
Submitted:
23 December 2023
Posted:
25 December 2023
You are already at the latest version
Abstract
A series of polynuclear, dinuclear, and mononuclear Mo(VI) complexes were synthesized with the hydrazonato ligands derived from 5-methoxysalicylaldehyde and the corresponding hydrazides (isonicotinic hydrazide (H2L1), nicotinic hydrazide (H2L2), 2-aminobenzhydrazide (H2L3) or 4-aminobenzhydrazide (H2L4)). The metallosupramolecular compounds obtained from non-coordinating solvents, [MoO2(L1,2)]n (1 and 2) and [MoO2(L3,4)]2 (3 and 4), formed infinite structures and metallacycles, respectively. By blocking two coordination sites with cis-dioxo ligands, the molybdenum centers have three coordination sites occupied by the ONO donor atoms from the rigid hydrazone ligands and one by N atom of pyridyl or amine-functionalized ligand subcomponents from the neighbouring Mo building units. The reaction in methanol afforded the mononuclear analogues [MoO2(L1-4)(MeOH)] (1a-4a) with additional monodentate MeOH ligand. All isolated complexes were tested as catalysts for cyclooctene epoxidation using tBuOOH (TBHP) as an oxidant in water. The impact of the structure and ligand lability on the catalytic efficiency in homogeneous cyclooctene epoxidation was elucidated through theoretical considerations. Thus, dinuclear assemblies exhibited better catalytic activity than mononuclear or polynuclear complexes.
Keywords:
molybdenum
; hydrazone
; metallosupramolecular
; dinuclear
; metallacycles
; polymers
; epoxidation
; cyclooctene
; catalysis
; DFT
1. Introduction
Metallosupramolecular compounds are species formed from metal ions and organic ligands in coordination-driven self-assembly processes [1,2,3,4,5]. Aesthetically fascinating architectures can be self-assembled from the same building blocks depending on metal coordination geometry, ligand structure, metal-to-ligand ratio, solvent, concentration, and presence of guests. The formation of zero-dimensional, polyhedral, or infinite structures reflects a complex interplay between kinetic and thermodynamic contributions [6,7,8,9].
The interest in this class of compounds comes not only from the challenges related to the development of reliable synthetic strategies and rational design but also from their wide-ranging potential. Thus, those assemblies can find application in catalysis [10,11], optics [12,13,14], magnetism [15,16,17], gas storage separation [18,19], and as sensors [20,21]. Considering the importance to integrate green chemistry principles and sustainability into research, development of novel catalysts is essential, particularly those that could exhibit high activity and selectivity at mild operating conditions, is of high interest.
The catalytic properties of molybdenum coordination compounds [22,23,24,25,26,27] have led to an increased interest in their metallosupramolecular chemistry in years. As a result, the investigation has grown from essential design principles enabling the formation of the first coordination polymers and metallocycles [28,29,30,31,32,33], to exploration of their structural and compositional fine-tuning in order to obtain catalysts with excellent properties [34,35,36,37,38,39]. Accordingly, constrained cyclic Mo(VI) metallosupramolecular complexes were recently found to be among the most effective catalysts for olefin epoxidation [37,38,39].
There is still a promising pool of catalysts based on metallosupramolecular or simple mononuclear (dioxo)Mo(VI) coordination assemblies. Such materials can utilize the advantages of organic components for fine structural tuning and, consequently, their properties. In particular, the compounds with hemilabile coordination bonds are of interest as they could serve as high-efficiency catalysts. For example, the high performance of hexacoordinated Mo(VI) (pre)catalysts closely correlates with their possibility of converting into the pentacoordinated counterparts [40,41].
To explore the (dioxo)Mo(VI) complexes and their catalytic properties, four hydrazone ligands have been selected (Scheme 1) where the subunit derived from 5-methoxysalicylaldehyde remained unchanged. It was expected that the catalytic activity could be modulated by the hydrazide moiety. Self-assembly of {MoO2}2+ units with ligands bearing (iso)nicotinoyl or aminobenzoyl moieties in dichloromethane yielded metallosupramolecular polymers and dimers, respectively, based on metal-nitrogen linkages. The reaction in methanol gave rise to mononuclear coordination compounds containing MeOH ancillary ligands with metal–oxygen bonds. Crystal and molecular structures of two hydrazones, one dinuclear and three mononuclear coordination compounds were determined by the single crystal X-ray diffraction method. All compounds were additionally characterized by elemental analysis, powered X-ray diffraction, thermogravimetric, and spectroscopic methods.
Molybdenum-catalyzed epoxidation of alkenes is an important process as it allows the conversion of simple hydrocarbon substrates into high-value fine chemicals. We, therefore, explored the effects of the Mo–N (N being a pyridyl or amine nitrogen atom) or Mo–O (O being a MeOH oxygen atom) bond lability on the catalytic performances of materials obtained in the oxidation of cyclooctene. As in our previous works we focused on catalytic reaction under environmentally-friendly conditions without organic solvents. The catalytic efficiency of the Mo-coordination entities being evaluated using tert-butyl hydroperoxide as the oxidant in water. Catalytic results are discussed in relation to the complex nuclearity, coordination environment, and ligand structure. The obtained results were compared to the previously published ones.
2. Results and Discussion
2.1. Synthesis and Structural Characterization
2.1.1. Hydrazones
Hydrazones H2L1-H2L4 (Scheme 1) were afforded by reacting equimolar amounts of 5-methoxysalicylaldehyde with the corresponding hydrazides (isonicotinic hydrazide, nicotinic hydrazide, 2-aminobenzhydrazide or 4-aminobenzhydrazide) in a methanolic solution. In the case of H2L1 and H2L2, the reagents were heated under reflux. The synthesis of H2L3 and H2L4 required lowering the reaction temperature primarily to avoid the secondary condensation reaction of the aldehyde with the amine functional group. All compounds, except H2L4, remained stable in the solid state for a longer time at room temperature. However, upon exposure to the air, hydrazone H2L4 transformed over time to the more stable hydrate H2L4·H2O.
Crystal structures of H2L1 and H2L1·H2O were reported previously [42,43]. Ligands H2L3 and H2L4 crystallize in orthorhombic Pna21 and triclinic P-1 space groups, respectively (Figure 1a, Supplementary Materials, Figure S1, Table S1). Crystal structure of H2L3 contains one molecule of ligand in the asymmetric unit, which pack in alternating zig-zag fashion (Supplementary Materials, Figure S2), as dictated by a singular intermolecular hydrogen bond between hydrazide N–H donor and aryl hydroxy group acceptor (Supplementary Materials, Figure S3a). Meanwhile, in the crystal structure of H2L4 one can find two symmetrically inequivalent ligand molecules, which through interactions between hydrazide and aryl amine donors, and amide keto and aryl hydroxy group acceptors form a complex hydrogen-bonded network (Supplementary Materials, Figure S3b). However, both ligands crystallize in keto-amino tautomeric form (Supplementary Materials, Scheme S1), as evidenced by C2–O2, C2–N2 and N2–N1 bond lengths (Supplementary Materials, Table S2), which correspond to lengths expected for double, single and double bond, respectively. Additionally, both ligands are in E conformation (Supplementary Materials, Table S3).
2.1.2. Metallosupramolecular Coordination Assemblies
The Mo(VI) metallosupramolecular complexes 1-4 were synthesized via reaction of [MoO2(acac)2] with H2L1-H2L4, respectively, in CH2Cl2 at room temperature. The formulae of [MoO2(L1,2)]n (1 and 2) and [MoO2(L3,4)]2 (3 and 4), containing the corresponding ligand in a doubly deprotonated form, are consistent with elemental, thermogravimetric analysis, spectroscopic, and X-ray diffraction data. Their formation is in accordance with the energy-related principle of “maximum site occupancy”, according to which species with all the coordination sites occupied are more stable than those with vacant sites [44].
Synthesis with ligands H2L1 or H2L2 bearing nitrogen containing heterocyclic ring resulted in the coordination polymers, comprising Mo coordinated by one hydrazone ligand through ONO donor atoms (phenolic-oxygen, azomethine-nitrogen, and hydrazidic-oxygen) and the second ligand through the isonycotinoyl N atom from the adjacent Mo complex unit. Although polymer 1 has been previously reported (CSD refcode ZILVIB) [45], it was prepared under different conditions. The polymer forms infinite chains, topology of which are dictated by the rigid geometry of the isonicotinoyl fragment and {MoO2}2+ core. The only degree of freedom in this chain is the Mo–Npyridyl bond length, which is slightly longer than expected for similar complexes with ONO ligand coordinated on the {MoO2}2+ core and pyridyl fragment coordinated on the free axial site. Particularly, the bond length in 1 is found to be 2.455 Å as opposed to average of 2.378±0.082 Å for 102 structures in CSD database [46]. This indicates significant steric impedance in the assembly of 1 polymer.
Ligands modified by incorporating the amino functionality, H2L3 and H2L4, gave rise the cyclic dimers [MoO2(L)3,4]2 (3 and 4). The molecular structure of 3 is shown in Figure 1c. Selected geometric data are given in Supplementary Materials, Table S2. The favoured formation of dimers over polymers is related to the preferred formation of smaller molecules containing fewer building blocks [47].
The structure of 3 (Supplementary Materials, Figures S1 and S2) exhibits a distorted octahedral geometry around the Mo(VI) metal center. The ONO ligand chelates in a meridional fashion occupying three coordination sites. Two cis oriented oxido ligands and the amino N atom from the adjacent Mo complex unit completes the coordination sphere. Similar to 1, one can analyze the oligomerization “propensity” based on observed Mo–Namine length. In 3, that length is 2.536(2) Å, whereas for the 11 known examples in the CSD database the mean length is 2.534±0.098 Å. One must observe that complexes derived from H2L3 and H2L4 have greater conformational flexibility when considering coordination of the amine nitrogen atom, compared to the coordination of pyridyl nitrogen atom in 1 and 2. The PXRD patterns of 1 and 3 were in good agreement with the simulated patterns of the corresponding crystal structures (Figure 2).
2.1.3. Mononuclear Coordination Assemblies
In the reaction of [MoO2(acac)2] complex with hydrazone ligands in methanol, coordination of the pyridyl/amine moiety was not observed. Instead, [MoO2(L1-4)(MeOH)] (1a-4a) compounds crystallized containing additional monodentate MeOH ligand (Figure 1b, Supplementary Materials, Figure S1). Complexes 1a-4a were obtained in higher yield (64-89%) than the metallosupramolecular species 1-4 (31-38%). A single-crystal X-ray study of 1a, 2a, and 3a, confirmed the structures to be mononuclear. Experimental powder diffractograms of all obtained samples matched well with the calculated ones (Figure 3). Complexes 1a-4a were stable in the solid state at room temperature. However, their quantitative conversion to polynuclear 1 and 2 or dinuclear forms 3 and 4 was observed upon heating in acetone or acetonitrile.
The crystal structures of 1a, 2a and 3a (Supplementary Materials, Figures S1, S2) show features already well established for the majority of [MoO2(L)(MeOH)] complexes. The {MoO2}2+ core has the same coordinative environment as in polymers (see chapter 2.1.2.), except for the axial coordination site occupied with oxygen atom from methanol molecule. Sterically undemanding methanol ligand has well-defined coordinative bond length of 2.343±0.035 Å based on 206 examples in the CSD database, and complexes 1a, 2a and 3a have this length in the same ballpark, albeit a bit shorter (2.313(2), 2.295(7) and 2.287(4) Å, respectively).
Crystal packing (Supplementary Materials, Figure S2), and supramolecular interactions in prepared methanol complexes are dictated by a small number of hydrogen bond donors (namely, methanol OH and amine NH2 moieties), forming relatively simple supramolecular chains. However, due to presence of amino group donor, supramolecular chain in 3a is built upon supramolecular dimers, akin to coordinative dimers of 3, whereas supramolecular chains in 1a and 2a are made of monomers interacting solely through methanol OH∙∙∙Npyridyl hydrogen bond (Supplementary Materials, Figure S4).
2.2. Spectroscopic Characterization
The infrared spectra confirmed the presence of the keto tautomer. The intense absorptions in the range 1695-1653 cm−1 and 1654−1626 cm−1 are attributed to the C=O and C=N imine bond vibrations, respectively. All hydrazones exhibit bands at approximately 3200 cm–1 assigned to –O–H and =N–NH. The medium intensity band observed at 1163 cm−1− 1115 cm−1 is assigned to N−N stretching vibrations [48].
The stretch bands of C=O, –O–H, and =N–NH characteristic of free hydrazones disappeared upon metal complexation. The presence of bands in 1-4 belonging to C−Ohydrazone (at 1320-1345 cm–1), C−Ophenolic (at 1260-1275 cm–1), and C=N (at 1600-1610 cm–1) indicated hydrazone functionality tautomerization N–NH–C=O→N–N=C–OH, deprotonation, and the molybdenum atom coordination through hydrazone ONO donor atoms. Strong absorption bands characteristic for {MoO2}2+ core in the range 935-908 cm–1 and weaker intensity bands for Mo−N bonds below 900 cm-1 corroborated the metallosupramolecular complex formation [38]. The methanol coordination in 1a-4a is supported by a new absorption band typical for C−OMeOH around 1030 cm−1. Additionally, the mononuclear complexes exhibited stretching frequencies O=Mo−OMeOH around 900 cm−1. The –NH2 bond vibrations of the mononuclear complexes present as two medium-intensity bands (3356-3263 cm–1) are higher in energy than those of the dinuclear complexes (3334-3227 cm–1).
Proton and carbon NMR chemical shifts of H2L1-H2L4 (Supplementary Materials, Tables S9-S12, Schemes S2-S5, Figures S5-S8) are assigned by 1H, APT, HMQC, and HMBC experiments in DMSO-d6. Downfield shifts in the range of 11.02-10.66 ppm and 12.37-11.88 ppm suggested the presence of –OH and =N–NH groups, respectively, thus indicating the amide form =N−NH−(C=O)− in solution. Singlets at 6.54 and 5.89 confirmed the presence of −NH2 protons in the spectra of H2L3 and H2L4, respectively. The OH, N=NH, and NH2 signals are broadened to some extent due to their involvement in hydrogen bonding. Singlets at 8.75-8.63 ppm and 3.77-3.73 ppm are assigned to CH=N and OCH3 groups, respectively. The aromatic moieties gave signals in the range of 9.19-6.65 ppm. In the 13C NMR spectra of H2L1-H2L4, the py and phenyl hydrazone moiety signals C5-C10 are observed in the range of 151.90-121.06 ppm and 152.03-112.09 ppm, respectively. The signals in the range 164.59-160.94 ppm and 148.07-145.95 ppm are attributed to C4 of the C=O group and C1 carbon of the CH=N group, respectively.
Due to donor properties of the solvent DMSO-d6, the metallosupramolecular nature of complexes 1-4 and the presence of ancillary ligand in the mononuclear complexes 1a-4a are disrupted, and complexes [(MoO2)2(L1-4)(DMSO)] appeared instead. The signals for the (iso)nicotinoyl and aminobenzoyl protons do not exhibit any appreciable change in chemical shift. In the 1H NMR spectra, the absence of −OH and =N–NH protons in the low field region indicated their deprotonation upon complexation. Multiple groups of signals in the range of 6.57−9.15 ppm are due to aromatic ring protons. The most significant coordination-induced difference between signals is noticed for imine CH=N, up to 0.3 ppm. In the 13C NMR spectra, the complexation-induced differences in chemical shifts are the most pronounced for C1 and C4 carbons which experience deshielding effects up to 9.28 ppm and 6.74 ppm, respectively. The chemical shifts for phenolic C12 carbons differed less noticeably, up to 2.6 ppm.
2.3. Thermal Analysis DSC
The first mass loss in the TG curves of H2L4∙H2O is related to the removal of a water molecule from a crystalline hydrate (starting at 105 °C). The data from the DSC measurement shows a broad endothermic peak due to evaporation of the water during the dehydration process. From DSC measurements, the melting onset point temperature for anhydrous hydrazone H2L1 is at 197 °C, H2L2 at 111 °C, H2L3 at 159 °C, and H2L4 at 191 °C at 10 °C/min scanning rate. They start to decompose at 270 °C, 283 °C, 211 °C, and 289 °C, respectively.
Thermal behaviour of the complexes was studied in the temperature region from 25 to 600 °C, under the oxygen atmosphere. The polynuclear and dinuclear complexes 1-4 demonstrated thermal stability up to 300 °C. Afterwards, the decomposition of complexes occurred in one step. The TG curves of 1a-4a showed two distinct mass losses: the first one due to MeOH removal at temperatures around 140 °C, and the second once due to decomposition (with onset temperatures of 333 °C, 317 °C, 286 °C, and 281 °C, respectively).
2.4. Cyclooctene Epoxidation with TBHP
Cyclooctene epoxidation reactions were done with TBHP in water as an oxidizing agent. The idea was to test the prepared molybdenum complexes as catalysts, following the protocol previously established, to compare the activity and selectivity parameters with the results already published [49]. For that reason, the green chemistry protocol was applied. Even though TBHP is commercially available in decane and water, and the catalytic results with the assistance of TBHP in decane are very often much better, the choice of TBHP in water justifies the green catalytic concept. All the complexes were tested as catalysts with 0.25 % of catalyst loading. The obtained results are collected in Table 1 and catalytic profiles are presented in Figure 4.
All the tested catalysts showed very good activity, with cyclooctene conversion being more than 85 %, with the exception for the dinuclear catalyst 4 (67 %). Further, the selectivity toward corresponding epoxide is larger than 80 %, for all the compounds tested, as usually observed. TOF20 min parameter is the highest for dinuclear catalyst 3, meaning that it converts to the catalytically active species in the shortest time and the lowest for catalyst 2. It can be also noticed that the catalytic profiles of the catalysts 1a and 2a had a very similar pattern, directly implying that the position of N atom on the hydrazide part of the ligand (3rd or 4th atom) had almost no effect on the catalytic behaviour. Furthermore, a similar trend was also noticed for catalysts 3a and 4a, meaning the position of the amino group (on the 2nd or 4th C atom) did not influence the final activity outcome. What is quite interesting is the comparison of the TOF parameter. It is obvious that the dinuclear compounds 3 and 4, with the TOF20 min values of 415 and 279, respectively, are more easily converted to the catalytically active species than the polynuclear compounds 1 and 2. On the contrary, the mononuclear complexes 1a and 2a have very similar TOF20 min values (around 350), almost twice higher than the TOF20 min values for 3a and 4a.
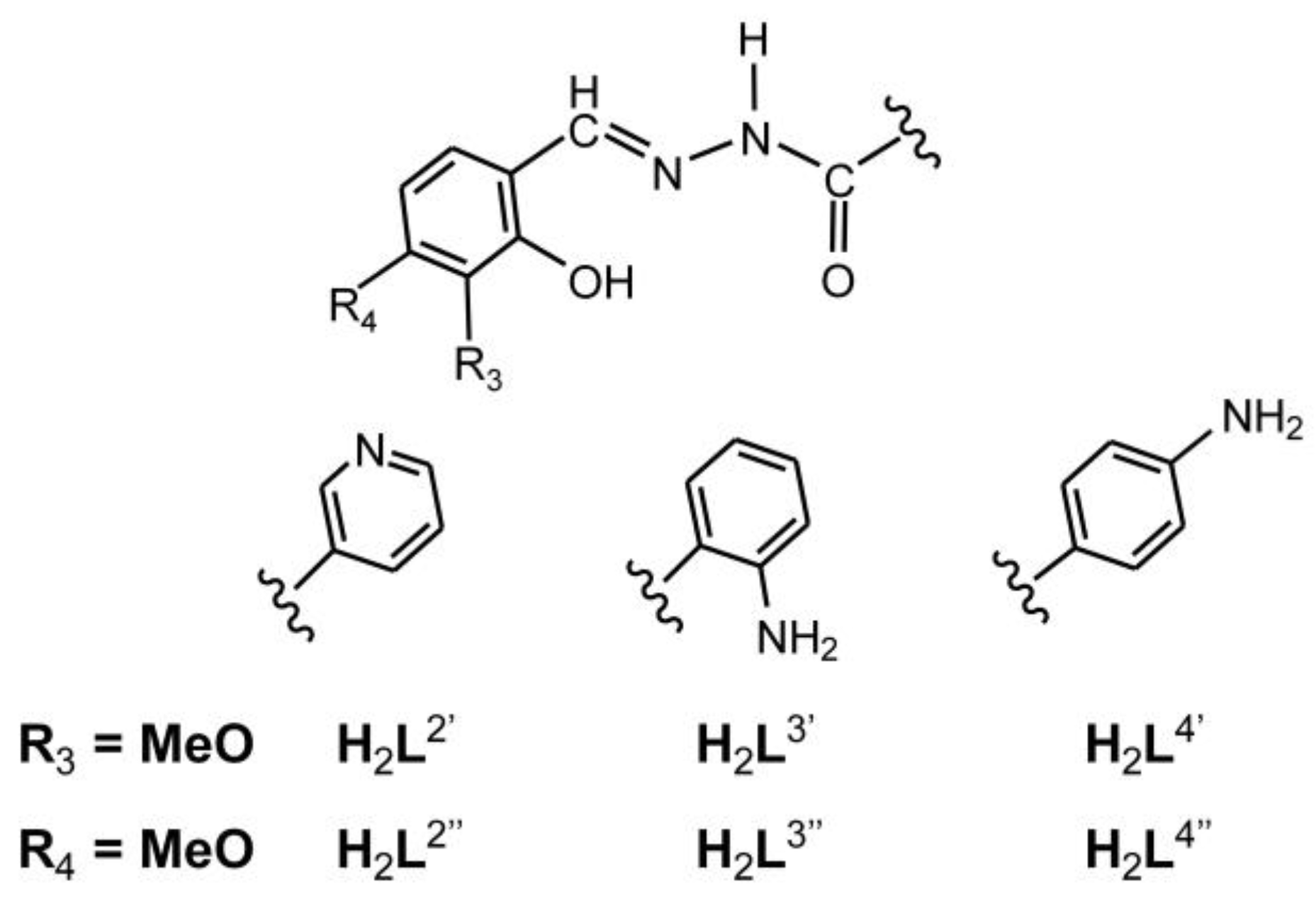
All the obtained results can be compared to the similar published results, compiled in Table 2. The hydrazone used for the coordination to the molybdenum center are presented in Scheme 2.
As noted, complex 2 can be compared to the complexes [MoO2(L2’)]n and [MoO2(L2’’)]n, showing the activity trend 2 > [MoO2(L2’’)]n > [MoO2(L2’)]n. Furthermore, the selectivity parameter followed the same trend, meaning that the position of the –OMe group on the aldehyde part of the ligands has a strong impact on the catalytic performances, favouring the 5th position on the benzene ring.
On the other side, the influence of the –OMe group on the aldehyde part of the ligands was not so pronounced when comparing catalysts 3 and 3a with [MoO2(L3’)]2, [MoO2(L3’’)(MeOH)] and [MoO2(L3’’)]2. For the TOF and thus activation energy, there is a bigger difference between 3 (the highest TOF) and 3a (the lowest one) compared to the three other complexes with relatively similar TOF for both dimers and slightly higher for the methanol-coordinated species. The behaviour seems to indicate that the possibility of decoordination and formation of the pentacoordinated active species has a strong impact within the mechanisms.
However, the –OMe position impacts the activity of the complexes 4 and 4a when correlated to [MoO2(L4’)(MeOH)], [MoO2(L4’’)(MeOH)], [MoO2(L4’)]2 and [MoO2(L4’’)]2. Methanol coordinated compounds, [MoO2(L4’)(MeOH)] and [MoO2(L4’’)(MeOH)]are lower conversion and TOF than the catalyst 4a, while the cyclooctene conversion for dinuclear catalysts has an unusual trend [MoO2(L4’’)]2 > [MoO2(L4’)]2 > 4 but with very close TOF value. In this case, we can conclude that there is an effect of methanol that has to be investigated in order to explain the mechanism. However, in general, it can be concluded that the catalysts bearing –OMe group on the 5th C atom of the benzene ring show better catalytic performances, except in the case whan NH2 is in 4th position on its aromatic ring, complex 4.
2.5. Density Functional Theory (DFT) Calculations.
It is interesting to see if the experimental results can be validated by theoretical calculations to understand the mechanisms and the effect of the ligand on the catalytic activity. The main points responsible for such activity, and correlation of catalytic activity to the structure are possible to be answered when the ligand functionalization is very different. However, subtle changes as in the presented research are sometimes difficult to analyze through DFT calculations.
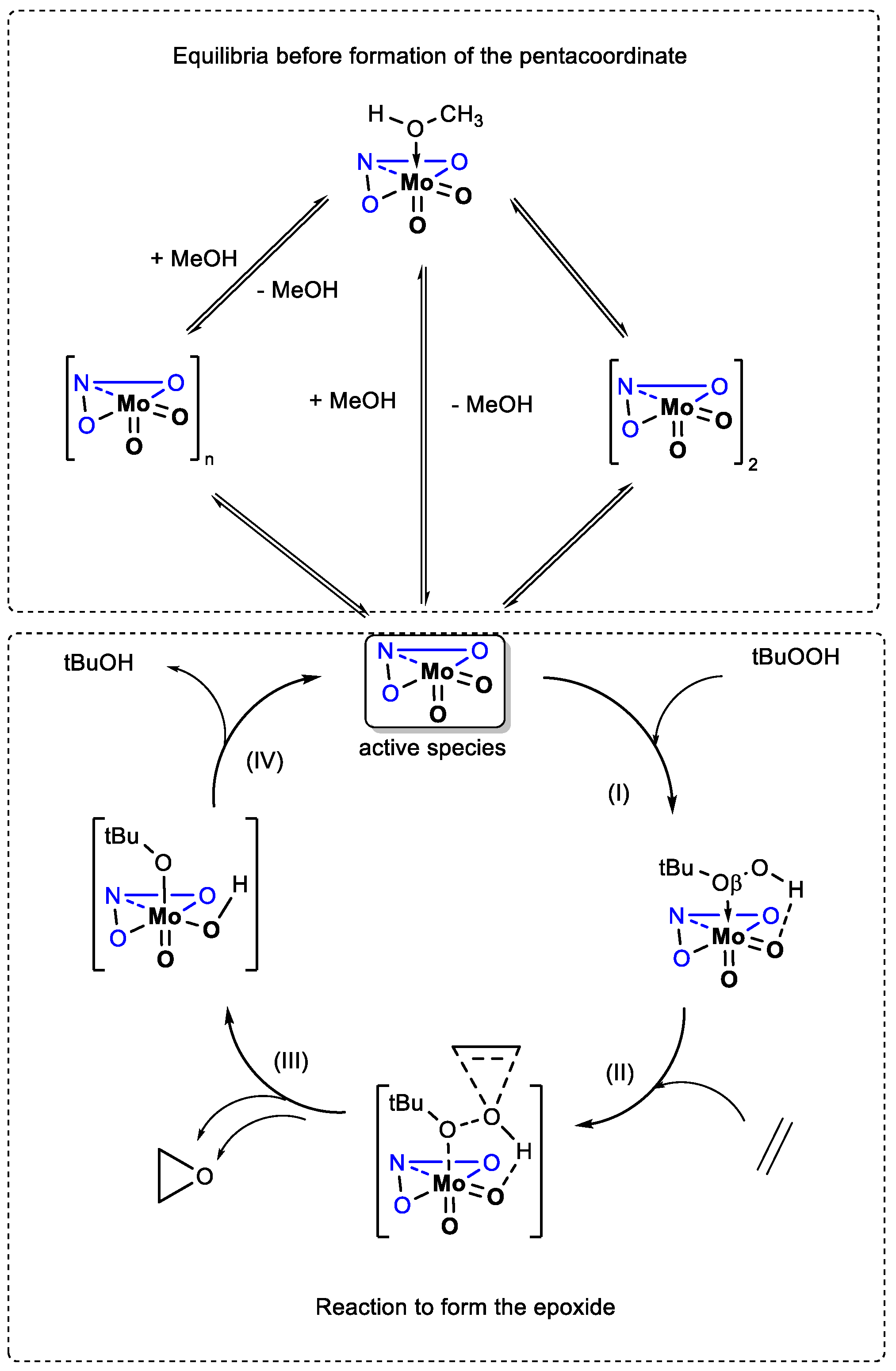
The mechanism proposed several years ago [40] postulated that [MoO2(L)(D)]or [MoO2(L)]n species turned into a pentacoordinated species [MoO2(L)], before reacting with TBHP to create the transient species [MoO2(L)(TBHP)], the latter permitting an easier epoxide formation through the Oβ of the epoxide. It has been shown that presence of substituents with strong electronic effects on ligands had dramatic effects on the catalysis [40]. Those results were linked to different electronic effects that could even be assessed through geometrical features for the calculated species.
In the present case, the differences between four ligands lie in the hydrazide nature: N atom being within the aromatic ring (in the case of complexes with L1– and L2– ligands) or bonded on the aromatic ring as NH2 (in the case of complexes with L3– and L4 ligands). In the case of coordination polymers 1 and 2, an insight for the decoordination through DFT would be too complicated. Therefore, we limited discussion about the decoordination to the complexes 3 and 4. Crystal structure of compound 3 was determined as dimer by X-ray diffraction method, and therefore the calculations for compound 4 were adopted according to the known structure. Further calculations included mononuclear complexes 1a-4a.
We limited calculations to the gas phase since trends can be observed from those conditions [51], and data are gathered in Table 3. All other calculated steps were indicated in Scheme 3. Optimized geometries are collected in Supplementary Materials Tables S14.
Experimentally, MeOH coordination does not lower the activity in case of [MoO2(L1)(MeOH)] (1a) and [MoO2(L2)(MeOH)] (2a), but lowers it in the case of [MoO2(L3)(MeOH)] (3a), and [MoO2(L4)(MeOH)] (4a). It has to be considered that, once the methanol decoordinated, the resulting pentacoordinated complex [MoO2(L)] can have several options: (i) turning back into the mononuclear complex [MoO2(L)(D)], (ii) oligomerize or (iii) interact with TBHP. From absolute enthalpies values obtained by calculations for each step, Table 3, the decoordination of the methanol (step 0) lies in a range of 10.3 to 11.1 kcal and those values do not provide clear conclusion. In identical manner, in the case of the stabilization of the formed pentacoordinate species [MoO2(L)] with TBHP on Mo center, (step I), values are also very close for all four ligands. On the other side, TS enthalpies (step II) were ligand dependent and the trend observed is TS2 < TS1 < TS3 < TS4. The lower the TS, the more easily are the active species formed and consequently TOF20 min should be higher. This is accordance with the experimentally obtained TOF values for the complexes 1a-4a, TOF2a ≈ TOF1a > TOF3a >TOF4a.
In contrary, for the dinuclear complexes 3 and 4, the order of experimental TOFs, TOF4 <TOF3 cannot follow calculated TS values, TS3 > TS4. It is expected that dimers 3 and 4 are transformed more easily than polymers 1 and 2. Even if it is not simple to conclude between dimers and polymers, the comparison between dimers and methanol-coordinated monomers 3 and 3a (and 4 and 4a) can be done. At this point, TOF values are directly linked to the TS shift (in absolute values) considering the energy of the starting molecule. Energy to maintain those dimers is weaker than for the methanol coordinated ones and this difference provokes a relative TS around 5 kcal lower for the polymers. This seems to be relevant enough in this case to conclude of the TOF difference.
It is interesting to see if those activities can be deduced from calculated geometrical data. Relevant distances and dihedral angle in absolute values (related to ligand planarity) around the molybdenum atom have been collected in Table 4. Experimental results showed that the [MoO2(L3)]2 (3) species seemed to be the best catalyst. Thus, as seen in energy diagram, Mo–Oβ interatomic distance parameter influenced the TS path mainly (step II). As explained previously, the lower TS value (higher activity) is linked to the shorter distance, as seen if. It is the case if we compare the analogue complexes with L3 and L4 ligands. Interesting facts appeared with the N1 bonded to Mo, the shorter Mo-N1 distances in the TS being for the least active compounds.
The ligand distortion seemed to have an influence. While [MoO2(L)(MeOH)] dihedral angle lies between 9.5 and 11.3°, the removal of MeOH molecule favors the planarity (5.6 to 6.3) addition of TBHP increase the dihedral angle and, in TS, distortion is the highest.
3. Materials and Methods
3.1. Preparative part
Elemental analyses were provided by the Analytical Services Laboratory of the Ruđer Bošković Institute, Zagreb. NMR data are given in Tables S9-S12 and Figures S5-S8 Supplementary Materials.
3.2. Synthesis of H2L1 and H2L2
A mixture of 5-methoxysalicylaldehyde (0.76 g, 5 mmol) and isonicotinic or nicotinic hydrazide (0.69, 5 mmol) in 100 mL of methanol was heated at reflux temperature with continuous stirring for 3 h. The resulting solution was concentrated under reduced pressure to one quarter of its volume and left at room temperature for several days. The obtained product H2L1 or H2L2 was filtered and dried up to a constant weight. Yield: 1.05 g, 77% (H2L1); 1.14 g, 84% (H2L2).
3.3. Synthesis of H2L3 and H2L4
5-methoxysalicylaldehyde (0.76 g, 5 mmol) dissolved in 40 mL of methanol was added dropwise to a solution of 2- or 4-aminobenzhydrazide (0.75 g, 5 mmol in 75 mL of methanol) and stirred at room temperature for 30 min and then heated at 40 °C for 4 h. The resulting solution was concentrated to 10 mL in vacuo and left at room temperature for several days. The obtained product was filtered and dried in a desiccator up to a constant weight. Yield: 0.99 mg, 70% (H2L3); 1.15 mg, 81% (H2L4).
3.4. Synthesis of 1-4
[MoO2(acac)2] (0.05 g, 0.15 mmol) was dissolved in dichloromethane (50 mL) and H2L1-4 (0.042 g H2L1,2 or 0.087g H2L3,4; 0.15 mmol) was added. Each suspension was shaken (at 50 rpm) for 6 hours at room temperature and left overnight. The resulting red product was collected by filtration and washed with a small amount of dichloromethane.
[MoO2(L1)]n (1): Yield: 42 mg, 34%. Anal. Calcd. for C14H11MoN3O5 (397.194): C, 42.33; H, 2.79; N, 10.58. Found: C, 42.19; H, 2.57; N, 10.39%. TG: MoO3, 35.94% (Calcd. 36.05%). Selected IR data (cm−1): 1600 (C=N), 1344 (C−Ohydrazone), 1270 (C−Ophenolate), 935, 917 (MoO2), 904 (Mo−N).
[MoO2(L2)]n (2): Yield: 38 mg, 31%. Anal. Calcd. for C14H11MoN3O5 (397.194): C, 42.33; H, 2.79; N, 10.58. Found: C, 42.14; H, 2.55; N, 10.36%. TG: MoO3, 35.94% (Calcd. 36.02%). Selected IR data (cm−1): 1615, 1601 (C=N), 1334 (C−Ohydrazone), 1265 (C−Ophenolate), 934, 915 (MoO2), 901 (Mo−N).
[MoO2(L3)]2 (3): Yield: 48 mg, 38%. Anal. Calcd. for C30H26Mo2N6O10 (822.440): C, 43.81; H, 3.19; N, 10.22. Found: C, 43.65; H, 3.02; N, 10.03%. TG: calcd. for MoO3, 35.00%, found 35.18%. Selected IR data (cm−1): 3285, 3227 (NH2), 1610, 1598 (C=N), 1332 (C−Ohydrazone), 1270 (C−Ophenolate), 925, 917 (MoO22+), 887, 861 (Mo−N).
[MoO2(L4)]2 (4): Yield: 46 mg, 37%. Anal. Calcd. for C30H26Mo2N6O10 (822.440): C, 43.81; H, 3.19; N, 10.22. Found: C, 43.58; H, 2.89; N, 10.05%. TG: calcd. for MoO3, 35.00%, found 34.74%. Selected IR data (cm−1): 3334, 3257 (NH2), 1613, 1602 (C=N), 1324 (C−Ohydrazone), 1266 (C−Ophenolate), 919, 908, (MoO22+), 887, 868 (Mo−N).
3.5. Synthesis of 1a-4a
Hydrazone H2L1-4 (0.083 g H2L1,2 or 0.087g H2L3,4; 0.31 mmol) was dissolved in 20 mL methanol and [MoO2(acac)2] (0.1 g, 0.31 mmol) was added to the resulting solution. In the case of reaction with H2L1,2 the reaction mixture was refluxed for two hours, whereas in the case of H2L3,4 it was heated at 40 °C. The resulting red product was collected by filtration and washed with small amount of cold methanol.
[MoO2(L1)(MeOH)] (1a). Yield: 85 mg, 64%. Anal. Calcd. for C15H15MoN3O6 (429.241): C, 41.97; H, 3.52; N, 9.79. Found C, 41.72; H, 3.29; N, 9.63%. TG: calcd. for MoO3, 33.53%, found: 33.42%; calcd. for CH3OH, 7.46%, found: 7.34%. Selected IR data (cm−1): 1621 (C=N)py, 1601 (C=N), 1341 (C−Ohydrazone), 1275 (C−Ophenolate), 1117 (C−OMeOH), 925, 913 (MoO2), 897(O=Mo–OMeOH).
[MoO2(L2)(MeOH)] (2a). Yield: 108 mg, 82%. Anal. Calcd. for C15H15MoN3O6 (429.241): C, 41.97; H, 3.52; N, 9.79. Found C, 41.76; H, 3.30; N, 9.65%. TG: calcd. for MoO3, 33.53%, found: 33.17%; calcd. for CH3OH, 7.46%, found: 7.62%. Selected IR data (cm−1): 1620 (C=N)py, 1607 (C=N), 1333 (C−Ohydrazone), 1267 (C−Ophenolate), 1115 (C−OMeOH), 923, 913 (MoO2), 900 (O=Mo–OMeOH).
[MoO2(L3)(MeOH)] (3a). Yield: 120 mg, 89%. Anal. Calcd. for C16H17MoN3O6 (443.262): C, 43.35; H, 3.87; N, 9.48. Found: C, 43.18; H, 3.66; N, 9.28%. TG: calcd. for MoO3, 32.47%, found: 32.65%; calcd. for CH3OH, 7.23%, found 6.89%. Selected IR data (cm−1): 3344, 3267 (NH2), 1611, 1599 (C=N), 1328 (C−Ohydrazone), 1263 (C−Ophenolate), 1163 (C−OMeOH), 926, 914 (MoO22+), 895, 874 (O=Mo–OMeOH).
[MoO2(L4)(MeOH)] (4a). Yield: 110 mg, 81%. Anal. Calcd. for C16H17MoN3O6 (443.262): C, 43.35; H, 3.87; N, 9.48. Found: C, 43.12; H, 3.63; N, 9.24%. TG: calcd. for MoO3, 32.47%, found 32.69%; calcd. for CH3OH, 7.23%, found 6.95%. Selected IR data (cm−1): 3456, 3356 (NH2), 1628, 1600 (C=N), 1321 (C−Ohydrazone), 1275 (C−Ophenolate), 1155 (C−OMeOH), 923, 914 (MoO22+), 896, 868 (O=Mo–OMeOH).
3.6. Physical Methods
Thermogravimetric (TG) analyses were carried out with a Mettler-Toledo TGA/SDTA851e thermobalance with alumina crucibles under oxygen flow (10 mL min-1) in the temperature range from 25 °C to 600 °C, with a heating rate of 10 °C min−1. Differential scanning calorimetry (DSC) measurements were performed under the nitrogen stream (10 mL min-1) on the TA Discovery DSC 25 instrument in the temperature range from 25 to 400 °C using Tzero aluminium pans and lids. Heating rates of 10 K min−1 were used for all investigations. The results of TG and DSC measurements were evaluated using the Mettler STARe Software (version 16.10) and TA Instruments Trios (v5.1.1.46572), respectively.
1H, 13C, and 2D NMR spectra were acquired at 298 K on Bruker Avance III HD 400 spectrometer operating at 400 MHz. The spectra were recorded in DMSO-d6 with a sample concentration of 20 mg mL−1 with TMS as an internal standard. The Bruker Topspin™ software was used for data analysis and signal integrations.
Attenuated Total Reflectance Infrared (ATR-IR) spectra were acquired on a Perkin Elmer Spectrum One spectrometer.
The powder X-ray diffraction (PXRD) for qualitative phase analysis data were collected using a Malvern Panalytical Aeris diffractometer in the Bragg-Brentano geometry with a PIXcel1D detector, using CuKα radiation (λ = 1.5406 Å). Samples were contained on a Si sample holder. Powder patterns were collected at room temperature in the range of 5–40° (2θ) and visualized utilizing the Malvern Panalytical HighScore Software Suite [52]
All reagents and solvents were commercially available (Alpha-Aesar, Sigma-Aldrich), and used as without further purification. [MoO2(acac)2] was prepared according to the published procedure [53].
Single crystals of ligands H2L3 and H2L4 and complexes 1a, 2a, 3a and 3 of suitable quality were selected for the diffraction experiments. Data were collected using via ω-scans: (1) for ligands H2L3 and H2L4 on an Oxford Xcalibur diffractometer equipped with 4-circle kappa geometry goniometer, CCD Sapphire 3 detector and graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) at 150(2) K, (2) for complexes 1a, 2a, 3a and 3 on a Rigaku XtaLAB Synergy-S diffractometer armed with a Dualflex source (Cu Kα radiation, λ = 1.54184 Å) and a HyPix detector at 170 K. Data were processed with the CrysAlis program package [54]. A summary of the general crystal data, along with the data collection and structure refinement parameters, is presented in Supplementary Materials, Tables S1 and S5. The structures were solved via dual-space methods using SHELXT [55]. The refinement procedure was accomplished via full-matrix least-squares methods based on F2 values against all reflections, including the anisotropic displacement parameters for all non-H atoms. Hydrogen atoms bound to carbon atoms were placed in geometrically idealized positions and refined by using the riding model, with Uiso = 1.2 Ueq of the connected carbon atom, or as ideal CH3 groups, with Uiso = 1.5 Ueq. Hydrogen atoms attached to heteroatoms were located in the different Fourier maps in the final stages of the refinement procedure. Their coordinates were refined freely but with restrained N−H distances of 0.86(2) and O–H distances of 0.82(2) A. All refinements were conducted using SHELXL [56]. The SHELX programs were operated within the Olex2 suite [57]. Geometrical calculations were performed and molecular graphics were produced using Mercury [46].
Chromatograms were obtained using Agilent 7820A chromatograph with FID detector and HP5-MS capillary column (30 m × 0.32 mm × 0.25 µm). The GC parameters were quantified with authentic samples of the reactants and products. Conversion of olefins and formation of corresponding epoxides were calculated from calibration curves relative to acetophenone as an internal standard.
3.7. Theoretical Calculations
The geometries of all species under investigation were optimized without any symmetry constraint with the Gaussian 09 rev. D01 program suite [58], with the DFT approach using the B3LYP three-parameter functional [59,60,61] in conjunction with the 6-31G* basis set [62,63,64,65] for the light atoms (O, N, C, H) and the CEP-31G set for the Mo atom [66,67]. The geometries of all complexes and intermediates were optimized from starting geometries determined or inspired by X-ray diffraction without any symmetry constraint. All coordinates have been listed in Supplementary Materials (Tables S14). Frequency analysis confirmed that the optimized geometries of all the stable compounds and intermediates were local minima. The transition states were optimized using a preliminary scan of a relevant internal coordinate, followed by full optimization of the TS guided by the knowledge of such coordinates. All optimized geometries were confirmed to be stationary points and local minima (for stable molecules or reaction intermediates) or first-order saddle points (for the TSs) by frequency analyses. For all the TSs, analysis of the imaginary frequency confirmed the expected motion along the reaction coordinate. Those values and the relative schemes have been added Table S13). The calculated frequencies were also used to derive the thermochemical parameters at 298 K according to the standard approximations (ideal gas, rigid rotor and harmonic oscillator).
4. Conclusions
The structural formations of molybdenum complexes can vary based on the specific hydrazide employed. Depending on this hydrazide moiety (aminobenzohydrazide vs. (iso)nicotinic hydrazide) structural assemblies as dinuclear or polynuclear molybdenum complexes, can be attained. Furthermore, the selection of solvent plays a pivotal role as it determines whether a mononuclear (from methanol) or dinuclear/polynuclear (from dichloromethane) complex can be isolated. The catalytic performances of the synthesized complexes were evaluated under environmentally friendly, green chemistry conditions. Overall, all examined compounds exhibited remarkable catalytic capabilities concerning cyclooctene conversion and selectivity towards epoxide production. Moreover, dinuclear compounds demonstrated a superior rate of conversion to the catalytically active species compared to polynuclear catalysts. Conversely, mononuclear catalysts derived from (iso)nicotinic hydrazide displayed a higher propensity for conversion into the pentacoordinated active species compared to complexes derived from aminobenzohydrazides. Future investigations will focus on catalytic oxidations of bio-derived substrates, particularly emphasizing the exploration of dinuclear catalysts.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Scheme S1: Hydrazidato and hydrazonato tautomeric forms; Figure S1: Molecular structures; Figure S2: Crystal packings; Figures S3 and S4: Supramolecular environment of ligand and complex molecules; Tables S1 and S5: Experimental and crystallographic data; Tables S2, S3, S6, S7: Selected bond lengths and angles; Tables S4, S8. Hydrogen bond parameters; Schemes S2-S5: The structural formula of ligands with the NMR numbering scheme; Tables S9-S12: 1H and 13C chemical shifts of compounds in dmso-d6; Figures S5-S8 NMR spectra in dmso-d6. Table S13: Imaginary frequency (cm−1) of the calculated transition states shown in Table S14. Table S14: DFT Coordinates of all species.
Author Contributions
Conceptualization, supervision, V.V., J.P., D.A; investigation, formal analysis, writing– original draft preparation, M.M., E.T. D.A., J.P., V.V., visualization, E.T., J.P., V.V., writing– review and editing, J.P. D.A. V.V. All authors have read and agreed to the published version of the manuscript.
Funding
This work has been fully supported by Croatian Science Foundation under the project (IP-2016-06-4221) as well as Chemistry Dept of IUT Paul Sabatier and LCC-CNRS.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Crystallographic data sets for the structures H2L3, H2L4, 1a, 2a, 3a and 3 are available through the Cambridge Structural Database with deposition numbers CCDC 2310499-2310504. These data can be obtained free of charge via https://www.ccdc.cam.ac.uk/ conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: deposit@ccdc.cam.ac.uk) (accessed on November 27, 2023).
Acknowledgments
We acknowledge the support of project CIuK co-financed by the Croatian Government and the European Union through the European Regional Development Fund-Competitiveness and Cohesion Operational Programme (Grant KK.01.1.1.02.0016.). The work of doctoral student Mirna Mandarić has been supported by the “Young researchers’ career development project—training of doctoral students” of the Croatian Science Foundation funded by the European Union from the European Social Fund. LCC CNRS and IUT Chem Dept are acknowledged for equipment for the catalysis experiments. LCC and Calmip are acknowledged for the facilities in terms of calculation time.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Chen, L.; Chen, Q.H.; Wu, M.Y.; Jiang, F.L.; Hong, M.C. Structure Switching and Modulation of the Magnetic Properties in Diarylethene-Bridged Metallosupramolecular Compounds by Controlled Coordination-Driven Self-Assembly. Acc. Chem. Res. 2015, 48, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Lescop, C. Coordination-Driven Syntheses of Compact Supramolecular Metallacycles toward Extended Metallo-organic Stacked Supramolecular Assemblies. Acc. Chem. Res. 2017, 50, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Northrop, B.H.; Zheng, Y.R.; Chi, K.W.; Stang, P.J. Coordination-Driven Syntheses of Compact Supramolecular Metallacycles toward Extended Metallo-organic Stacked Supramolecular Assemblies. Acc. Chem. Res. 2009, 42, 1554–1563. [Google Scholar] [CrossRef] [PubMed]
- Cook, T.R.; Stang, P. J. Recent Developments in the Preparation and Chemistry of Metallacycles and Metallacages via Coordination. Chem. Rev. 2015, 115, 7001–7045. [Google Scholar] [CrossRef] [PubMed]
- Cook, T.R.; Zheng, Y.R.; Stang, P.J. Metal–Organic Frameworks and Self-Assembled Supramolecular Coordination Complexes: Comparing and Contrasting the Design, Synthesis, and Functionality of Metal–Organic Materials. Chem. Rev. 2013, 113, 734–777. [Google Scholar] [CrossRef] [PubMed]
- Giuseppone, N.; Schmitt, J.L.; Lehn, J.M. Driven Evolution of a Constitutional Dynamic Library of Molecular Helices Toward the Selective Generation of [2 × 2] Gridlike Arrays under the Pressure of Metal Ion Coordination. J. Am. Chem. Soc. 2006, 128, 16748–16763. [Google Scholar] [CrossRef]
- Dietrich-Buchecker, C.; Colasson, B.; Fujita, M.; Hori, A.; Geum, N.; Sakamoto, S.; Yamaguchi, K.; Sauvage, J.P. Quantitative Formation of [2]Catenanes Using Copper(I) and Palladium(II) as Templating and Assembling Centers: The Entwining Route and the Threading Approach. J. Am. Chem. Soc. 2003, 125, 5717–5725. [Google Scholar] [CrossRef]
- Berl, V.; Huc, I.; Khoury, R.G.; Krische, M.J.; Lehn, J.M. Interconversion of single and double helices formed from synthetic molecular strands. Nature 2000, 407, 720–723. [Google Scholar] [CrossRef]
- Levchenko, A.A.; Yee, C.K.; Parikh, A.N.; Navrotsky, A. Energetics of Self-Assembly and Chain Confinement in Silver Alkanethiolates: Enthalpy−Entropy Interplay. Chem. Mater. 2005, 17, 5428–5438. [Google Scholar] [CrossRef]
- Kunz, V.; Lindner, J.O.; Schulze, M.; Röhr, M. I.S.; Schmidt, D.; Mitrić, R.; Würthner, F. Cooperative water oxidation catalysis in a series of trinuclear metallosupramolecular ruthenium macrocycles. Energy Environ. Sci. 2017, 10, 2137–2153. [Google Scholar] [CrossRef]
- Kim, S.; Cho, K.-B.; Lee, Y.-M.; Chen, J.; Fukuzumi, S.; Nam, W. Factors Controlling the Chemoselectivity in the Oxidation of Olefins by Nonheme Manganese(IV)-Oxo Complexes. J. Am. Chem. Soc. 2016, 138, 10654–10663. [Google Scholar] [CrossRef]
- Liu, Y.; Xuan, W.; Zhang, H.; Cui, Y. Chirality- and Threefold-Symmetry-Directed Assembly of Homochiral Octupolar Metal−Organoboron Frameworks. Inorg. Chem. 2009, 48, 10018–10023. [Google Scholar] [CrossRef]
- Liang, L.-L.; Ren, S.-B.; Zhang, J.; Li, Y.-Z.; Du, H.-B.; You, X.-Z. Two Thermostable Three-Dimensional Homochiral Metal−Organic Polymers with Quartz Topology. Cryst. Growth Des. 2010, 10, 1307–1311. [Google Scholar] [CrossRef]
- Bark, T.; von Zelewsky, A.; Rappoport, D.; Neuburger, M.; Schaffner, S.; Lacour, J.; Jodry, J. Synthesis and Stereochemical Properties of Chiral Square Complexes of Iron(II). Chem. Eur. J. 2004, 10, 4839–4845. [Google Scholar] [CrossRef] [PubMed]
- Demadis, K.D.; Papadaki, M.; Aranda, M.A.G.; Cabeza, A.; Olivera-Pastor, P.; Sanakis, Y. Stepwise Topotactic Transformations (1D to 3D) in Copper Carboxyphosphonate Materials: Structural Correlations. Cryst. Growth Des. 2010, 10, 357–364. [Google Scholar] [CrossRef]
- Li, Z.-Y.; Dai, J.-W.; Damjanović, M.; Shiga, T.; Wang, J.-H.; Zhao, J.; Oshio, H.; Yamashita, M.; Bu, X.-H. Structure Switching and Modulation of the Magnetic Properties in Diarylethene-Bridged Metallosupramolecular Compounds by Controlled Coordination-Driven Self-Assembly. Angew. Chem. Int. Ed. 2019, 58, 4339–4344. [Google Scholar] [CrossRef]
- Estrader, M.; Uber, J.S.; Barrios, L.A.; Garcia, J.; Lloyd-Williams, P.; Roubeau, O.; Teat, S.J.; Aromi, G. A Magneto-optical Molecular Device: Interplay of Spin Crossover, Luminescence, Photomagnetism, and Photochromism. Angew. Chem. Int. Ed. 2017, 56, 15622–15627. [Google Scholar] [CrossRef]
- Duan, J.; Jin, W.; Krishna, R. Natural Gas Purification Using a Porous Coordination Polymer with Water and Chemical Stability. Inorg. Chem. 2015, 54, 4279–4284. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.K.; Kim, E.J.; Bae, J.; Kim, K.S. Probing the origin and stability of bivalency in copper based porous coordination network and its application for H2S gas capture. Sci. Rep. 2022, 12, 15388. [Google Scholar] [CrossRef]
- Saha, M.L.; Yan, X.Z.; Stang, P.J. Photophysical Properties of Organoplatinum(II) Compounds and Derived Self-Assembled Metallacycles and Metallacages: Fluorescence and its Applications. Acc. Chem. Res. 2016, 49, 2527–2539. [Google Scholar] [CrossRef]
- Rösner, B.; Milek, M.; Witt, A.; Gobaut, B.; Torelli, P.; Fink, R.H.; Khusniyarov, M.M. Reversible Photoswitching of a Spin-Crossover Molecular Complex in the Solid State at Room Temperature. Angew. Chem. Int. Ed. 2015, 54, 12976–12980. [Google Scholar] [CrossRef] [PubMed]
- Pisk, J.; Agustin, D.; Vrdoljak, V.; Poli, R. Epoxidation Processes by Pyridoxal Dioxomolybdenum(VI) (Pre)Catalysts Without Organic Solvent. Adv. Synth. Catal. 2011, 353, 2910–2914. [Google Scholar] [CrossRef]
- Vrdoljak, V.; Pisk, J.; Agustin, D.; Novak, P.; Parlov Vuković, J.; Matković-Čalogović, D. Dioxomolybdenum(VI) and dioxotungsten(VI) complexes chelated with the ONO tridentate hydrazone ligand: synthesis, structure and catalytic epoxidation activity. New J. Chem. 2014, 38, 6176–6185. [Google Scholar] [CrossRef]
- Judmaier, M.E.; Holzer, C.; Volpe, M.; Mösch-Zanetti, N.C. Molybdenum(VI) dioxo complexes employing Schiff base ligands with an intramolecular donor for highly selective olefin epoxidation. Inorg. Chem. 2012, 51, 9956–9966. [Google Scholar] [CrossRef] [PubMed]
- Pisk, J.; Prugovečki, B.; Matković-Čalogović, D.; Poli, R.; Agustin, D.; Vrdoljak, V. Charged dioxomolybdenum (VI) complexes with pyridoxal thiosemicarbazone ligands as molybdenum(V) precursors in oxygen atom transfer process and epoxidation (pre) catalysts. Polyhedron 2012, 33, 441–449. [Google Scholar] [CrossRef]
- Xu, W.X.; Li, W.H. Synthesis, Crystal Structures, and Catalytic Property of Dioxomolybdenum(VI) Complexes with Hydrazones. Russ. J. Coord. Chem. 2012, 38, 92–98. [Google Scholar] [CrossRef]
- Bagherzadeh, M.; Zare, M.; Amani, V.; Ellern, A.; Woo, L.K. Dioxo and oxo-peroxo molybdenum (VI) complexes bearing salicylidene 2-picoloyl hydrazone: Structures and catalytic performances. Polyhedron 2013, 53, 223–229. [Google Scholar] [CrossRef]
- Vrdoljak, V.; Prugovečki, B.; Matković-Čalogović, D.; Dreos, R.; Siega, P.; Tavagnacco, C. Zigzag Chain, Square Tetranuclear, and Polyoxometalate-Based Inorganic−Organic Hybrid Compounds - Molybdenum vs Tungsten. Cryst. Growth Des. 2010, 10, 1373–1382. [Google Scholar] [CrossRef]
- Vrdoljak, V.; Prugovečki, B.; Matković-Čalogović, D.; Pisk, J.; Dreos, R.; Siega, P. Supramolecular Hexagon and Chain Coordination Polymer Containing the MoO22+ Core: Structural Transformation in the Solid State. Cryst. Growth Des. 2011, 11, 1244–1252. [Google Scholar] [CrossRef]
- Vrdoljak, V.; Prugovečki, B.; Matković-Čalogović, D.; Hrenar, T.; Dreos, R.; Siega, P. Three Polymorphic Forms of a Monomeric Mo(VI) Complex: Building Blocks for Two Metal–Organic Supramolecular Isomers. Intermolecular Interactions and Ligand Substituent Effects. Cryst. Growth Des. 2013, 13, 3773–3784. [Google Scholar] [CrossRef]
- Vrdoljak, V.; Prugovečki, B.; Pulić, I.; Cigler, M.; Sviben, D.; Parlov Vuković, J.; Novak, P.; Matković-Čalogović, D.; Cindrić, M. Dioxidomolybdenum(VI) Complexes with Isoniazid-Related Hydrazones: Solution-Based, Mechanochemical and UV-light Assisted Deprotonation. New J. Chem. 2015, 39, 7322–7332. [Google Scholar] [CrossRef]
- Sinha, S.; Chakraborty, M.; Pramanik, N.R.; Raychaudhuri, T.K.; Mondal, T.K.; Sarkar, D.; Drew, M.G.B.; Ghosh, S.; Mandal, S.S. Dimer Formation by Symbiotic Donor–Acceptor Interaction Between Two Molecules of a Specially Designed Dioxomolybdenum(VI) Complex Containing Both Donor and Acceptor Centers – A Structural, Spectroscopic and DFT Study. Polyhedron 2013, 55, 192–200. [Google Scholar] [CrossRef]
- McCormick, L.J.; Abrahams, B.F.; Boughton, B.A.; Grannas, M.J.; Hudson, T.A.; Robson, R. Synthesis, Structure and Cation-Binding Properties of Some [4 + 4] Metallocyclic MO22+ (M = Mo or W) Derivatives of 9-Phenyl-2,3,7-trihydroxyfluor-6-one. Inorg. Chem. 2014, 53, 1721–1728. [Google Scholar] [CrossRef]
- Bikas, R.; Lippolis, V.; Noshiranzadeh, N.; Farzaneh-Bonab, H.; Blake, A.J.; Siczek, M.; Hosseini-Monfared, H.; Lis, T. Electronic Effects of Aromatic Rings on the Catalytic Activity of Dioxidomolybdenum(VI)–Hydrazone Complexes. Eur. J. Inorg. Chem. 2017, 2017, 999–1006. [Google Scholar] [CrossRef]
- Maurya, M.R.; Rana, L.; Avecilla, F. Catalytic Oxidation of Internal and Terminal Alkenes by Oxidoperoxidomolybdenum(VI) and Dioxidomolybdenum(VI) Complexes. Inorg. Chim. Acta, 2015, 429, 138–147. [Google Scholar] [CrossRef]
- Biswal, D.; Pramanik, N.R.; Chakrabati, S.; Drew, M.G.B.; Sarkar, B.; Maurya, M.R.; Mukherjee, S.K.; Chowdhury, P. New Polymeric, Dimeric and Mononuclear Dioxidomolybdenum(VI) Complexes with an ONO Donor Ligand: Crystal Structures, DFT Calculations, Catalytic Performance and Protein Binding study of the Ligand. New J. Chem. 2017, 41, 4116–4137. [Google Scholar] [CrossRef]
- Pisk, J.; Rubčić, M.; Kuzman, D.; Cindrić, M.; Agustin, D.; Vrdoljak, V. Molybdenum(VI) complexes of hemilabile aroylhydrazone ligands as efficient catalysts for greener cyclooctene epoxidation: an experimental and theoretical approach. New J. Chem. 2019, 43, 5531–5542. [Google Scholar] [CrossRef]
- Cvijanović, D.; Pisk, J.; Pavlović, G.; Šišak-Jung, D.; Matković-Čalogović, D.; Cindrić, M.; Agustin, D.; Vrdoljak, V. Discrete Mononuclear and Dinuclear Compounds Containing a MoO22+ Core and 4-Aminobenzhydrazone Ligands: Synthesis, Structure and Organic-Solvent-Free Epoxidation Activity. New J. Chem. 2019, 43, 1791–1802. [Google Scholar] [CrossRef]
- Pisk, J.; Agustin, D.; Vrdoljak, V. Tetranuclear molybdenum(VI) hydrazonato epoxidation (pre)catalysts: Is water always the best choice? Catal Commun 2020, 142. [Google Scholar] [CrossRef]
- Morlot, J.; Uyttebroeck, N.; Agustin, D.; Poli, R. Solvent-Free Epoxidation of Olefins Catalyzed by “[MoO2(SAP)]”: A New Mode of tert-Butylhydroperoxide Activation. ChemCatChem 2013, 5, 601–611. [Google Scholar] [CrossRef]
- Cvijanović, D.; Pisk, J.; Pavlović, G.; Šišak-Jung, D.; Matković-Čalogović, D.; Cindrić, M.; Agustin, D.; Vrdoljak, V. Discrete mononuclear and dinuclear compounds containing a MoO22+ core and 4-aminobenzhydrazone ligands: synthesis, structure and organic-solvent-free epoxidation activity. New J. Chem. 2018, 43, 1791–1802. [Google Scholar] [CrossRef]
- Prasanna, M. K.; Sithambaresan, M.; Pradeepkumar, K.; Kurup, M. R. P. N’-[(E)-2-Hy droxy-5-meth oxy benzyl idene]pyridine-4-carbohydrazide monohydrate. Acta Cryst. 2013, E69, o881. [Google Scholar] [CrossRef]
- Kargar, H.; Kia, R.; Akkurt, M.; Buyukgungor, O. 5-Chloro-2-hy dr oxy benzaldehyde thio semicarbazone. Acta Cryst. 2010, E66, o2982. [Google Scholar] [CrossRef]
- Kramer, R.; Lehn, J.M.; Marquis-Rigault, A. Self-recognition in helicate self-assembly: spontaneous formation of helical metal complexes from mixtures of ligands and metal ions. Proc. Natl. Acad. Sci. USA 1993, 90, 5394–5398. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.-X.; Yuan, Y.-M.; Li, W.-H. Syntheses, crystal structures, and catalysis by polymeric dioxomolybdenum(VI) complexes with similar (iso)nicotinohydrazones. J. Coord. Chem. 2013, 66, 2726–2735. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Krämer, R.; Lehn, J.M.; Marquis-Rigault, A. Self-recognition in helicate self-assembly: Spontaneous formation of helical metal complexes from mixtures of ligands and metal ions. Proc. Natl. Acad. Sci. USA 1993, 90, 5394–5398. [Google Scholar] [CrossRef] [PubMed]
- Pisk, J.; Hrenar, T.; Rubčić, M.; Pavlović, G.; Damjanović, V.; Lovrić, J.; Cindrić, M.; Vrdoljak, V. Comparative studies on conventional and solvent free synthesis toward hydrazones: application of PXRD and chemometric data analysis in mechanochemical reaction monitoring. CrystEngComm. 2018, 20, 1804–1817. [Google Scholar] [CrossRef]
- Pisk, J.; Agustin, D. Molybdenum, Vanadium, and Tungsten-Based Catalysts for Sustainable (ep)Oxidation. Molecules 2022, 27, 6011. [Google Scholar] [CrossRef]
- Vrdoljak, V.; Mandarić, M.; Hrenar, T.; Đilović, I.; Pisk, J.; Pavlović, G.; Cindrić, M.; Agustin, D. Geometrically constrained molybdenum(VI) metallosupramolecular architectures: conventional Synthesis versus vapor and thermally induced solid-state structural transformations. Cryst. Growth. Des. 2019, 19, 3000–3011. [Google Scholar] [CrossRef]
- Bafti, A.; Razum, M.; Topić, E.; Agustin, D.; Pisk, J.; Vrdoljak, V. Implication of oxidant activation on olefin epoxidation catalysed by Molybdenum catalysts with aroylhydrazonato ligands: Experimental and theoretical studies. Mol. Catal. 2021, 512, 111764. [Google Scholar] [CrossRef]
- Degen, T.; Sadki, M.; Bron, E.; König, U.; Nénert, G. The High Score suite. Powder Diffr. 2014, 29, S13–S18. [Google Scholar] [CrossRef]
- Chen, J.-J.; McDonald, J.W.; Newton, W.E. Synthesis of molybdenum(IV) and molybdenum(V) complexes using oxo abstraction by phosphines. Mechanistic implications. Inorg. Chem. 1976, 15, 2612–2615. [Google Scholar] [CrossRef]
- CrysAlisPro. Software System. Version 1.171.43.90; Rigaku Oxford Diffraction, 2023. [Google Scholar]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXT. Acta Crystallogr. Sect. A Struct. Chem. 2015, 71, 3–8. [Google Scholar]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. Journal of Applied Crystallography. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- 58. Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H.P.; Izmaylov, A.F.; Bloino, J.; Zheng, G.; Sonnenberg, J.L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery Jr., J.A.; Peralta, J.E.; Ogliaro, F.; Bearpark, M.; Heyd, J.J.; Brothers, E.; Kudin, K.N.; Staroverov, V.N.; Keith, T.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J.C.; Iyengar, S.S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J.M.; Klene, M.; Knox, J.E.; Cross, J.B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Martin, R.L.; Morokuma, K.; Zakrzewski, V.G.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Dapprich, S.; Daniels, A.D.; Farkas, O.; Foresman, J.B.; Ortiz, J.V.; Cioslowski, J.; Fox., D.J. Gaussian, Inc.: Wallingford, CT, USA, 2013.
- Becke, D. Density–functional thermochemistry. III. The role of exact exchange. J.Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C. T.; Yang, W. T.; Parr, R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W. J.; Pople, J. A. Self–Consistent Molecular–Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Hehre, W.; Ditchfield, R.; Pople, J. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P. C.; Pople, J. The influence of polarization functions on molecular orbital hydrogenation energies. A. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Hariharan, P. C.; Pople, J. A. Accuracy of AHn equilibrium geometries by single determinant molecular orbital theory. Mol. Phys. 1974, 27, 209–214. [Google Scholar] [CrossRef]
- Stevens, W. J.; Basc, H.; Krauss, M. Compact effective potentials and efficient shared–exponent basis sets for the first- and second-row atoms. J. Chem. Phys. 1984, 81, 6026–6033. [Google Scholar] [CrossRef]
- Stevens, W. J.; Krauss, M.; Basch, H.; Jasien, P. G. Relativistic compact effective potentials and efficient, shared-exponent basis sets for the third-, fourth-, and fifth-row atoms. Can. J. Chem. 1992, 70, 612–630. [Google Scholar] [CrossRef]
Scheme 1.
Hydrazones derived from 5-methoxysalicylaldehyde and the corresponding hydrazides: isonicotinic hydrazide (H2L1), nicotinic hydrazide (H2L2), 2-aminobenzhydrazide (H2L3) or 4-aminobenzhydrazide (H2L4)
Scheme 1.
Hydrazones derived from 5-methoxysalicylaldehyde and the corresponding hydrazides: isonicotinic hydrazide (H2L1), nicotinic hydrazide (H2L2), 2-aminobenzhydrazide (H2L3) or 4-aminobenzhydrazide (H2L4)
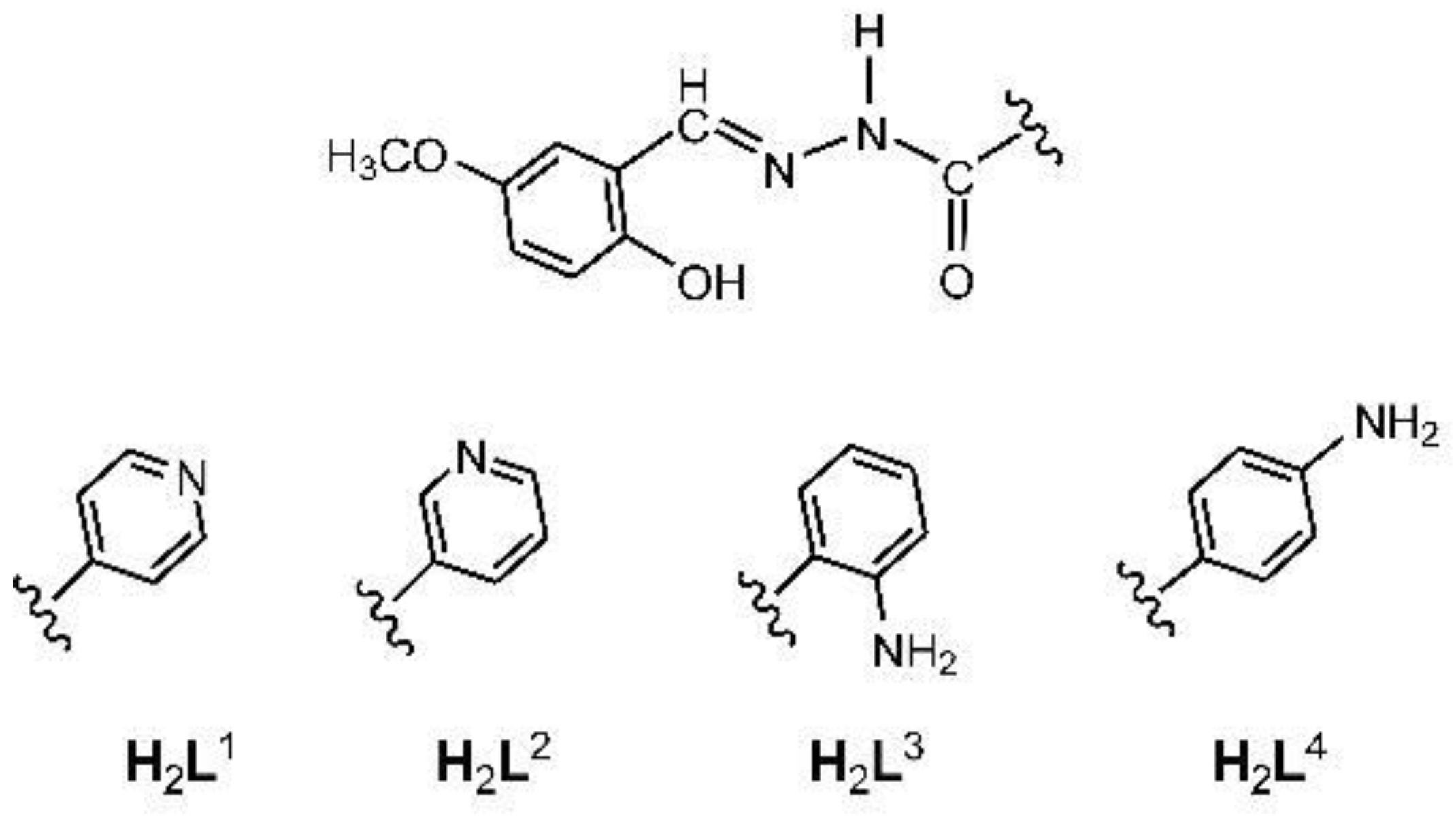
Figure 1.
Molecular structure of (a) H2L3, (b) 3a, (c) 3, with atoms represented as thermal ellipsoids at 50% probability level. Structural overlay of mononuclear complex 3a (green) and complex dimer 3 (purple) is shown in (d), showing the similarity of ligand conformation in both cases.
Figure 1.
Molecular structure of (a) H2L3, (b) 3a, (c) 3, with atoms represented as thermal ellipsoids at 50% probability level. Structural overlay of mononuclear complex 3a (green) and complex dimer 3 (purple) is shown in (d), showing the similarity of ligand conformation in both cases.
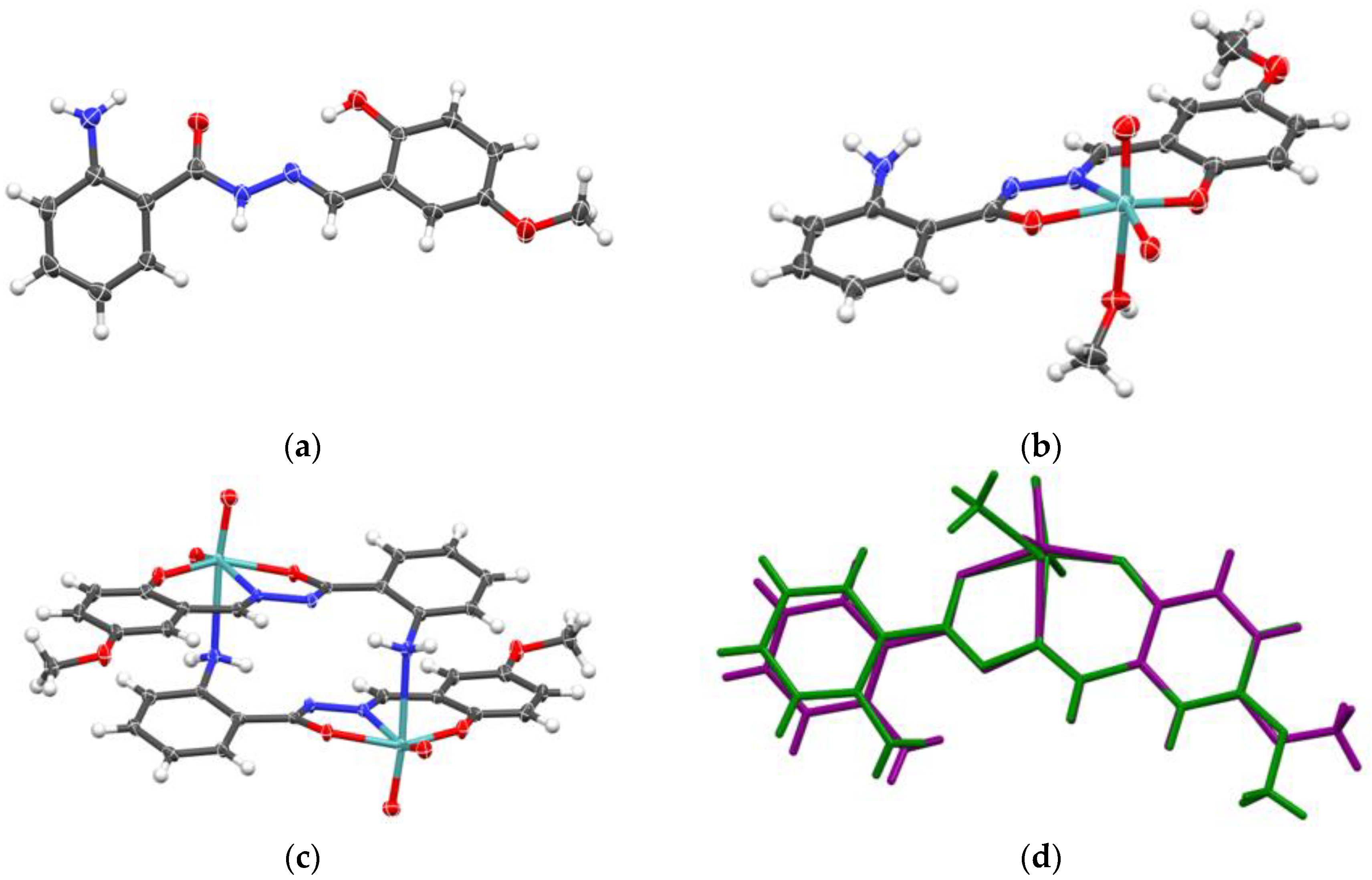
Figure 2.
Comparison of the measured PXRD patterns (blue) and patterns calculated from the X-ray single-crystal structure (black): (a and b) [MoO2(L1)]n, (c and d) [MoO2(L3)]2.
Figure 2.
Comparison of the measured PXRD patterns (blue) and patterns calculated from the X-ray single-crystal structure (black): (a and b) [MoO2(L1)]n, (c and d) [MoO2(L3)]2.
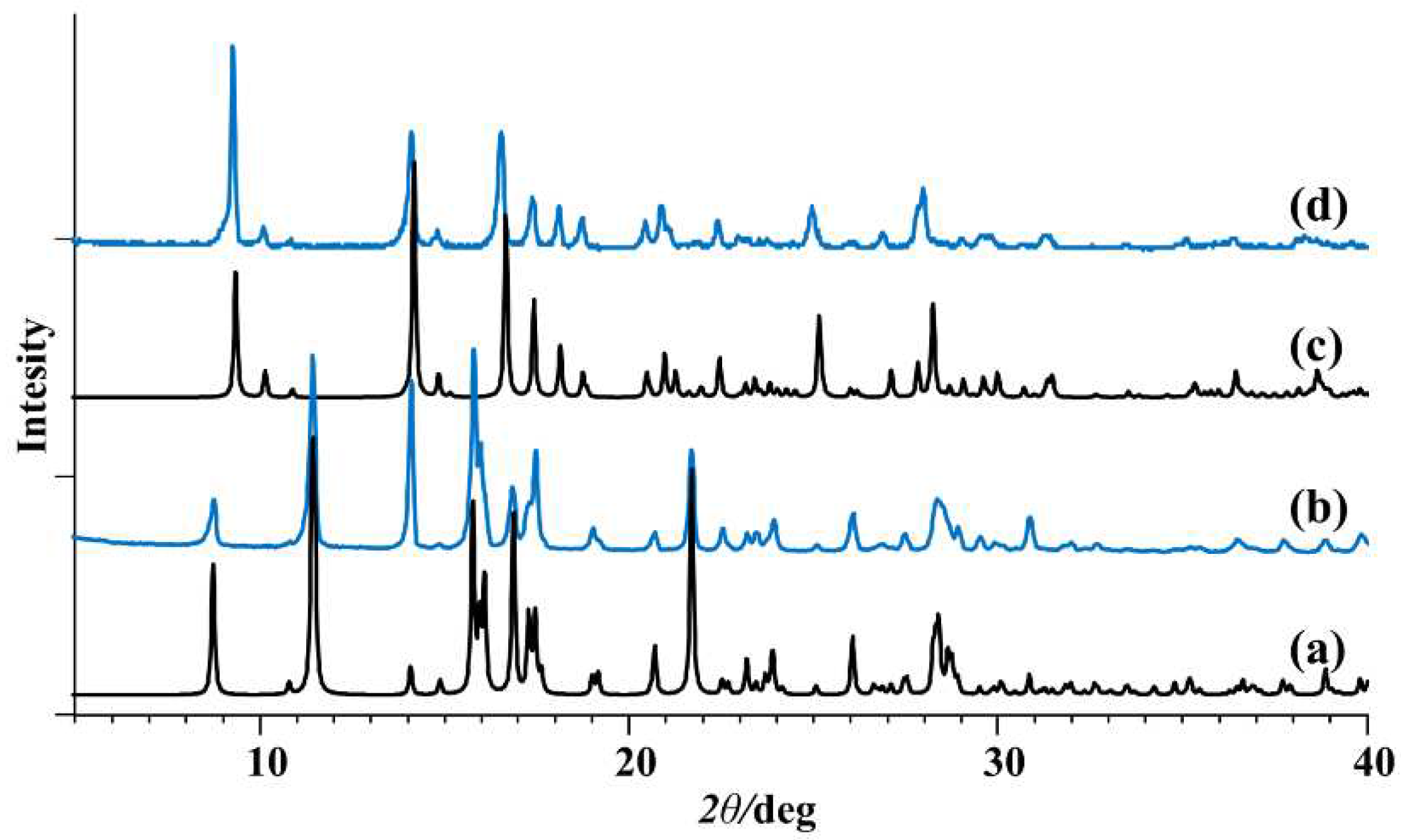
Figure 3.
Comparison of the measured PXRD patterns (blue) and patterns calculated from the X-ray single-crystal structure (black): (a and b) MoO2(L1)(MeOH)], (c and d) [MoO2(L2)(MeOH)], and (e and f) [MoO2(L3)(MeOH)].
Figure 3.
Comparison of the measured PXRD patterns (blue) and patterns calculated from the X-ray single-crystal structure (black): (a and b) MoO2(L1)(MeOH)], (c and d) [MoO2(L2)(MeOH)], and (e and f) [MoO2(L3)(MeOH)].
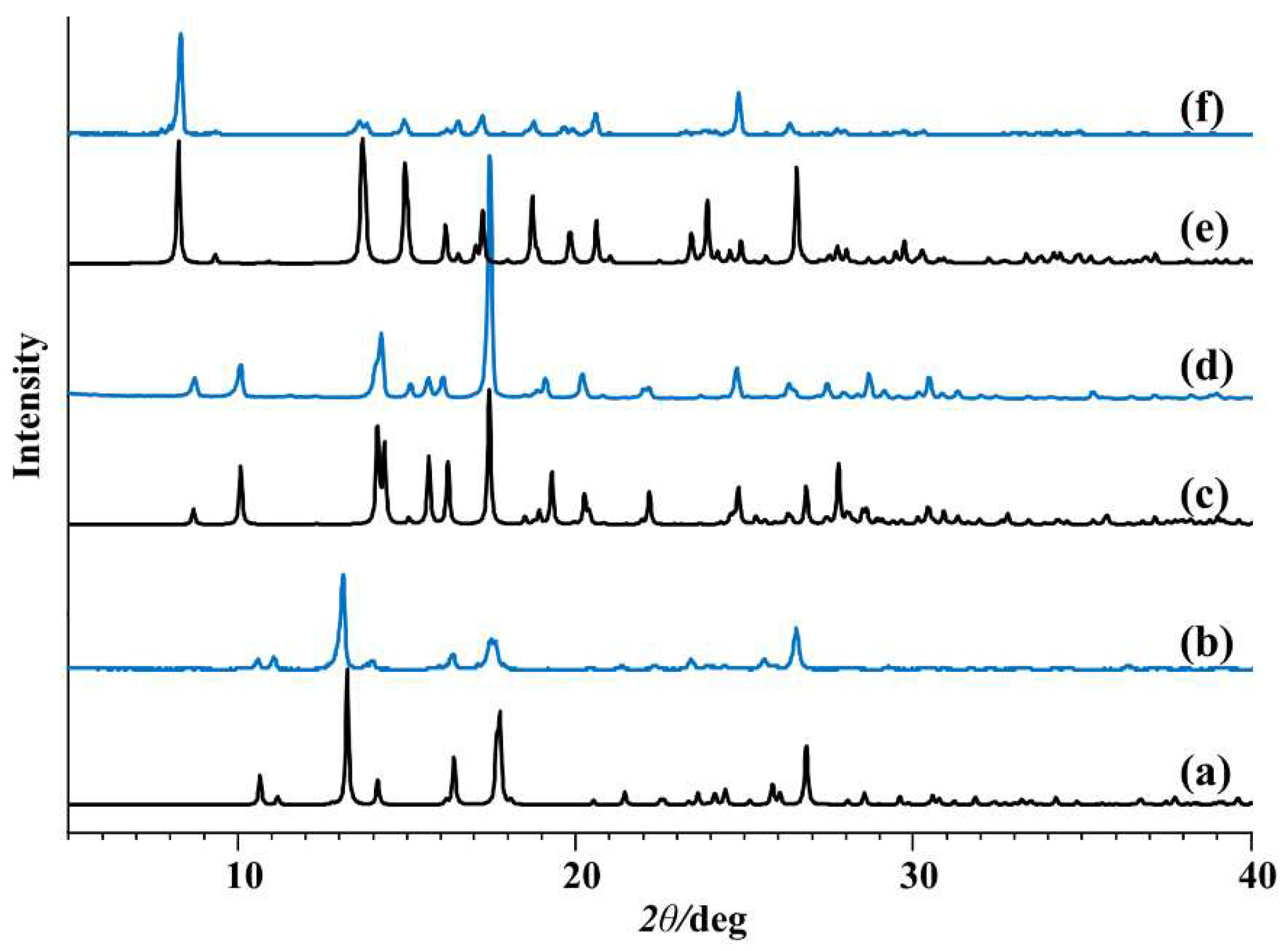
Figure 4.
Kinetic profiles of cyclooctene conversion with catalysts (a) 1-4 and (b) 1a-4a. The blue curve presents the reaction with catalyst 1 (left) and 1a (right), orange curve reaction with catalyst 2 (left) and 2a (right), grey curve reaction with catalyst 3 (left) and 3a (right), and yellow curve reaction with catalyst 4 (left) and 4a (right). TBHP in water was used as an oxidizing agent.
Figure 4.
Kinetic profiles of cyclooctene conversion with catalysts (a) 1-4 and (b) 1a-4a. The blue curve presents the reaction with catalyst 1 (left) and 1a (right), orange curve reaction with catalyst 2 (left) and 2a (right), grey curve reaction with catalyst 3 (left) and 3a (right), and yellow curve reaction with catalyst 4 (left) and 4a (right). TBHP in water was used as an oxidizing agent.
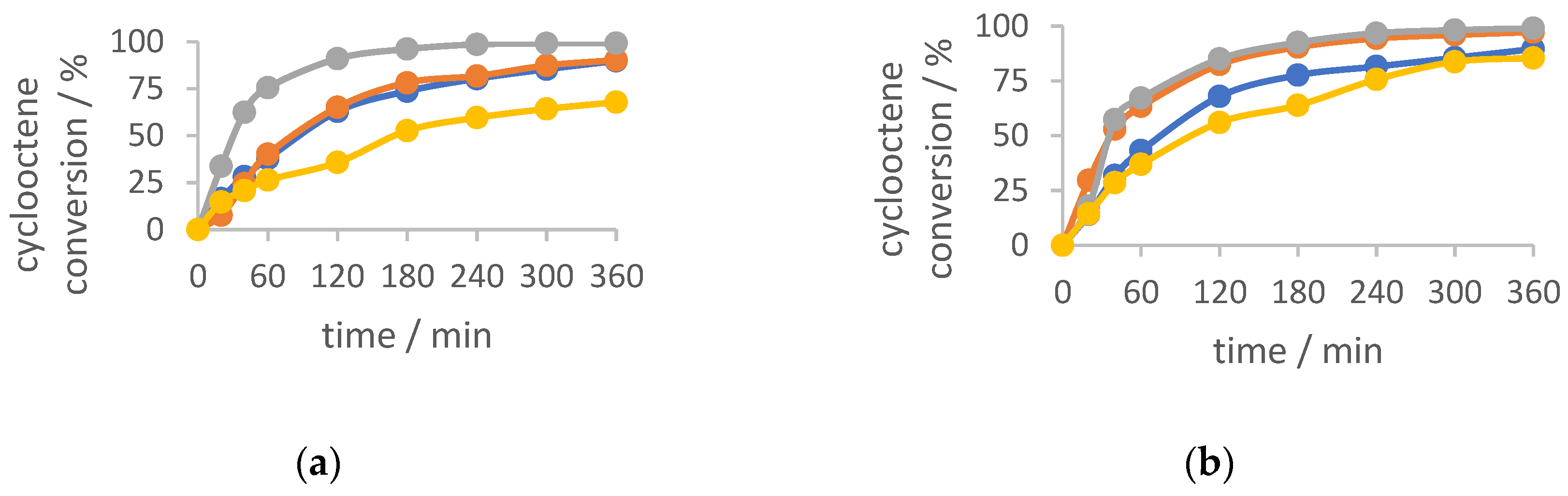
Scheme 2.
Hydrazones derived from 3- or 4-methoxysalicylaldehyde with the corresponding hydrazides (nicotinic hydrazide, 2-aminobenzhydrazide or 4-aminobenzhydrazide)
Scheme 2.
Hydrazones derived from 3- or 4-methoxysalicylaldehyde with the corresponding hydrazides (nicotinic hydrazide, 2-aminobenzhydrazide or 4-aminobenzhydrazide)
Scheme 3.
Presentation of the postulated mechanism for the epoxidation olefin. The ONO atoms (blue) and lines symbolizes the ligand. The different steps (I-IV) are indicated with numbers close to the arrows.
Scheme 3.
Presentation of the postulated mechanism for the epoxidation olefin. The ONO atoms (blue) and lines symbolizes the ligand. The different steps (I-IV) are indicated with numbers close to the arrows.
Scheme 4.
Representation of MoO2L complex with coordination sphere, highlighting the considered atoms taken into consideration for bonds lengths (bold black) and for dihedral angles (red). Oβ is indicated in Scheme 3.
Scheme 4.
Representation of MoO2L complex with coordination sphere, highlighting the considered atoms taken into consideration for bonds lengths (bold black) and for dihedral angles (red). Oβ is indicated in Scheme 3.
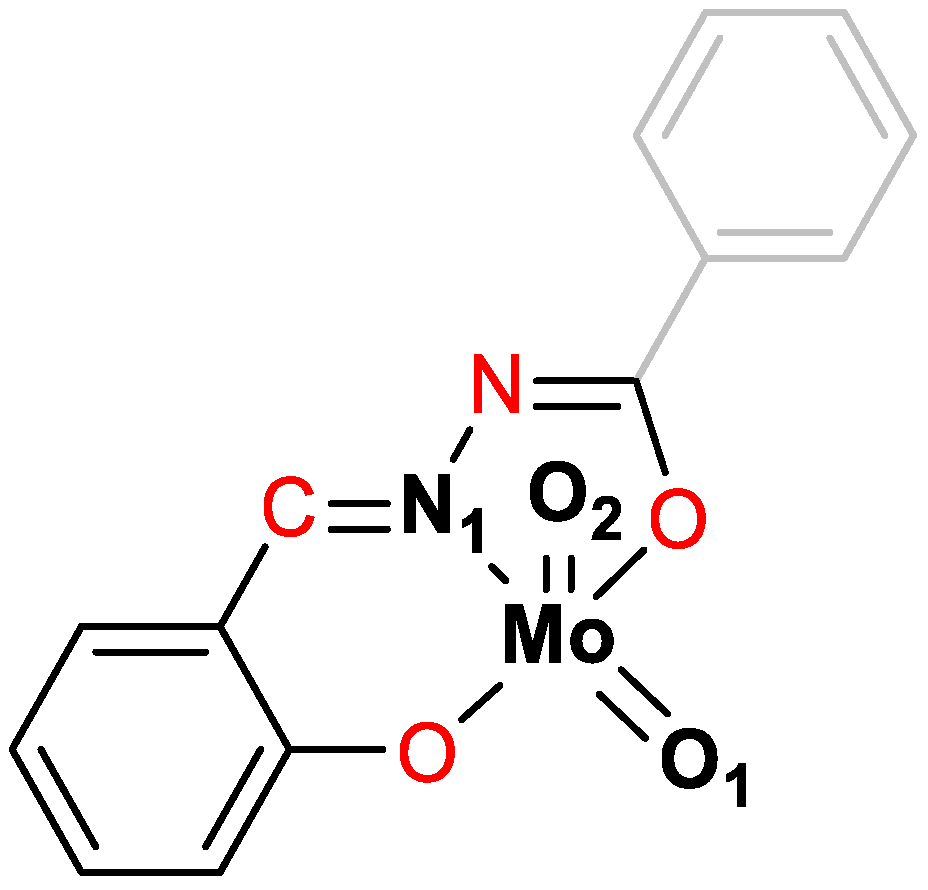

Table 1.
Catalytic results of cis-cyclooctene epoxidation. Reaction conditions: time, 6 h; temperature, 80 °C, n(catalyst)/n(cyclooctene)/n(oxidant) = 0.05 mmol/20 mmol/40 mmol.
Table 1.
Catalytic results of cis-cyclooctene epoxidation. Reaction conditions: time, 6 h; temperature, 80 °C, n(catalyst)/n(cyclooctene)/n(oxidant) = 0.05 mmol/20 mmol/40 mmol.
| Catalyst | Con.a / % | Sel.a / % | TONc | TOF20mind |
|---|---|---|---|---|
| 1 | 89 | 78 | 348 | 191 |
| 2 | 90 | 84 | 360 | 91 |
| 3 | 99 | 85 | 399 | 415 |
| 4 | 67 | 90 | 180 | 279 |
| 1a | 89 | 89 | 359 | 350 |
| 2a | 97 | 85 | 389 | 356 |
| 3a | 99 | 92 | 395 | 214 |
| 4a | 85 | 98 | 341 | 191 |
a cyclooctene consumed at the end of the reaction. b Formed epoxide per converted olefin at the end of the reaction. c n(cyclooctene) transformed/n(catalyst)/time(h) at 20 min. d n(cyclooctene) transformed/n(catalyst) at the end of reaction.
Table 2.
Comparison of the catalytic parameters (conversion, selectivity, TOF20min, TON) or the previously published catalysts.
Table 2.
Comparison of the catalytic parameters (conversion, selectivity, TOF20min, TON) or the previously published catalysts.
| Catalyst | Con. / % | Sel. / % | TON | TOF20min | Ref. |
|---|---|---|---|---|---|
| [MoO2(L2’)]n | 27 | 56 | 113 | 72 | [50] |
| [MoO2(L2’’)]n | 49 | 67 | 192 | 119 | [50] |
| [MoO2(L3’)]2 | 90 | 92 | 372 | 343 | [37] |
| [MoO2(L3’’)(MeOH)] | 85 | 90 | 349 | 383 | [37] |
| [MoO2(L3’’)]2 | 87 | 91 | 330 | 345 | [37] |
| [MoO2(L4’)(MeOH)] | 56 | 86 | 229 | 75 | [38] |
| [MoO2(L4’’)(MeOH)] | 63 | 85 | 257 | 97 | [38] |
| [MoO2(L4’)]2 | 84 | 90 | 380 | 295 | [38] |
| [MoO2(L4’’)]2 | 89 | 86 | 386 | 298 | [38] |
Table 3.
Enthalpy values (ΔH) (in kcal mol−1) for each step depicted in Scheme 3 (n.c. = non calculated).
Table 3.
Enthalpy values (ΔH) (in kcal mol−1) for each step depicted in Scheme 3 (n.c. = non calculated).
| Absolute (Relative) Values | |||||
|---|---|---|---|---|---|
| Steps | [MoO2(L1)] | [MoO2(L2)] | [MoO2(L3)] | [MoO2(L4)] | |
| (0) | Dimer decoordination | n.c. | n.c. | 5.9 | 5.5 |
| (0) | MeOH decoordination | 10.6 | 11.1 | 10.8 | 10.3 |
| (I) | TBHP coordination | -8.7 | -8.7 | -8.4 | -8.9 |
| (II) | TS barrier | 23.5 | 23.2 | 23.8 | 24.6 |
| (III) | Release of epoxide | -51.3 | -51.1 | -51.4 | -51.7 |
| (IV) | Regeneration of catalyst | -4.9 | -4.9 | -5.4 | -5.4 |
Table 4.
Interatomic distances (Å) and dihedral angle (°) for the calculated structures [MoO2Li(D)] (i=1-4) and related TS according to Scheme 4.
Table 4.
Interatomic distances (Å) and dihedral angle (°) for the calculated structures [MoO2Li(D)] (i=1-4) and related TS according to Scheme 4.
| [MoO2L(D)] | |||||
|---|---|---|---|---|---|
| D | MeOH | no | TBHP | TS | |
| Interatomic distances (Å) | |||||
| [Mo–Oβ] | L1 | 2.552 | - | 2.990 | 2.394 |
| L2 | 2.499 | - | 2.993 | 2.397 | |
| L3 | 2.504 | - | 3.018 | 2.412 | |
| L4 | 2.509 | - | 3.041 | 2.428 | |
| N1–Mo | L1 | 2.283 | 2.274 | 2.268 | 2.278 |
| L2 | 2.296 | 2.274 | 2.269 | 2.278 | |
| L3 | 2.293 | 2.273 | 2.268 | 2.275 | |
| L4 | 2.294 | 2.275 | 2.265 | 2.273 | |
| Mo=O1 (±plane) |
L1 | 1.709 | 1.697 | 1.714 | 1.733 |
| L2 | 1.705 | 1.697 | 1.714 | 1.733 | |
| L3 | 1.706 | 1.698 | 1.715 | 1.734 | |
| L4 | 1.708 | 1.699 | 1.716 | 1.735 | |
| Mo–O2 (⊥plane) |
L1 | 1.694 | 1.696 | 1.691 | 1.693 |
| L2 | 1.695 | 1.696 | 1.691 | 1.693 | |
| L3 | 1.696 | 1.697 | 1.692 | 1.693 | |
| L4 | 1.696 | 1.697 | 1.693 | 1.695 | |
| Interatomic angle (absolute values in °) | |||||
| OCNO Dihedral angle |
L1 | 9.99 | 6.16 | 11.05 | 15.69 |
| L2 | 10.36 | 6.28 | 11.18 | 17.10 | |
| L3 | 10.45 | 6.05 | 11.99 | 16.39 | |
| L4 | 10.63 | 6.10 | 12.19 | 15.71 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



