
Submitted:
22 December 2023
Posted:
25 December 2023
You are already at the latest version
Abstract
Pancreatic neuroendocrine tumors (PNETs) are characterized by dysregulated signaling pathways that are crucial for tumor formation and progression. The efficacy of traditional therapies is limited, particularly in treatment of PNETs at advanced stage. Epigenetic alterations profoundly impact the activity of signaling pathways in cancer development, offering potential opportunities for drug development. There is currently a lack of extensive research on epigenetic regulation in PNETs. To fill this gap, we first summarize major signaling events which are involved in PNET development. Then, we discuss the epigenetic regulation of these signaling pathways in the context of both PNET and commonly occurring, and therefore more extensively studied, malignancies. Finally, we will offer perspective on the future research direction of PNET epigenome and its potential applications in patient care.
Keywords:
Pancreatic neuroendocrine tumors
; signaling pathways
; epigenetic regulation
Contents
- Introduction
- Major signaling pathways in PNETs
-
Epigenetic regulation of PNET-related signaling pathways
- 1.1
- DNA methylation
- 1.2
- Histone modifications
- 1.3
- Non-coding RNAs
- Future directions for epigenetic research and clinical applications in PNET patient care
- Conclusion
1. Introduction
Pancreatic neuroendocrine tumors (PNETs) are a rare and heterogeneous group of neoplasms arising from pancreatic islet cells. They account for approximately 2% of all pancreatic malignancies and are characterized by their slow-growing nature and potential for metastasis [1]. Current treatment options for PNETs are limited, and the prognosis for patients with advanced disease remains poor, underscoring the urgent need for innovative therapeutic strategies [2]. The pathogenesis of PNETs is intricate, characterized by complex interactions among numerous signaling pathways. Each pathway plays a role in different facets of tumor development, encompassing cell proliferation, survival, migration, and angiogenesis. Together, these pathways constitute a complex network that, when disrupted, can result in uncontrolled tumor growth and metastasis [3]. Germline and somatic whole-genome sequencing provided a comprehensive analysis of PNET-related genetic variations [4]. Most PNETs occur sporadically, with only around 10% of the cases being associated with germline mutations. Germline mutations such as multiple endocrine neoplasia type 1 (MEN1), von Hippel-Lindau syndrome (VHL), neurofibromatosis type 1 (NF1), and occasionally tuberous sclerosis complex (TSC) are the most identified PNET-associated mutations [5,6]. For sporadically occurring PNETs, MEN1, DAXX (death domain associated protein), ATRX (α-thalassemia/mental retardation syndrome X-linked), and genes related to the mammalian target of rapamycin (mTOR) pathway harbor commonly identified somatic alterations [6].
While somatic and germline mutations remain of great significance for diagnosis and therapeutic treatment, emerging evidence highlights the pivotal role of epigenetic modifications in shaping the intricate landscape of PNET-related signaling [7]. Epigenetic modifications encompass a range of reversible alterations that modulate gene expression patterns without affecting the underlying DNA sequence, such as DNA methylation, histone modifications, and non-coding RNAs (ncRNAs) which coordinate together to govern cellular processes crucial for normal development and homeostasis [8]. In the context of PNETs, epigenetic dysregulation is starting to gain recognition as a significant player in the initiation and evolution of the disease [1,9,10]. Epigenetic alterations can lead to aberrant gene expression patterns, contributing to uncontrolled cell growth, evasion of cell death, and increased metastatic potential – hallmark characteristics of cancer [11]. This is particularly relevant in the case of PNET because PNETs have a low tumor mutational burden and are considered an epigenetic disorder, due to the fact that multiple high-frequency mutations, including MEN1, DAXX, and ATX, are all found involved in epigenetic regulation [7].
Several recent reviews have addressed the complicated signaling network in PNET development [1,3,12,13,14]. However, to the best of our knowledge, there is a deficiency in comprehensive studies that systematically review the epigenetic status of PNET-related signaling pathways, considering both PNET and other frequently encountered cancers simultaneously. Given that epigenetic mechanisms are common across various cancers [15,16], conducting a thorough examination of the epigenetic events within signaling pathways in the broader context of cancer research is certain to offer valuable insights. This approach will undoubtedly guide the identification of future directions for the relatively understudied field of epigenetic research in PNETs.
Because of our strong interest in enhancing the care of patients diagnosed with PNETs, we will concentrate on the epigenetic mechanisms that hold the highest clinical relevance. Presently, there are a total of eight FDA-approved anti-tumor epigenetic drugs primarily targeting DNA methylation and histone modifications [17]. Beyond their therapeutic applications, epigenetic markers, particularly in the realm of non-coding RNAs such as microRNAs (miRNAs), play a crucial role in cancer diagnosis [18]. This review will specifically delve into the three aforementioned epigenetic regulatory mechanisms: DNA methylation, histone modifications, and non-coding RNAs. This is by no means all-compassing, as we recognized that we will not discuss other epigenetic modifications including RNA methylation, histone ubiquitylation, phosphorylation, SUMOylation, ADP ribosylation, citrullination, and biotinylation at specific amino acidic residue [19].
2. Major Signaling Pathways and Molecules in PNETs
Various molecular alterations have been identified as correlated with the development of PNETs. These events manifest through diverse mechanisms, encompassing both genetic and epigenetic modifications, resulting in a complex regulatory network. Genetic variations of PNET include both familial and predominantly sporadic mutations [20]. Each of the hereditary syndromes, namely MEN1, VHL, NF1, and TSC, is characterized by distinct sets of signaling pathways and molecules [20]. Moreover, numerous signaling pathways exhibit sporadic mutations in PNET samples [20]. It is worth noting familial and sporadic mutations are not exclusive of each other in PNET. One such example is MEN1 signaling. MEN1 mutations play a pivotal role in the initiation and progression of pNETs, as over 40% of sporadic pNETs and all MEN1 patients exhibit somatic mutations in the MEN1 gene [21,22]. More interestingly, the status of MEN1 mutations and menin protein expression don’t always correlate well with each other. One study, which included the mutational analysis and immunohistochemistry results (IHC) of 169 PNET patients, showed that 80% of sporadic cases showed a loss of menin nuclear localization, while only 25% of the patients carried a mutation in the MEN1 gene itself [23]. This study clearly suggests that other regulatory mechanisms, besides genetic mutations, are involved in the altered level of signaling pathway in PNET pathology. The complicated regulatory mechanisms in PNET pathology have been nicely reviewed previously [1,3,12,13,14]. Here we adopt a different approach by discussing the PNET-related signaling events based on the major epigenetic regulatory mechanisms that they fall into, including DNA methylation, histone modifications, and ncRNAs (Figure 1). Dynamic alterations in DNA methylation, histone modifications, and ncRNA activity are integral aspects of the pathogenesis of other extensively studied cancers. However, the contributions of these epigenetic modifications to PNET pathology have yet to be fully explored.
Abbreviations: Acyl-CoA synthetase isoform 1(ACSL1); Secreted frizzled related protein 1 (SFRD1); WNT Inhibitory Factor 1(WIF1); Phosphatase and tensin homolog (PTEN); Receptor tyrosine kinase (RTK); Insulin receptor substrate 1 (IRS1); DEP domain containing 5 (DEPDC5); Hypoxia inducible factor 1 α (HIF1 α); Cyclin dependent kinase inhibitor 2A (CDKN2A); Wild-type p53-induced phosphatase 1 (WIP1); Mouse double minute 2 (MDM2); Pleckstrin Homology Like Domain Family A Member 3 (PHLDA3); Rab-like protein 6 (RABL6); Cyclin-dependent kinase 4 and cyclin-dependent kinase 6 (CDK4/6).
3. Epigenetic Regulation of PNET-Related Signaling Pathways
3.1. DNA Methylation
Methylation status has long been recognized as one of the primary mechanisms by which epigenetic modification is carried out within cells to regulate gene expression. DNA methyl transferases (DNMTs) cause DNA methylation by adding a methyl group to the cytosine in CpG DNA sequences (CpG islands) of promoter or enhancer. In cancer cells, dysregulated CpG methylation leads to promoter hypermethylation and downregulated gene expression, resulting in gene silencing of tumor suppressor genes. In addition, DNA demethylating enzymes might cause genome-wide hypomethylation, causing DNA instability. A variety of tumor suppressors as well as proto-oncogenes have been shown to be modified via methylation in the tumorigenesis [25].
3.1.1. MEN1
MEN1 is one of the most extensively studied genes in PNET. Germline mutations in this gene result in Multiple Endocrine Neoplasia Syndrome Type 1, causing different types of cancer including PNET, pituitary adenoma, and parathyroid hyperplasia. The genetic mutation responsible for MEN1 spans 9.8 kb of chromosome 11q13. The protein product of MEN1, termed menin, is a tumor suppressor protein ubiquitously expressed in the cell nucleus [26]. Considering menin’s crucial role as an epigenetic regulator, it is unsurprising that the analysis of DNA methylation in MEN1-associated PNETs revealed the promoter hypermethylation of numerous potential tumor suppressor genes [27]. Epigenetic modifications of MEN1 are also identified in non-endocrine tumors. Notably, in advanced breast cancer, the overexpression of the MEN1 gene is associated with promoter hypomethylation, which differs from the scenario observed in PNETs. [28].
3.1.2. mTOR-TSC
The mechanistic target of rapamycin (mTOR) is a kinase that in humans is encoded by the mTOR gene. It functions as a serine/threonine protein kinase that regulates a variety of cellular functions including cell growth and proliferation, motility, survival, protein synthesis, autophagy, gene transcription, and thus carcinogenesis. [29]. Activation of mTOR signaling can affect serine one-carbon metabolism enough to modulate DNA methylation and promote tumorigenesis [30]. Aberrant hypermethylation of the promoter of tumor suppressor genes followed by their silencing is important for aberrant mTOR-PI3K pathway activation in gastric cancer [31]. Tumor suppressor TRPM4, a calcium-activated nonselective cation channel, inhibits PI3K/Akt/mTOR signaling pathway to impede tumor migration and invasion. This gene has been shown to be methylated at its promoter with silenced or reduced expression in colorectal cancers, though this effect can be reversed with the calpain inhibitor calpeptin, providing possible therapeutic strategies for aggressive disease [32]. ZDHHC22, as a well-known member of the palmitoyltransferase family, regulates mTOR stability and activation of the AKT signaling pathway. Hypermethylation of the promoter CpG island of the ZDHHC22 gene is associated with poorer prognosis in breast cancer. Overexpression of ZDHHC22 inhibits breast cancer cell growth both in vitro and in vivo [33]. The major signaling molecules of mTOR pathway, including PTEN, PI3K, AKT, TSC and c-Myc, have been well documented in PNET pathology previously [34,35,36,37,38,39,40]. Promoter of PTEN gene has been shown to be hypermethylated while the methylation status of TSC promoter remains unchanged in PNET samples [41].
3.1.3. Hypoxia-Induced Factor 1α (HIF1α)-VHL
Hypoxia inducible factor1⍺ (HIF1⍺) is pro-tumorigenic factor that has found to lead to poor prognosis, metastasis, and recurrence in multiple cancer subtypes including breast, renal cell, gastric, and ovarian cancer. Its involvement in B-cell lymphoma, Chronic lymphocytic leukemia, colon cancer, small cell lung cancer and PNET have also been established [42]. HIF1⍺ is regulated by hypoxia. Under normoxic conditions, HIF1α interacts with von Hippel Lindau (VHL) protein (pVHL) resulting in its polyubiquitination and proteasomal degradation [43]. During cellular hypoxia, HIF1⍺ translocates to the nucleus where it becomes stabilized activating genes involved in cellular proliferation, angiogenesis, glucose homeostasis and metastasis [44]. In patients with von Hippel Lindau syndrome, the tumor suppressor gene is mutated and pVHL is absent limiting HIF1⍺ degradation. Patients with VHL are at increased risk for developing hemangioblastomas, retinal angiomas, renal cell carcinomas, pheochromocytoma, and pancreatic lesions including PNETs [45]. Methylation status of the HIF binding region within the hypoxia response element correlates with increased expression of epidermal growth factor receptor (EGFR) under hypoxic conditions in breast cancer. These changes can be reversed by treatment with DNA methyltransferase inhibitors such as azacytidine or decitabine, suggesting that patients with hypoxic breast tumors and hypomethylated EGFR status may benefit from EGFR inhibitors [46]. HIF1α driven transcriptional response in hypoxia in pediatric neuroblastoma is subject to epigenetic control via DNA methylation status of gene regulatory regions. Hypoxia exposure induces a global DNA hypermethylation in neuroblastoma cells and HIF1A itself might control DNA methylation [47]. In VHL disease, loss of VHL leads to activation of its binding partner HIF1α, causing multi-organ tumorigenesis including PNET [48,49]. The VHL gene promoter has been shown to be hypermethylated in a subset of familial PNET samples [50].
3.1.4. RAS-MAPK-NF1
The RAS-MAPK pathway mediates cellular responses to growth signals and is often dysregulated in cancer. The mitogen-activated protein kinase (MAPK) pathway involves extracellular signal regulated kinases 1&2 (ERK1/2) that mediates multiple cellular functions including proliferation, growth, and senescence [51]. The rat sarcoma (RAS) gene is an oncogene part of the large family of GTPases and acts as a control switch for the EKR1/2 pathway. In order to transmit signals to downstream effector proteins, RAS must properly bind to cellular membranes which requires posttranslational modifications such as DNA methylation and histone acetylation [52]. Global DNA hypermethylation correlated with the RAS and MAPK oncogenic pathways (among others) is significantly associated with high-grade tumors, platinum resistance, and poor prognosis in high-grade serous ovarian carcinoma (HGSOC). Treatment with demethylating agents shows significant growth retardation in ovarian cancer cells through differential inductions, such as cell apoptosis or G2/M cell cycle arrest [53]. There have been newly identified biomarkers for colorectal cancer metastasis that are related to MAPK, including Eye Absent Homolog 4 (EYA4) [54]. 5-methylcytosine (m5C) regulator status and expression is a robust predictor of prognosis and therapy response in pancreatic ductal adenocarcinoma (PDAC) [55]. Differentially expressed genes, both hypermethylated–low expression and hypomethylation–high expression, in the Ras/MAPK pathways are seen in neurofibromatosis type 2 vestibular schwannomas [56]. Genomes of acute myeloid leukemia (AML) cells resistant to the BCL-2–selective inhibitor venetoclax (VEN) show extensive differential methylation with activation of RAS/MAPK pathway, leading to increased stability and higher levels of MCL-1 protein. VEB sensitivity can then be at least partially restored by silencing or pharmacologic inhibition of MCL-1 [57]. RASSF1A is a negative regulator of RAS-MAPK signaling pathway. In addition, RASSF1A can mediate DNA repair through nucleotide excision repair [58]. RASSF1A might be the most frequently inactivated tumor suppressor identified in human cancers and has been found to be inactivated in more than 40 types of human malignancies [59]. The expression of RASSF1A is lower in bladder and prostate cancer patients due to promoter hypermethylation. If this is reversed and RASSF1 is overexpressed, enhanced cytotoxicity to chemotherapeutic drugs is observed [60]. Urinary methylation assays can even be used to help predict prostate tumor stage and grade, and related tests utilizing RASSF1A methylation in various contexts have been investigated in endometrial, breast, and colorectal cancers [61,62,63,64]. Methylation of the RASSF1 gene promoter is also significantly higher in lung tumor tissues and is associated with lymph node metastasis and clinical stage. Interestingly, it appears that associations exist between methylation levels and cigarette smoking-related parameters [65,66]. RASSF1 promoter methylation, under the impact of the methyltransferase genes DNMT1 and MGMT, can also be a papillary thyroid cancer genetic marker [67]. In PNET, the promoter of RASSF1 gene was found to be hypermethylated in 75% of NF-PNET where its inactivation results in the degradation of beta-catenin [68]. Inhibition of RAS-MAPK attenuate PNET cell growth [69]. Methylation has been associated with RASSF1A downregulation, and, interestingly, up-regulation of RASSF1C in PNET samples [70]. Neurofibromatosis type 1 (NF1) encodes a RAS GTPase activating protein called Neurofibromin, which has been found to be mutated in both sporadic cancers and in familial syndrome. Mutations of NF1 result in excessive RAS signaling. NF1-associated sporadic cancers include glioblastoma, neuroblastoma, AML, lung cancer, ovarian cancer, and breast cancer [71]. In the NF1 familial syndrome, patients with neurofibromatosis will present with benign cutaneous neurofibromas, and are predisposed to breast, gastric, and pancreatic neuroendocrine tumors [10,72]. Despite the significant importance of NF1 in PNET regulation, there is currently no reported information concerning the DNA methylation of NF1 in the progression of PNETs.
3.1.5. ATRX/DAXX
ATRX and DAXX form a histone chaperone complex that deposits histone variant H3.3 into specific genomic regions [73]. Mutations in ATRX constitute the most prevalent genetic abnormalities in gliomas and, interestingly, ATRX alterations are associated with favorable outcomes. CRISPR/Cas9 knockout ATRX glioblastoma cells demonstrated compromised H3K9 trimethylation, which led to decreased DNA repair by the tumor cells. In addition, increased response to DNA damage-inducing agent temozolomide was confirmed in these cells, suggesting that these mutations might serve as a prognostic maker in predicting chemosensitivity [74]. In high-grade meningiomas, there is a significant increase of ATRX expression at both gene and protein levels, particularly in the nuclear compartment, and an increased DAXX protein expression level. In these tumors, ATRX/DAXX is suggested to have a role in telomere maintenance due to low variability of telomere length [75]. In histopathologic evaluation of human pituitary adenomas, ATRX or DAXX protein loss was observed, suggesting either homozygous loss of the gene or the presence of another silencing mechanism such as promoter methylation [76]. Mutations in ATRX and DAXX have been identified in PNET tumor samples [34,35,77]. The DAXX gene has been shown to be hypermethylated at promoter region in a fraction of PNET tumor samples [78].
3.1.6. CDKN2A-RB1
Abnormal DNA methylation is found in the early stages of kidney cancers. Genistein, a potent antioxidant and demethylating agent in soybean-enriched foods, induces cell apoptosis and inhibits the cell proliferation of multiple types of kidney cancer cells. It achieves this through the expression of CDKN2a via decreased CDKN2a methylation [79]. CDKN2A promoter methylation correlates with a poor prognosis and progression-free survival in both ovarian and colorectal cancers [80,81]. Peroxisome proliferator-activated receptor alpha (PPARα), the molecular target of fibrates commonly used to treat dyslipidemia and diabetes, inhibits DNMT1 activity and abolishes methylation-mediated CDKN2A repression in colorectal cancer cells. It also appears to inhibit tumor growth and DNMT1 activity in vivo, indicating that it may be an applicable agent for epigenetic therapy of colon cancer patients [82]. In addition, methylation of the CDKN2A gene is recorded as an early epigenetic event in the progression of cervical tumorigenesis. There is a strong and significant correlation between CDKN2A gene methylation and risk of cervical cancer as well as pre-cancerous lesions [83]. Furthermore, the level of CDKN2A gene methylation in human gastric cancer (GC) samples is increased compared to noncancerous tissues. Variations in methylation patterns and different loci are associated with changes in overall survival [84]. Inducing methylation in GC cells increased their sensitivity to palbociclib, an anti-CDK4/6 chemical for cancer treatment, both in vitro and in vivo [85]. As part of the CDKN2A-CDK4/6-RB1 axis, retinoblastoma tumor suppressor protein1 (RB1) plays critical roles in tumor suppression. RB1 inhibits cellular proliferation through repressing the transcription of genes essential for cell cycle progression, serving as a negative regulator of the cell cycle [86]. Aberrant DNA methylation at the RB1 gene promoter has been reported in multiple types of tumor [87]. Members of the CDKN2A-CDK4/6-RB1 pathway, including CDKN2A, P14ARF, and RB1 have shown promoter hypermethylation in PNET tumor samples [41,88,89,90], suggesting that DNA methylation-mediated downregulation of CDKN2A-RB1 signaling might contribute to PNET development.
3.1.7. p53
p53, also known as tumor protein TP53, is a regulatory protein that is often mutated in human cancers. DNA hypermethylation of TP53 has been found associated with the pathogenesis of cervical cancer in Northeastern Indian patients [91]. Resveratrol, a chemical mostly found in red grapes and products made from these grapes, alters the methylation of histones in the TP53 promoter in human breast cancer cells and upregulates the expression of SET domain-containing lysine methyltransferase 7/9 (SET7/9) in colorectal cells to positively regulates p53 through its mono-methylation function [92,93]. Luteolin, a dietary flavone molecule, modulates various signaling pathways involved in carcinogenesis and has apoptotic effects mediated by DNA demethylation of the nuclear factor erythroid 2-related factor 2 (NRF2) promoter and the interaction of Nrf2 and p53 in human colon cancer cells [94]. DNMT3B has been shown to be elevated in nasopharyngeal carcinoma tissues and predicts a poor prognosis. In addition, ionizing radiation can induce DNMT3B, which might be one of the reasons for radio-resistance; silencing DNMT3B restores p53 via DNA demethylation, which leads to cell cycle arrest and apoptosis both in vitro and in vivo [95]. In gastric cancer, PBX/Knotted Homeobox 2 (PKNOX2) is a candidate tumor suppressor, which exerts its tumor suppressive effect by promoting the upregulation of p53. PKNOX2 mRNA expression is largely silenced in gastric cancer cell lines and primary gastric cancer via promoter methylation, which is associated with poor outcomes [96]. Genetic profiling of BON-1 and QGP-1 PNET cell lines showed homozygous TP53 mutations which suggests loss-of-function of p53 contributes to PNET pathology [97], which is consistent with the observation that advanced metastatic PNET patients often display mutations in TP53 and RB1 locus [77]. In addition, both P53 and its downstream effector PHLDA3 have been found to be hypermethylated at their gene promoter regions, respectively [41,98], suggesting that p53 signaling regulates PNET development at both genetic and epigenetic levels.
3.1.8. Notch Signaling
The Notch signaling pathway is a highly conserved cell signaling system present in most animals, whose proteins span the cell membrane [99]. Notch signaling is responsible for differentiation and tissue homeostasis, and its dysregulation has been attributed to development of multiple cancers including leukemias such as T-cell acute lymphoblastic leukemia (T-ALL) and chronic lymphocytic leukemia (CLL) [100]. In patients with breast cancer, elevated expression of histone methyltransferase NSD3 was associated with recurrence, distant metastasis, and poor survival. In mice, NSD3 promoted malignant transformation of mammary epithelial cells [101]. NSD3-induced methylation of H3K36 is crucial for Notch-dependent tumor initiation and metastasis, suggesting that Notch-related methylation factors may be an actionable therapeutic target in localized and metastatic breast cancer [102]. In colorectal cancer, the upregulation of oncogenic histone cluster 2 H2B family member F (HIST2H2BF) enhances malignancy aggressiveness in humans and increases liver metastasis in mice through the activation of Notch signaling. Reactivation of this cluster is found related to promoter CpG hypomethylation [103]. High expression of SETD1A, a histone methyltransferase that specifically methylates H3K4, acts as a key oncogene in ovarian cancer and is associated with a poor prognosis. Accordingly, overexpression of SETD1A augments cell proliferation, migration, and invasion in vitro via initiation of Notch signaling, and downregulation of SETD1A reduces tumorigenesis in vivo [104]. Among North Indian patients with cervical cancer, the methylation rate of Notch1 and Notch3 promoters is notably elevated compared to healthy tissues, accompanied by a downregulation in protein expression. In addition, Notch1 promoter methylation increases with age, severity of the disease, and HPV infection in patients with cervical cancer [105]. Low expression of the methyltransferase DNMT3A, a key regulator of DNA methylation, is associated with more aggressive types of chronic lymphocytic leukemia (CLL). Dnmt3a knockout mice consistently developed CLL and showed a general upregulation of Notch signaling genes with sensitivity to Notch inhibition using daptomycin [106]. In PNETs, the expression of Notch1 signaling was highly related to the cancer progression [107]. Activation of Notch 1 inhibits tumor growth, suggesting that Notch 1 serves as a tumor suppressor [108,109,110]. Accordingly, Notch1 activation has been shown to be induce stable disease in a fraction of PNET patients in a Phase II trial [111]. As of now, there is no documentation of DNA methylation occurring in the Notch1 signaling pathway in PNET tumor samples.
3.1.9. Wnt/β-catenin
Wnt/β-catenin signaling has been widely known to be closely related to tumor progression, metastasis, migration, and invasion [112]. The Wnt pathway includes both canonical (β-catenin dependent) and non-canonical signaling. The canonical Wnt signaling has been known to play a large role in the pathogenesis of colon cancer with greater than 90% of colorectal cancers carrying a mutation that activates Wnt signaling. When Wnt signaling is activated, Wnt molecules will bind to the FZD-LRP5/6 co-receptor complex. LRP6 requires acetylation by p300 to become activated for Wnt signaling [113]. In epithelial ovarian cancer (EOC) cells, alternations of DNA methylation regulate members of the Wnt/β-catenin pathway [114]. Interestingly, the combination of curcumin, commonly recognized as turmeric, with decitabine hinders the formation and migration of ovarian cancer cell colonies. This effect might be achieved, at least partially, by downregulation of DNMT3a protein which in turn contributes to inhibition of the epithelial-to-mesenchymal transition (EMT) process. [115]. Adherens Junctions Associated Protein 1 (AJAP) forms a complex with E-cadherin and β-catenin in the cytomembrane, thereby diminishing the nuclear translocation of β-catenin and impeding the Wnt/β-catenin signaling pathway. In salivary adenoid cystic carcinoma (SACC) tumors, AJAP has been found downregulated, primarily due to promoter hypermethylation which causes AJAP gene silencing. Silencing AJAP1 in SACC cells markedly amplifies both in vitro and in vivo processes of proliferation, invasion, and metastasis. Furthermore, it independently contributes as a risk factor to the prognosis of SACC (38). Zinc-finger protein 471 (ZNF471) exerts its tumor-suppressive functions through suppressing EMT, tumor cell stemness and inhibition Wnt/β-catenin signaling. In both breast cell lines and tissues, there is notable downregulation of the promoter CpG methylation of ZNF471, as observed in comparison to normal mammary epithelial cells and their corresponding surgical-margin tissues [116]. In colorectal cancer, SET and MYND domain-containing protein 2 (SMYD2) suppresses the expression of adenomatous polyposis coli 2 (APC2), an inhibitor of the Wnt/β-catenin pathway. The decreased APC2 expression in colorectal cancer cells is due to SMYD2-mediated DNA methylation, which requires synergism with DNMT1 [117]. Lysine-specific histone demethylase 1A (KDM1A) regulates the stemness of thyroid cancer and promotes thyroid cancer progression via the Wnt/β-catenin pathway. Mechanistically, KDM1A downregulates APC2 and DKK1, two antagonists of the canonical Wnt pathway, by demethylation. Additionally, GSK-LSD1, a highly selective inhibitor of KDM1A, significantly inhibits thyroid cancer progression and enhances the sensitivity of thyroid cancer to chemotherapy [118]. In the context of PNET development, precise regulation of the Wnt/β-catenin signaling pathway is essential for normal pancreas development [119]. However, the dysregulation of Wnt/β-catenin leads to elevated expression of β-catenin and the decreased expression of Wnt/β-catenin inhibitors, a phenomenon frequently observed in higher-grade pNET tumors [120]. In addition, aberrant methylation of SFP1, an inhibitor of Wnt/β-catenin pathway, caused downregulation of SFP1 protein expression and enhanced tumor growth. Conversely, overexpression of SFP1 and WIF-1, another Wnt/β-catenin inhibitor, resulted in inhibition of tumor growth both in vitro and in vivo [121]. Furthermore, there is a strong correlation between promoter CpG hypermethylation and decreased expression of O6-methylguanine-methyltransferase (MGMT), a downstream target of the Wnt/β-catenin pathway, in PNETs [122,123].
3.1.10. NF-κB
NF-κB signaling plays critical roles in cancer development by mediating the inflammatory tumor microenvironment [124]. There have been numerous reports of epigenetic regulation of NF-κB signaling in cancers. In colorectal carcinoma, protein arginine methyltransferase 5 (PRMT5) catalyzes methylation of the multifunctional protein Y-box binding protein 1 (YBX1) and causes NF-κB activation which in turn leads to increased cell proliferation in vitro [125]. In addition, NF-κB signaling has been involved in the regulation of fatty acid-binding proteins (FABPs), dysregulation of which have been shown in many types of cancer. FABP5 promoter hypomethylation correlates with its increased expression, which forms a positive feed-back loop with NF-κB to promote tumor metastasis [126]. On the contrary, DRD2, involved in restricting NF-κB signaling and EMT in breast cancer (BC), undergoes downregulation due to promoter hypermethylation. DRD2 demonstrates the ability to suppress tumorigenesis in both in vitro and in vivo settings. Notably, there exists a positive correlation between DRD2 expression and extended survival times in BC patients [127]. In non-small cell lung cancer (NSCLC), genetic deletion and methylation contribute to decreased expression of circadian gene hepatic leukemia factor (HLF). HLF inhibits NF-κB/p65 signaling via increasing activity of PPAR in malignant tissues [128]. In PNET samples, upregulation of NF-kB is positively correlated with tumors with higher grade. In addition, down-regulation NF-kB and STAT3 inhibits proliferation, viability, and spheroids growth of PNET cell lines [129]. Presently, there is no documentation on the DNA methylation status of NF-κB in PNET.
3.1.11. Somatostatin Receptor 2 (SSTR2)
Somatostatins are a family of peptide hormones which are secreted by pancreatic islet δ cells and binds to somatostatin receptor (SSTR) signaling to inhibit the release of a variety of enzymes including insulin, glucagon, pancreatic amylase, along with other hormones secreted by pancreas. Upon binding of SST, SSTR exerts anti-proliferative functions, thereby serving as a tumor suppressor in PNET [69]. Since SSTR2 has been found highly-expressed in a majority of neuroendocrine neoplasms [130], the antitumor activity of SSTR2 has been explored in treatment of NE including PNET [131]. Methylation profiling of PNET samples showed hypermethylated promoter of SSTR2 gene [132] and, not surprisingly, DNMT inhibition recovered SSTRs expression in cell cultures and facilitated delivery of novel peptide receptor radiotherapy against PNET [133].
3.1.12. Smad3
SMAD transcriptional factors, which are part of transforming growth factor-β (TGF-β) signaling pathway, regulate cell differentiation and proliferation [134,135]. Depending on the cell status, activated SMAD can lead to either induction or inhibition of cell growth [135]. In PNETs, TGF-β/SMAD signaling mainly functions to inhibit cell growth via the SMAD downstream effectors, p21WAF1/CIP1 tumor suppressor [135,136]. Loss of homozygosity (LOH) of the SMAD3 gene has been observed in a fraction of PNET patients [137,275]. In addition, inhibition of TGFβ/ SMAD by inhibition of SMAD7 causes over-proliferation of islet β cell proliferation in adult mice, suggesting that TGF-β/Smad signaling might plays a role in PNET progression [138]. Smad3 has been shown to be undergone promoter CpG methylation in colorectal cancer [139,140]. Nevertheless, there is currently no information available regarding the DNA methylation of Smad3 in PNET. A complete list of DNA methylation profiling of PNET-related pathways was shown in Table 1.
3.2. Histone Modifications
Histone modifications include both acetylation and methylation of histones which control the conformation of genomic DNA. Acetylation is a post-translational histone modification process that modulates the opening of the chromatin structure allowing for gene transcription. Through the opposing activities of histone deacetylase (HDACs) and histone acetyl transferases (HATs), the process of acetylation is reversible through either the addition or removal of acetyl groups from the amino- terminal ɛ-group of lysines on histones. HDACs catalyze the removal of the acetyl moieties from acetylated histones and are generally associated with transcriptional repression [144]. Whereas histone acetylation is linked to transcriptional activation, histone methylation can either be repressive or activating. This depends on which lysine residue (K) on which histone (H3, H4) is modified, and the extent of methylation, i.e. di- or tri-methylation (me2, me3). For example, there are markers for inhibitory histone methylation, such as H3K9me2/3 and H3K27me2/3. In contrast, H3K4me2/3, H2K36me3, and H3K79me3 serve as markers for activating histone methylation. The mediation of the histone methylation process involves histone lysine methyltransferases (HMTs) and histone demethylases (HDMs) [145].
Histone modifications have been shown to regulate many important biological functions including cell cycle progression, metabolism, differentiation, and development. They are found involved in various tumors and are involved in vital chromosomal translocation-mediated oncogenic protein fusion and carcinogenic events [146,147]. Histone modifications play a crucial role in the different stages of cancer and have been linked to a variety of malignancies including solid and hematologic tumors. For example, increased levels of HDACs are correlated with advanced diseases, and poor outcomes. For example, increased levels of HDAC 5 have been correlated with metastatic PNETs. Similarly, elevated expression of HDACs 1, 2, and 3 is linked to poor outcomes in gastric and ovarian cancers [148,149]. Both histone acetylation and methylation have been found involved in regulation of multiple signaling pathways in cancer development [147].
3.2.1. MEN1
Despite the lack of evidence to support a direct role for histone acetylation or deacetylation in MEN1-associated endocrine cells, there is indirect evidence through examining the effects of HDAC inhibitors on MEN1-associated cell lines. Menin protein regulates the cyclin B2 promoter region by modifying histone H3 acetylation and H3K4me3 methylation levels in mouse embryonic fibroblast cells (MEF) [150]. HDAC inhibitors such as TSA, thailandepsin-A (TDP-A), sodium butyrate (NaB), valproic acid (VPA), and BET protein bromodomain inhibitors (BETi) have been well studied in neuroendocrine tumor cell lines including BON-1, NCI-H720, NCI-H727 and QGP-1 [151,152,153,154,155]. HDAC inhibitors have been found to induce dose dependent growth inhibition on NET cells. The anti-tumor effects of HDAC inhibitors have been further demonstrated in a BON-1 xenograft model [153] and a pancreatic beta cell-specific MEN1 knockout mouse model [155]. In addition, menin itself can serve as part of a mixed-lineage leukemia histone methyltransferase complex that functions to promotes histone methylation [156]. Polycomb repressive complex 2 (PRC2)-mediated H3K27 tri-methylation regulates the genome-wide distribution of MLL1 and MEN1 in diffuse large B-cell lymphoma (DLBCL) cells. In cells with EZH2 gain-of-function mutations, combinatorial inhibition of MEN1 using MI-503 or VTP50469 with EZH2 inhibitor Tazemetostat (EPZ-6438) has been shown to decrease tumor burden and survivability in a xenograft model [157]. Aberrations of EZH2 gene, along with MEN1 and others, can cause an imbalance in the activities of histones via methylation, contributing to parathyroid tumorigenesis in parathyroid carcinoma as well [158]. MEN1 even appears to play a role as a tumor suppressor gene in colorectal cancer, with recent work finding a hotspot mutation in MEN1 that affects 4% of BRAF mutant cancers [159].
3.2.2. mTOR-TSC
PI3-AKT-mTOR signaling is the key mechanism that normal cells use to metabolize glucose. In glioblastoma, this pathway has been found to be acetylated resulting in the amplification of c-Myc and promotion of glutaminolysis. mTORC2 regulates c-Myc levels through Akt-independent phosphorylation of class II HDACs (4,5,7,9), which leads to the acetylation of FoxO1 and FoxO3 and release of c-Myc. Upregulation of mTORC2, c-Myc, and acetylated FoxO have been found to be attributed to worse prognosis in glioblastoma patients. Pharmacologic inhibition of PI3K and mTOR kinase have been found to suppress FoxO acetylation, decrease c-Myc levels, and result in tumor cell death [160]. mTORC1 and mTORC2 cooperatively regulate H3K27 causing hypermethylation which subsequently promotes glioblastoma tumor cell survival both in vitro and in vivo, suggesting that dynamic regulation of histone methylation by mTOR complexes could be exploitable as a novel therapeutic target against this deadly tumor [161]. Phosphatase and tensin homologue (PTEN) is a tumor suppressor gene that dephosphorylates phosphatidylinositol-3,4,5 trisphosphate (PIP3) to negatively regulate the PI3K-AKT signaling pathway. The PI3/AKT signaling pathway is responsible for upregulating protein synthesis, cell migration, and tumor induced angiogenesis [162]. Mutations of PTEN are responsible for many cancers including brain, breast, prostate, endometrial carcinoma, head and neck cancers, melanoma, and PNET [163]. PTEN is acetylated at K125 and K128 by the p300/CBP associated factor (PCAF), resulting in its inactivation and upregulation of the PI3K-AKT signaling pathway [164]. TSA and SAHA have been found to up-regulate PTEN by inducing PTEN membrane translocation through acetylation at K163 in 293T cells. Further studies have determined that HDAC6 inhibition, using the HDAC6 specific inhibitor tubastatin A, caused PTEN acetylation and activation [165]. TSC2 forms a complex with TSC1, which negatively regulates mTOR. Dissolution of the TSC2/TSC1 complex allows for the activation of mTOR. [166]. Mutations in either TSC1 or TSC2 lead to the onset of tuberous sclerosis complex, characterized by the formation of hamartomas, also called benign tumors, in the skin, brain, lungs, heart, and kidneys [167]. Patients with TSC are also at greater risk for developing malignancies, such as renal cell carcinoma, breast cancer, thyroid cancer, and PNETs [168]. There is currently no report of histone modifications of mTOR pathway regarding PNET.
3.2.3. HIF1α-VHL
Epigenetic regulation of HIF1⍺ signaling has been well established in a variety of cancers. P300 is a transcriptional component of HIF1⍺ which acetylates HIF1⍺ at Lys-709 increasing its stability as shown in both osteosarcoma and renal cell carcinoma cell lines [169]. HDAC inhibitor (HDACi) SAHA has been found to decrease HIF1⍺ levels in tumor cell lines via direct acetylation of heat shock protein 90 (Hip90), a HIF⍺ chaperone protein, using HDAC6 [170]. In addition, HIF1α R282 has been shown methylated by protein arginine methyltransferases 3 (PRMT3) in CRC, which is necessary for its stabilization and oncogene function. PRMT3 inhibition with a novel therapeutic molecule (MPG-peptide) decreases tumor growth and angiogenesis in vivo, potentially offering a future therapeutic strategy [171]. Presently, there is no available information on the histone modifications of the mTOR pathway regarding the HIF1α-VHL signaling in PNET.
3.2.4. RAS-MAPK-NF1
RAS has been found acetylated in human cancer lines on lysine 104 resulting in destabilization of its Switch II domain, which modulates the interaction between RAS and GEF as well as RAS and PI3K for GAP-induced GTP hydrolysis. Essentially, acetylation stands as a negative regulator of RAS function exerting anti-oncogenic effects [172]. Other studies have established a link between RAS signaling and H3K9ac modification, which is one of the most widely studied acetylation site of histone H3 tails and has been commonly observed in ovarian cancer, hepatocellular carcinoma, oral cancer, and cervical cancer [173]. Through binding to xeroderma pigmentosum A protein (XPA), RASSF1A regulates the acetylation and deacetylation cycle of XPA [174]. Not only does RASSF1A acts as a tumor suppressor through DNA repair, but RASSF1A has also been found to increase microtubule stability through HDAC6 which prevents tumor cell migration, invasion, and metastasis [59]. The effect of RASSF1A was also demonstrated in NSCLC xenograft model and in human bronchial cells [175]. Patients with NF1 familial syndrome sometimes develop benign plexiform neurofibromas, which can develop to form peripheral nerve sarcomas known as malignant peripheral sheath tumors (MPNSTs). Bromodomain containing protein 4 (BRD4), a histone acetyltransferase, binds to acetylated histone H3K27Ac in MPNSTs, and has been found to be one of the most highly unregulated genes in MPNSTs. The Bromodomain-containing inhibitor, JQ-1 has been demonstrated to decrease MPNST tumor growth in vitro and in vivo [176]. A phase II trial was conducted in patients with MPNST using JQ-1 (NCT02986919); however, the trial was discontinued due to lack of participants. Currently, there is a new Phase 1/2 Trial that is evaluating the efficacy of Bromodomain-containing inhibitor AZD5153 in combination with PL1 therapy and MEK inhibitor therapy for the treatment of neurofibromatosis [177]. In PNET, the only report of histone modifications in RAS-MAPK signaling comes from one of the receptor tyrosine kinases, IGF2, which shown aberrant H3K4 methylation in the pancreatic islets of MEN1-deficient mice [178].
3.2.5. ATRX/DAXX
The chromatin remodeler ATRX and the histone chaperone DAXX are mutated in a variety of cancers including glioblastoma multiforme, pediatric adrenocotical carcinoma, osteosarcoma, neuroblastoma, myelodysplastic syndrome, acute myeloid leukemia, and PNETs [179]. As mentioned in Section 3.1, ATRX and DAXX form a histone chaperone complex that deposits histone variant H3.3 into specific genomes. The first discovery of ATRX/DAXX mutations came from PNETs where they were found to be mutated in 23% of the tumors. These mutations are inactivating mutations and exhibit alternative lengthening of telomeres (ALT) phenotype. Recently, DAXX has been found to exist in two distinct H3.3. complexes, which include DAXX-ATRX complex and DAXX-SETB1-KAP1-HDAC1 complex [180]. The DAXX-SETB1-KAP1-HDAC1 represses endogenous retroviral (ERV) sequences. ERVs lead to genomic instability through insertion into protein coding regions or by promoting transcription of neighboring genes leading to tumorigenesis [181].
3.2.6. CDKN2A/RB1
RB1 inhibits G1 to S transition through the repression of E2F target genes which are involved in DNA synthesis and cell cycle progression. RB1 antagonizes E2F activity through binding to E2F transcription factors as well as multiple corepressor molecules including HDACs (HDAC1, 2, 3) [182,183]. HDAC inhibitors such as SAHA have been shown to cause RB1-mediated silencing of cell cycle oncogenes [184]. More recently, HDAC5 has been found to act as a corepressor for RB1 but can be inhibited by CDK 4/6. CDK 4/6 inhibition was found to specifically enhance HDAC5 binding to RB1. Resistance to CKD 4/6 inhibitor therapy such as palbociclib was found to be related to low HDAC levels in cancer cells, which means HDAC5 could act as a useful biomarker in guiding therapy [185]. HDAC inhibition has been shown to have anti proliferative effects by inducing cell cycle arrest in G1 via cycling-dependent kinase inhibitors or downregulation of cyclins and CDKs [186]. Inhibition of HDACs induced elevated expression of CDK inhibitors, resulting in the blockade of the cell cycle at the G1 phase and disruption of the G1/S transition. This occurred through the reactivation of RB1 function, leading to the inhibition of E2F, halt of G1 progression, and cell apoptosis [187,188]. HDAC inhibitors such as TSA, SAHA, and VPA have been shown to result in cell cycle arrest at the G2/M checkpoint [189]. HDACs not only play a transcriptional role in cell cycle progression, but also can act as a cell cycle regulator through modulating Aurora B kinase in mitotic progression [190]. Maggi et al. reported that histone demethylase retinoblastoma binding protein 2 (Rbp2) was found overexpressed in PNET tumors. In addition, aberrant expression of Rbp2 altered histone demethylation and contributed to PNET pathogenesis [191].
3.2.7. P53
One of the first recognized non-histone proteins affected by acetylation is p53 [192,193]. P53 is a tumor suppressor gene that is negatively regulated through MDM2 and transiently stabilized and activated by various intracellular processes. While the phosphorylation of serine residues was previously recognized as the primary stabilizer of p53, it has been discovered that acetylation also plays a role in stabilizing p53 during cellular stress and contributes to enhancing transcriptional activity of p53 [193]. MDM2 has been reported to negatively regulate p53 acetylation, but can be reversed by tumor suppressor p14 ARF [193]. In H1299 carcinoma cells, acetylation status regulated degradation of p53 through directly interacting with MDM2 E3 ligase [194]. HDAC inhibitors such as TSA and SIRT1 were found to reverse inhibitory effects of cAMP on DNA damage-induced p53 stabilization and apoptosis [195]. TSA has also been found to upregulate PUMA promoter of p53 in gastric cancer through inhibition of HDAC3 [196]. HDAC inhibitors may lead to p53 activation and pro-apoptotic events, but they do not need p53 for exerting anticancer effects. Rather, studies have shown HDAC inhibitors can independently act to upregulate p21 and BID as a backup mechanism for tumor suppression [197]. Recent evidence has found that p53 itself can be a driver of HDAC inhibition, which in turn leads to autophagy and programmed cell death [198]. HDAC inhibitors VPA and TSA were found to induce apoptosis in pancreatic cancer derived cell lines, Panc1 and PaCa44, through upregulation of p21 and Puma resulting in mutant p53 degradation [199]. Currently, there is no available information on histone modifications of p53 in PNET research.
3.2.8. Notch Signaling
In Notch signaling pathway, ligand and notch receptor binding releases Notch Intracellular Domain (NICD), which translocates to the nucleus to activate gene expression through assembling a coactivator complex which includes RBJP and histone acetyltransferase p300. During this process, HDAC3 increases NICD stability through reducing p300 mediated acetylation of NICD protein. HDAC3 expression was found to be significantly higher in T-ALL patients and in CLL B cell samples [100]. VPA has been found to activate Notch signaling in tumor cells, resulting in suppressed growth of carcinoid tumors in a mouse tumor xenograft [108,200]. These finds were further confirmed in a phase II pilot study that examined the effects of VPA on eight patients with low grade neuroendocrine tumors (carcinoid and pancreatic). Pretreatment tumor biopsies revealed low Notch1 levels, and a 10-fold increase in Notch1 activity on post VPA treatment tumor biopsy [111].
3.2.9. Wnt/β-catenin
Histone modifications have been shown to be involved in regulation of Wnt signaling via complicated mechanisms. Upon activation, Wnt molecules bind to the FZD-LRP5/6 co-receptor complex. LRP6 requires acetylation by p300 to become activated for Wnt signaling. During Wnt signaling, β-catenin is released from the destruction complex and can become acetylated by CBP, p300, and PCAF, which allows for upregulation of β-catenin activity. β-catenin will then undergo nuclear translocation to activate gene transcription [113]. When Wnt signaling is in its inactive state, beta-catenin becomes phosphorylated by the protein destruction complex and targeted for proteasomal degradation. Once degraded, a repressive complex containing T cell factor (TCF)/ lymphoid enhancer factor (LEF) and transducing like enhancer protein (TLE) utilizes HDACs to repress target genes [201]. HDACis such as TSA have been found to inhibit the Wnt/β-catenin pathway by increasing β-catenin phosphorylation, reducing β-catenin nuclear translocation, and inhibiting β-catenin/TCF complex formation in pituitary corticotrope tumor cells (AtT20) [202]. TSA and SAHA have demonstrated anti-tumor effects in colon cancer cell lines with β-catenin mutations by downregulating the Wnt transcription factor TCF7L2 [203]. Curcumin, as discussed in Section 3.1, serves as an inhibitor of acetyltransferase, specifically targeting p300 in the Wnt pathway. Currently, curcumin is undergoing Phase I clinical trials for the treatment of breast, colon, and pancreatic cancers [204]. Currently, there is no available information on histone modifications of Wnt/β-catenin in PNET research.
3.2.10. NFκB
Post translational modifications, including acetylation and methylation, play a major role in the regulation of NF-KB signaling. These modifications occur in the cytoplasm and allow for the activation of IκB kinases (IKKs) resulting in the degradation of IκBalpha, which allows for translocation of NFκB into the nucleus [205]. Within the nucleus, Re1A, a subunit of NFκB, undergoes acetylation by p300/CBP at lysine 310 which promotes transcriptional activity of NFκB [206]. Stat 3 is an oncogenic transcription factor that is commonly activated in cancer. Stat 3 has been found to maintain tumor NFκB activity through acetylation of Re1A. This has been studied in human A2058 melanoma cell lines and in DU145 prostate cancer cells [207]. To date, there have been no documented instances of histone modifications affecting NFκB in PNET.
3.2.11. SSTR2
In the PNET cell lines BON-1 and QGP-1, histone 3 acetylation was observed on SSTR2 [208]. This finding was subsequently corroborated by Veenstra et al. [209], suggesting that the regulation of SSTR2 expression is likely influenced by both histone acetylation and DNA methylation [132]. In addition, using an in vivo BON-1 xenograft mouse model, Kidd et al. showed that the combination treatment of HDACi (VPA) and camptothecin-somatostatin conjugate significantly reduced growth comparing to monotherapies, respectively [210], suggesting that combinatorial therapy might hold promise in PNET patient treatment.
3.2.12. Smad3
Epigenetic regulation of Smad3 via histone modifications plays important roles in cancer progression and metastasis [211]. In lung cancer, the actin binding protein profilin-2 binds to and thereby prevents HDAC1’s access to the promoters of Smad2 and Smad3, which causes activation and promotes EMT and angiogenesis in lung cancer cells [212]. Besides acetylation, histone methylation has also been observed in lung cancer cells. Smad3 mediates recruitment of the histone methyltransferase SETDB1, which controls the expression of SNAIL1 and EMT in breast cancer [213]. Despite its crucial regulatory role in PNET, as outlined in Section 3.1, there is presently no information on histone modifications of Smad3 in the pathology of PNET. A complete list of histone modifications in PNET-related signaling pathways are listed in Table 2.
3.3. Non-Coding RNAs
Alterations in both coding and ncRNAs have been widely implicated in cancer pathophysiology [218]. There are a few recent reviews which described the limited literature concerning the role of ncRNA in pancreatic adenocarcinoma cancers and PNET [219,220,221]. However, comparing to other heavily studied cancers, there is still lack of study of both miRNAs and long non-coding RNAs (lncRNAs) due to the low incidence of PNET cases.
3.3.1. MEN1
The regulatory landscape of the MEN1 signaling pathway in cancer has recently become a focal point of research, with ncRNAs emerging as influential players in this intricate network. NcRNAs exert nuanced control over gene expression, contributing to the dynamic modulation of the MEN1 pathway in cancer progression. For instance, multiple miRNAs, including miR-142-3p, let-7i, miR-125a-5p, miR-199b-5p, and miR-1274b_v16.0, and miR-193b have been found differentially expressed in MEN1 parathyroid tumors comparing to normal parathyroid tissues [222]. Furthermore, lncRNA NEAT1 has been implicated in the dysregulation of MEN1 signaling in multiple endocrine neoplasia type 1 (MEN1) syndrome, acting as a competing endogenous RNA to sequester miR-34a and alleviate its inhibitory effect on MEN1 [26,223,224]. Additionally, miR-29a has been identified as a negative regulator of MEN1 in parathyroid adenomas, influencing cell proliferation and apoptosis [225]. These examples underscore the intricate interplay between ncRNAs and the MEN1 pathway, highlighting the diverse regulatory roles of ncRNAs in shaping the landscape of cancer progression. Investigating these molecular interactions offers promising avenues for understanding the underlying mechanisms and identifying novel therapeutic targets in MEN1-associated cancers. Human maternally expressed gene 3 (MEG3) encodes an RNA transcript that exhibits tumor suppressive ncRNA functions (lncRNA MEG3) with an unknown contribution to cellular processes in its limited translated form [226,227,228]. MEG3 levels are downregulated in PNET through a variety of mechanisms. In murine embryonic stem cells undergoing differentiation into pancreatic islet-like endocrine cells (PILECs), a biallelic MEN1 mutation resulted in reduced menin protein levels, subsequently leading to decreased transcription of MEG3 [178]. In both human PNET samples and MIN6 murine insulinoma cells, hypermethylation of the MEG3 promoter are associated with downregulation of MEG3 compared to normal tissue [229]. Luzi et al. used BON-1 luciferase reporter cell line to show that menin acted as a negative regulator of miR-24-1, which in turn downregulated menin in a negative feedback loop [230]. In a subsequent investigation, it was demonstrated that reduced menin expression resulting from mutations in neuroendocrine cancers disrupted the feedback loop between menin and miR-24-1. This disruption further contributed to a decrease in menin levels due to unregulated expression of miR-24-1 [231]. These studies unequivocally demonstrate that diminished menin levels, with miR-24-1 as a driving factor, play a role in the pathogenesis and progression of neuroendocrine neoplasms [232].
3.3.2. mTOR
The intricate regulation of the mTOR signaling pathway in cancers has garnered substantial attention, with ncRNAs standing out as prominent players in modulating the mTOR pathway, influencing various facets of cancer progression. For example, miR-99a has been identified as a critical regulator by directly targeting mTOR, suppressing its expression and impeding cell proliferation in hepatocellular carcinoma [233]. In breast cancer, the lncRNA HOTAIR acts as a molecular scaffold to facilitate the assembly of the mTOR signaling complex, promoting tumor growth [234]. Additionally, the lncRNA TUG1 has been shown to modulate mTOR signaling in renal cell carcinoma, influencing cellular processes such as proliferation and apoptosis [235]. In MIN6 luciferase reporter cell line, overexpression of miR-144 downregulated PTEN which in turn caused upregulation of p-AKT pathway and a subsequent increase of beta cell proliferation [236]. This piece of evidence is in line with the expression pattern of p-AKT and miR-144 in a differential expression analysis of 29 human insulinoma samples in comparison with normal tissues [236]. In addition, overexpression of a lncRNA H19 plasmid activated VGF and PI3K/AKT pathway in QGP-1 cell line, suggesting a link between lncRNA H19 and an increase in proliferation and metastasis of neuroendocrine tumors [237].
3.3.3. HIF1α
The regulatory influence of ncRNAs has been recognized as pivotal players in modulating the dynamic HIF-1α signaling cascade, influencing critical aspects of cancer progression. Notably, miR-210 has been identified as a key regulator by directly targeting HIF-1α, promoting angiogenesis and metastasis in various cancers [235,238]. In renal cell carcinoma, the lncRNA H19 acts as a molecular sponge for miR-29a, derepressing HIF-1α expression and fostering tumor growth [239]. Additionally, miR-20a has been implicated in the regulation of HIF-1α in colorectal cancer, influencing cellular responses to hypoxia [240]. The lncRNA MALAT1 has also been associated with HIF-1α modulation in breast cancer, affecting tumor invasion and metastasis [241]. These examples underscore the intricate roles of ncRNAs in shaping the dynamics of HIF-1α signaling in cancers. There are a few reports regarding miRNA-mediated regulation of HIF1α signaling in PNET. Thorns et al. compared the differential expression of miRNAs in 37 PNET tissue samples and 9 non-neoplastic controls, from which they were able to identify two miRNAs. While levels of miR-642 were positively correlated with Ki-67 scores, levels of miR-210 expression were positively correlated with metastasis [242]. The connection between miR-210 and its targets has not been elucidated in PNET, although in liver metastasis samples from colorectal adenocarcinoma patients, miR-210 was shown to affect disease progression possibly through interaction with the HIFa pathway [243,244].
3.3.4. Ras-MAPK
The Ras-MAPK signaling pathway plays a pivotal role in regulating various cellular processes, and the influence of ncRNAs on this pathway has emerged as a significant area of exploration in cancer research. For instance, miR-21 has been implicated in the activation of Ras-MAPK signaling in pancreatic cancer, promoting cell proliferation and invasion [245]. Conversely, miR-143 functions as a tumor suppressor by targeting KRAS, a key component of the Ras pathway, and inhibiting its downstream signaling in colorectal cancer [246]. In melanoma, the lncRNA SPRY4-IT1 has been shown to activate the Ras-MAPK pathway by regulating SPRY4 expression [247]. Additionally, the lncRNA MALAT1 has been associated with Ras-MAPK signaling in lung cancer, influencing cell proliferation and migration [248]. Using both QGP-1 GFP reporter cell line and a murine xenograft model, Zhang et al. showed that miR-431 promoted EMT, invasion, and metastasis by silencing DAB2IP, a tumor suppressor protein whose downregulation resulted in the direct activation of the Ras pathway [249].
3.3.5. DAXX/ATRX
Recently, ncRNAs have emerged as key regulators of the DAXX-ATRX signaling pathway, contributing to the complexity of cancer progression. For example, miR-1269a has been identified as a regulator that targets ATRX, promoting glioma cell proliferation and invasion [250]. In addition, miR-21 is found upregulated in gastric cancer. Accordingly, overexpression of miR-21 promotes tumor growth by targeting DAXX in gastric cancer cell lines [251]. Furthermore, the lncRNA XIST has been implicated in the regulation of DAXX-ATRX in glioblastoma, influencing both chromatin remodeling and tumor progression [252]. In the context of PNET research, Gill et al. performed microarray differential expression profiling of miRNA in 37 PNET tissue samples including local and distant metastases and found significant upregulation of miR-3653 in the metastatic group. Following bioinformatic analysis of three miRNA databases, ATRX was revealed to be a negatively regulated target for miR-3653, suggesting a possible link between miR-3653 and PNET pathogenesis through dysregulation of the DAXX/ATRX pathway [253].
3.3.6. RB1
Recent investigations reveal that ncRNAs play intricate roles in modulating RB1 signaling, contributing to the complex landscape of cancer progression. For instance, miR-106b-5p has been identified as an oncogenic miRNA that target RB1 signaling, promoting cell cycle progression and proliferation in laryngeal carcinoma [254]. Conversely, miR-34a and miR-15a/16 act synergistically to induce cell cycle arrest and apoptosis in NSCLC cell lines in a RB1-dependent manner [255]. In addition, the H19 ncRNA controls cell proliferation and metastasis by regulating essential RB1-E2F signaling in colorectal cancer [256]. Furthermore, the lncRNA PVT1 has been shown to regulate RB1 in gastric cancer, contributing to tumor growth [257]. Despite its critical regulatory role in PNET as described in Section 3.1, there have been no documented instances of ncRNAs affecting RB1 in PNET.
3.3.7. p53
The p53 signaling pathway, a crucial guardian of genomic stability and a key player in tumor suppression, is intricately regulated by ncRNAs in the complex landscape of cancer biology. For instance, the miRNA miR-125b has been implicated as an oncomiR that targets p53, promotes cell proliferation, and inhibits apoptosis in leukemia. On the other hand, the miRNA miR-34a, a well-established tumor suppressor, directly targets p53, inducing cell cycle arrest and apoptosis in multiple cancer types, including breast and lung cancers. The lncRNA TP53TG1 also acts as a positive regulator of p53 by enhancing its stability and transcriptional activity in gastric cancer [258]. Additionally, the NEAT1 functions as tumor suppressor in hepatocellular carcinoma in a p53-dependent manner [259]. Furthermore, Zhou et al. showed that MEG3 caused downregulation of MDM2 expression and stabilize p53, suggesting that lncRNA MEG3 stimulates p53 transcription and is linked to p53 longevity and activity [260]. As of now, there is no available information on the regulation of p53 signaling in PNET through ncRNA mechanisms.
3.3.8. Notch
Both miRNAs and lncRNAs play crucial roles in modulating Notch signaling in cancer development. For instance, the expressions of Notch-1 and miR-21 were found to have a positive correlation with the development of colorectal cancer, suggesting that miR-21 may function to promote cancer progression [261]. Conversely, miR-34a has been identified as a key player in negatively regulating Notch signaling by targeting Notch1 and Jagged1, leading to suppressed proliferation and enhanced apoptosis in colorectal cancer [262]. In addition, miR-34a has been demonstrated to directly inhibit Notch-1/2 by binding directly to their 3’ UTR in glioma cells [263]. Moreover, the lncRNA HOTAIR regulates the expression of notch3 by competitively binding with miR-613, making the notch3-HOTAIR-miR-613 complex a potential drug target in pancreatic cancer [264]. In the context of PNET, by combining RNASeq and IHC expression analysis, He et al. showed that upregulation of lncRNA XLOC_221242 correlates with overexpression of DNER protein and activation of a variety of precursors in the Notch and Wnt signaling pathways, suggesting that lncRNA XLOC_221242 might interact with Notch signaling in the regulation of PNET progression [265].
3.3.9. Wnt/β-catenin
The Wnt/β-catenin signaling pathway is subject to intricate modulation by ncRNAs in the context of cancer progression. For instance, miR-21 has been identified as an oncomiRNA that promotes Wnt/β-catenin signaling activation by targeting negative regulators, contributing to enhanced cell proliferation and invasion in colorectal cancer [266]. Conversely, the tumor suppressor miRNA miR-34a directly targets Wnt/β-catenin pathway components, inhibiting cell growth and inducing apoptosis in hepatocellular carcinoma [267]. The lncRNA HOTAIR has been associated with Wnt/β-catenin pathway activation in breast cancer, fostering tumor progression [268]. Additionally, the lncRNA CCAT2 forms a feedback loop with Wnt/β-catenin signaling in colon cancer [269]. In the context of PNET, decrease of MEG3 caused upregulation of miR-183 in BON-1 cells. MiR-183 functions as a downstream target for MEG3 and is negatively regulated by MEG3. When miR-183 activity was increased, it upregulates BRI3, which positively relationship correlates with p38/ERK/AKT and Wnt/β-catenin pathways, suggesting a complicated tumor promoting mechanism which includes interaction among MEG3, miR-183, and BRI3 via p38/ERK/AKT and Wnt/β-catenin signaling [228,229]. In addition, in BON-1 and LCC-18 colonic neuroendocrine cell lines, overexpression of lncNEN885 caused a decrease of invasion and EMT by downregulating Wnt/β-catenin signaling. siRNA silencing of lncNEN885 restored markers of EMT and component of the Wnt/β-catenin pathway, suggesting that lncNEN885 might contribute to the regulation of neuroendocrine tumor metastasis through the Wnt/β-catenin pathway [270].
3.3.10. NFκB
The NFκB signaling pathway, a central regulator of inflammation, immunity, and cell survival, is intricately modulated by ncRNAs in the context of cancer. For example, miR-1892b has been identified as a key negative regulator of NFκB, thereby inhibiting inflammatory responses and tumor growth in breast cancer [271]. On the other hand, the oncogenic miRNA miR-21 promotes NFκB activation by targeting negative regulators in colon adenocarcinomas, as shown by evidence of diverse cell lines, xenograft mouse models, and gene expression profiling of patient tumor samples [272]. Additionally, the lncRNA MALAT1 has been associated with NFκB signaling in oral squamous cell carcinoma [273]. Furthermore, the lncRNA NEAT1 inhibits miRNA-216b and promotes colorectal cancer progression [274]. In the context of PNET, Huang et al. studied differential expression of miRNAs in 37 PNET samples and compared the differences between localized and metastatic tumors. Their findings indicated that MiR-196a is involved in NFκB signaling and exhibits a notable correlation with elevated tumor grade, advanced stage, and tissue invasion in PNET [275].
3.3.11. SSTR2
To date, there have been limited reports on the interplay between ncRNAs and SSTR2 signaling in cancers, with the existing literature mainly focusing on neuroendocrine neoplasms. For instance, the combination of SSTR2 with miR-7 and miR-148a caused inhibition of the growth of both lung and intestinal carcinoid cell lines [276]. In PNET, it has been reported that the combination of overexpression of miR-16-5p and the somatostatin analog octreotide resulted in the upregulation of SSTR2 expression in the INS-1 neuroendocrine cell line. This suggests that miRNA may play a role in advancing the development of combinatorial therapeutic approaches, particularly for patients who do not respond adequately to somatostatin analog treatment targeting SSTR2 alone [277].
3.3.12. Smad3
NcRNAs interact with TGF-β/Smad3 signaling to regulate important facets of cancer cell development, including EMT, invasion, migration, cancer cell stemness, and metastasis [278]. For example, miR-21 targets Smad7, an inhibitor of TGF-β/Smad3 signaling, which leads to enhanced cell proliferation and invasion in various cancers [279]. Conversely, the tumor-suppressive miRNA miR-145 directly targets Smad3, inhibiting TGF-β-induced EMT and metastasis in colorectal cancer [110]. In addition, miR-15a correlates with Smad3 expression NSCLC tissues. Not surprisingly, overexpression of miR-15a caused significantly downregulation of Smad3 expression and inhibited proliferation of A549 lung cancer cell line [280]. Furthermore, lncRNA H19 has been found associated with the regulation of TGF-β/Smad3 in hepatocellular carcinoma, impacting tumor growth [281]. Despite its important regulatory role in PNET pathology as described in Section 3.1, as of now, there are no reports on the miRNA-dependent regulation of Smad3 in research studies focusing on PNETs. A complete list of ncRNAs in PNET-related signaling pathways are listed in Table 3.
4. Future Directions for Epigenetic Research and Clinical Applications in PNET Patient Care
Up to this point, we have explored the epigenetic regulation of signaling pathways in the context of both PNET and other extensively studied cancers. It’s important to note that our intention is not to present exhaustive reviews that include detailed introductions to all the implicated signaling pathways. We acknowledge that providing such an in-depth review of the epigenetic regulation of even a single signaling pathway, like MEN1 signaling, would necessitate a comprehensive review in its own right [232,287,288]. Therefore, rather than aiming to cover all signaling pathways extensively, our focus is on discerning the disparities or gaps in our understanding of the epigenetic regulation of signaling pathways between PNET and other extensively studied cancers. The overarching goal is to utilize the understanding derived from existing knowledge in various cancers and translate this knowledge into advancements in research and clinical applications specific to PNET. This is particularly pertinent in the pursuit of more effective epigenetic drugs, akin to the successful cases observed in extensively studied cancers.
There has been notable advancement in comprehending the epigenetic control of cancers, although the available FDA-approved epigenetic therapies remain relatively scarce, with the majority targeting blood-borne cancers. Importantly, none of these approved therapies are directed at PNET. Although there have been endeavors to explore the use of FDA-approved drugs in clinical trials for PNET treatment, no successful reports have emerged thus far, as detailed in Table 4.
It is worth noting that currently approved FDA epigenetic drugs, such as DMTi and HDACi (Table 4), are lack of pathway specificity, thereby causing severe off-target effects and hindering their broad applications in the ever-growing field of precision medicine. In response to this limitation, ongoing efforts involve the utilization of CRISPR-guided systems to selectively activate or silence the promoter regions of either tumor suppressor or oncogenes, respectively [294]. However, this approach necessitates a more comprehensive understanding of the epigenetic regulation governing signaling pathways in PNETs.
As outlined in Section 3, it is evident that the epigenetic regulation of PNET remains an insufficiently explored domain in comparison to other extensively researched cancers, barring a few exceptions like the MEN1 and SSTR2 signaling pathways. This discrepancy is noticeable across various facets of epigenetic research, spanning from comprehensive genome profiling of PNET patient samples to laboratory-based animal studies and gene manipulation of cell lines (refer to Table 1, Table 2 and Table 3). For instance, in the field of DNA methylation research, there are lack of large-scale epigenetic profiling of PNET patient samples. As of 2022, there have been only nine studies reporting global-DNA methylation in human PNETs, which includes a total of 739 samples [295]. The lack of research samples could be, at least partially, due to the lack of overall PNET patient cases [1]. As discussed in section 1, PNETs make up around 2% of all pancreatic malignancies, characterized by their slow-growth tendencies and metastatic potential. These special characteristics resulted in a limited availability of patient tissue samples, particularly in the case of high-grade PNETs obtained from tumor autopsies [1]. Larger-scale studies and more advanced technologies are desperately needed to validate the existing results because identification of molecularly different NET subtypes will have a significant impact on clinical practice, given the high heterogeneity of PNET tumors. For example, in the research of some commonly occurring cancers, there have been reports of using advanced single-cell whole genome methylation profiling to obtain much more complicated information about of cancer epigenome [296].
The unique slowing growth characteristic of PNET also presents a practical challenge for research work in laboratories. For instance, STC-1, one of the most used mouse PNET cell lines, has a doubling time of 54 hours, which not only makes it hard to perform gene manipulation, but also creates a practical hurdle for creating syngenetic xenograft mouse models [297], which have been proven particularly suitable for studies of tumor immunity and immunotherapy response because of the presence of fully functional murine immune system [298]. Another challenge lies in the scarcity of PNET modeling systems. Currently, only a handful of human PNET cell lines, including the most widely used BON-1 and QGP-1, are accessible for research purposes. Consequently, there is a pressing need for further efforts to enhance and diversify the available tools for verifying both gain-of-function and loss-of-function in the continuously expanding list of genes associated with epigenetics research of PNET [299].
Despite the inherent challenges, some encouraging advancements have been achieved. Notably, recognizing the limited effectiveness of epigenetic cancer drugs has prompted investigations into combining these therapies with existing or emerging targeted treatments for a synergistic and combinatorial approach. This innovative strategy holds promise for improving clinical outcomes. For example, combination therapies that concurrently enhance SSTR expression using HDAC inhibitors and target SSTRs may demonstrate enhanced efficacy compared to individual therapies [10] (also refer to Section 3.2). Additionally, our laboratory has previously demonstrated that a combination of 5-azacytidine and chemotherapy can effectively reduce cell proliferation, activate silenced tumor suppressor genes, and diminish PNET tumors in vivo [300] .
Moreover, within the spectrum of diverse biomarkers, miRNAs display characteristics such as stability, relative abundance, and accessibility in blood samples. These qualities make miRNAs highly suitable as biomarkers for the diagnosis and monitoring of cancer [301,302]. Consequently, there is a call for more extensive global miRNA sequencing of PNET samples to establish a more dependable diagnostic platform for the optimal care of patients with PNETs.
5. Conclusion
In summary, the epigenetic regulation of PNETs exhibits similarities with broader cancer scenarios, providing valuable insights and therapeutic possibilities. Ongoing exploration of the specific mechanisms governing epigenetic changes in PNETs, alongside innovative strategies for drug development, holds considerable promise for enhancing the prognosis and quality of life for individuals grappling with this challenging malignancy. Notably, epigenetic changes play pivotal roles in immune surveillance and the development of drug resistance. As a result, the use of epigenetic drugs, including inhibitors targeting various enzymes like DNMTs, HMTs, HDMs, HATs, and HDACs, has the potential to effectively complement other treatments such as standard chemotherapy or immunotherapy in the care of PNET patients.
Funding
This research was supported by the Department of Surgery, Cooper University Health Care.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pipinikas, C.P.; Berner, A.M.; Sposito, T.; Thirlwell, C. The evolving (epi)genetic landscape of pancreatic neuroendocrine tumours. Endocrine-Related Cancer 2019, 26, R519–R544. [Google Scholar] [CrossRef]
- Nigri, G.; Petrucciani, N.; Debs, T.; Mangogna, L.M.; Crovetto, A.; Moschetta, G.; Persechino, R.; Aurello, P.; Ramacciato, G. Treatment options for PNET liver metastases: a systematic review. World J. Surg. Oncol. 2018, 16, 142. [Google Scholar] [CrossRef]
- Shen, X.; Wang, X.; Lu, X.; Zhao, Y.; Guan, W. Molecular biology of pancreatic neuroendocrine tumors: From mechanism to translation. Front. Oncol. 2022, 12, 967071. [Google Scholar] [CrossRef]
- Scarpa, A.; Chang, D.K.; Nones, K.; Corbo, V.; Patch, A.-M.; Bailey, P.; Lawlor, R.T.; Johns, A.L.; Miller, D.K.; Mafficini, A.; et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 2017, 543, 65–71. [Google Scholar] [CrossRef]
- Noë, M.; Pea, A.; Luchini, C.; Felsenstein, M.; Barbi, S.; Bhaijee, F.; Yonescu, R.; Ning, Y.; Adsay, N.V.; Zamboni, G.; et al. Whole-exome sequencing of duodenal neuroendocrine tumors in patients with neurofibromatosis type 1. Mod. Pathol. 2018, 31, 1532–1538. [Google Scholar] [CrossRef]
- Mohindroo, C.; McAllister, F.; De Jesus-Acosta, A. Genetics of Pancreatic Neuroendocrine Tumors. Hematol. Oncol. Clin. North Am. 2022, 36, 1033–1051. [Google Scholar] [CrossRef]
- Ciobanu, O.A.; Martin, S.C.; Herlea, V.; Fica, S. Insights into Epigenetic Changes Related to Genetic Variants and Cells-of-Origin of Pancreatic Neuroendocrine Tumors: An Algorithm for Practical Workup. Cancers 2022, 14, 4444. [Google Scholar] [CrossRef]
- Sahafnejad, Z.; Ramazi, S.; Allahverdi, A. An Update of Epigenetic Drugs for the Treatment of Cancers and Brain Diseases: A Comprehensive Review. Genes 2023, 14, 873. [Google Scholar] [CrossRef]
- Di Domenico, A.; et al. Epigenetic landscape of pancreatic neuroendocrine tumours reveals distinct cells of origin and means of tumour progression. Commun Biol, 2020, 3, 740. [Google Scholar] [CrossRef]
- Maharjan, C.K.; Ear, P.H.; Tran, C.G.; Howe, J.R.; Chandrasekharan, C.; Quelle, D.E. Pancreatic Neuroendocrine Tumors: Molecular Mechanisms and Therapeutic Targets. Cancers 2021, 13, 5117. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Mafficini, A.; Scarpa, A. Genetics and Epigenetics of Gastroenteropancreatic Neuroendocrine Neoplasms. Endocr. Rev. 2019, 40, 506–536. [Google Scholar] [CrossRef]
- Crabtree, J.S. Epigenetic Regulation in Gastroenteropancreatic Neuroendocrine Tumors. Front. Oncol. 2022, 12, 901435. [Google Scholar] [CrossRef]
- Maharjan, C.K.; Ear, P.H.; Tran, C.G.; Howe, J.R.; Chandrasekharan, C.; Quelle, D.E. Pancreatic Neuroendocrine Tumors: Molecular Mechanisms and Therapeutic Targets. Cancers 2021, 13, 5117. [Google Scholar] [CrossRef]
- Blair, L.P.; Yan, Q. Epigenetic Mechanisms in Commonly Occurring Cancers. DNA Cell Biol. 2012, 31 (Suppl. S1), S49–S61. [Google Scholar] [CrossRef]
- Tulsyan, S.; Aftab, M.; Sisodiya, S.; Khan, A.; Chikara, A.; Tanwar, P.; Hussain, S. Molecular basis of epigenetic regulation in cancer diagnosis and treatment. Front. Genet. 2022, 13, 885635. [Google Scholar] [CrossRef]
- Feehley, T.; O’donnell, C.W.; Mendlein, J.; Karande, M.; McCauley, T. Drugging the epigenome in the age of precision medicine. Clin. Epigenetics 2023, 15, 1–13. [Google Scholar] [CrossRef]
- Zhang, C.; Sun, C.; Zhao, Y.; Wang, Q.; Guo, J.; Ye, B.; Yu, G. Overview of MicroRNAs as Diagnostic and Prognostic Biomarkers for High-Incidence Cancers in 2021. Int. J. Mol. Sci. 2022, 23, 11389. [Google Scholar] [CrossRef]
- Lu, Y.; Chan, Y.-T.; Tan, H.-Y.; Li, S.; Wang, N.; Feng, Y. Epigenetic regulation in human cancer: the potential role of epi-drug in cancer therapy. Mol. Cancer 2020, 19, 79. [Google Scholar] [CrossRef]
- Soczomski, P.; Jurecka-Lubieniecka, B.; Krzywon, A.; Cortez, A.J.; Zgliczynski, S.; Rogozik, N.; Oczko-Wojciechowska, M.; Pawlaczek, A.; Bednarczuk, T.; Jarzab, B. A Direct Comparison of Patients With Hereditary and Sporadic Pancreatic Neuroendocrine Tumors: Evaluation of Clinical Course, Prognostic Factors and Genotype–Phenotype Correlations. Front. Endocrinol. 2021, 12, 681013. [Google Scholar] [CrossRef]
- Chatani, P.D.; Agarwal, S.K.; Sadowski, S.M. Molecular Signatures and Their Clinical Utility in Pancreatic Neuroendocrine Tumors. Front. Endocrinol. 2021, 11, 575620. [Google Scholar] [CrossRef]
- Ye, Z., et al., MEN1 promotes ferroptosis by inhibiting mTOR-SCD1 axis in pancreatic neuroendocrine tumors. Acta Biochim Biophys Sin (Shanghai), 2022, 54, 1599-1609.
- Corbo, V.; Dalai, I.; Scardoni, M.; Barbi, S.; Beghelli, S.; Bersani, S.; Albarello, L.; Doglioni, C.; Schott, C.; Capelli, P.; et al. MEN1 in pancreatic endocrine tumors: analysis of gene and protein status in 169 sporadic neoplasms reveals alterations in the vast majority of cases. Endocrine-Related Cancer 2010, 17, 771–783. [Google Scholar] [CrossRef]
- Ishida, N.; Miyazu, T.; Tamura, S.; Suzuki, S.; Tani, S.; Yamade, M.; Iwaizumi, M.; Osawa, S.; Hamaya, Y.; Shinmura, K.; et al. Tuberous sclerosis patient with neuroendocrine carcinoma of the esophagogastric junction: A case report. World J. Gastroenterol. 2020, 26, 7263–7271. [Google Scholar] [CrossRef]
- Tirosh, A.; Kebebew, E. Genetic and epigenetic alterations in pancreatic neuroendocrine tumors. J. Gastrointest. Oncol. 2020, 11, 567–577. [Google Scholar] [CrossRef]
- Chandrasekharappa, S.C.; Guru, S.C.; Manickam, P.; Olufemi, S.-E.; Collins, F.S.; Emmert-Buck, M.R.; Debelenko, L.V.; Zhuang, Z.; Lubensky, I.A.; Liotta, L.A.; et al. Positional Cloning of the Gene for Multiple Endocrine Neoplasia-Type 1. Science 1997, 276, 404–407. [Google Scholar] [CrossRef]
- Conemans, E.B.; Lodewijk, L.; Moelans, C.B.; A Offerhaus, G.J.; Pieterman, C.R.C.; Morsink, F.H.; Dekkers, O.M.; de Herder, W.W.; Hermus, A.R.; van der Horst-Schrivers, A.N.; et al. DNA methylation profiling in MEN1-related pancreatic neuroendocrine tumors reveals a potential epigenetic target for treatment. Eur. J. Endocrinol. 2018, 179, 153–160. [Google Scholar] [CrossRef]
- Massey, S.; Khan, M.A.; Rab, S.O.; Mustafa, S.; Khan, A.; Malik, Z.; Shaik, R.; Verma, M.K.; Deo, S.; Husain, S.A. Evaluating the role of MEN1 gene expression and its clinical significance in breast cancer patients. PLOS ONE 2023, 18, e0288482. [Google Scholar] [CrossRef]
- Brown, E.J.; Albers, M.W.; Shin, T.B.; Ichikawa, K.; Keith, C.T.; Lane, W.S.; Schreiber, S.L. A mammalian protein targeted by G1-arresting rapamycin–receptor complex. Nature 1994, 369, 756–758. [Google Scholar] [CrossRef]
- Zeng, J.-D.; Wu, W.K.; Wang, H.-Y.; Li, X.-X. Serine and one-carbon metabolism, a bridge that links mTOR signaling and DNA methylation in cancer. Pharmacol. Res. 2019, 149, 104352. [Google Scholar] [CrossRef]
- Fattahi, S.; Amjadi-Moheb, F.; Tabaripour, R.; Ashrafi, G.H.; Akhavan-Niaki, H. PI3K/AKT/mTOR signaling in gastric cancer: Epigenetics and beyond. Life Sci. 2020, 262, 118513. [Google Scholar] [CrossRef]
- Wang, C.; et al. A novel methylated cation channel TRPM4 inhibited colorectal cancer metastasis through Ca(2+)/Calpain-mediated proteolysis of FAK and suppression of PI3K/Akt/mTOR signaling pathway. Int. J. Biol. Sci. 2022, 18, 5575–5590. [Google Scholar] [CrossRef]
- Huang, J.; Li, J.; Tang, J.; Wu, Y.; Dai, F.; Yi, Z.; Wang, Y.; Li, Y.; Wu, Y.; Ren, G.; et al. ZDHHC22-mediated mTOR palmitoylation restrains breast cancer growth and endocrine therapy resistance. Int. J. Biol. Sci. 2022, 18, 2833–2850. [Google Scholar] [CrossRef]
- Zhuang, Z.; O Vortmeyer, A.; Pack, S.; Huang, S.; A Pham, T.; Wang, C.; Park, W.S.; Agarwal, S.K.; Debelenko, L.V.; Kester, M. Somatic mutations of the MEN1 tumor suppressor gene in sporadic gastrinomas and insulinomas. Cancer Res. 1997, 57, 4682–4686. [Google Scholar]
- Görtz, B.; Roth, J.; Krähenmann, A.; de Krijger, R.R.; Muletta-Feurer, S.; Rütimann, K.; Saremaslani, P.; Speel, E.J.; Heitz, P.U.; Komminoth, P. Mutations and Allelic Deletions of the MEN1 Gene Are Associated with a Subset of Sporadic Endocrine Pancreatic and Neuroendocrine Tumors and Not Restricted to Foregut Neoplasms. Am. J. Pathol. 1999, 154, 429–436. [Google Scholar] [CrossRef]
- Shan, L.; Nakamura, Y.; Nakamura, M.; Yokoi, T.; Tsujimoto, M.; Arima, R.; Kameya, T.; Kakudo, K. Somatic mutations of multiple endocrine neoplasia type 1 gene in the sporadic endocrine tumors. Lab Invest 1998, 78, 471–475. [Google Scholar]
- Lawrence, B.; Blenkiron, C.; Parker, K.; Tsai, P.; Fitzgerald, S.; Shields, P.; Robb, T.; Yeong, M.L.; Kramer, N.; James, S.; et al. Recurrent loss of heterozygosity correlates with clinical outcome in pancreatic neuroendocrine cancer. npj Genom. Med. 2018, 3, 18. [Google Scholar] [CrossRef]
- Ghayouri, M.; Boulware, D.; Nasir, A.; Strosberg, J.; Kvols, L.; Coppola, D. Activation of the serine/theronine protein kinase Akt in enteropancreatic neuroendocrine tumors. Anticancer Res. 2010, 30, 5063–5067. [Google Scholar]
- Shida, T.; Kishimoto, T.; Furuya, M.; Nikaido, T.; Koda, K.; Takano, S.; Kimura, F.; Shimizu, H.; Yoshidome, H.; Ohtsuka, M.; et al. Expression of an activated mammalian target of rapamycin (mTOR) in gastroenteropancreatic neuroendocrine tumors. Cancer Chemother. Pharmacol. 2009, 65, 889–893. [Google Scholar] [CrossRef]
- Chang, T.M.; et al. The regulatory role of aberrant Phosphatase and Tensin Homologue and Liver Kinase B1 on AKT/mTOR/c-Myc axis in pancreatic neuroendocrine tumors. Oncotarget 2017, 8, 98068–98083. [Google Scholar] [CrossRef]
- Stefanoli, M.; La Rosa, S.; Sahnane, N.; Romualdi, C.; Pastorino, R.; Marando, A.; Capella, C.; Sessa, F.; Furlan, D. Prognostic Relevance of Aberrant DNA Methylation in G1 and G2 Pancreatic Neuroendocrine Tumors. Neuroendocrinology 2014, 100, 26–34. [Google Scholar] [CrossRef]
- Peng, X.; Gao, H.; Xu, R.; Wang, H.; Mei, J.; Liu, C. The interplay between HIF-1α and noncoding RNAs in cancer. J. Exp. Clin. Cancer Res. 2020, 39, 1–19. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.-W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Carmeliet, P.; et al. Role of HIF-1α or in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998, 394, 485–490. [Google Scholar] [CrossRef]
- Nielsen, S.M.; Rhodes, L.; Blanco, I.; Chung, W.K.; Eng, C.; Maher, E.R.; Richard, S.; Giles, R.H. Von Hippel-Lindau Disease: Genetics and Role of Genetic Counseling in a Multiple Neoplasia Syndrome. J. Clin. Oncol. 2016, 34, 2172–2181. [Google Scholar] [CrossRef]
- Mamo, M.; Ye, I.C.; DiGiacomo, J.W.; Park, J.Y.; Downs, B.; Gilkes, D.M. Hypoxia Alters the Response to Anti-EGFR Therapy by Regulating EGFR Expression and Downstream Signaling in a DNA Methylation–Specific and HIF-Dependent Manner. Cancer Res 2020, 80, 4998–5010. [Google Scholar] [CrossRef]
- Cimmino, F.; Avitabile, M.; Lasorsa, V.A.; Montella, A.; Pezone, L.; Cantalupo, S.; Visconte, F.; Corrias, M.V.; Iolascon, A.; Capasso, M. HIF-1 transcription activity: HIF1A driven response in normoxia and in hypoxia. BMC Med Genet. 2019, 20, 37. [Google Scholar] [CrossRef]
- Perigny, M.; et al. Pancreatic endocrine microadenomatosis in patients with von Hippel-Lindau disease: characterization by VHL/HIF pathway proteins expression. Am. J. Surg. Pathol. 2009, 33, 739–748. [Google Scholar]
- Speisky, D.; Duces, A.; Bièche, I.; Rebours, V.; Hammel, P.; Sauvanet, A.; Richard, S.; Bedossa, P.; Vidaud, M.; Murat, A.; et al. Molecular Profiling of Pancreatic Neuroendocrine Tumors in Sporadic and Von Hippel-Lindau Patients. Clin. Cancer Res. 2012, 18, 2838–2849. [Google Scholar] [CrossRef]
- Schmitt, A.M.; Schmid, S.; Rudolph, T.; Anlauf, M.; Prinz, C.; Klöppel, G.; Moch, H.; Heitz, P.U.; Komminoth, P.; Perren, A. VHL inactivation is an important pathway for the development of malignant sporadic pancreatic endocrine tumors. Endocrine-Related Cancer 2009, 16, 1219–1227. [Google Scholar] [CrossRef]
- Chakraborty, J.; Chakraborty, S.; Chakraborty, S.; Narayan, M.N. Entanglement of MAPK pathways with gene expression and its omnipresence in the etiology for cancer and neurodegenerative disorders. Biochim. et Biophys. Acta (BBA) - Gene Regul. Mech. 2023, 1866, 194988. [Google Scholar] [CrossRef]
- Pudewell, S.; Wittich, C.; Jasemi, N.S.K.; Bazgir, F.; Ahmadian, M.R. Accessory proteins of the RAS-MAPK pathway: moving from the side line to the front line. Commun. Biol. 2021, 4, 1–10. [Google Scholar] [CrossRef]
- Chan, D.W.; et al. Genome-wide DNA methylome analysis identifies methylation signatures associated with survival and drug resistance of ovarian cancers. Clin. Epigenetics 2021, 13, 142. [Google Scholar] [CrossRef]
- Gutierrez, A.; Demond, H.; Brebi, P.; Ili, C.G. Novel Methylation Biomarkers for Colorectal Cancer Prognosis. Biomolecules 2021, 11, 1722. [Google Scholar] [CrossRef]
- Yun, D.; Yang, Z.; Zhang, S.; Yang, H.; Liu, D.; Grützmann, R.; Pilarsky, C.; Britzen-Laurent, N. An m5C methylation regulator-associated signature predicts prognosis and therapy response in pancreatic cancer. Front. Cell Dev. Biol. 2022, 10, 975684. [Google Scholar] [CrossRef]
- Shi, J.; Lu, D.; Gu, R.; Xie, J.; Yu, L.; Sun, X.; Zhang, Y. Integrated Analysis of Transcriptome and Differential Methylation of Neurofibromatosis Type 2 Vestibular Schwannomas. World Neurosurg. 2021, 157, e66–e76. [Google Scholar] [CrossRef]
- Zhang, Q.; et al. Activation of RAS/MAPK pathway confers MCL-1 mediated acquired resistance to BCL-2 inhibitor venetoclax in acute myeloid leukemia. Signal. Transduct. Target Ther. 2022, 7, 51. [Google Scholar] [CrossRef]
- Xiao, Y.; Dong, J. The Hippo Signaling Pathway in Cancer: A Cell Cycle Perspective. Cancers 2021, 13, 6214. [Google Scholar] [CrossRef]
- Dubois, F.; Bergot, E.; Zalcman, G.; Levallet, G. RASSF1A, puppeteer of cellular homeostasis, fights tumorigenesis, and metastasis—an updated review. Cell Death Dis. 2019, 10, 928. [Google Scholar] [CrossRef]
- Khandelwal, M.; Anand, V.; Appunni, S.; Seth, A.; Singh, P.; Mathur, S.; Sharma, A. RASSF1A–Hippo pathway link in patients with urothelial carcinoma of bladder: plausible therapeutic target. Mol. Cell. Biochem. 2019, 464, 51–63. [Google Scholar] [CrossRef]
- Bakavicius, A.; Daniunaite, K.; Zukauskaite, K.; Barisiene, M.; Jarmalaite, S.; Jankevicius, F. Urinary DNA methylation biomarkers for prediction of prostate cancer upgrading and upstaging. Clin. Epigenetics 2019, 11, 115. [Google Scholar] [CrossRef]
- Sangtani, A.; Wang, C.; Weaver, A.; Hoppman, N.L.; Kerr, S.E.; Abyzov, A.; Shridhar, V.; Staub, J.; Kocher, J.-P.A.; Voss, J.S.; et al. Combining copy number, methylation markers, and mutations as a panel for endometrial cancer detection via intravaginal tampon collection. Gynecol. Oncol. 2019, 156, 387–392. [Google Scholar] [CrossRef]
- Aibel, C.; De Peña, A.C.; Tripathi, A. An Optimized CoBRA Method for the Microfluidic Electrophoresis Detection of Breast Cancer Associated RASSF1 Methylation. BioTech 2023, 12, 7. [Google Scholar] [CrossRef]
- Raos, D.; Ulamec, M.; Bojanac, A.K.; Bulic-Jakus, F.; Jezek, D.; Sincic, N. Epigenetically inactivated RASSF1A as a tumor biomarker. Bosn. J. Basic Med Sci. 2020, 21, 386–397. [Google Scholar] [CrossRef]
- Mashayekhi, M.; Asadi, M.; Hashemzadeh, S.; Vahedi, A.; Shanehbandi, D.; Al-Omar, A.F.; Akbari, M.; Raeisi, M. Promoter methylation levels of RASSF1 and ATIC genes are associated with lung cancer in Iranian patients. Horm. Mol. Biol. Clin. Investig. 2023, 44, 145–152. [Google Scholar] [CrossRef]
- Daniunaite, K.; Sestokaite, A.; Kubiliute, R.; Stuopelyte, K.; Kettunen, E.; Husgafvel-Pursiainen, K.; Jarmalaite, S. Frequent DNA methylation changes in cancerous and noncancerous lung tissues from smokers with non-small cell lung cancer. Mutagenesis 2020, 35, 373–379. [Google Scholar] [CrossRef]
- Khatami, F.; Larijani, B.; Heshmat, R.; Nasiri, S.; Saffar, H.; Tavangar, S.M. Promoter Methylation of Four Tumor Suppressor Genes in Human Papillary Thyroid Carcinoma. Iran. J. Pathol. 2019, 14, 290–298. [Google Scholar] [CrossRef]
- House, M.G.; Herman, J.G.; Guo, M.Z.; Hooker, C.M.; Schulick, R.D.; Lillemoe, K.D.; Cameron, J.L.; Hruban, R.H.; Maitra, A.; Yeo, C.J. Aberrant Hypermethylation of Tumor Suppressor Genes in Pancreatic Endocrine Neoplasms. Ann. Surg. 2003, 238, 423–432, discussion 431–432. [Google Scholar] [CrossRef]
- Pyronnet, S.; Bousquet, C.; Najib, S.; Azar, R.; Laklai, H.; Susini, C. Antitumor effects of somatostatin. Mol. Cell. Endocrinol. 2008, 286, 230–237. [Google Scholar] [CrossRef]
- Malpeli, G.; et al. Methylation-associated down-regulation of RASSF1A and up-regulation of RASSF1C in pancreatic endocrine tumors. BMC Cancer 2011, 11, 351. [Google Scholar] [CrossRef]
- Ratner, N.; Miller, S.J. A RASopathy gene commonly mutated in cancer: the neurofibromatosis type 1 tumour suppressor. Nat. Rev. Cancer 2015, 15, 290–301. [Google Scholar] [CrossRef]
- Gauci, J.; et al. Neurofibromatosis Type 1 A Rare Predisposition for Gastrinomas and Other Neuroendocrine Tumors. Pancreas 2022, 51, 559–562. [Google Scholar] [CrossRef]
- Dyer, M.A.; Qadeer, Z.A.; Valle-Garcia, D.; Bernstein, E. ATRX and DAXX: Mechanisms and Mutations. Cold Spring Harb. Perspect. Med. 2017, 7, a026567. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Cai, J.; Gao, W.; Meng, X.; Gao, F.; Wu, P.; Duan, C.; Wang, R.; Dinislam, M.; Lin, L.; et al. Loss of ATRX suppresses ATM dependent DNA damage repair by modulating H3K9me3 to enhance temozolomide sensitivity in glioma. Cancer Lett. 2018, 419, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante, S.G.; Pereira, B.J.; Lerario, A.M.; Sola, P.R.; Oba-Shinjo, S.M.; Marie, S.K. The chromatin remodeler complex ATRX-DAXX-H3.3 and telomere length in meningiomas. Clin. Neurol. Neurosurg. 2021, 210, 106962. [Google Scholar] [CrossRef]
- Heaphy, C.M.; Bi, W.L.; Coy, S.; Davis, C.; Gallia, G.L.; Santagata, S.; Rodriguez, F.J. Telomere length alterations and ATRX/DAXX loss in pituitary adenomas. Mod. Pathol. 2020, 33, 1475–1481. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Vakiani, E.; White, C.M.B.; Zhong, Y.; Saunders, T.H.; Morgan, R.; de Wilde, R.F.; Maitra, A.M.; Hicks, J.B.; DeMarzo, A.M.; et al. Small Cell and Large Cell Neuroendocrine Carcinomas of the Pancreas are Genetically Similar and Distinct From Well-differentiated Pancreatic Neuroendocrine Tumors. Am. J. Surg. Pathol. 2012, 36, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Totoki, Y.; Noë, M.; Nakatani, Y.; Horie, M.; Kawasaki, K.; Nakamura, H.; Saito-Adachi, M.; Suzuki, M.; Takai, E.; et al. Comprehensive Genomic Profiling of Neuroendocrine Carcinomas of the Gastrointestinal System. Cancer Discov. 2022, 12, 692–711. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Huo, C.; Yang, P. Genistein inhibited the proliferation of kidney cancer cells via CDKN2a hypomethylation: role of abnormal apoptosis. Int. Urol. Nephrol. 2020, 52, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Xia, L., W. Zhang, and L. Gao, Clinical and prognostic effects of CDKN2A, CDKN2B and CDH13 promoter methylation in ovarian cancer: a study using meta-analysis and TCGA data. Biomarkers 2019, 24, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-C.; Kuo, Y.-C.; Hu, J.-M.; Chang, P.-K.; Sun, C.-A.; Yang, T.; Li, C.-W.; Chen, C.-Y.; Lin, F.-H.; Hsu, C.-H.; et al. MTNR1B polymorphisms with CDKN2A and MGMT methylation status are associated with poor prognosis of colorectal cancer in Taiwan. World J. Gastroenterol. 2021, 27, 5737–5752. [Google Scholar] [CrossRef]
- Kong, R.; Wang, N.; Han, W.; Bao, W.; Lu, J. Fenofibrate Exerts Antitumor Effects in Colon Cancer via Regulation of DNMT1 and CDKN2A. PPAR Res. 2021, 2021, 6663782. [Google Scholar] [CrossRef]
- Truong, P.K., T. D. Lao, and T.A.H. Le, Methylation - an Epigenetic Biomarker for Cervical Cancer Risk: A Meta-Analysis. Pharmacophore 2020, 11, 21–29. [Google Scholar]
- Xu, J.; Li, N.; Deng, W.; Luo, S. Discovering the mechanism and involvement of the methylation of cyclin-dependent kinase inhibitor 2A (CDKN2A) gene and its special locus region in gastric cancer. Bioengineered 2021, 12, 1286–1298. [Google Scholar] [CrossRef]
- Li, P.; et al. P16 methylation increases the sensitivity of cancer cells to the CDK4/6 inhibitor palbociclib. PLoS One 2019, 14, e0223084. [Google Scholar] [CrossRef]
- Hsu, J.Y.; et al. Clinical Utility of CDK4/6 Inhibitors in Sarcoma: Successes and Future Challenges. JCO Precis Oncol. 2022, 6, e2100211. [Google Scholar] [CrossRef]
- Anwar, S.L. and U. Lehmann, Detection of Aberrant DNA Methylation Patterns in the RB1 Gene. Methods Mol. Biol. 2018, 1726, 35–47. [Google Scholar]
- Bartsch, D.K.; Kersting, M.; Wild, A.; Ramaswamy, A.; Gerdes, B.; Schuermann, M.; Simon, B.; Rothmund, M. Low Frequency of p16INK4a Alterations in Insulinomas. Digestion 2000, 62, 171–177. [Google Scholar] [CrossRef]
- Cao, L.Q.; et al. Exosomal miR-21 regulates the TETs/PTENp1/PTEN pathway to promote hepatocellular carcinoma growth. Mol. Cancer, 2019, 18, 148. [Google Scholar] [CrossRef]
- Lindberg, D., G. Akerstrom, and G. Westin, Evaluation of CDKN2C/p18, CDKN1B/p27 and CDKN2B/p15 mRNA expression, and CpG methylation status in sporadic and MEN1-associated pancreatic endocrine tumours. Clin. Endocrinol. (Oxf), 2008, 68, 271–277. [Google Scholar] [CrossRef]
- Khan, M.A.; Tiwari, D.; Dongre, A.; Sadaf, *!!! REPLACE !!!*; Mustafa, S.; Das, C.R.; Massey, S.; Bose, P.D.; Bose, S.; Husain, S.A. Exploring the p53 connection of cervical cancer pathogenesis involving north-east Indian patients. PLoS ONE 2020, 15, e0238500. [Google Scholar] [CrossRef]
- Chatterjee, B.; Ghosh, K.; Kanade, S.R. Resveratrol modulates epigenetic regulators of promoter histone methylation and acetylation that restores BRCA1, p53, p21CIP1 in human breast cancer cell lines. BioFactors 2019, 45, 818–829. [Google Scholar] [CrossRef]
- Liu, Z.L.; et al. Resveratrol induces p53 in colorectal cancer through SET7/9. Oncol. Lett. 2019, 17, 3783–3789. [Google Scholar] [CrossRef]
- Kang, K.A.; Piao, M.J.; Hyun, Y.J.; Zhen, A.X.; Cho, S.J.; Ahn, M.J.; Yi, J.M.; Hyun, J.W. Luteolin promotes apoptotic cell death via upregulation of Nrf2 expression by DNA demethylase and the interaction of Nrf2 with p53 in human colon cancer cells. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef]
- Wu, C.; Guo, E.; Ming, J.; Sun, W.; Nie, X.; Sun, L.; Peng, S.; Luo, M.; Liu, D.; Zhang, L.; et al. Radiation-Induced DNMT3B Promotes Radioresistance in Nasopharyngeal Carcinoma through Methylation of p53 and p21. Mol. Ther. - Oncolytics 2020, 17, 306–319. [Google Scholar] [CrossRef]
- Zhang, L.; Li, W.; Cao, L.; Xu, J.; Qian, Y.; Chen, H.; Zhang, Y.; Kang, W.; Gou, H.; Wong, C.C.; et al. PKNOX2 suppresses gastric cancer through the transcriptional activation of IGFBP5 and p53. Oncogene 2019, 38, 4590–4604. [Google Scholar] [CrossRef]
- Vandamme, T.; Peeters, M.; Dogan, F.; Pauwels, P.; Van Assche, E.; Beyens, M.; Mortier, G.; Vandeweyer, G.; de Herder, W.; Van Camp, G.; et al. Whole-exome characterization of pancreatic neuroendocrine tumor cell lines BON-1 and QGP-1. J. Mol. Endocrinol. 2015, 54, 137–147. [Google Scholar] [CrossRef]
- Ohki, R.; Saito, K.; Chen, Y.; Kawase, T.; Hiraoka, N.; Saigawa, R.; Minegishi, M.; Aita, Y.; Yanai, G.; Shimizu, H.; et al. PHLDA3 is a novel tumor suppressor of pancreatic neuroendocrine tumors. Proc. Natl. Acad. Sci. 2014, 111, E2404–E2413. [Google Scholar] [CrossRef]
- Wharton, K.A.; Johansen, K.M.; Xu, T.; Artavanis-Tsakonas, S. Nucleotide sequence from the neurogenic locus Notch implies a gene product that shares homology with proteins containing EGF-like repeats. Cell 1985, 43, 567–581. [Google Scholar] [CrossRef]
- Ferrante, F.; Giaimo, B.D.; Bartkuhn, M.; Zimmermann, T.; Close, V.; Mertens, D.; Nist, A.; Stiewe, T.; Meier-Soelch, J.; Kracht, M.; et al. HDAC3 functions as a positive regulator in Notch signal transduction. Nucleic Acids Res. 2020, 48, 3496–3512. [Google Scholar] [CrossRef]
- Jeong, G.-Y.; Park, M.K.; Choi, H.-J.; An, H.W.; Park, Y.-U.; Choi, H.-J.; Park, J.; Kim, H.-Y.; Son, T.; Lee, H.; et al. NSD3-Induced Methylation of H3K36 Activates NOTCH Signaling to Drive Breast Tumor Initiation and Metastatic Progression. Cancer Res 2021, 81, 77–90. [Google Scholar] [CrossRef]
- Yousefi, H.; Bahramy, A.; Zafari, N.; Delavar, M.R.; Nguyen, K.; Haghi, A.; Kandelouei, T.; Vittori, C.; Jazireian, P.; Maleki, S.; et al. Notch signaling pathway: a comprehensive prognostic and gene expression profile analysis in breast cancer. BMC Cancer 2022, 22, 1282. [Google Scholar] [CrossRef]
- Qiu, L.; Yang, X.; Wu, J.; Huang, C.; Miao, Y.; Fu, Z. HIST2H2BF Potentiates the Propagation of Cancer Stem Cells via Notch Signaling to Promote Malignancy and Liver Metastasis in Colorectal Carcinoma. Front. Oncol. 2021, 11, 677646. [Google Scholar] [CrossRef]
- Chai, H.; Pan, C.; Zhang, M.; Huo, H.; Shan, H.; Wu, J. Histone methyltransferase SETD1A interacts with notch and promotes notch transactivation to augment ovarian cancer development. BMC Cancer 2023, 23, 96. [Google Scholar] [CrossRef]
- Kadian, L.K.; Gulshan, G.; Ahuja, P.; Singhal, G.; Sharma, S.; Nanda, S.; Yadav, R. Aberrant promoter methylation of NOTCH1 and NOTCH3 and its association with cervical cancer risk factors in North Indian population. 2020, 12, 2814–2826.
- Biran, A.; et al. Activation of Notch and Myc Signaling via B-cell-Restricted Depletion of Dnmt3a Generates a Consistent Murine Model of Chronic Lymphocytic Leukemia. Cancer Res. 2021, 81, 6117–6130. [Google Scholar] [CrossRef]
- Wang, H.; Chen, Y.; Castillo, C.F.-D.; Yilmaz, O.; Deshpande, V. Heterogeneity in signaling pathways of gastroenteropancreatic neuroendocrine tumors: a critical look at notch signaling pathway. Mod. Pathol. 2013, 26, 139–147. [Google Scholar] [CrossRef]
- Greenblatt, D.Y.; Vaccaro, A.M.; Jaskula-Sztul, R.; Ning, L.; Haymart, M.; Kunnimalaiyaan, M.; Chen, H. Valproic Acid Activates Notch-1 Signaling and Regulates the Neuroendocrine Phenotype in Carcinoid Cancer Cells. Oncol. 2007, 12, 942–951. [Google Scholar] [CrossRef]
- Kunnimalaiyaan, M.; Traeger, K.; Chen, H. Conservation of the Notch1 signaling pathway in gastrointestinal carcinoid cells. Am. J. Physiol. Liver Physiol. 2005, 289, G636–G642. [Google Scholar] [CrossRef]
- Adler, J.T.; Hottinger, D.G.; Kunnimalaiyaan, M.; Chen, H. Histone deacetylase inhibitors upregulate Notch-1 and inhibit growth in pheochromocytoma cells. Surgery 2008, 144, 956–962, discussion 961–962. [Google Scholar] [CrossRef]
- Mohammed, T.A.; Holen, K.D.; Jaskula-Sztul, R.; Mulkerin, D.; Lubner, S.J.; Schelman, W.R.; Eickhoff, J.; Chen, H.; LoConte, N.K. A Pilot Phase II Study of Valproic Acid for Treatment of Low-Grade Neuroendocrine Carcinoma. Oncol. 2011, 16, 835–843. [Google Scholar] [CrossRef]
- Liu, J.; et al. Wnt/beta-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal. Transduct. Target Ther. 2022, 7, 3. [Google Scholar] [CrossRef]
- You, H.; Li, Q.; Kong, D.; Liu, X.; Kong, F.; Zheng, K.; Tang, R. The interaction of canonical Wnt/β-catenin signaling with protein lysine acetylation. Cell. Mol. Biol. Lett. 2022, 27, 1–14. [Google Scholar] [CrossRef]
- Mehdi, S.; et al. LY75 Ablation Mediates Mesenchymal-Epithelial Transition (MET) in Epithelial Ovarian Cancer (EOC) Cells Associated with DNA Methylation Alterations and Suppression of the Wnt/beta-Catenin Pathway. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Yen, H.Y.; et al. Regulation of carcinogenesis and modulation through Wnt/beta-catenin signaling by curcumin in an ovarian cancer cell line. Sci. Rep. 2019, 9, 17267. [Google Scholar] [CrossRef]
- Tao, C.; et al. The tumor suppressor Zinc finger protein 471 suppresses breast cancer growth and metastasis through inhibiting AKT and Wnt/beta-catenin signaling. Clin. Epigenetics 2020, 12, 173. [Google Scholar] [CrossRef]
- Meng, F.; et al. SMYD2 suppresses APC2 expression to activate the Wnt/beta-catenin pathway and promotes epithelial-mesenchymal transition in colorectal cancer. Am. J. Cancer Res. 2020, 10, 997–1011. [Google Scholar]
- Zhang, W.; et al. KDM1A promotes thyroid cancer progression and maintains stemness through the Wnt/beta-catenin signaling pathway. Theranostics 2022, 12, 1500–1517. [Google Scholar] [CrossRef]
- Papadopoulou, S.; Edlund, H. Attenuated Wnt Signaling Perturbs Pancreatic Growth but Not Pancreatic Function. Diabetes 2005, 54, 2844–2851. [Google Scholar] [CrossRef]
- Weiss, V.; et al. Immunohistochemical analysis of the Wnt/beta-catenin signaling pathway in pancreatic neuroendocrine neoplasms. World J. Gastrointest Oncol. 2016, 8, 615–622. [Google Scholar] [CrossRef]
- Kim, J.T.; Li, J.; Jang, E.R.; Gulhati, P.; Rychahou, P.G.; Napier, D.L.; Wang, C.; Weiss, H.L.; Lee, E.Y.; Anthony, L.; et al. Deregulation of Wnt/β-catenin signaling through genetic or epigenetic alterations in human neuroendocrine tumors. Carcinog. 2013, 34, 953–961. [Google Scholar] [CrossRef]
- de Mestier, L.; et al. Critical appraisal of MGMT in digestive NET treated with alkylating agents. Endocr Relat. Cancer 2020, 27, R391–R405. [Google Scholar] [CrossRef]
- Brighi, N.; Lamberti, G.; Andrini, E.; Mosconi, C.; Manuzzi, L.; Donati, G.; Lisotti, A.; Campana, D. Prospective Evaluation of MGMT-Promoter Methylation Status and Correlations with Outcomes to Temozolomide-Based Chemotherapy in Well-Differentiated Neuroendocrine Tumors. Curr. Oncol. 2023, 30, 1381–1394. [Google Scholar] [CrossRef]
- Chauhan, A.; et al. Phytochemicals targeting NF-kappaB signaling: Potential anti-cancer interventions. J. Pharm. Anal. 2022, 12, 394–405. [Google Scholar] [CrossRef]
- Hartley, A.V.; et al. PRMT5-mediated methylation of YBX1 regulates NF-κB activity in colorectal cancer. Sci. Rep. 2020, 10. [Google Scholar]
- Kawaguchi, K.; et al. Aberrant DNA methylation-mediated NF-icB/fatty acid-binding protein 5 (FABP5) feed-forward loop promotes malignancy of colorectal cancer cells. Biochim. Et Biophys. Acta-Mol. Cell Biol. Lipids, 2023; 1868. [Google Scholar]
- Tan, Y.Q.; et al. Tumor suppressor DRD2 facilitates M1 macrophages and restricts NF-κB signaling to trigger pyroptosis in breast cancer. Theranostics 2021, 11, 5214–5231. [Google Scholar] [CrossRef]
- Chen, J.; Liu, A.; Lin, Z.; Wang, B.; Chai, X.; Chen, S.; Lu, W.; Zheng, M.; Cao, T.; Zhong, M.; et al. Downregulation of the circadian rhythm regulator HLF promotes multiple-organ distant metastases in non-small cell lung cancer through PPAR/NF-κb signaling. Cancer Lett. 2020, 482, 56–71. [Google Scholar] [CrossRef]
- Vitali, E.; et al. Pancreatic neuroendocrine tumor progression and resistance to everolimus: the crucial role of NF-kB and STAT3 interplay. J Endocrinol Invest, 2023.
- Lehman, J.M.; Hoeksema, M.D.; Staub, J.; Qian, J.; Harris, B.; Callison, J.C.; Miao, J.; Shi, C.; Eisenberg, R.; Chen, H.; et al. Somatostatin receptor 2 signaling promotes growth and tumor survival in small-cell lung cancer. Int. J. Cancer 2018, 144, 1104–1114. [Google Scholar] [CrossRef]
- Si, Y.; Kim, S.; Ou, J.; Lu, Y.; Ernst, P.; Chen, K.; Whitt, J.; Carter, A.M.; Markert, J.M.; Bibb, J.A.; et al. Anti-SSTR2 antibody-drug conjugate for neuroendocrine tumor therapy. Cancer Gene Ther. 2020, 28, 799–812. [Google Scholar] [CrossRef]
- Torrisani, J.; Hanoun, N.; Laurell, H.; Lopez, F.; Maoret, J.-J.; Souque, A.; Susini, C.; Cordelier, P.; Buscail, L. Identification of an Upstream Promoter of the Human Somatostatin Receptor, hSSTR2, Which Is Controlled by Epigenetic Modifications. Endocrinology 2008, 149, 3137–3147. [Google Scholar] [CrossRef]
- Evans, J.S.; Beaumont, J.; Braga, M.; Masrour, N.; Mauri, F.; Beckley, A.; Butt, S.; Karali, C.S.; Cawthorne, C.; Archibald, S.; et al. Epigenetic potentiation of somatostatin-2 by guadecitabine in neuroendocrine neoplasias as a novel method to allow delivery of peptide receptor radiotherapy. Eur. J. Cancer 2022, 176, 110–120. [Google Scholar] [CrossRef]
- Zhao, M., L. Mishra, and C.X. Deng, The role of TGF-beta/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef]
- Kidd, M.; et al. EGFR/TGFalpha and TGFbeta/CTGF Signaling in Neuroendocrine Neoplasia: Theoretical Therapeutic Targets. Neuroendocrinology 2013, 97, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Wimmel, A., B. Wiedenmann, and S. Rosewicz, Autocrine growth inhibition by transforming growth factor beta-1 (TGFbeta-1) in human neuroendocrine tumour cells. Gut 2003, 52, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Shattuck, T.M.; et al. Mutational analysis of Smad3, a candidate tumor suppressor implicated in TGF-beta and menin pathways, in parathyroid adenomas and enteropancreatic endocrine tumors. J. Clin. Endocrinol. Metab. 2002, 87, 3911–3914. [Google Scholar] [PubMed]
- Sehrawat, A.; et al. SMAD7 enhances adult beta-cell proliferation without significantly affecting beta-cell function in mice. J. Biol. Chem. 2020, 295, 4858–4869. [Google Scholar] [CrossRef] [PubMed]
- Norollahi, S.E.; et al. DNA Methylation Profiling of MYC, SMAD2/3 and DNMT3A in Colorectal Cancer. Oman Med. J. 2021, 36, e315. [Google Scholar] [CrossRef] [PubMed]
- Ansar, M.; Wang, C.-J.; Wang, Y.-H.; Shen, T.-H.; Hung, C.-S.; Chang, S.-C.; Lin, R.-K. SMAD3 Hypomethylation as a Biomarker for Early Prediction of Colorectal Cancer. Int. J. Mol. Sci. 2020, 21, 7395. [Google Scholar] [CrossRef] [PubMed]
- Arnold, C.N.; Sosnowski, A.; Schmitt-Gräff, A.; Arnold, R.; Blum, H.E. Analysis of molecular pathways in sporadic neuroendocrine tumors of the gastro-entero-pancreatic system. Int. J. Cancer 2007, 120, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Gao, Z.; Li, L.; Jiang, X.; Shan, A.; Cai, J.; Peng, Y.; Li, Y.; Jiang, X.; Huang, X.; et al. Whole exome sequencing of insulinoma reveals recurrent T372R mutations in YY1. Nat. Commun. 2013, 4, 2810. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.O.-O.; Kim, S.G.; Bedeir, A.; Issa, J.-P.; Hamilton, S.R.; Rashid, A. CpG island methylation in carcinoid and pancreatic endocrine tumors. Oncogene 2003, 22, 924–934. [Google Scholar] [CrossRef]
- Li, G.; Tian, Y.; Zhu, W.-G. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 576946. [Google Scholar] [CrossRef]
- Klomp, M.; Dalm, S.; de Jong, M.; Feelders, R.; Hofland, J.; Hofland, L. Epigenetic regulation of somatostatin and somatostatin receptors in neuroendocrine tumors and other types of cancer. Rev. Endocr. Metab. Disord. 2020, 22, 495–510. [Google Scholar] [CrossRef] [PubMed]
- Hess, L.; Moos, V.; Lauber, A.A.; Reiter, W.; Schuster, M.; Hartl, N.; Lackner, D.; Boenke, T.; Koren, A.; Guzzardo, P.M.; et al. A toolbox for class I HDACs reveals isoform specific roles in gene regulation and protein acetylation. PLOS Genet. 2022, 18, e1010376. [Google Scholar] [CrossRef] [PubMed]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef] [PubMed]
- Weichert, W.; Denkert, C.; Noske, A.; Darb-Esfahani, S.; Dietel, M.; Kalloger, S.E.; Huntsman, D.G.; Köbel, M. Expression of Class I Histone Deacetylases Indicates Poor Prognosis in Endometrioid Subtypes of Ovarian and Endometrial Carcinomas. Neoplasia 2008, 10, 1021–1027. [Google Scholar] [CrossRef]
- Mori, M.; Sudo, T.; Mimori, K.; Nishida, N.; Kogo, R.; Iwaya, T.; Tanaka, F.; Shibata, K.; Fujita, H.; Shirouzu, K. Histone deacetylase 1 expression in gastric cancer. Oncol. Rep. 2011, 26, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; et al. Regulation of cyclin B2 expression and cell cycle G2/m transition by menin. J. Biol. Chem. 2010, 285, 18291–18300. [Google Scholar] [CrossRef]
- Baradari, V.; Huether, A.; Höpfner, M.; Schuppan, D.; Scherübl, H. Antiproliferative and proapoptotic effects of histone deacetylase inhibitors on gastrointestinal neuroendocrine tumor cells. Endocrine-Related Cancer 2006, 13, 1237–1250. [Google Scholar] [CrossRef]
- Adler, J.T.; Hottinger, D.G.; Kunnimalaiyaan, M.; Chen, H. Combination Therapy with Histone Deacetylase Inhibitors and Lithium Chloride: A Novel Treatment for Carcinoid Tumors. Ann. Surg. Oncol. 2008, 16, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.; Laddha, S.V.; Tang, L.; Vosburgh, E.; Levine, A.J.; Normant, E.; Sandy, P.; Harris, C.R.; Chan, C.S.; Xu, E.Y. The bromodomain and extra-terminal inhibitor CPI203 enhances the antiproliferative effects of rapamycin on human neuroendocrine tumors. Cell Death Dis. 2014, 5, e1450. [Google Scholar] [CrossRef]
- Arvidsson, Y.; Johanson, V.; Pfragner, R.; Wängberg, B.; Nilsson, O. Cytotoxic Effects of Valproic Acid on Neuroendocrine Tumour Cells. Neuroendocrinology 2015, 103, 578–591. [Google Scholar] [CrossRef]
- Lines, K.E.; Vas Nunes, R.P.; Frost, M.; Yates, C.J.; Stevenson, M.; Thakker, R.V. A MEN1 pancreatic neuroendocrine tumour mouse model under temporal control. Endocr. Connect. 2017, 6, 232–242. [Google Scholar] [CrossRef]
- Yokoyama, A.; Wang, Z.; Wysocka, J.; Sanyal, M.; Aufiero, D.J.; Kitabayashi, I.; Herr, W.; Cleary, M.L. Leukemia Proto-Oncoprotein MLL Forms a SET1-Like Histone Methyltransferase Complex with Menin To Regulate Hox Gene Expression. Mol. Cell. Biol. 2004, 24, 5639–5649. [Google Scholar] [CrossRef]
- Chen, X.; Li, Y.; Zhu, F.; Xu, X.; Estrella, B.; Pazos, M.A.; McGuire, J.T.; Karagiannis, D.; Sahu, V.; Mustafokulov, M.; et al. Context-defined cancer co-dependency mapping identifies a functional interplay between PRC2 and MLL-MEN1 complex in lymphoma. Nat. Commun. 2023, 14, 4259. [Google Scholar] [CrossRef] [PubMed]
- Silva-Figueroa, A.M.; Perrier, N.D. Epigenetic processes in sporadic parathyroid neoplasms. Mol. Cell. Endocrinol. 2018, 469, 54–59. [Google Scholar] [CrossRef]
- Fennell, L.J.; Kane, A.; Liu, C.; McKeone, D.; Fernando, W.; Su, C.; Bond, C.; Jamieson, S.; Dumenil, T.; Patch, A.-M.; et al. APC Mutation Marks an Aggressive Subtype of BRAF Mutant Colorectal Cancers. Cancers 2020, 12, 1171. [Google Scholar] [CrossRef] [PubMed]
- Masui, K.; Tanaka, K.; Akhavan, D.; Babic, I.; Gini, B.; Matsutani, T.; Iwanami, A.; Liu, F.; Villa, G.R.; Gu, Y.; et al. mTOR Complex 2 Controls Glycolytic Metabolism in Glioblastoma through FoxO Acetylation and Upregulation of c-Myc. Cell Metab. 2013, 18, 726–739. [Google Scholar] [CrossRef]
- Harachi, M.; Masui, K.; Honda, H.; Muragaki, Y.; Kawamata, T.; Cavenee, W.K.; Mischel, P.S.; Shibata, N. Dual Regulation of Histone Methylation by mTOR Complexes Controls Glioblastoma Tumor Cell Growth via EZH2 and SAM. Mol. Cancer Res. 2020, 18, 1142–1152. [Google Scholar] [CrossRef] [PubMed]
- Leslie, N.R.; et al. The regulation of cell migration by PTEN. Biochem Soc. Trans. 2005, 33, 1507–1508. [Google Scholar] [CrossRef] [PubMed]
- Fusco, N.; et al. Alterations and Their Role in Cancer Management: Are We Making Headway on Precision Medicine? Genes 2020, 11. [Google Scholar]
- Okumura, K.; Mendoza, M.; Bachoo, R.M.; DePinho, R.A.; Cavenee, W.K.; Furnari, F.B. PCAF Modulates PTEN Activity. J. Biol. Chem. 2006, 281, 26562–26568. [Google Scholar] [CrossRef]
- Meng, Z.; Jia, L.-F.; Gan, Y.-H. PTEN activation through K163 acetylation by inhibiting HDAC6 contributes to tumour inhibition. Oncogene 2015, 35, 2333–2344. [Google Scholar] [CrossRef]
- Zhu, Q.-Y.; He, Z.-M.; Cao, W.-M.; Li, B. The role of TSC2 in breast cancer: a literature review. Front. Oncol. 2023, 13. [Google Scholar] [CrossRef]
- Uysal, S.P.; Şahi̇n, M. Tuberous sclerosis: a review of the past, present, and future. Turk. J. Med Sci. 2020, 50, 1665–1676. [Google Scholar] [CrossRef]
- Sauter, M.; Belousova, E.; Benedik, M.P.; Carter, T.; Cottin, V.; Curatolo, P.; Dahlin, M.; D’amato, L.; D’augères, G.B.; de Vries, P.J.; et al. Rare manifestations and malignancies in tuberous sclerosis complex: findings from the TuberOus SClerosis registry to increAse disease awareness (TOSCA). Orphanet J. Rare Dis. 2021, 16, 301. [Google Scholar] [CrossRef]
- Geng, H.; Liu, Q.; Xue, C.; David, L.L.; Beer, T.M.; Thomas, G.V.; Dai, M.-S.; Qian, D.Z. HIF1α Protein Stability Is Increased by Acetylation at Lysine 709. J. Biol. Chem. 2012, 287, 35496–35505. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, C.; Feldman, M.J.; Wang, H.; Pang, Y.; Maggio, D.M.; Zhu, D.; Nesvick, C.L.; Dmitriev, P.; Bullova, P.; et al. Vorinostat suppresses hypoxia signaling by modulating nuclear translocation of hypoxia inducible factor 1 alpha. Oncotarget 2017, 8, 56110–56125. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, K.; Feng, X.; Wang, J.; Chu, Y.; Jia, C.; He, Q.; Chen, C. PRMT3 promotes tumorigenesis by methylating and stabilizing HIF1α in colorectal cancer. Cell Death Dis. 2021, 12, 1066. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; et al. Regulation of RAS oncogenicity by acetylation. Proc. Natl. Acad. Sci. USA 2012, 109, 10843–10848. [Google Scholar] [CrossRef]
- Hałasa, M.; Wawruszak, A.; Przybyszewska, A.; Jaruga, A.; Guz, M.; Kałafut, J.; Stepulak, A.; Cybulski, M. H3K18Ac as a Marker of Cancer Progression and Potential Target of Anti-Cancer Therapy. Cells 2019, 8, 485. [Google Scholar] [CrossRef] [PubMed]
- Donninger, H.; Clark, J.; Rinaldo, F.; Nelson, N.; Barnoud, T.; Schmidt, M.L.; Hobbing, K.R.; Vos, M.D.; Sils, B.; Clark, G.J. The RASSF1A Tumor Suppressor Regulates XPA-Mediated DNA Repair. Mol. Cell. Biol. 2015, 35, 277–287. [Google Scholar] [CrossRef]
- Dubois, F.; et al. RASSF1A Suppresses the Invasion and Metastatic Potential of Human Non-Small Cell Lung Cancer Cells by Inhibiting YAP Activation through the GEF-H1/RhoB Pathway. Cancer Res. 2016, 76, 1627–1640. [Google Scholar] [CrossRef]
- Patel, A.J.; Liao, C.-P.; Chen, Z.; Liu, C.; Wang, Y.; Le, L.Q. BET Bromodomain Inhibition Triggers Apoptosis of NF1-Associated Malignant Peripheral Nerve Sheath Tumors through Bim Induction. Cell Rep. 2013, 6, 81–92. [Google Scholar] [CrossRef]
- Brosseau, J.-P.; Liao, C.-P.; Le, L.Q. Translating current basic research into future therapies for neurofibromatosis type 1. Br. J. Cancer 2020, 123, 178–186. [Google Scholar] [CrossRef]
- Agarwal, S.K.; Jothi, R. Genome-Wide Characterization of Menin-Dependent H3K4me3 Reveals a Specific Role for Menin in the Regulation of Genes Implicated in MEN1-Like Tumors. PLOS ONE 2012, 7, e37952. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Qi, Y.; Fowlkes, N.; Lazic, N.; Su, X.; Lozano, G.; Wasylishen, A.R. The histone chaperone function of Daxx is dispensable for embryonic development. Cell Death Dis. 2023, 14, 1–9. [Google Scholar] [CrossRef]
- Hoelper, D.; Huang, H.; Jain, A.Y.; Patel, D.J.; Lewis, P.W. Structural and mechanistic insights into ATRX-dependent and -independent functions of the histone chaperone DAXX. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Mahmud, I.; Liao, D. DAXX in cancer: phenomena, processes, mechanisms and regulation. Nucleic Acids Res. 2019, 47, 7734–7752. [Google Scholar] [CrossRef] [PubMed]
- Brehm, A.; Miska, E.A.; McCance, D.J.; Reid, J.L.; Bannister, A.J.; Kouzarides, T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature 1998, 391, 597–601. [Google Scholar] [CrossRef]
- Puri, P.L.; Iezzi, S.; Stiegler, P.; Chen, T.-T.; Schiltz, R.; Muscat, G.E.; Giordano, A.; Kedes, L.; Wang, J.Y.; Sartorelli, V. Class I Histone Deacetylases Sequentially Interact with MyoD and pRb during Skeletal Myogenesis. Mol. Cell 2001, 8, 885–897. [Google Scholar] [CrossRef]
- Huang, H.; Fu, Y.; Zhang, Y.; Peng, F.; Lu, M.; Feng, Y.; Chen, L.; Chen, Z.; Li, M.; Chen, Y. Dissection of Anti-tumor Activity of Histone Deacetylase Inhibitor SAHA in Nasopharyngeal Carcinoma Cells via Quantitative Phosphoproteomics. Front. Cell Dev. Biol. 2020, 8, 577784. [Google Scholar] [CrossRef]
- Zhou, Y.K.; et al. HDAC5 Loss Impairs RB Repression of Pro-Oncogenic Genes and Confers CDK4/6 Inhibitor Resistance in Cancer. Cancer Res. 2021, 81, 1486–1499. [Google Scholar] [CrossRef] [PubMed]
- Chun, P. Histone deacetylase inhibitors in hematological malignancies and solid tumors. Arch. Pharmacal Res. 2015, 38, 933–949. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; et al. Trichostatin A, a histone deacetylase inhibitor, suppresses proliferation and epithelial-mesenchymal transition in retinal pigment epithelium cells. J. Cell Mol. Med. 2014, 18, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Mrakovcic, M.; Kleinheinz, J.; Fröhlich, L.F. p53 at the Crossroads between Different Types of HDAC Inhibitor-Mediated Cancer Cell Death. Int. J. Mol. Sci. 2019, 20, 2415. [Google Scholar] [CrossRef] [PubMed]
- Juengel, E.; Nowaz, S.; Makarevi, J.; Natsheh, I.; Werner, I.; Nelson, K.; Reiter, M.; Tsaur, I.; Mani, J.; Harder, S.; et al. HDAC-inhibition counteracts everolimus resistance in renal cell carcinoma in vitro by diminishing cdk2 and cyclin A. Mol. Cancer 2014, 13, 152. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kao, G.D.; Garcia, B.A.; Shabanowitz, J.; Hunt, D.F.; Qin, J.; Phelan, C.; Lazar, M.A. A novel histone deacetylase pathway regulates mitosis by modulating Aurora B kinase activity. Genes Dev. 2006, 20, 2566–2579. [Google Scholar] [CrossRef] [PubMed]
- Maggi, E.C.; Trillo-Tinoco, J.; Struckhoff, A.P.; Vijayaraghavan, J.; Del Valle, L.; Crabtree, J.S. Retinoblastoma-binding protein 2 (RBP2) is frequently expressed in neuroendocrine tumors and promotes the neoplastic phenotype. Oncogenesis 2016, 5, e257. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Roeder, R.G. Activation of p53 Sequence-Specific DNA Binding by Acetylation of the p53 C-Terminal Domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; et al. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. Embo J. 2001, 20, 1331–1340. [Google Scholar] [CrossRef]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Indispensable for p53 Activation. Cell 2008, 133, 612–626. [Google Scholar] [CrossRef]
- Kloster, M.M.; Naderi, E.H.; Haaland, I.; Gjertsen, B.T.; Blomhoff, H.K.; Naderi, S. cAMP signalling inhibits p53 acetylation and apoptosis via HDAC and SIRT deacetylases. Int. J. Oncol. 2013, 42, 1815–1821. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Pan, M.; Sun, J.; Lu, H.; Shen, Q.; Zhang, S.; Jiang, T.; Liu, L.; Jin, W.; Chen, Y.; et al. Histone deacetylase 3 inhibits expression of PUMA in gastric cancer cells. J. Mol. Med. 2012, 91, 49–58. [Google Scholar] [CrossRef]
- Chi, X.Z.; et al. RUNX3 suppresses gastric epithelial cell growth by inducing p21(WAF1/Cip1) expression in cooperation with transforming growth factor beta-activated SMAD. Mol. Cell Biol. 2005, 25, 8097–8107. [Google Scholar] [CrossRef]
- Fröhlich, L.F.; Mrakovcic, M.; Smole, C.; Zatloukal, K. Molecular mechanism leading to SAHA-induced autophagy in tumor cells: evidence for a p53-dependent pathway. Cancer Cell Int. 2016, 16, 68. [Google Scholar] [CrossRef]
- Montani, M.S.G.; Granato, M.; Santoni, C.; Del Porto, P.; Merendino, N.; D’orazi, G.; Faggioni, A.; Cirone, M. Histone deacetylase inhibitors VPA and TSA induce apoptosis and autophagy in pancreatic cancer cells. Cell. Oncol. 2017, 40, 167–180. [Google Scholar] [CrossRef]
- Jang, S.; Jin, H.; Roy, M.; Ma, A.L.; Gong, S.; Jaskula-Sztul, R.; Chen, H. Antineoplastic effects of histone deacetylase inhibitors in neuroendocrine cancer cells are mediated through transcriptional regulation of Notch1 by activator protein 1. Cancer Med. 2017, 6, 2142–2152. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T., N. Rindtorff, and M. Boutros, Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-Y.; Huang, G.-D.; Chen, L.; Zhang, C.; Chen, B.-D.; Li, Q.-Z.; Wang, X.; Zhang, X.-J.; Li, W.-P. Tanshinone IIA induces apoptosis via inhibition of Wnt/β-catenin/MGMT signaling in AtT-20 cells. Mol. Med. Rep. 2017, 16, 5908–5914. [Google Scholar] [CrossRef] [PubMed]
- Götze, S.; Coersmeyer, M.; Müller, O.; Sievers, S. Histone deacetylase inhibitors induce attenuation of Wnt signaling and TCF7L2 depletion in colorectal carcinoma cells. Int. J. Oncol. 2014, 45, 1715–1723. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y.; Vallée, J.-N. Curcumin: a therapeutic strategy in cancers by inhibiting the canonical WNT/β-catenin pathway. J. Exp. Clin. Cancer Res. 2019, 38, 1–16. [Google Scholar] [CrossRef]
- Chen, J. and L.F. Chen, Methods to detect NF-kappaB acetylation and methylation. Methods Mol. Biol. 2015, 1280, 395–409. [Google Scholar]
- Chen, L.F., Y. Mu, and W.C. Greene, Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J. 2002, 21, 6539–6548. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; et al. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell 2009, 15, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Wanek, J.; Gaisberger, M.; Beyreis, M.; Mayr, C.; Helm, K.; Primavesi, F.; Jäger, T.; Di Fazio, P.; Jakab, M.; Wagner, A.; et al. Pharmacological Inhibition of Class IIA HDACs by LMK-235 in Pancreatic Neuroendocrine Tumor Cells. Int. J. Mol. Sci. 2018, 19, 3128. [Google Scholar] [CrossRef] [PubMed]
- Veenstra, M.J.; van Koetsveld, P.M.; Dogan, F.; Farrell, W.E.; Feelders, R.A.; Lamberts, S.W.; de Herder, W.W.; Vitale, G.; Hofland, L.J. Epidrug-induced upregulation of functional somatostatin type 2 receptors in human pancreatic neuroendocrine tumor cells. Oncotarget 2016, 9, 14791–14802. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; et al. Valproic acid induces NET cell growth arrest and enhances tumor suppression of the receptor-targeted peptide-drug conjugate via activating somatostatin receptor type II. J. Drug Target. 2016, 24, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Ryu, T.Y.; Jung, E.; Han, T.-S.; Lee, J.; Kim, S.-K.; Na Roh, Y.; Lee, M.-S.; Jung, C.-R.; Lim, J.H.; et al. Epigenetic regulation of SMAD3 by histone methyltransferase SMYD2 promotes lung cancer metastasis. Exp. Mol. Med. 2023, 55, 952–964. [Google Scholar] [CrossRef]
- Tang, Y.-N.; Ding, W.-Q.; Guo, X.-J.; Yuan, X.-W.; Wang, D.-M.; Song, J.-G. Epigenetic regulation of Smad2 and Smad3 by profilin-2 promotes lung cancer growth and metastasis. Nat. Commun. 2015, 6, 8230. [Google Scholar] [CrossRef]
- Du, D.; et al. Smad3-mediated recruitment of the methyltransferase SETDB1/ESET controls Snail1 expression and epithelial-mesenchymal transition. EMBO Rep. 2018, 19, 135–155. [Google Scholar] [CrossRef]
- Lin, W.; Watanabe, H.; Peng, S.; Francis, J.M.; Kaplan, N.; Pedamallu, C.S.; Ramachandran, A.; Agoston, A.; Bass, A.J.; Meyerson, M. Dynamic Epigenetic Regulation by Menin During Pancreatic Islet Tumor Formation. Mol. Cancer Res. 2015, 13, 689–698. [Google Scholar] [CrossRef]
- Wang, F.; Xu, X.; Ye, Z.; Qin, Y.; Yu, X.; Ji, S. Prognostic Significance of Altered ATRX/DAXX Gene in Pancreatic Neuroendocrine Tumors: A Meta-Analysis. Front. Endocrinol. 2021, 12, 691557. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.W.; Elsaesser, S.J.; Noh, K.-M.; Stadler, S.C.; Allis, C.D. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. 2010, 107, 14075–14080. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.J.; Hostetter, G.; Macfarlane, A.W.; Madaj, Z.; Ross, E.A.; Hinoue, T.; Kulchycki, J.R.; Burgos, R.S.; Tafseer, M.; Alpaugh, R.K.; et al. A Phase II Trial of Guadecitabine plus Atezolizumab in Metastatic Urothelial Carcinoma Progressing after Initial Immune Checkpoint Inhibitor Therapy. Clin. Cancer Res. 2023, 29, 2052–2065. [Google Scholar] [CrossRef] [PubMed]
- Goodall, G.J. and V.O. Wickramasinghe, RNA in cancer. Nat. Rev. Cancer 2021, 21, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Moschovis, D. " Long non-coding RNA in pancreatic adenocarcinoma and pancreatic neuroendocrine tumors". Ann. Gastroenterol. 2017, 30, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Moschovis, D.; Vasilaki, E.; Tzouvala, M.; Karamanolis, G.; Katifelis, H.; Legaki, E.; Vezakis, A.; Aravantinos, G.; Gazouli, M. Association between genetic polymorphisms in long non-coding RNAs and pancreatic cancer risk. Cancer Biomarkers 2019, 24, 117–123. [Google Scholar] [CrossRef]
- Ji, M.; et al. Long noncoding RNA-mRNA expression profiles and validation in pancreatic neuroendocrine neoplasms. Clin. Endocrinol. 2020, 92, 312–322. [Google Scholar] [CrossRef]
- Hwang, S.; Jeong, J.J.; Kim, S.H.; Chung, Y.J.; Song, S.Y.; Lee, Y.J.; Rhee, Y. Differential expression of miRNA199b-5p as a novel biomarker for sporadic and hereditary parathyroid tumors. Sci. Rep. 2018, 8, 12016. [Google Scholar] [CrossRef]
- Prinz, F.; Kapeller, A.; Pichler, M.; Klec, C. The Implications of the Long Non-Coding RNA NEAT1 in Non-Cancerous Diseases. Int. J. Mol. Sci. 2019, 20, 627. [Google Scholar] [CrossRef]
- Ding, N.; Wu, H.; Tao, T.; Peng, E. NEAT1 regulates cell proliferation and apoptosis of ovarian cancer by miR-34a-5p/BCL2. OncoTargets Ther. 2017, ume 10, 4905–4915. [Google Scholar] [CrossRef]
- Ouyang, M.; Su, W.; Xiao, L.; Rao, J.N.; Jiang, L.; Li, Y.; Turner, D.J.; Gorospe, M.; Wang, J.-Y. Modulation by miR-29b of intestinal epithelium homoeostasis through the repression of menin translation. Biochem. J. 2015, 465, 315–323. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, Y.; Mehta, K.R.; Danila, D.C.; Scolavino, S.; Johnson, S.R.; Klibanski, A. A Pituitary-Derived MEG3 Isoform Functions as a Growth Suppressor in Tumor Cells. J. Clin. Endocrinol. Metab. 2003, 88, 5119–5126. [Google Scholar] [CrossRef]
- Zhang, Y.; et al. Long noncoding RNA MEG3 inhibits breast cancer growth via upregulating endoplasmic reticulum stress and activating NF-kappaB and p53. J. Cell Biochem. 2019, 120, 6789–6797. [Google Scholar] [CrossRef]
- Zhang, Y.-Y.; Feng, H.-M. MEG3 Suppresses Human Pancreatic Neuroendocrine Tumor Cells Growth and Metastasis by Down-Regulation of Mir-183. Cell. Physiol. Biochem. 2017, 44, 345–356. [Google Scholar] [CrossRef]
- Modali, S.D.; Parekh, V.I.; Kebebew, E.; Agarwal, S.K. Epigenetic Regulation of the lncRNA MEG3 and Its Target c-MET in Pancreatic Neuroendocrine Tumors. Mol. Endocrinol. 2015, 29, 224–237. [Google Scholar] [CrossRef]
- Luzi, E.; Marini, F.; Giusti, F.; Galli, G.; Cavalli, L.; Brandi, M.L. The Negative Feedback-Loop between the Oncomir Mir-24-1 and Menin Modulates the Men1 Tumorigenesis by Mimicking the “Knudson’s Second Hit”. PLOS ONE 2012, 7, e39767. [Google Scholar] [CrossRef] [PubMed]
- Luzi, E.; Marini, F.; Ciuffi, S.; Galli, G.; Brandi, M.L. An autoregulatory network between menin and pri-miR-24-1 is required for the processing of its specific modulator miR-24-1 in BON1 cells. Mol. Biosyst. 2016, 12, 1922–1928. [Google Scholar] [CrossRef]
- Marini, F.; Brandi, M.L. Role of miR-24 in Multiple Endocrine Neoplasia Type 1: A Potential Target for Molecular Therapy. Int. J. Mol. Sci. 2021, 22, 7352. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Xue, J.; Yang, S.; Chen, Y.; Wang, Y.; Zhu, Y.; Wang, X.; Kuang, D.; Ruan, Q.; Duan, Y.; et al. Co-targeting of IGF1R/mTOR pathway by miR-497 and miR-99a impairs hepatocellular carcinoma development. Oncotarget 2017, 8, 47984–47997. [Google Scholar] [CrossRef]
- Li, Z.; et al. Knockdown of lncRNA-HOTAIR downregulates the drug-resistance of breast cancer cells to doxorubicin via the PI3K/AKT/mTOR signaling pathway. Exp. Ther. Med. 2019, 18, 435–442. [Google Scholar]
- Yang, Y.; Sun, D.; Yu, J.; Zhang, M.; Yi, C.; Yang, R.; Dan, B.; Li, A. Long noncoding RNA TUG1 promotes renal cell carcinoma cell proliferation, migration and invasion by downregulating microRNA-196a. Mol. Med. Rep. 2018, 18, 5791–5798. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; et al. miR-144/451 Promote Cell Proliferation via Targeting PTEN/AKT Pathway in Insulinomas. Endocrinology 2015, 156, 2429–2439. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.; Yao, Y.; Liu, A.; Shi, L.; Chen, D.; Tang, L.; Yang, G.; Liang, X.; Peng, J.; Shao, C. lncRNA H19 binds VGF and promotes pNEN progression via PI3K/AKT/CREB signaling. Endocrine-Related Cancer 2019, 26, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Wei, N.; Ma, R.; Jiang, S.; Song, D. A miR-210-3p regulon that controls the Warburg effect by modulating HIF-1α and p53 activity in triple-negative breast cancer. Cell Death Dis. 2020, 11, 731. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.; et al. Transcription factor HIF1alpha promotes proliferation, migration, and invasion of cholangiocarcinoma via long noncoding RNA H19/microRNA-612/Bcl-2 axis. Transl. Res. 2020, 224, 26–39. [Google Scholar] [CrossRef]
- Derry, M.M.; Raina, K.; Balaiya, V.; Jain, A.K.; Shrotriya, S.; Huber, K.M.; Serkova, N.J.; Agarwal, R.; Agarwal, C. Grape Seed Extract Efficacy against Azoxymethane-Induced Colon Tumorigenesis in A/J Mice: Interlinking miRNA with Cytokine Signaling and Inflammation. Cancer Prev. Res. 2013, 6, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Shih, C.-H.; Chuang, L.-L.; Tsai, M.-H.; Chen, L.-H.; Chuang, E.Y.; Lu, T.-P.; Lai, L.-C. Hypoxia-Induced MALAT1 Promotes the Proliferation and Migration of Breast Cancer Cells by Sponging MiR-3064-5p. Front. Oncol. 2021, 11, 658151. [Google Scholar] [CrossRef] [PubMed]
- Thorns, C.; Schurmann, C.; Gebauer, N.; Wallaschofski, H.; Kümpers, C.; Bernard, V.; Feller, A.C.; Keck, T.; Habermann, J.K.; Begum, N.; et al. Global microRNA profiling of pancreatic neuroendocrine neoplasias. . 2014, 34, 2249–2254. [Google Scholar] [PubMed]
- Huang, X.; Ding, L.; Bennewith, K.L.; Tong, R.T.; Welford, S.M.; Ang, K.K.; Story, M.; Le, Q.-T.; Giaccia, A.J. Hypoxia-Inducible mir-210 Regulates Normoxic Gene Expression Involved in Tumor Initiation. Mol. Cell 2009, 35, 856–867. [Google Scholar] [CrossRef]
- Pizzini, S.; Bisognin, A.; Mandruzzato, S.; Biasiolo, M.; Facciolli, A.; Perilli, L.; Rossi, E.; Esposito, G.; Rugge, M.; Pilati, P.; et al. Impact of microRNAs on regulatory networks and pathways in human colorectal carcinogenesis and development of metastasis. BMC Genom. 2013, 14, 589. [Google Scholar] [CrossRef]
- Zhao, Q.; Chen, S.; Zhu, Z.; Yu, L.; Ren, Y.; Jiang, M.; Weng, J.; Li, B. miR-21 promotes EGF-induced pancreatic cancer cell proliferation by targeting Spry2. Cell Death Dis. 2018, 9, 1157. [Google Scholar] [CrossRef] [PubMed]
- Sugito, N.; Heishima, K.; Akao, Y. Chemically modified MIR143-3p exhibited anti-cancer effects by impairing the KRAS network in colorectal cancer cells. Mol. Ther. - Nucleic Acids 2022, 30, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Tang, X.; Duan, S. Interference from LncRNA SPRY4-IT1 restrains the proliferation, migration, and invasion of melanoma cells through inactivating MAPK pathway by up-regulating miR-22-3p. Int. J. Clin. Exp. Pathol. 2019, 12, 477–487.
- Feichtenschlager, V.; Zheng, Y.J.; Ho, W.; Chen, L.; Callanan, C.; Chen, C.; Lee, A.; Ortiz, J.; Rappersberger, K.; Ortiz-Urda, S. Deconstructing the role of MALAT1 in MAPK-signaling in melanoma: insights from antisense oligonucleotide treatment. Oncotarget 2023, 14, 543–560. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; et al. miR-431 Promotes Metastasis of Pancreatic Neuroendocrine Tumors by Targeting DAB2 Interacting Protein, a Ras GTPase Activating Protein Tumor Suppressor. Am. J. Pathol. 2020, 190, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Q.; Luo, N.; Liu, J.; Ren, H.; Shao, X.; Zhang, L.; Yu, Y. MicroRNA-1269a Promotes Proliferation and Arrest of Apoptosis of Glioma Cells by Directly Targeting ATRX. Front. Oncol. 2020, 10, 563901. [Google Scholar] [CrossRef]
- Liu, X.; Abraham, J.M.; Cheng, Y.; Wang, Z.; Wang, Z.; Zhang, G.; Ashktorab, H.; Smoot, D.T.; Cole, R.N.; Boronina, T.N.; et al. Synthetic Circular RNA Functions as a miR-21 Sponge to Suppress Gastric Carcinoma Cell Proliferation. Mol. Ther. - Nucleic Acids 2018, 13, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Medeiros, N.; Warneford-Thomson, R.; Wulfridge, P.; Yan, Q.; Bian, J.; Sidoli, S.; Garcia, B.A.; Skordalakes, E.; Joyce, E.; et al. Disruption of ATRX-RNA interactions uncovers roles in ATRX localization and PRC2 function. Nat. Commun. 2020, 11, 2219. [Google Scholar] [CrossRef]
- Gill, P.; Kim, E.; Chua, T.C.; Clifton-Bligh, R.J.; Nahm, C.B.; Mittal, A.; Gill, A.J.; Samra, J.S. MiRNA-3653 Is a Potential Tissue Biomarker for Increased Metastatic Risk in Pancreatic Neuroendocrine Tumours. Endocr. Pathol. 2019, 30, 128–133. [Google Scholar] [CrossRef]
- Cai, K.; Wang, Y.; Bao, X. MiR-106b promotes cell proliferation via targeting RB in laryngeal carcinoma. J. Exp. Clin. Cancer Res. 2011, 30, 73. [Google Scholar] [CrossRef]
- Bandi, N.; Vassella, E. miR-34a and miR-15a/16 are co-regulated in non-small cell lung cancer and control cell cycle progression in a synergistic and Rb-dependent manner. Mol. Cancer 2011, 10, 55–55. [Google Scholar] [CrossRef]
- Ohtsuka, M.; et al. H19 Noncoding RNA, an Independent Prognostic Factor, Regulates Essential Rb-E2F and CDK8-beta-Catenin Signaling in Colorectal Cancer. EBioMedicine 2016, 13, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-Z.; Cui, H.-P.; Lv, H.-J.; Feng, L. Knockdown of lncRNA PVT1 inhibits retinoblastoma progression by sponging miR-488-3p. Biomed. Pharmacother. 2019, 112, 108627. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; Ou, X.; Sun, K.; Zhou, X.; Li, Y.; Shi, P.; Zhao, Z.; He, Y.; Peng, J.; Xu, J. m6A modification–mediated lncRNA TP53TG1 inhibits gastric cancer progression by regulating CIP2A stability. Cancer Sci. 2022, 113, 4135–4150. [Google Scholar] [CrossRef] [PubMed]
- Idogawa, M.; et al. Prognostic Effect of Long Noncoding RNA NEAT1 Expression Depends on p53 Mutation Status in Cancer. J. Oncol. 2019, 2019, 4368068. [Google Scholar] [PubMed]
- Zhou, Y.; Zhong, Y.; Wang, Y.; Zhang, X.; Batista, D.L.; Gejman, R.; Ansell, P.J.; Zhao, J.; Weng, C.; Klibanski, A. Activation of p53 by MEG3 Non-coding RNA. J. Biol. Chem. 2007, 282, 24731–24742. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Zhang, Y.-Y.; Wu, Y.-Y.; Wang, X.-D.; Wan, L.-H.; Li, L.; Zhou, L.-M. Correlation of over-expressions of miR-21 and Notch-1 in human colorectal cancer with clinical stages. Life Sci. 2014, 106, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ai, F.; Li, X.; Tian, L.; Wang, X.; Shen, S.; Liu, F. MicroRNA-34a suppresses colorectal cancer metastasis by regulating Notch signaling. Oncol. Lett. 2017, 14, 2325–2333. [Google Scholar] [CrossRef] [PubMed]
- Guessous, F.; Zhang, Y.; Kofman, A.; Catania, A.; Li, Y.; Schiff, D.; Purow, B.; Abounader, R. microRNA-34a is tumor suppressive in brain tumors and glioma stem cells. Cell Cycle 2010, 9, 1031–1036. [Google Scholar] [CrossRef]
- Cai, H.; Yao, J.; An, Y.; Chen, X.; Chen, W.; Wu, D.; Luo, B.; Yang, Y.; Jiang, Y.; Sun, D.; et al. LncRNA HOTAIR acts as competing endogenous RNA to control the expression of Notch3 via sponging miR-613 in pancreatic cancer. Oncotarget 2017, 8, 32905–32917. [Google Scholar] [CrossRef]
- He, R.; Zhang, W.; Chen, S.; Liu, Y.; Yang, W.; Li, J. Transcriptional Profiling Reveals the Regulatory Role of DNER in Promoting Pancreatic Neuroendocrine Neoplasms. Front. Genet. 2020, 11, 587402. [Google Scholar] [CrossRef]
- Yu, Y.; et al. MicroRNA-21 induces stemness by downregulating transforming growth factor beta receptor 2 (TGFbetaR2) in colon cancer cells. Carcinogenesis 2012, 33, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; et al. miR-34a screened by miRNA profiling negatively regulates Wnt/beta-catenin signaling pathway in Aflatoxin B1 induced hepatotoxicity. Sci. Rep. 2015, 5, 16732. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; et al. Overexpression of long non-coding RNA HOTAIR leads to chemoresistance by activating the Wnt/beta-catenin pathway in human ovarian cancer. Tumour Biol. 2016, 37, 2057–2065. [Google Scholar] [CrossRef]
- Ling, H.; Spizzo, R.; Atlasi, Y.; Nicoloso, M.; Shimizu, M.; Redis, R.S.; Nishida, N.; Gafà, R.; Song, J.; Guo, Z.; et al. CCAT2, a novel noncoding RNA mapping to 8q24, underlies metastatic progression and chromosomal instability in colon cancer. Genome Res. 2013, 23, 1446–1461. [Google Scholar] [CrossRef]
- Wei, Y.L.; et al. LncNEN885 inhibits epithelial-mesenchymal transition by partially regulation of Wnt/beta-catenin signalling in gastroenteropancreatic neuroendocrine neoplasms. Cancer Sci. 2018, 109, 3139–3148. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; et al. miR-892b Silencing Activates NF-kappaB and Promotes Aggressiveness in Breast Cancer. Cancer Res. 2016, 76, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Jaeger, S.A.; Hirsch, H.A.; Bulyk, M.L.; Struhl, K. STAT3 Activation of miR-21 and miR-181b-1 via PTEN and CYLD Are Part of the Epigenetic Switch Linking Inflammation to Cancer. Mol. Cell 2010, 39, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liu, S.; Cai, G.; Kong, L.; Zhang, T.; Ren, Y.; Wu, Y.; Mei, M.; Zhang, L.; Wang, X. Long Non Coding RNA MALAT1 Promotes Tumor Growth and Metastasis by inducing Epithelial-Mesenchymal Transition in Oral Squamous Cell Carcinoma. Sci. Rep. 2015, 5, 15972. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, X.; Zheng, L.; Li, D.; Liu, Z.; Teng, L. The lncRNA NEAT1 Inhibits miRNA-216b and Promotes Colorectal Cancer Progression by Indirectly Activating YY1. J. Oncol. 2022, 2022, 8130132. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, H.; Kim, H.W.; Lee, J.-C.; Paik, K.-H.; Kang, J.; Kim, J.; Yoon, Y.-S.; Han, H.-S.; Sohn, I.; et al. High Expression of MicroRNA-196a Indicates Poor Prognosis in Resected Pancreatic Neuroendocrine Tumor. Medicine 2015, 94, e2224. [Google Scholar] [CrossRef]
- Døssing, K.B.V.; Kjær, C.; Vikeså, J.; Binderup, T.; Knigge, U.; Culler, M.D.; Kjær, A.; Federspiel, B.; Friis-Hansen, L. Somatostatin Analogue Treatment Primarily Induce miRNA Expression Changes and Up-Regulates Growth Inhibitory miR-7 and miR-148a in Neuroendocrine Cells. Genes 2018, 9, 337. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.; Park, Y.; Kim, J.; Kwon, H.; Kim, T.; Lee, J.; Pyun, J.-C.; Lee, M.; Yun, M. Elevated miR-16-5p induces somatostatin receptor 2 expression in neuroendocrine tumor cells. PLOS ONE 2020, 15, e0240107. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.N.; et al. MiRNAs and LncRNAs: Dual Roles in TGF-beta Signaling-Regulated Metastasis in Lung Cancer. Int. J. Mol. Sci. 2020, 21. [Google Scholar]
- Li, Q.; et al. MiR-21/Smad 7 signaling determines TGF-beta1-induced CAF formation. Sci. Rep. 2013, 3, 2038. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Li, M.; Li, J.; Lv, Y. Inhibition mechanism of lung cancer cell metastasis through targeted regulation of Smad3 by miR-15a. Oncol. Lett. 2019, 19, 1516–1522. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; et al. A Transforming Growth Factor-beta and H19 Signaling Axis in Tumor-Initiating Hepatocytes That Regulates Hepatic Carcinogenesis. Hepatology 2019, 69, 1549–1563. [Google Scholar] [CrossRef] [PubMed]
- Vijayaraghavan, J.; Maggi, E.C.; Crabtree, J.S. miR-24 regulates menin in the endocrine pancreas. Am. J. Physiol. Metab. 2014, 307, E84–E92. [Google Scholar] [CrossRef]
- Yan, X., H. Jia, and J. Zhao, LncRNA MEG3 attenuates the malignancy of retinoblastoma cells through inactivating PI3K /Akt/mTOR signaling pathway. Exp. Eye Res. 2023, 226, 109340. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; et al. miR-144/451 Promote Cell Proliferation via Targeting PTEN/AKT Pathway in Insulinomas. Endocrinology 2015, 156, 2429–2439. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Ding, L.; Bennewith, K.L.; Tong, R.T.; Welford, S.M.; Ang, K.K.; Story, M.; Le, Q.-T.; Giaccia, A.J. Hypoxia-Inducible mir-210 Regulates Normoxic Gene Expression Involved in Tumor Initiation. Mol. Cell 2009, 35, 856–867. [Google Scholar] [CrossRef]
- Blázquez-Encinas, R.; Moreno-Montilla, M.T.; García-Vioque, V.; Gracia-Navarro, F.; Alors-Pérez, E.; Pedraza-Arevalo, S.; Ibáñez-Costa, A.; Castaño, J.P. The uprise of RNA biology in neuroendocrine neoplasms: altered splicing and RNA species unveil translational opportunities. Rev. Endocr. Metab. Disord. 2022, 24, 267–282. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Ma, J.; Hua, X. Epigenetic regulation by the menin pathway. Endocrine-Related Cancer 2017, 24, T147–T159. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Hua, X.; Jin, G. Menin regulates endocrine diseases by controlling histone modification and gene transcription. Ann. d’Endocrinologie 2008, 69, 426–432. [Google Scholar] [CrossRef]
- Ehsanullah, S.; A Trikalinos, N. Synchronous AML and pancreatic neuroendocrine neoplasm, both successfully treated with somatostatin analogs and decitabine. Endocr. Oncol. 2022, 2, K1–K4. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.H.; Menda, Y.; Zamba, K.; Madsen, M.; O’Dorisio, M.S.; O’Dorisio, T.; Bushnell, D. Potential for Increasing Uptake of Radiolabeled 68Ga-DOTATOC and 123I-MIBG in Patients with Midgut Neuroendocrine Tumors Using a Histone Deacetylase Inhibitor Vorinostat. Cancer Biotherapy Radiopharm. 2021, 36, 632–641. [Google Scholar] [CrossRef]
- Doss, H.H.; Jones, S.F.; Infante, J.R.; Spigel, D.R.; Willcutt, N.; Lamar, R.; Barton, J.; Keegan, M.; Burris, H.A. A phase I trial of romidepsin in combination with gemcitabine in patients with pancreatic and other advanced solid tumors. J. Clin. Oncol. 2008, 26, 2567–2567. [Google Scholar] [CrossRef]
- Jin, N.; Lubner, S.J.; Mulkerin, D.L.; Rajguru, S.; Carmichael, L.; Chen, H.; Holen, K.D.; LoConte, N.K. A Phase II Trial of a Histone Deacetylase Inhibitor Panobinostat in Patients With Low-Grade Neuroendocrine Tumors. Oncol. 2016, 21, 785–786g. [Google Scholar] [CrossRef]
- Balasubramaniam, S.; Redon, C.E.; Peer, C.J.; Bryla, C.; Lee, M.-J.; Trepel, J.B.; Tomita, Y.; Rajan, A.; Giaccone, G.; Bonner, W.M.; et al. Phase I trial of belinostat with cisplatin and etoposide in advanced solid tumors, with a focus on neuroendocrine and small cell cancers of the lung. Anti-Cancer Drugs 2018, 29, 457–465. [Google Scholar] [CrossRef]
- Katti, A.; Diaz, B.J.; Caragine, C.M.; Sanjana, N.E.; Dow, L.E. CRISPR in cancer biology and therapy. Nat. Rev. Cancer 2022, 22, 259–279. [Google Scholar] [CrossRef]
- A English, K.; Thakker, R.V.; E Lines, K. The role of DNA methylation in human pancreatic neuroendocrine tumours. Endocr. Oncol. 2023, 3, e230003. [Google Scholar] [CrossRef]
- O’neill, H.; Lee, H.; Gupta, I.; Rodger, E.J.; Chatterjee, A. Single-Cell DNA Methylation Analysis in Cancer. Cancers 2022, 14, 6171. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, T.; et al. STC-1 Cells, in The Impact of Food Bioactives on Health: in vitro and ex vivo models, K. Verhoeckx, et al., Editors. 2015: Cham (CH). p. 211-220.
- Zeng, Z.; Wong, C.J.; Yang, L.; Ouardaoui, N.; Li, D.; Zhang, W.; Gu, S.; Zhang, Y.; Liu, Y.; Wang, X.; et al. TISMO: syngeneic mouse tumor database to model tumor immunity and immunotherapy response. Nucleic Acids Res. 2021, 50, D1391–D1397. [Google Scholar] [CrossRef]
- Ney, A.; Canciani, G.; Hsuan, J.J.; Pereira, S.P. Modelling Pancreatic Neuroendocrine Cancer: From Bench Side to Clinic. Cancers 2020, 12, 3170. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Sandilos, G.; Williamson, J.; Emery, R.; Platoff, R.; Joneja, U.; Acharya, N.K.; Lin, A.; Badach, J.; Zilberman, B.; et al. Novel treatment strategy of targeting epigenetic dysregulation in pancreatic neuroendocrine tumors. Surgery 2023, 173, 1045–1051. [Google Scholar] [CrossRef]
- Wang, H.; Peng, R.; Wang, J.; Qin, Z.; Xue, L. Circulating microRNAs as potential cancer biomarkers: the advantage and disadvantage. Clin. Epigenetics 2018, 10, 59. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; et al. A novel circulating miRNA-based signature for the early diagnosis and prognosis prediction of non-small-cell lung cancer. J. Clin. Lab. Anal. 2020, 34, e23505. [Google Scholar] [CrossRef]
Figure 1.
Schematic of epigenetic modifications of signaling pathways in PNET pathogenesis. Pathways are color-coded based on whether they are tumor-suppressor or oncogenic. Additional colors were added to indicate whether the regulatory mechanism of the signal molecules belong to DNA methylation, histone modifications, or ncRNAs in PNETs. For the enhancement of clarity and ease of understanding, specific protein molecules were organized with canonical pathways, even when these molecules are not typically mentioned alongside the canonical pathways. One such example is the inclusion of TSC in the mTOR pathway, based on correlations identified in previously published studies [24].
Figure 1.
Schematic of epigenetic modifications of signaling pathways in PNET pathogenesis. Pathways are color-coded based on whether they are tumor-suppressor or oncogenic. Additional colors were added to indicate whether the regulatory mechanism of the signal molecules belong to DNA methylation, histone modifications, or ncRNAs in PNETs. For the enhancement of clarity and ease of understanding, specific protein molecules were organized with canonical pathways, even when these molecules are not typically mentioned alongside the canonical pathways. One such example is the inclusion of TSC in the mTOR pathway, based on correlations identified in previously published studies [24].
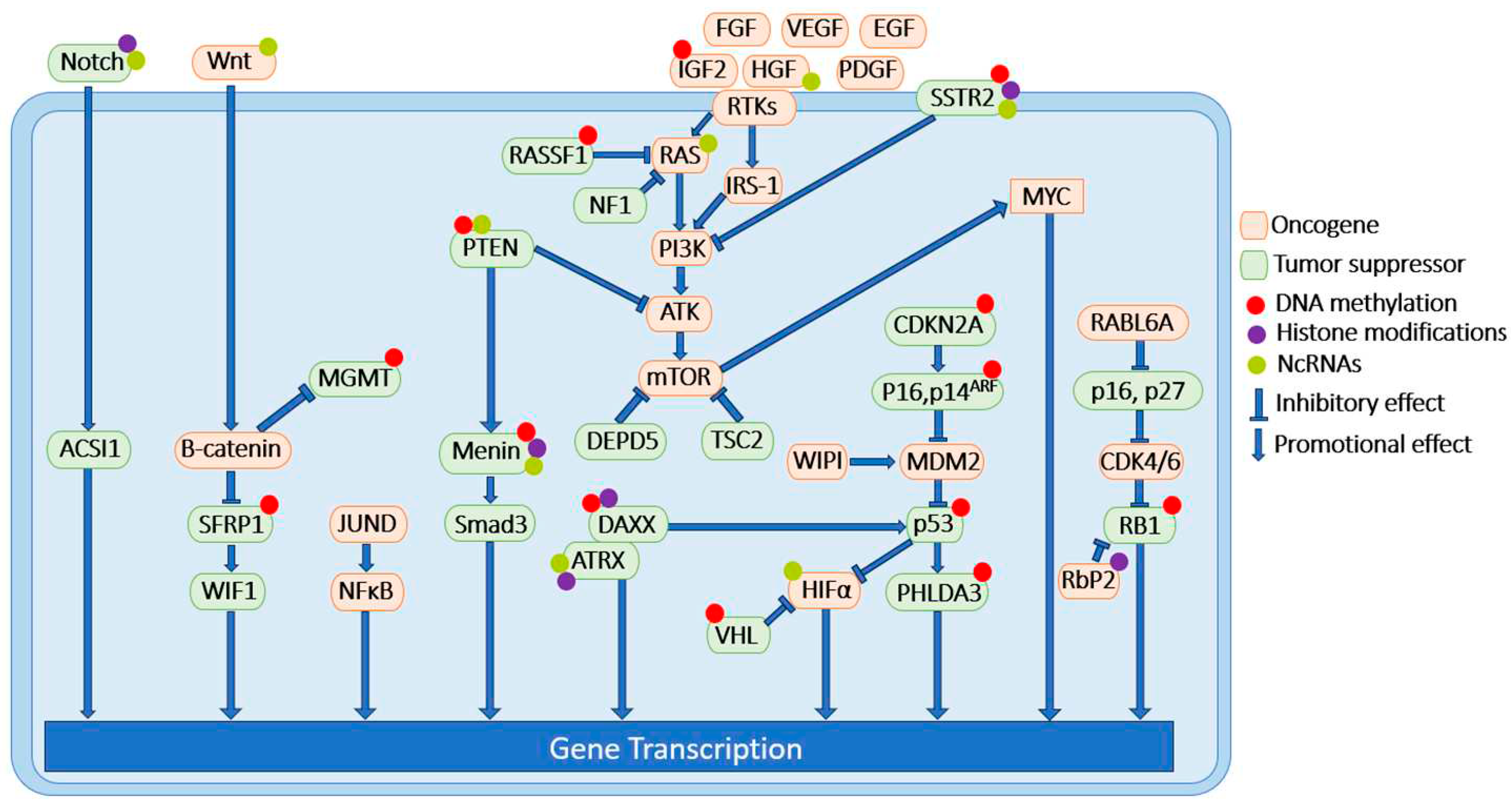

Table 1.
DNA methylation modifications in PNET. 1 RTKs include IGF, FGF, VEGF, EGF, HGF, PDGF.
| Signaling pathway | Signaling molecules | DNA methylation status of promoter region | Experimental systems |
|---|---|---|---|
| MEN1 | MEN1 | Hypermethylated | human PNET samples [141] |
| PTEN/PI3K/AKT/mTOR/c-Myc/TSC/RTK1 | PTEN | Hypermethylated | human PNET samples [41] |
| TSC | no change | human PNET samples [41] | |
| IGF2 | hypermethylated | human PNET samples [142] | |
| HIF-1α/VHL | VHL | Hypermethylated | human PNET samples [50] |
| RAS/MAPK/NF1 | RASSF1 | Hypermethylated | human PNET samples [70] |
| ALT/DAXX/ATRX | DAXX | Hypermethylated | human PNET samples [78] |
| CDKN2A/RB1 | CDKN2A | Hypermethylated | human PNET samples [88] |
| P16, P14ARF | Hypermethylated | human PNET samples [143] | |
| P27 | no change | human PNET samples [90] | |
| RB1 | Hypermethylated | human PNET samples [41] | |
| P53 | P53 | Hypermethylated | human PNET samples [41] |
| PHLDA3 | Hypermethylated | human PNET samples [98] | |
| Wnt/β Catenin/MGMT | SFRP1 | Hypermethylated | BON-1, and QGP-1 cell lines [121] |
| WIF1 | no change | BON-1, and QGP-1 cell lines [121] | |
| MGMT | MGMT-promoter methylation status correlates with chemoresistance in well-differentiated PNET. | PNET patient samples[122,123] | |
| SSTR | SSTR2 | SSTR2 promoter is hypermethylated in PNETs comparing to non-NET tissue and is inversely correlated with SSTR2 protein expression. | human PNET samples BON-1, and QGP-1 cell lines xenograft mouse model [133] |
Table 2.
Histone modification status in PNET. 1 RTKs include IGF, FGF, VEGF, EGF, HGF, PDGF.
| Signaling pathway | Signaling molecules | Histone modification status | Experimental systems |
|---|---|---|---|
| MEN1 | MEN1 | Loss of menin causes H3K4me3 loss and sporadic PNET tumors. | PNET patient samples [178] |
| PTEN/PI3K/AKT/mTOR/ c-Myc/TSC/RTK1 |
IGF2 | Genome-wide studies of H3K4 methylation in pancreatic islets indicate that loss of MEN1 alters the epigenetic landscape of its target genes such as insulin like growth factor binding protein 2 (Igf2bp2), p18ink4c (CDKN2C) and p27kip1 (CDKN1B). | Pancreatic islets from MEN1-deficient mice [214] |
| DAXX/ATRX/ALT |
DAXX ATRX |
DAXX and TRX form a histone chaperone complex to deposit histone variant H3.3 at the telomeres and pericentric heterochromatin regions of the genome. They are frequently mutated in PNET samples. | Human PNET samples, Hela cells [215,216] |
| CDKN2A/RB1 | RB1 | Histone demethylase retinoblastoma binding protein 2 (Rbp2) was found overexpressed in PNET tumors. Aberrant expression of Rbp2 altered histone demethylation and contributed to PNET pathogenesis. | PNET patient samples, βlox5 cell, H727 cell, QGP-1 cell [191] |
| Notch | Notch1 | HDAC inhibitor causes increased Notch 1 expression in tumor cells and mouse tumor xenograft[108,217] | BON-1 cells [217], carcinoid cancer cells and mouse tumor xenograft [108] |
| SSTR2 | SSTR2 | Histone acetylation present on SSTR2. In addition, the combination treatment of HDACi (VPA) and camptothecin-somatostatin conjugate significantly reduced tumor growth comparing to monotherapies. | BON-1 and QGP-1 cells [208] [209], BON-1 xenograft mouse model [210] |
Table 3.
NcRNAs in PNET.1 RTKs include IGF, FGF, VEGF, EGF, HGF, PDGF.
| Signaling pathway | Signaling molecules | Non-coding RNA status | Experimental systems |
|---|---|---|---|
| MEN1 | MEN1 | Menin negatively regulates miR-24-1 in a negative feedback loop manner. | BON-1 cells [230,231] |
| MiR-24 negatively regulates menin in endocrine pancreas. | MIN6 cells, βlox5 cells; floxed MEN1 mouse model [282] | ||
| Menin upregulates expression of MEG3. | Mouse insulinoma cells [229] | ||
| PTEN/PI3K/AKT/mTOR/c-Myc/ TSC/PRK1 | PTEN | MEG3 causes decreased p-PI3K, p-AKT, p-mTOR, and smaller tumor size. | human retinoblastoma cells [283] |
| PI3K | miR-144 causes decreased PTEN. | and xenograft mouse model [284] | |
| AKT | MiR-144 correlated with increased p-AKT. | MIN6 cells [236] | |
| mTOR | IncRNA H19 causes increased PI3K-AKT and PNET progression. | Human insulinoma samples, QGP-1, PNET primary Cells.QGP-1 xenograft model [237] | |
| HGF/MET | MEG3 downregulates c-MET in PNET. | MIN6 cells, mouse, PNET patient samples [229]. | |
| HIF-1α/VHL | HIF-1α | MiR-210 expression is positively correlated with PNET progression and was shown to regulate colorectal adenocarcinoma progression through HIF1α. | PNET patient samples [242] |
| FaDu head & neck cancer cell line, SU86.76 pancreatic cancer cell line, Xenograft mouse model [244,285]. | |||
| RAS/MAPK/NF1 | RAS | MiR-431 promotes PNET progression by silencing DAB21P, resulting in the activation of RAS pathway. | QGP-1 cell line, and xenograft mouse model [249]. |
| ALT/DAXX/ATRX | ATRX | ATRX negatively regulates miR-3653, which might serve as a risk factor of metastatic disease in PNET. | Microarray differential expression of human PNET tissue samples [253]. |
| Notch | Notch1,2,3, ASCL1 | LncRNA XLOC_221242 is positively correlated with Notch/Wnt signaling. | PNET patient samples [265,286]. |
| Wnt/β Catenin |
Wnt, β-Catenin, SFRP1, WIF1 |
LncNEN885 negatively regulates Wnt/β-catenin signaling, leading to reduction of EMT in PNET. LncNEN885 is negatively correlated with PNET progression. | BON-1 cells, and PNET patient samples [270]. |
| SSTR | SSTR2 | Upregulation of miR-16-5p induces SSTR2 expression. | INS-1 cell line [277] |
Table 4.
FDA-approved anti-tumor epigenetic drugs and trials in PNET treatment. (DNMTi = DNA methyltransferase inhibitor; HDMi = Histone lysine demethylase inhibitor; HDACi = Histone deacetylase inhibitor; AML = acute myeloid leukemia; CML = chronic myelogenous leukemia; MDS = myelodysplastic syndromes; CTCL = cutaneous T-cell lymphoma; PTCL = peripheral T-cell lymphoma; MM = multiple myeloma; NR = no report; NA = not applicable).
Table 4.
FDA-approved anti-tumor epigenetic drugs and trials in PNET treatment. (DNMTi = DNA methyltransferase inhibitor; HDMi = Histone lysine demethylase inhibitor; HDACi = Histone deacetylase inhibitor; AML = acute myeloid leukemia; CML = chronic myelogenous leukemia; MDS = myelodysplastic syndromes; CTCL = cutaneous T-cell lymphoma; PTCL = peripheral T-cell lymphoma; MM = multiple myeloma; NR = no report; NA = not applicable).
| Drug name | Drug target | Targeted disease | Trial in PNET | Status of trial |
|---|---|---|---|---|
| Azacitidine | DNMTi | AML, CML and MDS | NR | NA |
| 5-Aza-2’-deoxycytidine | DNMTi | AML, CML and MDS | Trial on synchronous AML and PNET | Treatment was successful with combination of somatostatin analogs and decitabine, but with severe side effects [289]. |
| Tazemetostat | HDMi | Advance epithelioid sarcoma | NR | NA |
| Enasidenib | HDMi | AML | NR | NA |
| Vorinostat | HDACi | CTCL | Pilot-imaging study to test efficacy of vorinostat on radionuclide uptake. | Statistically significant increase of radionuclide uptake was observed [290]. |
| Romidepsin | HDACi | CTCL and PTCL | Phase I trial of romidepsin in patients with pancreatic and other advanced solid tumors. | Stable disease status was observed when combined with treatment of gemcitabine [291]. |
| Panobinostat | HDACi | MM | Phase II-trial against low-Grade PNET | 15 patients were in the trial. No response was observed [292]. |
| Belinostat | HDACi | PTCL | Phase I-trial against NET and small cell lung cancer | Partial response was observed when patients were treated with belinostat combined with cisplatin and etoposide [293]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



