Submitted:
23 December 2023
Posted:
25 December 2023
You are already at the latest version
Abstract
Both scleroderma and immunoglobulin G4-related disease (IgG4-RD) are systemic fibro-inflammatory diseases characterised by lymphoplasmacytic infiltrates. IgG4-RD and systemic Sclerosis (SSc) may share common pathophysiological mechanisms, but no examples of co-occurrence of the diseases have been found. Autologous haematopoietic stem cell transplantation (AHSCT) is implemented in selected rapidly progressive SSc with high risk of organ failure. However, existing guidelines are based on clinical trials that do not represent the entire patient population and exclude critically ill patients with no therapeutic alternatives. Examples of AHSCT in IgG4-RD are absent. We report the case of AHSCT in a female patient with overlapping progressive diffuse SSc and sinonasal IgG4-RD. Despite immunosuppressive therapy, IgG4-RD developed and clinically manifested shortly before AHSCT. Lacking therapeutic alternatives, after 11 years of SSc therapy, the 44-year-old successfully underwent AHSCT. The 63-month follow-up showed regression of symptoms. Only after surgical treatment (bilateral ethmoidectomy, sphenoidotomy, intranasal buccal antrostomy) the initially progressive clinical course of IgG4-RD remained indolent.
Keywords:
IgG4-related disease
; systemic sclerosis
; overlap syndrome
; autologous hematopoietic stem cell transplantation
1. Introduction
Systemic sclerosis (SSc) is a heterogeneous, autoimmune connective tissue disease characterized by immune response changes, vasculopathy and progressive fibrosis [1]. Commonly, Raynaud's phenomenon and skin fibrosis are early signs of the disease. Later, internal organs such as the lungs, heart or kidneys may be affected. In 48% to 68% of cases, disease-related complications lead to a fatal outcome, with interstitial lung disease (ILD) being the most common [2,3]. Currently, cyclophosphamide (CYC) is the drug of choice for SSc-associated ILD. However, the data on CYC is contradictory. The treatment effect is often only partial, the disease progresses after discontinuation of the drug and long-term use is highly toxic [4,5,6]. In contrast, autologous hematopoietic stem cell transplantation (AHSCT) is more effective but associated with a higher risk [7,8,9].
IgG4-RD is a rare systemic fibro-inflammatory disease first described in 2003. It can affect any organ causing a variety of symptoms [10,11]. Organ dysfunction develops through infiltration of tissue by lymphocytes and IgG4-secreting plasma cells, leading to inflammation, hypertrophy and progressive tissue fibrosis [10]. The aetiology of IgG4-RD disease is still unclear. Given the association with certain HLA variants, the overlap with other autoimmune diseases and the sensitivity to antirheumatic drugs, it is reasonable to consider IgG4-RD as a systemic autoimmune disease [12].
IgG4-RD of the head and neck with sinonasal involvement is very rare and is not included in the 2019 EULAR (European League Against Rheumatism) and ACR (American College of Rheumatology) IgG4-RD classification criteria[13]. Sinonasal disease is increasingly recognized, with the first case presented in 2009 [14]. The clinical and radiological features of sinonasal IgG4-RD are non-specific. Pathological IgG4-RD masses in the sinuses can cause swelling and pain, and less commonly visual disturbances. The maxillary sinuses are mostly affected with only local progression [14,15]. On CT, IgG4-RD appears as an irregular, heterogeneous mass with soft tissue density. Destruction of the sinus bone wall with infiltration of the surrounding structures and thickening of the nasal septum can also occur. In MRI, hypointense changes in the T2 sequence are disease specific [15].
Overlap syndrome (OS) is an autoimmune disease in which the classification criteria of at least two connective tissue diseases are met. 20-30 % of SSc patients develop OS [16]. Cases of IgG4-RD and overlapping systemic autoimmune connective tissue diseases are known, but no examples of comorbidity of SSc and IgG4-RD have been reported [12,17].
2. Case Presentation
In January 2003, a 33-year-old female patient presented with one-year lasting Raynaud’s phenomenon, finger numbness and swelling. Her medical history included allergic rhinitis, obstructive bronchitis, bronchial asthma, septal and adenoid surgery. Elevated antinuclear antibody (ANA) titres and a skin biopsy led to the diagnosis of SSc. Discrete signs of fibrotic lung changes were seen on chest X-ray.
Initial treatment included aspirin and pentoxifylline followed by additional penicillamine and prednisolone. After an allergic reaction to penicillamine, azathioprine (AZA) was initiated. Despite three years of outpatient therapy, the disease manifested as CREST-Syndrome (Calcinosis; Raynaud's phenomenon; Esophageal dysfunction; Sclerodactyly with ulceration (Figure 1) ; Telangiectasia). The patient worked as a community nurse, the cold working conditions during flood season aggravated the disease progression and led to disability.
September 2006 follow-up showed whole body skin induration with reduced range of motion, microstomia with prominent labial wrinkles (Figure 2) and acro-osteolysis in the radiographs, chest X-ray was normal. Under daily therapy with AZA 150 mg, prednisolone 10 mg and regular alprostadil 20-40 μg a day infusions patient developed dyspnoea on exertion. In February 2007, pulmonary function tests revealed moderate airway obstruction. After CYC pulse therapy (12 infusions of 1 g), skin induration decreased slightly, poor digital perfusion and exertional dyspnoea persisted, osteolytic changes progressed. High-resolution computed tomography (HRCT) showed signs of pulmonary fibrosis. Methotrexate (MTX) 10-15 mg weekly was started in July 2009. In 2012, the patient developed supraclavicular tumorous calcinosis (5 cm Ø).
The condition continued to deteriorate, and in January 2014 the multidisciplinary team decided to perform AHSCT. At the time of hospitalisation, the patient's overall condition was satisfactory (Table 1). The anti-topoisomerase I antibodies (anti-Scl-70) and anti-double stranded DNA antibodies (anti-dsDNA) where positive. The echocardiogram showed moderate left atrial dilatation, first degree tricuspid regurgitation, sclerodegenerative mitral valve, delayed left ventricular relaxation, ejection fraction >55 % and no pulmonary arterial hypertension (PAH). The MRI of the heart showed no focal lesions and no signs of pleural effusion.
During AHSCT, the haematopoietic progenitor cells were mobilised with filgrastim. A total dose of 200 mg/kg CYC (50 mg/kg on days -5, -4, -3, -2) and antithymocytic globulin (0.5 mg/kg on day -5 and 1.5 mg/kg on days -4, -3, -2, -1) in combination with methylprednisolone were used for conditioning. A complication-free transplantation of cumulative 4.95 × 106 / kg CD34 + cells was performed. After stimulation with filgrastim, granulopoiesis onset was on day 11 after transplantation. The results of the treatment are presented in Table 1.
Before AHSCT, the patient reported hoarseness, bilateral tingling and numbness in the postauricular area. CT revealed chronic frontitis, ethmoiditis, sphenoiditis and protruding masses in the maxillary sinuses on both sides. An outpatient follow-up was recommended. In spring 2018, discomfort and swelling developed in the right cheek. MRI showed moderate progression of the lesions. Cystic, irregular, 4-7 cm2 masses with peripheral accumulation of contrast and heterogeneous signal intensity were observed in both maxillary sinuses. Bilateral ethmoidectomy, sphenoidotomy, intranasal buccal antrostomy were performed. Histologically, abundant mucosal infiltration with IgG and IgG4 positive plasmocytes and tissue fibrosis were found. Nine months follow-up was without dynamics. In IgG fraction analysis, IgG4 levels were elevated.
3. Discussion
The guidelines recommend AHSCT for selected patients with severe, rapidly progressive, refractory SSc in the first 4 to 5 years after diagnosis [18,19,20,21]. The approach is based on the results of cohort studies and three randomized controlled trials comparing different AHSCT regimens with CYC pulse therapy. The mean disease duration before transplantation in the randomized controlled trials was 18.5 months, the longest duration was 38 months. In the cohort study (NISSC1), the mean disease duration was longer (23.8 months), with the longest period before transplantation of 103.7 months [7,8,9,22]. The presented patient underwent effective AHSCT almost 12 years after the onset of first SSc symptoms and 11 years (133 months) after diagnosis. This is an example of effective AHSCT in late-stage SSc. However, some important advances have been made in recent years, new therapies (e.g., mycophenolate mofetil, nintedanib, rituximab (RTX) and tocilizumab for the treatment of the key disease manifestations) have become available and accepted by regulatory authorities. Further research will be needed to investigate the role of AHSCT in the emerging context [23].
In the case presented, the context of SSc led to a degree of uncertainty in the IgG4-RD diagnosis. The patient developed nonspecific but characteristic symptoms of sinonasal IgG4-RD. CT imaging was consistent with IgG4-RD, but nonspecific. MRI images also showed nonspecific cystic changes with heterogeneous signal intensity, without characteristic hypointense changes found in the T2 sequence [15]. Two signs necessary for confirmation of the histopathologic diagnosis were observed: fibrous tissue and a highly pronounced IgG- and IgG4-positive plasmocytic infiltration. Obliterative phlebitis, immunohistochemical IgG4+ counts, and IgG4+ to total IgG+ cell ratios were not described [24]. However, the prior knowledge that both IgG4-RD and scleroderma are characterized by lymphoplasmacytic infiltrates and fibrosis complicated the clinical interpretation of the histopathological findings. In addition, IgG4-positive plasma cell infiltrates were also detected in SSc skin samples, although the number of IgG4-positive cells and the ratio of IgG to IgG4 were insignificant [25]. Nevertheless, the role of IgG4 in the pathogenesis of IgG4-RD is still unclear, as this IgG subtype is anti-inflammatory and has little ability to activate the complement system [12]. Observation of the clinical course increased certainty that the clinical symptoms were expressions of two different nosological entities. After transplantation, progression of sinus lesions occurred, whereas treatment was effective concerning SSc-specific lesions. However, after surgical removal of the sinus lesions and without IgG4-RD-specific treatment, no progression of the disease was observed within 9 months.
A plausible pathogenetic pathway for the manifestation of the overlapping diseases may be that both diseases are driven by an interplay of T and B cells, with subsets of T cells and CD4+ cytotoxic T lymphocytes (CTLs) playing a dominant role. T helper 2 cells in SSc and T follicular helper cells in IgG4-RD cells have overlapping phenotypic and functional characteristics. They appear to utilize regulation of interleukin-4 expression to induce B-cell maturation. Meanwhile, CTLs secreting interferon-γ, interleukin-1β, and tumor growth factor-β play important roles in inducing tissue damage and fibrosis [1,12,26]. T lymphocytes are stimulated by microorganisms in the gut. The intestinal microflora of IgG4-RD and SSc patients is remarkably similar and different from the healthy population (e.g., more opportunistic pathogens and pro-inflammatory bacteria) [27]. Further studies on the gut microbiome are needed to confirm this hypothesis.
The fact that infiltrative sinonasal lesions developed during monotherapy with immunosuppressive drugs (MTX, AZA, CYC) and progressed after transplantation may suggest that the given treatment is ineffective in sinonasal IgG4-RD. However, a potentially more aggressive course without immunosuppressive treatment cannot be excluded. Further monitoring of the patient is required to confirm to confirm progression free survival. Although sinonasal IgG4-RD is not malignant, systemic evaluation and timely treatment are necessary to prevent irreversible fibrous lesions. Commonly, steroids are effective. RTX or AZA are suggested in case of inadequate response or relapse [14]. In addition, RTX could be the agent of choice in systemic sclerosis after transplantation [28]. In this regard, the patient might have benefited from the additional RTX after AHSCT.
4. Conclusions
This is an instance of overlapping syndrome of systemic sclerosis and sinonasal IgG4-RD, with successful autologous hematopoietic stem cell transplantation in late-stage progressive diffuse systemic sclerosis. IgG4-RD is rather not part of the scleroderma spectrum disorders, however, a possible etiopathogenetic microbiomic link of diseases has been identified. Standard immunosuppressive therapy for systemic sclerosis an autologous haematopoietic stem cell transplantation have little effect in IgG4-RD but may have contributed to the indolent course of IgG4-RD. Rituximab might be suggested in a patient with systemic sclerosis after transplantation and overlapping active IgG4-RD. Further monitoring of the patient is needed to confirm the validity of the conclusions.
Informed Consent Statement
Written informed consent has been obtained from the patient to publish this paper.
References
- Denton, C.P.; Khanna, D.; Systemic sclerosis’; Engl, L.L.; vol.; no. 10103, pp. 1685–1699, Oct. 2017. [CrossRef]
- Poudel, D.R.; Jayakumar, D.; Danve, A.; Sehra, S.T.; Derk, C.T., ‘Determinants of mortality in systemic sclerosis: a focused review’, Rheumatol. Int., vol. 38, no. 10, pp. 1847–1858, Oct. 2018. [CrossRef]
- Rm, P.A.S; Av, A.; Kb, H., ‘Systemic sclerosis-associated interstitial lung disease’, Lancet Respir. Med., vol. 8, no. 3, Mar. 2020. [CrossRef]
- Zhao, M.; Wu, J.; Wu, H.; Sawalha, A.H.; Lu, Q., ‘Clinical Treatment Options in Scleroderma: Recommendations and Comprehensive Review’, Clin. Rev. Allergy Immunol., vol. 62, no. 2, pp. 273–291, Apr. 2022. [CrossRef]
- Shouval, R.; Furie, N.; Raanani, P.; Nagler, A.; Gafter-Gvili, A., ‘Autologous Hematopoietic Stem Cell Transplantation for Systemic Sclerosis: A Systematic Review and Meta-Analysis’, Biol. Blood Marrow Transplant. J. Am. Soc. Blood Marrow Transplant., vol. 24, no. 5, pp. 937–944, May 2018. 20 May. [CrossRef]
- Kowal-Bielecka, O. et al., ‘Update of EULAR recommendations for the treatment of systemic sclerosis’, Ann. Rheum. Dis., vol. 76, no. 8, pp. 1327–1339, Aug. 2017. [CrossRef]
- Burt, R.K. et al., ‘Autologous non-myeloablative haemopoietic stem-cell transplantation compared with pulse cyclophosphamide once per month for systemic sclerosis (ASSIST): an open-label, randomised phase 2 trial’, Lancet Lond. Engl., vol. 378, no. 9790, pp. 498–506, Aug. 2011. [CrossRef]
- van Laar, J.M. et al., ‘Autologous hematopoietic stem cell transplantation vs intravenous pulse cyclophosphamide in diffuse cutaneous systemic sclerosis: a randomized clinical trial’, JAMA, vol. 311, no. 24, pp. 2490–2498, Jun. 2014. [CrossRef]
- Sullivan, K.M. et al., ‘Myeloablative Autologous Stem-Cell Transplantation for Severe Scleroderma’, N. Engl. J. Med., vol. 378, no. 1, pp. 35–47, Jan. 2018. [CrossRef]
- Lanzillotta, M.; Mancuso, G.; Della-Torre, E., ‘Advances in the diagnosis and management of IgG4 related disease’, BMJ, vol. 369, p. m1067, Jun. 2020. [CrossRef]
- Wallace, Z.S. et al., ‘Clinical phenotypes of IgG4-related disease: an analysis of two international cross-sectional cohorts’, Ann. Rheum. Dis., vol. 78, no. 3, pp. 406–412, Mar. 2019. [CrossRef]
- Peyronel, F.; Fenaroli, P.; Maritati, F.; Schleinitz, N.; Vaglio, A., ‘IgG4-related disease: advances in pathophysiology and treatment’, Expert Rev. Clin. Immunol., vol. 19, no. 5, pp. 537–547, May 2023. 20 May. [CrossRef]
- Wallace, Z.S. et al., ‘The 2019 American College of Rheumatology/European League Against Rheumatism Classification Criteria for IgG4-Related Disease’, Arthritis Rheumatol. Hoboken NJ, vol. 72, no. 1, pp. 7–19, Jan. 2020. [CrossRef]
- Kaur, K. et al., ‘Sinonasal IgG4-related disease: a rare and emerging entity broadening the differential diagnosis in the sinonasal universe’, Eur. Arch. Oto-Rhino-Laryngol. Off. J. Eur. Fed. Oto-Rhino-Laryngol. Soc. EUFOS Affil. Ger. Soc. Oto-Rhino-Laryngol. - Head Neck Surg., vol. 278, no. 8, pp. 2883–2890, Aug. 2021. [CrossRef]
- Fujita, A.; Sakai, O.; Chapman, M.N.; Sugimoto, H., ‘IgG4-related disease of the head and neck: CT and MR imaging manifestations’, Radiogr. Rev. Publ. Radiol. Soc. N. Am. Inc, vol. 32, no. 7, pp. 1945–1958, 2012. [CrossRef]
- Wielosz, E.; Majdan, M.; Dryglewska, M.; Targońska-Stępniak, B., ‘Overlap syndromes in systemic sclerosis’, Adv. Dermatol. Allergol. Dermatol. Alergol., vol. 35, no. 3, pp. 246–250, Jun. 2018. [CrossRef]
- Naramala, S.; Biswas, S.; Adapa, S.; Gayam, V.; Konala, V.M.; Bose, S., ‘An Overlapping Case of IgG4-Related Disease and Systemic Lupus Erythematosus’, J. Investig. Med. High Impact Case Rep., vol. 7, p. 2324709619862297, 2019. [CrossRef]
- Sullivan, K.M. et al., ‘Systemic Sclerosis as an Indication for Autologous Hematopoietic Cell Transplantation: Position Statement from the American Society for Blood and Marrow Transplantation’, Biol. Blood Marrow Transplant. J. Am. Soc. Blood Marrow Transplant., vol. 24, no. 10, pp. 1961–1964, Oct. 2018. [CrossRef]
- Kowal-Bielecka et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann. Rheum. Dis. 76, 1327–1339 (2017). - „Google“ paieška’. Available online: https://www.google.com/search?q=Kowal-Bielecka%2C+O.+et+al.+Update+of+EULAR+recommendations+for+the+treatment+of+systemic+sclerosis.+Ann.+Rheum.+Dis.+76%2C+1327%E2%80%931339+(2017).&oq=Kowal-Bielecka%2C+O.+et+al.+Update+of+EULAR+recommendations+for+the+treatment+of+systemic+sclerosis.+Ann.+Rheum.+Dis.+76%2C+1327%E2%80%931339+(2017).&aqs=chrome..69i57.606j0j4&sourceid=chrome&ie=UTF-8 (accessed on 18 July 2023).
- Snowden, J.A. et al., ‘Haematopoietic SCT in severe autoimmune diseases: updated guidelines of the European Group for Blood and Marrow Transplantation’, Bone Marrow Transplant., vol. 47, no. 6, pp. 770–790, Jun. 2012. [CrossRef]
- Farge, D. et al., ‘Cardiopulmonary assessment of patients with systemic sclerosis for hematopoietic stem cell transplantation: recommendations from the European Society for Blood and Marrow Transplantation Autoimmune Diseases Working Party and collaborating partners’, Bone Marrow Transplant., vol. 52, no. 11, pp. 1495–1503, Nov. 2017. [CrossRef]
- Henes, J. et al., ‘Autologous stem cell transplantation for progressive systemic sclerosis: a prospective non-interventional study from the European Society for Blood and Marrow Transplantation Autoimmune Disease Working Party’, Haematologica, vol. 106, no. 2, pp. 375–383, Feb. 2021. [CrossRef]
- Galdo, F.D. et al., ‘Op0234 2023 Update of Eular Recommendations for the Treatment of Systemic Sclerosis’, Ann. Rheum. Dis., vol. 82, no. Suppl 1, pp. 154–155, Jun. 2023. [CrossRef]
- Deshpande, V. et al., ‘Consensus statement on the pathology of IgG4-related disease’, Mod. Pathol. Off. J. U. S. Can. Acad. Pathol. Inc, vol. 25, no. 9, pp. 1181–1192, Sep. 2012. [CrossRef]
- Reddi, D.M.; Cardona, D.M.; Burchette, J.L.; Puri, P.K.; Scleroderma; IgG4-related disease’; Dermatopathol, A.J.; vol.; no.; pp.; Jun. 2013. [CrossRef]
- Jin, W.; Zheng, Y.; Zhu, P., ‘T cell abnormalities in systemic sclerosis’, Autoimmun. Rev., vol. 21, no. 11, p. 103185, Nov. 2022. [CrossRef]
- Plichta, D.R. et al., ‘Congruent microbiome signatures in fibrosis-prone autoimmune diseases: IgG4-related disease and systemic sclerosis’, Genome Med., vol. 13, no. 1, p. 35, Feb. 2021. [CrossRef]
- Gressenberger, P. et al., ‘Rituximab as a Treatment Option after Autologous Hematopoietic Stem Cell Transplantation in a Patient with Systemic Sclerosis’, J. Pers. Med., vol. 11, no. 7, p. 600, Jun. 2021. [CrossRef]
Figure 1.
Sclerodactyly with shortened distal phalanges, ulceration.
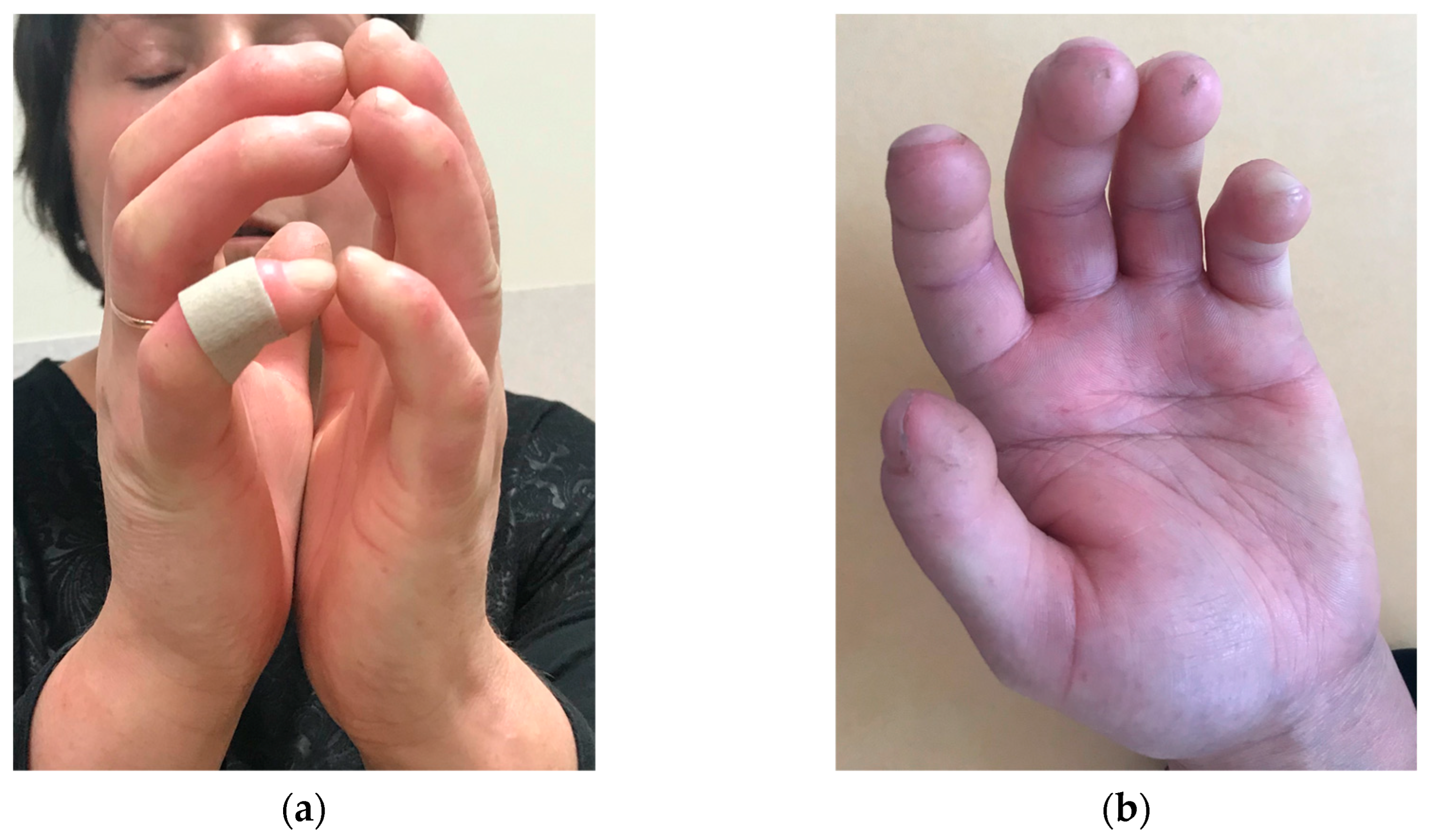
Figure 2.
Microstomia (a) with prominent labial wrinkles (b).
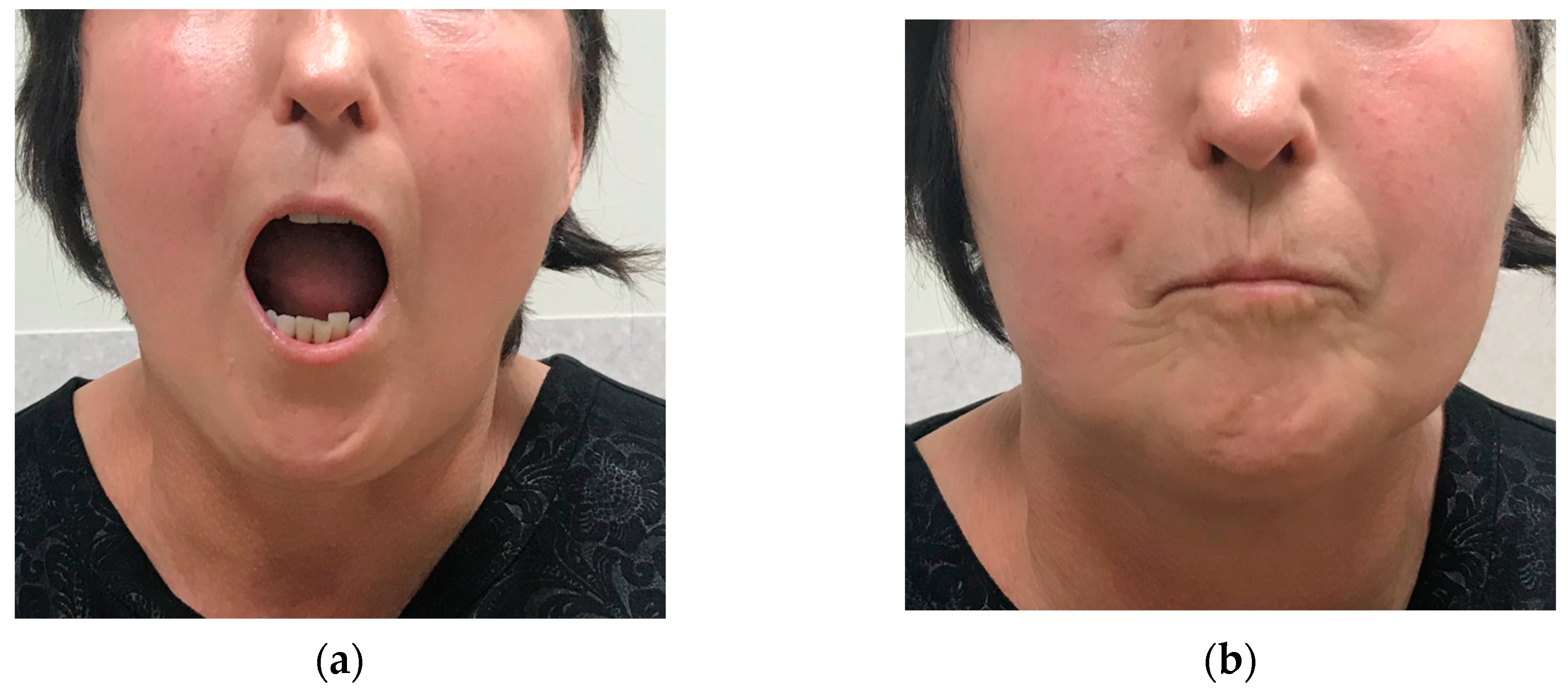

Table 1.
Pre- and post-transplant assessment results.
| Before AHSCT | After AHSCT | |||
|---|---|---|---|---|
| Date (duration after AHSCT) |
2014 01 | 2014 09 (8 mo.) |
2015 04 (15 mo.) |
2019 04 (63 mo.) |
| Hand injury, Raynaud's syndrome, skin conditions | advanced sclerodactyly with flexural contractures, osteolysis, deep scarring, cutaneous fibrosis over the whole body, restricted movement | softer hands and body skin, greater range of movement | skin induration is reduced all over the body, acral contractures are reduced, wrinkles appear on the face, the amplitude of the yawning increases | Raynaud’s syndrome persistence, mild cutaneous induration; sclerodactyly with flexural contractures, multiple scars, osteolysis of the fingertips (overall positive dynamic) |
| DLCO, %, diffusing capacity for carbon monoxide |
67 % mild reduction |
61 % mild reduction |
63 % mild reduction |
73 % mild reduction |
| Pulmonary function tests assessment | moderate airway obstruction | moderate airway obstruction | moderate airway obstruction | moderate airway obstruction |
| The patient subjectively assessed her condition | worsening | 50–60 % improvement | 90 % improvement | 30 % improvement |
| VAS | 3 | 1 | 1 | 3 |
| mRSS | 40 | 34 | 30 | 23 |
| Assessment of fibrotic lesions in the lungs on HRCT scan (semi-quantitative analysis according to Warrick, 0-15 points) | 14 | 13 (slightly less frosted glass zones) | 12 (areas of opaque glass in the lungs, pleural thickenings, subpleural lines and isolated subpleural cysts) | semi-quantitative analysis according to Warrick was not performed (lungs uniformly airy on both sides, no interstitial lesions observed) |
| Capillaroscopy | abundant avascular zones, isolated giant capillaries, signs of angiogenesis, marked fibrosis | increased capillary density and isolated haemorrhages are observed as ischaemia and fibrosis decrease | in dynamics, increasing capillary density, decreasing areas of avascular zones, increasing number of giant capillaries | |
| CRP/ESR | CRP 5,1 mg/l ESR 34 mm/h |
ESR 2 mm/h | ESR 17 mm/h | |
AHSCT - autologous haematopoietic stem cell transplantation; DLCO - diffusing capacity for carbon monoxide (normal range 77-100%); VAS - visual analog scale (0-10 cm, 0 cm corresponds to the lowest pain intensity); mRSS - modified Rodnan skin score (0-51, 0 corresponds to the condition with the fewest skin lesions); HRCT - high-resolution computed tomography; CRP - C-reactive protein (norm ≤ 5 mg/l); ESR - erythrocyte sedimentation rate (norm in women ≤ 20 mm/h).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



