
Submitted:
23 December 2023
Posted:
25 December 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2) utilizes Angiotensin Converting Enzyme 2 (ACE2) as its main receptor for cell entry. We have bioengineered a soluble ACE2 protein termed ACE2 618-DDC-ABD that has increased binding to SARS-CoV-2 and prolonged duration of action. Here we investigated the protective effect of this protein when administered intranasally to k18-hACE2 mice infected with the aggressive SARS-CoV-2 delta variant. k18-hACE2 mice were infected with the SARS-CoV-2 delta variant by inoculation of a lethal dose (2x104 PFU) and followed for up to 14 days. ACE2-618-DDC-ABD (10mg/kg) or PBS was administered intranasally six hours prior and 24 and 48 hours post viral inoculation. All animals in the PBS-control group had succumbed to the disease on day 7 post infection (0% survival) whereas, by contrast, there was only one casualty in the group that received ACE2-618-DDC-ABD (90% survival). Mice in the ACE2-618-DDC-ABD group had minimal disease as assessed by a clinical score and stable weight and both brain and lung viral titers were markedly reduced. These findings demonstrate the efficacy of a bioengineered soluble ACE2 decoy with extended duration of action in protecting against the aggressive delta SARS-CoV-2 variant. Together with previous work these findings demonstrate the universal protective potential against current and future emerging SARS-CoV-2 variants.
Keywords:
SARS-CoV-2
; COVID-19
; Angiotensin-Converting-Enzyme 2
; ACE2
1. Introduction
Since its first appearance in Wuhan, China in December 2019, severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2) spread worldwide posing a threat to public health [1,2]. Four years later and despite massive improvements largely because of the vaccination efforts, new cases are frequently reported [3,4]. Omicron variants currently are the most commonly variants seen worldwide [5,6,7]. SARS-CoV-2, like other coronaviruses including its predecessor SARS-CoV-1, uses membrane bound Angiotensin Converting Enzyme 2 (ACE2) as its primary cell entry receptor [8,9,10,11]. The complex multistep process of SARS-CoV-2 cell entry starts with interaction of the receptor binding domain (RBD) of the viral S1 spike protein with ACE2 on the cell surface [12,13,14,15]. Conformational changes then lead to priming of the viral spike protein by proteases like transmembrane protease serine 2 (TMPRSS2) which results in fusion pore formation and ultimately cell infection [9,16,17,18].
At the beginning of the COVID-19 pandemic in early 2020 our group proposed the use of soluble ACE2 proteins to intercept the binding of SARS-CoV-2 to its receptor, membrane bound ACE2, via the so called “decoy effect” [19,20]. We had bioengineered a human soluble ACE2 protein that was shortened from 740 to 618 amino acids and fused with an albumin binding domain (ABD) to increase its duration of action [21]. Later a dodecapeptide (DDC) motif that contains four cysteine residues was introduced to enhance binding affinity for SARS-CoV-2 via formation of a disulfide bonded dimeric protein [21,22]. In human kidney organoids and the k18-hACE2 mouse model of lethal SARS-CoV-2 infection we demonstrated the potential of our bioengineered soluble ACE2 proteins to neutralize SARS-CoV-2, markedly improve survival and provide in-vivo lung, brain and kidney protection [21,22]. We established preventative, intranasal delivery, starting one hour before viral inoculation followed by two additional doses at 24 and 48 hours post viral inoculation, as an effective mode of ACE2-618-DDC-ABD delivery achieving optimal protection in terms of survival and organ injury in the k18-hACE2 mouse model infected with ancestral SARS-CoV-2 [23].
As new variants of SARS-CoV-2 emerge, mutational escape and resistance to treatment are critical issues and both antibodies and antiviral drugs have been shown to be associated with the development of mutational escape of SARS-CoV-2 [24,25]. Mutations of SARS-CoV-2 that would decrease ACE2 decoy affinity while simultaneously maintaining efficacious binding of SARS-CoV-2 to the membrane bound ACE2 receptor have not been described and are unlikely to occur [26,27]. This highlights an advantage of soluble ACE2 proteins over monoclonal antibodies [28]. Studies in permissive cells have shown that soluble ACE2 proteins moreover are effective against different SARS-CoV-2 variants [10,21,22,23,29,30,31]. Here we investigated intranasal administration of ACE2-618-DDC-ABD to k18-hACE2 mice infected with the SARS-CoV-2 delta variant which causes severe disease in humans to further demonstrate the universal efficacy of our unique soluble ACE2 protein. Moreover, ACE2-618-DDC-ABD was applied as early as six hours prior viral inoculation to further determine its lasting protective effect and extended duration of action.
2. Materials and Methods
2.1. In-vivo infectivity studies
All in-vivo infectivity studies were performed at the BSL-3 facility of the Ricketts Regional Biocontainment Laboratory in accordance with a protocol approved by the Institutional Animal Care and Use Committees of Northwestern University (IS00004795) and the University of Chicago (72642).
k18-hACE2 mice were purchased from The Jackson Laboratory at 6-9 weeks of age. k18-hACE2 mice express full-length human ACE2 (hACE2) with an estimated hACE2 gene copy number of 8 and are susceptible to infection with SARS-CoV-2 [22,23,32,33,34,35,36,37]. Animals were infected by intranasal inoculation of 2×104 plaque-forming units (PFU) SARS-CoV-2 delta variant (Isolate hCoV-19/USA/MD-HP05647/2021 Lineage B.1.617.2; Delta variant) in a volume of 20 μl per animal. Disease severity in k18-hACE2 mice infected with the SARS-CoV-2 delta variant has been reported to be similar to or greater than infection with the ancestral virus, where animals typically succumb to disease by days 5-9 post viral inoculation [22,23,32,33,34,35,38,39].
2.2. Administration of ACE2-618-DDC-ABD
For the following experiments two groups of k18-hACE2 mice (n=10 per group, five male and five female each) administered with either ACE2-618-DDC-ABD or phosphate buffered saline (PBS) as vehicle control were investigated. ACE2 618-DDC-ABD was administered intranasally (10 mg/kg BW in a volume of 30 μl per animal) for a total of three doses. The first dose was given six hours before inoculation of the animal with the SARS-CoV-2 delta variant followed by the same dose 24 and 48 hours after viral inoculation. PBS as vehicle control was administered following the same protocol at the same time points.
2.3. Evaluation of body weight and clinical score
Following inoculation of the animals with the SARS-CoV-2 delta variant all mice were monitored for health two to three times a day using a clinical scoring system as previously reported [22,23]. Additionally, animals were weighed once daily throughout the 14-day study period. Mice that lost more than 20% of their initial body weight and did not regain it within two days or had a severely worsened clinical score were humanely euthanized. This was considered a fatal event as per study protocol.
To compare viral titers at similar time points, randomly selected animals of each sex from both groups were euthanized on day six post viral inoculation together with the animals that were euthanized because of disease severity. All remaining mice were monitored for up to 14 days at the BSL-3 facility of the Ricketts Regional Biocontainment Laboratory and euthanized at day 14 at the end of the study protocol if not previously deceased. Mice were euthanized by CO2 forced inhalation. After the last breathing movement, cervical dislocation was performed and lung, brain and kidney tissue were collected for plaque assay analysis (see below).
2.4. Plaque Assay
Lung, brain, and kidney tissue was removed from all euthanized animals and used for viral load measurements by plaque assay. Organs from one mouse in the PBS-control group could not be collected, and the animal was therefore not included in the plaque assay analysis.
Tissue samples for plaque assay analysis were collected in Dulbecco’s Modified Eagle Medium (DMEM) with 2% Fetal Bovine Serum (FBS). Samples were then homogenized and centrifuged at 1000g for five minutes. The supernatant was serially diluted 10-fold and used to infect Vero E6 cells for one hour. Inoculum was removed and 1.25% methylcellulose DMEM solution was added to the cells followed by incubation for three days. Plates were then fixed in 1:10 formalin for one hour, stained with crystal violet for one hour, and counted to determine PFU expressed as PFU/ml.
2.5. Statistical Analysis
Statistical analysis and figure preparation were performed using GraphPad Prism v. 9.5.1. Normal distribution was tested using the Shapiro-Wilk test. When data was normally distributed, differences between two groups were analyzed by unpaired t-test. Differences between groups with non-normally distributed data were analyzed by Mann-Whitney test. Differences in survival were analyzed by log-rank (Mantel-Cox) test. A p-value < 0.05 was considered significant.
3. Results:
3.1. Survival of k18-hACE2 mice infected with SARS-CoV-2 delta variant
All infected k18-hACE2 mice that received PBS either died or had to be humanely euthanized by day seven post viral inoculation as per the study protocol (Figure 1, black). In contrast, survival of the mice that received ACE2-618-DDC-ABD was 90% on day six post viral inoculation. Only one mouse in this group had to be humanely euthanized on day six post viral inoculation (Figure 1, purple). To compare viral titers at similar time points, five healthy mice from the ACE2 618-DDC-ABD treated group were sacrificed on day six post viral inoculation. The remaining four mice in this group all survived until day 14, the end of the study period. This marked difference in survival was statistically significant (P=0.0004, by log-rank (Mantel-Cox) test).
3.2. Clinical score of k18-hACE2 mice infected with SARS-CoV-2 delta variant
All infected animals were evaluated for health by a clinical score two to three times a day (see material and methods section). Infected k18-hACE2 mice that received PBS as vehicle control developed severe disease and clinical scores increased until day six and seven post viral inoculation (Figure 2A, black). At that point all animals in the PBS-vehicle group had either died or had to be humanely euthanized as per the study protocol (Figure 1). Animals that received ACE2-618-DDC-ABD developed less severe disease and clinical scores were lower (Figure 2A, purple). By the end of the 14-day study period all remaining mice in the ACE2-618-DDC-ABD group had recovered.
3.3. Weight loss of k18-hACE2 mice infected with SARS-CoV-2 delta variant
All infected mice were weighed once daily throughout the 14-day study period. Infected mice that received PBS lost up to ~20% of their initial bodyweight until days six and seven post viral inoculation. At this point all mice from the PBS-control group either died or had to be humanly euthanized (Figure 2B, black). Mice that received ACE2-618-DDC-ABD lost on average less than ~10% of their initial body weight by day six post viral inoculation and the remaining mice in this group recovered stable body weight towards the end of the 14-day study period (Figure 2B, purple).
3.4. Lung virial titers of k18-hACE2 mice infected with SARS-CoV-2 delta variant
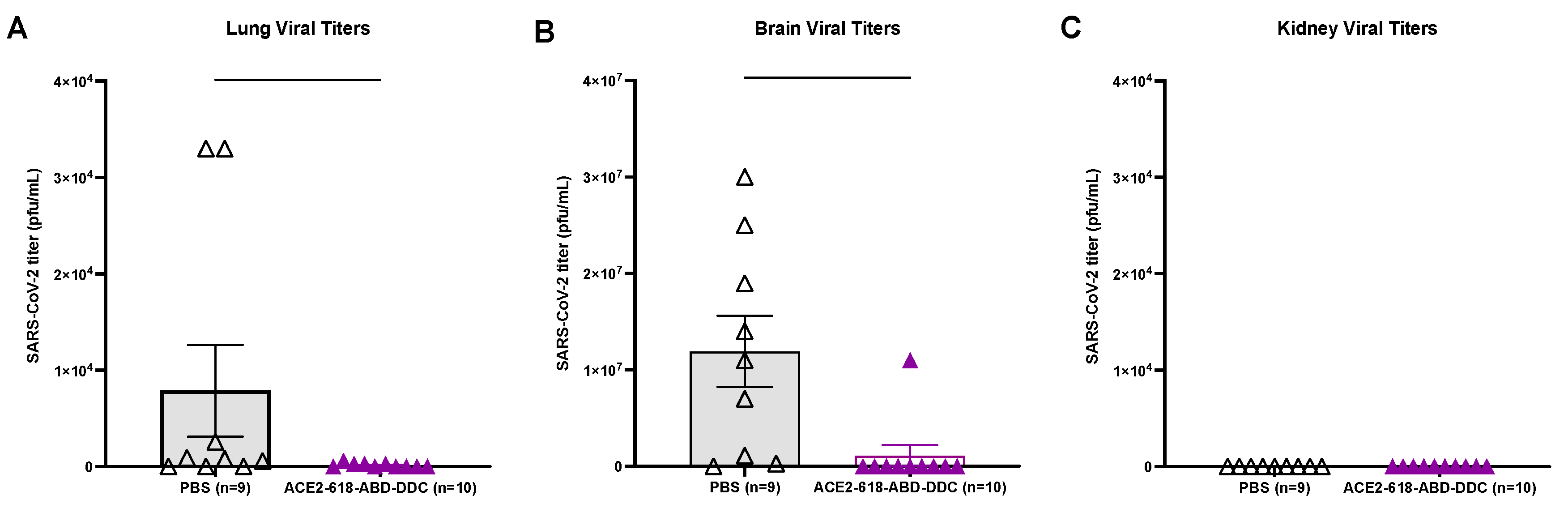
Lung viral titers were assessed in all ten mice that received ACE2-618-DDC-ABD and in nine mice that received PBS. Lung viral titers in infected animals that received ACE2-618-DDC-ABD were reduced as compared to infected mice in the PBS-control group (1.5x102 ± 0.7x102 vs. 7.8x103 ± 4.7x103 PFU/ml, P=0.04) (Figure 3A). When only comparing lung viral titers in animals sacrificed at the same time point on day six post viral inoculation (n=6 per group), a marked reduction in the ACE2-618-DDC-ABD treated group was seen as well (2.5x102 ± 0.9x102 vs. 1.18x104 ± 6.7x103 PFU/ml, P=0.004). Of note, lung viral titers were completely undetectable in all remaining animals of the ACE2-618-DDC-ABD treated group at the end of the 14-day study period.
3.5. Brain viral titers of k18-hACE2 mice infected with SARS-CoV-2 delta variant
Brain viral titers were assessed in all ten mice that received ACE2-618-DDC-ABD and nine mice that received PBS. Infected animals in the PBS-control group had very high brain viral titers, whereas brain viral titers were markedly reduced in infected mice that received ACE2-618-DDC-ABD (1.1x106 ± 1.1x106 vs. 1.2x107 ± 3.7x106 PFU/ml, P=0.0007) (Figure 3B). When only comparing brain viral titers in animals sacrificed at the same time point on day six post viral inoculation (n=6 per group), a marked reduction in the ACE2-618-DDC-ABD treated group was seen as well (1.8x106 ± 1.8x106 vs. 1.4x107 ± 5.0x106 PFU/ml, P=0.008). Similar to the data for lung viral titers, brain viral titers were completely undetectable in all remaining animals of the ACE2-618-DDC-ABD treated group at the end of the 14-day study period.
Of note, when directly comparing viral titers in brains to lungs from the PBS-control group, brain viral titers were about 1,000-fold higher than lung viral titers (1,2x107 ± 3.7x106 vs. 7.8x103 ± 4.7x103 PFU/ml, P=0.004). In fact, several of the infected, untreated mice had only slightly increased lung viral titers (see Figure 3A).
3.6. Kidney viral titers of k18-hACE2 mice infected with SARS-CoV-2 delta variant
Kidney viral titers were assessed in all ten mice that received ACE2-618-DDC-ABD and in nine mice that received PBS. No kidney viral titers could be detected in either the PBS-control or the ACE2-618-DDC-ABD treated group (Figure 3C). The limit of detection for plaque assay is 102 PFU/ml.
4. Discussion
This study demonstrates a dramatic protective effect of the soluble ACE2 decoy protein termed ACE2-618-DDC-ABD in the lethal k18-hACE2 mouse model of SARS-CoV-2 infection caused by the SARS-CoV-2 delta variant. Animals that received ACE2-618-DDC-ABD six hours prior to as well as 24 and 48 hours after viral inoculation had 90% survival as compared to uniform lethality in the PBS-control group. Disease severity as evaluated by a clinical score and weight loss were markedly improved in the ACE2 618-DDC-ABD treated group. Lung and brain viral titers moreover were markedly reduced in animals receiving ACE2-618-DDC-ABD as compared to the PBS-control group. At the end of the study period on day 14 post viral inoculation all remaining animals in the ACE2-618-DDC-ABD treated group had no detectable lung and brain viral titers Taken together with our previous studies in the same mouse model infected with the ancestral SARS-CoV-2 variant (Washington isolate) [22,23] our data demonstrates remarkable efficacy of ACE2-618-DDC-ABD in preventing mortality and organ infectivity in the aggressive delta variant. Furthermore, we also show that intranasal administration of ACE2-618-DDC-ABD prior to viral inoculation results in a protective effect when administered at least six hours prior to viral inoculation which expands our previous work when ACE2-618-DDC-ABD was administered close to viral inoculation with the ancestral SARS-CoV-2 variant (one hour before).
The SARS-CoV-2 delta variant first emerged in October 2020 and was shown to cause severe disease in humans [40,41,42,43]. Thirteen amino acid mutations have been found in the viral spike protein of the SARS-CoV-2 delta variant as compared to the ancestral virus genome [38]. Of particular interest are the mutations L452R, T478K, P681R, and D614G, which have been reported to promote infectivity and immune escape by increasing the stability of the spike protein and RBD-ACE2 interaction, as well as facilitating cleavage of the spike precursor protein and promoting viral replication and transmission [44,45,46,47,48,49,50,51,52]. Infection of k18-hACE2 mice with the SARS-CoV-2 delta variant induces distinct pathogenic patterns of respiratory disease [38,39]. As compared to infection with the SARS-CoV-2 alpha variant significantly more inflammation, increased production of antiviral cytokines and a greater number of activated immunological pathways have been demonstrated which underlines the uniqueness of the host response against different SARS-CoV-2 variants [39]. Of note, we found that in k18-hACE2 mice infected with the SARS-CoV-2 delta variant brain viral titers were markedly higher than lung viral titers. This is in agreement with our findings in the same model infected with the ancestral virus variant and strongly suggests that the high lethality is due to brain viral invasion [23].
We have previously reported that ACE2-618-DDC-ABD, administered to k18-hACE2 mice both intranasally and systemically one hour before as well as 24 and 48 hours after viral inoculation with the wildtype SARS-CoV-2 variant markedly improved survival and mitigated disease severity as assessed by clinical score and weight [22]. ACE2-618-DDC-ABD also provided in-vivo lung and kidney protection and reduced lung and brain viral titers [22]. As recently demonstrated by Hassler et al. the best protective effect can be achieved when administering ACE2-618-DDC-ABD intranasally to k18-hACE2 mice both before and after viral inoculation which is superior to systemic administration at the same time-points [23]. Administration of ACE2 618-DDC-ABD only post viral inoculation provided only partial protection as compared to the untreated control group and was markedly less efficacious than administration of ACE2 618-DDC-ABD both before and after viral inoculation [23]. Specifically, intranasal administration of ACE2-618-DDC-ABD before and after viral inoculation resulted in 90% survival on day 6 post viral inoculation, brain viral titers were undetectable in all mice, and lung and brain histopathology were essentially normal [23]. In the present study that followed the same protocol of intranasal ACE2-618-DDC-ABD administration brain viral titers were similarly undetectable in the ACE2-618-DDC-ABD treated group except for the one animal that had to be humanly euthanized and considered a fatality on day six post viral inoculation. This further supports the idea that brain invasion is likely the main cause of lethality in this model.
The present study was aimed at evaluating the protective effect of ACE2-618-DDC-ABD in k18-hACE2 mice infected with the SARS-CoV-2 delta variant to establish the universality of the in-vivo protective effect of this ACE2 decoy protein. In previous in-vitro studies ACE2-618-DDC-ABD exerted a neutralizing effect on infection of Vero E6 cells by the SARS-CoV-2 wildtype, delta and gamma variants at high concentrations [10,22,23]. Infection of A549 cells with the SARS-CoV-2 omicron BA.1 variant was neutralized by 20-fold lower concentrations of ACE2 618-DDC-ABD than those required to neutralize the wildtype variant [23]. Some SARS-CoV-2 variants, like omicron BA.1, may therefore be neutralized by markedly lower concentrations of ACE2 618-ABD-DDC.
The previous in-vitro cell studies together with the in-vivo data in this study show a universal protective effect of ACE2-618-DDC-ABD against SARS-CoV-2 variants. Mutational changes during viral evolution may lead to the development of resistance to current treatment approaches [24]. Strategies that have been employed to combat SARS-CoV-2 include monoclonal antibodies and specific antiviral drugs [53,54,55]. For those approaches applied in clinical practice the development of mutations that compromise treatment efficacy has been described. In-vitro studies have demonstrated the development of resistance towards the antiviral drugs Remdesivir and Paxlovid [56,57,58]. The emergence of resistant mutations has moreover been shown when in-vitro passing the virus in the presence of SARS-CoV-2 specific monoclonal antibodies highlighting the potential of rapidly occurring mutational changes [28,54,59]. Further studies have shown the development of resistant mutations in patients infected with the SARS-CoV-2 delta and omicron variants that received the monoclonal antibody Sotrovimab [60,61,62]. Mutations causing resistance against the antibody cocktails Casirivimab/Imdevimab (REGN-CoV-2) and Bamlanivimab/Etesevimab (LY-CoV555/LY-CoV016), moreover, have also been shown [63,64]. These drugs consists of two monoclonal antibodies targeting distinct epitopes [65,66,67], which is particularly interesting as combination therapies are typically less prone to mutational escape and decreased treatment efficiency [68,69,70,71]. The possibility of mutational escape has also been described for polyclonal antibodies, although unlike monoclonal antibodies a variety of different epitopes is being targeted [72,73].
In contrast to monoclonal antibodies, in-vitro passaging of SARS-CoV-2 in the presence of a soluble ACE2 decoy protein did not lead to the development of resistant mutations [28]. Importantly, saturation mutagenesis of the SARS-CoV-2 RBD followed by in vitro selection with full-length ACE2 and an engineered ACE2 decoy protein did not show any RBD mutations that discriminated in favor of full-length ACE2 as compared to the decoy protein [26]. Although the development of mutations that compromise the affinity for ACE2 decoy binding is theoretically possible, ACE2 decoy proteins clearly have less susceptibility to resistance caused by SARS-CoV-2 mutations [26,27]. Mutations that decrease decoy affinity would simultaneously decrease affinity for full-length membrane bound ACE2 which is essential for cell entry and infection of all currently known SARS-CoV-2 variants [8,9,10]. In fact, mutational escape from the neutralizing effect of ACE2 decoy proteins would therefore come at the expense of diminished viral infectivity and virulence [26,27].
During the viral evolution different mutations have been described that increase affinity of the SARS-CoV-2 RBD for ACE2 receptor binding as for example the amino acid exchanges N501Y in the SARS-CoV-2 alpha and beta variants as well as L452R in the SARS-CoV-2 delta variant [44,74,75]. The currently dominant circulating SARS-CoV-2 omicron variants also harbor various spike protein mutations leading to increased ACE2 receptor binding while efficiently evading neutralizing antibodies [76,77,78]. Consistent with this, as we have previously shown and noted above, concentrations of ACE2-618-DDC-ABD required to neutralize the SARS-CoV-2 omicron BA.1 variant in-vitro are significantly lower than those needed to neutralize the wild-type variant [23]. Of note, while this might be explained by higher binding affinity of the SARS-CoV-2 RBD to ACE2, in-vitro studies with SARS-CoV-2 omicron variants have also shown decreased fusion and syncytium formation which may in turn result in lower concentrations being effective for neutralization of the SARS-CoV-2 omicron variant than those needed for other SARS-CoV-2 variants [79,80,81].
The present study also shows the absence of kidney SARS-CoV-2 titers in k18-hACE2 mice infected with the SARS-CoV-2 delta variant. While renal involvement has been recognized as a frequent complication of COVID-19 with increased occurrence of acute kidney injury and associated higher mortality in hospitalized COVID-19 patients, the contributing molecular mechanisms remain unclear [82,83,84,85]. Direct SARS-CoV-2 infection and viral replication in the kidney parenchyma has been discussed as ACE2 is highly expressed in many cell types present in the kidney like proximal tubular cells, podocytes, mesangial and endothelial cells [86,87,88]. SARS-CoV-2 has been shown to directly infect human kidney organoids and the presence of SARS-CoV-2 in kidney parenchyma of patients with COVID-19 has been demonstrated by methods like immunohistochemistry (IHC), immunofluorescence (IF), real-time PCR, and single cell RNA sequencing [89]. This data, however, originates mainly from autopsies where the postmortem interval from death to autopsy ranges from several hours to days during which SARS-CoV-2 could spread to and within the kidney post-mortem [89]. Many studies have shown negative findings regarding the presence of SARS-CoV-2 in the kidney including a large series of kidney biopsy samples (n=284) of patients with COVID-19 where no direct kidney SARS-CoV-2 infection could be demonstrated [89,90]. Moreover, results from our group in the k18-hACE2 mouse model infected with the ancestral SARS-CoV-2 variant also showed lack of evidence for kidney invasion [22,91]. Kidney tissue of k18hACE2 mice infected with the ancestral SARS-CoV-2 variant showed no detectable viral titers by plaque assay as well as no evidence for viral spike and nucleoprotein by IHC and IF staining [22,91]. Further analysis using single molecule fluorescence in situ hybridization was also negative for SARS-CoV-2 RNA [91]. The absence of kidney viral titers in k18-hACE2 mice infected with the delta SARS-CoV-2 variant supports the findings from our previous study. Considering conflicting data throughout the literature, the question whether direct kidney invasion by SARS-CoV-2 occurs remains not fully elucidated. Multiple factors, such as timing between confirmation of SARS-CoV-2 infection and obtaining of the kidney sample, the type of sample obtained (biopsy or autopsy) as well as sensitivity of the methods to detect potential presence of SARS-CoV-2 may influence the detection of SARS-CoV-2 in the kidney [89,92].
In conclusion, the present study demonstrates the efficacy of ACE2-618-DDC-ABD to protect k18-hACE2 mice from lethal infection with the aggressive SARS CoV-2 delta variant. Animals that received ACE2-618-DDC-ABD before and after viral inoculation had significantly improved survival, mitigated weight loss and improved clinical scores. Lung and brain SARS-CoV-2 titers, moreover, were significantly reduced in the ACE2 618-DDC-ABD treated group. Importantly, brain viral titers were much higher than lung viral titers and appear much more likely to be the cause of lethality in the k18-hACE2 model. Taken together with previous in-vivo experiments with wildtype SARS-CoV-2 and in-vitro data on the neutralizing effect on various variants including wildtype, gamma, delta, and omicron BA.1 [22,23], this study demonstrates the universality of the protective effect of ACE2-618-DDC-ABD against infection with SARS-CoV-2 variants. Its therapeutic and preventive potential in humans should be investigated for current and future emerging coronaviruses that use ACE2 as their main receptor for cell entry.
Author Contributions
Grant support to D Batlle from NIH (1R21 AI166940-01) and a gift from the Joseph and Bessie Feinberg Foundation. L Hassler and C Cianfarini were supported by the Biomedical Education Program during a part of their stay in Chicago. A/BSL3 research management at the Howard T. Ricketts Laboratory is supported by NIAID award UC7AI180312 under the leadership of Dr. D. Missiakas. Isolate hCoV-19/USA/MD-HP05647/2021 (Lineage B.1.617.2; Delta variant), NR-55672, was obtained through BEI Resources, NIAID, NIH: SARS-Related Coronavirus 2, and contributed by Dr. Andrew S. Pekos.
Acknowledgments
Grant support to D Batlle from NIH (1R21 AI166940-01) and a gift from the Joseph and Bessie Feinberg Foundation. L Hassler and C Cianfarini were supported by the Biomedical Education Program during a part of their stay in Chicago. A/BSL3 research management at the Howard T. Ricketts Laboratory is supported by NIAID award UC7AI180312 under the leadership of Dr. D. Missiakas. Isolate hCoV-19/USA/MD-HP05647/2021 (Lineage B.1.617.2; Delta variant), NR-55672, was obtained through BEI Resources, NIAID, NIH: SARS-Related Coronavirus 2, and contributed by Dr. Andrew S. Pekos.
Conflicts of Interest
D Batlle and J Wysocki are coinventors of patents entitled “Active Low Molecular Weight Variants of Angiotensin Converting Enzyme 2 (ACE2),” and “Soluble ACE2 Variants and Uses therefor.” D Batlle is founder of Angiotensin Therapeutics Inc. D Batlle has received consulting fees from Advicenne unrelated to this work and received unrelated research support from a grant from AstraZeneca; G Randall reports consultancy agreements with Optikira. J Wysocki and J Henkin report scientific advisor capacity for Angiotensin Therapeutics Inc. All remaining authors have nothing to disclose related to this publication.
References
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Barouch, D.H. Covid-19 Vaccines - Immunity, Variants, Boosters. N. Engl. J. Med. 2022, 387, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Tregoning, J.S.; Flight, K.E.; Higham, S.L.; Wang, Z.; Pierce, B.F. Progress of the COVID-19 vaccine effort: viruses, vaccines and variants versus efficacy, effectiveness and escape. Nat. Rev. Immunol. 2021, 21, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Karim, S.S.A.; Karim, Q.A. Omicron SARS-CoV-2 variant: a new chapter in the COVID-19 pandemic. Lancet 2021, 398, 2126–2128. [Google Scholar] [CrossRef]
- Tegally, H.; Moir, M.; Everatt, J.; Giovanetti, M.; Scheepers, C.; Wilkinson, E.; Subramoney, K.; Makatini, Z.; Moyo, S.; Amoako, D.G.; et al. Emergence of SARS-CoV-2 Omicron lineages BA.4 and BA.5 in South Africa. Nat. Med. 2022, 28, 1785–1790. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J. Virol. 2020, 94, e00127–00120. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Batlle, D.; Monteil, V.; Garreta, E.; Hassler, L.; Wysocki, J.; Chandar, V.; Schwartz, R.E.; Mirazimi, A.; Montserrat, N.; Bader, M.; Penninger, J.M. Evidence in favor of the essentiality of human cell membrane-bound ACE2 and against soluble ACE2 for SARS-CoV-2 infectivity. Cell 2022, 185, 1837–1839. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; Wang, X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Glowacka, I.; Bertram, S.; Muller, M.A.; Allen, P.; Soilleux, E.; Pfefferle, S.; Steffen, I.; Tsegaye, T.S.; He, Y.; Gnirss, K.; et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 2011, 85, 4122–4134. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Nagata, N.; Shirato, K.; Kawase, M.; Takeda, M.; Taguchi, F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J. Virol. 2010, 84, 12658–12664. [Google Scholar] [CrossRef]
- Shulla, A.; Heald-Sargent, T.; Subramanya, G.; Zhao, J.; Perlman, S.; Gallagher, T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J. Virol. 2011, 85, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Batlle, D.; Wysocki, J.; Satchell, K. Soluble angiotensin-converting enzyme 2: a potential approach for coronavirus infection therapy? Clin. Sci. 2020, 134, 543–545. [Google Scholar] [CrossRef]
- Davidson, A.M.; Wysocki, J.; Batlle, D. Interaction of SARS-CoV-2 and Other Coronavirus With ACE (Angiotensin-Converting Enzyme)-2 as Their Main Receptor: Therapeutic Implications. Hypertension 2020, 76, 1339–1349. [Google Scholar] [CrossRef]
- Wysocki, J.; Ye, M.; Hassler, L.; Gupta, A.K.; Wang, Y.; Nicoleascu, V.; Randall, G.; Wertheim, J.A.; Batlle, D. A novel soluble ACE2 variant with prolonged duration of action neutralizes SARS-CoV-2 infection in human kidney organoids. J. Am. Soc. Nephrol. 2021, 32, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Hassler, L.; Wysocki, J.; Gelarden, I.; Sharma, I.; Tomatsidou, A.; Ye, M.; Gula, H.; Nicoleascu, V.; Randall, G.; Pshenychnyi, S.; et al. A Novel Soluble ACE2 Protein Provides Lung and Kidney Protection in Mice Susceptible to Lethal SARS-CoV-2 Infection. J. Am. Soc. Nephrol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Hassler, L.; Wysocki, J.; Ahrendsen, J.T.; Ye, M.; Gelarden, I.; Nicolaescu, V.; Tomatsidou, A.; Gula, H.; Cianfarini, C.; Forster, P.; et al. Intranasal soluble ACE2 improves survival and prevents brain SARS-CoV-2 infection. Life Sci. Alliance 2023, 6. [Google Scholar] [CrossRef] [PubMed]
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; Consortium, C.-G.U.; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J.; Robertson, D.L. SARS-CoV-2 variant biology: immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef]
- Chan, K.K.; Tan, T.J.C.; Narayanan, K.K.; Procko, E. An engineered decoy receptor for SARS-CoV-2 broadly binds protein S sequence variants. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Qian, K.; Zhang, S.; Fu, W.; Zhao, J.; Lei, C.; Hu, S. Engineered soluble ACE2 receptor: Responding to change with change. Front. Immunol. 2022, 13, 1084331. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, Y.; Suzuki, T.; Arimori, T.; Ikemura, N.; Mihara, E.; Kirita, Y.; Ohgitani, E.; Mazda, O.; Motooka, D.; Nakamura, S.; et al. Engineered ACE2 receptor therapy overcomes mutational escape of SARS-CoV-2. Nat. Commun. 2021, 12, 3802. [Google Scholar] [CrossRef]
- Hofmann, H.; Geier, M.; Marzi, A.; Krumbiegel, M.; Peipp, M.; Fey, G.H.; Gramberg, T.; Pohlmann, S. Susceptibility to SARS coronavirus S protein-driven infection correlates with expression of angiotensin converting enzyme 2 and infection can be blocked by soluble receptor. Biochem. Biophys. Res. Commun. 2004, 319, 1216–1221. [Google Scholar] [CrossRef]
- Lei, C.; Qian, K.; Li, T.; Zhang, S.; Fu, W.; Ding, M.; Hu, S. Neutralization of SARS-CoV-2 spike pseudotyped virus by recombinant ACE2-Ig. Nat. Commun. 2020, 11, 2070. [Google Scholar] [CrossRef]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkruys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Hurtado Del Pozo, C.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.S.; Bailey, A.L.; Kafai, N.M.; Nair, S.; McCune, B.T.; Yu, J.; Fox, J.M.; Chen, R.E.; Earnest, J.T.; Keeler, S.P.; et al. SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat. Immunol. 2020, 21, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Wong, L.R.; Li, K.; Verma, A.K.; Ortiz, M.E.; Wohlford-Lenane, C.; Leidinger, M.R.; Knudson, C.M.; Meyerholz, D.K.; McCray, P.B., Jr.; Perlman, S. COVID-19 treatments and pathogenesis including anosmia in K18-hACE2 mice. Nature 2021, 589, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Golden, J.W.; Cline, C.R.; Zeng, X.; Garrison, A.R.; Carey, B.D.; Mucker, E.M.; White, L.E.; Shamblin, J.D.; Brocato, R.L.; Liu, J.; et al. Human angiotensin-converting enzyme 2 transgenic mice infected with SARS-CoV-2 develop severe and fatal respiratory disease. JCI Insight 2020, 5, e142032. [Google Scholar] [CrossRef]
- Oladunni, F.S.; Park, J.G.; Pino, P.A.; Gonzalez, O.; Akhter, A.; Allue-Guardia, A.; Olmo-Fontanez, A.; Gautam, S.; Garcia-Vilanova, A.; Ye, C.; et al. Lethality of SARS-CoV-2 infection in K18 human angiotensin-converting enzyme 2 transgenic mice. Nat. Commun. 2020, 11, 6122. [Google Scholar] [CrossRef] [PubMed]
- Moreau, G.B.; Burgess, S.L.; Sturek, J.M.; Donlan, A.N.; Petri Jr, W.A.; Mann, B.J. Evaluation of K18-hACE2 mice as a model of SARS-CoV-2 infection. Am. J. Trop. Med. Hyg. 2020, 103, 1215–1219. [Google Scholar] [CrossRef]
- Rathnasinghe, R.; Strohmeier, S.; Amanat, F.; Gillespie, V.L.; Krammer, F.; García-Sastre, A.; Coughlan, L.; Schotsaert, M.; Uccellini, M.B. Comparison of transgenic and adenovirus hACE2 mouse models for SARS-CoV-2 infection. Emerg. Microbes Infect. 2020, 9, 2433–2445. [Google Scholar] [CrossRef]
- Liu, X.; Mostafavi, H.; Ng, W.H.; Freitas, J.R.; King, N.J.C.; Zaid, A.; Taylor, A.; Mahalingam, S. The Delta SARS-CoV-2 Variant of Concern Induces Distinct Pathogenic Patterns of Respiratory Disease in K18-hACE2 Transgenic Mice Compared to the Ancestral Strain from Wuhan. mBio 2022, 13, e0068322. [Google Scholar] [CrossRef]
- Lee, K.S.; Wong, T.Y.; Russ, B.P.; Horspool, A.M.; Miller, O.A.; Rader, N.A.; Givi, J.P.; Winters, M.T.; Wong, Z.Y.A.; Cyphert, H.A.; et al. SARS-CoV-2 Delta variant induces enhanced pathology and inflammatory responses in K18-hACE2 mice. PLoS ONE 2022, 17, e0273430. [Google Scholar] [CrossRef]
- Singh, J.; Rahman, S.A.; Ehtesham, N.Z.; Hira, S.; Hasnain, S.E. SARS-CoV-2 variants of concern are emerging in India. Nat. Med. 2021, 27, 1131–1133. [Google Scholar] [CrossRef]
- McCrone, J.T.; Hill, V.; Bajaj, S.; Pena, R.E.; Lambert, B.C.; Inward, R.; Bhatt, S.; Volz, E.; Ruis, C.; Dellicour, S.; et al. Context-specific emergence and growth of the SARS-CoV-2 Delta variant. Nature 2022, 610, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Fisman, D.N.; Tuite, A.R. Evaluation of the relative virulence of novel SARS-CoV-2 variants: a retrospective cohort study in Ontario, Canada. CMAJ 2021, 193, E1619–E1625. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.; McMenamin, J.; Taylor, B.; Robertson, C.; Public Health, S.; the, E.I.I.C. SARS-CoV-2 Delta VOC in Scotland: demographics, risk of hospital admission, and vaccine effectiveness. Lancet 2021, 397, 2461–2462. [Google Scholar] [CrossRef] [PubMed]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Saito, A.; Nasser, H.; Tan, T.S.; Ngare, I.; Kimura, I.; Uriu, K.; Kosugi, Y.; et al. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe 2021, 29, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; VanBlargan, L.A.; Bloyet, L.M.; Rothlauf, P.W.; Chen, R.E.; Stumpf, S.; Zhao, H.; Errico, J.M.; Theel, E.S.; Liebeskind, M.J.; et al. Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe 2021, 29, 477–488. [Google Scholar] [CrossRef]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182, 1284–1294. [Google Scholar] [CrossRef]
- Callaway, E. The mutation that helps Delta spread like wildfire. Nature 2021, 596, 472–473. [Google Scholar] [CrossRef] [PubMed]
- Di Giacomo, S.; Mercatelli, D.; Rakhimov, A.; Giorgi, F.M. Preliminary report on severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Spike mutation T478K. J. Med. Virol. 2021, 93, 5638–5643. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H., 3rd; Leist, S.R.; Schafer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2020, 370, 1464–1468. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827. [Google Scholar] [CrossRef]
- Gobeil, S.M.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Manne, K.; Stalls, V.; Kopp, M.F.; Henderson, R.; Edwards, R.J.; et al. D614G Mutation Alters SARS-CoV-2 Spike Conformation and Enhances Protease Cleavage at the S1/S2 Junction. Cell Rep. 2021, 34, 108630. [Google Scholar] [CrossRef] [PubMed]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2021, 592, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Murakami, N.; Hayden, R.; Hills, T.; Al-Samkari, H.; Casey, J.; Del Sorbo, L.; Lawler, P.R.; Sise, M.E.; Leaf, D.E. Therapeutic advances in COVID-19. Nat. Rev. Nephrol. 2023, 19, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Focosi, D.; McConnell, S.; Casadevall, A.; Cappello, E.; Valdiserra, G.; Tuccori, M. Monoclonal antibody therapies against SARS-CoV-2. Lancet Infect. Dis. 2022, 22, e311–e326. [Google Scholar] [CrossRef]
- Tao, K.; Tzou, P.L.; Nouhin, J.; Bonilla, H.; Jagannathan, P.; Shafer, R.W. SARS-CoV-2 Antiviral Therapy. Clin. Microbiol. Rev. 2021, 34, e0010921. [Google Scholar] [CrossRef] [PubMed]
- Szemiel, A.M.; Merits, A.; Orton, R.J.; MacLean, O.A.; Pinto, R.M.; Wickenhagen, A.; Lieber, G.; Turnbull, M.L.; Wang, S.; Furnon, W.; et al. In vitro selection of Remdesivir resistance suggests evolutionary predictability of SARS-CoV-2. PLoS Pathog. 2021, 17, e1009929. [Google Scholar] [CrossRef] [PubMed]
- Stevens, L.J.; Pruijssers, A.J.; Lee, H.W.; Gordon, C.J.; Tchesnokov, E.P.; Gribble, J.; George, A.S.; Hughes, T.M.; Lu, X.; Li, J.; et al. Mutations in the SARS-CoV-2 RNA-dependent RNA polymerase confer resistance to remdesivir by distinct mechanisms. Sci. Transl. Med. 2022, 14, eabo0718. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Gammeltoft, K.A.; Ryberg, L.A.; Pham, L.V.; Tjornelund, H.D.; Binderup, A.; Duarte Hernandez, C.R.; Fernandez-Antunez, C.; Offersgaard, A.; Fahnoe, U.; et al. Nirmatrelvir-resistant SARS-CoV-2 variants with high fitness in an infectious cell culture system. Sci. Adv. 2022, 8, eadd7197. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Arora, P.; Gross, R.; Seidel, A.; Hornich, B.F.; Hahn, A.S.; Kruger, N.; Graichen, L.; Hofmann-Winkler, H.; Kempf, A.; et al. SARS-CoV-2 variants B.1.351 and P.1 escape from neutralizing antibodies. Cell 2021, 184, 2384–2393. [Google Scholar] [CrossRef]
- Birnie, E.; Biemond, J.J.; Appelman, B.; de Bree, G.J.; Jonges, M.; Welkers, M.R.A.; Wiersinga, W.J. Development of Resistance-Associated Mutations After Sotrovimab Administration in High-risk Individuals Infected With the SARS-CoV-2 Omicron Variant. JAMA 2022, 328, 1104–1107. [Google Scholar] [CrossRef]
- Rockett, R.; Basile, K.; Maddocks, S.; Fong, W.; Agius, J.E.; Johnson-Mackinnon, J.; Arnott, A.; Chandra, S.; Gall, M.; Draper, J.; et al. Resistance Mutations in SARS-CoV-2 Delta Variant after Sotrovimab Use. N. Engl. J. Med. 2022, 386, 1477–1479. [Google Scholar] [CrossRef] [PubMed]
- Vellas, C.; Tremeaux, P.; Del Bello, A.; Latour, J.; Jeanne, N.; Ranger, N.; Danet, C.; Martin-Blondel, G.; Delobel, P.; Kamar, N.; Izopet, J. Resistance mutations in SARS-CoV-2 omicron variant in patients treated with sotrovimab. Clin. Microbiol. Infect. 2022, 28, 1297–1299. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.N.; Greaney, A.J.; Addetia, A.; Hannon, W.W.; Choudhary, M.C.; Dingens, A.S.; Li, J.Z.; Bloom, J.D. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science 2021, 371, 850–854. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.N.; Greaney, A.J.; Dingens, A.S.; Bloom, J.D. Complete map of SARS-CoV-2 RBD mutations that escape the monoclonal antibody LY-CoV555 and its cocktail with LY-CoV016. Cell Rep. Med. 2021, 2, 100255. [Google Scholar] [CrossRef] [PubMed]
- Baum, A.; Fulton, B.O.; Wloga, E.; Copin, R.; Pascal, K.E.; Russo, V.; Giordano, S.; Lanza, K.; Negron, N.; Ni, M.; et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 2020, 369, 1014–1018. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.; Baum, A.; Pascal, K.E.; Russo, V.; Giordano, S.; Wloga, E.; Fulton, B.O.; Yan, Y.; Koon, K.; Patel, K.; et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science 2020, 369, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, R.L.; Nirula, A.; Chen, P.; Boscia, J.; Heller, B.; Morris, J.; Huhn, G.; Cardona, J.; Mocherla, B.; Stosor, V.; et al. Effect of Bamlanivimab as Monotherapy or in Combination With Etesevimab on Viral Load in Patients With Mild to Moderate COVID-19: A Randomized Clinical Trial. JAMA 2021, 325, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Hammond, J.; Leister-Tebbe, H.; Gardner, A.; Abreu, P.; Bao, W.; Wisemandle, W.; Baniecki, M.; Hendrick, V.M.; Damle, B.; Simon-Campos, A.; et al. Oral Nirmatrelvir for High-Risk, Nonhospitalized Adults with Covid-19. N. Engl. J. Med. 2022, 386, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.H.; Chokkakula, S.; Min, S.C.; Kim, B.K.; Choi, W.S.; Oh, S.; Yun, Y.S.; Kang, D.H.; Lee, O.J.; Kim, E.G.; et al. Combination therapy with nirmatrelvir and molnupiravir improves the survival of SARS-CoV-2 infected mice. Antiviral Res. 2022, 208, 105430. [Google Scholar] [CrossRef]
- Clavel, F.; Hance, A.J. HIV drug resistance. N. Engl. J. Med. 2004, 350, 1023–1035. [Google Scholar] [CrossRef]
- Dunning, J.; Baillie, J.K.; Cao, B.; Hayden, F.G.; International Severe Acute, R.; Emerging Infection, C. Antiviral combinations for severe influenza. Lancet Infect. Dis. 2014, 14, 1259–1270. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Starr, T.N.; Barnes, C.O.; Weisblum, Y.; Schmidt, F.; Caskey, M.; Gaebler, C.; Cho, A.; Agudelo, M.; Finkin, S.; et al. Mapping mutations to the SARS-CoV-2 RBD that escape binding by different classes of antibodies. Nat. Commun. 2021, 12, 4196. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.E.; Zhang, X.; Case, J.B.; Winkler, E.S.; Liu, Y.; VanBlargan, L.A.; Liu, J.; Errico, J.M.; Xie, X.; Suryadevara, N.; et al. Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat. Med. 2021, 27, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.D.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310. [Google Scholar] [CrossRef]
- Zahradnik, J.; Marciano, S.; Shemesh, M.; Zoler, E.; Harari, D.; Chiaravalli, J.; Meyer, B.; Rudich, Y.; Li, C.; Marton, I.; et al. SARS-CoV-2 variant prediction and antiviral drug design are enabled by RBD in vitro evolution. Nat. Microbiol. 2021, 6, 1188–1198. [Google Scholar] [CrossRef] [PubMed]
- Mannar, D.; Saville, J.W.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Tuttle, K.S.; Marquez, A.C.; Sekirov, I.; Subramaniam, S. SARS-CoV-2 Omicron variant: Antibody evasion and cryo-EM structure of spike protein-ACE2 complex. Science 2022, 375, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Zhang, L.; Pohlmann, S. Omicron: Master of immune evasion maintains robust ACE2 binding. Signal Transduct. Target. Ther. 2022, 7, 118. [Google Scholar] [CrossRef]
- Meng, B.; Abdullahi, A.; Ferreira, I.; Goonawardane, N.; Saito, A.; Kimura, I.; Yamasoba, D.; Gerber, P.P.; Fatihi, S.; Rathore, S.; et al. Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature 2022, 603, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Meng, B.; Abdullahi, A.; Ferreira, I.A.; Goonawardane, N.; Saito, A.; Kimura, I.; Yamasoba, D.; Gerber, P.P.; Fatihi, S.; Rathore, S. Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature 2022, 603, 706–714. [Google Scholar] [CrossRef]
- Suzuki, R.; Yamasoba, D.; Kimura, I.; Wang, L.; Kishimoto, M.; Ito, J.; Morioka, Y.; Nao, N.; Nasser, H.; Uriu, K. Attenuated fusogenicity and pathogenicity of SARS-CoV-2 Omicron variant. Nature 2022, 603, 700–705. [Google Scholar] [CrossRef]
- Sun, C.; Wang, H.; Yang, J.; Kong, D.; Chen, Y.; Wang, H.; Sun, L.; Lu, J.; Teng, M.; Xie, L. Mutation N856K in spike reduces fusogenicity and infectivity of Omicron BA. 1. Signal Transduct. Target. Ther. 2023, 8, 75. [Google Scholar] [CrossRef]
- Batlle, D.; Soler, M.J.; Sparks, M.A.; Hiremath, S.; South, A.M.; Welling, P.A.; Swaminathan, S.; Covid; Ace2 in Cardiovascular, L. ; Kidney Working, G. Acute Kidney Injury in COVID-19: Emerging Evidence of a Distinct Pathophysiology. J. Am. Soc. Nephrol. 2020, 31, 1380–1383. [Google Scholar] [CrossRef] [PubMed]
- Kunutsor, S.K.; Laukkanen, J.A. Renal complications in COVID-19: a systematic review and meta-analysis. Ann. Med. 2020, 52, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, J.S.; Ng, J.H.; Ross, D.W.; Sharma, P.; Shah, H.H.; Barnett, R.L.; Hazzan, A.D.; Fishbane, S.; Jhaveri, K.D.; Northwell, C.-R.C.; Northwell Nephrology, C.-R.C. Acute kidney injury in patients hospitalized with COVID-19. Kidney Int. 2020, 98, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Luo, R.; Wang, K.; Zhang, M.; Wang, Z.; Dong, L.; Li, J.; Yao, Y.; Ge, S.; Xu, G. Kidney disease is associated with in-hospital death of patients with COVID-19. Kidney Int. 2020, 97, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Hikmet, F.; Mear, L.; Edvinsson, A.; Micke, P.; Uhlen, M.; Lindskog, C. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 2020, 16, e9610. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Wysocki, J.; William, J.; Soler, M.J.; Cokic, I.; Batlle, D. Glomerular localization and expression of Angiotensin-converting enzyme 2 and Angiotensin-converting enzyme: implications for albuminuria in diabetes. J. Am. Soc. Nephrol. 2006, 17, 3067–3075. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Hassler, L.; Reyes, F.; Sparks, M.A.; Welling, P.; Batlle, D. Evidence For and Against Direct Kidney Infection by SARS-CoV-2 in Patients with COVID-19. Clin. J. Am. Soc. Nephrol. 2021, 16, 1755–1765. [Google Scholar] [CrossRef]
- May, R.M.; Cassol, C.; Hannoudi, A.; Larsen, C.P.; Lerma, E.V.; Haun, R.S.; Braga, J.R.; Hassen, S.I.; Wilson, J.; VanBeek, C.; et al. A multi-center retrospective cohort study defines the spectrum of kidney pathology in Coronavirus 2019 Disease (COVID-19). Kidney Int 2021, 100, 1303–1315. [Google Scholar] [CrossRef]
- Syed, M.; Cianfarini, C.; Hassler, L.; Ye, M.; Wysocki, J.; Kanwar, Y.; Missiakas, D.; Randall, G.; Batlle, D. Lack of Evidence for Kidney Invasion by SARS-CoV-2 in a Lethal Mouse Model. J. Am. Soc. Nephrol. 2022, 331. [Google Scholar]
- Hassler, L.; Batlle, D. Potential SARS-CoV-2 kidney infection and paths to injury. Nat. Rev. Nephrol. 2022, 18, 275–276. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Survival of k18-hACE2 mice infected with the SARS-CoV-2 delta variant that received either ACE2-618-DDC-ABD or PBS. Infected animals in the PBS-control group (black) showed uniform lethality and survival by day seven post viral inoculation was 0%. By contrast, in the ACE2-618-DDC-ABD treated group (purple), survival was 90% by day six post viral inoculation with only one mouse having to be humanely euthanized on day six. Five mice from the ACE2-618-DDC-ABD treated group that were healthy by clinical score and weight were sacrificed on day six post viral inoculation for comparison of viral titers at similar time points. The remaining four mice in the ACE2 618-DDC-ABD treated group all survived until the end of the study protocol at day 14 post viral inoculation. The difference in survival was statistically significant (P=0.0004 by log-rank (Mantel-Cox) test).
Figure 1.
Survival of k18-hACE2 mice infected with the SARS-CoV-2 delta variant that received either ACE2-618-DDC-ABD or PBS. Infected animals in the PBS-control group (black) showed uniform lethality and survival by day seven post viral inoculation was 0%. By contrast, in the ACE2-618-DDC-ABD treated group (purple), survival was 90% by day six post viral inoculation with only one mouse having to be humanely euthanized on day six. Five mice from the ACE2-618-DDC-ABD treated group that were healthy by clinical score and weight were sacrificed on day six post viral inoculation for comparison of viral titers at similar time points. The remaining four mice in the ACE2 618-DDC-ABD treated group all survived until the end of the study protocol at day 14 post viral inoculation. The difference in survival was statistically significant (P=0.0004 by log-rank (Mantel-Cox) test).
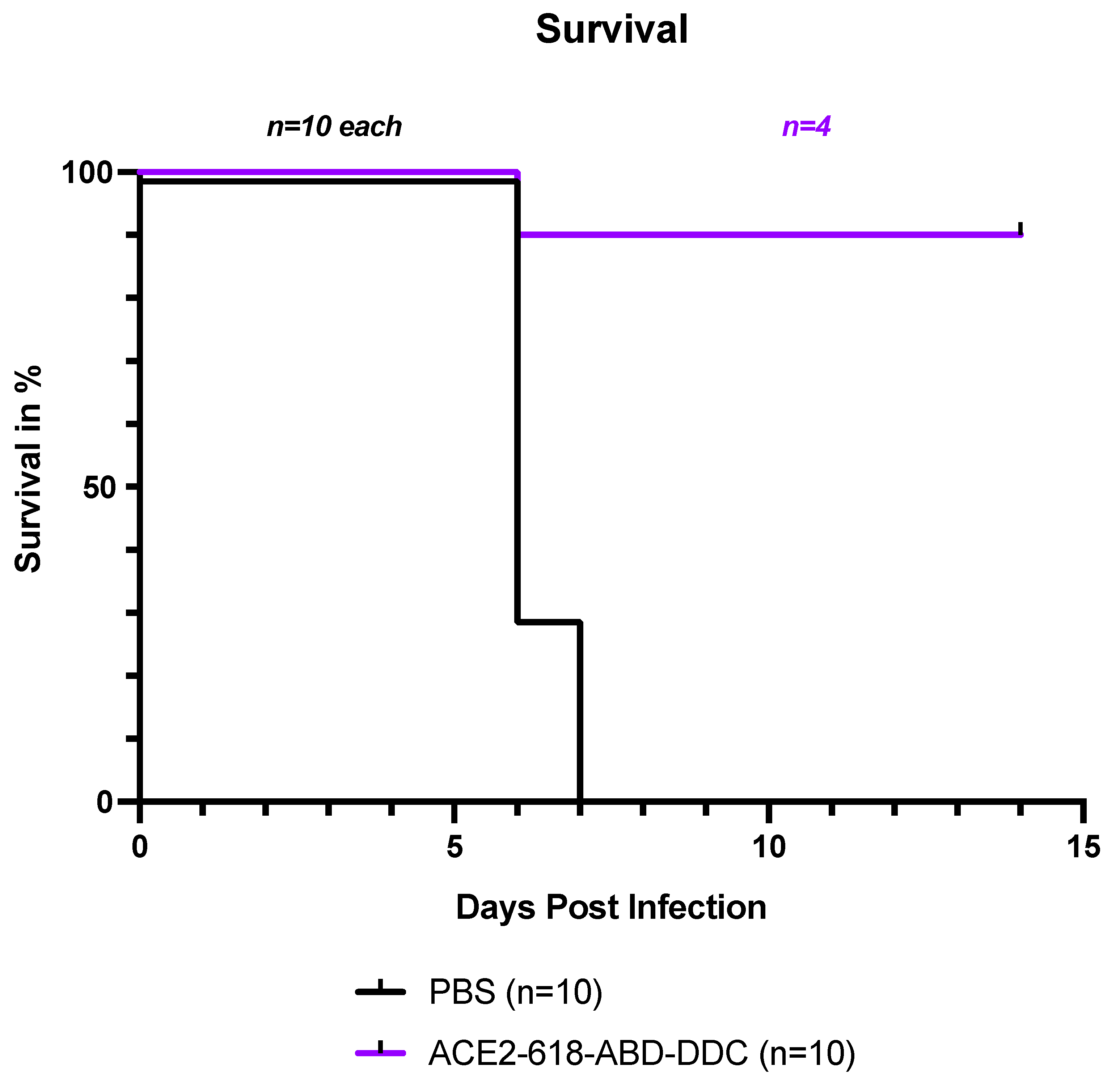
Figure 2.
Clinical score and body weight of k18-hACE2 mice infected with the SARS-CoV-2 delta variant that received either ACE2-618-DDC-ABD or PBS. Panel A. Infected animals in the PBS-control group (black) developed severe disease and clinical scores increased accordingly until days six and seven post viral inoculation by which time point all mice had either died or had to be humanely euthanized. By contrast, animals in the ACE2-618-DDC-ABD treated group (purple) developed less severe disease and the clinical score was lower as compared to the PBS-control group. All remaining animals from the ACE2-618-DDC-ABD treated group had recovered by the end of the 14-day study period. Panel B. Infected animals in the PBS-control group (black) lost up to ~20% of their initial bodyweight until days six and seven post viral inoculation by which time point all mice had either died or had to be humanely euthanized. By contrast, animals in the ACE2-618-DDC-ABD treated group (purple) lost on average less than ~10% of their initial body weight by days six and seven post viral inoculation. All remaining animals from the ACE2-618-DDC-ABD treated group recovered stable body weight by the end of the 14-day study period.
Figure 2.
Clinical score and body weight of k18-hACE2 mice infected with the SARS-CoV-2 delta variant that received either ACE2-618-DDC-ABD or PBS. Panel A. Infected animals in the PBS-control group (black) developed severe disease and clinical scores increased accordingly until days six and seven post viral inoculation by which time point all mice had either died or had to be humanely euthanized. By contrast, animals in the ACE2-618-DDC-ABD treated group (purple) developed less severe disease and the clinical score was lower as compared to the PBS-control group. All remaining animals from the ACE2-618-DDC-ABD treated group had recovered by the end of the 14-day study period. Panel B. Infected animals in the PBS-control group (black) lost up to ~20% of their initial bodyweight until days six and seven post viral inoculation by which time point all mice had either died or had to be humanely euthanized. By contrast, animals in the ACE2-618-DDC-ABD treated group (purple) lost on average less than ~10% of their initial body weight by days six and seven post viral inoculation. All remaining animals from the ACE2-618-DDC-ABD treated group recovered stable body weight by the end of the 14-day study period.
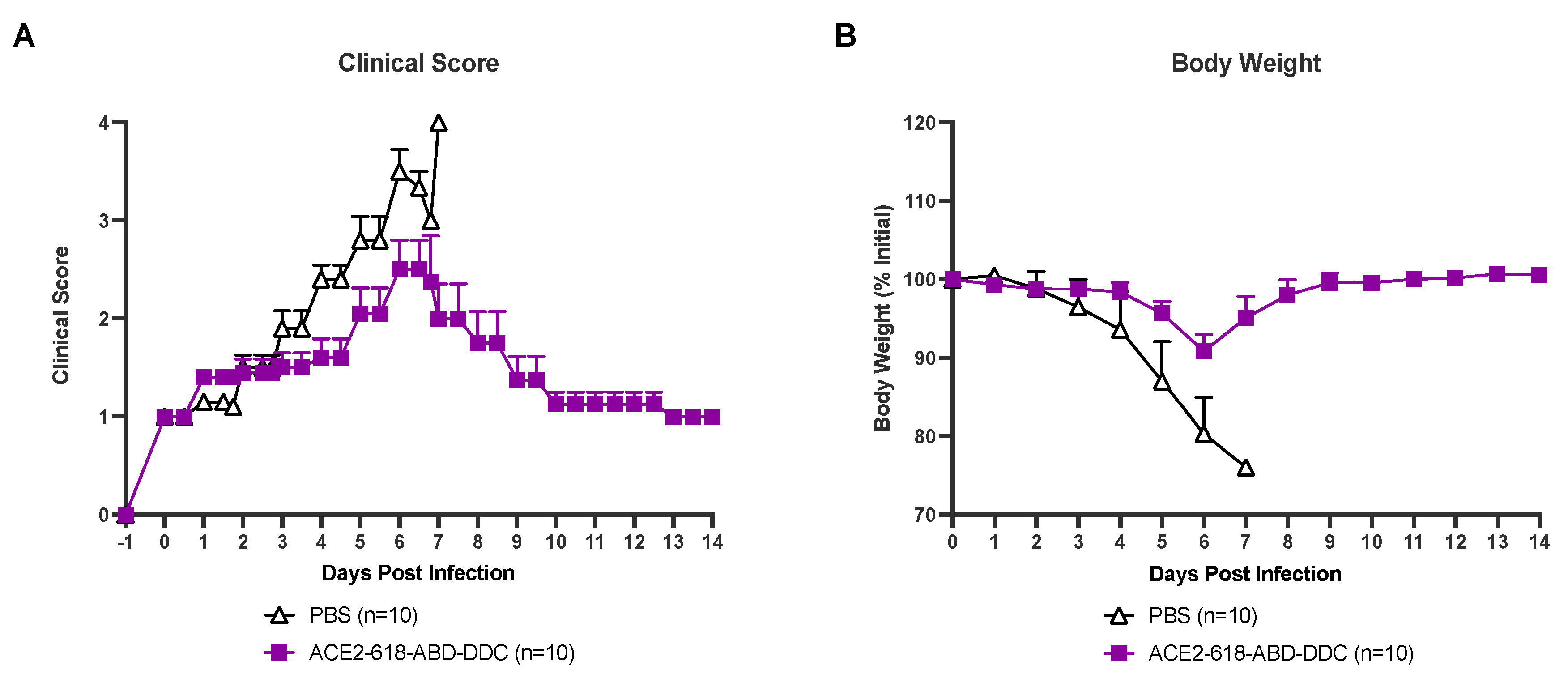
Figure 3.
Lung, brain and kidney SARS-CoV-2 titers of k18-hACE2 mice infected with the SARS-CoV-2 delta variant that received either ACE2-618-DDC-ABD or PBS. Panel A. Lung viral titers in infected animals from the ACE2-618-DDC-ABD treated group (purple) were markedly reduced as compared to lung viral titers in infected animals that received PBS (black) (1.5 × 102 ± 0.7 × 102 vs. 7.8 × 103 ± 4.7 × 103 PFU/ml, P=0.04). Panel B. Brain viral titers were also markedly reduced in mice that received ACE2-618-DDC-ABD (purple) as compared to brain viral titers in the PBS-control group (black) (1.1 × 106 ± 1.1 × 106 vs. 1.2 × 107 ± 3.7 × 106 PFU/ml, P=0.0007). Panel C. Kidney viral titers could not be detected in any of the animals from either the ACE2-618-DDC-ABD treated (purple) or the PBS-control group (black).
Figure 3.
Lung, brain and kidney SARS-CoV-2 titers of k18-hACE2 mice infected with the SARS-CoV-2 delta variant that received either ACE2-618-DDC-ABD or PBS. Panel A. Lung viral titers in infected animals from the ACE2-618-DDC-ABD treated group (purple) were markedly reduced as compared to lung viral titers in infected animals that received PBS (black) (1.5 × 102 ± 0.7 × 102 vs. 7.8 × 103 ± 4.7 × 103 PFU/ml, P=0.04). Panel B. Brain viral titers were also markedly reduced in mice that received ACE2-618-DDC-ABD (purple) as compared to brain viral titers in the PBS-control group (black) (1.1 × 106 ± 1.1 × 106 vs. 1.2 × 107 ± 3.7 × 106 PFU/ml, P=0.0007). Panel C. Kidney viral titers could not be detected in any of the animals from either the ACE2-618-DDC-ABD treated (purple) or the PBS-control group (black).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



