol. Cell Endocrinol. 2018, 472, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Valkovic, A.L.; Bathgate, R.A.; Samuel, C.S.; Kocan, M. Understanding relaxin signalling at the cellular level. Mol. Cell Endocrinol. 2019, 487, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Aragón-Herrera, A.; Feijóo-Bandín, S.; Rodríguez-Penas, D.; Roselló-Lletí, E.; Portolés, M.; Rivera, M.; Bigazzi, M.; Bani, D.; Gualillo, O.; González-Juanatey, J.R.; Lago, F. Relaxin activates AMPK-AKT signaling and increases glucose uptake by cultured cardiomyocytes. Endocrine 2018, 60, 103–111. [Google Scholar] [CrossRef] [PubMed]
- D'Onofrio, N.; Servillo, L.; Balestrieri, M.L. SIRT1 and SIRT6 Signaling Pathways in Cardiovascular Disease Protection Antioxid. Redox Signal. 2018, 28, 711–732. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhou, M.; Ge, Y.; Wang, X. SIRT1 and aging related signaling pathways. Mech. Ageing Dev. 2020, 187, 111215. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Ubaid, S. Role of Silent Information Regulator 1 (SIRT1) in Regulating Oxidative Stress and Inflammation. Inflammation 2020, 43, 1589–1598. [Google Scholar] [CrossRef]
- Hsu, C.P.; Zhai, P.; Yamamoto, T.; Maejima, Y.; Matsushima, S.; Hariharan, N.; Shao, D.; Takagi, H.; Oka, S.; Sadoshima, J. Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation 2010, 122, 2170–2182. [Google Scholar] [CrossRef]
- Becatti, M.; Taddei, N.; Cecchi, C.; Nassi, N.; Nassi, P.A.; Fiorillo, C. SIRT1 modulates MAPK pathways in ischemic-reperfused cardiomyocytes. Cell Mol. Life Sci. 2012, 69, 2245–2260. [Google Scholar]
- Yu, L.; Li, Q.; Yu, B.; Yang, Y.; Jin, Z.; Duan, W.; Zhao, G.; Zhai, M.; Liu, L.; Yi, D.; Chen, M.; Yu, S. Berberine attenuates myocardial ischemia/reperfusion injury by reducing oxidative stress and inflammation response: Role of silent information regulator 1. Oxid. Med. Cell. Longev. 2016, 1689602. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Cao, W.; Yue, R.; Yuan, Y.; Guo, X.; Qin, D.; Xing, J.; Wang, X. Pretreatment with Tilianin improves mitochondrial energy metabolism and oxidative stress in rats with myocardial ischemia/reperfusion injury via AMPK/SIRT1/PGC-1 alpha signaling pathway. J. Pharmacol. Sci. 2019, 139, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Chen, X. , Liu, X.; Li, Z.; Shi, A.; Tang, X.; Xia, P.; Zhang, J.; Yu, P. AMPK: The key to ischemia-reperfusion injury. J. Cell Physiol. 2022, 237, 4079–4096. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliot, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Hescheler, J.; Meyer, R.; Plant, S.; Krautwurst, D.; Rosenthal, W.; Schultz, G. Morphological, biochemical, and electrophysiological characterization of a clonal cell (H9c2) line from rat heart. Circ. Res. 1991, 69, 1476–1486. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, C.; Zhang, L.; Lv, J.; Ni, H. Rutin alleviates hypoxia/reoxygenation-induced injury in myocardial cells by up-regulating SIRT1 expression. Chem. Biol. Interact. 2019, 297, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Feng, J.; Zhang, J.; Kang, X.; Qian, D. Quercetin modulates AMPK/SIRT1/NF-κB signaling to inhibit inflammatory/oxidative stress responses in diabetic high fat diet-induced atherosclerosis in the rat carotid artery. Experimental and Therapeutic Medicine 2020, 20, 280. [Google Scholar] [CrossRef] [PubMed]
- Cen, Y.; Liao, W.; Wang, T.; Zhang, D. APPL1 ameliorates myocardial ischemia-reperfusion injury by regulating the AMPK signaling pathway. Exp. Ther. Med. 2022, 23, 157. [Google Scholar] [CrossRef]
- Pini, A.; Boccalini, G.; Baccari, M.C.; Becatti, M.; Garella, R.; Fiorillo, C.; Calosi, L.; Bani, D.; Nistri, S. Protection from cigarette smoke-induced vascular injury by recombinant human relaxin-2 (serelaxin). J. Cell. Mol. Med. 2016, 20, 891–902. [Google Scholar] [CrossRef]
- Pini, A.; Boccalini, G.; Lucarini, L.; Catarinicchia, S.; Guasti, D.; Masini, E.; Bani, D.; Nistri, S. Protection from Cigarette Smoke-Induced Lung Dysfunction and Damage by H2 Relaxin (Serelaxin). J. Pharmacol. Exp. Ther. 2016, 357, 451–458. [Google Scholar] [CrossRef]
- Chen, L.; Li, S.; Zhu, J.; You, A.; Huang, X.; Yi, X.; Xue, M. Mangiferin prevents myocardial infarction-induced apoptosis and heart failure in mice by activating the Sirt1/FoxO3a pathway. J. Cell. Mol. Med. 2021, 25, 2944–2955. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Cui, M.; Lin, H.; Zhao, L.; Wang, J.; Chen, S.; Shao, Z. Melatonin resists oxidative stress-induced apoptosis in nucleus pulposus cells. Life Sci. 2018, 199, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Chilian, W.; Crea, F. , Davidson, S.M.; Ferdinandy, P.; Garcia-Dorado, D.; van Royen, N.; Schulz, R.; Heusch, G. The coronary circulation in acute myocardial ischaemia/reperfusion injury: a target for cardioprotection. Cardiovasc. Res. 2019, 115, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Quan, N.; Sun, W.; Chen, X.; Cates, C.; Rousselle, T.; Zhou, X.; Zhao, X.; Li, J. Cardiomyocyte-specific deletion of Sirt1 gene sensitizes myocardium to ischaemia and reperfusion injury. Cardiovasc. Res. 2018, 114, 805–821. [Google Scholar] [CrossRef]
- Duan, J.; Guan, Y.; Mu, F.; Guo, C.; Zhang, E.; Yin, Y.; Wei, G.; Zhu, Y.; Cui, J.; Cao, J.; Weng, Y.; Wang, Y.; Xi, M.; Wen, A. Protective effect of butin against ischemia/reperfusion-induced myocardial injury in diabetic mice: Involvement of the AMPK/GSK-3beta/Nrf2 signaling pathway. Scientific Reports 2017, 7, 41491. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, X.; Zhang, W.; He, J.; Xu, B.; Lei, B.; Wang, Z.; Cates, C.; Rousselle, T.; Li, J. Activation of AMPK inhibits inflammatory response during hypoxia and reoxygenation through modulating JNK-mediated NF-κB pathway. Metabolism. 2018, 83, 56–270. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Hong, J.; Hu, C.; Huang, C.; Gao, J.; Huang, J.; Wang, D.; Geng, Q.; Dong, Y. Palmatine protects against cerebral ischemia/reperfusion injury by activation of the AMPK/Nrf2 pathway. Oxidative Medicine and Cellular Longevity 2021, 6660193. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, A.; Suuronen, T.; Ojala, J.; Kaarniranta, K.; Salminen, A. Antagonistic crosstalk between NF-κB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal. 2013, 25, 1939–1948. [Google Scholar] [CrossRef]
- Sung, B.; Chung, J.W.; Bae, H.R.; Choi, J.S.; Kim, C.M.; Kim, N.D. Humulus japonicus extract exhibits antioxidative and anti-aging effects via modulation of the AMPK-SIRT1 pathway. Exp. Ther. Med. 2015, 9, 1819–1826. [Google Scholar] [CrossRef]
- Han, X.; Tai, H.; Wang, X.; Wang, Z.; Zhou, J.; Wei, X.; Ding, Y.; Gong, H.; Mo, C.; Zhang, J.; Qin, J.; Ma, Y.; Huang, N.; Xiang, R.; Xiao, H. AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD(+) elevation. Aging Cell. 2016, 15, 416–427. [Google Scholar] [CrossRef]
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J. Biol. Chem. 2008, 283, 27628–27635. [Google Scholar] [CrossRef]
- Yang, M.; Lv, H.; Liu, Q.; Zhang, L.; Zhang, R.; Huang, X.; Wang, X.; Han, B.; Hou, S.; Liu, D.; Wang, G.; Hou, J.; Yu, B. Colchicine Alleviates Cholesterol Crystal-Induced Endothelial Cell Pyroptosis through Activating AMPK/SIRT1 Pathway. Oxid. Med. Cell Longev. 2020, 2020, 9173530. [Google Scholar] [CrossRef]
- Lu, C.; Jiang, B.; Xu, J.; Zhang, X.; Jiang, N. Neferine protected cardiomyocytes against hypoxia/oxygenation injury through SIRT1/Nrf2/HO-1 signaling. J. Biochem. Mol. Toxicol. 2023, 37, e23398. [Google Scholar] [CrossRef]
- Harvey, C.J.; Thimmulappa, R.K.; Singh, A.; Blake, D.J.; Ling, G.; Wakabayashi, N.; Fujii, J.; Myers, A.; Biswal, S. Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic. Biol. Med. 2009, 46, 443–453. [Google Scholar]
Figure 1.
RLX upregulates SIRT1 expression in H9c2 cells exposed to H/R. Representative images of Western blotting and quantitative analysis showing SIRT1 and β actin expression in H9c2 cells under normoxia (A,B) and upon H/R challenge (C,D). Significance of differences (B: Student’s unpaired t-test; D: one-way ANOVA and Student-Newman-Keuls multiple comparison test): ***p<0.001 vs. controls (Ctrl); ##p<0.01 vs. H/R.
Figure 1.
RLX upregulates SIRT1 expression in H9c2 cells exposed to H/R. Representative images of Western blotting and quantitative analysis showing SIRT1 and β actin expression in H9c2 cells under normoxia (A,B) and upon H/R challenge (C,D). Significance of differences (B: Student’s unpaired t-test; D: one-way ANOVA and Student-Newman-Keuls multiple comparison test): ***p<0.001 vs. controls (Ctrl); ##p<0.01 vs. H/R.
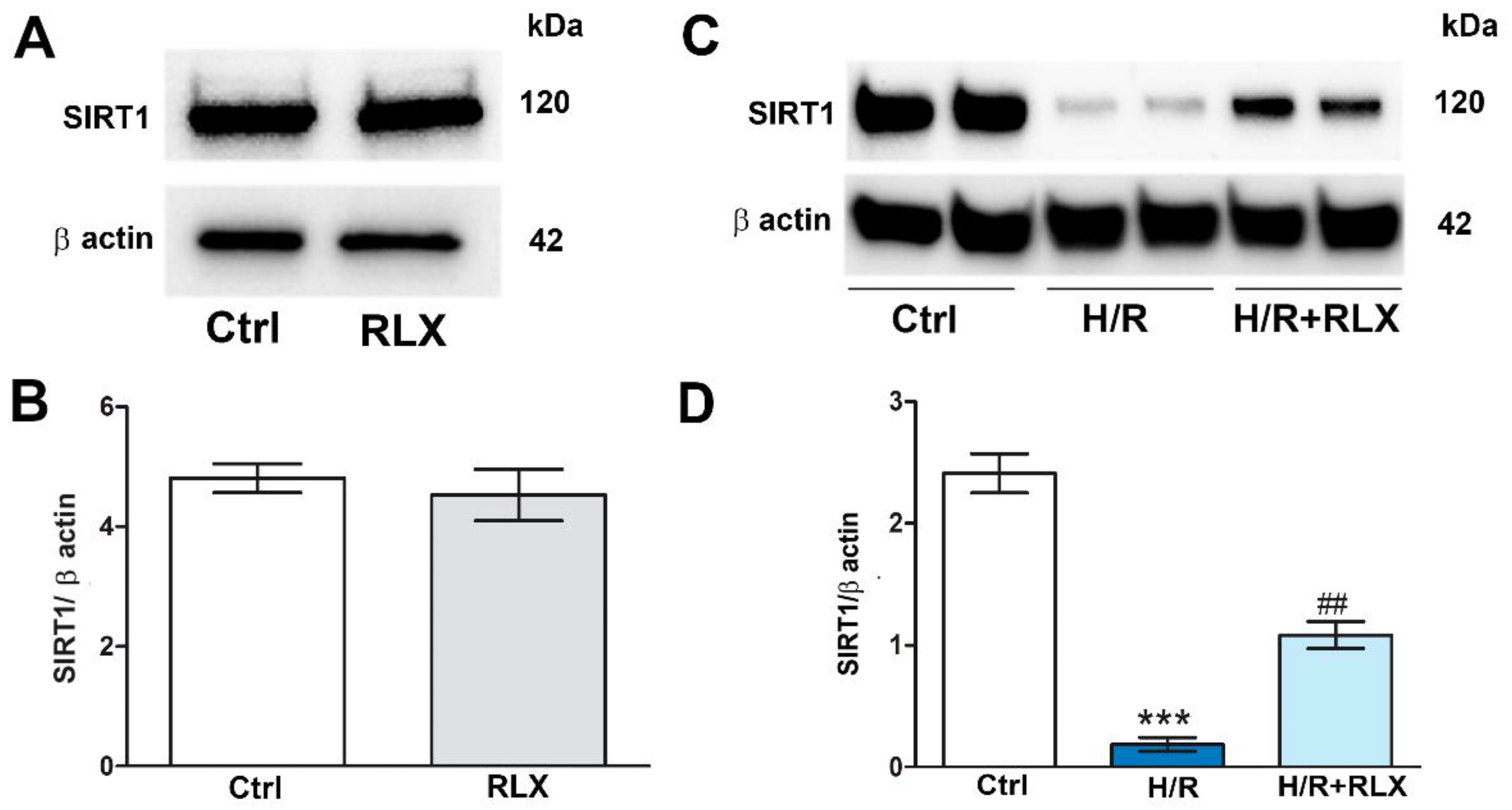
Figure 2.
RLX upregulates SIRT1 activity in H9c2 cells exposed to H/R. Representative diagrams showing SIRT1 activity in H9c2 in normoxia (A) and upon H/R challenge (B). Significance of differences (A: Student’s unpaired t-test; B: one-way ANOVA and Student-Newman-Keuls multiple comparison test): ***p<0.001 vs. controls (ctrl); ###p<0.001 vs. H/R.
Figure 2.
RLX upregulates SIRT1 activity in H9c2 cells exposed to H/R. Representative diagrams showing SIRT1 activity in H9c2 in normoxia (A) and upon H/R challenge (B). Significance of differences (A: Student’s unpaired t-test; B: one-way ANOVA and Student-Newman-Keuls multiple comparison test): ***p<0.001 vs. controls (ctrl); ###p<0.001 vs. H/R.
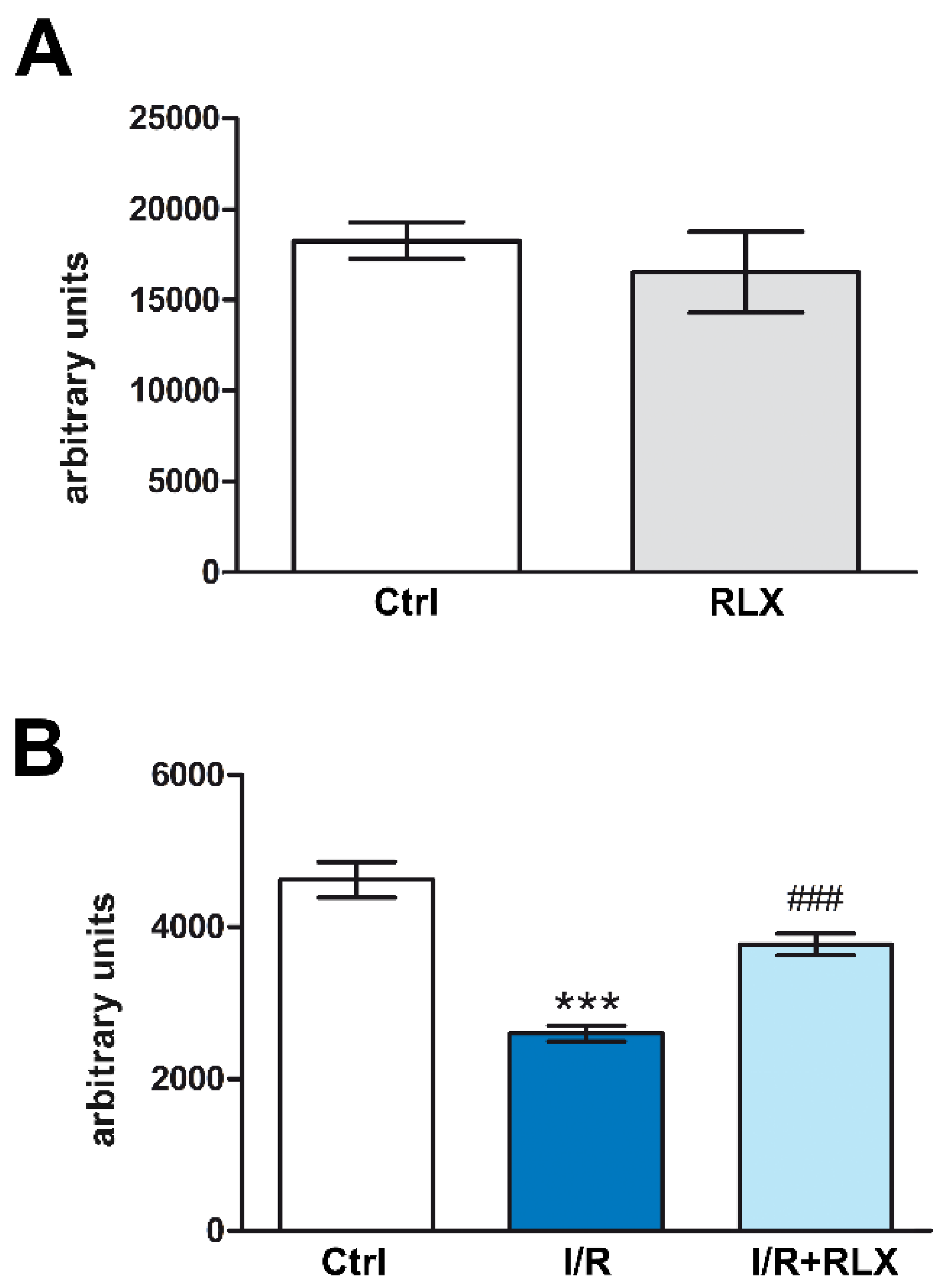
Figure 3.
Inhibition of SIRT1 signaling counteracts the beneficial effects of RLX on mitochondrial respiratory chain efficiency in H9c2 cells exposed to H/R. Representative diagram showing mitochondrial activity, assayed by MTT, of H9c2 cells in normoxia (Ctrl) and under H/R in the presence or absence of the selective SIRT1 inhibitor, EX527 (1 and 10 µmol L−1). Significance of differences (One-way ANOVA and Student-Newman-Keuls multiple comparison test): ***p<0.001 vs. controls (Ctrl); #p<0.05 vs. H/R; §p<0.05 vs. H/R+RLX.
Figure 3.
Inhibition of SIRT1 signaling counteracts the beneficial effects of RLX on mitochondrial respiratory chain efficiency in H9c2 cells exposed to H/R. Representative diagram showing mitochondrial activity, assayed by MTT, of H9c2 cells in normoxia (Ctrl) and under H/R in the presence or absence of the selective SIRT1 inhibitor, EX527 (1 and 10 µmol L−1). Significance of differences (One-way ANOVA and Student-Newman-Keuls multiple comparison test): ***p<0.001 vs. controls (Ctrl); #p<0.05 vs. H/R; §p<0.05 vs. H/R+RLX.
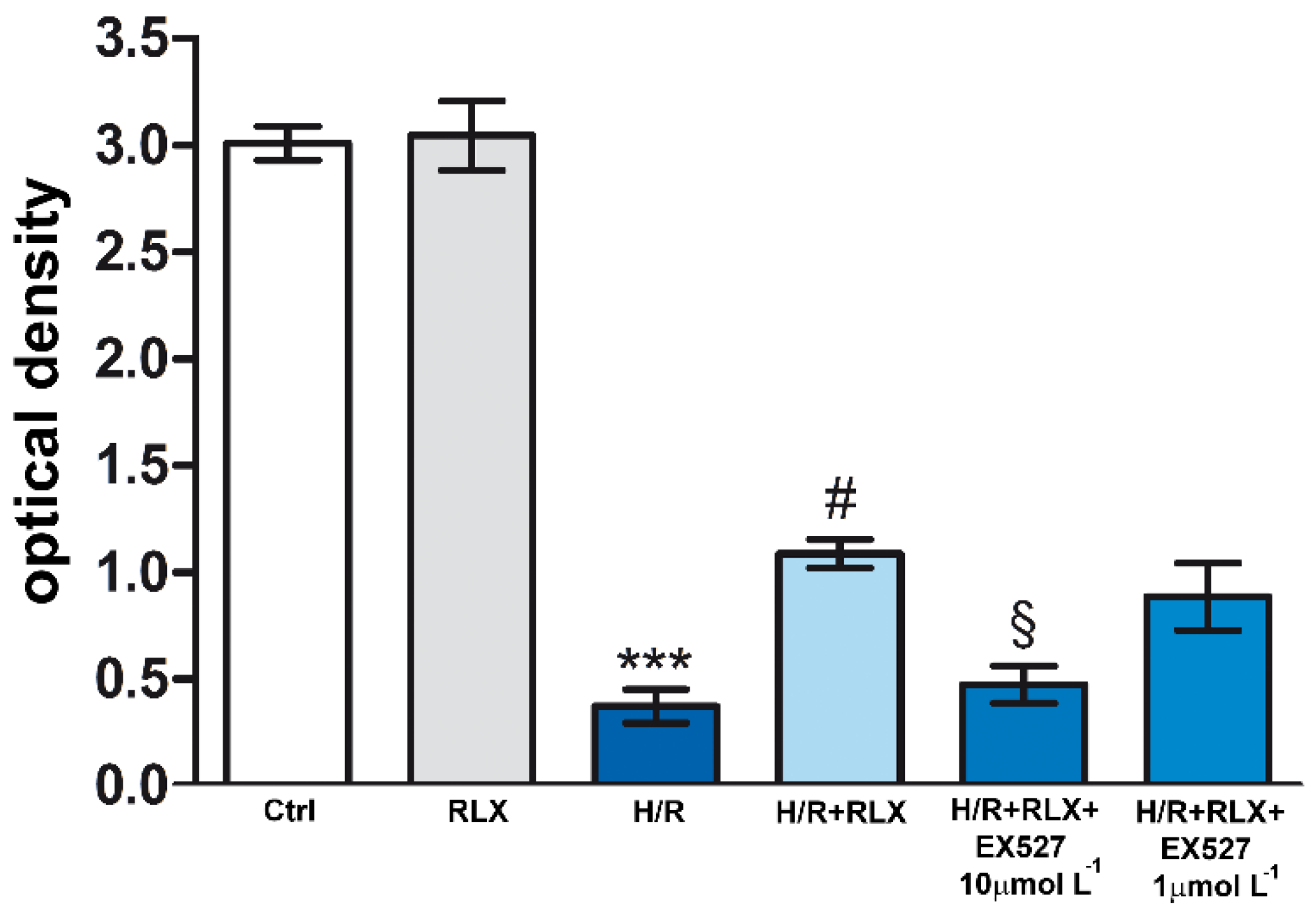
Figure 4.
Inhibition of SIRT1 signaling counteracts the beneficial effects of RLX on apoptosis of H9c2 cells exposed to H/R. Representative image of Western blotting (A) and quantitative analysis (B) showing the expression of cleaved and full-length caspase 3 and β actin in H9c2 cells under normoxia (Ctrl) and upon H/R challenge in the presence or absence of the selective SIRT1 inhibitor EX527 (1 and 10 µmol L−1). Significance of differences (One-way ANOVA and Student-Newman-Keuls multiple comparison test): ###p<0.001 vs. H/R; §§p<0.01 vs. H/R+RLX.
Figure 4.
Inhibition of SIRT1 signaling counteracts the beneficial effects of RLX on apoptosis of H9c2 cells exposed to H/R. Representative image of Western blotting (A) and quantitative analysis (B) showing the expression of cleaved and full-length caspase 3 and β actin in H9c2 cells under normoxia (Ctrl) and upon H/R challenge in the presence or absence of the selective SIRT1 inhibitor EX527 (1 and 10 µmol L−1). Significance of differences (One-way ANOVA and Student-Newman-Keuls multiple comparison test): ###p<0.001 vs. H/R; §§p<0.01 vs. H/R+RLX.
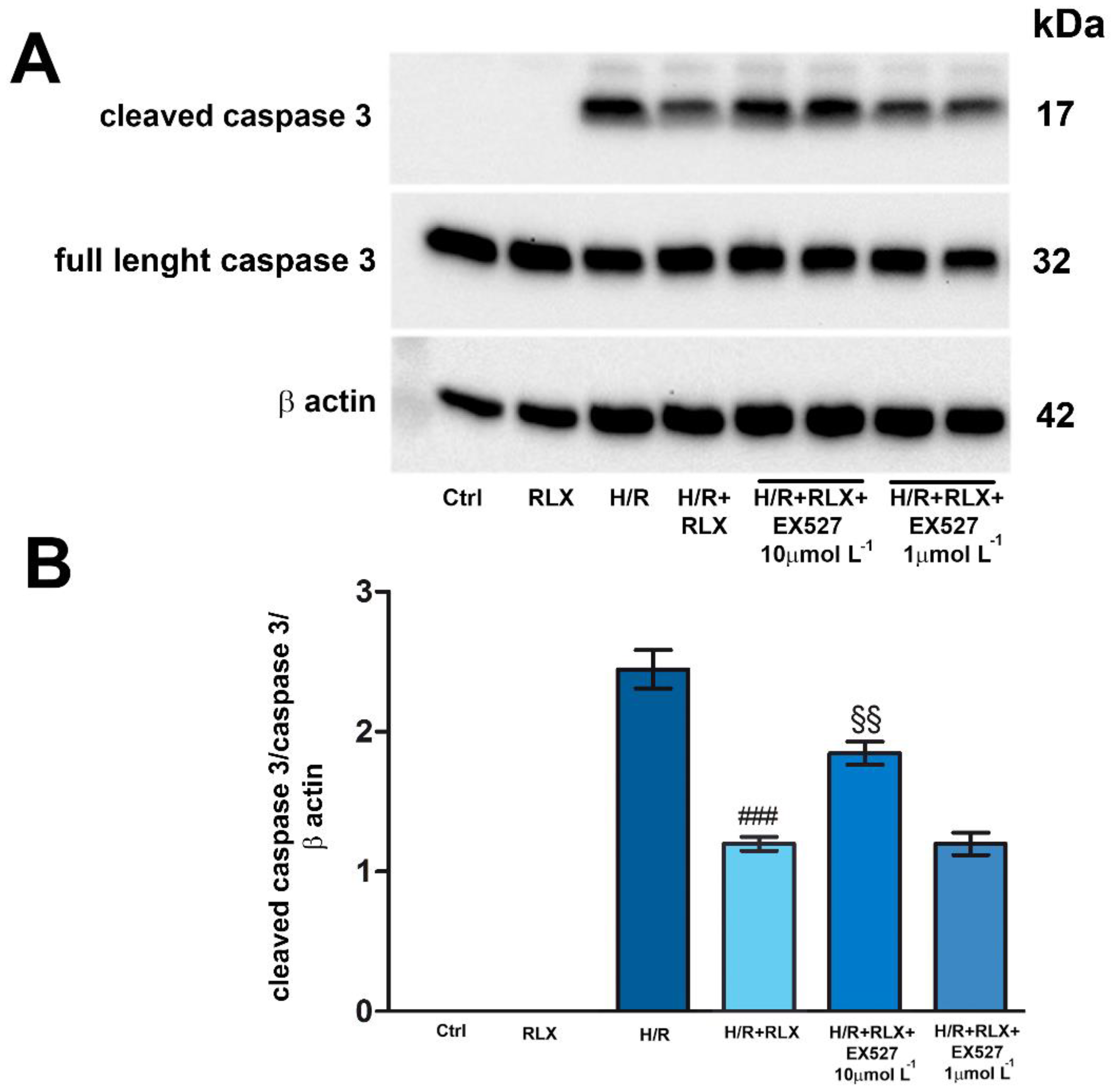
Figure 5.
RLX treatment activates AMPK signaling in H9c2 cells exposed to H/R. Representative images of Western blotting (A) and quantitative analysis (B) showing phospho-AMPK, AMPK and β actin expression in H9c2 cells upon H/R challenge. Significance of differences (One-way ANOVA and Student-Newman-Keuls multiple comparison test): ***p<0.001 vs. controls (Ctrl); ##p<0.01 vs. H/R.
Figure 5.
RLX treatment activates AMPK signaling in H9c2 cells exposed to H/R. Representative images of Western blotting (A) and quantitative analysis (B) showing phospho-AMPK, AMPK and β actin expression in H9c2 cells upon H/R challenge. Significance of differences (One-way ANOVA and Student-Newman-Keuls multiple comparison test): ***p<0.001 vs. controls (Ctrl); ##p<0.01 vs. H/R.
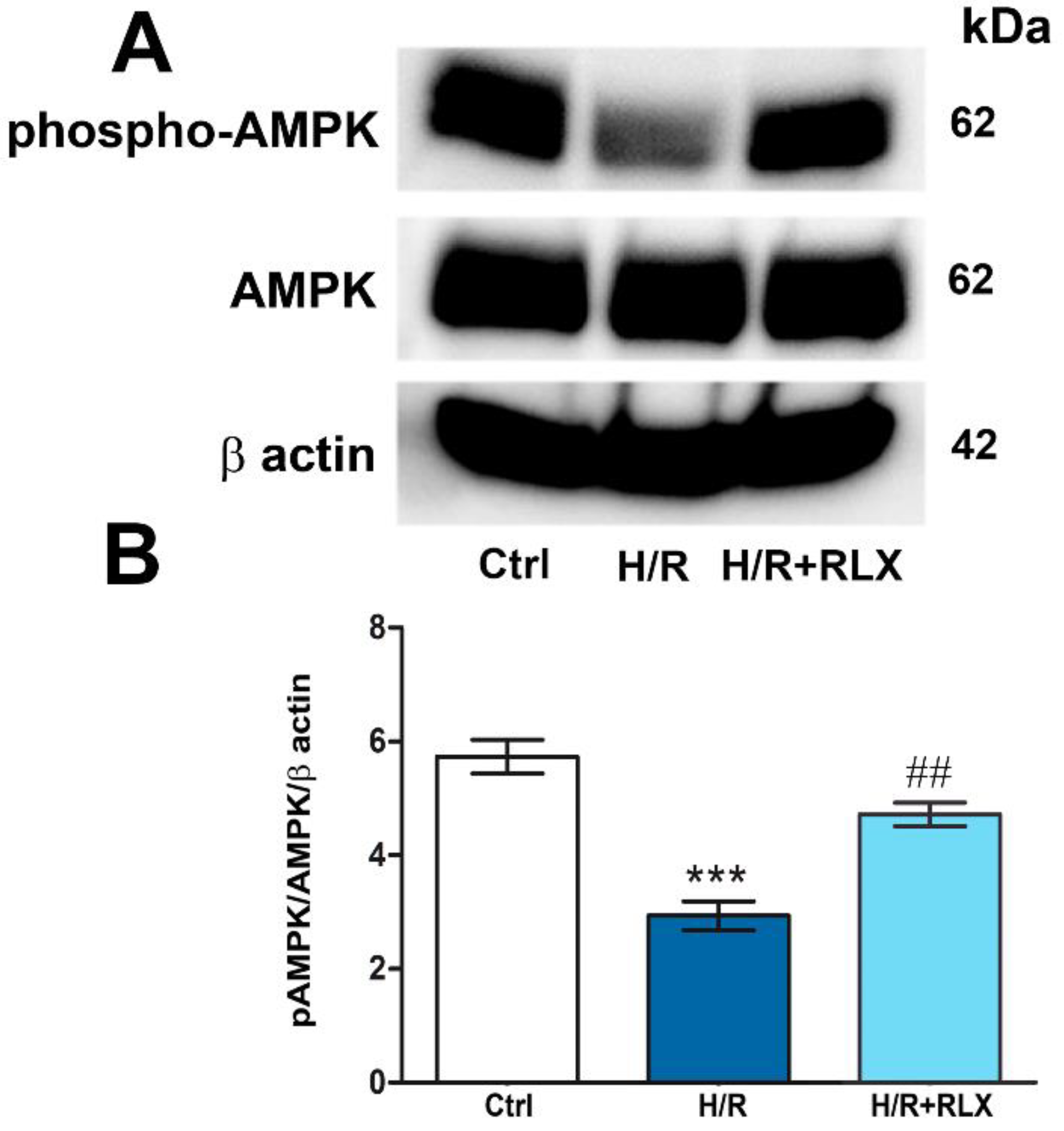
Figure 6.
Inhibition of AMPK signaling counteracts the up-regulation of SIRT1 expression induced by RLX in H9c2 cells exposed to H/R. Representative images of Western Blotting (A) and quantitative analysis (B) showing SIRT1 and β actin expression in H9c2 cells upon H/R challenge in the presence or absence of the selective AMPK inhibitor Compound C (10 and 20 µmol L−1). Significance of differences (One-way ANOVA and Student-Newman-Keuls multiple comparison test): ***p<0.001 vs. controls (Ctrl); ###p<0.001 vs. H/R; §§§p<0.001 vs. H/R+RLX.
Figure 6.
Inhibition of AMPK signaling counteracts the up-regulation of SIRT1 expression induced by RLX in H9c2 cells exposed to H/R. Representative images of Western Blotting (A) and quantitative analysis (B) showing SIRT1 and β actin expression in H9c2 cells upon H/R challenge in the presence or absence of the selective AMPK inhibitor Compound C (10 and 20 µmol L−1). Significance of differences (One-way ANOVA and Student-Newman-Keuls multiple comparison test): ***p<0.001 vs. controls (Ctrl); ###p<0.001 vs. H/R; §§§p<0.001 vs. H/R+RLX.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



