
Submitted:
23 March 2024
Posted:
25 March 2024
You are already at the latest version
Abstract
The presence of antibiotic residues in honey has recently become a growing public health concern due to the overuse of these products in beekeeping and their transfer to the food industry. The aim of this study was to develop and validate a simple, selective and sensitive method for the simultaneous identification and quantification of residues of twenty-two sulfonamides, trimethoprim and dapsone in honey using a rapid and reliable liquid chromatography–tandem mass spectrometry (LC–MS/MS) method. The developed method showed good linearity, specificity, precision (repeatability and intra-laboratory reproducibility) and recovery. The decision limit (CCalpha) ranged between 2.16-22.12 µg/kg for CCα, while the detection capability (CCbeta) was between 2.16- 2.67-27.36 µg/kg. The method proved to be rapid, reliable and effective for the determination of sulfonamides, trimethoprim and dapsone from honey samples. The average recoveries were estimated to be between 70 and 106%. The relative standard deviations (RSDs) were between 6-18%. In practice, the method is essential to check the honey sold on the market and ensure its safety before it reaches the consumer.
Keywords:
analytical method
; antibiotic residues
; sulfonamides
; trimethoprim
; dapsone
; honey
; liquid chromatography-tandem mass spectrometry
1. Introduction
Sulfonamides, sometimes also known as sulfa drugs, are antibiotics specifically targeting bacteria that can cause infections. These antibiotics are usually broad-spectrum antibiotics that act against a wide range of bacteria and are used to treat a variety of bacterial infections. Sulfonamides account for 11% of total sales of antimicrobial veterinary medicines in Europe in 2011 [1]. Trimethoprim and dapsone, which have a similar effect to sulfonamides, are often administered together with some sulfonamides in pharmaceutical products. When used in combination with trimethoprim and dapsone, their bacteriostatic effect is synergistic and is enhanced by the inhibition of bacterial dihy-dropteroate synthetase and dihydrofolate reductase. It is known that high levels of sulfonamides, dapsone and trimethoprim in food can have various adverse effects on human health, including increased bacterial resistance to antimicrobial agents, allergic reactions or possible carcinogenicity [2,3,4].
Bees produce and refine honey through the collection and processing of plant nectar and honeydew, encompassing natural sugars like glucose and fructose, along with amino acids, enzymes, minerals, and antioxidants [3]. Throughout history, honey has played a dual role as a fundamental dietary component and a medicinal resource. Bee-derived products are commonly regarded as natural and health-promoting, believed to possess the ability to combat infections and facilitate wound healing [4]. Nevertheless, the production of beekeeping products occurs in natural settings, potentially exposing them to contamination. The sources of contamination include the immediate surroundings and the practices involved in beekeeping. Xenobiotics present in the environment can stem from industrial activities, agriculture, or individual treatments. These include organic contaminants (polychlorinated biphenyls – PCBs), heavy metals (Pb, Cd), insecticides (organochlorines - OC, organophosphorus pesticides -OP, carbamates), herbicides (asulam), bactericides (streptomycin), fungicides (vinclozolin, iprodione and methyl tiophanate, captan and difenoconazole), and radioactive isotopes e (40K and 137Cs) [5].
In order to protect consumers against potential risks and to prevent the development of bacterial resistance, it becomes essential to routinely assess whether honey contains drug residues. In some regions of the world, sulfonamides (SAs), such as sulfathiazole or sulfamonomethoxine, are the preferred choice. Aminoglycosides, macrolides, quinolones, nitrofurans and the sulfonamide family are used in various countries in the treatment of infected urticaria, even for prophylactic purposes [6]. The tetracycline family of antibiotics has historically dominated the treatment of urticaria disease, but resistance has gradually developed in American Foolbrood (AFB) [7] and European Foolbrood (EFB) bacteria. Among the many treatments used in beekeeping, antibiotic therapy is a common approach to treat the major bacterial diseases AFB, EFB, Nosema and Varroa protozoan diseases [8]. Topical treatments with their active ingredients spread quickly and bees carry them into the hive. Targeted contamination from beekeeping practices varies by outcome and region. To date, no maximum residue limits for SAs in honey have been set in the European Union. This lack of regulation for the honey matrix could change in the future, especially if studies showing the presence of veterinary drug residues in beehives complete the picture [9] and lead to the establishment of MRLs for honey as for other foodstuffs. In the European Union, only recommended concentrations for minimum performance limits for confirmatory methods have been proposed, set at 50 µg/kg for trimethoprim and all sulfonamides. Dapsone is banned for use in agricultural food production, but no maximum residue limits (MRLs) have been set for sulfonamides and trimethoprim, although this family of veterinary medicines is covered by Commission Regulation (EU) No 37/2010 and 470/2009/EC (formerly 2377/90/EC) [10,11,12].
A receptor test named Sulfasensor has the capability to identify SA residues in honey or other spices [13], while a swift screening technique for sulfonamides in honey utilizes a flow injection system linked with a liquid waveguide capillary cell [14]. However, reliable and highly sensitive methods are needed to unequivocally detect and confirm residues in honey. Numerous methods have been described to ascertain the presence of one or more SAs, including UV-HPLC [8,15,16], fluorescence detection [17,18,19,20], and liquid chromatography coupled with mass spectrometry [21,22,23,24]. Occasionally, multifamily methods [25,26,27,28,29] identify and quantify one or more SAs alongside other molecules. Notably, there is currently no published LC-MS/MS confirmation method, as per the 2001/2003 2002/657/CE [30], that encompasses all of the 22 SAs together with trimethoprim and dapsone proposed in our method.
The primary aim of this article was to develop and validate a method that is simple, selective, reliable, and sensitive for concurrently identifying and quantifying residues of twenty-two sulfonamides, trimethoprim, and dapsone in honey using LC-MS/MS. This article illustrates our efforts to identify such a combination of target substances within this matrix, following the guidelines outlined in Decision 2002/657/EC [31] and its amended Directive SANCO 2726/2004 [32], regarding residues of “prohibited and unauthorized” substances.
In this context, the determination of antibiotic residues in honey is considered important, while the development of accurate, selective, rapid and multi-screening methods for the determination of such residues is still urgently needed.
No such analytical method has been previously reported in Romania. Moreover, the method has been implemented and is currently successfully applied to simultaneously analyse the above-mentioned veterinary drug residues in Romanian veterinary laboratories within the framework of the national residue control plan for honey products.
2. Materials and Methods
2.1. Chemicals and Reagents
Certified standards of sulfacetamide, sulfaguanidine, sulfanilamide, sulfisomidine, sulfadiazine, sulfathiazole, sulfapyridine, sulfamerazine, sulfamethazine, sulfamethizole, sulfamethoxypyridazine, sulfamethoxydiazine, sulfamonomethoxine, sulfamoxol, sulfadoxine, sulfamethoxazole, sulfisoxazole, sulfabenzamide, sulfadimethoxine, sulfaquinoxaline, sulfanitran, dapsone and trimethoprim and the internal standards sulfadimidine-13C6 and sulfamethoxazole-13C6 were purchased from Sigma-Aldrich (St. Quentin Fallavier, France), while sulfachloropyridazine and sulfaphenazole were obtained from Dr. Ehrenstorfer (Augsburg, Germany).
LC-MS grade acetonitrile, n-hexane, methanol, formic acid and acetone together with hydrochloric acid (0.1 N) and ammonium acetate buffer (0.2 mol/l, pH 5.3) were obtained from Sigma-Aldrich (St. Quentin Fallavier, France). A Millipore Simplicity 185 purification system (Millipore, USA) was used in order to obtain de-ionized double distilled water.
All chemicals and reagents used were of analytical grade, unless otherwise specified.
For the solid-phase extraction StrataTM-X (500mg/ 6ml) cartridges from Fenomenex were used.
2.2. Standards Solutions
Individual standard stock solutions of the analytes and of the internal standards-sulfadimidine-13C6 and sulfamethoxazole-13C6 (100µg/ml) were prepared in acetonitrile and further stored in glass ambered bottles. Individual working solutions of the analytes dapsone and trimethoprim (1µg/ml), as well as a mixed working solution of the analysed sulfonamides (1µg/ml) were prepared by the appropriate dilution of the standard stock solutions with hydrochloric acid. Individual working solutions of the internal standards (10µg/ml) were also prepared by the appropriate dilution of the individual standard stock solutions with hydrochloric acid and appropriately stored.
Eight mixed calibration solutions containing the SAs and trimethoprim analytes in the concentration range 5-150 ng/ml and dapsone in the range 2.5-25 ng/ml, as well as the internal standards at a fixed concentration of 20 ng/ml were prepared daily, by serial dilution of the mixed working solution of sulfonamides, the individual working solution of dapsone, the individual working solution of trimethoprim and of the individual working solutions of the internal standards in the aqueous mobile phase (0.1% v/v HCOOH in water).
2.3. Matrix Calibrations
Eight matrix-matched calibration solutions containing all the SAs and trimethoprim analytes in the range of 5-150 µg/kg and in the range of 2.5-25 µg/kg for dapsone and the internal standards at a fixed concentration of 20 µg/kg, were prepared by subjecting “blank” honey samples (5 g) to solid-phase extraction and further spiking of the extract with the appropriate volumes of the mixed sulfonamides working solution, the individual working solutions of dapsone and trimethoprim, as well as the individual working solutions of the internal standards.
2.4. Traceability
Honey samples were gathered from various botanical origins of north central and east Romania and analysed in a laboratory which is overseen by The National Veterinary Health and Food Safety Authority. The method presented in this work was developed, validated and accredited in order to offer a rigorous procedure for the determination and quantitative confirmation of sulfonamides, trimetroprim and dapsone residues in honey matrices at low levels of concentrations to meet the requirements.
The blank material was a blend of 20 different types of honey (varying in colour and texture), all of which were examined for residues before being used in the validation process, none of them contained detectable levels of SAs, trimethoprim and dapsone. 24 different honey samples were utilized to examine matrix effects.
2.5. Sample Preparation and Extraction
Testing Sample
Approximately 50g of “blank” honey was heated at 45⁰C in a water bath for 1h. A quantity of 5 g of “blank” honey were accurately weighted in a 50 ml polytetrafluoroethylene (PTFE) centrifuge tube and then spiked with 100 µl of each individual working solution of internal standards- sulfadimidine-13C6 and sulfamethoxazole-13C6 of concentration 1 µg/ml. 10 ml of a 0.1 N hydrochloric acid (HCl) were added to the previously obtained mixture, which was further vortexed for 15 minutes and then sonicated for 15 minutes, until complete dissolution. Then, the testing sample was centrifuged at 3500 rpm for 10 minutes and stored over night at room temperature for a better static extraction. The next day it is applied to the solid phase extraction (SPE) cartridges.
Fortified (spiked) Sample
The fortified (spiked) sample was prepared by accurately weighing 5g of honey in a 50 ml PTFE centrifuge tube. 250 µl of mixed sulfonamides working solution (1µg/ml), 25 µl of dapsone working solution (1 g/ml), 250 µl of trimethoprim working solution (1 µg/ml), as well as 100 µl of each working solution of internal standards (1 µg/ml) were added to the sample. 10 ml of HCl (0.1N) were further added in the centrifuge tube. The probe was vortexed for 15 minutes and then sonicated for 15 minutes. The sample was afterwards centrifuged at 3500 rpm for 10 minutes and stored over night at room temperature. The next day it is applied to the SPE cartridges.
Blank
The blank was prepared of 10 ml HCl (0.1 N) and 100 µl of each working solution of internal standards (1 µg/ml), vortexed for 15 minutes and then sonicated for 15 minutes until complete dissolution and then centrifuged at 3500 rpm for 10 minutes and stored over night at room temperature. The next day it is applied to the SPE cartridges.
Samples Purification
The solid-phase extraction (SPE) was performed on StrataTM-X cartridges. The cartridges were pre-conditioned with 10 ml of methanol and 10 ml of ammonium acetate (0.2 mol/l, pH 5.3). The samples were percolated through the cartridges, these being afterwards rinsed with 10 ml of water and vacuum dried for 5 minutes. The retained analytes were further eluted with 10 ml of methanol. The eluate was concentrated on a water bath at 50°C to near dryness under a nitrogen stream. The solution was reconstituted with 1000 µl of HCl (0.1 N).
2.6. Instrumentation
The LC–tandem MS system included a Bruker UHPLC advance pump, a PAL HTC-xt autosampler, and a Bruker EVOQ Elite triple-quadrupole mass spectrometer with an electrospray ionization (ESI) interface. Data acquisition was done with Bruker MSWS software version 8.
A Pursuit XRs Ultra C18 column (100 mm x 2 mm/ 2.8 µm) for chromatographic separation (Agilent Technology, Netherlands). The temperature of the column was kept constant at 35°C. Two mobile phases were used in a multi-step binary elution gradient: phase A: water containing 0.1 % (v/v) HCOOH, and phase B: acetonitrile containing 0.1 % (v/v) HCOOH.
For both the standard and sample solutions, the flow rate was 200 µL/min and a volume of 30 µL was injected. The gradient is presented in Supplementary Material Table S1.
2.7. Method Validation
The method validation was performed according to Commission Decision 2002/657/EC [31] and its amending guideline SANCO/2006/3228 [32] considering the requirements for a quantitative confirmatory method. In order to determinate the identification criteria for each analyte, the relative retention time and relative ion intensities of internal standard solutions were assessed (Supplementary Table S1).
Specificity, sensitivity, linearity, precision (repeatability and within-laboratory reproducibility), trueness (in terms of recovery), decision limit (CCα), and detection capability (CCβ) were the performance criteria examined in the validation study [31,32,33,34] (Supplementary Table S2). Twenty organically cultivated honey samples of various origins were analyzed to determine the specificity.
2.7.1. Linearity
Method linearity was assessed by conducting regression analysis on solvent calibration and matrix-matched calibration solutions. This analysis involved plotting the ratio of the standard area (obtained from the most intense transition) to the internal standard area against analyte concentrations. For each analyte, a linear calibration curve in the matrix was obtained. The linear regression coefficient and relative standard deviation (RSD) are determined individually for each curve using the software Workstation MS Bruker v. 8.2, (Bruker Corporation, Billerica, MA, USA) which is used in assessing the performance of the curve.
2.7.2. Recovery, Trueness and Accuracy
For the recovery determination, “blank” honey samples (5 g) were spiked with appropriate amounts of the standard working mixture of analytes and the working mixture of deuterated standards, resulting in concentration levels of 25, 50, 75, 100 µg/kg for SAs and trimethroprim, of 2.5, 5, 7.5, 10 µg/kg for dapsone and 20 µg/kg for the internal standards. To allow the sulfonamides to sufficiently bind to the sugars in the honey, the spiked samples were kept at room temperature for at least 1 hour [28,35]. Six replicates were analyzed at each concentration level and the recovery was calculated by comparing the concentration of the extracted sulfonamide with those from the matrix-matched calibration curve.. The chromatograms were obtained for internal standards, for “blank” honey sample spiked with 20 µg/kg of the internal standards and blank honey samples spiked with 20 µg/kg of the internal standards and 50 µg/kg of each compound (5 µg/kg for dapsone), as shown in Figure 1 and Figure 2.
Each series, which included a matrix calibration curve and 24 spiked samples, was prepared on two distinct days for a total of 48 spiked samples with various time, operator, and LC–MS/MS calibration/operation status. These tests determined trueness (in terms of percent recoveries) and accuracy (in terms of repeatability and within-laboratory reproducibility in terms of respective percent relative standard deviations). Six repetitions were conducted for each level of analysis. Subsequently, the samples underwent analysis, and the concentration present in each sample was identified. Mean concentration, standard deviation, relative standard deviation (RSDr %), and repeatability limit were calculated for the results obtained from each set of analyses.
2.7.3. Decision Limit (CCα) and Detection Capability (CCβ)
The decision limit (CCα) and detection capability (CCβ) were determined in accordance with the ISO Standard 11843, linear regression method, as proposed in European Decision No 2002/657/EC [31]. Both critical limits, CCα and CCβ were calculated using calibration curves obtained from “blank” honey samples spiked at four concentration levels of 25, 50, 75, 100 µg/kg for SAs and trimethroprim and of 2.5, 5, 7.5, 10 µg/kg for dapsone – six replicates per level – and processed via solid-phase extraction (SPE), using the ratio of the signal of the less intense transition of the target analyte to that of the internal standard applicable in each case.
3. Results and Discussions
3.1. LC-MS/MS Method Development
Sulfonamides are banned as antibiotics in honey in the European Union, as no maximum residue levels have been set for these foods. However, with SANCO/2006/3228 [36] a guideline has been established that sets recommended levels for substances for which there are no legal MRLs or minimum performance levels (MRPLs). The performance of analytical methods in relation to CCα must be below these values, even if these values are only analytical limits without regulatory significance. Non-authorized substances are monitored with two MRM (Multiple Reaction Monitoring) transitions. Since SAs contain a nitrogen atom, optimization in ESI mode was quite simple and the responses obtained in positive mode were high. It is known that the signals are weaker in negative ionization [21]. With the exception of some sulfonamides, the usual fragment ions with values of m/z 156, 108 and 92 were used.
Optimization of the liquid chromatography settings with focus on the signals of the analytes and selection of the optimal separation conditions for the mobile phase, especially for the isomers SDX-SDM (m/z 311) and SMM-SMP-SME (m/z 281). A comparison of 0.1 % formic acid and 0.1 % PFPA in the aqueous mobile phase was investigated. PFPA had the advantage of successfully retaining the two most polar SAM and SGD analytes, but it interfered with the analytes’ signals, so formic acid was finally chosen. Two columns were tested to observe the performance of the analytical columns: Pursuit XRs Ultra C18 column (100 mm x 2 mm/ 2.8 µm particle size, Agilent Technology) and Phenomenex Aqua C18 column (150 mm × 2.0 mm/ 3 µm particle size, Waters). The first column was chosen because it offers high reproducibility for the analytes investigated in this work.
Most published methods for measuring SAs in foods of various origins include at least one solid-phase extraction (SPE) step using polar [37], non-polar [38,39,40] or strongly cation-exchanging sorbents [41]. The advantage of an SPE step prior to LC–MS/MS analysis is that suppression by matrix components is reduced under certain circumstances and the detection limit of the method is lowered in most cases.
Simple extractions with HCl (0.1 N) in water versus trichloroacetic acid (TCA) (0.03 N) in water were both tested while improving the conditions for sample preparation. As a result, simple extraction procedures are required for the analysis of samples from national monitoring plans. TCAs show an inability to sufficiently break the bonds between the sulfonamides and the honey sugars. As the extraction yield and sensitivity were insufficient when using TCA, HCl was chosen to separate the sulfonamides from the honey components.
3.2. Sample Extraction Procedure
Three types of samples were obtained:
- -
- Sample to be analyzed: 5 g of honey sample mixed with 10 µl of internal standard solutions with concentration of 10 µg/ ml.
- -
- Fortified sample for the determination of recovery: 5 ± 0.5 g honey sample mixed with 250 µl solution of mixed sulfonamides with concentration of 1 µg/ ml, 50 µl solution of dapsone with concentration of 1 µg/ ml, 250 µl solution of trimethoprime with concentration of 1 µg/ ml and 10 µl of internal standard solutions with concentration of 10 µg/ ml.
- -
- Blank sample: contains 5 g of blank honey and 10 µl of internal standard solutions with concentration of 10 µg/ ml.
The acid hydrolysis with 10 ml HCl 0.1 N of the sulfonamides bound to the sugar was followed by an SPE purification The recovery of “blank” honey samples spiked with the target chemicals was used to evaluate the efficacy of the different sample extraction procedures. The influence of the pH of the honey sample after acid hydrolysis was investigated in the range of 2–7. The best sulfonamide recoveries occurred when the sample was adjusted to a pH of 5.3During our initial experiments, it was discovered that the cartridges were frequently clogged by undissolved material – most likely wax in the honey sample – resulting in low recovery values; as a result, the cartridges were conditioned and the sample solution was diluted to 10 mL with HCl (0.1 N) to reduce the likelihood of the sulfonamides re-binding to sugars and to avoid blocking of the SPE cartridges. The SPE cartridges were conditioned with methanol followed by acetate buffer 0.2 mol/L, pH 5.3, and extraction was carried out by gravitational flow. When compared to methanol, acetonitrile was used to elute the target chemicals since it yielded greater recoveries. The eluate was concentrated on a water bath at 50°C to near dryness under a nitrogen stream. The solution was reconstituted with 1000 µl of HCl (0.1 N).
3.3. Method Validation
3.3.1. Specificity, Linearity, Sensitivity
The method’s specificity was testedby structural analysis of the compounds of interest and internal standards which can be isolated from the analytes by mass spectrometry..The precusor ion was determined for each standard compound and then the majority ions were chosen after dividing the precusor ion and the collision energy was optimized for each individual ion. Blank samples of honey were analyzed and the presence of any interferences (peaks/signals) were checked. Each compound was separated without other interferences No interfering peaks overlapping with the analyte peaks were seen in the SRM chromatograms in these “blank” sample experiments, confirming appropriate specificity for the detection of the target compounds. Figure 1a,b show SRM chromatograms of a “blank” honey sample and a “blank” honey sample spiked with 50 µg/kg sulfamide and 5 µg/kg for dapsone, respectively. The matrix-matched calibration curves for each compound were built using blank samples spiked at eight concentration levels between 5µg/kg and 150µg/kg. For each compound, the correlation coefficients ≥ 0.99 and RSD<15% were obtained.
For each compound, the previously mentioned software was used to determinate the signal/noise ratio at a well-known concentration (MRPL). The concentration corresponding to a signal-to-noise ratio equal to 3/1 represents the limit of detection (LOD).The limit of quantification was calculated with the formula:
LOQ = 3 x LOD
S3.3.2. Accuracy
Recovery studies utilizing fortified “blank” honey samples spiked with the target components were used to assess method accuracy – trueness and precision. The blank sample was analyzed and no residues of sulfonamides, dapsone and trimethoprim were detected. A total of 24 parts of blank sample were selected: six parts each were fortified at 0.5 * minimum required performance limit (MRPL), at 1 * MRPL, at 1.5 *MRPL and 2 * MRPL.
The percent recovery, R percent, was computed using the following formula:
Where CP+Et – concentration of the compound in the fortified samples CP – concentration of the compound in the sample
CEt - the theoretical concentration of the compound in the fortified sample.
The mean recovery was calculated. The percentage of mean recovery obtained for each component ranged between 80-120%, meeting the requirements for trueness in the CD 2002/657/EC [42] (Supplementary Table S2).
Also, the mean concentration, the standard deviation, RSDr (%) and repeatability limit were calculated for the results of each series.
For all the compounds, the method’s mean repeatability (expressed as percent relative standard deviation, %RSDr percent) and within-laboratory reproducibility (expressed as percent relative standard deviations, RSDR %) were lower then 20 % at the different concentration levels (data shown in Supplementary Material Table S2), meeting the CD 2002/657/EC criteria [42].
3.3.3. Uncertainty
The measurement uncertainty can be estimated based on the repeatability and reproducibility studies carried out during the verification of the method (internal validation).
The uncertainties associated with the recoveries were computed using relative standard deviations, uRm=Rm.) The results of uncertainties are presented in Table 1.
3.3.4. Decision limits (CCalpha) and detection capabilities (CCbeta)
The decision limit (CCalpha), where α is 1% and the detection capability (CCbeta), where β is 5%, were calculated for this method. To establish the presence of residues of sulfonamides, trimethoprim, and dapsone (prohibited compounds), the decision limit was computed following the guidelines outlined in the ISO 11843 standard [33,34].
where: sR = standard deviation of the intra-laboratory reproducibility of the intercept
CCα = cy + 2,33 x sR
cy = the concentration corresponding to the y-intercept;
For every compound, CCβ was determined by equating it to the concentration associated with the decision limit value plus 1.64 times the standard deviation of reproducibility for the mean content measured at the decision limit (β=5%), following the guidelines outlined in the ISO 11843 standard [33,34]:
CCβ = CCα + 1,64 x sR
The values of CCα and CCbeta are shown in Table 2.
5. Conclusions
After finalizing the implementation and validation of the method in the laboratory, the method was verified by participating in a Proeficiency Testing scheme organised by Fapas T02353 in which 59 international laboratories attended. Considering the z-scores obtained by the laboratory for this method, we can conclude that the method is adequate, rigorous and efficient for the correct identification and quantification of sulfonamides in honey. The method has low limits of determination which is very important considering the prohibition of sulfonamides in honey. Our method is used currently for the surveillance of antibiotic residues in honey, serving as an initial effective assessment tool.
Romania is an important honey producer, exporting honey on the European market, and therefore, this method is essential for investigating how safe this product is before reaching a large number of consumers from both national and European level.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Fragmentation conditions for the target compounds, Table S2: The values of performance parameters obtained through the internal validation of the method.
Author Contributions
Conceptualization, M.M., G.V.V.; methodology, A.V.-L., G.V.V., M.-M.P.; formal analysis, G.V.V.; investigation, L.M.C., A.V.-L., G.V.V., A.I.G.-H., A.-A.C.; writing—original draft preparation, L.M.C., A.V.-L., G.V.V., A.I.G.-H., A.-A.C., M.-M.P.; writing—review and editing, G.V.V., A.I.G.-H., A.-A.C.; supervision, M.M.. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
The authors would like to thank the D.S.V.S.A. Laboratory from Cluj Napoca for their support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Grave, K.; Torren-Edo, J.; Muller, A.; Greko, C.; Moulin, G.; Mackay, D. Variations in the sales and sales patterns of veterinary antimicrobial agents in 25 European countries. J Antimicrob Chemother 2014, 69, 2284–2291. [Google Scholar] [CrossRef]
- Chang, H.; Hu, J.; Asami, M.; Kunikane, S. Simultaneous analysis of 16 sulfonamide and trimethoprim antibiotics in environmental waters by liquid chromatography–electrospray tandem mass spectrometry. Journal of Chromatography A 2008, 1190, 390–393. [Google Scholar] [CrossRef] [PubMed]
- Stolker, A.A.; Brinkman, U.A. Analytical strategies for residue analysis of veterinary drugs and growth-promoting agents in food-producing animals--a review. J Chromatogr A 2005, 1067, 15–53. [Google Scholar] [CrossRef] [PubMed]
- Gentili, A.; Perret, D.; Marchese, S. Liquid chromatography-tandem mass spectrometry for performing confirmatory analysis of veterinary drugs in animal-food products. TrAC Trends in Analytical Chemistry 2005, 24, 704–733. [Google Scholar] [CrossRef]
- Bogdanov, S. Contaminants of bee products. Apidologie 2005, 37. [Google Scholar] [CrossRef]
- Reybroeck, W.; Daeseleire, E.; De Brabander, H.F.; Herman, L. Antimicrobials in beekeeping. Veterinary Microbiology 2012, 158, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Genersch, E.; Evans, J.D.; Fries, I. Honey bee disease overview. Journal of Invertebrate Pathology 2010, 103, S2–S4. [Google Scholar] [CrossRef] [PubMed]
- Dubreil-Chéneau, E.; Pirotais, Y.; Verdon, E.; Hurtaud-Pessel, D. Confirmation of 13 sulfonamides in honey by liquid chromatography–tandem mass spectrometry for monitoring plans: Validation according to European Union Decision 2002/657/EC. Journal of Chromatography A 2014, 1339, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.J.; Fussell, R.J.; Dickinson, M.; Wilkins, S.; Sharman, M. Study of the depletion of lincomycin residues in honey extracted from treated honeybee (Apis mellifera L.) colonies and the effect of the shook swarm procedure. Analytica Chimica Acta 2009, 637, 315–320. [Google Scholar] [CrossRef]
- Commission, E. Commission Regulation (EU) No 37/2010 of 22 December 2009 on pharmacologically active substances and their classification regarding maximum residue limits in foodstuffs of animal origin (Text with EEA relevance); Luxembourg, 2010; Vol. L 15, pp. 1–72.
- Commission, E. Council Regulation (EEC) No 2377/90 of 26 June 1990 laying down a Community procedure for the establishment of maximum residue limits of veterinary medicinal products in foodstuffs of animal origin; The Publications Office of the European Union: Luxembourg, 1990; Vol. 224, pp. 1–8. [Google Scholar]
- Commission, E. Regulation (EC) No 470/2009 of the European Parliament and of the Council of 6 May 2009 laying down Community procedures for the establishment of residue limits of pharmacologically active substances in foodstuffs of animal origin, repealing Council Regulation (EEC) No 2377/90 and amending Directive 2001/82/EC of the European Parliament and of the Council and Regulation (EC) No 726/2004 of the European Parliament and of the Council (Text with EEA relevance ); The Publications Office of the European Union: Luxembourg, 2009; Vol. 152, pp. 1–11. [Google Scholar]
- Gaudin, V.; Rault, A.; Verdon, E. Validation of a commercial receptor kit Sulfasensor® Honey for the screening of sulfonamides in honey according to Commission Decision 2002/657/EC. Food Additives & Contaminants: Part A 2012, 29, 942–950. [Google Scholar] [CrossRef]
- Catelani, T.A.; Tóth, I.V.; Lima, J.L.F.C.; Pezza, L.; Pezza, H.R. A simple and rapid screening method for sulfonamides in honey using a flow injection system coupled to a liquid waveguide capillary cell. Talanta 2014, 121, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Granja, R.H.M.M.; Niño, A.M.M.; Rabone, F.; Salerno, A.G. A reliable high-performance liquid chromatography with ultraviolet detection for the determination of sulfonamides in honey. Analytica Chimica Acta 2008, 613, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.-H.; Wang, J.; Wang, X.-F.; Liu, Y.; Han, J.; Hou, F.; Xie, M.-X. Application of Matrix Solid-Phase Dispersion and High-Performance Liquid Chromatography for Determination of Sulfonamides in Honey. Journal of AOAC INTERNATIONAL 2008, 91, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Maudens, K.E.; Zhang, G.-F.; Lambert, W.E. Quantitative analysis of twelve sulfonamides in honey after acidic hydrolysis by high-performance liquid chromatography with post-column derivatization and fluorescence detection. Journal of Chromatography A 2004, 1047, 85–92. [Google Scholar] [CrossRef]
- Tölgyesi, Á.; Berky, R.; Békési, K.; Fekete, S.; Fekete, J.; Sharma, V.K. ANALYSIS OF SULFONAMIDE RESIDUES IN REAL HONEY SAMPLES USING LIQUID CHROMATOGRAPHY WITH FLUORESCENCE AND TANDEM MASS SPECTROMETRY DETECTION. Journal of Liquid Chromatography & Related Technologies 2013, 36, 1105–1125. [Google Scholar] [CrossRef]
- Tsai, W.-H.; Chuang, H.-Y.; Chen, H.-H.; Wu, Y.-W.; Cheng, S.-H.; Huang, T.-C. Application of sugaring-out extraction for the determination of sulfonamides in honey by high-performance liquid chromatography with fluorescence detection. Journal of Chromatography A 2010, 1217, 7812–7815. [Google Scholar] [CrossRef] [PubMed]
- Economou, A.; Petraki, O.; Tsipi, D.; Botitsi, E. Development of a liquid chromatography–tandem mass spectrometry method for the determination of sulfonamides, trimethoprim and dapsone in honey and validation according to Commission Decision 2002/657/EC for banned compounds [corrected]. Talanta 2012, 97, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Verzegnassi, L.; Savoy-Perroud, M.C.; Stadler, R.H. Application of liquid chromatography–electrospray ionization tandem mass spectrometry to the detection of 10 sulfonamides in honey. Journal of Chromatography A 2002, 977, 77–87. [Google Scholar] [CrossRef]
- Tamošiūnas, V.; Padarauskas, A. Comparison of LC and UPLC Coupled to MS–MS for the Determination of Sulfonamides in Egg and Honey. Chromatographia 2008, 67, 783–788. [Google Scholar] [CrossRef]
- Thompson, T.S.; Noot, D.K. Determination of sulfonamides in honey by liquid chromatography–tandem mass spectrometry. Analytica Chimica Acta 2005, 551, 168–176. [Google Scholar] [CrossRef]
- Krivohlavek, A.; Šmit, Z.; Baštinac, M.; Žuntar, I.; Plavšc-Plavšic, F. The determination of sulfonamides in honey by high performance liquid chromatography – mass spectrometry method (LC/MS). Journal of Separation Science 2005, 28, 1434–1439. [Google Scholar] [CrossRef] [PubMed]
- Hammel, Y.-A.; Mohamed, R.; Gremaud, E.; LeBreton, M.-H.; Guy, P.A. Multi-screening approach to monitor and quantify 42 antibiotic residues in honey by liquid chromatography–tandem mass spectrometry. Journal of Chromatography A 2008, 1177, 58–76. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.I.; Pettis, J.S.; Smith, I.B.; Chu, P.-S. Multiclass Determination and Confirmation of Antibiotic Residues in Honey Using LC-MS/MS. Journal of Agricultural and Food Chemistry 2008, 56, 1553–1559. [Google Scholar] [CrossRef] [PubMed]
- Vidal, J.L.M.; Aguilera-Luiz, M.d.M.; Romero-González, R.; Frenich, A.G. Multiclass Analysis of Antibiotic Residues in Honey by Ultraperformance Liquid Chromatography−Tandem Mass Spectrometry. Journal of Agricultural and Food Chemistry 2009, 57, 1760–1767. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, R.; Policastro, B.; Thomas, S.; Rice, D. Analysis and Occurrence of 14 Sulfonamide Antibacterials and Chloramphenicol in Honey by Solid-Phase Extraction Followed by LC/MS/MS Analysis. Journal of Agricultural and Food Chemistry 2008, 56, 3509–3516. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Gao, L.; Zhao, Y.; Peng, W.; Chen, Z. Simultaneous determination of metronidazole, chloramphenicol and 10 sulfonamide residues in honey by LC–MS/MS. Anal. Methods 2013, 5, 1283–1288. [Google Scholar] [CrossRef]
- Commission, E. Council Directive 96/23/EC of 29 April 1996; 1996Vol. L125, pp. 10–32.
- Commission, E. Consolidated text: Commission Decision of 14 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results (notified under document number C(2002) 3044) (Text with EEA relevance) (2002/657/EC)Text with EEA relevance. Official Journal 2002, L 221, 8. [Google Scholar]
- Commission, E. SANCO/2004/2726-rev 4-December 2008. GUIDELINES FOR THE IMPLEMENTATION OF DECISION 2002/657/EC 2 Availabe online: https://food.ec.europa.eu/system/files/2016-10/cs_vet-med-residues_cons_2004-2726rev4_en.pdf.
- ISO. ISO-11843-1:1997.Capability of detection. Part1: terms and definitions. International Organization for Standardization (ISO): 1997.
- ISO. ISO-11843-2:2000. Capability of detection.Part 2: methodology in the linear calibration case. International Standard Organization (ISO): 2000.
- Kaufmann, A.; Roth, S.; Ryser, B.; Widmer, M.; Guggisberg, D. Quantitative LC/MS-MS determination of sulfonamides and some other antibiotics in honey. J AOAC Int 2002, 85, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Commission, E. SANCO 2006/3228; 2006.
- Shao, B.; Dong, D.; Wu, Y.; Hu, J.; Meng, J.; Tu, X.; Xu, S. Simultaneous determination of 17 sulfonamide residues in porcine meat, kidney and liver by solid-phase extraction and liquid chromatography–tandem mass spectrometry. Analytica Chimica Acta 2005, 546, 174–181. [Google Scholar] [CrossRef]
- Heller, D.N.; Ngoh, M.A.; Donoghue, D.; Podhorniak, L.; Righter, H.; Thomas, M.H. Identification of incurred sulfonamide residues in eggs: methods for confirmation by liquid chromatography–tandem mass spectrometry and quantitation by liquid chromatography with ultraviolet detection. Journal of Chromatography B 2002, 774, 39–52. [Google Scholar] [CrossRef]
- Zotou, A.; Vasiliadou, C. Selective Determination of Sulfonamide Residues in Honey by SPE-RP-LC with UV Detection. Chromatographia 2006, 64, 307–311. [Google Scholar] [CrossRef]
- Mitrowska, K.; Posyniak, A.; Zmudzki, J. Determination of malachite green and leucomalachite green in carp muscle by liquid chromatography with visible and fluorescence detection. J Chromatogr A 2005, 1089, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Oka, H.; Ikai, Y.; Matsumoto, H.; Miyazaki, Y.; Nagase, H. Application of ion-exchange cartridge clean-up in food analysis. V. Simultaneous determination of sulphonamide antibacterials in animal liver and kidney using high-performance liquid chromatography with ultraviolet and mass spectrometric detection. J Chromatogr A 2000, 898, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Commission, E. Commission Decision 2002/657/EC of 12 August 2002; Off. J. Eur. Commun.: 2002; Vol. L 221.
Figure 1.
Chromatograms of “blank” honey samples with 20 µg/kg of the internal standards (a) and honey samples with 20 µg/kg of the internal standards and spiked with 50 µg/kg (5 µg/kg for dapsone) of each of the compounds of interest (b).
Figure 1.
Chromatograms of “blank” honey samples with 20 µg/kg of the internal standards (a) and honey samples with 20 µg/kg of the internal standards and spiked with 50 µg/kg (5 µg/kg for dapsone) of each of the compounds of interest (b).
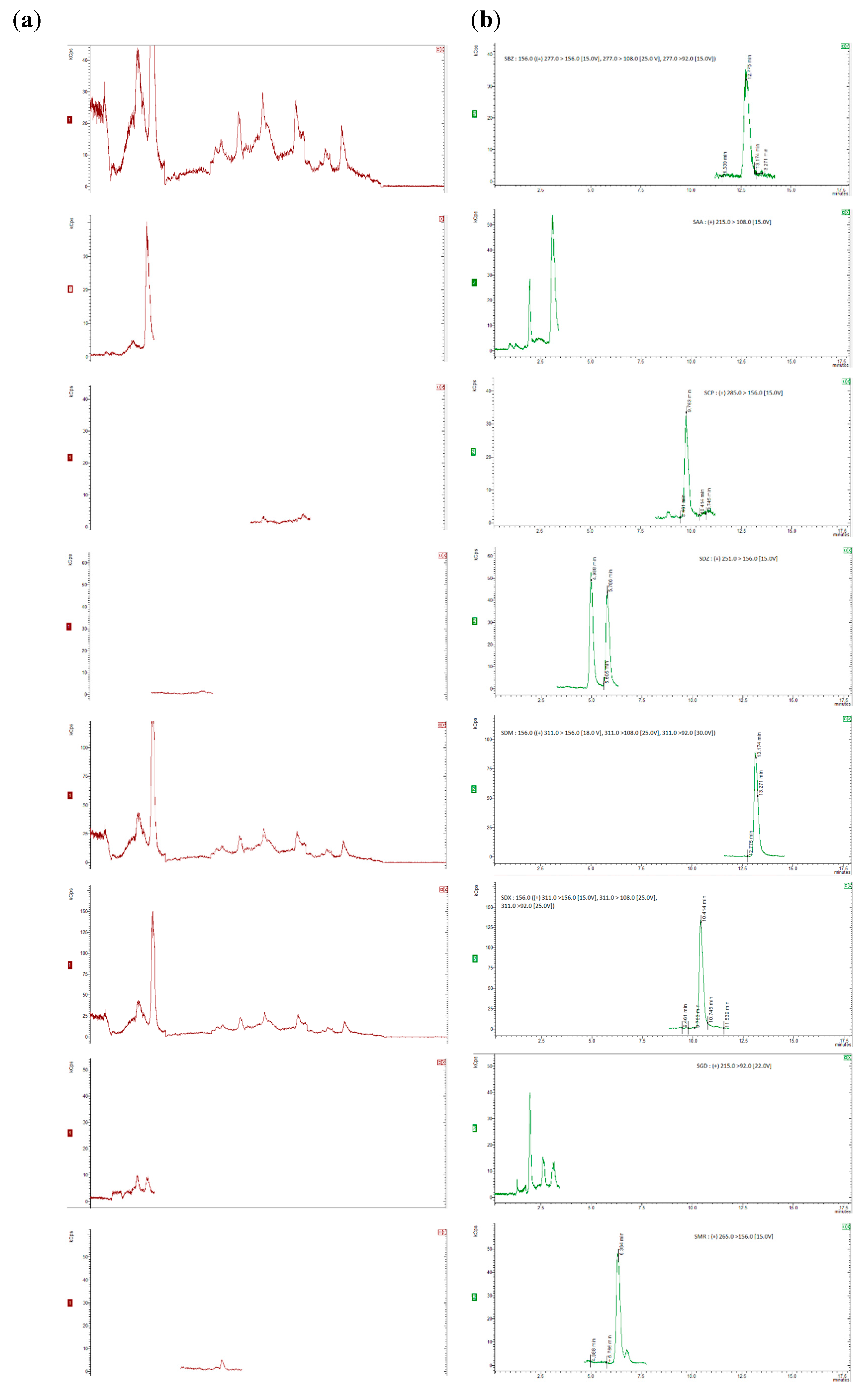
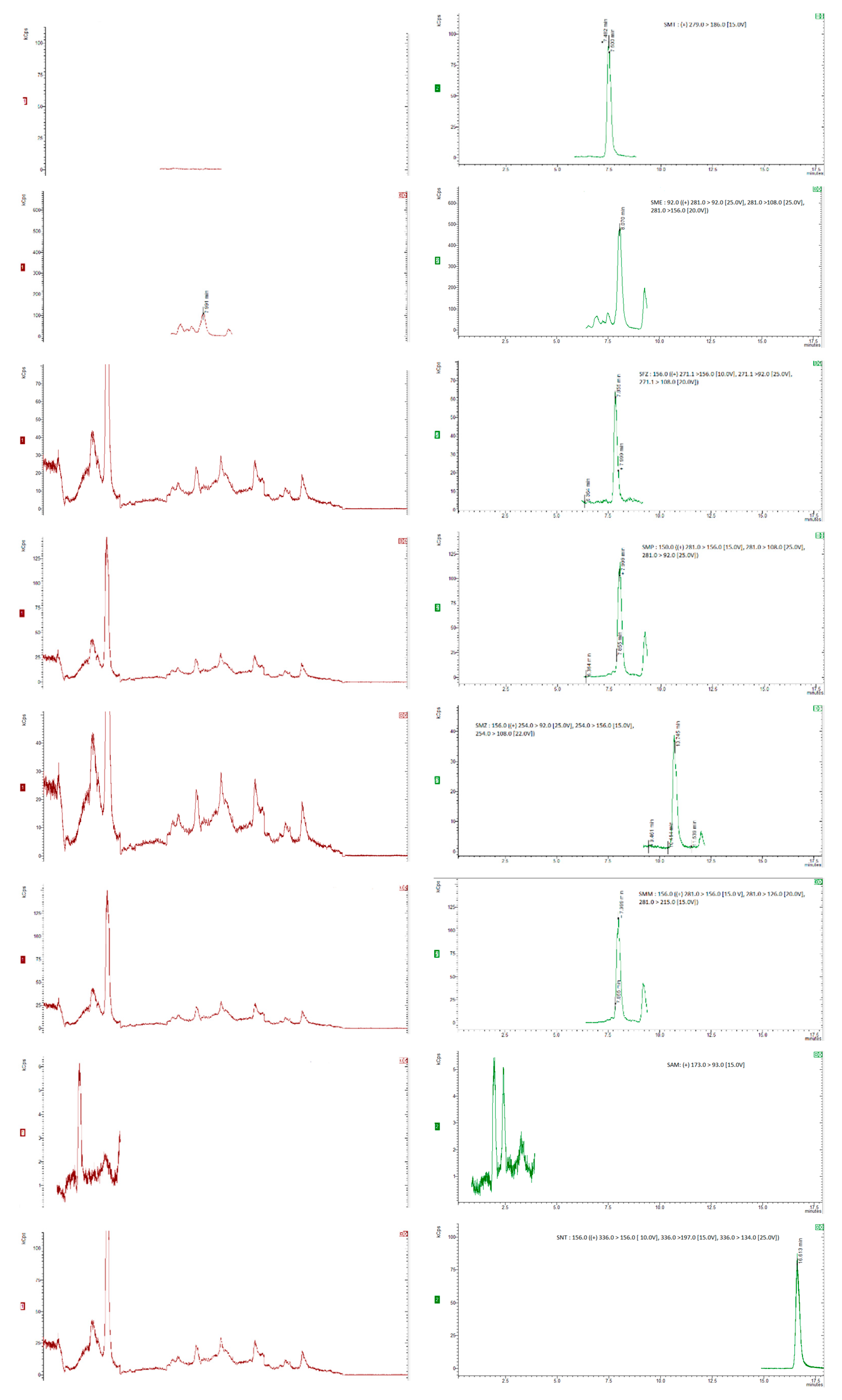
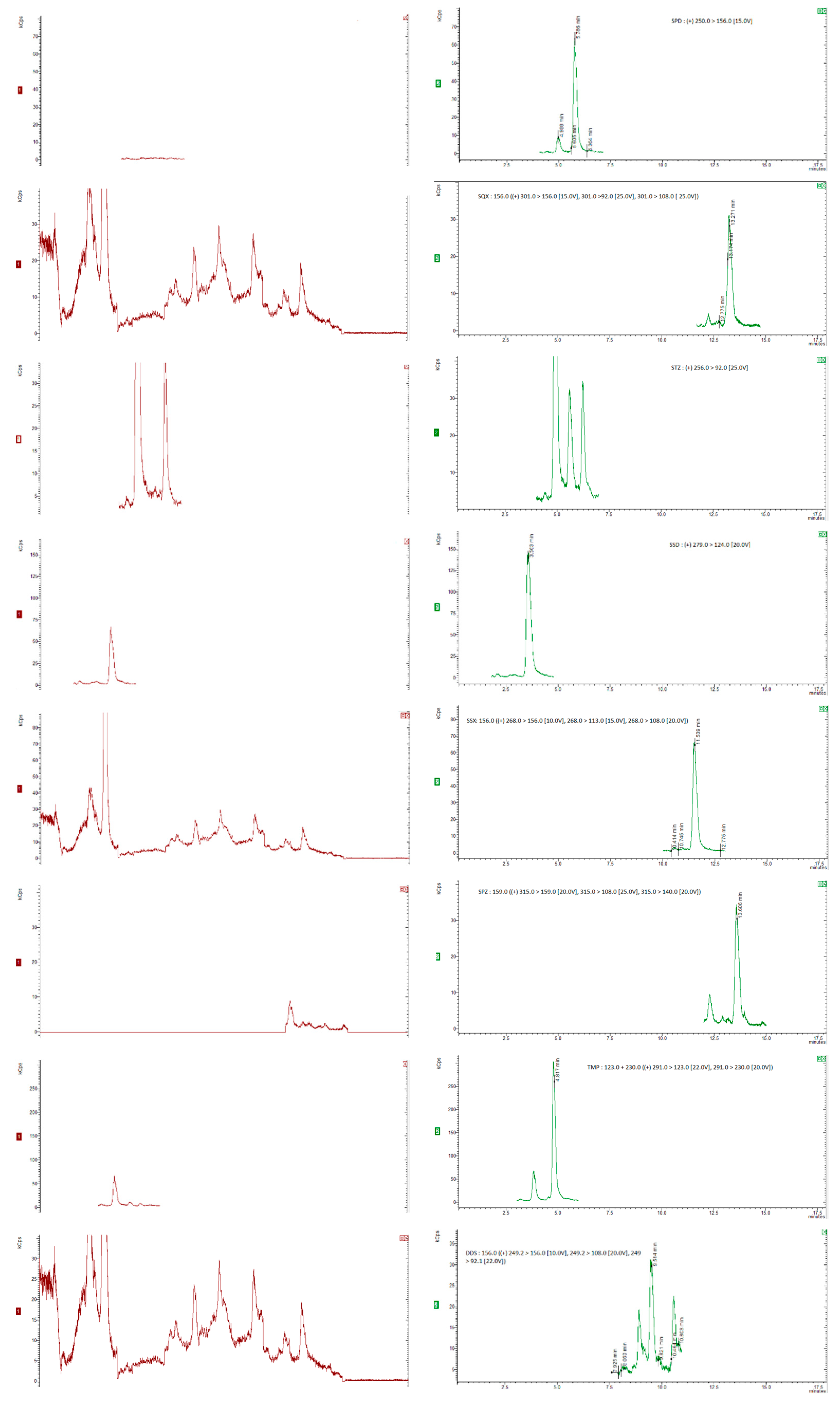
Figure 2.
Chromatograms of “blank” honey samples with 20 µg/kg internal standards (a) and honey samples with 20 µg/kg internal standards and spiked with 50 µg/kg of each of the compounds of interest (b).
Figure 2.
Chromatograms of “blank” honey samples with 20 µg/kg internal standards (a) and honey samples with 20 µg/kg internal standards and spiked with 50 µg/kg of each of the compounds of interest (b).
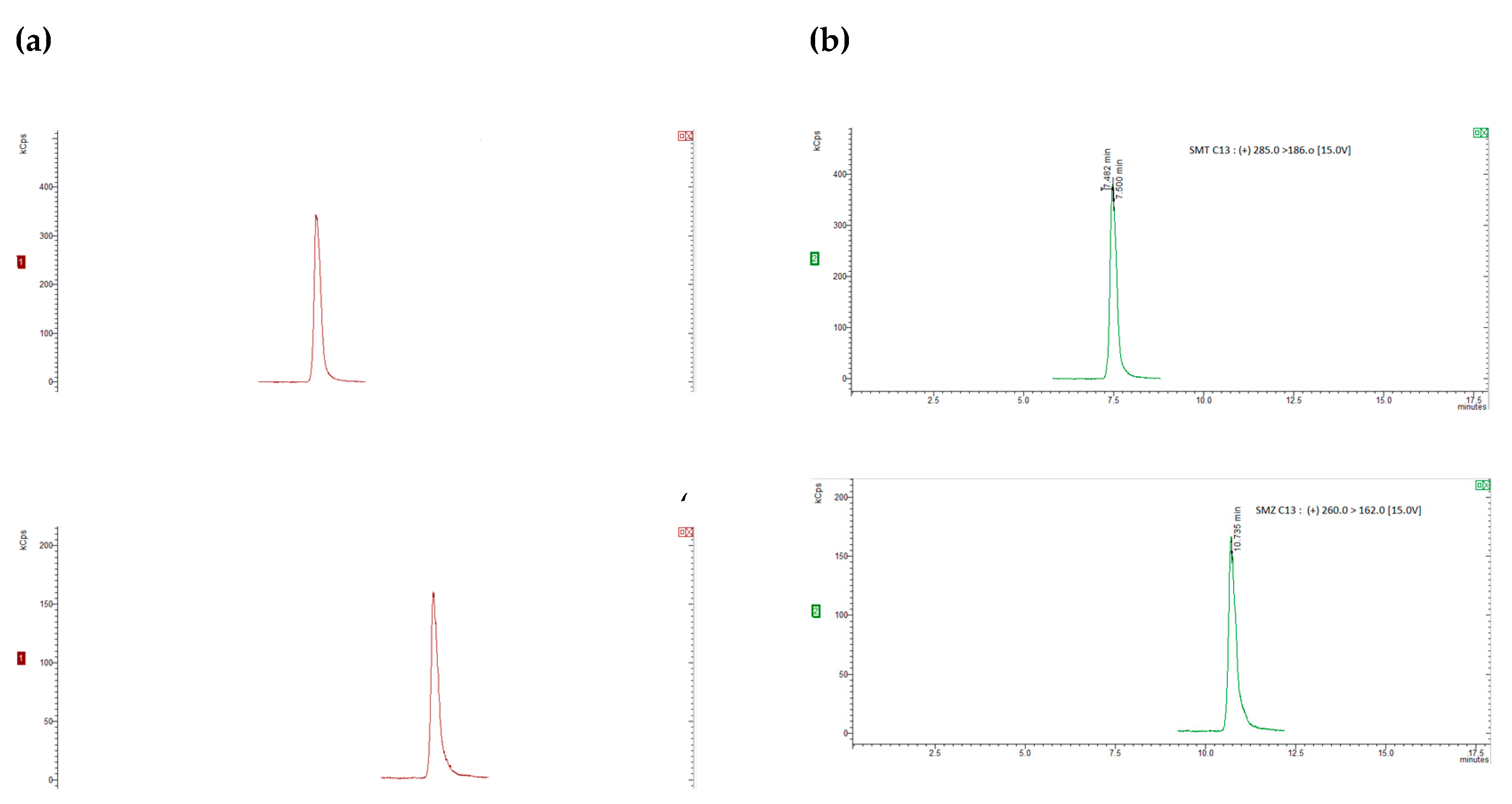

Table 1.
The values of uncertainties for the target compounds (µg/kg).
| Compounds | Standard Uncertainty uc (µg/kg) |
Expanded Uncertainty ue (k=2) (µg/kg) |
Final results |
|---|---|---|---|
| Sulfaguanidine | 4,386 | 8,772 | 50 ± 8,772 |
| Sulfacetamide | 4,309 | 8,618 | 50 ± 8,618 |
| Sulfanilamide | 6,155 | 12,310 | 50 ± 12,310 |
| Sulfisomidine | 4,157 | 8,314 | 50 ± 8,314 |
| Sulfadiazine | 3,184 | 6,368 | 50 ± 6,368 |
| Sulfathiazole | 4,030 | 8,060 | 50 ± 8,060 |
| Sulfapyridine | 3,468 | 6,936 | 50 ± 6,936 |
| Sulfamerazine | 3,330 | 6,660 | 50 ± 6,660 |
| Sulfamethazine | 2,773 | 5,546 | 50 ± 5,546 |
| Sulfamethizole | 3,433 | 6,866 | 50 ± 6,866 |
| Sulfamethoxypyridazine | 2,434 | 4,868 | 50 ± 4,868 |
| Sulfamonomethoxine | 3,498 | 6,996 | 50 ± 6,996 |
| Sulfameter | 3,012 | 6,024 | 50 ± 6,024 |
| Sulfachloropyridazine | 3,309 | 6,618 | 50 ± 6,618 |
| Sulfadoxine | 3,613 | 7,226 | 50 ± 7,226 |
| Sulfamethoxazol | 3,167 | 6,334 | 50 ± 6,334 |
| Sulfisoxazole | 3,195 | 6,390 | 50 ± 6,390 |
| Sulfabenzamide | 2,853 | 5,706 | 50 ± 5,706 |
| Sulfadimethoxine | 2,995 | 5,990 | 50 ± 5,990 |
| Sulfaquinoxaline | 3,053 | 6,106 | 50 ± 6,106 |
| Sulfaphenazole | 3,060 | 6,120 | 50 ± 6,120 |
| Sulfanitran | 4,484 | 8,968 | 50 ± 8,968 |
| Trimethoprim | 4,985 | 9,970 | 50 ± 9,970 |
| Dapsone | 0,421 | 0,842 | 5 ± 0,842 |
Table 2.
Decision limits (CCα) and detection capabilities (CCβ) of the method (µg/kg).
| Compounds | CCα (µg/kg) | CCβ (µg/kg) |
|---|---|---|
| Sulfaguanidine | 3,50 | 4,33 |
| Sulfacetamide | 2,44 | 3,02 |
| Sulfanilamide | 22,12 | 27,36 |
| Sulfisomidine | 7,78 | 9,62 |
| Sulfadiazine | 2,30 | 2,85 |
| Sulfathiazole | 7,64 | 9,45 |
| Sulfapyridine | 5,58 | 6,90 |
| Sulfamerazine | 4,37 | 5,40 |
| Sulfamethazine | 2,16 | 2,67 |
| Sulfamethizole | 3,53 | 4,37 |
| Sulfamethoxypyridazine | 5,59 | 6,91 |
| Sulfamonomethoxine | 5,94 | 7,35 |
| Sulfameter | 9,36 | 11,57 |
| Sulfachloropyridazine | 7,20 | 8,91 |
| Sulfadoxine | 7,35 | 9,09 |
| Sulfamethoxazol | 7,89 | 9,76 |
| Sulfisoxazole | 7,87 | 9,74 |
| Sulfabenzamide | 7,66 | 9,48 |
| Sulfadimethoxine | 7,61 | 9,42 |
| Sulfaquinoxaline | 8,22 | 10,16 |
| Sulfaphenazole | 8,34 | 10,32 |
| Sulfanitran | 9,52 | 11,77 |
| Trimethoprim | 7,67 | 9,48 |
| Dapsone | 2,45 | 3,03 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



