
Submitted:
22 December 2023
Posted:
22 December 2023
You are already at the latest version
Abstract
Alzheimer’s disease (AD) and type 2 diabetes mellitus (T2DM) are comorbidities that result from the sharing of common genes. The molecular background of comorbidities can provide clues for the development of treatment and management strategies. Here, the common genes involved in the development of the two diseases and in memory and cognitive function are reviewed. Network clustering based on protein-protein interaction networks identified tightly connected gene clusters that have an impact on memory and cognition among the comorbidity genes of AD and T2DM. Genes with functional implications were intensively reviewed, and relevant evidence was summarized. Gene information will be useful in the discovery of biomarkers and the identification of therapeutic targets for AD.
Keywords:
Alzheimer’s disease
; type 2 diabetes
; comorbidity
; memory
; cognition.
1. Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by memory and cognitive impairment. The main pathology of AD is the accumulation of beta-amyloid (Aβ), which is believed to cause the main symptoms of the disease [1]. Clearing Aβ or tau proteins that are alleged to induce AD has been the main strategy in the development of therapeutic agents; however, the results of clinical studies have been unsatisfactory, and there has been no definite treatment for AD [2]. This may be due to the fact that a correlation between Aβ and tau protein accumulation and clinical outcomes has not been clearly established.
It is well known that type 2 diabetes (T2DM) co-occurs with AD [3]. There are a lot of evidences that two diseases develop concomitantly, and the comorbidity relationship is based on the shared molecular mechanisms between AD and T2DM [4]. Moreover, genes involved in comorbidity can be valuable resource for drug repurposing [4,5]. Therefore, it is desirable that identification of comorbidity genes for AD and T2DM provides clues for further development of AD drugs or management strategies.
In this review, disease genes gathered from previous studies were used and protein-protein interaction network-based clustering (Markov clustering algorithm) was applied for identification of comorbidity genes of AD and T2DM that are related to memory and cognitive function. For this purpose, genes that involved in memory and cognitive functions were also collected and intersection of these genes and the comorbidity genes were applied to the clustering.
2. Biological mechanisms of Alzheimer’s disease and type 2 diabetes
AD and T2DM genes collected from the DisGeNet database were tentatively associated with biological processes involved in the pathogenesis of both diseases by functional annotation. For this purpose, overrepresentation analysis (ORA) with Fisher’s exact test was performed. In the enrichment test, those genes common to AD and T2DM were used as input genes (n = 1381, Table S1).
Further, in the enrichment analysis of 7763 Gene Ontology (GO) biological processes (GOBPs), 2857 statistically significant terms were identified (Table 1 and Table S2), being “RESPONSE_TO_OXYGEN_CONTAINING_COMPOUND” was the most significant one (odds ratio = 16.06, P = 1.97E-301). Because the GO database has a hierarchical structure, GOBP terms are linked to a superset or subset of common genes. Therefore, we also analyzed those GOBPs associated with similar concepts. The “REGULATION_OF_CELL_DEATH” was among the most highly ranked GOBPs (odds ratio = 11.97, P = 5.53E-222).
3. Gene clusters of common genes that are associated with AD, T2DM, and memory function
Among the 1381 common genes of AD and T2DM, 361 genes overlapped with the memory-associated genes in the DisGeNet database (Table S1). Using the information on protein interactions in the STRING database, Markov clustering of the interaction network was performed using the default parameters, and 93 clusters with different numbers of genes, ranging from 1 to 13, were obtained. Table S4 lists these clusters and their associated proteins. The cluster numbers were determined according to the average local clustering coefficient of the network-based clustering method. Therefore, the first cluster (Cluster 1) had the highest average local clustering coefficient, indicating tighter connections between the proteins within the cluster compared to the other clusters.
3.1. Cluster 1 (CL1)
CL1 included 13 genes (Figure 1). PI3K and PDGF-related genes were frequent in this cluster.
PI3K is a well-known enzyme involved in various cellular functions, including apoptosis, glucose uptake, and neuroprotection [6]. Many PI3K family members (PI3K subtypes) function in the Akt and mTOR pathway [6,7]. In AD, the PI3K pathway is inhibited by Aβ, which has been linked to increased apoptosis of neurons [8]. Moreover, the PI3K/Akt signaling pathway is involved in tau phosphorylation, dysregulated insulin signaling, suppression of autophagy through the activation of mTOR, and altered responses to oxidative stress in patients with AD [8,9]. PI3K plays a role in glucose uptake by muscle and adipose cells [10], and abnormal PI3K signaling causes insulin resistance in animal models [11]. The PI3K-related pathways, including Akt and mTOR, are associated with neuronal development and brain memory function [12,13,14]. PI3K subtypes PIK3CA, PIK3CB, PIK3CD, PIK3CG, and PIK3R1 were all included in CL1.
PIK3CA was a hub gene in CL1; it is predicted to be involved in the immune-related phenomena of AD development [15]. In an AD zebrafish model, 20S-protopanaxatriol (PPT) facilitates neurogenesis of neural stem cells (NSCs) and reduces NSC apoptosis and cell cycle arrest by Aβ (which might hinder PIK3CA and PPT binding) [16]. Bioinformatics analysis of molecular docking and identification of network modules revealed that PIK3CA was one of the target genes for Byu dMar 25 (BM25), a molecule known to have therapeutic potential in AD [17]. When frog skin peptide, which is a stimulant of insulin release, was administered to a T2DM mouse model, the expression of Pik3ca (the mouse ortholog) increased in skeletal muscles [18,19]. PIK3CB has been associated with insulin resistance and hepatic glucose production according to promoter variants [20,21,22]. The expression of PIK3CB is downregulated in patients with AD and linked to the apoptosis and axon guidance pathways [23]. PIK3CB is also genetically associated with mild cognitive impairment (MCI) showing abnormalities in temporal lesions that modulate memory function [24]. PIK3CD mRNA in peripheral leukocytes is upregulated in patients with gestational diabetes, whereas it is downregulated in patients with T2DM treated with sitagliptin [25,26]. Similar to PIK3CB, PIK3CD is also genetically associated with MCI [24]. PIK3R1 is well known for its relationship with T2DM and insulin resistance [27,28]. Mutations in PIK3R1 cause SHORT (short stature, hyperextensibility of joints and/or inguinal hernia, ocular depression, Rieger anomaly, and teething delay) syndrome and accompanying T2DM [29,30,31]. Moreover, the analysis of exome sequencing data from over 10,000 subjects in the Alzheimer’s Disease Sequencing Project showed evidence of a functional variant of PIK3R1 [32]. Coexpression network analysis has revealed that PIK3R1 is one of the core immune genes involved in AD and that it is associated with Aβ and tau protein pathology [33].
CL1 included two PDGF-related proteins, PDGFB and PDGFRB. PDGF is associated with vascular complications in T2DM [34] and cell death caused by Alzheimer-associated neuronal thread protein [35]. PDGFB and PDGFRB also involves in vascular complications of T2DM [36,37]. In AD, PDGFRB activation has a mitogenic effect that is blocked by Aβ, preventing the neuroprotective effects of PDGF-BB [38]. Mutations in these two genes cause brain calcifications [39,40], which can be observed in patients with AD [41].
3.2. Cluster 2 (CL2)
In CL2, P53 acted as a hub gene by showing the strongest connectivity (Figure 1). P53 has a neuroprotective effect by repressing BACE1 and thus the Aβ production cascade. Interestingly, Aβ may also repress P53 expression in AD [42]. Moreover, MCI is affected by conformational changes in P53 [16,43]. It is well known that cancer and AD have an inverse correlation in incidence, and the underlying molecular mechanisms seem to involve P53 and related genes [44,45]. Phosphorylated forms, genetic variations, and unfolded P53 have been proposed as biomarkers for AD [44,46,47]. P53-related novel mechanisms, including mitochondrial dysfunction and overexpression of CDK5 in AD and other neurodegenerative diseases, have also been proposed as biomarkers [46,48]. In previous studies, genetic variants of P53 have also been associated with T2DM [49,50,51]. Therefore, P53 has been identified as one of the hub genes involved in the pathogenesis of AD and T2DM [52]. Notably, P53 also regulates pancreatic cell survival and glucose homeostasis [53].
BRCA1 plays a role in repairing DNAs under stress, including the stresses caused by ultraviolet light and reactive oxygen species, and failures of this mechanism in neurons may be related to AD [54,55]. Downregulation of BRCA1 and other DNA repair genes has been observed in patients with clinically evident AD [56]. BRCA1 depletion was shown to impair cognitive function in mice [57]. In addition, abnormal accumulation of P53 occurs in AD and other tauopathies [58,59], and may be caused by hypomethylation of the promoter region of P53 [60]. BRCA1 is known to interact with acetyl coenzyme A (CoA) carboxylase α (ACCA), which results in lipogenesis [61]. Hypermethylation of BRCA1 was observed in patients with T2DM [62].
S100B is well known for its role in AD. S100B is involved in gliosis and inflammatory reactions, and suppresses the neurodegeneration of cholinergic neurons in mouse models of AD [63,64]. Besides, S100B is associated with memory and cognition. The inhibition of IL-1, for example, decreases S100B, leading to an alleviation of cognitive deficits and tau production [65]. Neutralization of S100B in a rat sepsis model increased cognitive performance scores [66], and pharmaceutical suppression of S100B reduced gliosis and neuronal loss [67]. Besides, it has been shown that S100B and receptor for advanced glycation products (RAGE) affect learning and memory impairment by interacting with IL-1, IL-6, and TNF-α [68]. Serum S100B levels positively correlate with cognitive performance tests in a healthy elderly population [69]. In contrast, they also show a positive correlation with AD severity [70]. S100B is also associated with the pathophysiology of T2DM. In a mouse model, S100B induced beta cell apoptosis [71]. Serum S100B levels were elevated in patients with T2DM with peripheral neuropathy [72], and S100B levels correlated with cognitive performance in patients with T2DM [73]. In the coronary arterioles of a mouse model, S100B suppresses the vasodilatation effect of acetylcholine [74].
DNMT1 is an enzyme that catalyzes the transfer of methyl groups to DNA CpG sites, and previous research in animal models has shown that aberrant DNMT1 expression is associated with memory impairment [75,76,77,78]. In a high methionine-induced AD rat model, DNMT1 was downregulated and tyrosine receptor kinase-induced memory impairment was observed [79,80]. In humans, DNMT1 has been associated with both AD [81,82,83,84], and T2DM, and increased DNMT1 expression has been observed in beta islet cells from patients with T2DM [85]. IL-6, which is a major inflammatory mediator, induces insulin resistance and reduces DNMT1 protein levels in endothelial cells [86]. In CL2, PARP1 was not directly connected to P53, but linked to it via DNMT1.
In diabetic mice, NF-kB inhibition improves vascular function and increases cleaved PARP1 [87,88]. The role of PARP1 in T2DM was discovered through the modulation of PARP1 by diverse inhibitors. PARP1-inhibition reduces cardiac ischemia and inflammation in diabetic rats [89], and prolongs the lifespan of Caenorhabditis elegans under hyperglycemic conditions —probably via TCF7L2— [90]. PARP1 is associated with the vascular complications of T2DM, and has treatment potential for this condition [91,92,93,94]. Angiotensin II-treated heart muscles of diabetic mice showed elevated PARP1 activity, cardiac hypertrophy, and inflammation, which were reversed by PARP1 inhibition [91]. Mendelian randomization identified a causal relationship between genetic variants of PARP1 and obstructive coronary arterial disease in patients with T2DM [92]. When bromocriptine is used for the treatment of prolactinomas, it controls glucose and lipid profiles in diabetic rats, leading to changes in p-AKT, followed by changes in Nf1 and PARP1 [93]. Cholesterol-induced lipotoxicity, which is related to beta cell dysfunction in obese patients with T2DM, has been shown to be controlled by the inhibition of PARP1 by GLP-1 administration [94].
3.3. Cluster 3
CL3 had well-known AD-associated genes, whose relatedness to T2DM has been less reported (Figure 1). Amyloid precursor protein (APP) is probably the most frequently studied molecule in AD research. Therefore, only APP studies related to memory or cognitive impairment were included in this review. For this purpose, a PubMed search was performed with using “APP gene and Alzheimer’s disease and brain memory” as keywords; the results included many studies on APP and their impact on memory function. JNK inhibition, for example, was shown to eliminate memory impairment and long-term potentiation deficits in a mouse model of AD in which APP phosphorylation was inhibited [95]. CRTC1 is a CREB coactivator whose expression is suppressed by APP [96]. When all-trans-retinoic acid was administered to APP/PS1 transgenic mice, improved spatial learning and memory were observed, compared with those of the control group, together with downregulation of CDK5 (a major kinase for APP and tau phosphorylation) [97]. According with a mouse model, low-density lipoprotein receptor-related protein 6 (LRP6) is involved in memory deficits via Wnt signaling, and the downregulation of this process is linked to the phosphorylation of APP and increased production of Aβ [98]. Besides, APP haploinsufficiency prevents memory deficits in Familial British Dementia mouse models [99], and PTEN-induced putative kinase 1 (PINK1) is associated with memory impairment induced by APP PP [16]. Moreover, increased APP intracellular domain (AICD) production in hippocampal neurons disrupts spatial memory [100]. Meanwhile, the role of APP in T2DM pathophysiology remains unclear, given that there is limited molecular evidence. However, it has been suggested that APP is the main regulator of insulin secretion in pancreatic islets [101]. Moreover, BACE2 (β-site APP-cleaving enzyme 2), a protease that is related to AD, is associated with insulin secretion in pancreatic islet cells [102].
APOE is a well-known AD biomarker. Moreover, the functional relationship between APOE and memory has been reported in many studies. When a proteomic analysis was applied to an AD mouse model, APOE was found to be differentially expressed in the hippocampus, which is related to memory function [103]. APOE is a transcriptional regulator of APP [104,105,106], and is involved in various biological pathways, such as the PGC-1alpha/sirtuin 3 axis, which alters mitochondrial function and, eventually, memory performance [107]. Multi-omics data analysis has revealed APOE haplotype-specific molecular alterations in both at gene and protein expression levels [108]. APOE4 genotype induces an increase in unsaturated fatty acids and accumulation of lipid droplets [109], and single-cell sequencing of postmortem human samples identified that some signaling pathways of cholesterol metabolism were altered in APOE4 carriers, resulting in reduced myelination [110]. The effects of APOE on brain function were confirmed using clinical data and imaging analyses. Using functional MRI analyses, APOE4 carriers performing moderate or severe working memory tasks showed less brain activation than non-APOE4 carriers [111]; APOE4 carriers also showed worse CA1 apical neuropil atrophy and episodic memory function [112]. APOE genotypes were found to be related to lower memory testing scores in patients with amnestic MCI and AD [113], lower memory performance in the normal elderly population [114], and reduced white matter connectivity [115], and gray matter volume [116]. APOE is associated with cardiovascular complications in patients with T2DM [117,118]. In particular, atherosclerosis and nephropathy are the most frequently reported complications associated with APOE genotypes [119,120,121,122,123,124]. Mechanistically, APOE has been associated with insulin resistance in the muscles of mouse models [125], islet amyloidosis [126], and adipocyte enlargement in atherosclerosis [127].
Clusterin (CLU) is a core protein in CL3; it is concurrently linked to APP and APOE. Studies of CLU gene variants and plasma protein levels have consistently revealed that CLU is associated with AD [128,129,130,131,132,133,134,135]. Molecular biology studies have identified the role of CLU in the pathophysiology of AD. In a CLU knockout mouse model, amyloid plaques were sparse in the cerebral parenchyma but prevalent in cerebral vessels, indicating that Aβ clearance had shifted to perivascular drainage [136]. CLU affects the lysosome pathway and Aβ processing in stem cell-derived neurons [137]. Additionally, overexpression of CLU in astrocytes ameliorates amyloid accumulation and gliosis [138]. It has also been found that the C allele of CLU is expressed at higher levels than other allelic variants and that C allele expression leads to exacerbation of inflammation and to an eventual inhibition of oligodendrocyte progenitor cell proliferation and myelination [139]. CLU is also associated with memory function. In a young population, working memory performance differed between CLU genotypes [140], and methylation around SNPs rs9331888 and rs9331896 in the CLU gene was associated with episodic verbal memory in patients with schizophrenia [141]. In patients with AD, delayed word recall test scores significantly correlated with rs11136000, one of the CLU gene SNPs [142]. Interestingly, the reduced episodic memory function that is associated with some CLU genotypes is attenuated by physical activity [143]. CLU protein levels increase in exercised mice, increasing memory performance and reducing brain inflammation [144].
4. Clusters of cognitive function-associated genes
Cognition-related genes were downloaded from the DisGeNet website and used for PPI network analysis. In total, 308 genes were at the intersection of AD, T2DM, and memory function genes, and were applied to the STRING database for a new round of analysis (Table S1). In total, 61 clusters were identified using the Markov clustering algorithm. Table S5 contains the list of the clusters and their proteins. As in Section 3, these clusters were sorted according to the average local clustering coefficient.
4.1. Cluster 1
Cluster 1 contained 10 tightly interconnected genes (Figure 2). EP300 was the hub gene of the cluster. Mutational studies have shown that EP300 is associated with cognitive function. Mutations in EP300 have been reported in patients with Rubinstein-Taybi syndrome, which is characterized by cognitive impairment [145,146]. Fragile X syndrome protein (FMRP) is associated with EP300, and the loss of FMRP increases EP300 and HDAC1 levels in adult NSCs, resulting in age-related NSC depletion and cognitive impairment in mouse models [147]. EP300 expression is not activated when PS1 is mutated, and EP300 is involved in histone acetylation of PS1 and BACE1, which are key genes in AD pathogenesis [148]. It has been reported that EP300 and IL-17A are activated in SH-SY5Y cells, and that inhibition of EP300 improves cognitive impairment [149]. An elevated EP300 activity is associated with an aberrant accumulation of immature autophagy markers, and blocking EP300 increases autophagy flux, reduces tau production, and decreases tau propagation [150]. In T2DM, overactivation of EP300 has also been identified; it is related to muscle atrophy by autophagy inhibition [151].
FOXO1 is a transcription factor involved in gluconeogenesis via insulin signaling [152]. Therefore, FOXO1 is closely linked to T2DM. Previous studies have reported that FOXO1 is involved in various mechanisms, including oxidative stress and cytokine induction, that cause beta cell dysfunction [153,154]. Autophagy and FOXO1 are associated with beta cell viability, apoptosis, and insulin resistance [155]. Furthermore, FOXO1 is considered a potential therapeutic target for T2DM [156]. Reduced insulin receptor and insulin-like growth factor-1 receptor signaling decreased Aβ toxicity in a rodent model, which might be induced by FOXOs, especially by FOXO1 and FOXO3 [157]. FOXO1 is involved in the autophagy of neurons [158], and the rs7981045 SNP variant of FOXO1 is associated with poor responses to acetylcholine esterase inhibitor treatment in patients with AD [159]. MiR-181a is an miRNA associated with cognitive function in pentylenetetrazol-induced epileptic rats [160], and miR-181a expression is reduced in APP-/PS1- mice. MiR-181a has a protective effect against Aβ accumulation, but this effect is suppressed by FOXO1 [161]. When blood miRNA profiling was used to build a model for predicting the conversion from MCI to AD, FOXO1 was one of the four hub genes revealed by a network-based meta-analysis of microRNA expression quantitative trait loci target genes (involving expression variations) [162].
Notch1 is a transmembrane receptor that interacts with APP [163]. Besides, the proteolytic cleavage of Notch1 is affected by PS1 and Rac1 [164], and alterations of this process by gamma secretase may cause AD [165]. Furthermore, Notch1 affects neuronal progenitor cell differentiation [166]. It has been observed that elevated transcription of the intracellular domain of Notch1 restores the self-renewal activity of murine neuronal progenitor cells induced by PSEN1 mutations [166,167,168]. In addition, folic acid was shown to stimulate hippocampal neurogenesis of adult rat brains after ischemic injury [167], and Notch1 expression is reduced in the subventricular zone of ischemic aged brains of rats [168]. Downstream signaling of Notch1 is mediated by HES-1 and Hey-1, which bind to insulin-degrading enzyme (IDE), a protein involved in the proteolytic cleavage of Aβ protein [169]. Moreover, IDE levels decreased when the intracellular domain of Notch was transfected into cell lines expressing human APP. In humans, immunohistochemistry identified Notch1 accumulation in brain tissues of patients with sporadic AD [170]. Notch1 signaling is associated with cognitive function in AD [171], and several agents, including a hormone (melatonin) and a variety of chemicals (such as asiatic acid, risperidone, and valproic acid), affect cognitive function via Notch1 [172,173,174]. In diabetic rats and high glucose induced HepG2 cells, Notch1 is downregulated. When an miR-363 inhibitor was applied to HepG2 cells, glucose consumption and uptake increased, while lipid droplet accumulation decreased [175]. Additionally, salsalate is an anti-inflammatory drug with an antidiabetic effect, and its protective effect is diminished by the suppression of Notch1 [176].
4.2. Cluster 2
In cluster 2, JAK2 was an obvious hub gene linked to all the other genes in the cluster (Figure 2). JAK2 is associated with Aβ-induced hepatic insulin resistance. When Aβ is injected into the peritoneum of AD mouse models, it activates the hepatic Jak2/STAT3/SOCS-1 pathway, resulting in elevated fasting glucose and impaired insulin tolerance and hepatic insulin signaling [177]. When SH2B1 was knocked down, insulin expression and glucose-stimulated insulin levels decreased, and the reverse phenomena were observed with the overexpression of SH2B1 in rat beta cells [178]. Egr2 represses the expression of SOCS-1 and the phosphorylation of JAK2 and STAT3 in HepG2 cells following palmitate treatment, and Egr2 upregulation induces insulin resistance in HepG2 cells [179]. A high-fat diet is known to induce lipotoxicity in islet beta cells, which is associated with reduced PDX-1 expression, while the glucagon receptor agonist liraglutide induces the expression of PDX-1, JAK2, and STAT3, restoring insulin capacity and increasing the number of islet beta cells [180]. The antidiabetic effects of bromocriptine and the renoprotective effects of baricitinib, together with recombinant anti-IL-6 receptor proteins, were found to be associated with JAK2 inhibition [93,181,182]. IL-3 activates JAK2 and STAT3 in microglia, and this activation is associated with AD [183]. Inhibition of JAK2/STAT3 induced loss of spatial working memory by reduced choline esterase and desensitizing acetylcholine receptor [184]. Beta-amyloid downregulated IGF-1 expression by inhibiting JAK2/STAT5 pathway in adult rabbit hippocampus [185], and JAK2 inhibitors decrease PGE2 release and microglial phagocytosis [186]. When BDNF/TrkB activity is repressed, the JAK2/STAT3 axis activates, resulting in upregulation of C/EBPβ. This process is associated with increased δ-secretase and APP levels and tau fragmentation [187]. The JAK2/STAT3 cascade plays a crucial role in astrocyte reactivity, a hallmark of AD pathology [188].
IRS2 mediates the activation of the PI3K/Akt and MAPK pathways in insulin target tissues, and IRS2 knockout induces insulin resistance and beta cell degeneration [189]. Furthermore, IRS2 is involved in the autocrine regulation of insulin gene expression in beta cells [190]. In addition, beta cell survival is regulated by IRS2 expression and calcium ions [191], and the calmodulin-dependent kinase 4 (CaMKK)/CREB/IRS2 cascade stimulates beta cell survival in mice [192]. Calcineurin/NFAT signaling controls glucose-induced IRS2 expression in rat beta cells [193]. Notably, IRS2 mediates hepatic gluconeogenesis suppression by HIF2α and VEGF-induced inhibition effects on glucose tolerance [194]. Prolyl hydroxylase domain-containing protein isoforms, including Phd1, Phd2, and Phd3, regulate the anabolic effect of insulin, and deletion of hepatic Phd3 improves insulin sensitivity by increasing Irs2 transcription and Akt activation [195]. IRS2 is closely associated with amyloid pathology in AD. In amyloid overexpressing mice, deletion of Irs2 reduced Aβ deposition by increasing clearance [196]. This finding was replicated in another study showing that the beneficial effect of Irs2 deletion was associated with IGF1 signaling alterations in AD mice [197]. Moreover, premature death of AD mice was prevented by Irs2 deletion [197]. In contrast, decreased levels of IRS1 and IRS2 have been observed in the neurons of AD patients with aberrant IGF1R distributions [198]. Pathological changes in IGF1, IRS1, and IRS2 seemed to precede amyloid accumulation in an AD mouse model [199]. Recently, IRS2 was shown to play a predominant role in the brain insulin/IGF1 signaling pathway [200]. and abscisic acid was found to affect hippocampal BDNF, TNFα, and IRS2, showing protective effects against AD [201].
IL-6R, which has a tight connection with JAK2, was a hub gene of cluster 2. In the Chinese Han population, IL-6R gene polymorphisms have been associated with the onset of sporadic AD [202]. In contrast, Asp homozygotes of functional polymorphisms in IL-6R (Asp358Ala) were associated with higher cognitive performance [203]. Moreover, an IL-6R-responsive gene signature increases in the presence of IL-6R variant rs2228145, indicating the functional implications of IL-6R [204]. Moreover, the Asp358Ala variant of rs2228145 and elevated soluble IL-6R levels were associated with lower scores in modified preclinical Alzheimer’s cognitive composite and Montreal cognitive assessment [205]. When tocilizumab, an anti-IL-6R receptor, was administered to streptozotocin-induced AD mice, learning and spatial memory significantly improved [206]. In a human study, genetic variants of IL-6R were associated with the development of T2DM [207,208,209]. Inhibition of IL-6R by miR-22 augmented the viability of pancreatic cells and reduced the expression of apoptosis-related proteins [210].
4.3. Cluster 3
In cluster 3, purinergic receptors were tightly connected (Figure 2). Purinergic receptors are involved in ATP-mediated signaling pathways [211]. There are three subtypes: P1, P2X, and P2Y. These receptors play different roles in a variety of biological processes, and cluster 3 contains all types of P2RXs (P2RX1–P2RX7), which are ligand-gated ion channel receptors [211]. P2RX4 appears to be a hub gene of this cluster; however, few studies have reported an association between P2RX4 and AD or T2DM. Microglial P2XR4 regulates cathepsin B activity and promotes ApoE degradation, and deletion of P2XR4 recovers spatial memory impairment in mouse models [212]. OXYS rats, an advanced AD murine model, showed increased expression of p2xr4 [213]. Aβ fragment 1-42-induced neuronal death in rodents is enhanced by an upregulation of P2XR4 expression [214]. Not a single study reporting a relationship between P2XR4 and T2DM was found.
Among the P2RXs, P2RX7 is the most frequently studied receptor. P2RX7 knockout mice shows rapid postprandial hyperglycemia and increased beta cell apoptosis [215]. Besides, the fibroblasts of patients with T2DM show increased expression of P2XR7 and accompanying cellular responses —such as enhanced fibronectin and IL-6 secretion—, and activation of apoptosis[216]. The genetic variant rs1718119 of P2XR7 is associated with insulin sensitivity and secretion [217], increased beta-cell function, and the release of IL-1Ra in patients with T2DM [218]. P2XR7 is associated with ATP-mediated pathophysiology of AD. In rats, when ATP is administered to primary microglia, P2XR7 mediates the stimulation of superoxide production, and microglia-induced cortical cell death occurs [219]. P2XR7 is also involved in the secretion of cytokines in microglia [220], and the activation of microglia by Aβ is accomplished by the upregulation of P2XR7, as observed in a transgenic mouse model of AD [221]. Furthermore, protein expression of P2XR7 in postmortem human brain samples was observed; it modulated the NLRP3 inflammasome pathway [222]. P2XR7 activation is associated with neuronal autophagy and cognitive and memory impairment after traumatic brain injury [223]. In tau transgenic mice, P2XR7 induces exosome secretion by microglia, and blockade of P2XR7 reverses cognitive deficits in the Y-maze, prepulse inhibition, and contextual fear conditioning tests [224].
VSNL1 is located at the periphery of the P2 receptor network in this cluster; however, its role as a biomarker of AD is well known. Visinin-like protein 1 (VILIP-1) is encoded by the VSNL1 gene; it acts as a neuronal calcium sensor protein, and is involved in intracellular neuronal signaling [225]. VILIP-1 enhances tau protein hyperphosphorylation in P12 cells [226]. The VSNL1 SNP variant rs4038131 is associated with psychotic symptoms in patients with AD, who are more prone to rapid cognitive decline [227]. VILIP-1 levels in the cerebrospinal fluid (CSF) have been shown to predict AD [228,229,230,231]. In addition, VILIP-1 levels predict the cognitive decline rates of patients with AD (measured by clinical dementia ratings and other scores) [231]. VILIP-1 levels in the CSF also discriminates between patients with AD and patients with Lewy bodies —which are difficult to diagnose based on clinical symptoms—[230], and have a predictive power for the differential diagnosis of AD and MCI, especially in conjunction with conventional biomarkers, such as p-tau181 and Aβ(1-42) [229]. This finding was replicated in a meta-analysis of the association between VILIP-1 levels in CSF and AD [228]. While VSNL1 and VILIP-1 have implications in the pathophysiology of AD, relatively few connections have been found between VSNL1 and T2DM. VILIP-1 expression, for example, has an impact on the secretion of cyclic-AMP (cAMP) and insulin in MIN6 cells and mouse islets [232]. Genetic fine mapping of quantitative expression traits using islet cell transcriptomics data revealed that VSNL1 is a candidate T2DM risk gene [233]. However, no clinical studies have found an association between VSNL1 and T2DM development, which should be investigated in future studies.
5. Discussion
In this study, the genes related to AD and T2DM comorbidity were reviewed. Common comorbidity genes and genes affecting memory and cognition were used for PPI-based network clustering, and tightly-connected gene clusters were obtained. Since common genes were detected with respect to different phenotypes, they were unlikely to be a randomly identified group. Moreover, instead of using comorbidity genes directly, the memory and cognition gene subset was used in the analysis; therefore, the genes of the clusters are most likely involved in the pathophysiology of AD. Although the overall impact of the cluster genes on the entire genetic network of AD brain cells should be assessed for an accurate estimation of their roles in AD, these genes provide valuable guidelines for future research.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org., Supplementary Methods; Table S1: common genes of AD, T2DM, memory and cognition; Table S2: total result of ORA with GOBP; Table S3: total result of ORA with KEGG pathways, Table S4: cluster proteins from results of network-based clustering of common genes of AD, T2DM and memory-associated genes; Table S5: cluster proteins from results of network-based clustering of common genes of AD, T2DM and cognition-associated genes.
Funding
This research was funded by Gil hospital (grant number: FRD2021-18).
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in Supplementary Materials.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer's disease. Mol Neurodegener 2020, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, H.; Li, R.; Sterling, K.; Song, W. Amyloid beta-based therapy for Alzheimer's disease: challenges, successes and future. Signal Transduct Target Ther 2023, 8, 248. [Google Scholar] [CrossRef] [PubMed]
- Barbagallo, M.; Dominguez, L.J. Type 2 diabetes mellitus and Alzheimer's disease. World J Diabetes 2014, 5, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Kodamullil, A.T.; Hofmann-Apitius, M. Comorbidity Analysis between Alzheimer's Disease and Type 2 Diabetes Mellitus (T2DM) Based on Shared Pathways and the Role of T2DM Drugs. J Alzheimers Dis 2017, 60, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Diogo, D.; Tian, C.; Franklin, C.S.; Alanne-Kinnunen, M.; March, M.; Spencer, C.C.A.; Vangjeli, C.; Weale, M.E.; Mattsson, H.; Kilpelainen, E.; et al. Phenome-wide association studies across large population cohorts support drug target validation. Nat Commun 2018, 9, 4285. [Google Scholar] [CrossRef] [PubMed]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol 2012, 4, a011189. [Google Scholar] [CrossRef] [PubMed]
- Jean, S.; Kiger, A.A. Classes of phosphoinositide 3-kinases at a glance. J Cell Sci 2014, 127, 923–928. [Google Scholar] [CrossRef]
- Razani, E.; Pourbagheri-Sigaroodi, A.; Safaroghli-Azar, A.; Zoghi, A.; Shanaki-Bavarsad, M.; Bashash, D. The PI3K/Akt signaling axis in Alzheimer's disease: a valuable target to stimulate or suppress? Cell Stress Chaperones 2021, 26, 871–887. [Google Scholar] [CrossRef] [PubMed]
- Curtis, D.; Bandyopadhyay, S. Mini-review: Role of the PI3K/Akt pathway and tyrosine phosphatases in Alzheimer's disease susceptibility. Ann Hum Genet 2021, 85, 1–6. [Google Scholar] [CrossRef]
- Maffei, A.; Lembo, G.; Carnevale, D. PI3Kinases in Diabetes Mellitus and Its Related Complications. Int J Mol Sci 2018, 19. [Google Scholar] [CrossRef]
- Luo, J.; Sobkiw, C.L.; Hirshman, M.F.; Logsdon, M.N.; Li, T.Q.; Goodyear, L.J.; Cantley, L.C. Loss of class IA PI3K signaling in muscle leads to impaired muscle growth, insulin response, and hyperlipidemia. Cell Metab 2006, 3, 355–366. [Google Scholar] [CrossRef]
- Giese, K.P.; Mizuno, K. The roles of protein kinases in learning and memory. Learn Mem 2013, 20, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Bockaert, J.; Marin, P. mTOR in Brain Physiology and Pathologies. Physiol Rev 2015, 95, 1157–1187. [Google Scholar] [CrossRef]
- Dyer, A.H.; Vahdatpour, C.; Sanfeliu, A.; Tropea, D. The role of Insulin-Like Growth Factor 1 (IGF-1) in brain development, maturation and neuroplasticity. Neuroscience 2016, 325, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Jia, J. Immune-Related Hub Genes and the Competitive Endogenous RNA Network in Alzheimer's Disease. J Alzheimers Dis 2020, 77, 1255–1265. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, X.; Liu, J.; Song, J.; Zhang, S.; Chen, L.; Zhang, M. 20S-Protopanaxatriol improves cognitive function of Alzheimer's disease by promoting endogenous neurogenesis. Food Funct 2023, 14, 4191–4203. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Guo, J.; Zhou, Y.; Yan, S.; Xu, B.; Wang, Y.; Lu, D.; Ma, Z.; Chen, Q.; Tang, Q.; et al. Revealing the Mechanisms of Byu dMar 25 in the Treatment of Alzheimer's Disease through Network Pharmacology, Molecular Docking, and In Vivo Experiment. ACS Omega 2023, 8, 25066–25080. [Google Scholar] [CrossRef]
- Owolabi, B.O.; Ojo, O.O.; Srinivasan, D.K.; Conlon, J.M.; Flatt, P.R.; Abdel-Wahab, Y.H. Glucoregulatory, endocrine and morphological effects of [P5K]hymenochirin-1B in mice with diet-induced glucose intolerance and insulin resistance. Naunyn Schmiedebergs Arch Pharmacol 2016, 389, 769–781. [Google Scholar] [CrossRef]
- Musale, V.; Moffett, R.C.; Conlon, J.M.; Flatt, P.R.; Abdel-Wahab, Y.H. Beneficial actions of the [A14K] analog of the frog skin peptide PGLa-AM1 in mice with obesity and degenerative diabetes: A mechanistic study. Peptides 2021, 136, 170472. [Google Scholar] [CrossRef]
- Le Stunff, C.; Dechartres, A.; Mariot, V.; Lotton, C.; Trainor, C.; Miraglia Del Giudice, E.; Meyre, D.; Bieche, I.; Laurendeau, I.; Froguel, P.; et al. Association analysis indicates that a variant GATA-binding site in the PIK3CB promoter is a Cis-acting expression quantitative trait locus for this gene and attenuates insulin resistance in obese children. Diabetes 2008, 57, 494–502. [Google Scholar] [CrossRef]
- Clement, K.; Le Stunff, C.; Meirhaeghe, A.; Dechartres, A.; Ferrieres, J.; Basdevant, A.; Boitard, C.; Amouyel, P.; Bougneres, P. In obese and non-obese adults, the cis-regulatory rs361072 promoter variant of PIK3CB is associated with insulin resistance not with type 2 diabetes. Mol Genet Metab 2009, 96, 129–132. [Google Scholar] [CrossRef]
- Ribel-Madsen, R.; Poulsen, P.; Holmkvist, J.; Mortensen, B.; Grarup, N.; Friedrichsen, M.; Jorgensen, T.; Lauritzen, T.; Wojtaszewski, J.F.; Pedersen, O.; et al. Impact of rs361072 in the phosphoinositide 3-kinase p110beta gene on whole-body glucose metabolism and subunit protein expression in skeletal muscle. Diabetes 2010, 59, 1108–1112. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Bai, J.; Zhong, S.; Zhang, R.; Kang, K.; Zhang, X.; Xu, Y.; Zhao, C.; Zhao, M. Downregulation of PIK3CB Involved in Alzheimer's Disease via Apoptosis, Axon Guidance, and FoxO Signaling Pathway. Oxid Med Cell Longev 2022, 2022, 1260161. [Google Scholar] [CrossRef]
- Su, F.; Shu, H.; Ye, Q.; Wang, Z.; Xie, C.; Yuan, B.; Zhang, Z.; Bai, F. Brain insulin resistance deteriorates cognition by altering the topological features of brain networks. Neuroimage Clin 2017, 13, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, M.; Mac-Marcjanek, K.; Wozniak, L.A.; Nadel, I.; Lewinski, A.; Cypryk, K. The association of leukocyte phosphatidylinositol 3-kinase delta overexpression with gestational diabetes mellitus (GDM). Endokrynol Pol 2014, 65, 17–24. [Google Scholar] [CrossRef]
- Ma, R.; Deng, X.L.; Aleteng, Q.Q.; Li, L.; Zhu, J. Genome-Wide Transcriptome Analysis in Type 2 Diabetes Patients Treated by Sitagliptin. Diabetes Metab Syndr Obes 2022, 15, 1761–1770. [Google Scholar] [CrossRef] [PubMed]
- Malodobra, M.; Pilecka, A.; Gworys, B.; Adamiec, R. Single nucleotide polymorphisms within functional regions of genes implicated in insulin action and association with the insulin resistant phenotype. Mol Cell Biochem 2011, 349, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.; Ueki, K.; Takahashi, N.; Hashimoto, S.; Okamoto, M.; Awazawa, M.; Okazaki, Y.; Ohsugi, M.; Inabe, K.; Umehara, T.; et al. Class IA phosphatidylinositol 3-kinase in pancreatic beta cells controls insulin secretion by multiple mechanisms. Cell Metab 2010, 12, 619–632. [Google Scholar] [CrossRef]
- Zhang, Y.; Ji, B.; Li, J.; Li, Y.; Zhang, M.; Ban, B. SHORT syndrome in two Chinese girls: A case report and review of the literature. Mol Genet Genomic Med 2020, 8, e1385. [Google Scholar] [CrossRef]
- Masunaga, Y.; Fujisawa, Y.; Muramatsu, M.; Ono, H.; Inoue, T.; Fukami, M.; Kagami, M.; Saitsu, H.; Ogata, T. Insulin resistant diabetes mellitus in SHORT syndrome: case report and literature review. Endocr J 2021, 68, 111–117. [Google Scholar] [CrossRef]
- Chung, B.K.; Gibson, W.T. Autosomal dominant PIK3R1 mutations cause SHORT syndrome. Clin Genet 2014, 85, 228–229. [Google Scholar] [CrossRef]
- Curtis, D.; Bakaya, K.; Sharma, L.; Bandyopadhyay, S. Weighted burden analysis of exome-sequenced late-onset Alzheimer's cases and controls provides further evidence for a role for PSEN1 and suggests involvement of the PI3K/Akt/GSK-3beta and WNT signalling pathways. Ann Hum Genet 2020, 84, 291–302. [Google Scholar] [CrossRef]
- Qian, X.H.; Liu, X.L.; Chen, S.D.; Tang, H.D. Identification of Immune Hub Genes Associated With Braak Stages in Alzheimer's Disease and Their Correlation of Immune Infiltration. Front Aging Neurosci 2022, 14, 887168. [Google Scholar] [CrossRef]
- Shen, S.; Wang, F.; Fernandez, A.; Hu, W. Role of platelet-derived growth factor in type II diabetes mellitus and its complications. Diab Vasc Dis Res 2020, 17, 1479164120942119. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M.; Wands, J.R. Alzheimer-associated neuronal thread protein mediated cell death is linked to impaired insulin signaling. J Alzheimers Dis 2004, 6, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Yeboah, J.; Sane, D.C.; Crouse, J.R.; Herrington, D.M.; Bowden, D.W. Low plasma levels of FGF-2 and PDGF-BB are associated with cardiovascular events in type II diabetes mellitus (diabetes heart study). Dis Markers 2007, 23, 173–178. [Google Scholar] [CrossRef]
- Bessa, S.S.; Hussein, T.A.; Morad, M.A.; Amer, A.M. Urinary platelet-derived growth factor-BB as an early marker of nephropathy in patients with type 2 diabetes: an Egyptian study. Ren Fail 2012, 34, 670–675. [Google Scholar] [CrossRef]
- Liu, H.; Saffi, G.T.; Vasefi, M.S.; Choi, Y.; Kruk, J.S.; Ahmed, N.; Gondora, N.; Mielke, J.; Leonenko, Z.; Beazely, M.A. Amyloid-beta Inhibits PDGFbeta Receptor Activation and Prevents PDGF-BBInduced Neuroprotection. Curr Alzheimer Res 2018, 15, 618–627. [Google Scholar] [CrossRef]
- Keller, A.; Westenberger, A.; Sobrido, M.J.; Garcia-Murias, M.; Domingo, A.; Sears, R.L.; Lemos, R.R.; Ordonez-Ugalde, A.; Nicolas, G.; da Cunha, J.E.; et al. Mutations in the gene encoding PDGF-B cause brain calcifications in humans and mice. Nat Genet 2013, 45, 1077–1082. [Google Scholar] [CrossRef] [PubMed]
- Arts, F.A.; Velghe, A.I.; Stevens, M.; Renauld, J.C.; Essaghir, A.; Demoulin, J.B. Idiopathic basal ganglia calcification-associated PDGFRB mutations impair the receptor signalling. J Cell Mol Med 2015, 19, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Tsolaki, E.; Csincsik, L.; Xue, J.; Lengyel, I.; Bertazzo, S. Nuclear and cellular, micro and nano calcification in Alzheimer's disease patients and correlation to phosphorylated Tau. Acta Biomater 2022, 143, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Abate, G.; Frisoni, G.B.; Bourdon, J.C.; Piccirella, S.; Memo, M.; Uberti, D. The pleiotropic role of p53 in functional/dysfunctional neurons: focus on pathogenesis and diagnosis of Alzheimer's disease. Alzheimers Res Ther 2020, 12, 160. [Google Scholar] [CrossRef] [PubMed]
- Buizza, L.; Cenini, G.; Lanni, C.; Ferrari-Toninelli, G.; Prandelli, C.; Govoni, S.; Buoso, E.; Racchi, M.; Barcikowska, M.; Styczynska, M.; et al. Conformational altered p53 as an early marker of oxidative stress in Alzheimer's disease. PLoS One 2012, 7, e29789. [Google Scholar] [CrossRef] [PubMed]
- Rosas, I.; Martinez, C.; Coto, E.; Clarimon, J.; Lleo, A.; Illan-Gala, I.; Dols-Icardo, O.; Borroni, B.; Almeida, M.R.; van der Zee, J.; et al. Genetic variation in APOE, GRN, and TP53 are phenotype modifiers in frontotemporal dementia. Neurobiol Aging 2021, 99, 99.e15–99.e22. [Google Scholar] [CrossRef] [PubMed]
- Lanni, C.; Masi, M.; Racchi, M.; Govoni, S. Cancer and Alzheimer's disease inverse relationship: an age-associated diverging derailment of shared pathways. Mol Psychiatry 2021, 26, 280–295. [Google Scholar] [CrossRef]
- Kazmierczak, A.; Czapski, G.A.; Adamczyk, A.; Gajkowska, B.; Strosznajder, J.B. A novel mechanism of non-Abeta component of Alzheimer's disease amyloid (NAC) neurotoxicity. Interplay between p53 protein and cyclin-dependent kinase 5 (Cdk5). Neurochem Int 2011, 58, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Amor-Gutierrez, O.; Costa-Rama, E.; Arce-Varas, N.; Martinez-Rodriguez, C.; Novelli, A.; Fernandez-Sanchez, M.T.; Costa-Garcia, A. Competitive electrochemical immunosensor for the detection of unfolded p53 protein in blood as biomarker for Alzheimer's disease. Anal Chim Acta 2020, 1093, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.Q.; Luo, T.T.; Luo, S.C.; Wang, J.Q.; Wang, S.M.; Bai, Y.H.; Yang, Y.L.; Wang, Y.Y. p53 and mitochondrial dysfunction: novel insight of neurodegenerative diseases. J Bioenerg Biomembr 2016, 48, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Qu, L.; He, B.; Pan, Y.; Xu, Y.; Zhu, C.; Tang, Z.; Bao, Q.; Tian, F.; Wang, S. Association between polymorphisms in RAPGEF1, TP53, NRF1 and type 2 diabetes in Chinese Han population. Diabetes Res Clin Pract 2011, 91, 171–176. [Google Scholar] [CrossRef]
- Guo, D.; Fang, L.; Yu, X.; Wang, C.; Wang, Y.; Guo, W. Different Roles of TP53 Codon 72 Polymorphism in Type 2 Diabetes and Its Complications: Evidence from a Case-Control Study on a Chinese Han Population. Int J Gen Med 2021, 14, 4259–4268. [Google Scholar] [CrossRef]
- Burgdorf, K.S.; Grarup, N.; Justesen, J.M.; Harder, M.N.; Witte, D.R.; Jorgensen, T.; Sandbaek, A.; Lauritzen, T.; Madsbad, S.; Hansen, T.; et al. Studies of the association of Arg72Pro of tumor suppressor protein p53 with type 2 diabetes in a combined analysis of 55,521 Europeans. PLoS One 2011, 6, e15813. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Wang, H.; Zhang, F.; Zhang, M.; Wang, Q.; Wang, J. The common genes involved in the pathogenesis of Alzheimer's disease and type 2 diabetes and their implication for drug repositioning. Neuropharmacology 2023, 223, 109327. [Google Scholar] [CrossRef] [PubMed]
- Kung, C.P.; Murphy, M.E. The role of the p53 tumor suppressor in metabolism and diabetes. J Endocrinol 2016, 231, R61–R75. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, A.; Minami, A.; Kitagishi, Y.; Ogura, Y.; Matsuda, S. BRCA1 and p53 tumor suppressor molecules in Alzheimer's disease. Int J Mol Sci 2015, 16, 2879–2892. [Google Scholar] [CrossRef] [PubMed]
- Wezyk, M.; Zekanowski, C. Role of BRCA1 in Neuronal Death in Alzheimer's Disease. ACS Chem Neurosci 2018, 9, 870–872. [Google Scholar] [CrossRef]
- Silva, A.R.; Santos, A.C.; Farfel, J.M.; Grinberg, L.T.; Ferretti, R.E.; Campos, A.H.; Cunha, I.W.; Begnami, M.D.; Rocha, R.M.; Carraro, D.M.; et al. Repair of oxidative DNA damage, cell-cycle regulation and neuronal death may influence the clinical manifestation of Alzheimer's disease. PLoS One 2014, 9, e99897. [Google Scholar] [CrossRef] [PubMed]
- Suberbielle, E.; Djukic, B.; Evans, M.; Kim, D.H.; Taneja, P.; Wang, X.; Finucane, M.; Knox, J.; Ho, K.; Devidze, N.; et al. DNA repair factor BRCA1 depletion occurs in Alzheimer brains and impairs cognitive function in mice. Nat Commun 2015, 6, 8897. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Kaneko, S.; Dickson, D.W.; Kusaka, H. Aberrant Accumulation of BRCA1 in Alzheimer Disease and Other Tauopathies. J Neuropathol Exp Neurol 2020, 79, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, M.; Mano, T.; Saito, Y.; Murayama, S.; Toda, T.; Iwata, A. Colocalization of BRCA1 with Tau Aggregates in Human Tauopathies. Brain Sci 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Mano, T.; Nagata, K.; Nonaka, T.; Tarutani, A.; Imamura, T.; Hashimoto, T.; Bannai, T.; Koshi-Mano, K.; Tsuchida, T.; Ohtomo, R.; et al. Neuron-specific methylome analysis reveals epigenetic regulation and tau-related dysfunction of BRCA1 in Alzheimer's disease. Proc Natl Acad Sci U S A 2017, 114, E9645–E9654. [Google Scholar] [CrossRef]
- Brunet, J.; Vazquez-Martin, A.; Colomer, R.; Grana-Suarez, B.; Martin-Castillo, B.; Menendez, J.A. BRCA1 and acetyl-CoA carboxylase: the metabolic syndrome of breast cancer. Mol Carcinog 2008, 47, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Karachanak-Yankova, S.; Dimova, R.; Nikolova, D.; Nesheva, D.; Koprinarova, M.; Maslyankov, S.; Tafradjiska, R.; Gateva, P.; Velizarova, M.; Hammoudeh, Z.; et al. Epigenetic alterations in patients with type 2 diabetes mellitus. Balkan J Med Genet 2015, 18, 15–24. [Google Scholar] [CrossRef]
- Mori, T.; Asano, T.; Town, T. Targeting S100B in Cerebral Ischemia and in Alzheimer's Disease. Cardiovasc Psychiatry Neurol 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Serbinek, D.; Ullrich, C.; Pirchl, M.; Hochstrasser, T.; Schmidt-Kastner, R.; Humpel, C. S100b counteracts neurodegeneration of rat cholinergic neurons in brain slices after oxygen-glucose deprivation. Cardiovasc Psychiatry Neurol 2010, 2010, 106123. [Google Scholar] [CrossRef]
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal beta-catenin pathway function in an Alzheimer's disease model. J Immunol 2011, 187, 6539–6549. [Google Scholar] [CrossRef] [PubMed]
- Rocha, M.; Vieira, A.; Michels, M.; Borges, H.; Goulart, A.; Fernandes, F.; Dominguini, D.; Ritter, C.; Dal-Pizzol, F. Effects of S100B neutralization on the long-term cognitive impairment and neuroinflammatory response in an animal model of sepsis. Neurochem Int 2021, 142, 104906. [Google Scholar] [CrossRef]
- Cirillo, C.; Capoccia, E.; Iuvone, T.; Cuomo, R.; Sarnelli, G.; Steardo, L.; Esposito, G. S100B Inhibitor Pentamidine Attenuates Reactive Gliosis and Reduces Neuronal Loss in a Mouse Model of Alzheimer's Disease. Biomed Res Int 2015, 2015, 508342. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, E.; Sturchler, E.; Vetter, S.W. The S100B/RAGE Axis in Alzheimer's Disease. Cardiovasc Psychiatry Neurol 2010, 2010, 539581. [Google Scholar] [CrossRef]
- Lam, V.; Albrecht, M.A.; Takechi, R.; Giles, C.; James, A.P.; Foster, J.K.; Mamo, J.C. The Serum Concentration of the Calcium Binding Protein S100B is Positively Associated with Cognitive Performance in Older Adults. Front Aging Neurosci 2013, 5, 61. [Google Scholar] [CrossRef]
- Chaves, M.L.; Camozzato, A.L.; Ferreira, E.D.; Piazenski, I.; Kochhann, R.; Dall'Igna, O.; Mazzini, G.S.; Souza, D.O.; Portela, L.V. Serum levels of S100B and NSE proteins in Alzheimer's disease patients. J Neuroinflammation 2010, 7, 6. [Google Scholar] [CrossRef]
- Lee, B.W.; Chae, H.Y.; Kwon, S.J.; Park, S.Y.; Ihm, J.; Ihm, S.H. RAGE ligands induce apoptotic cell death of pancreatic beta-cells via oxidative stress. Int J Mol Med 2010, 26, 813–818. [Google Scholar] [PubMed]
- Afarideh, M.; Zaker Esteghamati, V.; Ganji, M.; Heidari, B.; Esteghamati, S.; Lavasani, S.; Ahmadi, M.; Tafakhori, A.; Nakhjavani, M.; Esteghamati, A. Associations of Serum S100B and S100P With the Presence and Classification of Diabetic Peripheral Neuropathy in Adults With Type 2 Diabetes: A Case-Cohort Study. Can J Diabetes 2019, 43, 336–344.e332. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Li, H.; Liu, X.; Du, X.; Deng, B. Levels of serum S100B are associated with cognitive dysfunction in patients with type 2 diabetes. Aging (Albany NY) 2020, 12, 4193–4203. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhang, H.; Schmidt, A.M.; Zhang, C. AGE/RAGE produces endothelial dysfunction in coronary arterioles in type 2 diabetic mice. Am J Physiol Heart Circ Physiol 2008, 295, H491–498. [Google Scholar] [CrossRef] [PubMed]
- Kesner, R.P.; Hui, X.; Sommer, T.; Wright, C.; Barrera, V.R.; Fanselow, M.S. The role of postnatal neurogenesis in supporting remote memory and spatial metric processing. Hippocampus 2014, 24, 1663–1671. [Google Scholar] [CrossRef] [PubMed]
- Benoit, J.D.; Rakic, P.; Frick, K.M. Prenatal stress induces spatial memory deficits and epigenetic changes in the hippocampus indicative of heterochromatin formation and reduced gene expression. Behav Brain Res 2015, 281, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zhou, Y.; Campbell, S.L.; Le, T.; Li, E.; Sweatt, J.D.; Silva, A.J.; Fan, G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci 2010, 13, 423–430. [Google Scholar] [CrossRef]
- Mitchnick, K.A.; Creighton, S.; O'Hara, M.; Kalisch, B.E.; Winters, B.D. Differential contributions of de novo and maintenance DNA methyltransferases to object memory processing in the rat hippocampus and perirhinal cortex--a double dissociation. Eur J Neurosci 2015, 41, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Pi, T.; Wei, S.; Jiang, Y.; Shi, J.S. High Methionine Diet-Induced Alzheimer's Disease like Symptoms Are Accompanied by 5-Methylcytosine Elevated Levels in the Brain. Behav Neurol 2021, 2021, 6683318. [Google Scholar] [CrossRef]
- Duan, S.; Li, C.; Gao, Y.; Meng, P.; Ji, S.; Xu, Y.; Mao, Y.; Wang, H.; Tian, J. The tyrosine kinase inhibitor LPM4870108 impairs learning and memory and induces transcriptomic and gene-specific DNA methylation changes in rats. Arch Toxicol 2022, 96, 845–857. [Google Scholar] [CrossRef]
- Tannorella, P.; Stoccoro, A.; Tognoni, G.; Petrozzi, L.; Salluzzo, M.G.; Ragalmuto, A.; Siciliano, G.; Haslberger, A.; Bosco, P.; Bonuccelli, U.; et al. Methylation analysis of multiple genes in blood DNA of Alzheimer's disease and healthy individuals. Neurosci Lett 2015, 600, 143–147. [Google Scholar] [CrossRef]
- Wang, S.C.; Oelze, B.; Schumacher, A. Age-specific epigenetic drift in late-onset Alzheimer's disease. PLoS One 2008, 3, e2698. [Google Scholar] [CrossRef] [PubMed]
- Pezzi, J.C.; Ens, C.M.; Borba, E.M.; Schumacher-Schuh, A.F.; de Andrade, F.M.; Chaves, M.L.; Fiegenbaum, M.; Camozzato, A.L. DNA methyltransferase haplotype is associated with Alzheimer's disease. Neurosci Lett 2014, 579, 70–74. [Google Scholar] [CrossRef]
- Mastroeni, D.; Grover, A.; Delvaux, E.; Whiteside, C.; Coleman, P.D.; Rogers, J. Epigenetic changes in Alzheimer's disease: decrements in DNA methylation. Neurobiol Aging 2010, 31, 2025–2037. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.T.; Dayeh, T.A.; Volkov, P.A.; Kirkpatrick, C.L.; Malmgren, S.; Jing, X.; Renstrom, E.; Wollheim, C.B.; Nitert, M.D.; Ling, C. Increased DNA methylation and decreased expression of PDX-1 in pancreatic islets from patients with type 2 diabetes. Mol Endocrinol 2012, 26, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, A.; Guruprasad, K.P.; Satyamoorthy, K.; Joshi, M.B. Interleukin-6 determines protein stabilization of DNA methyltransferases and alters DNA promoter methylation of genes associated with insulin signaling and angiogenesis. Lab Invest 2018, 98, 1143–1158. [Google Scholar] [CrossRef]
- Kassan, M.; Choi, S.K.; Galan, M.; Bishop, A.; Umezawa, K.; Trebak, M.; Belmadani, S.; Matrougui, K. Enhanced NF-kappaB activity impairs vascular function through PARP-1-, SP-1-, and COX-2-dependent mechanisms in type 2 diabetes. Diabetes 2013, 62, 2078–2087. [Google Scholar] [CrossRef]
- Kassan, M.; Choi, S.K.; Galan, M.; Trebak, M.; Belmadani, S.; Matrougui, K. Nuclear factor kappa B inhibition improves conductance artery function in type 2 diabetic mice. Diabetes Metab Res Rev 2015, 31, 39–49. [Google Scholar] [CrossRef]
- Zakaria, E.M.; El-Bassossy, H.M.; El-Maraghy, N.N.; Ahmed, A.F.; Ali, A.A. PARP-1 inhibition alleviates diabetic cardiac complications in experimental animals. Eur J Pharmacol 2016, 791, 444–454. [Google Scholar] [CrossRef]
- Xia, Q.; Lu, S.; Ostrovsky, J.; McCormack, S.E.; Falk, M.J.; Grant, S.F.A. PARP-1 Inhibition Rescues Short Lifespan in Hyperglycemic C. Elegans And Improves GLP-1 Secretion in Human Cells. Aging Dis 2018, 9, 17–30. [Google Scholar] [CrossRef]
- Waldman, M.; Nudelman, V.; Shainberg, A.; Abraham, N.G.; Kornwoski, R.; Aravot, D.; Arad, M.; Hochhauser, E. PARP-1 inhibition protects the diabetic heart through activation of SIRT1-PGC-1alpha axis. Exp Cell Res 2018, 373, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Cui, N.H.; Yang, J.M.; Liu, X.; Wang, X.B. Poly(ADP-Ribose) Polymerase Activity and Coronary Artery Disease in Type 2 Diabetes Mellitus: An Observational and Bidirectional Mendelian Randomization Study. Arterioscler Thromb Vasc Biol 2020, 40, 2516–2526. [Google Scholar] [CrossRef] [PubMed]
- Reda, E.; Hassaneen, S.; El-Abhar, H.S. Novel Trajectories of Bromocriptine Antidiabetic Action: Leptin-IL-6/ JAK2/p-STAT3/SOCS3, p-IR/p-AKT/GLUT4, PPAR-gamma/Adiponectin, Nrf2/PARP-1, and GLP-1. Front Pharmacol 2018, 9, 771. [Google Scholar] [CrossRef]
- Li, R.; Sun, X.; Li, P.; Li, W.; Zhao, L.; Zhu, L.; Zhu, S. GLP-1-Induced AMPK Activation Inhibits PARP-1 and Promotes LXR-Mediated ABCA1 Expression to Protect Pancreatic beta-Cells Against Cholesterol-Induced Toxicity Through Cholesterol Efflux. Front Cell Dev Biol 2021, 9, 646113. [Google Scholar] [CrossRef]
- Sclip, A.; Antoniou, X.; Colombo, A.; Camici, G.G.; Pozzi, L.; Cardinetti, D.; Feligioni, M.; Veglianese, P.; Bahlmann, F.H.; Cervo, L.; et al. c-Jun N-terminal kinase regulates soluble Abeta oligomers and cognitive impairment in AD mouse model. J Biol Chem 2011, 286, 43871–43880. [Google Scholar] [CrossRef] [PubMed]
- Espana, J.; Valero, J.; Minano-Molina, A.J.; Masgrau, R.; Martin, E.; Guardia-Laguarta, C.; Lleo, A.; Gimenez-Llort, L.; Rodriguez-Alvarez, J.; Saura, C.A. beta-Amyloid disrupts activity-dependent gene transcription required for memory through the CREB coactivator CRTC1. J Neurosci 2010, 30, 9402–9410. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Qiao, A.; Wang, Z.; Goodwin, J.S.; Lee, E.S.; Block, M.L.; Allsbrook, M.; McDonald, M.P.; Fan, G.H. Retinoic acid attenuates beta-amyloid deposition and rescues memory deficits in an Alzheimer's disease transgenic mouse model. J Neurosci 2008, 28, 11622–11634. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Tsai, C.W.; Deak, F.; Rogers, J.; Penuliar, M.; Sung, Y.M.; Maher, J.N.; Fu, Y.; Li, X.; Xu, H.; et al. Deficiency in LRP6-mediated Wnt signaling contributes to synaptic abnormalities and amyloid pathology in Alzheimer's disease. Neuron 2014, 84, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Tamayev, R.; D'Adamio, L. Memory deficits of British dementia knock-in mice are prevented by Abeta-precursor protein haploinsufficiency. J Neurosci 2012, 32, 5481–5485. [Google Scholar] [CrossRef]
- Pousinha, P.A.; Mouska, X.; Bianchi, D.; Temido-Ferreira, M.; Rajao-Saraiva, J.; Gomes, R.; Fernandez, S.P.; Salgueiro-Pereira, A.R.; Gandin, C.; Raymond, E.F.; et al. The Amyloid Precursor Protein C-Terminal Domain Alters CA1 Neuron Firing, Modifying Hippocampus Oscillations and Impairing Spatial Memory Encoding. Cell Rep 2019, 29, 317–331.e315. [Google Scholar] [CrossRef]
- Tu, Z.; Keller, M.P.; Zhang, C.; Rabaglia, M.E.; Greenawalt, D.M.; Yang, X.; Wang, I.M.; Dai, H.; Bruss, M.D.; Lum, P.Y.; et al. Integrative analysis of a cross-loci regulation network identifies App as a gene regulating insulin secretion from pancreatic islets. PLoS Genet 2012, 8, e1003107. [Google Scholar] [CrossRef] [PubMed]
- Alcarraz-Vizan, G.; Casini, P.; Cadavez, L.; Visa, M.; Montane, J.; Servitja, J.M.; Novials, A. Inhibition of BACE2 counteracts hIAPP-induced insulin secretory defects in pancreatic beta-cells. FASEB J 2015, 29, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Neuner, S.M.; Wilmott, L.A.; Hoffmann, B.R.; Mozhui, K.; Kaczorowski, C.C. Hippocampal proteomics defines pathways associated with memory decline and resilience in normal aging and Alzheimer's disease mouse models. Behav Brain Res 2017, 322, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.C.; Goh, M.Q.L.; Koo, E.H. Transcriptional regulation of APP by apoE: To boldly go where no isoform has gone before: ApoE, APP transcription and AD: Hypothesised mechanisms and existing knowledge gaps. Bioessays 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Gezen-Ak, D.; Atasoy, I.L.; Candas, E.; Alaylioglu, M.; Dursun, E. The Transcriptional Regulatory Properties of Amyloid Beta 1-42 may Include Regulation of Genes Related to Neurodegeneration. Neuromolecular Med 2018, 20, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.; Feldman, H.M.; Lu, T.; Drake, D.; Lim, E.T.; Ling, K.H.; Bishop, N.A.; Pan, Y.; Seo, J.; Lin, Y.T.; et al. REST and Neural Gene Network Dysregulation in iPSC Models of Alzheimer's Disease. Cell Rep 2019, 26, 1112–1127.e1119. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Nielsen, M.; Carcione, T.; Li, S.; Shi, J. Apolipoprotein E regulates mitochondrial function through the PGC-1alpha-sirtuin 3 pathway. Aging (Albany NY) 2019, 11, 11148–11156. [Google Scholar] [CrossRef]
- Madrid, L.; Moreno-Grau, S.; Ahmad, S.; Gonzalez-Perez, A.; de Rojas, I.; Xia, R.; Martino Adami, P.V.; Garcia-Gonzalez, P.; Kleineidam, L.; Yang, Q.; et al. Multiomics integrative analysis identifies APOE allele-specific blood biomarkers associated to Alzheimer's disease etiopathogenesis. Aging (Albany NY) 2021, 13, 9277–9329. [Google Scholar] [CrossRef] [PubMed]
- Sienski, G.; Narayan, P.; Bonner, J.M.; Kory, N.; Boland, S.; Arczewska, A.A.; Ralvenius, W.T.; Akay, L.; Lockshin, E.; He, L.; et al. APOE4 disrupts intracellular lipid homeostasis in human iPSC-derived glia. Sci Transl Med 2021, 13. [Google Scholar] [CrossRef]
- Blanchard, J.W.; Akay, L.A.; Davila-Velderrain, J.; von Maydell, D.; Mathys, H.; Davidson, S.M.; Effenberger, A.; Chen, C.Y.; Maner-Smith, K.; Hajjar, I.; et al. APOE4 impairs myelination via cholesterol dysregulation in oligodendrocytes. Nature 2022, 611, 769–779. [Google Scholar] [CrossRef]
- Chen, C.J.; Chen, C.C.; Wu, D.; Chi, N.F.; Chen, P.C.; Liao, Y.P.; Chiu, H.W.; Hu, C.J. Effects of the apolipoprotein E epsilon4 allele on functional MRI during n-back working memory tasks in healthy middle-aged adults. AJNR Am J Neuroradiol 2013, 34, 1197–1202. [Google Scholar] [CrossRef]
- Kerchner, G.A.; Berdnik, D.; Shen, J.C.; Bernstein, J.D.; Fenesy, M.C.; Deutsch, G.K.; Wyss-Coray, T.; Rutt, B.K. APOE epsilon4 worsens hippocampal CA1 apical neuropil atrophy and episodic memory. Neurology 2014, 82, 691–697. [Google Scholar] [CrossRef]
- Mikos, A.E.; Piryatinsky, I.; Tremont, G.; Malloy, P.F. The APOE epsilon4 allele is associated with increased frontally mediated neurobehavioral symptoms in amnestic MCI. Alzheimer Dis Assoc Disord 2013, 27, 109–115. [Google Scholar] [CrossRef]
- Luck, T.; Then, F.S.; Luppa, M.; Schroeter, M.L.; Arelin, K.; Burkhardt, R.; Thiery, J.; Loffler, M.; Villringer, A.; Riedel-Heller, S.G. Association of the apolipoprotein E genotype with memory performance and executive functioning in cognitively intact elderly. Neuropsychology 2015, 29, 382–387. [Google Scholar] [CrossRef]
- Chang, P.; Li, X.; Ma, C.; Zhang, S.; Liu, Z.; Chen, K.; Ai, L.; Chang, J.; Zhang, Z. The Effects of an APOE Promoter Polymorphism on Human White Matter Connectivity during Non-Demented Aging. J Alzheimers Dis 2017, 55, 77–87. [Google Scholar] [CrossRef]
- Salvado, G.; Ferreira, D.; Operto, G.; Cumplido-Mayoral, I.; Arenaza-Urquijo, E.M.; Cacciaglia, R.; Falcon, C.; Vilor-Tejedor, N.; Minguillon, C.; Groot, C.; et al. The protective gene dose effect of the APOE epsilon2 allele on gray matter volume in cognitively unimpaired individuals. Alzheimers Dement 2022, 18, 1383–1395. [Google Scholar] [CrossRef] [PubMed]
- El-Lebedy, D.; Raslan, H.M.; Mohammed, A.M. Apolipoprotein E gene polymorphism and risk of type 2 diabetes and cardiovascular disease. Cardiovasc Diabetol 2016, 15, 12. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, J.; Weng, R.; Gu, X.; Zhong, Z. Apolipoprotein E gene polymorphism and the risk of cardiovascular disease and type 2 diabetes. BMC Cardiovasc Disord 2019, 19, 213. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.W.; Benzie, I.F.; Yeung, V.T. Type 2 diabetes mellitus and its renal complications in relation to apolipoprotein E gene polymorphism. Transl Res 2008, 152, 134–142. [Google Scholar] [CrossRef]
- Perron, P.; Brisson, D.; Santure, M.; Blackburn, P.; Bergeron, J.; Vohl, M.C.; Despres, J.P.; Gaudet, D. Apolipoprotein E and lipoprotein lipase gene polymorphisms interaction on the atherogenic combined expression of hypertriglyceridemia and hyperapobetalipoproteinemia phenotypes. J Endocrinol Invest 2007, 30, 551–557. [Google Scholar] [CrossRef]
- Guangda, X.; Linshuang, Z.; Jie, H.; Ling, Y.; Huijuan, X. Apo e4 allele is associated with endothelium-dependent arterial dilation in women with type 2 diabetes. Diabetes Res Clin Pract 2006, 72, 155–161. [Google Scholar] [CrossRef]
- Monastiriotis, C.; Papanas, N.; Trypsianis, G.; Karanikola, K.; Veletza, S.; Maltezos, E. The epsilon4 allele of the APOE gene is associated with more severe peripheral neuropathy in type 2 diabetic patients. Angiology 2013, 64, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Lumsden, A.L.; Mulugeta, A.; Zhou, A.; Hypponen, E. Apolipoprotein E (APOE) genotype-associated disease risks: a phenome-wide, registry-based, case-control study utilising the UK Biobank. EBioMedicine 2020, 59, 102954. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Fu, X.; Chu, Q.; Li, J.; Shu, G. Relationship Between the ApoE Gene Polymorphism and Type 2 Diabetes Mellitus Complications. Genet Test Mol Biomarkers 2021, 25, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, Y.; Chen, J.; Sun, H.; Lann, D.; Hajjar, R.J.; Yakar, S.; Leroith, D. Apolipoprotein E deficiency abrogates insulin resistance in a mouse model of type 2 diabetes mellitus. Diabetologia 2009, 52, 1434–1441. [Google Scholar] [CrossRef]
- Guan, J.; Zhao, H.L.; Sui, Y.; He, L.; Lee, H.M.; Lai, F.M.; Tong, P.C.; Chan, J.C. Histopathological correlations of islet amyloidosis with apolipoprotein E polymorphisms in type 2 diabetic Chinese patients. Pancreas 2013, 42, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Tomono, Y.; Iwai, M.; Inaba, S.; Mogi, M.; Horiuchi, M. Blockade of AT1 receptor improves adipocyte differentiation in atherosclerotic and diabetic models. Am J Hypertens 2008, 21, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Jun, G.; Naj, A.C.; Beecham, G.W.; Wang, L.S.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Ertekin-Taner, N.; Fallin, M.D.; Friedland, R.; et al. Meta-analysis confirms CR1, CLU, and PICALM as alzheimer disease risk loci and reveals interactions with APOE genotypes. Arch Neurol 2010, 67, 1473–1484. [Google Scholar] [CrossRef] [PubMed]
- Thambisetty, M.; Simmons, A.; Velayudhan, L.; Hye, A.; Campbell, J.; Zhang, Y.; Wahlund, L.O.; Westman, E.; Kinsey, A.; Guntert, A.; et al. Association of plasma clusterin concentration with severity, pathology, and progression in Alzheimer disease. Arch Gen Psychiatry 2010, 67, 739–748. [Google Scholar] [CrossRef]
- Carrasquillo, M.M.; Belbin, O.; Hunter, T.A.; Ma, L.; Bisceglio, G.D.; Zou, F.; Crook, J.E.; Pankratz, V.S.; Dickson, D.W.; Graff-Radford, N.R.; et al. Replication of CLU, CR1, and PICALM associations with alzheimer disease. Arch Neurol 2010, 67, 961–964. [Google Scholar] [CrossRef]
- Corneveaux, J.J.; Myers, A.J.; Allen, A.N.; Pruzin, J.J.; Ramirez, M.; Engel, A.; Nalls, M.A.; Chen, K.; Lee, W.; Chewning, K.; et al. Association of CR1, CLU and PICALM with Alzheimer's disease in a cohort of clinically characterized and neuropathologically verified individuals. Hum Mol Genet 2010, 19, 3295–3301. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet 2009, 41, 1094–1099. [Google Scholar] [CrossRef]
- Xing, Y.Y.; Yu, J.T.; Cui, W.Z.; Zhong, X.L.; Wu, Z.C.; Zhang, Q.; Tan, L. Blood clusterin levels, rs9331888 polymorphism, and the risk of Alzheimer's disease. J Alzheimers Dis 2012, 29, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Schrijvers, E.M.; Koudstaal, P.J.; Hofman, A.; Breteler, M.M. Plasma clusterin and the risk of Alzheimer disease. JAMA 2011, 305, 1322–1326. [Google Scholar] [CrossRef]
- Schjeide, B.M.; Schnack, C.; Lambert, J.C.; Lill, C.M.; Kirchheiner, J.; Tumani, H.; Otto, M.; Tanzi, R.E.; Lehrach, H.; Amouyel, P.; et al. The role of clusterin, complement receptor 1, and phosphatidylinositol binding clathrin assembly protein in Alzheimer disease risk and cerebrospinal fluid biomarker levels. Arch Gen Psychiatry 2011, 68, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Wojtas, A.M.; Kang, S.S.; Olley, B.M.; Gatherer, M.; Shinohara, M.; Lozano, P.A.; Liu, C.C.; Kurti, A.; Baker, K.E.; Dickson, D.W.; et al. Loss of clusterin shifts amyloid deposition to the cerebrovasculature via disruption of perivascular drainage pathways. Proc Natl Acad Sci U S A 2017, 114, E6962–E6971. [Google Scholar] [CrossRef]
- Robbins, J.P.; Perfect, L.; Ribe, E.M.; Maresca, M.; Dangla-Valls, A.; Foster, E.M.; Killick, R.; Nowosiad, P.; Reid, M.J.; Polit, L.D.; et al. Clusterin Is Required for beta-Amyloid Toxicity in Human iPSC-Derived Neurons. Front Neurosci 2018, 12, 504. [Google Scholar] [CrossRef]
- Wojtas, A.M.; Sens, J.P.; Kang, S.S.; Baker, K.E.; Berry, T.J.; Kurti, A.; Daughrity, L.; Jansen-West, K.R.; Dickson, D.W.; Petrucelli, L.; et al. Astrocyte-derived clusterin suppresses amyloid formation in vivo. Mol Neurodegener 2020, 15, 71. [Google Scholar] [CrossRef]
- Liu, Z.; Chao, J.; Wang, C.; Sun, G.; Roeth, D.; Liu, W.; Chen, X.; Li, L.; Tian, E.; Feng, L.; et al. Astrocytic response mediated by the CLU risk allele inhibits OPC proliferation and myelination in a human iPSC model. Cell Rep 2023, 42, 112841. [Google Scholar] [CrossRef]
- Stevens, B.W.; DiBattista, A.M.; William Rebeck, G.; Green, A.E. A gene-brain-cognition pathway for the effect of an Alzheimer׳s risk gene on working memory in young adults. Neuropsychologia 2014, 61, 143–149. [Google Scholar] [CrossRef]
- Alfimova, M.; Kondratyev, N.; Golov, A.; Golimbet, V. Relationship between Alzheimer's disease-associated SNPs within the CLU gene, local DNA methylation and episodic verbal memory in healthy and schizophrenia subjects. Psychiatry Res 2019, 272, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.H.; Heng Mak, T.S.; Fan, Y.; Yin Ho, D.T.; Sham, P.C.; Chu, L.W.; Song, Y.Q. Associations between CLU polymorphisms and memory performance: The role of serum lipids in Alzheimer's disease. J Psychiatr Res 2020, 129, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Ferencz, B.; Laukka, E.J.; Welmer, A.K.; Kalpouzos, G.; Angleman, S.; Keller, L.; Graff, C.; Lovden, M.; Backman, L. The benefits of staying active in old age: physical activity counteracts the negative influence of PICALM, BIN1, and CLU risk alleles on episodic memory functioning. Psychol Aging 2014, 29, 440–449. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, Z.; Khoury, N.; Betley, M.J.; Lehallier, B.; Willoughby, D.; Olsson, N.; Yang, A.C.; Hahn, O.; Lu, N.; Vest, R.T.; et al. Exercise plasma boosts memory and dampens brain inflammation via clusterin. Nature 2021, 600, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, O.; Labonte, J.; Albrecht, B.; Wieczorek, D.; Lechno, S.; Zechner, U.; Haaf, T. Two patients with EP300 mutations and facial dysmorphism different from the classic Rubinstein-Taybi syndrome. Am J Med Genet A 2010, 152A, 181–184. [Google Scholar] [CrossRef]
- Van Gils, J.; Magdinier, F.; Fergelot, P.; Lacombe, D. Rubinstein-Taybi Syndrome: A Model of Epigenetic Disorder. Genes (Basel) 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Stockton, M.E.; Eisinger, B.E.; Zhao, Y.; Miller, J.L.; Bhuiyan, I.; Gao, Y.; Wu, Z.; Peng, J.; Zhao, X. Reducing histone acetylation rescues cognitive deficits in a mouse model of Fragile X syndrome. Nat Commun 2018, 9, 2494. [Google Scholar] [CrossRef]
- Lu, X.; Deng, Y.; Yu, D.; Cao, H.; Wang, L.; Liu, L.; Yu, C.; Zhang, Y.; Guo, X.; Yu, G. Histone acetyltransferase p300 mediates histone acetylation of PS1 and BACE1 in a cellular model of Alzheimer's disease. PLoS One 2014, 9, e103067. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Tan, B.; Cheng, Y. P300 Inhibition Improves Cell Apoptosis and Cognition Impairment Induced by Sevoflurane Through Regulating IL-17A Activation. World Neurosurg 2021, 154, e566–e571. [Google Scholar] [CrossRef]
- Chen, X.; Li, Y.; Wang, C.; Tang, Y.; Mok, S.A.; Tsai, R.M.; Rojas, J.C.; Karydas, A.; Miller, B.L.; Boxer, A.L.; et al. Promoting tau secretion and propagation by hyperactive p300/CBP via autophagy-lysosomal pathway in tauopathy. Mol Neurodegener 2020, 15, 2. [Google Scholar] [CrossRef]
- Fan, Z.; Wu, J.; Chen, Q.N.; Lyu, A.K.; Chen, J.L.; Sun, Y.; Lyu, Q.; Zhao, Y.X.; Guo, A.; Liao, Z.Y.; et al. Type 2 diabetes-induced overactivation of P300 contributes to skeletal muscle atrophy by inhibiting autophagic flux. Life Sci 2020, 258, 118243. [Google Scholar] [CrossRef] [PubMed]
- Long, H.Z.; Cheng, Y.; Zhou, Z.W.; Luo, H.Y.; Wen, D.D.; Gao, L.C. PI3K/AKT Signal Pathway: A Target of Natural Products in the Prevention and Treatment of Alzheimer's Disease and Parkinson's Disease. Front Pharmacol 2021, 12, 648636. [Google Scholar] [CrossRef]
- Talchai, C.; Lin, H.V.; Kitamura, T.; Accili, D. Genetic and biochemical pathways of beta-cell failure in type 2 diabetes. Diabetes Obes Metab 2009, 11 Suppl 4, 38–45. [Google Scholar] [CrossRef]
- Kitamura, T. The role of FOXO1 in beta-cell failure and type 2 diabetes mellitus. Nat Rev Endocrinol 2013, 9, 615–623. [Google Scholar] [CrossRef]
- Li, X.; Wan, T.; Li, Y. Role of FoxO1 in regulating autophagy in type 2 diabetes mellitus (Review). Exp Ther Med 2021, 22, 707. [Google Scholar] [CrossRef] [PubMed]
- Benchoula, K.; Arya, A.; Parhar, I.S.; Hwa, W.E. FoxO1 signaling as a therapeutic target for type 2 diabetes and obesity. Eur J Pharmacol 2021, 891, 173758. [Google Scholar] [CrossRef] [PubMed]
- Moll, L.; Schubert, M. The Role of Insulin and Insulin-Like Growth Factor-1/FoxO-Mediated Transcription for the Pathogenesis of Obesity-Associated Dementia. Curr Gerontol Geriatr Res 2012, 2012, 384094. [Google Scholar] [CrossRef]
- Xu, P.; Das, M.; Reilly, J.; Davis, R.J. JNK regulates FoxO-dependent autophagy in neurons. Genes Dev 2011, 25, 310–322. [Google Scholar] [CrossRef]
- Paroni, G.; Seripa, D.; Fontana, A.; D'Onofrio, G.; Gravina, C.; Urbano, M.; Cascavilla, L.; Pellegrini, F.; Greco, A.; Pilotto, A. FOXO1 locus and acetylcholinesterase inhibitors in elderly patients with Alzheimer's disease. Clin Interv Aging 2014, 9, 1783–1791. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, X.; Liao, Y.; Luo, C.; Zou, D.; Wei, X.; Huang, Q.; Wu, Y. MiR-181a influences the cognitive function of epileptic rats induced by pentylenetetrazol. Int J Clin Exp Pathol 2015, 8, 12861–12868. [Google Scholar]
- Wu, Q.; Yuan, X.; Bai, J.; Han, R.; Li, Z.; Zhang, H.; Xiu, R. MicroRNA-181a protects against pericyte apoptosis via directly targeting FOXO1: implication for ameliorated cognitive deficits in APP/PS1 mice. Aging (Albany NY) 2019, 11, 6120–6133. [Google Scholar] [CrossRef] [PubMed]
- Shigemizu, D.; Akiyama, S.; Higaki, S.; Sugimoto, T.; Sakurai, T.; Boroevich, K.A.; Sharma, A.; Tsunoda, T.; Ochiya, T.; Niida, S.; et al. Prognosis prediction model for conversion from mild cognitive impairment to Alzheimer's disease created by integrative analysis of multi-omics data. Alzheimers Res Ther 2020, 12, 145. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.Y.; Ellenstein, A.; Chen, C.D.; Hinman, J.D.; Berg, E.A.; Costello, C.E.; Yamin, R.; Neve, R.L.; Abraham, C.R. Amyloid precursor protein interacts with notch receptors. J Neurosci Res 2005, 82, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Boo, J.H.; Sohn, J.H.; Kim, J.E.; Song, H.; Mook-Jung, I. Rac1 changes the substrate specificity of gamma-secretase between amyloid precursor protein and Notch1. Biochem Biophys Res Commun 2008, 372, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.N.; Park, J.S.; Gwon, A.R.; Arumugam, T.V.; Jo, D.G. Alzheimer's disease and Notch signaling. Biochem Biophys Res Commun 2009, 390, 1093–1097. [Google Scholar] [CrossRef] [PubMed]
- Veeraraghavalu, K.; Choi, S.H.; Zhang, X.; Sisodia, S.S. Presenilin 1 mutants impair the self-renewal and differentiation of adult murine subventricular zone-neuronal progenitors via cell-autonomous mechanisms involving notch signaling. J Neurosci 2010, 30, 6903–6915. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Huang, G.; Liu, H.; Chang, H.; Wilson, J.X. Folic acid enhances Notch signaling, hippocampal neurogenesis, and cognitive function in a rat model of cerebral ischemia. Nutr Neurosci 2012, 15, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Mao, X.; Xie, L.; Ding, M.; Shao, B.; Jin, K. Notch1 signaling modulates neuronal progenitor activity in the subventricular zone in response to aging and focal ischemia. Aging Cell 2013, 12, 978–987. [Google Scholar] [CrossRef] [PubMed]
- Leal, M.C.; Surace, E.I.; Holgado, M.P.; Ferrari, C.C.; Tarelli, R.; Pitossi, F.; Wisniewski, T.; Castano, E.M.; Morelli, L. Notch signaling proteins HES-1 and Hey-1 bind to insulin degrading enzyme (IDE) proximal promoter and repress its transcription and activity: implications for cellular Abeta metabolism. Biochim Biophys Acta 2012, 1823, 227–235. [Google Scholar] [CrossRef]
- Brai, E.; Alina Raio, N.; Alberi, L. Notch1 hallmarks fibrillary depositions in sporadic Alzheimer's disease. Acta Neuropathol Commun 2016, 4, 64. [Google Scholar] [CrossRef]
- Kapoor, A.; Nation, D.A. Role of Notch signaling in neurovascular aging and Alzheimer's disease. Semin Cell Dev Biol 2021, 116, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, P.; Ren, L.; Hu, C.; Bi, J. Protective effect of melatonin on soluble Abeta1-42-induced memory impairment, astrogliosis, and synaptic dysfunction via the Musashi1/Notch1/Hes1 signaling pathway in the rat hippocampus. Alzheimers Res Ther 2016, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Sirichoat, A.; Chaijaroonkhanarak, W.; Prachaney, P.; Pannangrong, W.; Leksomboon, R.; Chaichun, A.; Wigmore, P.; Welbat, J.U. Effects of Asiatic Acid on Spatial Working Memory and Cell Proliferation in the Adult Rat Hippocampus. Nutrients 2015, 7, 8413–8423. [Google Scholar] [CrossRef] [PubMed]
- Xue, F.; Chen, Y.C.; Zhou, C.H.; Wang, Y.; Cai, M.; Yan, W.J.; Wu, R.; Wang, H.N.; Peng, Z.W. Risperidone ameliorates cognitive deficits, promotes hippocampal proliferation, and enhances Notch signaling in a murine model of schizophrenia. Pharmacol Biochem Behav 2017, 163, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.H.; Wang, P.; He, X.Q.; Hong, M.Z.; Liu, F. Micro ribonucleic acid-363 regulates the phosphatidylinositol 3-kinase/threonine protein kinase axis by targeting NOTCH1 and forkhead box C2, leading to hepatic glucose and lipids metabolism disorder in type 2 diabetes mellitus. J Diabetes Investig 2022, 13, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Li, X.; Yang, J.; Liu, H.; Zhang, Y.; Yang, X.; Yang, S.; Chang, B.; Chen, L.; Chang, B. Salsalate Prevents beta-Cell Dedifferentiation in OLETF Rats with Type 2 Diabetes through Notch1 Pathway. Aging Dis 2019, 10, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, B.; Deng, B.; Zhang, F.; Wu, J.; Wang, Y.; Le, Y.; Zhai, Q. Amyloid-beta induces hepatic insulin resistance in vivo via JAK2. Diabetes 2013, 62, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Morris, D.L.; Jiang, L.; Liu, Y.; Rui, L. SH2B1 in beta-cells promotes insulin expression and glucose metabolism in mice. Mol Endocrinol 2014, 28, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Ye, X.; Yao, Q.; Lu, A.; Zhao, Z.; Ding, Y.; Meng, C.; Yu, W.; Du, Y.; Cheng, J. Egr2 enhances insulin resistance via JAK2/STAT3/SOCS-1 pathway in HepG2 cells treated with palmitate. Gen Comp Endocrinol 2018, 260, 25–31. [Google Scholar] [CrossRef]
- Hao, T.; Zhang, H.; Li, S.; Tian, H. Glucagon-like peptide 1 receptor agonist ameliorates the insulin resistance function of islet beta cells via the activation of PDX-1/JAK signaling transduction in C57/BL6 mice with high-fat diet-induced diabetes. Int J Mol Med 2017, 39, 1029–1036. [Google Scholar] [CrossRef]
- Tuttle, K.R.; Brosius, F.C., 3rd; Adler, S.G.; Kretzler, M.; Mehta, R.L.; Tumlin, J.A.; Tanaka, Y.; Haneda, M.; Liu, J.; Silk, M.E.; et al. JAK1/JAK2 inhibition by baricitinib in diabetic kidney disease: results from a Phase 2 randomized controlled clinical trial. Nephrol Dial Transplant 2018, 33, 1950–1959. [Google Scholar] [CrossRef]
- Zhang, N.; Zheng, Q.; Wang, Y.; Lin, J.; Wang, H.; Liu, R.; Yan, M.; Chen, X.; Yang, J.; Chen, X. Renoprotective Effect of the Recombinant Anti-IL-6R Fusion Proteins by Inhibiting JAK2/STAT3 Signaling Pathway in Diabetic Nephropathy. Front Pharmacol 2021, 12, 681424. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, C.; Sriram, S.; Muthian, G.; Bright, J.J. Signaling through JAK2-STAT5 pathway is essential for IL-3-induced activation of microglia. Glia 2004, 45, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Yamada, M.; Sasabe, J.; Terashita, K.; Shimoda, M.; Matsuoka, M.; Aiso, S. Amyloid-beta causes memory impairment by disturbing the JAK2/STAT3 axis in hippocampal neurons. Mol Psychiatry 2009, 14, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Marwarha, G.; Prasanthi, J.R.; Schommer, J.; Dasari, B.; Ghribi, O. Molecular interplay between leptin, insulin-like growth factor-1, and beta-amyloid in organotypic slices from rabbit hippocampus. Mol Neurodegener 2011, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- He, G.L.; Luo, Z.; Shen, T.T.; Li, P.; Yang, J.; Luo, X.; Chen, C.H.; Gao, P.; Yang, X.S. Inhibition of STAT3- and MAPK-dependent PGE(2) synthesis ameliorates phagocytosis of fibrillar beta-amyloid peptide (1-42) via EP2 receptor in EMF-stimulated N9 microglial cells. J Neuroinflammation 2016, 13, 296. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.H.; Xiang, J.; Liu, X.; Yu, S.P.; Manfredsson, F.P.; Sandoval, I.M.; Wu, S.; Wang, J.Z.; Ye, K. Deficiency in BDNF/TrkB Neurotrophic Activity Stimulates delta-Secretase by Upregulating C/EBPbeta in Alzheimer's Disease. Cell Rep 2019, 28, 655–669.e655. [Google Scholar] [CrossRef] [PubMed]
- Ceyzeriat, K.; Ben Haim, L.; Denizot, A.; Pommier, D.; Matos, M.; Guillemaud, O.; Palomares, M.A.; Abjean, L.; Petit, F.; Gipchtein, P.; et al. Modulation of astrocyte reactivity improves functional deficits in mouse models of Alzheimer's disease. Acta Neuropathol Commun 2018, 6, 104. [Google Scholar] [CrossRef]
- Oliveira, J.M.; Rebuffat, S.A.; Gasa, R.; Gomis, R. Targeting type 2 diabetes: lessons from a knockout model of insulin receptor substrate 2. Can J Physiol Pharmacol 2014, 92, 613–620. [Google Scholar] [CrossRef]
- Muller, D.; Huang, G.C.; Amiel, S.; Jones, P.M.; Persaud, S.J. Identification of insulin signaling elements in human beta-cells: autocrine regulation of insulin gene expression. Diabetes 2006, 55, 2835–2842. [Google Scholar] [CrossRef]
- Lingohr, M.K.; Briaud, I.; Dickson, L.M.; McCuaig, J.F.; Alarcon, C.; Wicksteed, B.L.; Rhodes, C.J. Specific regulation of IRS-2 expression by glucose in rat primary pancreatic islet beta-cells. J Biol Chem 2006, 281, 15884–15892. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Barbosa-Sampaio, H.; Jones, P.M.; Persaud, S.J.; Muller, D.S. The CaMK4/CREB/IRS-2 cascade stimulates proliferation and inhibits apoptosis of beta-cells. PLoS One 2012, 7, e45711. [Google Scholar] [CrossRef]
- Demozay, D.; Tsunekawa, S.; Briaud, I.; Shah, R.; Rhodes, C.J. Specific glucose-induced control of insulin receptor substrate-2 expression is mediated via Ca2+-dependent calcineurin/NFAT signaling in primary pancreatic islet beta-cells. Diabetes 2011, 60, 2892–2902. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.; Piecewicz, S.M.; McGinnis, L.M.; Taniguchi, C.M.; Wiegand, S.J.; Anderson, K.; Chan, C.W.; Mulligan, K.X.; Kuo, D.; Yuan, J.; et al. A liver Hif-2alpha-Irs2 pathway sensitizes hepatic insulin signaling and is modulated by Vegf inhibition. Nat Med 2013, 19, 1331–1337. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, C.M.; Finger, E.C.; Krieg, A.J.; Wu, C.; Diep, A.N.; LaGory, E.L.; Wei, K.; McGinnis, L.M.; Yuan, J.; Kuo, C.J.; et al. Cross-talk between hypoxia and insulin signaling through Phd3 regulates hepatic glucose and lipid metabolism and ameliorates diabetes. Nat Med 2013, 19, 1325–1330. [Google Scholar] [CrossRef] [PubMed]
- Killick, R.; Scales, G.; Leroy, K.; Causevic, M.; Hooper, C.; Irvine, E.E.; Choudhury, A.I.; Drinkwater, L.; Kerr, F.; Al-Qassab, H.; et al. Deletion of Irs2 reduces amyloid deposition and rescues behavioural deficits in APP transgenic mice. Biochem Biophys Res Commun 2009, 386, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Freude, S.; Hettich, M.M.; Schumann, C.; Stohr, O.; Koch, L.; Kohler, C.; Udelhoven, M.; Leeser, U.; Muller, M.; Kubota, N.; et al. Neuronal IGF-1 resistance reduces Abeta accumulation and protects against premature death in a model of Alzheimer's disease. FASEB J 2009, 23, 3315–3324. [Google Scholar] [CrossRef] [PubMed]
- Moloney, A.M.; Griffin, R.J.; Timmons, S.; O'Connor, R.; Ravid, R.; O'Neill, C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer's disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging 2010, 31, 224–243. [Google Scholar] [CrossRef] [PubMed]
- Chua, L.M.; Lim, M.L.; Chong, P.R.; Hu, Z.P.; Cheung, N.S.; Wong, B.S. Impaired neuronal insulin signaling precedes Abeta42 accumulation in female AbetaPPsw/PS1DeltaE9 mice. J Alzheimers Dis 2012, 29, 783–791. [Google Scholar] [CrossRef]
- Ochiai, T.; Sano, T.; Nagayama, T.; Kubota, N.; Kadowaki, T.; Wakabayashi, T.; Iwatsubo, T. Differential involvement of insulin receptor substrate (IRS)-1 and IRS-2 in brain insulin signaling is associated with the effects on amyloid pathology in a mouse model of Alzheimer's disease. Neurobiol Dis 2021, 159, 105510. [Google Scholar] [CrossRef]
- Alves-Borba, L.; Espinosa-Fernandez, V.; Canseco-Rodriguez, A.; Sanchez-Perez, A.M. ABA Supplementation Rescues IRS2 and BDNF mRNA Levels in a Triple-Transgenic Mice Model of Alzheimer's Disease. J Alzheimers Dis Rep 2023, 7, 1007–1013. [Google Scholar] [CrossRef]
- Wang, M.; Song, H.; Jia, J. Interleukin-6 receptor gene polymorphisms were associated with sporadic Alzheimer's disease in Chinese Han. Brain Res 2010, 1327, 1–5. [Google Scholar] [CrossRef]
- Sasayama, D.; Hori, H.; Teraishi, T.; Hattori, K.; Ota, M.; Matsuo, J.; Kawamoto, Y.; Kinoshita, Y.; Amano, N.; Kunugi, H. Association of cognitive performance with interleukin-6 receptor Asp358Ala polymorphism in healthy adults. J Neural Transm (Vienna) 2012, 119, 313–318. [Google Scholar] [CrossRef]
- Haddick, P.C.; Larson, J.L.; Rathore, N.; Bhangale, T.R.; Phung, Q.T.; Srinivasan, K.; Hansen, D.V.; Lill, J.R.; Alzheimer's Disease Genetic Consortium, A.s.D.N.I.; Pericak-Vance, M.A.; et al. A Common Variant of IL-6R is Associated with Elevated IL-6 Pathway Activity in Alzheimer's Disease Brains. J Alzheimers Dis 2017, 56, 1037–1054. [Google Scholar] [CrossRef]
- Quillen, D.; Hughes, T.M.; Craft, S.; Howard, T.; Register, T.; Suerken, C.; Hawkins, G.A.; Milligan, C. Levels of Soluble Interleukin 6 Receptor and Asp358Ala Are Associated With Cognitive Performance and Alzheimer Disease Biomarkers. Neurol Neuroimmunol Neuroinflamm 2023, 10. [Google Scholar] [CrossRef]
- Elcioglu, H.K.; Aslan, E.; Ahmad, S.; Alan, S.; Salva, E.; Elcioglu, O.H.; Kabasakal, L. Tocilizumab's effect on cognitive deficits induced by intracerebroventricular administration of streptozotocin in Alzheimer's model. Mol Cell Biochem 2016, 420, 21–28. [Google Scholar] [CrossRef]
- Qi, L.; Rifai, N.; Hu, F.B. Interleukin-6 receptor gene variations, plasma interleukin-6 levels, and type 2 diabetes in U.S. Women. Diabetes 2007, 56, 3075–3081. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, Z.; Chu, W.; Hale, T.; Cooper, J.J.; Elbein, S.C. Molecular screening and association analyses of the interleukin 6 receptor gene variants with type 2 diabetes, diabetic nephropathy, and insulin sensitivity. J Clin Endocrinol Metab 2005, 90, 1123–1129. [Google Scholar] [CrossRef]
- Hamid, Y.H.; Urhammer, S.A.; Jensen, D.P.; Glumer, C.; Borch-Johnsen, K.; Jorgensen, T.; Hansen, T.; Pedersen, O. Variation in the interleukin-6 receptor gene associates with type 2 diabetes in Danish whites. Diabetes 2004, 53, 3342–3345. [Google Scholar] [CrossRef]
- Wu, X.; Yu, T.; Ji, N.; Huang, Y.; Gao, L.; Shi, W.; Yan, Y.; Li, H.; Ma, L.; Wu, K.; et al. IL6R inhibits viability and apoptosis of pancreatic beta-cells in type 2 diabetes mellitus via regulation by miR-22 of the JAK/STAT signaling pathway. Diabetes Metab Syndr Obes 2019, 12, 1645–1657. [Google Scholar] [CrossRef]
- Burnstock, G. Purine and purinergic receptors. Brain Neurosci Adv 2018, 2, 2398212818817494. [Google Scholar] [CrossRef] [PubMed]
- Hua, J.; Garcia de Paco, E.; Linck, N.; Maurice, T.; Desrumaux, C.; Manoury, B.; Rassendren, F.; Ulmann, L. Microglial P2X4 receptors promote ApoE degradation and contribute to memory deficits in Alzheimer's disease. Cell Mol Life Sci 2023, 80, 138. [Google Scholar] [CrossRef] [PubMed]
- Stefanova, N.A.; Maksimova, K.Y.; Rudnitskaya, E.A.; Muraleva, N.A.; Kolosova, N.G. Association of cerebrovascular dysfunction with the development of Alzheimer's disease-like pathology in OXYS rats. BMC Genomics 2018, 19, 75. [Google Scholar] [CrossRef] [PubMed]
- Varma, R.; Chai, Y.; Troncoso, J.; Gu, J.; Xing, H.; Stojilkovic, S.S.; Mattson, M.P.; Haughey, N.J. Amyloid-beta induces a caspase-mediated cleavage of P2X4 to promote purinotoxicity. Neuromolecular Med 2009, 11, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Glas, R.; Sauter, N.S.; Schulthess, F.T.; Shu, L.; Oberholzer, J.; Maedler, K. Purinergic P2X7 receptors regulate secretion of interleukin-1 receptor antagonist and beta cell function and survival. Diabetologia 2009, 52, 1579–1588. [Google Scholar] [CrossRef]
- Solini, A.; Chiozzi, P.; Morelli, A.; Adinolfi, E.; Rizzo, R.; Baricordi, O.R.; Di Virgilio, F. Enhanced P2X7 activity in human fibroblasts from diabetic patients: a possible pathogenetic mechanism for vascular damage in diabetes. Arterioscler Thromb Vasc Biol 2004, 24, 1240–1245. [Google Scholar] [CrossRef] [PubMed]
- Todd, J.N.; Poon, W.; Lyssenko, V.; Groop, L.; Nichols, B.; Wilmot, M.; Robson, S.; Enjyoji, K.; Herman, M.A.; Hu, C.; et al. Variation in glucose homeostasis traits associated with P2RX7 polymorphisms in mice and humans. J Clin Endocrinol Metab 2015, 100, E688–696. [Google Scholar] [CrossRef]
- Uresti-Rivera, E.E.; Garcia-Jacobo, R.E.; Mendez-Cabanas, J.A.; Gaytan-Medina, L.E.; Cortez-Espinosa, N.; Portales-Perez, D.P.; Gonzalez-Amaro, R.; Enciso-Moreno, J.A.; Garcia-Hernandez, M.H. The presence of the 1068 G>A variant of P2X7 receptors is associated to an increase in IL-1Ra levels, insulin secretion and pancreatic beta-cell function but not with glycemic control in type 2 diabetes patients. Gene 2018, 652, 1–6. [Google Scholar] [CrossRef]
- Parvathenani, L.K.; Tertyshnikova, S.; Greco, C.R.; Roberts, S.B.; Robertson, B.; Posmantur, R. P2X7 mediates superoxide production in primary microglia and is up-regulated in a transgenic mouse model of Alzheimer's disease. J Biol Chem 2003, 278, 13309–13317. [Google Scholar] [CrossRef]
- Rampe, D.; Wang, L.; Ringheim, G.E. P2X7 receptor modulation of beta-amyloid- and LPS-induced cytokine secretion from human macrophages and microglia. J Neuroimmunol 2004, 147, 56–61. [Google Scholar] [CrossRef]
- Sanz, J.M.; Chiozzi, P.; Ferrari, D.; Colaianna, M.; Idzko, M.; Falzoni, S.; Fellin, R.; Trabace, L.; Di Virgilio, F. Activation of microglia by amyloid beta requires P2X7 receptor expression. J Immunol 2009, 182, 4378–4385. [Google Scholar] [CrossRef]
- Beltran-Lobo, P.; Hughes, M.M.; Troakes, C.; Croft, C.L.; Rupawala, H.; Jutzi, D.; Ruepp, M.D.; Jimenez-Sanchez, M.; Perkinton, M.S.; Kassiou, M.; et al. P2X(7)R influences tau aggregate burden in human tauopathies and shows distinct signalling in microglia and astrocytes. Brain Behav Immun 2023, 114, 414–429. [Google Scholar] [CrossRef]
- Sun, L.; Gao, J.; Zhao, M.; Cui, J.; Li, Y.; Yang, X.; Jing, X.; Wu, Z. A novel cognitive impairment mechanism that astrocytic p-connexin 43 promotes neuronic autophagy via activation of P2X7R and down-regulation of GLT-1 expression in the hippocampus following traumatic brain injury in rats. Behav Brain Res 2015, 291, 315–324. [Google Scholar] [CrossRef]
- Ruan, Z.; Delpech, J.C.; Venkatesan Kalavai, S.; Van Enoo, A.A.; Hu, J.; Ikezu, S.; Ikezu, T. P2RX7 inhibitor suppresses exosome secretion and disease phenotype in P301S tau transgenic mice. Mol Neurodegener 2020, 15, 47. [Google Scholar] [CrossRef]
- Groblewska, M.; Muszynski, P.; Wojtulewska-Supron, A.; Kulczynska-Przybik, A.; Mroczko, B. The Role of Visinin-Like Protein-1 in the Pathophysiology of Alzheimer's Disease. J Alzheimers Dis 2015, 47, 17–32. [Google Scholar] [CrossRef]
- Schnurra, I.; Bernstein, H.G.; Riederer, P.; Braunewell, K.H. The neuronal calcium sensor protein VILIP-1 is associated with amyloid plaques and extracellular tangles in Alzheimer's disease and promotes cell death and tau phosphorylation in vitro: a link between calcium sensors and Alzheimer's disease? Neurobiol Dis 2001, 8, 900–909. [Google Scholar] [CrossRef]
- Hollingworth, P.; Sweet, R.; Sims, R.; Harold, D.; Russo, G.; Abraham, R.; Stretton, A.; Jones, N.; Gerrish, A.; Chapman, J.; et al. Genome-wide association study of Alzheimer's disease with psychotic symptoms. Mol Psychiatry 2012, 17, 1316–1327. [Google Scholar] [CrossRef]
- Hu, X.; Yang, Y.; Gong, D. A meta-analysis of cerebrospinal fluid visinin-like protein-1 in alzheimers disease patients relative to healthy controls and mild cognitive impairment patients. Neurosciences (Riyadh) 2017, 22, 94–101. [Google Scholar] [CrossRef]
- Babic Leko, M.; Borovecki, F.; Dejanovic, N.; Hof, P.R.; Simic, G. Predictive Value of Cerebrospinal Fluid Visinin-Like Protein-1 Levels for Alzheimer's Disease Early Detection and Differential Diagnosis in Patients with Mild Cognitive Impairment. J Alzheimers Dis 2016, 50, 765–778. [Google Scholar] [CrossRef]
- Mroczko, B.; Groblewska, M.; Zboch, M.; Muszynski, P.; Zajkowska, A.; Borawska, R.; Szmitkowski, M.; Kornhuber, J.; Lewczuk, P. Evaluation of visinin-like protein 1 concentrations in the cerebrospinal fluid of patients with mild cognitive impairment as a dynamic biomarker of Alzheimer's disease. J Alzheimers Dis 2015, 43, 1031–1037. [Google Scholar] [CrossRef]
- Tarawneh, R.; Lee, J.M.; Ladenson, J.H.; Morris, J.C.; Holtzman, D.M. CSF VILIP-1 predicts rates of cognitive decline in early Alzheimer disease. Neurology 2012, 78, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Dai, F.F.; Zhang, Y.; Kang, Y.; Wang, Q.; Gaisano, H.Y.; Braunewell, K.H.; Chan, C.B.; Wheeler, M.B. The neuronal Ca2+ sensor protein visinin-like protein-1 is expressed in pancreatic islets and regulates insulin secretion. J Biol Chem 2006, 281, 21942–21953. [Google Scholar] [CrossRef] [PubMed]
- Atla, G.; Bonas-Guarch, S.; Cuenca-Ardura, M.; Beucher, A.; Crouch, D.J.M.; Garcia-Hurtado, J.; Moran, I.; Consortium, T.D.; Irimia, M.; Prasad, R.B.; et al. Genetic regulation of RNA splicing in human pancreatic islets. Genome Biol 2022, 23, 196. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The result of network clustering with common genes of Alzheimer’s disease (AD), type 2 diabetes, and memory-associated genes: (a) total result, (b) In Cluster 1, PIK3C genes constitute hub proteins of the cluster; (c) TP53 is the hub protein of Cluster 2, (d) APP, well-known for its roles in AD, is the hub gene of Cluster 3. Nodes with the same colors indicate the same clusters.
Figure 1.
The result of network clustering with common genes of Alzheimer’s disease (AD), type 2 diabetes, and memory-associated genes: (a) total result, (b) In Cluster 1, PIK3C genes constitute hub proteins of the cluster; (c) TP53 is the hub protein of Cluster 2, (d) APP, well-known for its roles in AD, is the hub gene of Cluster 3. Nodes with the same colors indicate the same clusters.
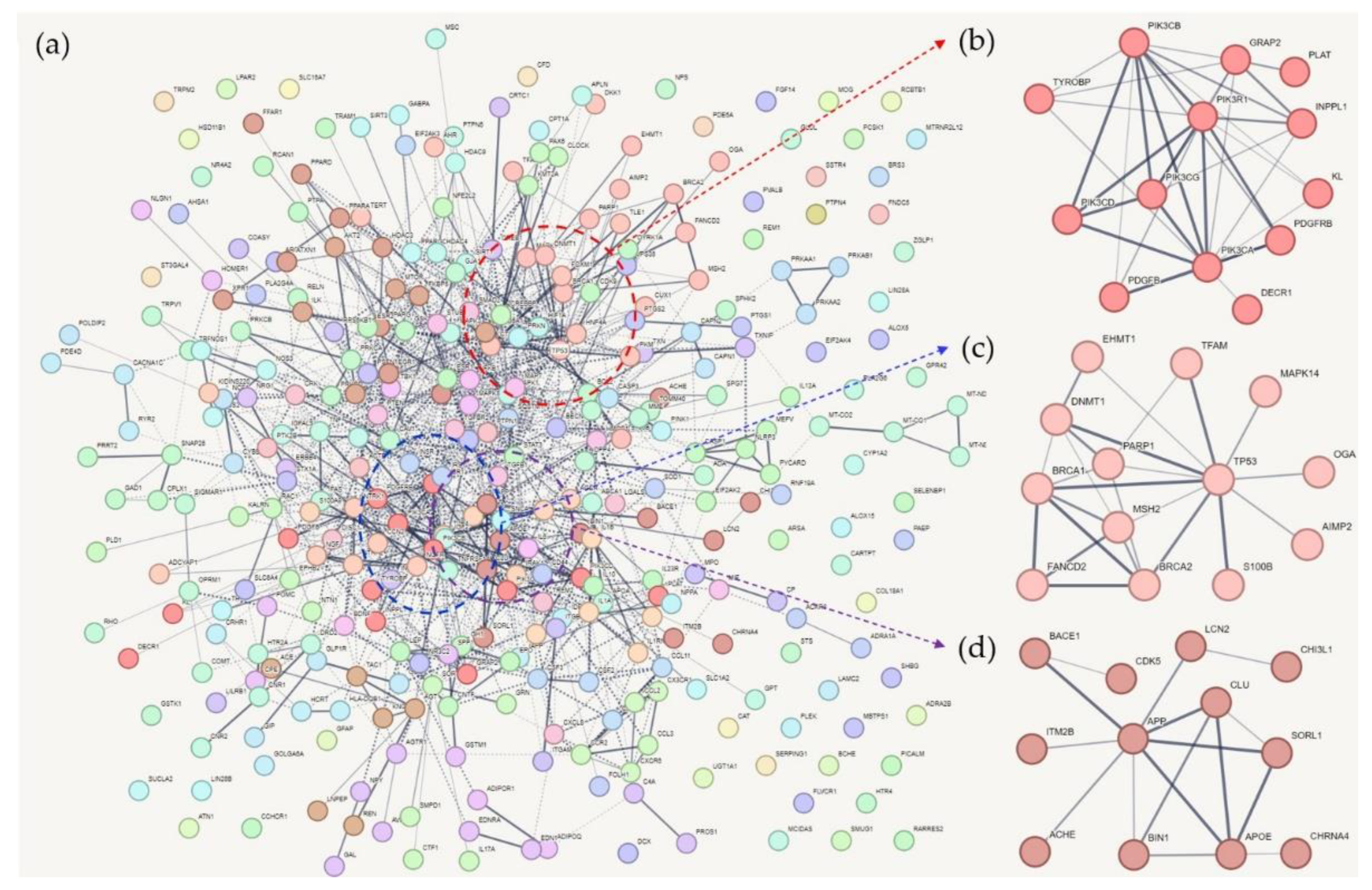
Figure 2.
Network-based clustering of common genes of Alzheimer’s disease, type 2 diabetes, and cognition-associated genes: (a) Several clusters were distinctly detected. In Cluster 1 (a), Cluster 2 (b), and Cluster 3 (c), EP300, JAK2, and P2RX4 were hub proteins, respectively.
Figure 2.
Network-based clustering of common genes of Alzheimer’s disease, type 2 diabetes, and cognition-associated genes: (a) Several clusters were distinctly detected. In Cluster 1 (a), Cluster 2 (b), and Cluster 3 (c), EP300, JAK2, and P2RX4 were hub proteins, respectively.
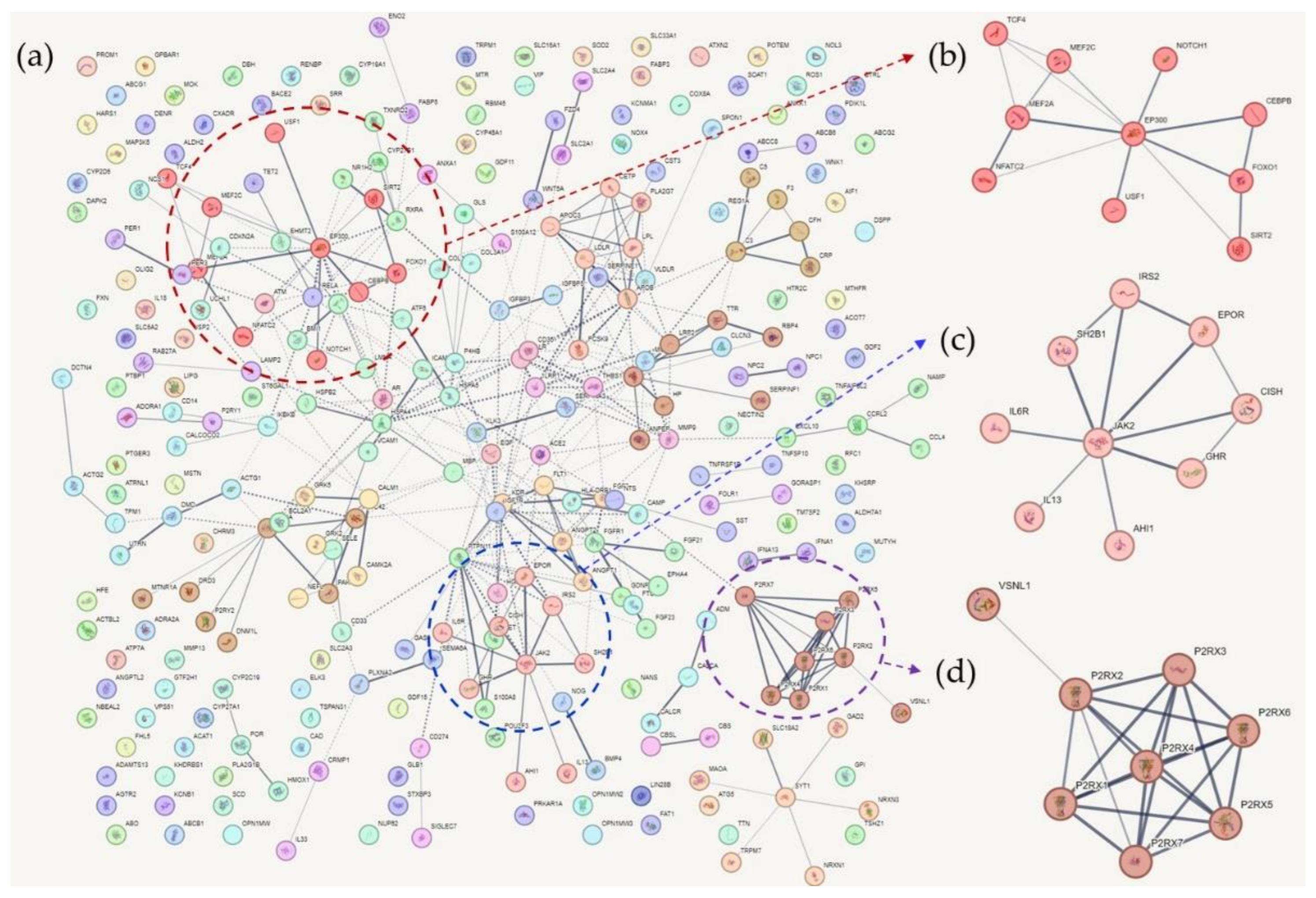

Table 1.
Over-representation analysis result with gene ontology.
| GOBP1 | Odds ratio | P value |
|---|---|---|
| RESPONSE TO OXYGEN CONTAINING COMPOUND | 16.06 | 1.97E-301 |
| POSITIVE REGULATION OF MULTICELLULAR ORGANISMAL PROCESS | 13.616 | 1.24E-240 |
| RESPONSE TO ENDOGENOUS STIMULUS | 12.81 | 2.40E-234 |
| CELLULAR RESPONSE TO OXYGEN CONTAINING COMPOUND | 15.04 | 3.76E-224 |
| POSITIVE REGULATION OF SIGNALING | 11.70 | 1.76E-222 |
| REGULATION OF TRANSPORT | 11.53 | 3.81E-222 |
| REGULATION OF CELL DEATH | 11.97 | 5.53E-222 |
| APOPTOTIC PROCESS | 10.91 | 1.28E-219 |
| HOMEOSTATIC PROCESS | 11.63 | 1.17E-216 |
| REGULATION OF CELL POPULATION PROLIFERATION | 11.11 | 6.84E-212 |
1 GOBP; Gene Ontology Biological Process.
Table 2.
This is a table. Tables should be placed in the main text near to the first time they are cited.
Table 2.
This is a table. Tables should be placed in the main text near to the first time they are cited.
| KEGG1 Pathway | Odds ratio | P value |
|---|---|---|
| PATHWAYS IN CANCER | 21.62 | 9.22E-41 |
| NEUROTROPHIN SIGNALING PATHWAY | 36.62 | 9.14E-32 |
| LEISHMANIA INFECTION | 59.33 | 2.17E-30 |
| TOLL LIKE RECEPTOR SIGNALING PATHWAY | 41.26 | 1.18E-29 |
| CYTOKINE CYTOKINE RECEPTOR INTERACTION | 18.45 | 1.46E-28 |
| FOCAL ADHESION | 22.04 | 2.64E-27 |
| MAPK SIGNALING PATHWAY | 17.61 | 3.42E-27 |
| COLORECTAL CANCER | 59.67 | 1.10E-26 |
| APOPTOSIS | 42.63 | 1.43E-26 |
| PROSTATE CANCER | 41.36 | 2.49E-26 |
1 KEGG; Kyoto Encyclopedia of Genes and Genomes.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



