
Submitted:
21 December 2023
Posted:
22 December 2023
You are already at the latest version
Abstract
In the contest of childhood epilepsies, the concept of continuous spike-waves during slow sleep (CSWS) includes several childhood-onset heterogeneous conditions, sharing an electroencephalogram (EEG) characterized by a high frequency of paroxysmal abnormalities during sleep which are attributed negative effects on the cognitive development and behavior of the child. These negative effects may have the characteristics of a clear regression or of a slowdown in development. Seizures are very often present, but not constantly. The above makes it clear why CSWS have been included in epileptic encephalopathies, in which by definition the frequent EEG paroxysmal abnormalities have an unfavorable impact on the cognitive functions, including socio-communicative skills causing autistic features, even regardless of the presence of clinically overt seizures. Although several decades have passed since the original descriptions of the electroclinical condition of CSWS, there are still many areas that are little known and deserve to be further studied, including the EEG diagnostic criteria, the most effective electrophysiological parameter for monitoring the evolution, the role of the thalamus in the CSWS pathogenesis, the long-term evolution, the nosographic location of Landau-Kleffner syndrome, the standardized neuropsychological and behavioral assessment, the pharmacological and non-pharmacological therapies.
Keywords:
epilepsy
; epileptic encephalopathies
; CSWS
; ESES
; Landau-Kleffner syndrome
1. Introduction
Within the concept of continuous spike-waves during slow sleep (CSWS) we can find several childhood-onset heterogeneous conditions sharing an electroencephalogram (EEG) characterized by a high frequency of paroxysmal abnormalities during sleep that are attributed negative effects on the cognitive development and behavior of the child. These negative effects may have the characteristics of a clear regression or of a slowdown in development. Seizures are very often present, but not constantly. The above makes it clear why CSWS have been included in epileptic encephalopathies, in which by definition the frequent EEG paroxysmal abnomalities have an unfavorable impact on the cognitive functions, including socio-communicative skills causing autistic features [1,2,3,4,5,6], even regardless of the presence of clinically overt seizures. This unfavorable impact has been confirmed by brain functional imaging studies [7]. In other words, in epileptic encephalopathies epileptic activity itself (evidenced by the EEG paroxysmal abnormalities) contributes to the development of serious cognitive and behavioral disorders, regardless of what might be expected due to the underlying pathology (if any) alone [8]. However, there have also been authors who aimed at diminishing the importance of CSWS in the development of cognitive deficits [9]. The first report that pointed out the connection between continuous EEG paroxysmal abnormalities (spike and wave complexes) and the presence of cognitive disorders should be attributed to Kennedy and Hill, who in 1942 described a child affected by “dementia dysrhythmica infantum” [10]. Between the 1970s and 1980s Tassinari et al. described in a very detailed and modern way the electroclinical entity characterized by an EEG with diffuse spike-and-waves lasting for at least 85% of slow sleep, clinically associated with cognitive decline, seizures, and motor impairment [11]. This is the typical form of the “electrical status epilepticus during slow-wave sleep” (ESES) that has been included among the epileptic encephalopathies according to the International Classification of the Epilepsies, due to the marked EEG abnormalities leading to a cognitive impairment, that may cause a severe degree disability [12]. Over the years, it has been found that also when EEG paroxysmal abnormalities recur less frequently than typical ESES, cognitive disturbances can appear, but more heterogeneous and tending to be less severe than typical ESES. According to several authors, the current trend is to consider a spike and wave index (SWI) > 60% as suggestive of CSWS [13]. However, defining features and diagnostic criteria of CSWS and in particular of ESES are still a matter of debate. Further, for a long time the terms CSWS and ESES have been used interchangeably, but currently ESES mainly refers to the EEG pattern, while CSWS refers to the syndromic picture associated with ESES: even on this point, however, there is no unanimity of opinion [14]. In this paper for simplicity we will use these two terms interchangeably. The main purpose of therapy for CSWS should be to prevent or reduce the cognitive deficits [15]. Etiopathogenesis, when known, is heterogeneous. To give an idea, Sonnek et al. [2021] [16] in a large restrospective monocentric study (95 children), including also “near CSWS” patients (SWI = 40-85%), found a structural/metabolic etiology in 43.2% of cases and genetic alterations in 17.9%, while etiology was unknown in 38.9%. Regarding the pathogenetic mechanisms, it has been hypothesized that the negative effects of the EEG continuous paroxysmal abnormalities on cognitive functions and behavior may be related to the spike-induced damage of the synaptic homeostasis processes that occur physiologically in sleep and that are crucial during development. During sleep, the homeostasis is favored by synaptic weakening/elimination following the synaptic strength increase during wakefulness. Alterations in synaptic strength are shown by EEG through alterations of sleep slow wave activity (SWA). During CSWS there are no sleep SWA changes, which occur again after CSWS remission: this finding suggests a spike-related impairment of the synaptic homeostasis during sleep. All of this may impair cortical wiring causing the neuropsychological dysfuncions typical of CSWS [17,18].
The CSWS phenomenon is generally considered as a childhood-onset condition affecting the age of development and which tends to disappear during adolescence [19], but exceptionally may persist into older adulthood [20]. However, cognitive long-term outcome is often poor particularly in cases with a longer CSWS duration [19].
Landau-Kleffner syndrome (LKS), otherwise known as acquired epileptic aphasia, deserves a separate discussion, although there is a current of thought that considers LKS as an electro-clinical framework within ESES [21]. In this nosographic entity, the core symptom is an acquired aphasia associated with continuous EEG paroxysmal abnomalities during sleep localized bilaterally in temporal regions, and not a global cognitive deterioration as in ESES [22]; seizures are not present in all cases. The possibility of a switch from LKS to ESES suggests a rather close link between these two nosographic entities which, however, in our opinion should remain separate.
In this review we will describe the clinic and EEG of typical and atypical ESES, the etiology, the pathogenesis, the pharmacological and non-pharmacological treatments, and the prognosis; we will do the same for LKS.
2. Materials and Methods
We carried out a narrative review based on a paper search using PubMed database (U.S. National Library of Medicine). We utilized two combinations of terms joined by the Boolean operator “AND”: “CSWS” AND “epilepsy”; “ESES” AND “epilepsy”; and also the term “Landau-Kleffner syndrome”. First, we considered all the articles found through these search terms that were published between June 2012 and May 2023. We found 131, 166, and 135 articles, respectively: after we removed the duplicates, the total articles were 326. We read all these papers and excluded those that addressed scantly or not at all the CSWS topic or that were mere comments on other articles. Both authors of this paper contributed to the article selection. Finally, we included 273 papers in this review (Figure 1). We considered also some additional significant references, mentioned in the papers found using PubMed or known in some way by the authors. Both authors read all references included in this review.
3. Results
3.1. EEG and clinics of typical and atypical ESES
The core features of typical ESES, as described by Tassinari et al. [11], are: 1) electrical status epilepticus lasting for ≥ 85% of NREM (slow) sleep, documented by more than 2 EEG recordings during a period of ≥ 1 month; 2) different degrees of global cognitive deterioration, with decrease of Full-Scale IQ and of Performance IQ [23]; 3) behavioral disorders, including attention deficit, hyperactivity, aggressiveness, difficulty in social interaction, and (more rarely) psychosis; also autistic behavior is possible [11,24,25,26]; 4) epileptic seizures, focal or apparently generalized, with a heterogeneous semeiology including: absences; epileptic falls; clonic, tonic-clonic, partial motor, and complex partial seizures; and negative myoclonus (see atonic component for example at the upper limbs, showed by polygraphic recordings or restricted to lower limbs) can be associated in atypical absences [27,28,29]; it is possible that there are no overt clinical seizures [30], but this is a rare occurrence; 5) motor signs, including dyspraxia, ataxia, and dystonia. The described clinical EEG characteristics have substantially remained the same over the years [18]. Overnight video EEG is considered as the gold standard for ESES diagnosis [31] because an ESES pattern may be recorded during the first sleep cycle of the night and may appear fragmented in subsequent ones. But in clinical practice, even an ambulatory EEG can be effective, according to several authors. For example, Nagyova et al. studied retrospectively 199 consecutive pediatric patients who underwent an ambulatory EEG. Suspected ESES was the motivation in 38.6% of cases and this examination was useful in 97.5% of cases [32].
In the atypical forms of ESES, there is a great heterogeneity in the reported clinical features depending on the frequency and localization of the EEG paroxysmal abnormalities [33]. In fact, the localization of prevailing paroxysmal abnormalities is more heterogeneous than in the typical forms: focal, multifocal, unilateral, asymmetric or symmetric bilateral, and diffuse paroxysmal abnormalities have been reported [34]. Figure 2 shows an example of atypical ESES.
Among the neuropsychiatric clinical features in atypical ESES, ADHD-like symptoms are prominent and seem to be related positively to SWI [35]. Acquired visual agnosia may be found in CSWS, as suggested by the case reported by Van Iterson et al., a male aged almost 7 years presenting CSWS (SWI = 100%), which prevailed in the left posterior regions, who lost the skill to recognize familiar faces as well as to name pictures [36]. Kuki et al. reported acquired Kanji (morphograms) dysgraphia associated with visual processing impairment in two children with CSWS prevailing in the occipito-temporal region, in which functional neuroimaging showed a dysfunction located in the left posterior temporal lobe [37]. According to Tassinari et al., also in individual cases, an adequate neurophysiological study aiming at localizing the “functional lesion”, associated with a detailed neuropsychological assessment, could help to analyze the cortical networks responsible for specific cognitive functions [38]. However, not all authors agree on the existence of a clear correlation between EEG picture and neuropsychological impairment, such as Pavlidis et al. who studied 24 children with idiopathic encephalopathy related to ESES (three had LKS). They found a lack of correlation between severity/type of cognitive impairment and SWI and/or topography of EEG paroxysmal abnormalities, suggesting the possible role of further factors during sleep (including an altered sleep homeostasis) in the neurobehavioral disorders [39]. It should not be forgotten that any structural brain abnormalities, if they are not the primary cause of the CSWS picture, can still influence its characteristics. For example, Mohammadi et al. described one child with hemi-ESES (that is, limited to one cerebral hemisphere) related to congenital corpus callosum absence inhibiting the CSWS propagation from the right hemisphere to the left one. Their findings point out the focal origin of ESES as well as the corpus callosum role in bilateral synchronous expression of EEG paroxysmal abnormalities [40]. On the other hand, the 85% SWI limit that separates typical forms of ESES from atypical ones, may be of relative importance. Caraballo et al. in a multicenter, retrospective long-term follow-up study including 117 CSWS cases found similar electroclinical features comparing patients with a > 85% SWI and patients with a < 85% SWI [41]. Gencpinar et al. analyzed retrospectively the EEG recording of 44 ESES/CSWS cases with a follow-up of at least two years in order to study: (a) the SWI during NREM sleep EEG: > 85% in 33 cases (typical ESES) and = 50-85% in 11 (atypical ESES); and (b) the maximum amplitude area of CSWS: anterior in 33 cases and posterior in 11. The authors found that the SWI rate (typical versus atypical ESES) as well as the maximum amplitude area of CSWS (anterior versus posterior) were not significantly related to clinical and brain imaging features or to the response to treatment (see seizure control and SWI reduction) [42]. Particularly in atypical forms of ESES, the technique called magnetoencephalography, utilizing superconducting quantum interference devices, can be useful in precisely localizing the spike source [43]. Still today, epileptic encephalopathy related to ESES is underdiagnosed and this, delaying the initiation of an effective drug therapy, could have negative repercussions on cognitive evolution since the duration of ESES is probably the most important prognostic factor (see later) [44]. The diagnosis of CSWS, on the electrophysiological level, is therefore based on qualitative and quantitative criteria whose detection in the past was entrusted solely to the human eye. Over the years, increasingly precise and reliable automatic systems have been developed for the detection and quantification of spike-waves. These systems are very useful in CSWS, where EEG paroxysmal abnormalities are particularly frequent and their classic visual quantification can be very tiring [45]. Yu et al. proposed a SWI quantification neural network (SQNN) using a pre-labeling algorithm in order to quantify SWI automatically in children with ESES. The authors found that it would be possible to quantify accurately and reliably SWI through the SQNN with a processing speed 100 times faster than that of experts [46]. However, to quantify the number of EEG paroxysmal abnormalities, the SWI computing is not the only method: in fact, also the number of spikes per unit of time can be calculated. It should be borne in mind that “time occupied by epileptiform activities” (i.e. SWI) and the “number of spikes per unit of time”, otherwise known as “spike wave frequency” (SWF), differ susbstantially. Using the SWF, we can avoid the ceiling effect that is intrinsic to SWI calculation, especially when EEG paroxysmal abnormalities are more than 60 per minute, because SWI cannot exceed the limit of 100% while SWF has not a defined upper limit. Unfortunately, it is unclear whether the SF or the SWI is more related to the ESES clinical features [47,48]. In any case, the SWI is still today the most used method to quantify the number of EEG paroxysmal abnormalities. Not using the same method does not help with data sharing among CSWS researchers. Apart from this last important aspect, in general the lack in literature of a shared terminology concerning CSWS can be an obstacle to the reciprocal communication among clinicians and researchers dealing with this condition [49]. Still regarding the diagnosis on the electrophysiological level, Carvalho et al. showed that a wearable EEG device with only 2 bipolar channels for 24 hours, through a semiautomatic template match spike search, could be a good alternative, less expensive and better tolerated, to the full 10-20 long-term ambulatory EEG that is the classic tool for interictal spike quantification. In a clinical context, this alternative could be useful particularly for the spike activity follow-up [50].
CSWS syndrome can begin as such, but very often it is the electroclinical evolution of another, more or less severe, epilepsy. For example, it can follow Ohtahara syndrome, a severe early-onset form of epileptic encephalopathy [51], but the most frequent occurrence is CSWS as the evolution of Rolandic epilepsy otherwise known as benign epilepsy with centro-temporal spikes (BECTS) [33] (see also later). This is very important to keep in mind, as it shows us the possibility of changing from one apparently benign form of epilepsy to another with a less favorable prognosis. Further, CSWS may be the atypical electroclinical evolution of Panayiotopoulos syndrome, as suggested by two cases reported by Oguni et al. [52]. The recurrence of CSWS has been described also in a rare epileptic condition, the sunflower epilepsy, that is a photosensitive and usually drug-resistant reflex epilepsy with seizures characterized by head turning towards light, similarly to a sunflower, and hand waving in front of the eyes [53]. Neuropsychiatric outcome is often disappointing [47,54,55]. Margari et al. carried out a long-term follow-up study in 25 CSWS children. At the onset of CSWS, 96% of the cases showed one or more neuropsychiatric disorders, including behavioral problems in 54%, intellectual disability in 37.5%, learning disorders in 33%, developmental coordination disorder in 17%, language disorder in 12.5%, and autism spectrum disorder in 8%. In the course of the follow-up, neuropsychiatric disorders persisted unaltered in 52% of the cases, worsened in 24%, and improved in 24%. Most of the cases without improvement in the follow-up had symptomatic CSWS [56].
How can CSWS interfere with cognitive functions? We know that spikes showed by EEG correspond topographically to focal cortical areas of hypermetabolism according to the results of the positron emission tomography (PET) [57]: however, these metabolic alterations are not sufficient to answer our question. In healthy individuals, the physiologic overnight decrease of the slow wave slope in NREM sleep is closely related to the sleep recovery function that is necessary for optimal cognitive performance. Starting from this assumption, Bölsterli Heinzle et al. analyzed retrospectively 14 CSWS patients, identifying the spike wave “focus” as the area of highest spike amplitude and the frequency of spike waves as the SWI. They found no overnight change of the slow wave slope in the “focus”, while in “nonfocal” areas the slow wave slope decreased significantly. Spike wave density (see: SWI) was correlated with the overnight slope decrease impairment: the higher the SWI, the more impaired the slope decrease. The authors concluded proposing that the overnight slope decrease impairment is a possible mechanism leading to CSWS neuropsychological deficits [58]. In another study, Bölsterli et al. carried out a retrospective analysis of overnight EEG in 10 children with idiopathic ESES. Across the night, the slope of slow waves did not diminish significantly during ESES, especially at the focus of paroxysmal abnormalities, while after ESES remission the slope diminished significantly. Compared to healthy controls, cases after ESES remission showed no significant difference in overnight slope decrease. The best cognitive outcome after ESES remission has been found in three cases who had some degree of slope decrease during ESES. These findings suggest that alterations of NREM-sleep slow waves induced by ESES are reversible, and the severity of cognitive impairment might be related to the severity of slow wave impairment during ESES. Therefore, slow wave analysis might be a prognostic factor for cognitive outcome [59]. The longitudinal course of the slow wave slope overnight change could be an objective EEG marker related to the cognitive function course [60]. Through a retrospective cross-sectional study considering 22 CSWS cases, Bölsterli Heinzle et al. found that the maximal spike wave location, corresponding to the epileptic focus, was age-related and followed a posterior-anterior trajectory, similarly to various focal epilepsies. Therefore, younger cases should be more likely to show posterior foci than older ones. The authors hypothesized that this posterior-anterior trajectory of CSWS maximal spike waves could be related to maturational modifications of maximal expression of sleep slow waves [61]. Moreover, EEG paroxysmal abnormalities during NREM sleep causing both an activation in epileptogenic zones (particularly in perisylvian and in prefrontal areas) and a deactivation of default mode network (DMN) located beyond the epileptogenic area may produce neuropsychological impairments [62]. Halász and Szűcs proposed for perisylvian network epilepsies, including idiopathic focal childhood epilepsies, ESES, and LKS, that a sleep-related alteration of physiological neural networks may underlie epileptogenesis. Homeostatic plasticity, which is a compensative process for cell loss recovery, may be associated with an excitability increase up to an epileptic level. In this way, physiological functioning derails to a pathological (epileptic) working mode. NREM sleep heightens epileptic processes, which in turn impair sleep functions. The vicious circle between sleep and epilepsy works every night, altering brain functions, particularly during the development. Type and degree of cognitive impairment should be related to the involved network’s function [63]. Ng and Hodges found that in the comparison with patients with other types of epilepsy, children with recently diagnosed ESES turned out less impaired on the neurocognitive level than children with generalized epilepsy. This finding suggests that neuropsychological differences between ESES and other epileptic syndromes may develop as a long-term consequence of the neurological disorder and/or of pharmacological treatment [64]. However, within generalized epilepsies there can be very heterogeneous situations in terms of severity and clinical characteristics, so much so as to make it difficult to interpret the data obtained by these authors [65]. Table 1 summarizes clinical and EEG features of typical and atypical ESES.
3.2. Predictive factors of the evolution into CSWS
Considering the relevant impact of CSWS on cognitive neurodevelopment, it is very important to recognize, if possible, what the predictive factors of an evolution into CSWS are. Rolandic epilepsy is the prototype of benign epilepsies in childhood, but in some cases it may have an atypical development including CSWS. A retrospective study by Pesántez-Ríos et al. comprised 9 patients with BECTS, all of whom showed atypical clinical features and CSWS. The average age at onset of Rolandic seizures was 5 years, while clinical and EEG deterioration occurred, on average, one and a half years later. Electroclinical features suggesting an atypical development BECTS were: early onset of seizures; appearance of new seizures with increased frequency; and the presence of EEG fronto-centrotemporal focus, with increasing frequency, both in wakefulness and in sleep [66]. Porat Rein et al. carried out a retrospective cohort study involving 104 cases with BECTS. They identified the following risk factors for the development in these patients of severe atypical variants including LKS and ESES: EEG spike-waves; EEG without evidence of left lateralization; and centro-temporal, frontal, or fronto-temporal EEG localization [67]. Subsequently, Porat Rein et al. developed a predictive model of transformation of BECTS into epileptic encephalopathy with CSWS or LKS collecting data from a cohort of 91 BECTS cases, of which 18 showed an encephalopathic transformation. The most important risk factors were: localization of EEG paroxysmal abnormalities on fronto-temporal and temporo-parietal areas, and seizure semiology including dysarthria or somatosensory auras [68]. But other studies were not limited to investigating the possible evolution into CSWS of BECTS alone. Desprairies et al. carried out a retrospective case-control study to search for any clinical or EEG feature indicative of later CSWS development at the time of the first seizure. They included 10 CSWS cases with available EEG at the time of the first seizure compared with 10 matched controls. They found no clinical or EEG features suggesting later CSWS development. However, during the follow-up, the occurrence of multiple types of seizures and seizure worsening were significantly more frequent in the CSWS cases [69]. According to Caraballo et al., clinical parameters predicting an evolution into CSWS were: early-onset seizures, appearance of new seizures, and a relevant increase in seizure frequency, while EEG parameters predicting an evolution into CSWS may be: increased frequency of the interictal EEG paroxysmal abnormalities during wakefulness and sleep, and the presence of bilateral spike-and-waves [70]. Aeby et al. through a retrospective study compared the awake EEG of 15 CSWS patients (including 1 with LKS: see later) and of 15 matched cases with self-limited focal epilepsy (SFE). They found that, at the time of cognitive regression, CSWS patients were more likely than SFE cases to have a slow-wave index (SLWI) > 6%, a SWI > 10%, a cluster of spike-waves (CLSW) of ≥ 1 second, a SWF of > 11%, and an EEG score of ≥ 3 (qualitative assessment: from grade 0 = normal to grade 4 = maximally pathological) [71]. Table 2 summarizes the clinical and EEG predictive factors of the evolution into CSWS.
3.3. Electrophysiology and functional neuroimaging
Classic quantification of EEG paroxysmal abnormalities through SWI is still useful and widely used today, but it has long had alternatives that have advantages over it (see paragraph 3.1). Furthermore, spike index semiautomatic quantification in CSWS is available today as a reliable alternative to the classic quantification based on visual scoring [72]. Peltola et al., searching and averaging spikes in pre- and postoperative EEG recordings of 10 CSWS cases who underwent surgical treatment, found that calculating an integrated root mean square using multiple scalp electrodes during steady NREM sleep might provide a reliable parameter for the spike strength evaluation in CSWS [73]. Ouyang et al., assuming that to explain the transition of brain activity to ESES is a challenge, used an S-estimator method (see the evaluation of regression coefficient) for analyzing multichannel EEG data in 11 ESES cases. The results confirmed the presence of a significant spike and global synchronization increase from wake to sleep. Further, global synchronization was strongly correlated with spikes [74]. Balaram et al. studied the overnight EEGs of 30 ESES children (SWI: ≥ 50%) through the analysis of spikes utilizing spatio-temporal mapping and electrical source analysis, coming to question literature misconceptions. They subdivided “generalised” (80%) ESES and focal (20%) ESES patterns. According to Balaram et al., the bisynchronous ESES subtype included in the “generalised” pattern (found in 10 cases) was due to the apparently synchronous bilateral spike bilateral activation with a tangential/oblique dipole (spikes were localized around the peri-Rolandic cortex). They considered the classic description of “diffuse” ESES spikes prevailing in the bilateral frontal areas as a misinterpretation related to the use of the 10-20 EEG system. Utilizing voltage mapping and source analysis, they identified an activation in the Rolandic cortex. The authors classified focal ESES as parietal, occipital, and temporo-occipital patterns, while (note!) they did not find a frontal ESES pattern [75]. De Tiège et al. studied the neurophysiological correlate of regional cerebral glucose metabolism in 6 children with CSWS (three with LKS and three with atypical Rolandic epilepsy) through a technique associating time-sensitive magnetic source imaging and positron emission tomography (PET) using fluorodeoxyglucose. In all cases, spike-wave onsets were associated with focal hypermetabolism, while the PA propagation to other regions was associated with focal hypermetabolism (five cases), hypometabolism (one case) or the absence of significant metabolic change (one case). Probably, most of the hypometabolic areas were related to a remote inhibition mechanism [76]. Japaridze et al. studied in 15 CSWS patients the neuronal networks underlying background EEG oscillations of this epileptic encephalopathy using two types of EEG analyses: dynamic imaging of coherent sources and renormalized partial directed coherence. Regardless of epilepsy etiology, background EEG pattern in CSWS patients was associated with a very complex network of coherent sources involving many cortical areas, thalamus, and cerebellum. Within this great network, medial parietal cortex, precuneus, and thalamus play the role of central hubs that drive the information flow to other areas, in particular to the temporal and frontal cortex. This hierarchical network organization has ceased after successful treatment and therefore seems to be CSWS specific [77]. Magara et al. using magnetoencephalography (MEG) in 16 non-lesional CSWS children found dipole clusters located on heterogeneous cortical areas: right Rolandic area (4 cases), right supramarginal gyrus (3 cases), left Rolandic area (2 cases), left supramarginal gyrus (2 cases), bilateral Rolandic area (3 cases), multiple anatomical areas (2 cases). Earlier age at onset of epilepsy had the strongest negative prognostic effect on the intellectual level. In 12 cases, long-term prognosis of the intellectual level was related to the fact that it was assessed at the time of CSWS diagnosis [78]. Li et al. using magnetoencephalography assessed magnetic source activity in 55 children: 14 with self-limited epilepsy with centro-temporal spikes (SeLECTS) and SWI ≥ 50%; 21 with SeLECTS and SWI < 50%; and 20 healthy controls. More severe cognitive impairment was present in cases with SeLECTS and SWI ≥ 50% compared to those with SeLECTS and SWI < 50% and to controls. Comparing the three groups, the authors found results suggesting that deactivation of magnetic source activity located in the posterior cingulate cortex and in the medial frontal cortex could lead to the cognitive impairment in SeLECTS [79]. Moeller et al. studied neuronal networks in 10 children with atypical benign partial epilepsy and SWI = 10-70% who underwent simultaneous EEG - functional magnetic resonance imaging (fMRI) recording. Several types of EEG paroxysmal abnormalities were recorded in all 10 children. The authors studied the individual paroxysmal abnormalities-associated blood oxygen level-dependent (BOLD) signal changes in a single subject analysis for each paroxysmal abnormality type. They found focal BOLD signal changes that were concordant with the spike field (patterns similar to BECTS) as well as distant cortical and subcortical BOLD signal changes (similar to the patterns detected in CSWS). This finding suggests that childhood idiopathic focal epilepsies constitute a spectrum of overlapping conditions. Further, the group analysis showed a thalamic activation (see later regarding the hypothesis of a pathogenetic role of the thalamus) [80]. According to the review of Siniatchkin and Van Bogaert, in the last decades fMRI have contributed much to the understanding of the pathogenetic mechanisms underlying CSWS. MEG and EEG source reconstruction showed sources of pathological brain activity associated with paroxysmal abnormalities in the perisylvian region. PET showed hypermetabolism in perisylvian, superior temporal, inferior parietal, and in central cortex areas, which were related to paroxysmal abnormalities. The diffuse hypometabolism found in regions belonging to the DMN (see prefrontal and posterior cingulate cortices, parahippocampal gyrus and precuneus) could be due to a remote inhibition following epileptic activity. The aforementioned metabolic changes disappeared after CSWS recovery. EEG-fMRI technique showed characteristic findings of epileptic encephalopathy: positive changes of blood-oxygen-level-dependent (BOLD) signal were found in the perisylvian regions, prefrontal cortex, anterior cingulate, and thalamus, while negative changes of BOLD signal were found in the DMN regions. The activation pattern represents a diffusion of epileptic activity, which is not dependent on etiology and associated seizure type [81].
CSWS-associated high-frequency oscillations (HFOs) are hypothesized to be related to alterations in higher brain functions. Toda et al. studied the mechanisms generating HFOs in CSWS by analyzing the effects of diazepam (DZP) intravenous (IV) injection in three cases with CSWS previously treated with IV DZP. They performed time-frequency power spectral analysis on the spikes and compared peak power and frequency of HFOs before and after IV DZP. The recovery of the HFOs peak power levels lagged behind the recovery of the spike amplitudes. Also, within 25’ after IV DZP the HFOs power levels were lower than the baseline levels. The authors found no consistent modifications of the HFOs spectral frequencies. They hypothesized that dissociation in terms of recovery after IV DZP between spike amplitudes and HFOs power might be due to different pathophysiological mechanisms generating spikes and HFOs [82]. Ohuchi et al., through a semi-automatic tool, examined ripple-band (80-200 Hz) HFOs in the EEG of 94 children with various epilepsies characterized by spikes activated by sleep, including CSWS. They found in the initial EEG that the median ratio of ripples per spike was significantly higher in the idiopathic CSWS cases than in those with BECTS, Panayiotopoulos syndrome, other focal epilepsies, and also focal spikes without clinical seizures. The authors concluded hypothesizing a close relation between the ripples’ dense generation and the CSWS pathophysiology [83]. HFOs could be an electrophysiological parameter useful from a clinical perspective: according to Cao et al., the persistence of interictal scalp EEG HFOs after steroid therapy in CSWS children can predict seizure relapse and an unfavorable cognitive outcome [84]. According to Gong et al., HFOs prevalence might somehow reflect epileptic activity. In fact, they found that CSWS cases with HFOs (n = 12) tended to show more often epileptic negative myoclonus, atonies, myoclonus, and atypical absences than cases (n = 9) without HFOs [85]. Further, HFOs might be considered as markers of seizure-onset zone from a surgery perspective and at the same time they seem to be related to functional disruption of brain networks in CSWS [86].
In general, while NREM sleep facilitates the spread and propagation of paroxysmal EEG abnormalities, REM sleep has a suppressive effect on them. The inhibitory effect on paroxysmal abnormalities is mainly due to the phasic REM (PREM) microstate with respect to the tonic REM (TREM) microstate. All this also applies to ESES, as demonstrated by the study by Giacomini et al. involving eight patients with ESES. Finding out the reason for the protective effect of PREM sleep on paroxysmal abnormalities could be very important for the development of new therapeutic approaches [87].
Auditory discrimination is very important for language development and for school learning. In a retrospective study including 14 children with typical BECTS and 9 children with atypical BECTS, Filippini et al. showed that CSWS impairs central auditory discrimination, assessed through mismatch negativity (central evoked potential considered as the most sensitive auditory sensory accuracy marker), as well as may produce long-lasting negative effects on academic skills [88].
Table 3 summarizes the electrophysiology and functional neuroimaging findings in CSWS.
3.4. Pharmacological and non-pharmacological therapies
Little evidence guides ESES treatment as in literature almost exclusively retrospective studies and case reports can be found concerning this topic [89]. Treatment options for ESES are basically represented by: antiepileptic drugs, steroids (corticosteroids and ACTH), immunoglobulins, ketogenic diet, and (in selected cases) surgery [90]. In these conditions, conventional antiseizure drugs (ASDs) prove to be ineffective in most cases and in particular corticosteroid treatment, also combined with antiepileptic drugs [91], has caught on widely [44,54,92,93,94], leading to cognitive improvements [95]. There is no full agreement on the best treatments to be administered [96]. The drug resistance of this condition can lead to the use of several ASDs at the same time, but this represents a further risk factor as the use of polytherapy appears to be related to cognitive deterioration [97].
Indeed, sometimes, unpredictable worsening of the clinical picture can occur, as demonstrated by the case described by Samanta et al., who presented an absence status following oral intake of clobazam [98]. Among the various ASDs, one that has shown to be more effective on ESES (with favorable effects on seizures and with behavioral and cognitive improvement) is undoubtedly sulthiame (STM) as an add-on treatment [99,100,101]. Unfortunately, the frequent and severe adverse reactions of this drug (including ataxia, headache, anorexia, behavioral disorders, gastrointestinal disorders, metabolic acidosis, and so on) have greatly limited its use. The choice of how to treat epilepsy with CSWS should be considered on a case-by-case basis. According to Kanemura et al., in CSWS epilepsy, characterized by secondary bilateral synchrony (SBS), the suppression of this EEG phenomenon might be necessary to avoid the progressive neuropsychological dysfunction. The authors studied levetiracetam (LEV) efficacy on SBS, seizure frequency, and neuropsychological deficits in 11 children affected by drug-resistant epilepsy with SBS including CSWS. Eight cases (72.7%) were responders for SBS. All these 8 cases were responders also for clinical seizures and 7 of these 8 cases showed reduced hyperactivity and impulsivity [102]. Not only the high frequency of drug resistance in CSWS should be taken into account, but also the possibility of a relapse after a remission phase. Yuan et al. analyzed retrospectively 8 CSWS children (follow-up: 6 months to 4 years), 5 of which showed brain lesions. After treatment with valproate (8 cases), clonazepam (8 cases), lamotrigine (4 cases), and hormone (8 cases: continuous pulse intravenous methylprednisolone for 3 days, followed by oral prednisone) for 3 months, 7 cases showed a significant clinical and EEG improvement. Unfortunately, at 6 months 2 cases relapsed [103]. Chen et al. analyzed retrospectively LEV efficacy in the treatment of 71 ESES patients. In 35 out of 50 cases (70%) showing seizures there was a > 50% decrease in seizure frequency. In the first 3-4 months of LEV treatment there was a favorable response on EEG in 32 out of 71 (45%) cases, with EEG normalization in 5. Relapse occurred in 8 out of 32 (25%) initial EEG responders. At the last follow-up, 47 cases (66%) still showed ESES and only 13 cases (18%) regained their baseline function level. The authors concluded that the LEV efficacy on EEG and on neuropsychological level was limited on the whole [104]. Zhang performed a meta-analysis about the effects of hormones associated with ASDs versus ASDs alone in children with encephalopathy related to ESES. The study included 403 cases treated with hormones plus ASDs and 402 cases treated with only ASDs. Zhang found that the association of hormones and ASDs shows better effects than only ASDs as regards seizures, EEG, and cognition, and is relatively safe; this meta-analysis, which took into consideration a large population of patients, suggests that treatment with common ASDs is often ineffective in CSWS [105]. Fine et al., in a retrospective study, found in six children (four with ESES and two with LKS) that acetazolamide (AZM), an antiseizure drug with pharmacologic properties similar to sulthiame, may be effective in CSWS. After AZM, three cases out of six showed resolution of CSWS, while all cases showed decreased clinical seizures as well as subjective improvement in communication abilities and in school performance. In five cases out of six, a subjective improvement in hyperactivity and in deficit of attention was reported [106]. Fatema et al. found favorable effects of midazolam drip in eight out of 10 patients with CSWS [107]. According to Su et al. in 15 children with BECTS and ESES, LEV combined with short-term clonazepam can be effective both on EEG and on seizures, with few side effects [108]. But in this study, as in most of the others we are mentioning here, data relating to the long-term effects of drug therapy are lacking. And unfortunately, as suggested by the study of Wiwattanadittakul et al. involving 33 children, the likelihood of a relapse of ESES for both steroids and benzodiazepines is high [109]. Also for a relatively new ASD such as topiramate (TPM) relapse rates in CSWS are high: Vrielynck et al. retrospectively analyzed the results of TPM in 21 children with CSWS. At 3 months after TPM introduction, sleep EEG was improved in 14 cases and normalized in 4: 16 out of these 18 patients showed cognitive or behavioural improvement. One year after TPM introduction, 20 cases were still on TPM and 10 showed persistent EEG improvement, most of them with clinical benefits, while relapse occurred in nearly half of patients [110].
Prospective studies about pharmacological therapy of CSWS are very lacking: one exception is the paper of Chen et al., who carried out a prospective study about continuous oral dexamethasone effects in 15 CSWS cases that were refractory (not responding to several ASDs and prednisolone). Based on clinical and EEG evaluations, 7 out of 15 cases were considered responders. No serious or life-threatening side effects were reported [111].
In recent years, new methods of administering steroid therapy have been developed. Bast et al. analyzed through a retrospective study the effects of methylprednisolone pulse therapy in a sample including also CSWS or LKS cases. The treatment included 4 pulses with oral or intravenous methylprednisolone administered every week during 3 consecutive days. After this phase, the intervals between the pulses increased. After 4 pulses, according to clinical and EEG criteria, 11 of 15 (73%) patients with CSWS or LKS were responders. Adverse effects were mild and transient [112]. Hempel et al. studied the pulse-dose prednisone effects on language and behavior in 17 ESES children. An improvement was found in most cases (10 out of 17: around 59%) in either language or behavior, with a higher likelihood of low IQ cases to show improvements [113]. Jauhari et al. performed a retrospective study about children with neurobehavioral deterioration showing EEG paroxysmal abnormalities during sleep. Cases were classified as: 1) ESES, when SWI during sleep was ≥ 50%; or 2) sleep-induced epileptiform activity (SIEA) when SWI during sleep was ≥ 25% and <50%. Outcome was assessed at a follow-up of 3 months. Eighteen children were included: 7 with SIEA and 11 with ESES. All patients were administered intravenous-methylprednisolone pulse and after oral steroids for 8 weeks. In SIEA cases median SWI was 40%, with paroxysmal abnormalities prevailing in anterior regions, while in ESES cases median SWI was 80%. SIEA and ESES patients presented a similar neurobehavioral profile. Post-steroid behavior scores improved in 6/8 cases with ESES and in 5/7 with SIEA. Median SWI improved in both groups: respectively to < 5% in SIEA and to 45% in ESES. IQ/social quotient mildly improved both in SIEA and in ESES groups. The authors concluded that SWI > 50% during sleep should not be considered as a limiting factor for steroid therapy [114]. Meng and Dai analyzed retrospectively the effects of pulse methylprednisolone for three courses in 56 children with drug-resistant ESES. Each course lasted 3 days and was followed by oral prednisone for 3 days. Pulse methylprednisolone showed a global response rate of 73% on seizures and of 70% on EEG SWI. Significant improvements were found in verbal performance, and full IQ as well as in learning and behavior. Unfortunately, after 1 year the overall recurrence rate was high, i.e. 29% [115]. Chen et al. evaluated the effects of methylprednisolone in 82 children with electrical status epilepticus during sleep: 49 with BECTS variants, 27 with epilepsy with CSWS, and 6 with LKS. All patients received three courses, each including intravenous methylprednisolone for 3 days, followed by oral prednisone for 4 days. After three courses, prednisone was taken for 6 months. On EEG total effective rate was 83%: respectively 82% for BECTS variants, 81% for CSWS, and 100% for LKS. Seizures improved rapidly in all 3 groups. Unfortunately, after 1-year follow-up the recurrence rates were quite high: 47% for BECTS variants, 59% for CSWS, and 50% for LKS [116]. Studies comparing the effect of various drug classes on CSWS offer interesting insights. Through a retrospective USA multicenter study, Baumer et al. evaluated in 81 children the clinical and EEG effects of current CSWS treatments, classified as follows: benzodiazepines (BZDs), steroids, other ASDs, and other therapies. The most frequently used treatments as first line therapy were BZDs and ASDs (respectively 62% and 27%). Patients showed significantly greater odds of clinical improvement using BZDs or steroids than using ASDs and significantly greater odds of EEG improvement with steroids than with ASDs. The authors concluded suggesting an erlier use of BZDs and steroids in these patients [117]. Kılıç et al. studied retrospectively 33 ESES children with a follow-up of at least one year. At first access, 90% of cases had seizures, while 10% showed only school failure. Heterogeneous etiologies were detected: asphyxia (6 cases), hydrocephalus (2), polymicrogyria (1), and mesial temporal sclerosis (1). All children received at least two ASDs except one who received only one. BZDs proved to be the most effective medications. At the end of the follow-up, 72.7% of patients were seizure free, while 57.5% of patients showed complete SWI recovery during NREM sleep. However, the authors did not consider patients receiving steroids [118]. Good effects of steroids suggest a pathogenetic role of inflammation, but prolonged treatments with corticosteroids may lead to undesirable side effects, so that alternative therapies targeting inflammation are necessary. Jyonouchi and Geng described a patient with ESES successfuly treated using a combination of immunomodulating agents regardless of oral corticosteroids that have been discontinued due to side effects. The patient showed 30%, 50%, and 100% reduction in the ESES with the sequential addition of 3 immune-modulating agents selected on the basis of monocyte cytokine profiles: respectively anakinra, intravenous immunoglobulin, and sirolimus. Also speech and behavior gradually improved [119]. Ville et al. performed a monocenter retrospective study about 42 patients with steroid-resistant or -dependent epileptic encephalopathies, including 13 with CSWS, in whom they associated oral steroids with the ketogenic diet (KD). For at least 6 months, 8/13 cases with CSWS responded to the KD addition, which allowed the discontinuation of steroids in the responders. Cases with steroid-dependent CSWS seemed to be the best candidates for KD used in combination with steroids [120]. The KD, already used for a long time with often satisfactory results in epileptic encephalopathies, may theoretically be considered as a treatment for ESES due to its influence on the GABA systems and its inflammation-reducing action. Results of the KD in ESES were heterogeneous: 38 children were reported in 6 papers. Overall, 53% cases showed EEG improvement but only in 9% EEG normalized, 41% cases had > 50% seizure decrease, and 45% showed cognitive improvement [121,122]. KD can be effective also in cases with structural etiology [123]. What is the best approach when treating CSWS? According to Kotagal, CSWS requires prompt diagnosis and aggressive therapy. Close follow-up including overnight EEG is useful to evaluate treatment effects. Cases who do not respond to BZDs at high doses and/or to valproate should receive prednisone for 3 months. For cases who do not respond to steroids or who show steroid dependence, IV immunoglobulin could be another option. Drug-resistant cases should be assessed for a possible surgery treatment (focal resection). KD as well as vagus nerve stimulation are other possible therapeutic options [124]. However, the choice of when to start treating CSWS is not always easy. A suggestion regarding this comes from Uliel-Sibony and Kramer, who carried out a retrospective study including 17 children with BECTS, SWI >30% (mean: 60%), and ADHD/attention deficit disorder. They found data suggesting that the most important parameter leading to the use of steroids or high-dose diazepam is a neuropsychological evaluation showing cognitive deterioration [125]. This underlines once again the importance of carrying out serial neuropsychological and neurobehavioral assessments in patients with CSWS.
The lack of effective pharmacological therapies that are at the same time free of severe side effects has led to research into the effects of innovative drugs on CSWS. Wilson et al. studied retrospectively the effects of amantadine in 20 patients with refractory ESES. The use of this drug was suggested by some reports of amantadine efficacy in refractory absences. Treatment median duration was 11.5 months; it was generally well-tolerated. Median post-amantadine SWI during slow-wave sleep (53%) was significantly lower than baseline SWI (76%). Six (30%) cases showed ESES resolution. Most cases showed subjective cognitive, linguistic, or behavioral improvement [126]. Kanemura et al. studied the effects of perampanel (PER) on EEG secondary bilateral synchrony (SBS) in adolescents with epilepsy that was resistant to ASDs including levetiracetam. They considered as responders for seizures and EEG those who showed a ≥ 50% decrease respectively from the baseline seizure frequency and SBS on EEG. All four CSWS cases included in the study were responders for seizures, while three of the four CSWS cases were responders for SBS on EEG [127]. More recently, Yu et al. studied retrospectively the effects of PER add-on treatment in 54 children with focal epilepsy and ESES. After 6 months of this treatment, ESES resolved in 29 cases (53.7%). A longer ESES duration increased the risk of PER failure [128]. Li et al. in a restrospective study about PER treatment in the epilepsy of children found an effective rate of 72.73% on seizures in BECTS combined with ESES, but the effects on the EEG are not specified [129]. Grosso et al. studied the effects of lacosamide add-on therapy in eight children with drug-resistant CSWS. A 24h EEG was performed every 6 months in all patients. Neuropsychological assessment was performed before lacosamide introduction and after at least 12 months of therapy. After 6 month of therapy, six out of eight cases were considered as responders, one case as partial responder and one case as non-responder. In three out of eight cases EEG normalized. After at least 12 months of therapy, five out of eight cases were considered as responders, while neuropsychological assessment showed a slight improvement in two out of eight cases [130].
The studies mentioned mostly consider numerically limited patient series. There would be a need for studies on larger populations. In this regard we cite the paper by van den Munckhof et al.: their pooled analysis of literature data, considering 575 cases, suggested a higher efficacy (i.e. improvement on cognition or on EEG) of steroids (81%) and neurosurgery (90%) versus ASDs (49%) and BZDs (68%) [131]. Sonnek et al. in their large restrospective monocentric study (95 children), including also “near CSWS” patients (SWI = 40-85%), found that CSWS was drug-resistant in over 70% of cases and that the most effective treatments were steroids and neurosurgery [16]. In a systematic review about the effects of pharmacological interventions in children with CSWS syndrome, Moresco et al. found no complete studies to include. They concluded that there was no evidence to support or refute the pharmacological treatment use for CSWS syndrome and therefore well-designed randomized controlled trials are necessary [132]. RESCUE ESES* is a European multicenter randomized controlled trial, which is still ongoing, aimed at comparing the effects of corticosteroids (monthly IV methylprednisolone pulses or daily oral prednisolone) versus oral clobazam on cognitive functioning in epileptic encephalopathy with ESES. The study also aims at examining whether treatment response can be predicted by blood levels of inflammatory mediators and autoantibodies [133].
Regarding specifically the neurosurgical treatment of CSWS, it should be borne in mind that, contrary to what was once thought, the presence of generalized seizures in ESES is not an absolute contraindication for hemispherectomy [134]. Gröppel et al. compared 11 children with ESES before surgery with 21 age-matched controls with epilepsy but without ESES. They calculated language quotients before and after surgery. Before surgery severe developmental delay prevailed significantly in the ESES group (n=9) compared with controls. In the first group after surgery ESES remitted promptly in 10 cases out of 11, and a significant improvement of language was found. While children with ESES had significantly worse preoperative language skills than those without ESES, at the last follow-up after surgery, no significant differences were found compared to controls [135]. Wang et al. carried out a retrospective study about 11 children with ESES who underwent resective (seven cases) or disconnective (four cases) neurosurgery. Etiologies, when known, included cortical malformations, encephalomalacia and gliosis, porencephaly, and Rasmussen’s encephalitis. Before surgery, all cases showed developmental delay, nine out of 11 cases had motor deficits, seven out of 11 showed language delay, and three out of 11 had visual field defects. After neurosurgery, nine out of 11 children showed reduced seizure frequency, eight out of 11 showed neuropsychological improvement, and nine out of 11 had ESES resolution. Children with poor outcomes after surgery showed, in addition to ESES, more severe preoperative comorbidities [136]. Marashly et al. studied 14 carefully selected ESES cases who underwent resective neurosurgery for epilepsy and ESES; 12 with magnetic resonance imaging (MRI) suggestive of perinatal suffering and two with normal MRI. In ten out of 14 cases surgical procedure was hemispherotomy, while in the other cases was: temporo-parieto-occipital disconnection, frontal lobectomy, parieto-occipital resection, and limited corticectomy. The authors found that resective neurosurgery was effective in most patients leading to long-term seizure freedom, ESES resolution, and cognitive/behavioral functioning stabilization [137]. It should also be kept in mind that also the presence of a genetic etiology nowadays no longer represents an absolute contraindication to a neurosurgical treatment [138]. Alawadhi et al. reported five patients who showed CSWS resolution after surgical resection of an inciting focal lesion. Remarkably, in three cases out of five MRI showed no visible epileptogenic structural abnormalities of the brain. The authors concluded that in drug-resistant CSWS cases an epilepsy presurgical workup should be considered [139]. Yokosako et al. studied retrospectively three cases with drug-resistant ESES without structural abnormalities who underwent corpus callosotomy. After surgery, one patient showed complete ESES resolution and an IQ improvement, while in the other two cases EEG paroxysmal abnormalities were lateralized to one hemisphere and SWI decreased with moderate development and seizure improvement. In these last two cases from 6 months after surgery SWI increased again, but without developmental regression [140]. Jeong et al. analyzed retrospectively the outcomes in 9 children with drug-resistant ESES and a unilateral structural lesion, treated with functional hemispherotomy. After neurosurgery, ESES was finished in all 6 cases with available postoperative EEG during sleep. Developmental regression stopped after hemispherotomy, even if no patient returned to the pre-ESES baseline [141]. The case report described by Carosella et al. suggests that also vagus nerve stimulation could be considered in some cases: a 12-year-old girl with drug-resistant CSWS who, after the implantation of the vagus nerve stimulator, stayed seizure free for more than 1 year, had the resolution of CSWS EEG pattern, and showed significant cognitive improvement [142]. Faria et al., based on promising preliminary results in two cases with drug-resistant CSWS, suggested that transcranial direct current stimulation (tDCS), using stimulation with 1 mA, could be efficacious and well tolerated in CSWS. But this therapeutic approach did not develop in the following years [143]. As demonstrated by the case described by Jellinek et al., ESES can be clinically associated with very complex symptoms consisting of intellectual disability, autism spectrum disorder, and ADHD, and it should never be forgotten that these situations cannot be treated solely with drugs, but also using a behavioral intervention. In this boy the authors used, via telemedicine, a structured parent training program, obtaining a decrease of challenging behaviors and an improvement in adaptive skills [144].
Table 4 summarizes the pharmacological and non-pharmacological effective treatments for CSWS.
3.5. Evolution and prognostic factors
In general, the evolution of CSWS is often unsatisfactory, with a complete recovery on the cognitive-behavioral level after the cessation of the CSWS which is observed only in a minority of cases [6,39]. Yilmaz et al. studied 14 ESES patients followed up for at least 2 years. During ESES 12 patients showed a cognitive impairment involving also school performance. After ESES, seven cases had intellectual disability of heterogeneous severity. At the end of follow-up, globally 12 cases were seizure free, of which seven were still taking antiseizure treatment, while two cases still showed seizures despite antiseizure therapy; ESES was stopped in all cases and EEG was completely normal in seven patients [145].
As regards the prognostic factors, long CSWS duration and the presence of a cerebral lesion seem to be the most important factors related to an unfavorable outcome for cognition. In cases with normal brain MRI, clinical phenotype at onset seems to be very important for cognitive outcome: when the presentation is that of an atypical BECTS, the patients usually will show no major cognitive deficits [6]. According to van den Munckhof et al., who included 575 cases in their pooled analysis of literature data, a normal development before ESES onset, as well as normal neuroimaging findings may be predictors of improved outcome concerning cognition and/or EEG [131]. Research regarding long-term follow-up of this condition is not very rich. Pera et al. studied the long-term cognitive outcome in 25 CSWS children (mean follow-up: 13.5 years). Seven cases (28%) with nonlesional epilepsy showed a positive outcome; three cases (12%) had persisting motor deficit without cognitive impairment; and seven patients (28%) with a long-lasting CSWS (mean: 28.1 months) showed a negative cognitive outcome. Cognitive outcomes did not change in six cases with structural or metabolic pathologies preceding the CSWS onset; and finally two patients (8%) showed a negative outcome regardless of the CSWS duration or presence of other neurologic disorders preceding the CSWS onset. The authors concluded that CSWS syndrome long-term outcome seems to be influenced by treatment response, CSWS duration, and underlying etiology [146]. Hegyi et al. carried out a long-term follow-up (average time: 7.5 years) retrospective study including 33 ESES children (15 non-lesional versus 18 lesional), with bilateral discharges lasting ≥ 75% of NREM sleep. Seizure type variability, seizure frequency, and status epilepticus frequency were higher in lesional cases. Cognitive function impairment was more severe in the lesional cases [147]. Maltoni et al., in a retrospective study including 61 CSWS patients with a SWF during sleep ≥ 25/min., found that SWF was inversely correlated with full and performance IQ during CSWS. Further, longer-lasting SWF ≥ 25/min was related to worse verbal IQ and performance IQ results after CSWS disappearance. But also other variables could play a role in the neuropsychological outcome, including earlier age at SWF ≥ 25/min first recording, perinatal distress, pathologic neurologic examination, and drug resistance to ASDs [148]. De Giorgis et al. in a retrospective study including 16 children with idiopathic CSWS, found that an intellectual impairment was significantly higher in children with a CSWS early-onset (before 6 years) and lower in those with a CSWS later onset (from 8 years onwards) [149].
According to Caraballo et al., in non-lesional cases, cognitive recovery after the CSWS end depends on the initial regression severity and duration. CSWS duration appears as the most important prognostic factor of cognitive outcome. However, etiology plays a main role in the outcome [70]. Escobar Fernández et al., in their retrospective study including 25 children, found that CSWS remission was longer when CSWS onset was later. Cognitive and behavioral outcome was poorer in “secondary” cases (that is with abnormal neuroimaging or psychomotor delay) and in those with earlier onset and with longer lasting CSWS [150]. Saraf et al. conducted a retrospective study including 52 patients with CSWS (idiopathic: 19; symptomatic: 33). They found that at 1-year follow-up immune-modulating treatments (steroids and IV immunoglobulin) was effective on seizures and on language irrespective of whether cases were idiopathic or symptomatic. They found some favorable predictive factors for language at 1-year follow-up based on EEG: normal background on baseline EEG, presence of generalized spikes, absence of frontal-negative spikes, and relative low SWF during sleep [151]. Öztoprak et al. studied retrospectively 48 children with ESES/CSWS, 21 with typical ESES (SWI > 85-100% in the NREM sleep) and 27 with atypical ESES (SWI ≥ 50 % and < 85 % in the NREM sleep); median follow-up duration after ESES diagnosis: 57 months. Neurocognitive outcome was unfavorable in 50% of cases. Unfavorable neurocognitive outcome seemed to be related to: symptomatic/structural etiology, SWI ≥ 85 %, earlier age at ESES diagnosis, longer ESES duration, and the longer interval between epilepsy onset and ESES [152]. As suggested by Gardella et al., the EEG features should be correlated with the cognitive and behavioral assessment, possibly considering not only the SWI during NREM sleep, but also other EEG parameters that may affect the clinical picture, including: topography, patterns of spread, and fluctuations of the paroxysmal abnormalities, epileptiform activity during wakefulness, possible presence of focal slowing, EEG background organization, and a sleep architecture derangement [153]. Let us never forget that not only the evolution of EEG and neuropsychological tests administered to the affected individual, but also behavioral observations and parental reports in ESES children can play an important role in determining the evolution of the clinical picture (see the initial cognitive deterioration and the subsequent improvement) [154]. Ucar et al. evaluated in 33 BCETS children who developed ESES clinical and EEG parameters related to the prognosis. They found that particularly among cases with age ≤ 8 years, shorter time to ESES resolution, presence of ESES remission, and seizure control were associated with a good prognosis [155]. In conclusion, according to Arzimanoglou and Cross, the issue of cognitive and behavioral outcome should take into account the great complexity of this condition, considering not only the EEG evolution but also the underlying etiology and age at diagnosis [55].
Table 5 summarizes the main prognostic factors of CSWS.
3.6. Clinics and EEG of LKS
LKS is characterized by acquired mixed (comprehension and production) aphasia, which begins at the age of 3-5 years, with poor decoding of verbal and/or non-verbal sounds. In a minority of cases there is a previous mild speech delay. EEG recording shows spikes and spike-waves prevailing in the posterior temporal regions, bilaterally, much more diffuse and frequent during non-REM sleep when they become continuous or subcontinuous. Background activity during wakefulness and sleep is normal [156,157,158]. As suggested by the three cases reported by van Bogaert et al., EEG during sleep may be normal in the early language regression phase, so that it should be repeated when the clinical suspicion of SLK is strong [159]. Even today, the diagnostic process of SLK may not be easy and this can cause a delay in diagnosis [160]. Clinical, neurophysiological, and brain glucose metabolism findings suggest that EEG paroxysmal abnormalities play a crucial role in the cognitive impairment by meddling in the neuronal networks both at the epileptic focus site and at connected (also far) areas. Consequently, therapy should have as target the suppression of EEG paroxysmal abnormalities [161]. The core neuropsychological dysfunction in LKS seems to be a deficit of phonological decoding, involving not only verbal language. In fact, according to Lévêque et al., also music perception skills seem to be impaired in the form of high pitch discrimination thresholds and inadequate short-term memory for melody and rhythm. These findings suggest that in LKS, beyond verbal impairments, brain networks implicated in sound processing and encoding are severely altered [162]. Discriminating frequency modulation changes within speech is crucial for phoneme detection, and therefore, for language comprehension. The frequency modulated auditory evoked response (FMAER) technique assesses the processing of rapid frequency modulation within an auditory stream performed by the superior temporal gyri and contiguous cortex. Patients with language receptive deficits, including individuals with LKS, show absent left or bilateral responses [163]. Also the nondominant hemisphere may play an important role in language comprehension (see speech processing). Language acquisition would need a process of understanding the meaning of words by integrating visual, auditory, and contextual information. It has been hypothesized that the nondominant hemisphere works mainly through this integrating role [164]. Pullens et al., through the fMRI of a recovered adult LKS female (no longer showing epileptic discharges on EEG), studied audiovisual multi-sensory processing, because LKS individuals are often skilled in reading, but fail in speech perception. They found data compatible with undamaged temporal lobe processing and with impaired connectivity between temporal and frontal lobe. This impaired connectivity could be behind the observed deficits of short-term verbal memory, sound-motor interaction and online speech feedback, being one pathogenetic mechanism underlying LKS [165]. Seizures are present in the majority of LKS cases (around two-thirds of patients) and are semeiologically heterogeneous: focal motor, tonic-clonic seizures, and atypical absences (e.g., with chewing gestures or with lip-smacking). Behavioral symptoms are frequently associated: hyperactivity, attention deficit, impulsiveness (leading up to an ADHD picture), irritability, aggressive behavior, in some cases autistic-like symptoms, anxiety, and depression [24,26,158,166,167,168,169,170,171,172,173]. In LKS autism could be due to the epilepsy itself, but the presence of genetic factors common to LKS and autism should not be overlooked [25]. Learning disorders, frequent also in mathematics, are the rule in patients with LKS [169]. Sleep disorders may be associated [158]. Also difficulty in maintaining posture has been reported [174]. Development is not always completely regular before the onset of aphasia: according to a study of Caraballo et al., before aphasia onset, developmental language disorders were present in 19 out of 29 LKS cases while behavior disorders were present in 14 out of 29 LKS cases [167]. Although epidemiological data are lacking, the condition is undoubtedly rare. According to Kaga et al., in Japan the incidence of LKS individuals aged 5-14 years was about 1 in 1.000.000, while the prevalence of LKS individuals aged 5-19 years and under medical care was about 1 in 300,000-410,000 [175]. Furthermore, it cannot be ruled out that the real incidence of LKS is underestimated due to incomplete awareness of this condition. Yet, for a better outcome, the awareness of LKS is very important, as it favors an early diagnosis and the prompt start of a treatment [176,177]. Etiology is not known in most cases. Brain lesions are rare and aspecific. Genetic factors have recently been called into question. In particular, mutations of GRIN2A gene (16p13.2) have been found in some cases with LKS [178]. GRIN2A gene encodes for the GluN2A protein, a subunit of the N-methyl-D-aspartate (NMDA) receptor. High concentrations of GluN2A protein are present in cerebral regions critical for language. In the last years, the concept of common genetic factors for BECTS, CSWS, and LKS has caught on [179,180], further indicating a link between these nosographic entities. As regards the pathophysiology of LKS, literature data suggest that aphasia derives from EEG paroxysmal abnormalities located in cortical areas critical for language. Probably, these abnormalities and not seizures are related to aphasia [158]. Using 99mTc-ECD SPECT imaging, asymmetrical temporo-parietal perfusion seems to be a common finding in LKS [181]. As suggested by the case described by Datta et al., a right-handed child with BECTS that evolved to LKS and (after adequate treatment) returned to BECTS, in LKS there may be a cerebral reorganization of language network to the right (non-dominant) hemisphere, therefore leading to an atypical language representation, demonstrated by fMRI studies. This phenomenon, however, is also present in other epileptic syndromes that involve the cortical areas responsible for language [182]. The topic of the etiopathogenetic factors of LKS will be further addressed later, together with ESES (see paragraph 3.8).
A multidisciplinary approach appears to be the most proper for the diagnosis and for the treatment [183]. Concerning differential diagnosis, it is crucial to rule out in children with aphasia a large group of heterogeneous pathological conditions including primarily hearing loss, autism spectrum disorder, or ADHD [158,184]. In particular for ASD the differential diagnosis could be difficult, due to the early language regression reported in a large number of ASD children and to the frequent finding of EEG paroxysmal abnormalities in ASD. On the other hand, it should be borne in mind that aphasia can be due to various organic causes including space-occupying brain lesions, post-traumatic sequelae, and infections [158,185]. A differential diagnosis has to be carried out also with other epileptic syndromes characterized by marked increase of EEG paroxysmal abnormalities during sleep, including ESES (typical and atypical) and Lennox-Gastaut syndrome [158]. Finally, also metabolic disorders such as mucopolysaccharidosis type III, due to a lysosomal enzyme deficit in the heparan sulfate catabolism, that is characterized by behavioral disorders, decreased verbal communication, and only subtle somatic signs, should be considered as differential diagnosis [186]. Evidently, for a correct differential diagnosis, EEG during sleep is of crucial importance.
Outcome of LKS is very heterogeneous. According to a study of Caraballo et al., at the end of a mean follow-up of 12 years, only eight out of 29 LKS cases recovered language completely, while 21 cases still showed language deficits of different degrees [167]. Patients with LKS could need for a life-long support, due to persisting and potentially debilitating deficits of verbal communication [187]. In a retrospective study involving 11 children, four cases showed no language problems after adolescence, four cases had moderate language problems, and three cases presented a severe language impairment > 10 years after the diagnosis [188]. A positive outcome was associated with aphasia late-onset, short-lived initial receptive manifestations, and speech performances characterized by marked fluctuations. Seizure prognosis is good as they remit within the adolescence [158].
Table 6 summarizes the clinical and EEG features of LKS.
3.7. Treatments for LKS
In the above-mentioned systematic review of Moresco et al., also concerning pharmacological treatments for LKS, no completed studies to include were found and therefore well-designed randomized controlled trials are necessary [132]. In order to improve the prognosis, treatment should start as soon as possible. If seizures are present, several ASDs may be effective and usually seizure control is easy to achieve. Drugs such as valproate, clobazam, levetiracetam, and ethosuximide, considederd as “spike-suppressing”, are preferred. Instead, there are some drugs such as carbamazepine, oxcarbazepine, phenytoin, and phenobarbital that in LKS should not be used due to a possible exacerbation of EEG paroxysmal abnormalities [158]. The most effective treatments on EEG picture, and consequently on language and behavior, are corticosteroids (e.g., oral prednisone or oral prednisolone) [93,158], suggesting an underlying inflammatory pathogenetic mechanism. On the other hand, there are insufficient elements in favor of the use of IV immunoglobulin in these patients [158]. Immunotherapy (oral steroids and IV immunoglobulin) should be a therapeutic option particularly in LKS patients with GRIN2A mutations [189]. Combined treatment with pulse BZD and corticosteroids could be very useful both on the clinical and EEG level in LKS according to Devinsky et al. [190]. There are limited data in favor of the favorable effects of a KD in individuals with LKS [122]. Also, AZM might be an effective treatment: see above the paper of Fine et al. [106].
Multiple subpial transections (MST) (that is the transection of only the horizontal corticocortical fibers and not of the vertical cortico-subcortical ones) in the posterior dominant temporal lobe is a surgical technique aiming at interrupting epileptic seizures without damaging the eloquent cortex. According to some authors, MST might lead to the recovery of language skills in LKS children [191]. However, this surgical technique should be restricted to drug-resistant patients and in cases with steroid dependency or toxicity. But unfortunately, there are relatively few reliable data concerning the behavioral outcome of surgery for epilepsy (in general, not only for LKS) [192]. Downes et al. studied the effects on long-term outcome of MST in children with drug-resistant LKS or other ESES-related regressive epilepsies. Further, they explored predictors of outcome. They included 14 cases who underwent MST of the posterior temporal lobe compared with 21 cases who underwent only presurgical investigations and not surgery. At follow-up, no significant differences were found between the two groups with regard to language, nonverbal skills, adaptive behavior, or quality of life. The authors concluded that there is insufficient evidence for significant benefits of MST in these conditions [193]. Fine and Nickels reported a girl with drug-resistant LKS, in which the seizure focus was localized in the right parietal cortex. The child underwent right temporo-parietal resection after which she remained seizure free, EEG normalized, and language was recovered [194]. According to the recent systematic review of Hajtovic et al., there is limited evidence regarding the effects of vagus nerve stimulation (VNS) on seizure, cognitive, and behavioral outcomes in children with LKS [195].
Obviously, also speech therapy is indicated in children with LKS: it consists of several patient-centered interventions that usually include augmentative and alternative communication techniques aimed at improving speech. In younger patients, a treatment plan should include also psychomotor therapy [158]. In particular for older children, cognitive linguistic treatment could be useful to improve spoken language. However, we need more research necessary to optimize speech therapy in children with LKS [196]. Literature data about rehabilitation or speech therapy in LKS are even more sparse than those about pharmacological or surgical treatment [197].
Table 7 summarizes the effective treatments for LKS (pharmacological and not pharmacological).
3.8. Etiopathogenetic factors of CSWS (including ESES and LKS)
CSWS is a very heterogeneous condition that may be related to genetic factors as well as to congenital or acquired cerebral lesions [139]. A large number of heterogeneous brain structural alterations have been reported in subjects with CSWS, including polymicrogyria (in particular unilateral), migration disorders, perinatal hypoxic-ischemic encephalopathy, hydrocephalus, schizencephaly, porencephalic lesions, encephalitis, and intracranial hemorrhage [198,199]. Note that an EEG pattern closely resembling CSWS has been reported also in a mouse model of focal cortical dysplasia [200]. EEG pattern of ESES has been reported even in patients with congenital Zika virus syndrome [201]. He et al. studied resting-state functional fMRI in 9 cases with BECTS and ESES, in 17 with BECTS but without ESES, and in 36 healthy subjects. They found that cases with BECTS and ESES, compared with others, showed decreased whole-brain functional connectivity in the salience network (otherwise known as the midcingulo-insular network) or in the central executive network (otherwise known as the lateral frontoparietal network), but not in the DMN (otherwise known as the medial frontoparietal network). These findings may lead to better understand the underlying pathogenetic mechanisms in cases with BECTS with ESES [202].
Growing data from the literature suggest an involvement of the thalamus in the pathogenesis of CSWS [203,204,205]. Neonatal thalamic lesions are not rarely described in CSWS. Losito et al., studying a sample of 60 CSWS children with early thalamic injury, found that cases evolving into typical ESES (that is occupying > 85% of slow sleep) had more often a damage in the mediodorsal thalamus and a bilateral cortico-subcortical injury; moreover, cases with typical ESES showed in the acute stage the most severe behavior and cognitive impairment. Ischemic stroke, hemorrhagic infarction and hydrocephalus shunting were associated with behavior disorders [206]. Leal et al. reported nine cases with unilateral neonatal lesions of the thalamus that evolved into CSWS. Loss of thalamic volume prevailed on medial and dorsal nuclei. Thalamic lesions were associated with white matter loss and ventricle enlargement in the same hemisphere. Cortical thickness quantification showed no hemispheric asymmetries. The impact on physiological EEG rhythms was mild. CSWS were lateralized to the lesioned hemisphere. Unilateral selective thalamic-cortical disconnection was therefore associated with a focal pattern of CSWS in patients with unilateral neonatal thalamic lesions [207]. Also neonates with thalamic hemorrhage due to straight sinus thrombosis, without widespread cerebral damage, are predisposed to develop typical or atypical ESES disorder [208]. Öztürk et al. studied subcortical gray matter volumes in a sample of 15 children with ESES, all with MRI considered normal. They found a total relative volume of thalamus significantly lower in ESES cases than in 30 healthy controls and than in 15 cases with BECTS. Both right and left relative volumes of thalamus were lower in ESES cases than in healthy controls and than in cases with BECTS. This study suggests the possible involvement of the thalamus in the ESES pathogenesis, even in the absence of detectable lesions on MRI [209]. A pathogenetic role of the thalamus can also be hypothesized in cases of CSWS with cortical malformation. Bartolini et al. carried out a prospective follow-up study involving 27 cases with polymicrogyria and CSWS. Using comparative volumetric analysis, they found significantly smaller volumes of ipsilateral talami and polymicrogyric hemispheres in patients with polymicrogyria and CSWS compared to cases with polymicrogyria without CSWS, cases with BECTS, and controls with headache [210]. Van den Munckhof et al. studied neuroimaging findings in a sample of individuals with perinatal thalamic injury. They found in the prospective cohort (23 cases) that perinatal thalamic injury was followed by ESES in the majority (about 83%) of individuals after a mean follow-up of 8 years. Thalamic volume at 3 months was correlated to neurodevelopmental outcome (see IQ/developmental quotient) [211]. Carvalho et al. found in 25 out of 53 CSWS patients structural lesions, which included an early thalamic injury in almost all cases (24/25). Most thalamic lesions were localized in one hemisphere, corresponding in all patients to the CSWS lateralization [212]. Sánchez Fernández et al. found small relative thalamic volume in 18 ESES children with normal neuroimaging in comparison with 29 subjects with refractory epilepsy and with 51 healthy individuals, controlling for age and for total brain volume [213].
Functional thalamic abnormalities may be present also in cases with normal thalamic structures. Agarwal et al. analyzed thalamic glucose metabolism in 23 CSWS children (with normal thalami based on MRI) through F-18-fluorodeoxyglucose (FDG)-positron emission tomography (PET), considering the standardized uptake value normalized to whole brain (nSUV). Abnormalities of thalamic glucose metabolism were found in 18 cases (78.3%). Thalamic nSUV was lower (six cases) or higher (one case) bilaterally in seven children without asymmetries. Abnormal thalamic symmetry was found in 11 cases of which six showed a unilateral thalamic abnormality (three with higher metabolism, and three with lower metabolism), while five children showed abnormal asymmetry index with bilaterally normal (four cases) or higher (one case) thalamic metabolism [214]. Using magnetic resonance spectroscopy, Kilic et al. found in 21 patients with ESES a significantly low NAA/Creatine ratio, which is indicative of neuronal cell loss/dysfunction, bilaterally in the thalamus, structure that seems to be normal on MRI [215]. Leal, through high resolution EEG carried out in five CSWS children with perinatal thalamic hemorrhages localized in one hemisphere, found that perinatal thalamic lesions may produce, years later, a focal onset of paroxysmal abnormalities with characteristics of ESES in a cortex without demonstrable lesions, leading also to a prominent secondary propagation of paroxysmal abnormalities [216].
But there are also data that do not seem to be in line with an involvement of the thalamus in the pathogenesis of CSWS. Ligot et al. performed positron emission tomography through [18F]-fluorodeoxyglucose during wakefulness in 17 children affected by cryptogenic CSWS, finding hypermetabolism in bilateral perisylvian areas and hypometabolism in the following areas: prefrontal cortex, precuneus, posterior cingulate cortex, and parahippocampus. Functional connectivity between hyper- and hypometabolic areas was altered. They found no thalamic metabolic alterations, suggesting a primary cortex role in CSWS genesis [217].
Even apart from lesions specifically of the thalamus, CSWS may occur after a perinatal stroke. In a cross-sectional study including globally 43 patients, Azeem et al. found that higher EEG spike frequency and delta power during NREM sleep may be EEG biomarkers of the ESES developing risk in subjects with perinatal stroke [218]. ESES can also be the long-term consequence of neonatal cerebral sinovenous thrombosis [219]. Symptomatic CSWS cases include also those with early-onset hydrocephalus; in these patients, EEG paroxysmal abnormalities would hypothetically be related to the shunt or to the possible presence of associated cortical or subcortical lesions [220]. Taskin et al. described the case of an 8-year-old girl with a previous history of treated Herpes simplex virus (HSV)-1 encephalitis at the age of 13 months. Focal epilepsy developped at the age of 2 years. At the age of 5 years, although the girl was free of clinical seizures, she showed behavioral and cognitive impairment, which was attributed to the onset of EEG focal seizures and to an EEG worsening leading to ESES. After a right temporal lobectomy, the diagnosis of chronic granulomatous encephalitis was made. Her clinical course improved with seizure and EEG abnormalities’ resolution [221]. Dedeoglu et al., using brain MRI, found no significant difference as regards corpus callosum thickness between children affected by SeLECTS respectively with (N = 15) and without (N = 53) ESES [222].
The case reported by Hu et al. suggests a possible involvement of onconeuronal antibodies in the pathogenesis of CSWS. They described a boy aged 8 years with a previous history of neuroblastoma with opsoclonus-myoclonus successfully treated. The boy showed intellectual disability, drug-resistant epilepsy, and CSWS. An extensive workup did not reveal a tumor relapse, but testing for onconeuronal antibodies showed a positivity for anti-Ma2 and anti-CV2/CRMP5 antibodies. The boy was successfully treated with 2 cycles of IV methylprednisolone with disappearance of CSWS, while onconeuronal antibodies were not detectable anymore [223]. Inflammation could play an important role in the pathogenesis of CSWS. Van den Munckhof et al., studying 11 ESES cases, found that serum inflammatory mediators, and in particular interleukin-6 (increased during ESES, decreased after immunomodulating treatment), could be related with disease activity. Decrease of interleukin-6 was associated with clear EEG and neuropsychological improvement [224].
Ayça et al. [2019] found that in CSWS children 9:00 a.m. melatonin levels were significantly lower than in patients with epilepsy and in healthy controls [225]. Instead, Tarcin et al. found no significant differences in median basal melatonin levels between 91 children with epileptic seizures and 21 children with ESES; however, median basal melatonin levels were significantly lower both in children with epileptic seizures and in those with ESES compared with the controls [226]. ESES has been be associated also to fetal alcohol syndrome [227,228]. In children with benign focal eplepsy, ESES, associated with a cognitive deterioration, can be induced by old ASDs, such as phenytoin, carbamazepine, and phenobarbital, but also by a relatively new antiepileptic drug such as oxcarbazepine. In these cases, the therapy change is followed by a prompt electroclinical improvement [229].
Over the last few years, an increasing number of genetic abnormalities, each different from the other, associated with CSWS have been gradually described in patients with CSWS. Among the reported genetic conditions with a high recurrence of CSWS is the dup15q (copy number gains of 15q11-q13): Arkilo et al. retrospectively found that EEG showed CSWS in 15 out of 42 children (35.7%) with neurodevelopmental disorders due to dup15q; 13 of these 15 children showed also clinical seizures [230]. This anomaly is identifiable using array-CGH, as well as 22q11.2 microduplication syndrome, another genetic condition frequently associated with CSWS [231]. Many other copy number variants, detected through array-CGH, affecting several other chromosomes, have been described in CSWS (3q25 deletion, 3q29 duplication, 8p23.3 deletion, 10q21.3 deletion, 11p13 duplication, 16p13 deletion, and Xp22.12 deletion) [19]: we do not deem it useful to dwell on them. But frequently mutations of single genes are involved in the etiopathogenesis of CSWS. In a minority of patients with LKS and CSWS (no more than 20% of cases), de novo or inherited in an autosomal dominant manner, heterozygous pathogenic mutations of GRIN2A gene have been found. This gene encodes the GluN2A, that is the α2 subunit of the N-methyl-D-aspartate (NMDA) excitatory glutamate receptor, which plays a very important role in the control of synaptic plasticity [39,232,233,234,235,236,237,238,239,240,241,242,243,244,245,246,247]. Also mutations of several other genes have been implicated in the etiopathogenesis of CSWS: for example connector enhancer of kinase suppressor of Ras-2 (CNKSR2), which is located on the short arm of X chromosome (Xp22.12) [248,249]; cyclin-dependent kinase-like 5 (CDKL5) gene, whose mutations are usually involved in an early onset epileptic encephalopathy with a severe neurodevelopmental disorder [250]; Cysteinyl-tRNA synthetase 2 (CARS2) gene [251], which has been reported to be associated with severe myoclonic epilepsy, neuroregression, and complex movement disorders; KCNQ2 gene, involved in benign familial neonatal seizures [252]. According to Gong et al., pathogenic or likely pathogenic mutations also of other genes such as KCNA2, SLC9A6, HIVEP2, and RARS2 are involved in some cases with developmental and/or epileptic encephalopathy with ESES [246,253]. The most common phenotype reported in DYNC1H1-related epilepsy is represented by West syndrome, but also one case with CSWS has been described [254]. Also cases with a genetic basis usually show neurocognitive and behavioral worsening associated with CSWS, which however is often difficult to identify because they usually have a neurodevelopmental impairment that precedes the onset of CSWS. Knowing the genetic diagnosis can sometimes suggest the most effective treatment. An example of this is given by the case recently reported by Russo et al., a girl showing encephalopathy with ESES and cerebellar signs, who presented a de novo KCNA1 variant in the Kv-specific Pro-Val-Pro motif. Remarkably, this case had a persisting noteworthy electroclinical response to ACTH treatment [255]. The pathogenetic relationships between involved gene mutations and ESES are not always clear. For example, SCN2A gene encodes Nav1.2 that is a voltage-gated sodium channel expressed in the brain. SCN2A gene mutations are associated with several epileptic syndromes including ESES. Interestingly, ESES can be associated with both gain and loss of functional variants of the SCN2A gene [256]. ESES can develop also in patients with Christianson syndrome, an X-linked condition due to SLC9A6 gene mutations characterized by a complex clinical picture including intellectual disability, lack of speech, autistic behavior, ataxia, acquired microcephaly, hyperactivity, and drug-resistant epilepsy [257,258,259,260]. Bhat et al. reported CSWS in one patient with Rett syndrome (with MECP2 gene mutation) and in another with both neurofibromatosis 1 and Lhermitte-Duclos syndrome [261]. We would mention another possible genetic etiology of CSWS, that is a mutation of WAC (WW Domain Containing Adaptor With Coiled-Coil) gene (located on 10p12.1). WAC gene mutations are considered to be related to the DeSanto–Shinawi syndrome, characterized by intellectual disability, behavioral disorders, and speech impairment [262]. An increased risk of epilepsy, including ESES, has been reported in Prader-Willi syndrome (PWS), a genomic imprinting disorder due to an absent expression of paternal genes located on chromosome 15q11.2-q13 [263]. Koolen-de Vries syndrome (KdVS), due to 17q21.31 microdeletions or to pathogenic KANSL1 gene mutations, is clinically characterized by: congenital malformations, neonatal hypotonia, developmental delay, and facial dysmorphysms. About half of cases show epilepsy. Khan et al. reported six KdVS children with CSWS: four of them received a diagnosis of epileptic encephalopathy with CSWS, while two of them were diagnosed with LKS. Two cases underwent a KD variation, and for both a clinical improvement was reported. The authors concluded that a prolonged EEG during sleep should be performed in all KdVS children showing a regression or plateau of development, especially when there is a previous history of seizures [264]. Mowat-Wilson syndrome is a genetic rare disease due to a heterozygous deletion or function loss of the ZEB2 gene located on chromosome 2. In a retrospective study, Bonanni et al. found anterior ESES with SWI > 85% in all five cases with overnight sleep EEG and in three out of five cases there was a related cognitive and motor regression. In two cases a marked cognitive and motor improvement was observed when EEG paroxysmal abnormalities during sleep decreased. The clinical significance of ESES is difficult to assess in these cases due to severe intellectual disability [265]. CSWS has been reported even in β-Propeller protein-associated neurodegeneration, a neurodegeneration with brain iron accumulation due to a de novo WDR45 deletion at Xp11.23 [266]. Sager et al. reported one boy with a de novo loss-of-function mutation in TET3 gene located on chromosome 2p13 (Beck-Fahrner syndrome), revealed by whole-exome sequencing. The boy showed an early-onset neurodevelopmental delay, autistic behavior, ADHD, and learning disabilities. At 5 years of age he began to have generalized tonic-clonic seizures and clinical regression. EEG showed ESES. He showed a clinical improvement, while taking valproic acid and immunotherapy, regardless of EEG, that then improved with clobazam [267]. The CSWS EEG pattern has been found also in ferric chelate reductase 1 like (FRRS1L) encephalopathy. Recognizing ESES in these patients could be very important for a proper management, although there are insufficient data regarding the real efficacy of the therapy [268]. The case with FRRS1L encephalopathy reported by Hadi responded well to the therapy with sulthiame for seizures, but not for CSWS [269]. Ünalp et al. described a child with Ehlers-Danlos syndrome and an epileptic encephalopathy as ‘ESES’ related to a STXBP1 (9q34.11) gene mutation, whose drug-resistant seizures stopped with KD [270]. We reiterate here the importance of paying particular attention to any cognitive and behavioral worsening that may be related to the CSWS EEG pattern in individuals with genetic pathologies, that are most often characterized by intellectual and behavioral disorders already present before the onset of CSWS.
Advances in genetics also help us find commonalities between clinically similar but distinct nosographic entities. The concept of epilepsy-aphasia spectrum includes some epileptic syndromes [BECTS, atypical benign partial epilepsy (ABPE), LKS, and epileptic encephalopathy with CSWS] that have in common the association between epilepsy, language disorders and centro-temporal spikes showed by EEG [242]. The theory of a spectrum that unites these conditions, suggested by the shared morphology of EEG paroxysmal abnormalities and by the possible evolution from one into another syndrome (e.g.: BECTS into CSWS), is present in many works of literature, albeit with some differentiations among authors [271,272,273,274,275,276,277,278]. Results obtained through genetic tecniques such as copy number variation, exome sequencing, and linkage suggest for these conditions a genetic etiology [273,279,280]. In particular, mutations of the GRIN2A gene have been found in patients with focal epilepsy with speech disorder including LKS [242]. There are data in the literature suggesting that patients with GRIN2A mutations are predisposed to a good response to immunotherapy, in particular using IV immunoglobulins [281]. According to Samanta, the discovery of GRIN2A as the main monogenic etiology of epilepsy-aphasia spectrum led to hypothesize the possibility of precision therapies. Pathogenic variants of GRIN2A induce gain or loss of NMDAR function; possible therapies for the former are uncompetitive NMDAR antagonists (see in particular memantine), while a possible therapy for the latter is the NMDAR co-agonist serine. Precision therapies for GRIN2A-related disorders could benefit these patients [282].
Table 8 summarizes the etiopathogenetic factors of CSWS.
4. Discussion
CSWS are one of the most fascinating and complex topics in childhood epileptology of recent decades due to the various implications that characterize them: the role of epileptic seizures and epileptiform EEG paroxysmal abnormalities in determining a neuropsychological/behavioral disorder, the role of sleep in promoting learning, the search for effective therapies but at the same time without significant harmful effects on neurocognitive development and behavior.
First of all, we intend to underline the importance of performing, and possibly repeating over time, an EEG tracing, particularly in sleep, in a child with neurodevelopmental problems (autistic behavior, language impairments, cognitive deterioration) even in the absence of overt epileptic seizures [283,284]. The earliness of a correct diagnosis of these cases is crucial, since in these situations the neurocognitive impairment of the child can be treated effectively. Note that CSWS have also been found in subjects with epilepsy at least apparently in the absence of cognitive and behavioral deterioration during a long-term follow-up (mean duration: 14 years) [285], but also in these cases we cannot exclude that CSWS had a negative impact on the neurocognitive development, as we do not know what results these individuals would obtain on neuropsychological tests in the absence of CSWS. When faced with CSWS, it appears fundamental to carry out a series of tests, particularly genetic and brain imaging, which can better outline the prognosis of the condition and can sometimes give suggestions regarding the treatment to be carried out.
ESES diagnosis should be considered in all children with unexplained developmental regression or stagnation with or without overt seizures. In these cases, sleep EEG should be mandatory [145]. Particularly in patients who already have an impairment of intellectual functioning due to the underlying pathology, it may not be easy to identify the signs/symptoms that are indicative of a cognitive deterioration related to the ESES picture. In these cases the administration of standardized neuropsychological and behavioral tests is fundamental in order to have an objective evaluation of the individual’s situation. In fact, the clinical impression that can be obtained during an outpatient visit and the anamnestic data reported by caregivers are not sufficient and are sometimes even misleading.
It is still to be ascertained whether the SWI is the best electrophysiological parameter to monitor the situation. Other parameters have also been studied and appear promising, in particular the HFOs, but they have so far had little use in clinical practice.
Concerning the treatment, CSWS is frequently a drug-resistant condition and also possible side effects of ASDs have to be carefully considered. The question relating to treatment still appears widely controversial: when to start it? What kind of treatment? How long? In the face of a developmental arrest or even more so of cognitive decline that occurs concomitantly with a picture of CSWS, an adequate pharmacological treatment appears necessary. However, everything should be evaluated in the single case we have before us. If, for example, we are dealing with a patient who has already suffered from epilepsy, we must first rule out a possible iatrogenic cause (in particular the introduction of a drug that potentially favors the onset of CSWS). In this case it may be sufficient to change medication. In the other cases, which are clearly the most numerous, it is necessary to undertake a pharmacological treatment whose aim is the resolution of the EEG picture of CSWS and the improvement of cognitive skills in the absence of significant side effects. First, treatment with a benzodiazepine may be attempted, the effects of which on the EEG can be assessed quite quickly. In case of failure, or in case of relapse of CSWS, the best option seems to be a course of steroid therapy, which requires particularly careful medical monitoring. From a research perspective, we find interesting the point of view of Shao and Stafstrom who reviewed CSWS or LKS animal models, underlining that they can reveal pathogenetic mechanisms potentially treatable with targeted therapies [286].
5. Conclusions
In conclusion, although several decades have passed since the original descriptions of the electroclinical condition of CSWS, there are still many areas that are little known and deserve to be further studied. We do not yet have a complete knowledge of the natural evolution of these conditions. There is not yet a shared approach among researchers regarding the nosographic classification of LKS, which according to some authors should be considered separately from ESES, while according to others it represents a variant of it. It has not been fully ascertained whether the SWI is the most effective electrophysiological parameter for monitoring the evolution of the disorder. There is not yet a neuropsychological evaluation protocol that is shared internationally among the Centers dealing with these problems. There is still no consensus as to whether to treat and if so when exactly to start drug treatment. We do not yet know a drug therapy that is effective for all cases of CSWS and at the same time free from significant side effects. We still know little about the non-pharmacological (medical, neurosurgical, and rehabilitative) therapies of these conditions. Finally, we believe what Halasz [2017] underlined is still today valid at least in part, namely we lack detailed knowledge of the nature (i.e.: generalized or focal/regional?) of the EEG paroxysmal abnormalities in CSWS, and most EEG paroxysmal abnormalities show a morphology, spatial localization, and functional properties different from the generalized spike-wave pattern of typical ESES [287]. This is one of the main reasons for the great heterogeneity and complexity of these conditions in terms of diagnosis, prognosis, and treatment.
Author Contributions
Conceptualization, A.P. and P.V.; methodology, A.P.; validation, A.P. and P.V.; investigation, A.P.; data curation, A.P.; writing—original draft preparation, A.P.; writing—review and editing, A.P.; visualization, A.P. and P.V.; supervision, P.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors would like to thank Cecilia Baroncini for help in editing the text.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Schmitt, B. Sleep and epilepsy syndromes. Neuropediatrics. 2015, 46, 171–180. [Google Scholar] [CrossRef]
- Shbarou, R.; Mikati, M.A. The expanding clinical spectrum of genetic pediatric epileptic encephalopathies. Semin. Pediatr. Neurol. 2016, 23, 134–142. [Google Scholar] [CrossRef]
- Besag, F.M. Epilepsy in patients with autism: Links, risks and treatment challenges. Neuropsychiatr. Dis. Treat. 2017, 14, 1–10. [Google Scholar] [CrossRef]
- Appendino, J.P.; Appendino, J.I. Encefalopatías epilépticas determinadas genéticamente [Genetically determined epileptic encephalopathies]. Medicina (BAires). 2019, 79 (Suppl. 3), 42–47. [Google Scholar]
- Specchio, N.; Wirrell, E.C.; Scheffer, I.E.; Nabbout, R.; Riney, K.; Samia, P.; Guerreiro, M.; Gwer, S.; Zuberi, S.M.; Wilmshurst, J.M.; et al. International League Against Epilepsy classification and definition of epilepsy syndromes with onset in childhood: Position paper by the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022, 63, 1398–1442. [Google Scholar] [CrossRef] [PubMed]
- Van Bogaert, P. Long-term outcome of developmental and epileptic encephalopathies. Rev. Neurol. (Paris). 2022, 178, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Van Bogaert, P. Epilepsy syndromes of childhood with sleep activation: Insights from functional imaging. Eur. J. Paediatr. Neurol. 2020, 24, 58–60. [Google Scholar] [CrossRef] [PubMed]
- Auvin, S.; Cilio, M.R.; Vezzani, A. Current understanding and neurobiology of epileptic encephalopathies. Neurobiol. Dis. 2016, 92, 72–89. [Google Scholar] [CrossRef] [PubMed]
- Japaridze, N.; Menzel, E.; von Ondarza, G.; Steinmann, E.; Stephani, U. Risk factors of cognitive outcome in patients with atypical benign partial epilepsy/pseudo-Lennox syndrome (ABPE/PLS) and continues spike and wave during sleep (CSWS). Eur. J. Paediatr. Neurol. 2014, 18, 368–375. [Google Scholar] [CrossRef]
- Kennedy, A.; Hill, D. Dementia infantilis with cortical dysrhythmia. Arch. Dis. Child. 1942, 17, 122–129. [Google Scholar] [CrossRef]
- Tassinari, C.A.; Rubboli, G.; Volpi, L.; Meletti, S.; d’Orsi, G.; Franca, M.; Sabetta, A.R.; Riguzzi, P.; Gardella, E.; Zaniboni, A.; et al. Encephalopathy with electrical status epilepticus during slow sleep or ESES syndrome including the acquired aphasia. Clin. Neurophysiol. 2000, 111 (Suppl. 2), 94–102. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.T.; Berkovic, S.F.; Brodie, M.J.; Buchhalter, J.; Cross, J.H.; van Emde Boas, W.; Engel, J.; French, J.; Glauser, T.A.; Mathern, G.W.; et al. Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005-2009. Epilepsia. 2010, 51, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Azcona, G.; Gurtubay, I.G.; Mosquera, A.; Ibanez, B.; Cambra, K.; Aguilera-Albesa, S.; Yoldi-Petri, M.E. Estudio comparativo entre tres sistemas de cuantificacion del indice de punta-onda en pacientes con punta-onda continua del sueño lento [A comparative study of three systems for quantifying the spike and wave index in patients with continuous spikes and waves during slow sleep]. Rev. Neurol. 2017, 65, 439–446. [Google Scholar] [PubMed]
- Hirsch, E.; Caraballo, R.; Dalla Bernardina, B.; Loddenkemper, T.; Zuberi, S.M. Encephalopathy related to status epilepticus during slow sleep: From concepts to terminology. Epileptic. Disord. 2019, 21, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Jansen, F.E.; Nikanorova, M.; Peltola, M. Current treatment options for encephalopathy related to status epilepticus during slow sleep. Epileptic Disord. 2019, 21, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Sonnek, B.; Döring, J.H.; Mütze, U.; Schubert-Bast, S.; Bast, T.; Balke, D.; Reuner, G.; Schuler, E.; Klabunde-Cherwon, A.; Hoffmann, G.F.; et al. Clinical spectrum and treatment outcome of 95 children with continuous spikes and waves duringsleep (CSWS). Eur J Paediatr Neurol. 2021, 30, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Rubboli, G.; Huber, R.; Tononi, G.; Tassinari, C.A. Encephalopathy related to status epilepticus during slow sleep: A link with sleep homeostasis? Epileptic Disord. 2019, 21 (Suppl. S1), 62–70. [Google Scholar] [CrossRef] [PubMed]
- Rubboli, G.; Gardella, E.; Cantalupo, G.; Tassinari, C.A. Encephalopathy related to status epilepticus during slow sleep (ESES). Pathophysiological insights and nosological considerations. Epilepsy Behav. 2023, 140, 109105. [Google Scholar] [CrossRef]
- Samanta, D.; Al Khalili, Y. Electrical status epilepticus in sleep. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Nath, A.; Whitworth, E.; Bretz, D.; Davila-Williams, D.; McIntosh, L. Electrical status epilepticus in sleep (ESES) in an elderly adult: A case report. Cureus. 2022, 14, e26372. [Google Scholar] [CrossRef]
- Rubboli, G.; Tassinari, C.A. Linking epilepsy, sleep disruption and cognitive impairment in encephalopathy related to status epilepticus during slow sleep (ESES). Epileptic Disord. 2019, 21 (Suup. 1), 1–2. [Google Scholar] [CrossRef]
- Camfield, P.; Camfield, C. Regression in children with epilepsy. Neurosci Biobehav Rev. 2019, 96, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Dorris, L.; O’Regan, M.; Wilson, M.; Zuberi, S.M. Progressive intellectual impairment in children with encephalopathy related to status epilepticus during slow sleep. Epileptic Disord. 2019, 21 (Suppl. S1), 88–96. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J. Cortical interneuron dysfunction in epilepsy associated with autism spectrum disorders. Epilepsia. 2016, 57, 182–193. [Google Scholar] [CrossRef]
- Besag, F.; Aldenkamp, A.; Caplan, R.; Dunn, D.W.; Gobbi, G.; Sillanpää, M. Psychiatric and behavioural disorders in children with epilepsy (ILAE Task Force Report): Epilepsy and autism. Epileptic Disord. 2016, 18 (Suppl. 1), 16–23. [Google Scholar] [CrossRef]
- Besag, F.; Gobbi, G.; Aldenkamp, A.; Caplan, R.; Dunn, D.W.; Sillanpää, M. Psychiatric and behavioural disorders in children with epilepsy (ILAE Task Force Report): Behavioural and psychiatric disorders associated with epilepsy syndromes. Epileptic Disord. 2016, 18 (Suppl. 1), 37–48. [Google Scholar] [CrossRef]
- Gong, P.; Xue, J.; Qian, P.; Yang, H.; Liu, X.; Zhang, Y.; Jiang, Y.; Yang, Z. Epileptic negative myoclonus restricted to lower limbs in benign childhood focal epilepsy with vertex spikes. Eur. J. Neurol. 2019, 26, 1318–1326. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Xue, J.; Qian, P.; Yang, H.P.; Zhang, Y.H.; Jiang, Y.W.; Yang, Z.X. [Electroclinical characteristics of epilepsy children with midline epileptiform discharges related epileptic negative myoclonus as the first symptom]. Zhonghua Er Ke Za Zhi. 2019, 57, 943–949. [Google Scholar] [PubMed]
- Aoun, M.A.; Eisermann, M.; Chemaly, N.; Losito, E.; Desguerre, I.; Nabbout, R.; Kaminska, A. Jerking during absences: Video-EEG and polygraphy of epileptic myoclonus associated with two paediatric epilepsy syndromes. Epileptic Disord. 2021, 23, 191–200. [Google Scholar] [CrossRef]
- Kalscheur, E.J.; Farias-Moeller, R.; Koop, J. Role of neuropsychology in identification of CSWS in a school-aged child with a remote neurological insult. Epilepsy Behav. Rep. 2021, 18, 100514. [Google Scholar] [CrossRef]
- Bennett-Back, O.; Uliel-Siboni, S.; Kramer, U. The yield of video-EEG telemetry evaluation for non-surgical candidate children. Eur. J. Paediatr. Neurol. 2016, 20, 848–854. [Google Scholar] [CrossRef]
- Nagyova, R.; Horsburgh, G.; Robertson, A.; Zuberi, S.M. The clinical utility of ambulatory EEG in childhood. Seizure. 2019, 64, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Tassinari, C.A.; Rubboli, G. Encephalopathy related to status epilepticus during slow sleep: Current concepts and future directions. Epileptic Disord. 2019, 21, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Belousova, E.D.; Ermakov, A. Épilepticheskaia éntsefalopatiia s prodolzhennoĭ spaĭk-vol-no-voĭ aktivnost’iu vo sne: Obzor literatury [Epileptic encephalopathy with continuous spikes and waves during sleep (CSWS): A review]. Zh. Nevrol. Psikhiatr. Im. S. S. Korsakova. 2014, 114, 52–58. [Google Scholar] [PubMed]
- Altunel, A.; Altunel, E.Ö.; Sever, A. Response to adrenocorticotropic in attention deficit hyperactivity disorder-like symptoms in electrical status epilepticus in sleep syndrome is related to electroencephalographic improvement: A retrospective study. Epilepsy Behav. 2017, 74, 161–166. [Google Scholar] [CrossRef] [PubMed]
- van Iterson, L.; Vrij, S.; Sie, L.T.L.; Augustijn, P.B.; Rooze, A.C.S.; Jansen, F.E. Acquired visual agnosia as an uncommon presentation of epileptic encephalopathy in a 6-year-old boy with CSWS. Epilepsy Behav. Rep. 2021, 16, 100465. [Google Scholar] [CrossRef]
- Kuki, I.; Kawawaki, H.; Okazaki, S.; Ikeda, H.; Tomiwa, K. Epileptic encephalopathy with continuous spikes and waves in the occipito-temporal region during slow-wave sleep in two patients with acquired Kanji dysgraphia. Epileptic Disord. 2014, 16, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Tassinari, C.A.; Cantalupo, G.; Rubboli, G. Focal ESES as a selective focal brain dysfunction: A challenge for clinicians, an opportunity for cognitive neuroscientists. Epileptic Disord. 2015, 17, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Pavlidis, E.; Møller, R.S.; Nikanorova, M.; Kölmel, M.S.; Stendevad, P.; Beniczky, S.; Tassinari, C.A.; Rubboli, G.; Gardella, E. Idiopathic encephalopathy related to status epilepticus during slow sleep (ESES) as a “pure” model of epileptic encephalopathy. An electroclinical, genetic, and follow-up study. Epilepsy Behav. 2019, 97, 244–252. [Google Scholar] [CrossRef]
- Mohammadi, M.; Kowkabi, S.; Asadi-Pooya, A.A.; Malamiri, R.A.; Badv, R.S. Hemi-ESES associated with agenesis of the corpus callosum and normal cognition. Epilepsy Behav. Case Rep. 2019, 11, 96–98. [Google Scholar] [CrossRef]
- Caraballo, R.H.; Veggiotti, P.; Kaltenmeier, M.C.; Piazza, E.; Gamboni, B.; Lopez Avaria, M.F.; Noli, D.; Adi, J.; Cersosimo, R. Encephalopathy with status epilepticus during sleep or continuous spikes and waves during slow sleep syndrome: A multicenter, long-term follow-up study of 117 patients. Epilepsy Res. 2013, 105, 164–173. [Google Scholar] [CrossRef]
- Gencpinar, P.; Dundar, N.O.; Tekgul, H. Electrical status epilepticus in sleep (ESES)/continuous spikes and waves during slow sleep (CSWS) syndrome in children: An electroclinical evaluation according to the EEG patterns. Epilepsy Behav. 2016, 61, 107–111. [Google Scholar] [CrossRef]
- Shiraishi, H.; Haginoya, K.; Nakagawa, E.; Saitoh, S.; Kaneko, Y.; Nakasato, N.; Chan, D.; Otsubo, H. Magnetoencephalography localizing spike sources of atypical benign partial epilepsy. Brain Dev. 2014, 36, 21–27. [Google Scholar] [CrossRef]
- RamachandranNair, R. Encephalopathy associated with electrical status epilepticus of sleep (ESES): A practical approach. Indian J. Pediatr. 2020, 87, 1057–1061. [Google Scholar] [CrossRef]
- Nonclercq, A.; Foulon, M.; Verheulpen, D.; De Cock, C.; Buzatu, M.; Mathys, P.; Van Bogaert, P. Cluster-based spike detection algorithm adapts to interpatient and intrapatient variation in spike morphology. J. Neurosci. Methods. 2012, 210, 259–265. [Google Scholar] [CrossRef]
- Yu, Y.; Chen, Y.; Li, Y.; Gao, Z.; Gai, Z.; Zhou, Y. SQNN: A spike-wave index quantification neural network with a pre-labeling algorithm for epileptiform activity identification and quantification in children. J. Neural Eng. 2022, 19, 016040. [Google Scholar] [CrossRef] [PubMed]
- Sánchez Fernández, I.; Peters, J.M.; Hadjiloizou, S.; Prabhu, S.P.; Zarowski, M.; Stannard, K.M.; Takeoka, M.; Rotenberg, A.; Kothare, S.V.; Loddenkemper, T. Clinical staging and electroencephalographic evolution of continuous spikes and waves during sleep. Epilepsia. 2012, 53, 1185–1195. [Google Scholar] [CrossRef] [PubMed]
- Cantalupo, G.; Pavlidis, E.; Beniczky, S.; Avanzini, P.; Gardella, E.; Larsson, P.G. Quantitative EEG analysis in encephalopathy related to status epilepticus during slow sleep. Epileptic Disord. 2019, 21 (Suppl. S1), 31–40. [Google Scholar] [CrossRef]
- Sánchez Fernández, I.; Chapman, K.E.; Peters, J.M.; Kothare, S.V.; Nordli, D.R. Jr; Jensen, F.E.; Berg, A.T.; Loddenkemper, T. The tower of Babel: Survey on concepts and terminology in electrical status epilepticus in sleep and continuous spikes and waves during sleep in North America. Epilepsia. 2013, 54, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, D.; Mendes, T.; Dias, A.I.; Leal, A. Interictal spike quantification in continuous spike-wave of sleep (CSWS): Clinical usefulness of a wearable EEG device. Epilepsy Behav. 2020, 104, 106902. [Google Scholar] [CrossRef]
- Poothrikovil, R.P.; Koul, R.L.; Mani, R.; Al Futaisi, A. Evolution of Ohtahara syndrome to continuous spikes and waves during slow sleep in an infant. Neurodiagn J. 2012, 52, 261–274. [Google Scholar]
- Oguni, H.; Hirano, Y.; Nagata, S. Encephalopathy related to status epilepticus during slow sleep (ESES) as atypical evolution of Panayiotopoulos syndrome: An EEG and neuropsychological study. Epileptic Disord. 2020, 22, 67–72. [Google Scholar]
- Nava, E.; Mori, A.C.; Striano, P.; Ramantani, G. Atypical presentation of sunflower epilepsy featuring an EEG pattern of continuous spike waves during slow-wave sleep. Epileptic Disord. 2021, 23, 927–932. [Google Scholar] [CrossRef]
- Sánchez Fernández, I.; Chapman, K.E.; Peters, J.M.; Harini, C.; Rotenberg, A.; Loddenkemper, T. Continuous spikes and waves during sleep: Electroclinical presentation and suggestions for management. Epilepsy Res. Treat. 2013, 2013, 583531. [Google Scholar] [CrossRef]
- Arzimanoglou, A.; Cross, H.J. Cognitive impairment and behavioral disorders in encephalopathy related to status epilepticus during slow sleep: Diagnostic assessment and outcome. Epileptic Disord. 2019, 21 (Suppl. S1), 71–75. [Google Scholar] [CrossRef]
- Margari, L.; Buttiglione, M.; Legrottaglie, A.R.; Presicci, A.; Craig, F.; Curatolo, P. Neuropsychiatric impairment in children with continuous spikes and waves during slow sleep: A long-term follow-up study. Epilepsy Behav. 2012, 25, 558–562. [Google Scholar] [CrossRef]
- Curnow, S.R.; Vogrin, S.J.; Barton, S.; Bailey, C.A.; Harvey, A.S. Focal cortical hypermetabolism in atypical benign rolandic epilepsy. Epilepsy Res. 2020, 161, 106288. [Google Scholar] [CrossRef]
- Bölsterli Heinzle, B.K.; Fattinger, S.; Kurth, S.; Lebourgeois, M.K.; Ringli, M.; Bast, T.; Critelli, H.; Schmitt, B.; Huber, R. Spike wave location and density disturb sleep slow waves in patients with CSWS (continuous spike waves during sleep). Epilepsia. 2014, 55, 584–591. [Google Scholar] [CrossRef]
- Bölsterli, B.K.; Gardella, E.; Pavlidis, E.; Wehrle, F.M.; Tassinari, C.A.; Huber, R.; Rubboli, G. Remission of encephalopathy with status epilepticus (ESES) during sleep renormalizes regulation of slow wave sleep. Epilepsia. 2017, 58, 1892–1901. [Google Scholar] [CrossRef]
- Oser, N.; Hubacher, M.; Nageleisen-Weiss, A.; van Mierlo, P.; Huber, R.; Weber, P.; Bölsterli, B.K.; Datta, A.N. 6-year course of sleep homeostasis in a case with epilepsy-aphasia spectrum disorder. Epilepsy Behav. Rep. 2021, 16, 100488. [Google Scholar] [CrossRef]
- Bölsterli Heinzle, B.K.; Bast, T.; Critelli, H.; Huber, R.; Schmitt, B. Age-dependency of location of epileptic foci in “continuous spike-and-waves during sleep”: A parallel to the posterior-anterior trajectory of slow wave activity. Neuropediatrics. 2017, 48, 36–41. [Google Scholar]
- Filippini, M.; Arzimanoglou, A.; Gobbi, G. Neuropsychological approaches to epileptic encephalopathies. Epilepsia. 2013, 54 (Suppl. 8), 38–44. [Google Scholar] [CrossRef]
- Halász, P.; Szűcs, A. Sleep and epilepsy link by plasticity. Front Neurol. 2020, 11, 911. [Google Scholar] [CrossRef]
- Ng, R.; Hodges, E. Neurocognitive profiles of pediatric patients with ESES, generalized epilepsy, or focal epilepsy. Epilepsy Res. 2020, 167, 106351. [Google Scholar] [CrossRef]
- Panda, P.K.; Natarajan, V.; Sharawat, I.K. Neurocognitive profile of children with ESES, generalized, and focal epilepsy: Is there any difference? Epilepsy Res. 2021, 172, 106451. [Google Scholar] [CrossRef]
- Pesántez-Ríos, G.; Martínez-Bermejo, A.; Arcas, J.; Merino-Andreu, M.; Ugalde-Canitrot, A. Las evoluciones atipicas de la epilepsia rolandica son complicaciones predecibles [The atypical developments of rolandic epilepsy are predictable complications]. Rev. Neurol. 2015, 61, 106–113. [Google Scholar]
- Porat Rein, A.; Kramer, U.; Mitelpunkt, A. Development of ontology for self-limited epilepsy with centrotemporal spikes and application of data mining algorithms to identify new subtypes. Isr. Med. Assoc. J. 2019, 21, 503. [Google Scholar]
- Porat Rein, A.; Kramer, U.; Hausman Kedem, M.; Fattal-Valevski, A.; Mitelpunkt, A. Early prediction of encephalopathic transformation in children with benign epilepsy with centro-temporal spikes. Brain Dev. 2021, 43, 268–279. [Google Scholar] [CrossRef]
- Desprairies, C.; Dozières-Puyravel, B.; Ilea, A.; Bellavoine, V.; Nasser, H.; Delanöe, C.; Auvin, S. Early identification of epileptic encephalopathy with continuous spikes-and-waves during sleep: A case-control study. Eur. J. Paediatr. Neurol. 2018, 22, 837–844. [Google Scholar] [CrossRef]
- Caraballo, R.; Pavlidis, E.; Nikanorova, M.; Loddenkemper, T. Encephalopathy with continuous spike-waves during slow-wave sleep: Evolution and prognosis. Epileptic Disord. 2019, 21 (Suppl. S1), 15–21. [Google Scholar] [CrossRef]
- Aeby, A.; Santalucia, R.; Van Hecke, A.; Nebbioso, A.; Vermeiren, J.; Deconinck, N.; DeTiège, X.; Van Bogaert, P. A qualitative awake EEG score for the diagnosis of continuous spike and waves during sleep (CSWS) syndrome in self-limited focal epilepsy (SFE): A case-control study. Seizure. 2021, 84, 34–39. [Google Scholar] [CrossRef]
- Peltola, M.E.; Palmu, K.; Liukkonen, E.; Gaily, E.; Vanhatalo, S. Semiautomatic quantification of spiking in patients with continuous spikes and waves in sleep: Sensitivity to settings and correspondence to visual assessment. Clin. Neurophysiol. 2012, 123, 1284–1290. [Google Scholar] [CrossRef]
- Peltola, M.E.; Sairanen, V.; Gaily, E.; Vanhatalo, S. Measuring spike strength in patients with continuous spikes and waves during sleep: Comparison of methods for prospective use as a clinical index. Clin. Neurophysiol. 2014, 125, 1639–1646. [Google Scholar] [CrossRef]
- Ouyang, G.; Wang, Y.; Yang, Z.; Li, X. Global synchronization of multichannel EEG in patients with electrical status epilepticus in sleep. Clin. EEG Neurosci. 2015, 46, 357–363. [Google Scholar] [CrossRef]
- Balaram, N.; Jose, J.; Gafoor, A.V.; Balachandran, S. Classification of electrical status epilepticus in sleep based on EEG patterns and spatiotemporal mapping of spikes. Epileptic Disord. 2022, 24, 1060–1072. [Google Scholar] [CrossRef]
- De Tiège, X.; Trotta, N.; Op de Beeck, M.; Bourguignon, M.; Marty, B.; Wens, V.; Nonclercq, A.; Goldman, S.; Van Bogaert, P. Neurophysiological activity underlying altered brain metabolism in epileptic encephalopathies with CSWS. Epilepsy Res. 2013, 105, 316–325. [Google Scholar] [CrossRef]
- Japaridze, N.; Muthuraman, M.; Dierck, C.; von Spiczak, S.; Boor, R.; Mideksa, K.G.; Anwar, R.A.; Deuschl, G.; Stephani, U.; Siniatchkin, M. Neuronal networks in epileptic encephalopathies with CSWS. Epilepsia. 2016, 57, 1245–1255. [Google Scholar] [CrossRef]
- Magara, S.; Komatsubara, T.; Hojo, M.; Kobayashi, Y.; Yoshino, M.; Saitoh, A.; Tohyama, J. The association of epileptic focus estimated by magnetoencephalography with cognitive function in non-lesional epilepsy with continuous spikes and waves during slow wave sleep (ECSWS) children. Brain Dev. 2019, 41, 163–172. [Google Scholar] [CrossRef]
- Li, Y.; Li, Y.; Sun, J.; Niu, K.; Wang, P.; Xu, Y.; Wang, Y.; Chen, Q.; Zhang, K.; Wang, X. Relationship between brain activity, cognitive function, and sleep spiking activation in new-onset self-limited epilepsy with centrotemporal spikes. Front. Neurol. 2022, 13, 956838. [Google Scholar] [CrossRef]
- Moeller, F.; Moehring, J.; Ick, I.; Steinmann, E.; Wolff, S.; Jansen, O.; Boor, R.; Stephani, U.; Siniatchkin, M. EEG-fMRI in atypical benign partial epilepsy. Epilepsia. 2013, 54, e103–e108. [Google Scholar] [CrossRef]
- Siniatchkin, M.; Van Bogaert, P. Pathophysiology of encephalopathy related to continuous spike and waves during sleep: The contribution of neuroimaging. Epileptic Disord. 2019, 21 (Suppl. S1), 48–53. [Google Scholar] [CrossRef]
- Toda, Y.; Kobayashi, K.; Hayashi, Y.; Inoue, T.; Oka, M.; Ohtsuka, Y. Effects of intravenous diazepam on high-frequency oscillations in EEGs with CSWS. Brain Dev. 2013, 35, 540–547. [Google Scholar] [CrossRef]
- Ohuchi, Y.; Akiyama, T.; Matsuhashi, M.; Kobayashi, K. High-frequency oscillations in a spectrum of pediatric epilepsies characterized by sleep-activated spikes in scalp EEG. Clin. Neurophysiol. 2019, 130, 1971–1980. [Google Scholar] [CrossRef]
- Cao, D.; Chen, Y.; Liao, J.; Nariai, H.; Li, L.; Zhu, Y.; Zhao, X.; Hu, Y.; Wen, F.; Zhai, Q. Scalp EEG high frequency oscillations as a biomarker of treatment response in epileptic encephalopathy with continuous spike-and-wave during sleep (CSWS). Seizure. 2019, 71, 151–157. [Google Scholar] [CrossRef]
- Gong, P.; Yang, Z.X.; Xue, J.; Qian, P.; Yang, H.P.; Liu, X.Y.; Bian, K.G. [Application of scalp-recorded high-frequency oscillations in epileptic encephalopathy with continuous spike-and-wave during sleep]. Beijing Da Xue Xue Bao Yi Xue Ban. 2018, 50, 213–220. [Google Scholar]
- Gong, P.; Xue, J.; Qian, P.; Yang, H.; Liu, X.; Cai, L.; Bian, K.; Yang, Z. Scalp-recorded high-frequency oscillations in childhood epileptic encephalopathy with continuous spike-and-wave during sleep with different etiologies. Brain Dev. 2018, 40, 299–310. [Google Scholar] [CrossRef]
- Giacomini, T.; Luria, G.; D’Amario, V.; Croci, C.; Cataldi, M.; Piai, M.; Nobile, G.; Bruni, O.; Consales, A.; Mancardi, M.M.; et al. On the role of REM sleep microstructure in suppressing interictal spikes in electrical status epilepticus during sleep. Clin. Neurophysiol. 2022, 136, 62–68. [Google Scholar] [CrossRef]
- Filippini, M.; Boni, A.; Giannotta, M.; Pini, A.; Russo, A.; Musti, M.A.; Guerra, A.; Lassonde, M.; Gobbi, G. Comparing cortical auditory processing in children with typical and atypical benign epilepsy with centrotemporal spikes: Electrophysiologic evidence of the role of non-rapid eye movement sleep abnormalities. Epilepsia. 2015, 56, 726–734. [Google Scholar] [CrossRef]
- Arican, P.; Gencpinar, P.; Olgac Dundar, N.; Tekgul, H. Electrical status epilepticus during slow-wave sleep (ESES): Current perspectives. J. Pediatr. Neurosci. 2021, 16, 91–96. [Google Scholar]
- Zhang, K.; Yan, Y.; Su, T. Treatment strategies for encephalopathy related to status epilepticus during slow sleep, a narrative review of the literature. Rev. Neurosci. 2020, 31, 793–802. [Google Scholar] [CrossRef]
- Carreño, M.; Fernández, S. Sleep-related epilepsy. Curr. Treat. Options Neurol. 2016, 18, 23. [Google Scholar] [CrossRef]
- Raha, S.; Shah, U.; Udani, V. Neurocognitive and neurobehavioral disabilities in epilepsy with electrical status epilepticus in slow sleep (ESES) and related syndromes. Epilepsy Behav. 2012, 25, 381–385. [Google Scholar] [CrossRef]
- McTague, A.; Cross, J.H. Treatment of epileptic encephalopathies. CNS Drugs. 2013, 27, 175–184. [Google Scholar] [CrossRef]
- Hani, A.J.; Mikati, M.A. Current and emerging therapies of severe epileptic encephalopathies. Semin. Pediatr. Neurol. 2016, 23, 180–186. [Google Scholar] [CrossRef]
- van den Munckhof, B.; Alderweireld, C.; Davelaar, S.; van Teeseling, H.C.; Nikolakopoulos, S.; Braun, K.P.J.; Jansen, F.E. Treatment of electrical status epilepticus in sleep: Clinical and EEG characteristics and response to 147 treatments in 47 patients. Eur. J. Paediatr. Neurol. 2018, 22, 64–71. [Google Scholar] [CrossRef]
- Sánchez Fernández, I.; Chapman, K.; Peters, J.M.; Klehm, J.; Jackson, M.C.; Berg, A.T.; Loddenkemper, T. Treatment for continuous spikes and waves during sleep (CSWS): Survey on treatment choices in North America. Epilepsia. 2014, 55, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Kessi, M.; Yan, F.; Pan, L.; Chen, B.; Olatoutou, E.; Li, D.; He, F.; Rugambwa, T.; Yang, L.; Peng, J.; et al. Treatment for the benign childhood epilepsy with centrotemporal spikes: A monocentric study. Front. Neurol. 2021, 12, 670958. [Google Scholar] [CrossRef]
- Samanta, D.; Willis, E.; Sharp, G.B. Absence status after starting clobazam in a patient with syndrome of continuous spike and wave during slow sleep (CSWS). Neurol. India. 2014, 62, 685–687. [Google Scholar] [CrossRef]
- Fejerman, N.; Caraballo, R.; Cersósimo, R.; Ferraro, S.M.; Galicchio, S.; Amartino, H. Sulthiame add-on therapy in children with focal epilepsies associated with encephalopathy related to electrical status epilepticus during slow sleep (ESES). Epilepsia. 2012, 53, 1156–1161. [Google Scholar] [CrossRef]
- Kanmaz, S.; Simsek, E.; Serin, H.M.; Yilmaz, S.; Aktan, G.; Tekgul, H.; Gokben, S. Sulthiame add-on treatment in children with epileptic encephalopathy with status epilepticus: An efficacy analysis in etiologic subgroups. Neurol. Sci. 2021, 42, 183–191. [Google Scholar] [CrossRef]
- Topçu, Y.; Kılıç, B.; Tekin, H.G.; Aydın, K.; Turanlı, G. Effects of sulthiame on seizure frequency and EEG in children with electrical status epilepticus during slow sleep. Epilepsy Behav. 2021, 116, 107793. [Google Scholar] [CrossRef]
- Kanemura, H.; Sano, F.; Sugita, K.; Aihara, M. Effects of levetiracetam on seizure frequency and neuropsychological impairments in children with refractory epilepsy with secondary bilateral synchrony. Seizure. 2013, 22, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Li, F.; Zhong, H. Early diagnosis, treatment and prognosis of epilepsy with continuous spikes and waves during slow sleep. Int. J. Clin. Exp. Med. 2015, 8, 4052–4058. [Google Scholar] [PubMed]
- Chen, J.; Cai, F.; Jiang, L.; Hu, Y.; Feng, C. Levetiracetam efficacy in children with epilepsy with electrical status epilepticus in sleep. Epilepsy Behav. 2015, 44, 73–77. [Google Scholar] [CrossRef]
- Zhang, J. The effectiveness and safety of hormonal combinations of antiepileptic drugs in the treatment of epileptic electrical continuity in children during sleep: A meta-analysis. Comput. Intell. Neurosci. 2022, 2022, 5395383. [Google Scholar] [CrossRef] [PubMed]
- Fine, A.L.; Wirrell, E.C.; Wong-Kisiel, L.C.; Nickels, K.C. Acetazolamide for electrical status epilepticus in slow-wave sleep. Epilepsia. 2015, 56, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Fatema, K.; Rahman, M.M.; Begum, S. Characteristics and management of children with continuous spikes and waves during slow sleep. Mymensingh Med. J. 2015, 24, 806–812. [Google Scholar] [PubMed]
- Su, T.F.; Xu, S.Q.; Chen, L. [Efficacy of levetiracetam combined with short-term clonazepam in treatment of electrical status epilepticus during sleep in children with benign childhood epilepsy with centrotemporal spikes]. Zhongguo Dang Dai Er Ke Za Zhi. 2014, 16, 829–833. [Google Scholar] [PubMed]
- Wiwattanadittakul, N.; Depositario-Cabacar, D.; Zelleke, T.G. Electrical status epilepticus in sleep (ESES) - Treatment pattern and EEG outcome in children with very high spike-wave index. Epilepsy Behav. 2020, 105, 106965. [Google Scholar] [CrossRef]
- Vrielynck, P.; Marique, P.; Ghariani, S.; Lienard, F.; de Borchgrave, V.; van Rijckevorsel, K.; Bonnier, C. Topiramate in childhood epileptic encephalopathy with continuous spike-waves during sleep: A retrospective study of 21 cases. Eur. J. Paediatr. Neurol. 2017, 21, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cai, F.; Jiang, L.; Hu, Y.; Feng, C. A prospective study of dexamethasone therapy in refractory epileptic encephalopathy with continuous spike-and-wave during sleep. Epilepsy Behav. 2016, 55, 1–5. [Google Scholar] [CrossRef]
- Bast, T.; Richter, S.; Ebinger, F.; Rating, D.; Wiemer-Kruel, A.; Schubert-Bast, S. Efficacy and tolerability of methylprednisolone pulse therapy in childhood epilepsies other than infantile spasms. Neuropediatrics. 2014, 45, 378–385. [Google Scholar]
- Hempel, A.; Frost, M.; Agarwal, N. Language and behavioral outcomes of treatment with pulse-dose prednisone for electrical status epilepticus in sleep (ESES). Epilepsy Behav. 2019, 94, 93–99. [Google Scholar] [CrossRef]
- Jauhari, P.; Madaan, P.; Sirolia, V.; Sharma, S.; Chakrabarty, B.; Gulati, S. Neurobehavioral deterioration associated with sleep-augmented epileptiform abnormalities: A steroid responsive state in children. Epilepsy Behav. 2022, 129, 108505. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.P.; Dai, Y.Y. [A clinical analysis of electrical status epilepticus during sleep in children and a follow-up study of methylprednisolone pulse therapy]. Zhongguo Dang Dai Er Ke Za Zhi. 2019, 21, 348–353. [Google Scholar] [PubMed]
- Chen, J.; Yang, Z.; Liu, X.; Ji, T.; Fu, N.; Wu, Y.; Xiong, H.; Wang, S.; Chang, X.; Zhang, Y.; et al. [Efficacy of methylprednisolone therapy for electrical status epilepticus during sleep in children]. Zhonghua Er Ke Za Zhi. 2014, 52, 678–682. [Google Scholar]
- Baumer, F.M.; McNamara, N.A.; Fine, A.L.; Pestana-Knight, E.; Shellhaas, R.A.; He, Z.; Arndt, D.H.; Gaillard, W.D.; Kelley, S.A.; Nagan, M.; et al. Treatment practices and outcomes in continuous spike and wave during slow wave sleep: A multicenter collaboration. J. Pediatr. 2021, 232, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Kılıç, B.; Acar, M.; Topçu, Y.; Turanlı, G. Epileptic encephalopathy with electrical status epilepticus during slow sleep: Evaluation of treatment response from a tertiary center. Turk. J. Pediatr. 2022, 64, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Jyonouchi, H.; Geng, L. Resolution of EEG findings and clinical improvement in a patient with encephalopathy and ESES with a combination of immunomodulating agents other than corticosteroids: A case report. Epilepsy Behav. Rep. 2020, 14, 100379. [Google Scholar] [CrossRef]
- Ville, D.; Chiron, C.; Laschet, J.; Dulac, O. The ketogenic diet can be used successfully in combination with corticosteroids for epileptic encephalopathies. Epilepsy Behav. 2015, 48, 61–65. [Google Scholar] [CrossRef]
- Reyes, G.; Flesler, S.; Armeno, M.; Fortini, S.; Ariela, A.; Cresta, A.; Mestre, G.; Caraballo, R.H. Ketogenic diet in patients with epileptic encephalopathy with electrical status epilepticus during slow sleep. Epilepsy Res. 2015, 113, 126–131. [Google Scholar] [CrossRef]
- Kelley, S.A.; Kossoff, E.H. How effective is the ketogenic diet for electrical status epilepticus of sleep? Epilepsy Res. 2016, 127, 339–343. [Google Scholar] [CrossRef]
- Pasca, L.; Caraballo, R.H.; De Giorgis, V.; Reyes, J.G.; Macasaet, J.A.; Masnada, S.; Armeno, M.; Musicco, M.; Tagliabue, A.; Veggiotti, P. Ketogenic diet use in children with intractable epilepsy secondary to malformations of cortical development: A two-centre experience. Seizure. 2018, 57, 34–37. [Google Scholar] [CrossRef]
- Kotagal, P. Current status of treatments for children with electrical status in slow-wave sleep (ESES/CSWS). Epilepsy Curr. 2017, 17, 214–216. [Google Scholar] [CrossRef] [PubMed]
- Uliel-Sibony, S.; Kramer, U. Benign childhood epilepsy with centro-temporal spikes (BCECTSs), electrical status epilepticus in sleep (ESES), and academic decline--how aggressive should we be? Epilepsy Behav. 2015, 44, 117–120. [Google Scholar] [CrossRef]
- Wilson, R.B.; Eliyan, Y.; Sankar, R.; Hussain, S.A. Amantadine: A new treatment for refractory electrical status epilepticus in sleep. Epilepsy Behav. 2018, 84, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Kanemura, H.; Sano, F.; Hoshino, H.; Takayama, K.; Aihara, M. Effects of perampanel on secondary bilateral synchrony and behavioral problems in adolescents with epilepsy showing insufficient response with levetiracetam. Seizure. 2020, 80, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Teng, Z.T.; Liu, X.Y.; Wang, H. Effectiveness of perampanel in the treatment of pediatric patients with focal epilepsy and ESES: A single-center retrospective study. Front. Pharmacol. 2022, 13, 1026836. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Huang, S.; Wang, X.; Yang, L.; Song, T. Efficacy and adverse reactions of perampanel in the treatment of epilepsy in children. Front. Neurol. 2022, 13, 924057. [Google Scholar] [CrossRef] [PubMed]
- Grosso, S.; Parisi, P.; Giordano, L.; di Bartolo, R.; Balestri, P. Lacosamide efficacy in epileptic syndromes with continuous spike and waves during slow sleep (CSWS). Epilepsy Res. 2014, 108, 1604–1608. [Google Scholar] [CrossRef]
- van den Munckhof, B.; van Dee, V.; Sagi, L.; Caraballo, R.H.; Veggiotti, P.; Liukkonen, E.; Loddenkemper, T.; Sánchez Fernández, I.; Buzatu, M.; Bulteau, C.; et al. Treatment of electrical status epilepticus in sleep: A pooled analysis of 575 cases. Epilepsia. 2015, 56, 1738–1746. [Google Scholar] [CrossRef]
- Moresco, L.; Bruschettini, M.; Calevo, M.G.; Siri, L. Pharmacological treatment for continuous spike-wave during slow wave sleep syndrome and Landau-Kleffner syndrome. Cochrane Database Syst. Rev. 2020, 11, CD013132. [Google Scholar] [CrossRef]
- van den Munckhof, B.; Arzimanoglou, A.; Perucca, E.; van Teeseling, H.C.; Leijten, F.S.S.; Braun, K.P.J.; Jansen, F.E.; RESCUE ESES study group. Corticosteroids versus clobazam in epileptic encephalopathy with ESES: A European multicentre randomised controlled clinical trial (RESCUE ESES*). Trials. 2020, 21, 957. [Google Scholar] [CrossRef]
- Liu, M.; Ji, T.; Liu, Q.; Wang, S.; Wu, Y.; Wang, W.; Cheng, W.; Wang, R.; Yu, G.; Liu, X.; et al. Generalized seizures presurgically in a cohort of children with hemispherectomy: Predictors and a potential link to surgical outcome? Seizure. 2018, 58, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Gröppel, G.; Dorfer, C.; Dressler, A.; Mühlebner, A.; Porsche, B.; Hainfellner, J.A.; Czech, T.; Feucht, M. Immediate termination of electrical status epilepticus in sleep after hemispherotomy is associated with significant progress in language development. Dev. Med. Child Neurol. 2017, 59, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Weil, A.G.; Ibrahim, G.M.; Fallah, A.; Korman, B.; Ragheb, J.; Bhatia, S.; Duchowny, M. Surgical management of pediatric patients with encephalopathy due to electrical status epilepticus during sleep (ESES). Epileptic Disord. 2020, 22, 39–54. [Google Scholar] [CrossRef]
- Marashly, A.; Koop, J.; Loman, M.; Lee, Y.W.; Lew, S.M. Examining the utility of resective epilepsy surgery in children with electrical status epilepticus in sleep: Long term clinical and electrophysiological outcomes. Front. Neurol. 2020, 10, 1397. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.T. Increasing blurriness of the borders between focal and generalized as well as cryptogenic and idiopathic epilepsies in defining the role for focal epilepsy surgery. Semin. Pediatr. Neurol. 2014, 21, 104–105. [Google Scholar] [CrossRef]
- Alawadhi, A.; Appendino, J.P.; Hader, W.; Rosenblatt, B.; Moreau, J.T.; Dubeau, F.; Dudley, R.W.R.; Myers, K.A. Surgically remediable secondary network epileptic encephalopathies with continuous spike wave in sleep: Lesions may not be visible on brain magnetic resonance imaging (MRI). J Child Neurol. 2022, 37, 992–1002. [Google Scholar] [CrossRef]
- Yokosako, S.; Muraoka, N.; Watanabe, S.; Kosugi, K.; Takayama, Y.; Iijima, K.; Kimura, Y.; Kaneko, Y.; Sumitomo, N.; Saito, T.; et al. Corpus callosotomy in pediatric patients with non-lesional epileptic encephalopathy with electrical status epilepticus during sleep. Epilepsy Behav. Rep. 2021, 16, 100463. [Google Scholar] [CrossRef]
- Jeong, A.; Strahle, J.; Vellimana, A.K.; Limbrick, D.D. Jr; Smyth, M.D.; Bertrand, M. Hemispherotomy in children with electrical status epilepticus of sleep. J. Neurosurg. Pediatr. 2017, 19, 56–62. [Google Scholar] [CrossRef]
- Carosella, C.M.; Greiner, H.M.; Byars, A.W.; Arthur, T.M.; Leach, J.L.; Turner, M.; Holland, K.D.; Mangano, F.T.; Arya, R. Vagus nerve stimulation for electrographic status epilepticus in slow-wave sleep. Pediatr. Neurol. 2016, 60, 66–70. [Google Scholar] [CrossRef]
- Faria, P.; Fregni, F.; Sebastião, F.; Dias, A.I.; Leal, A. Feasibility of focal transcranial DC polarization with simultaneous EEG recording: Preliminary assessment in healthy subjects and human epilepsy. Epilepsy Behav. 2012, 25, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Jellinek, E.R.; Duda, T.A.; Fein, R.H. The RUBI parent training for disruptive behavior in a child with electrical status epilepticus in sleep (ESES): A case report. J. Clin. Psychol. Med. Settings. 2023, 30, 770–779. [Google Scholar] [CrossRef]
- Yilmaz, S.; Serdaroglu, G.; Akcay, A.; Gokben, S. Clinical characteristics and outcome of children with electrical status epilepticus during slow wave sleep. J. Pediatr. Neurosci. 2014, 9, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Pera, M.C.; Brazzo, D.; Altieri, N.; Balottin, U.; Veggiotti, P. Long-term evolution of neuropsychological competences in encephalopathy with status epilepticus during sleep: A variable prognosis. Epilepsia. 2013, 54 (Suppl. 7), 77–85. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, M.; Siegler, Z.; Fogarasi, A.; Barsi, P.; Halász, P. Long term follow-up of lesional and non-lesional patients with electrical status epilepticus in slow wave sleep. Ideggyogy. Sz. 2016, 69, 21–28. [Google Scholar] [CrossRef]
- Maltoni, L.; Posar, A.; Parmeggiani, A. Long-term follow-up of cognitive functions in patients with continuous spike-waves during sleep (CSWS). Epilepsy Behav. 2016, 60, 211–217. [Google Scholar] [CrossRef]
- De Giorgis, V.; Filippini, M.; Macasaet, J.A.; Masnada, S.; Veggiotti, P. Neurobehavioral consequences of continuous spike and waves during slow sleep (CSWS) in a pediatric population: A pattern of developmental hindrance. Epilepsy Behav. 2017, 74, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Escobar Fernández, L.; Coccolo Góngora, A.; Vázquez López, M.; Polo Arrondo, A.P.; Miranda Herrero, M.C.; Barredo Valderrama, E.; Castro de Castro, P. Patrón punta-onda continua en el sueño lento: Nuestra experiencia durante 20 años [Continuous spike-waves during slow-wave sleep: Experience during 20 years]. An. Pediatr. (Engl. Ed.). 2019, 91, 180–188. [Google Scholar] [CrossRef]
- Saraf, U.U.; Asranna, A.; Menon, R.N.; Mohan, P.M.; Vp, V.; Radhakrishnan, A.; Cherian, A.; Thomas, S.V. Predictors of one-year language and seizure outcomes in children with epileptic encephalopathy with continuous spike-and-wave during sleep (CSWS). Seizure. 2020, 81, 315–324. [Google Scholar] [CrossRef]
- Öztoprak, Ü.; Yayici Köken, Ö.; Aksoy, E.; Yüksel, D. Spike-wave index assessment and electro-clinical correlation in patients with encephalopathy associated with epileptic state during slow sleep (ESES / CSWS); Single-center experience. Epilepsy Res. 2021, 170, 106549. [Google Scholar] [CrossRef] [PubMed]
- Gardella, E.; Cantalupo, G.; Larsson, P.G.; Fontana, E.; Bernardina, B.D.; Rubboli, G.; Darra, F. EEG features in encephalopathy related to status epilepticus during slow sleep. Epileptic Disord. 2019, 21, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Pulsipher, D.T.; Stanford, L.D. Serial neuropsychological testing before and after hemispherectomy in a child with electrical status epilepticus in slow wave sleep. Epilepsy Behav. Rep. 2022, 18, 100539. [Google Scholar] [CrossRef]
- Ucar, H.K.; Arhan, E.; Aydin, K.; Hirfanoglu, T.; Serdaroglu, A. Electrical status epilepticus during sleep (ESES) in benign childhood epilepsy with centrotemporal spikes (BCECTS): Insights into predictive factors, and clinical and EEG outcomes. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 1885–1896. [Google Scholar] [PubMed]
- Tuft, M.; Årva, M.; Bjørnvold, M.; Wilson, J.A.; Nakken, K.O. Landau-Kleffner syndrome. Tidsskr. Nor. Laegeforen. 2015, 135, 2061–2064. [Google Scholar] [CrossRef]
- Lo, H.Y.; Ke, D.S.; Chaou, W.T. Landau-Kleffner syndrome: An acquired epileptic aphasia. Acta Neurol. Taiwan. 2015, 24, 34–35. [Google Scholar] [PubMed]
- Muzio, M.R.; Cascella, M.; Al Khalili, Y. Landau-Kleffner Syndrome. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Van Bogaert, P.; King, M.D.; Paquier, P.; Wetzburger, C.; Labasse, C.; Dubru, J.M.; Deonna, T. Acquired auditory agnosia in childhood and normal sleep electroencephalography subsequently diagnosed as Landau-Kleffner syndrome: A report of three cases. Dev. Med. Child Neurol. 2013, 55, 575–579. [Google Scholar] [CrossRef]
- Chowdhury, N.; Bansal, A.R.; Goyal, R.; Nikhila, G. Cerebral dominance in an unusual case of Landau-Kleffner syndrome. BMJ Case Rep. 2021, 14, e246696. [Google Scholar] [CrossRef]
- Van Bogaert, P. Epileptic encephalopathy with continuous spike-waves during slow-wave sleep including Landau-Kleffner syndrome. Handb. Clin. Neurol. 2013, 111, 635–640. [Google Scholar]
- Lévêque, Y.; Roulet-Perez, E.; Deonna, T.; Moulin, A.; Fornoni, L.; Mayor-Dubois, C.; Caclin, A.; Tillmann, B. Music processing deficits in Landau-Kleffner syndrome: Four case studies in adulthood. Cortex. 2020, 129, 99–111. [Google Scholar] [CrossRef]
- Duffy, F.H.; Eksioglu, Y.Z.; Rotenberg, A.; Madsen, J.R.; Shankardass, A.; Als, H. The frequency modulated auditory evoked response (FMAER), a technical advance for study of childhood language disorders: Cortical source localization and selected case studies. BMC Neurol. 2013, 13, 12. [Google Scholar] [CrossRef] [PubMed]
- Kawai, M.; Abe, Y.; Yumoto, M.; Kubota, M. Aphasia and a dual-stream language model in a 4-year-old female with Landau-Kleffner syndrome. Neuropediatrics. 2022, 53, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Pullens, P.; Pullens, W.; Blau, V.; Sorger, B.; Jansma, B.M.; Goebel, R. Evidence for normal letter-sound integration, but altered language pathways in a case of recovered Landau-Kleffner syndrome. Brain Cogn. 2015, 99, 32–45. [Google Scholar] [CrossRef]
- Giovanardi Rossi, P.; Parmeggiani, A.; Posar, A.; Scaduto, M.C.; Chiodo, S.; Vatti, G. Landau-Kleffner syndrome (LKS): Long-term follow-up and links with electrical status epilepticus during sleep (ESES). Brain Dev. 1999, 21, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Caraballo, R.H.; Cejas, N.; Chamorro, N.; Kaltenmeier, M.C.; Fortini, S.; Soprano, A.M. Landau-Kleffner syndrome: A study of 29 patients. Seizure. 2014, 23, 98–104. [Google Scholar] [CrossRef]
- Uğur, C.; Saday Duman, N.; Bektaş, O.; Kağan Gürkan, C. Antiepileptic treatment in a child with Landau Kleffner syndrome: A case report. Turk Psikiyatri Derg. 2014, 25, 282–286. [Google Scholar]
- Riccio, C.A.; Vidrine, S.M.; Cohen, M.J.; Acosta-Cotte, D.; Park, Y. Neurocognitive and behavioral profiles of children with Landau-Kleffner syndrome. Appl. Neuropsychol. Child. 2017, 6, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Besag, F.M.C.; Vasey, M.J. Social cognition and psychopathology in childhood and adolescence. Epilepsy Behav. 2019, 100, 106210. [Google Scholar] [CrossRef]
- Ewen, J.B.; Marvin, A.R.; Law, K.; Lipkin, P.H. Epilepsy and autism severity: A study of 6,975 children. Autism Res. 2019, 12, 1251–1259. [Google Scholar] [CrossRef]
- Sigafoos, J.; O’Reilly, M.F.; Ledbetter-Cho, K.; Lim, N.; Lancioni, G.E.; Marschik, P.B. Addressing sequelae of developmental regression associated with developmental disabilities: A systematic review of behavioral and educational intervention studies. Neurosci. Biobehav. Rev. 2019, 96, 56–71. [Google Scholar] [CrossRef]
- Peng, B.W.; Zhu, H.X.; Wang, X.Y.; Li, X.J.; Liang, H.C.; Li, J.L.; Zhang, F.Q.; Ning, S.Y.; Zhong, Y.Y.; Chen, W.X. A follow-up study in children with status epilepticus during sleep: From clinical spectrum to outcome. Epilepsy Behav. 2021, 117, 107843. [Google Scholar] [CrossRef]
- Ahmed, M.; Saleem, A.; Nasir, S.; Ariff, M.; Iftikhar, P. Landau-Kleffner syndrome: A diagnostic challenge. Cureus. 2020, 12, e7182. [Google Scholar] [CrossRef] [PubMed]
- Kaga, M.; Inagaki, M.; Ohta, R. Epidemiological study of Landau-Kleffner syndrome (LKS) in Japan. Brain Dev. 2014, 36, 284–286. [Google Scholar] [CrossRef]
- Plyler, E.; Harkrider, A.W. Serial auditory-evoked potentials in the diagnosis and monitoring of a child with Landau-Kleffner syndrome. J. Am. Acad. Audiol. 2013, 24, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Motwani, N.; Afsar, S.; Dixit, N.S.; Sharma, N. Landau-Kleffner syndrome: An uncommon dealt with case in Southeast Asia. BMJ Case Rep. 2015, 2015, bcr2015212333. [Google Scholar] [CrossRef]
- Nieh, S.E.; Sherr, EH. Epileptic encephalopathies: New genes and new pathways. Neurotherapeutics. 2014, 11, 796–806. [Google Scholar] [CrossRef]
- Tsai, M.H.; Vears, D.F.; Turner, S.J.; Smith, R.L.; Berkovic, S.F.; Sadleir, L.G.; Scheffer, IE. Clinical genetic study of the epilepsy-aphasia spectrum. Epilepsia. 2013, 54, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Zavadenko, N.N.; Kholin, A.A.; Zavadenko, A.N.; Michurina, E.S. Narusheniia razvitiiarechi pri épilepsii: Patofiziologicheskie mekhanizmy i terapevticheskie podkhody [Speech and language neurodevelopmental disorders in epilepsy: Pathophysiologic mechanisms and therapeutic approaches]. Zh. Nevrol. Psikhiatr. Im. S. S. Korsakova. 2018, 118, 118–125. [Google Scholar] [CrossRef]
- Nemati, R.; Nabipour, I.; Javadi, H.; Chabi, N.; Assadi, M. Regional cerebral blood-flow with 99mTc-ECD brain perfusion SPECT in Landau-Kleffner syndrome: Report of two cases. Case Rep. Radiol. 2014, 2014, 617343. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.N.; Oser, N.; Ramelli, G.P.; Gobbin, N.Z.; Lantz, G.; Penner, I.K.; Weber, P. BECTS evolving to Landau-Kleffner syndrome and back by subsequent recovery: A longitudinal language reorganization case study using fMRI, source EEG, and neuropsychological testing. Epilepsy Behav. 2013, 27, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Klimova, B.; Valis, M.; Hort, J.; Kuca, K. Selected rare paediatric communication neurological disorders. J. Appl. Biomed. 2019, 17, 33. [Google Scholar] [CrossRef] [PubMed]
- De Lima, T.A.; Zuanetti, P.A.; Nunes, M.E.N.; Hamad, A.P.A. Differential diagnosis between autism spectrum disorder and other developmental disorders with emphasis on the preschool period. World J. Pediatr. 2023, 19, 715–726. [Google Scholar] [CrossRef]
- Murugesan, B.G.; Jafroodifar, A.; Anilkumar, A.C.; Leontieva, L. Differential diagnosis of Landau-Kleffner syndrome versus post encephalitis syndrome in a 13-year-old boy with autism spectrum disorder. Cureus. 2020, 12, e9385. [Google Scholar] [CrossRef] [PubMed]
- Rezayi, A.; Feshangchi-Bonab, M.; Taherian, R. An uncommon presentation of mucopolysaccharidosis type IIIb. Iran. J. Child Neurol. 2019, 13, 105–111. [Google Scholar] [PubMed]
- Jokel, R.; Meloff, K. Acquired epileptiform aphasia: 44 years after diagnosis. Epilepsy Behav. Rep. 2020, 14, 100388. [Google Scholar] [CrossRef] [PubMed]
- Cockerell, I.; Bølling, G.; Nakken, KO. Landau-Kleffner syndrome in Norway: Long-term prognosis and experiences with the health services and educational systems. Epilepsy Behav. 2011, 21, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Fainberg, N.; Harper, A.; Tchapyjnikov, D.; Mikati, M.A. Response to immunotherapy in a patient with Landau-Kleffner syndrome and GRIN2A mutation. Epileptic Disord. 2016, 18, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Devinsky, O.; Goldberg, R.; Miles, D.; Bojko, A.; Riviello, J. Jr. Episodic epileptic verbal auditory agnosia in Landau Kleffner syndrome treated with combination diazepam and corticosteroids. J. Child Neurol. 2014, 29, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- Grote, C.L.; Van Slyke, P.; Hoeppner, J.A. Language outcome following multiple subpial transection for Landau-Kleffner syndrome. Brain. 1999, 122, 561–566. [Google Scholar] [CrossRef]
- Besag, F.; Caplan, R.; Aldenkamp, A.; Dunn, D.W.; Gobbi, G.; Sillanpää, M. Epileptic Disord. 2016, 18 (Suppl. 1), 68–76. Psychiatric and behavioural disorders in children with epilepsy (ILAE Task Force Report): Behavioural effects of epilepsy surgery.
- Downes, M.; Greenaway, R.; Clark, M.; Helen Cross, J.; Jolleff, N.; Harkness, W.; Kaliakatsos, M.; Boyd, S.; White, S.; Neville, B.G. Outcome following multiple subpial transection in Landau-Kleffner syndrome and related regression. Epilepsia. 2015, 56, 1760–1766. [Google Scholar] [CrossRef]
- Fine, A.; Nickels, K. Temporoparietal resection in a patient with Landau-Kleffner syndrome. Semin. Pediatr. Neurol. 2014, 21, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Hajtovic, S.; LoPresti, M.A.; Zhang, L.; Katlowitz, K.A.; Kizek, D.J.; Lam, S. The role of vagus nerve stimulation in genetic etiologies of drug-resistant epilepsy: A meta-analysis. J. Neurosurg. Pediatr. 2022, 29, 667–680. [Google Scholar] [CrossRef] [PubMed]
- van der Meulen, I.; Pangalila, R.F.; van de Sandt-Koenderman, W.M.E. Cognitive linguistic treatment in Landau Kleffner syndrome: Improvement in daily life communication. Child Neurol. Open. 2021, 8, 2329048X211022196. [Google Scholar] [PubMed]
- Pangalila, R.; van der Meulen, I. What is the effect of pharmacological treatment for continuous spike-wave during slow wave sleep syndrome and Landau-Kleffner syndrome? A Cochrane review summary with commentary. Dev. Med. Child Neurol. 2022, 64, 411–412. [Google Scholar] [CrossRef]
- Caraballo, R.H.; Cersósimo, R.O.; Fortini, P.S.; Ornella, L.; Buompadre, M.C.; Vilte, C.; Princich, J.P.; Fejerman, N. Congenital hemiparesis, unilateral polymicrogyria and epilepsy with or without status epilepticus during sleep: A study of 66 patients with long-term follow-up. Epileptic Disord. 2013, 15, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Q.; Zhang, W.N.; Hu, L.Y.; Liu, M.J.; Zou, L.P. Syndrome of electrical status epilepticus during sleep: Epileptic encephalopathy related to brain development. Pediatr. Neurol. 2016, 56, 35–41. [Google Scholar] [CrossRef]
- Sun, Q.Q.; Zhou, C.; Yang, W.; Petrus, D. Continuous spike-waves during slow-wave sleep in a mouse model of focal cortical dysplasia. Epilepsia. 2016, 57, 1581–1593. [Google Scholar] [CrossRef] [PubMed]
- Krueger, M.B.; Magalhães, S.C.; Pessoa, A.; Bueno, C.; Masruha, M.R.; Sobreira-Neto, M.A. Electrical status epilepticus during sleep in patients with congenital Zikavirus syndrome: An unprecedented clinical finding. Seizure. 2020, 81, 250–253. [Google Scholar] [CrossRef]
- He, W.; Liu, H.; Liu, Z.; Wu, Q. Electrical status epilepticus in sleep affects intrinsically connected networks in patients with benign childhood epilepsy with centrotemporal spikes. Epilepsy Behav. 2020, 106, 107032. [Google Scholar] [CrossRef]
- Issa, N.P. Neurobiology of continuous spike-wave in slow-wave sleep and Landau-Kleffner syndromes. Pediatr. Neurol. 2014, 51, 287–296. [Google Scholar] [CrossRef]
- Singhal, N.S.; Sullivan, J.E. Continuous spike-wave during slow wave sleep and related conditions. ISRN Neurol. 2014, 2014, 619079. [Google Scholar] [CrossRef]
- Gibbs, S.A.; Nobili, L.; Halász, P. Interictal epileptiform discharges in sleep and the role of the thalamus in encephalopathy related to status epilepticus during slow sleep. Epileptic Disord. 2019, 21, 54–61. [Google Scholar] [CrossRef]
- Losito, E.; Battaglia, D.; Chieffo, D.; Raponi, M.; Ranalli, D.; Contaldo, I.; Giansanti, C.; De Clemente, V.; Quintiliani, M.; Antichi, E.; et al. Sleep-potentiated epileptiform activity in early thalamic injuries: Study in a large series (60 cases). Epilepsy Res. 2015, 109, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.; Calado, E.; Vieira, J.P.; Mendonça, C.; Ferreira, J.C.; Ferreira, H.; Carvalho, D.; Furtado, F.; Gomes, R.; Monteiro, J.P. Anatomical and physiological basis of continuous spike-wave of sleep syndrome after early thalamic lesions. Epilepsy Behav. 2018, 78, 243–255. [Google Scholar] [CrossRef]
- Kersbergen, K.J.; de Vries, L.S.; Leijten, F.S.; Braun, K.P.; Nievelstein, R.A.; Groenendaal, F.; Benders, M.J.; Jansen, F.E. Neonatal thalamic hemorrhage is strongly associated with electrical status epilepticus in slow wave sleep. Epilepsia. 2013, 54, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Öztürk, Z.; Karalok, Z.S.; Güneş, A. Reduced thalamic volume is strongly associated with electrical status epilepticus in sleep. Acta Neurol. Belg. 2021, 121, 211–217. [Google Scholar] [CrossRef]
- Bartolini, E.; Falchi, M.; Zellini, F.; Parrini, E.; Grisotto, L.; Cosottini, M.; Posar, A.; Parmeggiani, A.; Ambrosetto, G.; Ferrari, A.R.; et al. The syndrome of polymicrogyria, thalamic hypoplasia, and epilepsy with CSWS. Neurology. 2016, 86, 1250–1259. [Google Scholar] [CrossRef] [PubMed]
- van den Munckhof, B.; Zwart, A.F.; Weeke, L.C.; Claessens, N.H.P.; Plate, J.D.J.; Leemans, A.; Kuijf, H.J.; van Teeseling, H.C.; Leijten, F.S.S.; Benders, M.J.N.; et al. Perinatal thalamic injury: MRI predictors of electrical status epilepticus in sleep and long-term neurodevelopment. Neuroimage Clin. 2020, 26, 102227. [Google Scholar] [CrossRef]
- Carvalho, D.; Mendonça, C.; Carvalho, J.; Martins, A.; Leal, A. High incidence of early thalamic lesions in the continuous spike-wave related with slow sleep (CSWS). Epilepsy Behav. 2023, 138, 109031. [Google Scholar] [CrossRef]
- Sánchez Fernández, I.; Peters, J.M.; Akhondi-Asl, A.; Klehm, J.; Warfield, S.K.; Loddenkemper, T. Reduced thalamic volume in patients with electrical status epilepticus in sleep. Epilepsy Res. 2017, 130, 74–80. [Google Scholar] [CrossRef]
- Agarwal, R.; Kumar, A.; Tiwari, V.N.; Chugani, H. Thalamic abnormalities in children with continuous spike-wave during slow-wave sleep: An F-18-fluorodeoxyglucose positron emission tomography perspective. Epilepsia. 2016, 57, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Kilic, H.; Yilmaz, K.; Asgarova, P.; Kizilkilic, O.; Hatay, G.H.; Ozturk-Isik, E.; Yalcinkaya, C.; Saltik, S. Electrical status epilepticus in sleep: The role of thalamus in etiopathogenesis. Seizure. 2021, 93, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Leal, A. Spatial and temporal dynamics of epileptic activity at sleep onset in the encephalopathy with status epilepticus during slow sleep (ESES) after unilateral thalamic lesions. Clin. Neurophysiol. 2021, 132, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Ligot, N.; Archambaud, F.; Trotta, N.; Goldman, S.; Van Bogaert, P.; Chiron, C.; De Tiège, X. Default mode network hypometabolism in epileptic encephalopathies with CSWS. Epilepsy Res. 2014, 108, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Azeem, A.; Kirton, A.; Appendino, J.P.; Kozlik, S.; Mineyko, A. Automated quantification of spike-wave activity may be used to predict the development of electrical status epilepticus in sleep (ESES) in children with perinatal stroke. Clin. Neurophysiol. 2021, 132, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Brandhoff, J.; de Vries, L.S.; Smal, J.C. Neonatale convulsies: Denk aan eensinustrombose [Neonatal seizures: Consider sinus thrombosis]. Ned. Tijdschr. Geneeskd. 2015, 159, A8055. [Google Scholar] [PubMed]
- Posar, A.; Parmeggiani, A. Neuropsychological impairment in early-onset hydrocephalus and epilepsy with continuous spike-waves during slow-wave sleep: A case report and literature review. J. Pediatr Neurosci. 2013, 8, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Taskin, B.D.; Tanji, K.; Feldstein, N.A.; McSwiggan-Hardin, M.; Akman, C.I. Epilepsy surgery for epileptic encephalopathy as a sequela of herpes simplex encephalitis: Case report. J. Neurosurg. Pediatr. 2017, 20, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Dedeoglu, Ö.; Altaş, H.; Yılmaz, D.; Gürkaş, E.; Gülleroğlu, B.; Ekşioğlu, S.; Çıtak Kurt, N. Corpus callosum thickness: A predictive factor for the first drug efficiency of self-limited epilepsy with centrotemporal spikes (selects)? Epilepsy Res. 2023, 190, 107072. [Google Scholar] [CrossRef]
- Hu, L.Y.; Shi, X.Y.; Feng, C.; Wang, J.W.; Yang, G.; Lammers, S.H.; Yang, X.F.; Ebrahimi-akhari, D.; Zou, L.P. An 8-year old boy with continuous spikes and waves during slow sleep presenting with positive onconeuronal antibodies. Eur. J. Paediatr. Neurol. 2015, 19, 257–261. [Google Scholar] [CrossRef]
- van den Munckhof, B.; de Vries, E.E.; Braun, K.P.; Boss, H.M.; Willemsen, M.A.; van Royen-Kerkhof, A.; de Jager, W.; Jansen, F.E. Serum inflammatory mediators correlate with disease activity in electrical status epilepticus in sleep (ESES) syndrome. Epilepsia. 2016, 57, e45–e50. [Google Scholar] [CrossRef] [PubMed]
- Ayça, S.; Aksoy, H.U.; Taştan, İ.; Polat, M. Levels of melatonin in continuous spikes and waves during sleep. J. Child Neurol. 2019, 34, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Tarcin, G.; Aksu Uzunhan, T.; Kacar, A.; Kucur, M.; Saltik, S. The relationship between epileptic seizure and melatonin in children. Epilepsy Behav. 2020, 112, 107345. [Google Scholar] [CrossRef]
- Nicita, F.; Verrotti, A.; Pruna, D.; Striano, P.; Capovilla, G.; Savasta, S.; Spartà, M.V.; Parisi, P.; Parlapiano, G.; Tarani, L.; et al. Seizures in fetal alcohol spectrum disorders: Evaluation of clinical, electroencephalographic, and neuroradiologic features in a pediatric case series. Epilepsia. 2014, 55, e60–e66. [Google Scholar] [CrossRef]
- Boronat, S.; Vicente, M.; Lainez, E.; Sánchez-Montañez, A.; Vázquez, E.; Mangado, L.; Martínez-Ribot, L.; Del Campo, M. Seizures and electroencephalography findings in 61 patients with fetal alcohol spectrum disorders. Eur. J. Med. Genet. 2017, 60, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Pavlidis, E.; Rubboli, G.; Nikanorova, M.; Kölmel, M.S.; Gardella, E. Encephalopathy with status epilepticus during sleep (ESES) induced by oxcarbazepine in idiopathic focal epilepsy in childhood. Funct. Neurol. 2015, 30, 139–141. [Google Scholar] [PubMed]
- Arkilo, D.; Devinsky, O.; Mudigoudar, B.; Boronat, S.; Jennesson, M.; Sassower, K.; Vaou, O.E.; Lerner, J.T.; Jeste, S.S.; Luchsinger, K.; et al. Electroencephalographic patterns during sleep in children with chromosome 15q11.2-13.1 duplications (Dup15q). Epilepsy Behav. 2016, 57, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Valvo, G.; Novara, F.; Brovedani, P.; Ferrari, A.R.; Guerrini, R.; Zuffardi, O.; Sicca, F. 22q11.2 microduplication syndrome and epilepsy with continuous spikes and waves during sleep (CSWS). A case report and review of the literature. Epilepsy Behav. 2012, 25, 567–572. [Google Scholar] [CrossRef]
- Lesca, G.; Rudolf, G.; Labalme, A.; Hirsch, E.; Arzimanoglou, A.; Genton, P.; Motte, J.; de Saint Martin, A.; Valenti, M.P.; Boulay, C.; et al. Epileptic encephalopathies of the Landau-Kleffner and continuous spike and waves during slow-wave sleep types: Genomic dissection makes the link with autism. Epilepsia. 2012, 53, 1526–1538. [Google Scholar] [CrossRef]
- Lemke, J.R.; Lal, D.; Reinthaler, E.M.; Steiner, I.; Nothnagel, M.; Alber, M.; Geider, K.; Laube, B.; Schwake, M.; Finsterwalder, K.; et al. Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes. Nat. Genet. 2013, 45, 1067–1072. [Google Scholar] [CrossRef]
- Lesca, G.; Rudolf, G.; Bruneau, N.; Lozovaya, N.; Labalme, A.; Boutry-Kryza, N.; Salmi, M.; Tsintsadze, T.; Addis, L.; Motte, J.; et al. GRIN2A mutations in acquired epileptic aphasia and related childhood focal epilepsies and encephalopathies with speech and language dysfunction. Nat. Genet. 2013, 45, 1061–1066. [Google Scholar] [CrossRef] [PubMed]
- Carvill, G.L.; Regan, B.M.; Yendle, S.C.; O’Roak, B.J.; Lozovaya, N.; Bruneau, N.; Burnashev, N.; Khan, A.; Cook, J.; Geraghty, E.; et al. GRIN2A mutations cause epilepsy-aphasia spectrum disorders. Nat. Genet. 2013, 45, 1073–1076. [Google Scholar] [CrossRef] [PubMed]
- Kingwell, K. Epilepsy: GRIN2A mutations identified as key genetic drivers of epilepsy-aphasia spectrum disorders. Nat. Rev. Neurol. 2013, 9, 541. [Google Scholar] [CrossRef] [PubMed]
- Conroy, J.; McGettigan, P.A.; McCreary, D.; Shah, N.; Collins, K.; Parry-Fielder, B.; Moran, M.; Hanrahan, D.; Deonna, T.W.; Korff, C.M.; et al. Towards the identification of a genetic basis for Landau-Kleffner syndrome. Epilepsia. 2014, 55, 858–865. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.J.; Morgan, A.T.; Perez, E.R.; Scheffer, I.E. New genes for focal epilepsies with speech and language disorders. Curr. Neurol. Neurosci. Rep. 2015, 15, 35. [Google Scholar] [CrossRef] [PubMed]
- Myers, K.A.; Scheffer, I.E. GRIN2A-related speech disorders and epilepsy. In GeneReviews [Internet]; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2016.
- Gao, K.; Tankovic, A.; Zhang, Y.; Kusumoto, H.; Zhang, J.; Chen, W.; XiangWei, W.; Shaulsky, G.H.; Hu, C.; Traynelis, S.F.; et al. A de novo loss-of-function GRIN2A mutation associated with childhood focal epilepsy and acquired epileptic aphasia. PLoS ONE. 2017, 12, e0170818. [Google Scholar] [CrossRef] [PubMed]
- Sculier, C.; Tilmant, A.S.; De Tiège, X.; Giurgea, S.; Paquier, P.; Rudolf, G.; Lesca, G.; Van Bogaert, P. Acquired epileptic opercular syndrome related to a heterozygous deleterious substitution in GRIN2A. Epileptic Disord. 2017, 19, 345–350. [Google Scholar] [CrossRef]
- Yang, X.; Qian, P.; Xu, X.; Liu, X.; Wu, X.; Zhang, Y.; Yang, Z. GRIN2A mutations in epilepsy-aphasia spectrum disorders. Brain Dev. 2018, 40, 205–210. [Google Scholar] [CrossRef]
- Qian, P.; Yang, X.; Xu, X.; Liu, X.; Zhang, Y.; Yang, Z. [Study of GRIN2A mutation in epilepsy-aphasia spectrum disorders]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2018, 35, 314–318. [Google Scholar]
- Lesca, G.; Møller, R.S.; Rudolf, G.; Hirsch, E.; Hjalgrim, H.; Szepetowski, P. Update on the genetics of the epilepsy-aphasia spectrum and role of GRIN2A mutations. Epileptic Disord. 2019, 21 (Suppl. S1), 41–47. [Google Scholar] [CrossRef]
- Li, X.; Xie, L.L.; Han, W.; Hong, S.Q.; Ma, J.N.; Wang, J.; Jiang, L. Clinical forms and GRIN2A genotype of severe end of epileptic-aphasia spectrum disorder. Front. Pediatr. 2020, 8, 574803. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Xue, J.; Jiao, X.; Zhang, Y.; Yang, Z. Genetic etiologies in developmental and/or epileptic encephalopathy with electrical status epilepticus during sleep: Cohort study. Front. Genet. 2021, 12, 607965. [Google Scholar] [CrossRef] [PubMed]
- Stanley, K.; Hostyk, J.; Tran, L.; Amengual-Gual, M.; Dugan, P.; Clark, J.; Choi, H.; Tchapyjnikov, D.; Perucca, P.; Fernandes, C.; et al. Genomic analysis of “microphenotypes” in epilepsy. Am. J. Med. Genet. A. 2022, 188, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Bonardi, C.M.; Mignot, C.; Serratosa, J.M.; Giraldez, B.G.; Moretti, R.; Rudolf, G.; Reale, C.; Gellert, P.M.; Johannesen, K.M.; Lesca, G.; et al. Expanding the clinical and EEG spectrum of CNKSR2-related encephalopathy with status epilepticus during slow sleep (ESES). Clin. Neurophysiol. 2020, 131, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Higa, L.A.; Wardley, J.; Wardley, C.; Singh, S.; Foster, T.; Shen, J.J. CNKSR2-related neurodevelopmental and epilepsy disorder: A cohort of 13 new families and literature review indicating a predominance of loss of function pathogenic variants. BMC Med. Genomics. 2021, 14, 186. [Google Scholar] [CrossRef]
- Arican, P.; Gencpinar, P.; Olgac Dundar, N. A new cause of developmental and epileptic encephalopathy with continuous spike-and-wave during sleep: CDKL5 disorder. Neurocase. 2019, 25, 59–61. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, D.; Majethia, P.; Anand, A.; Shukla, A.; Sharma, S. Expanding the electro-clinical phenotype of CARS2 associated neuroregression. Epilepsy Behav. Rep. 2021, 16, 100485. [Google Scholar] [CrossRef]
- Lee, I.C.; Yang, J.J.; Li, S.Y. A KCNQ2 E515D mutation associated with benign familial neonatal seizures and continuous spike and waves during slow-wave sleep syndrome in Taiwan. J. Formos. Med. Assoc. 2017, 116, 711–719. [Google Scholar] [CrossRef]
- Gong, P.; Xue, J.; Jiao, X.R.; Zhang, Y.H.; Yang, Z.X. [Genotype and phenotype of children with KCNA2 gene related developmental and epileptic encephalopathy]. Zhonghua Er Ke Za Zhi. 2020, 58, 35–40. [Google Scholar]
- Liu, W.; Cheng, M.; Zhu, Y.; Chen, Y.; Yang, Y.; Chen, H.; Niu, X.; Tian, X.; Yang, X.; Zhang, Y. DYNC1H1-related epilepsy: Genotype-phenotype correlation. Dev. Med. Child Neurol. 2023, 65, 534–543. [Google Scholar] [CrossRef]
- Russo, A.; Gobbi, G.; Pini, A.; Møller, R.S.; Rubboli, G. Encephalopathy related to status epilepticus during sleep due to a de novo KCNA1 variant in the Kv-specific Pro-Val-Pro motif: Phenotypic description and remarkable electroclinical response to ACTH. Epileptic Disord. 2020, 22, 802–806. [Google Scholar] [CrossRef]
- Miao, P.; Tang, S.; Ye, J.; Tang, J.; Wang, J.; Zheng, C.; Li, Y.; Feng, J. Differential functional changes of Nav1.2 channel causing SCN2A-related epilepsy and status epilepticus during slow sleep. Front Neurol. 2021, 12, 653517. [Google Scholar] [CrossRef]
- Zanni, G.; Barresi, S.; Cohen, R.; Specchio, N.; Basel-Vanagaite, L.; Valente, E.M.; Shuper, A.; Vigevano, F.; Bertini, E. A novel mutation in the endosomal Na+/H+exchanger NHE6 (SLC9A6) causes Christianson syndrome with electrical status epilepticus during slow-wave sleep (ESES). Epilepsy Res. 2014, 108, 811–815. [Google Scholar] [CrossRef]
- Coorg, R.; Weisenberg, J.L. Successful treatment of electrographic status epilepticus of sleep with felbamate in a patient with SLC9A6 mutation. Pediatr. Neurol. 2015, 53, 527–531. [Google Scholar] [CrossRef]
- Mathieu, M.L.; de Bellescize, J.; Till, M.; Flurin, V.; Labalme, A.; Chatron, N.; Sanlaville, D.; Chemaly, N.; des Portes, V.; Ostrowsky, K.; et al. Electrical status epilepticus in sleep, a constitutive feature of Christianson syndrome? Eur. J. Paediatr. Neurol. 2018, 22, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, A.; Yamamoto, A.; Ichikawa, K.; Tsuyusaki, Y.; Tsuji, M.; Iai, M.; Enomoto, Y.; Murakami, H.; Kurosawa, K.; Miyatake, S.; et al. Epilepsy in Christianson syndrome: Two cases of Lennox-Gastaut syndrome and a review of literature. Epilepsy Behav. Rep. 2019, 13, 100349. [Google Scholar] [CrossRef]
- Bhat, S.; Ming, X.; Dekermenjian, R.; Chokroverty, S. Continuous spike and wave in slow-wave sleep in a patient with Rett syndrome and in a patient with Lhermitte-Duclos syndrome and neurofibromatosis 1. J. Child Neurol. 2014, 29, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, E.; Bellini, M.; Aspromonte, M.C.; Polli, R.; Mercante, A.; Ciaccio, C.; Granocchio, E.; Bettella, E.; Donati, I.; Cainelli, E.; et al. A novel WAC loss of function mutation in an individual presenting with encephalopathy related to status epilepticus during sleep (ESES). Genes (Basel). 2020, 11, 344. [Google Scholar] [CrossRef] [PubMed]
- Ergun-Longmire, B.; Nguyen, M.H.N.; Com, G. Electrical status epilepticus during sleep in a child with Prader-Willi syndrome: A case report. AME Case Rep. 2022, 6, 7. [Google Scholar] [CrossRef]
- Khan, A.Q.; Coorg, R.K.; Gill, D.; Marini, C.; Myers, K.A. Koolen-de Vries syndrome associated with continuous spike-wave in sleep. Epileptic Disord. 2022, 24, 928–933. [Google Scholar] [CrossRef]
- Bonanni, P.; Negrin, S.; Volzone, A.; Zanotta, N.; Epifanio, R.; Zucca, C.; Osanni, E.; Petacchi, E.; Fabbro, F. Electrical status epilepticus during sleep in Mowat-Wilson syndrome. Brain Dev. 2017, 39, 727–734. [Google Scholar] [CrossRef]
- Christoforou, S.; Christodoulou, K.; Anastasiadou, V.; Nicolaides, P. Early-onset presentation of a new subtype of β-Propeller protein-associated neurodegeneration (BPAN) caused by a de novo WDR45 deletion in a 6 year-old female patient. Eur. J. Med. Genet. 2020, 63, 103765. [Google Scholar] [CrossRef] [PubMed]
- Sager, S.G.; Turkyilmaz, A.; Gunbey, H.P.; Karatoprak, E.Y.; Aslan, E.S.; Akın, Y. A novel de novo TET3 loss-of-function variant in a Turkish boy presenting with neurodevelopmental delay and electrical status epilepticus during slow-wave sleep. Brain Dev. 2023, 45, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Mir, A.; Amer, F.; Ali, M.; Alotaibi, W.; Alotaibi, M.; Hedaithy, A.; Aldurayhim, F.; Hussain, F.; Bashir, S.; Housawi, Y. Continuous spikes and waves during sleep (CSWS), severe epileptic encephalopathy, and choreoathetosis due to mutations in FRRS1L. Clin. EEG Neurosci. 2023, 54, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Hadi, D.A.; Mohamed, A.R.; Rethanavelu, K.; Khoo, T.B. Clonic seizures, continuous spikes-and-waves during slow sleep, choreoathetosis and response to sulthiame in a child with FRRS1L encephalopathy. Brain Dev. 2022, 44, 44–49. [Google Scholar] [CrossRef]
- Ünalp, A.; Gazeteci Tekin, H.; Karaoğlu, P.; Akışın, Z. Benefits of ketogenic diet in a pediatric patient with Ehlers-Danlos syndrome and STXBP1-related epileptic encephalopathy. Int. J. Neurosci. 2022, 132, 950–952. [Google Scholar] [CrossRef] [PubMed]
- Sánchez Fernández, I.; Loddenkemper, T.; Peters, J.M.; Kothare, S.V. Electrical status epilepticus in sleep: Clinical presentation and pathophysiology. Pediatr. Neurol. 2012, 47, 390–410. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.M.; Conroy, J.; Deonna, T.; McCreary, D.; McGettigan, P.; Madigan, C.; Carter, I.; Ennis, S.; Lynch, S.A.; Shahwan, A.; et al. Atypical benign partial epilepsy of childhood with acquired neurocognitive, lexical semantic, and autistic spectrum disorder. Epilepsy Behav. Case Rep. 2016, 6, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Pal, D.K.; Ferrie, C.; Addis, L.; Akiyama, T.; Capovilla, G.; Caraballo, R.; de Saint-Martin, A.; Fejerman, N.; Guerrini, R.; Hamandi, K.; et al. Idiopathic focal epilepsies: The “lost tribe”. Epileptic Disord. 2016, 18, 252–288. [Google Scholar] [CrossRef]
- Lee, Y.J.; Hwang, S.K.; Kwon, S. The clinical spectrum of benign epilepsy with centro-temporal spikes: A challenge in categorization and predictability. J. Epilepsy Res. 2017, 7, 1–6. [Google Scholar] [CrossRef]
- Dryżałowski, P.; Jóźwiak, S.; Franckiewicz, M.; Strzelecka, J. Benign epilepsy with centrotemporal spikes - Current concepts of diagnosis and treatment. Neurol. Neurochir. Pol. 2018, 52, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Halász, P.; Kelemen, A.; Rosdy, B.; Rásonyi, G.; Clemens, B.; Szűcs, A. Perisylvian epileptic network revisited. Seizure. 2019, 65, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Fortini, S.; Espeche, A.; Galicchio, S.; Cersósimo, R.; Chacon, S.; Gallo, A.; Gamboni, B.; Adi, J.; Fasulo, L.; Semprino, M.; et al. More than one self-limited epilepsy of childhood in the same patient: A multicenter study. Epilepsy Res. 2021, 177, 106768. [Google Scholar] [CrossRef] [PubMed]
- Sable, S.; Sable, R.; Tamhankar, P.; Tamhankar, V. Clinical profile of patients with rolandic epilepsy at a clinic in rural Maharashtra. J. Family Med. Prim. Care. 2021, 10, 1263–1266. [Google Scholar] [CrossRef] [PubMed]
- Vercueil, L. GRIN2A, a green semaphore on the lumping route to idiopathic focal epilepsy in childhood. Rev. Neurol. (Paris). 2013, 169, 921–922. [Google Scholar] [CrossRef] [PubMed]
- Rudolf, G.; de Bellescize, J.; de Saint Martin, A.; Arzimanoglou, A.; Valenti Hirsch, M.P.; Labalme, A.; Boulay, C.; Simonet, T.; Boland, A.; Deleuze, J.F.; et al. Exome sequencing in 57 patients with self-limited focal epilepsies of childhood with typical or atypical presentations suggests novel candidate genes. Eur. J. Paediatr. Neurol. 2020, 27, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Hausman-Kedem, M.; Menascu, S.; Greenstein, Y.; Fattal-Valevski, A. Immunotherapy for GRIN2A and GRIN2D-related epileptic encephalopathy. Epilepsy Res. 2020, 163, 106325. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D. GRIN2A-related epilepsy and speech disorders: A comprehensive overview with a focus on the role of precision therapeutics. Epilepsy Res. 2023, 189, 107065. [Google Scholar] [CrossRef] [PubMed]
- Imataka, G.; Arisaka, O. Serial EEG study in a girl with Landau-Kleffner syndrome associated with continuous spikes and waves during slow sleep. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 2145–2147. [Google Scholar]
- Posar, A.; Visconti, P. Some considerations about the association between autism spectrum disorder and epilepsy. Turk. Pediatri Ars. 2020, 55, 331–332. [Google Scholar] [CrossRef]
- Bebek, N.; Gürses, C.; Baykan, B.; Gökyiğit, A. Lack of prominent cognitive regression in the long-term outcome of patients having electrical status epilepticus during sleep with different types of epilepsy syndromes. Clin. EEG Neurosci. 2015, 46, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.R.; Stafstrom, C.E. Pediatric epileptic encephalopathies: Pathophysiology and animal models. Semin. Pediatr. Neurol. 2016, 23, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Halász, P. Comment on neuronal networks in epileptic encephalopathies with CSWS. Epilepsia. 2017, 58, 1296–1297. [Google Scholar] [CrossRef]
Figure 1.
Paper selection flow chart.
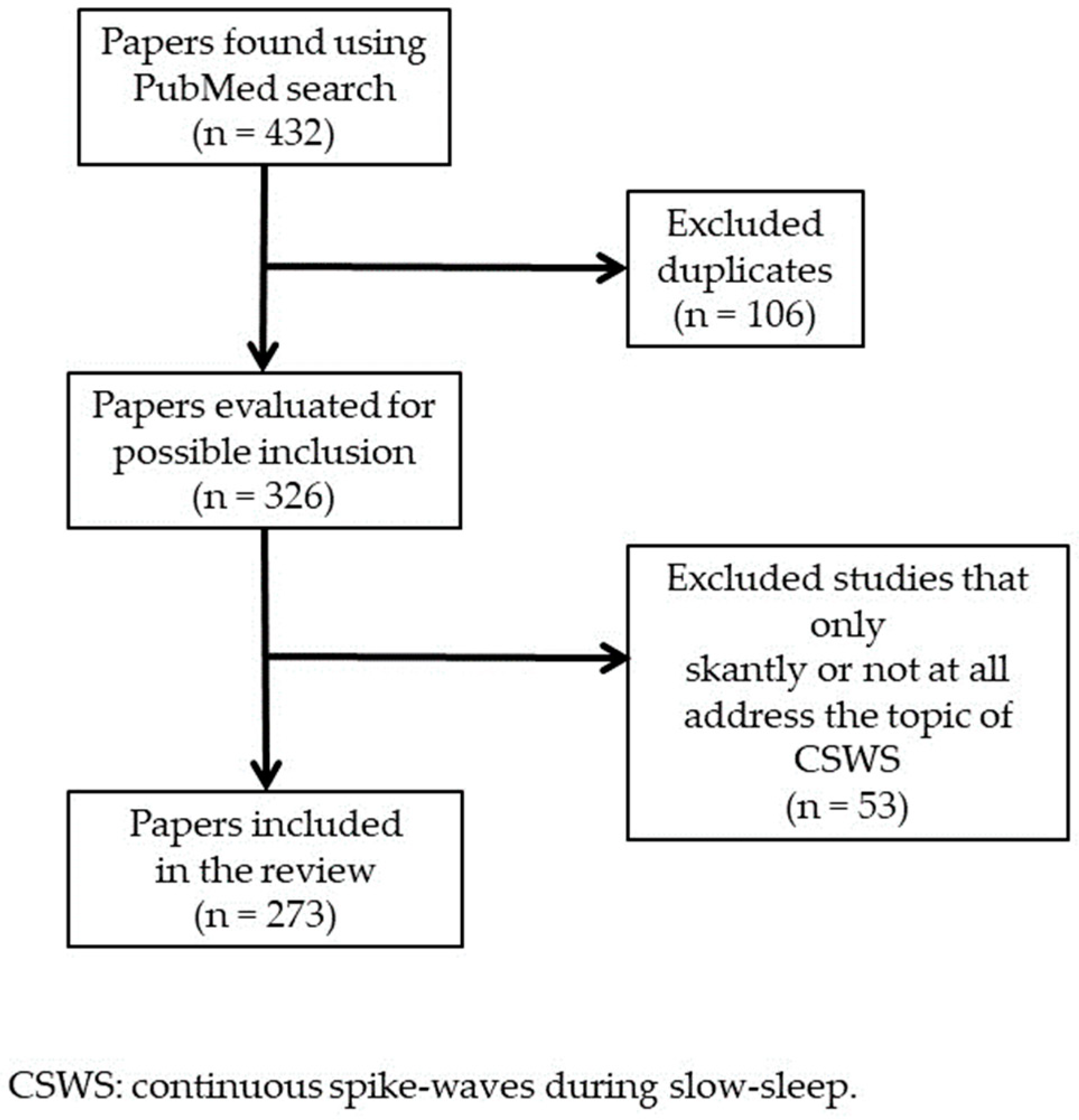
Figure 2.
NREM sleep EEG in a boy aged 5 years 7 months with autism spectrum disorder, intellectual disability, and focal epilepsy, showing subcontinuous diffuse spike-waves (lasting < 85% of NREM sleep), prevailing on the right posterior regions. NREM: nonrapid eye movement. EEG: electroencephalogram.
Figure 2.
NREM sleep EEG in a boy aged 5 years 7 months with autism spectrum disorder, intellectual disability, and focal epilepsy, showing subcontinuous diffuse spike-waves (lasting < 85% of NREM sleep), prevailing on the right posterior regions. NREM: nonrapid eye movement. EEG: electroencephalogram.
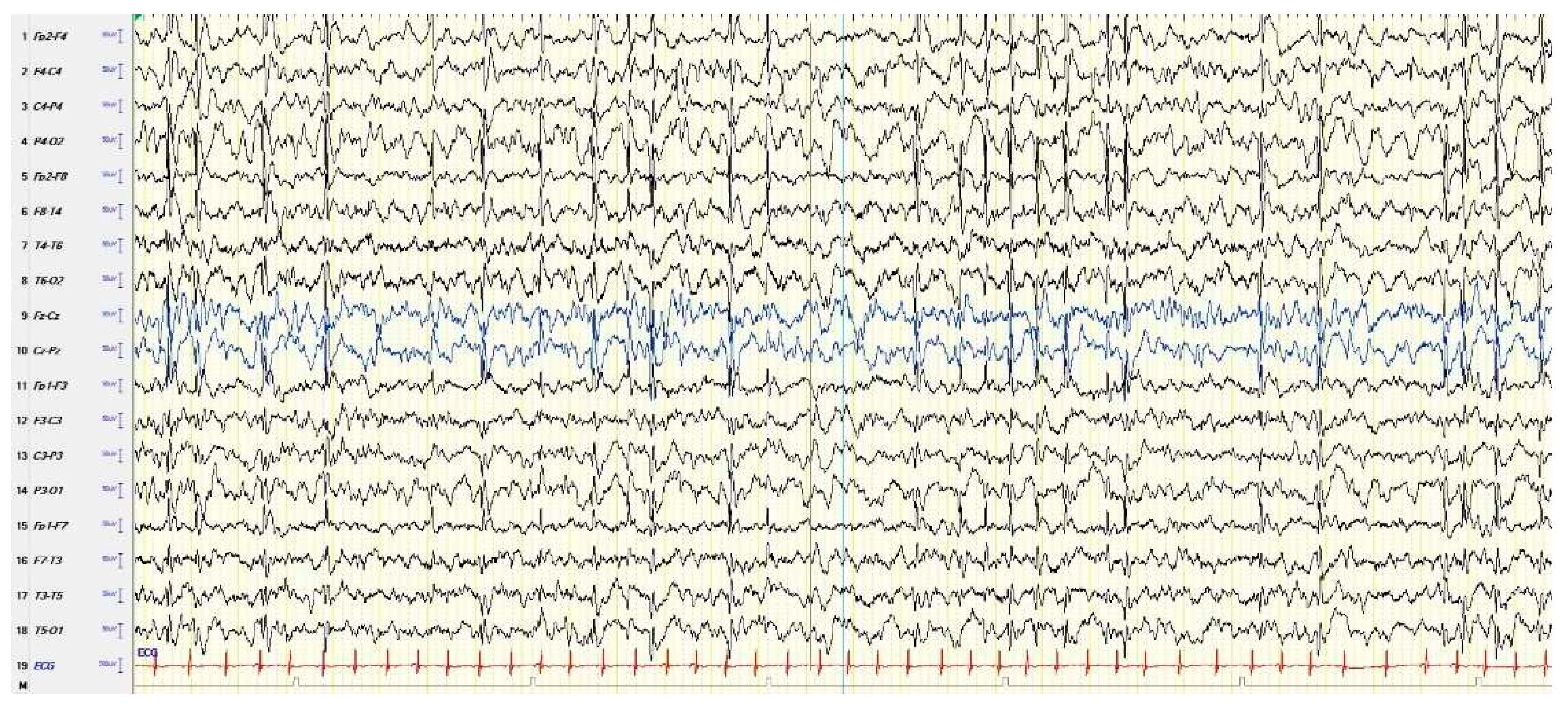

Table 1.
Clinical and EEG features of typical and atypical ESES.
| Clinics | EEG | |
|---|---|---|
| Typical ESES |
Cognitive deterioration. Behavioral disorders, including attention deficit, hyperactivity, aggressiveness, difficulty in social interaction, and (more rarely) psychosis; also autistic behavior is possible. Epileptic seizures, focal or apparently generalized, with a heterogeneous semeiology. Motor signs, including dyspraxia, ataxia, and dystonia. |
Electrical status epilepticus lasting for ≥ 85% of N-REM (slow) sleep, documented by more than 2 EEG recordings during a period of ≥ 1 month. |
| Atypical ESES |
Great heterogeneity in the reported clinical features depending on the frequency and localization of the EEG paroxysmal abnormalities: a cognitive deterioration has been reported, but less frequently than typical ESES. Behavioral disorders, in particular ADHD-like symptoms. Epileptic seizures, focal or apparently generalized, with a heterogeneous semeiology. |
Paroxysmal abnormalities lasting for < 85% of N-REM sleep (usually > 50% < 85%), with a localization more heterogeneous than typical forms: focal, multifocal, unilateral, asymmetric or symmetric bilateral, and diffuse. |
EEG: electroencephalogram. ESES: electrical status epilepticus during slow-wave sleep. ADHD: Attention-Deficit/Hyperactivity Disorder.
Table 2.
Predictive factors of the evolution into CSWS.
| Clinical | Early onset of seizures; multiple types of seizures; appearance of new seizures with an increased frequency; seizure semiology including dysarthria or somatosensory auras. | |
|---|---|---|
| EEG | Fronto-centro-temporal focus, with increasing frequency, both in wakefulness and in sleep; pattern EEG of spike-waves. |
CSWS: continuous spike-waves during slow sleep. EEG: electroencephalogram.
Table 3.
Electrophysiology and functional neuroimaging in CSWS.
| Semiautomatic quantification of paroxysmal abnormalities in CSWS is a reliable alternative to the classic quantification based on visual scoring. |
|---|
| Global synchronization increase from awake to sleep is strongly correlated with spikes. |
| Associating time-sensitive magnetic source imaging and PET, spike-waves onset is associated with focal hypermetabolism. |
| Magnetoencephalography in non-lesional CSWS children showed dipole clusters located on heterogeneous cortical areas: right Rolandic area, right supramarginal gyrus, left Rolandic area, left supramarginal gyrus, bilateral Rolandic area, multiple anatomical areas. |
| PET showed hypermetabolism in perisylvian, superior temporal, inferior parietal, and in central cortex areas that were related to paroxysmal abnormalities. The diffuse hypometabolism found in regions belonging to the DMN (see prefrontal and posterior cingulate cortices, parahippocampal gyrus and precuneus) could be due to remote inhibition following epileptic activity. |
| EEG-fMRI showed characteristic findings of epileptic encephalopathy: positive changes of BOLD signal in the perisylvian regions, prefrontal cortex, anterior cingulate, and thalamus, while negative changes of BOLD signal were found in the DMN regions. The activation pattern represents a diffusion of epileptic activity. |
| HFOs are hypothesized to be related to alterations of higher brain functions. They seem to be related to functional disruption of brain networks in CSWS. |
CSWS: continuous spike-waves during slow sleep. PET: positron emission tomography. DMN: default mode network. EEG-fMRI: electroencephalogram-functional magnetic resonance imaging. BOLD: blood-oxygen-level-dependent. HFOs: high-frequency oscillations.
Table 4.
Effective treatments for CSWS.
| Pharmacological | Antiepileptic drugs: sulthiame (++), levetiracetam (+), acetazolamide (+), benzodiazepines (++), topiramate (++), perampanel (+), lacosamide (+). Steroids (corticosteroids and ACTH) (+++). Immunoglobulins (+). Amantadine (+). |
|---|---|
| Not pharmacological |
Ketogenic diet (+). Neurosurgery (feasible only in selected cases) (+++) |
| Comments about the literature concerning these topics: almost only retrospective studies were performed, with small or very small samples of patients; there is a lack of information about the long-term effects of the drugs. Often cognitive and behavioral findings reported do not derive from standardized objective neuropsychological assessments. | |
CSWS: continuous spike-waves during slow-sleep. (+): limited data. (++): consistent data. (+++): very consistent data.
Table 5.
Main prognostic factors of CSWS.
| Unfavorable | Long duration of CSWS. Presence of a cerebral lesion. High frequency of EEG paroxysmal abnormalities. CSWS early onset (before 6 years). |
| Favorable | Clinical phenotype of an atypical BECTS at onset. Normal development before CSWS. CSWS later onset (from 8 years onwards). Normal EEG background organization. |
CSWS: continuous spike-waves during slow-sleep. EEG: electroencephalogram. BECTS: benign epilepsy with centro-temporal spikes.
Table 6.
Clinical and EEG features of LKS.
| Clinical | Acquired mixed aphasia, with onset at 3-5 years of age, with poor decoding of verbal and/or non-verbal sounds. Seizures are present in around two-thirds of cases and are semeiologically heterogeneous: focal motor, tonic-clonic seizures, and atypical absences. Behavioral symptoms are frequently associated: ADHD symptoms, irritability, aggressive behavior, in some cases autistic-like symptoms, anxiety, and depression. Learning disorders are the rule. Outcome is very heterogeneous. Only in a minority of cases language recover is complete. |
|---|---|
| EEG | Spikes and spike-waves prevailing in the posterior temporal regions, bilaterally, much more diffuse and frequent during non-REM sleep becoming continuous or subcontinuous. Background activity during wakefulness and sleep is normal. |
EEG: electroencephalogram. LKS: Landau-Kleffner syndrome. ADHD: Attention-Deficit/Hyperactivity Disorder.
Table 7.
Effective treatments for LKS.
| Pharmacological | Antiepileptic drugs: valproate (+), benzodiazepines (+), levetiracetam (+), ethosuximide (+), acetazolamide (+). Corticosteroids (+++). Immunoglobulins in cases with GRIN2A mutations (++). |
|---|---|
| Not pharmacological |
Ketogenic diet (+). Multiple subpial transections in selected cases (+). Vagus nerve stimulation (+). Speech therapy (++?). |
LKS: Landau-Kleffner syndrome. (+): limited data. (++): consistent data. (+++): very consistent data.
Table 8.
Etiopathogenetic factors of CSWS.
| Brain structural alterations |
Polymicrogyria (in particular unilateral), migration disorders, perinatal hypoxic-ischemic encephalopathy, hydrocephalus, schizencephaly, porencephalic lesions, encephalitis, intracranial hemorrhage, and thalamic lesions. |
|---|---|
| Inflammation | Altered levels of interleukin-6. |
| Acquired factors | Fetal alcohol syndrome. Iatrogenic (in particular old antiepileptic drugs). |
| Genetics | Dup15q (copy number gains of 15q11-q13), 22q11.2 microduplication, and many other copy number variants, affecting several other chromosomes, including 3q25 deletion, 3q29 duplication, 8p23.3 deletion, 10q21.3 deletion, 11p13 duplication, 16p13 deletion, and Xp22.12 deletion. Mutations of GRIN2A gene (16p13.2). Mutations also of a lot of other genes: CNKSR2 (Xp22.12), CDKL5 (Xp22.13), CARS2 (13q34), KCNQ2 (20q13.33), KCNA2 (1P13.3), SLC9A6 (Xq26.3), HIVEP2 (6q24.2), RARS2 (6q15), DYNC1H1 (14q32.31), FRRS1L (9q31.3), KCNA1 (12p13.32), SLC9A6 (Xq26.3), STXBP1 (9q34.11), MECP2 (Xq28), WAC (10p12.1), KANSL1 (17q21.31), TET3 (2p13), ZEB2 (2q22.3), WDR45 (Xp11.23). |
CSWS: continuous spike-waves during slow-sleep.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



