
Submitted:
22 December 2023
Posted:
25 December 2023
You are already at the latest version
Abstract
High incidence of epithelial malignancies in HIV-1 infection is associated with co-infection with oncogenic viruses, such as high-risk human papillomaviruses (HR HPVs), most often HPV16. A collaboration between HIV-1 and HR HPVs in the malignant transformation of epithelial cells exposed to both viruses have long been anticipated. Here, we delineated the effects on in vitro and in vivo properties of HPV16-infected cervical cancer cells of HIV-1 reverse transcriptase. Epidermoid carcinoma Ca Ski cells were made to express RT of HIV-1 clade A (RT_A) or Green Fluorescence Protein (GFP) by lentiviral transduction, subclones with one or six copies of RT_A and six copies of GFP DNA were generated. Expression by subclones of GFP was assessed by flow cytometry, and of RT_A, by Western blotting. Cells were assessed for the levels of mRNA of the RT_A, E6 and its isoforms, and cellular factors characterizing oxidative stress, and EMT by the real-time PCR. Parameters of glycolysis and mitochondrial respiration were assessed by Seahorse technology. Subclones were surveyed for the changes in cell cycle, doubling time, migration capacity, clonogenic activity and for the capacity to form tumors in nude mice. Ca Ski RT_A produced 20-55 fg of RT_A per cell. Expression of RT_A caused a proportional increase in the expression of E6*I isoform. In Ca Ski, RT_A suppressed mitochondrial respiration and oxygen consumption, and increased glycolysis. Compared to parental cells and GFP control, transcription in Ca Ski RT_A of epithelial (E-CADHERIN) state marker was increased, and of mesenchymal (VIMENTIN) decreased, indicating acquisition by cells of the hybrid epithelial/mesenchymal (E/M) phenotype. While Ca Ski GFP and Ca Ski RT_A with one gene insert had reduced migration rate, decreased clonogenic activity in vitro, and diminished capacity to form tumors in nude mice, increase in RT_A copicity/expression mitigated these effects. Altogether, expression of HIV-1 RT_A gave Ca Ski cells the plasticity required to overcome negative effects of lentiviral transduction and potentially increased their tumorigenicity.
Keywords:
HIV-1
; reverse transcriptase
; HPV16-positive cells
; E6*I isoform expression
; glycolysis
; mitochondrial respiration
1. Introduction
People living with human immunodeficiency virus (HIV-1) (PLWH) are at increased risk of developing cancer affecting epithelial cells, even with long-term successful antiretroviral therapy. One of the driving mechanisms was suggested to be the direct tumorigenic effect(s) of some HIV-1 proteins, specifically their ability to cause malignant transformation of the epithelial cells [1]. HIV proteins tat, nef, gp120, matrix protein p17, reverse transcriptase (RT) induce oxidative stress with serious consequences in the form of DNA, protein and lipid damage, as well as changes in the intracellular signaling and have a direct carcinogenic potential in vivo [2,3,4]. Another potent driver are co-infections with the oncogenic viruses, such as HBV, HCV, EBV, or CMV [5]. Individuals with HIV, especially with acquired immunodeficiency syndrome (AIDS), are at specifically high risk to develop cancers associated with infection with human papilloma viruses (HPV) [6,7,8,9,10,11]. Infection with high risk HPVs (HR HPVs) is responsible for most of the cases of cervical cancer, and a subset of cancers of the anus, oropharynx, penis, vagina, and vulva [12,13,14]. Women living with HIV are 6 times more likely to develop cervical cancer compared to women without HIV, an estimated 5% of new cervical cancer cases are attributable to HIV [15]. Cervical cancer in HIV-infected women shows specific features: it occurs at a younger age than in general population, reveals advanced stages at presentation, develops metastases in unusual locations, demonstrates poor response to treatment, higher recurrence rate, and shorter time interval to death [16]. Anal cancer is also more common in immunocompromised individuals, specifically PLWH [17,18]. HIV-infected women had an anal cancer rate of 30/100 000 person-years, while no anal cancer cases were observed for HIV-noninfected women. Even in HIV patients with CD4 cell counts stably over 500/μL, the risk to develop anal cancer was shown to be more than 20-fold higher than among general population [19]. Anal infections HR HPVs are very common among HIV-infected men who have sex with men (MSM). The unadjusted anal cancer incidence rates per 100,000 person-years were reported to be 2 for noninfected men, 46 for other HIV+ men and 131 for HIV+ MSM [20,21,22,23].
Cooperation between HIV-1 and HR HPVs, specifically, HPV16 in malignant transformation of epithelial cells has long been anticipated, with HR HPVs infecting epithelial cells [24,25], and HIV-1 entering these cells by multiple un-conventional mechanisms as transversion of the epithelial lining or “natural pseudotyping” [26,27,28,29]. HIV-1 can also destroy antigens affecting the “innocent” bystander cells [1,30]. This enables an interaction between HIV-1 and HPV viral proteins in HPV-infected epithelial cells. Possibility of the molecular interactions between these viruses and/or their antigens leading to enhanced risk(s) to develop cancer have been independently addressed in several studies. As an example, HIV-1 nef is transported from HIV-1-infected cells to the neighboring target cells via filopodia and/or exosomes [31,32,33] and interacts with ubiquitin (Ub)-protein ligase E3A (UBE3A/E6AP) complex [34] targeting the tumor suppressor p53 for ubiquitination and degradation, thereby contributing to HPV-induced cervical carcinogenesis [35,36,37]. The effects of HIV-1 and HR HPVs can be independent but unidirectional, potentiating one another.
As of today, the data on direct interaction(s) between HPV and other HIV-1 proteins is missing. However, HIV-1 tat was shown to increase the expression of HPV E6 and E7 oncoproteins, enhancing the E6 and E7 mediated oncogenic effects of HPV [38,39,40,41] as well as increase E2 transcription, which modulated HPV replication [42]. НIV-1 rev was found to upregulate the expression of HPV L1 [43] (which remains a mystery given its nuclear localization in HIV-1-infected cells). The same applies to other HIV-1 antigens: the nuclear protein vpr involved in cell cycle regulation [44], the envelope protein gp120 and the reverse transcriptase (RT). Vpr was shown to interact with E6 in cervical cancer cells [45]. Both gp120 and RT increase the expression of HPV16 E6 in HPV16 infected immortalized and/or completely transformed epithelial cells, while other HIV-1 proteins, such as capsid protein p24 had no effect [1]. Furthermore, prolonged interaction of HIV-1 proteins gp120 and tat and cell-free HIV-1 virions with HPV16-immortalized anal, cervical, and oral epithelial cells stimulated EMT and increased the invasiveness of the cells [46,47]. Overall, these data indicate that presence of HIV-1 and/or its antigens in HPV16-infected neoplastic cells may potentiate HPV-associated tumorigenicity, making HIV-1 infection an integral part/a player in the process of HPV-associated tumorigenesis.
Earlier, we have shown that expression of HIV-1 RT in murine adenocarcinoma cells induces production of ROS and genetic instability, increases the expression of factors promoting the epithelial-mesenchymal transition (EMT). Furthermore, RT expressing cancer cells acquire an enhanced capacity to grow and metastasize in immunocompetent mice [1,4]. The aim of this study was to evaluate the effect of HIV-RT on the HPV16-infected human epithelial cancer cells to test if RT can, as in murine cells, modulate their phenotypical characteristics, metabolic activity, gene expression patterns and in vivo tumorigenicity.
2. Materials and Methods
2.1. Lentiviral Transduction of Ca Ski Cells and Isolation of Clones Expressing RT_A and GFP Variants
The coding sequence for the consensus HIV-1 RT_A (RT_A) was synthesized by Evrogen (Moscow, Russia). Coding sequence for RT_A was recloned from p6HRT_A into the lentiviral vector pRRLSIN.cPPT.PGK (Addgene, Watertown, Massachusetts, USA) generating the lentiviral vector pLV-RT_A as described earlier [4]. The production of lentiviral particles was carried out in a HEK293T culture cells by transfection of the pLV-RT_A in the presence of a polyethyleneimine (PEI) (MW 25000, Polysciences, United States), as described earlier [48]. 48 hours after transfection, cell culture medium containing viral particles was harvested, clarified by low-speed centrifugation and filtered through a 0.45 μm styrile filter, followed by concentration using Amicon Ultra 100K centrifuge concentrators (Merck-Millipore, Darmstadt, Germany).
Infectious titers of the lentiviral particles were determined in HeLa cells by quantitative real-time PCR using standard samples of HeLa DNA with a known number of copies of the provirus [49]. Transduction of HPV16 infected human cervical squamous cell carcinoma Ca Ski cells (CRL-1550) was performed with the multiplicity of lentiviral infection (MOI) of 5 and 10 transducing units per cell without the addition of polybrene. Monoclonal populations of Ca Ski derivative clones were generated by limiting dilution in 96-well plates. Four subclones containing RT_A DNA were re-cloned by limiting dilution, generating three derivatives from the first-round subclone B8 (MOI 5; B8B5, B8D5, B8D2) and three, from the subclone H6 (MOI 10; H6G11, H6D7, H6F8). Resulting Ca Ski derivatives were cultured in the full RPMI-1640 (Paneco, Moscow, Russia) medium with 100 mg/mL penicillin/streptomycin (Paneco) mix and 12% FBS (HyClone, United States) (due to reduced re-cloning efficiency and slow growth rate) at 37 °C with 5% CO2 and split every 2–3 days.
Control cell lines expressing a fluorescent marker were obtained with the help of the lentiviral vector pRRLSIN.PGK.EGFP (Addgene) encoding GFP under the control of the polyglycerate kinase (PGK) promoter. The transduced cells were cloned into 96-well plates by the limiting dilution. One individual subclone was selected based on the presence of a fluorescent signal. The stability of the GFP gene expression after two weeks in culture was confirmed using a flow cytometer (Beckman Coulter CytoFLEX, San Jose, CA, USA).
2.2. Determination of the Number of Copies of the Provirus in the Genome of the Transduced Lines
A comparative analysis of the number of copies of the lentiviral genome in the DNA of the Ca Ski derivatives was carried out using real-time PCR according to the protocol used to determine the infectious titer [49]. The ∆Ct was defined as the difference between the threshold values of two amplification reactions with primers to the housekeeping gene (beta-actin) and to the 5’-region of the provirus genome (Suppl., Table S1). The number of copies of the provirus was determined using a calibration curve using control DNA samples of HeLa cells with a known number of copies of the provirus [49].
2.3. Confirmation of RT Production by Western Blotting
Production of RT_A protein was analyzed by Western blot of cell lysates. Lysates of 105 cells of each RT variant were resolved by PAGE and subjected to Western blotting using polyclonal anti-RT rabbit antibodies [50] and anti-ß-actin monoclonal antibodies (AC-15, Invitrogen). Lysate of the parental Ca Ski cell line was used as a negative control. Recombinant RT_A obtained as described earlier [4] loaded on the gel in fixed amounts from 1 to 27 ng per well was used as a positive control, to build a calibration curve. Using this curve, we assessed the amount of RT in the aliquots of cell lysates loaded onto the gel, and by dividing this value by the number of cells used to prepare the lysate, we determined the levels of RT expression per cell for each derivative clone. Signals from the bands were quantified by ImageJ software (http://rsb.info.nih.gov/ij).
2.4. Isolation of Nucleic Acids, Reverse Transcription, and Semiquantitative PCR
Cell culture medium was discarded; cells were detached using 0.05% Trypsin with EDTA (Paneco) and centrifuged 600g. RNA was isolated using the GeneJET RNA Purification Kit (ThermoFisher scientific, cat. K0731) according to the manufacturer’s instructions and reverse transcription was performed using the MMLV RT kit (Evrogen).
Gene-specific PCRs were performed on a RotorGene 6000 (Qiagen, Hilden, Germany) cycler using primers specific to RT_A, E6FL, E6*I, E6*II, E7, NRF2, NQO1, GCLC, A-TUBULIN, Y-TUBULIN, N-CADHERIN and VIMENTIN, E-CADHERIN, TWIST1, SNAI1 and SNAI2 genes and SYBR Green (Evrogen) (Suppl., Table S1). Levels of mRNAs were measured relative to GAPDH or GUSB mRNA. Relative gene expression levels were calculated using the ddCt method [51].
2.5. Cell Culture and Microscopic Quantitation of Proliferation
Ca Ski and their derivatives cells were maintained in RPMI-1640 (Paneco) with 10% FBS (HyClone). Doubling time of derivative clones was estimated as described previously [4]. Briefly, adherent cells were washed with PBS and detached with 0.05% Trypsin with EDTA (Paneco). Thereafter, 10 μl of the resulting suspension was transferred to a Gorjaev’s count chamber and cells were counted (× 5). Scoring of the proliferation rate was performed by counting cells in 20 fields of the counting grid.
The population doubling time, or the time required for a culture to double in number were calculated with the following formula: DT=T ln2/ln(Xe/Xb), where T is the incubation time in a given time unit; Xe is the cell number at the end of the incubation time; Xb is the cell number at the beginning of the incubation time (https://www.atcc.org/resources/culture-guides/animal-cell-culture-guide).
2.6. Measurement of Glycolysis and Mitochondrial Respiration
Glycolysis and mitochondrial respiration were assessed by the Seahorse technology on a XFe24 analyzer (Agilent Technologies, Santa-Clara, CA, USA) according to manufacturer’s instructions with a number of modifications [52]. Briefly, 24 h prior to analysis the cells were seeded via XF24 Cell Culture Microplate into (3 × 104 cells/well) in RPMI-1640 (Paneco) with 10%FBS (HyClone) with four replicates.
For the MitoStress test, 45 min before analysis the media was changed to DMEM (Gibco, Thermo Fisher, Waltham, MA, USA) lacking phenol red dye and bicarbonate and supplemented with 2 mM glutamine, and the plate was kept at 37°C at normal atmosphere. To evaluate respiration-linked ATP production, maximum respiratory capacity, and non-mitochondrial respiration, ATP-synthase inhibitor oligomycin, uncoupler FCCP, and a mixture of complex I and III inhibitors rotenone and antimycin were added to final concentrations of 1 µM (Oligomycin), 0,75 µM and 1,5 µM (FCCP), and 1 µM each (rotenone/antimycin). For each condition, three readings were performed at 3 min intervals.
In case of the GlycoStress test, 30 min prior to analysis, the medium was changed to DMEM lacking phenol red dye, bicarbonate and glucose and supplemented with 2 mM glutamine. During the assay 11 mM and 30 mM Glycose, 1 µM oligomycin and 50 mM 2-deoxyglucose were added to access basal glycolysis, maximal glycolytic capacity, and non-glycolytic acidification, respectively.
After the analysis, the cells were lysed, and the total protein concentration was measured using the PierceBCA Protein Assay Kit (Thermo Fisher Scientific, USA) according to the manufacturer’s instructions. Mitostress and Glycostress test results were normalized to the total protein levels. The raw data were processed by Seahorse Wave Desktop software (Agilent Technologies) and further analyzed by GraphPad Prism (GraphPad Software, La Jolla, CA, USA).
2.7. Assessment of the Production of Reactive Oxygen Species (ROS)
Production of reactive oxygen species (ROS) was accessed using redox-sensitive fluorescent probes: dihydroethidium (DHE) and 2′,7′-dichlorodihydrofluoresceine diacetate (DCFH2-DA). The cells were stained as described earlier [53]. Stained cells were incubated at room temperature for 30 min in a fresh medium containing 25 μM DCFH2-DA, then washed 10 times with 0.5 mL PBS, and resuspended in 200 μL PBS. The fluorescence intensities (FLI) were recorded on a Plate CHAMELEON V reader (Hidex Ltd., Turku, Finland). Specific levels of superoxide anion were measured after treating cells with 25 μM dihydroethidium (DHE) using the protocol given above for DCFH2-DA. In case of DHE, FLI were measured with excitation at 510 nm and emission at 590 nm, and in case of DCFH2-DA, with excitation at 485 nm and emission at 535 nm.
2.8. Wound Healing Assay
The effect of RT_A and GFP expression on Ca Ski cell migration was assessed using the wound healing assay [54]. Cells were grown in 96-well plates until 100% monolayer was achieved. The surface of the well was scratched with a pipette tip. Cells, eight repeats per each subclone, were washed with PBS followed by medium change and continuously imaged using CELENA® X High Content Imaging System (Gyeonggi-do, South Korea) at 4× magnification for 24 hours. The images were processed using the ImageJ/Fiji® module, quantifying the entire area of the wound, the average width of the wound and the standard deviation of the width in all images. Data reflecting the change in the area values (size of the wound) for each individual repeat during each hour of monitoring was used to build a slope-intercept from y=mx+b, where “m” is the slope (or steepness) of a line, “b” is the y-intercept where the line crosses the y-axis, and therefrom calculate the slope parameter. Setting y = b/2 (ie: the point at which the gap is half the original area) and solving for x. The speed of migration (U) was calculated as U = |slope, m| / 2*L, where L is the length of the wound [54].
2.9. Clonogenic assay
To analyze the effect of RT on the clonogenic activity, cells were plated in 60 mm dishes in an amount of 200 cells and incubated under the standard conditions, after which single cells formed colonies. The mean size and number of colonies were measured at 21-days post seeding, colonies were washed three times with PBS, fixed with 100% methanol, stained with 0.5% crystal violet, thereafter their number and mean size were quantified using the ImageJ software (https://imagej.net/ij/download.html). Each assay was performed in triplicate.
2.10. Cell Cycle Analysis
The representation of Ca Ski subclones in different stages of the cell cycle was assessed by flow cytometry as described cytometric analysis was carried out as described [55]. In brief, cells were cultured in a T25 culture flask, detached, sedimented, washed twice with PBS. Thereafter, cells were fixed with chilled 70% ethanol, incubated on ice for 30 minutes, washed again with cold PBS and stained with propidium iodide (PI) dye (25 µg/mL) and treated with the solution of RNase A (50 µg/mL) followed by incubation at room temperature for 30 min. Finally, cell cycle distribution of cells was carried out by using a flow cytometer (Beckman Coulter CytoFLEX, San Jose, CA, USA). Distribution of cells in G1/G0, S, and G2/M areas was assessed using FlowJo software (https://www.flowjo.com).
2.11. Assessment of Tumorigenicity of Ca Ski Derivative Clones
The capacity of the derivative cell lines to form tumors was tested by ectopic implantation into 8-week-old female Nu/J mice. Animal experiments were performed in the animal facilities of the NF Gamaleya Research Center of Epidemiology and Microbiology (GRCEM), Ministry of Health of the Russian Federation (Moscow, Russia). The experiments were carried out in accordance with the bioethical principles adopted by the European Convention for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes (Strasbourg, 1986). Experimental procedures were approved by the ethical committee of the Gamaleya National Research Center for Epidemiology and Microbiology (protocol N39, March 24, 2023). Six-week-old 17 g Nu/J mice from the nursery of the Center for Collective Use “SPF-vivarium” of the Federal Research Center Institute of Cytology and Genetics, Siberian Branch of Russian Academy of Sciences (Novosibirsk, Russia) were housed under a 12 h/12 h light-dark cycle with ad libitum access to water and food and used for experiments after two weeks of adaptation.
Prior to injection, Ca Ski and derivative clones grown in the RPMI-1640 (Paneco) with 10% FBS (HyClone) were detached, sedimented, washed with serum-free medium, stained for viability with Trypan Blue dye (Life Technologies, Carlsbad, CA), then counted in a Gorjaev’s count chamber and aliquoted 105 in 50 µl and 106 in 100 µl of RPMI-1640 was placed in sterile tubes. Aliquots were injected subcutaneously into two sites, to the right and to the left of the base of the tail with a 25G needle mounted on an insulin syringe (B Braun, Germany) (for injection scheme, see Figure 1).
Tumor size was assessed by morphometric measurements done at regular intervals on days 3, 5, 8, 11, 15, 18, 22, 25, 29 after injection using calipers; tumor volume was calculated using the standard formula for xenograft volume [56]: V = L×W2/2, where V is the tumor volume, L is the length (the longest dimension), W is the width (the distance perpendicular to and in same plane as the length). The humane end point of the experiment was considered when one of the parameters (length and/or width) of the tumor reached 1 cm. At the end of the experiment, 29 days after implantation, mice were anesthetized with Zoletil 100 (Virbac) and humanely euthanized by cervical dislocation. Tumors and spleens were surgically excised and weighed.
2.12. Statistical Analysis
Data were analyzed using nonparametrical and parametrical statistics (GraphPad Prism 9, manufacturer, city, country). Continuous but not normally distributed variables, such as doubling time, cell cycle, count and size of colonies, tumor volume and tumor weight were compared in groups using Dunn’s test for pairwise multiple comparisons of the ranked data as the post hoc test following Kruskal-Wallis test and pairwise by Mann-Whitney U test with Bonferroni correction. Normally distributed variables were analyzed by Unpaired t test with Bonferroni correction. Correlation analysis was done using the Spearman Rank Correlation test. For all types of analysis, p< 0.05 were considered significant.
3. Results
3.1. Lentivirally Transduced Ca Ski Cells Express HIV-1 Reverse Transcriptase
For human cervical squamous carcinoma cell line Ca Ski with integrated HPV16 genome (CRL-1550; [57]), supplementation of HIV-1 RT variants into the culture medium led to increase in the expression of HPV16 E6 [1]. Here, we aimed to assess if RT has a systemic effect on Ca Ski cells. Cells were transduced with lentiviral particles expressing HIV-1 RT under the control of human phosphoglycerate kinase gene (hPGK) promoter. After two rounds of single cell cloning, we obtained a panel of subclones, three derived from subclone B8 with one (B8B5, B8D5, B8D2; Table 1), and three derived from subclone H6 with six copies of RT_A coding sequence per cell genome (H6G11, H6D7, H6F8; Table 1). Expression of RT mRNA was confirmed by RT PCR, and of RT_A protein, by Western blotting (Table 1; Figure 2 A, B). All RT_A Ca Ski subclones produced a protein with the expected molecular mass of 66 kDa, specifically recognized by anti-RT antibodies [58] (Figure 2A). Subclones with six RT_A DNA inserts were characterized by higher level of RT_A production compared to subclones with one insert (Table 1; Figure 2B). The level of RT_A expression per cell (determined based on the relative levels of RT_A and calibration curve built using recombinant RT, see Materials and Methods and [4]) varied from 20 to 55 fg per cell (Table 1).
An effective strategy for mitigating nonspecific effects of viral transduction on cell lime properties is to generate control cells expressing unrelated proteins. It enables to assess both the effects of lentiviral transduction per se, and of the metabolic burden due to the overexpression of exogenic protein [59,60]. Here, we obtained control cells by transduction of Ca Ski with lentivirus carrying GFP coding sequence under the control of the PGK promoter, as was done for RT_A. Respective Ca Ski GFP G9 subclone carried six copies of GFP gene per genome. Expression of GFP was confirmed by flow cytometry (Suppl., Figure S1).
3.2. RT Increases the Expression of HPV16 E6*I Isoform
Of the cell line properties that RT_A expression can modulate, we firstly evaluated the levels expression of mRNA of the full-length E6 (E6FL), its constitutive isoforms [61,62], and E7. Expression levels of E6FL (full-length), E6*II and E7 were increased in single subclones, the effect was not associated with the expression of RT_A and could have been due to the lentiviral transduction (Suppl., Figure S2). E6*I isoform expression was increased in Ca Ski GFP control cells as compared to Ca Ski, the difference attributable to the effect of lentiviral transduction (Figure 3A). However, all RT-expressing subclones, with one as well as with six RT_A DNA inserts, demonstrated an increased expression of E6*I isoform over that observed in the Ca Ski GFP control (Figure 3A, B). Interestingly, we observed a decrease in E6*I expression in clones with six copies of RT_A DNA insert compared to one copy, as if overexpression of RT_A was mitigating an unspecific effect of lentiviral transduction (Figure 3B). The effect was not related to the level of RT_A expression, as there was no difference in the levels of E6*I in Ca Ski subclones with high and low level of RT_A expression (Figure 3B).
Thus, the constitutive expression of HIV-1 RT in Ca Ski cells increased the level of expression of E6*I mRNA but had little effect on the level of transcripts of E6*II isoform or of full length E6, or E7. Since E6*I isoform plays a crucial role in HPV16-driven carcinogenesis [63], we have launched a study of the features Ca Ski subclones associated with their malignancy (tumorigenicity in vitro and in vivo).
3.3. HIV-1 RT Expression Increases the Extracellular Acidification Rate and Decreases the Oxygen Consumption rate of Ca SKI CELLS
We sought to find out if RT, when expressed in Ca Ski, caused changes in their metabolism, specifically, glycolysis and mitochondrial respiration. Changes in glycolysis were quantified by assessing the extracellular acidification rate (ECAR) of the culture medium, and changes in the substrate utilization in mitochondrial respiration, by measuring the oxygen consumption rate (OCR).
3.3.1. HIV-1 RT Expression in Ca Ski Cells Increases the Glycolysis
During glycolysis, glucose is converted to pyruvate, which is transported into mitochondria by the subsequent incorporation into the Krebs cycle or secreted in the form of lactate. Enhanced aerobic glycolysis constitutes an additional source of energy for tumor proliferation and metastasis [64]. The efficiency of glycolysis was evaluated using the analyzer Seahorse, which allows to evaluate the levels of acidification of the culture medium using the Glycostress test method. Summary is presented in Figure 4A. Firstly, acidification was assessed in absence of glucose (the level of non-glycolytic acidification, which is caused by cell processes other than glycolysis; Figure 4A), and thereafter, in the presence of glucose in low (11 mM; Figure 4A, B) and high-glucose DMEM medium (30 mM, Figure 4A, C). Further, we assessed acidification in cells treated with mitochondrial ATPase inhibitor oligomycin which activates glycolysis to the maximum possible level to compensate for abrupted ATP synthesis [65] (Figure 4A, D), and lastly, in the cells treated with the inhibitor of the first stage of glycolysis, 2-deoxy-D-glucose (2-DG) (Figure 4A). Glycolytic reserve was calculated as the difference between the maximum glycolytic capacity and the level of glycolysis at 30 mM glucose (Figure 4E).
Ca Ski subclones with six RT_A gene inserts demonstrated an increased acidification at the low and high glucose levels, reflecting an increase in the basal and maximal glycolysis, respectively (Figure 4B, C), and an increased maximum glycolytic capacity (Figure 4D). No changes were observed for Ca Ski subclone expressing GFP (Figure 4B, C, D). Thus, the above changes were not associated with either lentiviral transduction, or metabolic burden on the cells due to (over)expression of a foreign protein. Thus, constitutive expression of RT_A in Ca Ski cells caused an increase in the intensity of glycolysis under aerobic conditions. A decrease in the glycolytic reserve was observed for both 6 GFP and RT-expressing subclones and could have been due to the lentiviral transduction (Figure 4E).
3.3.2. HIV-1 RT Expression in Ca Ski Cells Suppresses Mitochondrial Respiration
Mitochondrial respiration shows the activity of the oxidative phosphorylation system and related processes of the Krebs cycle, which provides the respiratory system with substrates.
The influence of HIV-1 RT_A on the respiratory activity of mitochondria was assessed on a Seahorse analyzer using the Mitostress method (Figure 5A). Firstly, we determined the basic level of respiration (Figure 5A, B), then the decrease in respiratory activity after treatment with oligomycin, which inhibits ATP synthase, the data obtained reflecting the level of ATP synthesis by the oxidative phosphorylation system (Figure 5A, C). Thereafter, the respiration was induced to the maximum possible level by the addition of the proton transport uncoupler carbonyl cyanide-4-trifluoromethoxyphenylhydrazone (FCCP) (Figure 5A, D). The difference between the levels of basal and maximum respiration defined the “spare capacity” [66] (Figure 5E). At the final stage, we assessed the residual level of respiration (non-mitochondrial respiration) by adding the inhibitors of mitochondrial complexes I and III, rotenone and antimycin (Figure 5A, F). The difference between the maximum level of oxygen uptake and the level after the addition of oligomycin reflected the intensity of proton leakage (Suppl., Figure S4).
Ca Ski GFP subclone demonstrated a decrease in the basal respiration, ATP production, maximal respiration with low FCCP and in the spare capacity compared to Ca Ski (Figure 5B, C D, F), the phenomena attributable to the lentiviral transduction. For maximal respiration with high FCCP there was no statistical differences due to non-normal distribution of the data (Figure 5E). As most of the mitochondrial respiration parameters in Ca Ski subclones with both one and six RT_A gene inserts (1 RT_A and 6 RT_A) did not differ from each other, these subgroups were pooled and compared to the parameters of Ca Ski GFP. Overall, RT_A expressing Ca Ski subclones had decreased levels of ATP production, maximal respiration with high FCCP and spare capacity as compared to Ca Ski GFP (Figure C, E, F). The level of proton leak did not differ (Suppl., Figure S4). Thus, despite lentiviral transduction led to decrease in mitochondrial respiration parameters, expression of RT_A suppressed mitochondrial respiration in Ca Ski cells as compared to the GFP control.
3.4. Expression of HIV-1 RT Did Not Lead to the Changes in the Cytoskeleton
Changes in the mitochondrial homeostasis can be due to the changes in the cell cytoskeleton. To check if this is the case, we have tested if expression of RT affects the structure of microtubule (MT) structure by assessing the expression of A-tubulin, and if RT modulates the MT-supported mitochondrial network by assessing the expression of Y-tubulin. No difference was observed between the RT_A and GFP expressing Ca Ski subclones in the levels of mRNA of either A-TUBULIN, or Y-TUBULIN. Single RT_A subclones and GFP subclone demonstrated a tendency for the decreased levels of Y-TUBULIN mRNA, but it did not reach the level of significance (Suppl., Figure S5). These data implies that the observed changes in the glycolysis and mitochondrial respiration of RT_A expressing Ca Sci subclones were not due to the RT_A induced rearrangements of the cytoskeleton.
3.5. Expression of HIV-1 RT in Epithelial Ca Ski Cells Does Not Induce Oxidative Stress
Switching to a more glycolytic metabolism is associated with a decrease in ROS [67]. To test if this is the case, we assessed the levels of ROS in Ca Ski and in Ca Ski subclones. The levels of ROS production in Ca Ski derivatives were assessed using an intracellular fluorescent probes: 2′,7′-dichlorodihydrofluoresceine diacetate (DCFH2-DA) and dihydroethidium (DHE). DCFH2-DA reacts to various types of ROS, including H2O2, O2•− and hydroxyl radical, yielding a fluorescent product which allows to assess the general redox status of cell, whereas DHE senses only superoxide (O2•−) [68,69].
Technically, we could not use the GFP subclone as the control due to the overlap of the fluorescence emission spectrum for the DCFH2-DA and GFP. A significant decrease in the overall level of ROS production, compared to that in the parental Ca Ski cells, was observed for four out of six RT_A expressing subclones except for B8D2 and H6D7 (Table 1) (Figure 6A). At the same time, staining with DHE demonstrated no change in the levels of production of superoxide in either in RT_A or control GFP expressing Ca Ski cells (Figure 6B). Thus, the expression of both RT_A and GFP in Ca Ski cells caused a reduction in the production of ROS (H2O2, hydroxyl radical), but not of superoxide O2•−. Due to inability to use GFP subclones as the assay control, we could not distinguish if this effect was due to the lentiviral transduction with (over)expression of the foreign protein, or specifically to (over)expression of RT_A.
To circumvent the limitation of the assays with fluorescent probes, we turned to the assessment of the expression of the transcription factors involved in cellular redox balance Nrf2, enzymes of glutathione biosynthesis GCLС, and of the enzyme of Phase II of oxidative stress response NQO1. The levels of mRNA of NRF2, GCLC and NQO1 in RT-expressing Ca Ski subclones were compared to their levels on the GFP control and the parental Ca Ski cells. For all subclones, we observed a significant decrease in the expression of mRNA NRF2, and NQO1, a decrease in the levels of GCLC mRNA did not reach the level of significance (Figure 7A-C). No difference was observed between RT_A and GFP expressing subclones, indicating that the effect was due to the lentivirus transduction.
On contrary tour observations for RT_A expressing mammary gland adenocarcinoma 4T1 cells [70], stable expression of HIV-1 RT in Ca Ski cells did not increase the production of ROS, or the expression of transcription factors associated with oxidative stress, or of the enzymes of the Phase II protection from oxidative stress, i.e. had no effect on the redox balance of Ca Ski cells.
3.6. Lentivirus-Transduced Ca Ski Express Reduced Levels of Factors Associated with Epithelial-Mesenchymal Transition
As we have shown that expression of HIV-1 RT causes metabolic shift towards glycolysis, we have next tested if RT_A expression modulates the epithelial-to-mesenchymal transition (EMT), since glucose metabolism and EMT have previously been linked. Control GFP Ca Ski subclone demonstrated an increased expression of mRNA of the factors associated with EMT, namely VIMENTIN and N-CADHERIN, the two markers of the mesenchymal state which promote the acquisition of the spindle-shaped mesenchymal cell morphology [71], while levels of E-CADHERIN mRNA were unchanged compared to the parental cells (Figure 8 A-C). On contrary to Ca Ski GFP, Ca Ski RT_A demonstrated an increase in the expression of E-CADHERIN mRNA (p<0.05 for 1 RT_A, and p<0.1 for 6 RT_A subclones; Figure 8A), manifesting a loss of the typical heterogeneous morphology of epithelial cells. At the same time, Ca Ski RT_A subclones did not differ from the GFP subclones in the levels of N-CADHERIN and demonstrated decreased levels of VIMENTIN mRNA (Figure 8 B, C). Also, the expression of, SNAI1 and SNAI2 in Ca Ski RT_A was reduced compared Ca Ski GFP control (Suppl., Figure S6), altogether indicating a shift towards the epithelial phenotype. Expression of TWIST1 did not change compared to GFP control (Suppl., Figure S6).
3.7. Effects of HIV-1 RT Expression on Phenotypical Properties of Ca Ski Cells—Motility and Clonogenic Activity
Further, we assessed if expression of RT_A had any effect on the phenotypic characteristics of Ca Ski cells, that shape the metastatic activity of tumor cells, such as cell cycle progression, doubling time, and cell migration rate. Collective cell migration is the coordinated movement of a group of cells that maintain intracellular connections and is crucial to various biological processes including embryo development, the immune response, and cancer metastasis [72]. Another important criterion for the malignancy of tumor cells is a decrease in the need for growth factors and cytokines secreted by microenvironmental cells, which can be revealed by the analysis of the clonogenic activity.
3.7.1. Expression of RT_A Causes an Increase in Cell Doubling Time, but Does Not Affect Cell Cycle Progression
Five out of six Ca Ski RT_A subclones as well as Ca Ski GFP had increased doubling time compared to the control Ca Ski cells over a month of consecutive observations (Kruskal - Wallis, p˂0.05, n = 12) (Table 2, Suppl., Figure S7), insignificant for B8D5 subclone (Table 1). While Ca Ski GFP cells demonstrated a decrease in the percentage of cells in the G1/G0 phase of cell cycle as compared to the parental cells (Table 2, Suppl., Figure S8), RT_A subclones showed no difference from the parental cells in distribution of cells in all phases of cell cycle (Table 2, Suppl., Figure S8). Thus, the expression of RT_A had no effect on cell cycle progression but might have served to overcome the G1/G0 stop caused by the lentiviral transduction.
3.7.2. Overexpression of HIV-1 RT Reverses Inhibitory Effect of Lentiviral Transduction on Cell Mobility
The effect of RT_A expression at on Ca Ski cell motility was assessed using a wound healing assay.
Lentiviral transduction decreased cell motility for Ca Ski GFP subclone (Suppl., Figure S9). Ca Ski 1 RT_A subclones had a discrepant effect on cell motility, B8D5 and B8D2 subclones was characterized by an increased cell motility compared to the GFP control, whereas B8B5 did not change motility (for description of clones, see Table 1). Interestingly, Ca Ski RT_A subclones with six inserts (6 RT_A) demonstrated partial restoration of cell motility, at least for all three subclones the motility was significantly higher than for the GFP subclone (Table 2, Suppl., Figure S9). Thus, in Ca Ski the expression of RT_A partially compensated a decrease in cell motility caused by the lentiviral transduction and/or overexpression of the foreign protein.
The phenotypic parameters, cell doubling time, distribution of cells by the phases of the cell cycle, and migration rate, were not correlated to each other, or to the levels of RT_A mRNA or protein production by the subclones (all p>0.1).

Table 2.
Phenotypic characteristics of Ca Ski expressing consensus RT of HIV-1 clade A FSU_A strain (RT_A) and fluorescent marker GFP: doubling time, % of cells in the phases of the cell cycle, and migration rate for assessed using wound healing assay (WHA).
Table 2.
Phenotypic characteristics of Ca Ski expressing consensus RT of HIV-1 clade A FSU_A strain (RT_A) and fluorescent marker GFP: doubling time, % of cells in the phases of the cell cycle, and migration rate for assessed using wound healing assay (WHA).
| Cells | Doubling time, h (n=12) * | % of cells in cell cycle phase (n=3) ** | Migration rate in WHA, μm/h (n=6) *** | ||
| G1/G0, % | S, % | G2/M, % | |||
| Ca Ski | 25,90±3,26 | 50±3,90 | 24,43±4,90 | 18,47±1,25 | 23,84±1,56 |
| GFP | 35,95±7,78 | 38,57±2,63 | 27,67±3,62 | 22,3±3,60 | 11,61±2,06 |
| B8B5 | 32,59±6,05 | 45,57±1,01 | 28,17±5,92 | 16,67±4,88 | 13,57±1,42 |
| B8D5 | 31,05±7,80 | 50,23±0,94 | 24,1±2,68 | 18,17±1,97 | 15,87±0,91 |
| B8D2 | 31,04±7,57 | 47,8±4,07 | 23,87±6,53 | 19,53±4,96 | 31,12±3,35 |
| H6G11 | 35,04±7,83 | 41,03±3,27 | 27,53±2,87 | 23,03±1,79 | 20,71±2,61 |
| H6D7 | 30,47±6,17 | 43,77±0,25 | 23,77±1,15 | 24,27±1,50 | 27,55±1,04 |
| H6F8 | 38,60±9,45 | 44,37±3,73 | 26,33±1,59 | 21,93±1,80 | 15,84±1,72 |
*, **, *** - Number of assay repeats.
A multiple regression analysis was then performed to dissect if the rate of cell migration in wound healing assay depends on any of the above studied parameters (expression of RT_A as a protein, expression of E6*I mRNA, metabolic characteristics, and expression of EMT factors). Six parameters were found to predict the motility, namely, the expression of RT_A protein, expression of E-CADHERIN and TWIST1 mRNAs and ECAR at high glucose, which stimulated, and expression of E6*I and of N-CADHERIN, which inhibited cell migration (Table 3). Some of the effects, such as stimulating effect of E-cadherin, and suppressive effect of N-cadherin, contradicted the accepted role of these proteins in cell motility.
3.7.3. Loss of Clonogenic Activity after Retroviral Transduction Is Compensated in Ca Ski Cells Expressing High Levels of HIV-1 RT
Next, we performed the clonogenic analysis of Ca Ski subclones, as the formation of clones characterize the ability of cancer cells to initiate the formation of tumors. By day 21 following the lentiviral transduction, control subclone Ca Ski GFP demonstrated a significant decrease in both colony counts (Figure 9A) and mean size of the colonies (Figure 9B) as compared to the parental Ca Ski cells. Ca Ski 1 RT_A also demonstrated a decrease in the number of colonies compared to the GFP control, however, Ca Ski 6 RT_A were indistinguishable from the parental Ca Ski cells (Figure 9A). Also, Ca Ski 1 RT_A subclones had decreased colony size as the GFP control, while Ca Ski 6 RT_A subclones were undistinguishable from the parental cells (Figure 9B). Thus, while lentiviral transduction caused suppressed the clonogenic activity, significantly decreasing the number and size of the colonies, (over)expression of RT_A reversed these effects, restoring both colony counts and mean size of the colonies to the levels characteristic to the parental Ca Ski cells.
3.8. The Relationship between the Levels of mRNA/Protein Production RT_A and E6*I and Various Specific Cell Characteristics
Finally, we asked if in vitro properties of Ca Ski and their subclones, their metabolic and phenotypic characteristics, are correlated to the expression of RT_A and/or of E6*I isoform. Results of the series of linear correlation tests of these parameters with the expression levels of RT_A and E6*I are presented in Table 4.
RT_A was found to have an independent from HPV16 E6 effect on the metabolism of Ca Ski cells by increasing the glycolysis (ECAR at low and high glucose). The latter was specifically sensitive to RT_A at the low levels of glucose. Expression of E6*I had a positive effect, but it did not reach the level of significance (Table 4). At the same time, RT_A inhibited mitochondrial respiration (basal, maximal respiration at low and high FCCP, ATP production). Interestingly, on contrary to glycolysis, for these parameters, there was a coherent negative association with E6*I expression, either highly significant (p<0.01), or at a low significance level (R in the range of -0,498 to -0,585; p<0.05), indicating that inhibition of mitochondrial respiration was determined by both the presence and the levels of RT_A and the levels of E6*I. Since significance (by R value) was higher for RT_A, one can assume that RT_A may have played a leading role by inducing E6*I expression. Further, we found that the production of ROS and expression of E-CADHERIN correlated to the levels of E6*I mRNA, but not on the levels of expression of RT_A (Table 4). Namely, expression of E6*I mRNA increased the expression of E-CADHERIN mRNA but affected the production of ROS (Table 4).
We further analyzed if expression of RT_A as mRNA and as a protein, and of E6*I mRNA had any effect on the phenotypic characteristics of Ca Ski subclones and found a correlation of the colony forming capacity of the cells with the levels of E6*I mRNA, but not on the levels of expression of RT_A (Table 4). Specifically, expression of E6*I mRNA interfered with colony formation, reducing both the number of colonies and their size (Table 4). There was no quantitative dependence of these processes on the amount of RT stimulating E6*I expression (despite the RT-mediated increase in the production of E6*I isoform).
3.9. Overexpression of RT Rescues Tumorigenic Activity of Ca SKI SUBCLONES AFFECTED by the Retroviral Transduction
The effect of HIV-1 RT expression on the tumorigenic activity of Ca Ski was assessed by their well characterized ability to form tumors in the immunosuppressed mice [73]. The experiment was done on two Ca Ski subclones, one carrying one, and the other – six copies of RT_A DNA (B8B5 and H6G11, respectively; Table 1). The parental Ca Ski and its GFP derivative were used as controls. Cells were implanted subcutaneously at a dose of 105 and 106 in one injection site [74].
Tumor growth in the Ca Ski control group implanted at 106 cells was observed starting from day 11, in Ca Ski GFP group, from day 15, and for 6 RT_A subclone H6G11 from day 22 post the injection (Figure 10A). Subclone B8B5 produced no tumors by the experimental endpoint, which was consistent with its reduced clonogenic activity in vitro (Suppl., Figure S9). Interestingly, by the experimental endpoint on day 29, a significant difference in tumor growth rate was noted only between the parental Ca Ski and Ca Ski GFP control, as the latter was growing much slower (p=0.0195; Figure 10D). In terms of the ability to form solid tumors in immunosuppressed mice, the H6G11 subclone did not differ from the parental Ca Ski cells (p>0.1; Figure 10D), although not all injections resulted in the palpable tumors. The late on-start of tumor growth and late entry into the exponential growth phase did not allow us to evaluate the full effect of the restoration for RT-expressing subclone of the tumorigenicity of Ca Ski cells affected by the lentivirus transduction, since according to the ethical approval experiment had to be terminated when the first tumor formed in mice in the experiment reaches 1 mm3 in size (was observed in the control Ca Ski group). Implantation of 105 cells did not give tumors by day 29 post injections for any of the subclones.
By experiment end point mice were euthanized, tumors were excised and weighed. Significant difference was observed between the parental Ca Ski and Ca Ski GFP controls, the latter were much smaller, whereas no difference was observed in the volumes of tumors formed by Ca Ski and Ca Ski 6 RT_A subclone (Figure 10E), which was consistent with the growth kinetics assessment by day 29 (Figure 10D, E).
In terms of the ability to form solid tumors in immunosuppressed mice, the 6 RT_A Ca Ski subclone did not differ from the parental cells (p>0.1), while expression of GFP led to significant reduction of tumorigenicity. This indicated that, although, lentiviral transduction and further synthesis of by tumor cells of a non-oncogenic exogenous protein such as GFP decreases their tumorigenicity, overexpression of HIV reverse transcriptase has restored the compromised tumorigenic activity to the levels observed in the parental Ca Ski cells.
4. Discussion
We have an overall goal to uncover the mechanisms by which HIV-1 promotes malignant transformation of the epithelial cells, on its own and/or in interaction with other oncoviruses, or their proteins. Candidate number one causing the bulk of non-AIDS related cancer cases in HIV-1 infection is HPV16. In this study, we concentrated on the interplay between HPV16 and HIV-1 antigens. Major globally recognized oncoproteins of HPV16 are early proteins E6 and E7, the main actors in HR HPV - associated malignant transformation. E6 acts by targeting tumor suppressor protein p53 for proteasomal degradation, and E7, by inactivation of another tumor suppressor protein, pRb [75]. Besides, both proteins are involved in wide panel of regulatory activities imposed on the intracellular machinery of infected and/or E6 and E7 exposed cells that gradually lead to transformation of the affected epithelial cells.
HIV-1 antigens with the capacity to induce ROS, be exported out of the cells (into extracellular space) and taken by other innocent bystander cells and induce transformation or enhance tumorigenic and metastatic activities of tumor cells, play active part in this process [1]. We hypothesized that HIV-1 reverse transcriptase (RT) is one of the players in this team.
Reverse transcriptase is a multifunctional enzyme that converts the viral RNA genome into dsDNA in the cytoplasm, shortly after virus entry into the cell. This enzyme displays a DNA polymerase activity that can copy either DNA or RNA templates, and a ribonuclease H (RNase H) activity that cleaves the RNA strand of RNA-DNA heteroduplexes in a partially processive 3’ to 5’ endonucleasic mode and is essential for HIV replication [76]. It has been shown that RT was detected in the exosomes in the uterine of PLWH [77] and capable to secrete into cell culture fluids of cells transiently expressing RT [78]. Early studies demonstrated that expression of different RT variants, including drug resistant variants, and variants retargeted for lysosomal processing and secretion induced ROS production [78,79]. We have shown that constitutive expression of HIV-1 RT in murine mammary gland adenocarcinoma 4T1 cells leads to ROS – dependent activation of the transcription factor Twist, involved in EMT [4], which regulates the expression of transcription factors involved in cellular redox balance Nrf2, which stimulating the expression of Phase II detoxifying enzymes HO-1 and Nqo-1 [4].
Mitochondria are considered a major contributor to intracellular ROS production in most types of cells. Expression of HIV-1 RT led to an increase in mitochondrial respiration and the level of ATP synthesis in the cell, which was associated with restoration of the mitochondrial network. In fact, we have shown that HIV-1 RT induces an increased tolerance to stress in breast adenocarcinoma cells due to change cancer cell metabolism towards oxidative phosphorylation (OXPHOS) [70].
Induction of E6 (FL) expression – property of a broad panel of RT variants, property of extracellularly added protein, indicating that it enters into the cell. Altogether, these properties provide for RT capacity to enhance growth and metastatic activity of adenocarcinoma cells in mice.
Although this proves active role of HIV-1 RT in modifying the in vitro and in vivo properties of cancer cells of epithelioid origins, it does not prove that it can make an input into the properties of human epithelial cells, specifically epithelioid cancer cells infected with HPV16. The latter represent an adequate model to check of HIV-1 RT modulates their properties, and if so, does this modulation involve HPV16 antigens, namely, if RT works alone/on its own, alongside with, or in collaboration with HPV16 oncoproteins.
For this, we have selected a model cell line – Ca Ski cells (HPV-16 infected human cervical squamous carcinoma, [57]). Ca Ski cells were transduced with lentivirus encoding consensus RT of HIV-1 clade A FSU_A strain (RT_A) [4]. We generated subclones with one and six genomic inserts of RT_A coding sequence expressing from 20 to 55 fg protein per cell.
One HIV-1 virion contains approximately 50 molecules of RT [80] as a complex of a polypeptides of 66 kDa and its proteolytic digestion product 51 kDa that lacks the carboxy-terminal sequences present in the 66-kDa form [81]. For HIV-1, a single productively infected CD4+ T cells should carries a viral load of approximately 500 virions [82]. Another estimation gives an average of 3900 (range 3162–5011) viral RNA copies per infected cell, with two RNA molecules per virion this accounts for approximately 2000 virions per cell [83], ie four times more. After recalculation from kDa into fg, the total amount of RT in CD4+ T cells could be estimated as 2,75 fg of p66 and 2,12 fg of p51 polypeptide, on total 5 fg per infected cell based on the first, and 20 fg per cell based on the second estimation. Thus, Ca Ski subclones produced an adequate amount of HIV-1 RT corresponding to the levels of protein in natural infection (although in the epithelial cells, not CD4+ T lymphocytes).
Lentiviral infection has been reported to change properties of the transduced cells [58,59]. There are indications of the effects of lentiviral transduction as such, such as altered expression of JUN [84] as well as restricted in vivo growth of lentivirally transduced tumor cell lines - due to activation of type I IFNs [85]. Furthermore, (over) expression of foreign proteins overloads protein synthesis machinery and can grossly affect properties of the cells. An effective strategy for mitigating nonspecific effects of viral transduction on cell line properties is to generate control cells expressing unrelated proteins. It enables to account for both effects (lentiviral transduction and overexpression of exogenic protein/metabolic burden). For this end, we have created a control Ca Ski cell line transduced by lentivirus encoding GFP, with multiplicity of infection similar to the highest one used to generate Ca Ski RT_A subclones. All in vitro and in vivo properties of Ca Ski RT_A subclones were further compared to the Ca Ski GFP control.
Firstly, we assessed the effect of HIV-1 RT on expression of HPV16 oncoproteins, checking the level of their transcription in Ca Ski cells. A bicistronic pre-mRNA encodes HPV-16 E6 and E7 oncoproteins and alternatively spliced transcripts encoding E6*I, E6*II [86]. RT had no effect on the expression of mRNA of E7, full-length E6, or E6*II isoform, but increased the expression of E6*I isoform. The effect was only partly due to the lentiviral transduction, as it was also observed for the Ca Ski GFP subclone, but at a significantly lower level. Interestingly, the phenomenon was observed for all RT-expressing subclones, with one as well as with six RT_A DNA inserts and was not related to the level of RT_A expression (as there was no difference in the levels of E6*I in subclones expressing high and low level of RT_A). The latter indicated that RT_A was not the direct mediator of E6*I expression, but rather an inducer of the expression in concentration independent manner.
We next investigated the effects of HIV-1 RT on cell metabolism. The alteration of glucose metabolism is the basic property of cancer cells registered as the increases glucose consumption and glycolytic rates, also in the presence of oxygen, so called “aerobic glycolysis” or the Warburg effect. Oncoproteins E6 and E7 favor the Warburg effect through the increase in the activity of glycolytic enzymes, as well as the inhibition of the Krebs cycle and the respiratory chain [87]. With this, HPV16 infected Ca Ski cells employ the metabolic pathway of aerobic glycolysis. Furthermore, artificial overexpression of HPV16 E6/E7 in Ca Ski cells significantly upregulates the glycolysis pathway [88]. One can expect that any factor leading to an increase of E6 expression would tint these cells further into the glycolytic pathway. Indeed, the constitutive expression of RT_A in Ca Ski cells inducing an increase in the levels of E6*I isoform led to an increase in the intensity of glycolysis under aerobic conditions. Ca Ski subclones with six RT_A gene inserts demonstrated an increase in the basal and maximal glycolysis, and an increased maximum glycolytic capacity, while no changes in glycolytic pathway were observed for Ca Ski GFP subclone. At the same time, expression of RT_A suppressed mitochondrial respiration as compared to the GFP control. An increase (no reduction in our case) in proton leak and a decrease in basal or maximal respiration are indicators of mitochondrial dysfunction [89]. Linear correlation analysis demonstrated a direct correlation of ECAR at low and high glucose, and an inverse correlation of the parameters of mitochondrial respiration with the levels on RT_A mRNA and of RT_A as a protein, as well as with the levels of E6*I mRNA, although the significance for the latter was lower (had lower R values). This pointed at unidirectional effect(s) of HIV-1 RT and of E6*I, with RT as a primary factor modifying cell metabolism by altering the amounts of E6*I isoform. Thus, expressed in Ca Ski cells HIV-1 RT changed their metabolism by enhancing glycolysis and inhibiting mitochondrial respiration, suppressive effect possibly mediated through the modulation of the levels of E6*I isoform. It was also shown that HIV-1 tat protein revealed decreased maximal respiration and reduced spare respiratory capacity in lund human mesencephalic (LUHMES) cells [90]. On contrary, in other HPV associated cancers, namely head and neck squamous cell carcinoma (HNSCC) cells positive for human papillomavirus were found to favor mitochondrial metabolism mediated by HPV16 and HPV18 E6 over the glucose metabolism [91]. Difference in the effects observed for Ca Ski RT_A subclones and HNSCC could have been due to HIV-1 RT interference.
One has to also consider an E6-independent effect of HIV-1 RT. We have already shown it to be able to alter metabolism of cancer cells. Interestingly, in adenocarcinoma cells characterized by the high levels of ROS, HIV-1 RT enhanced mitochondrial respiration (OXPHOS), not the glycolysis [70]. The direction of the metabolic changes is tightly linked to the redox status of the cell. ROS are involved in glycolysis (dys)regulation. ROS inhibit multiple glycolytic enzymes, including glyceraldehyde 3-phosphate dehydrogenase, pyruvate kinase M2, and phosphofructokinase-1 [92]. Vice versa, shift to glycolysis inhibits production of ROS. Consistently, glycolytic inhibition promotes flux into the oxidative arm (of the pentose phosphate pathway to generate NADPH) [93]. The enhancement of OXPHOS on the background of high ROS reflected this process with HIV-1 RT adapting the adenocarcinoma cells to the high-ROS environment by shifting their metabolism towards mitochondrial respiration.
To see if this could be the case for Ca Ski subclones, we assessed their overall levels of ROS using a sensor dyes, DCFH2-DA sensing H2O2, hydroxyl radical and superoxide O2•−, and DHE sensing only superoxide O2•−. The majority of Ca Ski subclones demonstrated decreased levels of total ROS, while the levels of superoxide production remained unchanged compared to the parental cells. We could not delineate a specific effect of RT_A on the total ROS due to the overlap of the signals of DCFH2-DA and GFP. To compensate for this, we assessed the levels of transcription factors induced in response to the oxidative stress Nrf2, enzymes of glutathione biosynthesis GCLС, and of the enzyme of Phase II of oxidative stress response NQO1, which follow the changes in the levels of ROS [94]. The levels of mRNA of NRF2, GCLC and NQO1 in RT-expressing Ca Ski subclones did not differ from that in Ca Ski GFP indicating that expression of HIV-1 RT in human epithelioid cells infected with HPV16 is not associated with the induction of oxidative stress. Thus, the shift towards glycolysis in Ca Ski expressing HIV-1 RT was not associated with the change of the redox balance of the cells.
Metabolic changes often reflect changes in the mitochondrial dynamics and functionality caused by the changes in the cell cytoskeleton [95,96]. We addressed this option by assessing the levels of expression in Ca Ski subclones of A-tubulin as the parameter reflecting the state of microtubules (MT) and of Y-tubulin as the parameter reflecting the condition of the MT-supported mitochondrial network [97]. Lentiviral transduction and (over)expression of RT_A and of GFP had no effect on the expression of either A-TUBULIN, or Y-TUBULIN genes which could have affected the cytoskeleton of Ca Ski subclones and influence their choice of metabolic pathway. Furthermore, earlier we have shown that overexpression of HIV-1 RT by murine adenocarcinoma 4T1luc2 actually restores the mitochondrial networks disrupted in the original tumor cells [70]. Altogether, this spoke against metabolic changed being caused by HIV-1 RT (or cumulative HIV-1 RT & E6*I) induced disruption of cytoskeleton of Ca Ski cells.
Which properties of HIV-1 RT determine its capacity to regulate cell metabolism, apart from inducing the expression of E6*I isoform, remains unclear. One can hypothesize that these are certain cell factors that interact with HIV-1 RT and through this, fail to carry their functions in the cells causing a metabolic shift. Warren et al. described a number of cellular factors which can directly or indirectly bind to RT [98]. Specific interest represents the kinase anchor protein 121 (AKAP121), also referred to as AKAP1 or AKAP149 (human homologue). AKAP121 is an essential regulator of the mitochondrial respiration. Decreased AKAP1 expression was detected in the glycolytic metabolism dependent migrating cells found in invasive populations of breast cancer cells [99]. Molecular, cellular, and in silico analyses of breast cancer cell lines confirmed that AKAP1 depletion is associated with the impaired mitochondrial function and dynamics, concomitant with the increased glycolytic potential and invasiveness [100]. One can hypothesize that interaction of HIV-1 RT with AKAP1 (AKAP121) may “deplete” functional protein from the cells, causing a shift towards glycolytic pathway.
Glucose metabolism is tightly linked to the epithelial-to-mesenchymal transition (EMT) [101]. Cancer cells employ EMT to acquire the ability to migrate, resist therapeutic agents and escape immunity [102]. Metabolic dysregulation is known to trigger EMT [103,104,105,106,107], which in turn modulates cell migration [108]. The process of EMT is characterized by the profiles of expression of a number of cellular factors, including cadherins E and N, and transcriptional factors Twist1, Snai1 (Snail) and Snai2 (Slug). Data show increase of expression level of N-CADHERIN, TWIST1, SNAI1, SNAI2 and VIMENTIN mRNA in lentiviral transduction control cell expressing GFP. In early study, transfection of a murine colon adenocarcinoma CT26 cells with empty lentivirus vector (GFP-vector group) did no lead to changes in associated with EMT factors [109]. It is likely that transfection of cells does not have such a strong effect, but the genomic damage caused by transduction leads to changes in the expression of epithelial-mesenchymal factors. Despite, Ca Ski RT_A subclones demonstrated an increase in the expression of E-CADHERIN manifesting a loss of the typical heterogeneous morphology of epithelial cells. Besides, Ca Ski RT_A exhibited decreased levels of expression of VIMENTIN, a type III intermediate filament expressed in the mesenchymal cells and upregulated during cancer metastasis [102], as well as of the transcription factors Snai1 (Snail) and Snai2 (Slug) (but not for all subclones), master regulatory factors for organogenesis and wound healing, tightly involved in EMT of cancer cells [110,111,112]. These data indicated that Ca Ski RT_A subclones acquired features of the hybrid epithelial/mesenchymal (E/M) phenotype where cells simultaneously demonstrate the epithelial traits of cell-to-cell adhesion and mesenchymal characteristics of migration and invasion [113]. Hybrid E/M cells express both epithelial markers (E-cadherin, cytokeratins, claudins, occludins) and mesenchymal markers (N-cadherin, vimentin, fibronectins), and they may display phenotypic characteristics of both cell types [114]. Hybrid E/M has been associated with an increased malignancy of tumor cells [115], indicating the potential consequences of the presence of HIV-1 RT for Ca Ski cells.
We further assessed the effect of the biochemical & molecular characteristics of Ca Ski subclones described above on their phenotypic features, such as doubling time, cell cycle progression, cell migration and clonogenic activity. Lentiviral transduction and (over)expression of RT_A or GFP control increase cell doubling time and significantly decreased cell motility in wound healing assay (WHA). The expression of RT_A had no effect on cell cycle progression but might have served to overcome the G1/G0 decreased caused by the lentiviral transduction (for GFP control).
As for the behavior of Ca Ski RT_A and GFP subclones in the clonogenic assay, the lentiviral transduction was found to suppress the clonogenic activity, significantly decreasing the number and size of the colonies. Notably, as in the case of cell migration, (over)expression of RT_A (six RT_A inserts) reversed these effects, restoring the colony counts and mean size of the colonies to the levels characteristic to the parental Ca Ski cells. Both observations on cell motility in WHA and results of the clonogenic analysis indicated that overexpression of HIV-1 RT was able to compensate for the adverse effect(s) of lentiviral transduction and/or foreign protein overexpression in Ca Ski cells.
Multiple regression analysis revealed that cell motility in WHA could be predicted using the levels of expression of RT_A and of RT_A dependent parameters, including the expression levels of E-cadherin and Twist1 mRNA and ECAR at high glucose, E6*I and of N-cadherin. The first three were positive, and the last two, negative predictors of cell migration. While Twist1 as well as the shift to glycolysis are known to stimulate cell motility, the effects of E-cadherin (E-cad) and N-cadherin (N-cad) were unexpected, contradicting the accepted role of these proteins. E-cadherin is the major homophilic cell-cell adhesion molecule that inhibits motility of individual cells on matrix [116], while N-cad is a key regulator of collective cell migration [117,118]. Although E-cad has been implicated as suppressor of tumor metastasis, some data indicate that it can also serve as the promoter of metastasis growth and spread [119]. Specifically, expression of E-cad in the epithelial cancer cells increases their motility through the mechanical feedback as E-cad promotes direction sensing during collective cell migration [120]. N-cadherin, on contrary, promotes cell migration by cell-to-cell adhesion, but the opposite to E-cad, N-cad function is cell-context-dependent, as it can mediate strong cell to cell adhesion in muscles, such as the heart, and in bones, but induce changes in cell behavior in favor of a migratory phenotype in the epithelial cells in the context of EMT [121,122,123]. Thus, functions of both largely depend on the cellular context. Expression of HIV-1 RT in Ca Ski cells led to an increase of the expression of E-cad, possibly mimicking the stimulating effect observed in E-cad overexpressing epithelial cancer cells [120]. Lentiviral transduction (but not the expression of RT_A) caused also a simultaneous increase in the expression of N-cadherin. The latter would increase the levels of N-cadherin-expressing cellular substrates, their adhesion to the migrating cells could decrease cell migration through fibroblast growth factor receptor-n-cadherin crosstalk [124]. Interestingly, HPV16 E6 had the same effect, E6 expression by immortalized human keratinocytes increased the proportion of cells attached to the cell culture dishes [125]. Capacity of E6 isoform(s) to promote cell adhesion was listed among the known “contravercial” anti-tumor properties of E6 [63]. Our data indicated that N-cad and HPV16 E6 can cooperate in enhancing cell adhesion, resulting in the decreased cell motility in WHA.
All above would have an effect on the aggressiveness of these cells in the body. To see the biological outcomes, we characterized the ability of Ca Ski RT_A subclones to form tumors in the immunosuppressed mice, the property well defined for the parental Ca Ski cells [73].Lentiviral transduction with expression of GFP grossly compromised the ability of Ca Ski cells to form tumors in nude mice, tumorigenicity of highly expressing Ca Ski RT_A was partially restored, coming to the level observed for the parental Ca Ski cells.
Altogether, the analysis of motility/migration, clonogenic and tumorigenic activities of HPV16 infected epithelial cells in the presence of HIV-1 RT revealed that: (i) these process depend on both HIV-1 RT and HPV16 E6; (ii) although HIV-1 RT increases the expression of E6*I isoform, the effects of HIV-1 RT and of HPV16 E6 are not aligned and often not cooperative, specifically in clonogenic activity test with HIV-1 RT partially mitigating the effect(s) of E6; (iii) direct or indirect interaction(s) of HIV-1 RT and HPV16 E6 dysregulated the cells switching on the “non-classical” traits and pathways.
5. Conclusions
To summarize, HIV-1 RT expressed in epithelioid cells infected with HPV16 conferred the cells the plasticity required to overcome negative effects of lentiviral transduction, retain basic properties including the capacity to form tumors in nude mice, and also acquire features, such as the increased expression of E6*I isoform, or shift to glycolysis, associated with an increased tumorigenicity.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Supplementary Table S1 – Table of primers. Primers used for conformation of expression of RT HIV-1 in Ca Ski derivatives and for relative quantification of mRNA expression of HPV16 E7 and E6 oncoproteins and its constitutive isoforms, transcription factors associated with oxidative stress, or of the enzymes of the Phase II protection from oxidative stress, factors associated with changes in the cell cytoskeleton and epithelial-mesenchymal transition markers. Supplementary Figure S1 - Confirmation of GFP expression in Ca Ski subclone by flow cytometry. Histogram of events count recorded in FITC-A channel confirms GFP expression in Ca Ski GFP G9 subclone. Supplementary Figure S2 - Relative mRNA expression levels of E6FL, E6*II isoforms and E7 HPV16 in Ca Ski subclones expressing consensus HIV-1 FSU_A reverse transcriptase. Graphs demonstrate no specific effect of RT_A production on mRNA expression level of HPV16 full-length E6, E6*II transcript and E7. Changes noted only for single subclones. Expression level was normalized to GAPDH expression. Supplementary Figure S3. The efficiency of glycolysis in Ca Ski subclones expressing HIV-1 reverse transcriptase (RT_A). Graphs demonstrate determination of extracellular acidification rate (ECAR) reflecting changes in glycolysis in individual Ca Ski subclones using a Seahorse analyzer according to the Glycostress test method. Glycostress test results were normalized to the total protein levels. Supplementary Figure S4. The efficiency of mitochondrial respiration in Ca Ski subclones expressing HIV-1 reverse transcriptase (RT_A). Graphs demonstrate measuring the oxygen consumption rate (OCR) reflecting changes in the substrate utilization in mitochondrial respiration in individual Ca Ski subclones using a Seahorse analyzer according to the Mitostress test method. Mitostress test results were normalized to the total protein levels. Supplementary Figure S5. Relative mRNA expression levels of A-TUBULIN and Y-TUBULIN in the derivatives of Ca Ski cells expressing variants of consensus HIV-1 FSU_A reverse transcriptase. Graphs showing no effect of RT_A production on cell cytoskeleton. There are no RT-associating changes in A-tubulin defining changes in structure of microtubule (MT) and Y-tubulin modulating the MT-supported mitochondrial network. Expression level was normalized to GUSB expression. Supplementary Figure S6. Relative mRNA expression levels of factors, associated with epithelial-mesenchymal transition in individual derivatives of Ca Ski cells expressing variants of consensus HIV-1 FSU_A reverse transcriptase. Graphs show increase of expression level of N-CADHERIN, TWIST1, SNAI1, SNAI2 and VIMENTIN mRNA in lentiviral transduction control cell expressing GFP. All RT_A expressing Ca Ski subclones demonstrate no changes in TWIST1 expression and decrease in level of VIMENTIN expression. For N-CADHERIN, SNAI1 and SNAI2 expression level observed decrease expression only for individual RT_A expressing subclones and this effect was not associated with RT. Expression level was normalized to GAPDH expression. Supplementary Figure S7. Graphs showing increase in doubling time in the derivatives of Ca Ski cells expressing variants of consensus HIV-1 FSU_A reverse transcriptase in contrast to the parental cells over month of consecutive observations. Scoring of the proliferation rate was performed by detach of cells using Trypsin/EDTA, transferred to a Gorjaev’s count chamber and counted (× 5) in 20 fields of the counting grid. Supplementary Figure S8. Cell cycle distribution in G1/G0 phase, S phase, and G2/M phase for subclones of Ca Ski expressing RT. The representation of Ca Ski subclones in different stages of the cell cycle was assessed by flow cytometry with using FlowJo software. Graphs showing decrease in G1/G0 phase for GFP control cells. There are no changes in distribution of RT-expressing subclones in all phases of cell cycle might have served to overcome the G1/G0 stop caused by the lentiviral transduction. Supplementary Figure S9. Average migration rate of Ca Ski-derived subclones assessment by wound healing assay. Graph showing decrease cell motility in lentiviral transduction control cells expressing GFP and restoration effect for RT-expressing subclones (except B8B5). The surface of the well was scratched with a pipette tip and continuously imaged using CELENA® X High Content Imaging System for 24 hours. The images were processed using the ImageJ/Fiji® module, quantifying the entire area of the wound, the average width of the wound and the standard deviation of the width. Supplementary Figure S10. Evaluation of clonogenic activity at days 21 in individual Ca Ski derivatives subclones. Graphs demonstrate decrease in count and mean size of colonies in GFP control cells and Ca Ski subclones with one RT_A inserts, which have restorative effect for Ca Ski subclones with six RT_A inserts. Cells were plated in an amount of 200 cells and incubated under the standard conditions during 21-days followed by fixation. The mean size and number of colonies were quantified using the ImageJ software.
Author Contributions
AZ: EB experiment – conceived the study, acquired the data, analyzed the collected material, wrote the manuscript; DA, GF, JJ, AI - acquired the data, analyzed the collected material; IG – acquired the data, discussed the results, secured funding; SC – discussed the results, reviewed the manuscript; JP – generated the concept, planned the experiment, discussed the results, secured funding, discussed the results, reviewed the manuscript; MI – generated the concept, planned the experiment, secured funding, discussed the results, wrote and reviewed the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
Twinning on DNA-Based Cancer Vaccines (VACTRAIN) project No.692293; Initiation Project INNOVIMMUNE, Swedish Institute 2017-2018. RFBR 17-54-30002 and R01 CA217715/CA/NCI NIH HHS/United States to JP and MI. M.I. and JJ were supported by the Latvian Science Fund, project LZP 2021/1-0484. Metabolic analysis was supported by Russian Ministry of Science and Higher Education of the Russian Federation (agreement 075-15-2019-1660).
Conflicts of Interest
The authors declare no conflict of interests.
References
- Isaguliants, M.; Bayurova, E.; Avdoshina, D.; Kondrashova, A.; Chiodi, F.; Palefsky, J. Oncogenic Effects of HIV-1 Proteins, Mechanisms Behind. Cancers 2021, 13, 305. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Ivanova, O.N.; Kochetkov, S.N.; Starodubova, E.S.; Bartosch, B.; Isaguliants, M.G. Oxidative Stress during HIV Infection: Mechanisms and Consequences. Oxidative Medicine and Cellular Longevity 2016, 2016, e8910396. [Google Scholar] [CrossRef]
- El-Amine, R.; Germini, D.; Zakharova, V.V.; Tsfasman, T.; Sheval, E.V.; Louzada, R.A.N.; Dupuy, C.; Bilhou-Nabera, C.; Hamade, A.; Najjar, F.; et al. HIV-1 Tat Protein Induces DNA Damage in Human Peripheral Blood B-Lymphocytes via Mitochondrial ROS Production. Redox Biol 2017, 15, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Bayurova, E.; Jansons, J.; Skrastina, D.; Smirnova, O.; Mezale, D.; Kostyusheva, A.; Kostyushev, D.; Petkov, S.; Podschwadt, P.; Valuev-Elliston, V.; et al. HIV-1 Reverse Transcriptase Promotes Tumor Growth and Metastasis Formation via ROS-Dependent Upregulation of Twist. Oxidative Medicine and Cellular Longevity 2019, 2019, 1–28. [Google Scholar] [CrossRef]
- Riedel, D.J.; Tang, L.S.; Rositch, A.F. The Role of Viral Co-Infection in HIV-Associated Non-AIDS Related Cancers. Curr HIV/AIDS Rep 2015, 12, 362–372. [Google Scholar] [CrossRef]
- Frisch, M. Association of Cancer With AIDS-Related Immunosuppression in Adults. JAMA 2001, 285, 1736. [Google Scholar] [CrossRef]
- Clifford, G.M.; Polesel, J.; Rickenbach, M.; on behalf of the Swiss HIV Cohort Study; Dal Maso, L.; Keiser, O.; Kofler, A.; Rapiti, E.; Levi, F.; Jundt, G.; et al. Cancer Risk in the Swiss HIV Cohort Study: Associations With Immunodeficiency, Smoking, and Highly Active Antiretroviral Therapy. JNCI Journal of the National Cancer Institute 2005, 97, 425–432. [CrossRef]
- Palefsky, J.M.; Holly, E.A. Chapter 6: Immunosuppression and Co-Infection with HIV. JNCI Monographs 2003, 2003, 41–46. [Google Scholar] [CrossRef] [PubMed]
- International Collaboration on HIV and Cancer Highly Active Antiretroviral Therapy and Incidence of Cancer in Human Immunodeficiency Virus-Infected Adults. Journal of the National Cancer Institute 2000, 92, 1823–1830. [CrossRef] [PubMed]
- Frisch, M. Human Papillomavirus-Associated Cancers in Patients With Human Immunodeficiency Virus Infection and Acquired Immunodeficiency Syndrome. Journal of the National Cancer Institute 2000, 92, 1500–1510. [Google Scholar] [CrossRef]
- Palefsky, J.M. Human Papillomavirus Infection and Anogenital Neoplasia in Human Immunodeficiency Virus-Positive Men and Women. JNCI Monographs 1998, 1998, 15–20. [Google Scholar] [CrossRef]
- Gillison, M.L.; Shah, K.V. Chapter 9: Role of Mucosal Human Papillomavirus in Nongenital Cancers. J Natl Cancer Inst Monogr 2003, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Parkin, D.M.; Bray, F. Chapter 2: The Burden of HPV-Related Cancers. Vaccine 2006, 24, S11–S25. [Google Scholar] [CrossRef] [PubMed]
- de Martel, C.; Plummer, M.; Vignat, J.; Franceschi, S. Worldwide Burden of Cancer Attributable to HPV by Site, Country and HPV Type. Int J Cancer 2017, 141, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Stelzle, D.; Tanaka, L.F.; Lee, K.K.; Ibrahim Khalil, A.; Baussano, I.; Shah, A.S.V.; McAllister, D.A.; Gottlieb, S.L.; Klug, S.J.; Winkler, A.S.; et al. Estimates of the Global Burden of Cervical Cancer Associated with HIV. Lancet Glob Health 2021, 9, e161–e169. [Google Scholar] [CrossRef]
- Dryden-Peterson, S.; Bvochora-Nsingo, M.; Suneja, G.; Efstathiou, J.A.; Grover, S.; Chiyapo, S.; Ramogola-Masire, D.; Kebabonye-Pusoentsi, M.; Clayman, R.; Mapes, A.C.; et al. HIV Infection and Survival Among Women With Cervical Cancer. J Clin Oncol 2016, 34, 3749–3757. [Google Scholar] [CrossRef]
- Shiels, M.S.; Cole, S.R.; Kirk, G.D.; Poole, C. A Meta-Analysis of the Incidence of Non-AIDS Cancers in HIV-Infected Individuals. J Acquir Immune Defic Syndr 2009, 52, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Khandwala, P.; Singhal, S.; Desai, D.; Parsi, M.; Potdar, R. HIV-Associated Anal Cancer. Cureus 13, e14834. [CrossRef]
- Piketty, C.; Selinger-Leneman, H.; Grabar, S.; Duvivier, C.; Bonmarchand, M.; Abramowitz, L.; Costagliola, D.; Mary-Krause, M.; Co 4, on behalf of the F.-A. Marked Increase in the Incidence of Invasive Anal Cancer among HIV-Infected Patients despite Treatment with Combination Antiretroviral Therapy. AIDS 2008, 22, 1203. [CrossRef]
- Pereira, A.C.C.; Lacerda, H.R. de; Barros, R.C. do R. Diagnostic Methods for Prevention of Anal Cancer and Characteristics of Anal Lesions Caused by HPV in Men with HIV/AIDS. Braz J Infect Dis 2008, 12, 293–299. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, F.; Ceccarelli, M.; Rullo, E.V.; Facciolà, A.; D’Aleo, F.; Cacopardo, B.; Iacobello, C.; Costa, A.; Altavilla, G.; Pellicanò, G.F.; et al. Cancer Screening in HIV-Infected Patients- Early Diagnosis in a High-Risk Population.
- Ceccarelli, M.; Condorelli, F.; Rullo, E.V.; Pellicanò, G.F. Editorial – Improving Access and Adherence to Screening Tests for Cancers.
- Tanaka, L.F.; Latorre, M. do R.D.O.; Gutierrez, E.B.; Curado, M.P.; Dal Maso, L.; Herbinger, K.-H.; Froeschl, G.; Heumann, C. Cancer Survival in People with AIDS: A Population-Based Study from São Paulo, Brazil. Int J Cancer 2018, 142, 524–533. [Google Scholar] [CrossRef]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human Papillomavirus Molecular Biology and Disease Association. Reviews in Medical Virology 2015, 25, 2–23. [Google Scholar] [CrossRef]
- Schiller, J.T.; Day, P.M.; Kines, R.C. Current Understanding of the Mechanism of HPV Infection. Gynecologic Oncology 2010, 118, S12–S17. [Google Scholar] [CrossRef]
- Carias, A.M.; McCoombe, S.; McRaven, M.; Anderson, M.; Galloway, N.; Vandergrift, N.; Fought, A.J.; Lurain, J.; Duplantis, M.; Veazey, R.S.; et al. Defining the Interaction of HIV-1 with the Mucosal Barriers of the Female Reproductive Tract. J Virol 2013, 87, 11388–11400. [Google Scholar] [CrossRef]
- Visualization of HIV-1 Interactions with Penile and Foreskin Epithelia: Clues for Female-to-Male HIV Transmission | PLOS Pathogens. [CrossRef]
- Joag, S.V.; Adany, I.; Li, Z.; Foresman, L.; Pinson, D.M.; Wang, C.; Stephens, E.B.; Raghavan, R.; Narayan, O. Animal Model of Mucosally Transmitted Human Immunodeficiency Virus Type 1 Disease: Intravaginal and Oral Deposition of Simian/Human Immunodeficiency Virus in Macaques Results in Systemic Infection, Elimination of CD4+ T Cells, and AIDS. J Virol 1997, 71, 4016–4023. [Google Scholar] [CrossRef]
- Girard, M.; Mahoney, J.; Wei, Q.; van der Ryst, E.; Muchmore, E.; Barré-Sinoussi, F.; Fultz, P.N. Genital Infection of Female Chimpanzees with Human Immunodeficiency Virus Type 1. AIDS Res Hum Retroviruses 1998, 14, 1357–1367. [Google Scholar] [CrossRef]
- Yasen, A.; Herrera, R.; Rosbe, K.; Lien, K.; Tugizov, S.M. HIV Internalization into Oral and Genital Epithelial Cells by Endocytosis and Macropinocytosis Leads to Viral Sequestration in the Vesicles. Virology 2018, 515, 92–107. [Google Scholar] [CrossRef]
- Lenassi, M.; Cagney, G.; Liao, M.; Vaupotic, T.; Bartholomeeusen, K.; Cheng, Y.; Krogan, N.J.; Plemenitas, A.; Peterlin, B.M. HIV Nef Is Secreted in Exosomes and Triggers Apoptosis in Bystander CD4+ T Cells. Traffic 2010, 11, 110–122. [Google Scholar] [CrossRef]
- Nobile, C.; Rudnicka, D.; Hasan, M.; Aulner, N.; Porrot, F.; Machu, C.; Renaud, O.; Prévost, M.-C.; Hivroz, C.; Schwartz, O.; et al. HIV-1 Nef Inhibits Ruffles, Induces Filopodia, and Modulates Migration of Infected Lymphocytes. J Virol 2010, 84, 2282–2293. [Google Scholar] [CrossRef]
- Xu, W.; Santini, P.A.; Sullivan, J.S.; He, B.; Shan, M.; Ball, S.C.; Dyer, W.B.; Ketas, T.J.; Chadburn, A.; Cohen-Gould, L.; et al. HIV-1 Evades Virus-Specific IgG2 and IgA Responses by Targeting Systemic and Intestinal B Cells via Long-Range Intercellular Conduits. Nat Immunol 2009, 10, 1008–1017. [Google Scholar] [CrossRef]
- Pyeon, D.; Rojas, V.K.; Price, L.; Kim, S.; Singh, M.; Park, I.-W. HIV-1 Impairment via UBE3A and HIV-1 Nef Interactions Utilizing the Ubiquitin Proteasome System. Viruses 2019, 11, 1098. [Google Scholar] [CrossRef] [PubMed]
- Kelley, M.L.; Keiger, K.E.; Lee, C.J.; Huibregtse, J.M. The Global Transcriptional Effects of the Human Papillomavirus E6 Protein in Cervical Carcinoma Cell Lines Are Mediated by the E6AP Ubiquitin Ligase. J Virol 2005, 79, 3737–3747. [Google Scholar] [CrossRef]
- Talis, A.L.; Huibregtse, J.M.; Howley, P.M. The Role of E6AP in the Regulation of P53 Protein Levels in Human Papillomavirus (HPV)-Positive and HPV-Negative Cells*. Journal of Biological Chemistry 1998, 273, 6439–6445. [Google Scholar] [CrossRef]
- Massimi, P.; Shai, A.; Lambert, P.; Banks, L. HPV E6 Degradation of P53 and PDZ Containing Substrates in an E6AP Null Background. Oncogene 2008, 27, 1800–1804. [Google Scholar] [CrossRef]
- Tornesello, M.L.; Buonaguro, F.M.; Beth-Giraldo, E.; Giraldo, G. Human Immunodeficiency Virus Type 1 Tat Gene Enhances Human Papillomavirus Early Gene Expression. Intervirology 2008, 36, 57–64. [Google Scholar] [CrossRef]
- Buonaguro, F.M.; Tornesello, M.L.; Buonaguro, L.; Del Gaudio, E.; Beth-Giraldo, E.; Giraldo, G. Role of HIV as Cofactor in HPV Oncogenesis: In Vitro Evidences of Virus Interactions. Antibiot Chemother (1971) 1994, 46, 102–109. [Google Scholar] [CrossRef]
- Kim, R.H.; Yochim, J.M.; Kang, M.K.; Shin, K.-H.; Christensen, R.; Park, N.-H. HIV-1 Tat Enhances Replicative Potential of Human Oral Keratinocytes Harboring HPV-16 Genome. Int J Oncol 2008, 33, 777–782. [Google Scholar]
- Barillari, G.; Palladino, C.; Bacigalupo, I.; Leone, P.; Falchi, M.; Ensoli, B. Entrance of the Tat Protein of HIV-1 into Human Uterine Cervical Carcinoma Cells Causes Upregulation of HPV-E6 Expression and a Decrease in P53 Protein Levels. Oncol Lett 2016, 12, 2389–2394. [Google Scholar] [CrossRef] [PubMed]
- Vernon, S.D.; Hart, C.E.; Reeves, W.C.; Icenogle, J.P. The HIV-1 Tat Protein Enhances E2-Dependent Human Papillomavirus 16 Transcription. Virus Research 1993, 27, 133–145. [Google Scholar] [CrossRef]
- Tan, W.; Felber, B.K.; Zolotukhin, A.S.; Pavlakis, G.N.; Schwartz, S. Efficient Expression of the Human Papillomavirus Type 16 L1 Protein in Epithelial Cells by Using Rev and the Rev-Responsive Element of Human Immunodeficiency Virus or the Cis-Acting Transactivation Element of Simian Retrovirus Type 1. J Virol 1995, 69, 5607–5620. [Google Scholar] [CrossRef]
- Nuclear Import and Cell Cycle Arrest Functions of the HIV-1 Vpr Protein Are Encoded by Two Separate Genes in HIV-2/SIV(SM). | The EMBO Journal. Available online: https://www.embopress.org/doi/abs/10.1002/j.1460-2075.1996.tb01003.x (accessed on 7 June 2023).
- Toy, E.P.; Rodríguez-Rodríguez, L.; McCance, D.; Ludlow, J.; Planelles, V. Induction of Cell-Cycle Arrest in Cervical Cancer Cells by the Human Immunodeficiency Virus Type 1 Viral Protein R11Named a Searle-Donald F. Richardson Prize Paper after Presentation at the ACOG District II Junior Fellows Annual Meeting, New York, October 1998, and Annual Clinical Meeting of ACOG, Philadelphia, May 1999. Obstetrics & Gynecology 2000, 95, 141–146. [Google Scholar] [CrossRef]
- Tugizov, S.M.; Herrera, R.; Veluppillai, P.; Greenspan, D.; Palefsky, J.M.; Tugizov, S.M.; Herrera, R.; Veluppillai, P.; Greenspan, D.; Palefsky, J.M. 46. HIV-Induced Epithelial–Mesenchymal Transition in Mucosal Epithelium Facilitates HPV Paracellular Penetration. Sex. Health 2013, 10, 592–592. [Google Scholar] [CrossRef]
- Lien, K.; Mayer, W.; Herrera, R.; Padilla, N.T.; Cai, X.; Lin, V.; Pholcharoenchit, R.; Palefsky, J.; Tugizov, S.M. HIV-1 Proteins Gp120 and Tat Promote Epithelial-Mesenchymal Transition and Invasiveness of HPV-Positive and HPV-Negative Neoplastic Genital and Oral Epithelial Cells. Microbiol Spectr 2022, 10, e0362222. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Garson, K.; Li, L.; Vanderhyden, B.C. Optimization of Lentiviral Vector Production Using Polyethylenimine-Mediated Transfection. Oncol Lett 2015, 9, 55–62. [Google Scholar] [CrossRef]
- Giry-Laterrière, M.; Verhoeyen, E.; Salmon, P. Lentiviral Vectors. Methods Mol Biol 2011, 737, 183–209. [Google Scholar] [CrossRef]
- Isaguliants, M.G.; Belikov, S.V.; Starodubova, E.S.; Gizatullin, R.Z.; Rollman, E.; Zuber, B.; Zuber, A.K.; Grishchenko, O.I.A.; Rytting, A.-S.; Källander, C.F.R.; et al. Mutations Conferring Drug Resistance Affect Eukaryotic Expression of HIV Type 1 Reverse Transcriptase. AIDS Res Hum Retroviruses 2004, 20, 191–201. [Google Scholar] [CrossRef]
- Zhang, J.; Ruschhaupt, M.; Biczok, R. ddCt Method for qRT–PCR Data Analysis.; 2010.
- Golikov, M.V.; Karpenko, I.L.; Lipatova, A.V.; Ivanova, O.N.; Fedyakina, I.T.; Larichev, V.F.; Zakirova, N.F.; Leonova, O.G.; Popenko, V.I.; Bartosch, B.; et al. Cultivation of Cells in a Physiological Plasmax Medium Increases Mitochondrial Respiratory Capacity and Reduces Replication Levels of RNA Viruses. Antioxidants 2022, 11, 97. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Smirnova, O.A.; Ivanova, O.N.; Masalova, O.V.; Kochetkov, S.N.; Isaguliants, M.G. Hepatitis C Virus Proteins Activate NRF2/ARE Pathway by Distinct ROS-Dependent and Independent Mechanisms in HUH7 Cells. PLOS ONE 2011, 6, e24957. [Google Scholar] [CrossRef]
- Jonkman, J.E.N.; Cathcart, J.A.; Xu, F.; Bartolini, M.E.; Amon, J.E.; Stevens, K.M.; Colarusso, P. An Introduction to the Wound Healing Assay Using Live-Cell Microscopy. Cell Adh Migr 2014, 8, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Khan, F. Jab1 Inhibition by Methanolic Extract of Moringa Oleifera Leaves in Cervical Cancer Cells: A Potent Targeted Therapeutic Approach. Nutr Cancer 2021, 73, 2411–2419. [Google Scholar] [CrossRef]
- Tomayko, M.M.; Reynolds, C.P. Determination of Subcutaneous Tumor Size in Athymic (Nude) Mice. Cancer Chemother. Pharmacol. 1989, 24, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Pattillo, R.A.; Hussa, R.O.; Story, M.T.; Ruckert, A.C.F.; Shalaby, M.R.; Mattingly, R.F. Tumor Antigen and Human Chorionic Gonadotropin in CaSki Cells: A New Epidermoid Cervical Cancer Cell Line. Science 1977, 196, 1456–1458. [Google Scholar] [CrossRef]
- Isaguliants, M.G.; Gudima, S.O.; Ivanova, O.V.; Levi, M.; Hinkula, J.; Garaev, M.M.; Kochetkov, S.N.; Wahren, B. Immunogenic Properties of Reverse Transcriptase of HIV Type 1 Assessed by DNA and Protein Immunization of Rabbits. AIDS Res Hum Retroviruses 2000, 16, 1269–1280. [Google Scholar] [CrossRef] [PubMed]
- Shearer, R.F.; Saunders, D.N. Experimental Design for Stable Genetic Manipulation in Mammalian Cell Lines: Lentivirus and Alternatives. Genes Cells 2015, 20, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rowe, D.C.D.; Summers, D.K. The Quiescent-Cell Expression System for Protein Synthesis in Escherichia Coli. Appl Environ Microbiol 1999, 65, 2710–2715. [Google Scholar] [CrossRef]
- Filippova, M.; Johnson, M.M.; Bautista, M.; Filippov, V.; Fodor, N.; Tungteakkhun, S.S.; Williams, K.; Duerksen-Hughes, P.J. The Large and Small Isoforms of Human Papillomavirus Type 16 E6 Bind to and Differentially Affect Procaspase 8 Stability and Activity. Journal of Virology 2007, 81, 4116–4129. [Google Scholar] [CrossRef] [PubMed]
- Pim, D.; Tomaić, V.; Banks, L. The Human Papillomavirus (HPV) E6* Proteins from High-Risk, Mucosal HPVs Can Direct Degradation of Cellular Proteins in the Absence of Full-Length E6 Protein. Journal of Virology 2009, 83, 9863–9874. [Google Scholar] [CrossRef] [PubMed]
- Olmedo-Nieva, L.; Muñoz-Bello, J.O.; Contreras-Paredes, A.; Lizano, M. The Role of E6 Spliced Isoforms (E6*) in Human Papillomavirus-Induced Carcinogenesis. Viruses 2018, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Liu, T.; Han, C.; Xuan, Y.; Jiang, D.; Sun, Y.; Zhang, X.; Zhang, W.; Xu, Y.; Liu, Y.; et al. HPV E6/E7 Promotes Aerobic Glycolysis in Cervical Cancer by Regulating IGF2BP2 to Stabilize m6A-MYC Expression. Int J Biol Sci 2022, 18, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Mookerjee, S.A.; Gerencser, A.A.; Nicholls, D.G.; Brand, M.D. Quantifying Intracellular Rates of Glycolytic and Oxidative ATP Production and Consumption Using Extracellular Flux Measurements. Journal of Biological Chemistry 2017, 292, 7189–7207. [Google Scholar] [CrossRef]
- Petrovskaya, A.V.; Tverskoi, A.M.; Barykin, E.P.; Varshavskaya, K.B.; Dalina, A.A.; Mitkevich, V.A.; Makarov, A.A.; Petrushanko, I.Y. Distinct Effects of Beta-Amyloid, Its Isomerized and Phosphorylated Forms on the Redox Status and Mitochondrial Functioning of the Blood–Brain Barrier Endothelium. International Journal of Molecular Sciences 2023, 24, 183. [Google Scholar] [CrossRef]
- Rodic, S.; Vincent, M.D. Reactive Oxygen Species (ROS) Are a Key Determinant of Cancer’s Metabolic Phenotype. International Journal of Cancer 2018, 142, 440–448. [Google Scholar] [CrossRef]
- LeBel, C.P.; Ischiropoulos, H.; Bondy, S.C. Evaluation of the Probe 2′,7′-Dichlorofluorescin as an Indicator of Reactive Oxygen Species Formation and Oxidative Stress. Chem Res Toxicol 1992, 5, 227–231. [Google Scholar] [CrossRef]
- Robinson, J.P.; Carter, W.O.; Narayanan, P.K. Chapter 28 Oxidative Product Formation Analysis by Flow Cytometry. In Methods in Cell Biology; Darzynkiewicz, Z., Paul Robinson, J., Crissman, H.A., Eds.; Flow Cytometry Second Edition, Part A; Academic Press, 1994; Vol. 41, pp. 437–447.
- Zakirova, N.F.; Kondrashova, A.S.; Golikov, M.V.; Ivanova, O.N.; Ivanov, A.V.; Isaguliants, M.G.; Bayurova, E.O. [Expression of HIV-1 Reverse Transcriptase in Murine Cancer Cells Increases Mitochondrial Respiration]. Mol Biol (Mosk) 2022, 56, 795–807. [Google Scholar] [CrossRef]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Developmental Cell 2019, 49, 361–374. [Google Scholar] [CrossRef]
- Mishra, A.K.; Campanale, J.P.; Mondo, J.A.; Montell, D.J. Cell Interactions in Collective Cell Migration. Development 2019, 146, dev172056. [Google Scholar] [CrossRef]
- Yoysungnoen, B.; Bhattarakosol, P.; Changtam, C.; Patumraj, S. Effects of Tetrahydrocurcumin on Tumor Growth and Cellular Signaling in Cervical Cancer Xenografts in Nude Mice. BioMed Research International 2016, 2016, e1781208. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Chen, R.; Chen, J.; Qi, Q.; Pan, Y.; Du, L.; Xiao, G.; Jiang, S. Combining Metformin and Nelfinavir Exhibits Synergistic Effects against the Growth of Human Cervical Cancer Cells and Xenograft in Nude Mice. Sci Rep 2017, 7, 43373. [Google Scholar] [CrossRef] [PubMed]
- Yim, E.-K.; Park, J.-S. The Role of HPV E6 and E7 Oncoproteins in HPV-Associated Cervical Carcinogenesis. Cancer Res Treat 2005, 37, 319–324. [Google Scholar] [CrossRef]
- Purohit, V.; Balakrishnan, M.; Kim, B.; Bambara, R.A. Evidence That HIV-1 Reverse Transcriptase Employs the DNA 3′ End-Directed Primary/Secondary RNase H Cleavage Mechanism during Synthesis and Strand Transfer*. Journal of Biological Chemistry 2005, 280, 40534–40543. [Google Scholar] [CrossRef]
- Anyanwu, S.I.; Doherty, A.; Powell, M.D.; Obialo, C.; Huang, M.B.; Quarshie, A.; Mitchell, C.; Bashir, K.; Newman, G.W. Detection of HIV-1 and Human Proteins in Urinary Extracellular Vesicles from HIV+ Patients. Adv Virol 2018, 2018, 7863412. [Google Scholar] [CrossRef]
- Latanova, A.; Petkov, S.; Kuzmenko, Y.; Kilpeläinen, A.; Ivanov, A.; Smirnova, O.; Krotova, O.; Korolev, S.; Hinkula, J.; Karpov, V.; et al. Fusion to Flaviviral Leader Peptide Targets HIV-1 Reverse Transcriptase for Secretion and Reduces Its Enzymatic Activity and Ability to Induce Oxidative Stress but Has No Major Effects on Its Immunogenic Performance in DNA-Immunized Mice. J Immunol Res 2017, 2017, 7407136. [Google Scholar] [CrossRef]
- Isaguliants, M.; Smirnova, O.; Ivanov, A.V.; Kilpelainen, A.; Kuzmenko, Y.; Petkov, S.; Latanova, A.; Krotova, O.; Engström, G.; Karpov, V.; et al. Oxidative Stress Induced by HIV-1 Reverse Transcriptase Modulates the Enzyme’s Performance in Gene Immunization. Hum Vaccin Immunother 2013, 9, 2111–2119. [Google Scholar] [CrossRef]
- Hu, W.-S.; Hughes, S.H. HIV-1 Reverse Transcription. Cold Spring Harb Perspect Med 2012, 2, a006882. [Google Scholar] [CrossRef]
- Ferris, A.L.; Hizi, A.; Showalter, S.D.; Pichuantes, S.; Babe, L.; Craik, C.S.; Hughes, S.H. Immunologic and Proteolytic Analysis of HIV-1 Reverse Transcriptase Structure. Virology 1990, 175, 456–464. [Google Scholar] [CrossRef]
- Boer, R.J.D.; Ribeiro, R.M.; Perelson, A.S. Current Estimates for HIV-1 Production Imply Rapid Viral Clearance in Lymphoid Tissues. PLOS Computational Biology 2010, 6, e1000906. [Google Scholar] [CrossRef] [PubMed]
- Hockett, R.D.; Michael Kilby, J.; Derdeyn, C.A.; Saag, M.S.; Sillers, M.; Squires, K.; Chiz, S.; Nowak, M.A.; Shaw, G.M.; Bucy, R.P. Constant Mean Viral Copy Number per Infected Cell in Tissues Regardless of High, Low, or Undetectable Plasma HIV RNA. J Exp Med 1999, 189, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhang, C.; Wang, B.; Wang, F.; Pei, B.; Cheng, C.; Yang, W.; Zhao, Z. Transduction with Lentiviral Vectors Altered the Expression Profile of Host MicroRNAs. Journal of Virology 2018, 92, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Bhuniya, A.; Pattarayan, D.; Yang, D. Lentiviral Vector Transduction Provides Nonspecific Immunogenicity for Syngeneic Tumor Models. Molecular Carcinogenesis 2022, 61, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Filippova, M.; Evans, W.; Aragon, R.; Filippov, V.; Williams, V.M.; Hong, L.; Reeves, M.E.; Duerksen-Hughes, P. The Small Splice Variant of HPV16 E6, E6⁎, Reduces Tumor Formation in Cervical Carcinoma Xenografts. Virology 2014, 450–451, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Ramírez, I.; Carrillo-García, A.; Contreras-Paredes, A.; Ortiz-Sánchez, E.; Cruz-Gregorio, A.; Lizano, M. Regulation of Cellular Metabolism by High-Risk Human Papillomaviruses. International Journal of Molecular Sciences 2018, 19, 1839. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Huang, Y.; Song, S. Inhibiting the HPV16 Oncogene-Mediated Glycolysis Sensitizes Human Cervical Carcinoma Cells to 5-Fluorouracil. Onco Targets Ther 2019, 12, 6711–6720. [Google Scholar] [CrossRef]
- Brand, M.D.; Nicholls, D.G. Assessing Mitochondrial Dysfunction in Cells. Biochem J 2011, 435, 297–312. [Google Scholar] [CrossRef]
- Arjona, S.P.; Allen, C.N.S.; Santerre, M.; Gross, S.; Soboloff, J.; Booze, R.; Sawaya, B.E. Disruption of Mitochondrial-Associated ER Membranes by HIV-1 Tat Protein Contributes to Premature Brain Aging. CNS Neuroscience & Therapeutics 2023, 29, 365–377. [Google Scholar] [CrossRef]
- Cruz-Gregorio, A.; Aranda-Rivera, A.K.; Aparicio-Trejo, O.E.; Coronado-Martínez, I.; Pedraza-Chaverri, J.; Lizano, M. E6 Oncoproteins from High-Risk Human Papillomavirus Induce Mitochondrial Metabolism in a Head and Neck Squamous Cell Carcinoma Model. Biomolecules 2019, 9, 351. [Google Scholar] [CrossRef] [PubMed]
- Liemburg-Apers, D.C.; Willems, P.H.G.M.; Koopman, W.J.H.; Grefte, S. Interactions between Mitochondrial Reactive Oxygen Species and Cellular Glucose Metabolism. Arch Toxicol 2015, 89, 1209–1226. [Google Scholar] [CrossRef] [PubMed]
- Mullarky, E.; Cantley, L.C. Diverting Glycolysis to Combat Oxidative Stress. In Innovative Medicine: Basic Research and Development; Nakao, K., Minato, N., Uemoto, S., Eds.; Springer: Tokyo, 2015 ISBN 978-4-431-55650-3.
- Wu, W.; Papagiannakopoulos, T. The Pleiotropic Role of the KEAP1/NRF2 Pathway in Cancer. Annual Review of Cancer Biology 2020, 4, 413–435. [Google Scholar] [CrossRef]
- Liu, A.; Lv, Z.; Yan, Z.; Wu, X.; Yan, L.; Sun, L.; Yuan, Y.; Xu, Q. Association of Mitochondrial Homeostasis and Dynamic Balance with Malignant Biological Behaviors of Gastrointestinal Cancer. Journal of Translational Medicine 2023, 21, 27. [Google Scholar] [CrossRef]
- ABEL, E.D. MITOCHONDRIAL DYNAMICS AND METABOLIC REGULATION IN CARDIAC AND SKELETAL MUSCLE. Trans Am Clin Climatol Assoc 2018, 129, 266–278.
- Binarová, P.; Tuszynski, J. Tubulin: Structure, Functions and Roles in Disease. Cells 2019, 8, 1294. [Google Scholar] [CrossRef] [PubMed]
- Warren, K.; Warrilow, D.; Meredith, L.; Harrich, D. Reverse Transcriptase and Cellular Factors: Regulators of HIV-1 Reverse Transcription. Viruses 2009, 1, 873. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Merrill, R.A.; Strack, S. A-Kinase Anchoring Protein 1: Emerging Roles in Regulating Mitochondrial Form and Function in Health and Disease. Cells 2020, 9, 298. [Google Scholar] [CrossRef]
- Aggarwal, S.; Gabrovsek, L.; Langeberg, L.K.; Golkowski, M.; Ong, S.-E.; Smith, F.D.; Scott, J.D. Depletion of dAKAP1–Protein Kinase A Signaling Islands from the Outer Mitochondrial Membrane Alters Breast Cancer Cell Metabolism and Motility. J Biol Chem 2019, 294, 3152–3168. [Google Scholar] [CrossRef]
- Schwager, S.C.; Mosier, J.A.; Padmanabhan, R.S.; White, A.; Xing, Q.; Hapach, L.A.; Taufalele, P.V.; Ortiz, I.; Reinhart-King, C.A. Link between Glucose Metabolism and Epithelial-to-Mesenchymal Transition Drives Triple-Negative Breast Cancer Migratory Heterogeneity. iScience 2022, 25, 105190. [Google Scholar] [CrossRef] [PubMed]
- Usman, S.; Waseem, N.H.; Nguyen, T.K.N.; Mohsin, S.; Jamal, A.; Teh, M.-T.; Waseem, A. Vimentin Is at the Heart of Epithelial Mesenchymal Transition (EMT) Mediated Metastasis. Cancers (Basel) 2021, 13, 4985. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Aboukameel, A.; Kong, D.; Wang, Z.; Sethi, S.; Chen, W.; Sarkar, F.H.; Raz, A. Phosphoglucose Isomerase/Autocrine Motility Factor Mediates Epithelial-Mesenchymal Transition Regulated by miR-200 in Breast Cancer Cells. Cancer Res 2011, 71, 3400–3409. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zhang, Y.; Liang, J.; Li, W.; Zhu, Y.; Zhang, M.; Wang, C.; Hou, J. Deregulation of Hexokinase II Is Associated with Glycolysis, Autophagy, and the Epithelial-Mesenchymal Transition in Tongue Squamous Cell Carcinoma under Hypoxia. BioMed Research International 2018, 2018, e8480762. [Google Scholar] [CrossRef]
- Funasaka, T.; Hogan, V.; Raz, A. Phosphoglucose Isomerase/Autocrine Motility Factor Mediates Epithelial and Mesenchymal Phenotype Conversions in Breast Cancer. Cancer Res 2009, 69, 5349–5356. [Google Scholar] [CrossRef]
- Kang, H.; Kim, H.; Lee, S.; Youn, H.; Youn, B. Role of Metabolic Reprogramming in Epithelial−Mesenchymal Transition (EMT). Int J Mol Sci 2019, 20, 2042. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Tang, Z.; Huang, A.; Chen, P.; Liu, P.; Yang, J.; Lu, W.; Liao, J.; Sun, Y.; Wen, S.; et al. Glyceraldehyde-3-Phosphate Dehydrogenase Promotes Cancer Growth and Metastasis through Upregulation of SNAIL Expression. International Journal of Oncology 2017, 50, 252–262. [Google Scholar] [CrossRef]
- Bhattacharya, S. Metabolic Reprogramming and Cancer: 2022. Qeios 2022. [Google Scholar] [CrossRef]
- Song, G.-L.; Jin, C.-C.; Zhao, W.; Tang, Y.; Wang, Y.-L.; Li, M.; Xiao, M.; Li, X.; Li, Q.-S.; Lin, X.; et al. Regulation of the RhoA/ROCK/AKT/β-Catenin Pathway by Arginine-Specific ADP-Ribosytransferases 1 Promotes Migration and Epithelial-Mesenchymal Transition in Colon Carcinoma. International Journal of Oncology 2016, 49, 646–656. [Google Scholar] [CrossRef]
- Nieto, M. Nieto MA.. The Snail Superfamily of Zinc-Finger Transcription Factors. Nat Rev Mol Cell Biol 3: 155-166. Nature reviews. Molecular cell biology 2002, 3, 155–166. [Google Scholar] [CrossRef]
- Elloul, S.; Bukholt Elstrand, M.; Nesland, J.M.; Tropé, C.G.; Kvalheim, G.; Goldberg, I.; Reich, R.; Davidson, B. Snail, Slug, and Smad-Interacting Protein 1 as Novel Parameters of Disease Aggressiveness in Metastatic Ovarian and Breast Carcinoma. Cancer 2005, 103, 1631–1643. [Google Scholar] [CrossRef] [PubMed]
- Storci, G.; Sansone, P.; Trere, D.; Tavolari, S.; Taffurelli, M.; Ceccarelli, C.; Guarnieri, T.; Paterini, P.; Pariali, M.; Montanaro, L.; et al. The Basal-like Breast Carcinoma Phenotype Is Regulated by SLUG Gene Expression. J Pathol 2008, 214, 25–37. [Google Scholar] [CrossRef]
- Canciello, A.; Cerveró-Varona, A.; Peserico, A.; Mauro, A.; Russo, V.; Morrione, A.; Giordano, A.; Barboni, B. “In Medio Stat Virtus”: Insights into Hybrid E/M Phenotype Attitudes. Frontiers in Cell and Developmental Biology 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Sample, R.A.; Nogueira, M.F.; Mitra, R.D.; Puram, S.V. Epigenetic Regulation of Hybrid Epithelial-Mesenchymal Cell States in Cancer. Oncogene 2023, 42, 2237–2248. [Google Scholar] [CrossRef]
- Shah, S.; Philipp, L.-M.; Giaimo, S.; Sebens, S.; Traulsen, A.; Raatz, M. Understanding and Leveraging Phenotypic Plasticity during Metastasis Formation. NPJ Syst Biol Appl 2023, 9, 48. [Google Scholar] [CrossRef]
- Cai, D.; Chen, S.-C.; Prasad, M.; He, L.; Wang, X.; Choesmel-Cadamuro, V.; Sawyer, J.K.; Danuser, G.; Montell, D.J. Mechanical Feedback through E-Cadherin Promotes Direction Sensing during Collective Cell Migration. Cell 2014, 157, 1146–1159. [Google Scholar] [CrossRef]
- Nieman, M.T.; Prudoff, R.S.; Johnson, K.R.; Wheelock, M.J. N-Cadherin Promotes Motility in Human Breast Cancer Cells Regardless of Their E-Cadherin Expression. J Cell Biol 1999, 147, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Shih, W.; Yamada, S. N-Cadherin as a Key Regulator of Collective Cell Migration in a 3D Environment. Cell Adh Migr 2012, 6, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Na, T.-Y.; Schecterson, L.; Mendonsa, A.M.; Gumbiner, B.M. The Functional Activity of E-Cadherin Controls Tumor Cell Metastasis at Multiple Steps. Proceedings of the National Academy of Sciences 2020, 117, 5931–5937. [Google Scholar] [CrossRef]
- Park, S.-Y.; Jang, H.; Kim, S.-Y.; Kim, D.; Park, Y.; Kee, S.-H. Expression of E-Cadherin in Epithelial Cancer Cells Increases Cell Motility and Directionality through the Localization of ZO-1 during Collective Cell Migration. Bioengineering 2021, 8, 65. [Google Scholar] [CrossRef]
- Derycke, L.D.M.; Bracke, M.E. N-Cadherin in the Spotlight of Cell-Cell Adhesion, Differentiation, Embryogenesis, Invasion and Signalling. Int J Dev Biol 2004, 48, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Mrozik, K.M.; Blaschuk, O.W.; Cheong, C.M.; Zannettino, A.C.W.; Vandyke, K. N-Cadherin in Cancer Metastasis, Its Emerging Role in Haematological Malignancies and Potential as a Therapeutic Target in Cancer. BMC Cancer 2018, 18, 939. [Google Scholar] [CrossRef]
- Radice, G.L. N-Cadherin-Mediated Adhesion and Signaling from Development to Disease: Lessons from Mice. Prog Mol Biol Transl Sci 2013, 116, 263–289. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Duchesne, L.; Sankara Narayana, G.H.N.; Boggetto, N.; Fernig, D.D.; Uttamrao Murade, C.; Ladoux, B.; Mège, R.-M. Enhanced Cell-Cell Contact Stability and Decreased N-Cadherin-Mediated Migration upon Fibroblast Growth Factor Receptor-N-Cadherin Cross Talk. Oncogene 2019, 38, 6283–6300. [Google Scholar] [CrossRef]
- Epshtein, A.; Jackman, A.; Gonen, P.; Sherman, L. HPV16 E6 Oncoprotein Increases Cell Adhesion in Human Keratinocytes. Arch Virol 2009, 154, 55–63. [Google Scholar] [CrossRef]
Figure 1.
Mice were marked with a special label and placed in 4 groups of 6 animals per cage. Group 1 - control group implanted with Ca Ski; Group 2 implanted with Ca Ski GFP G9 subclone; Group 3, mice implanted with Ca Ski RT_A B8B5 subclone; Group 4, mice implanted with Ca Ski RT_A H6G11 subclone. In each group, mice nn 1-3 were implanted with 105 cells on the left, and 105 cells on the right, and mice nn 4-6, with 106 cells on the left, 106 cells on the right from the back of the tail.
Figure 1.
Mice were marked with a special label and placed in 4 groups of 6 animals per cage. Group 1 - control group implanted with Ca Ski; Group 2 implanted with Ca Ski GFP G9 subclone; Group 3, mice implanted with Ca Ski RT_A B8B5 subclone; Group 4, mice implanted with Ca Ski RT_A H6G11 subclone. In each group, mice nn 1-3 were implanted with 105 cells on the left, and 105 cells on the right, and mice nn 4-6, with 106 cells on the left, 106 cells on the right from the back of the tail.
Figure 2.
Relative expression levels of the transgene in the Ca Ski subclones with genomic inserts of coding sequence of the consensus reverse transcriptase of HIV-1 FSU_A (RT_A). (A) Western blot analysis of lysates of Ca Ski subclones stained with rabbit polyclonal anti-RT antibodies [58] (Panel I), and re-stained anti-β-actin monoclonal antibodies (Panel II), the parental Ca Ski cell line and GFP control serves as a reference. Recombinant RT_A (9 ng) resolved in PAGE after Western blotting (Panel III). Position of the weight mass markers is shown on the left. (B) Quantification of RT_A expression using ImageJ software. Signal in the lane corresponding to a given derivative clone was quantified using calibration curve built with the help of recombinant RT_A. Total amount of the expressed RT_A variant was divided by the number of cells used to make the lysate (see Materials and Methods for description). Results are presented as mean ± SD. Statistical significance was assessed using by Unpaired t test with Bonferroni correction. ns: not significant; ∗p < 0.05; ∗∗p < 0.01.
Figure 2.
Relative expression levels of the transgene in the Ca Ski subclones with genomic inserts of coding sequence of the consensus reverse transcriptase of HIV-1 FSU_A (RT_A). (A) Western blot analysis of lysates of Ca Ski subclones stained with rabbit polyclonal anti-RT antibodies [58] (Panel I), and re-stained anti-β-actin monoclonal antibodies (Panel II), the parental Ca Ski cell line and GFP control serves as a reference. Recombinant RT_A (9 ng) resolved in PAGE after Western blotting (Panel III). Position of the weight mass markers is shown on the left. (B) Quantification of RT_A expression using ImageJ software. Signal in the lane corresponding to a given derivative clone was quantified using calibration curve built with the help of recombinant RT_A. Total amount of the expressed RT_A variant was divided by the number of cells used to make the lysate (see Materials and Methods for description). Results are presented as mean ± SD. Statistical significance was assessed using by Unpaired t test with Bonferroni correction. ns: not significant; ∗p < 0.05; ∗∗p < 0.01.
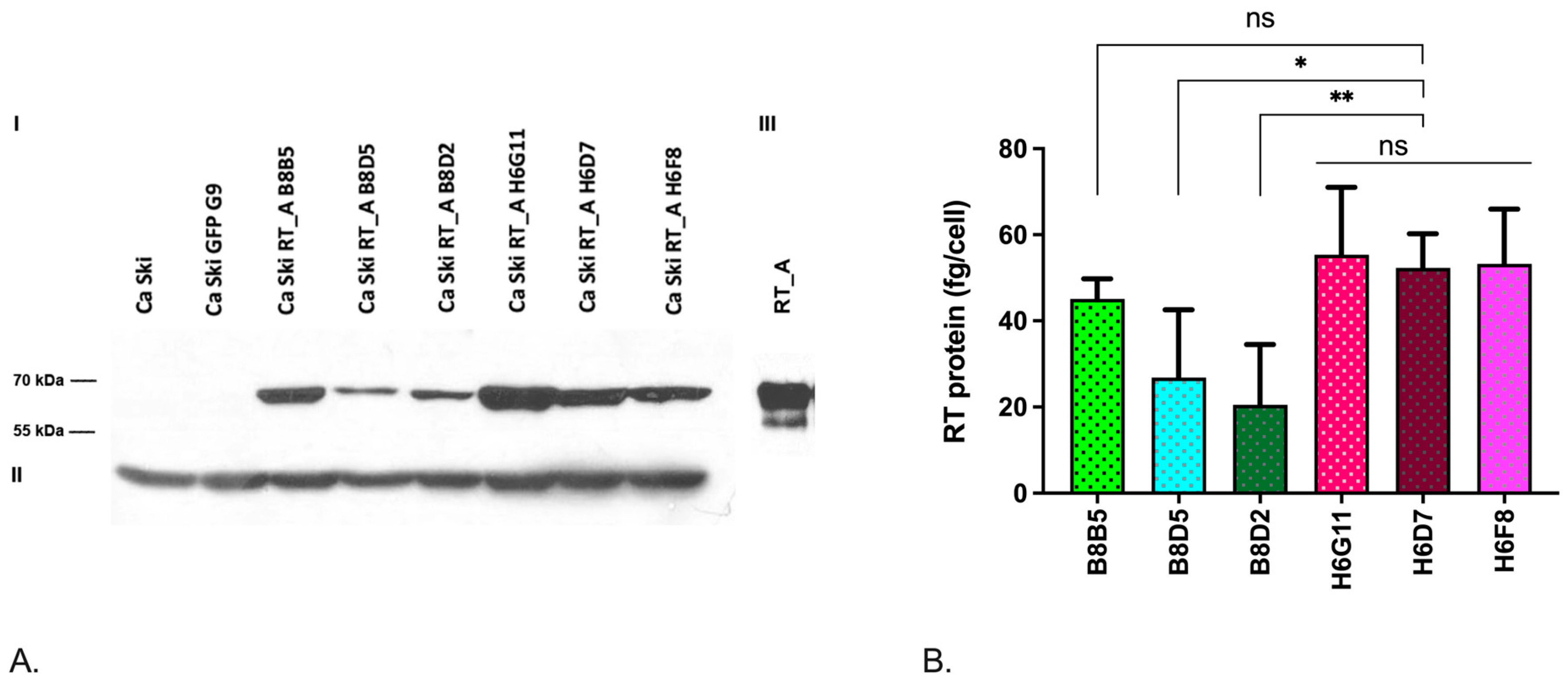
Figure 3.
Relative mRNA expression levels of isoform I of HPV16 E6 (E6*I) in Ca Ski subclones expressing consensus HIV-1 FSU_A reverse transcriptase compared Ca Ski expressing GFP and parental Ca Ski cells. (A) Levels of mRNA of E6*I in individual subclones and parental Ca Ski. *Significant difference from the value of the gene expression level in the GFP subclone (p <0.05; Unpaired t test with Bonferroni correction). (B) Comparison of the levels of E6*I mRNA in Ca Ski subclones groups according the number of RT_A DNA inserts: 1 RT_A (B8B5, B8D5, B8D2) and 6 RT_A (H6G11, H6D7, H6F8), and level of RT_A expression determined by Western blot (<40 fg/cell, low, B8D5 and B8D2 subclones; >41 fg/cell, high, B8B5, H6G11, H6D7, H6F8 subclones) (Table 1). #Significant difference from the value of the gene expression level in the GFP subclone (#p < 0.05; ##p < 0.01; ###p < 0.001; ####p < 0.0001; Unpaired t test with Bonferroni correction). Expression level was normalized to GAPDH expression and calculated as fold change compared to the parental Ca Ski line. Results are presented as mean ± SD, ns: not significant; ∗p < 0.05; ∗∗p < 0.01.
Figure 3.
Relative mRNA expression levels of isoform I of HPV16 E6 (E6*I) in Ca Ski subclones expressing consensus HIV-1 FSU_A reverse transcriptase compared Ca Ski expressing GFP and parental Ca Ski cells. (A) Levels of mRNA of E6*I in individual subclones and parental Ca Ski. *Significant difference from the value of the gene expression level in the GFP subclone (p <0.05; Unpaired t test with Bonferroni correction). (B) Comparison of the levels of E6*I mRNA in Ca Ski subclones groups according the number of RT_A DNA inserts: 1 RT_A (B8B5, B8D5, B8D2) and 6 RT_A (H6G11, H6D7, H6F8), and level of RT_A expression determined by Western blot (<40 fg/cell, low, B8D5 and B8D2 subclones; >41 fg/cell, high, B8B5, H6G11, H6D7, H6F8 subclones) (Table 1). #Significant difference from the value of the gene expression level in the GFP subclone (#p < 0.05; ##p < 0.01; ###p < 0.001; ####p < 0.0001; Unpaired t test with Bonferroni correction). Expression level was normalized to GAPDH expression and calculated as fold change compared to the parental Ca Ski line. Results are presented as mean ± SD, ns: not significant; ∗p < 0.05; ∗∗p < 0.01.
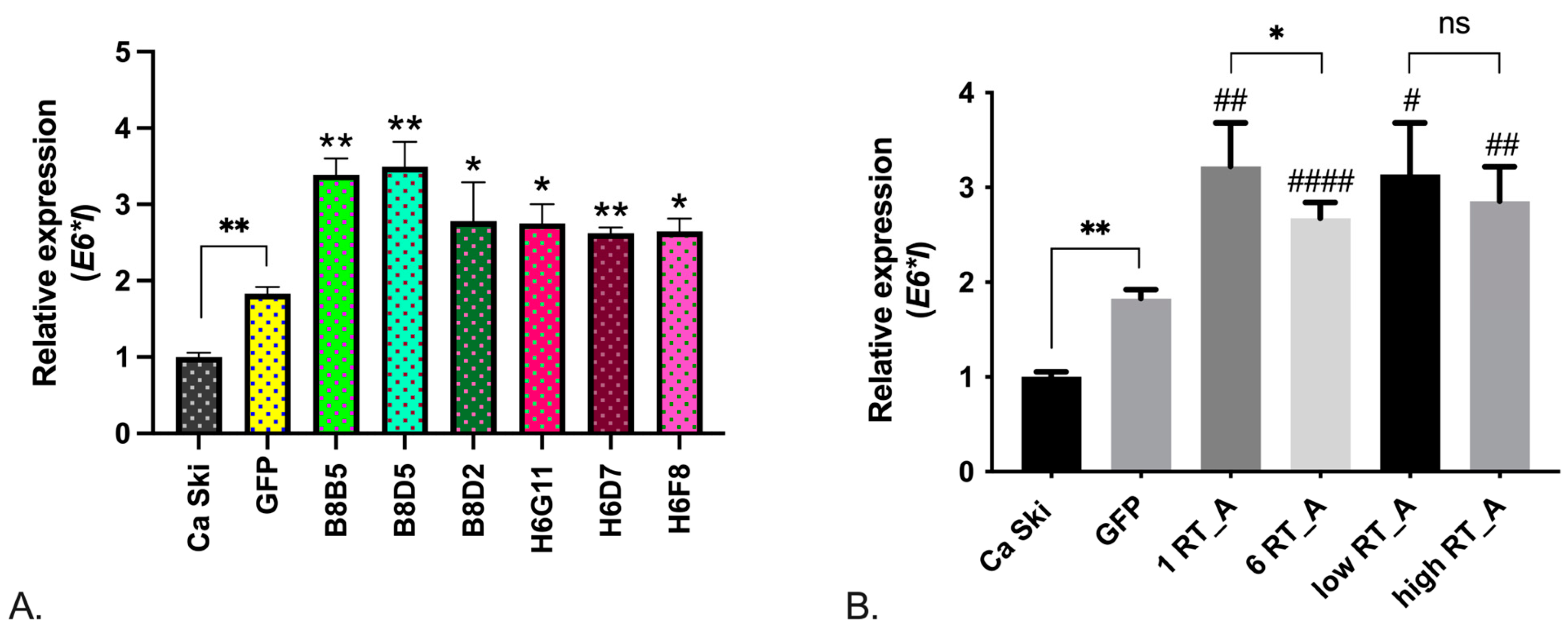
Figure 4.
Expression of HIV-1 reverse transcriptase (RT_A) in Ca Ski cells causes an increase in the intensity of glycolysis. Glycolysis efficiency of Ca Ski derivatives was evaluated using a Seahorse analyzer according to the Glycostress test method (A). Determination of ECAR in medium with low, 11 mM (B), and high, 30 mM glucose concentration (C); Maximum ECAR values upon stimulation with 1 µM oligomycin (D); The difference between the maximum glycolytic capacity and the level of glycolysis at 30 mM glucose (E). ECAR values are expressed in units of mpH/min and normalized to 1 mg of total cellular protein (mpH/min/Norm. Unit). Ca Ski subclones with one genomic insert of RT_A coding sequence (B8B5, B8D5, B8D2) are designated as “1 RT_A”, and with six inserts as “6 RT_A” (H6G11, H6D7, H6F8), Ca Ski with six genomic inserts of GFP is given as a control. The efficiency of glycolysis in individual subclones was presented in Supplements Figure S3. Histograms are presented as mean ± SD for the analysis performed in quadruplicate. Significant difference from the value of the gene expression level in the GFP subclone (p <0.05; Unpaired t test with Bonferroni correction). ns: not significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Figure 4.
Expression of HIV-1 reverse transcriptase (RT_A) in Ca Ski cells causes an increase in the intensity of glycolysis. Glycolysis efficiency of Ca Ski derivatives was evaluated using a Seahorse analyzer according to the Glycostress test method (A). Determination of ECAR in medium with low, 11 mM (B), and high, 30 mM glucose concentration (C); Maximum ECAR values upon stimulation with 1 µM oligomycin (D); The difference between the maximum glycolytic capacity and the level of glycolysis at 30 mM glucose (E). ECAR values are expressed in units of mpH/min and normalized to 1 mg of total cellular protein (mpH/min/Norm. Unit). Ca Ski subclones with one genomic insert of RT_A coding sequence (B8B5, B8D5, B8D2) are designated as “1 RT_A”, and with six inserts as “6 RT_A” (H6G11, H6D7, H6F8), Ca Ski with six genomic inserts of GFP is given as a control. The efficiency of glycolysis in individual subclones was presented in Supplements Figure S3. Histograms are presented as mean ± SD for the analysis performed in quadruplicate. Significant difference from the value of the gene expression level in the GFP subclone (p <0.05; Unpaired t test with Bonferroni correction). ns: not significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
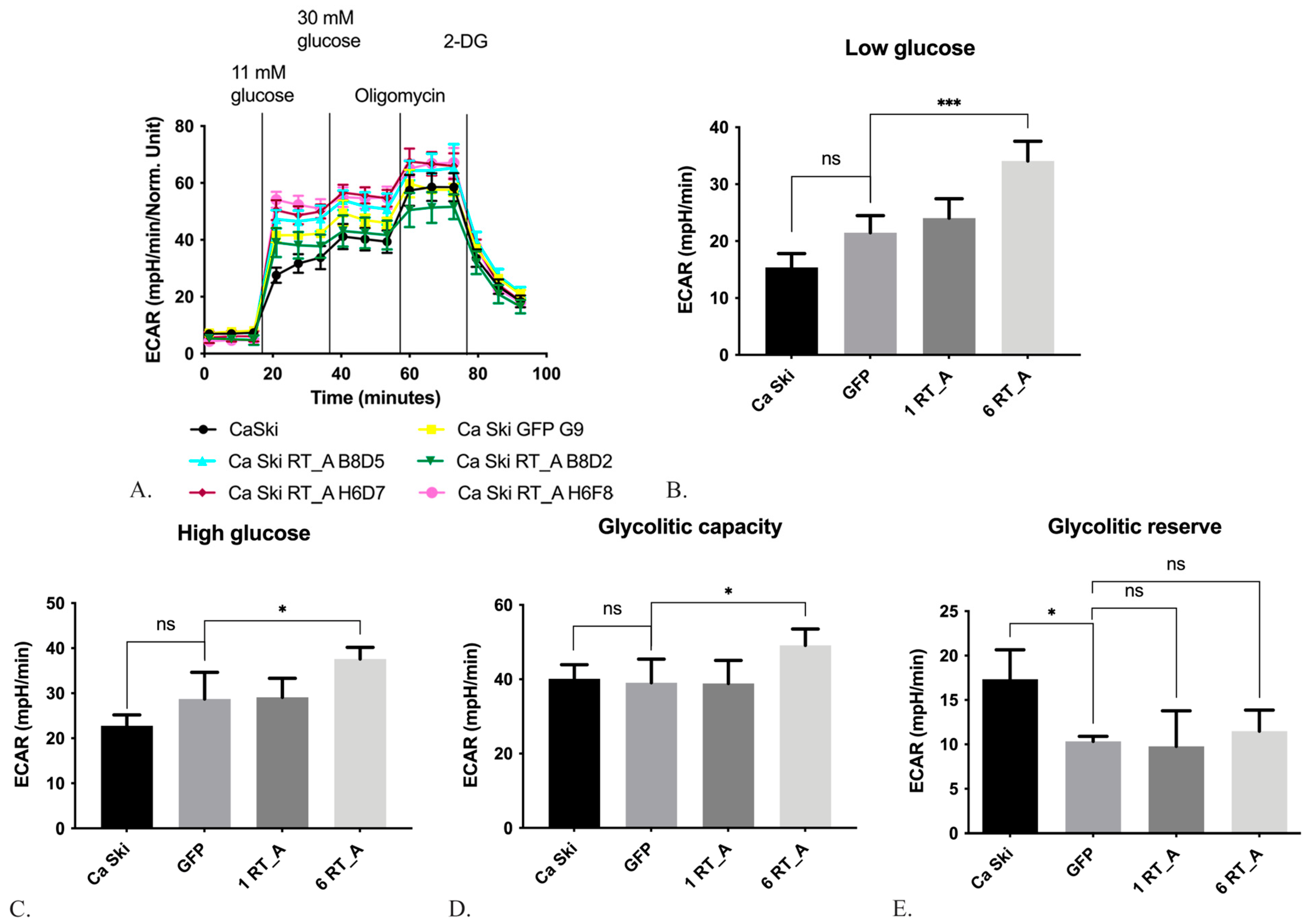
Figure 5.
Expression of HIV-1 reverse transcriptase (RT_A) in Ca Ski cells decreases the respiratory activity of mitochondria. Respiratory activity of Ca Ski derivatives was analyzed using Seahorse technology and the Mitostress reagent kit (A). Basal respiration rate determined as the difference between baseline and non-mitochondrial OCR values (B). ATP-bound OCR determined as the difference between the basal OCR and OCR inhibited by antimycin A (C); Maximum OCR value stimulated by the addition of low FCCP at concentrations of 0.75 μM (D) and high 1.5 μM (E); The difference between the levels of basal and maximum respiration (F). OCR values are expressed in units of pmol/min and normalized to 1 mg of total cellular protein (pmol/min/Norm. Unit). Ca Ski subclones with one genomic insert of RT_A coding sequence (B8B5, B8D5, B8D2) are designated as “1 RT_A”, and with six inserts as “6 RT_A” (H6G11, H6D7, H6F8), Ca Ski with six genomic inserts of GFP is given as a control. The respiratory activity of mitochondria in individual subclones was presented in Supplements Figure S4. Histograms are presented as mean ± SD for the analysis performed in quadruplicate. Significant difference from the value of the gene expression level in the GFP subclone (p <0.05; Unpaired t test with Bonferroni correction). ns: not significant; ∗p < 0.05; ∗∗p < 0.01.
Figure 5.
Expression of HIV-1 reverse transcriptase (RT_A) in Ca Ski cells decreases the respiratory activity of mitochondria. Respiratory activity of Ca Ski derivatives was analyzed using Seahorse technology and the Mitostress reagent kit (A). Basal respiration rate determined as the difference between baseline and non-mitochondrial OCR values (B). ATP-bound OCR determined as the difference between the basal OCR and OCR inhibited by antimycin A (C); Maximum OCR value stimulated by the addition of low FCCP at concentrations of 0.75 μM (D) and high 1.5 μM (E); The difference between the levels of basal and maximum respiration (F). OCR values are expressed in units of pmol/min and normalized to 1 mg of total cellular protein (pmol/min/Norm. Unit). Ca Ski subclones with one genomic insert of RT_A coding sequence (B8B5, B8D5, B8D2) are designated as “1 RT_A”, and with six inserts as “6 RT_A” (H6G11, H6D7, H6F8), Ca Ski with six genomic inserts of GFP is given as a control. The respiratory activity of mitochondria in individual subclones was presented in Supplements Figure S4. Histograms are presented as mean ± SD for the analysis performed in quadruplicate. Significant difference from the value of the gene expression level in the GFP subclone (p <0.05; Unpaired t test with Bonferroni correction). ns: not significant; ∗p < 0.05; ∗∗p < 0.01.
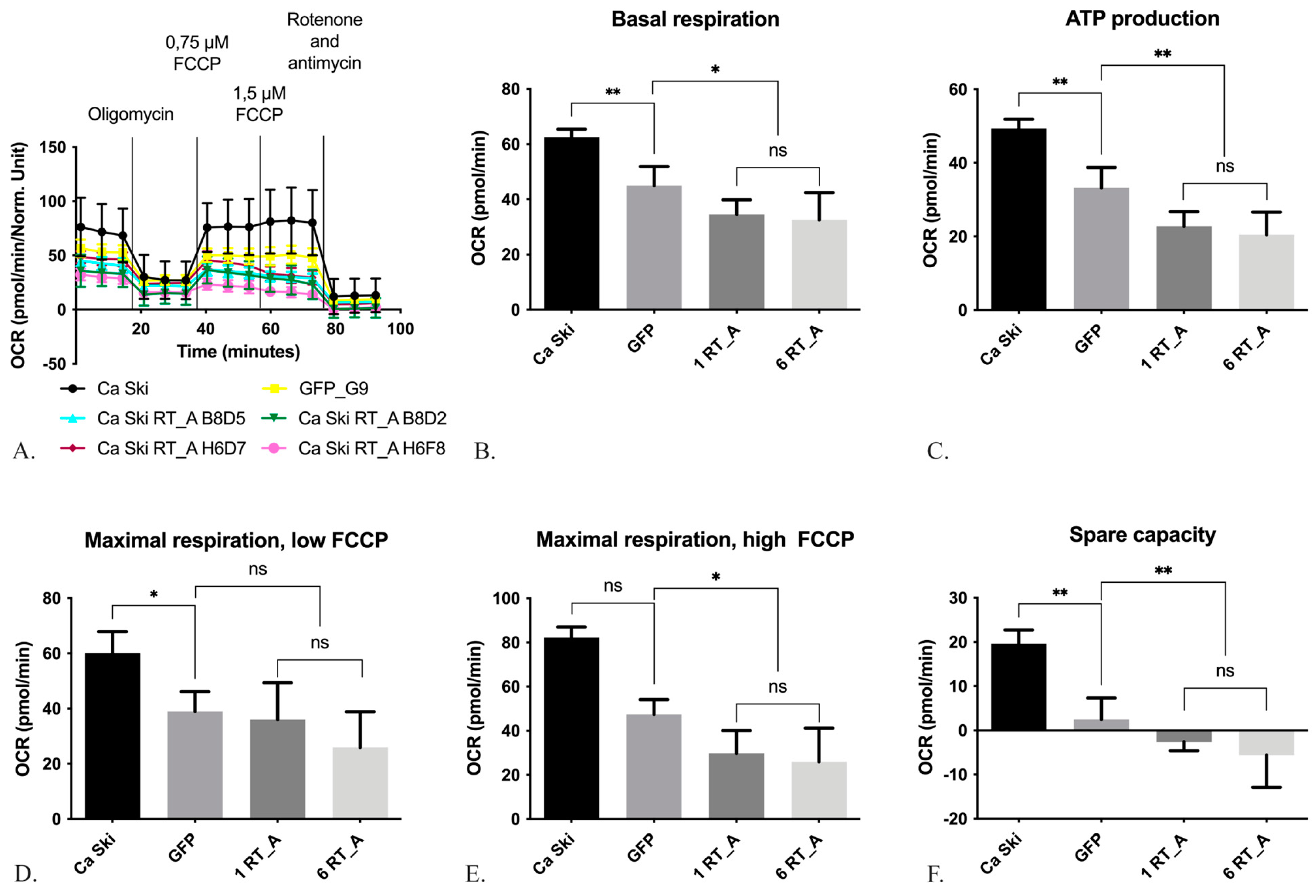
Figure 6.
Derivatives of Ca Ski cells expressing HIV-1 FSU_A consensus reverse transcriptase variants show no ROS production. ROS production was measured using the fluorescent dyes DCFH2-DA (a) and DHE (b). Values are the mean ± standard deviation of two independent analyzes performed in duplicate. ns: not significant; ∗р < 0.05; ∗∗p < 0.01 by Unpaired t test with Bonferroni correction.
Figure 6.
Derivatives of Ca Ski cells expressing HIV-1 FSU_A consensus reverse transcriptase variants show no ROS production. ROS production was measured using the fluorescent dyes DCFH2-DA (a) and DHE (b). Values are the mean ± standard deviation of two independent analyzes performed in duplicate. ns: not significant; ∗р < 0.05; ∗∗p < 0.01 by Unpaired t test with Bonferroni correction.
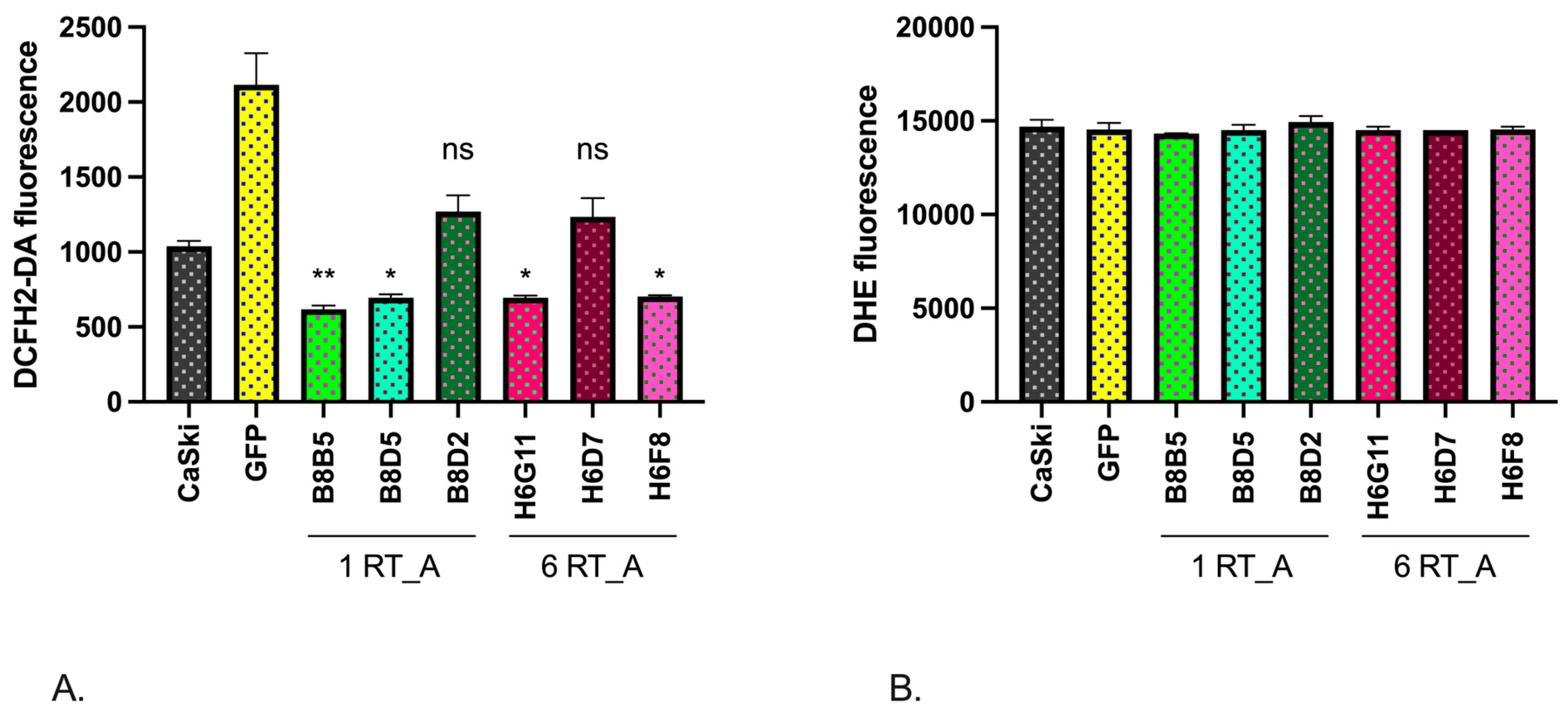
Figure 7.
Relative expression levels of mRNA of NRF2 (A), GCLC (B), NQO1(C) in the derivatives of Ca Ski cells expressing variants of consensus HIV-1 FSU_A reverse transcriptase. Expression level was normalized to the expression levels of GUSB and calculated as fold change compared to the parental Ca Ski line. Results are presented as mean ± SD, ns: not significant; ∗р < 0.05; ∗∗p < 0.01 by Unpaired t test with Bonferroni correction.
Figure 7.
Relative expression levels of mRNA of NRF2 (A), GCLC (B), NQO1(C) in the derivatives of Ca Ski cells expressing variants of consensus HIV-1 FSU_A reverse transcriptase. Expression level was normalized to the expression levels of GUSB and calculated as fold change compared to the parental Ca Ski line. Results are presented as mean ± SD, ns: not significant; ∗р < 0.05; ∗∗p < 0.01 by Unpaired t test with Bonferroni correction.
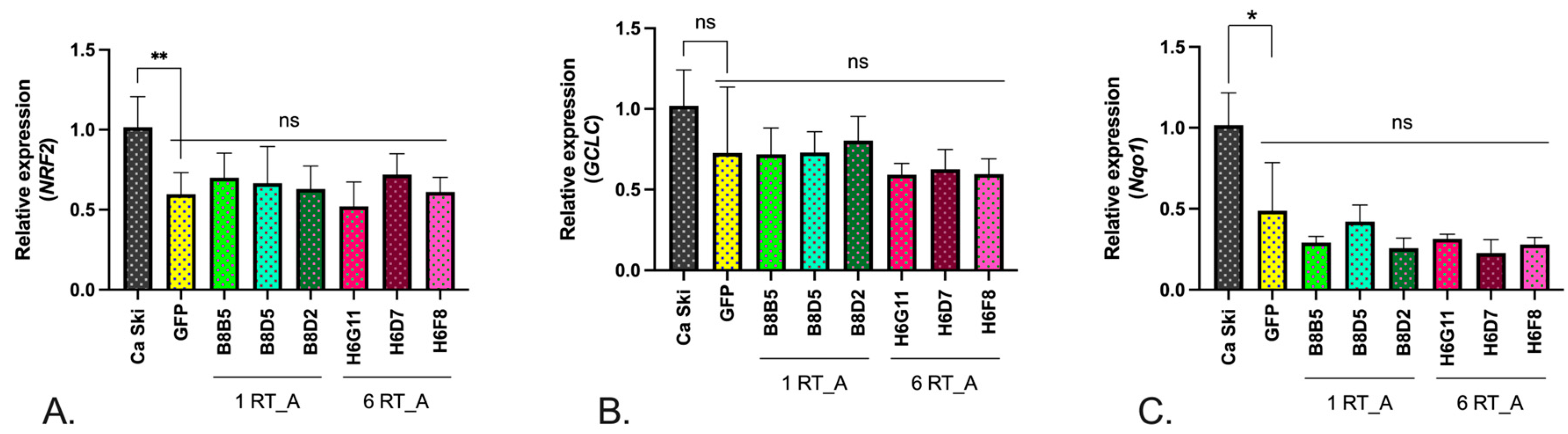
Figure 8.
Relative mRNA expression levels of E-CADHERIN (a), VIMENTIN (b), N-CADHERIN (c) in the derivatives of Ca Ski cells expressing HIV-1 RT_A. Ca Ski subclones with one genomic insert of RT_A coding sequence (B8B5, B8D5, B8D2) are designated as “1 RT_A”, and with six inserts as “6 RT_A” (H6G11, H6D7, H6F8), Ca Ski with six genomic inserts of GFP is given as a control. Expression level was normalized to GAPDH expression and calculated as fold change compared to the parental Ca Ski cell line. Results are presented as mean ± SD, ns: not significant; ∗р < 0.05; ∗∗p < 0.01; ∗∗∗р < 0.001∗∗∗∗p < 0.0001 by Unpaired t test with Bonferroni correction.
Figure 8.
Relative mRNA expression levels of E-CADHERIN (a), VIMENTIN (b), N-CADHERIN (c) in the derivatives of Ca Ski cells expressing HIV-1 RT_A. Ca Ski subclones with one genomic insert of RT_A coding sequence (B8B5, B8D5, B8D2) are designated as “1 RT_A”, and with six inserts as “6 RT_A” (H6G11, H6D7, H6F8), Ca Ski with six genomic inserts of GFP is given as a control. Expression level was normalized to GAPDH expression and calculated as fold change compared to the parental Ca Ski cell line. Results are presented as mean ± SD, ns: not significant; ∗р < 0.05; ∗∗p < 0.01; ∗∗∗р < 0.001∗∗∗∗p < 0.0001 by Unpaired t test with Bonferroni correction.
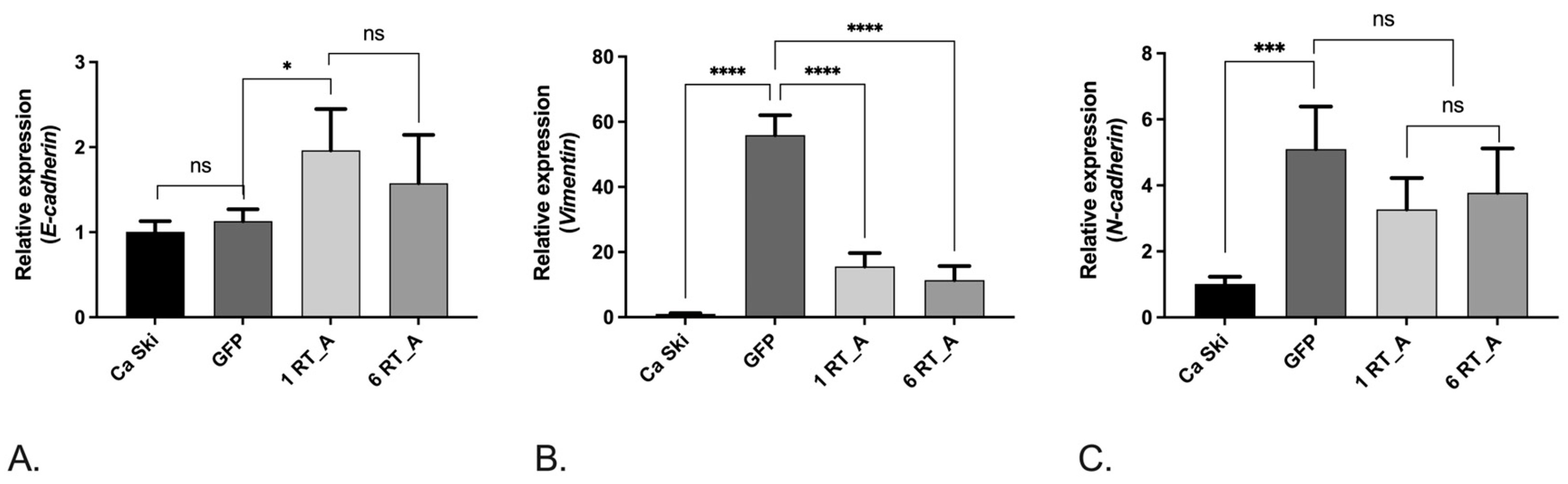
Figure 9.
Colony counts (A) and mean colony size (B) of Ca Ski cells, its subclone with six inserts of GFP DNA (GFP) and subclones with one and six inserts of RT_A DNA (B8B5, B8D5, B8D2, and H6G11, H6D7, H6F8, respectively; Table 1) at day 21 of the assay. Evaluation of clonogenic growth in individual subclones was presented in Supplements Figure S10. Statistical significance was assessed using the Kruskal-Wallis test with Dunn’s multiple comparison test and in pairs using the Mann-Whitney test (∗р < 0.05; ∗∗p < 0.01; ∗∗∗р < 0.001).
Figure 9.
Colony counts (A) and mean colony size (B) of Ca Ski cells, its subclone with six inserts of GFP DNA (GFP) and subclones with one and six inserts of RT_A DNA (B8B5, B8D5, B8D2, and H6G11, H6D7, H6F8, respectively; Table 1) at day 21 of the assay. Evaluation of clonogenic growth in individual subclones was presented in Supplements Figure S10. Statistical significance was assessed using the Kruskal-Wallis test with Dunn’s multiple comparison test and in pairs using the Mann-Whitney test (∗р < 0.05; ∗∗p < 0.01; ∗∗∗р < 0.001).
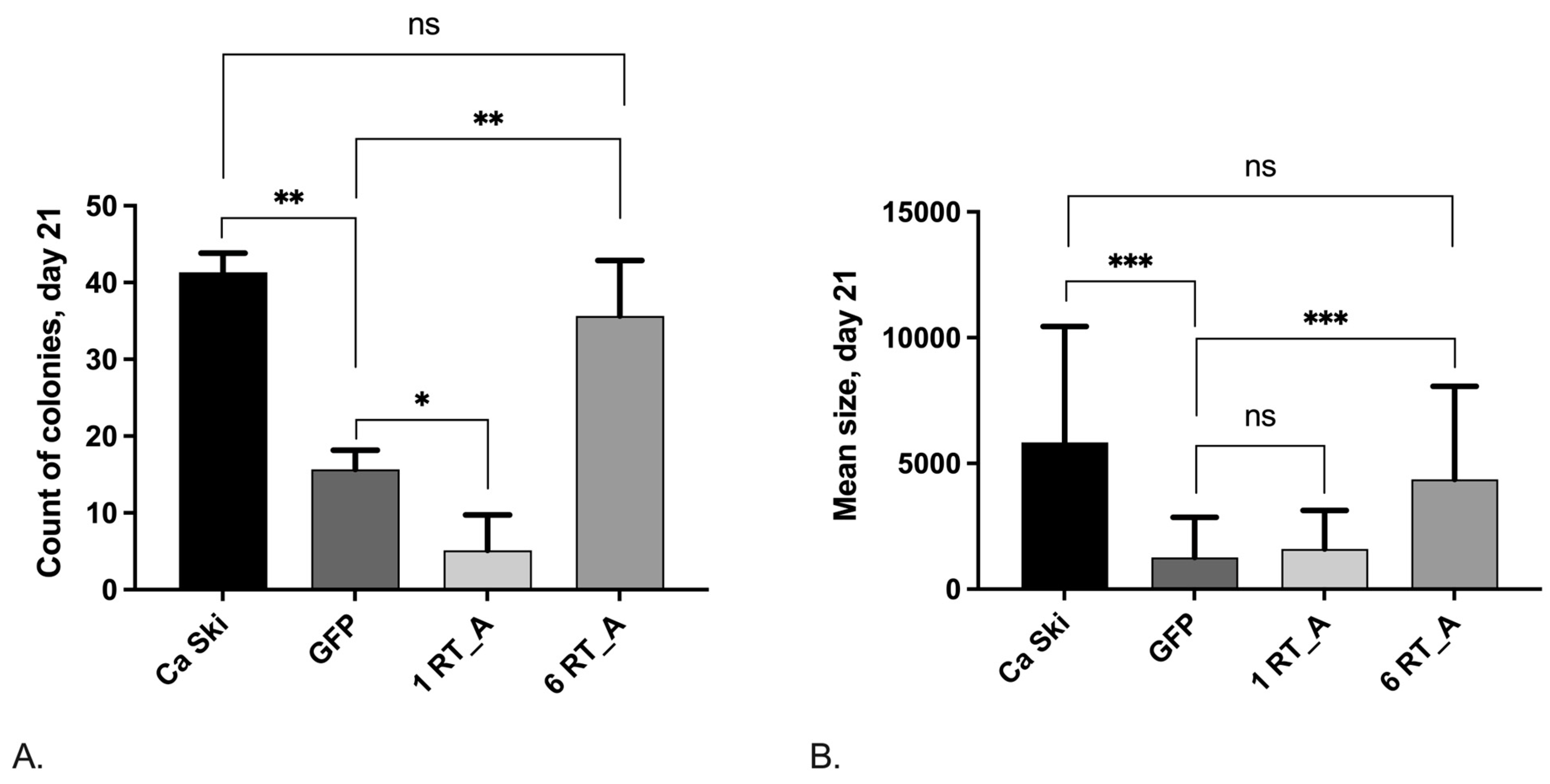
Figure 10.
Formation of solid tumors by Ca Ski derived clones expressing HIV-1 FSU_A reverse transcriptase (RT_A) variants and EGFP fluorescent marker on days 11, 18, and 25 after ectopic implantation of 106 cells in Nu/J mice (A). Ex vivo Ca Ski, Ca Ski GFP G9 and Ca Ski RT_A H6G11 tumor size on day 29 (B). General kinetics of tumor growth (C). Comparison of the sizes of palpable tumors on days 29 (D) and weight of tumors at the end point of the experiment (E). Statistical significance was assessed using the Kruskal-Wallis test with Dunn’s multiple comparison test and in pairs using the Mann-Whitney test (*p < 0.05).
Figure 10.
Formation of solid tumors by Ca Ski derived clones expressing HIV-1 FSU_A reverse transcriptase (RT_A) variants and EGFP fluorescent marker on days 11, 18, and 25 after ectopic implantation of 106 cells in Nu/J mice (A). Ex vivo Ca Ski, Ca Ski GFP G9 and Ca Ski RT_A H6G11 tumor size on day 29 (B). General kinetics of tumor growth (C). Comparison of the sizes of palpable tumors on days 29 (D) and weight of tumors at the end point of the experiment (E). Statistical significance was assessed using the Kruskal-Wallis test with Dunn’s multiple comparison test and in pairs using the Mann-Whitney test (*p < 0.05).
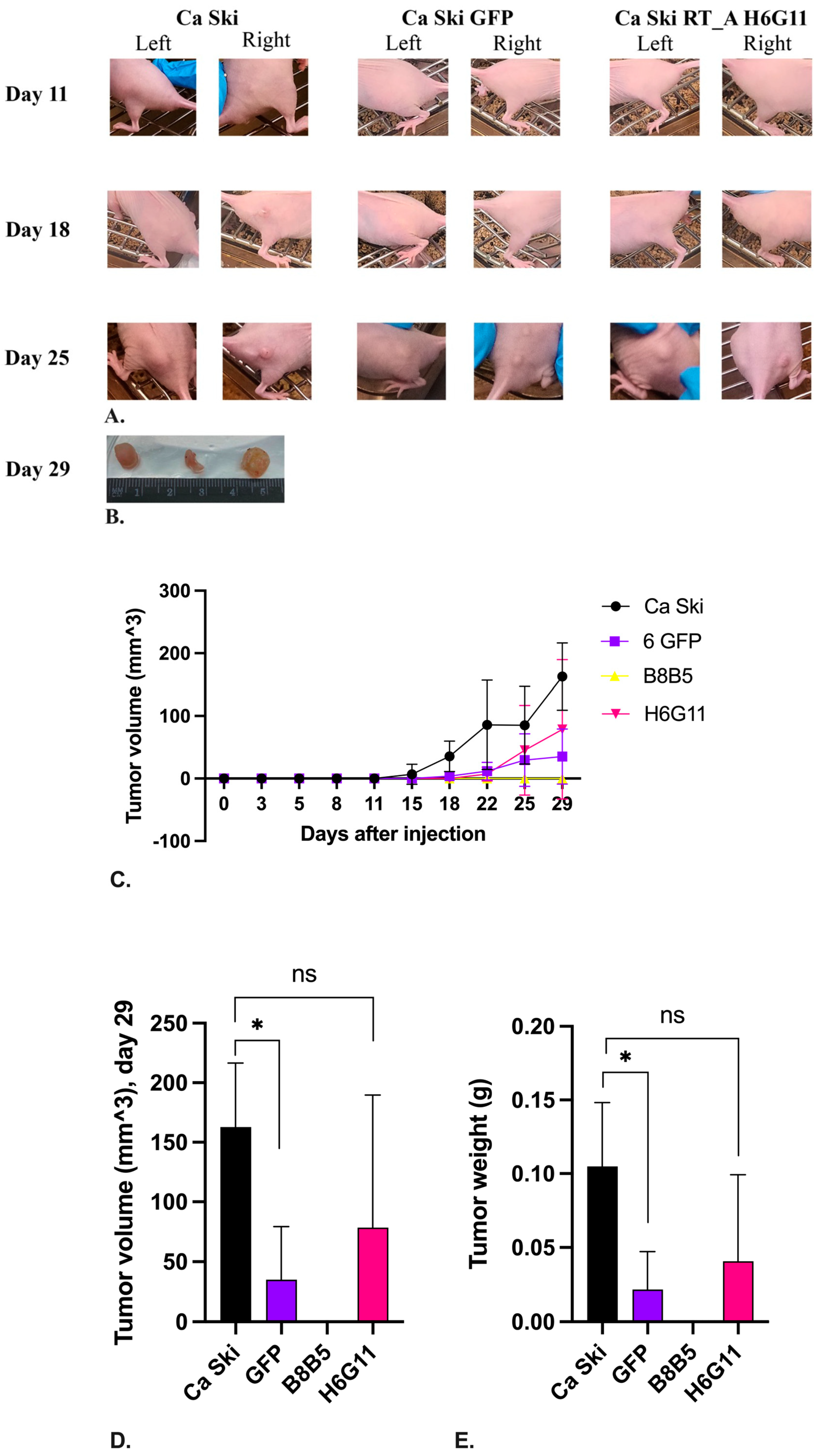
Table 1.
Subclones of Ca Ski expressing the consensus RT of HIV-1 clade A FSU_A strain (RT_A) obtained by lentiviral transduction of Ca Ski cells at different multiplicities of infection (MOI).
Table 1.
Subclones of Ca Ski expressing the consensus RT of HIV-1 clade A FSU_A strain (RT_A) obtained by lentiviral transduction of Ca Ski cells at different multiplicities of infection (MOI).
| Transgene | Monoclonal cell line | MOI | The number of copies per genome | Series | Relative expression of mRNA (RT), 2^-ddCt |
Quantification of RT_A protein expression (fg/cell) |
|---|---|---|---|---|---|---|
| B8B5 | 5 | 1 | 2,40±0,19 | 45,13±4,0 | ||
| B8D5 | 5 | 1 | 1 RT_A | 1±0,53 | 26,82±13,66 | |
| RT_A | B8D2 | 5 | 1 | 1,99±0,19 | 20,50±12,13 | |
| H6G11 | 10 | 6 | 12,61±0,09 | 55,35±12,82 | ||
| H6D7 | 10 | 6 | 6 RT_A | 12,64±0,13 | 52,30±6,87 | |
| H6F8 | 10 | 6 | 14,25±0,32 | 53,26±10,99 |
Table 3.
Multiple regression analysis of the phenotypic parameters of Ca Ski and its GFP, RT_A 1 copy, RT_A 6 subclones determining their rate of migration in wound healing assay. Regression Summary for Dependent Variables “Migration rate” (Ca Ski, 6 GFP, RT_A): R=0.94578193 R2=0.89450345, Adjusted R2=0.81538104, F(6,8) = 11.305, p < 0.00156 Std. Error of estimate: 3.1149.
Table 3.
Multiple regression analysis of the phenotypic parameters of Ca Ski and its GFP, RT_A 1 copy, RT_A 6 subclones determining their rate of migration in wound healing assay. Regression Summary for Dependent Variables “Migration rate” (Ca Ski, 6 GFP, RT_A): R=0.94578193 R2=0.89450345, Adjusted R2=0.81538104, F(6,8) = 11.305, p < 0.00156 Std. Error of estimate: 3.1149.
| B* | Standard Error of b* |
b | Standard Error of b |
T(8) | p-Value | |
|---|---|---|---|---|---|---|
| Intercept | 26,5005 | 9,009944 | 2,94125 | 0,018674 | ||
| RT, fg/cell | 0,50138 | 0,181762 | 9,2949 | 3,369607 | 2,75845 | 0,024734 |
| mRNA E-cadherin, 2DDCt | 2,09612 | 0,277794 | 29,0282 | 3,847040 | 7,54560 | 0,000066 |
| mRNA Twist1, 2DCt | 1,02590 | 0,217805 | 7,0439 | 1,495474 | 4,71017 | 0,001521 |
| mRNA N-cadherin, 2DCt | -1,67859 | 0,273251 | -10,5204 | 1,712571 | -6,14302 | 0,000276 |
| mRNA E6*I, 2DCt | -2,12317 | 0,323863 | -25,7346 | 3,925501 | -6,55575 | 0,000177 |
| ECAR at high glucose, mpH/min | 0,71069 | 0,190328 | 0,9383 | 0,251297 | 3,73403 | 0,005754 |
Table 4.
Correlation of HIV-1 RT_A mRNA expression and protein production and expression of E6*I mRNA with the metabolic and phenotypic characteristics of Ca Ski and Ca Ski subclones (Table 1) (Spearman’s rank correlation test). **Correlations significant with *p < 0.01 (red), *with p<0.05 (blue).
Table 4.
Correlation of HIV-1 RT_A mRNA expression and protein production and expression of E6*I mRNA with the metabolic and phenotypic characteristics of Ca Ski and Ca Ski subclones (Table 1) (Spearman’s rank correlation test). **Correlations significant with *p < 0.01 (red), *with p<0.05 (blue).
| ECAR, low glucose, mpH/min | ECAR, high glucose, mpH/min | Basal respiration, pmol/min | ATP production, pmol/min | Maximal respiration, low FCCP, pmol/min | Maximal respiration, high FCCP, pmol/min | Relative intensity of DCFH2-DA | mRNA E-cadherin, 2DCt | Colony nn, day 21 | Colony area, day 21 |
|
|---|---|---|---|---|---|---|---|---|---|---|
| RT_A mRNA, 2DCt | 0,802027 | 0,696912 | -0,677992 | -0,798874 | -0,651713 | -0,778902 | -0,385992 | 0,408336 | 0,218884 | 0,216436 |
| RT_A, fg/cell | 0,804130 | 0,762084 | -0,604411 | -0,737907 | -0,561314 | -0,673787 | -0,447330 | 0,263754 | 0,163998 | 0,166489 |
| mRNA E6*I, 2DCt | 0,467492 | 0,399381 | -0,585139 | -0,603715 | -0,498452 | -0,669763 | -0,674783 | 0,757391 | -0,655339 | -0,413913 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



