
Submitted:
25 December 2023
Posted:
26 December 2023
You are already at the latest version
Abstract
Active vitamin D derivatives (VDDs) – 1α,25-dihydroxyvitamin D3/D2 and their synthetic analogs – are well-known inducers of cell maturation with the potential for differentiation therapy of acute myeloid leukemia (AML). However, their dose-limiting calcemic activity is a significant obstacle to using VDDs as an anticancer treatment. We have shown that different activators of the NF-E2-related factor-2/Antioxidant Response Element (Nrf2/ARE) signaling pathway, such as the phenolic antioxidant carnosic acid (CA) or the multiple sclerosis drug monomethyl fumarate (MMF), synergistically enhance the antileukemic effects of various VDDs applied at low concentrations in vitro and in vivo. This study aimed to investigate whether glutathione, the major cellular antioxidant and the product of the Nrf2/ARE pathway, can mediate the Nrf2-dependent differentiation-enhancing activity of CA and MMF in HL60 human AML cells. We report that glutathione depletion using L-buthionine sulfoximine attenuated the enhancing effects of both Nrf2 activators concomitant with downregulating vitamin D receptor (VDR) target genes and the activator protein-1 (AP-1) family protein c-Jun levels and phosphorylation. On the other hand, adding reduced glutathione ethyl ester to dominant negative Nrf2-expressing cells re-stored both the suppressed differentiation responses and the downregulated expression of VDR protein, VDR target genes, as well as c-Jun and P-c-Jun levels. Finally, using the transcription factor decoy strategy, we demonstrated that AP-1 is necessary for the enhancement by CA and MMF of 1α,25-dihydroxyvitamin D3-induced VDR and RXRα protein expression, transactivation of the vitamin D response element, and cell differentiation. Collectively, our findings suggest that glutathione mediates, at least in part, the potentiating effect of Nrf2 activators on VDDs-induced differentiation of AML cells, likely through the positive regulation of AP-1.
Keywords:
acute myeloid leukemia
; buthionine sulfoximine
; carnosic acid
; monomethyl fumarate
; vitamin D receptor
; activator protein-1.
1. Introduction
Acute myeloid leukemia (AML) is one of the most aggressive hematologic malignancies, primarily targeting older adults aged ≥ 65. It is characterized by genetic and epigenetic defects that block the development of myeloid progenitor cells in the bone marrow at early stages of differentiation and promote the uncontrolled growth of leukemic blasts. Combination chemotherapy with cytosine β-D-arabinofuranoside (cytarabine) and an anthracycline antibiotic (e.g., daunorubicin) has been the frontline treatment for AML for more than 40 years and is relatively successful for younger patients. However, older individuals are mainly unfit for intensive chemotherapy and their treatment options remain sparse, resulting in a very short median overall survival (6-25 months) [1,2]. Although several novel targeted AML drugs are currently available, their impact on long-term patient survival is yet to be determined [1,3,4]. Since maturation block is the primary feature of AML blasts, differentiation therapy presents an attractive alternative option for treating this disease [5]. One AML subtype, acute promyelocytic leukemia (APL), has been successfully treated with the combination of the natural differentiation inducer all-trans-retinoic acid (ATRA) and arsenic trioxide [6]. However, no differentiation therapy is currently available for non-APL AML.
The hormonal form of vitamins D3 and D2, 1α,25-dihydroxyvitamin D3/D2 (1,25D3/2), is a well-known inducer of myeloid differentiation in various non-APL AML cell types [7,8]. 1,25D3 is the physiological ligand of the vitamin D receptor (VDR), a member of the nuclear receptor subfamily type II that includes retinoid X receptors (RXRs) and retinoic acid receptors. Upon binding 1,25D3, VDR interacts with RXRα and/or other transcription factors, such as purine-rich box-1 (PU.1) or CCAAT enhancer binding protein alpha (CEBPα), to form protein complexes that act as ligand-activated transcription factors [9,10].
A major obstacle to the clinical development of vitamin D derivatives (VDDs) - 1,25D3/2 and their synthetic analogs - for AML therapy is their dose-limiting calcemic toxicity. Clinical trials of different VDDs conducted so far have reported low anticancer efficiency at safe plasma levels of the compounds [7,8,11]. Furthermore, since some AML subtypes showed resistance to VDDs in ex vivo studies, only those patients who are likely to respond would probably benefit from VDD-based differentiation therapy [7]. A possible way of managing VDD toxicity is combining these compounds at tolerated doses with other agents that would potentiate their anticancer effect but not the calcemic activity.
We and others have shown that different plant antioxidants, such as carotenoids [12,13] and polyphenols, e.g., carnosic acid (CA), silibinin and curcumin [12,14,15,16,17,18,19], can synergistically enhance the differentiation-inducing and antiproliferative effects of various VDDs applied at low (sub)nanomolar concentrations on human and murine AML cell lines and patient-derived AML blasts. Notably, combined treatment with CA-rich rosemary extract and low-calcemic VDDs resulted in cooperative antileukemic effects in syngeneic mouse models of AML without inducing hypercalcemia [16,20]. The VDD/CA-induced differentiation was associated with lowered intracellular levels of reactive oxygen species (ROS), upregulated expression of antioxidant enzymes, such as NAD(P)H quinone oxidoreductase-1 (NQO1) and the rate-limiting glutathione-synthesizing enzyme γ-glutamylcysteine synthetase (γGCS), and increased total glutathione content in AML cells [20,21,22]. On the other hand, depletion of cellular glutathione reduced the extent of differentiation [21].
These findings suggested the role of redox-related mechanisms in the differentiation-enhancing effects of polyphenols. Indeed, we have shown that the CA enhancement is mediated by activation of the Nuclear factor erythroid 2-related factor 2 (Nrf2) transcription factor [22], a major regulator of the cytoprotective response to electrophilic agents and oxidative stress [23,24]. This was demonstrated by manipulating Nrf2 activity and expression in U937 human AML cells stably expressing a dominant-negative Nrf2 mutant (dnNrf2) lacking the transactivation domain and those overexpressing the wild-type Nrf2 [22]. Further, we found that besides CA, other structurally distinct Nrf2 activators, including the multiple sclerosis drugs dimethyl fumarate (DMF) and monomethyl fumarate (MMF) [25], synergistically potentiated the antileukemic effects of several VDDs on different AML cell types [26]. Notably, combined treatment with DMF and the highly potent low-calcemic vitamin D2 analog PRI-5202 cooperatively inhibited leukemia progression in a xenograft model of AML [26].
The cooperation between VDDs and Nrf2 activators was associated with a mutual upregulation of VDR, RXRα, and Nrf2 protein levels and activation of VDR and Nrf2 signaling [22,26]. Other transcriptional pathways are likely to contribute to this synergy. For instance, we have demonstrated a cooperative upregulation of several Activator Protein 1 (AP-1) family members and augmented DNA binding and transcriptional activity of AP-1 [15,22,27]. A marked upregulation of the Early Growth Response protein 1 (EGR-1) transcription factor was also observed [15,27]. Stable expression of the wild-type Nrf2 or dnNrf2 in U937 cells resulted in an enhanced or reduced AP-1 upregulation and activation, respectively [22], suggesting that Nrf2 may serve as an upstream regulator of AP-1 in AML cells.
The present study was designed to investigate whether glutathione, the most abundant cellular antioxidant and the product of the Nrf2/antioxidant response element (Nrf2/ARE) signaling pathway [24], may mediate the enhancing effects of Nrf2 activators on VDD-induced differentiation of AML cells. To this end, we employed two approaches: 1) glutathione depletion in nontransfected HL60 cells using BSO and 2) repletion of reduced glutathione (GSH) levels in dnNrf2-expressing HL60 cells by adding membrane-permeant GSH ethyl ester (GEE). We found that co-treatment with BSO attenuated the potentiating effect of both CA and MMF on 1,25D3-induced differentiation. This was paralleled by the downregulation of VDR-responsive genes, the AP-1 family protein c-Jun, and its phosphorylation. On the other hand, the addition of GEE partially reversed the suppressing effects of dnNrf2 on cell differentiation, vitamin D- and Nrf2-related gene and/or protein expression, and c-Jun and P-c-Jun levels. Finally, using the transcription factor decoy strategy [22,28], we demonstrated that cell loading with AP-1-specific oligodeoxynucleotide (ODN) markedly inhibited the enhancing effects of CA and MMF on 1,25D3-induced expression of myeloid differentiation markers and VDR and RXRα proteins, and transactivation of the vitamin D response element (VDRE).
2. Results
2.1. Glutathione depletion attenuates the differentiation-enhancing effects of Nrf2 activators
To characterize the effect of glutathione depletion on 1,25D3-induced differentiation and its enhancement by Nrf2 activators, HL60 cells were preincubated with either vehicle or BSO (30 µM) for 24 h, followed by exposure to 1 nM 1,25D3, 10 µM CA or 50 µM MMF alone, or the combinations of 1,25D3 with either CA or MMF for another 48 h. The extent of myeloid differentiation was assessed by measuring the surface expression of the specific monocytic marker CD14 and the general myeloid marker CD11b using flow cytometry. In accordance with our previous data (e.g., [15,26]), we found that at the non-cytotoxic concentrations used, CA and MMF markedly potentiated the expression of CD14 and CD11b induced by a low concentration of 1,25D3 (1 nM) in a synergistic manner. Co-treatment with BSO only slightly influenced 1,25D3-induced cell differentiation but significantly attenuated the enhancing effects of the two Nrf2 activators (Figure 1a,b).
To document the inhibition of γGCS activity by BSO in our system, we determined changes in the total cellular glutathione levels using the glutathione reductase recycling assay. As expected, incubation with either CA or MMF significantly elevated the glutathione content (Figure 1c), which was accompanied by lowering the cytosolic ROS levels measured using the DCFH-DA fluorescence probe (Figure 1d). Co-incubation with BSO resulted in a marked decrease in both the basal and the treatment-induced glutathione levels, abrogating the CA and MMF stimulation compared to the control cells (Figure 1c). Predictably, this was associated with a marked elevation of ROS levels (Figure 1d). The above results support the notion that glutathione mediates, at least in part, the differentiation-enhancing effects of Nrf2-activating compounds and that this enhancement may involve the ability of glutathione to maintain reducing conditions in AML cells.
2.2. Effects of glutathione depletion on mRNA and protein levels of molecular regulators involved in cell differentiation
To explore the role of glutathione in the enhancement of 1,25D3-induced differentiation of HL60 cells by Nrf2 activators, we examined the effects of glutathione depletion on the mRNA and protein expression of the transcription factors VDR, RXRα and Nrf2 and their target genes. This was carried out using cell samples collected after incubation with 1,25D3, CA, MMF, 1,25D3/CA, and 1,25D3/MMF in the absence or presence of BSO for 48 h, as described in Section 2.1 above.
2.2.1. Glutathione depletion differentially affects mRNA expression of vitamin D- and Nrf2-related genes
Using quantitative RT-PCR (qPCR), we analyzed mRNA expression of the vitamin D-related genes VDR, RXRA (RXRα), CAMP (cathelicidin antimicrobial peptide), CYP24A1 (1,25D 24-hydroxylase), CD14, and ITGAM (CD11b) as well as NFE2L2 (Nrf2) and its target genes NQO1, HMOX1 (heme oxygenase 1), TXNRD1 (thioredoxin reductase 1), GCLC (catalytic subunit of γGCS) and GCLM (modifier subunit of γGCS).
As shown in Figure 2(a), single or combined treatments with 1,25D3, CA or MMF did not affect or even slightly reduced VDR mRNA levels and moderately elevated RXRA expression (Figure 2b). On the other hand, a marked induction of all the VDR target genes tested was detected in 1,25D3-treated cells, while neither CA nor MMF alone had noticeable effects. However, combining 1,25D3 with either Nrf2 activator resulted in a substantial synergistic upregulation of these genes (Figure 2c-f). Of note, the upregulated CD14 and ITGAM expression strongly correlated with the elevated cell surface levels of CD14 and CD11b, respectively (see Figure 1a,b). Similar to VDR, the expression of NFE2L2 was practically unaltered by our treatments (Figure 2g). Yet, CA or MMF significantly upregulated the Nrf2 target genes NQO1, TXNRD1, and GCLM (Figure 2h,j,l). 1,25D3 did not potentiate these effects, nor did it significantly induce any of the Nrf2-responsive genes tested when applied alone. However, both CA and MMF synergistically cooperated with 1,25D3 in upregulating HMOX1 (Figure 2i). Unlike GCLM, the expression of GCLC was unresponsive to any treatment (Figure 2k).
Glutathione depletion with BSO slightly affected VDR and RXRA expression (Figure 2a,b,g). Nonetheless, it markedly suppressed the induction of the vitamin D-responsive genes in cells exposed to 1,25D3, alone or together with CA or MMF (Figure 2c-f). This suppression correlated with the suppression of cell differentiation in BSO-treated cells (see Figure 1a,b). In contrast, BSO treatment further enhanced the induction of all the Nrf2 target genes, except TXNRD1, without affecting NFE2L2 expression (Figure 2g-l).
2.2.2. Glutathione depletion differentially affects the expression of proteins encoded by vitamin D- and Nrf2-related genes
Western blot analysis was used to determine protein levels of VDR and RXRα as well as Nrf2 and the proteins encoded by its target genes: NQO1, heme oxygenase 1 (HO-1), thioredoxin reductase 1 (TrxR1), and the catalytic (ɣ-GCSc) and modifier (ɣ-GCSm) subunits of γGCS. As shown in Figure 3(a,b), treatment with 1,25D3 strongly increased VDR levels, while CA or MMF had only slight effects. However, both Nrf2 activators dramatically enhanced the impact of 1,25D3 in a synergistic manner. Unlike VDR, RXRα expression was more responsive to CA or MMF than to 1,25D3, but still, combined treatments produced a synergistic effect (Figure 3a,c). As expected, protein levels of Nrf2 and most of its target gene products (NQO1, TrxR1, ɣ-GCSc and ɣ-GCSm) were significantly upregulated by CA and/or MMF (Figure 3a,d,e,g-i). While 1,25D3 alone had a moderate or no effect, it was capable of potentiating the impact of one (Figure 3a,d,h) or both (Figure 3a,f,g,i) Nrf2 activators in most cases. Interestingly, HO-1 expression was insensitive to single agents, but it was highly evident in combination-treated cells (Figure 3a,f). Likewise, ɣ-GCSc levels were elevated by the 1,25D3/CA combination, while neither 1,25D3 nor CA alone had any significant effect (Figure 3a,h).
As BSO markedly inhibited VDR target gene induction without consistently affecting VDR and RXRA mRNA levels (see Figure 2a-f), it also had no effect on VDR and RXRα protein levels in cells treated with 1,25D3 and Nrf2 activators separately or the 1,25D3/CA combination. The effect of 1,25D3 + MMF on RXRα expression was even potentiated under these conditions (Figure 3a-c). The enhanced induction of most Nrf2-regulated genes in glutathione-depleted cells (see Figure 2h,i,k,l) was generally paralleled by increased upregulation of the corresponding proteins (Figure 3e,f,h,i). The Nrf2 protein levels also tended to increase following some of the treatments (Figure 3d), even though the NFE2L2 gene expression was unaffected by BSO (Figure 2g).
We have previously shown that the transcription factor AP-1 is activated by concerted actions of 1,25D3 and plant polyphenols in HL60 and U937 AML cells. This was characterized by increased AP-1 DNA binding and transcriptional activity and associated with upregulation and phosphorylation of the AP-1 family proteins c-Jun and ATF-2 [21,22,27]. Consistent with these findings, both 1,25D3/CA and 1,25D3/MMF combinations synergistically upregulated c-Jun at comparable magnitudes. Interestingly, when added alone, MMF was a more potent inducer than 1,25D3 or CA (Figure 3j,k). The levels of the phosphorylated (activated) form of c-Jun (P-c-Jun) were similarly elevated by the above treatments, MMF alone being as effective as its combination with 1,25D3 (Figure 3j,l). Notably, the increases in both c-Jun and P-c-Jun levels caused by MMF, with or without 1,25D3, were dramatically reduced following co-incubation with BSO, whereas the effects of CA ± 1,25D3 were slightly affected (Figure 3j,k,l).
In summary, the above data indicate that glutathione depletion in HL60 cells resulted in a marked inhibition of VDR target gene expression induced by 1,25D3, particularly in combination with Nrf2 activators. This occurred without significantly reducing VDR mRNA and protein levels, while RXRα expression even tended to increase. On the other hand, the induction of Nrf2 and its target genes by Nrf2 activators and their combinations with 1,25D3 was generally augmented in glutathione-depleted cells. Our results also suggest that cellular glutathione is necessary for upregulating c-Jun protein levels and phosphorylation induced by MMF, alone and combined with 1,25D3, and is less critical for c-Jun regulation by 1,25D3 ± CA.
2.3. Introduction of exogenous GSH partially reverses the suppressing effect of a dominant-negative Nrf2 mutant on myeloid differentiation of HL60 cells
To further explore the role of glutathione in VDD-induced differentiation and its enhancement by Nrf2 activation, we employed HL60 cells stably expressing a dominant-negative Nrf2 mutant (dnNrf2), which lacks the transactivation domain [22,29], and those transfected with either an empty vector (pEF). These experiments were performed using the clinically approved vitamin D2 analog paricalcitol [30] as a VDD and CA as an Nrf2 activator. We have reported that 1,25D3 and paricalcitol display comparable differentiation-inducing potencies in the absence or presence of Nrf2 activators [26]. Similar to our data obtained in dnNrf2-expressing U937 cells [22], the extent of myeloid differentiation induced by paricalcitol and enhanced by CA was substantially lower in dnNrf2-HL60 cells than in pEF-HL60 cells (Figure 4a,b).
Since dnNrf2 inhibits Nrf2 transcriptional activity, we hypothesized that dnNrf2-HL60 cells produce less glutathione than pEF-HL60 controls. Indeed, the basal and CA-induced total glutathione production in dnNrf2-HL60 was significantly lower than in pEF-HL60 cells (Figure 4c). Thus, we suggested that re-introducing GSH to dnNrf2-HL60 cells would improve the differentiation response to paricalcitol and its combination with CA. Since GSH is a well-known antioxidant, we also examined if the potential improvement of differentiation would specifically be attributed to the molecular features of GSH or would be due to its general antioxidant effect. To this end, we compared the effects of a membrane-permeable GSH ethyl ester (GEE) and an unrelated antioxidant, Trolox, on pEF-HL60 and dnNrf2-HL60 cell differentiation. Cells were pre-incubated with a vehicle, 250 µM GEE or 300 µM Trolox for 1 h, followed by treatment with 2.5 nM paricalcitol, 10 µM CA or their combination, for another 48 h, followed by the flow cytometric CD14 and CD11b assay.
The results demonstrated that Trolox did not affect either the basal or induced cell differentiation of dnNrf2-HL60 and pEF-HL60 cells (Figure 5a,b). In contrast, the addition of GEE essentially reversed the inhibitory effect of dnNrf2 on cell differentiation, restoring the responsiveness of dnNrf2-HL60 cells to paricalcitol ± CA nearly up to the levels detected in pEF-HL60 cells. In pEF-HL60 cells, the augmenting effect of GEE was relatively less pronounced compared to dnNrf2-HL60 cells (Figure 5a,b). N-acetyl cysteine (1000 µM), a precursor of L-cysteine, which is the rate-limiting factor in cellular glutathione biosynthesis [31], also tended to enhance the differentiation of dnNrf2-HL60 cells; however, it was less effective than GEE (Supplementary Figure S1b). There was no enhancement in NAC-treated pEF-HL60 cells and even a small reduction in CD14 and CD11b surface expression was observed (Supplementary Figure S1a).
We then examined whether the difference between GEE and Trolox in affecting cell differentiation correlated with their possible differential influence on intracellular ROS levels. Thus, the abilities of the two compounds to counteract H2O2-induced ROS generation in pEF-HL60 and dnNrf2-HL60 cells were determined. Cells were incubated with vehicle, 250 µM GEE, or 300 µM Trolox for 48 h, followed by exposure to 10 µM H2O2 for an additional 15 min. Cytosolic ROS levels were then measured by flow cytometry using the fluorescent probe DCFH2-DA. The data demonstrated that in pEF-HL60 cells, the basal ROS level was unaffected by either GEE or Trolox (Figure 5c), while in dnNrf2-HL60 cells, it was surprisingly elevated by GEE, but not by Trolox (Figure 5d). Incubation of vehicle-treated cells of both types with 10 µM H2O2 resulted in similar increases in ROS levels (Figure 5c,d). The addition of H2O2 to GEE-treated pEF-HL60 cells had approximately the same effect as in the vehicle-treated cells (Figure 5c); however, in GEE-treated dnNrf2-HL60 cells, the prooxidant effect of H2O2 was markedly enhanced (Figure 5d). In contrast, Trolox treatment significantly prevented H2O2-induced ROS generation in both cell types (Figure 5c,d).
Collectively, the results demonstrate that the inhibitory effect of dnNrf2 on paricalcitol ± CA-induced differentiation of HL60 cells correlated with decreased glutathione production (Figure 4 and Figure 5), whereas adding GEE reversed this inhibition. Notably, the rescuing effect of GEE was opposite to that of the glutathione-depleting agent BSO, which suppressed differentiation of intact HL60 cells (Figure 1a,b), even though both compounds appeared to act as prooxidants (compare Figure 1d and Figure 5d). Combined with the fact that the antioxidant Trolox did not affect the surface expression of CD14 and CD11b, the above opposite effects of GEE and BSO imply that in our system, the differentiation-promoting action of glutathione is not mediated by cytosolic ROS.
2.4. Effects of exogenous GSH on mRNA and protein levels of molecular regulators of cell differentiation in vector-transfected and dominant-negative Nrf2-expressing HL60 cells
We next determined changes in gene and protein expression associated with restoring the paricalcitol ± CA-induced differentiation of dnNrf2-expressing HL60 cells by GEE. For this purpose, pEF-HL60 and dnNrf2-HL60 cells were exposed to vehicle, 2.5 nM paricalcitol, 10 µM CA, with or without 250 µM GEE, for 48 h followed by qPCR and Western blot analyses, as described above (Section 2.2).
2.4.1. Regulation of vitamin D- and Nrf2-related gene expression by GSH ethyl ester
As shown in Figure 6, basal mRNA levels of the vitamin D- and Nrf2-related genes tested were similar in pEF-HL60 and dnNrf2-HL60 cells. Consistent with the data obtained in nontransfected HL60 cells (Figure 2), in the vector-transfected cells, the expression of VDR was unaffected by paricalcitol ± CA treatments (Figure 6a), and RXRA was moderately induced by CA and its combination with paricalcitol (Figure 6b). The expression of VDR target genes (CAMP, CYP24A1, CD14, and ITGAM) was upregulated to a varying extent by the VDD and further substantially enhanced by adding the polyphenol (Figure 6c-f). Notably, stable transfection of dnNrf2 in HL60 cells generally reduced the responsiveness of the above target genes to paricalcitol and markedly suppressed the enhancing effects of CA relative to those detected in pEF-HL60 cells (Figure 6c-f). This inhibition correlated with relatively lower CD14 and CD11b surface levels in paricalcitol ± CA-treated dnNrf2-HL60 cells, as determined by flow cytometry (Figure 4 and Figure 5).
The introduction of exogenous GSH in the form of GEE was generally without a significant effect on VDR and RXRA mRNA levels, except moderately upregulating these genes in paricalcitol/CA-treated dnNrf2-HL60 cells, but not in pEF-HL60 cells (Figure 6b). In contrast to the inhibitory effect of glutathione depletion on the expression of VDR target genes (Figure 2c-f), co-incubation with GEE mostly had a positive impact, depending on the treatment and transfectant type. For instance, GEE did not significantly affect CAMP and CYP24A1 induction by paricalcitol alone in either pEF-HL60 or dnNrf2-HL60 cells while tending to augment the effect of the paricalcitol/CA combination to some degree (Figure 6c, d). On the other hand, paricalcitol alone-induced expression of CD14 and ITGAM was enhanced in the presence of GEE in both cell types. In pEF-HL60 cells, GEE did not further augment paricalcitol/CA-induced CD14 and ITGAM upregulation. However, GEE addition to dnNrf2-HL60 cells completely restored the synergistic induction of the two genes to the levels established in the pEF-HL60 controls (Figure 6e, f).
Similar to nontransfected HL60 cells, CA or paricalcitol did not induce NFE2L2 in either cell type (Figure 6g). As expected, CA significantly upregulated most of the tested genes known to be driven by Nrf2 (NQO1, HMOX1, TXNRD1, and GCLM) in pEF-HL60 cells. Paricalcitol alone had no effect and even tended to attenuate the CA induction of NQO1 and GCLM, but it could positively cooperate with CA in inducing HMOX1 (Figure 6h-l). Surprisingly, none of these genes was significantly repressed by dnNrf2 transfection. Instead, there was even greater upregulation of NFE2L2, HMOX1, TXNRD1, and GCLC in dnNrf2-HL60 cells relative to pEF-HL60 cells (Figure 6g-l).
Interestingly, similar to BSO-treated HL60 cells (Figure 2h,i,k,l), co-incubation with GEE led to a significant potentiation of CA- and/or paricalcitol/CA-induced expression of Nrf2-related genes either in both transfectant types (NQO1, HMOX1, and GCLM) or just in dn-Nrf2-HL60 cells (NFE2L2, TXNRD1, and GCLC), the latter transfectant displaying more robust responses (Figure 6g-l).
2.4.2. Regulation of vitamin D- and Nrf2-related protein expression by GSH ethyl ester
Western blot analysis demonstrated that in pEF-HL60 cells, both paricalcitol and, to a lesser extent, CA upregulated VDR protein levels, and their combination produced a synergistic effect (Figure 7a,b). RXRα expression was induced CA while paricalcitol was ineffective either in the absence or presence of the polyphenol (Figure 7a, c). Stable transfection of dnNrf2 slightly affected VDR upregulation by single compounds but practically abolished their synergistic activity. Likewise, RXRα induction by CA ± paricalcitol was significantly weaker in dnNrf2-HL60 cells compared to their empty vector-transfected counterparts (Figure 7a-c). CA significantly upregulated Nrf2 and its target gene products NQO1, HO-1, TrxR1, γ-GCSc, and γ-GCSm in pEF-HL60 cells, and this effect was predictably less pronounced in dnNrf2-HL60 cells. Paricalcitol did not significantly affect the levels of these proteins or their upregulation by CA in any of the two cell types (Figure 7a,d-i).
Interestingly, co-treatment with GEE increased the basal levels of most vitamin D- and Nrf2-related proteins tested in pEF-HL60 and dnNrf2-HL60 cells (Figure 7a-i). GEE further augmented VDR upregulation by paricalcitol ± CA in both cell types and largely restored its synergistic induction by the VDD/CA combination in dnNrf2-HL60 cells (Figure 7a,b). Likewise, the upregulation of Nrf2 and the related proteins by CA and/or its combination with paricalcitol was generally enhanced by GEE, to a varying extent, in one or both cell types (Figure 7a,d-i). On the other hand, CA-induced RXRα upregulation was surprisingly suppressed by GEE in these samples, while the effects of the paricalcitol/CA combination did not change significantly (Figure 7a,c).
The protein levels of c-Jun and P-c-Jun increased to some extent following single treatments with paricalcitol and CA in both pEF-HL60 and dnNrf2-HL60 cells, but a cooperative effect of the combination was clearly seen only in pEF-HL60 cells (Figure 7j,k,l). Adding GEE moderately increased the basal and paricalcitol-induced expression of c-Jun and its phosphorylated form. However, it dramatically potentiated the effects of CA and its combination with paricalcitol, restoring the synergy between the two agents in dnNrf2-HL60 cells (Figure 7j,k,l).
In summary, co-treatment with GEE enhanced the induction of VDR (Figure 7a,b) and its target genes by paricalcitol and its combination with CA (Figure 6c-f). A similar enhanced upregulation of c-Jun and its phosphorylated form was also observed (Figure 7j-l). These findings directly correlated with augmenting paricalcitol ± CA-induced myeloid differentiation, especially in dnNrf2-expressing cells (Figure 5b,c). The induction of Nrf2 (Figure 6g and Figure 7d) and its target genes (Figure 6h-l and Figure 7e-i) by CA ± paricalcitol increased in the presence of exogenous GSH. However, similar increases were observed in glutathione-depleted cells (see Figure 2h,i,k,l and Figure 3d-f,h,i), in which the induction of differentiation was suppressed (Figure 1a,b). A complex relationship between glutathione and the Nrf2/ARE pathway in AML cells induced to differentiate by VDDs and their combination with Nrf2 activators will be addressed in the Discussion.
2.5. Involvement of AP-1 in the regulation of VDR/RXRα protein expression and transcriptional activity and the enhancement of 1,25D3-induced cell differentiation by Nrf2 activators
As demonstrated above, glutathione depletion resulted in a downregulation of c-Jun protein expression, whereas the introduction of external GSH produced the opposite effect. These data suggest that glutathione may regulate AP-1 expression and activity, which in turn may influence VDD-induced myeloid differentiation of AML cells. We, thus, employed the AP-1 decoy strategy to explore the involvement of AP-1 in the differentiation of HL60 cells, VDR and RXRα protein expression, and VDRE transactivation induced by 1,25D3 and its combinations with Nrf2 activators.
Cells were preincubated with either vehicle, 10 µM TRE-ODN or mutant TRE (mTRE)-ODN, for 24 h followed by treatment with 1 nM 1,25D3, 10 µM CA or 50 µM MMF, alone or in combination, for an additional 48 h. The results demonstrated that neither TRE-ODN nor mTRE-ODN affected CD14 and CD11b surface expression induced by 1,25D3 alone; however, the differentiation-enhancing effects of CA and MMF were practically abolished by TRE-ODN, but not by mTRE-ODN (Figure 8a). Notably, this was paralleled by a marked reduction in VDR and RXRα protein levels, as determined by Western blotting (Figure 8b,c). Consistently, the VDR/RXRα transcriptional activity was also profoundly reduced by TRE-ODN, as measured by the VDRE-luciferase reporter assay (Figure 8d).
These results strongly suggest that AP-1 is essential for the synergistic enhancement of 1,25D3-induced differentiation of AML cells by CA and MMF, likely through the positive regulation of VDR/RXRα protein levels and transcriptional activity.
3. Discussion
Accumulating evidence indicates that the transcription factors Nrf2 [32,33,34,35,36] and AP-1 [28,37,38,39,40,41,42] play significant roles in the differentiation of various normal and cancer cell types, including hematopoietic cells. We have previously reported that the antileukemic synergy between VDDs and Nrf2 activators is associated with a mutual upregulation of VDR and Nrf2 signaling [22,26] and that Nrf2 may function as an upstream regulator of VDR, RXRα and AP-1 protein levels in AML cells [22]. However, the mode of the interaction between the Nrf2, AP-1 and VDR/RXRα pathways remains unclear. The present study was designed to explore the role of glutathione as the potential mediator of the differentiation-enhancing effects of Nrf2 activators in this system.
Glutathione is a ubiquitous thiol tripeptide formed by glutamic acid, cysteine, and glycine. It is synthesized in the cytosol by consecutive action of two enzymes, γ-GCS (the rate-limiting enzyme) and glutathione synthetase, and reaches millimolar intracellular concentrations [24,31]. Nrf2, AP-1, and nuclear factor kappa B (NFκB) are among the key transcription factors that regulate the expression of these enzymes [31]. The reduced form of glutathione (GSH) functions as the principal cellular reducing agent and antioxidant and participates in various regulatory processes, including cytoprotection, cell signaling, metabolism of xenobiotics, gene expression, protein synthesis and modification, cell cycle, apoptosis, and immunomodulation [24,43,44,45,46].
Glutathione depletion has been shown to impair the differentiation of various cell types. For instance, Esposito et al. [47] reported that the glutathione-conjugating compound diethyl maleate inhibited 12-O-tetradecanoylphorbol-13-acetate (TPA)-driven differentiation of HL60 and KG-1 AML cells. We showed that the specific γGCS inhibitor BSO attenuates 1,25D3- and 1,25D3/CA-induced differentiation of HL60 cells [21]. Similar results were obtained in other cell types, such as T cells [48], dendritic cells [49], macrophages [50], C2C12 skeletal muscle cells [51], osteoblasts [52], and osteoclasts [53]. These data indicate an essential role of glutathione in cell maturation.
The main findings of the present study are summarized below and schematically represented in Figure 9.
3.1. Involvement of glutathione in the regulation of VDR signaling and myeloid differentiation
We found that glutathione depletion and repletion had opposite impacts on the induction of monocytic differentiation of HL60 cells. Although BSO minimally attenuated the 1,25D3-induced surface expression of CD14 and CD11b, it significantly suppressed the enhancing effects of both Nrf2 activators. This correlated with decreased induction of CD14 and ITGAM and two other known vitamin D-responsive genes, CAMP and CYP24A1. Notably, target gene expression was inhibited without noticeably reducing mRNA and protein levels of VDR and RXRα, suggesting that glutathione depletion primarily inhibited VDR transcriptional activity in our system.
In contrast to BSO, exogenous GSH (as GEE) or, to a lesser extent, its precursor NAC augmented cell differentiation induced by paricalcitol and its combination with CA. This effect was especially pronounced in dnNrf2-expressing HL60 cells, which exhibited a relatively lower level of differentiation and glutathione content than vector-transfected cells. The boosted differentiation was accompanied by the increased expression of both VDR protein and all the vitamin D target genes tested. The above results indicate that glutathione positively regulates VDR transcriptional activity and can mediate, at least in part, the enhancing effect of Nrf2 activators on VDD-induced differentiation of HL60 cells.
Interestingly, using similar approaches, Fujita et al. [53] have shown that BSO suppresses TNFα-stimulated osteoclast differentiation in vitro, while exogenous GSH promotes it both in cell culture and a mouse model of lipopolysaccharide-induced osteoclastogenesis. These opposite effects were associated with corresponding changes in the nuclear localization of the Nuclear Factor of Activated T cells c1 (NFATc1), a master regulator of osteoclastogenesis, and the expression of osteoclast-specific genes [53].
3.2. Modulation of glutathione levels and Nrf2/ARE signaling: Role of the intracellular ROS accumulation
Cellular Nrf2 levels are primarily regulated by Kelch-like ECH-associated protein 1 (Keap-1), a subunit of Cullin 3 (CUL3)-based E3 ubiquitin ligase. Under physiological conditions, Keap-1 physically binds Nrf2 and promotes its proteasomal degradation. ROS and electrophilic compounds, e.g., the quinone form of CA [54] and fumaric acid esters, react with cysteine SH-groups of Keap-1, triggering dissociation and cellular accumulation of Nrf2 [23,55,56].
Accordingly, treating intact or vector-transfected HL60 cells with CA or MMF increased Nrf2 protein levels and the expression of Nrf2 target genes and encoded proteins, including γGCS subunits. This correlated with an increase in the total glutathione levels. As expected, both dnNrf2-expressing and BSO-treated cells had lower basal and induced glutathione levels than the corresponding reference cells. However, only in dnNrf2-HL60 cells was this associated with impaired induction of Nrf2 and its target gene products. In contrast, BSO-treated cells exhibited enhanced induction of most tested genes and encoded proteins attributed to the Nrf2/ARE pathway. The latter could be due to the compensatory upregulation of this and other redox-sensitive regulatory pathways in response to ROS accumulation caused by pharmacological inhibition of γGCS enzymatic activity.
Unexpectedly, GEE was also found to act as a prooxidant, but only in dnNrf2-HL60 cells with impaired antioxidant defense, even though we used the compound at a much lower concentration (0.25 mM) compared to other studies (1.0-5.0 mM) [57,58,59,60]. This effect might be due to reductive stress, which can result in excess ROS generation [61,62,63]. Nonetheless, both dnNrf2-HL60 cells and pEF-HL60 cells exhibited enhanced induction of Nrf2-related genes and proteins when co-treated with GEE. Therefore, GSH appears to promote Nrf2/ARE activation by electrophilic agents independently of the cytosolic ROS levels, but ROS accumulation in GEE-treated dnNrf2-HL60 cells may have an additional positive effect on this pathway.
3.3. Modulation of c-Jun by glutathione and the role of AP-1 in differentiation enhancement
The transcription factor AP-1 is a dimeric protein complex composed of transcription factors belonging to the Jun, Fos, ATF, and Maf families, which controls the expression of various genes regulating cell proliferation, cell cycle, apoptosis, and differentiation [37,64,65]. It has also been established that AP-1 can be upregulated and activated by ROS to induce the expression of antioxidant and detoxifying enzymes [66,67]. Furthermore, c-Jun binding to the NFE2L2 promoter was found to transcriptionally upregulate Nrf2, leading to an antioxidant effect [68]. Additionally, c-Jun can dimerize with Nrf2 and activate Nrf2/ARE-induced transcription [69].
Here, we demonstrated that, consistent with our previous findings [22,27], VDDs and CA strongly cooperated in increasing c-Jun protein expression and phosphorylation. On the other hand, MMF was quite active alone, particularly in elevating P-c-Jun levels. Interestingly, another fumaric acid ester, DMF, was shown to differentially affect c-Jun in a cell type- and treatment-dependent manner. For instance, DMF upregulated c-Jun and P-c-Jun levels in macrophage migration inhibitory factor-stimulated human keratinocytes [70] but inhibited hypoxia-induced c-Jun phosphorylation in endothelial cells [71].
It was previously reported that inhibition of TPA-induced differentiation of AML cells by glutathione depletion was associated with a reversible reduction in DNA binding of AP-1 [47]. In line with these data, we found that both BSO treatment and dnNrf2 expression reduced c-Jun and P-c-Jun levels in our experimental system. Although both BSO and GEE induced ROS generation and promoted Nrf2 signaling in HL60 cells, glutathione depletion and repletion had opposite effects on c-Jun levels and phosphorylation. These results suggest that it is glutathione, and probably not other Nrf2/ARE activities, that positively regulates AP-1. By exploiting the transcription factor decoy strategy [22,28,72], we obtained evidence supporting the mediatory function of AP-1 in the differentiation-enhancing effects of the Nrf2 activators, probably via upregulating VDR/RXRα levels and transcriptional activity.
3.4. A possible role of proteasome inhibition in a cooperative upregulation of VDR and Nrf2 protein expression by VDDs and Nrf2 activators
It has been reported that in 1,25D3-treated HL60 cells, VDR protein levels are elevated without significant changes in VDR gene expression [73,74]. Here, we observed a similar lack of VDR induction by VDDs alone and also when VDR protein levels were synergistically increased by adding Nrf2 activators. These data indicate that VDR upregulation occurred at posttranscriptional or posttranslational levels. It was previously suggested that liganded VDR undergoes conformational changes, which prevent its proteolysis [75,76]. Indeed, several studies have demonstrated that 1,25D3 upregulates VDR by protecting it from proteasomal degradation [77,78,79]. Interestingly, 1,25D3 was also found to promote Nrf2 accumulation in bone marrow mesenchymal stem cells by inhibiting its degradation via transcriptionally repressing Keap-1 [80].
Various natural polyphenolic compounds have been shown to inhibit or activate the ubiquitin/proteasome pathway depending on multiple factors [81]. For instance, resveratrol acted as a proteasome inhibitor in breast cancer cells, inducing the accumulation of the ∆16HER2 splice variant of HER-2 [82], but promoted proteasomal degradation of Nanog in glioma stem cells [83]. These effects are also dose-dependent, e.g., curcumin increases proteasome activity at low concentrations (≤1 µM) but inhibits it at high concentrations (≥10 µM) [84].
Although both CA and MMF can activate the Nrf2/ARE pathway by inducing Keap-1-Nrf2 dissociation, which results in Nrf2 protein stabilization, CA has been shown to promote proteasomal degradation of other proteins in cancer cells (e.g., [85,86]). The major oxidized CA metabolite, carnosol, was found to target several proteins to proteasome degradation in breast cancer cells [87,88] and to directly inhibit proteasome activity in colon cancer cells [89]. DMF was shown to promote protein degradation in fibroblasts [71,90]. However, both DMF and MMF enhanced the cytotoxic effect of proteasome inhibitors in other cell types [91,92].
To the best of our knowledge, there have been no reports on regulating VDR protein proteolysis by polyphenols and fumaric acid esters. Still, CA (or its oxidized metabolites) and MMF could potentially inhibit VDR proteasomal degradation in VDD-treated HL60 cells, which enhanced VDR protein, but not mRNA, expression following combined treatments. Conversely, VDDs might cooperate with the electrophilic compounds to stabilize Nrf2. In addition, our data demonstrate that treatment with exogenous GSH alone upregulated most proteins tested in this study, suggesting a contribution of a general activation of protein synthesis or stabilization under these conditions.
4. Materials and Methods
4.1. Materials
Carnosic acid (>98%) was obtained from ShenZhen Ipure Biological Import and Export Co., Ltd. (Shenzhen, China). 1,25D3 was purchased from Selleck Chemicals (Houston, TX). Monomethyl fumarate (MMF) and DMSO were obtained from Sigma Chemical Co. (St. Louis, MO). Paricalcitol and glutathione ethyl ester (GEE) were from Cayman Chemical (Ann Arbor, MI, USA). The antibodies against VDR (D-6 and C-20), RXRα (D-20), NQO-1 (A-5), ɣ-GCSc (H-5), ɣ-GCSm (E-4), TrxR1 (B-2), and 2’,7’-dichlorofluorescein-diacetate (DCFH-DA) were procured from Santa Cruz Biotechnology Inc. (Dallas, TX). Antibodies against P-c-Jun (54B3), c-Jun (60AB), and HO-1 (D60611) were acquired from Cell Signaling Technology (Danvers, MA, USA). Antibodies against Nrf2 (MAB3925) and calreticulin (PA3-900) were purchased from R&D Systems (Minneapolis, MN, USA) and Thermo Fisher Scientific (Waltham, MA, USA), respectively. Peroxidase-conjugated AffiniPure donkey anti-rabbit and sheep anti-mouse IgG antibodies were bought from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA). L-buthionine-sulfoximine (BSO) was obtained from Merck-Sigma-Aldrich (Rehovot, Israel). Hanks’ buffered salt solution (HBSS), penicillin, streptomycin, and HEPES buffer were from IMBH (Beth Haemek, Israel). RPMI 1640 medium and heat-inactivated fetal bovine serum (FBS) were purchased from Gibco-Invitrogen (Carlsbad, CA, USA). Stock solutions of 1,25D3 and paricalcitol (2.4 mM), CA (10 mM), and MMF (50 mM) were prepared in absolute ethanol. The precise concentration of 1,25D3 in ethanol was verified spectrophotometrically at 264 nm (ε=19,000).
4.2. Cell culture and stable transfection
HL60 myeloblastic leukemia cells (ATCC-CCL-240) were grown in RPMI 1640 medium supplemented with 10% fetal calf serum (FCS), penicillin (100 U/ml), streptomycin (0.1 mg/ml), and 10 mM HEPES (pH = 7.4) in a humidified atmosphere of 95% air and 5% CO2 at 37°C. The expression vector for dnNrf2, which lacks the transactivation domain residues 1-392 in the NH2-terminal portion of the protein, and the empty vector (pEF) were generously provided by Dr. Jawed Alam (Louisiana State University Medical Center, New Orleans, LA). Both plasmids carried the neomycin (neoR) resistance gene. Stable nucleofection was performed by Dr. Irene Bobilev (Ben Gurion University of the Negev), as described previously [22]. Briefly, HL60 cells (1 × 107 cells/ml) were mixed with 1 µg plasmid in Cell Line Nucleofector Solution V and transfected in an Amaxa Nucleofector (Lonza, Cologne, Germany) according to the manufacturer’s protocol (program T-19).
4.3. Determination of cell differentiation markers
Cells were seeded at 1 × 105 cells/ml and treated with test agents or vehicle (<0.2% ethanol) for 48 h. Cell numbers and viability were determined using the trypan blue exclusion assay by enumeration in a Vi-Cell XR cell viability analyzer (Beckman Coulter Inc., Fullerton, CA, USA). Aliquots of 5 × 105 cells were harvested, washed with PBS, and incubated for 45 min at room temperature with 0.3 µl MO1-FITC and 0.3 µl MY4-RD1 (Beckman Coulter) to determine the expression of myeloid surface antigens CD11b and CD14, respectively, by flow cytometry as described previously [18,21]. For each analysis, 10,000 events were recorded, and the data were processed using Kaluza software, version 2.1 (Beckman Coulter).
4.4. Determination of intracellular levels of reactive oxygen species
Cytosolic ROS levels were determined using the oxidation-sensitive fluorescent probe DCFH-DA. Intracellular ROS oxidize this probe to a highly fluorescent compound, DCF. Following treatments with the specified compounds at the indicated time points, cells were washed with HBSS containing 10 mM HEPES buffer (pH=7.4). Subsequently, cells were stained with 5 μM DCFH-DA for 15 min at 37°C in the dark using a shaking water bath and washed with HEPES-buffered HBSS. For the positive control, DCFH-DA-loaded cells were treated with 0.5 mM H2O2 for 15 min. Untreated and unstained cells served as the negative control. In the experiment reported in Figure 5(c,d), DCFH-DA-loaded cells were divided into two groups and incubated with vehicle (HBSS) or 10 µM of H2O2 for 15 min, followed by washing with HEPES-buffered HBSS. The DCF fluorescence intensity was measured by flow cytometry, recording 10,000 events for each analysis. Data were analyzed using Kaluza Analysis Software version 2.1 (Beckman Coulter) [57].
4.5. Preparation of whole cell lysates and western blotting
Cells were seeded at 1 × 105 cells/ml and incubated with test agents or vehicle (<0.2% ethanol) for 48 h. Preparation of whole cell lysates and Western blotting analysis were performed as described previously [18]. Briefly, cells were lysed in a buffer containing 1% (v/v) Triton X-100 at 4°C, subjected to SDS-PAGE, and electroblotted into nitrocellulose membranes. The membranes were exposed to primary antibodies overnight at 4°C. Blots were washed and incubated with horse-radish peroxidase-conjugated secondary antibodies. Membranes were stripped and reprobed for calreticulin, the internal loading control. The protein bands were visualized using the WESTAR ANTARES Chemiluminescent Substrate for Western blotting (Cyanagen, Bologna, Italy). The absorbance of each band was determined using the Image Quant LAS 4000 system (GE Healthcare, Little Chalfont, UK).
4.6. RNA extraction, cDNA synthesis, and RT-qPCR
Total RNA was purified from cell cultures according to the manufacturer’s instructions using an RNA extraction kit (GENEzolTM TriRNA Pure Kit+DNASE I; Geneaid, New Taipei City, Taiwan). A micro-volume spectrophotometer (NanoDrop; Wilmington, DE, USA) was used for RNA quantification. First-strand cDNA was generated by reverse transcriptase kit (qScript cDNA synthesis kit; QUANTA Biosciences; Gaithersburg, MD, USA) using random oligo (dT) after a sample concentration was normalized. Quantitative cDNA amplification was performed by real-time PCR (StepOne Real-Time PCR System; Thermo Fisher Scientific; Wilmington, DE, USA) using qPCRBIO Fast qPCR SyGreen Blue Mix from Tamar Laboratory Supplies Ltd (Mevaseret Zion, Israel). Relative mRNA expression levels were determined using the 2-(ΔΔCt) formula, where ΔCt is Ct (target gene) - mean of Ct (reference genes). The reference gene used in this study was GAPDH. Each experiment was performed using three biological replicates, each assayed in triplicate. Primers for qPCR were synthesized by Hy Laboratories Ltd. (Rehovot, Israel) and Tamar Laboratory Supplies Ltd. (Mevaseret Zion, Israel).
The primer sequences used in this study were as follows:
| CAMP FR | GCTAACCTCTACCGCCTCCT |
| CAMP REV | GGTCACTGTCCCCATACACC |
| CD14 FR | CAACCTAGAGCCGTTTCTAAAGC |
| CD14 REV | GCGCCTACCAGTAGCTGAG |
| CYP24A1 FR | GGAAGTGATGAAGCTGGACAACA |
| CYP24A1 REV | CTCATACAACACGAGGCAGATAC |
| GAPDH FR | CATGAGAAGTATGACAACAGCCT |
| GAPDH REV | AGTCCTTCCACGATACCAAAGT |
| GCLC FR | GGAGGAAACCAAGCGCCAT |
| GCLC REV | CTTGACGGCGTGGTAGATGT |
| GCLM FR | GGAAGAAGTGCCCGTCCA |
| GCLM REV | CTGAACAGGCCATGTCAACT |
| HO-1 FR | AAGACTGCGTTCCTGCTCAA |
| HO-1 REV | GGTCCTTGGTGTCATGGGTC |
| ITGAM FR | CTGTCTGCCAGAGAATCCAGTG |
| ITGAM REV | GAGGTGGTTATGCGAGGTCTTG |
| NQO1 FR | AAAGAAGGCCATCTGAGCCC |
| NQO1 REV | CCAGGCGTTTCTTCCATCCT |
| Nrf2 FR | CCTTGTCACCATCTCAGGGG |
| Nrf2 REV | TGGGGTTTTCCGATGACCAG |
| TXNRD1 FR | ACGTTACTTGGGCATCCCTG |
| TXNRD1 REV | AGAAATCCAGCGCACTCCAA |
| VDR FR | GACCTGTGGCAACCAAGACT |
| VDR REV | AATCAGCTCCAGGCTGTGTC |
4.7. Total glutathione assay
Cells (2 × 106) were collected by centrifugation (1,000×g for 5 min), washed with ice-cold PBS, and resuspended in 200 µl of 5% 5-sulfosalicylic acid. After 15 min on ice with intermittent vortexing, the suspension was centrifuged at 16,000×g for 5 min to remove protein precipitates. Total glutathione was determined in the supernatants by the glutathione reductase recycling assay [21,22]. The rates of 5-thio-2-nitrobenzoic acid (TNB) formation were measured kinetically at 412 nm for 30 min using VersaMax microplate spectrophotometer (Molecular Devices).
4.8. Cell treatment with AP-1 decoy oligodeoxynucleotides
HL60 cells were preincubated for 24 h with 10 µM double-stranded phosphorothioate oligodeoxynucleotide containing the proximal binding site for AP-1 (TPA-response element; TRE) from the promoter region of hVDR (-77 to -97 relative to the transcription start site; 5′- CTG GCA AGA GAG GAC TGG ACC-3′) or its mutant (5′-CTG GCA AGA GAG TGC TGG ACC-3′) in the complete culture medium, followed by treatment with test agents for an additional 48 h [22]. The oligonucleotides were synthesized by Integrated DNA Technologies, lnc. (Coralville, IA).
4.9. Transient transfection and reporter gene assay
Cells were harvested after 48 h of culture, washed once in a preheated growth medium at 37°C, and resuspended at 5 × 105 cells/ml. Four hundred μl of the cell suspension were transferred to 24-well plates, followed by co-transfection with 0.8 µg VDREx6-Luc luciferase reporter plasmid and 0.2 µg Renilla luciferase (pRL-null) reporter plasmid (internal control) using jetPEI reagent (Polyplus Transfection, Illkrich, France). Four hours later, cells were pre-incubated for 24 h with either a vehicle, 10 µM TRE-ODN, or mutant TRE (mTRE)-ODN. Subsequently, they were treated for an additional 48 h with 1 nM 1,25D3, 10 µM CA or 50 µM MMF, alone or in combination. Firefly and Renilla luciferase activities were then measured using the Dual-Luciferase Reporter Assay system (Promega, Madison, WI, USA) in accordance with the manufacturer’s instructions. Luminescence was determined using a Turner 20/20 luminometer (Turner BioSystems, Sunnyvale, CA, USA). Data are expressed as firefly luciferase-to-Renilla luciferase ratios (RLU) [22]. The VDREx6-Luc reporter plasmid was gifted by Dr. David G. Garner (University of California, San Francisco, CA). The pRL-null vector was purchased from Promega (Madison, WI, USA).
4.10. Statistical analysis
All experiments were performed at least three times. Statistically significant differences between the two experimental groups were assessed using one-way ANOVA. Data are presented as the mean ± SD. p < 0.05 was considered statistically significant. The synergy between the effects of two compounds (A and B) was assumed if the effect of their combination (AB) was larger than the sum of their individual effects (AB>A+B), the data being compared after subtraction of the respective control values from A, B, and AB [15]. The statistical analyses were performed using GraphPad Prism 6.0 software (Graph-Pad Software, San Diego, CA, USA).
5. Conclusions
This study addressed the mechanism of the synergy between VDDs and Nrf2 activators in inducing monocytic differentiation of AML cells. Using glutathione depletion and repletion approaches as well as impairment of Nrf2 activity, we obtained evidence that glutathione mediates, at least in part, the interplay between the Nrf2/ARE and vitamin D signaling pathways, likely through the positive regulation of AP-1. Upregulated AP-1 appears to be essential for enhancing VDD-induced differentiation by Nrf2 activators via increasing VDR/RXRα protein levels and transcriptional activity. Glutathione may also promote Nrf2/ARE activity, which appears to contribute to potentiating VDR signaling.
High expression and persisted activation of Nrf2 in AML blasts promote a more malignant phenotype and resistance to chemotherapy [93,94,95]. Still, our results support the notion that a dose-sparing combination therapy with low-calcemic VDDs and Nrf2 activators may be advantageous for a subset of AML patients with upregulated Nrf2 signaling. This mild treatment strategy may also be explored as part of therapeutic regimens in older patients who are unfit for standard intensive chemotherapy.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org., Figure S1: N-acetylcysteine partially reverses the inhibitory effect of dominant-negative Nrf2 on the differentiation of HL60 cells.
Author Contributions
Conceptualization, M.D.; methodology, Y.J.-S.; validation, Y.J.-S. and M.D.; investigation, Y.J.-S.; resources, M.D.; writing—original draft preparation, Y.J.-S. and M.D.; writing—review and editing, M.D.; visualization, Y.J.-S. and M.D.; supervision, M.D.; funding acquisition, M.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the US-Israel Binational Science Foundation grant number 2017112 and ISF-IPMP-Israel Precision Medicine Program grant number 3480/19.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All datasets generated for this study are included in the manuscript and Supplementary Materials.
Acknowledgments
The authors thank Ms. Victoria Novik for her valuable contribution to studies involving AP-1 decoy oligonucleotides and reporter gene assays.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lazarevic, V.L. Acute myeloid leukaemia in patients we judge as being older and/or unfit. J Intern Med 2021, 290, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Recher, C.; Rollig, C.; Berard, E.; Bertoli, S.; Dumas, P.Y.; Tavitian, S.; Kramer, M.; Serve, H.; Bornhauser, M.; Platzbecker, U.; et al. Long-term survival after intensive chemotherapy or hypomethylating agents in AML patients aged 70 years and older: a large patient data set study from European registries. Leukemia 2022, 36, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Kayser, S.; Levis, M.J. Updates on targeted therapies for acute myeloid leukaemia. Br J Haematol 2022, 196, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Short, N.J.; Ong, F.; Ravandi, F.; Nogueras-Gonzalez, G.; Kadia, T.M.; Daver, N.; DiNardo, C.D.; Konopleva, M.; Borthakur, G.; Oran, B.; et al. Impact of type of induction therapy on outcomes in older adults with AML after allogeneic stem cell transplantation. Blood Adv 2023, 7, 3573–3581. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Koeffler, H.P. Differentiation therapy of myeloid leukemia: four decades of development. Haematologica 2021, 106, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Kantarjian, H.; Ravandi, F. Acute promyelocytic leukemia current treatment algorithms. Blood Cancer J 2021, 11, 123. [Google Scholar] [CrossRef] [PubMed]
- Marcinkowska, E. Vitamin D Derivatives in Acute Myeloid Leukemia: The Matter of Selecting the Right Targets. Nutrients 2022, 14. [Google Scholar] [CrossRef]
- Cao, H.; Xu, Y.; de Necochea-Campion, R.; Baylink, D.J.; Payne, K.J.; Tang, X.; Ratanatharathorn, C.; Ji, Y.; Mirshahidi, S.; Chen, C.S. Application of vitamin D and vitamin D analogs in acute myelogenous leukemia. Exp Hematol 2017, 50, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rochel, N. Vitamin D and Its Receptor from a Structural Perspective. Nutrients 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, C. Genomic signaling of vitamin D. Steroids 2023, 198, 109271. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Murray, A.; Synnott, N.C.; O’Donovan, N.; Crown, J. Vitamin D analogues: Potential use in cancer treatment. Crit Rev Oncol Hematol 2017, 112, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Sokoloski, J.A.; Hodnick, W.F.; Mayne, S.T.; Cinquina, C.; Kim, C.S.; Sartorelli, A.C. Induction of the differentiation of HL-60 promyelocytic leukemia cells by vitamin E and other antioxidants in combination with low levels of vitamin D3: possible relationship to NF-kappaB. Leukemia 1997, 11, 1546–1553. [Google Scholar] [CrossRef] [PubMed]
- Amir, H.; Karas, M.; Giat, J.; Danilenko, M.; Levy, R.; Yermiahu, T.; Levy, J.; Sharoni, Y. Lycopene and 1,25-dihydroxyvitamin D3 cooperate in the inhibition of cell cycle progression and induction of differentiation in HL-60 leukemic cells. Nutr Cancer 1999, 33, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chang, R.L.; Cui, X.X.; Newmark, H.L.; Conney, A.H. Synergistic effects of curcumin on all-trans retinoic acid- and 1 alpha,25-dihydroxyvitamin D3-induced differentiation in human promyelocytic leukemia HL-60 cells. Oncol Res 1997, 9, 19–29. [Google Scholar] [PubMed]
- Danilenko, M.; Wang, X.; Studzinski, G.P. Carnosic acid and promotion of monocytic differentiation of HL60-G cells initiated by other agents. J Natl Cancer Inst 2001, 93, 1224–1233. [Google Scholar] [CrossRef] [PubMed]
- Sharabani, H.; Izumchenko, E.; Wang, Q.; Kreinin, R.; Steiner, M.; Barvish, Z.; Kafka, M.; Sharoni, Y.; Levy, J.; Uskokovic, M.; et al. Cooperative antitumor effects of vitamin D3 derivatives and rosemary preparations in a mouse model of myeloid leukemia. Int J Cancer 2006, 118, 3012–3021. [Google Scholar] [CrossRef]
- Kang, S.N.; Lee, M.H.; Kim, K.M.; Cho, D.; Kim, T.S. Induction of human promyelocytic leukemia HL-60 cell differentiation into monocytes by silibinin: involvement of protein kinase C. Biochem Pharmacol 2001, 61, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Nachliely, M.; Sharony, E.; Kutner, A.; Danilenko, M. Novel analogs of 1,25-dihydroxyvitamin D2 combined with a plant polyphenol as highly efficient inducers of differentiation in human acute myeloid leukemia cells. J Steroid Biochem Mol Biol 2016, 164, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Harrison, J.S.; Uskokovic, M.; Danilenko, M.; Studzinski, G.P. Silibinin can induce differentiation as well as enhance vitamin D3-induced differentiation of human AML cells ex vivo and regulates the levels of differentiation-related transcription factors. Hematol Oncol 2010, 28, 124–132. [Google Scholar] [CrossRef]
- Shabtay, A.; Sharabani, H.; Barvish, Z.; Kafka, M.; Amichay, D.; Levy, J.; Sharoni, Y.; Uskokovic, M.R.; Studzinski, G.P.; Danilenko, M. Synergistic antileukemic activity of carnosic acid-rich rosemary extract and the 19-nor Gemini vitamin D analogue in a mouse model of systemic acute myeloid leukemia. Oncology 2008, 75, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Danilenko, M.; Wang, Q.; Wang, X.; Levy, J.; Sharoni, Y.; Studzinski, G.P. Carnosic acid potentiates the antioxidant and prodifferentiation effects of 1alpha,25-dihydroxyvitamin D3 in leukemia cells but does not promote elevation of basal levels of intracellular calcium. Cancer Res 2003, 63, 1325–1332. [Google Scholar] [PubMed]
- Bobilev, I.; Novik, V.; Levi, I.; Shpilberg, O.; Levy, J.; Sharoni, Y.; Studzinski, G.P.; Danilenko, M. The Nrf2 transcription factor is a positive regulator of myeloid differentiation of acute myeloid leukemia cells. Cancer Biol Ther 2011, 11, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: a Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol Rev 2018, 98, 1169–1203. [Google Scholar] [CrossRef]
- Hecht, F.; Zocchi, M.; Alimohammadi, F.; Harris, I.S. Regulation of antioxidants in cancer. Mol Cell, 2023. [Google Scholar] [CrossRef]
- Berger, A.A.; Sottosanti, E.R.; Winnick, A.; Izygon, J.; Berardino, K.; Cornett, E.M.; Kaye, A.D.; Varrassi, G.; Viswanath, O.; Urits, I. Monomethyl Fumarate (MMF, Bafiertam) for the Treatment of Relapsing Forms of Multiple Sclerosis (MS). Neurol Int 2021, 13, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Nachliely, M.; Trachtenberg, A.; Khalfin, B.; Nalbandyan, K.; Cohen-Lahav, M.; Yasuda, K.; Sakaki, T.; Kutner, A.; Danilenko, M. Dimethyl fumarate and vitamin D derivatives cooperatively enhance VDR and Nrf2 signaling in differentiating AML cells in vitro and inhibit leukemia progression in a xenograft mouse model. J Steroid Biochem Mol Biol 2019, 188, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Salman, H.; Danilenko, M.; Studzinski, G.P. Cooperation between antioxidants and 1,25-dihydroxyvitamin D3 in induction of leukemia HL60 cell differentiation through the JNK/AP-1/Egr-1 pathway. J Cell Physiol 2005, 204, 964–974. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Studzinski, G.P. The requirement for and changing composition of the activating protein-1 transcription factor during differentiation of human leukemia HL60 cells induced by 1,25-dihydroxyvitamin D3. Cancer Res 2006, 66, 4402–4409. [Google Scholar] [CrossRef]
- Alam, J.; Stewart, D.; Touchard, C.; Boinapally, S.; Choi, A.M.; Cook, J.L. Nrf2, a Cap'n'Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem 1999, 274, 26071–26078. [Google Scholar] [CrossRef] [PubMed]
- Bover, J.; Dasilva, I.; Furlano, M.; Lloret, M.J.; Diaz-Encarnacion, M.M.; Ballarin, J.; Cozzolino, M. Clinical Uses of 1,25-dihydroxy-19-nor-vitamin D2 (Paricalcitol). Curr Vasc Pharmacol 2014, 12, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Glutathione synthesis. Biochim Biophys Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S.; Motohashi, H. Roles of Nrf2 in cell proliferation and differentiation. Free Radic Biol Med 2015, 88, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wang, Y.; Gao, Y.; Ju, Y.; Zhao, Y.; Wu, Z.; Gao, S.; Zhang, B.; Pang, X.; Zhang, Y.; et al. The PRAK-NRF2 axis promotes the differentiation of Th17 cells by mediating the redox homeostasis and glycolysis. Proc Natl Acad Sci U S A 2023, 120, e2212613120. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, P.; Russo, M.; D’Amico, J.; Petrillo, S.; Aquilano, K.; Lettieri-Barbato, D.; Turchi, R.; Bertini, E.S.; Piemonte, F. Nrf2 Induction Re-establishes a Proper Neuronal Differentiation Program in Friedreich’s Ataxia Neural Stem Cells. Front Cell Neurosci 2019, 13, 356. [Google Scholar] [CrossRef] [PubMed]
- Hinoi, E.; Fujimori, S.; Wang, L.; Hojo, H.; Uno, K.; Yoneda, Y. Nrf2 negatively regulates osteoblast differentiation via interfering with Runx2-dependent transcriptional activation. J Biol Chem 2006, 281, 18015–18024. [Google Scholar] [CrossRef] [PubMed]
- Yagishita, Y.; Chartoumpekis, D.V.; Kensler, T.W.; Wakabayashi, N. NRF2 and the Moirai: Life and Death Decisions on Cell Fates. Antioxid Redox Signal 2023, 38, 684–708. [Google Scholar] [CrossRef]
- Liebermann, D.A.; Gregory, B.; Hoffman, B. AP-1 (Fos/Jun) transcription factors in hematopoietic differentiation and apoptosis. Int J Oncol 1998, 12, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Wurm, S.; Zhang, J.; Guinea-Viniegra, J.; Garcia, F.; Munoz, J.; Bakiri, L.; Ezhkova, E.; Wagner, E.F. Terminal epidermal differentiation is regulated by the interaction of Fra-2/AP-1 with Ezh2 and ERK1/2. Genes Dev 2015, 29, 144–156. [Google Scholar] [CrossRef]
- Li, J.; King, I.; Sartorelli, A.C. Differentiation of WEHI-3B D+ myelomonocytic leukemia cells induced by ectopic expression of the protooncogene c-jun. Cell Growth Differ 1994, 5, 743–751. [Google Scholar]
- Grant, S.; Freemerman, A.J.; Birrer, M.J.; Martin, H.A.; Turner, A.J.; Szabo, E.; Chelliah, J.; Jarvis, W.D. Effect of 1-beta-D-arabinofuranosylcytosine on apoptosis and differentiation in human monocytic leukemia cells (U937) expressing a c-Jun dominant-negative mutant protein (TAM67). Cell Growth Differ 1996, 7, 603–613. [Google Scholar]
- Novoszel, P.; Drobits, B.; Holcmann, M.; Fernandes, C.S.; Tschismarov, R.; Derdak, S.; Decker, T.; Wagner, E.F.; Sibilia, M. The AP-1 transcription factors c-Jun and JunB are essential for CD8alpha conventional dendritic cell identity. Cell Death Differ 2021, 28, 2404–2420. [Google Scholar] [CrossRef]
- Comandante-Lou, N.; Baumann, D.G.; Fallahi-Sichani, M. AP-1 transcription factor network explains diverse patterns of cellular plasticity in melanoma cells. Cell Rep 2022, 40, 111147. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.C.; Mieyal, J.J. Glutathione and Glutaredoxin-Key Players in Cellular Redox Homeostasis and Signaling. Antioxidants (Basel) 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Di Giacomo, C.; Malfa, G.A.; Tomasello, B.; Bianchi, S.; Acquaviva, R. Natural Compounds and Glutathione: Beyond Mere Antioxidants. Antioxidants (Basel) 2023, 12. [Google Scholar] [CrossRef]
- Franco, R.; Cidlowski, J.A. Apoptosis and glutathione: beyond an antioxidant. Cell Death Differ 2009, 16, 1303–1314. [Google Scholar] [CrossRef] [PubMed]
- Fraternale, A.; Brundu, S.; Magnani, M. Glutathione and glutathione derivatives in immunotherapy. Biol Chem 2017, 398, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Esposito, F.; Agosti, V.; Morrone, G.; Morra, F.; Cuomo, C.; Russo, T.; Venuta, S.; Cimino, F. Inhibition of the differentiation of human myeloid cell lines by redox changes induced through glutathione depletion. Biochem J 1994, 301 Pt 3, 649–653. [Google Scholar] [CrossRef]
- Lian, G.; Gnanaprakasam, J.R.; Wang, T.; Wu, R.; Chen, X.; Liu, L.; Shen, Y.; Yang, M.; Yang, J.; Chen, Y.; et al. Glutathione de novo synthesis but not recycling process coordinates with glutamine catabolism to control redox homeostasis and directs murine T cell differentiation. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Barajas, B.; Chan, R.C.; Nel, A.E. Glutathione depletion inhibits dendritic cell maturation and delayed-type hypersensitivity: implications for systemic disease and immunosenescence. J Allergy Clin Immunol 2007, 119, 1225–1233. [Google Scholar] [CrossRef]
- Kim, J.M.; Kim, H.; Kwon, S.B.; Lee, S.Y.; Chung, S.C.; Jeong, D.W.; Min, B.M. Intracellular glutathione status regulates mouse bone marrow monocyte-derived macrophage differentiation and phagocytic activity. Biochem Biophys Res Commun 2004, 325, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Ardite, E.; Barbera, J.A.; Roca, J.; Fernandez-Checa, J.C. Glutathione depletion impairs myogenic differentiation of murine skeletal muscle C2C12 cells through sustained NF-kappaB activation. Am J Pathol 2004, 165, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.H.; Kim, Y.S.; Noh, K.; Lee, Y.M.; Chang, S.W.; Kim, E.C. Deferoxamine promotes osteoblastic differentiation in human periodontal ligament cells via the nuclear factor erythroid 2-related factor-mediated antioxidant signaling pathway. J Periodontal Res 2014, 49, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Ochi, M.; Ono, M.; Aoyama, E.; Ogino, T.; Kondo, Y.; Ohuchi, H. Glutathione accelerates osteoclast differentiation and inflammatory bone destruction. Free Radic Res 2019, 53, 226–236. [Google Scholar] [CrossRef]
- Satoh, T.; Kosaka, K.; Itoh, K.; Kobayashi, A.; Yamamoto, M.; Shimojo, Y.; Kitajima, C.; Cui, J.; Kamins, J.; Okamoto, S.; et al. Carnosic acid, a catechol-type electrophilic compound, protects neurons both in vitro and in vivo through activation of the Keap1/Nrf2 pathway via S-alkylation of targeted cysteines on Keap1. J Neurochem 2008, 104, 1116–1131. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Canning, P. Keap1, the cysteine-based mammalian intracellular sensor for electrophiles and oxidants. Arch Biochem Biophys 2017, 617, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Lipton, S. Recent advances in understanding NRF2 as a druggable target: development of pro-electrophilic and non-covalent NRF2 activators to overcome systemic side effects of electrophilic drugs like dimethyl fumarate. F1000Res 2017, 6, 2138. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Dawod, A.; Nachliely, M.; Harrison, J.S.; Danilenko, M.; Studzinski, G.P. Differentiation agents increase the potential AraC therapy of AML by reactivating cell death pathways without enhancing ROS generation. J Cell Physiol 2020, 235, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Leite de Oliveira, R.; Huijberts, S.; Bosdriesz, E.; Pencheva, N.; Brunen, D.; Bosma, A.; Song, J.Y.; Zevenhoven, J.; Los-de Vries, G.T.; et al. An Acquired Vulnerability of Drug-Resistant Melanoma with Therapeutic Potential. Cell 2018, 173, 1413–1425. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Z.; Poore, B.; Alt, J.; Price, A.; Allen, S.J.; Hanaford, A.R.; Kaur, H.; Orr, B.A.; Slusher, B.S.; Eberhart, C.G.; et al. Unbiased Metabolic Profiling Predicts Sensitivity of High MYC-Expressing Atypical Teratoid/Rhabdoid Tumors to Glutamine Inhibition with 6-Diazo-5-Oxo-L-Norleucine. Clin Cancer Res 2019, 25, 5925–5936. [Google Scholar] [CrossRef] [PubMed]
- Tsai-Turton, M.; Luong, B.T.; Tan, Y.; Luderer, U. Cyclophosphamide-induced apoptosis in COV434 human granulosa cells involves oxidative stress and glutathione depletion. Toxicol Sci 2007, 98, 216–230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tew, K.D. Reductive stress in cancer. Adv Cancer Res 2021, 152, 383–413. [Google Scholar] [CrossRef] [PubMed]
- Korge, P.; Calmettes, G.; Weiss, J.N. Increased reactive oxygen species production during reductive stress: The roles of mitochondrial glutathione and thioredoxin reductases. Biochim Biophys Acta 2015, 1847, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Maity, S.; Rajkumar, A.; Matai, L.; Bhat, A.; Ghosh, A.; Agam, G.; Kaur, S.; Bhatt, N.R.; Mukhopadhyay, A.; Sengupta, S.; et al. Oxidative Homeostasis Regulates the Response to Reductive Endoplasmic Reticulum Stress through Translation Control. Cell Rep 2016, 16, 851–865. [Google Scholar] [CrossRef] [PubMed]
- Bejjani, F.; Evanno, E.; Zibara, K.; Piechaczyk, M.; Jariel-Encontre, I. The AP-1 transcriptional complex: Local switch or remote command? Biochim Biophys Acta Rev Cancer 2019, 1872, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Shaulian, E. AP-1—The Jun proteins: Oncogenes or tumor suppressors in disguise? Cell Signal 2010, 22, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol 2014, 2, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, R.; Jaiswal, A.K. Nrf2 and Nrf1 in association with Jun proteins regulate antioxidant response element-mediated expression and coordinated induction of genes encoding detoxifying enzymes. Oncogene 1998, 17, 3145–3156. [Google Scholar] [CrossRef] [PubMed]
- Gesser, B.; Rasmussen, M.K.; Raaby, L.; Rosada, C.; Johansen, C.; Kjellerup, R.B.; Kragballe, K.; Iversen, L. Dimethylfumarate inhibits MIF-induced proliferation of keratinocytes by inhibiting MSK1 and RSK1 activation and by inducing nuclear p-c-Jun (S63) and p-p53 (S15) expression. Inflamm Res 2011, 60, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Grzegorzewska, A.P.; Seta, F.; Han, R.; Czajka, C.A.; Makino, K.; Stawski, L.; Isenberg, J.S.; Browning, J.L.; Trojanowska, M. Dimethyl Fumarate ameliorates pulmonary arterial hypertension and lung fibrosis by targeting multiple pathways. Sci Rep 2017, 7, 41605. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Wang, Q.; Wang, X.; Studzinski, G.P. Up-regulation of Egr1 by 1,25-dihydroxyvitamin D3 contributes to increased expression of p35 activator of cyclin-dependent kinase 5 and consequent onset of the terminal phase of HL60 cell differentiation. Cancer Res 2004, 64, 5425–5433. [Google Scholar] [CrossRef] [PubMed]
- Gocek, E.; Kielbinski, M.; Wylob, P.; Kutner, A.; Marcinkowska, E. Side-chain modified vitamin D analogs induce rapid accumulation of VDR in the cell nuclei proportionately to their differentiation-inducing potential. Steroids 2008, 73, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Gocek, E.; Marchwicka, A.; Baurska, H.; Chrobak, A.; Marcinkowska, E. Opposite regulation of vitamin D receptor by ATRA in AML cells susceptible and resistant to vitamin D-induced differentiation. J Steroid Biochem Mol Biol 2012, 132, 220–226. [Google Scholar] [CrossRef] [PubMed]
- van den Bemd, G.C.; Pols, H.A.; Birkenhager, J.C.; van Leeuwen, J.P. Conformational change and enhanced stabilization of the vitamin D receptor by the 1,25-dihydroxyvitamin D3 analog KH1060. Proc Natl Acad Sci U S A 1996, 93, 10685–10690. [Google Scholar] [CrossRef] [PubMed]
- Jaaskelainen, T.; Ryhanen, S.; Mahonen, A.; DeLuca, H.F.; Maenpaa, P.H. Mechanism of action of superactive vitamin D analogs through regulated receptor degradation. J Cell Biochem 2000, 76, 548–558. [Google Scholar] [CrossRef]
- Li, X.Y.; Boudjelal, M.; Xiao, J.H.; Peng, Z.H.; Asuru, A.; Kang, S.; Fisher, G.J.; Voorhees, J.J. 1,25-Dihydroxyvitamin D3 increases nuclear vitamin D3 receptors by blocking ubiquitin/proteasome-mediated degradation in human skin. Mol Endocrinol 1999, 13, 1686–1694. [Google Scholar] [CrossRef]
- Kongsbak, M.; von Essen, M.R.; Boding, L.; Levring, T.B.; Schjerling, P.; Lauritsen, J.P.; Woetmann, A.; Odum, N.; Bonefeld, C.M.; Geisler, C. Vitamin D up-regulates the vitamin D receptor by protecting it from proteasomal degradation in human CD4+ T cells. PLoS One 2014, 9, e96695. [Google Scholar] [CrossRef] [PubMed]
- Peleg, S.; Nguyen, C.V. The importance of nuclear import in protection of the vitamin D receptor from polyubiquitination and proteasome-mediated degradation. J Cell Biochem 2010, 110, 926–934. [Google Scholar] [CrossRef]
- Yang, R.; Zhang, J.; Li, J.; Qin, R.; Chen, J.; Wang, R.; Goltzman, D.; Miao, D. Inhibition of Nrf2 degradation alleviates age-related osteoporosis induced by 1,25-Dihydroxyvitamin D deficiency. Free Radic Biol Med 2022, 178, 246–261. [Google Scholar] [CrossRef]
- Golonko, A.; Pienkowski, T.; Swislocka, R.; Lazny, R.; Roszko, M.; Lewandowski, W. Another look at phenolic compounds in cancer therapy the effect of polyphenols on ubiquitin-proteasome system. Eur J Med Chem 2019, 167, 291–311. [Google Scholar] [CrossRef]
- Andreani, C.; Bartolacci, C.; Wijnant, K.; Crinelli, R.; Bianchi, M.; Magnani, M.; Hysi, A.; Iezzi, M.; Amici, A.; Marchini, C. Resveratrol fuels HER2 and ERalpha-positive breast cancer behaving as proteasome inhibitor. Aging (Albany NY) 2017, 9, 508–523. [Google Scholar] [CrossRef]
- Sato, A.; Okada, M.; Shibuya, K.; Watanabe, E.; Seino, S.; Suzuki, K.; Narita, Y.; Shibui, S.; Kayama, T.; Kitanaka, C. Resveratrol promotes proteasome-dependent degradation of Nanog via p53 activation and induces differentiation of glioma stem cells. Stem Cell Res 2013, 11, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Hasima, N.; Aggarwal, B.B. Targeting proteasomal pathways by dietary curcumin for cancer prevention and treatment. Curr Med Chem 2014, 21, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Petiwala, S.M.; Li, G.; Bosland, M.C.; Lantvit, D.D.; Petukhov, P.A.; Johnson, J.J. Carnosic acid promotes degradation of the androgen receptor and is regulated by the unfolded protein response pathway in vitro and in vivo. Carcinogenesis 2016, 37, 827–838. [Google Scholar] [CrossRef]
- Cortese, K.; Daga, A.; Monticone, M.; Tavella, S.; Stefanelli, A.; Aiello, C.; Bisio, A.; Bellese, G.; Castagnola, P. Carnosic acid induces proteasomal degradation of Cyclin B1, RB and SOX2 along with cell growth arrest and apoptosis in GBM cells. Phytomedicine 2016, 23, 679–685. [Google Scholar] [CrossRef]
- Alsamri, H.; Alneyadi, A.; Muhammad, K.; Ayoub, M.A.; Eid, A.; Iratni, R. Carnosol Induces p38-Mediated ER Stress Response and Autophagy in Human Breast Cancer Cells. Front Oncol 2022, 12, 911615. [Google Scholar] [CrossRef]
- Alsamri, H.; Hasasna, H.E.; Baby, B.; Alneyadi, A.; Dhaheri, Y.A.; Ayoub, M.A.; Eid, A.H.; Vijayan, R.; Iratni, R. Carnosol Is a Novel Inhibitor of p300 Acetyltransferase in Breast Cancer. Front Oncol 2021, 11, 664403. [Google Scholar] [CrossRef] [PubMed]
- Valdes, A.; Garcia-Canas, V.; Artemenko, K.A.; Simo, C.; Bergquist, J.; Cifuentes, A. Nano-liquid Chromatography-orbitrap MS-based Quantitative Proteomics Reveals Differences Between the Mechanisms of Action of Carnosic Acid and Carnosol in Colon Cancer Cells. Mol Cell Proteomics 2017, 16, 8–22. [Google Scholar] [CrossRef] [PubMed]
- Toyama, T.; Looney, A.P.; Baker, B.M.; Stawski, L.; Haines, P.; Simms, R.; Szymaniak, A.D.; Varelas, X.; Trojanowska, M. Therapeutic Targeting of TAZ and YAP by Dimethyl Fumarate in Systemic Sclerosis Fibrosis. J Invest Dermatol 2018, 138, 78–88. [Google Scholar] [CrossRef]
- Booth, L.; Cruickshanks, N.; Tavallai, S.; Roberts, J.L.; Peery, M.; Poklepovic, A.; Dent, P. Regulation of dimethyl-fumarate toxicity by proteasome inhibitors. Cancer Biol Ther 2014, 15, 1646–1657. [Google Scholar] [CrossRef]
- Valesky, E.M.; Hrgovic, I.; Doll, M.; Wang, X.F.; Pinter, A.; Kleemann, J.; Kaufmann, R.; Kippenberger, S.; Meissner, M. Dimethylfumarate effectively inhibits lymphangiogenesis via p21 induction and G1 cell cycle arrest. Exp Dermatol 2016, 25, 200–205. [Google Scholar] [CrossRef]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-kappaB and underlies its chemo-resistance. Blood 2012, 120, 5188–5198. [Google Scholar] [CrossRef] [PubMed]
- Ganan-Gomez, I.; Wei, Y.; Yang, H.; Boyano-Adanez, M.C.; Garcia-Manero, G. Oncogenic functions of the transcription factor Nrf2. Free Radic Biol Med 2013, 65, 750–764. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.; Irwin, M.E.; Herbrich, S.M.; Cheng, T.; Patterson, L.L.; Aitken, M.J.L.; Bhalla, K.; You, M.J.; Konopleva, M.; Zweidler-McKay, P.A.; et al. Targeting the NRF2/HO-1 Antioxidant Pathway in FLT3-ITD-Positive AML Enhances Therapy Efficacy. Antioxidants (Basel) 2022, 11. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Glutathione depletion by buthionine sulfoximine inhibits the potentiating effects of Nrf2 activators on 1,25D3-induced differentiation and elevates ROS levels in HL60 cells. Cells were preincubated with vehicle (water) or 30 μM BSO for 24 h, followed by treatment with the indicated concentrations of 1,25D3, carnosic acid (CA), or monomethyl fumarate (MMF), alone or in combination, for another 48 h. (a) Representative flow cytometric data showing the enhancing effect of CA on 1,25D3-induced surface expression of CD14 and CD11b and the inhibitory effect of BSO on this enhancement. (b) Summarized CD14 and CD11b expression data, as exemplified in panel a. (c) Changes in the total glutathione content, as determined by the glutathione reductase recycling assay following 24 h of preincubation with BSO and 16 h of treatment with CA or MMF. (d) Averaged ROS levels measured as DCF geometric mean fluorescence intensity (MFI) units (% of control). Cells were preincubated with BSO for 24 h and treated with CA or MMF for 48 h. The data are means ± SD of at least 3 independent experiments performed in duplicate. *, p < 0.05; **, p < 0.01; ****, p < 0.0001 vs. untreated control group; ##, p < 0.01; ###, p < 0.001 vs. sum of the effects of single agents; $$, p < 0.01; $$$, p < 0.001; $$$$, p < 0.0001 vs. corresponding BSO-untreated group.
Figure 1.
Glutathione depletion by buthionine sulfoximine inhibits the potentiating effects of Nrf2 activators on 1,25D3-induced differentiation and elevates ROS levels in HL60 cells. Cells were preincubated with vehicle (water) or 30 μM BSO for 24 h, followed by treatment with the indicated concentrations of 1,25D3, carnosic acid (CA), or monomethyl fumarate (MMF), alone or in combination, for another 48 h. (a) Representative flow cytometric data showing the enhancing effect of CA on 1,25D3-induced surface expression of CD14 and CD11b and the inhibitory effect of BSO on this enhancement. (b) Summarized CD14 and CD11b expression data, as exemplified in panel a. (c) Changes in the total glutathione content, as determined by the glutathione reductase recycling assay following 24 h of preincubation with BSO and 16 h of treatment with CA or MMF. (d) Averaged ROS levels measured as DCF geometric mean fluorescence intensity (MFI) units (% of control). Cells were preincubated with BSO for 24 h and treated with CA or MMF for 48 h. The data are means ± SD of at least 3 independent experiments performed in duplicate. *, p < 0.05; **, p < 0.01; ****, p < 0.0001 vs. untreated control group; ##, p < 0.01; ###, p < 0.001 vs. sum of the effects of single agents; $$, p < 0.01; $$$, p < 0.001; $$$$, p < 0.0001 vs. corresponding BSO-untreated group.
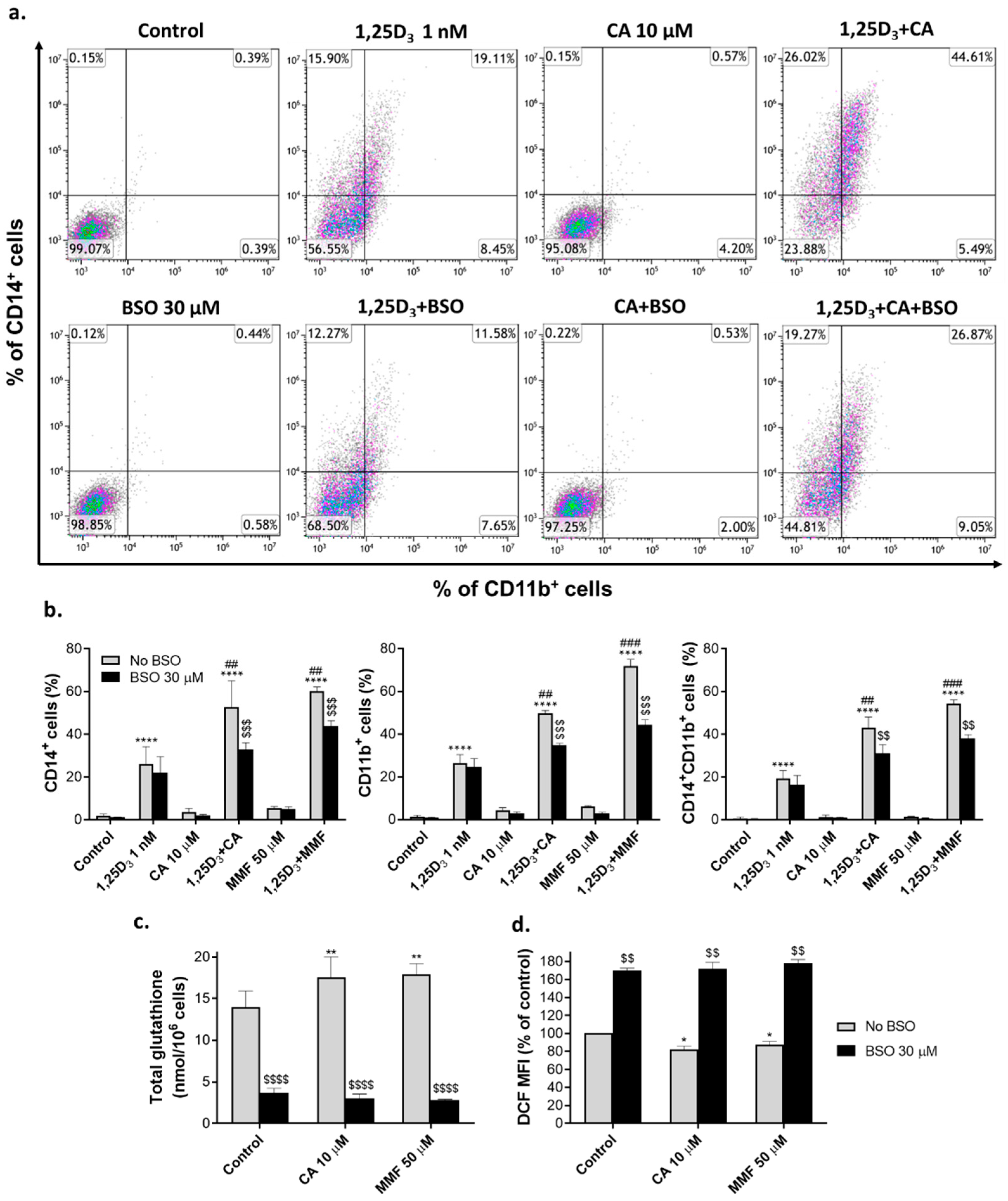
Figure 2.
Effects of glutathione depletion on mRNA levels of vitamin D- and Nrf2-related genes. HL60 cells were incubated with the indicated agents for 48 h, as described in the legend to Figure 1. Cell samples were then analyzed for mRNA levels of the indicated genes using quantitative RT-PCR. The expression of specific genes was normalized by the CT value of the internal reference gene (GAPDH). ). The data are means ± SD of 3 experiments performed in triplicate. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 vs. untreated control group; #, p < 0.05; ###, p < 0.001; ####, p < 0.0001 vs. sum of the effects of single agents; $, p < 0.05; $$, p < 0.01; $$$, p < 0.001 vs. corresponding BSO-untreated group.
Figure 2.
Effects of glutathione depletion on mRNA levels of vitamin D- and Nrf2-related genes. HL60 cells were incubated with the indicated agents for 48 h, as described in the legend to Figure 1. Cell samples were then analyzed for mRNA levels of the indicated genes using quantitative RT-PCR. The expression of specific genes was normalized by the CT value of the internal reference gene (GAPDH). ). The data are means ± SD of 3 experiments performed in triplicate. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 vs. untreated control group; #, p < 0.05; ###, p < 0.001; ####, p < 0.0001 vs. sum of the effects of single agents; $, p < 0.05; $$, p < 0.01; $$$, p < 0.001 vs. corresponding BSO-untreated group.
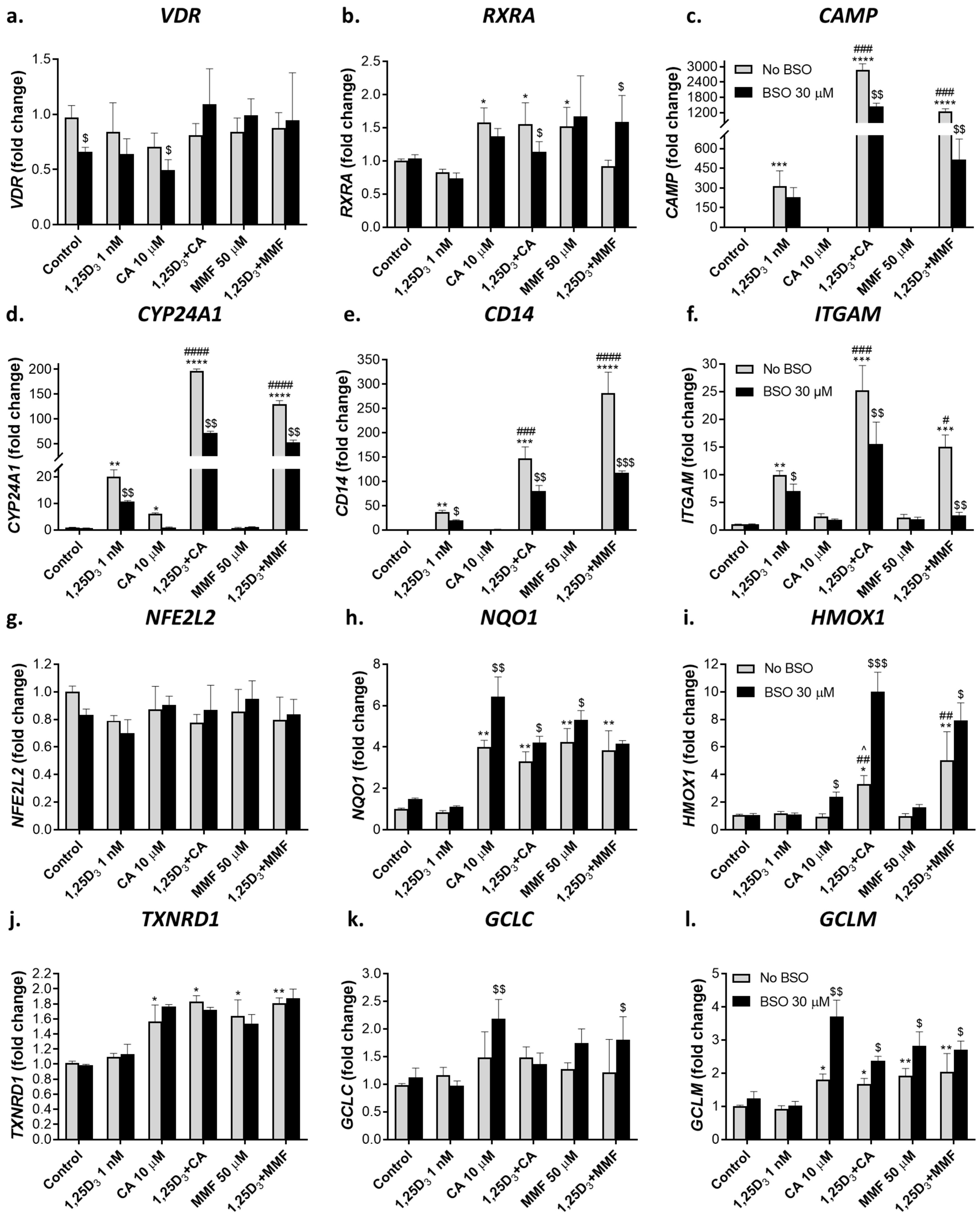
Figure 3.
Effects of glutathione depletion on the levels of vitamin D- and Nrf2-related proteins, c-Jun and its phosphorylation. HL60 cells were incubated with the indicated agents, as described in the legend to Figure 1. Cell samples were analyzed by Western blotting. Calreticulin was used as the protein loading control. (a, j) Representative Western blot images. (b-i, k, l) Absorbance values for specific proteins were normalized to those of calreticulin and expressed in the bar graphs as fold change relative to the corresponding untreated controls. The data are means ± SD of 3 experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001 vs. untreated control cells; #, p < 0.05; ##, p < 0.01; ###, p < 0.001 vs. sum of the effects of single agents; $, p < 0.05; $$, p < 0.01; $$$, p < 0.001 vs. corresponding BSO-untreated group.
Figure 3.
Effects of glutathione depletion on the levels of vitamin D- and Nrf2-related proteins, c-Jun and its phosphorylation. HL60 cells were incubated with the indicated agents, as described in the legend to Figure 1. Cell samples were analyzed by Western blotting. Calreticulin was used as the protein loading control. (a, j) Representative Western blot images. (b-i, k, l) Absorbance values for specific proteins were normalized to those of calreticulin and expressed in the bar graphs as fold change relative to the corresponding untreated controls. The data are means ± SD of 3 experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001 vs. untreated control cells; #, p < 0.05; ##, p < 0.01; ###, p < 0.001 vs. sum of the effects of single agents; $, p < 0.05; $$, p < 0.01; $$$, p < 0.001 vs. corresponding BSO-untreated group.
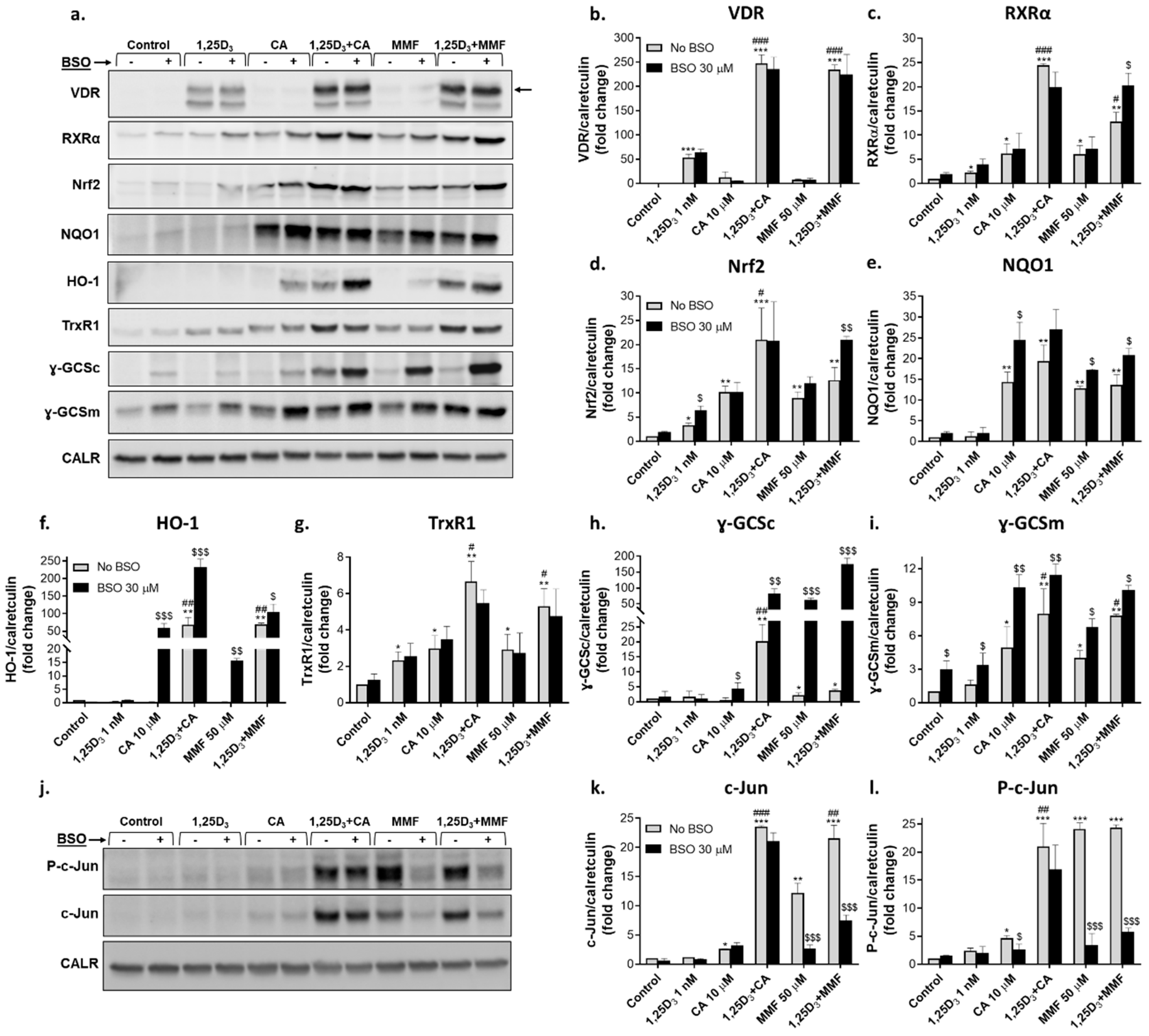
Figure 4.
Stable expression of dominant-negative Nrf2 suppresses the differentiation induced by paricalcitol and its combination with carnosic acid and reduces glutathione levels. Cells stably transfected with either empty vector (pEF-HL60) or dominant-negative Nrf2 (dnNrf2-HL60) were incubated with the indicated concentrations of paricalcitol (19-D2) and carnosic acid (CA), alone or combination, for 48 h. (a) Representative flow cytometric data showing changes in CD14 and CD11b surface expression. (b) Summarized CD14 and CD11b expression data exemplified in panel a. (c) pEF-HL60 and dnNrf2-HL60 cells differ in the total glutathione content. Cells were treated with vehicle or the indicated concentrations of CA for 48 h. The total cellular glutathione (GSH+GSSG) concentration was determined by the glutathione reductase recycling assay. The data are means ± SD of at least 3 independent experiments performed in duplicate. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 vs. corresponding control group; ##, p < 0.01; ###, p < 0.001 vs. corresponding sum of the effects of single agents; ^, < p < 0.05; ^^, <p < 0.01, dnNrf2-HL60 cells vs. pEF-HL60 cells.
Figure 4.
Stable expression of dominant-negative Nrf2 suppresses the differentiation induced by paricalcitol and its combination with carnosic acid and reduces glutathione levels. Cells stably transfected with either empty vector (pEF-HL60) or dominant-negative Nrf2 (dnNrf2-HL60) were incubated with the indicated concentrations of paricalcitol (19-D2) and carnosic acid (CA), alone or combination, for 48 h. (a) Representative flow cytometric data showing changes in CD14 and CD11b surface expression. (b) Summarized CD14 and CD11b expression data exemplified in panel a. (c) pEF-HL60 and dnNrf2-HL60 cells differ in the total glutathione content. Cells were treated with vehicle or the indicated concentrations of CA for 48 h. The total cellular glutathione (GSH+GSSG) concentration was determined by the glutathione reductase recycling assay. The data are means ± SD of at least 3 independent experiments performed in duplicate. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 vs. corresponding control group; ##, p < 0.01; ###, p < 0.001 vs. corresponding sum of the effects of single agents; ^, < p < 0.05; ^^, <p < 0.01, dnNrf2-HL60 cells vs. pEF-HL60 cells.
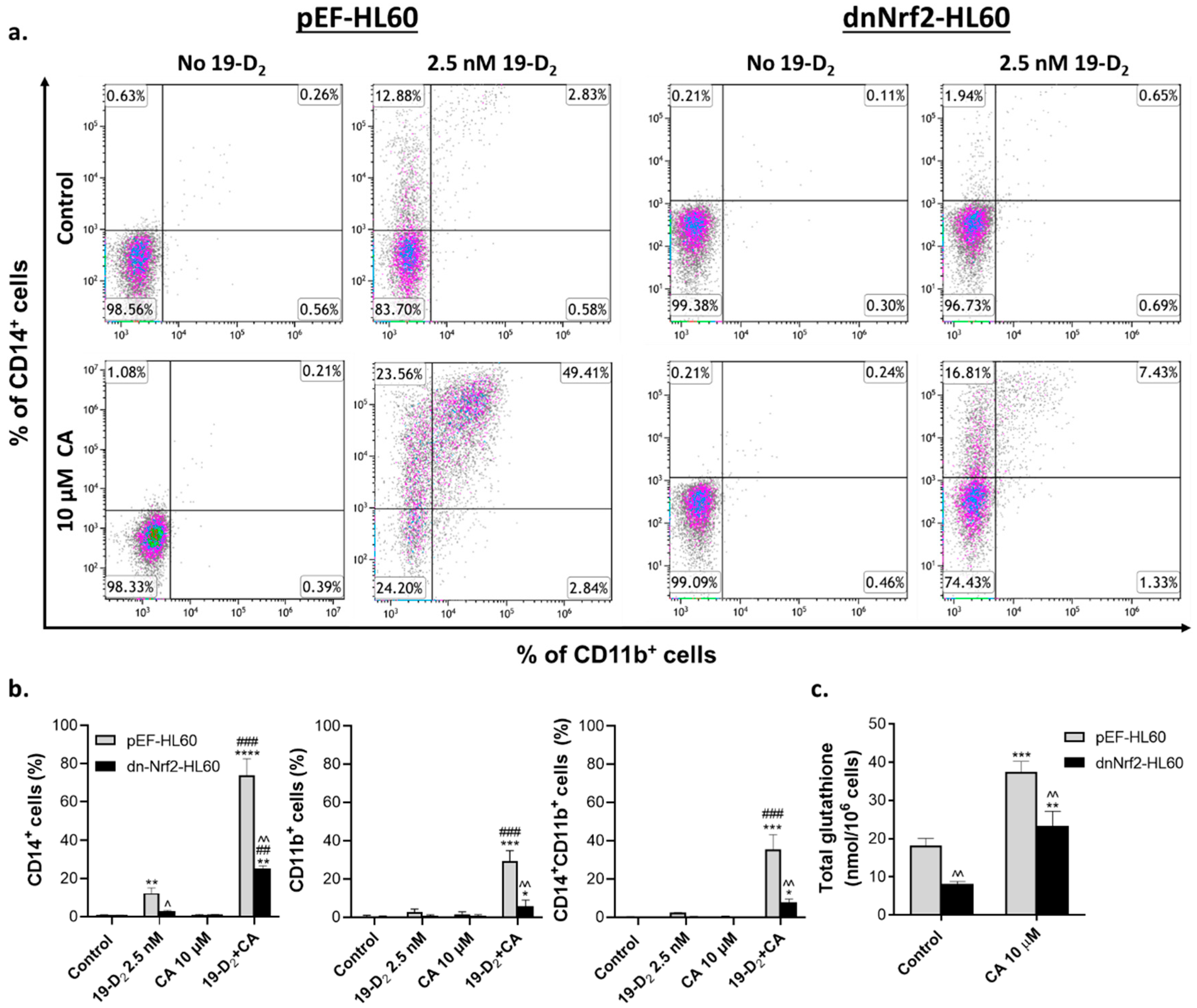
Figure 5.
GSH ethyl ester, but not Trolox, reverses the inhibitory effect of dominant-negative Nrf2 on the differentiation of HL60 cells while increasing cytosolic ROS levels. (a, b) Effects of GEE or Trolox on cell differentiation. Cells were pre-incubated with vehicle, GEE or Trolox for 1 h, followed by adding paricalcitol, CA or their combination and incubating for another 48 h. The expression of CD14 and CD11b was determined by flow cytometry. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001, vs. corresponding control group; #, p < 0.05; ##, p < 0.01; ###, p < 0.001, vs. corresponding sum of the effects of single agents; $, p < 0.05; $$, p < 0.01, GEE-treated vs. corresponding GEE-untreated group. (c, d) Changes in cytosolic ROS levels. Cells were incubated with vehicle, GEE or Trolox for 48 h, followed by exposure to 10 µM H2O2 for an additional 15 min. The results are expressed as DCF geometric mean fluorescence intensity (MFI) units (% of control). The data are means ± SD of 3 independent experiments. **, p < 0.01; ***, p < 0.001; ****, p < 0.0001, vs. corresponding H2O2-untreated group; $, p < 0.05; $$, p < 0.01; $$$, p < 0.001 vs. corresponding GEE- or Trolox-untreated group.
Figure 5.
GSH ethyl ester, but not Trolox, reverses the inhibitory effect of dominant-negative Nrf2 on the differentiation of HL60 cells while increasing cytosolic ROS levels. (a, b) Effects of GEE or Trolox on cell differentiation. Cells were pre-incubated with vehicle, GEE or Trolox for 1 h, followed by adding paricalcitol, CA or their combination and incubating for another 48 h. The expression of CD14 and CD11b was determined by flow cytometry. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001, vs. corresponding control group; #, p < 0.05; ##, p < 0.01; ###, p < 0.001, vs. corresponding sum of the effects of single agents; $, p < 0.05; $$, p < 0.01, GEE-treated vs. corresponding GEE-untreated group. (c, d) Changes in cytosolic ROS levels. Cells were incubated with vehicle, GEE or Trolox for 48 h, followed by exposure to 10 µM H2O2 for an additional 15 min. The results are expressed as DCF geometric mean fluorescence intensity (MFI) units (% of control). The data are means ± SD of 3 independent experiments. **, p < 0.01; ***, p < 0.001; ****, p < 0.0001, vs. corresponding H2O2-untreated group; $, p < 0.05; $$, p < 0.01; $$$, p < 0.001 vs. corresponding GEE- or Trolox-untreated group.
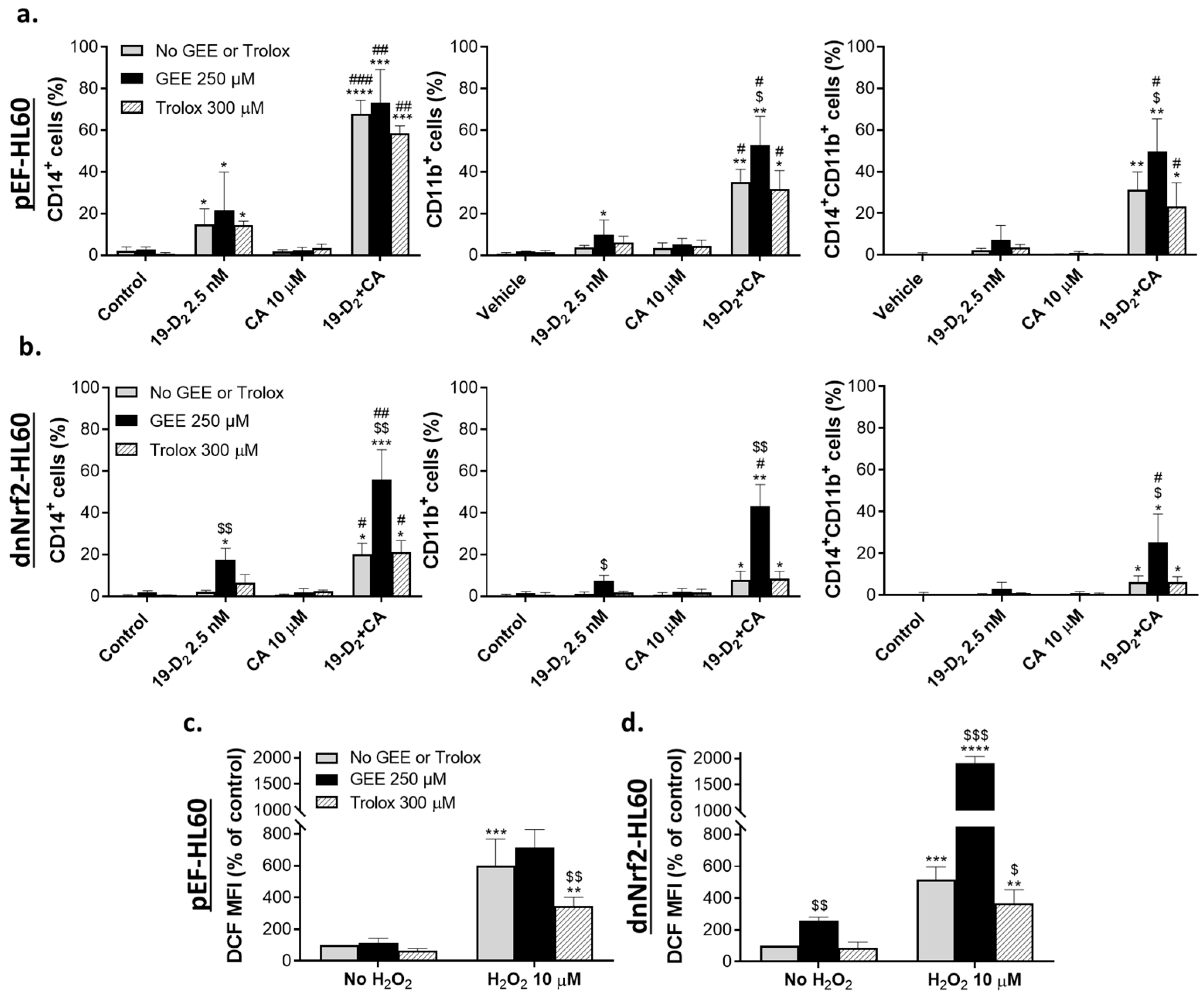
Figure 6.
Effects of GSH ethyl ester on mRNA levels of vitamin D- and Nrf2-related genes. pEF-HL60 and dnNrf2-HL60 cells were incubated with the indicated agents for 48 h. Samples were analyzed for mRNA levels of the indicated genes using quantitative RT-PCR. The expression of specific genes is normalized by the CT value of the internal reference gene (GAPDH). The data are means ± SD of 3 experiments performed in triplicate. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 vs. corresponding untreated control group; #, p < 0.05; ##, p < 0.01; ###, p < 0.001; ####, p < 0.0001 vs. corresponding sum of the effects of single agents. $, p < 0.05; $$, p < 0.01; $$$, p < 0.001; $$$$, p < 0.0001, GEE-treated vs. corresponding GEE-untreated group. ^, p < 0.05; ^^, p < 0.01; ^^^, p < 0.001, dnNrf2-HL60 cells vs. pEF-HL60 cells.
Figure 6.
Effects of GSH ethyl ester on mRNA levels of vitamin D- and Nrf2-related genes. pEF-HL60 and dnNrf2-HL60 cells were incubated with the indicated agents for 48 h. Samples were analyzed for mRNA levels of the indicated genes using quantitative RT-PCR. The expression of specific genes is normalized by the CT value of the internal reference gene (GAPDH). The data are means ± SD of 3 experiments performed in triplicate. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 vs. corresponding untreated control group; #, p < 0.05; ##, p < 0.01; ###, p < 0.001; ####, p < 0.0001 vs. corresponding sum of the effects of single agents. $, p < 0.05; $$, p < 0.01; $$$, p < 0.001; $$$$, p < 0.0001, GEE-treated vs. corresponding GEE-untreated group. ^, p < 0.05; ^^, p < 0.01; ^^^, p < 0.001, dnNrf2-HL60 cells vs. pEF-HL60 cells.
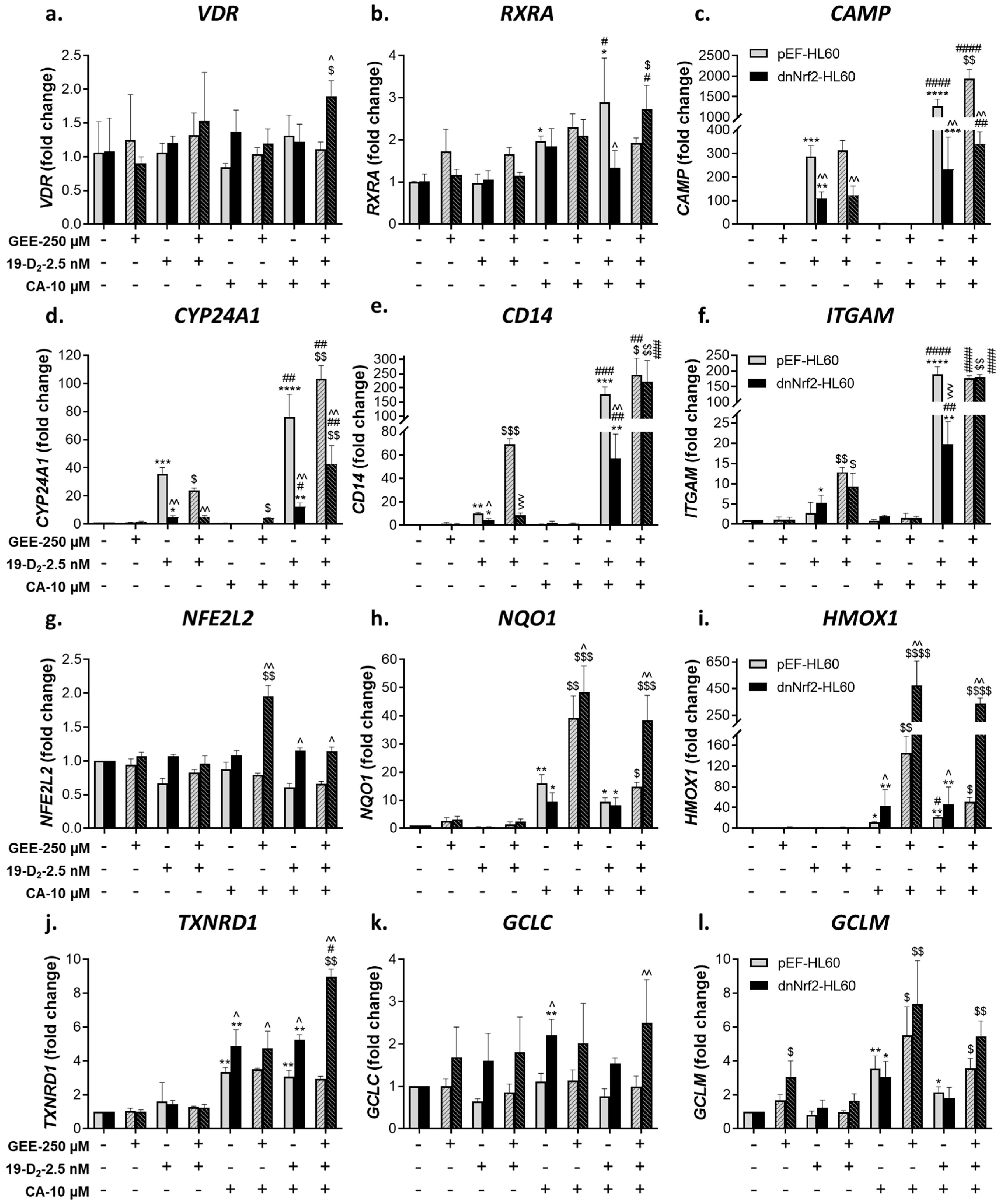
Figure 7.
Effects of GSH ethyl ester on the levels of vitamin D- and Nrf2-related proteins, c-Jun and its phosphorylation. pEF-HL60 and dnNrf2-HL60 cells were incubated with the indicated agents for 48 h. Samples were analyzed for the expression of the indicated proteins by Western blotting. Calreticulin was used as a protein loading control. (a, j) Representative Western blot images. (b-i,k,l) Absorbance values for specific proteins normalized to those of calreticulin and expressed in the bar graphs as fold change relative to the corresponding untreated controls. The data are means ± SD of 3 experiments. *, p < 0.05; **, p < 0.01 vs. corresponding control group; #, p < 0.05; ##, p < 0.01; ###, p < 0.001 vs. corresponding sum of the effects of single agents; $, p < 0.05; $$, p < 0.01; $$$, p < 0.001, GEE-treated vs. corresponding GEE-untreated group; ^, p < 0.05; ^^, p < 0.01, dnNrf2-HL60 cells vs. pEF-HL60 cells.
Figure 7.
Effects of GSH ethyl ester on the levels of vitamin D- and Nrf2-related proteins, c-Jun and its phosphorylation. pEF-HL60 and dnNrf2-HL60 cells were incubated with the indicated agents for 48 h. Samples were analyzed for the expression of the indicated proteins by Western blotting. Calreticulin was used as a protein loading control. (a, j) Representative Western blot images. (b-i,k,l) Absorbance values for specific proteins normalized to those of calreticulin and expressed in the bar graphs as fold change relative to the corresponding untreated controls. The data are means ± SD of 3 experiments. *, p < 0.05; **, p < 0.01 vs. corresponding control group; #, p < 0.05; ##, p < 0.01; ###, p < 0.001 vs. corresponding sum of the effects of single agents; $, p < 0.05; $$, p < 0.01; $$$, p < 0.001, GEE-treated vs. corresponding GEE-untreated group; ^, p < 0.05; ^^, p < 0.01, dnNrf2-HL60 cells vs. pEF-HL60 cells.
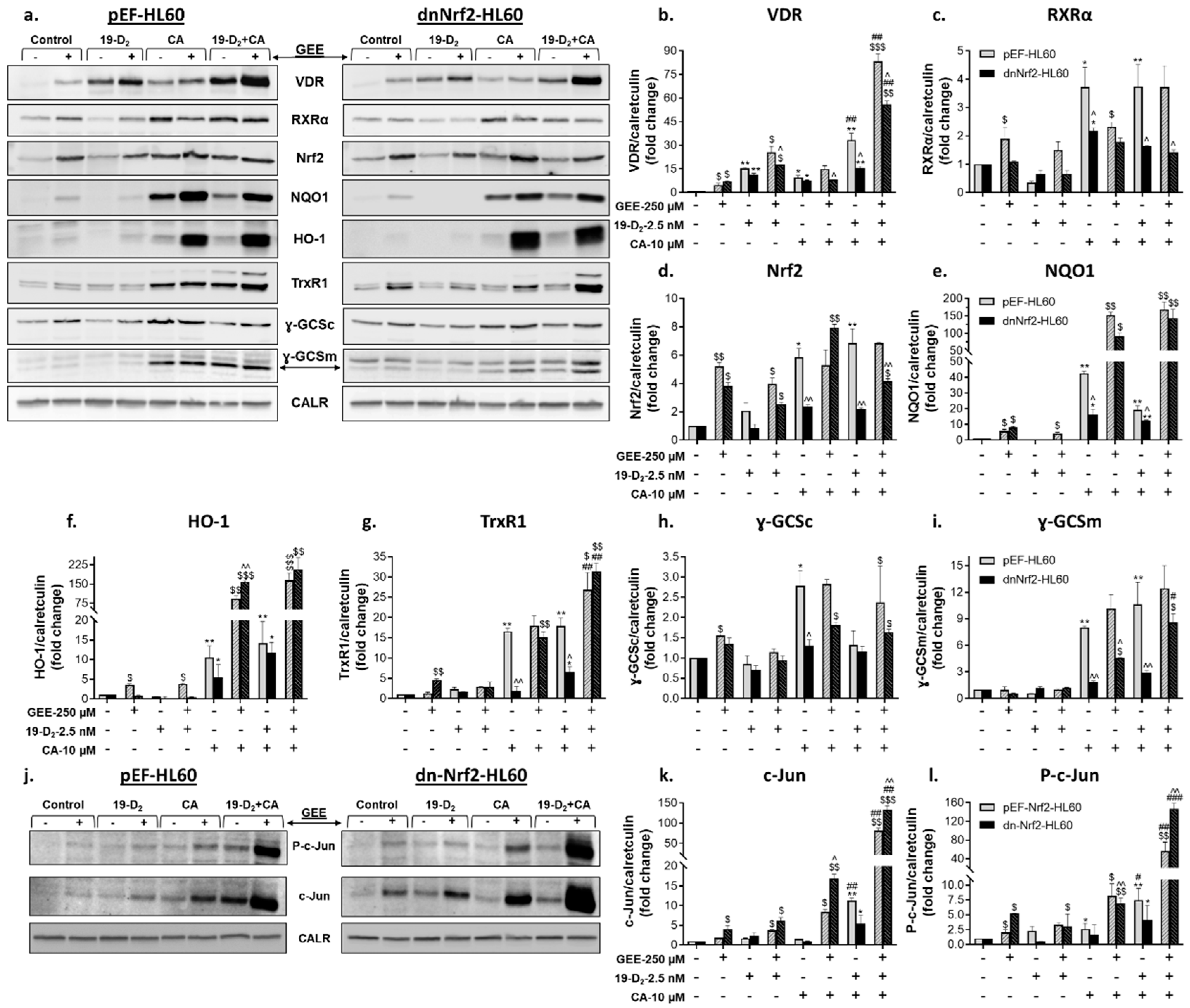
Figure 8.
Involvement of AP-1 in the enhancing effects of Nrf2 activators on 1,25D3-induced cell differentiation, VDR and RXRα protein expression, and VDRE transactivation.HL60 cells were preincubated for 24 h either without oligodeoxynucleotide (ODN) or with 10 μM TRE-ODN or mTRE-ODN followed by treatment with 1,25D3, CA, MMF, or their combinations at the indicated concentrations for an additional 48 h. (a) Averaged CD14 and CD11b surface expression, as measured by flow cytometry. (b) Representative Western blots showing changes in VDR and RXRα protein levels. Calreticulin was used as a protein loading control. (c) Cells were transiently transfected with VDREx6-Luc and Renilla luciferase plasmids, followed by pretreatment with or without TRE-ODN or mTRE-ODN for 24 h. Cells were then incubated with or without the indicated test agents for another 24 h, followed by measuring luciferase activity. The data are means ± SD of 3 experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 vs. corresponding control group; #, p < 0.05; ##, p < 0.01; ###, p < 0.001; combination vs. corresponding sum of the effects of single agents; $$, p < 0.01, TRE-ODN-treated group vs. corresponding TRE-ODN untreated group.
Figure 8.
Involvement of AP-1 in the enhancing effects of Nrf2 activators on 1,25D3-induced cell differentiation, VDR and RXRα protein expression, and VDRE transactivation.HL60 cells were preincubated for 24 h either without oligodeoxynucleotide (ODN) or with 10 μM TRE-ODN or mTRE-ODN followed by treatment with 1,25D3, CA, MMF, or their combinations at the indicated concentrations for an additional 48 h. (a) Averaged CD14 and CD11b surface expression, as measured by flow cytometry. (b) Representative Western blots showing changes in VDR and RXRα protein levels. Calreticulin was used as a protein loading control. (c) Cells were transiently transfected with VDREx6-Luc and Renilla luciferase plasmids, followed by pretreatment with or without TRE-ODN or mTRE-ODN for 24 h. Cells were then incubated with or without the indicated test agents for another 24 h, followed by measuring luciferase activity. The data are means ± SD of 3 experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 vs. corresponding control group; #, p < 0.05; ##, p < 0.01; ###, p < 0.001; combination vs. corresponding sum of the effects of single agents; $$, p < 0.01, TRE-ODN-treated group vs. corresponding TRE-ODN untreated group.
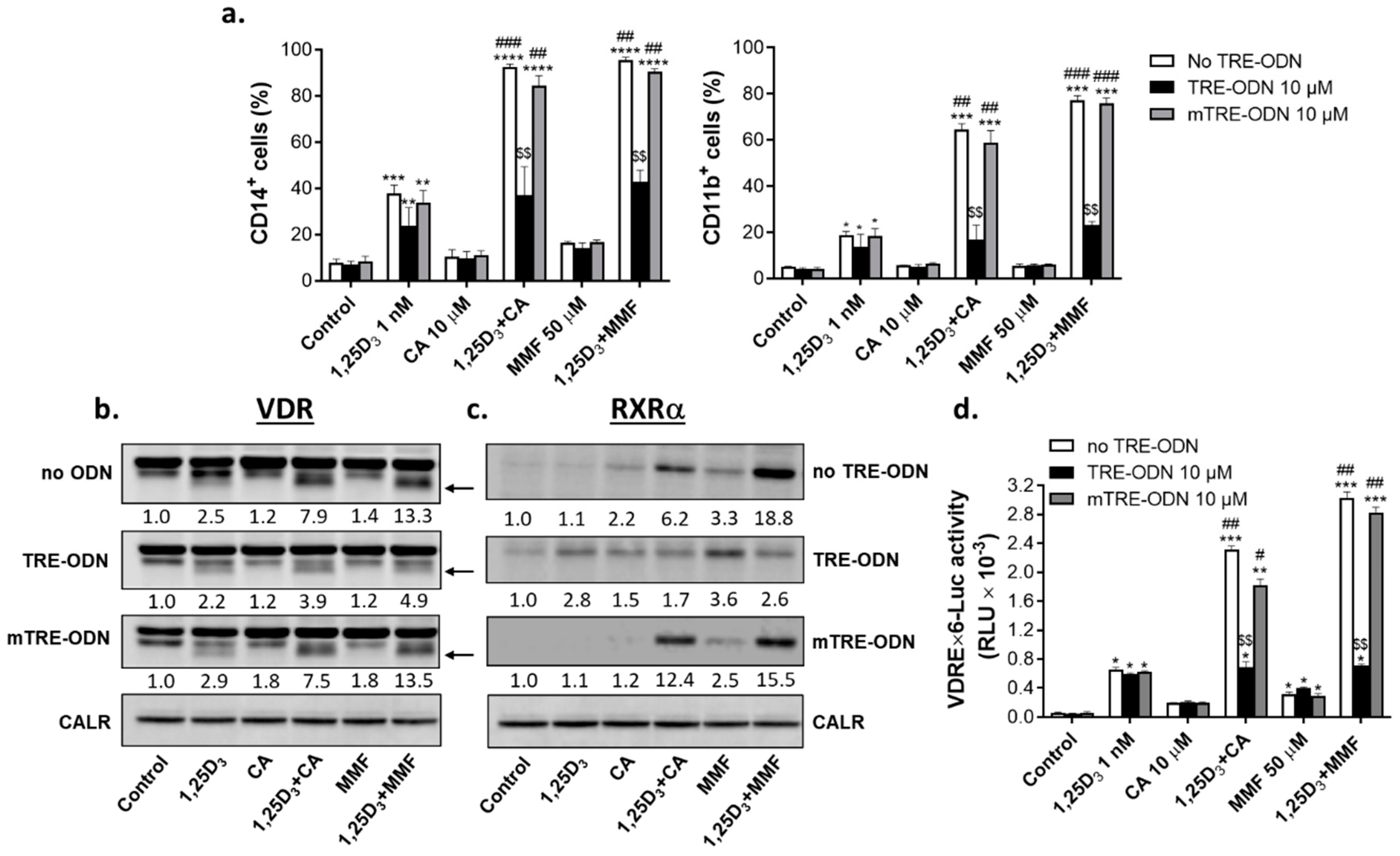
Figure 9.
Putative roles of glutathione and AP-1 in the cooperation between VDDs and Nrf2 activators in inducing differentiation of AML cells. See explanations in the text.
Figure 9.
Putative roles of glutathione and AP-1 in the cooperation between VDDs and Nrf2 activators in inducing differentiation of AML cells. See explanations in the text.
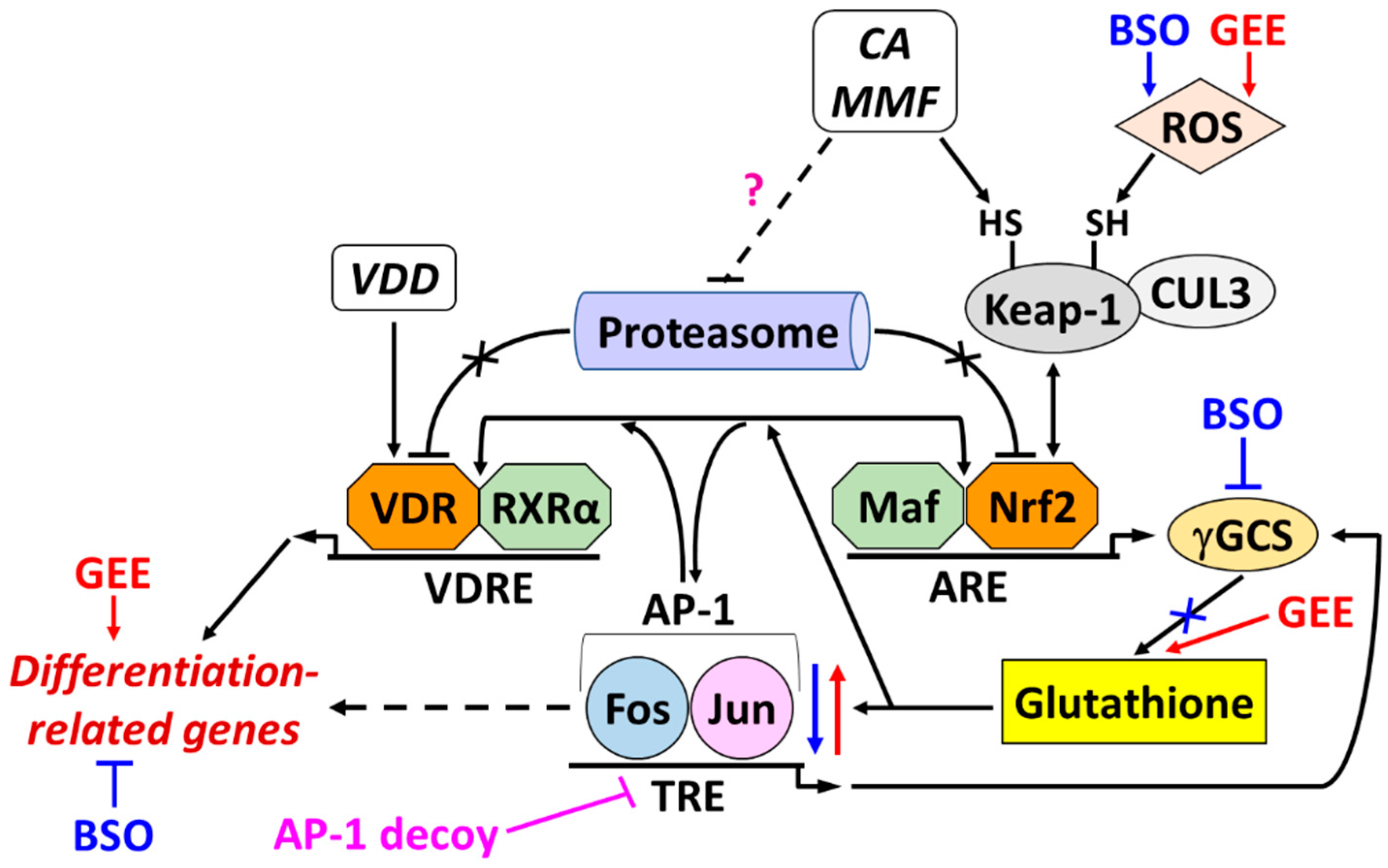
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



