
Submitted:
15 December 2023
Posted:
19 December 2023
You are already at the latest version
Abstract
Introduction: Maturity-onset diabetes of the young (MODY) represents the most frequent form of monogenic diabetes mellitus (DM), currently classified in 14 distinct subtypes according to single gene mutations involved in the differentiation and function of pancreatic β-cells.
Case report: The female patient at the age of 16 years old was diagnosed with non-autoimmune DM with glycated haemoglobin (HbA1c) value of 6.5%. After 6 years of dietary and lifestyle interventions due to the worsening of the glycometabolic profile, oral hypoglicemic therapy was initiated including at first only metformin, and subsequently also sulfonylureas and DPP-IV inhibitor. 11 years after diagnosis glargine insulin was started, associated 3 years later with Lispro insulin because of the worsening of the post-prandial glycaemia. According to the genetic testing a compound heterozygosis of two novel variants of uncertain/unknown clinical significance of BLK and RFX6 genes, respectively c.1013T>C (Ile338Thr) and c.1072G>A (Val358Ile), was identified. The RFX6 variant was also detected in the patient’s mother genome, while the BLK variant in her father’s.
Discussion: Up to 11% of MODY has unknown etiology, suggesting that the genetic landscape is still to be explored. Recently, novel causal genes, involved in the differentiation and function of β-cells, have been identified such as RFX6, NKX2.2, NKX6.1, WFS1 and PCBD1. Genetic and clinical features of MODY variants remain highly heterogeneous, with no direct genotype-phenotype correlation especially in the low-penetrant subtypes. Thus, the aim of our paper is to present additionally to such rare case of a compound heterozygosity BLK/RFX6 underlying MODY, also a brief review of literature focused on the rare and reduced-penetrant variants, as well as on some controversies regarding this subtype of monogenic diabetes.
Conclusions: Due to the limited data available, the assessment of MODY related-genes pathogenicity remains challenging especially in the setting of rare and low-penetrant subtypes. In consideration of the crucial importance of an accurate diagnosis, prognosis and management of MODY, more studies are warranted to further investigate its genetic landscape, the genotype-phenotype correlation, as well as the pathogenetic contribution of the non-genetic modifiers in this cohort of patients.
Keywords:
maturity-onset diabetes of the young
; MODY
; monogenic diabetes
; diabetes mellitus
1. Introduction
Maturity-onset diabetes of the young (MODY) represents the most frequent form of monogenic diabetes mellitus (DM) with an estimated prevalence of 108 cases per million. (1) The diagnostic criteria of the Practice Guideline for MODY in 2008 comprise early onset of diabetes before 25 years of age, history of diabetes in two consecutive generations, absence of pancreatic β-cell autoimmunity, and preserved β-cell function defined by the lack of insulin treatment or serum C-peptide levels > 200 pmol/L after 3 years of insulin therapy (2) To date, at least 14 distinct MODY subtypes have been identified according to single gene mutations inherited by autosomal dominant pattern, and involved in the differentiation, development and function of β-cells. (3, 4) Such heterogeneity significantly impacts diabetes pathophysiology, clinical course and treatment response, which highly differ among all MODY forms. (5) Ranging from 80% to 95% of MODY cases the underlying cause involves the pathogenetic variants of hepatocyte nuclear factor-1 homeobox A (HNF1A), hepatocyte nuclear factor-4 homeobox A (HNF4A), hepatocyte nuclear factor-1 homeobox B (HNF1B) and glucokinase (GCK) genes. (3, 6) However, despite the molecular evidence, up to 11% of MODY has unknown etiology, which suggests that the genetic landscape is still to be explored. (8) Genetic and clinical characterization of MODY variants is highly heterogeneous, with no direct genotype-phenotype correlation, especially in the rare and low-penetrant subtypes. Recently, novel causal genes, involved in the differentiation and function of β-cells, have been detected such as RFX6, NKX2.2, NKX6.1, WFS1 and PCBD1. (7, 8, 9, 10, 11, 12) This paper aims to present a rare case of the compound heterozygosity BLK/RFX6 as an underlying genetic cause of MODY. In addition, a brief review of the literature is conducted on the rare and low-penetrant MODYs, as well as on the controversies associated with this subtype of monogenic diabetes.
2. Case Report
The patient, a single daughter of a non-consanguineous couple, was diagnosed with diabetes mellitus at the age of 16 years old in 2007. At presentation, her glycated haemoglobin (HbA1c) and body mass index (BMI) were about 6.5% and 21 kg/m2, respectively. Glutamic acid decarboxylase antibody (GAD-Ab) and islet cell antibody (ICA) were negative. Screening for autoimmune thyroid and celiac disease resulted also negative. At diagnosis, there was no retinopathy or neuropathy. Our patient has been an active smoker since 2005 (10 cigarettes/die). Her family history was positive for type II diabetes mellitus (T2DM) in both grandmothers (Figure 1) For the first six years after diagnosis, our patient underwent only dietary and physical activity interventions obtaining an adequate glycaemic control with HbA1c about 6.5%. In 2013 due to the worsening of the glycometabolic profile (HbA1c 8.8%) and ponderal growth of 7 kg in 2 years (body weight 58 kg, BMI 24 kg/m2), also subsequent to the low adherence to the lifestyle changes, pharmacological treatment with metformin was initiated. Concurrently, our patient was diagnosed with familial hypercholesterolemia (LDL cholesterol 190 mg/dL, triglycerides 362 mg/dL) and started hypocholesterolemic therapy. For the following two years, an improvement of both glyco-lipid status (HbA1c 6.6-7.3%) and weight (50 kg, BMI 20 kg/m2) was achieved. However, since 2015 due to a progressive worsening of the glycaemic control, the oral hypoglycaemic therapy was subsequently modified: gliclazide combined with metformin (2016), metformin associated with sitagliptin (2017), glipizide (2020). Moreover, since 2018 insulin therapy was initiated with glargine insulin, and since 2021 due to the increase of the post-prandial glycemia also Lispro insulin was introduced. In consideration of the young age at diabetes onset, the absence of pancreatic autoantibodies, the familial history for diabetes mellitus, the lack of obesity and the variable clinical presentation, in 2020 our patient underwent genetic testing. The genomic Next-Generation Sequencing (NGS) identified a compound heterozygosis of two novel variants of uncertain/unknown clinical significance located in the genes BLK and RFX6, respectively c.1013T>C (Ile338Thr) and c.1072G>A (Val358Ile). Moreover, RFX6 variant c.1072G>A was detected in her mother’s genome, meanwhile BLK variant c.1013T>C in her father’s. Up to now, neither of the parents has presented a history of altered glucose metabolism. During the follow-up period, our patient had an overall variable clinical course and non-optimal compliance to dietary, lifestyle and pharmacological interventions.
The genetic pedigree of the patient is illustrated in Figure 1.
3. A Brief Review of Rare and Reduced-Penetrant Modys
MODY is defined as a single gene defective diabetes, encompassing highly genetically and clinically heterogenous variants, where genotype is not always predictive of a specific phenotype. (12) To assess the pathogenicity of the causal mutations, scoring systems have been implemented, nonetheless, statistical evidence is well-established only for some major high-penetrating genes such as GCK, HNF1A, HNF4A, NEUROD1, KCNJ11 and HNF1B. (12) In rare MODYs, mutations appear to be associated with reduced penetrance and small mutagenic impact. (13) Therefore, it is plausible that the diversity observed in the penetrance rate of MODY may be likely related to both genetic and non-genetic modifiers. (14) Molecular factors could comprise epigenetic modulation, genetic variant factors, or allele expression patterns. Moreover, similarly to T2DM, other modifiers such as environmental conditions, intrauterine growth, or ethnicity could also contribute to the development of diabetes in this cohort of patients. (13) However, additional studies are warranted to further investigate the molecular mechanisms and the role of non-genetic factors in MODY pathophysiology. (8) Considering the evaluation bias related to clinical assessment, accurate data of prevalence and penetrance regarding rare and low-penetrant MODYs is generally not well-known. (14) Using the latest sequencing techniques, novel MODY causal genes have been recently reported in the literature such as RFX6, RFX6 protein-truncating variant (RFX6 PTV), NKX2.2, NKX6.1, WFS1, PCBD1. (7, 8, 9, 10, 11) In these rare variants no direct genotype-phenotype correlation is observed most likely due to their incomplete penetrance. (7)
Information regarding the identities of these genes, their function and pathophysiology is presented in Table 1.
3.1. RFX6 (Regulatory factor X6)
RFX6 (6q22.1) encodes RFX6, a component of the RFX family of winged-helix transcription factors (TFs) involved in pancreatic islet cell differentiation and function. (11) RFX6, initially expressed in the endoderm, is limited at mid-gestation to gut and pancreatic bud, and then becomes progressively and ultimately restricted to the endocrine lineage in the mature pancreas. (8, 15) Genetic studies in both mice and humans showed that during embryogenesis, RFX6 contributes to direct the β-cell and other islet cells development downstream of the transcription factor neurogenin 3 (NEUROG3), as well as upstream of several islet transcription regulators such as NeuroD, Pax4, Arx. (15, 16) Moreover, RFX6 participates also in modulating insulin secretion in human β-cells by upregulating the expression of the insulin gene and of other genes involved in insulin secretion such as glucokinase and voltage-dependent calcium channel. (17) Hence, RFX6 may contribute to both pancreatic islet maturation and insulin production in distinct and independent pathways. (18) Biallelic mutations of RFX6 are associated with Mitchey-Rilley syndrome, a rare autosomal recessive disorder comprising neonatal diabetes, pancreatic hypoplasia, gallbladder agenesis, duodenal and jejunal atresia. In such context, the severe diabetes phenotype is a consequence of the aberrant pancreatic islet maturation and function, including insulin production. (16, 18, 19) Heterozygous mutations of RFX6 and RFX6 TPV, instead, have been linked to isolated MODY with reduced penetrance. (7, 20) In this cohort of patients, diabetes pathogenesis is supported by insulin deficiency associated, however, with normal development of the pancreatic islets. (18, 21) Phenotypically, RFX6 TPV-related MODY was described in the study of Patel et al. enrolling 27 patients, as mild and with a median age at diagnosis of 32 years old. At recruitment endogenous insulin levels were significant, but insulin treatment was required after 10 years of diabetes in 69% of patients. No relevant sensitivity to sulphonylurea was documented. (8) Moreover, differently from other forms of diabetes comprising T2DM, T1DM and HFN1A-MODY, RFX-6 MODY was associated with a decreased glucose-dependent insulinotropic peptide (GIP) secretion. (8) In the case report of Imaki et al. regarding a case of MODY caused by a novel heterozygous RFX6 mutation p.R652X, the patient presented in addition to a reduced insulin and GIP response, also a reduced glucagon-like peptide 1 (GLP-1) response. Such findings appear in accordance with the lower GIP and GLP-1 levels detected in RFX6-deficient murine models, and may be suggestive of the potential role of GLP-1 receptor agonist therapy in this subset of patients. (11) Furthermore, also a good response to dipeptidyl peptidase-4 (DDP-4) inhibitors was evidenced in other RFX6-MODY patients. (22) Therefore, clinical research is necessary to better explore the potential efficacy of GLP-1 receptor agonists and DPP-4 inhibitors in this subset of patients.
3.2. NK2.2 (NK2 Homeobox 2)
NKX2.2 (20p11.22) encodes NKX-2.2, a member of the mammalian Nk2 homeobox transcriptional regulators, which plays a crucial role in the pancreatic islet cell differentiation, as well as in the morphogenesis of the ventral central nervous system and of the epithelial enteroendocrine cells (23) Within the pancreas, NKX2.2 is early expressed in progenitor cells during embryogenesis, and then progressively limited to α-, β- and pancreatic polypeptide (PP) cells. (24) Murine studies provide evidence of the essential NKX2.2 involvement not only during the initial islet cell specification, but also in the maintenance of mature β-cell function and in the establishment of proper islet architecture. (24) Consistently to the role of NKX2.2 documented in mice, in humans recessive loss-of-function mutations of NKX2.2 are associated with development delay, neonatal diabetes and obesity. (25) Regarding obesity, its pathogenesis may be linked to higher levels of ghrelin, an orexigenic hormone, most likely subsequent to the change of pancreatic islet cell differentiation, due to the absent NKX2.2 function, in favor of ghrelin-producing cells rather than the α-, β- and PP cells. (23, 25) Pathogenetic variants of NKX2.2 have been detected also in extremely rare MODY cases when transmitted by heterozygous inheritance pattern. (7) So far, in consideration of the limited data in the literature and of the limited analysis of phenotypes among the family members of these patients, it results difficult to understand precisely the impact of such variants on MODY development, as well as the specific genotype-phenotype correlations (7)
3.3. NKX6.1 (NK6 Homeobox 1)
NKX6.1 (4q21.23) encodes homeobox protein NKX6.1, a transcriptional factor (TF) involved in the pancreatic β-cell differentiation and function, as well as in the neural development and moto neuron specification. (26, 27) In particular, NKX6.1 expression is essential both in initial and late stages of pancreatic development for the specification of multipotent cells (MPC) into functional β-cell lineage. In the early pancreatic progenitor phase, NKX6.1 especially with PTF1A (pancreas transcription factor 1 α), an important regulator of acinar gene transcription, control pancreatic cell fate by coordinating the balance in differentiation, proliferation and maturation of both endocrine and exocrine cells. Moreover, in mature β-cells NKX6.1 plays a crucial role in the regulation of cell functional properties, including insulin production, glucose uptake and metabolism, and cell proliferation. Despite the fact that in human pancreatic islets NKX6.1 is exclusively limited to β-cells and finalized to maintain cell identity, however, NKX6.1 contributes also to the suppression of α-cell development by regulating glucagon gene (GCG) expression. (26, 28) Generally reduced expression of crucial TFs, comprising Nkx6.1, is associated with destabilized β-cell homeostasis and function. From accumulating evidence in literature decreased levels of NKX6.1 appear to contribute to T2DM pathogenesis both in humans and mice. (26, 29) NKX6.1 deficient β-cells present severely reduced levels of Pcsk1 (PC1), which converts proinsulin to insulin, and low expression of zinc transporter Slc30a8 (oxidoreductase Ero1lb), both mechanisms leading to decreased insulin biosynthesis and secretion. (29) Moreover, also aberrant activation of δ-cell genes in NKX6.1 deficient β-cells could be observed and contribute furthermore to β-cell identity alterations. (26) It has also been evidenced an association between NKX6 mutations and MODY development. In particular, NKX6.1 plays a crucial role in modulating HNF1A expression, which is the causative gene of MODY3. Therefore, it is plausible to consider that NKX6.1 variants could participate in MODY3 pathogenesis by leading to HNF1A altered levels. (30) Furthermore, heterogeneous pathogenic variants of NKX6.1 have been recently associated also with extremely rare cases of MODY. (7)
3.4. WFS1 (Wolframin ER Transmembrane Glycoprotein)
WFS1 (4p16.1) encodes wolframin, a transmembrane protein of the endoplasmatic reticulum expressed ubiquitously, but mostly in pancreatic β-cells, brain and heart. Wolframin modulates endoplasmic reticulum calcium homeostasis and calcium signal transduction processes involved in cellular apoptosis. (31) Regarding pancreas, it is documented that WFS1 aberrations lead to progressive β-cell loss, altered glucose metabolism and impaired insulin secretion. (32) There is data in the literature of WFS1 variants associated with different diabetes phenotypes. Several studies have documented a relevant association of WFS1 and type 2 diabetes mellitus, suggesting a possible contribution of WFS1 gene in T2DM pathogenesis. (33, 34, 35, 36, 37, 38) Moreover, a missense alteration of WFS1 (Arg456His) has been also described in one case of T1DM, which could be interpreted as a potential genetic risk factor for type 1 diabetes. (39) WFS1 mutations are also responsible for diabetes development in the setting of Wolfram Syndrome 1 (WS1), a rare neurodegenerative disorder with other classical features comprising central diabetes insipidus, optic atrophy and neurosensorial deafness. (33) WS1 diabetes presents MODY-like features. (12, 40) In particular, it is early-onset and non auto-immune insulin-dependent. WS1 diabetic patients present an average onset age of 6 years old, ranging from 3 weeks to 16 years, and can be easily misdiagnosed as T1DM. In comparison with type 1 diabetes mellitus, WS1 patients are usually characterized by inferior values of HbA1c, lower doses of insulin requirement, less cases of ketoacidosis at diagnosis and a longer remission period. (33)
3.5. PCBD1 (Pterin-4 alpha-carbinolamine dehydratase 1)
PCBD1 (10q22.1), encodes a dual-functional protein of the PCBD family, expressed differently in several tissues including pancreas, and predominantly in liver and kidney. (41) In the cytoplasm PCBD1 participates in the tetrahydrobiopterin (BH4) biosynthesis, a cofactor for aromatic amino acid hydroxylases. whereas in the nucleus it acts as a dimerization co-activator of both HNF1A and HNF1B by enhancing their transcriptional activity, which is crucial for adequate β-cell differentiation and function. (41) Therefore, PCBD1 by stabilizing the above-mentioned HFN1 factors plays a relevant role both in modulating the progenitor pool during early pancreatic development stages, as well as in maintaining proper homeostasis and function in mature β-cells. (10) Recessive loss-of-function mutations of PCBD1 have been recently associated with early-onset non-autoimmune diabetes with MODY HFN1A-like features, which could benefit from oral antidiabetic treatments such as sulphonylureas or glinides. Thus, in order to optimize the clinical management in this subset of patients, screening for biallelic mutations in PCBD1 has been suggested in case of HNF1A-like diabetes occurrence with absent alterations in HNF1A and HNF1B. Heterozygous variants of PCBD1, instead, may participate in the development of type 2 diabetes particularly when present additionally to other risk factors such as excess weight and age. (10) More studies are warranted to further investigate the pathogenetic mechanisms of PCBD1, the associated diabetes clinical course and the response to treatment.
In addition to the novel single genes above-mentioned, another underlying mechanism of MODY could be represented by the co-inheritance pattern of the compound heterozygosis especially involving low-penetrant variants. In this paper, we document the first case to our knowledge of the linkage between BLK/RFX6 compound mutations and early-onset non-autoimmune diabetes. Unlike RFX6, BLK (B lymphocyte kinase) is already linked to the development of MODY 11. BLK gene (8p23), a member of the SRC tyrosine kinase family is mainly expressed in β-cells and B-lymphocytes. BLK protein is involved in β-cell development, and by upregulating the transcriptional activity of PDX1 and NKX6.1, also controls insulin biosynthesis and secretion. (42) Phenotypically, patients affected by MODY 11 tend to present diabetes in the third to fourth decade of life, are generally overweight or obese, and usually require insulin therapy. Nevertheless, among 21 subjects studied from three families almost 30% presented an adequate response to dietary intervention and oral hypoglycemic treatment. (43) The clinical course in our patient was characterized by an initial response to only dietary and lifestyle interventions for about 6 years, subsequent initiation of oral hypoglycemic therapy, and after 11 years from diagnosis introduction of insulin treatment due to the worsening diabetes control, however in a setting of non-optimal compliance to therapy and follow-up evaluations. Another case of compound heterozygosis was also described by Ivanoshchuk et al., who identified the co-inheritance of variant c.1562G>A (p.Arg521Gln) in the ABCC8 gene (ATP-binding cassette transporter subfamily C member 8) and of variant c.160C>T (p.Arg54*) in the HNF1A gene, respectively associated with MODY 12 and MODY 3, in a diabetic patient and his mother in Western Siberia. While c.160C>T in HFN1A is already defined as pathogenic, Arg521Gln in ABCC8, instead, is characterized by “conflicting interpretations of pathogenicity” or “uncertain significance” (44)
4. Controversial Data of Mody-Related Genetic Background
Moreover, in the literature there is ambiguous data about the etiological role of several genetic aberrations currently linked to MODY. In particular, some variants such as PDX1 (pancreatic duodenal homeobox 1) variant P33T, E224, P242L (MODY 4), HNF4A V169I (MODY 1), BLK A71T (MODY11) and NEUROD1 (neurogenic differentiation factor 1) H241Q (MODY 6) were reported in the study of Mohan et al. to have almost no impact in diabetes development. In fact their prevalence in both MODY patients and the general population of South India was similar. (9) The controversy could be attributed to the limited population-specific data and subsequently, to its challenging interpretation. In addition, considering the distinct diabetes phenotype in the Southern Asian population, a different pattern of gene variation and inheritance of MODY may be considered plausible in this cohort of patients. (9)
Furthermore, a reanalysis of published variants of BLK (MODY 11), PAX4 (Paired Box Gene 4, MODY 9) and KLF11 (Krüppel-like factor 11, MODY 7), failed to demonstrate variant- and gene-level evidence of a causal correlation between these genes and MODY. (1) Thus, based on such results, it was suggested not to include BLK, PAX4 and KLF11 in the diagnostic genetic panels. (1) Accordingly, another study conducted on a European cohort did not document any relevant linkage between BLK gene and MODY development. BLK mutations, instead, resulted slightly associated with T2DM, especially in obese individuals. (45) Additionally, there is evidence in the literature supporting the role of some of the above-mentioned genes not only in MODY pathogenesis, but also in other forms of diabetes. (43) The underlying molecular mechanisms are still to be investigated. In particular, common variants of KCNJ11 (Potassium Inwardly Rectifying Channel Subfamily J Member 11) and ABCC8 have been related to a major susceptibility to type 2 diabetes and neonatal diabetes. (46) KCNJ11 and ABCC8 mutations both determine a compromised insulin secretory response. The underlying mechanism in the first case is the reduction of KATP channel sensitivity to ATP, while in the latter is the membrane hyperpolarization caused by Mg-nucleotide binding to nucleotide-binding domains of SUR1. (47, 48) In addition, more than 90% of MODY patients affected by KCNJ11 or ABCC8 pathogenetic variants present a favorable response to oral treatment with sulfonylureas. (46) Similarly, mutations of PDX1 gene (MODY 4), which plays a crucial role in β-cell differentiation and function, are also associated with neonatal diabetes, T2DM, as well as ketosis-prone diabetes (KPD). (46, 49, 50, 51, 52) Also some PAX4 (MODY 9) pathogenic variants have been linked to T2DM and KPD. In the patients carrying such variants there is evidence of insulin deficiency due to the impairment of β-cell development and hyperglucagonemia subsequent to the altered transcriptional PAX4 repression of glucagon (53, 54, 55) Polymorphisms or mutations of NEUROD1 (MODY 6), essential for the development and maintenance of mature β-cell activity, were reported also in individuals affected by permanent neonatal diabetes and T2DM (43, 56, 57) INS (preproinsulin gene) mutations, which cause hyperinsulinemia by promoting the production of structurally defective insulin, have been linked to type 1 and 2 diabetes, and neonatal diabetes, other than MODY 10. (58, 59) Furthermore, variants of the most common causal MODY genes HNF1A, HNF4A, HNF1B and GCK have been found to be associated with an increased predisposition of T2DM in several populations. (60, 61, 62, 63)
5. Molecular Advances and Future Prospectives
Since the first description of MODY cases in the 1960s and the coining of “MODY” abbreviation in 1974, there have been remarkable advances in our understanding of the maturity-onset diabetes of the young spacing from phenotypic spectrum and therapeutic approaches to molecular diagnostics. (64, 65) In particular, after the first genetic mutations identified in the 1990s regarding GCK (MODY 2), HFN1A (MODY 3) and HFN14A (MODY 1), the development of DNA sequencing methods has significantly enhanced the exploration of novel causal variants. (5, 66, 67, 68, 69, 70) Sanger sequencing represents the conventional genetic testing for single-gene genetic disorders, leading, however, to a relevant rate of unclear MODY diagnosis (also known as MODY-X) ranging from 46.2-73.9%. (70) Unlike the candidate gene approach of the Sanger method, the advent of Next-Generation Sequencing (NGS) technologies, such as whole genome, exome and targeted sequencing, currently offer a pivotal role in providing valuable insights into the genetic architecture of MODY. By providing a highly sensitive and specific technique, NGS enables the detection of both common and rare pathogenetic variants associated with the maturity-onset diabetes of the young. (70) Additionally, the efficacy of NGS is also highlighted by its accelerated turnaround time of comprehensive genetic profiling, which has expedited the diagnostic algorithm, enabling subsequently a prompt and tailored clinical management of the patient. In the era of precision medicine NGS affordability, however, differs widely among diverse socio-geographical settings also due to financial considerations. (71) According to the 2008 Practice Guidelines for genetic diagnosis of MODY, molecular analysis is recommended only for individuals fulfilling the clinical criteria of MODY. (2) Therefore, the limitations of the advanced genetic investigations, as well as the high prevalence of MODY misdiagnosis, have enhanced the exploration of biomarkers aimed at optimizing diagnostic accuracy. (5) Bonner et al. documented a correlation between the overexpression of miR-103 and miR-224 and HFN1A-MODY, suggesting that the detection of such miRNAs in the serum of HNF1A-MODY patients could be useful for differential diagnosis. (72) Alternative biomarkers could also be epigenetic signatures, such as DNA methylation or histone acetylation, of genes impacting β-cell development and function. It was documented that aberrant DNA methylation patterns, which mediate a major risk of diabetes in subjects exposed to a diabetic intrauterine environment, concern promoter regions of genes associated with MODY, other than with type 2 diabetes and Notch signaling pathway. (73) Indeed, accumulating evidence in epigenetics could provide a deeper understanding of the complex interplay between genetic and environmental factors in MODY pathogenesis. Furthermore, the identification of potential epigenetic biomarkers could represent an exciting prospect for advancing precision medicine for both diagnosis and management of MODY patients, finally offering a more effective and personalized therapy, including any future implementation of epigenetic targeted strategies such as demethylating agents or histone deacetylase inhibitors in the affected individuals.
6. Conclusions
Due to the limited data available, the small-scale population-specific studies, and the unclear causal mechanisms of a significant proportion of MODY cases, the assessment of the related genes pathogenicity remains challenging, especially in the context of rare and low-penetrant variants. Monogenic and polygenic diabetes classically represent two distinguishable conditions. The first one is generally associated with single high-penetrating gene mutations, whereas the latter with the contribution of both several genetic and environmental factors. The controversial data regarding the above-mentioned genes, as well as the identification of other inheritance patterns, underscore the relevant variability of MODY and the difficult interpretation of the specific pathogenetic contribution, in particular of the rare and reduced-penetrant genetic variants. As exemplified by our case report, the compound heterozygosis of BLK variant c.1013T>C (Ile338Thr) and RFX6 variant c.1072G>A (Val358Ile) challenged the traditional monogenic paradigm of MODY. Mutations in both genes are involved in the disruption of critical regulatory pathways of β-cells development and function. However, so far no clear causal correlation between the BLK or RFX6 variants alone and MODY was documented, since the single mutations separately have not led yet to an altered glucose metabolism in the patient’s parents. Only when inherited simultaneously by the patient, instead, their combined contribution to pathogenesis was enhanced and evidenced by the occurrence of MODY. Therefore, it could be potentially reasonable to question the definition of the early-onset non-autoimmune diabetes of the young as exclusively monogenic, particularly in this subset of rare and peculiar cases. In conclusion, more studies are warranted to further explore the genetic landscape of MODY, its correlation with the phenotype, and the impact of the non-genetic modifiers in the diabetes pathophysiology, considering the crucial importance of an accurate diagnosis and treatment in this cohort of patients, as well as of the genetic counseling for the family members.
References
- Laver, T. W., Wakeling, M., Knox, O., Colclough, K., Wright, C. F., Ellard, S., Hattersley, A. T., Weedon, M. N., & Patel, K. (2022). Evaluation of Evidence for Pathogenicity Demonstrates That BLK, KLF11, and PAX4 Should Not Be Included in Diagnostic Testing for MODY. Diabetes, 71(5), 1128–1136. [CrossRef]
- Ellard, S., Bellanné-Chantelot, C. and Hattersley, A.T. (2008). Best practice guidelines for the molecular genetic diagnosis of maturity-onset diabetes of the young. Diabetologia, 51(4), pp.546–553. [CrossRef]
- Tshivhase, A., Matsha, T., & Raghubeer, S. (2021). Diagnosis and Treatment of MODY: An Updated Mini Review. Applied Sciences, 11(20), 9436. Li, J., Wang, X., Mao, H., Li, W., Deng, A. [CrossRef]
- Li, Y., Zhang, H., & Liu, C. (2022). Precision therapy for three Chinese families with maturity-onset diabetes of the young (MODY12). Frontiers in Endocrinology, 13. [CrossRef]
- Urakami, T. (2019). Maturity-onset diabetes of the young (MODY): current perspectives on diagnosis and treatment. Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy, Volume 12, 1047–1056. [CrossRef]
- Yahaya, T. O., & Ufuoma, S. B. (2020). Genetics and Pathophysiology of Maturity-onset Diabetes of the Young (MODY): A Review of Current Trends. Oman Medical Journal, 35(3), e126–e126. [CrossRef]
- Płoszaj, T., Antosik, K., Jakiel, P., Zmysłowska, A., & Borowiec, M. (2021). Screening for extremely rare pathogenic variants of monogenic diabetes using targeted panel sequencing. Endocrine. [CrossRef]
- Patel, K., Kettunen, J., Laakso, M., Stančáková, A., Laver, T. W., Colclough, K., Johnson, M., Abramowicz, M., Groop, L., Miettinen, P. J., Shepherd, M., Flanagan, S. E., Ellard, S., Inagaki, N., Hattersley, A. T., Tuomi, T., Cnop, M., & Weedon, M. N. (2017). Heterozygous RFX6 protein truncating variants are associated with MODY with reduced penetrance. Nature Communications, 8(1). [CrossRef]
- Mohan, V., Radha, V., Nguyen, T. T., Stawiski, E. W., Pahuja, K. B., Goldstein, L. D., Tom, J., Anjana, R. M., Kong-Beltran, M., Bhangale, T., Jahnavi, S., Chandni, R., Gayathri, V., George, P., Zhang, N., Murugan, S., Phalke, S., Chaudhuri, S., Gupta, R., & Zhang, J. (2018). Comprehensive genomic analysis identifies pathogenic variants in maturity-onset diabetes of the young (MODY) patients in South India. BMC Medical Genetics, 19(1). [CrossRef]
- Simaite, D., Kofent, J., Gong, M., Rüschendorf, F., Jia, S., Arn, P., Bentler, K., Ellaway, C., Kühnen, P., Hoffmann, G. F., Blau, N., Spagnoli, F. M., Hübner, N., & Raile, K. (2014). Recessive Mutations in PCBD1 Cause a New Type of Early-Onset Diabetes. Diabetes, 63(10), 3557–3564. [CrossRef]
- Imaki S., Iizuka, K., Horikawa, Y., Yasuda, M., Kubota, S., Kato, T., Liu, Y., Takao, K., Mizuno, M., Hirota, T., Suwa, T., Kazuyoshi Hosomichi, Tajima, A., Fujiwara, Y., Yamazaki, Y., Kuwata, H., Seino, Y., & Daisuke Yabe. (2021). A novel RFX6 heterozygous mutation (p.R652X) in maturity-onset diabetes mellitus: A case report. Journal of Diabetes Investigation, 12(10), 1914–1918. [CrossRef]
- Billings, L. K., Shi, Z., Resurreccion, W. K., Wang, C., Wei, J., Pollin, T. I., Udler, M. S., & Xu, J. (2022). Statistical evidence for high-penetrance MODY-causing genes in a large population-based cohort. Endocrinology, Diabetes & Metabolism, 5(6), e372. [CrossRef]
- Horikawa, Y. (2018). Maturity-onset diabetes of the young as a model for elucidating the multifactorial origin of type 2 diabetes mellitus. Journal of Diabetes Investigation, 9(4), 704–712. [CrossRef]
- Mirshahi, U. L., Colclough, K., Wright, C. F., Wood, A. R., Beaumont, R. N., Tyrrell, J., Laver, T. W., Stahl, R., Golden, A., Goehringer, J. M., Frayling, T. F., Hattersley, A. T., Carey, D. J., Weedon, M. N., & Patel, K. A. (2022). Reduced penetrance of MODY-associated HNF1A/HNF4A variants but not GCK variants in clinically unselected cohorts. The American Journal of Human Genetics, 109(11), 2018–2028. [CrossRef]
- Soyer, J., Flasse, L., Raffelsberger, W., Beucher, A., Orvain, C., Peers, B., Ravassard, P., Vermot, J., Voz, M. L., Mellitzer, G., & Gradwohl, G. (2010). Rfx6 is an Ngn3-dependent winged helix transcription factor required for pancreatic islet cell development. Development, 137(2), 203–212. [CrossRef]
- Smith, S. B., Qu, H.-Q., Taleb, N., Kishimoto, N. Y., Scheel, D. W., Lu, Y., Patch, A.-M., Grabs, R., Wang, J., Lynn, F. C., Miyatsuka, T., Mitchell, J., Seerke, R., Désir, J., Eijnden, S. V., Abramowicz, M., Kacet, N., Weill, J., Renard, M.-È., & Gentile, M. (2010). Rfx6 directs islet formation and insulin production in mice and humans. Nature, 463(7282), 775–780. [CrossRef]
- Chandra, V., Albagli-Curiel, O., Hastoy, B., Piccand, J., Randriamampita, C., Vaillant, E., Cavé, H., Busiah, K., Froguel, P., Vaxillaire, M., Rorsman, P., Polak, M., & Scharfmann, R. (2014). RFX6 Regulates Insulin Secretion by Modulating Ca2+ Homeostasis in Human β Cells. Cell Reports, 9(6), 2206–2218. [CrossRef]
- Lu, J., Cheng, C., Cheng, Z.-C., Wu, Q., Shen, H., Yuan, M., Zhang, B., & Yang, J.-K. (2021). The dual role of RFX6 in directing β cell development and insulin production. Journal of Molecular Endocrinology, 66(2), 129–140. [CrossRef]
- Pearl, E. J., Jarikji, Z., & Horb, M. E. (2011). Functional analysis of Rfx6 and mutant variants associated with neonatal diabetes. Developmental Biology, 351(1), 135–145. [CrossRef]
- Piorno, A.G., Gata, I.L., Anez, R., Maricel, A., Collado Gonzalez, G., Gomez-Gordo Hernanz, M., & González Albarrán, O. (2023). Maturity Onset Diabetes of the Young (MODY) associated with mutations in the Regulatory factor X6 (RFX6) gene: A case report. Endocrine Abstracts. [CrossRef]
- Piccand, J., Strasser, P., Hodson, David J., Meunier, A., Ye, T., Keime, C., Birling, M.-C., Rutter, Guy A., & Gradwohl, G. (2014). Rfx6 Maintains the Functional Identity of Adult Pancreatic β Cells. Cell Reports, 9(6), 2219–2232. [CrossRef]
- Artuso, R., Provenzano, A., Mazzinghi, B., Giunti, L., Palazzo, V., Andreucci, E., Blasetti, A., Chiuri, R. M., Gianiorio, F. E., Mandich, P., Monami, M., Mannucci, E., & Giglio, S. (2014). Therapeutic implications of novel mutations of the RFX6 gene associated with early-onset diabetes. The Pharmacogenomics Journal, 15(1), 49–54. [CrossRef]
- Auerbach, A., Cohen, A., Ofek Shlomai, N., Weinberg-Shukron, A., Gulsuner, S., King, M.-C., Hemi, R., Levy-Lahad, E., Abulibdeh, A., & Zangen, D. (2020). NKX2-2 Mutation Causes Congenital Diabetes and Infantile Obesity With Paradoxical Glucose-Induced Ghrelin Secretion. The Journal of Clinical Endocrinology & Metabolism, 105(11), 3486–3495. [CrossRef]
- Doyle, M. J., & Sussel, L. (2007). Nkx2.2 Regulates β-Cell Function in the Mature Islet. Diabetes, 56(8), 1999–2007. [CrossRef]
- Mio, C., Baldan, F., & Damante, G. (2022). NK2 homeobox gene cluster: Functions and roles in human diseases. Genes & Diseases. [CrossRef]
- Aigha, I. I., & Abdelalim, E. M. (2020). NKX6.1 transcription factor: a crucial regulator of pancreatic β cell development, identity, and proliferation. Stem Cell Research & Therapy, 11(1). [CrossRef]
- Sander, M. (2000). Ventral neural patterning by Nkx homeobox genes: Nkx6.1 controls somatic motor neuron and ventral interneuron fates. Genes & Development, 14(17), 2134–2139. [CrossRef]
- Schisler, J. C., Per Bo Jensen, Taylor, D. G., Becker, T., Knop, F. K., Takekawa, S., German, M. S., Weir, G. C., Lu, D., Mirmira, R. G., & Newgard, C. B. (2005). The Nkx6.1 homeodomain transcription factor suppresses glucagon expression and regulates glucose-stimulated insulin secretion in islet beta cells. Proceedings of the National Academy of Sciences of the United States of America, 102(20), 7297–7302. [CrossRef]
- Taylor, B. L., Liu, F.-F., & Sander, M. (2013). Nkx6.1 Is Essential for Maintaining the Functional State of Pancreatic Beta Cells. Cell Reports, 4(6), 1262–1275. [CrossRef]
- Donelan, W., Koya, V., Li, S., & Li, Y. (2010). Distinct Regulation of Hepatic Nuclear Factor 1α by NKX6.1 in Pancreatic Beta Cells. Journal of Biological Chemistry, 285(16), 12181–12189. [CrossRef]
- Chapla, A., Johnson, J., Sophy Korula, Mohan, N., Ahmed, A., Varghese, D., Parthiban Rangasamy, Ravichandran, L., Jebasingh, F., Agrawal, K., Somasundaram, N., Asha Hesarghatta Shyamasunder, Mathai, S., Simon, A., Jha, S., Subhankar Chowdry, Venkatesan Radha, P. Raghupathy, & Nicolaï, T. (2022). WFS1 Gene–associated Diabetes Phenotypes and Identification of a Founder Mutation in Southern India. The Journal of Clinical Endocrinology & Metabolism, 107(5), 1328–1336. [CrossRef]
- Ishihara, H. (2004). Disruption of the WFS1 gene in mice causes progressive -cell loss and impaired stimulus-secretion coupling in insulin secretion. Human Molecular Genetics, 13(11), 1159–1170. [CrossRef]
- Serbis, A., Rallis, D., Giapros, V., Galli-Tsinopoulou, A., & Siomou, E. (2023). Wolfram Syndrome 1: A Pediatrician’s and Pediatric Endocrinologist’s Perspective. International Journal of Molecular Sciences, 24(4), 3690. [CrossRef]
- Minton, J. A. L., Hattersley, A. T., Owen, K., McCarthy, M. I., Walker, M., Latif, F., Barrett, T., & Frayling, T. M. (2002). Association Studies of Genetic Variation in the WFS1 Gene and Type 2 Diabetes in U.K. Populations. Diabetes, 51(4), 1287–1290. [CrossRef]
- Domènech, E., Gómez-Zaera, M., & Nunes, V. (2002). WFS1 mutations in Spanish patients with diabetes mellitus and deafness. European Journal of Human Genetics, 10(7), 421–426. [CrossRef]
- Sandhu, M. S., Weedon, M. N., Fawcett, K. A., Wasson, J., Debenham, S. L., Daly, A., Lango, H., Frayling, T. M., Neumann, R. J., Sherva, R., Blech, I., Pharoah, P. D., Palmer, C. N. A., Kimber, C., Tavendale, R., Morris, A. D., McCarthy, M. I., Walker, M., Hitman, G., & Glaser, B. (2007). Common variants in WFS1 confer risk of type 2 diabetes. Nature Genetics, 39(8), 951–953. [CrossRef]
- Franks, P. W., Rolandsson, O., Debenham, S. L., Fawcett, K. A., Payne, F., Dina, C., Froguel, P., Mohlke, K. L., Willer, C., Olsson, T., Wareham, N. J., Hallmans, G., Barroso, I., & Sandhu, M. S. (2007). Replication of the association between variants in WFS1 and risk of type 2 diabetes in European populations. Diabetologia, 51(3), 458–463. [CrossRef]
- Cheurfa, N., Brenner, G. M., Reis, A. F., Dubois-Laforgue, D., Roussel, R., Tichet, J., Lantieri, O., Balkau, B., Fumeron, F., Timsit, J., Marre, M., & Velho, G. (2010). Decreased insulin secretion and increased risk of type 2 diabetes associated with allelic variations of the WFS1 gene: the Data from Epidemiological Study on the Insulin Resistance Syndrome (DESIR) prospective study. Diabetologia, 54(3), 554–562. [CrossRef]
- Awata, T., Inoue, K., Kurihara, S., Ohkubo, T., Inoue, I., Abe, T., Takino, H., Kanazawa, Y., & Katayama, S. (2000). Missense Variations of the Gene Responsible for Wolfram Syndrome (WFS1/wolframin) in Japanese: Possible Contribution of the Arg456His Mutation to Type 1 Diabetes as a Nonautoimmune Genetic Basis. Biochemical and Biophysical Research Communications, 268(2), 612–616. [CrossRef]
- Bansal, V., Boehm, B. O., & Darvasi, A. (2018). Identification of a missense variant in the WFS1 gene that causes a mild form of Wolfram syndrome and is associated with risk for type 2 diabetes in Ashkenazi Jewish individuals. Diabetologia, 61(10), 2180–2188. [CrossRef]
- Ferrè, S., de Baaij, J. H. F., Ferreira, P., Germann, R., de Klerk, J. B. C., Lavrijsen, M., van Zeeland, F., Venselaar, H., Kluijtmans, L. A. J., Hoenderop, J. G. J., & Bindels, R. J. M. (2013). Mutations in PCBD1 Cause Hypomagnesemia and Renal Magnesium Wasting. Journal of the American Society of Nephrology, 25(3), 574–586. [CrossRef]
- Taha, D., Rajeev Thirunagari, & Adhikari, A. (2023). SAT081 Maturity Onset Diabetes Of The Young (MODY) Due To A New BLK Gene Mutation. Journal of the Endocrine Society, 7(Supplement_1). [CrossRef]
- Antal, Z. (2021). Maturity-Onset Diabetes of the Young (MODY): Genetic Causes, Clinical Characteristics, Considerations for Testing, and Treatment Options. Endocrines, 2(4), 485–501. [CrossRef]
- Ivanoshchuk, D.; Shakhtshneider, E.; Mikhailova, S.; Ovsyannikova, A.; Rymar, O.; Valeeva, E.; Orlov, P.; Voevoda, M. (2023). The Mutation Spectrum of Rare Variants in the Gene of Adenosine Triphosphate (ATP)-Binding Cassette Subfamily C Member 8 in Patients with a MODY Phenotype in Western Siberia. Journal of Personalized Medicine, 13 (2), 172-172. [CrossRef]
- Bonnefond, A.; Yengo, L.; Philippe, J.; Dechaume, A.; Ezzidi, I.; Vaillant, E.; Gjesing, A. P., Andersson, E. A., Sébastien Czernichow, Serge Herçberg, Samy Hadjadj, Charpentier, G., Lantieri, O., Balkau, B., Marre, M., Pedersen, O., Hansen, T., Philippe Froguel, & Vaxillaire, M. (2012). Reassessment of the putative role of BLK-p.A71T loss-of-function mutation in MODY and type 2 diabetes. Diabetologia, 56(3), 492–496. [CrossRef]
- Yang, Y., & Chan, L. (2016). Monogenic Diabetes: What It Teaches Us on the Common Forms of Type 1 and Type 2 Diabetes. Endocrine Reviews, 37(3), 190–222. [CrossRef]
- Chen, Y., Hu, X., Cui, J., Zhao, M., & Yao, H. (2021). A novel mutation KCNJ11 R136C caused KCNJ11-MODY. Diabetology & Metabolic Syndrome, 13(1). [CrossRef]
- Timmers, M., Dirinck, E., Lauwers, P., Wuyts, W., & De Block, C. (2021). ABCC8 variants in MODY12: Review of the literature and report of a case with severe complications. Diabetes/Metabolism Research and Reviews, 37(8). [CrossRef]
- Forero-Castro, N., Ramirez, L., Je, C., Henao, F., & Valencia, F. (2023). Clinical and molecular description of two cases of neonatal diabetes secondary to mutations in PDX1. Endocrinology, Diabetes & Metabolism Case Reports, 2023(3). [CrossRef]
- Wang, X., Sterr, M., Ansarullah, Burtscher, I., Böttcher, A., Beckenbauer, J., Siehler, J., Meitinger, T., Häring, H.-U., Staiger, H., Cernilogar, F. M., Schotta, G., Irmler, M., Beckers, J., Wright, C. V. E., Bakhti, M., & Lickert, H. (2019). Point mutations in the PDX1 transactivation domain impair human β-cell development and function. Molecular Metabolism, 24, 80–97. [CrossRef]
- Wang, N., Tong, R., Xu, J., Tian, Y., Pan, J., Cui, J., Chen, H., Peng, Y., Fei, S., Yang, S., Wang, L., Yao, J., & Cui, W. (2021). PDX1 and MC4R genetic polymorphisms are associated with type 2 diabetes mellitus risk in the Chinese Han population. BMC Medical Genomics, 14, 249. [CrossRef]
- Boike, S., Mir, M., Rauf, I., Jama, A. B., Shaleen Sunesara, Mushtaq, H., Anwar Khedr, Jain Nitesh, Surani, S., & Syed Anjum Khan. (2022). Ketosis-prone diabetes mellitus: A phenotype that hospitalists need to understand. World Journal of Clinical Cases, 10(30), 10867–10872. [CrossRef]
- Lau, H. H., Krentz, N. A. J., Abaitua, F., Perez-Alcantara, M., Chan, J.-W., Ajeian, J., Ghosh, S., Lee, Y., Yang, J., Thaman, S., Champon, B., Sun, H., Jha, A., Hoon, S., Tan, N. S., Gardner, D. S.-L., Kao, S. L., Tai, E. S., Gloyn, A. L., & Teo, A. K. K. (2023). PAX4 loss of function increases diabetes risk by altering human pancreatic endocrine cell development. Nature Communications, 14(1), 6119. [CrossRef]
- Ang, S. F., Tan, C. S. H., Wang, L., Dorajoo, R., Fong, J. C. W., Kon, W. Y. C., Lian, J. X., Ang, K., Rahim, J. B., Jeevith, B., Lee, S. B. M., Tang, W. E., Subramanium, T., Sum, C. F., Liu, J. J., & Lim, S. C. (2019). PAX4 R192H is associated with younger onset of Type 2 diabetes in East Asians in Singapore. Journal of Diabetes and Its Complications, 33(1), 53–58. [CrossRef]
- Mauvais-Jarvis, F., Smith, S. B., May, C. L., Leal, S. M., Gautier, J.-F., Molokhia, M., Riveline, J.-P., Rajan, A. S., Kevorkian, J.-P., Zhang, S., Vexiau, P., German, M. S., & Vaisse, C. (2004). PAX4 gene variations predispose to ketosis-prone diabetes. Human Molecular Genetics, 13(24), 3151–3159. [CrossRef]
- Demirbilek, H., Hatipoglu, N., Gul, U., Tatli, Z. U., Ellard, S., Flanagan, S. E., De Franco, E., & Kurtoglu, S. (2018). Permanent neonatal diabetes mellitus and neurological abnormalities due to a novel homozygous missense mutation in NEUROD1. Pediatric Diabetes, 19(5), 898–904. [CrossRef]
- Li, Y.-Y., Wang, H., & Zhang, Y.-Y. (2021). Neuronal Differentiation 1 gene Ala45Thr polymorphism and type 2 diabetes mellitus: A meta-analysis of 7,940 subjects. Nutrition, Metabolism and Cardiovascular Diseases, 31(6), 1809–1821. [CrossRef]
- Støy, J., De Franco, E., Ye, H., Park, S.-Y., Bell, G. I., & Hattersley, A. T. (2021). In celebration of a century with insulin – Update of insulin gene mutations in diabetes. Molecular Metabolism, 52, 101280. [CrossRef]
- Kamel, A. M., Mira, M. F., Gamal Ebid, Kassem, S., Radwan, E. R., Mamdouh, M., Amin, M. M., Badawy, N., Hafez Bazaraa, Ibrahim, A., & Salah, N. (2019). Association of insulin gene VNTR INS -23/Hph1 A>T (rs689) polymorphism with type 1 diabetes mellitus in Egyptian children. Egyptian Journal of Medical Human Genetics. [CrossRef]
- Li, L.-M., Jiang, B.-G., & Sun, L.-L. (2022). HNF1A:From Monogenic Diabetes to Type 2 Diabetes and Gestational Diabetes Mellitus. Frontiers in Endocrinology, 13. [CrossRef]
- Azizi, S. M., Sarhangi, N., Afshari, M., Abbasi, D., Aghaei Meybodi, H. R., & Hasanzad, M. (2019). Association Analysis of the HNF4A Common Genetic Variants with Type 2 Diabetes Mellitus Risk. International Journal of Molecular and Cellular Medicine, 8(Suppl1), 56–62. [CrossRef]
- Amaral, S., Palha, A., Bogalho, P., & José Silva-Nunes. (2023). Maturity-onset diabetes of the young secondary to HNF1B variants (HNF1B-MODY): a series of 10 patients from a single diabetes center. Diabetology & Metabolic Syndrome, 15(1). [CrossRef]
- Li, C., Yang, Y., Liu, X., Li, Z., Liu, H., & Tan, Q. (2020). Glucose metabolism-related gene polymorphisms as the risk predictors of type 2 diabetes. Diabetology & Metabolic Syndrome, 12(1). [CrossRef]
- Fajans, S. S., & Bell, G. I. (2011). MODY. Diabetes Care, 34(8), 1878–1884. [CrossRef]
- Tattersall, R. B., & Fajans, S. S. (1975). A difference between the inheritance of classical juvenile-onset and maturity-onset type diabetes of young people. Diabetes, 24(1), 44–53. [CrossRef]
- Velho, G. (1992). Primary pancreatic beta-cell secretory defect caused by mutations in glucokinase gene in kindreds of maturity onset diabetes of the young. The Lancet, 340(8817), 444–448. [CrossRef]
- Stoffel, M., Froguel, P., Takeda, J., Zouali, H., Vionnet, N., Nishi, S., Weber, I. T., Harrison, R. W., Pilkis, S. J., & Lesage, S. (1992). Human glucokinase gene: isolation, characterization, and identification of two missense mutations linked to early-onset non-insulin-dependent (type 2) diabetes mellitus. Proceedings of the National Academy of Sciences, 89(16), 7698–7702. [CrossRef]
- Yamagata, K., Furuta, H., Oda, N., Kaisaki, P. J., Menzel, S., Cox, N. J., Fajans, S. S., Signorini, S., Stoffel, M., & Bell, G. I. (1996). Mutations in the hepatocyte nuclear factor-4α gene in maturity-onset diabetes of the young (MODY1). Nature, 384(6608), 458–460. [CrossRef]
- Yamagata, K., Oda, N., Kaisaki, P. J., Menzel, S., Furuta, H., Vaxillaire, M., Southam, L., Cox, R. D., Lathrop, G. M., Boriraj, V. V., Chen, X., Cox, N. J., Oda, Y., Yano, H., Le Beau, M. M., Yamada, S., Nishigori, H., Takeda, J., Fajans, S. S., & Hattersley, A. T. (1996). Mutations in the hepatocyte nuclear factor-1α gene in maturity-onset diabetes of the young (MODY3). Nature, 384(6608), 455–458. [CrossRef]
- Maltoni G, Franceschi R, Di Natale V, Al-Qaisi R, Greco V, Bertorelli R, De Sanctis V, Quattrone A, Mantovani V, Cauvin V, Zucchini S. (2022). Next Generation Sequencing Analysis of MODY-X Patients: A Case Report Series. Journal of Personalized Medicine, 12(10), 1613–1613. [CrossRef]
- Tosur, M., & Philipson, L. H. (2022). Precision diabetes: Lessons learned from maturity-onset diabetes of the young (MODY). Journal of Diabetes Investigation. [CrossRef]
- Bonner, C., Nyhan, K. C., Bacon, S., Kyithar, M. P., Schmid, J., Concannon, C. G., Bray, I. M., Stallings, R. L., Prehn, J. H. M., & Byrne, M. M. (2013). Identification of circulating microRNAs in HNF1A-MODY carriers. Diabetologia, 56(8), 1743–1751. [CrossRef]
- del Rosario, M. C., Ossowski, V., Knowler, W. C., Bogardus, C., Baier, L. J., & Hanson, R. L. (2014). Potential epigenetic dysregulation of genes associated with MODY and type 2 diabetes in humans exposed to a diabetic intrauterine environment: An analysis of genome-wide DNA methylation. Metabolism, 63(5), 654–660. [CrossRef]
Figure 1.
Genetic pedigree of proband.
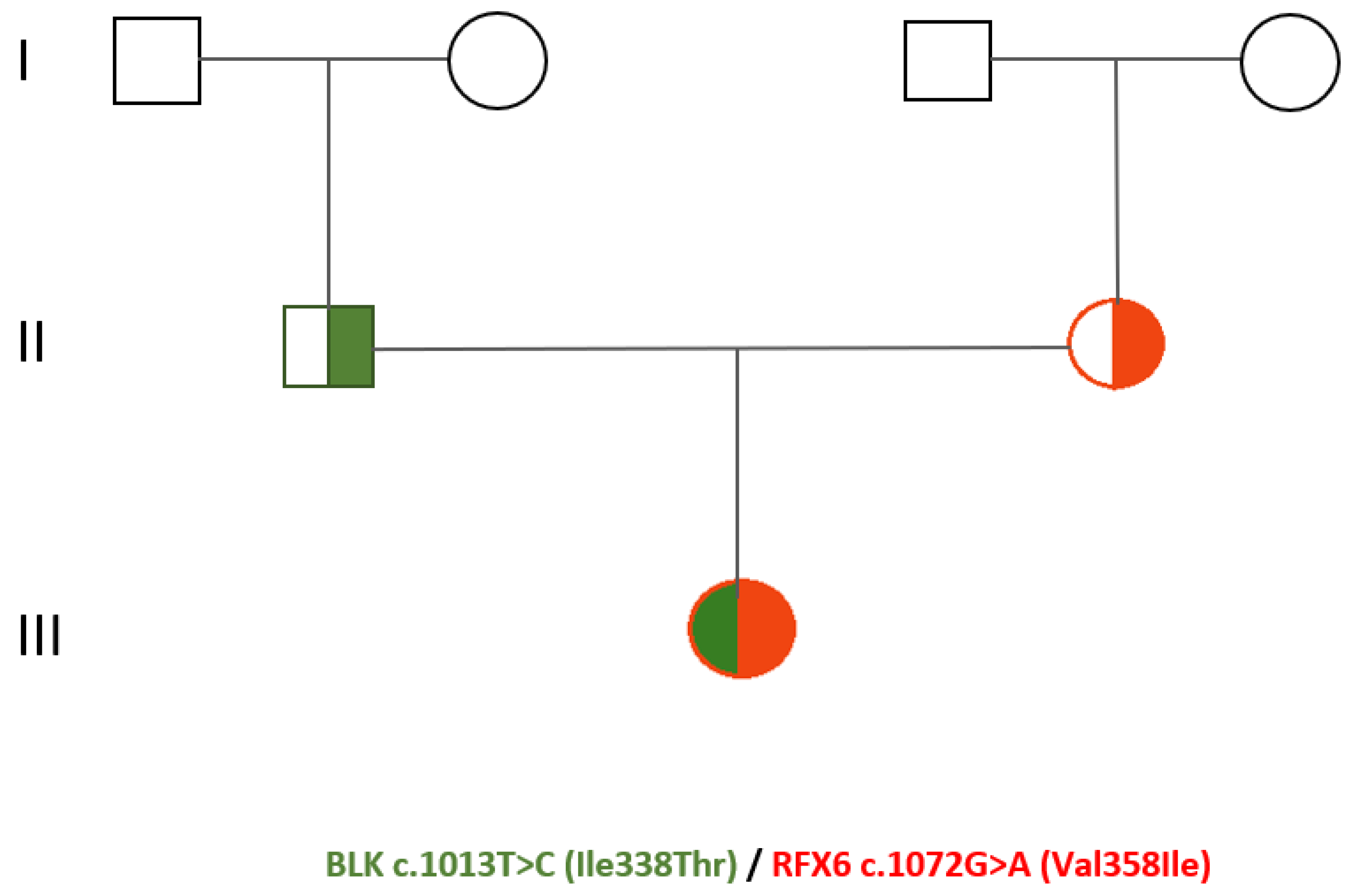

Table 1.
Identity and function of low-penetrant MODY-related genes.
| Gene | Locus | Function |
|---|---|---|
| RFX6 (regulatory factor X6) | 6q22.1 | regulates pancreatic islet cell differentiation and function, including β-cell insulin production |
| NK2.2 (NK2 Homeobox 2) | 20p11.22 | modulates pancreatic islet cell differentiation during embryogenesis and mature β-cell function, regulates pancreatic islet architecture participates in the morphogenesis of the ventral central nervous system and of the epithelial enteroendocrine cells |
| NKX6.1 (NK6 Homeobox 1) | 4q21.23 | regulates pancreatic β-cell differentiation and function, including insulin production, glucose uptake and metabolism, and cell proliferation regulates glucagon gene (GCG) expression, may induce suppression of pancreatic α-cell development contributes to the neural development and moto neuron specification |
| WFS1 (Wolframin ER Transmembrane Glycoprotein) | 4p16.1 | modulates endoplasmic reticulum calcium homeostasis and calcium signal transduction processes involved in cellular apoptosis regulates β-cell function, glucose metabolism and insulin secretion |
| PCBD1 (Pterin-4 alpha-carbinolamine dehydratase 1) | 10q22.1 | in nucleus acts as a dimerization cofactor of HNF1A and HNF1B, and enhances HNF1A/B-mediated transcription in cytoplasm modulates the tetrahydrobiopterin (BH4) biosynthesis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



