
Submitted:
20 December 2023
Posted:
20 December 2023
You are already at the latest version
Abstract
The new diprotic ligand 3,5-di-tert-butylsalicylaldehyde 4-ethyl-3-thiosemicarbazone, abbreviated H2(3,5-t-Bu2)-sal4eT, exists as the thio-keto tautomer and adopts the E-configuration with respect to the imine double bond as evidenced by single-crystal X-ray analysis and corroborated by spectroscopic characterisation. Upon treatment with Cu(OAc)2·H2O in the presence of either 2,9-dimethyl-1,10-phenanthroline (2,9-Me2-phen) or 1,10-phenanthroline (phen) as a co-ligand in MeOH, this thiosemicarbazone undergoes conformational transformation (relative donor-atom orientations: syn,anti → syn,syn) concomitantly with tautomerization and double deprotonation to afford the ternary copper(II) complexes [Cu{(3,5-t-Bu2)-sal4eT}(2,9-Me2-phen)] (1) and [Cu2{3,5-t-Bu2)-sal4eT}2(phen)] (2). Crystallographic elucidation has revealed that complex 1 is a centrosymmetric dimer of mononuclear copper(II) complex molecules brought about by intermolecular H-bonding. The coordination geometry at the copper(II) centre is best described as distorted square pyramidal in accord with the trigonality index (τ = 0.14). The co-ligand adopts an axial-equatorial coordination mode; hence there is a disparity between its two Cu–N coordinate bonds arising from weakening of the apical one as a consequence of the tetragonal distortion. The axial X-band ESR spectrum of complex 1 is consistent with retention of this structure in solution. Complex 2 is a centrosymmetric dimer of dinuclear copper(II) complex molecules exhibiting intermolecular H-bonding and π-π-stacking interactions. The two copper(II) centres, which are 4.8067(18) Å apart and bridged by the thio-enolate nitrogen of the quadridentate thiosemicarbazonate ligand, display two different coordination geometries, one distorted square planar (τ4 = 0.082) and the other distorted square pyramidal (τ5 = 0.33). Such dinuclear copper(II) thiosemicarbazone complexes, which are crystallographically characterised, are extremely rare. In vitro, complexes 1 and 2 outperform cisplatin as antiproliferative agents in terms of potency and selectivity towards HeLa and MCF-7 cancer cell lines.
Keywords:
mononuclear and dinuclear ternary copper(II) complexes
; thiosemicarbazone
; X-ray structures
; tetragonal distortion
; intermolecular forces
; spectroscopy
; selective potent cytotoxicity
1. Introduction
Thiosemicarbazones are multi-purpose hydrazone ligands of considerable interest in coordination chemistry [1]. Their characteristic functionality feature R1R2C=N–N(H)–C(=S)–NR3R4 is derived from the straightforward single-step Schiff-base condensation reaction between an aldehyde or a ketone and a thiosemicarbazide. Amongst their fascinating structural attributes is their coordination versatility arising from their propensity to undergo concomitant base-/metal-assisted tautomeric transformation and deprotonation as a means to meet charge-neutrality requirements. Moreover, a given thiosemicarbazone is capable of exhibiting different denticities and coordination modes as demanded by the metal centre [2,3,4,5,6,7,8,9]. By strategically employing aldehydes or ketones with moieties bearing donor atoms of interest in appropriate coordination positions, they can be tailor-designed for specially desired hetero-donor environments, coordination spheres [10], physicochemical features and biological properties [3,8,9,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27]. Substituent groups can impart electronic effects to thiosemicarbazone complexes with interesting impact on physicochemical and pharmacological properties. The literature is replete with examples of thiosemicarbazone complexes exhibiting a broad spectrum of pharmacological properties including antitumour [3,8,9,11,12,13,14,15,16,17,18,19,20,21,22], antibacterial [20,21,23], antiviral [24,25], antifungal [23,26] and antimalarial [22,27].
Thiosemicarbazones stabilize predominantly metal ions from the p-, d- and f -blocks [1]; some of the transition-metal thiosemicarbazone complexes exhibit fascinating magneto-structural [28] and catalytic [29] properties amongst other features. Electrochemically, for copper (Z = 29; [Ar]4s13d10), which is the subject of this paper, Schiff-base complexes shuttle between +1 and +2 oxidation states (CuII/CuI redox couple) [8,15,18,20,30]. Thiosemicarbazone complexes whose cyclic voltammograms exhibit these redox couples with potentials lying within the biologically accessible redox potential window ranging from –0.4 to +0.8 V vs. NHE [30], are of pharmacological importance in that they have the ability to generate intracellular reactive oxygen species (ROS) [8,15,18,20,30] desirable for apoptotic cytotoxicity. Ternary mononuclear complexes of copper(II) of the type [CuL(N,N-donor-L)]n+ [where L represents a neutral or anionic polydentate primary ligand and N,N-donor-L stands for a heterocyclic bidentate N,N-donor chelating co-ligand such as 1,10-phenanthroline (phen), 2,2´-bipyridine (bipy), dipyridoquinoxaline (dpq), dipyridophenazine (dppz) or derivatives of these] abound [31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58]. Such coordination compounds are of considerable interest partly because of their potential to exhibit DNA binding/cleavage [34,39,41,42,43,44,50,51,53,54,55], anticancer [39,43,44,49,53,54] and antimicrobial [35,36,37,40] activities. Some have been explored as models for metallo-enzymes [33,45] while others have been designed to investigate structural and spectroscopic features of interest [4,32,38,46,47,48,52,56,57,58]. There is a paucity of crystallographically characterised ternary copper(II) thiosemicarbazone complexes of this type [4,31,32,33,34,35,36,37,38,39].
In this work, we have synthesized and structurally characterised the ligand 3,5-di-tert-butylsalicylaldehyde 4-ethyl-3-thiosemicarbazone, H2(3,5-t-Bu2)-sal4eT. Reaction of copper(II) ion with this ligand and the co-ligand 2,9-dimethyl-1,10-phenanthroline (2,9-Me2-phen) or 1,10-phenanthroline (phen) in equimolar amounts produced the ternary copper(II) complex [Cu{(3,5-t-Bu2)-sal4eT}(2,9-Me2-phen)] (1) or [Cu2{3,5-t-Bu2)-sal4eT}2(phen)] (2), respectively. The 3-D structures have been determined by single-crystal X-ray analyses. Whereas complex 1 is mononuclear, complex 2 is dinuclear. In the crystal lattice each exists as a dimer arising from two symmetrically-related intermolecular H-bonding interactions. To the best of our knowledge, [Cu2{3,5-t-Bu2)-sal4eT}2(phen)] (2) is one of only two examples of crystallographically characterised dinuclear copper(II) thiosemicarbazone complexes of this kind. The other is [Cu2(sal4eT)2(bipy)], previously designated [Cu2(L2)2(bipy) (bipy = 2,2´-bipyridine) [4]. While complex 2 is a centrosymmetric dimer of dinuclear complexes, the crystallographic asymmetric unit of [Cu2(sal4eT)2(bipy)] consists of two independent intramolecularly hydrogen-bonded dinuclear complex molecules. In both 2 and [Cu2(sal4eT)2(bipy)], the two copper(II) ions are in different coordination spheres. These two complexes differ in the five-coordinate geometry. The antiproliferative activity of complexes 1 and 2 along with H2(3,5-t-Bu2)-sal4eT has been tested against the cancer cell lines MCF-7 (human breast adenocarcinoma) and HeLa (human cervical carcinoma). Whereas the ligand is inactive against HeLa and MCF-7 cancer cells, the complexes are highly potent and selective.
2. Results and Discussion
2.1. Synthesis and Chemical Identification of the Thiosemicarbazone Ligand
The thiosemicarbazone H2(3,5-t-Bu2)-sal4eT was synthesised from equimolar amounts of 3,5-di-tert -butylsalicylaldehyde and 4-ethyl-3-thiosemicarbazide by the usual single-step Schiff-base condensation reaction in refluxing absolute ethanol. The resultant light yellow solution afforded long lustrous colourless needles upon slow evaporation of the solvent under ambient conditions over a span of several days. Unlike the synthesis of pyridyl-based thiosemicarbazones [8,13,14,15], this phenolic thiosemicarbazone was produced straightforwardly in high yield without requiring acid-catalysis (Scheme 1). The chemical identity of this ligand was ascertained by microanalysis along with electrospray ionization (ESI) mass spectrometry. The ESI mass spectrum exhibits a parent peak at m/z = 334.3 [M – H+]– in the negative-ion mode [Figure 1] and at m/z = 336.3 [M + H+]+ in the positive-ion mode, consistent with the molecular mass (M = 335.51 amu).
2.2. FT-IR and NMR Spectroscopic Characterization of the Thiosemicarbazone Ligand
That H2(3,5-t-Bu2)-sal4eT exists as the thio-keto (thione) tautomer in the solid state is demonstrated by the prominent IR absorption band at 3157 cm–1 indicative of the thio-amide ν(N–H) (Figure 2). The associated thio-carbonyl bond is characterised by the vibrational band with a stretching frequency of 1032 cm–1. Indeed, the absence of a vibrational band around 2600 cm–1 due to ν(S–H) [10] excluded the possibility of the occurrence of the thio-enol tautomer. At 3320 cm–1 in the IR spectrum occurs an absorption band ascribable to the N–H stretch of the terminal secondary amino group of the thiosemicarbazone. A characteristic feature of Schiff bases is the imine bond whose presence in H2(3,5-t-Bu2)-sal4eT is evidenced by the absorption at 1609 cm–1 typifying ν(C=N). The tert-buyl C–H stretches are conspicuous by their characteristic pattern of absorption bands in the range 2867–2960 cm–1 [59]. Contributing to the intensity of these absorptions are the C–H vibrations of the N-ethyl substituent group. The sharp absorption band with the wavenumber 3013 cm–1 is attributable to the aromatic ν(C–H). Finally, the broad band around 3500 cm–1 is typical of phenolic C–O vibrations.
The 1H-NMR spectrum of H2(3,5-t-Bu2)-sal4eT was recorded in DMSO-d6 at a radiofrequency of 700 MHz with TMS as an internal reference standard (δ = 0). The broad peak at δ 9.98 assignable to the hydrazinic proton reveals that the thione tautomer of this thiosemicarbazone remains intact in solution. The phenolic proton, which is represented by the singlet at δ 11.27, is the most deshielded on account of its intramolecular interaction with the imine nitrogen atom. The aldimine proton is associated with the sharp singlet at δ 8.28. A broad resonance shaped like an unresolved triplet occurs at δ 8.48 and is attributable to the proton of the amino group between the thio-carbonyl and ethyl groups. The aromatic protons in positions 4 and 6 resonate as doublets at δ 7.13 and 7.29, respectively, with identical coupling constants (J = 2.38 Hz). The N-ethyl group is characterised by partially overlapping quartet signals at δ 3.59 (J = 6.55 Hz) and a triplet resonance at δ 1.16 (J = 7.14 Hz) corresponding to the methylene protons in non-equivalent environments and the methyl protons, respectively. Finally, the protons of the 3-tert-butyl and 5-tert-butyl substituent groups have singlet signals with the chemical shifts δ 1.41 and 1.27, respectively.
2.3. Single-Crystal X-ray Structure Determination of the Thiosemicarbazone Ligand
Definitive evidence for the solid-state 3-D structure of the ligand was provided by single-crystal X-ray analysis. A colourless needle amenable to X-ray diffraction was grown from a solution of H2(3,5-t-Bu2)-sal4eT in EtOH at room temperature. X-ray data collection was performed at 100 K. Crystal data, details of data collection and parameters for structure solution and refinement are compiled in Table 1. Evidently, this thiosemicarbazone ligand crystallized in the monoclinic space group P21/c with four molecules in the unit cell. The X-ray crystal structure is depicted in Figure 3 while selected bond distances and angles are presented in Table 2. The distance of the Schiff-base bond C=N [C(15)–N(1) = 1.2904(19) Å] lies within the range observed for normal imine bonds [1.26–1.30 Å] [13,14,15,19,20,60,61,62,63,64,65,66,67,68,69] in non-coordinated ligands. Upon reduction of the Schiff base, the imine double bond becomes a single bond (C–N) with a distance of ~1.47 Å [65]. The C(16)–S(1) distance of 1.7029(14) Å verifies the occurrence of the thione tautomer. Literature values for the distance of the thio-carbonyl bond in free thiosemicarbazone ligands range typically from 1.65 to 1.70 Å [13,14,15,19,20,60,61,62,63,64,65,66,67,68,69] (even longer if involved in bifurcated H-bonding) [67]. Both imine nitrogen and thio-carbonyl carbon are sp2-hybridised and the angles around them reflect the angular (with a lone pair) and trigonal planar geometries about these two atoms. The hydrazinic N–N bond [N(1)–N(2) = 1.3854(16) Å] is somewhat longer than most of those reported for other non-coordinated thiosemicarbazones and is consistent with single-bond character.
One of the prominent structural features of interest is the intramolecular H-bonding interaction between the phenolic –O–H group and the imine nitrogen atom [O(1)–H(1)···N(1): O1–H1 = 0.84 Å, H1···N1 = 1.98 Å, O1···N1 = 2.7221(15) Å, O1–H1···N1 = 147.4º]. Indeed, the vast majority of Schiff bases derived from 2-hydroxybenzaldehydes, 2-hydroxyacetophenones, 2-hydroxybenzophenones, 2-hydroxypropiophenones, etc. exhibit this intramolecular electrostatic force. It is well-established that pyridyl-/phenol-based thiosemicarbazones can adopt an E- or Z-configuration with respect to the imine double bond. Moreover, they can also orient themselves in different conformations as a consequence of free rotation about the C(py/phenol)–C(imine) [i.e. C(6)–C(15) in this structure] single bond and the amide N(H)–C(=S) [i.e. N(2)–((16) in this structure] single bond. Thus the potential donor atoms can be positioned anti or syn relative to each other. Examples of crystallographically observed orientations of phenolic thiosemicarbazones, viz. E(syn,anti) [60,62,63], E(syn,syn) [61,64] and E(ant,anti) [63], are shown in Figure 3. The structure of H2(3,5-t-Bu2)-sal4eT is consistent with the E-configuration; the phenolic –OH group and the imine nitrogen are positioned syn to each other while the thione sulfur points to the opposite side in an anti-orientation relative to the imine nitrogen.
2.4. Synthesis and Chemical Identification of the Copper(II) Thiosemicarbazone Complexes
Reaction of H2(3,5-t-Bu2)-sal4eT with a molar equivalent of Cu(OAc)2·H2O in refluxing MeOH, followed immediately by addition of a stoichiometric amount of 2,9-dimethyl-1,10-phenanthroline (2,9-Me2-phen) or 1,10-phenathroline (phen) with brief heating of the resultant dark olive green solution, afforded the mononuclear copper(II) complex [Cu{(3,5-t-Bu2)-sal4eT}(2,9-Me2-phen)] (1) or the dinuclear copper(II) complex [Cu2{(3,5-t-Bu2)-sal4eT}2(phen)] (2), respectively. The chemical formulations of these two ternary complexes were established by elemental analyses. The positive-ion ESI mass spectrum of complex 1 presented in Figure 4(a) shows a molecular peak at m/z = 605.4 in agreement with the molecular mass of this complex (605.25 amu). As regards the dinuclear complex (2), the parent ion was not detected; however, the ESR spectrum revealed important structural information from the fragmentation pattern. In the negative mode, the spectrum shows a minor peak at m/z = 792.5 consistent with the loss of the phen co-ligand. At m/z = 730.5 occurs a major peak ascribable to the fragment [Cu{(3,5-Bu2)-sal4eT}2]2– indicative of a loss of [Cu(phen)]2+ [Figure 4(b)]. On the other hand, the positive-ion ESI spectrum exhibits a peak at m/z = 732.6 attributable to the fragment [Cu{(3,5-t-Bu2)-sal4eT}2]+. Further dissociation affords the fragment [Cu{(3,5-t-Bu2)-sal4eT}]+ observed at m/z = 397.2, signifying the loss of one of the thiosemicarbazonate ligands. That complexes 1 and 2 are molecular has been demonstrated by the negligible value of the molar electrical conductivity (ΛM ~3 Ω–1 cm2 mol–1) of their nonelectrolyte solutions in MeOH, EtOH and DMF at room temperature [70].
2.5. FT-IR Spectroscopy and Magnetic Susceptibility Measurements
A comparison of the IR spectra of the thiosemicarbazone ligand and its copper(II) complexes (1 and 2) in Figure 3 shows clearly the absence of the vibrational band of the hydrazinic N–H bond from the spectra of the complexes. The disappearance of the hydrazinic proton coupled with the shift in the stretching frequency of the absorption band of the carbon-sulfur bond from 1032 cm–1 for the ligand to 842 and 858 cm–1 for 1 and 2, respectively, is indicative of tautomerisation and deprotonation of the thiosemicarbazone upon coordination to the copper(II) ion, as is indeed necessary for charge-neutrality of the resultant complexes. The presence of the N–H group attached to the terminal ethyl group is proven by the occurrence of sharp absorption bands at 3398 and 3342 cm–1 in the spectra of 1 and 2, respectively. The wavenumbers of the imine bond for 1 and 2 complexes are somewhat lower than that of the free ligand [ν(C=N): 1598 and 1599 cm–1 vs. 1609 cm–1)] consistent with coordination of the imine donor atom. Interestingly, the ν(N–N) absorptions for the ligand and complexes 1 and 2 virtually coincide (1172, 1170 and 1169 cm–1, respectively), implying minimal delocalisation of π-electrons, if any, along the ligand backbone in the complexes. Finally, the other ligand IR absorption patterns, especially those of the tert-butyl C–H bonds (2850–2960 cm–1), are retained.
Complexes 1 and 2 are paramagnetic with a single unpaired electron at the metal centre in the ground state. The room-temperature effective magnetic moment [µeff = (8χM)½] of the mononuclear complex (1) is 1.83 µB. It is comparable with the spin-only value [µS = {4S(S + 1)}½, where S = ½] and lies within the range of literature values [45,50,54,56,57,58,59]. In contrast, for the dinuclear complex (2), µeff = 2.38 µB at room temperature, which is close to the spin-only value for two magnetically uncoupled d9 paramagnetic centres [{4S1(S1 + 1) + 4S2(S2 + 1)}½, where S1 = S2 = ½].
2.6. Single-Crystal X-ray Analyses of the Ternary Copper(II) Complexes
For each of the complexes [Cu{(3,5-t-Bu2)-sal4eT}(2,9-Me2-phen)] (1) and [Cu2{(3,5-t-Bu2)-sal4eT}2(phen)] (2), X-ray diffraction data were collected on a single crystal at 100 K employing Cu-Kα radiation (λ = 1.54178 Å). Crystal data together with details of data collection and structure refinement are presented in Table 1. Selected bond distances and angles are given in Table 3. Whereas complex 1 crystallised in the monoclinic space group P21/n with Z = 4, complex 2 did so in the triclinic space group P with two complex molecules in the unit cell. Both complexes 1 and 2 do not possess solvent molecules of crystallisation.
The crystal structure of [Cu{(3,5-t-Bu2)-sal4eT}(2,9-Me2-phen)], depicted in Figure 5, reveals that this complex exists as a centrosymmetric dimer of mononuclear molecular ternary complexes of copper(II). The dimerisation occurs by two intermolecular hydrogen-bonding interactions involving the –N4–H group of one complex molecule and the thio-enolate sulfur of the other complex molecule [Figure 5(b)] [N(3)–H(3)···S(1): N–H = 0.88 Å, H···S = 2.64 Å, N···S = 3.482(3) Å, N–H–S = 159.9º (symmetry code: 1–x, 1–y, –z)]. The charge-neutrality of this ternary complex implies that the thiosemicarbazone been doubly deprotonated upon complexation. Indeed transformation of the ligand from the thio-keto tautomer to the thio-enolate anion is demonstrated by the changes to the lengths of the pertinent bonds of the thio-amide. The thio-amide N–C bond [N(2)–C(16): 1.3499(18) Å] in the free ligand has shortened considerably upon complexation [N(2)–C(8): 1.313(4) Å in complex 1] while the thio-carbonyl (C=S) bond [C(16)–S(1): 1.7029(14) Å] has converted to thio-enolate C–S– bond in the complex [C(8)–S(1) = 1.739(3) Å]. Carbon-nitrogen bonds with double-bond character have been reported to have distances in the range 1.27–1.32 Å [66,71,72,73] when the N donor atom is coordinated to a central metal ion. On the other hand, typical lengths of carbon-sulfur bonds with single-bond character in thio-enolate complexes are in the range 1.72–1.77 Å [66,71,72,73]. The distances of the imine C=N [C(7)–N(1) = 1.296(4) Å] and the hydrazinic N–N [N(1)–N(2) = 1.400(4) Å] in 1 are normal with respect to their respective bond orders.
The five-coordinate geometry at the copper(II) centre arises from the tridentate coordination of the thiosemicarbazonate ligand with the donor atoms, namely phenolate oxygen, imine nitrogen and thio-enolate sulfur, arranged meridionally and the bidentate coordination of the 2,9-Me2-phen co-ligand oriented nearly perpendicularly relative to the primary ligand. The axial-equatorial coordination mode of pyridyl nitrogen atoms of 2,9-Me2-phen leads to the construction of a coordination sphere best described as distorted square pyramidal in accord with the trigonality index [τ = (β – α)/60º] [74] of ~0.14, the largest two angles β and α being in the basal plane. The axial Cu–N bond is considerably longer than the one lying equatorially [Cu–Nax (co-ligand) = 2.308(3) Å vs. Cu–Neq (co-ligand) = 2.057(3) Å]. This elongation of the axial coordinate bond is attributable to the tetragonal distortion at the metal centre. Invariably, square pyramidal [31,32,33,34,35,36,37,38,39,40,41,42,43,45,46,47,48,49,50,51,52] and octahedral [44,53] ternary complexes with axial-equatorial coordination of bidentate co-ligand (bipy, phen or their derivatives) are subject to the Jahn-Teller effect evidenced by this structural feature. In some cases, even binary bis(chelate) copper(II) complexes [75] with two potentially tridentate ligands experience this effect, causing one of the ligands to coordinate bidentately due to considerable weakening of one of the axial Cu–L bonds. The magnitude of the disparity in the Cu–N distances of the asymmetrically coordinated N,N-donor co-ligand is in the range ~0.22–0.32 Å. Moreover, the complex cation of tris(1,10-phenanthroline)copper(II) perchlorate [76] exhibits Jahn-Teller distortion (Cu–N bond averages: Cu–Nax ~2.33 Å vs. Cu–Neq ~2.04 Å) whereby the axial Cu–Nphen bonds are elongated to the same extent as those in the above-mentioned square pyramidal copper(II) ternary complexes. In contrast, it has been crystallographically proven that the two Cu–N bonds of N,N-donor co-ligands in square pyramidal and octahedral complexes where they lie on the equatorial plane are virtually equivalent as neither is subject to the Jahn-Teller effect [54,55,56,57,58]. In addition, in the complex [Cu{N(CN)2}(phen)2]+ [77] with a distorted trigonal bipyramidal geometry at the metal centre the phen Cu–N distances are virtually indistinguishable from each other as the Jahn-Teller effect does not apply. The copper(II) ion in [Cu{(3,5-t-Bu2)-sal4eT}(2,9-Me2-phen)] (1) is displaced out of the mean basal plane [(N(1), S(1), O(1), N(4)] towards the apical phen N(5) donor atom by 0.1998(12) Å. Finally, the magnetostructural behaviour of this complex is consistent with half occupancy of the dx2–y2 orbital in the ground state.
As can be seen from Figure 6, [Cu2{(3,5-t-Bu2)-sal4eT}2(phen)] (2) exists in the crystal lattice as a centrosymmetric dimer of dinuclear molecular ternary complexes of copper(II) stabilized mainly by two types of intermolecular forces. The linkage of two dinuclear complex molecules occurs through two H-bonds between the N4–H group of one dinuclear molecule and the imine nitrogen of another dinuclear molecule [N(3)–H(3)···N(7): N–H = 0.88 Å, H···N = 2.17 Å, N···N = 2.964(11) Å, N–H···N = 150.5º (symmetry code: 2–x, –y, 1–z)]. Moreover, this complex exhibits π-π stacking interactions involving the plane N(5), C(24)–C(28) of the phen co-ligand in two complex molecules (symmetry code: 1–x, 1–y, 1–z) [angle of interaction of the two planes = 0.0(7)º, centroid-to-centroid distance = 6.603(8) Å, shift distance = 1.218(17) Å] (Figure S1).
The two copper(II) centres are 4.8067(18) Å apart and display different coordination numbers, one four-coordinate and the other five-coordinate, the respective coordination geometries being distorted square planar [τ4 = {360º – (α + β)}/141º = 0.082, α = 176.3º and β = 172.1º] [78] and distorted square pyramidal [τ5 = (β – α)/60º = 0.33, β = 172.4º and α = 152.4º] [74]. The two associated thiosemicarbazonate ligands exhibit different denticities: one coordinates in a tridentate fashion to the metal centre, [Cu(2) in Figure 6] with coordination number 4 whereas the other adopts the relatively unusual quadridentate coordination mode to bridge the two metal centres with the thio-enolate nitrogen atom, N(2), and coordinate meridionally to the other metal centre [Cu(1) in Figure 6]. For the tridentate ligand, coordinated to Cu(2), the distance of the newly-formed thio-enolate N=C bond is virtually indistinguishable from that of the imine C=N bond (cf. N(7)–C(38) = 1.316(12) Å vs. C(37)–N(6) = 1.310(12) Å, respectively] and the distance of C(32)–S(2) [1.746(9) Å] lies within the range reported for such thio-enolate bonds. Similarly, for the quadridentate ligand, the distances of the thio-enolate N=C and imine C=N bonds compare favourably [cf. N(2)–C(8) = 1.322(13) Å and C(7)–N(1) = 1.301(13) Å, respectively] [66,71,72,73]. The distance of the thio-enolate C–S– bond [C(8)–S(1) = 1.737(9) Å is normal for complexed thiosemicarbazonate ligands [66,71,72,73]. It is noteworthy that the lengths of the hydrazinic N–N bonds in this dinuclear complex [N(1)–N(2) = 1.381(11) Å and N(6)–N(7) = 1.391(11) Å] are very similar to that observed in the free ligand as a thio-keto (thione) tautomer [1.3854(16) Å], suggesting that there is no delocalization of electrons involving this chemical bond in the thiosemicarbazonate backbone.
The five-coordinate geometry at Cu(1) is similar to that described for the mononuclear ternary complex (1). The bidentate phen co-ligand adopts the axial-equatorial coordination mode. Consequently, the axial Cu–Nphen bond is tetragonally elongated, even longer than the Cu–S bond [Cu(1)–S(1) = 1.266(3) Å], causing asymmetric coordination of this co-ligand [Cu(1)–N(4)eq = 2.039(8) Å vs. Cu(1)–N(5) = 2.276(9) Å]. The copper(II) ion , Cu(1), resides 0.177(4) Å above the mean basal plane [N(1), S(1), O(1), N(4)] in the direction of the axial N(5)phen atom. Both copper(II) centres [Cu(1) and Cu(2)], regardless of the differences in the coordination geometries, have a dx2–y2 ground state. The literature has witnessed a number of examples of crystallographically characterised dinuclear thiosemicarbazone complexes of copper(II), but these tend to have the same coordination geometry at the two metal centres [8,9,10,72]. To the best of our knowledge, the dinuclear complex [Cu2{(3,5-t-Bu2)-sal4eT}2(phen)] (2) is one of only two of its kind. The other structurally characterised dinuclear thiosemicarbazone complex of copper(II) featuring two different coordination spheres is [Cu2(sal4eT)2(bipy)] (Scheme 1), reported as [Cu2(L2)2(bipy)] [4]. Beyond the superficial similarities, there is a sharp distinction between the structures of [Cu2{(3,5-t-Bu2)-sal4eT}2(phen)] (2) and [Cu2(sal4eT)2(bipy)] as regards the orientation of the ligands, intermolecular forces and the five-coordinate geometry at one of the two copper(II) centres. Unlike 2, [Cu2(sal4eT)2(bipy)] exists as two independent complex molecules which are similar but not identical and the thiosemicarbazonate ligands are oriented (differently from 2) such that intramolecular hydrogen bonding occurs between the phenolate oxygen atom bonded to the copper(II) ion in the distorted square planar geometry and the N4–H group of the bridging quadridentate ligand. Moreover, the geometries of the five-coordinate copper(II) centres in the two molecules of [Cu2(sal4eT)2(bipy)] were reported as distorted trigonal bipyramidal. We have calculated their trigonality indices, τ5 [74], to compare them with that of our dinuclear complex (2). For [Cu2(sal4eT)2(bipy)], the values of τ5 are ~0.50 and ~0.51 (intermediate between square pyramidal and trigonal bipyramidal); in contrast, for 2, τ5 = 0.33, clearly pointing to greater distortion towards square pyramidal.
2.7. X-Band ESR and UV-Visible Spectroscopic Characterisation
The X-band ESR spectrum of [Cu{(3,5-t-Bu2)-sal4eT}(2,9-Me2-phen)] (1) in frozen MeOH solution at 77 K, displayed in Figure 7, is axial. The ESR parameters gǁ = 2.20, gꞱ = 2.05, Aǁ = 19.3 mT and AꞱ = 3 mT (gǁ > gꞱ > 2.00; Aǁ > AꞱ) are consistent with the dx2–y2 ground state [75,79,80,81]. Hence ESR spectroscopy demonstrated that the crystallographically determined distorted square pyramidal geometry at the metal centre is retained in solution. Figure 8 shows the electronic absorption spectra of H2(3,5-t-Bu2)-sal4eT, [Cu{(3,5-t-Bu2)-sal4eT}(2,9-Me2-phen)] (1) and [Cu2{(3,5-t-Bu2)-sal4eT}2(phen)] (2). The ligand is colourless; accordingly, its spectrum exhibits UV absorptions only. These hugely intense absorption bands are assignable to π → π* (280–305 nm) and n → π* (~338 nm) electronic transitions [4]. This pattern of UV absorptions is also observed in the spectra of 1 and 2, albeit at somewhat higher energies. In the visible region of the electronic spectra of 1 and 2 are intense broad bands centred at 405 nm (εmax ~ 17000 dm3 mol–1 cm–1) and 401 nm (εmax ~ 20100 dm3 mol–1 cm–1), respectively, attributable to phenolate/thio-enolate-to-copper(II) charge-transfer electronic transitions [4]. Spin-allowed, but Laporte-forbidden, ligand-field transitions were observed as weak broad absorption bands at ~590 and ~570 nm for 1 and 2, respectively. For distorted square-pyramidal copper(II) complexes, such d-d electronic transitions have been assigned previously as dxz,dyz → dx2–y2 in nature [79].
2.8. In Vitro Cytotoxicity of the Thiosemicarbazone Ligand and the Ternary Copper(II) Complexes
The antiproliferative activity of H2(3,5-t-Bu2)-sal4eT, [Cu{(3,5-t-Bu2)-sal4eT}(2,9-Me2-phen)] (1) and [Cu2{(3,5-t-Bu2)-sal4eT}2(phen)] (2) was investigated in two cancer cell lines, namely human cervical carcinoma (HeLa) and human breast adenocarcinoma (MCF-7) using the MTT cell viability assay [MTT = 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide]. The values of the 50% inhibitory concentrations (IC50) of these substances together with the positive controls (docetaxel and paclitaxel) were determined as exemplified for complexes 1 and 2 in the HeLa and MCF-7 cancer cells, respectively (Figure 9). The in vitro antiproliferative potential of each substance was tested within the 0.01–100-μM range of concentrations; the results of these cytotoxicity measurements are presented in Table 4. IC50 values for cisplatin were obtained from the literature [82,83].
In striking contrast to the marked potent and selective antiproliferative activity of naphthol- and pyridyl-based thiosemicarbazones [11,12], together with their corresponding metal complexes, against tumour cells, the phenolic thiosemicarbazone H2(3,5-t-Bu2)-sal4eT is nontoxic towards both cancer cells in this investigation. However, as is often the case with hydrazones, complexation with metal ions induces phamarcological activity as can be seen from Table 4. Intriguingly, complex 1 exhibits selective potency towards the Hela cancer cells over the MCF-7 cancer cells. Conversely, the antiproliferative activity of complex 2 is specific towards MCF-7. Although this behaviour has only been observed from tests carried out in vitro, these results show that these copper(II) thiosemicarbazone complexes have potential applications as metallo-drugs in targeted cancer treatment.
Their selectivity over non-cancerous cells such as the human breast epithelial cell line (MCF-10A), have yet to be determined. It is noteworthy that in vitro complexes 1 and 2 are more efficacious as antiproliferative agents than cisplatin. Moreover, cisplatin lacks the cancer-specificity which these complexes possess. They also exhibit higher potent anticancer activity than the standards. Although the study of the mode of action of complexes 1 and 2 as anticancer agents is beyond the scope of this work, it has been amply demonstrated previously for a diverse range of copper(II) complexes, including those of thiosemicarbazones, that the potentials of the CuII/CuI redox couple [8,15,18,20,30] lie within the biologically accessible redox potential window leading to the generation of reactive oxygen species (ROS) which cause apoptotic cell death.
3. Conclusion
The thiosemicarbazone H2(3,5-t-Bu2)-sal4eT and its ternary copper(II) complexes [Cu{(3,5-t-Bu2)-sal4eT}(2,9-Me2-phen)] (1) and [Cu2{(3,5-t-Bu2)-sal4eT}2(phen)] (2) have been synthesised and their chemical identities ascertained by microanalyses, mass spectrometry and vibrational spectroscopy. Definitive evidence for their 3-D structures was provided by single-crystal X-ray analyses. As is commonly the case with phenolic thiosemicarbazones, this ligand was isolated as the thio-keto (thione) tautomer and in the E-configuration with respect to the Schiff-base imine bond. 1H-NMR spectroscopy showed that this isomeric form is maintained in solution. The X-ray structures of complexes [Cu{(3,5-t-Bu2)-sal4eT}(2,9-Me2-phen)] (1) and [Cu2{(3,5-t-Bu2)-sal4eT}2(phen)] (2) show that the thiosemicarbazone underwent base/metal-assisted tautomerization upon coordination to copper(II); moreover, this ligand also demonstrated coordination versatility in that in complex 2 it employed two different denticities, namely tridentate and quadridentate. The planarity and rigidity of the thiosemicarbazonate ligand and the co-ligand 2,9-dimethyl-1,10-phenanthroline imposed a distorted square-pyramidal geometry at the metal centre of complex 1 (τ = 0.14) whereby the donor atoms of the tridentate primary ligand are arranged meridionally whereas the bidentate co-ligand adopts an axial-equatorial coordination mode. As always observed in X-ray structures of this type of ternary copper(II) complexes, there is considerable elongation of the axial coordinate bond in conformity with the tetragonal distortion. The ESR spectrum of 1 in frozen solution is axial (gǁ > gꞱ > 2.00; Aǁ > AꞱ) and indicative of half occupancy of the dx2–y2 orbital. The dinuclear complex (2) exhibits different coordination geometries at the copper(II) centres, namely distorted square planar and distorted square pyramidal. Such dinuclear ternary copper(II) complexes are few and far between. Intriguingly, the cytotoxicity activities of [Cu{(3,5-t-Bu2)-sal4eT}(2,9-Me2-phen)] and [Cu2{(3,5-t-Bu2)-sal4eT}2(phen)] in the HeLa and MCF-7 cancer cell lines are vastly different. Whereas the former is highly antiproliferative against HeLa cancer cells but non-toxic towards MCF-7 cancer cells, the converse is true for the latter. Such selective cytotoxicity of 1 and 2 towards these cancer cells is not shown by cisplatin.
4. Experimental
4.1. Materials and Physical Techniques
All pertinent chemicals, reagents and solvents (HPLC/AR-grade) were purchased from Sigma-Aldrich and used as received. Microanalyses (CHN) were performed on a EuroVector elemental analyser. ESI mass spectra were measured with an Agilent 6460 Triple Quadrupole mass spectrometer using MeOH as a matrix. Electrical conductivities of the complexes were determined with a JENWAY 4520 conductivity meter at room temperature using freshly prepared solutions (1 mM) in MeOH. FT-IR spectra were recorded on a Perkin-Elmer spectrophotometer (4000–400 cm–1) with the samples compressed as KBr discs using a Specac press. 1H NMR spectroscopic measurements were carried out at room temperature in DMSO-d6 on a Bruker ASCEN 700 spectrometer operating at radiofrequencies of 700 MHz, respectively; the chemical shifts are referenced to TMS as an internal standard (δ = 0). X-band ESR spectra of the copper(II) complexes were recorded on a Bruker ELEXSYS E580X FT CW spectrometer (ν ~ 9.4 GHz). Electronic absorption spectra were measured with a Shimadzu 2450 UV-visible spectrophotometer (190–1000 nm) using freshly prepared solutions. Magnetic susceptibility measurements were carried out at room temperature using a Sherwood Scientific magnetic susceptibility balance. The magnetic data were corrected for diamagnetism using Pascal’s constants the usual way (). Single-crystal X-ray structure determinations were carried out on a Bruker APEX-II CCD area-detector diffractometer or a Bruker D8 Venture CMOS Photon 100 diffractometer. The crystals were mounted in fomblin oil and cooled in a stream of cold N2. Data were corrected for absorption using empirical methods (SADABS) [84] based upon symmetry equivalent reflections combined with measurements at different azimuthal angles [85]. The crystal structures were solved and refined against F2 values using ShelXT [86] for solution and ShelXL [87] for refinement (using least squares minimisation) accessed via the Olex2 programme [88].
4.2. Synthesis of H2(3,5-t-Bu2)-sal4eT
A sample of 3,5-di-tert-butylsalicylaldehyde (2.3433 g, 10.00 mmol) was dissolved in EtOH (30 mL). Separately, 4-ethyl-3-thiosemicarbazide (1.1919 g, 10.00 mmol) was dissolved in EtOH (30 mL). Then these two solutions were mixed together and the resultant solution heated under reflux over a period of three hours. On standing at room temperature for four days, the solution deposited shiny colourless crystals. This product was filtered off on the Büchner funnel, washed with ice-cold EtOH and dried in air (yield: 2.2419 g, 66.82%). Characterization: calcd for C18H29N3OS (M = 335.50 g/mol): C, 64.44%; H, 8.71%; N, 12.52%. Found: C, 64.17%; H, 8.55%; N, 12.63%; m.p. 218–219⁰C.
4.3. Synthesis of [Cu{(3,5-t-Bu2)-sal4eT}(2,9-Me2-phen)] (1)
To a hot solution of H2(3,5-t-Bu2)-sal4eT (0.1342 g, 0.40 mmol) in MeOH (30 mL) were added Cu(OAc)2·H2O (0.0799 g, 0.40 mmol) and 2,9-dimethyl-1,10-phenanthroline (0.0833 g, 0.40 mmol) consecutively with vigorous swirling. The resultant reaction mixture was heated under reflux for 15 minutes and then filtered and kept at room temperature. Upon slow solvent evaporation from the solution under ambient conditions over a period of one week, shiny black blocks were formed. This crystalline product was isolated by decantation of the mother liquor, washed with ice-cold EtOH and then left to dry in air (yield: 0.2160 g, 89.23%). Characterization: calcd for C32H39CuN5OS (M = 605.28 g/mol): C, 63.50%; H, 6.49%; N, 11.57%. Found: C, 63.37%; H, 6.50%; N, 11.58%; m.p. 248−249⁰C.
4.4. Synthesis of [Cu2{(3,5-t-Bu2)-sal4eT}2(phen)] (2)
This complex was synthesised as described for complex 1 above except that 1,10-phenanthroline monohydrate (0.0793 g, 0.4000 mmol) was used as a co-ligand instead of 2,9-dimethyl-1,10-phenanthroline. The resultant dark green solution was heated under reflux for 15 minutes and then filtered. Large shiny black crystals were obtained from the solution after one week of standing at room temperature. After removal of the supernatant, the crystals were washed with ice-cold EtOH and kept in air (yield: 0.1576 g, 80.86%). Characterization: calcd for C48H62Cu2N8O2S2 (MM = 974.25 g/mol): C, 59.17%; H, 6.41%; N, 11.50%. Found: C, 59.53%; H, 6.29%; N, 11.38%; m.p. 222−224⁰C.
4.5. Investigation of Anticancer Activity
4.5.1. Cell Lines and Cell Culture
Two cancerous cell lines, namely MCF-7 (human breast adenocarcinoma) and HeLa (human cervical carcinoma) were obtained from the Pasteur Institute (Tehran, Iran). The cells were cultured in RPMI-1640 medium (Sigma) supplemented with 10% heat-inactivated foetal bovine serum (FBS; Gibco, USA), penicillin (100 U/mL) and streptomycin (100 μg/mL) (Roche, Germany) at 37°C in a humidified incubator with 5% CO2.
4.5.2. Cytotoxicity Evaluation by the MTT Assay
All the final compounds were investigated for their antiproliferative potential at 0.01–100 μM concentrations. Three controls were prepared within each 96-well plate: a solvent [dimethyl sulfoxide (DMSO)] control and a positive controls (paclitaxel & Docetaxel). Briefly, appropriate cells were seeded in 96-well plates (Nunc, Roskilde, Denmark) at a density of 10 000 viable cells per well and incubated for 24 h to allow for cell attachment. Solutions of the compounds were prepared by serial dilution from the stock solution and added to each well. Cells were then incubated with the compounds for another 48 h. The response of cells to compounds was evaluated by determining the cell survival using 3-(4,5-dimethylthiazoyl- 2-yl) 2,5-diphenyl tetrazolium bromide (MTT, Carl Roth, Karlsruhe, Germany). At first, cells were washed with phosphate buffered saline (PBS) and then 20 μL of the MTT reagent (5 mg/mL) in PBS was added to each well. After 4 h of incubation at 37°C, the medium was discarded, DMSO (100 μL) was added to each well and the plates were vigorously shaken to dissolve the purple tetrazolium crystals in DMSO. The absorbance of each well was measured using a plate reader (Anthous 2020; Austria) at a test wavelength of 550 nm against a standard reference solution at 690 nm. Assays were performed in triplicate in three independent experiments, and the concentration required for 50% inhibition of cell viability (IC50) was determined from a plot of the percentage cytotoxicity versus the concentration on a logarithmic graph.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lobana, T.S.; Sharma, R.; Bawa, G.; Khanna, S. Bonding and structure trends of thiosemicarbazone derivatives of metals – an overview. Coord. Chem. Rev. 2009, 253, 977–1055. [Google Scholar] [CrossRef]
- Papathanasis, L.; Demertzis, M.A.; Yadav, P.N.; Kovala-Demertzi, D.; Prentjas, C.; Castiñeiras, A.; Skoulika, S.; West, D.X. Palladium(II) and platinum(II) complexes of 2-hydroxyacetophenone N(4)-ethylthiosemicarbazone – crystal structure and description of bonding properties. Inorg. Chim. Acta 2004, 357, 4113–4120. [Google Scholar] [CrossRef]
- Zhang, Z.; Gou, Y.; Wang, J.; Yang, K.; Qi, J.; Zhou, Z.; Liang, S.; Liang, H.; Yang, F. Four copper(II) compounds synthesized by anion regulation: structure, anticancer function and anticancer mechanism. Eur. J. Med. Chem, 2016, 121, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Lobana, T.S.; Kumari, P.; Bitcher, R.; Jasinski, J.P.; Golen, J.A. Metal derivatives of thiosemicarbazones: crystal and molecular structures of mono- and dinuclear copper(II) complexes with N1-substituted salicylaldehyde rhiosemicarbazones. Z. Anorg. Allg Chem. 2012, 638, 1861–1867. [Google Scholar] [CrossRef]
- Lobana, T.S.; Kumari, P.; Castiñeiras, A.; Butcher, R.J. The effect of C-2 substituents of salicyladehyde-based thiosemicarbazones on the synthesis, spectroscopy, structures, and fluorescence of nickel(II) complexes. Eur. J. Inorg. Chem. 2013, 3557–3566. [Google Scholar] [CrossRef]
- Kumari, P.; Lobana, S.T.; Butcher, R.J.; Castiñeiras, A.; Zeller, M. The effect of substituents at C2/N1 atoms of salicylaldehyde and 2-hydroxyacetophenone-based thiosemicarbazones on the nature of nickel(II) complexes with 1,10-phenanthroline and terpyridine as co-ligands. Inorg. Chim. Acta 2018, 482, 268–274. [Google Scholar] [CrossRef]
- Pereiras-Gabián, G.; Vázquez-López, M.; Abram, U. Dimeric rhenium(I) carbonyl complexes with thiosemicarbazone backbone. Z. Anorg. Allg. Chem. 2004, 630, 1665–1670. [Google Scholar] [CrossRef]
- Qi, J.; Yao, Q.; Tian, L.; Wang, Y. Piperidylthiosemicarbazones Cu(II) complexes with a high anticancer activity by catalysing hydrogen peroxide to degrade DNA and promote apoptosis. Eur. J. Med. Chem. 2018, 158, 853–862. [Google Scholar] [CrossRef]
- Qi, J.; Liang, S.; Gou, Y.; Zhang, Z.; Zhou, Z.; Yang, F.; Liang, H. Synthesis of four binuclear copper(II) complexes: structure, anticancer properties and anticancer mechanism. Eur. J. Med. Chem. 2015, 96, 360–368. [Google Scholar] [CrossRef]
- Duan, C.-Y.; Wu, B.-M.; Mak, T.C.W. ; Synthesis and structural characterization of new quadridentate N3S-compound di-2-pyridyl ketone thiosemicarbazone and its binuclear copper(II) complexes. J. Chem. Soc., Dalton Trans. 1996, 3485–3490. [Google Scholar] [CrossRef]
- Lovejoy, D.B.; Richardson, D.R. Novel “hybrid” iron chelators derived from aroylhydrazones and thiosemicarbazones demonstrate selective antiproliferative activity against tumor cells. Blood 2002, 100, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Lovejoy, D.B.; Richardson, D.R. Novel di-pyridyl-derived iron chelators with marked and selective antitumor activity: in vitro and in vivo assessment. Blood, 2004, 104, 1450–1458. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.R.; Sharpe, P.C.; Lovejoy, D.B.; Senaratne, D.; Kalinowski, D.S.; Islam, M.; Bernhardt, P.V. Dipyridyl thiosemicarbazone chelators with potent and selective antitumour activity form iron complexes with redox activity. J. Med. Chem. 2006, 49, 6510–6521. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, D.S.; Yu, Y.; Sharpe, P.C.; Islam, M.; Liao, Y.-T.; Lovejoy, D.B.; Kumar, N.; Bernhardt, P.V.; Richardson, D.R. Design, synthesis, and characterization of novel iron chelators: structure-activity relationships of the 2-benzoylpyridine thiosemicarbazone series and their 3-nitrobenzoyl analogues as potent antitumor agents. J. Med. Chem. 2007, 50, 3716–3729. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.R.; Kalinowski, D.S.; Richardson, V.; Sharpe, P.C.; Lovejoy, D.B.; Islam, M.; Bernhardt, P.V. 2-Acetylpyridine thiosemicarbazones are potent iron chelators and antiproliferative agents: redox activity, iron complexation and characterization of their antitumor activity. J. Med. Chem. 2009, 52, 1459–1470. [Google Scholar] [CrossRef] [PubMed]
- Jansson, P.J.; Sharpe, P.C.; Bernhardt, P.V.; Richardson, D.R. Novel thiosemicarbazones of the ApT and DpT series and their copper complexes: identification of pronounced redox activity and characterization of their antitumor activity. J. Med. Chem. 2010, 53, 5759–5769. [Google Scholar] [CrossRef]
- Stefani, C.; Punnia-Moorthy, G.; Lovejoy, D.B.; Jansson, P.J.; Kalinowski, D.S.; Sharpe, P.C.; Bernhardt, P.V.; Richardson, D.R. Halogenated 2´-benzoylpyridine thiosemicarbazone (XBpT) chelators with potent and selective anti-neoplastic activity: relationship to intracellular redox activity. J. Med. Chem. 2011, 54, 6936–6948. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, D.B.; Sharp, D.M.; Seebacher, N.; Obeidy, P.; Prichard, T.; Stefani, C.; Basha, M.T.; Sharpe, P.C.; Jansson, P.J.; Kalinowski, D.S.; Bernhardt, P.V.; Richardson, D.R. Novel second-generation di-2-pyridylketone thiosemicarbazones show synergism with standard chemotherapeutics and demonstrate potent activity against lung cancer xenografts after oral and intravenous administration in vivo. J. Med. Chem. 2012, 55, 7230–7244. [Google Scholar] [CrossRef] [PubMed]
- Stefani, C.; Jansson, P.J.; Gutierrez, E.; Bernhardt, P.V.; Richardson, D.R.; Kalinowski, D.S. Alkyl substituted 2´-benzoylpyridine thiosemicarbazone chelators with potent and selective anti-neoplastic activity: novel ligands that limit methemoglobin formation. J. Med. Chem. 2013, 56, 357–370. [Google Scholar] [CrossRef]
- Ohui, K.; Afanasenko, E.; Bacher, F.; Ting, R.L.X.; Zafar, A.; Blanco-Cabra, N.; Torrents, E.; Dömötör, O.; May, N.V.; Darvasiova, D.; Enyedy, E.A.; Popović-Bijelić, A.; Reynisson, J.; Rapta, P.; Babak, M.V.; Pastorin, G.; Arion, V.B. New water-soluble copper(II) complexes with morpholine-thiosemicarbazone hybrids: insights into the anticancer and antibacterial mode of action. J. Med. Chem. 2019, 62, 512–530. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Z.-M.; Li, M.-X. Synthesis, characterization, and biological activity of cadmium(II) and antimony(III) complexes based on 2-acetylpyrazine thiosemicarbazones. Inorg. Chim. Acta 2022, 530, 120671. [Google Scholar] [CrossRef]
- Savir, S.; Liew, J.W.K.; Vythilingam, I.; Lim, Y.A.L.; Tan, C.H.; Sim, K.S.; Lee, V.S.; Maah, M.J.; Tan, K.W. Nickel(II) complexes with plyhydroxybenzaldehyde and O,N,S tridentate thiosemicarbazone ligands: synthesis, cytotoxicity, antimalarial activity, and molecular docking studies. J. Mol. Struct. 2021, 1242, 130815. [Google Scholar] [CrossRef]
- Rodríguez-Argüelles, M.C.; López-Silva, E.C.; Sanmartín, J.; Pelagatti, P.; Zani, F. Copper complexes of imidazole-2-, pyrrole-2- and indol-3-carbaldehyde thiosemicarbazones: inhibitory activity against fungi and bacteria. J. Inorg. Biochem. 2005, 99, 2231–2239. [Google Scholar] [CrossRef] [PubMed]
- Genova, P.; Varadinova, T.; Matesanz, A.I.; Marinova, D.; Souza, P. Toxic effects of bis(thiosemicarbazone) compounds and its palladium(II) complexes on herpes simplex virus growth. Toxicol. Appl. Pharmacol. 2004, 197, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Karaküҫük-İyidoğan, A.; Taşdemir, D.; Oruҫ-Emre, E.E.; Balzarini, J. Novel platinum(II) and palladium(II) complexes of thiosemicarbazones derived from 5-substitutedthiophene-2-carbaldehydes and their antiviral and cytotoxicity activities. Eur. J. Med. Chem. 2011, 46, 5616–5624. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, K.; Buchanan, R.M.; Grapperhaus, C.A. Antifungal activity of thiosemicarbazones, bis(thiosemicarbazones), and their metal complexes. J. Inorg. Biochem. 2021, 225, 111620. [Google Scholar] [CrossRef] [PubMed]
- Khanye, S.D.; Smith, G.S.; Lategan, C.; Smith, P.J.; Gut, J.; Rosenthal, P.J.; Chibale, K. Synthesis and in vitro evaluation of gold(I) thiosemicarbazone complexes for antimalarial activity. J. Inorg. Biochem. 2010, 104, 1079–1083. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Shiota, Y.; Kariyazaki, A.; Kanegawa, S.; Yoshizawa, K.; Sato, O. Heterometallic FeIII/K coordination polymer with a wide thermal hysteretic spin transition at room temperature. Chem. Eur. J. 2016, 22, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Kovala-Demertzi, D.; Yadav, P.N.; Demertzis, M.A.; Jasiski, J.P.; Andreadaki, F.J.; Kostas, I.D. First use of a palladium complex with a thiosemicarbazone ligand as catalyst precursor for the Heck reaction. Tetrahedron Lett. 2004, 45, 2923–2926. [Google Scholar] [CrossRef]
- Jungwirth, U.; Kowol, C.R.; Keppler, B.K.; Hartinger, C.G.; Berger, W.; Heffeter, P. Anticancer Activity of Metal Complexes: Involvement of Redox Processes. Antioxid. Redox Signal. 2011, 15, 1085–1127. [Google Scholar] [CrossRef]
- Mohan, B.; Chaudhary., M. Synthesis, crystal structure, computational study and anti-virus effect of mixed ligand copper(II) complex with ONS donor Schiff base and 1,10-phenanthroline. J. Mol. Struct. 2021, 1246, 131246. [Google Scholar] [CrossRef] [PubMed]
- Ambalavanan, P.; Palani, K.; Ponnuswamy, M.N. Crystal structure of [1-(2-phenyloxyN[N-cyclohexylthiouryl)] ethylamine-(1,10-phenanthrolinyl)]copper(II). Cryst. Res. Technol. 2002, 37, 1249–1254. [Google Scholar] [CrossRef]
- Naik, A.D.; Reddy, P.A.N.; Nethaji, M.; Chakravarty, A.R. Ternary copper(II) complexes of thiosemicarbazones and heterocyclic bases showing N3OS coordination as models for the type-2 centers of copper monooxygenases. Inorg. Chim. Acta 2003, 349, 149–158. [Google Scholar] [CrossRef]
- Thomas, A.M.; Naik, A.D.; Nethaji, M.; Chakravarty, A.R. Synthesis, crystal structure and photo-induced DNA cleavage activity of ternary copper(II)-thiosemicarbazone complexes having heterocyclic bases. Inorg. Chim. Acta 2004, 357, 2315–2323. [Google Scholar] [CrossRef]
- Lobana, T.S.; Indoria, S.; Jassal, A.K.; Kaur, H.; Arora, D.S.; Jasinski, J.P. Synthesis, structures, spectroscopy and antimicrobial properties of complexes of copper(II) with salicylaldehyde N-substituted thiosemicarbazones and 2,2´-bipyridine or 1,10-phenanthroline. Eur. J. Med. Chem. 2014, 76, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Indoria, S.; Lobana, T.S.; Kauer, H.; Arora, D.S.; Randhawa, B.S.; Jassal, A.K.; Jasinski, J.P. Synthesis and structures of 5-methoxysalicyladehyde thiosemicarbazonates of copper(II): molecular spectroscopy, ESI-mass studies and antimicrobial activity. Polyhedron, 2016, 107, 9–18. [Google Scholar] [CrossRef]
- Lobana, T.S.; Indoria, S.; Sood, S.; Arora, D.S.; Randhawa, B.S.; Garcia-Santos, I.; Smolinski, V.A.; Jasinski, J.P. Synthesis of 5-nitrosalicylaldehyde-N-substituted thiosemicarbazonates of copper(II): molecular structures, spectroscopy, ESI-mass studies and antimicrobial activity. Inorg. Chim Acta 2017, 46, 248–260. [Google Scholar] [CrossRef]
- Ainscough, E.W.; Brodie, A.M.; Ranford, J.D.; Waters, J.M. Reaction of nitrogen and sulphur donor ligands with the antitumour complex [{CuL(MeCO2)}2] (HL = 2-formylpyridine thiosemicarbazone) and the single-crystal X-ray structureof [CuL(bipy)]ClO4 (bipy = 2,2´-bipyridine). J. Chem. Soc., Dalton Trans. 1991, 1737–1742.
- Kartikeyan, R.; Murugan, D.; Ajaykamal, T.; Varadhan, M.; Rangasamy, L.; Velusamy, M.; Palaniandavar, M.; Rajendiran, V. Mixed ligand copper(II)-diimine complexes of 2-formylpyridine-N4-phenylthiosemicarbazone: diimine co-ligands tune the in vitro nanomolar cytotoxicity. Dalton Trans. 2023, 52, 9148–9169. [Google Scholar] [CrossRef] [PubMed]
- Sreeja, P.B.; Kurup, M.R.P.; Kishore, A.; Jasmin, C. Spectral characterization, X-ray structure and biological investigations of copper(II) ternary complexes of 2-hydroxyacetophenone 4-hydroxybenzoic acid hydrazone and heterocyclic bases, Polyhedron 2004, 23, 575–581.
- Reddy, P.A.N.; Santra, B.K.; Nethji, M.; Chakravarty, A.R. Metal-assisted light-induced DNA cleavage activity of 2-(methylthio)phenylsalicyladimine Shiff base copper(II) complexes having planar heterocyclic bases. J. Inorg, Biochem. 2004, 98, 377–386. [Google Scholar] [CrossRef]
- Reddy, P.R.; Shilpa, A.; Raju, N.; Raghavaiah, P. Synthesis, structure, DNA binding and cleavage properties of ternary amino acid Schiff base-phen/bipy Cu(II) complexes. J. Inorg. Biochem. 2011, 105, 1603–1612. [Google Scholar] [CrossRef]
- Li. A.; Liu, Y.-H.; Yuan, L.-Z.; Ma, Z.-Y.; Zhao, C.-L.; Xie, C.-Z.; Bao, W.-G.; Xu, J.-Y. Association of structural modifications with bioactivity in three new copper(II) complexes of Schiff base ligands derived from 5-chlorosalicylaldehyde and amino acids. J. Inorg. Biochem. 2015, 146, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Rajendiran. ; karthik, R.; Palaniandavar, M.; Stoeckli-Evans, H.; Periasamy, V.S.; Akbarsha, M.A.; Srinag, B.S.; Krishnamurthy, H. Mixed-ligand copper(II)-phenolate complexes: effect of coligand on enhanced DNA and protein binding, DNA cleavage, and anticancer activity. Inorg. Chem. 2007, 46, 8208–8221. [Google Scholar]
- Reddy, P.A.N.; Nethaji, M.; Chakravarty, A.R. Synthesis, crystal structures and properties of ternary copper(II) complexes having 2,2´-bipyridine and amino acid salicylaldiminates as models for the type-2 sites in copper oxidases. Inorg. Chim. Acta 2002, 337, 450–458. [Google Scholar] [CrossRef]
- Mathew, N.; Sithambaresan, M.; Kurup, M.R.P. Spectral studies of copper(II) complexes of tridentate acylhdrazone ligands with heterocyclic compounds as coligands: X-ray crystal structure of one acylhydrazone copper(II) complex. Spectrochim. Acta Part A, 2011, 79, 1154–1161. [Google Scholar] [CrossRef] [PubMed]
- Koh, L.L.; Ranford, J.O.; Robinson, W.T.; Svensson, J.O.; Tan, A.L.C.; Wu, D. Model for the reduced Schiff base intermediate between amino acids and pyridoxal: copper(II) complexes of N-(2-hdroxybenzyl)amino acids with nonpolar side chains and the crystal structures of [Cu(N-(2-hydroxybenzyl)-D,L-alanine)(phen·H2 O and [Cu(N-(2-hydroxybenzyl)-D,L-alanine)(imidazole)]. Inorg. Chem. 1996, 35, 6466–6472. [Google Scholar] [PubMed]
- Bernal, M.; García-Vázquez, J.A.; Romero, J.; Gómez, C.; Durán, M.L.; Sousa, A.; Sousa-Pedrares, A.; Rose, D.J.; Maresca, K.P.; Zubieta, J. Electrochemical synthesis of cobalt, nickel, zinc and cadmium complexes with N[(2-hydroxyphenyl)methylidine]-N´-tosylbenzene-1,2-diamine. The crystal structures of {1,10-phenanthroline)[N-2-oxophenyl)methylidine]-N-tosylbenzene-1,2-diaminato}nickel(II) and {1,10-phenanthroline)[N-2-oxophenyl)methylidine]-N´-tosylbenzene-1,2-diaminato}copper(II). Inorg. Chim. Acta 1999, 295, 39–47. [Google Scholar]
- Wang, M.-Z.; Meng, Z.-X.; Liu, B.L.; Cai, G.-L.; Zhang, C.-L.; Wang, X.-Y. Novel tumor chemotherapeutic agents and tumor radio-imaging agents: potential tumor pharmaceuticals of ternary copper(II) complexes. Inorg. Chem. Commun. 2005, 8, 368–371. [Google Scholar] [CrossRef]
- Reddy, P.A.N.; Nethaji, M.; Chakravarty, A.R. Hydrolytic cleavage of DNA by ternary amino acid Schiff base copper(II) complexes having planar heterocyclic ligands. Eur. J. Inorg. Chem. 2004, 1440–1446. [Google Scholar] [CrossRef]
- Dong, J.; Li, L.; Liu, G.; Xu, T.; Wang, D. Synthesis, crystal structure and DNA-binding properties of a new copper(II) complex with L-valine Schiff base and 1,10-phenanthroline. J. Mol. Struct. 2011, 986, 57–63. [Google Scholar] [CrossRef]
- Labisbal, E.; Garcia-Vazquez, Romero, J.; Picos, S.; Sousa, A.; Castiñeiras, A.; Maichle-Mössmer. Electrochemical synthesis and structural characterization of nickel(II) and copper(II) complexes of tridentate Schiff bases: molecular structure and the five-coordinated copper(II) complex: 1,10-phenanthroline {2-[2-oxyphenyl)iminomethyl]phenolato}copper(II). Polyhedron 1995, 14, 663–670.
- Sharma, M.; Ganeshpandian, M.; Majumder, M.; Tamilarasan, A.; Sharma, M.; Mukhopadhyay, R.; Islam, N.S.; Palaniandavar, M. Octahedral copper(II)-diimine complexes of triethylenetetramine: effect of stereochemical fluxionality and ligand hydrophobicity on CuII/CuI redox, DNA binding and cleavage, cytotoxicity and apoptosis-inducing ability. Dalton Trans. 2020, 49, 8282–8297. [Google Scholar] [CrossRef]
- Ng, C.H.; Kong, K.C.; Von,S.T.; Balraj, P.; Jensen, P.; Thirthagiri, E.; Hamada, H.; Chikira, M. Synthesis, characterization, DNA-binding stuídy and anticancer properties of ternary metal(II) complexes of edda and an intercalating ligand. Dalton Trans. 2008, 447–454.
- Selvakumar, B.; Rajendiran, V.; Maheswari, P.U.; Stoeckli-Evans, H.; Palaniandavar, M. Structures, spectra, and DNA-binding properties of mixed ligand copper(II) complexes of iminodiacetic acid: the novel role of diamine co-ligands on DNA conformation and hydrolytic and oxidative double strand DNA cleavage. J. Inorg. Biochem. 2006, 100, 316–330. [Google Scholar] [CrossRef]
- Yang, C.-T.; Moubaraki, B.; Murray, K.S.; Vittal, J.J. Synthesis, characterization and properties of ternary copper(II) complexes containing reduced Schiff base N-(2-hdroxybenzyl)-amino acids and 1,10-phenanthroline. Dalton Trans. 2003, 880–890. [Google Scholar] [CrossRef]
- Yang, C.T.; Moubaraki, B.; Murray, K.S.; Ranford, J.D.; Vittal, J.J. Interconversion of a monomer and two coordination polymers of a copper(II)-reduced Schiff base ligand-1,10-phenanthroline complex based on hydrogen- and coordinative-bonding. Inorg. Chem. 2001, 40, 5934–5941. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.A.N.; Nethaji, M.; Chakravarty, A.R. Ternary mononuclear and ferromagnetically coupled dinuclear copper(II) complexes of 1,10-phenanthroline and N-salicylidine-2-methoxyaniline that show supramolecular self-organization. Eur. J. Inorg. Chem. 2003, 2318–2324. [Google Scholar] [CrossRef]
- Matoga, D.; Szklarzewicz, J.; Stadnicka, K.; Shongwe, M.S. Iron(III) complexes with a biologically relevant aroylhydrazone: crystallographic evidence for coordination versatility. Inorg. Chem. 2007, 46, 9042–9044. [Google Scholar] [CrossRef]
- Ali, M.S.; El-Saied, F.A.; Shakdofa, M.M.E.; Karnik, S.; Jaragh-Alhadad, L.A. Synthesis and characterization of thiosemicarbazone metal complexes: crystal structure, and proliferation activity against breast (MCF7) and lung (A549) cancers. J. Mol. Struct. 2023, 1274, 134485. [Google Scholar] [CrossRef]
- Latheef, L.; Manoj, E.; Kurup, M.R.P. Salicylaldehyde 4,4’-(hexane-1,6-diyl)-thiosemicarbazone. Acta Cryst. 2006, C62, o16–o18. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, D.; Mazumdar, S.K.; Banerjee, T.; Ghosh, S.; Mak, T.C.W. Structure of salicylaldehyde thiosemicarbazone. Acta Cryst. 1988, C44, 1025–1028. [Google Scholar] [CrossRef]
- Vrdoljak, V.; Cindrić, M.; Milić, D.; Matcović-Ćalogović, D.; Novak, P.; Kamenar, B. Synthwsis of five new molybdenum(VI) thiosemicarbazonato complexes. Crystal structures of salicylaldehyde and 3-methoxysalicylaldehyde 4-methylthiosemicarbazones and their molybdenum(VI) complexes. Polyhedron 2005, 24, 1717–1726. [Google Scholar] [CrossRef]
- Labisbal, E.; Haslow; K. D.; Sousa-Pedrares, Valdés-Martínez, J.; Hernández-Ortega, S.; West, D.X. Copper(II) and nickel(II) complexes of 5-methyl-2-hydroacetophenone N(4)-substituted thiosemicarbazones. Polyhedron 2003, 22, 2831–2837. [Google Scholar] [CrossRef]
- Kowol, C.R.; Reisner, E.; Chiorescu, I.; Arion, V.B.; galanski, M.; Deubel, D.V.; Keppler, B.K. An electrochemical study of antineoplatic gallium, iron, and ruthenium with redox noninnocent α-N-heterocyclic chalcogensemicarbazones. Inorg. Chem. 2008, 47, 11032–11047. [Google Scholar] [CrossRef]
- West, D.X.; Bain, G.A.; Butcher, R.J.; Jasinski, J.P.; Li, Y.; Pozdniakiv, R.Y.; Valdés-Martínez, J.; Toscano, R.A.; Hernández-Ortega, S. Structural studies of three isomeric forms of heterocyclic N(4)-substituted thiosemicarbazones and two nickel(II) complexes. Polyhedron 1996, 15, 665–674. [Google Scholar] [CrossRef]
- Kowol, C.R.; Eichinger, R.; Jakupec, M.A.; Galanski, M.S.; Arion, V.B.; Keppler, B.K. Effect of metal ion complexation and chalcogen donor identity on the antiproliferative activity of 2-acetylpyridine N,N-dimethyl(chalcogen)semicarbazones. J. Inorg. Biochem. 2007, 101, 1946–1957. [Google Scholar] [CrossRef] [PubMed]
- Usman, A.; Abdul Razak, I. , Chantrapromma, S.; Fun, H.-K.; Philip, V.; Sreekanth, A.; Kurup, M.R.P. Di-2-pyridyl ketone N4,N4-(butane-1,4-diyl)thiosemicarbazone. Acta Cryst. 2002, C58, 0652–o654. [Google Scholar]
- Philip, V.; Suni, V.; Kurup, M.R.P. Di-2-pyridyl ketone 4-methyl-4-phenylthiosemicarbazone. Acta Cryst. 2004, C60, o856–o858. [Google Scholar] [CrossRef] [PubMed]
- Geary, W.J. The use of conductivity measurements in organic solvents for the characterisation of coordination compounds. Coord. Chem. Rev. 1971, 7, 81–122. [Google Scholar] [CrossRef]
- Philip, V.; Manoj, E.; Kurup, M.R.P.; Nethaji, M. [Di-2-pyridyl ketone N4,N4-(butane-1,4-diyl)thiosemicarbazonato-κ3N,N´S]dioxovanadium(V). Acta Cryst. 2005, C61, m488–m490. [Google Scholar]
- Gómez-Saiz, P.; García-Tojal, J.; Mendia, A.; Donnadieu, B.; Lezama, L.; Pizarro, J.L.; Arriortua, M.I.; Rojo, T. Coordination modes in a tridentate NNS (thiosemicarbazonato)copper(II) system containing oxygen-donor coligands – structures of [{Cu(L)(X)}2] (X = formato, propionato, nitrito). Eur. J. Inorg. Chem. 2003, 518–527. [Google Scholar] [CrossRef]
- Shongwe, M.S.; Al-Kharousi, H.N.R.; Adams, H.; Morris, M.J.; Bill, E. Unprecedented [V2O]6+ Core of a centrosymmetric thiosemicarbazonato dimer: spontaneous deoxygenation of oxovanadium(IV). Inorg. Chem. 2006, 45, 1103–1107. [Google Scholar] [CrossRef]
- Addison, A.W.; Rao, T.N.; Reedijk, J.; van Rijn, J. , Verschoor, G.C. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen-sulphur donor ligands: the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2´-yl)-2,6-dithiaheptane]copper(II) perchlorate. J. Chem. Soc., Dalton Trans. 1984, 1349–1356. [Google Scholar] [CrossRef]
- Bernhardt, P.V.; Sharpe, P.C.; Islam, M.; Lovejoy, D.B.; Kalinowski, D.S.; Richardson, D.R. Iron chelators, of the dipyridylketone thiosemicarbazone class: precomplexation, and transmetalation effects on anticancer activity. J. Med. Chem. 2009, 52, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Anderson, O.P. Crystal and molecular structure of tris-(1,10-phenanthroline)copper(II) perchlorate. J. Chem. Soc., Dalton, 1973; 1237–1241. [Google Scholar]
- Burčák,M. ; Potočňák, I.; Baran, P.; Jäger, L. Low-dimensional compounds containing cyano groups. X. (Dicyanamido-κN1)bis(1,10-phenanthroline-κ2N,N´)copper(II) perchlorate. Acta Cryst. 2004, C60, m601–m604. [Google Scholar]
- Yang, L.; Powell, D.R.; Houser, R.P. Structural variation in copper(I) complexes with pyridylmethylamide ligands: structural analysis with a new four-coordinate geometry index, τ4. Dalton Trans., 2007, 955–964.
- Tei, L.; Blake, A.J.; Lippolis, V.; Wilson, C.; Schröder, M. Methanolysis of nitrile-functionalised pendant arm derivatives of 1,4,7-triazacyclononane upon coordination to CuII. Dalton Trans. 2003, 304–310. [Google Scholar] [CrossRef]
- Su, S.-Y.; Liao, S.; Wanner, M.; Fiedler, J.; Zhang, C.; Kang, B.-S.; Kaim, W. The copper(I)/copper(II) transition in complexes with 8-alkylthioquinoline based multidentate ligands. Dalton Trans. 2003, 189–202. [Google Scholar] [CrossRef]
- Tubbs, K.J.; Fuller, A.L.; Bennett, B.; Arif, A.M.; Makowska-Grzyska, M.M.; Berreau, L.M. Evaluation of the influence of a thioether substituent on the solid state and solution properties of N3S-ligated copper(II) complexes. Dalton Trans. 2003, 3111–3116. [Google Scholar] [CrossRef]
- Liu, Y.H.; Li, A.; Shao, J.; Xie, C.-Z.; Song, X.-Q.; Bao, W.-G.; Xu, J.-Y. Four Cu(II) complexes based on antitumour chelators: synthesis, structure, DNA binding/damage, HSA interaction and enhanced cytotoxicity. Dalton Trans. 2016, 45, 8036–8049. [Google Scholar] [CrossRef] [PubMed]
- Ying, P.; Zeng, P.; Lu, J.; Chen, H.; Liao, X.; Yang, N. New oxidovanadium complexes incorporating thiosemicarbazones and 1,10-phenanthroline dervatives as DNA cleavage, potential anticancer agents, and hydroxyl radical scavenger. Chem. Biol. Drug. Des. 2015, 86, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Bruker, SADABS, Bruker Axs Inc., Madison, Wisconsin, USA 2016.
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; D. Stalke, D. J. Appl. Crystallogr., 2015, 48, 3–10. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT - Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71 (Md), 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42(2), 339–341. [Google Scholar] [CrossRef]
Scheme 1.
(a) Illustration of synthetic routes to H2(3,5-t-Bu2)-sal4eT and complexes 1 and 2; (b) comparative structural representation of [Cu2(sal4eT)2(bipy)] [4].
Scheme 1.
(a) Illustration of synthetic routes to H2(3,5-t-Bu2)-sal4eT and complexes 1 and 2; (b) comparative structural representation of [Cu2(sal4eT)2(bipy)] [4].
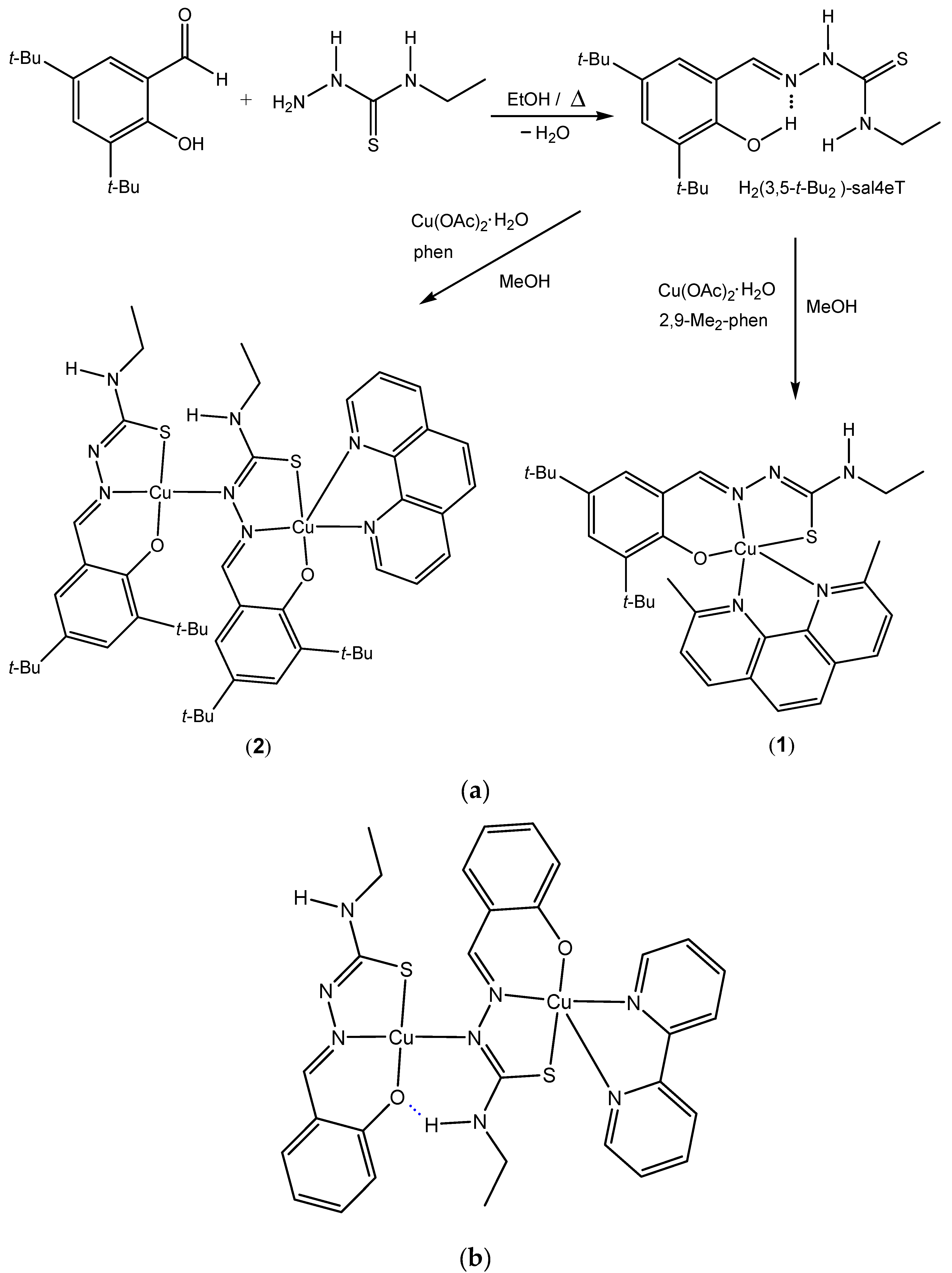
Figure 1.
ESI mass spectrum of H2(3,5-t-Bu2)-sal4eT in the negative mode.
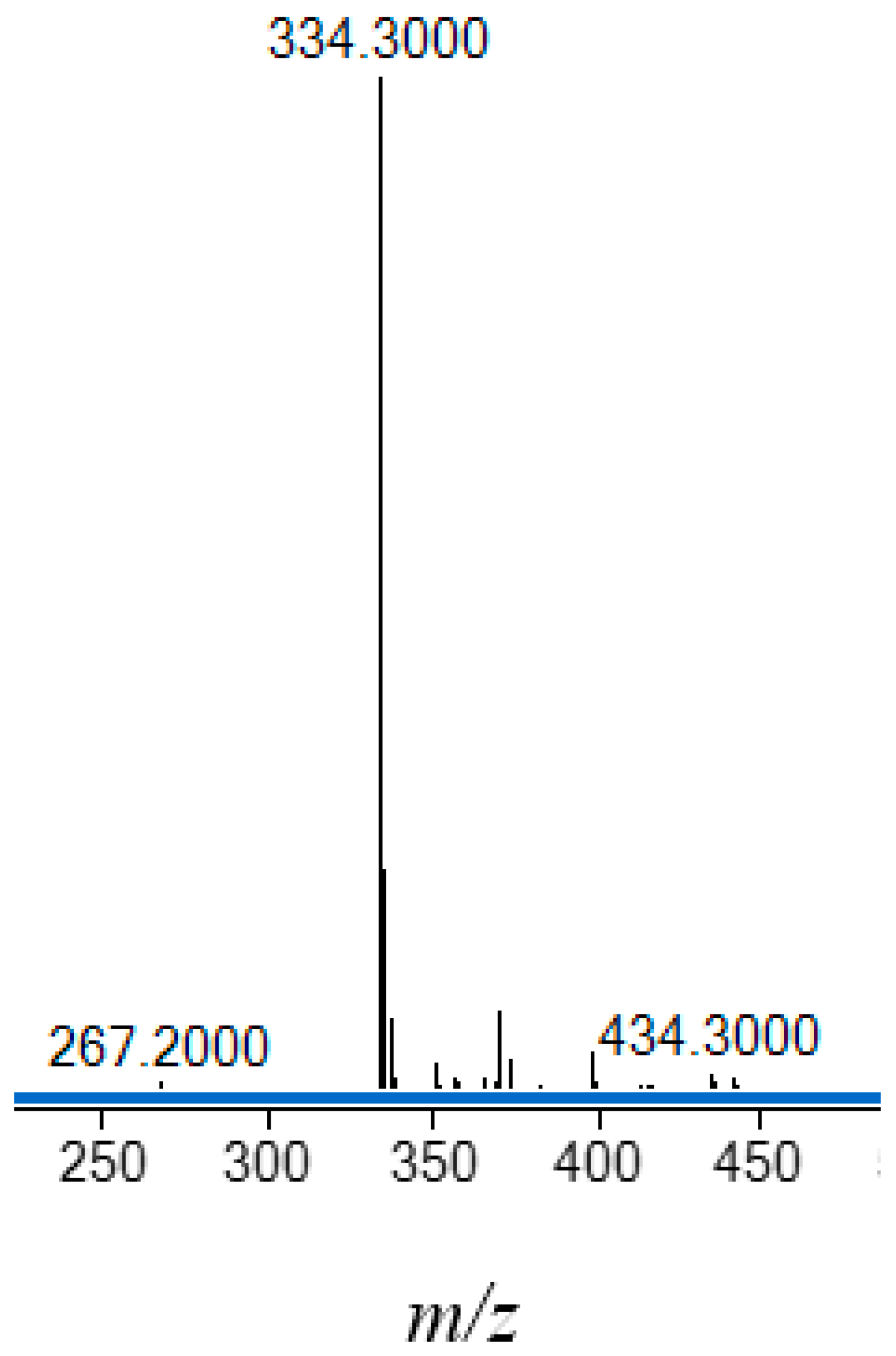
Figure 2.
FT-IR spectra of (a) H2(3,5-t-Bu2)-sal4eT (black line) and (b) [Cu{3,5-t-Bu2)-sal4eT}(2,9-Me2-phen)] (1) (blue line) and [Cu2{3,5-t-Bu2)-sal4eT}2(phen)] (2) (red line).
Figure 2.
FT-IR spectra of (a) H2(3,5-t-Bu2)-sal4eT (black line) and (b) [Cu{3,5-t-Bu2)-sal4eT}(2,9-Me2-phen)] (1) (blue line) and [Cu2{3,5-t-Bu2)-sal4eT}2(phen)] (2) (red line).
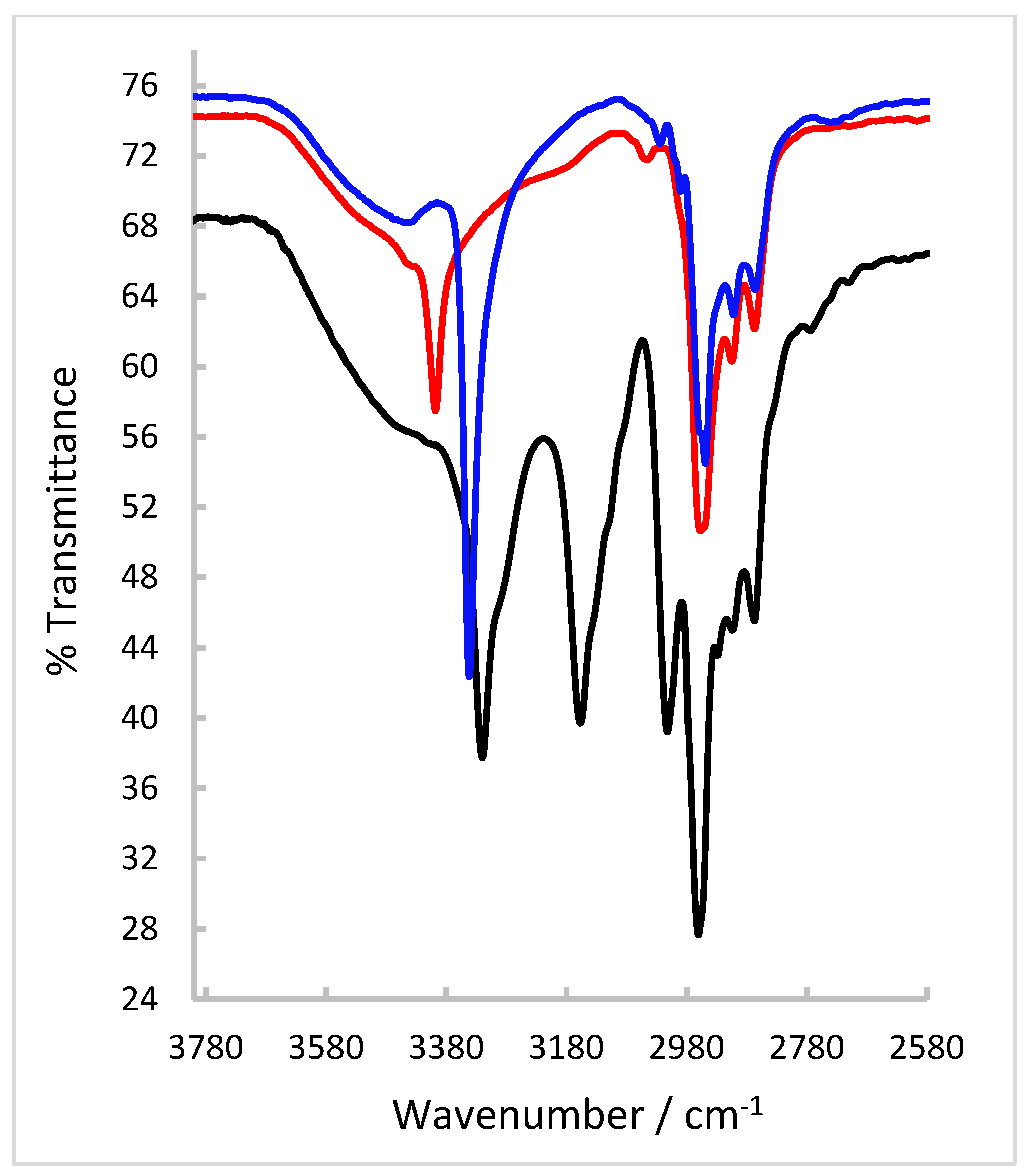
Figure 3.
(a) X-ray crystal structure of H2(3,5-t-Bu2)-sal4eT and (b) illustrations of crystallographically observed different orientations of phenolic thiosemicarbazones.
Figure 3.
(a) X-ray crystal structure of H2(3,5-t-Bu2)-sal4eT and (b) illustrations of crystallographically observed different orientations of phenolic thiosemicarbazones.
Figure 4.
ESI mass spectra of (a) [Cu{(3,5-t-Bu2)-sal4eT}(2,9-Me2-phen)] (1) in the positive-ion mode and (b) [Cu2{(3,5-t-Bu2)-sal4eT}2(phen)] (2) in the negative-ion mode.
Figure 4.
ESI mass spectra of (a) [Cu{(3,5-t-Bu2)-sal4eT}(2,9-Me2-phen)] (1) in the positive-ion mode and (b) [Cu2{(3,5-t-Bu2)-sal4eT}2(phen)] (2) in the negative-ion mode.
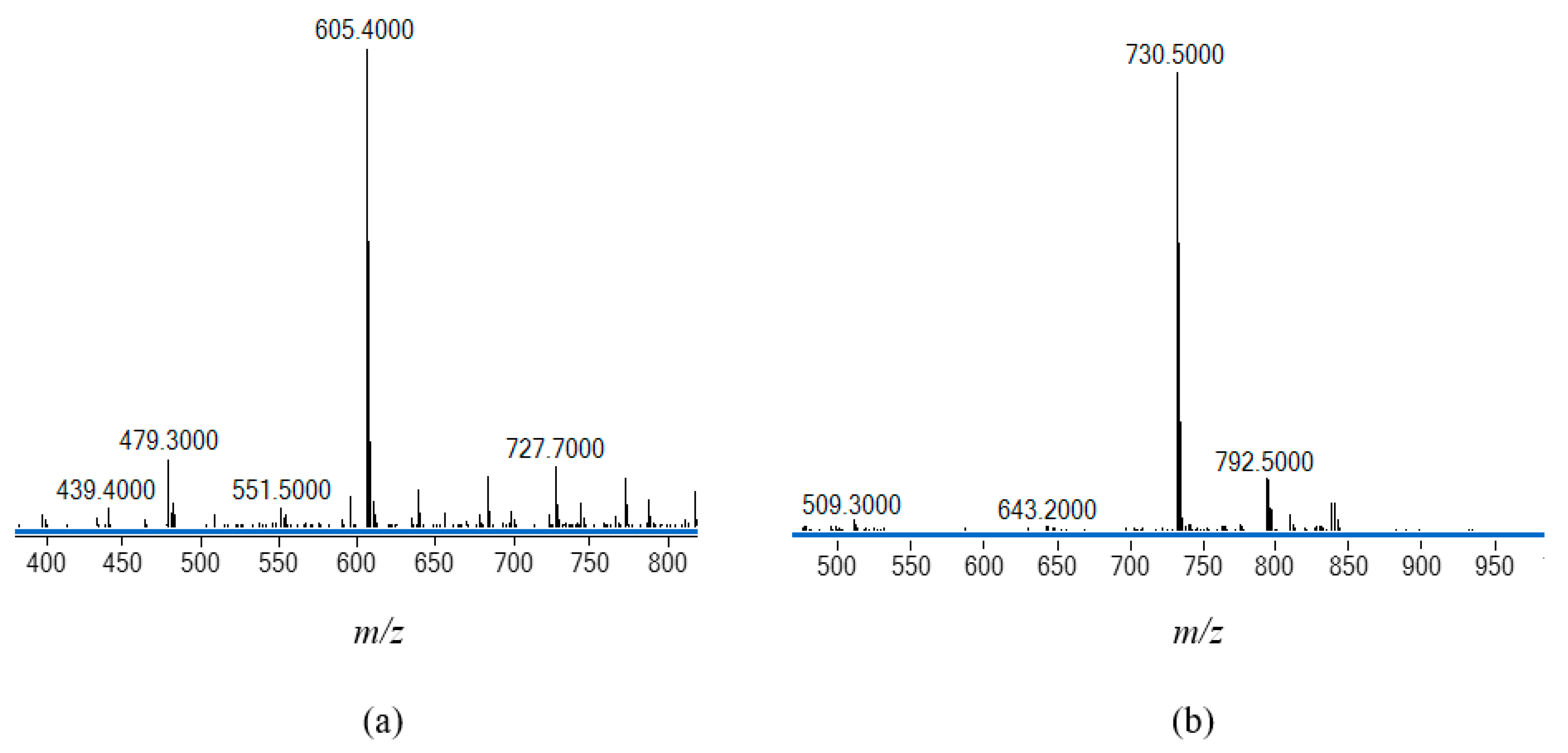
Figure 5.
(a) Molecular structure of [Cu{(3,5-t-Bu2)-sal4eT}(2,9-Me2-phen)] (1) and (b) illustration of dimerization of the mononuclear complex molecules.
Figure 5.
(a) Molecular structure of [Cu{(3,5-t-Bu2)-sal4eT}(2,9-Me2-phen)] (1) and (b) illustration of dimerization of the mononuclear complex molecules.
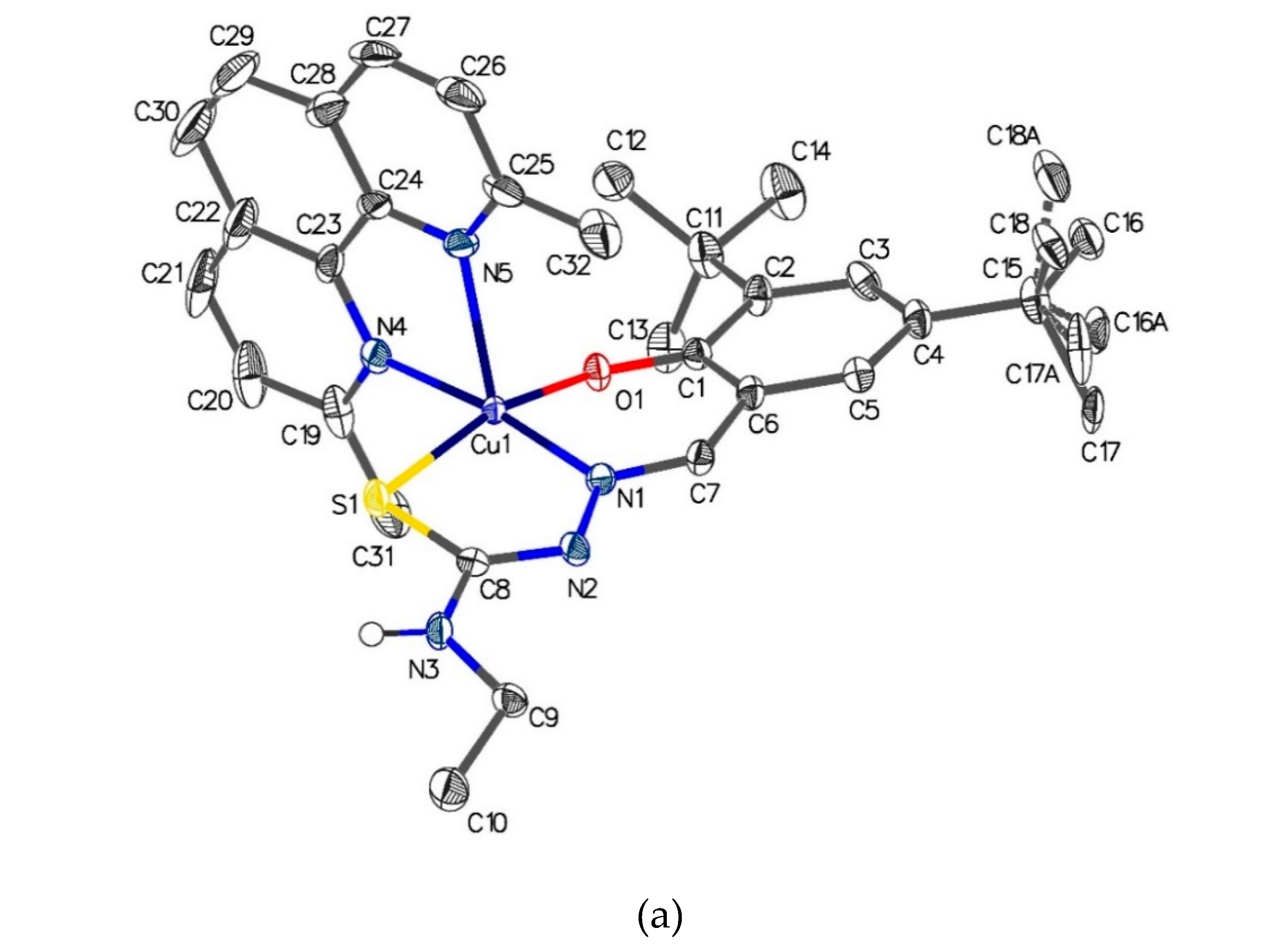
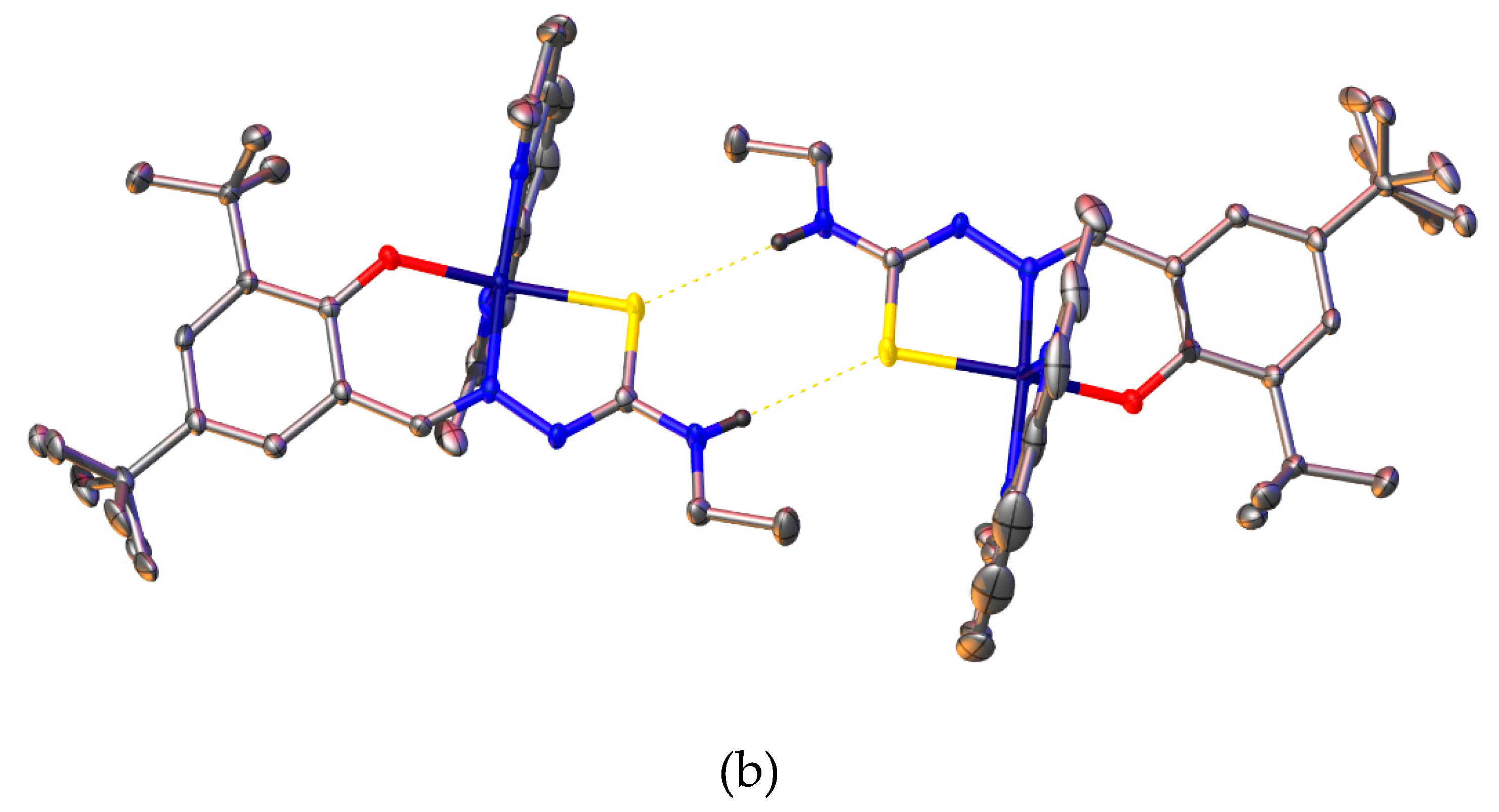
Figure 6.
(a) Molecular structure of [Cu2{(3,5-t-Bu2)-sal4eT}2(phen)] (2) and (b) illustration of dimerization of the dinuclear complex molecules.
Figure 6.
(a) Molecular structure of [Cu2{(3,5-t-Bu2)-sal4eT}2(phen)] (2) and (b) illustration of dimerization of the dinuclear complex molecules.
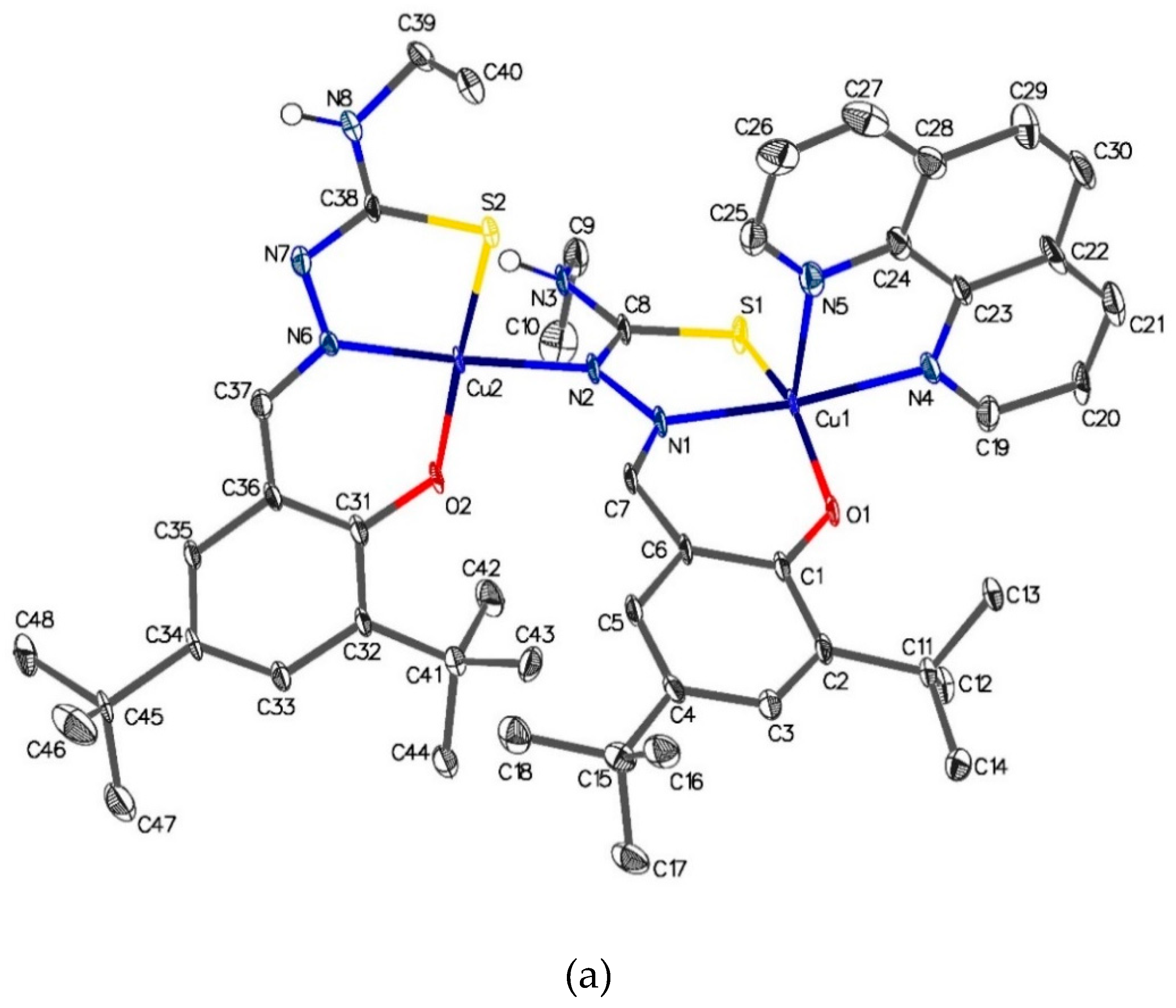
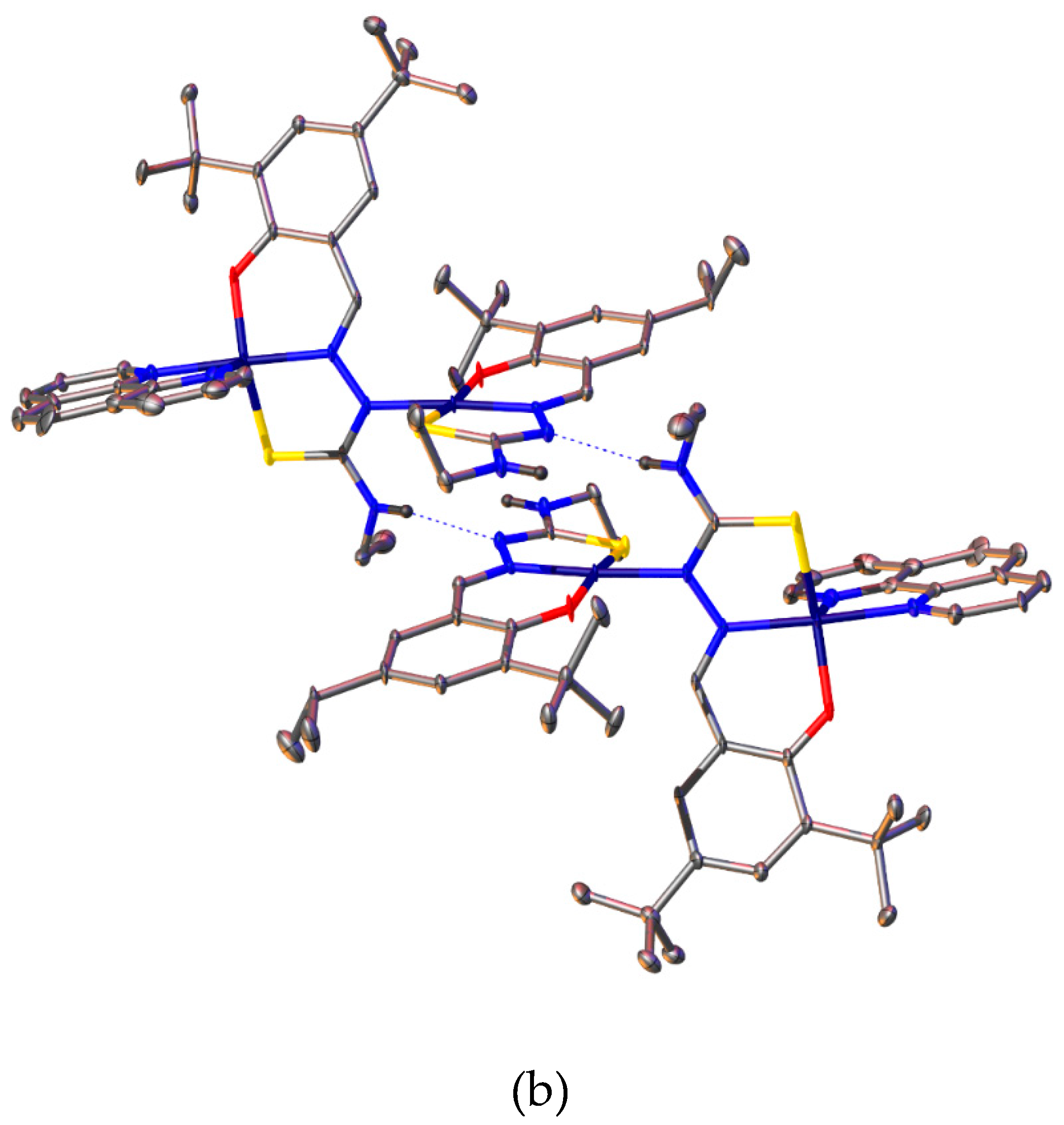
Figure 7.
X-band ESR spectrum of [Cu{(3,5-t-Bu2)-sal4eT}(2,9-Me2-phen)] (1) recorded in frozen MeOH solution at 77 K (ν = 9.3641 GHz).
Figure 7.
X-band ESR spectrum of [Cu{(3,5-t-Bu2)-sal4eT}(2,9-Me2-phen)] (1) recorded in frozen MeOH solution at 77 K (ν = 9.3641 GHz).
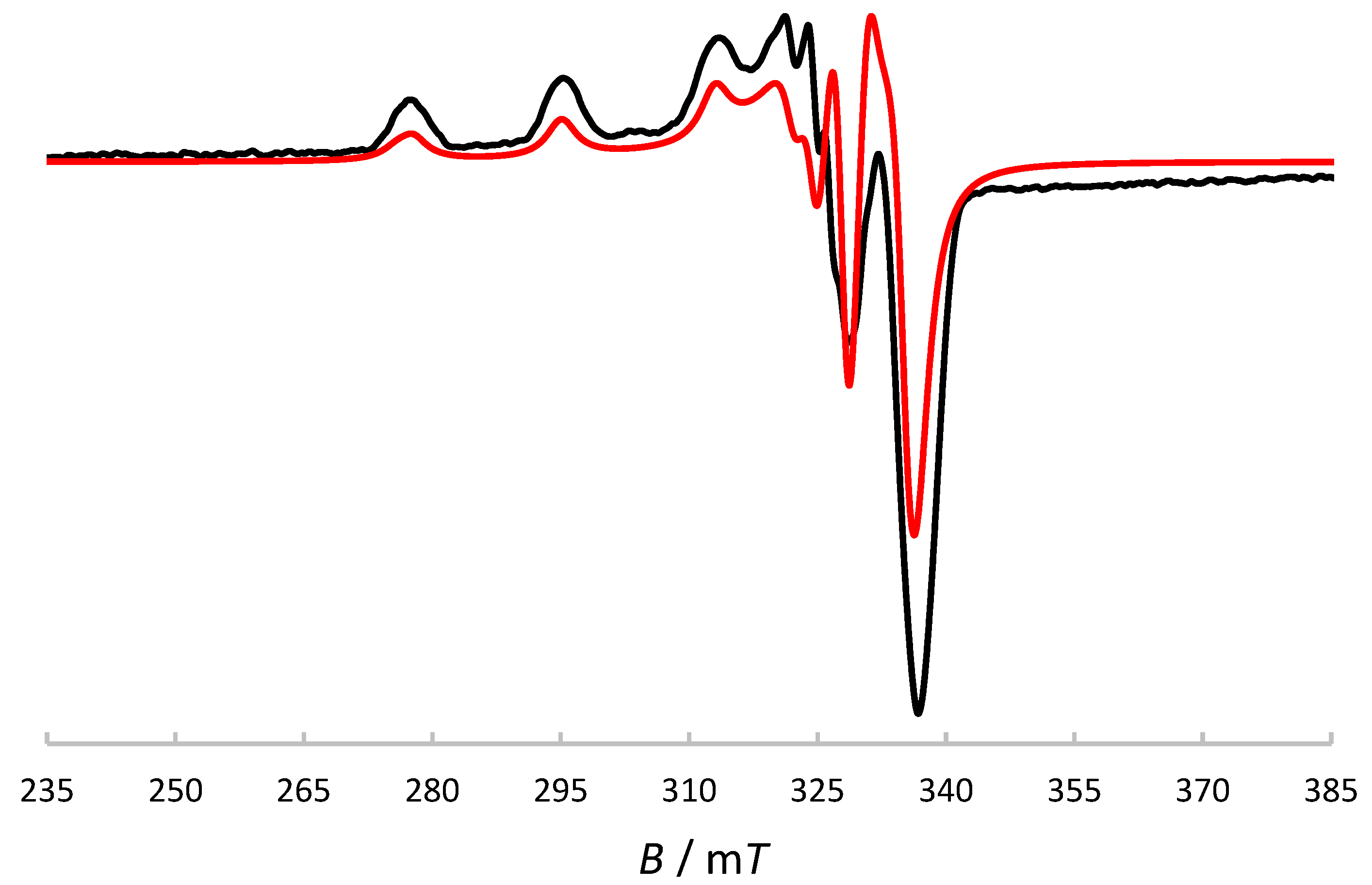
Figure 8.
Electronic absorption spectra of H2(3,5-t-Bu2)-sal4eT, [Cu{(3,5-t-Bu2)-sal4eT}(2,9-Me2-phen)] (1) and [Cu2{(3,5-t-Bu2)-sal4eT}2(phen)] (2) in MeOH.
Figure 8.
Electronic absorption spectra of H2(3,5-t-Bu2)-sal4eT, [Cu{(3,5-t-Bu2)-sal4eT}(2,9-Me2-phen)] (1) and [Cu2{(3,5-t-Bu2)-sal4eT}2(phen)] (2) in MeOH.
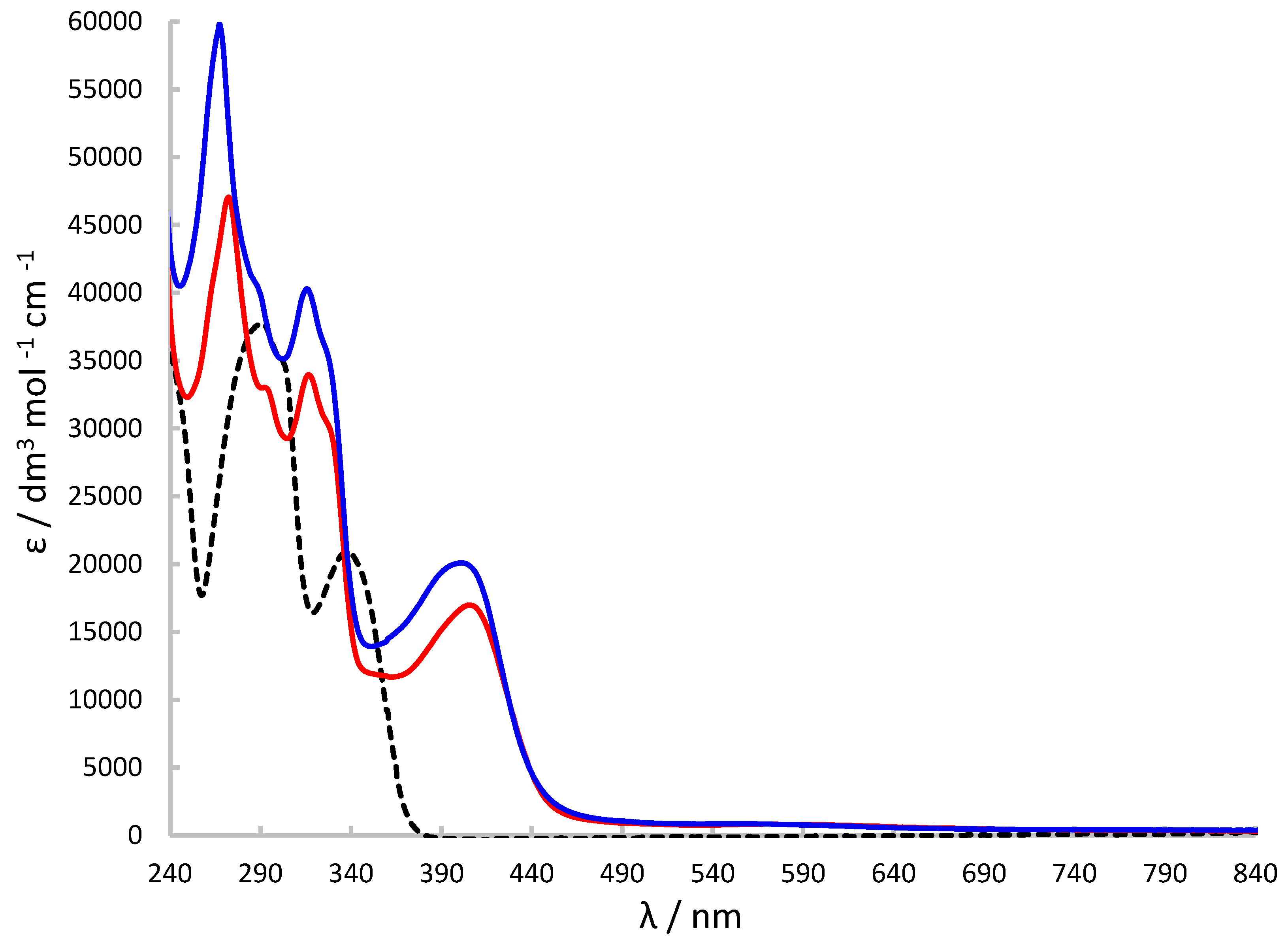
Figure 9.
Cytotoxicity of [Cu{(3,5-t-Bu2)-sal4eT}(2,9-Me2-phen)] (1) towards HeLa cells (a) and [Cu2{(3,5-t-Bu2)-sal4eT}2(phen)] (2) towards MCF-7 cells (b).
Figure 9.
Cytotoxicity of [Cu{(3,5-t-Bu2)-sal4eT}(2,9-Me2-phen)] (1) towards HeLa cells (a) and [Cu2{(3,5-t-Bu2)-sal4eT}2(phen)] (2) towards MCF-7 cells (b).
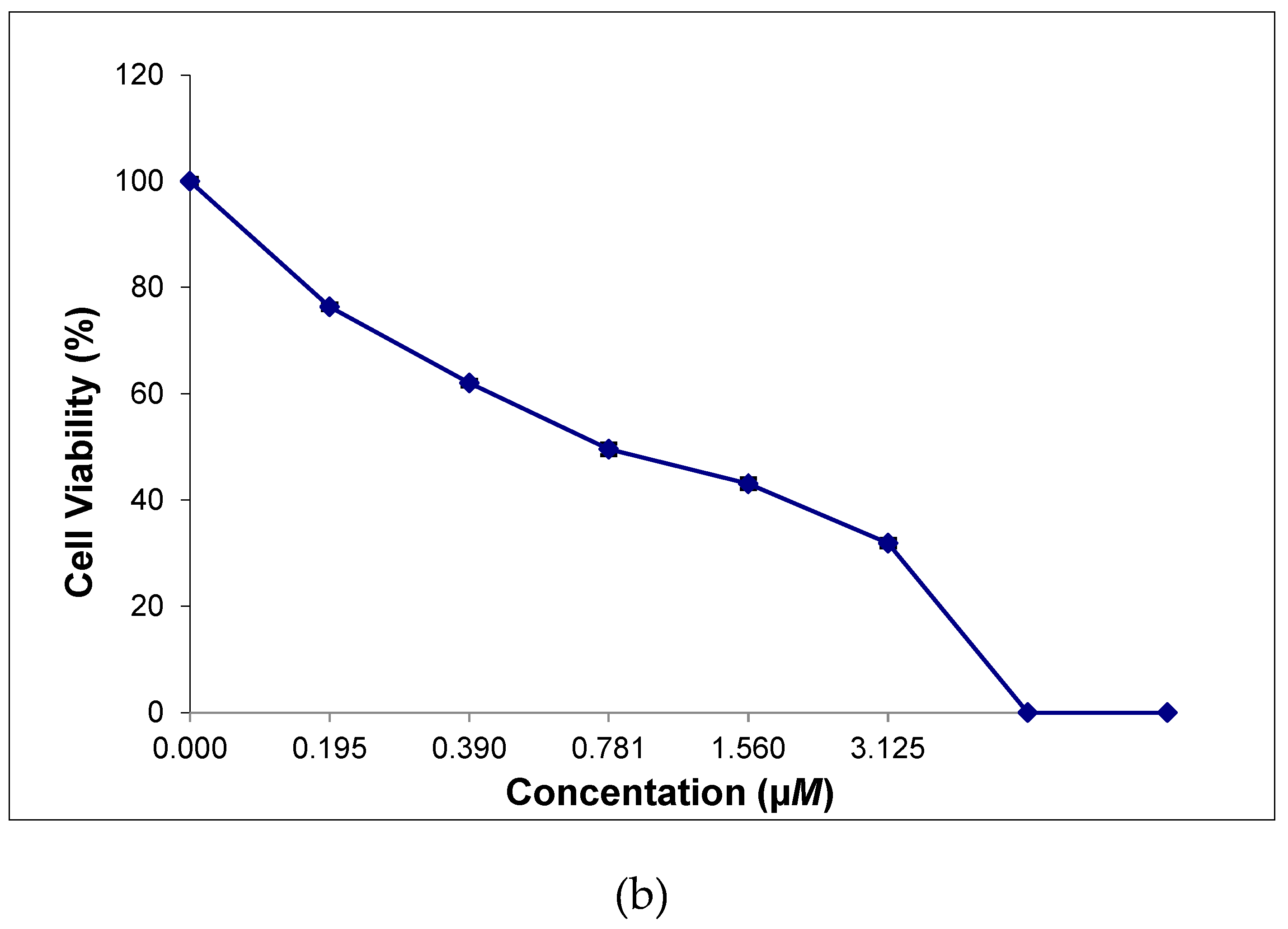

Table 1.
Selected crystallographic data for H2(3,5-t-Bu2)-sal4eT and complexes 1 and 2.
| Compound | H2(3,5-t-Bu2)-sal4eT | 1 | 2 |
|---|---|---|---|
| Chemical formula | C18H29N3OS | C32H39CuN5OS | C48H62Cu2N8O2S2 |
| Molar mass (g mol–1) | 335.50 | 605.28 | 974.25 |
| T (K) | 100 | 99.99 | 100.01 |
| Crystal system | monoclinic | monoclinic | triclinic |
| Space group | P21/c | P21/n | P |
| a (Å) | 18.4754(14) | 8.5924(3) | 11.3898(6) |
| b (Å) | 9.2040(7) | 18.5809(6) | 13.1195(8) |
| c (Å) | 11.5714(9) | 19.4132(6) | 16.6701(10) |
| α (°) | 90 | 90 | 78.980(2) |
| β (°) | 95.517(2) | 100.451(2) | 89.824(2) |
| γ (°) | 90 | 90 | 83.661(3) |
| V (Å3) | 1958.6(3) | 3047.99(17) | 2429.7(2) |
| Z | 4 | 4 | 2 |
| ρcalc (g cm–3) | 1.138 | 1.319 | 1.332 |
| μ (mm–1) | 0.173 | 1.904 | 2.241 |
| F(000) | 728.0 | 1276.0 | 1024.0 |
| Crystal size (mm) | 0.500 × 0.200 × 0.150 | 0.240 × 0.120 × 0.080 | 0.508 × 0.207 × 0.040 |
| Radiation (λ/ Å) | MoKα (λ = 0.71073) | CuKα (λ = 1.54178) | CuKα (λ = 1.54178) |
| 2Θ range (°) | 4.43 – 57.282 | 6.638 – 133.168 | 5.402 – 133.766 |
| Reflections collected | 37828 | 19602 | 30501 |
| Rint | 0.0661 | 0.0918 | 0.0708 |
| GOF on F2 | 1.051 | 1.044 | 1.189 |
| R1, wR2 [I≥2σ (I)] | 0.0403, 0.0909 | 0.0513, 0.1235 | 0.1083, 0.3568 |
| R1, wR2 [all data] | 0.0607, 0.1010 | 0.0749, 0.1378 | 0.1180, 0.3692 |
Table 2.
Selected bond distances (Å) and angles (°) for H2(3,5-t-Bu2)-sal4eT.
| C16-S1 | 1.7029(14) |
| C1-O1 | 1.3605(16) |
| N1-N2 | 1.3854(16) |
| C15-N1 | 1.2904(19) |
| C16-N2 | 1.3499(18) |
| C15-N1-N2 | 113.66(12) |
| N2-C16-N3 | 118.40(13) |
| N2-C16-S1 | 117.87(11) |
| N3-C16-S1 | 123.72(11) |
Table 3.
Selected bond distances (Å) and angles (°) for 1 and 2.
| [Cu{(3,5-t -Bu2)-sal4eT}(2,9-Me2-1,10-phen)] (1) | |||
|---|---|---|---|
| Cu(1)-S(1) | 2.2823(9) | S(1)-C(8) | 1.739(3) |
| Cu(1)-O(1) | 1.934(2) | N(2)-C(8) | 1.313(4) |
| Cu(1)-N(1) | 1.959(3) | N(1)-N(2) | 1.400(4) |
| Cu(1)-N(4) | 2.057(3) | N(1)-C(7) | 1.296(4) |
| Cu(1)-N(5) | 2.308(3) | N(3)-C(8) | 1.357(4) |
| O(1)-Cu(1)-N(1) | 91.55(10) | N(1)-Cu(1)-N(4) | 172.64(11) |
| N(1)-Cu(1)-S(1) | 84.77(8) | N(1)-Cu(1)-N(5) | 109.81(11) |
| O(1)-Cu(1)-N(4) | 90.08(10) | N(4)-Cu(1)-S(1) | 91.76(7) |
| O(1)-Cu(1)-N(5) | 98.14(10) | N(5)-Cu(1)-S(1) | 97.38(7) |
| O(1)-Cu(1)-S(1) | 164.38(8) | N(4)-Cu(1)-N(5) | 77.04(11) |
| [Cu2{(3,5-t-Bu2)-sal4eT}2(phen)] (2) | |||
| Cu(1)-S(1) | 2.266(3) | Cu(2)-S(2) | 2.231(3) |
| Cu(1)-O(1) | 1.908(7) | Cu(2)-O(2) | 1.888(7) |
| Cu(1)-N(1) | 1.952(8) | Cu(2)-N(2) | 2.017(8) |
| Cu(1)-N(4) | 2.039(8) | Cu(2)-N(6) | 1.940(8) |
| Cu(1)-N(5) | 2.276(9) | N(6)-C(37) | 1.310(12) |
| S(1)-C(8) | 1.737(9) | S(2)-C(38) | 1.746(9) |
| N(2)-C(8) | 1.322(13) | N(7)-C(38) | 1.316(12) |
| N(1)-C(7) | 1.301(13) | N(6)-N(7) | 1.391(11) |
| N(1)-N(2) | 1.381(11) | N(8)-C(38) | 1.353(13) |
| O(1)-Cu(1)-N(1) | 93.7(3) | N(5)-Cu(1)-S(1) | 102.9(2) |
| N(1)-Cu(1)-S(1) | 85.2(2) | N(4)-Cu(1)-N(5) | 77.7(3) |
| O(1)-Cu(1)-N(4) | 89.7(3) | O(2)-Cu(2)-N(2) | 88.1(3) |
| O(1)-Cu(1)-N(5) | 104.6(3) | O(2)-Cu(2)-N(6) | 94.2(3) |
| O(1)-Cu(1)-S(1) | 152.4(2) | N(2)-Cu(2)-N(6) | 176.3(3) |
| N(1)-Cu(1)-N(4) | 172.2(4) | S(2)-Cu(2)-N(2) | 91.8(2) |
| N(1)-Cu(1)-N(5) | 94.6(3) | S(2)-Cu(2)-N(6) | 86.3(2) |
| N(4)-Cu(1)-S(1) | 95.1(2) | S(2)-Cu(2)-O(2) | 172.1(3) |
Table 4.
Cytotoxicity evaluation of H(3,5-t-Bu2)-sal4eT, 1 and 2 by MTT assay in cancer cell lines over a 24-hour incubation period.
Table 4.
Cytotoxicity evaluation of H(3,5-t-Bu2)-sal4eT, 1 and 2 by MTT assay in cancer cell lines over a 24-hour incubation period.
| IC50 / µM | ||
|---|---|---|
| Compound | HeLa | MCF-7 |
| H(3,5-t-Bu2)-sal4eT | >100 | >100 |
| [Cu{3,5-t-Bu2)-sal4eT}(2,9-Me2-phen)] (1) | 1.35 ± 0.11 | >100 |
| [Cu2{3,5-t-Bu2)-sal4eT}2(phen)] (2) | >100 | 0.73 ± 0.06 |
| Docetaxel | 60.70 ± 5.13 | 92.54 ± 7.99 |
| Paclitaxel | 15.84±1.28 | >100 |
| Cisplatin | 13.28 ± 3.84 [82] | 13.36 ± 1.25 [83] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



