
Submitted:
18 December 2023
Posted:
19 December 2023
You are already at the latest version
Abstract
Aerobic glycolysis in cancer cells, originally observed by Warburg 100 years ago, which involves production of lactate as the end product of glucose breakdown even in the presence of adequate oxygen, is the foundation for the current interest in cancer cell-specific reprograming of metabolic pathways. The renewed interest in cancer cell metabolism has now gone well beyond the original Warburg effect related to glycolysis to other metabolic pathways to include amino acid metabolism, one-carbon metabolism, pentose phosphate pathway, nucleotide synthesis, antioxidant machinery, etc. Since glucose and amino acids constitute the primary nutrients that fuel the altered metabolic pathways in cancer cells, the transporters that mediate the transfer of these nutrients and their metabolites not only across the plasma membrane but also across the mitochondrial and lysosomal membranes have become an integral component of expansion of the Warburg effect. In this review, we focus on the interplay between these transporters and metabolic pathways that facilitates the metabolic reprogramming which has become the hallmark of cancer cells. The beneficial outcome of this recent understanding of the unique metabolic signature surrounding the Warburg effect is the identification of novel drug targets for development of a new generation of therapeutics to treat cancer.
Keywords:
oncogenes
; aerobic glycolysis
; lactate receptors
; nutrient transporters
; glutamine addiction
; one-carbon metabolism
; glutaminolysis
; reductive carboxylation
; oncometabolites
; tumor microenvironment
1. Introduction
About 100 years ago Otto Warburg made an interesting observation: cancer cells when cultured in vitro under normal oxygen levels (i.e., 20% or 160 mm Hg) as in physiological conditions or tumors growing in the body in vivo consumed glucose much more than normal cells but converted this glucose predominantly into lactic acid which was released into the culture medium [1,2]. This was unexpected because lactic acid is the end product of glucose breakdown only under conditions of oxygen deficit (i.e., hypoxia), a pathway known as “anerobic glycolysis”. In contrast to this normal process, cancer cells converted glucose into lactic acid in the presence of oxygen, thus leading to the coining of the term “aerobic glycolysis” to describe the observation made by Warburg in cancer cells. Though interesting and unexpected, the importance of this observation was not recognized for several decades. A part of the reason for this was the discovery of oncogenes and tumor suppressor genes and the domination of the idea in the field of cancer biology that the protein products of these genes are the principal drivers of cancer growth. Surprisingly however, investigations into the molecular targets of these oncogenes and tumor suppressor genes began to underscore the importance of metabolic pathways as the likely mediators of these genes in cancer growth. As a result, the interest in the field of cancer biology has shifted in the past couple of decades to cancer cell-specific metabolism. Naturally the starting point for this shift was the Warburg effect which represented the first metabolic pathway to be discovered that is specific to cancer cells. What followed in subsequent years in this field is simply a logical extension of the original observation by Warburg.
The greater than normal consumption of glucose in cancer cells brought attention to glucose transporters which delivered glucose into these cells. Since lactic acid is generated at high levels, cancer cells must find ways to prevent cellular acidification and get rid of lactic acid. This shifted the focus to lactate transporters that mediate the transfer of lactate and H+ across the plasma membrane [3,4]. Then came the discovery that lactate controls the proteasomal degradation of hypoxia-inducible factor-1α (HIF-1α), thus increasing HIF-1α protein levels in cancer cells even in the presence of normal oxygen, a condition now described as “pseudohypoxia” [5]. Since cancer cells release massive amounts of lactic acid into the tumor microenvironment which results in extracellular acidification, investigations began to explore the role of acidic pH in the tumor microenvironment in cancer growth. This led to the focus of H+-coupled transporters for various nutrients such as amino acids, peptides, citrate, folate and iron which might use the extracellular acidic pH to effectively transfer these important nutrients to cancer cells from the extracellular medium [6]. With the increased rate of glycolysis came increased levels of metabolic intermediates in the pathway. This brought attention to the use of these intermediates for the anabolic pathways to generate amino acids such as serine and glycine which are obligatory as the source of one-carbon moieties for one-carbon metabolism involved in the synthesis of purines, pyrimidines and thymidine monophosphate (TMP) [7,8].
Cancer cells must have a greater need for metabolic energy to support their high rate of proliferation, but the glucose-lactic acid pathway that occurs in these cells generates only a fraction of energy compared to complete oxidation of glucose (2 ATP versus 32 ATP). This raised the possibility of other metabolic pathways to generate ATP which led to the discovery of the obligate dependence of cancer cells on extracellular glutamine (glutamine addiction). Subsequent research showed that glutamine is used in cancer cells not only for ATP generation but also for lactic acid production (glutaminolysis) and fatty acid synthesis (reductive carboxylation) [9,10,11]. This also brought attention to amino acid transporters in the plasma membrane of cancer cells which deliver not only glutamine to satisfy the “glutamine addiction” but also serine and glycine to fuel the one-carbon metabolism in addition to serine and glycine that is synthesized from the glycolytic intermediates [12,13,14,15]. Cancer cells obtain amino acids not only from extracellular medium but also from lysosomal degradation of intracellular proteins via autophagy and extracellular proteins via macropinocytosis [16,17]. This necessitated studies on amino acid transporters in the lysosomal membrane in cancer cells that transfer amino acids from lysosomes into cytoplasm for subsequent use in metabolic pathways [18,19]. This also integrated amino acid nutrition to mTORC1 signaling because of the close association of mTORC1-associated proteins with the lysosomal membrane, a critical signaling pathway necessary for cancer cell proliferation and growth [18,19].
Cancer cells are also addicted to iron and heme because of their essential role in multitude of metabolic processes involved in energy production as well as catabolic and anabolic processes [20,21]. Some of the critical steps in heme synthesis occur within the mitochondrial matrix including the first regulatory step in the pathway which combines glycine with succinyl-CoA to generate δ-aminolevulinate. In addition, one-carbon metabolism also participates in important biochemical processes within the mitochondrial matrix that requires serine. This brings attention to transporters in the mitochondrial membrane that deliver serine and glycine from cytoplasm to the mitochondrial matrix [22]. Furthermore, with the accumulation of excess iron in cancer cells comes the risk of oxidative stress and iron-dependent cell death process (ferroptosis) [23,24,25]. Therefore, cancer cells must enhance their antioxidant machinery to protect themselves from these detrimental processes. This led to the focus on glutathione/glutathione peroxidase system and the pentose phosphate pathway that produces the reducing equivalent NADPH necessary for the antioxidant machinery [26]. In addition, since cysteine is the rate-limiting amino acid for glutathione synthesis, studies began on amino acid transporters in the plasma membrane and lysosomal membrane that provide this critical amino acid to cancer cells to support the enhanced glutathione production [27,28,29].
The original explanation for “aerobic glycolysis” thought by Warburg was that mitochondria are damaged in cancer cells and therefore the oxygen-dependent metabolism of pyruvate in mitochondria is impaired. This meant that almost all the metabolic energy needed for the cancer cells comes from the conversion of glucose to lactic acid. This view has now been revised considerably based on glutamine metabolism within the mitochondria in cancer cells. The source of energy production simply shifts to a significant extent from glycolysis to glutaminolysis with intact mitochondrial function necessary for the latter process. To support their growth and proliferation, cancer cells use the intermediates in the revved up glycolytic pathway in other metabolic pathways instead for energy production.
What follows in this review is a detailed description of these various components of cancer cell-specific metabolic pathways and the transporters related to them. Since these pathways and the transporters have been reprogrammed in cancer cells as a consequence and in conjunction with the “aerobic glycolysis”, they represent the metabolic signature of the Warburg effect.
2. Hypoxia and anaerobic glycolysis in normal cells
Glucose can be metabolized completely into CO2 and H2O in cells with mitochondria when oxygen is available. This complete oxidation of glucose involves glycolysis (glucose → pyruvate) in cytoplasm and pyruvate dehydrogenase (PDH) and citric acid cycle (pyruvate → CO2) in mitochondrial matrix (Figure 1A). However, neither the glycolysis nor the PDH/citric acid cycle involves oxygen. When glucose goes through glycolysis and PDH/citric acid cycle, it generates reducing equivalents NADH and FADH2 which enter the electron transport chain and oxidative phosphorylation where oxygen is used to convert these reducing equivalents back to NAD+ and FAD with concomitant production of ATP. The entire process results in the generation of 32 ATP per glucose. This “aerobic glycolysis” associated with complete oxidation of glucose in normal cells is subject to negative feedback regulation by ATP which inhibits phosphofructokinase-1 (PFK-1), the most important rate-limiting enzyme in glycolysis. In other words, the aerobic glycolysis in normal cells is self-limiting, controlled by the energy status of the cell.
When oxygen is deficient in normal cells, mitochondrial oxidation of pyruvate that is generated in glycolysis in the cytoplasm is impaired due to suppression of electron transport chain (ETC) and oxidative phosphorylation (OXPHOS). Even though most of NADH from glucose oxidation is produced within the mitochondria, the step mediated by glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in glycolysis also generates NADH. If this NADH cannot be oxidized back to NAD+ due to defective ETC/OXPHOS as under hypoxic conditions, the reaction mediated by GAPDH cannot continue because of the lack of NAD+. This forces the conversion of pyruvate to lactate in the cytoplasm by lactate dehydrogenase (LDH), a reaction that substitutes for ETC/OXPHOS to convert NADH to NAD+ but without O2 consumption and ATP production (Figure 1B). Now glycolysis can continue because of the functional coupling between GAPDH and LDH via NADH/NAD+ recycling, the entire process occurring in cytoplasm. This process where glucose gets converted to lactic acid in normal cells under hypoxic conditions is called “anaerobic glycolysis”. Interestingly, the same process occurs in erythrocytes even in the presence of oxygen because of the absence of mitochondria. Consequently, lactic acid is the end product of glycolysis in normal cells only under hypoxic conditions whereas erythrocytes generate lactic acid in glycolysis all the time.
3. Aerobic glycolysis in cancer cells
3.1. Mechanisms to facilitate “aerobic glycolysis” in cancer cells
As mentioned above, glycolysis in normal cells in the presence of oxygen is self-limiting and pyruvate gets converted to CO2. Conversion of glucose to lactic acid occurs in normal cells only under hypoxic conditions. In contrast, cancer cells metabolize glucose predominantly into lactic acid even in the presence of oxygen. The functional coupling between the reactions mediated by GAPDH and LDH is necessary for this process. Why doesn’t pyruvate get oxidized to CO2 in cancer cells when oxygen is available? This is mostly due to defective transport of pyruvate from cytoplasm into mitochondrial matrix because of the cancer-associated downregulation of the pyruvate-carrier components MPC1 and MPC2 in the inner mitochondrial membrane [30] and the decreased catalytic activity of PDH due to increased expression of PDH kinase-1 (PDK-1) and PDH kinase-3 (PDK-3) and consequent increased phosphorylation of PDH [31]. As a result, pyruvate is prevented from mitochondrial oxidation, thus getting diverted to lactate production in the cytoplasm (pyruvic acid + NADH + H+ → lactic acid + NAD+) (Figure 1C). This drives aerobic glycolysis in cancer cells by providing NAD+ for the reaction mediated by GAPDH.
Since aerobic glycolysis in cancer cells generates only 2 ATP per glucose instead of 32 ATP when glucose gets converted to CO2, it raises the question as to the energy status of the cancer cells. The unrestricted growth and proliferation of cancer cells cannot occur when the cells are energy deficient. The energy status to support rapid growth is maintained in cancer cells by two mechanisms, one by accelerating “aerobic glycolysis” with increased conversion of glucose to lactic acid and the other by generating energy from metabolism of amino acids, primarily glutamine (Figure 1C). Even though glucose → lactic acid in cancer cells generates only 2 ATP per glucose, more ATP can be produced if the pathway is revved up to metabolize more glucose. This metabolic switch together with glutamine-derived ATP maintains the energy status of the cancer cells to fuel their growth and proliferation.
Since glycolysis is subject to negative feedback regulation by ATP at the level of PFK-1, how can cancer cells rev up glycolysis and at the same time generate ATP at levels even higher than in normal cells? This is achieved by increased production of fructose-2,6-bisphosphate, another regulatory molecule for PFK-1. While ATP is an inhibitor of PFK-1, fructose-2,6-bisphosphate is an activator that annuls the inhibition by ATP (Figure 1C). The cellular levels of this activator are controlled by the bifunctional enzyme phosphofructokinase-2/fructose-2,6-bisphosphatase (PFKFB). Two isoforms of this enzyme, namely PFKFB3 and PFKFB4, are upregulated in cancer cells, leading to increased levels of fructose-2,6-bisphosphate to rescue glycolysis from the negative feedback regulation by ATP [32,33]. The levels of fructose-2,6-bisphosphate in cancer cells are also regulated by another mechanism involving the protein TIGAR (TP53-induced glycolysis and apoptosis inhibitor) [34]. The expression of TIGAR is downregulated in p53-mutant tumors. TIGAR possesses the catalytic activity of fructose-2,6-bisphosphatase and hence has the ability to degrade fructose-2,6-bisphosphate. This results in a reciprocal relationship between the levels of TIGAR and frunctose-2,6-bisphosphate. Since the expression of TIGAR is decreased in p53-deficient tumors, fructose-2,6-bisphosphate levels go up to maintain “aerobic glycolysis” by keeping PFK-1 active even in the presence of ATP.
There are two structurally distinct genes coding for LDH: LDH-A and LDH-B. Since the holoenzyme is a tetramer, LDH exists in five different isoforms depending on the composition of the two gene products in the tetramer. LDH1 consists of all four subunits being LDH-B whereas LDH5 consists of all four subunits being LDH-A. LDH2, LDH3, and LDH4 consist of varying mixtures of both LDH-A and LDH-B. The relative affinities of LDH-A and LDH-B for lactate and pyruvate make LDH-A more amenable to facilitate the conversion of pyruvate to lactate and make LDH-B more amenable to facilitate the conversion of lactate to pyruvate [35]. The expression of LDH-A is upregulated in all cancers whereas the expression of LDH-B is downregulated in most cancers. This shift in the relative amounts of the two isoforms facilitates “aerobic glycolysis” in cancer cells to convert pyruvate to lactate.
3.2. Lactic acidosis and tumor microenvironment
Cancer cells produce massive amounts of lactic acid as a result of not only “aerobic glycolysis” but also acceleration of the pathway. Unless this lactic acid is removed from the cells, cellular pH will become acidic to interfere with normal biological and metabolic processes, thus being detrimental to survival of cancer cells. To escape from such a consequence, cancer cells export lactic acid across the plasma membrane and release it into the extracellular medium. This results in severe lactic acidosis in the tumor microenvironment, increasing the levels of lactate as well as H+. Lactate levels in tumor microenvironment has been shown to be as high as 40 mM compared to normal levels of 1.5-2.5 mM in circulation [36,37]. Releasing lactic acid from cancer cells into the extracellular medium is critical to support “aerobic glycolysis” since acidic pH in cytosol will inhibit the activities of glycolytic enzymes. Additionally, a slightly basic intracellular pH is required for the activity of LDH, facilitation of cell proliferation, and escape from apoptosis [36]. At the same time, acidic extracellular pH limits normal cell growth and survival. Cancer cells are more adaptive to the acidic environment than normal cells, so they can survive by adjusting their metabolism. The death of normal cells in the tumor microenvironment clears the space for tumor to grow. Moreover, lower pH in the tumor microenvironment provides a favorable environment for cancer cells to suppress the immune cells and degrade the extracellular matrix (ECM), which facilitates cancer cells to survive, invade, and metastasize.
3.3. Lactate and pseudohypoxia
When oxygen availability is low in cells, the protein level of hypoxia-inducible factor-1α (HIF-1α), a transcription factor, increases to reprogram the transcriptional profile in such a way that the cells can adapt and try to survive under the hypoxic conditions. The underlying mechanism for the reciprocal changes in HIF-1α protein in response to oxygen levels involves oxygen-dependent regulation of HIF-1α protein degradation via proteasomes. This process is controlled by prolyl hydroxylases (PHDs) which hydroxylates certain prolyl residues in HIF-1α as a prerequisite for proteasomal degradation. PHDs use molecular oxygen for the hydroxylation step and also uses α-ketoglutarate and Fe2+ as additional cofactors [38]. Therefore, in the presence of oxygen, HIF-1α gets degraded. When oxygen supply is less, the catalytic activity of PHDs is impaired, thus leading to decreased prolyl hydroxylation of HIF-1α and consequent prevention of proteasomal degradation. As such, increase in HIF-1 protein is a hallmark of hypoxia. Interestingly, one of the isoforms of PHDs, namely PHD2, is inhibitable by lactate via blockade of the binding of α-ketoglutarate to the enzyme [39]. This inhibition occurs in the presence of normal oxygen supply. Accordingly, cancer cells which accumulate lactate due to “aerobic glycolysis” have reduced PHD2 activity, reduced proteasomal degradation of HIF-1α, and hence increased levels of HIF-1α protein. Normally HIF-1α levels go up only under hypoxic conditions whereas in cancer cells HIF-1α levels go up under normoxic conditions. In other words, cancer cells behave as if they are under hypoxic conditions even in the presence of adequate oxygen supply. This situation is described as “pseudohypoxia” and it is the consequence of increased production of lactate in cancer cells. As detailed subsequently in this review, the “pseudohypoxia” with the associated increase in HIF-1α is beneficial for the growth and proliferation of cancer cells since this transcription factor plays a critical role in the reprogramming of various metabolic pathways and in the expression of multiple nutrient transporters.
3.4. Oncogenic transcription factors HIF-1α and c-Myc and their relevance to “aerobic glycolysis”
HIFs are heterodimeric proteins with two subunits: an oxygen sensitivity α subunit and a constitutively expressed β subunit [40]. There are three paralogs of the HIF-1α subunit, including HIF-1α, HIF-2α, and HIF-3α, and two paralogs of the HIF-1β subunit (ARNT and ARNT2) in humans [41]. Among the three paralogs of the α subunit, HIF-1α is mostly expressed in normal tissues throughout the human body, whereas HIF-2α is specific tissue expression [42]. HIF-1α and HIF-2α are overexpressed in different cancer cells, which supports tumor growth by upregulating genes participating in tumor invasion and angiogenesis. HIF-3α has been studied less than HIF-1α and HIF-2α. The biological functions of HIF-3α have not been completely elucidated.
HIF-1α comprises an inhibitory domain (ID) (residues 576-785) inserted in between two transactivation domains, TAD-N (residues 531-575) and TAD-C (residues 786-826) [43,44]. These transactivation domains bind to coactivators, including (CBP)/p300, SRC-1, and TIF-2, to induce HIF-1α mRNA expression [45]. HIF-1α has a short half-life, approximately about 5-10 minutes, and is rapidly degraded in the normoxic condition [46]. HIF-1α mRNA, protein, and its activity are tightly regulated by O2 [45]. Especially, the ubiquitin-mediated process regulating the degradation of HIF-1α by activating the tumor suppressor protein pVHL (von Hippel-Lindau) is the O2-dependent process, whereas HIF-1β expression is independent of the presence of O2 [42]. Prolyl hydroxylase domain (PHD) enzymes use oxygen to hydroxylate conserved proline residues (Pro402 and Pro564) in the oxygen-dependent degradation domain (ODDD) region of the α subunit and acetyltransferase arrest defective 1 (ARD-33 1) enzyme acetylates lysine (Lys532) under the normoxic condition [47]. VHL, a component of E3 ubiquitin, selectively binds to the hydroxylated HIF1α at TAD-N and subsequently induces HIF1α degradation [48]. In addition to this proteasomal degradation process that controls the protein levels of HIF-1α, a different mechanism participates in regulating the transcriptional activity of HIF-1α. A factor inhibiting HIF (FIH) is an Fe (II)-dependent enzyme that interacts with ID and hydroxylates HIF-1α via residue asparagine (Asn803) at TAD-C [45]; this hydroxylation interferes with the transcriptional activity of HIF-1α. Together, VHL and FIH prevent coactivators from binding to HIF-1α transactivation domains as well as destabilize HIF-1α.
HIF-1α levels are elevated in cancer cells even under normoxic conditions because of the inhibition of PHD2 by lactate (pseudohypoxia). The resultant increase in the transcriptional activity of HIF-1α fuels aerobic glycolysis in cancer cells. The targets for HIF-1α include LDH-A, PFKFB3, PDK-1 and PDK-3; therefore, the expression and activities of these proteins are increased in cancer cells. LDH-A favors the conversion of pyruvate to lactate; PFKFB3 increases the levels of fructose-2,6-bisphosphate which then prevents ATP-dependent inhibition of PFK-1; PDK-1 and PDK-3 inactivate PDH and hence interfere with mitochondrial oxidation of pyruvate.
The Myc gene family is well-known to play roles in tumorigenesis and tumor progression. This family has several gene members. c-Myc has been well studied compared to other members and was first discovered as a homolog of v-Myc oncogenes of the avian myelocytomatosis virus [49]. c-Myc contains a basic (i.e., cationic) region that determines the sequence-specific DNA binding, followed by the helix-loop-helix-leucine zipper, known as DNA protein binding [49]. c-Myc interacts with Max to fully become an active transcription factor [50]. The active heterodimer binds to the consensus sequence CACGTG on c-Myc's target genes to activate their transcriptions [51,52]. Max can homodimerize itself, unlike c-Myc, but it prefers forming a heterodimer with c-Myc. The Max homodimer is not an active transcription factor. In normal cells, c-Myc undergoes ubiquitination and subsequently is degraded by the proteasome, so it is expressed at a low level in the cytoplasm [53]. c-Myc is overexpressed in 40% of tumors due to increased transcription and protein stability [53]. Mutant KRAS G12V, one of the most common mutated oncogenes driving cancers, increases c-Myc protein levels in pancreatic cancer cells, primarily through translational and post-translational processes [54]. c-Myc has been demonstrated to function as a transcriptional factor activating several genes that take part in the Warburg effect, glutamine consumption, and lactate production in cancer cells. The c-Myc target genes that are relevant to “aerobic glycolysis” in cancer cells include almost all the enzymes in the glycolytic pathway as well as LDH-A [55].
3.5. Transporters integral to “aerobic glycolysis” in cancer cells
3.5.1. Glucose transporters
To support the unrestricted growth of cancer cells, several nutrient transporters are upregulated to promote the influx of essential nutrients to feed into various metabolic pathways that are reprogrammed in the cells [56,57,58]. Among them, glucose transporters are integral to Warburg hypothesis and the associated “aerobic glycolysis”. The flux of glucose through glycolysis is accelerated in cancer cells. This necessitates increased influx of glucose from circulation into these cells. The first glucose transporter that was found to be upregulated in the plasma membrane of cancer cells was GLUT1, also identified as SLC2A1 [59,60]. In some cancers, GLUT3 (SLC2A3) is also upregulated. SLC2A1 and SLC2A3 belong to the class of facilitative glucose transporters with no involvement of ion gradients in the transport process. The genes coding for these two transporters are transcriptional targets for HIF-1α [59,60,61]. The Michaelis constant for this transporter for glucose across the membrane is ~3 mM, which is close the normal plasma level of glucose (~5 mM). SLC2A1 is not sensitive to insulin. Even though there is no energy involved in the transport mechanism of SLC2A1 and SLC2A3, the increased density of these transporters in the plasma membrane due to increased HIF-1α-induced transcription results in increased influx of glucose from the circulation into cancer cells to feed into the glycolytic pathway.
The increased flux of glucose into cancer cells compared to normal cells forms the basis of positron emission tomography (PET) as a diagnostic tool to detect tumors in the body in vivo. PET scan is a method of noninvasive imaging that merges the biochemical energy utilization variance of different cell types to the diagnosis of pathology [62]. While it can be used for any tissues that have regional variation in glucose uptake, such as the brain, muscle, kidney, etc., this imaging technique has become an important diagnostic tool in oncology field not only to detect tumors but also to evaluate the efficacy of chemotherapy or immunotherapy [63]. The biochemical basis of the success of this technique is directly related to Warburg effect. The process of PET scan imaging uses the acceptance of 2-deoxyglucose as a substrate for SLC2A1. Since cancer cells express higher levels of this transporter than the normal cells in areas surrounding the tumor, 2-deoxy-D-glucose when injected into blood gets into cancer cells at several fold higher levels than into surrounding normal cells. Once inside the cells, this glucose analog gets phosphorylated by hexokinase in the first step of the glycolytic pathway to generate 2-deoxy-D-glucose-6-phosphate. However, unlike glucose-6-phosphate which goes through subsequent steps in glycolysis, 2-deoxy-D-glucose-6-phosphate cannot enter the second step in the pathway, thus leading to accumulation in cells. This preferential accumulation in cancer cells versus normal cells can be detected by PET scan if 2-deoxy-D-glusose is labeled with a positron emitter, hence the use of 18F-2-deoxy-D-glucose (18FDG) as the PET probe in which 18F is the positron emitter. This enables the PET scan to tag the exact location of the tumor in vivo.
For a long time, it was thought that only SLC2A1 is responsible for the increased influx of glucose into cancer cells. SLC2A3 may contribute to glucose uptake in cancer cells to some extent. However, it was discovered later that certain tumors also upregulate the Na+-coupled glucose transporters SGLT1 (SLC5A1) and SGLT2 (SLC5A3) [64]. At the functional level, the increased expression of SLC5A3 has been demonstrated unequivocally [64,65]. In contrast to SLC2A1, SLC5A2 is a concentrative transporter and thus has a greater efficiency than SLC2A1 in transporting glucose into cancer cells. Interestingly, 2-deoxy-D-glucose is not recognized as a substrate by SLC5A2. Therefore, the reliability of PET scan with 18FDG as the probe in cancer detection still depends solely on SLC2A1 (and SLC2A3). α-Methyl-D-glucopyranoside is a selective substrate for SLC5A2; it is not recognized by SLC2A1/3. 18F-Labeled α-methyl-4-deoxy-D-glucopyranoside has been shown to be effective as the PET probe to detect tumors that are positive for SLC5A2 [64,65]. This may have an advantage over 18FDG for SLC5A2-positive tumors to differentiate them from the surrounding normal tissues because SLC2A1 is ubiquitously expressed whereas SLC5A2 expression is restricted almost exclusively to kidney among normal tissues.
3.5.2. Lactate/H+ symporters (monocarboxylate/H+ cotransporters)
Lactic acid is the end product of “aerobic glycolysis” in cancer cells. If this acid is not removed from the cells, it will lead to intracellular acidification with consequent detrimental effects on cell growth and proliferation. The ideal candidate transporters for the removal of lactic acid from cells are the monocarboxylate transporters (MCTs) that mediate the symport of lactate and H+ in an electroneutral mechanism. These transporters belong to Solute carrier 16 gene family (SLC16). There are 14 members in this family; among them, four members, MCT1 (SLC16A1), MCT2 (SLC16A7), MCT3 (SLC16A8), and MCT4 (SLC16A3), function as lactate/H+ symporters [66]. The transport process mediated by these four MCTs is bidirectional, the direction of lactate/H+ movement being dictated simply by the direction of the gradient for lactate. Among these four MCTs, only MCT1 (SLC16A1) and MCT4 (SLC16A3) have been shown to be most relevant to cancer cells [3,4,67,68].
MCT1 (SLC16A1) and MCT4 (SLC16A3) are highly upregulated in cancers. c-Myc, Wnt signaling, NF-kB, and mutant p53 are transcriptional inducers of MCT1 [4,68,69]. Hypoxia induces the expression of MCT4 through a HIF-1α- dependent mechanism [4,68,70]. MCT1 exhibits higher affinity for lactate compared to MCT4 and kinetically more suited to mediate the influx of extracellular lactate into cells. In contrast, MCT4 which has a relatively lower affinity for lactate is more suited to mediate the efflux of lactate from cancer cells that generate massive amounts of this metabolite. Accordingly, these two transporters are not upregulated uniformly in all cancer cells within the tumor. Tumor cells consist of different subpopulations, such as oxidative and glycolytic tumor cells. Oxidative tumor cells are located close to blood vessels and receive enough oxygen, whereas glycolytic tumor cells are far from blood vessels and hence are exposed to hypoxic conditions. This represents a significant revision to the original Warburg hypothesis in which all cancer cells were assumed to uniformly undergo “aerobic glycolysis”. This recent discovery of heterogeneous populations of cancer cells in terms of oxygen exposure has introduced a new term “reverse Warburg effect” [71,72]. This term describes the use of extracellular lactate for energy production via mitochondrial oxidation in some populations of cancer cells that are exposed to normal oxygen. This does not necessarily mean that Warburg effect occurs only in hypoxic cancer cells; if that were the case, what happens in such cells will be “anaerobic glycolysis”, and not “aerobic glycolysis” as defined by Warburg effect. A significant fraction of oxygen-exposed cancer cells still undergo the Warburg effect with “aerobic glycolysis”. It is likely that among the oxygen-exposed cancer cells which are subject to Warburg effect and which to reverse Warburg effect is dependent upon the expression levels of the oncogenes c-Myc, HIF-1α and mutant p53, relative activities of LDH-A versus LDH-B, and the levels of other regulatory mechanisms that facilitate “aerobic glycolysis”. As a consequence, lactate is shuttled between the two different subpopulations of cancer cells within the tumor (Figure 2). In glycolytic tumor cells undergoing the Warburg effect, lactate is produced from pyruvate by LDH-A and exported into the tumor microenvironment by MCT4. In oxidative tumor cells undergoing the reverse Warburg effect, extracellular lactate is taken up by MCT1 and gets converted to pyruvate by LDH-B for subsequent energy generation in mitochondria. This lactate shuttling may not be limited to cancer cells within the tumor. Activated T cells perform “aerobic glycolysis” and produce lactate and release it into the external environment. However, this MCT4-mediated lactate-release mechanism is impaired in tumors because of the presence of high concentration of lactate in the tumor microenvironment (Figure 2). Consequently, proliferation of cytotoxic T cells is suppressed, hence leading to the tumor’s ability to evade attack by the immune system. In contrast, tumor-associated endothelial cells and macrophages take up lactate from the tumor microenvironment via MCT1 for energy production. This promote cell proliferation and tumor angiogenesis in endothelial cells and also induces macrophage polarization to generate pro-tumor M2 macrophages involved in immunosuppression and neovascularization (Figure 2).
3.5.3. Additional transporters for H+ export in cancer cells
Cancer cells utilize several other mechanisms to prevent intracellular acidification caused by lactic acid production. Many of these transporters export H+ out of the cells and their expression is induced by multiple oncogenic drivers [73]. Such transporters include Na+/H+ exchanger-1 (NHE-1/SLC9A1) [74], Na+/HCO3 cotransporter NBCn1 (SLC4A7) [75], and V-type H+ pump [76]. Cancer cells also use carbonic anhydrase IX in the regulation of intracellular pH. Recent studies have shown that the amino acid transporter SLC38A5 is upregulated in some cancers [77,78]. This is a unique amino acid transporter that functions as an amino acid-dependent Na+/H+ exchanger with intracellular alkalinization in the presence of extracellular amino acid substrates [79]. As such, the upregulation of SLC38A5 in cancer cells has a dual role, namely provision of amino acids to support cell growth and proliferation and export of H+ to prevent intracellular acidification.
3.6. Acidic pH in tumor microenvironment and its relevance to tumor growth
3.6.1. Non-specific effects of acid pH on tumor microenvironment
With the concentration of lactic acid in the range of 30-40 mM in the extracellular space, cancer cells and the stromal cells in the tumor microenvironment are bathed in a medium with an unphysiological acid pH (pH 6.0-6.5). Cancer cells survive this acid pH because they upregulate their biochemical machinery to protect themselves from the harmful effects of extracellular acid pH with the aid of oncogenes and consequent changes in the expression levels of various transporters and biochemical processes. In contrast, the stromal cells in the same acidic environment do not have this luxury and hence face the consequences. In addition, cancer cells and cancer cell-associated stromal cells also begin to secrete various proteases, primarily metalloproteinases, which start digesting the extracellular proteins such as collagen. As such, the negative effects of acidic pH on stromal cells and the clearance of extracellular connective tissue pave the way for cancer cells to grow further.
In addition, the acidic pH in the extracellular medium coupled with the efficient maintenance of intracellular pH in cancer cells creates an inwardly directed H+ gradient across the plasma membrane in these cells. There are several transporters for important nutrients whose transport process is fueled by a transmembrane H+ gradient [80,81,82] and most them are upregulated in cancer cells to exploit the now-naturally occurring pH gradient across their plasma membrane to energize the H+-coupled transporters and obtain these nutrients to support their growth and proliferation. The first example is MCT1 which has already been discussed. This transport activity of MCT1 to mediate the influx of extracellular lactate into oxidative cancer cells is activated by the inwardly directed H+ gradient. While lactate is the “waste” product of cellular metabolism in cancer cells with “aerobic glycolysis”, it is an energy-rich metabolite for oxidative cancer cells. The efficient uptake of lactate via MCT1 with subsequent mitochondrial oxidation generates ATP to fuel cancer growth. The substrates of other H+-coupled nutrient transporters are also very important for cell proliferation. This includes peptides, amino acids, folate, iron and citrate.
3.6.2. H+-Coupled peptide transporter PEPT1 (SLC15A1)
PEPT1 transports small peptides consisting of 2-3 amino acids and it represents the first H+-coupled nutrient transporter discovered in mammalian cell plasma membrane [83,84]. This transporter is upregulated in cancers [85,86,87,88]. Normal plasma contains small peptides only at small levels, but this is most likely not the case in the tumor microenvironment. The metalloproteinases secreted by tumor cells and stromal cells digest extracellular proteins and release small peptides in the local environment, and these peptides are the likely substrates for PEPT1 that is upregulated in cancer cells [88]. The naturally occurring pH gradient across the plasma membrane of these cells would energize the transport activity of PEPT1 to satisfy their amino acid nutrition.
3.6.3. H+-Coupled amino acid transporter PAT1 (SLC36A1)
PAT1 transports small amino acids such as proline and glycine [89]. Even though the expression of this transporter is not altered in cancer, its expression is evident [85]. This could be relevant to amino acid nutrition in cancer cells because proline and glycine are likely to be present in the tumor microenvironment at higher concentrations than in normal plasma because of the metalloproteinase-mediated degradation of extracellular collagen, a protein rich in these two amino acids. Recent studies have shown that the transport activity of PAT1 is coupled to activation of mTORC1 signaling [90], thus the transporter becoming relevant to tumor growth.
3.6.4. H+-Coupled folate transporter PCFT (SLC46A1)
Folic acid is an obligatory vitamin for the synthesis of purines, pyrimidines and nucleotides, and mammalian cells employ multiple transport mechanisms as well as receptor-mediated endocytosis to acquire this vitamin from circulation. One of the transport mechanisms involves the H+-coupled transporter PCFT (SLC46A1) [91,92]. This transporter is highly expressed in cancers and is being exploited to deliver cytotoxic folate analogs to antagonize the biological functions of folate in cancer chemotherapy [93].
3.6.5. H+-Coupled divalent metal ion transporter DMT1 (SLC11A2)
Iron is a micronutrient obligatory for many vital functions necessary for the survival and proliferation of cells. It is an integral component, both in the free form and also in the form of heme, in the electron transport chain for ATP production. It is also an obligate cofactor for numerous enzymes and for hemoglobin. Most cells acquire this micronutrient in the form of transferrin-bound iron via receptor-mediated endocytosis involving transferrin receptor. But free unbound iron is also transported into cells across the plasma membrane via the H+-coupled divalent metal ion transporter DMT1 (SLC11A2) [94]. Interestingly, the same transporter is required to deliver free iron from lysosomes into cytoplasm following the entry and lysosomal processing of transferrin-bound iron in receptor-mediated endocytosis [95]. In both cases, the transport process is dependent on a transmembrane H+ gradient. SLC11A2 is upregulated in cancers [96], thus providing an efficient mechanism for cancer cells to accumulate iron in support of their growth and proliferation.
3.6.6. Na+/H+-coupled citrate transporter NaCT (SLC13A5)
Citrate is a key metabolic intermediate at the junction of important metabolic pathways. It is at the center of the citric-acid cycle, functions as a regulator of glycolysis, and serves as the carbon source for fatty acid synthesis. For a long time, it was thought that mitochondrial generation is the sole source of citrate in cells. But with the discovery of a plasma membrane transporter selective for citrate [97,98], it has become clear that citrate in circulation could form a significant source of citrate for cells. Extracellular citrate fuels cancer growth [99,100]. This transporter is upregulated in some cancers, particularly liver cancer [101]. Even though it is a Na+-coupled transporter [97,98], its activity is markedly stimulated by extracellular acidic pH [102]. The transporter possesses more than one Na+-binding sites and it seems that one or more of these binding sites have much higher affinity for H+ than for Na+. As such, SLC13A5 actually functions as a Na+/H+-coupled citrate transporter. Citrate is present at significant levels in normal circulation (~200 μM); therefore, SLC13A5 has the ability to deliver citrate into cancer cells and its transport activity is stimulated by the acidic pH in the tumor microenvironment. This phenomenon might be even more relevant to tumors that grow in bone after metastasis because bone holds more than 60% of citrate in the body in its matrix. When tumors grow in bone, the bone matrix is degraded and release citrate in free form that could form an important source of this metabolite to fuel the growth of tumors.
3.7. Lactate as a signaling molecule: Lactate receptors
Lactate has always been considered as an interesting metabolite because of certain unique features. It is the sole end product of glycolysis in mature erythrocytes which use glucose as the principal energy source. Due to lack of mitochondria, pyruvate generated at the end of the glycolytic pathway has to be converted into lactate to recycle NAD+. It is also at the center of Cori cycle, a metabolic crosstalk between skeletal muscle and liver. During exercise, skeletal muscle needs more oxygen than what is readily available, thus being exposed to hypoxia. This results in anerobic glycolysis during exercise, thus generating lactate as the end product of glycolysis, which is then released into circulation. This explains why plasma levels of lactate rise during exercise. Then liver picks up this lactate to use it as a carbon source for gluconeogenesis, and the newly synthesized glucose is then released into circulation. This glucose-lactate-glucose cycle is called Cori cycle. Based on these biochemical features, lactate is always considered as the biomarker of oxygen deficit. This is further supported by the fact that ischemic tissues generate lactate as the end product of glucose metabolism. But recently this ubiquitous metabolite has come to the forefront of cancer biology as a signaling molecule. This elevation of lactate from the old status of “waste product of glycolysis” to the new status of “hormone” has renewed interest in this molecule in the field of cancer biology. When the outcomes of lactate signaling are considered, a rationale emerges as to why this metabolite is a biomarker of oxygen deficit. These outcomes include, among many other things, stabilization of HIF-1α protein and promotion of angiogenesis. These lactate-induced processes are directly related to promotion of tumor growth and metastasis, thus underscoring the biological importance of lactate as the principal metabolite of glucose metabolism in cancer cells.
3.7.1. Intracellular lactate receptor NDRG3
NDRG3 is one of the four members of the NDRG (N-Myc-downstream regulated genes) family of proteins. NDRG3 functions as a tumor promoter [103,104]. It binds to c-Raf and activates ERK1/2 signaling, which regulates genes transcription, cell proliferation, migration, differentiation, and cytoskeletal remodeling to support tumor growth in the late stage of hypoxia [105,106]. NDRG3 is subject to proteasomal degradation and this requires its binding to VHL. Interestingly, lactate interferes with the interaction of NDRG3 with VHL by directly binding to NDRG3, thus preventing its depletion of NDRG3 via proteasomal degradation [105]. Thus, NDRG3 functions as an intracellular receptor for lactate. This is in addition to the influence of lactate on prolyl hydroxylate-2 (PHD2) that hydroxylates NDRG3 for subsequent binding to VHL and consequent proteasomal degradation. It is also possible that the binding of lactate to NDRG3 is needed not only for the stabilization of the NDRG3 protein but also for enabling the binding of NDRG3 to c-Raf. This lactate-stimulated NDRG3-Raf-ERK signaling promotes angiogenesis and cell growth.
3.7.2. Cell-surface G-protein-coupled receptor GPR81 for lactate
Lactate also elicits its intracellular signaling by functioning in the extracellular milieu as an agonist for the G-protein-coupled receptor GPR81 on the plasma membrane [107,108]. This receptor was first identified in adipocytes where its activation by lactate reduces intracellular levels of cAMP which reduces the hydrolysis of triglycerides by inactivating the hormone-sensitive lipase. Of importance to the field of cancer are the findings that GPR81 is expressed at high levels in cancer and that the receptor functions as a tumor promoter [6,109]. This has direct relevance to tumor growth because cancer cells generate lactate and release it into the tumor microenvironment where it can function extracellularly as an agonist for GPR81. As many other cell types in the stroma surrounding the tumor also express this receptor, lactate present in the extracellular milieu functions as an GPR81 agonist not only in cancer cells but also in adjacent non-cancer cells.
3.7.3. Autocrine functions of GPR81/lactate in tumor growth
Autocrine signaling involves a hormone secreted by a given cell binds to a cell-surface receptor on the same cells to induce downstream effects. Cancer cells secrete lactate and also express the lactate receptor GPR81, thus paving the path for autocrine signaling (Figure 3). Downregulation of GPR81 in breast cancer cells decreases tumor invasion and migration [110,111]. This effect is associated with decreased expression of three critical enzymes related to aerobic glycolysis: hexokinase 2, PFK-1, and LDH-A. As such, lactate elicits a positive feedback regulation on its own production by acting on GPR81 in an autocrine manner. In addition, the intracellular signaling resulting from GPR81 activation by lactate leads to promotion of synthesis and secretion of pro-angiogenic factors amphiregulin, platelet-derived growth factor, serpin peptidase inhibitors Serpin E1 and Serpin F1, plasminogen activator and vascular endothelial growth factor. This most likely occurs via activation of the PI3K/ATK pathway. In pancreatic cancer cells, knockdown of GPR81 reduces cell survival, mitochondrial activity, and monocarboxylate transporters MCT1 and MCT4 [112]. Lactate stabilized HIF-1α via GPR81-mediated decrease in cAMP and consequent inhibition of protein kinase-A in B16-F10 and Hepa1-6 cells [113]. In addition to supporting tumorigenesis, GPR81 helps tumor cells to escape from cytotoxic immune cells. In human lung cancer cells, activated GPR81 reduces intracellular cAMP levels and inhibits protein kinase-A which results in the activation of the transcriptional coactivator TAZ [114]. TAZ then interacts with TEAD1 and the resultant complex causes the transcriptional activation of the gene coding for PD-L1 (programmed cell death ligand 1). PD1 is an inhibitory receptor expressed in T cells during activation. The binding of PD-1 (T cell) and PD-L1 (cancer cell) inhibits T cell activation and prevent T cells from killing cancer cells. Thus, inducing PD-L1 on cancer cells' surface facilitates cancer cells to escape the immune checkpoints. Moreover, GPR81 signaling induces DNA repair proteins such as BRCA1, nibrin, and DNA-dependent protein kinases in cervical cancer cells [115]. It also enhances doxorubicin chemoresistance by increasing the expression of ABCB1, a drug efflux transporter. Interestingly, a recent study has reported that lactate released by cancer cells initiates a signaling pathway via GPR81 to induce the expression of the same receptor to a higher level to promote the autocrine signaling even further [116]. This process involves induction of the expression of the transcription factor Snail and promotion of the formation of the Snail/EZH2/STAT3 complex which then directly binds to the promoter of GPR81 gene to induce expression. In another study, lactate generated by breast cancer cells enhance their growth and invasiveness in an autocrine manner through regulation of extracellular matrix properties and Notch signaling [117].
3.7.4. Paracrine functions of GPR81/lactate in tumor growth
Paracrine signaling involves the secretion of a hormone by a given cell with subsequent action of the hormone on a cell-surface receptor expressed on a different cell present in adjacent or distant location to elicit a signal. A recent report by Brown et al [118] constitutes a prime example for the paracrine function of lactate in breast cancer. Lactate generated and released by breast cancer cells activate GPR81 present in dendritic cells to suppress antigen presentation via downregulation of MHC-II. This aids tumor cells to evade immunosurveillance because the dendritic cells are now defective in handling and presenting tumor cell-specific antigens to cytotoxic T cells. The cancer cell-generated lactate also acts on intratumoral plasmacytoid dendritic cells to suppress the production of cytokines that are needed for proliferation of T cells [119]. At the same time, the secretion of the tryptophan metabolite kynurenine is also increased in these dendritic cells in response to GPR81 activation by lactate, which then induces the production of tumor-suppressive regulatory T cells. The combination of interference with T cell expansion and promotion of Treg production enhances the ability of tumor cells to escape immune cells. Collectively these studies show that lactate generated by cancer cells seems to promote immune evasion of the tumor [120]. Similarly, lactate released from inflammatory bone marrow neutrophils binds to GPR81 on endothelial cells and subsequently induces vascular endothelial cadherin that increases blood vessel permeability and neutrophil mobilization [121]. This may have relevance to inflammation-associated cancers such as colon cancer and even breast cancer if cancer cell-generated lactate elicits a similar effect on tumor-associated endothelial cells. Thus, the paracrine function of GPR81/lactate plays various roles in cell-cell communication relevant to cancer growth (Figure 3).
3.8. Downstream metabolic consequences of aerobic glycolysis in cancer cells
3.8.1. Pentose phosphate pathway and antioxidant machinery
A robust antioxidant machinery is important for the survival and also for chemoresistance of cancer cells. Catabolic metabolism is oxidative and mitochondrial electron transport chain generates reactive oxygen species all the time because a small portion of molecular oxygen that is used in this process does not go through reduction completely to water but gets out of the electron transport chain partially reduced. For complete oxidation of O2 to form water, four electrons need to be added. Addition of less than four electrons to O2 results in reactive oxygen species: superoxide resulting from one electron and peroxide resulting from two electrons. Unless these reactive oxygen species are removed, cells face the risk of oxidative damage and cell death. The removal of superoxide and peroxides involves the glutathione/glutathione peroxidase/glutathione reductase system, which needs NADPH as the cofactor. There are two major pathways that generate NADPH: pentose phosphate pathway and malic enzyme. Both pathways are activated in cancer [26]. When aerobic glycolysis occurs at a much faster rate in cancer cells, the cellular levels of various intermediates in the pathway also go up. Glucose-6-phosphate, one of the intermediates, is the starting point for the pentose phosphate pathway. The rate-limiting enzyme in this pathway is glucose-6-phosphate dehydrogenase, which is induced by c-Myc [122]. The increased availability of the starting material and the increased activity of the rate-limiting enzyme fuels the increased flux of glucose-6-phosphate through the pentose phosphate pathway to generate NADPH. Another product of this pathway is ribose-5-phosphate which promotes purine and pyrimidine synthesis because of the production of phosphoribose pyrophosphate, a substrate as well as an activator of nucleotide synthesis, which is necessary for rapidly proliferating cells such as cancer cells. Thus, the increased activity of aerobic glycolysis is tied to increased activity of the pentose phosphate pathway. Malic enzyme converts malate into pyruvate along with production of NADPH. This enzyme is also upregulated in cancer cells [123]. The resultant robust antioxidant machinery protects cancer cells from oxidative damage.
This machinery is also related to development of chemoresistance. Many chemotherapeutic agents (e.g., cisplatin) are inactivated by reactions that use glutathione. When cellular levels of NADPH are high, the levels of reduced glutathione are also kept high because of the efficient conversion of oxidized glutathione into reduced glutathione. Therefore, the robust antioxidant machinery detoxifies chemotherapeutic drugs, thus favoring drug resistance and protecting the cancer cells from cell death induced by such drugs.
Glutathione is an obligatory component of the antioxidant machinery in cancer cells. It is synthesized using three amino acids: glutamate, cysteine, and glycine. Among these three amino acids, cysteine is rate-limiting. This amino acid is provided to cancer cells in the form of cystine via the transporter xCT/SLC7A11 which is upregulated in cancer cells [27,28].
3.8.2. Serine biosynthesis and one-carbon metabolism
Serine is an important source of one-carbon moieties for one-carbon metabolism. This pathway begins with the generation of N5, N10-methylenetetrahydrofolate from the conversion of serine into glycine by the reaction mediated by serinehydroxymethyl transferase. N5, N10-methylene tetrahydrofolate can then be converted into N5-formyltetrahydrofolate and N5-methyltetrahydrofolate. These folate derivates serve as the source of one-carbon moieties in one-carbon transfer reactions that participate in multiple metabolic processes, including purine and pyrimidine synthesis, nucleotide synthesis, and homocysteine metabolism. Therefore, serine becomes an important amino acid for cancer cells. Even though many amino acid transporters that can transfer extracellular serine into cells are upregulated in cancer cells [12,13,14,15], the endogenous synthesis of serine is also activated to satisfy the increased demand for this amino acid. The starting material for serine synthesis is 3-phosphoglycerate, an intermediate in glycolysis. The first and the rate-limiting enzyme in serine biosynthesis is phosphoglycerate dehydrogenase, which is induced in cancer cells [124].
3.8.3. Glutaminolysis and glutamine transporters
Similar to serine, the need in cancer cells for glutamine is also very high. Glutamine serves as the nitrogen source for purine synthesis, provides glutamate for glutathione synthesis, and also activates mTORC1. In addition, it feeds into citric acid cycle to generate ATP and also the signaling metabolite lactate. The conversion of glutamine to lactate involves a series of reactions, collectively called “glutaminolysis” (Figure 4A). This pathway utilizes a truncated form of the citric acid cycle and generates NADH and FADH2 that can be used in ETC/OXPHOS to produce ATP, malate that can be used by malic enzyme to generate NADPH to boost the antioxidant machinery, and pyruvate that can be converted to lactate, the tumor-promoting signaling molecule. It has been estimated that almost one-half of total lactate produced in cancer cells originates from glutaminolysis, the remainder being generated in “aerobic glycolysis” [125]. A portion of this malate also undergoes the next step in the citric acid cycle to generate NADH which is then used in ETC/OXPHOS to produce additional ATP. This underscores the critical role of glutamine metabolism in cancer particularly when given the notion that existed for a long time that almost all of lactate generated in cancer cells comes from aerobic glycolysis.
3.8.4. Reductive carboxylation and fatty acid synthesis
One of the reactions in the citric acid cycle within mitochondria is the conversion of isocitrate to α-ketoglutarate which is mediated by isocitrate dehydrogenase-3 (IDH-3) and involves generation of CO2 and NADH. Since this reaction catalyzes decarboxylation (removal of CO2 from isocitrate) and oxidation (removal of electrons from isocitrate to convert NAD+ to NADH), it is called “oxidative decarboxylation”. Mitochondria also contain IDH-2, another isoform of the same enzyme. This enzyme can use NAD+/NADH pair or NADP+/NADPH pair as cofactors. When cellular levels of NADPH are high as in cancer cells, IDH-2 can perform the reverse reaction in which α-ketoglutarate gets converted to isocitrate. This reaction involves carboxylation as well as addition of electrons (NADPH gets converted to NADP+). Accordingly, this reaction is called “reductive carboxylation” (Figure 4B). Cancer cells perform this reaction to reverse the citric acid cycle to generate citrate from α-ketoglutarate. Glutamine is the source of this α-ketoglutarate (glutamine → glutamate → α-ketoglutarate). Citrate is then converted to acetyl-CoA by ATP-citrate lyase and the resulting acetyl-CoA is then used as the carbon source for fatty acid synthesis. Increased fatty acid synthesis is necessary for cancer cell proliferation, especially for membrane biogenesis.
4. Oncometabolites
Oncometabolites refer to small-molecule metabolites that are present at less levels in normal cells but are increased in cancer cells and also elicit tumor-promoting effects [132,133,134]. These metabolites are generated as a consequence of rewiring of metabolic pathways in cancer cells. Lactate is considered as an oncometabolite. Cancer cells generate this metabolite at high levels and lactate has several biological functions, GPR81-dependent as well as GPR81-independent, that are involved in promotion of tumor growth, thus conforming to the definition of an oncometabolite. Citrate may also qualify as an oncometabolite. Two other metabolites commonly included in this category are fumarate and succinate. These are generated in certain cancers at higher-than-normal levels when fumarate hydratase and/or succinate dehydrogenase are defective due to mutations. These metabolites inhibit α-ketoglutarate-dependent dioxygenases, which leads to DNA hypermethylation, thus leading epigenetic changes conducive for carcinogenesis and cancer growth. D-2-hydroxyglutarate is another oncometabolite which is produced by specific mutants of IDH-1 and IDH-2. These are gain-of-function mutations and the mutated enzymes gain the ability to convert α-ketoglutarate into D-2-hydroxyglutarate. This oncometabolite also functions similar to fumarate and succinate in inhibiting α-ketoglutarate-dependent dioxygenases, thus influencing epigenetics. However, recent studies have discovered an additional function of this oncometabolite [135]. Tumor cells release this metabolite which is then taken up by T cells where it inhibits LDH-A, thus reducing the conversion of pyruvate into lactate and consequently forcing oxidative metabolism of pyruvate within mitochondria. Normally, proliferating T cells perform “aerobic glycolysis” similar to cancer cells. When the metabolic phenotype is changed to oxidative metabolism, T-cell proliferation is suppressed, which enables tumors to evade immune surveillance.
5. Conclusions
Cancer cells reprogram metabolic pathways to suit their biological needs and the oncogenic proteins c-Myc, HIF-1α, and mutant p53 aid or initiate this process. Glucose metabolism and glutamine metabolism are the ones that are most affected. The reprogrammed metabolic pathways have led to coining of new words such as “aerobic glycolysis”, “glutaminolysis” “glutamine addiction”, “reductive carboxylation” “oncometabolites”, and “reverse Warburg effect” in cancer vocabulary. The entire field of cancer cell metabolism started with Warburg effect but in the course of time has seen considerable expansion and modifications, including the revision of the explanation for the Warburg effect itself. The new information gained in this field has already led to identification of new therapeutic targets for cancer therapy. This evolution and shifting of focus to metabolism represent an exciting and refreshing change in recent years in the field of cancer biology.
Author Contributions
Conceptualization, S.S., Y.D.B. and V.G.; methodology, M.M. and N.T.N.; data curation, V.G.; writing—original draft preparation, M.M., N.T.N. and V.G.; writing—review and editing, S.S. and Y.D.B.; supervision, V.G.; funding acquisition, V.G. and Y.D.B. All authors have read and agreed to the published version of the manuscript.
Funding
Please add: This work was funded by National Institutes of Health Grants R21CA277140 (V.G) and R01CA262420 (Y.D.B).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
This is a review and all the information included in this manuscript is available from published reports in the literature.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Warburg, O. The metabolism of carcinoma cells. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Marchiq, I.; Pouyssegur, J. Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H+ symporters. J. Mol. Med. (Berl) 2016, 94, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Payen, V.L.; Mina, E.; Van Hee, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate transporters in cancer. Mol. Metab. 2020, 33, 48–66. [Google Scholar] [CrossRef] [PubMed]
- Polet, F.; Feron, O. Endothelial cell metabolism and tumour angiogenesis: glucose and glutamine as essential fuels and lactate as the driving force. J. Intern. Med. 2013, 273, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.P.; Ganapathy, V. Lactate/GPR81 signaling and proton motive force in cancer: Role in angiogenesis, immune escape, nutrition, and Warburg phenomenon. Pharmacol. Ther. 2020, 206, 107451. [Google Scholar] [CrossRef] [PubMed]
- Reina-Campos, M.; Diaz-Meco, M.T.; Moscat, J. The complexity of the serine glycine one-carbon pathway in cancer. J. Cell Biol. 2020, 219, e201907022. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Fan, M.; Liu, Z.; Li, X.; Wang, H. Serine, glycine and one-carbon metabolism in cancer. Int. J. Oncol. 2021, 58, 158–170. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Mates, J.M.; Di Paola, F.J.; Campos-Sandoval, J.A.; Mazurek, S.; Marquez, J. Therapeutic targeting of glutaminolysis as an essential strategy to combat cancer. Semin. Cell Dev. Biol. 2020, 98, 34–43. [Google Scholar] [CrossRef]
- Alberghina, L.; Gaglio, D. Redox control of glutamine utilization in cancer. Cell Death Dis. 2014, 5, e1561. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, V.; Thangaraju, M.; Prasad, P.D. Nutrient transporters in cancer: Relevance to Warburg hypothesis and beyond. Pharmacol. Ther. 2009, 121, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, Y.D.; Babu, E.; Ramachandran, S.; Ganapathy, V. Amino acid transporters in cancer and their relevance to “glutamine addiction”: Novel targets for the design of a new class of anticancer drugs. Cancer Res. 2015, 75, 1782–1788. [Google Scholar] [CrossRef]
- Bhutia, Y.D.; Ganapathy, V. Glutamine transporters in mammalian cells and their functions in physiology and cancer. Biochim. Biophys. Acta 2016, 1863, 2531–2539. [Google Scholar] [CrossRef] [PubMed]
- Sniegowski, T.; Korac, K.; Bhutia, Y.D.; Ganapathy, V. SLC6A14 and SLC38A5 drive the glutaminolysis and serine-glycine-one-carbon pathways in cancer. Pharmaceuticals 2021, 14, 216. [Google Scholar] [CrossRef] [PubMed]
- White, E. Exploiting the bad eating habits of Ras-driven cancers. Genes Dev. 2013, 27, 2065–2071. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulou, E.; Bulusu, V.; Kamphorst, J.J. Metabolic scavenging by cancer cells: when the going gets tough, the tough keep eating. Br. J. Cancer 2016, 116, 635–640. [Google Scholar] [CrossRef]
- Goberdhan, D.C.I.; Wilson, C.; Harris, A.L. Amino acid sensing my mTORC1: Intracellular transporters mark the spot. Cell Metab. 2016, 23, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Joiner, A.M.N.; Jansen, R.M.; Hurley, J.H. Amino acid sensing and lysosomal signaling complexes. Curr. Opin. Struct. Biol. 2023, 79, 102544. [Google Scholar] [CrossRef]
- Torti, S.V.; Torti, F.M. Iron: The cancer connection. Mol. Aspects Med. 2020, 75, 100860. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Jaramillo-Martinez, V.; Mathew, M.; Suresh, V.V.; Sivaprakasam, S.; Bhutia, Y.D.; Ganapathy, V. Sigma receptors: Novel regulators of iron/heme homeostasis and ferroptosis. Int. J. Mol. Sci. 2023, 24, 14672. [Google Scholar] [CrossRef] [PubMed]
- Kory, N.; Wyant, G.A.; Prakash, G.; Uit de Bos, J.; Bottanelli, F.; Pacold, M.E.; et al. SFXN1 is a mitochondrial serine transporter required for one-carbon metabolism. Science 2018, 362, eaat9528. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Zhang, L.; Zheng, J.; Sun, H.; Shao, C. Ferroptosis: Biochemistry and biology in cancers. Front. Oncol. 2021, 11, 579286. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Guo, Q.; Shen, Z.; Yang, W.; Zhou, Y.; Sun, Z.; et al. Frontiers of ferroptosis research: An analysis from the top 100 most influential articles in the field. Front. Oncol. 2022, 12, 948389. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.S.; Cha, Y.H.; Kim, H.S.; Kim, N.H.; Yook, J.I. The pentose phosphate pathway as a potential target for cancer therapy. Biomol. Ther. (Seoul) 2018, 26, 29–38. [Google Scholar] [CrossRef]
- Jyotsana, N.; Ta, K.T.; DelGiorno, K.E. The role of cystine/glutamate antiporter SLC7A11/xCT in the pathophysiology of cancer. Front. Oncol. 2022, 12, 858462. [Google Scholar] [CrossRef]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 2021, 12, 599–620. [Google Scholar] [CrossRef]
- Armenta, D.A.; Laqtom, N.N.; Alchemy, G.; Dong, W.; Morrow, D.; Poltorack, C.D.; et al. Ferroptosis inhibition by lysosome-dependent catabolism of extracellular protein. Cell. Chem. Biol. 2022, 29, 1588–1600.e7. [Google Scholar] [CrossRef]
- Yiew, N.K.H.; Finck, B.N. The mitochondrial pyruvate carrier at the crossroads of intermediary metabolism. Am. J. Physiol. Endocrinol. Metab. 2022, 323, E33–E52. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.W.; Lin, S.C.; Chen, K.F.; Lai, Y.Y.; Tsai, S.J. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J. Biol. Chem. 2008, 283, 28106–28114. [Google Scholar] [CrossRef]
- Yi, M.; Ban, Y.; Tan, Y.; Xiong, W.; Li, G.; Xiang, B. 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 and 4: A pair of valves for fine-tuning of glucose metabolism in human cancer. Mol. Metab. 2019, 20, 1–13. [Google Scholar] [CrossRef]
- Bartrons, R.; Simon-Molas, H.; Rodriguez-Garcia, A.; Costano, E.; Navarro-Sabate, A.; Manzano, A.; Martinez-Outschoorn, U.E. Fructose 2,6-bisphospahte in cancer cell metabolism. Front. Oncol. 2018, 8, 331. [Google Scholar] [CrossRef] [PubMed]
- Madan, E.; Gogna, R.; Bhatt, M.; Pati, U.; Kuppusamy, P.; Ali Mahdi, A. Regulation of glucose metabolism by p53: Emerging new roles for the tumor suppressor. Oncotarget 2011, 2, 948–957. [Google Scholar] [CrossRef] [PubMed]
- Markert, C.L.; Shaklee, J.B.; Whitt, G.S. Evolution of a gene: Multiple genes for LDH isozymes provide a model of the evolution of gene structure, function and regulation. Science 1975, 189, 102–114. [Google Scholar] [CrossRef]
- Perez-Tomas, R.; Perez-Guillen, I. Lactate in the tumor microenvironment: An essential molecule in cancer progression and treatment. Cancers (Basel) 2020, 12, 3244. [Google Scholar] [CrossRef] [PubMed]
- San-Millan, I.; Martinez, J.L.; Pickard, S.L.; Yu, H.; Hirsch, F.R.; Rivard, C.J.; Brooks, G.A. Role of lactate in the regulation of transcriptional activity of breast cancer-related genes and epithelial-to-mesenchymal proteins: A comparison of MCF7 and MDA-MB-231 cell lines. bioRxiv 2023, 2023.03.23.533060. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr. Von Hippel-Lindau disease: insights into oxygen sensing, protein degradation and cancer. J. Clin. Invest. 2022, 132, e162480. [Google Scholar] [CrossRef]
- Feng, T.; Zhao, X.; Gu, P.; Yang, W.; Wang, C.; Guo, Q.; et al. Adipocyte-derived lactate is a signaling metabolite that potentiates adipose macrophage inflammation via targeting PHD2. Nat. Commun. 2022, 13, 5208. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Graham, A.M.; Presnell, J.S. Hypoxia inducible factor (HIF) transcription factor family expansion, diversification, divergence and selection in eukaryotes. PLoS One 2017, 12, e0179545. [Google Scholar] [CrossRef]
- Al Tameeni, W.; dale, T.P.; Al-Jumaily, R.M.K.; Forsyth, N.R. Hypoxia-modified cancer cell metabolism. Front. Cell Dev. Biol. 2019, 7, 4. [Google Scholar] [CrossRef]
- Jiang, B.H.; Zheng, J.Z.; Leung, S.W.; Roe, R.; Semenza, G.L. Transactivation and inhibitory domains of hypoxia-inducible factor 1 alpha. Modulation of transcriptional activity by oxygen tension. J. Biol. Chem. 1997, 272, 19253–19260. [Google Scholar] [CrossRef]
- Pugh, C.W.; O’Rourke, J.F.; Nagao, M.; Gleadle, J.M.; Ratcliffe, P.J. Activation of hypoxia-inducible factor-1: definition of regulatory domains within the alpha subunit. J. Biol. Chem. 1997, 272, 11205–11214. [Google Scholar] [CrossRef]
- Mahon, P.C.; Hirota, K.; Semenza, G.L. FIH-1: a novel protein that interacts with HIF-1α and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001, 15, 2675–2686. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef]
- Dengler, V.L.; Galbraith, M.D.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef]
- Kierans, S.; Taylor, C. Regulation of glycolysis by the hypoxia-inducible factor (HIF): implication for cellular physiology. J. Physiol. 2021, 599, 23–37. [Google Scholar] [CrossRef]
- Ryan, K.M.; Birnie, G.D. Myc oncogenes: the enigmatic family. Biochem. J. 1996, 314, 713–721. [Google Scholar] [CrossRef]
- Nair, S.K.; Burley, S.K. X-ray structures of Myc-Max and Mad-Max recognizing DNA: molecular bases of regulation by proto-oncogenic transcription factors. Cell 2003, 112, 193–205. [Google Scholar] [CrossRef]
- Solomon, D.L.; Amati, B.; Land, H. Distinct DNA binding preferences for the c-Myc/Max and Max/Max dimers. Nucleic Acids Res. 1993, 21, 5372–5376. [Google Scholar] [CrossRef]
- Hann, S.R.; Dixit, M.; Sears, R.C.; Sealy, L. The alternatively initiated c-Myc proteins differentially regulate transcription through a noncanonical DNA-binding site. Genes Dev. 1994, 8, 2441–2452. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef]
- Vaseva, A.V.; Blake, D.R.; Gilbert, T.S.K.; Ng, S.; Hostetter, G.; Azam, S.H.; et al. KRAS suppression-induced degradation of MYC is antagonized by a MEK5-ERK5 compensatory mechanism. Cancer Cell 2018, 34, 807–822 E7. [Google Scholar] [CrossRef]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, metabolism, and cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef]
- Ganapathy, V.; Thangaraju, M.; Prasad, P. D. Nutrient transporters in cancer: relevance to Warburg hypothesis and beyond. Pharmacol. Ther. 2009, 121, 29–40. [Google Scholar] [CrossRef]
- Ganapathy, V.; Haferkamp, S.; Parkinson, E.K.; Mycielska, M.E. Editorial: Metabolite and nutrient transporters in cancer-cell metabolism: Role in cancer progression and metastasis. Front. Cell Dev. Biol. 2022, 10, 885717. [Google Scholar] [CrossRef]
- Nwosu, Z.C.; Song, M.G.; di Magliano, M.P.; Lyssiotis, C.A.; Kim, S.E. Nutrient transporters: connecting cancer metabolism to therapeutic opportunities. Oncogene 2023, 42, 711–724. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Heydarzadeh, S.; Moshtaghie, A.A.; Daneshpoor, M.; Hedayati, M. Regulators of glucose uptake in thyroid cancer cell lines. Cell Commun. Signal. 2020, 18, 83. [Google Scholar] [CrossRef]
- Baumann, M.U.; Zamudio, S.; Illsley, N.P. Hypoxic upregulation of glucose transporters in BeWo choriocarcinoma cells is mediated by hypoxia-inducible factor-1. Am. J. Physiol. Cell Physiol. 2007, 293, C477–C485. [Google Scholar] [CrossRef] [PubMed]
- Abouzied, M.M.; Crawford, E.S.; Nabi, H.A. 18F-FDG imaging: pitfalls and artifacts. J. Nucl. Med. Technol. 2005, 33, 145–155. [Google Scholar] [PubMed]
- Berz, A.M.; Dromain, C.; Vietti-Violi, N.; Boughdad, S.; Duran, R. Tumor response assessment on imaging following immunotherapy. Front. Oncol. 2022, 12, 982983. [Google Scholar] [CrossRef]
- Scafoglio, C.; Hirayama, B.A.; Kepe, V.; Liu, J.; Ghezzi, C.; Satyamurthy, N.; et al. Functional expression of sodium-glucose transporters in cancer. Proc. Natl. Acad. Sci. USA 2015, 112, E4111–E4119. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.M. SGLT2 and cancer. Pflugers Arch. 2020, 472, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Felmlee, M.A.; Jones, R.S.; Rodriguez-Cruz, V.; Follman, K.E.; Morris, M.E. Monocarboxylate transporters (SLC16): Function, regulation, and role in health and disease. Pharmacol. Rev. 2020, 72, 466–485. [Google Scholar] [CrossRef] [PubMed]
- Duan, Q.; Zhang, S. , Wang, Y.; Lu, D.; Sun, Y.; Wu, Y. Proton-coupled monocarboxylate transporters in cancer: From metabolic crosstalk, immunosuppression and anti-apoptosis to clinical applications. Front. Cell Dev. Biol. 2022, 10, 1069555. [Google Scholar] [CrossRef]
- Liu, T.; Han, S.; Yao, Y.; Zhang, G. Role of human monocarboxylate transporter 1 (hMCT1) and 4 (hMCT4) in tumor cells and the tumor microenvironment. Cancer Manag. Res. 2023, 15, 957–975. [Google Scholar] [CrossRef] [PubMed]
- Kasiappan, R.; Shih, H.J.; Wu, M.H.; Choy, C.; Lin, T.D.; Chen, L.; Hsu, H.L. The antagonism between MCT1 and p53 affects the tumorigenic outcomes. Mol. Cancer 2010, 9, 311. [Google Scholar] [CrossRef]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is upregulated by hypoxia through a HIF-1α-dependent mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [PubMed]
- Wilde, L.; Roche, M.; Domingo-Vidal, M.; Tanson, K.; Philp, N.; Curry, J.; Martinez-Outschoorn, U. Metabolic coupling and the reverse Warburg effect in cancer: Implications for novel biomarker and anticancer agent development. Semin. Oncol. 2017, 44, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Spugnini, E.P.; Sonveaux, P.; Stock, C.; Perez-Sayans, M.; De Millito, A.; Avnet, S.; et al. Proton channels and exchangers in cancer. Biochim. Biophys. Acta 2015, 1848, 2715–2726. [Google Scholar] [CrossRef]
- Cardone, R.A.; Greco, M.R.; Zeeberg, K.; Zaccagnino, A.; Saccomano, M.; Bellizzi, A.; et al. R,; Zeeberg, K,; Zaccagnino, A,; Saccomano, M,; Bellizzi, A,; et al. A novel NHE1-centered signaling cassette drives epidermal growth factor receptor-dependent pancreatic tumor metastasis and is a target for combination therapy. Neoplasia 2015, 17, 155–166. [Google Scholar] [CrossRef]
- Boedtkjer, E. Na+,HCO3- cotransporter NBCn1 accelerates breast carcinogenesis. Cancer Metastasis Rev. 2019, 38, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Kang, R.; Liu, J.; Tang, D. The V-ATPases in cancer and cell death. Cancer Gene Ther. 2022, 29, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Sennoune, S.R.; Sharma, M.; Thangaraju, M.; Suresh, V.V.; Sniegowski, T.; et al. Expression and function of SLC38A5, an amino acid-coupled Na+/H+ exchanger, in triple-negative breast cancer and its relevance to macropinocytosis. Biochem. J. 2021, 478, 3957–3976. [Google Scholar] [CrossRef] [PubMed]
- Sniegowski, T.; Rajasekaran, D.; Sennoune, S.R.; Sunitha, S.; Chen, F.; Fokar, M.; et al. Amino acid transporter SLC38A5 is a tumor promoter and a novel therapeutic target for pancreatic cancer. Sci. Rep. 2023, 13, 16863. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Kekuda, R.; Fei, Y.J.; Hatanaka, T.; Sugawara, M.; Martindale, R.G.; et al. Cloning and functional characterization of a new subtype of the amino acid transport system N. Am. J. Physiol. Cell Physiol. 2001, 281, C1757–C1768. [Google Scholar] [CrossRef]
- Ganapathy, V.; Leibach, F.H. Proton-coupled solute transport in the animal cell plasma membrane. Curr. Opin. Cell Biol. 1991, 3, 695–701. [Google Scholar] [CrossRef]
- Anderson, C.M.; Thwaites, D.T. Hijacking solute carriers for proton-coupled drug transport. Physiology (Bethesda) 2010, 25, 364–377. [Google Scholar] [CrossRef] [PubMed]
- Thwaites, D.T.; Anderson, C.M. H+-coupled nutrient, micronutrient and drug transporters in the mammalian small intestine. Exp. Physiol. 2007, 92, 603–619. [Google Scholar] [CrossRef]
- Ganapathy, V.; Burckhardt, G.; Leibach, F.H. Characteristics of glycylsarcosine transport in rabbit intestinal brush-border membranes. J. Biol. Chem. 1984, 259, 8954–8959. [Google Scholar] [CrossRef]
- Kennedy, D.; Leibach, F.H.; Ganapathy, V.; Thwaites, D.T. Optimal absorptive transport of the dipeptide glycylsarcosine is dependent on functional Na+/H+ exchange activity. Pflugers Arch. 2002, 445, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.M.H.; Jevons, M.; Thangaraju, M.; Edwards, N.; Conlon, N.J.; Woods, S.; et al. Transport of the photodynamic therapy agent 5-aminolevulenic acid by distinct H+-coupled nutrient carriers coexpressed in the small intestine. J. Pharmacol. Exp. Ther. 2020, 332, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.; Chen, Z.; Cheng, K. Expression profile and functional activity of peptide transporters in prostate cancer cells. Mol. Pharm. 2013, 10, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.E.; Covitz, K.M.; Sadee, W.; Mrsny, R.J. An oligopeptide transporter is expressed at high levels in the pancreatic carcinoma cell lines AsPc-1 and Capan-2. Cancer Res. 1998, 58, 519–525. [Google Scholar]
- Schniers, B.K.; Rajasekaran, D.; Korac, K.; Sniegowski, T.; Ganapathy, V.; Bhutia, Y.D. PEPT1 is essential for the growth of pancreatic cancer cells: a viable drug target. Biochem. J. 2021, 478, 3757–3774. [Google Scholar] [CrossRef]
- Thwaites, D.T.; Anderson, C.M. The SLC36 family of proton-coupled amino acid transporters and their potential role in drug transport. Br. J. Pharmacol. 2011, 164, 1892–1816. [Google Scholar] [CrossRef]
- Heublein, S.; Kazi, S.; Ogmundsdottir, M.H.; Attwood, E.V.; Kala, S.; Boyd, C.A.; et al. Proton-assisted amino-acid transporters are conserved regulators of proliferation and amino-acid-dependent mTORC1 activation. Oncogene 2010, 29, 4068–4079. [Google Scholar] [CrossRef]
- Qiu, A.; Jansen, M.; Sakaris, A.; Min, S.H.; Chattopadhyay, S.; Tsai, E.; et al. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell 2006, 127, 917–928. [Google Scholar] [CrossRef]
- Umapathy, N.S.; Gnanaprakasam, J.P.; Martin, P.M.; Mysona, B.; Dun, Y.; Smith, S.B.; et al. Cloning and functional characterization of the proton-coupled electrogenic folate transporter and analysis of its expression in retinal cell types. Invest. Ophthalmol. Vis. Sci. 2007, 48, 5299–5305. [Google Scholar] [CrossRef]
- Hou, Z.; Gangjee, A.; Matherly, L.H. The evolving biology of the proton-coupled folate transporter: New insights into regulation, structure, and mechanism. FASEB J. 2022, 36, e22164. [Google Scholar] [CrossRef]
- Gunshin, H.; Mackenzie, B.; Berger, U.V.; Gunshin, Y.; Romero, M.F.; Boron, W.F.; et al. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 1997, 388, 482–488. [Google Scholar] [CrossRef]
- Skjorringe, T.; Burkhart, A.; Johnsen, K.B.; Moos, T. Divalent metal transporter 1 (DMT1) in the brain: implications for a role in iron transport at the blood-brain barrier, and neuronal and glial pathology. Front. Mol. Neurosci. 2015, 8, 19. [Google Scholar] [CrossRef]
- Tian, L.; Li, X.; Lai, H.; Sun, T.; Li, X.; Wu, L.; et al. SLC11A2: a promising biomarker and therapeutic target in ovarian cancer. Sic. Rep. 2023, 13, 1132. [Google Scholar] [CrossRef]
- Inoue, K.; Zhuang, L.; Maddox, D.M.; Smith, S.B.; Ganapathy, V. Structure, function, and expression pattern of a novel sodium-coupled citrate transporter (NaCT) cloned from mammalian brain. J. Biol. Chem. 2002, 277, 39469–39476. [Google Scholar] [CrossRef]
- Inoue, K.; Zhuang, L.; Ganapathy, V. Human Na+-coupled citrate transporter: primary structure, genomic organization, and transport function. Biochem. Biophys. Res. Commun. 2002, 299, 465–471. [Google Scholar] [CrossRef]
- Haferkamp, S.; Drexler, K.; Federlin, M.; Schlitt, H.J.; Berneburg, M.; Adamski, J.; et al. Extracellular citrate fuels cancer cell metabolism and growth. Front. Cell Dev. Biol. 2020, 8, 602476. [Google Scholar] [CrossRef]
- Parkinson, E.K.; Adamski, J.; Zahn, G.; Gaumann, A.; Flores-Borja, F.; Ziegler, C.; Mycielska, M.E. Extracellular citrate and metabolic adaptations of cancer cells. Cancer Metastasis Rev. 2021, 40, 1073–1091. [Google Scholar] [CrossRef]
- Li, Z.; Wang, H. Molecular mechanisms of the SLC13A5 gene transcription. Metabolites 2021, 11, 706. [Google Scholar] [CrossRef]
- Gopal, E.; Babu, E.; Ramachandran, S.; Bhutia, Y.D.; Prasad, P.D.; Ganapathy, V. Species-specific influence of lithium on the activity of SLC13A5 (NaCT): Lithium-induced activation is specific for the transporter in primates. J. Pharmacol. Exp. Ther. 2015, 353, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, Y.; Li, Y.; Hong, A.; Wang, J.; Lin, B.; Li, R. NDRG3 is an androgen regulated and prostate enriched gene that promotes in vitro and in vivo prostate cancer cell growth. Int. J. Cancer 2009, 124, 521–530. [Google Scholar] [CrossRef]
- Lee, G.Y.; Chun, Y.-S.; Shin, H.-W.; park, J.-W. Potential role of the N-MYC downstream-regulated gene family in reprogramming cancer metabolism under hypoxia. Oncotarget 2016, 7, 57442–57451. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.C.; Sohn, H.A.; Park, Z.-Y.; Oh, S.; Kang, Y.K.; Lee, K.-M.; et al. . A lactate-induced response to hypoxia. Cell 2015, 161, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Park, K.C.; Lee, D.C.; Yeom, Y.I. NDRG3-mediated lactate signaling in hypoxia. BMB Rep. 2015, 48, 301–302. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.Q.; Ren, N.; Jin, L.; Cheng, K.; Kash, S.; Chen, R.; et al. Biochem. Biophys. Res. Commun. 2008, 377, 987–991. [CrossRef]
- Liu, C.; Wu, J.; Zhu, J.; Kuei, C.; Yu, J.; Shelton, J.; et al. Lactate inhibits lipolysis in fat cells through activation of an orphan G-protein-coupled receptor, GPR81. J Biol. Chem. 2009, 284, 2811–2822. [Google Scholar] [CrossRef] [PubMed]
- Ristic, B.; Bhutia, Y.D.; Ganapathy, V. Cell-surface G-protein-coupled receptors for tumor-associated metabolites: A direct link to mitochondrial dysfunction in cancer. Biochim. Biophys. Acta 2017, 1868, 246–257. [Google Scholar] [CrossRef]
- Lee, Y.J. G-protein-coupled receptor 81 promotes a malignant phenotype in breast cancer through angiogenic factor secretion. Oncotarget 2016, 7, 70898–70911. [Google Scholar] [CrossRef]
- Ishihara, S. The lactate receptor GPR81 regulates glycolysis and tumor growth of breast cancer. Sci. Rep. 2022, 12, 6261. [Google Scholar] [CrossRef]
- Roland, C.L.; Arumugam, T.; Deng, D.; Liu, S.H.; Philip, B.; Gomez, S.; et al. Cell surface lactate receptor GPR81 is crucial for cancer cell survival. Cancer Res. 2014, 74, 5301–5310. [Google Scholar] [CrossRef]
- Luo, M.; Zhu, J.; Ren, J.; Tong, Y.; Wang, L.; Ma, S.; Wang, J. Lactate increases tumor malignancy by promoting tumor small extracellular vesicles production via the GPR81-cAMP-PKA-HIF-1α axis. Front. Oncol. 2022, 12, 1036543. [Google Scholar] [CrossRef]
- Feng, J.; Yang, H.; Zhang, Y.; Wei, H.; Zhu, Z.; Zhu, B.; et al. Tumor cell-derived lactate induces TAZ-dependent upregulation of PD-L1 through GPR81 in human lung cancer cells. Oncogene 2017, 36, 5829–5839. [Google Scholar] [CrossRef]
- Wagner, W.; Kania, K.D.; Blauz, A.; Ciszewski, W.M. The lactate receptor (HCAR1/GPR81) contributes to doxorubicin chemoresistance via ABCB1 transporter up-regulation in human cervical cancer HeLa cells. J. Physiol. Pharmacol. 2017, 68, 555–564. [Google Scholar]
- Xie, Q.; Zhu, Z.; He, Y.; Zhang, Z.; Zhang, Y.; Wang, Y.; et al. A lactate-induced Snail/STAT3 pathway drives GPR81 expression in lung cancer cells. Biochim. Biophys. Acta Mol. Basis. Dis. 2020, 1866, 165576. [Google Scholar] [CrossRef] [PubMed]
- Lundo, K.; Dmytriyeva, O.; Spohr, L.; Goncalves-Alves, E.; Yao, J.; Blasco, L.P.; et al. Lactate receptor GPR81 drives breast cancer growth and invasiveness through regulation of ECM properties and Notch ligand DLL4. BMC Cancer 2023, 23, 1136. [Google Scholar] [CrossRef]
- Brown, T.P.; Bhattacharjee, P.; Ramachandran, S.; Sivaprakasam, S.; ristic, B.; Sikder, M.O.F.; Ganapathy, V. The lactate receptor GPR81 promotes breast cancer growth via a paracrine mechanism involving antigen-presenting cells in the tumor microenvironment. Oncogene 2020, 39, 3292–3304. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, D.; Bhattacharya, R.; Sinha, B.P.; Liu, C.S.C.; Ghosh, A.R.; Rahaman, O.; et al. Lactate induces pro-tumor reprogramming in intratumoral plasmacytoid dendritic cells. Front. Immunol. 2019, 10, 1878. [Google Scholar] [CrossRef]
- Lundo, K.; Trauelsen, M.; Pedersen, S.F.; Schwartz, T.W. Why Warburg works: Lactate controls immune evasion through GPR81. Cell Metab. 2020, 31, 666–668. [Google Scholar] [CrossRef]
- Khatib-Massalha, E.; Bhattacharya, S.; Massalha, H.; Biram, A.; Golan, K.; Killet, O.; et al. Lactate released by inflammatory bone marrow neutrophils induces their mobilization via endothelial GPR81 signaling. Nat. Commun. 2020, 11, 3547. [Google Scholar] [CrossRef] [PubMed]
- Che, D.; Wang, M.; Sun, J.; Li, B.; Xu, T.; Lu, Y.; et al. KRT6A promotes lung cancer cell growth and invasion through MYC-regulated pentose phosphate pathway. Front. Cell Dev. Biol. 2021, 9, 694071. [Google Scholar] [CrossRef]
- Li, W.; Kou, J.; Zhang, Z.; Li, H.; Li, L.; Du, W. Cellular redox homeostasis maintained by malic enzyme 2 is essential for MYC-driven T cell lymphomagenesis. Proc. Natl. Acad. Sci. USA 2023, 120, e2217869120. [Google Scholar] [CrossRef]
- Zhao, X.; Fu, J.; Du, J.; Xu, W. The role of D-3-phosphoglycerate dehydrogenase in cancer. Int. J. Mol. Sci. 2020, 16, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Helmlinger, G.; Sckell, A.; Dellian, M.; Forbes, N.S.; Jain, R.K. Acid production in glycolysis-impaired tumors provides new insights into tumor metabolism. Clin. Cancer Res. 2002, 8, 1284–1291. [Google Scholar] [PubMed]
- Cormerais, Y.; Vucetic, M.; Parks, S.K.; Pouyssegur, J. Amino acid transporters are a vital focal point in the control of mTORC1 signaling and cancer. Int. J. Mol. Sci. 2020, 22, 23. [Google Scholar] [CrossRef] [PubMed]
- Lopes, C.; Pereira, C.; Medeiros, R. ASCT2 and LAT1 contribution to the hallmarks of cancer: From a molecular perspective in clinical translation. Cancers (Basel) 2021, 13, 203. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Miyauchi, S.; Martindale, R.G.; Herdman, A.V.; Podolsky, R.; Miyake, K.; et al. Upregulation of the amino acid transporter ATB0,+ (SLC6A14) in colorectal cancer and metastasis in humans. Biochim. Biophys. Acta 2005, 1741, 215–223. [Google Scholar] [CrossRef]
- Gupta, N.; Prasad, P.D.; Ghamande, S.; Martin, P.M.; Herdman, A.V.; Martindale, R.G.; et al. Up-regulation of the amino acid transporter ATB0,+ (SLC6A14) in carcinoma of the cervix. Gynecol. Oncol. 2006, 100, 8–13. [Google Scholar] [CrossRef]
- Karunakaran, S.; Ramachandran, S.; Coothankandaswamy, V.; Elangovan, S.; Babu, E.; Periyasamy-Thandavan, S.; et al. SLC6A14 (ATB0,+) protein, a highly concentrative and broad specific amino acid transporter, is a novel and effective drug target for treatment of estrogen receptor-positive breast cancer. J. Biol. Chem. 2011, 286, 31830–31838. [Google Scholar] [CrossRef]
- Coothankandaswamy, V.; Cao, S.; Xu, Y.; Prasad, P.D.; Singh, P.K.; Reynolds, C.P.; et al. Amino acid transporter SLC6A14 is a novel and effective drug target for pancreatic cancer. Br. J. Pharmacol. 2016, 173, 3292–3306. [Google Scholar] [CrossRef] [PubMed]
- Baryla, M.; Semeniuk-Wojtas, A.; Rog, I.; Kraj, L.; Malyszko, M.; Stec, R. Oncometabolites – A link between cancer cells and tumor microenvironment. Biology (Basel) 2022, 11, 270. [Google Scholar] [CrossRef] [PubMed]
- Chou, F.J.; Liu, Y.; Lang, F.; Yang, C. D-2-Hydroxyglutarate in glioma biology. Cells 2021, 10, 2345. [Google Scholar] [CrossRef] [PubMed]
- Beyoglu, D.; Idle, J.R. Metabolic rewiring and the characterization of oncometabolites. Cancers (Basel) 2021, 13, 2900. [Google Scholar] [CrossRef]
- Notarangelo, G.; Spinelli, J.B.; Perez, E.M.; Baker, G.J.; Kurmi, K.; Elia, I.; et al. Oncometabolite d-2-HG alters T cell metabolism to impair CD8+ T cell function. Science 2022, 377, 1519–1529. [Google Scholar] [CrossRef]
Figure 1.
Glycolysis in normal cells and in cancer cells in the presence and absence of oxygen. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; PFK1, phosphofructokinase-1; F-2,6-BP, fructose-2,6-bisphosphate.
Figure 1.
Glycolysis in normal cells and in cancer cells in the presence and absence of oxygen. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; PFK1, phosphofructokinase-1; F-2,6-BP, fructose-2,6-bisphosphate.
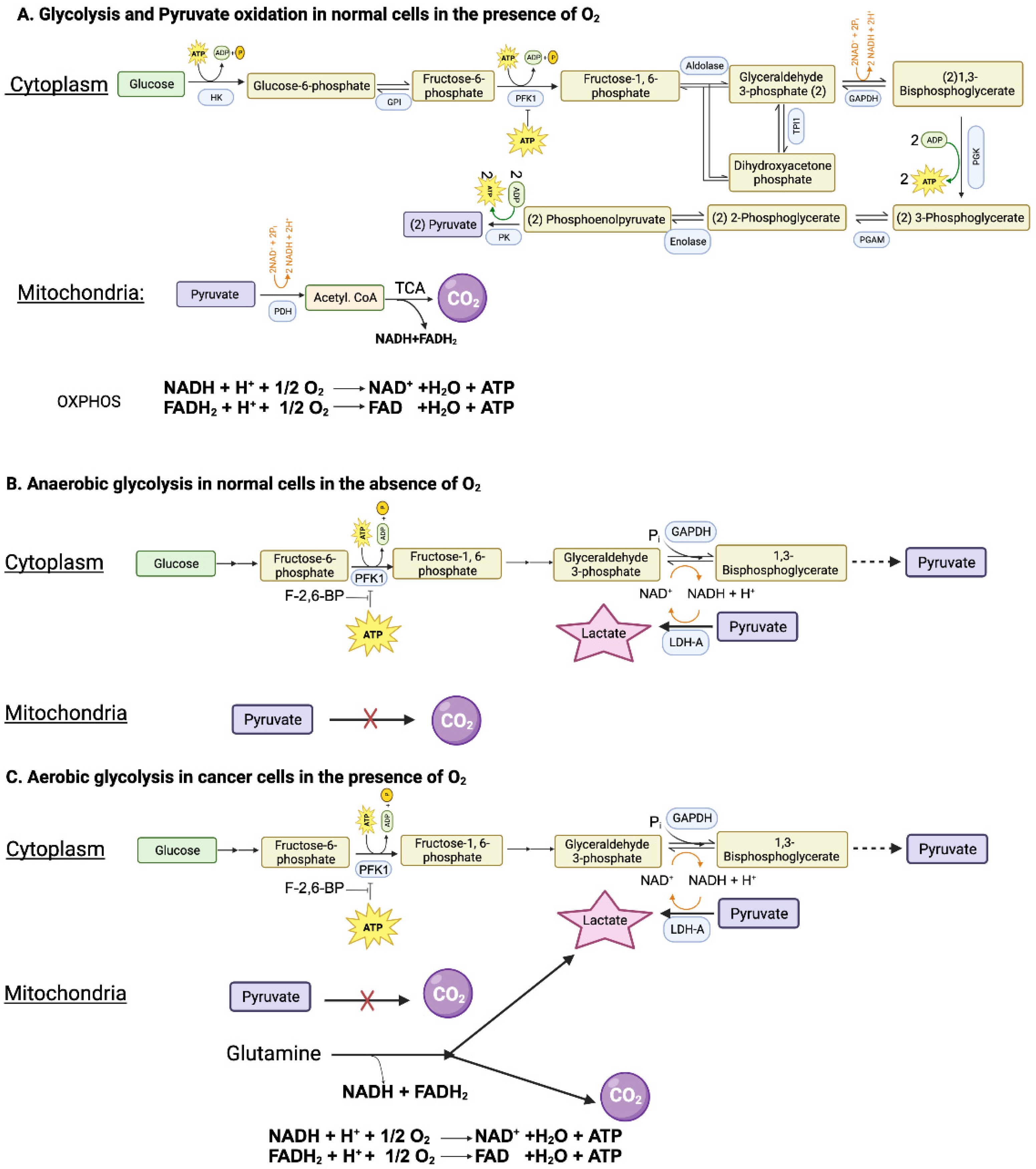
Figure 2.
Differential roles of MCT1 and MCT4 in tumors and in non-tumor cells in the tumor microenvironment. MCT1, monocarboxylate transporter 1 (SLC16A1); MCT4, monocarboxylate transporter 4 (SLC16A3); CD8+ cell, cytotoxic T cell. .
Figure 2.
Differential roles of MCT1 and MCT4 in tumors and in non-tumor cells in the tumor microenvironment. MCT1, monocarboxylate transporter 1 (SLC16A1); MCT4, monocarboxylate transporter 4 (SLC16A3); CD8+ cell, cytotoxic T cell. .
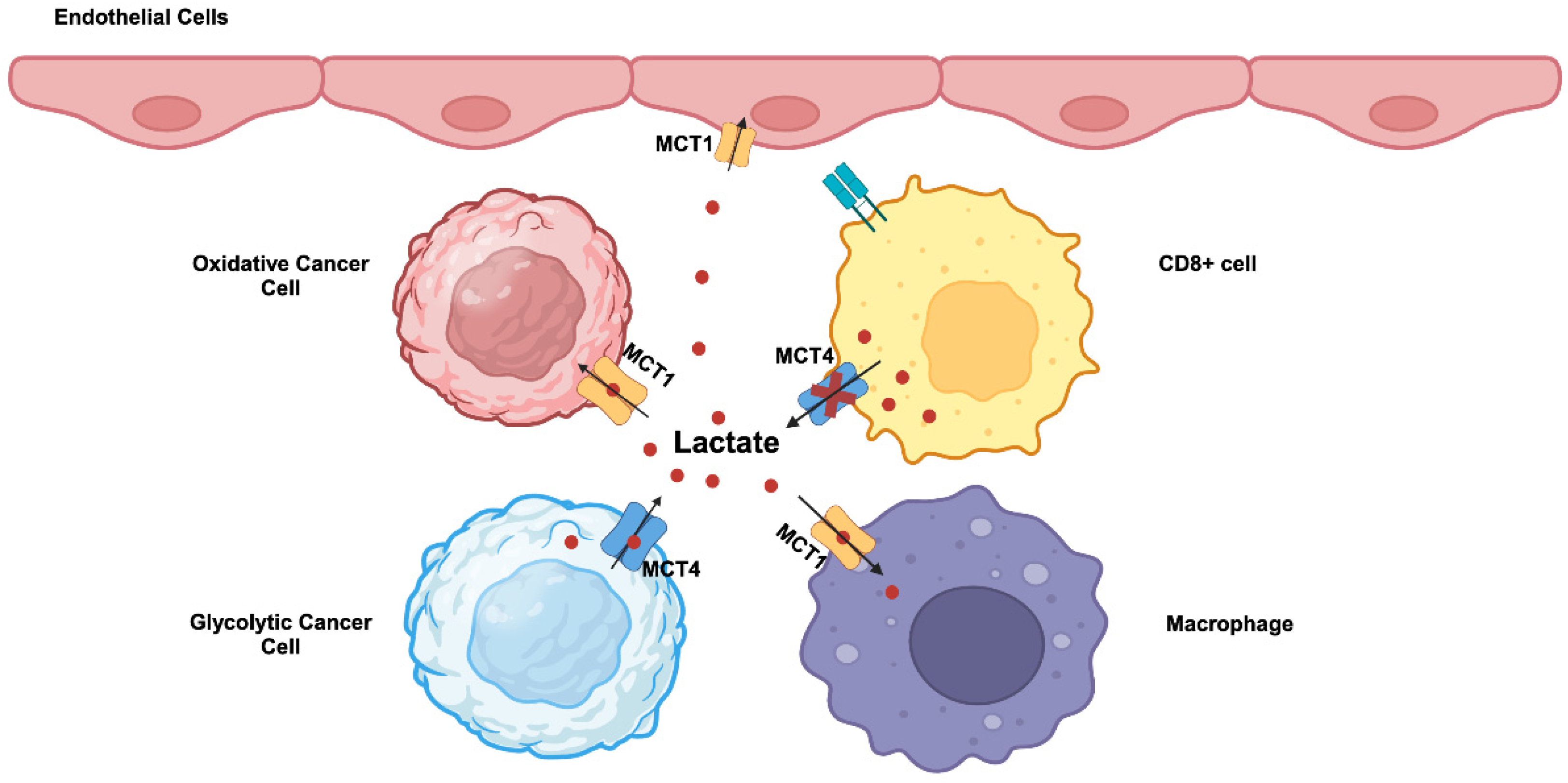
Figure 3.
Autocrine and paracrine functions of tumor cell-derived lactate. AREG, amphiregulin; ABCB1, ATP binding cassette transporter B1; PD1, programmed cell death protein 1; PD-L1, PD ligand 1; GPR81, lactate receptor; MCT4, monocarboxylate transporter 4 (SLC16A3); MHC, major histocompatibility complex.
Figure 3.
Autocrine and paracrine functions of tumor cell-derived lactate. AREG, amphiregulin; ABCB1, ATP binding cassette transporter B1; PD1, programmed cell death protein 1; PD-L1, PD ligand 1; GPR81, lactate receptor; MCT4, monocarboxylate transporter 4 (SLC16A3); MHC, major histocompatibility complex.
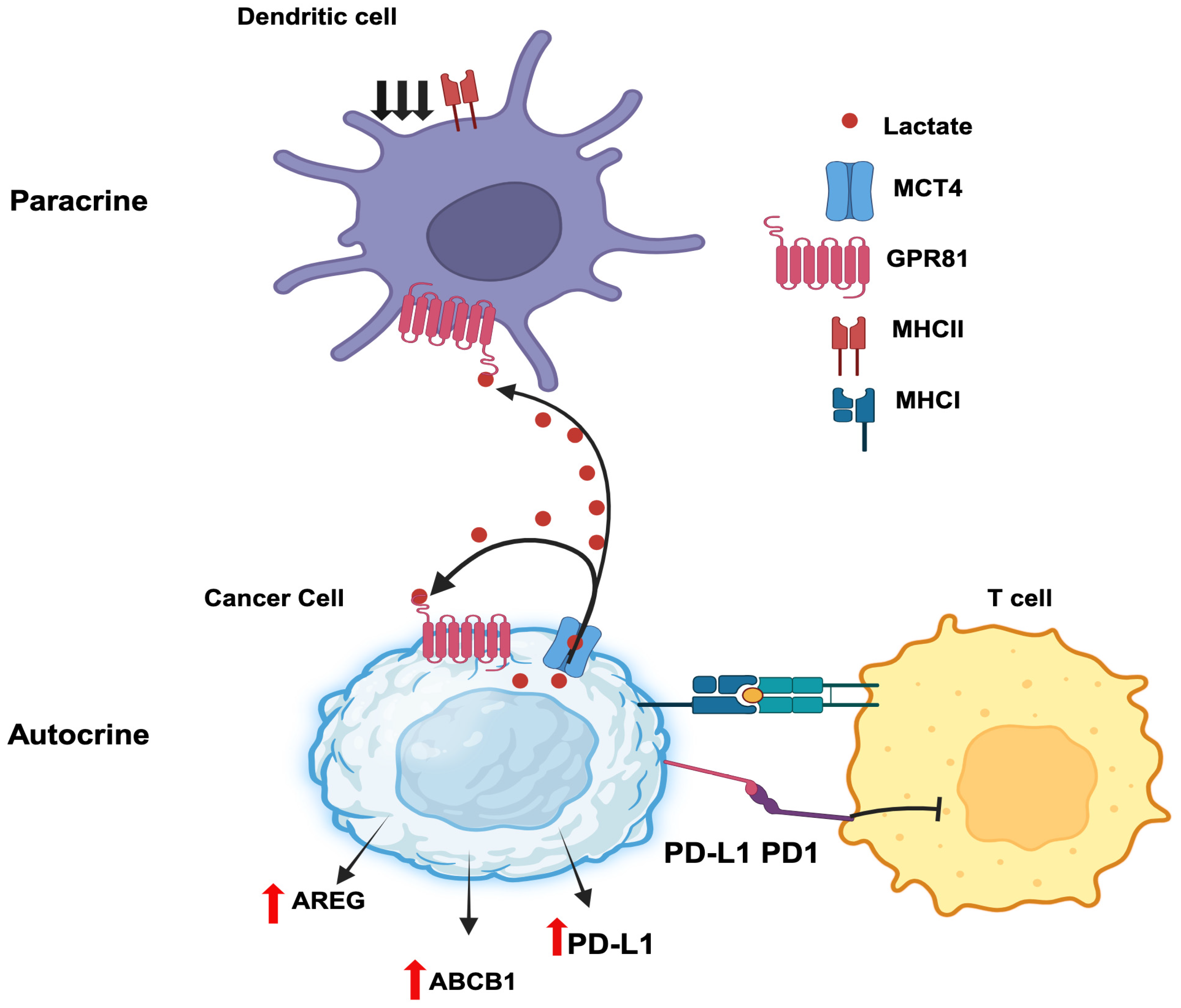
Figure 4.
Reactions involved in glutaminolysis and reductive carboxylation.
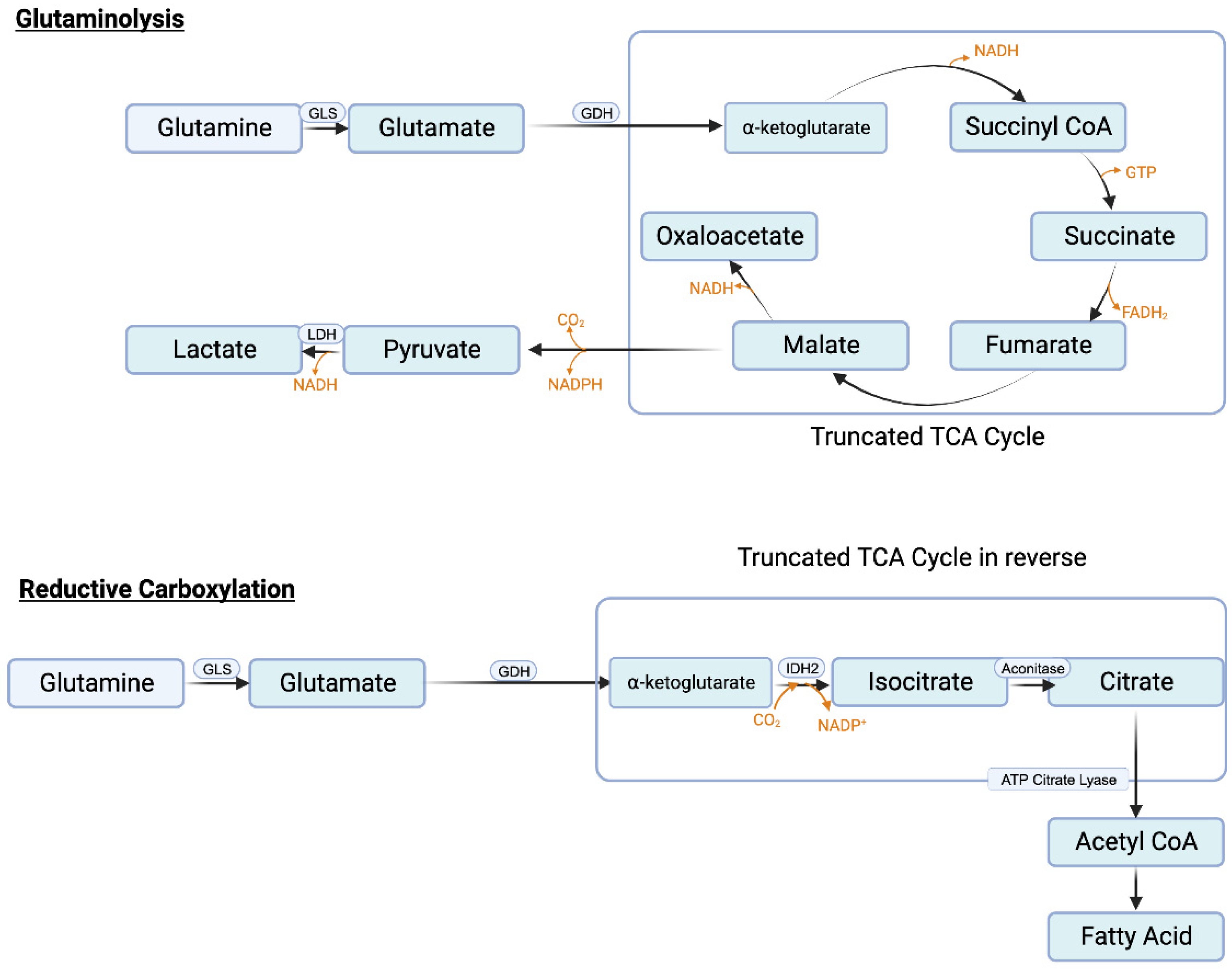
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



