
Submitted:
24 December 2023
Posted:
26 December 2023
You are already at the latest version
Abstract
The crosstalk between gut microbiota, intestinal epithelial cells, and innate and adaptive immune system governs the maintenance of the intestinal homeostasis. Any interference in this tight dialogue and in the processes preserving cellular homeostasis (e.g., autophagy) may dysregulate the immune response and impair the clearance of harmful bacteria favoring the dysbiotic alteration of the microbic flora that leads to chronic inflammation. Gut dysbiosis is strongly associated with gastrointestinal inflammatory disorders, among them the inflammatory bowel disease (IBD). This review discusses the current knowledge on IBD, from the genetic background of high-risk patients to the molecular mechanisms underlying the disease, the contribution of the microbic flora, and the role of autophagy in intestinal epithelial cells and provides the state of art regarding the targeted-nutritional approaches aimed to restore the beneficial crosstalk between an “anti-inflammatory” microbiota and the host. Analysis of the molecular pathogenesis of IBD will help identify genetic and diet-associated risk factors and thus suggest personalized strategies to prevent and manage the disease to improve quality of life with long-term maintenance of the remission phase.
Keywords:
IBD
; Crohn's disease
; ulcerative colitis
; inflammation
; immune system
; autophagy
; microbiota
; probiotics
; fecal microbiota transplantation
1. Introduction
The inflammatory bowel disease (IBD) comprises a variety of widespread gastrointestinal diseases whose incidence has been increasing in recent decades, especially in industrialized countries [1]. Epidemiologic data reporting the increased incidence of IBD are supported by the view that the worldwide adoption of a Western lifestyle may increase the occurrence of the disease [2].
IBD includes chronic inflammatory idiopathic disorders within the gastrointestinal tract, such as Crohn’s disease (CD) and ulcerative colitis (UC), characterized by the alternation of exacerbation and remission phases. CD and UC share the symptoms and the chronic inflammatory state, yet they differ in that CD may involve the whole gastrointestinal tract displaying a discontinuous pattern with the inflamed tissues alternated with non-inflamed tissues, whereas UC is localized in the mucosa and submucosa of the colonic region, partially or entirely, with a continuous inflamed pattern [3]. Both CD and UC may lead to the obstruction of the gastrointestinal tract and cause nausea, diarrhea with bleeding, vomiting, fever, fatigue and weakness, loss of weight, abdominal pain, cramps, as well as intestinal perforation, bloody ulceration, and crypt abscesses [4].
Although the etiopathogenesis is not fully understood, IBD is a multifactorial disease whose rise and progress rely on a complex interplay between genetic, environmental, and microbiological factors that leads to an uncontrolled immune system activation [5]. About 300 genes directly associated with IBD have been emerged by genome wide association studies (GWAS) and meta-analysis, and their polymorphisms may expose people to a high risk of developing the disease [6].
Recent GWAS have identified novel single nucleotide polymorphisms (SNPs) whose genetic variants may disrupt the intestinal homeostasis and increase host genetic susceptibility [7]. Of note, genetic variants in autophagy-related genes results in impaired clearance of intercellular bacteria and an increase in the release of inflammatory cytokines, promoting the onset of inflammatory chronic disease. This highlights how autophagy serves as a key mechanism in preventing the development of IBD, by modulating immune system functions, production of cytokines, and participating in pathogen clearance [8]. Besides, also epigenetic mechanisms may contribute to IBD development and progression. DNA methylation and miRNAs interfere in T-cell differentiation, cytokines regulation, Th17 molecular signaling, and autophagy [9].
Metagenomic studies reported that patients suffering from IBD present an altered balance in gut bacteria function and composition, a condition known as dysbiosis, compared to the healthy group. The composition of commensal bacteria results in a reduced bacterial biodiversity with a decrease of anaerobic strains, such as Firmicutes and Bacteroides, and predominant facultative anaerobic strains, such as Proteobacteria [10]. This may trigger primary inflammation that, if exacerbated, may evolve in chronic inflammation and impact on the permeability of intestinal epithelial barrier compromising gut mucosa structure [11].
2. The Intestinal Epithelial Barrier
The intestinal epithelial barrier plays a pivotal role in preserving the delicate balance between defense mechanisms and symbiotic interaction within the gut. Beyond its primary role in processing and absorbing food and nutrients, the intestinal epithelial barrier acts as biochemical and physical barrier against pathogens, toxins, and dietary antigens, establishing a primary defense line from the contents of the bowel lumen [12,13]. Simultaneously, it creates the proper microenvironment, preventing excessive colonization of commensal bacteria in the luminal compartment and facilitating the harmonious interaction between the host and the microbiota [14] (Figure 1).
The intestinal epithelium, composed of a single layer of columnar polarized cells, undergoes a self-renewal process approximately every 5-7 days. This renewal is facilitated by a population of intestinal pluripotent stem cells residing in specific niches within the inner part of the epithelium, called Lieberkühn crypts [15]. These stem cells actively proliferate, migrate to the upper surface along the crypt-luminal axis, differentiate into specialized cells and undergo apoptosis within a few days. The coordinated presence and activity of these stem cells ensures the continuous regeneration of columnar polarized cells, maintaining the structural integrity of the gut [16].
The differentiated cells deriving from intestinal stem cells (ISC) include enterocytes, Paneth cells, enteroendocrine cells, and goblet cells. Enterocytes, comprising approximately 80% of specialized cell types, play a crucial role in the absorption of dietary compounds by enhancing their adsorptive area with microvilli, particularly pronounced in the small intestine. Paneth cells support the intestinal stem cell population through signaling molecule secretion and regulate microbial flora with antimicrobial peptides and immunomodulating signals. A decreased expression of Paneth cell-derived α-defensins and defective anti-microbial peptides have been observed in patients with CD [17,18]. Enteroendocrine cells produce hormones governing food digestion, absorption, and appetite, also acting as communication molecules with the immune system [16,19]. Goblet cells, the specialized entities responsible for secretion, release a glycoprotein network which composes the mucus layer that covers the intestinal epithelium [20]. Mucin proteins produced by these cells create a gel-like structure that prevents the direct contact between the bowel lumen and epithelial cells representing the first barrier of defense against bacteria and inflammation in the intestine. This structure not only provides an adhesion substrate for the microbiota niche but also simultaneously impedes the transepithelial invasion of microorganisms into the systemic circulation [21]. The mucus layer exhibits antimicrobial properties by releasing secretory immunoglobulin A and antimicrobial molecules targeting viruses, fungi, and bacteria. The structural characteristics of the mucus, including its composition and thickness, maintain intestinal health. Several factors, including microbes, growth factors, neuropeptides, pro-inflammatory cytokines, and toxins, may affect the production of mucins and the architecture of mucus layer disrupting gut homeostasis and triggering an inflammatory response [22].
In the subepithelial layer, the gastrointestinal tract holds the gut-associated lymphoid tissue (GALT), which includes Peyer’s patches and specialized immune cells such as macrophages, dendritic cells, plasma cells, and lymphocytes. These cells ensure the integrity of the intestinal barrier and trigger immune responses in the presence of luminal antigens [23]. Intestinal macrophages, despite the diminished inflammatory activity in response to microbial antigens or host cytokines, promote the differentiation of regulatory T-cells. They also limit the Th1 and Th17 responses, while preserve their phagocytic and bactericidal functions [24]. Dendritic cells actively monitor the intestinal microenvironment and initiate adaptive immune responses through the release of molecular signals [25].
Adjacent epithelial cells are firmly connected to the apical surface through tight junctions, a transmembrane protein family that includes claudins and occluding. The multiprotein complexes, interacting with the actin-myosin cytoskeleton via scaffold molecules [26], maintains cell polarity and ensure the integrity of gut epithelial. Tight junctions also regulate paracellular transport by creating pores in the intercellular spaces, facilitating the transport of selected small molecules from the apical to the basolateral side [27]. Claudins are divided in two categories based on their function: some create pores facilitating selective ion passage, while others contribute to enhance barrier tightness. This dual functionality results in a distinctive expression pattern not only in the crypt and luminal surface, but also throughout the entire gastrointestinal tract [28]. Occludin is a component of the intracellular domain of tight junctions and is necessary for their assembly. The phosphorylation of specific residues affects occludin localization: low phosphorylation levels lead to cytoplasmic accumulation, while high phosphorylation levels induce positioning within the tight junction complex [29].
The integrity of intestinal epithelial barrier is guaranteed not only by tight junctions but also by anchoring junctions. Adherens junctions, a key component of anchoring junctions, participate in cell adhesion. They facilitate the connection between adjacent cells by engaging in cytoskeleton interaction through transmembrane adhesion molecules. The main actor in adherens junctions family is E-cadherin, which provides a calcium-dependent intercellular adhesion. On the cytoplasmic side, E-cadherin forms multiprotein complexes with members of the catenin superfamily fixing to the actin cytoskeleton [30,31]. Defective epithelial junctions and alterations in the genes encoding epithelial junctions have been identified as a significant cause of IBD. For instance, SNPs in CDH1, which encodes for E-cadherin, have been associated with UC [32]. Certain pro-inflammatory cytokines, such as TNF-α and IFN-γ, have been found to raise the permeability of tight junctions leading to the breakdown of the epithelial barrier function, pathogens entry and sustained inflammation associated with IBD [33,34].
3. Gut Microbiota
Human gut microbiota actively regulates host health, physiology, metabolism, and immune system, and prevents pathogens colonization by establishing a mutualistic relationship with the host, effectively overseeing the health status of the organism [35].
Microbiota includes up to 1000 different microbic species and the genomic material of these microorganisms, called microbiome, is one hundred times greater than human genome [36]. The composition and the abundance of commensal microorganisms is highly variable in term of species and is based on the anatomical location along the gastrointestinal tract. Microbiota population changes in each human and the biodiversity creates a unique microbiome profile that is considered a typical feature of each individual [37].
The microbic flora definition begins during childbirth and the natural or cesarean delivery, and its composition is further influenced by breast feeding or formula feeding [38]. Since the first years of age, commensal bacteria establish a relationship with the immune system enabling the maintenance of homeostasis with the host. This equilibrium remains stable unless external disturbances (e.g., aging, diet, antibiotics, and illnesses) disrupts this balance [39].
In healthy individuals, the intestinal microbiota consists of a large community of microbial species, represented by Bacteroidetes and Firmicutes as predominant phyla [36]. Commensal bacteria accomplish the fermentation of non-digestible carbohydrates from the diet producing short-chain fatty acids (SCFAs) that are then transported into the intestinal epithelial cells [40]. The SCFA butyrate is the most absorbed microbiota metabolite and represents the main source of energy for colonocytes, helping the proliferation and the differentiation of intestinal epithelial cells [41]. SCFAs possess anti-inflammatory properties by promoting T-cell differentiation, exert an anti-carcinogenic role, decrease oxidative stress, and participate in the maintenance of intestinal mucosa integrity and gut permeability. Collectively, they show the ability to modulate gene expression through the activation of G-protein coupled receptors (GPR41, GPR43, and GPR109a) and the inhibition of the histone deacetylase [42,43,44,45,46].
Within the intestinal microenvironment, the SCFAs decrease the release of pro-inflammatory cytokines, such as IL-13, IL-6, IL-12, and TNF-α, preventing interference with the expression of tight junction, consequently improving the permeability of the gut barrier [47]. The anti-inflammatory properties of SCFAs are mediated by the binding with GPR109a in the dendritic cells and macrophages. The activation of this receptor mediates the expansion of Treg cells with the increase of IL-10 and the reduction of Th17 and IL-6 [48]. The activation of GPR43 attenuates NLRP3 response and the consequent IL-18 secretion [49]. Through the inhibition of HDACs, commensal bacteria interfere with T-cell chemotaxis in the microenvironment and create an immunological tolerance that leads to an anti-inflammatory phenotype balancing immune homeostasis [50]. Mouse models lacking a eubiotic gut microbiota display immature lymphoid structures, low lymphocyte population, and reduced production of antimicrobial peptides [51].
Gut microbiota represents a natural defense barrier against pathogens infection [52]. Commensal microorganisms prevent pathogens invasion competing for nutrients within the microbial niche of the gastrointestinal tract. The fermentation of Bifidobacteria decreases the pH of the microenvironment and prevents the colonization of E. coli [53]. In addition, gut flora counteracts bacterial invasion through the release of molecules, such as lipopolysaccharides and flagellin, that stimulate the Toll-like receptor signaling triggering immune system activation [54].
Dietary habits considerably influence the composition of intestinal microbiota [55]. In addition to diet, the premature and excessive use of antibiotics contributes to intestinal dysbiosis, and this has been recognized over a long period to increase the risk of intestinal diseases [56].
Intestinal dysbiosis is defined as a pathogenic change in the functions and composition of gut microflora and is associated with intestinal inflammatory diseases [57,58]. A dysbiotic microbiota can overwhelm self-defense mechanisms, resulting in excessive oxidative stress and inflammasome formation. Through the release of toxins, the reduced production of beneficial metabolites, and the disruption of epithelial integrity, intestinal pathogens hyper-activate the immune system and stimulate a chronic inflammatory status that may evolve in a high-grade dysplasia. Long-standing colitis in IBD patients can lead to a particular subtype of colorectal cancer known as colitis-associated colorectal cancer (CAC) [59].
4. Inflammatory and Molecular Mechanisms in IBD
Despite the pathogenesis of IBD is not yet fully understood, it is thought to result from abnormal immune reactions towards microorganisms in genetically susceptible individuals [60]. The immune system in the intestine has a hard responsibility of rapidly and efficiently responding to harmful bacteria, while also tolerating beneficial microbes and food antigens.
The dynamic crosstalk between microbiota, intestinal epithelial cells, and local immune cells represents one of the fundamental features for maintaining intestinal homeostasis and for mounting protective immunity to pathogens [61]. Perturbations in this fine balance, due to genetic and/or external factors (e.g., lifestyle, diet, stress, anxiety, and depression) [62], may result in aberrant and chronic intestinal inflammation, tissue damage, and ultimately in the onset of IBD [63,64].
Several evidence indicates that the development of IBD is influenced by both dysfunctional innate and adaptive immune pathways within the intestine.
The innate immune response represents the primary defense mechanism against harmful microorganisms (Figure 2). Defective intestinal epithelial barrier and increased permeability are two of the major causes of intestinal inflammation and have long been observed in patients with both CD and UC [65,66].
Innate immune cells, such as macrophages, dendritic cells (DCs), neutrophils, natural killer (NK) cells, and innate lymphoid cells (ILCs), as well as epithelial cells, detect bacterial antigens by their pattern recognition receptors (PRRs) expressed both extra- and intra-cellularly [67]. PRRs include Toll-like receptors (TLRs), NOD-like receptors (NLRs), C-type lectin receptors (CLRs), and RIC-1-like receptors (RLRs) [61]. These receptors, by recognizing conserved structural motifs on microorganisms (known as pathogen-associated molecular patterns (PAMPs) lead to the activation of several signaling pathways and the production of pro-inflammatory cytokines, chemokines, and antimicrobial ensuring an effective innate response against pathogens [68]. Innate immune cells also activate effector cells, such as T helper lymphocytes, and inhibit the activation of T regulatory cells [69].
In the healthy intestine, basal PRR activation maintains barrier function and commensal composition, but aberrant PRR signaling may contribute to the pathophysiology of IBD.
Evidence suggests that the over-expression of certain TLRs and down-regulation of TLR antagonists may lead to an improper reaction to commensal bacteria playing a part in the predisposition and perpetuation of IBD [70]. For instance, TLR4 expression was reported to be elevated in colonic tissue of UC and CD patients [71]. The genetic polymorphism in the NOD2 gene determines a significantly higher incidence of CD associated with decreased functional integrity of the epithelium [72]. Other evidence suggests that loss-of-function mutations of NOD2 lead to reduced NF-ĸB activation and inadequate response which might result in reduced antibacterial agent production and pathogenic microbial invasion [73]. Loss-of-function in NOD2 may also results in the lack of inhibition of TLR2 stimulation, leading to the activation of inflammatory pathways and excessive Th-1 responses [74].
The inflammasome is considered one of the prominent mediators of innate immunity and recent studies have demonstrated that its activation plays an essential role in the pathogenesis of IBD. Inflammasome processes the precursor of IL-1β and IL-18 which regulate Th17 and Th1 cells, respectively, amplifying the immune response [75].
A significant increased activation of the IL-1 system was found in the intestinal mucosa of CD and UC patients compared with control group [76]. IL-1β is a pro-inflammatory cytokine mainly secreted by macrophages, which synergistically acts with TNF-α and IL-6, to induce intestinal inflammation [77].
IL-18 results overexpressed in intestinal lesions of patients with CD. IL-18 facilitates IFN-γ production by Th1 cells and Th2 cytokines, in conjunction with IL-12 and IL-2, respectively [78].
Some studies provide evidence that loss or over-activation of NLRP3, one of the most studied NOD-like receptors that form an inflammasome, may break down the immune balance leading to the onset of intestinal inflammation. Over-activation of NLRP3 caused by various genetic abnormalities lead to development of colitis [79], whereas deficiency of NLRP3 and inflammasome related genes may induce more severe colitis in mice [80].The inflamed intestinal mucosa recruits a large number of activated M1 macrophages characterized by secreting IL-1, IL-6, TNF-α, IL-12, and IL-23, as well as producing reactive oxygen species (ROS), and proteases that could degrade the extracellular matrix, triggering dramatic inflammation and phagocytosis of pathogens [61]. M1 macrophages also induce tight junction break down, epithelial barrier damage, epithelial cell apoptosis, and T helper responses, causing tissue damage and aggravating the inflammatory response [81].
The modulation of macrophage polarization by inhibiting the pro-inflammatory M1 subset and/or inducing the anti-inflammatory M2 subset may attenuate IBD. M2 macrophages promote resolving inflammation and remodeling tissue by producing several factors, especially IL-10 [82]. The lack of macrophage-derived IL-10 or inhibition of M2-like phenotype may result in macrophage hyper-responsiveness and in the exacerbation of colitis [83].
Gut DCs are activated with high levels of specific TLRs in IBD patients and lead to the production of high levels of IL-12 and IL-6, altering the mucosa and triggering inflammation. They further promote the inflammatory state by migrating to peripheral lymphoid tissue, where they generate antigen-specific T-cell responses [84].
ILCs are involved in the first line of immune response via the instant release of huge amounts of effector cytokines that orchestrate further immune reactions [85]. ILCs can be categorized into three main subgroups: ILC1s, ILC2s, and ILC3s. ILC1s, including cytotoxic NK cells, amplify immune responses against intracellular pathogens, via an extensive release of IFN-γ and TNF-α, whereas ILC2s and ILC3s are characterized by the secretion of effector cytokines IL-5, IL-13, IL-9 and IL-17A, IL-22, respectively. Tight control of ILCs number and their activation status guarantee barrier integrity and tissue homeostasis without the induction of chronic immune responses. Remarkable numerical changes in ILC populations have been found in inflamed intestine tissues. In particular, ILC1s are increased at sites of active inflammation in patients suffering from IBD [86].
Innate immune cells also promote antigen presentation and T cell activation participating in the crosstalk between innate and adaptive immune response [69] (Figure 3).
CD4+ T cells are the key players of the adaptive immune response, and they cooperate with the molecules and cells of the innate immune system to mount an effective immune response. The activation and the differentiation of Th0 cells into Th1, Th2 or Th17 is essential for the clearance of specific pathogens. IL-12/23 group is produced by activated antigen-presenting cells and promotes IBD by driving pathogenic T cell responses. In particular, IL-12 may promote the differentiation of naive CD4+ T cells into IFN-γ producing Th1 cells and the proliferation and effector functions of NK cells and cytotoxic T cells [87], whereas IL-23 reinforce Th17 cell response and antagonizes the anti-inflammatory responses of Treg cell responses to promote [88].
Abnormal development of activated T cells may lead to the onset of inflammation by an excessive release of cytokines and chemokines. Increased activity of Th1 producing higher amounts of IL-2, TNF-α and IFN-γ and Th2 characterized by an excessive production of IL-4, IL-5 and IL-13 have been observed in CD and UC, respectively [89,90]. Cytokines released by activated Th1 and Th2 are considered mediators of the lesions in inflamed mucosa of IBD patients. Th1 cells accumulate in the intestinal tract of individuals with CD and are directly associated with disease. Th1 cytokines activate the transcription factor STAT1 leading to up-regulation of transcription factor T-ß, and recruitment of macrophages, NK cells, and CD8+ T cells and thus the release of multiple downstream inflammatory cytokines, such as IL-6. In particular, IFN-γ is considered one of the major drivers of excessive immune response which leads to massive leukocyte infiltration and mucosal damage by inducing enterocyte apoptosis [89]. TNF-α, which level correlates with the clinical disease activity of CD patients, promotes the secretions of IL-1 and IL-6, and activates apoptosis pathway, JNK pathway, and NF-κB pathway stimulating the acute phase response [91,92].
IL-6 has been found increased in CD and UC patients. Augmented IL-6 levels are related to frequency of relapses and the severity of inflammation [93]. After binding to its receptor, IL-6 promotes the increased expression and nuclear translocation of STAT3, which results in the intestinal T-cell resistance to apoptosis through induction of the anti-apoptotic genes Bcl-2 and Bcl-xl [94]. The released IL-6 also results in the expression of some adhesion molecules and chemokines involved in neutrophils recruitment [95], in the differentiation of Th17 cells [96], and in the prevention of Treg differentiation, leading to chronic intestinal inflammation [97].
IL-13 released by Th2 cells has been shown to increase intestinal permeability and induce enterocyte differentiation and apoptosis [90,98].
Th17 cells are characterized by the production of IL-17A, IL-21, and IL-22 which exert an inflammatory role by activating STAT3. Clinical studies have found that the intestinal mucosa and lamina propria of IBD patients contain much higher levels of Th17 cells and IL-17 compared with the healthy controls [99,100]. The expression of IL-17 in peripheral blood mononuclear cells (PBMCs) of subjects with UC has been shown to correlate with the severity of the disease [101]. IL-17A induces neutrophil recruitment to the inflammatory site and mediates pro-inflammatory cytokine production by macrophages [102].
The activation and the effector function of Th cells is prevented by regulatory T (Treg) cells. Treg cells play a negative immunomodulatory role in immune tolerance and are involved in the maintenance of gut mucosal homeostasis by suppressing abnormal immune responses through the production of anti-inflammatory cytokines, such as IL-10 and TGF-β [103]. IL10 influences the renewal of intestinal stem cells and the regulation of the intestinal microflora promoting the proper functioning of the intestinal epithelial barrier [104], whereas TGF-β regulates immunological homeostasis [105].
IL-10 affects the activity of Th17 and inhibits the antigen presenting cells by reducing the expression of MHC. In particular, IL-10 inhibits the synthesis of pro-inflammatory cytokines, such as IL-1β, TNF-α, IL-6, and the Th2 cell-derived cytokines, chemokine release, as well as the expression of inflammatory enzymes in macrophages and the proliferation of CD4+ T. In addition, IL-10 increases the expression of several anti-inflammatory proteins, including IL-1 receptor antagonist, soluble TNF-α receptor, and tissue inhibitor of matrix metalloproteinases [104]. Loss-of-function mutations in the genes encoding for IL-10 and IL-10 receptors have been associated with a very early-onset form of IBD [106].
A main function of TGF-β signaling in T-cells is to suppress T-cell proliferation and activation through Treg differentiation. Impairment of TGF-β signaling increased colitis progression [107].
Unfortunately, Treg cells are depleted in peripheral blood of patients with active IBD compared to control group [108]. Therefore, the decrease in anti-inflammatory activity of Treg may be an important mechanism in contributing to IBD pathogenesis. Of note, it has been observed that Treg stimulated with IL-6 can express IL-17, thus acquiring the Th17-like cells phenotype. This phenomenon seems to be irreversible and could be important during the onset of chronic mucosal inflammation [109].
The study of interactions between cells and products of the innate and adaptive immune systems and their relationship with the intestinal microbiota, has led to new advancements in the development of novel strategies for the treatment of IBD. Given the fundamental role of cytokines in controlling mucosal inflammation in IBD, the utilization of recombinant anti-inflammatory cytokines or antibodies specific for pro-inflammatory cytokines [110,111], as well as the use of specific neutralizing antibody [112,113], may be an effective method for suppressing chronic intestinal inflammation in IBD suppressing patients.
5. Autophagy and the Maintenance of Intestinal Epithelial Barrier
Autophagy is deregulated in several diseases, including neurodegenerative and metabolic disorders, cancer, and inflammatory diseases, and contributes to the pathogenesis and disease progression [114]. Of note, autophagy may prevent the onset of the inflammatory chronic disease by modulating the immune system functions, the inflammasome formation and production of cytokines, and participating in pathogen clearance [115,116].
Autophagy is a highly conserved catabolic process in eukaryotic cells that physiologically participates in the maintenance of cell homeostasis through the lysosome-mediated degradation of damaged, aged, or redundant cellular components, as well as the destruction of intracellular pathogens. In response to several stress conditions, such as lack of nutrients, growth factor deprivation, infections, and hypoxia, cells induce autophagy as an adaptive mechanism to obtain amino acids during challenging environmental conditions. By contrast, growth factors, insulin, and amino acids induce the activation of the master negative regulator of autophagy, the mammalian target of rapamycin (mTOR), whose activation stimulates protein synthesis, cell survival, proliferation, and growth by inhibiting autophagy pathway.
Several studies have extensively reported the role of autophagy in the regulation of innate and adaptive immune cells, as well as in stromal cells. Recently, a new involvement of autophagy has been emerging in intestinal epithelial cells (Figure 4). In vivo and in vitro results, and clinical studies, show that autophagy is fundamental for intestinal homeostasis, immune response, and prevention from pathogen colonization.
GWAS found that SNPs in autophagy-related genes have been associated to genetic susceptibilities in developing inflammatory disease and, at last, colorectal cancer [117,118].
Paneth cells are considered the origin site of intestinal inflammation [119]. During pathogen infection in a cell where the Golgi apparatus is impaired in packaging and transporting vesicles containing antimicrobial peptides, Paneth cells trigger secretory autophagy as an alternative mechanism for the release of the lysozyme. ATG16L1 deficiency in intestinal epithelial cells, typical of CD patients, results in a defective morphology of Paneth cells, which fail in the release of antimicrobial peptides and in the mucin production [120,121]. Polymorphic variants in ATG16L1 sequence reduce the ability of dendritic cells to process antigens in the luminal compartment of gut and induce the expression of a pro-inflammatory phenotype in these cells [122].
Nucleotide binding oligomerization domain containing protein 2 (NOD2) induces autophagy by sequestering ATG16L1 on cell membrane. NOD2 frameshift mutations interfere with the homeostasis of the intestinal microbiota and excessively stimulate inflammatory responses in the gut. NOD2 polymorphisms prevent protein interaction with ATG16L1 in the plasma membrane, increasing susceptibilities to CD. This results in an impaired clearance of intercellular bacteria and increases the release of inflammatory cytokines [121]. NOD2 loss-of-function mutations hamper the activation of NF-ĸB against pathogens recognized by PRRs, leading to an exacerbated Th1 response [74].
Autophagy dysregulation in Paneth cells also affects the function of the whole intestinal cell populations. Paneth cells contribute, at least in part, to the molecular signals required for maintaining the stemness behavior of ISCs. Stem cells utilize the ATP generated through the oxidative phosphorylation, and autophagy in Paneth cells serves as protection mechanism against oxidative stress resulting from ROS production [123]. In case of chronic inflammation, defective Paneth cells are inefficient in supporting ISCs, making them more susceptible to ROS and lacking the proliferative response crucial for intestinal epithelial regeneration [124].
Crohn’s disease-associated polymorphisms in the sequence of MTMR3 (myotubularin related protein 3) and PTPN2 (protein tyrosine phosphatase, non-receptor type 2) genes compromise autophagy by interfering with autophagosome formation and exacerbate the secretion of inflammatory cytokines [125,126].
Besides polymorphic variants and mutations, also miRNAs participate in the regulation of autophagy-related genes [127,128,129,130]. miR-106b and miR-93 interfere with the clearance of Crohn’s disease-associated bacteria by contrasting the formation of autophagosome [130].
Autophagy is also involved in the occurrence of UC, although to a lesser extent than what has been observed in CD. Genomic analysis of patients with UC reveals a reduced expression of the autophagy-related protein activating transcription factor 4 (ATF4) in the intestinal inflamed mucosa compared to the mucosa of the normal counterpart [131].
The anti-inflammatory role of autophagy is related to its interaction with the inflammasome complex, the central player in inflammation. As described above, NLRP3 stands out as the most studied multi-protein complex that takes charge of triggering inflammatory responses. NLRP3 function is connected to CASP1 activity, leading to the release of IL-1β and IL-18. In the inflammatory process, NLRP3 interacts with mTOR promoting its phosphorylation and subsequent activation. The accumulation of pro-inflammatory cytokines and mediators, due to NLRP3 inhibition of autophagy, intensifies inflammatory response. The accumulation of damaged mitochondria releasing NLRP3-activating signals, coupled with ROS production and Th17 responses, amplifies aberrant inflammasome activation thereby contributing to chronic inflammation [132]. To prevent the exacerbation of the inflammatory response, cells autonomously activate autophagy, promoting the NLRP3 autolysosome inclusion, and subsequent autophagic degradation [133]. Autophagy may degrade ubiquitinated inflammasome and the inactive precursor of IL-1β [134]. ATG16L1 deficiency impairs autophagy and results in the accumulation of IL-1β and IL-18 in serum under the stimulation with lipopolysaccharide in colitis-exposed mice [135].
The targeted deletion of the gene encoding the autophagy adaptor SQSTM1/p62, specifically in macrophages, results in the accumulation of damaged mitochondria, excessive inflammasome activation, accumulation of IL-1β, and macrophage death [136].
Autophagy regulates tight junction expression. Its induction induces the degradation of claudin 2 (CLDN2) allowing the selective cation passage. The lysosomal degradation of this tight junction reduces cell permeability in intestinal epithelial barrier. TNF positively regulated the turnover of claudin 2, increasing epithelial permeability and contributing to the onset of intestinal inflammatory disease [137,138]. The intestinal barrier permeability is regulated also by the interaction between occludin and BECLIN1. Their interaction on the plasma membrane leads to occludin endocytic internalization reducing the integrity of the intestinal barrier. The interaction between BECLIN1 and occludin occurs independently of autophagy. However, when autophagy is induced, it interferes with BECLIN1-occludin interaction. This interference restores the localization of occludin in plasma membrane and re-establishes the barrier integrity [139].
6. Gut Dysbiosis in the Inflamed Gut Microenvironment
The intestinal epithelial barrier constitutes the first line defense mechanism against external agents. Compromising this physical wall means mucosal layer reduction, increase of intestinal permeability, dysregulation of immune response, and gut translocation of microbial molecules and pathogens that overall lead to a systemic inflammatory status.
A growing body of evidence suggest a correlation between the composition of intestinal microbiota and the chronic intestinal inflammation. This emphasizes that the intestinal dysbiosis may act as both the cause and outcome of chronic inflammation [140]. Microbiome analysis report that the dysbiotic gut microbiota in IBD patients triggers chronic immune activation and inflammation in colon. However, the colonization of the intestinal mucosa of IBD patients by microbiota from healthy donors prevents colitis. This suggests that the occurrence of a dysbiotic microenvironment is a fundamental aspect of IBD pathogenesis [141] (Figure 5).
To examine the gut microflora in individuals potentially affected by IBD, microbiome studies focus on α and β diversity parameters. These parameters relate to the variety of bacterial species in the same sample or across different samples, respectively. The main difference in the microbiota between IBD patients and the healthy group results in the reduction of biodiversity [142]. Microbiome sequencing of CD- and UC-affected patients revealed an increase of Proteobacteria, Ruminococcus, and pro-inflammatory strains, including species from Enterobacteriaceae, particularly E. coli. Conversely, there is a reduction in butyrate-producing Firmicutes, Bacteroidetes, Lactobacillus, Eubacterium, Roseburia, and Faecalibacterium species, all of which are involved in the production of butyrate, along with a decrease of anti-inflammatory bacteria (e.g., Faecalibacterium prausnitzii) [143,144,145,146,147,148]. Specifically, in individuals with CD, there is a reported increase in Enterobacteriaceae, including E. coli and Fusobacterium, as well as Ruminococcus gnavus, accompanied by a decrease in Bifidobacterium adolescentis, Dialister invisus, and Faecalibacterium prausnitzii [149,150].
The gut microenvironment is influenced by the availability of nutrients and oxygen, determining the growth of various bacteria species. During inflammation, pathogenic bacteria take advantageous from the condition, engaging in competition with commensal bacteria for the mucosal compartment. Salmonella typhimurium and Citrobacter jejuni are two of the main pathogens associated with infection and inflammation in human [151,152]. The initiation of inflammation raises the level of oxygen in the gut, favoring the colonization and the proliferation of facultative anaerobes strains (E. coli, Salmonella, Shigella, Proteus, and Klebsiella), even though the obligate anaerobic bacteria are still producing butyrate [153]. This metabolic shift amplifies the gut oxygenation and increase the lactate level in the intestinal lumen [154]. When the commensal microbic flora decreases, impairing butyrate production, colonocytes fail to utilize SCFAs as their primary fuel source and to maintain normal levels of ATP necessary for the maintenance of the intestinal epithelial barrier. The absence of butyrate also favors the expression of the “leaky” tight junction protein Claudin-2 and the downregulation of the “tight” one [155]. This outcome creates an energy-deficient mucosa, defined as “starved”, which promotes bacterial translocation in the lamina propria and entry in the systemic circulation [156,157]. This observation is supported by serum analysis revealing that IBD patients display elevated levels of lipopolysaccharide compared to their healthy counterparts. Importantly, this condition persists even when symptoms are in remission among patients with inactive CD, suggesting a compromised intestinal homeostasis [158].
7. Therapeutic Strategies for Manipulating Intestinal Microbiota
The therapeutic protocols for managing IBD are designed to extend the remission phase and involve medical and surgical interventions focused on addressing symptoms. Pharmacological treatments for patients suffering from IBD include corticosteroids, immunosuppressant agents, antibiotics, monoclonal antibodies, and biologic therapies [159]. Clinical trials suggesting the potential use of current small molecules and biological drugs, such as interleukin inhibitors, integrin inhibitors, modulator of sphingosine-1 phosphate, and JAK inhibitors, have been recently presented in [160]. However, the effectiveness of these treatments differs among patients, and their prolonged administration is associated with a high risk of developing drug tolerance and side effects.
Given the recognized role of gut microbiota in preserving intestinal homeostasis, treatments aiming to induce or sustain remission in IBD patients by restoring a healthy microbial environment have been proposed and are still ongoing, showing promising success [161]. Manipulating the gut microbiota may be the best choice proposed as a weapon in the challenge of IBD. Current strategies include fecal microbiota transplantation (FMT) and the administration of living bacteria capable of re-colonizing the intestinal mucosa and restoring gut homeostasis balance.
7.1. Therapeutic Strategies for Manipulating Intestinal Microbiota
Fecal microbiota transplantation (FMT) is a procedure involving the transplantation of healthy donor feces into the gastrointestinal tract of patients with chronic intestinal diseases, aiming to reverse microbiota dysbiosis. This can be carried out through i) enema or endoscopy, both of which are invasive approaches often poorly tolerated by patients, or alternatively, through ii) the oral delivery of lyophilized and stable bacteria in capsules, a method that is more favored by the recipients [162,163].
The FMT approach is currently utilized for treating Clostridium difficile infections, which arise after prolonged use of antibiotics against this microorganism and result in disruptions to gut microbiota homeostasis [164].
Despite variations in FMT protocols across different clinical trials, including differences in donor selection criteria, patient treatment, and therapy administration, the success of the interventions is reported in more than 90% of cases [165]. This highlights the promising outcome of these therapies, suggesting the potential application of FMT in the management of other diseases associated with dysbiosis, addressing the attention on IBD. Recently, a large body of evidence has demonstrated that patients with IBD who undergo FMT exhibit positive clinical outcome with an increase in microbiota composition and biodiversity. Notably, the microbiome of the recipients closely resembles the donor microbiome profile [166]. These findings support the consideration of FMT as a primary treatment of IBD [167,168,169,170].
Studies conducted on IBD patients report that recipients of FMT exhibit a higher remission rate compared to the placebo group [171,172]. Specifically, in UC patients, FMT induces remission within 8 weeks of the intervention [173] and prevents relapse [166]. Additionally, FMT is linked to an increase in butyrate production [174]. Clinical symptoms are alleviated and lesions in the colonic mucosa decreased [175]. For CD patients, the transplantation reduces the severity of the disease and the expression of inflammatory markers, along with an improvement in symptoms such as diarrhea, fever, and abdominal pain. This results in significant changes in gut microbiota composition, with qualitative and quantitative modifications in the microbiome profile [176,177]. FMT appears to be a promising strategy in the challenge for IBD treatment, though further investigations and extensive studies on safety and long-term effects are needed.
7.2. Probiotic Supplementation
The employment of bacterial strains that modulate the composition of gut microbiota and restore homeostasis in host-microbe interactions holds potential in the management of IBD, similar to the FMT approach. The beneficial effects mediated by commensal bacteria on the intestinal mucosa and microenvironment are achieved through the administration of probiotics, defined by the Food and Drug Administration as “live microorganisms that confer a health benefit to the host when administered in adequate amounts” [178].
For the past three decades, probiotics have been utilized in the treatment of IBD [179]. Probiotics exhibit beneficial properties in murine and rat models of colitis. Several strains of Bifidobacterium, E. coli, and Lactobacillus demonstrate anti-inflammatory effects [180] and reduce the outgrowth of pathogens by competing for nutrients [181]. Supplementation with Bifidobacterium, Lactococcus acidophilus, and Enterococcus in mouse models of colitis positively regulates the expression of tight junctions and influences the T cells population, particularly increasing Treg cells [182].
Patients affected by UC and treated with supplementation of Bifidobacterium longum and Escherichia coli Nissle 1917, display improved outcome, and the probiotics acts as well as the therapy with mesalazina in keeping the remission of the disease [183,184]. Propionibacterium freudenreichii modulates host inflammation by regulating the release of the anti-inflammatory cytokine IL-10, similar to engineered Lactoccoccus lactis which shows protective in murine models of colitis [185,186]. Children and adolescents whit mild or moderate-grade UC treated with supplementation of Lactobacillus lactis show improvements in the restoration of intestinal mucosa compared to the control group [187].
The use of a probiotic mixture containing strains of Lactobacillus, Bifidobacteria, and Streptococcus, known as VSL#3, has demonstrated the induction and the maintenance of disease remission in UC patients [188]. VSL#3 is employed either as adjuvant therapy or as monotherapy [189]. This mixture reduced the expression of inflammatory cytokines by inhibiting the signaling of TLR4/NF-ĸB and downregulating the expression of NF-ĸB and TNF-α [180]. In UC patients, Lactobacillus rhamnosus, either as monotherapy or in combination with mesalazine, has shown a safer and more effective effect than the administration of mesalazine alone, maintaining remission from the disease [190].
Different outcomes have been reported in clinical trials conducted on CD patients, suggesting the need for well-designed trials to assess the efficacy of probiotics in the treatment of CD [191]. Lactobacillus rhamnosus does not demonstrate any beneficial effect on children with CD, and in some case, it may lead to the relapse; similarly, VSL#3 has shown a loss of effectiveness [192]. Other trails are exploring the role of non-bacterial probiotics, including the use of yeast, such as Saccharomyces boulardii, which reduces exacerbation in CD patients and improves intestinal barrier permeability when combined with other therapy [193,194].
Given their role in immune system modulation, probiotics may open up new therapeutic possibilities for IBD patients [195]. However, severe intestinal damage may not be reversible and the use of probiotics as a tool for gastrointestinal diseases could be associated with the risk of significant side effects (i.e., bacteremia or sepsis) in patients with compromised immune defenses. After 13 days of oral probiotics administration, a UC adult patient experienced a worsening in health status due to bacteremia [196]. Therefore, the therapy should be considered individually for each patient. Only specific probiotic strains that have been well-tested and administered in proper doses should be employed for managing patients.
8. Conclusions
Crohn’s disease and ulcerative colitis are chronic inflammatory disorders that increase the risk of colitis-associated colorectal cancer two to three times more than in healthy individuals. Due to the complexity and the heterogeneity of the disease, fully understanding the pathogenetic mechanisms is challenging. Existing therapeutic approaches focus on symptoms management to enhance and sustain the remission phase rather than targeting the removal of triggering event.
The manipulation of the gut microbiota through fecal microbiota transplantation or the administration of bacterial strains that restore and maintain intestinal microbiota homeostasis, has proven to be a beneficial strategy for treating IBD. This primarily involves modulating inflammatory pathways and promoting mucosal healing through the synergistic action of different strains. Probiotic therapy has demonstrated effectiveness in rat and mouse models, as well as in clinical trials, either in monotherapy or in combination with the current drugs. Probiotics restore the population of commensal bacteria and appear to have a protective role in patients, showing promise as a therapeutic strategy. However, additional studies are needed to confirm the safety and the effectiveness of this novel approach in the management of IBD. The current findings present some limitations that should not go unnoticed: i) the widely used in vivo models do not entirely replicate the human disease due to the distinct organization of the gastrointestinal tract; ii) the key players involved in the pathogenesis cannot be artificially reproduced; iii) potential side effects arising from the compromise immune system of IBD patients need to be considered. To overcome this last limit, one solution may be to exploit post-biotics (microbial components and metabolites released by live bacteria) as a mixture in combination with probiotics, which could be safer than living microorganism. Taken together, the future of gut bacteria-based therapy must be improved, further developed, and confirmed to clarify whether this approach may be an option in the treatment and resolution of IBD.
Author Contributions
Conceptualization, C.I., B.G., and L.V; writing—original draft preparation, B.G. and L.V; writing—review and editing, C.I., B.G., and L.V, A.A. M.P. A.F.; visualization, C.I., B.G., and L.V, A.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no funding.
Acknowledgments
B.G. is recipient of a PhD fellowship granted by Comoli, Ferrari & SpA (Novara, Italy). L.V. is recipient of a post-doctoral fellowship granted by the Università del Piemonte Orientale (Novara, Italy). A.F. is recipient of a post-doctoral fellowship granted by Fondazione Umberto Veronesi (FUV, Italy). We are grateful for the support of Probiotical SpA (Novara, Italy) and the Consorzio Interuniversitario per le Biotecnologie (CIB).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Kaplan, G.G.; Windsor, J.W. The four epidemiological stages in the global evolution of inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2021, 18, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, K.; Kamada, N. Diet-Microbiota Interactions in Inflammatory Bowel Disease. Nutrients. 2021, 13, 1533. [Google Scholar] [CrossRef] [PubMed]
- Maaser C, Sturm A, Vavricka SR, Kucharzik T, Fiorino G, et al; European Crohn’s and Colitis Organisation [ECCO] and the European Society of Gastrointestinal and Abdominal Radiology [ESGAR] ECCO-ESGAR Guideline for Diagnostic Assessment in IBD Part 1: Initial diagnosis, monitoring of known IBD, detection of complications. J Crohns Colitis. 2019, 13, 144–164. [Google Scholar] [CrossRef]
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011, 474, 307–317. [Google Scholar] [CrossRef]
- Ko, Y.; Butcher, R.; Leong, R.W. Epidemiological studies of migration and environmental risk factors in the inflammatory bowel diseases. World J Gastroenterol. 2014, 20, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012, 491, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Z.; Li, Y.Y. Inflammatory bowel disease: pathogenesis. World J Gastroenterol. 2014, 20, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Larabi, A.; Barnich, N.; Nguyen, H.T.T. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy. 2020, 16, 38–51. [Google Scholar] [CrossRef]
- Annese, V. Genetics and epigenetics of IBD. Pharmacol Res. 2020, 159, 104892. [Google Scholar] [CrossRef]
- Liu, S.; Zhao, W.; Lan, P.; Mou, X. The microbiome in inflammatory bowel diseases: from pathogenesis to therapy. Protein Cell. 2021, 12, 331–345. [Google Scholar] [CrossRef]
- Kostic, A.D.; Xavier, R.J.; Gevers, D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology. 2014, 146, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Buckley, A.; Turner, J.R. Cell Biology of Tight Junction Barrier Regulation and Mucosal Disease. Cold Spring Harb Perspect Biol. 2018, 10, a029314. [Google Scholar] [CrossRef]
- Neish, A.S. Microbes in gastrointestinal health and disease. Gastroenterology. 2009, 136, 65–80. [Google Scholar] [CrossRef]
- Barker, N. Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nat Rev Mol Cell Biol. 2014, 15, 19–33. [Google Scholar] [CrossRef] [PubMed]
- van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol. 2009, 71, 241–260. [Google Scholar] [CrossRef]
- Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, Petras RE, Shen B, Schaeffeler E, Schwab M, Linzmeier R, Feathers RW, Chu H, Lima H Jr, Fellermann K, Ganz T, Stange EF, Bevins CL. Reduced Paneth cell alpha-defensins in ileal Crohn's disease. Proc Natl Acad Sci U S A. 2005, 102, 18129–18134. [Google Scholar] [CrossRef]
- Arijs, I.; De Hertogh, G.; Lemaire, K.; Quintens, R.; Van Lommel, L.; Van Steen, K.; Leemans, P.; Cleynen, I.; Van Assche, G.; Vermeire, S.; Geboes, K.; Schuit, F.; Rutgeerts, P. Mucosal gene expression of antimicrobial peptides in inflammatory bowel disease before and after first infliximab treatment. PLoS One. 2009, 4, e7984. [Google Scholar] [CrossRef]
- Salzman, N.H.; Underwood, M.A.; Bevins, C.L. Paneth cells, defensins, and the commensal microbiota: a hypothesis on intimate interplay at the intestinal mucosa. Semin Immunol. 2007, 19, 70–83. [Google Scholar] [CrossRef]
- Johansson, M.E.; Ambort, D.; Pelaseyed, T.; Schütte, A.; Gustafsson, J.K.; Ermund, A.; Subramani, D.B.; Holmén-Larsson, J.M.; Thomsson, K.A.; Bergström, J.H.; van der Post, S.; Rodriguez-Piñeiro, A.M.; Sjövall, H.; Bäckström, M.; Hansson, G.C. Composition and functional role of the mucus layers in the intestine. Cell Mol Life Sci. 2011, 68, 3635–3641. [Google Scholar] [CrossRef]
- Schroeder, B.O. Fight them or feed them: how the intestinal mucus layer manages the gut microbiota. Gastroenterol Rep (Oxf). 2019, 7, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Kebouchi, M.; Hafeez, Z.; Le Roux, Y.; Dary-Mourot, A.; Genay, M. Importance of digestive mucus and mucins for designing new functional food ingredients. Food Res Int. 2020, 131, 108906. [Google Scholar] [CrossRef]
- Mörbe, U.M.; Jørgensen, P.B.; Fenton, T.M.; von Burg, N.; Riis, L.B.; Spencer, J.; Agace, W.W. Human gut-associated lymphoid tissues (GALT); diversity, structure, and function. Mucosal Immunol. 2021, 14, 793–802. [Google Scholar] [CrossRef]
- Denning, T.L.; Wang, Y.C.; Patel, S.R.; Williams, I.R.; Pulendran, B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol. 2007, 8, 1086–1094. [Google Scholar] [CrossRef]
- Rescigno, M. Intestinal dendritic cells. Adv Immunol. 2010, 107, 109–138. [Google Scholar] [CrossRef] [PubMed]
- Balda, M.S.; Matter, K. Tight junctions as regulators of tissue remodelling. Curr Opin Cell Biol. 2016, 42, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Schulzke, J.D.; Fromm, M. Tight junctions: molecular structure meets function. Ann N Y Acad Sci. 2009, 1165, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Tsukita, S.; Tanaka, H.; Tamura, A. The Claudins: From Tight Junctions to Biological Systems. Trends Biochem Sci. 2019, 44, 141–152. [Google Scholar] [CrossRef]
- Dörfel, M.J.; Huber, O. Modulation of tight junction structure and function by kinases and phosphatases targeting occludin. J Biomed Biotechnol. 2012, 2012, 807356. [Google Scholar] [CrossRef]
- Niessen, C.M.; Gottardi, C.J. Molecular components of the adherens junction. Biochim Biophys Acta. 2008, 1778, 562–571. [Google Scholar] [CrossRef]
- Hartsock, A.; Nelson, W.J. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. 2008, 1778, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Anderson CA, Boucher G, Lees CW, Franke A, D'Amato M, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet. 2011, 43, 246–252, Erratum in: Nat Genet. 2011, 43, 919. [CrossRef]
- Su L, Nalle SC, Shen L, Turner ES, Singh G, Breskin LA, Khramtsova EA, Khramtsova G, Tsai PY, Fu YX, Abraham C, Turner JR. TNFR2 activates MLCK-dependent tight junction dysregulation to cause apoptosis-mediated barrier loss and experimental colitis. Gastroenterology. 2013, 145, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.Y.; Boivin, M.A.; Ye, D.; Pedram, A.; Said, H.M. Mechanism of TNF-{alpha} modulation of Caco-2 intestinal epithelial tight junction barrier: role of myosin light-chain kinase protein expression. Am J Physiol Gastrointest Liver Physiol. 2005, 288, G422–G430. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhou, J.; Wang, L. Role and Mechanism of Gut Microbiota in Human Disease. Front Cell Infect Microbiol. 2021, 11, 625913. [Google Scholar] [CrossRef]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010, 464, 59–65. [Google Scholar] [CrossRef]
- Faith, J.J.; Guruge, J.L.; Charbonneau, M.; Subramanian, S.; Seedorf, H.; Goodman, A.L.; Clemente, J.C.; Knight, R.; Heath, A.C.; Leibel, R.L.; Rosenbaum, M.; Gordon, J.I. The long-term stability of the human gut microbiota. Science. 2013, 341, 1237439. [Google Scholar] [CrossRef] [PubMed]
- Penders, J.; Thijs, C.; Vink, C.; Stelma, F.F.; Snijders, B.; Kummeling, I.; van den Brandt, P.A.; Stobberingh, E.E. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics. 2006, 118, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms. 2019, 7, 14. [Google Scholar] [CrossRef]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes. 2016, 7, 189–200. [Google Scholar] [CrossRef]
- Panebianco, C.; Potenza, A.; Andriulli, A.; Pazienza, V. Exploring the microbiota to better understand gastrointestinal cancers physiology. Clin Chem Lab Med. 2018, 56, 1400–1412. [Google Scholar] [CrossRef]
- Carlsson, A.H.; Yakymenko, O.; Olivier, I.; Håkansson, F.; Postma, E.; Keita, A.V.; Söderholm, J.D. Faecalibacterium prausnitzii supernatant improves intestinal barrier function in mice DSS colitis. Scand J Gastroenterol. 2013, 48, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Laval, L.; Martin, R.; Natividad, J.N.; Chain, F.; Miquel, S.; Desclée de Maredsous, C.; Capronnier, S.; Sokol, H.; Verdu, E.F.; van Hylckama Vlieg, J.E.; Bermúdez-Humarán, L.G.; Smokvina, T.; Langella, P. Lactobacillus rhamnosus CNCM I-3690 and the commensal bacterium Faecalibacterium prausnitzii A2-165 exhibit similar protective effects to induced barrier hyper-permeability in mice. Gut Microbes. 2015, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Basso, P.J.; Câmara, N.O.S.; Sales-Campos, H. Microbial-Based Therapies in the Treatment of Inflammatory Bowel Disease - An Overview of Human Studies. Front Pharmacol. 2019, 9, 1571. [Google Scholar] [CrossRef]
- Khan, I.; Ullah, N.; Zha, L.; Bai, Y.; Khan, A.; Zhao, T.; Che, T.; Zhang, C. Alteration of Gut Microbiota in Inflammatory Bowel Disease (IBD): Cause or Consequence? IBD Treatment Targeting the Gut Microbiome. Pathogens. 2019, 8, 126. [Google Scholar] [CrossRef]
- van der Beek CM, Dejong CHC, Troost FJ, Masclee AAM, Lenaerts K. Role of short-chain fatty acids in colonic inflammation, carcinogenesis, and mucosal protection and healing. Nutr Rev. 2017, 75, 286–305. [Google Scholar] [CrossRef]
- Edelblum, K.L.; Turner, J.R. The tight junction in inflammatory disease: communication breakdown. Curr Opin Pharmacol. 2009, 9, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; Lee, J.R.; Offermanns, S.; Ganapathy, V. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. 2014, 40, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, H.; Chen, P.; Xie, H.; Tao, Y. Demystifying the manipulation of host immunity, metabolism, and extraintestinal tumors by the gut microbiome. Signal Transduct Target Ther. 2019, 4, 41. [Google Scholar] [CrossRef]
- Chung, H.; Pamp, S.J.; Hill, J.A.; Surana, N.K.; Edelman, S.M.; Troy, E.B.; Reading, N.C.; Villablanca, E.J.; Wang, S.; Mora, J.R.; Umesaki, Y.; Mathis, D.; Benoist, C.; Relman, D.A.; Kasper, D.L. Gut immune maturation depends on colonization with a host-specific microbiota. Cell. 2012, 149, 1578–1593. [Google Scholar] [CrossRef] [PubMed]
- Gagnière, J.; Raisch, J.; Veziant, J.; Barnich, N.; Bonnet, R.; Buc, E.; Bringer, M.A.; Pezet, D.; Bonnet, M. Gut microbiota imbalance and colorectal cancer. World J Gastroenterol. 2016, 22, 501–518. [Google Scholar] [CrossRef]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; Xavier, R.J.; Teixeira, M.M.; Mackay, C.R. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009, 461, 1282–1286. [Google Scholar] [CrossRef] [PubMed]
- Panwar, R.B.; Sequeira, R.P.; Clarke, T.B. Microbiota-mediated protection against antibiotic-resistant pathogens. Genes Immun. 2021, 22, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Hold, G.L. Western lifestyle: a 'master' manipulator of the intestinal microbiota? Gut. 2014, 63, 5–6. [Google Scholar] [CrossRef]
- Becattini, S.; Taur, Y.; Pamer, E.G. Antibiotic-Induced Changes in the Intestinal Microbiota and Disease. Trends Mol Med. 2016, 22, 458–478. [Google Scholar] [CrossRef] [PubMed]
- Gomaa, E.Z. Human gut microbiota/microbiome in health and diseases: a review. Antonie Van Leeuwenhoek. 2020, 113, 2019–2040. [Google Scholar] [CrossRef] [PubMed]
- Glassner, K.L.; Abraham, B.P.; Quigley, E.M.M. The microbiome and inflammatory bowel disease. J Allergy Clin Immunol. 2020, 145, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Keller, D.S.; Windsor, A.; Cohen, R.; Chand, M. Colorectal cancer in inflammatory bowel disease: review of the evidence. Tech Coloproctol. 2019, 23, 3–13. [Google Scholar] [CrossRef]
- Strober, W.; Fuss, I.; Mannon, P. The fundamental basis of inflammatory bowel disease. J Clin Invest. 2007, 117, 514–521. [Google Scholar] [CrossRef]
- Maloy, K.J.; Powrie, F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011, 474, 298–306. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Khalili, H.; Pan, A.; Higuchi, L.M.; de Silva, P.; Richter, J.M.; Fuchs, C.S.; Chan, A.T. Association between depressive symptoms and incidence of Crohn's disease and ulcerative colitis: results from the Nurses' Health Study. Clin Gastroenterol Hepatol. 2013, 11, 57–62. [Google Scholar] [CrossRef]
- Schett, G.; Neurath, M.F. Resolution of chronic inflammatory disease: universal and tissue-specific concepts. Nat Commun. 2018, 9, 3261. [Google Scholar] [CrossRef]
- Graham, D.B.; Xavier, R.J. Pathway paradigms revealed from the genetics of inflammatory bowel disease. Nature. 2020, 578, 527–539. [Google Scholar] [CrossRef]
- Odenwald, M.A.; Turner, J.R. The intestinal epithelial barrier: a therapeutic target? Nat Rev Gastroenterol Hepatol. 2017, 14, 9–21. [Google Scholar] [CrossRef]
- Salim, S.Y.; Söderholm, J.D. Importance of disrupted intestinal barrier in inflammatory bowel diseases. Inflamm Bowel Dis. 2011, 17, 362–381. [Google Scholar] [CrossRef]
- Rakoff-Nahoum, S.; Paglino, J.; Eslami-Varzaneh, F.; Edberg, S.; Medzhitov, R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004, 118, 229–241. [Google Scholar] [CrossRef]
- Wallace, K.L.; Zheng, L.B.; Kanazawa, Y.; Shih, D.Q. Immunopathology of inflammatory bowel disease. World J Gastroenterol. 2014, 20, 6–21. [Google Scholar] [CrossRef]
- Holleran, G.; Lopetuso, L.; Petito, V.; Graziani, C.; Ianiro, G.; McNamara, D.; Gasbarrini, A.; Scaldaferri, F. The Innate and Adaptive Immune System as Targets for Biologic Therapies in Inflammatory Bowel Disease. Int J Mol Sci. 2017, 18, 2020. [Google Scholar] [CrossRef]
- Kordjazy, N.; Haj-Mirzaian, A.; Haj-Mirzaian, A.; Rohani, M.M.; Gelfand, E.W.; Rezaei, N.; Abdolghaffari, A.H. Role of toll-like receptors in inflammatory bowel disease. Pharmacol Res. 2018, 129, 204–215. [Google Scholar] [CrossRef]
- Harris, G.; KuoLee, R.; Chen, W. Role of Toll-like receptors in health and diseases of gastrointestinal tract. World J Gastroenterol. 2006, 12, 2149–2160. [Google Scholar] [CrossRef] [PubMed]
- Inohara, N.; Ogura, Y.; Fontalba, A.; Gutierrez, O.; Pons, F.; Crespo, J.; Fukase, K.; Inamura, S.; Kusumoto, S.; Hashimoto, M.; Foster, S.J.; Moran, A.P.; Fernandez-Luna, J.L.; Nuñez, G. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn's disease. J Biol Chem. 2003, 278, 5509–5512. [Google Scholar] [CrossRef]
- Abraham, C.; Cho, J.H. Functional consequences of NOD2 (CARD15) mutations. Inflamm Bowel Dis. 2006, 12, 641–650. [Google Scholar] [CrossRef]
- Watanabe T, Kitani A, Murray PJ, Strober W. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat Immunol. 2004, 5, 800–808. [Google Scholar] [CrossRef]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature. 2012, 481, 278–286. [Google Scholar] [CrossRef]
- Strober, W.; Fuss, I.J.; Blumberg, R.S. The immunology of mucosal models of inflammation. Annu Rev Immunol. 2002, 20, 495–549. [Google Scholar] [CrossRef]
- Ligumsky, M.; Simon, P.L.; Karmeli, F.; Rachmilewitz, D. Role of interleukin 1 in inflammatory bowel disease--enhanced production during active disease. Gut. 1990, 31, 686–689. [Google Scholar] [CrossRef]
- Nakanishi, K. Unique Action of Interleukin-18 on T Cells and Other Immune Cells. Front Immunol. 2018, 9, 763. [Google Scholar] [CrossRef]
- Liu, L.; Dong, Y.; Ye, M.; Jin, S.; Yang, J.; Joosse, M.E.; Sun, Y.; Zhang, J.; Lazarev, M.; Brant, S.R.; Safar, B.; Marohn, M.; Mezey, E.; Li, X. The Pathogenic Role of NLRP3 Inflammasome Activation in Inflammatory Bowel Diseases of Both Mice and Humans. J Crohns Colitis. 2017, 11, 737–750. [Google Scholar] [CrossRef]
- Zaki, M.H.; Boyd, K.L.; Vogel, P.; Kastan, M.B.; Lamkanfi, M.; Kanneganti, T.D. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010, 32, 379–391. [Google Scholar] [CrossRef]
- Lissner, D.; Schumann, M.; Batra, A.; Kredel, L.I.; Kühl, A.A.; Erben, U.; May, C.; Schulzke, J.D.; Siegmund, B. Monocyte and M1 Macrophage-induced Barrier Defect Contributes to Chronic Intestinal Inflammation in IBD. Inflamm Bowel Dis. 2015, 21, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009, 27, 451–483. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Yu, J.; Nie, Y.; Shi, X.; Liu, Y.; Li, F.; Zhang, X.L. Disequilibrium of M1 and M2 macrophages correlates with the development of experimental inflammatory bowel diseases. Immunol Invest. 2014, 43, 638–652. [Google Scholar] [CrossRef] [PubMed]
- Hart, A.L.; Al-Hassi, H.O.; Rigby, R.J.; Bell, S.J.; Emmanuel, A.V.; Knight, S.C.; Kamm, M.A.; Stagg, A.J. Characteristics of intestinal dendritic cells in inflammatory bowel diseases. Gastroenterology. 2005, 129, 50–65. [Google Scholar] [CrossRef] [PubMed]
- Britanova, L.; Diefenbach, A. Interplay of innate lymphoid cells and the microbiota. Immunol Rev. 2017, 279, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Forkel, M.; van Tol, S.; Höög, C.; Michaëlsson, J.; Almer, S.; Mjösberg, J. Distinct Alterations in the Composition of Mucosal Innate Lymphoid Cells in Newly Diagnosed and Established Crohn's Disease and Ulcerative Colitis. J Crohns Colitis. 2019, 13, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003, 3, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Teng MW, Bowman EP, McElwee JJ, Smyth MJ, Casanova JL, Cooper AM, Cua DJ. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat Med. 2015, 21, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine. 2015, 74, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Heller, F.; Florian, P.; Bojarski, C.; Richter, J.; Christ, M.; Hillenbrand, B.; Mankertz, J.; Gitter, A.H.; Bürgel, N.; Fromm, M.; Zeitz, M.; Fuss, I.; Strober, W.; Schulzke, J.D. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005, 129, 550–564. [Google Scholar] [CrossRef]
- Aardoom, M.A.; Veereman, G.; de Ridder, L. A Review on the Use of Anti-TNF in Children and Adolescents with Inflammatory Bowel Disease. Int J Mol Sci. 2019, 20, 2529. [Google Scholar] [CrossRef] [PubMed]
- Armuzzi, A.; Bouhnik, Y.; Cummings, F.; Bettey, M.; Pieper, B.; Kang, T. Enhancing treatment success in inflammatory bowel disease: Optimising the use of anti-TNF agents and utilising their biosimilars in clinical practice. Dig Liver Dis. 2020, 52, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Pawłowska-Kamieniak, A.; Krawiec, P.; Pac-Kożuchowska, E. Interleukin 6: Biological significance and role in inflammatory bowel diseases. Adv Clin Exp Med. 2021, 30, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Atreya, R.; Mudter, J.; Finotto, S.; Müllberg, J.; Jostock, T.; Wirtz, S.; Schütz, M.; Bartsch, B.; Holtmann, M.; Becker, C.; Strand, D.; Czaja, J.; Schlaak, J.F.; Lehr, H.A.; Autschbach, F.; Schürmann, G.; Nishimoto, N.; Yoshizaki, K.; Ito, H.; Kishimoto, T.; Galle, P.R.; Rose-John, S.; Neurath, M.F. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo. Nat Med. 2000, 6, 583–588, Erratum in: Nat. Med. 2010 Nov;16(11):1341. [Google Scholar] [CrossRef] [PubMed]
- Fielding CA, McLoughlin RM, McLeod L, Colmont CS, Najdovska M, Grail D, Ernst M, Jones SA, Topley N, Jenkins BJ. IL-6 regulates neutrophil trafficking during acute inflammation via STAT3. J Immunol. 2008, 181, 2189–2195. [CrossRef]
- Chonov DC, Ignatova MMK, Ananiev JR, Gulubova MV. IL-6 Activities in the Tumour Microenvironment. Part 1. Open Access Maced J Med Sci. 2019, 7, 2391–2398. [CrossRef]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Ceponis, P.J.; Botelho, F.; Richards, C.D.; McKay, D.M. Interleukins 4 and 13 increase intestinal epithelial permeability by a phosphatidylinositol 3-kinase pathway. Lack of evidence for STAT 6 involvement. J Biol Chem. 2000, 275, 29132–29137. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi T, Okamoto S, Hisamatsu T, Kamada N, Chinen H, Saito R, Kitazume MT, Nakazawa A, Sugita A, Koganei K, Isobe K, Hibi T. IL23 differentially regulates the Th1/Th17 balance in ulcerative colitis and Crohn's disease. Gut. 2008, 57, 1682–1689. [Google Scholar] [CrossRef]
- Rovedatti, L.; Kudo, T.; Biancheri, P.; Sarra, M.; Knowles, C.H.; Rampton, D.S.; Corazza, G.R.; Monteleone, G.; Di Sabatino, A.; Macdonald, T.T. Differential regulation of interleukin 17 and interferon gamma production in inflammatory bowel disease. Gut. 2009, 58, 1629–1636. [Google Scholar] [CrossRef]
- Lee, S.H.; Kwon, J.E.; Cho, M.L. Immunological pathogenesis of inflammatory bowel disease. Intest Res. 2018, 16, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Kolls, J.K.; Lindén, A. Interleukin-17 family members and inflammation. Immunity. 2004, 21, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Valencia X, Stephens G, Goldbach-Mansky R, Wilson M, Shevach EM, Lipsky PE. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood. 2006, 108, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Neumann, C.; Scheffold, A.; Rutz, S. Functions and regulation of T cell-derived interleukin-10. Semin Immunol. 2019, 44, 101344. [Google Scholar] [CrossRef] [PubMed]
- Marek A, Brodzicki J, Liberek A, Korzon M. TGF-beta (transforming growth factor-beta) in chronic inflammatory conditions - a new diagnostic and prognostic marker? Med Sci Monit. 2002, 8, RA145–RA151. [Google Scholar]
- Glocker EO, Kotlarz D, Klein C, Shah N, Grimbacher B. IL-10 and IL-10 receptor defects in humans. Ann N Y Acad Sci. 2011, 1246, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Ihara S, Hirata Y, Koike K. TGF-β in inflammatory bowel disease: a key regulator of immune cells, epithelium, and the intestinal microbiota. J Gastroenterol. 2017, 52, 777–787. [Google Scholar] [CrossRef]
- Singh, B.; Read, S.; Asseman, C.; Malmström, V.; Mottet, C.; Stephens, L.A.; Stepankova, R.; Tlaskalova, H.; Powrie, F. Control of intestinal inflammation by regulatory T cells. Immunol Rev. 2001, 182, 190–200. [Google Scholar] [CrossRef]
- Lee, Y.K.; Mukasa, R.; Hatton, R.D.; Weaver, C.T. Developmental plasticity of Th17 and Treg cells. Curr Opin Immunol. 2009, 21, 274–280. [Google Scholar] [CrossRef]
- Rousseaux, C.; Lefebvre, B.; Dubuquoy, L.; Lefebvre, P.; Romano, O.; Auwerx, J.; Metzger, D.; Wahli, W.; Desvergne, B.; Naccari, G.C.; Chavatte, P.; Farce, A.; Bulois, P.; Cortot, A.; Colombel, J.F.; Desreumaux, P. Intestinal antiinflammatory effect of 5-aminosalicylic acid is dependent on peroxisome proliferator-activated receptor-gamma. J Exp Med. 2005, 201, 1205–1215. [Google Scholar] [CrossRef]
- Allgayer, H. Review article: mechanisms of action of mesalazine in preventing colorectal carcinoma in inflammatory bowel disease. Aliment Pharmacol Ther. 2003, 18 Suppl 2, 10–14. [Google Scholar] [CrossRef]
- Billmeier, U.; Dieterich, W.; Neurath, M.F.; Atreya, R. Molecular mechanism of action of anti-tumor necrosis factor antibodies in inflammatory bowel diseases. World J Gastroenterol. 2016, 22, 9300–9313. [Google Scholar] [CrossRef] [PubMed]
- Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, Wehkamp J, Feagan BG, Yao MD, Karczewski M, Karczewski J, Pezous N, Bek S, Bruin G, Mellgard B, Berger C, Londei M, Bertolino AP, Tougas G, Travis SP; Secukinumab in Crohn's Disease Study Group. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn's disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012, 61, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Klionsky, D.J. Autophagy and disease: unanswered questions. Cell Death Differ. 2020, 27, 858–871. [Google Scholar] [CrossRef] [PubMed]
- Hooper, K.M.; Barlow, P.G.; Henderson, P.; Stevens, C. Interactions Between Autophagy and the Unfolded Protein Response: Implications for Inflammatory Bowel Disease. Inflamm Bowel Dis. 2019, 25, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Iida, T.; Onodera, K.; Nakase, H. Role of autophagy in the pathogenesis of inflammatory bowel disease. World J Gastroenterol. 2017, 23, 1944–1953. [Google Scholar] [CrossRef] [PubMed]
- Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL,et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet. 2010, 42, 1118–1125. [Google Scholar] [CrossRef]
- Mokarram, P.; Albokashy, M.; Zarghooni, M.; Moosavi, M.A.; Sepehri, Z.; Chen, Q.M.; Hudecki, A.; Sargazi, A.; Alizadeh, J.; Moghadam, A.R.; Hashemi, M.; Movassagh, H.; Klonisch, T.; Owji, A.A.; Łos, M.J.; Ghavami, S. New frontiers in the treatment of colorectal cancer: Autophagy and the unfolded protein response as promising targets. Autophagy. 2017, 13, 781–819. [Google Scholar] [CrossRef]
- Adolph, T.E.; Tomczak, M.F.; Niederreiter, L.; Ko, H.J.; Böck, J.; Martinez-Naves, E.; Glickman, J.N.; Tschurtschenthaler, M.; Hartwig, J.; Hosomi, S.; Flak, M.B.; Cusick, J.L.; Kohno, K.; Iwawaki, T.; Billmann-Born, S.; Raine, T.; Bharti, R.; Lucius, R.; Kweon, M.N.; Marciniak, S.J.; Choi, A.; Hagen, S.J.; Schreiber, S.; Rosenstiel, P.; Kaser, A.; Blumberg, R.S. Paneth cells as a site of origin for intestinal inflammation. Nature. 2013, 503, 272–276. [Google Scholar] [CrossRef]
- Bel, S.; Pendse, M.; Wang, Y.; Li, Y.; Ruhn, K.A.; Hassell, B.; Leal, T.; Winter, S.E.; Xavier, R.J.; Hooper, L.V. Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science. 2017, 357, 1047–1052. [Google Scholar] [CrossRef]
- Lassen, K.G.; Kuballa, P.; Conway, K.L.; Patel, K.K.; Becker, C.E.; Peloquin, J.M.; Villablanca, E.J.; Norman, J.M.; Liu, T.C.; Heath, R.J.; Becker, M.L.; Fagbami, L.; Horn, H.; Mercer, J.; Yilmaz, O.H.; Jaffe, J.D.; Shamji, A.F.; Bhan, A.K.; Carr, S.A.; Daly, M.J.; Virgin, H.W.; Schreiber, S.L.; Stappenbeck, T.S.; Xavier, R.J. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc Natl Acad Sci U S A. 2014, 111, 7741–7746. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Lapaquette, P.; Bringer, M.A.; Darfeuille-Michaud, A. Autophagy and Crohn's disease. J Innate Immun. 2013, 5, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Levy, A.; Stedman, A.; Deutsch, E.; Donnadieu, F.; Virgin, H.W.; Sansonetti, P.J.; Nigro, G. Innate immune receptor NOD2 mediates LGR5+ intestinal stem cell protection against ROS cytotoxicity via mitophagy stimulation. Proc Natl Acad Sci U S A. 2020, 117, 1994–2003. [Google Scholar] [CrossRef] [PubMed]
- Lahiri A, Hedl M, Abraham C. MTMR3 risk allele enhances innate receptor-induced signaling and cytokines by decreasing autophagy and increasing caspase-1 activation. Proc Natl Acad Sci U S A. 2015, 112, 10461–10466. [Google Scholar] [CrossRef] [PubMed]
- Scharl, M.; Mwinyi, J.; Fischbeck, A.; Leucht, K.; Eloranta, J.J.; Arikkat, J.; Pesch, T.; Kellermeier, S.; Mair, A.; Kullak-Ublick, G.A.; Truninger, K.; Noreen, F.; Regula, J.; Gaj, P.; Pittet, V.; Mueller, C.; Hofmann, C.; Fried, M.; McCole, D.F.; Rogler, G. Crohn's disease-associated polymorphism within the PTPN2 gene affects muramyl-dipeptide-induced cytokine secretion and autophagy. Inflamm Bowel Dis. 2012, 18, 900–912. [Google Scholar] [CrossRef]
- Vidoni, C.; Ferraresi, A.; Secomandi, E.; Vallino, L.; Dhanasekaran, D.N.; Isidoro, C. Epigenetic targeting of autophagy for cancer prevention and treatment by natural compounds. Semin Cancer Biol. 2020, 66, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Vidoni, C.; Vallino, L.; Ferraresi, A.; Secomandi, E.; Salwa, A.; Chinthakindi, M.; Galetto, A.; Dhanasekaran, D.N.; Isidoro, C. Epigenetic control of autophagy in women's tumors: role of non-coding RNAs. J Cancer Metastasis Treat 2021, 7, 4. [Google Scholar] [CrossRef]
- Vallino, L.; Ferraresi, A.; Vidoni, C.; Secomandi, E.; Esposito, A.; Dhanasekaran, D.N.; Isidoro, C. Modulation of non-coding RNAs by resveratrol in ovarian cancer cells: In silico analysis and literature review of the anti-cancer pathways involved. J Tradit Complement Med. 2020, 10, 217–229. [Google Scholar] [CrossRef]
- Lu C, Chen J, Xu HG, Zhou X, He Q, Li YL, Jiang G, Shan Y, Xue B, Zhao RX, Wang Y, Werle KD, Cui R, Liang J, Xu ZX. MIR106B and MIR93 prevent removal of bacteria from epithelial cells by disrupting ATG16L1-mediated autophagy. Gastroenterology. 2014, 146, 188–199. [CrossRef]
- Hu X, Deng J, Yu T, Chen S, Ge Y, Zhou Z, Guo Y, Ying H, Zhai Q, Chen Y, Yuan F, Niu Y, Shu W, Chen H, Ma C, Liu Z, Guo F. ATF4 Deficiency Promotes Intestinal Inflammation in Mice by Reducing Uptake of Glutamine and Expression of Antimicrobial Peptides. Gastroenterology. 2019, 156, 1098–1111. [Google Scholar] [CrossRef] [PubMed]
- Cosin-Roger, J.; Simmen, S.; Melhem, H.; Atrott, K.; Frey-Wagner, I.; Hausmann, M.; de Vallière, C.; Spalinger, M.R.; Spielmann, P.; Wenger, R.H.; Zeitz, J.; Vavricka, S.R.; Rogler, G.; Ruiz, P.A. Hypoxia ameliorates intestinal inflammation through NLRP3/mTOR downregulation and autophagy activation. Nat Commun. 2017, 8, 98. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Levine, B. Autophagy balances inflammation in innate immunity. Autophagy. 2018, 14, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Shenderov, K.; Huang, N.N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; Tanaka, K.; Kawai, T.; Tsujimura, T.; Takeuchi, O.; Yoshimori, T.; Akira, S. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008, 456, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, He F, Boassa D, Perkins G, Ali SR, McGeough MD, Ellisman MH, Seki E, Gustafsson AB, Hoffman HM, Diaz-Meco MT, Moscat J, Karin M. NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell. 2016, 164, 896–910. [Google Scholar] [CrossRef]
- Nighot, P.K.; Hu, C.A.; Ma, T.Y. Autophagy enhances intestinal epithelial tight junction barrier function by targeting claudin-2 protein degradation. J Biol Chem. 2015, 290, 7234–7246. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yan, J.; Xiao, Y.; Shen, Y.; Wang, J.; Ge, W.; Chen, Y. Inhibition of Autophagic Degradation Process Contributes to Claudin-2 Expression Increase and Epithelial Tight Junction Dysfunction in TNF-α Treated Cell Monolayers. Int J Mol Sci. 2017, 18, 157. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Ganapathy, A.S.; Suchanec, E.; Laidler, L.; Ma, T.; Nighot, P. Intestinal epithelial tight junction barrier regulation by autophagy-related protein ATG6/beclin 1. Am J Physiol Cell Physiol. 2019, 316, C753–C765. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q. A Comprehensive Review and Update on the Pathogenesis of Inflammatory Bowel Disease. J Immunol Res. 2019, 2019, 7247238. [Google Scholar] [CrossRef]
- Britton, G.J.; Contijoch, E.J.; Mogno, I.; Vennaro, O.H.; Llewellyn, S.R.; Ng, R.; Li, Z.; Mortha, A.; Merad, M.; Das, A.; Gevers, D.; McGovern, D.P.B.; Singh, N.; Braun, J.; Jacobs, J.P.; Clemente, J.C.; Grinspan, A.; Sands, B.E.; Colombel, J.F.; Dubinsky, M.C.; Faith, J.J. Microbiotas from Humans with Inflammatory Bowel Disease Alter the Balance of Gut Th17 and RORγt+ Regulatory T Cells and Exacerbate Colitis in Mice. Immunity. 2019, 50, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Derwa, Y.; Gracie, D.J.; Hamlin, P.J.; Ford, A.C. Systematic review with meta-analysis: the efficacy of probiotics in inflammatory bowel disease. Aliment Pharmacol Ther. 2017, 46, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Vester-Andersen, M.K.; Mirsepasi-Lauridsen, H.C.; Prosberg, M.V.; Mortensen, C.O.; Träger, C.; Skovsen, K.; Thorkilgaard, T.; Nøjgaard, C.; Vind, I.; Krogfelt, K.A.; Sørensen, N.; Bendtsen, F.; Petersen, A.M. Increased abundance of proteobacteria in aggressive Crohn's disease seven years after diagnosis. Sci Rep. 2019, 9, 13473. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Siles, M.; Martinez-Medina, M.; Abellà, C.; Busquets, D.; Sabat-Mir, M.; Duncan, S.H.; Aldeguer, X.; Flint, H.J.; Garcia-Gil, L.J. Mucosa-associated Faecalibacterium prausnitzii phylotype richness is reduced in patients with inflammatory bowel disease. Appl Environ Microbiol. 2015, 81, 7582–7592. [Google Scholar] [CrossRef]
- Martinez-Medina, M.; Aldeguer, X.; Lopez-Siles, M.; González-Huix, F.; López-Oliu, C.; Dahbi, G.; Blanco, J.E.; Blanco, J.; Garcia-Gil, L.J.; Darfeuille-Michaud, A. Molecular diversity of Escherichia coli in the human gut: new ecological evidence supporting the role of adherent-invasive E. coli (AIEC) in Crohn's disease. Inflamm Bowel Dis. 2009, 15, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Nishino, K.; Nishida, A.; Inoue, R.; Kawada, Y.; Ohno, M.; Sakai, S.; Inatomi, O.; Bamba, S.; Sugimoto, M.; Kawahara, M.; Naito, Y.; Andoh, A. Analysis of endoscopic brush samples identified mucosa-associated dysbiosis in inflammatory bowel disease. J Gastroenterol. 2018, 53, 95–106. [Google Scholar] [CrossRef]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [PubMed]
- Ott, S.J.; Musfeldt, M.; Wenderoth, D.F.; Hampe, J.; Brant, O.; Fölsch, U.R.; Timmis, K.N.; Schreiber, S. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut. 2004, 53, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Lupp, C.; Robertson, M.L.; Wickham, M.E.; Sekirov, I.; Champion, O.L.; Gaynor, E.C.; Finlay, B.B. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe. 2007, 2, 204. [Google Scholar] [CrossRef] [PubMed]
- Joossens, M.; Huys, G.; Cnockaert, M.; De Preter, V.; Verbeke, K.; Rutgeerts, P.; Vandamme, P.; Vermeire, S. Dysbiosis of the faecal microbiota in patients with Crohn's disease and their unaffected relatives. Gut. 2011, 60, 631–637. [Google Scholar] [CrossRef]
- Stecher, B.; Robbiani, R.; Walker, A.W.; Westendorf, A.M.; Barthel, M.; Kremer, M.; Chaffron, S.; Macpherson, A.J.; Buer, J.; Parkhill, J.; Dougan, G.; von Mering, C.; Hardt, W.D. Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 2007, 5, 2177–2189. [Google Scholar] [CrossRef] [PubMed]
- O'Hara, J.R.; Feener, T.D.; Fischer, C.D.; Buret, A.G. Campylobacter jejuni disrupts protective Toll-like receptor 9 signaling in colonic epithelial cells and increases the severity of dextran sulfate sodium-induced colitis in mice. Infect Immun. 2012, 80, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Rigottier-Gois, L. Dysbiosis in inflammatory bowel diseases: the oxygen hypothesis. ISME J. 2013, 7, 1256–1261. [Google Scholar] [CrossRef] [PubMed]
- Nagao-Kitamoto H, Shreiner AB, Gillilland MG 3rd, Kitamoto S, Ishii C, Hirayama A, Kuffa P, El-Zaatari M, Grasberger H, Seekatz AM, Higgins PD, Young VB, Fukuda S, Kao JY, Kamada N. Functional Characterization of Inflammatory Bowel Disease-Associated Gut Dysbiosis in Gnotobiotic Mice. Cell Mol Gastroenterol Hepatol. 2016, 2, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Saeedi BJ, Kao DJ, Kitzenberg DA, Dobrinskikh E, Schwisow KD, Masterson JC, Kendrick AA, Kelly CJ, Bayless AJ, Kominsky DJ, Campbell EL, Kuhn KA, Furuta GT, Colgan SP, Glover LE. HIF-dependent regulation of claudin-1 is central to intestinal epithelial tight junction integrity. Mol Biol Cell. 2015, 26, 2252–2262. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, D.R.; Garge, N.; Zhang, X.; Sun, W.; O'Connell, T.M.; Bunger, M.K.; Bultman, S.J. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011, 13, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Gassler, N.; Rohr, C.; Schneider, A.; Kartenbeck, J.; Bach, A.; Obermüller, N.; Otto, H.F.; Autschbach, F. Inflammatory bowel disease is associated with changes of enterocytic junctions. Am J Physiol Gastrointest Liver Physiol. 2001, 281, G216–G228. [Google Scholar] [CrossRef] [PubMed]
- Pastor Rojo, O.; López San Román, A.; Albéniz Arbizu, E.; de la Hera Martínez, A.; Ripoll Sevillano, E.; Albillos Martínez, A. Serum lipopolysaccharide-binding protein in endotoxemic patients with inflammatory bowel disease. Inflamm Bowel Dis. 2007, 13, 269–277. [Google Scholar] [CrossRef]
- Neurath, M.F. Current and emerging therapeutic targets for IBD. Nat Rev Gastroenterol Hepatol. 2017, 14, 269–278, Erratum in: Nat Rev Gastroenterol Hepatol. 2017 Oct 11. [Google Scholar] [CrossRef]
- Bretto, E.; Ribaldone, D.G.; Caviglia, G.P.; Saracco, G.M.; Bugianesi, E.; Frara, S. Inflammatory Bowel Disease: Emerging Therapies and Future Treatment Strategies. Biomedicines. 2023, 11, 2249. [Google Scholar] [CrossRef]
- Lê, A.; Mantel, M.; Marchix, J.; Bodinier, M.; Jan, G.; Rolli-Derkinderen, M. Inflammatory bowel disease therapeutic strategies by modulation of the microbiota: how and when to introduce pre-, pro-, syn-, or postbiotics? Am J Physiol Gastrointest Liver Physiol. 2022, 323, G523–G553. [Google Scholar] [CrossRef]
- Gupta, A.; Khanna, S. Fecal Microbiota Transplantation. JAMA. 2017, 318, 102. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Salzman, N.H.; Acharya, C.; Sterling, R.K.; White, M.B.; Gavis, E.A.; Fagan, A.; Hayward, M.; Holtz, M.L.; Matherly, S.; Lee, H.; Osman, M.; Siddiqui, M.S.; Fuchs, M.; Puri, P.; Sikaroodi, M.; Gillevet, P.M. Fecal Microbial Transplant Capsules Are Safe in Hepatic Encephalopathy: A Phase 1, Randomized, Placebo-Controlled Trial. Hepatology. 2019, 70, 1690–1703, Erratum in: Hepatology. 2020 Oct;72(4):1501. [Google Scholar] [CrossRef] [PubMed]
- Wortelboer, K.; Nieuwdorp, M.; Herrema, H. Fecal microbiota transplantation beyond Clostridioides difficile infections. EBioMedicine. 2019, 44, 716–729. [Google Scholar] [CrossRef]
- Quraishi, M.N.; Widlak, M.; Bhala, N.; Moore, D.; Price, M.; Sharma, N.; Iqbal, T.H. Systematic review with meta-analysis: the efficacy of faecal microbiota transplantation for the treatment of recurrent and refractory Clostridium difficile infection. Aliment Pharmacol Ther. 2017, 46, 479–493. [Google Scholar] [CrossRef]
- Sood, A.; Mahajan, R.; Singh, A.; Midha, V.; Mehta, V.; Narang, V.; Singh, T.; Singh Pannu, A. Role of Faecal Microbiota Transplantation for Maintenance of Remission in Patients With Ulcerative Colitis: A Pilot Study. J Crohns Colitis. 2019, 13, 1311–1317. [Google Scholar] [CrossRef]
- Lopez, J.; Grinspan, A. Fecal Microbiota Transplantation for Inflammatory Bowel Disease. Gastroenterol Hepatol (N Y). 2016, 12, 374–379. [Google Scholar]
- Costello, S.P.; Soo, W.; Bryant, R.V.; Jairath, V.; Hart, A.L.; Andrews, J.M. Systematic review with meta-analysis: faecal microbiota transplantation for the induction of remission for active ulcerative colitis. Aliment Pharmacol Ther. 2017, 46, 213–224. [Google Scholar] [CrossRef]
- Caldeira, L.F.; Borba, H.H.; Tonin, F.S.; Wiens, A.; Fernandez-Llimos, F.; Pontarolo, R. Fecal microbiota transplantation in inflammatory bowel disease patients: A systematic review and meta-analysis. PLoS One. 2020, 15, e0238910. [Google Scholar] [CrossRef]
- Kong, L.; Lloyd-Price, J.; Vatanen, T.; Seksik, P.; Beaugerie, L.; Simon, T.; Vlamakis, H.; Sokol, H.; Xavier, R.J. Linking Strain Engraftment in Fecal Microbiota Transplantation With Maintenance of Remission in Crohn's Disease. Gastroenterology. 2020, 159, 2193–2202. [Google Scholar] [CrossRef]
- Moayyedi, P.; Surette, M.G.; Kim, P.T.; Libertucci, J.; Wolfe, M.; Onischi, C.; Armstrong, D.; Marshall, J.K.; Kassam, Z.; Reinisch, W.; Lee, C.H. Fecal Microbiota Transplantation Induces Remission in Patients With Active Ulcerative Colitis in a Randomized Controlled Trial. Gastroenterology. 2015, 149, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Costello, S.P.; Hughes, P.A.; Waters, O.; Bryant, R.V.; Vincent, A.D.; Blatchford, P.; Katsikeros, R.; Makanyanga, J.; Campaniello, M.A.; Mavrangelos, C.; Rosewarne, C.P.; Bickley, C.; Peters, C.; Schoeman, M.N.; Conlon, M.A.; Roberts-Thomson, I.C.; Andrews, J.M. Effect of Fecal Microbiota Transplantation on 8-Week Remission in Patients With Ulcerative Colitis: A Randomized Clinical Trial. JAMA. 2019, 321, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.G.; Liu, S.P.; Ma, T.H.; Yan, W.; Zhou, J.F.; Shi, Y.T.; Shen, P.; Yang, X.Z.; Wu, S.N. Fecal microbiota transplantation treatment for refractory ulcerative colitis with allergy to 5-aminosalicylic acid: A case report. Medicine (Baltimore). 2018, 97, e0675. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.M.; Huang, H.L.; Xu, J.; He, J.; Zhao, C.; Peng, Y.; Zhao, H.L.; Huang, W.Q.; Cao, C.Y.; Zhou, Y.J.; Zhou, Y.L.; Nie, Y.Q. Cross-Talk Between Butyric Acid and Gut Microbiota in Ulcerative Colitis Following Fecal Microbiota Transplantation. Front Microbiol. 2021, 12, 658292. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Zhou, Y.; Huang, S.; Li, J.; Zhao, K.; Li, X.; Wen, X.; Li, X.A. Fecal microbiota transplantation for ulcerative colitis: a prospective clinical study. BMC Gastroenterol. 2019, 19, 116. [Google Scholar] [CrossRef] [PubMed]
- Fehily, S.R.; Basnayake, C.; Wright, E.K.; Kamm, M.A. Fecal microbiota transplantation therapy in Crohn's disease: Systematic review. J Gastroenterol Hepatol. 2021, 36, 2672–2686. [Google Scholar] [CrossRef] [PubMed]
- Paramsothy, S.; Paramsothy, R.; Rubin, D.T.; Kamm, M.A.; Kaakoush, N.O.; Mitchell, H.M.; Castaño-Rodríguez, N. Faecal Microbiota Transplantation for Inflammatory Bowel Disease: A Systematic Review and Meta-analysis. J Crohns Colitis. 2017, 11, 1180–1199. [Google Scholar] [CrossRef] [PubMed]
- Food and Agriculture Organization and of the United Nations/World Health Organization. (2002). Guidelines for the evaluation of probiotics in foods, Available at: https://www.who.int>fs_management>probiotic_guidelines.
- Coqueiro, A.Y.; Raizel, R.; Bonvini, A.; Tirapegui, J.; Rogero, M.M. Probiotics for inflammatory bowel diseases: a promising adjuvant treatment. Int J Food Sci Nutr. 2019, 70, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Jakubczyk, D.; Leszczyńska, K.; Górska, S. The Effectiveness of Probiotics in the Treatment of Inflammatory Bowel Disease (IBD)-A Critical Review. Nutrients. 2020, 12, 1973. [Google Scholar] [CrossRef]
- Deriu, E.; Liu, J.Z.; Pezeshki, M.; Edwards, R.A.; Ochoa, R.J.; Contreras, H.; Libby, S.J.; Fang, F.C.; Raffatellu, M. Probiotic bacteria reduce salmonella typhimurium intestinal colonization by competing for iron. Cell Host Microbe. 2013, 14, 26–37. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, X.; Zhu, Y.; Ma, J.; Ma, H.; Zhang, H. Probiotic Mixture Protects Dextran Sulfate Sodium-Induced Colitis by Altering Tight Junction Protein Expressions and Increasing Tregs. Mediators Inflamm. 2018, 2018, 9416391. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, H.; Nakase, H.; Inoue, S.; Kawanami, C.; Itani, T.; Ohana, M.; Kusaka, T.; Uose, S.; Hisatsune, H.; Tojo, M.; Noda, T.; Arasawa, S.; Izuta, M.; Kubo, A.; Ogawa, C.; Matsunaka, T.; Shibatouge, M. Efficacy of probiotic treatment with Bifidobacterium longum 536 for induction of remission in active ulcerative colitis: A randomized, double-blinded, placebo-controlled multicenter trial. Dig Endosc. 2016, 28, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Kruis, W.; Fric, P.; Pokrotnieks, J.; Lukás, M.; Fixa, B.; Kascák, M.; Kamm, M.A.; Weismueller, J.; Beglinger, C.; Stolte, M.; Wolff, C.; Schulze, J. Maintaining remission of ulcerative colitis with the probiotic Escherichia coli Nissle 1917 is as effective as with standard mesalazine. Gut. 2004, 53, 1617–1623. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, S.M.; Mariadassou, M.; Nicolas, P.; Parayre, S.; Le Guellec, R.; Chuat, V.; Peton, V.; Le Maréchal, C.; Burati, J.; Loux, V.; Briard-Bion, V.; Jardin, J.; Plé, C.; Foligné, B.; Jan, G.; Falentin, H. Identification of proteins involved in the anti-inflammatory properties of Propionibacterium freudenreichii by means of a multi-strain study. Sci Rep. 2017, 7, 46409. [Google Scholar] [CrossRef] [PubMed]
- Steidler, L.; Hans, W.; Schotte, L.; Neirynck, S.; Obermeier, F.; Falk, W.; Fiers, W.; Remaut, E. Treatment of murine colitis by Lactococcus lactis secreting interleukin-10. Science. 2000, 289, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Oliva, S.; Di Nardo, G.; Ferrari, F.; Mallardo, S.; Rossi, P.; Patrizi, G.; Cucchiara, S.; Stronati, L. Randomised clinical trial: the effectiveness of Lactobacillus reuteri ATCC 55730 rectal enema in children with active distal ulcerative colitis. Aliment Pharmacol Ther. 2012, 35, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Sood, A.; Midha, V.; Makharia, G.K.; Ahuja, V.; Singal, D.; Goswami, P.; Tandon, R.K. The probiotic preparation, VSL#3 induces remission in patients with mild-to-moderately active ulcerative colitis. Clin Gastroenterol Hepatol. 2009, 7, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Tursi, A.; Brandimarte, G.; Papa, A.; Giglio, A.; Elisei, W.; Giorgetti, G.M.; Forti, G.; Morini, S.; Hassan, C.; Pistoia, M.A.; Modeo, M.E.; Rodino', S.; D'Amico, T.; Sebkova, L.; Sacca', N.; Di Giulio, E.; Luzza, F.; Imeneo, M.; Larussa, T.; Di Rosa, S.; Annese, V.; Danese, S.; Gasbarrini, A. Treatment of relapsing mild-to-moderate ulcerative colitis with the probiotic VSL#3 as adjunctive to a standard pharmaceutical treatment: a double-blind, randomized, placebo-controlled study. Am J Gastroenterol. 2010, 105, 2218–2227. [Google Scholar] [CrossRef] [PubMed]
- Zocco, M.A.; dal Verme, L.Z.; Cremonini, F.; Piscaglia, A.C.; Nista, E.C.; Candelli, M.; Novi, M.; Rigante, D.; Cazzato, I.A.; Ojetti, V.; Armuzzi, A.; Gasbarrini, G.; Gasbarrini, A. Efficacy of Lactobacillus GG in maintaining remission of ulcerative colitis. Aliment Pharmacol Ther. 2006, 23, 1567–1574. [Google Scholar] [CrossRef]
- Butterworth, A.D.; Thomas, A.G.; Akobeng, A.K. Probiotics for induction of remission in Crohn's disease. Cochrane Database Syst Rev. 2008, 2008, CD006634. [Google Scholar] [CrossRef]
- Miele, E.; Shamir, R.; Aloi, M.; Assa, A.; Braegger, C.; Bronsky, J.; de Ridder, L.; Escher, J.C.; Hojsak, I.; Kolaček, S.; Koletzko, S.; Levine, A.; Lionetti, P.; Martinelli, M.; Ruemmele, F.; Russell, R.K.; Boneh, R.S.; van Limbergen, J.; Veereman, G.; Staiano, A. Nutrition in Pediatric Inflammatory Bowel Disease: A Position Paper on Behalf of the Porto Inflammatory Bowel Disease Group of the European Society of Pediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr. 2018, 66, 687–708. [Google Scholar] [CrossRef] [PubMed]
- Bousvaros, A.; Guandalini, S.; Baldassano, R.N.; Botelho, C.; Evans, J.; Ferry, G.D.; Goldin, B.; Hartigan, L.; Kugathasan, S.; Levy, J.; Murray, K.F.; Oliva-Hemker, M.; Rosh, J.R.; Tolia, V.; Zholudev, A.; Vanderhoof, J.A.; Hibberd, P.L. A randomized, double-blind trial of Lactobacillus GG versus placebo in addition to standard maintenance therapy for children with Crohn's disease. Inflamm Bowel Dis. 2005, 11, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Garcia Vilela, E.; De Lourdes De Abreu Ferrari, M.; Oswaldo Da Gama Torres, H.; Guerra Pinto, A.; Carolina Carneiro Aguirre, A.; Paiva Martins, F.; Marcos Andrade Goulart, E.; Sales Da Cunha, A. Influence of Saccharomyces boulardii on the intestinal permeability of patients with Crohn's disease in remission. Scand J Gastroenterol. 2008, 43, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.J.; Sartor, R.B. Therapeutic Manipulation of the Microbiome in IBD: Current Results and Future Approaches. Curr Treat Options Gastroenterol. 2015, 13, 105–120. [Google Scholar] [CrossRef]
- Meini, S.; Laureano, R.; Fani, L.; Tascini, C.; Galano, A.; Antonelli, A.; Rossolini, G.M. Breakthrough Lactobacillus rhamnosus GG bacteremia associated with probiotic use in an adult patient with severe active ulcerative colitis: case report and review of the literature. Infection. 2015, 43, 777–781. [Google Scholar] [CrossRef]
Figure 1.
Intestinal epithelial barrier. The intestinal epithelial barrier is constituted by a single layer of specialized cells (enterocytes, goblet cells, Paneth cells, and intestinal stem cells) that create a physical wall against the external microenvironment. The covering mucus layer provides the adhesion substrate for the microbiota and prevents the transepithelial invasion of microorganisms in the systemic circulation. Immune cells (macrophages, dendritic cells, plasma cells, and lymphocytes) reside in the subepithelial layer and trigger the immune response in the presence of luminal antigen (created with BioRender).
Figure 1.
Intestinal epithelial barrier. The intestinal epithelial barrier is constituted by a single layer of specialized cells (enterocytes, goblet cells, Paneth cells, and intestinal stem cells) that create a physical wall against the external microenvironment. The covering mucus layer provides the adhesion substrate for the microbiota and prevents the transepithelial invasion of microorganisms in the systemic circulation. Immune cells (macrophages, dendritic cells, plasma cells, and lymphocytes) reside in the subepithelial layer and trigger the immune response in the presence of luminal antigen (created with BioRender).
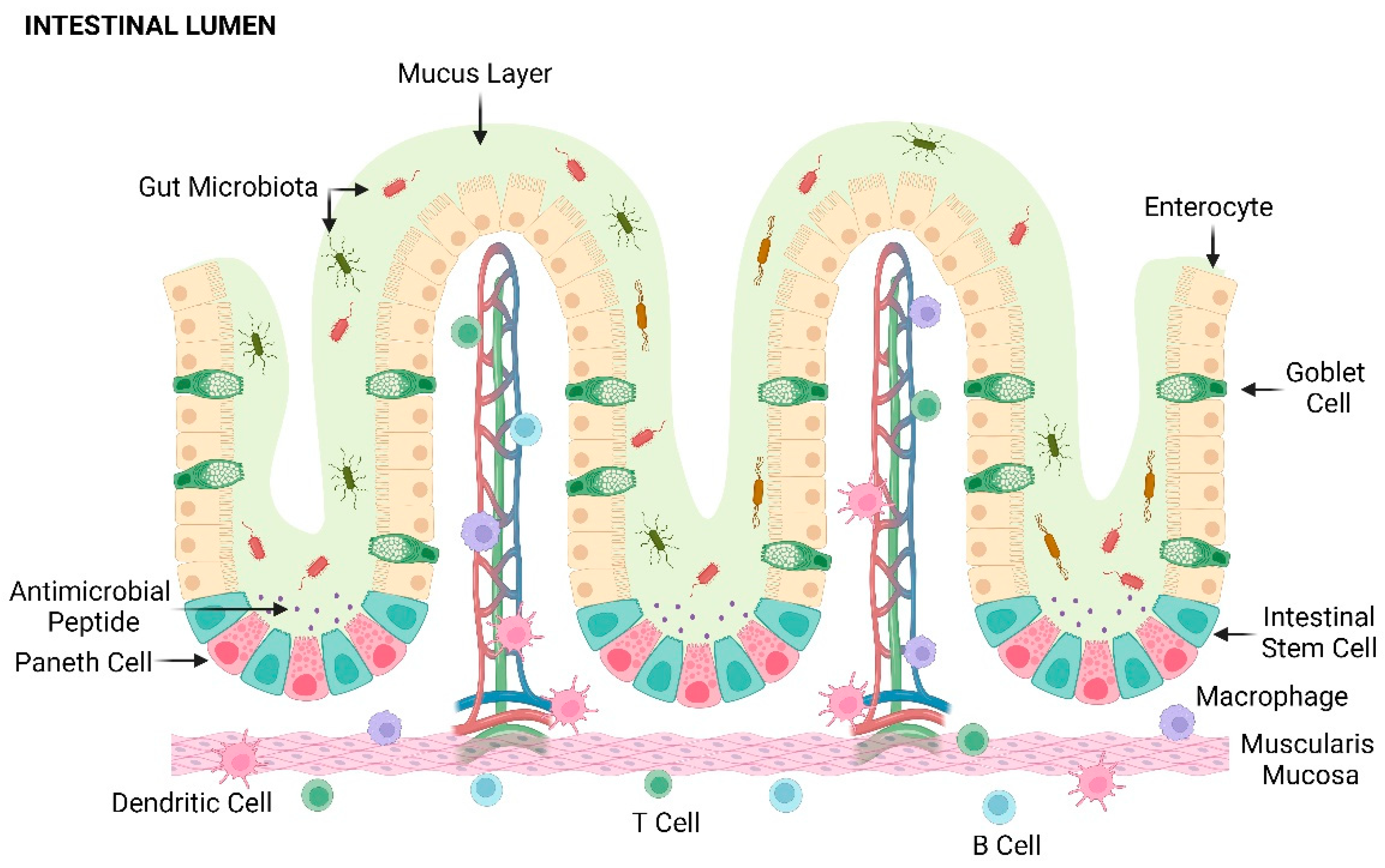
Figure 2.
Innate immune system. Macrophages, neutrophils, dendritic cells, natural killer, and innate lymphoid cells are the main actors of innate immunity. Innate immune response is fast and non-specific and provides an initial defense against infections after the recognition of non-self-components by the PRRs presenting on the membrane of the specialized cells. This leads to the neutralization or disruption of external antigens and to the activation of the adaptive immune system (created with BioRender).
Figure 2.
Innate immune system. Macrophages, neutrophils, dendritic cells, natural killer, and innate lymphoid cells are the main actors of innate immunity. Innate immune response is fast and non-specific and provides an initial defense against infections after the recognition of non-self-components by the PRRs presenting on the membrane of the specialized cells. This leads to the neutralization or disruption of external antigens and to the activation of the adaptive immune system (created with BioRender).
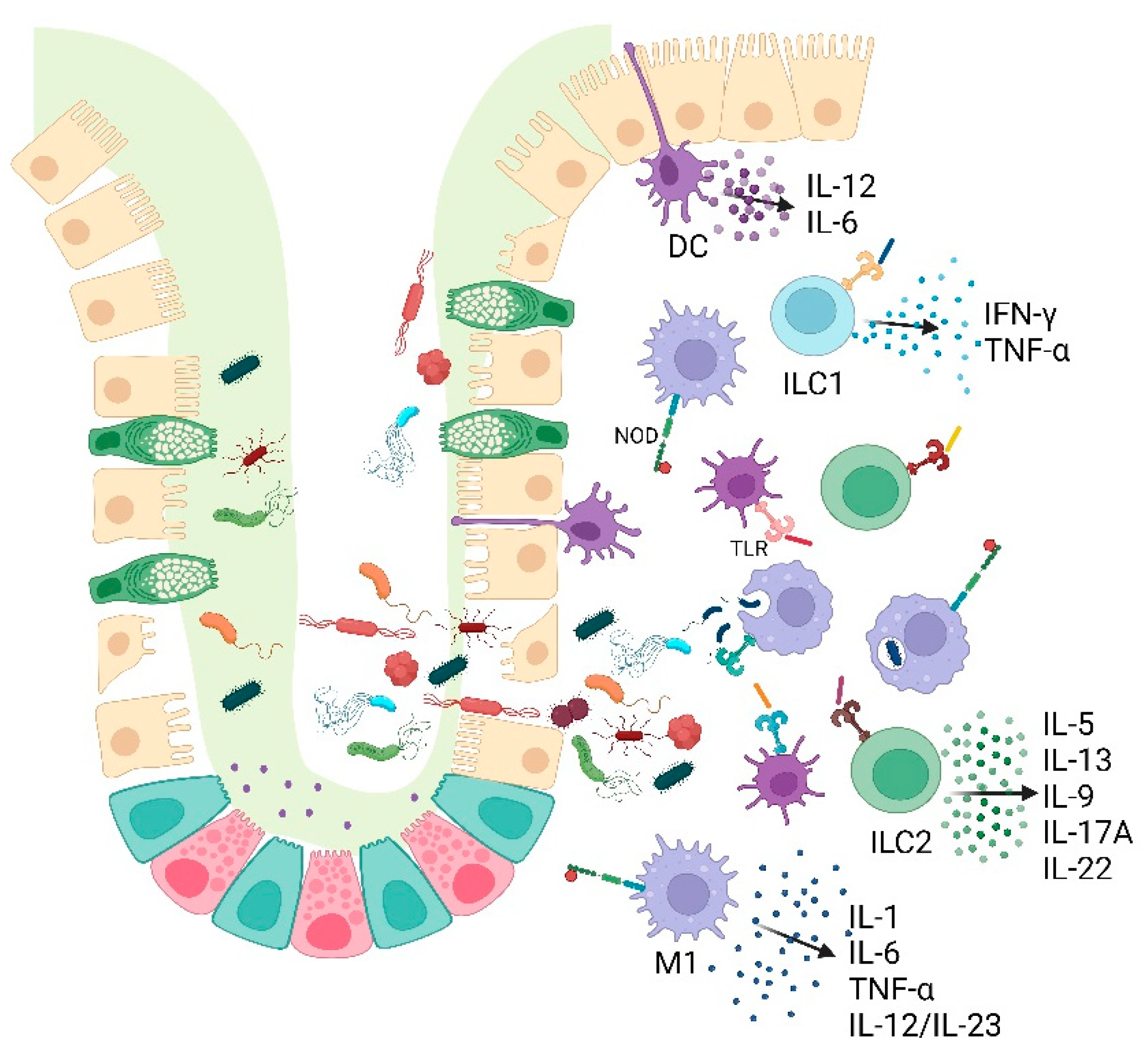
Figure 3.
Adaptive immune system. Adaptive immune response is secondary to the innate response and requires the activation of lymphocytes. The activation and the differentiation of Th0 cells into Th1, Th2 or Th17 is essential for the clearance of specific pathogens through the release cytokines and chemokines. Abnormal expansion of activated T cells is found in IBD patients (created with BioRender).
Figure 3.
Adaptive immune system. Adaptive immune response is secondary to the innate response and requires the activation of lymphocytes. The activation and the differentiation of Th0 cells into Th1, Th2 or Th17 is essential for the clearance of specific pathogens through the release cytokines and chemokines. Abnormal expansion of activated T cells is found in IBD patients (created with BioRender).
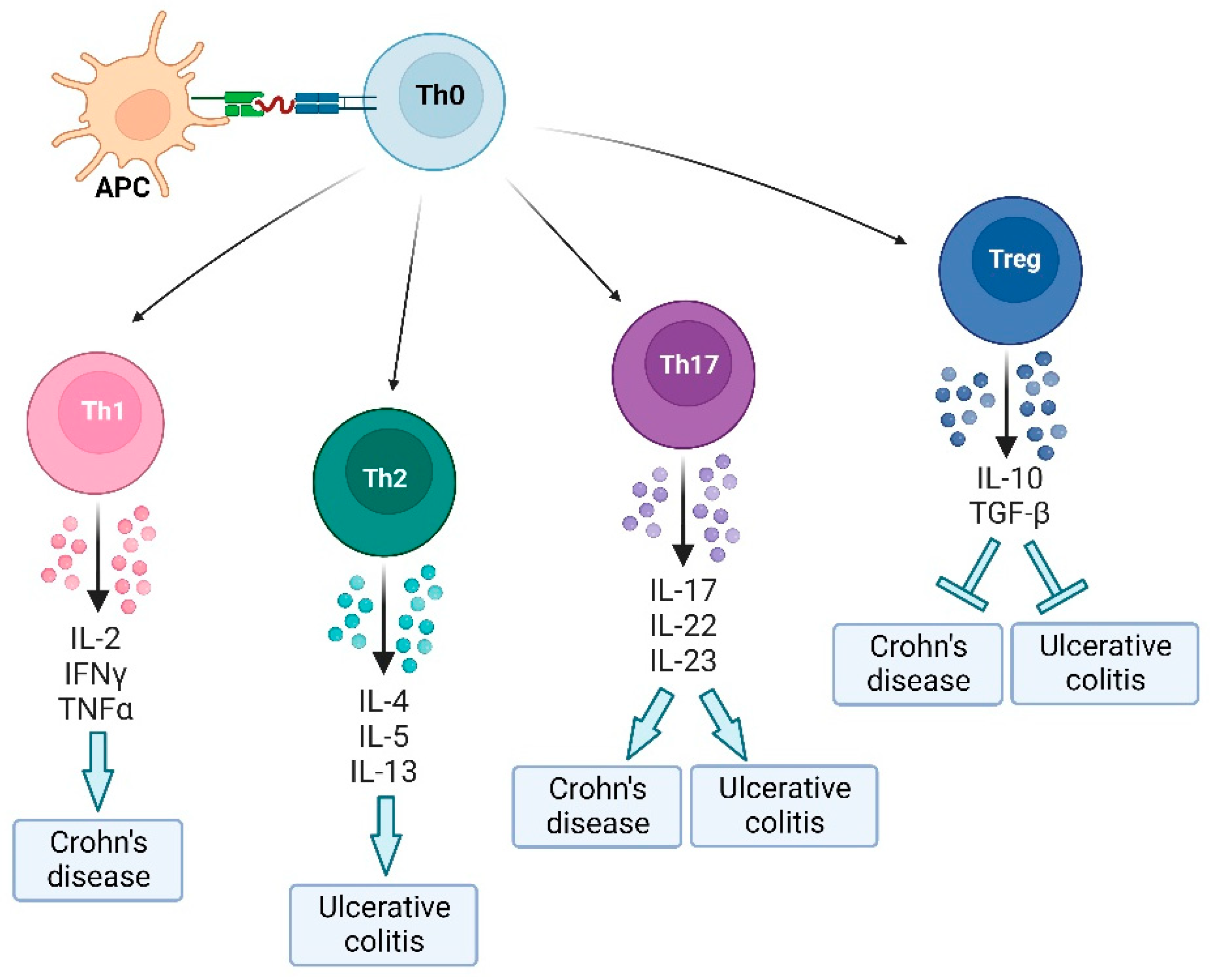
Figure 4.
Role of autophagy in the maintenance of the correct function of intestinal epithelial cells. In enterocytes, autophagy limits pathogen invasion and proliferation, enhances barrier integrity, and modulates the inflammatory response through the degradation of inflammasome complex. In Paneth cells and goblet cells, autophagy regulates the formation and the secretion of antimicrobial peptide and mucin granules, respectively. In intestinal stem cells, autophagy maintains stemness properties and protects the cells from oxidative stress (created with BioRender).
Figure 4.
Role of autophagy in the maintenance of the correct function of intestinal epithelial cells. In enterocytes, autophagy limits pathogen invasion and proliferation, enhances barrier integrity, and modulates the inflammatory response through the degradation of inflammasome complex. In Paneth cells and goblet cells, autophagy regulates the formation and the secretion of antimicrobial peptide and mucin granules, respectively. In intestinal stem cells, autophagy maintains stemness properties and protects the cells from oxidative stress (created with BioRender).
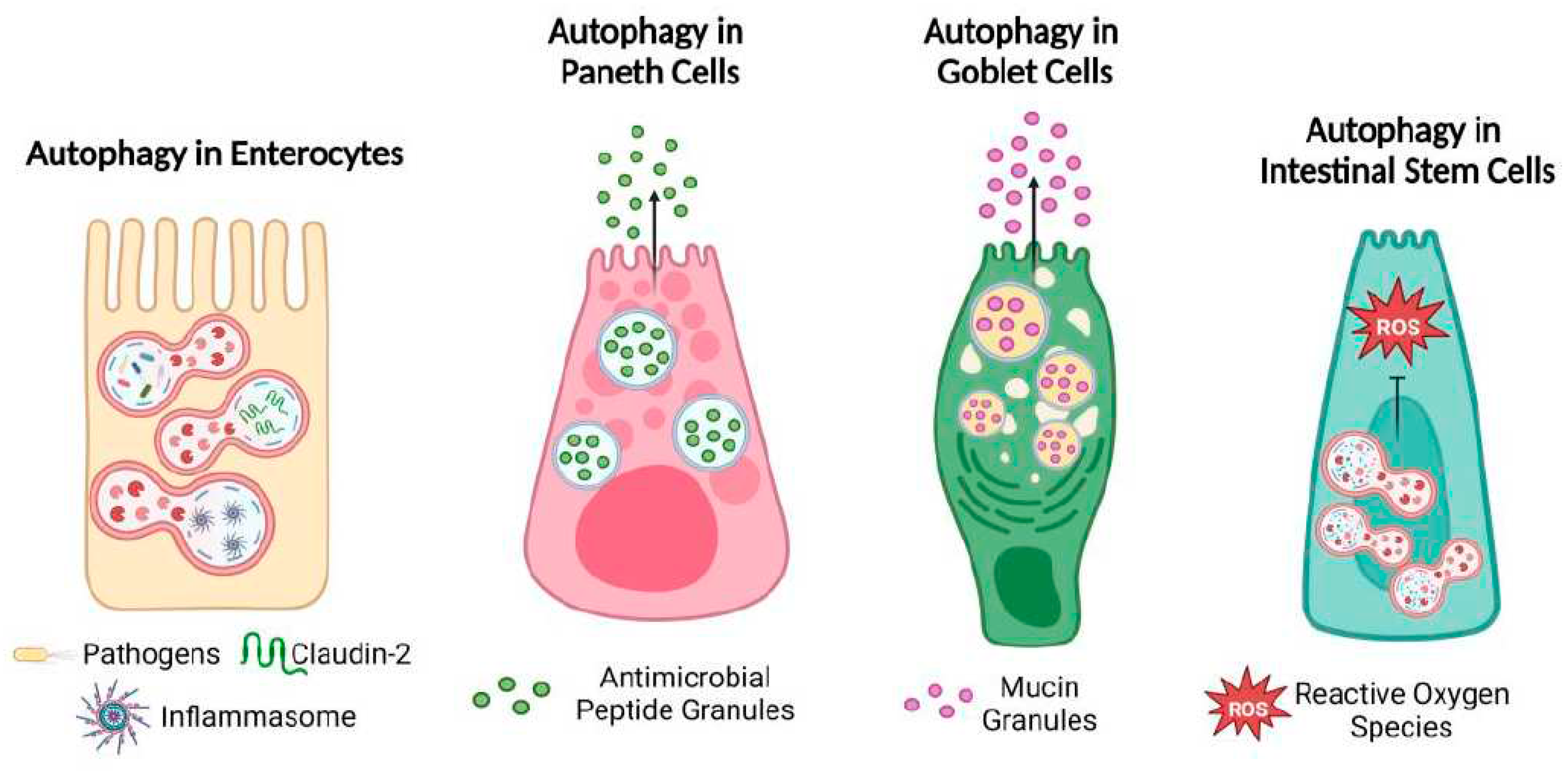
Figure 5.
The relationship between gut dysbiosis and inflammatory bowel disease. The condition of eubiosis, which defines the balance between the host and bacteria, is essential for proper intestinal homeostasis and host health. Alteration in the function and composition of gut microbiota due to environmental changes results in a condition defined as dysbiosis, which leads to the onset of IBD (created with BioRender).
Figure 5.
The relationship between gut dysbiosis and inflammatory bowel disease. The condition of eubiosis, which defines the balance between the host and bacteria, is essential for proper intestinal homeostasis and host health. Alteration in the function and composition of gut microbiota due to environmental changes results in a condition defined as dysbiosis, which leads to the onset of IBD (created with BioRender).
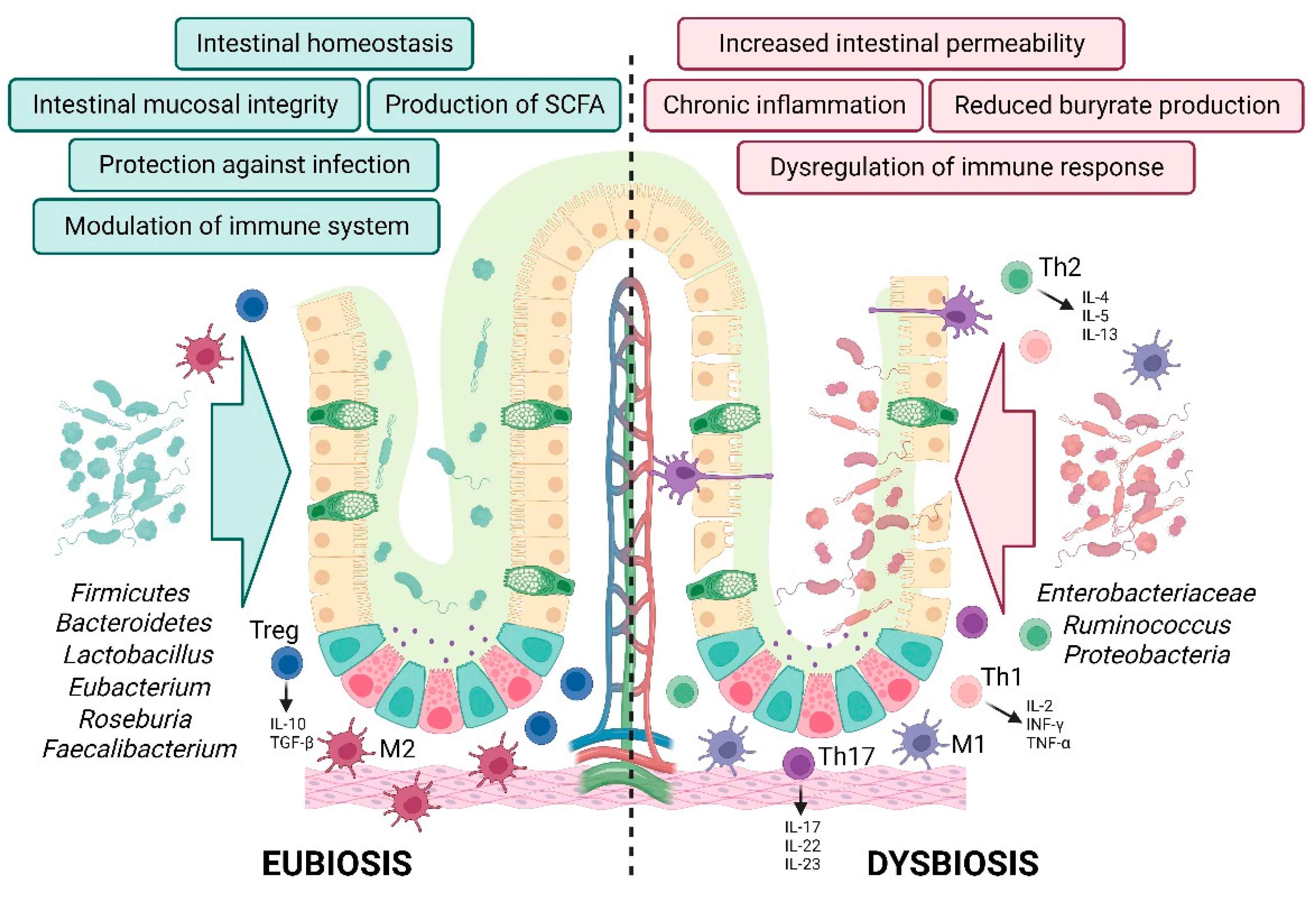
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



