
Submitted:
27 December 2023
Posted:
27 December 2023
You are already at the latest version
Abstract
Density functional theory (DFT) and coupled cluster theory (CCSD(T)) calculations are performed to investigate the geometric, electronic structures and chemical bonding of a series of Cu-doped zinc oxide clusters, CunZn3O3 (n = 1-4). The structural evolution of CunZn3O3 (n = 1-4) clusters are elaborated in our work. The planar seven-membered ring of CuZn3O3 cluster plays an important role in the structural evolution, that is, the Cu atom, Cu dimer (Cu2) and Cu trimer (Cu3) anchor on the CuZn3O3 cluster. The aggregate behavior of Cu atoms on Zn3O3 may offer insight into the study of Cu/ZnO-based catalysts. Additionally, it is found that the CunZn3O3 clusters become more stable as the Cu content (n) increases. Bader charge analysis points out that as the Cu atoms doping, the reducibility of Cu aggregation (Cun-1) on the CuZn3O3 cluster increases. Combined with the d-band centers and the surface electrostatic potential (ESP), the reactivity and the possible reaction sites of CunZn3O3 (n = 1-4) clusters are also illustrated.
Keywords:
Copper-doped zinc oxide clusters
; Density functional theory
; Structural evolution
; Reactivity
1. Introduction
Cu-based catalysts have played an important role in industry, such as electrocatalytic reduction of CO2, methanol steam reforming and water gas shift reaction.[1-4] But at the same time, Cu-based catalysts have still suffered some restrictions, such as thermal instabilities and low selectivity.[1,2] Cu-based catalysts are prone to sintering when the temperature rises above 300 ℃.[2] Cu/ZnO catalysts is one of the commonly used Cu-based catalysts, in which Cu usually acts as the main active component, and ZnO plays the dual role of promoter and support.[5] Specifically, Cu2+ ions would be reduced to the active Cu0/Cu+ species, and the addition of ZnO would increase the copper dispersion and reducibility which are related to the enhanced catalytic activity.[3,6] Meanwhile, the sizes and geometries of the metal nano-particles and the species formed at the Cu-ZnO interface also affect the catalytic properties of Cu/ZnO catalysts. To develop the Cu-based catalysts with superior performance, the precise structures and the structure-activity relationship of catalytic active sites and the interaction of the main active component with the additives including the supports are the fundamental issues that needs to be solved.[7] In this regard, clusters which may serve as the models at the atomic level and the highly active catalytic materials have attracted great research interest.
Recently, well-supported single/dual-atom and cluster catalysts have become the research hotspots.[8-12] Among these, copper oxide clusters have been considered as the catalytic active sites of copper-exchanged zeolites in the methane oxidation to methanol.[12] Herein, copper oxide clusters with the different stoichiometries and sizes were embedded in the channel of zeolites, the geometries, stabilities of these clusters and their underlying correlations with the catalytic activities were elaborated. Additionally, a fully-exposed Cu7 cluster anchored to the loop-like [6]cycloparaphenylene was found to be highly active and selective in the CO electro-reduction.[11] As for ZnO clusters, it is accepted that (ZnO)n (n < 8) clusters favored the Zn–O alternating ring structures. As the size (n) of (ZnO)n clusters increased to 8, a ring-to-cage transition occurred.[13-15] In these stoichiometric (ZnO)n (n = 1-13) clusters, (ZnO)3, (ZnO)9 and (ZnO)12 were found to possess relatively higher stability.[15] Moreover, the Zn3O3 six-membered ring can be found in the larger (ZnO)n (n > 8) clusters and the hexagonal wurtzite structures of ZnO crystals.[14,16,17] Thus, the relatively stable Zn3O3 cluster may be used as a molecular model for the wurtzite ZnO surface.
We have constantly strived to explore the novel chemical bonding and the cluster models to gain further insights into the active sites of complicated catalyst surfaces and the reaction mechanisms of catalytic processes.[18-21] In this work, we make an effort to reveal the evolution rule of the geometric, electronic structures and chemical bonding in the Cu-doped CunZn3O3 (n = 1-4) clusters. Bader charge analysis, d-band center theory and surface electrostatic potential (ESP) are used to analyze the reactivity and reaction sites of CunZn3O3 (n = 1-4) clusters. This work may enlighten the aggregation tendency of small Cu clusters on ZnO surface and the structures at the Cu-ZnO interface.
2. Methods
The initial geometries of CunZn3O3 (n = 1-4) clusters were constructed using the structural searches of the ABCluster program [22,23] in combination with the artificial constructions. These initial geometries were optimized using the B3LYP functional [24-26] in the Gaussian 09 program.[27] The Stuttgart small-core relativistic effective core potential (RECP) was used for the Cu and Zn atoms, whose corresponding basis sets are Cu: [6s,6p,4d,3f,2g], Zn: [6s,6p,4d,3f,2g].[28-31] As for O atoms, the aug-cc-pVTZ basis set was adopted.[32,33] In the structural optimizations, the vibrational frequencies were calculated to ensure that the optimized structures were free of imaginary frequencies. In order to verify the reliability of the above computational methods (denoted as B3LYP/BS), we compared the bond lengths, binding energies, vibrational frequencies and dipole moments of ZnO and CuO molecules from the available experiments with our calculations. As shown in Table S1, our calculations are in agreement with those data from experiments. The low-lying isomers within 0.50 eV at the B3LYP/BS level were then subjected to more accurate coupled cluster CCSD(T) single-point-energy calculations using the Molpro 2010 software.[34] Multiwfn program [35,36] was employed to analyze the Bader charge, surface electrostatic potential (ESP) and d-band centers of CunZn3O3 (n = 0-4) clusters.
3. Results
It is accepted that Zn3O3 cluster is a planar six-membered-ring structure with D3h symmetry (Figure 1a),[13] in which Zn and O atoms are alternately bonded (dZn-O = 1.817 Å). The distances between two Zn atoms are 2.654 Å.
3.1. Optimized Structures of CuZn3O3
CuZn3O3 clusters are constructed by adding a Cu atom to the most stable Zn3O3 cluster. The most stable structure of CuZn3O3 is shown in Figure 1b, which is consistent with the previous theoretical study.[15] It can be regard as inserting a Cu atom into the Zn-O bond of Zn3O3 cluster, leading to the planar seven-membered-ring structure. The second low-lying isomer (Figure 1c) lies 0.50 eV higher in energy, for which the six-membered Zn3O3 ring is broken. Other CuZn3O3 isomers are higher in energy by at least 0.50 eV. They are collected in the Supporting Information (Figure S1).
3.2. Optimized Structures of Cu2Zn3O3
To search for the ground state of Cu2Zn3O3 cluster, a Cu atom was added to the most stable CuZn3O3 cluster. The optimized Cu2Zn3O3 clusters with the relative energy below 0.50 eV are shown in Figure 2. The ground state of Cu2Zn3O3 cluster (Figure 2a) can be viewed as inserting a Cu atom into the Zn-Cu bond of CuZn3O3 cluster, resulting in a planar eight-membered-ring structure. It is consistent with the earlier finding of Cu2Zn3O3 cluster.[15] The Cu-Cu bond length is 2.366 Å, slightly longer than Cu-Zn bond length (2.343 Å). It is in agreement with the covalent radius of the Cu and Zn atoms (dCu = 1.52 Å, dZn =1.45 Å)[37] and suggests the metal-metal bonding of the inserting Cu atom with the neighboring Cu and Zn atoms. The second low-lying isomer (Figure 2b) which can be viewed as adding a bridged Cu to the CuZn3O3 ground state is 0.39 eV less stable than the ground state. Other isomers are found to be much higher in energy (ΔE > 0.50 eV) which are given in the Supporting Information (Figure S2).
3.3. Optimized Structures of Cu3Zn3O3
As the number of doped Cu atoms increases, more low-lying isomers appear for the Cu3Zn3O3 clusters (Figure 3). The most stable Cu3Zn3O3 cluster (Figure 3a) can be seen as adding a copper dimer (Cu2) between two bridged oxygen atoms of the CuZn3O3 ground state. At this point, the 3-fold coordinated bridged oxygen atom (μ3-O) begins to appear in the ground states of CunZn3O3 clusters. The Cu-Cu bond length of Cu dimer (Cu2) is 2.388 Å in Cu3Zn3O3 cluster which is much longer than that (2.232 Å) of isolated Cu2 molecule (D∞h 1Σg+) at the same calculation level. It is inferred that there are relatively strong interactions of the Cu2 moiety with the remaining fragment of Cu3Zn3O3. As shown in Figure 3, there are several isomers that have energies close to the ground state. To further distinguish the stability of these low-lying isomers, the single-point CCSD(T) calculations were performed using their B3LYP equilibrium geometries. The single-point CCSD(T) calculations still support the structure contained the Cu2 (Figure 3a) to be the most stable Cu3Zn3O3 cluster (Table S2). Other higher-energy isomers (ΔE > 0.50 eV) are shown in the Supporting Information (Figure S3).
3.4. Optimized Structures of Cu4Zn3O3
In our calculations, the most stable Cu4Zn3O3 cluster is shown in Figure 4a. It can be viewed as adding a copper trimer (Cu3) between two bridged oxygen atoms of the CuZn3O3 ground state. Meanwhile, several low-lying isomers within 0.50 eV were found (Figure 4). Among them, there is an isomer (Figure 4b) which contains the Cu4 moiety and is only 0.10 eV higher in energy. The relative energies of these isomers were further refined by the CCSD(T) single-point calculations (Table S2). The CCSD(T) results support the structure shown in Figure 4a to be the most stable one, and the isomer shown in Figure 4b is 0.28 eV less stable. Other higher-energy isomers (ΔE > 0.50 eV) are displayed in the Supporting Information (Figure S4).
4. Discussion
4.1. Structural Evolution in CunZn3O3 (n = 1-4) Clusters and Their Stability
It has been reported that the supported Cu2 and Cu3 clusters are the multi-atom cluster catalysts in specific reactions, and appropriate supports could improve their stability and dispersibility.[38,39] Zinc oxides as one of the most common promoter and support for Cu-based catalysts, the Zn3O3 six-membered ring is common in the larger (ZnO)n (n > 8) clusters and the wurtzite ZnO.[14,16,17] Studying the structural evolution of the CunZn3O3 (n = 1-4) clusters via sequential doping of Zn3O3 cluster with Cu atoms may help us gain insight into the aggregation behavior of small Cu clusters on ZnO surface.
For CuZn3O3, the Cu atom is inserted into the Zn-O bond of Zn3O3 cluster. For Cu2Zn3O3, the Cu atom is inserted into the Zn-Cu bond of CuZn3O3 cluster. As for Cu3Zn3O3 and Cu4Zn3O3 clusters, the planar seven-membered ring of CuZn3O3 cluster starts to play an important role in the subsequent aggregation of Cu atoms (Figure 5), that is, the Cu dimer (Cu2) and Cu trimer (Cu3) are attached to the CuZn3O3 cluster by two bridged oxygen atom (μ3-O). Herein, we found that at the low Cu content (n = 1,2), Cu atoms prefer to insert into the Zn-O bond of Zn3O3 first, then aggregate to form the ZnCu2 units. The six-membered ring of Zn3O3 gradually expanded to the eight-membered ring of Cu2Zn3O3. When the Cu content further increases (n = 3,4), the extra Cu atoms aggregate with each other to form Cun-1 units which is supported on CuZn3O3 cluster.
The relative stability of CunZn3O3 (n = 1-4) clusters is evaluated by the calculated atomization energy (Eb,1). The atomization energy (Eb,1) of CunZn3O3 clusters were calculated by the following formula:
Eb,1 = nE(Cu) + 3E(Zn) + 3E(O) – E(CunZn3O3)
E(CunZn3O3), E(Cu), E(Zn) and E(O) represents the energy of CunZn3O3 ground state, Cu, Zn and O atom, respectively. As seen in Table 1, the Eb,1 increases gradually with the increase of Cu content. It suggests the CunZn3O3 clusters become more stable as the Cu atoms doping (n = 0-4).
4.2. Chemical Bonding of CunZn3O3 (n = 1-4) Clusters
It is known that zinc has the electronic configuration of 3d104s2. Usually, its 3d electrons do not participate in bonding with other elements. So in zinc oxides, it almost shows the exclusively +2 oxidation state. But copper as the neighbor element of zinc, it has the 3d104s1 configuration, its 3d electrons participate in the bonding. So the oxidation state of Cu is more abundant (+1, +2 and +3).[40] To better understand the charge transfer in the sequential doping of Zn3O3 cluster with Cu atoms, we calculated the Bader charges of the CunZn3O3 (n = 1-4) clusters (Table 2). For Zn3O3 cluster, the Bader charge of Zn and O atom is +1.13 |e| and -1.13 |e|, respectively. Obviously, the oxidation state of Zn and O in Zn3O3 cluster is +2 and -2, respectively. Thus, the Bader charge of about ±0.5 |e| is indicative of a single-electron transfer, and the Bader charge of about ±1.0 |e| corresponds to two-electrons transfer.[41]
After inserting a Cu atom into the Zn-O bond of Zn3O3 cluster, the Bader charge of the Zn atom (denoted as Zn-1 in Figure 5) next to the newly added Cu atom (denoted as Cu-1) drops to +0.64 |e|, the charge of the Cu-1 atom is +0.43 |e|. In other words, the ZnCu unit in CuZn3O3 transfers 1.07 |e| to the nearby oxygen atoms (denoted as O-1 and O-3) in total. It is inferred that Zn-1 and Cu-1 atoms each transfer an electron to the adjacent oxygen atoms (O-1 and O-3). It leads to the metal-oxygen single bond. Meanwhile, the remaining 4s electron of Zn-1 forms the metal bond with Cu-1 atom. Continuing adding a Cu atom to the CuZn3O3 cluster, the added Cu atom (denoted as Cu-2) inserts into the Zn-Cu bond of CuZn3O3 cluster. As given in Table 2, the Bader charge of Cu-2 is approximately zero (+0.02 |e|). It could be understood by the formation of metal bonds between the Cu-2 atom and the Zn-1 and Cu-1 atoms. Herein, ZnCu2 unit in Cu2Zn3O3 cluster transfers 1.14 |e| to the adjacent oxygen atoms (O-1 and O-3) in total. Compared with Zn3O3 cluster, the charges on the other atoms do not change much.
For CunZn3O3 (n = 3,4) clusters, they can be viewed as adding the Cu2 (Cu-2 and Cu-3) and Cu3 (Cu-2, Cu-3 and Cu-4) to the CuZn3O3 cluster linked by two 3-fold coordinated oxygen atoms (O-1 and O-3). Compared with the ZnCu diatom of CuZn3O3 cluster, the Bader charge of Zn-1 reduces from +0.64 |e| to +0.37 |e|, and the charge of Cu-1 also decreases from +0.43 |e| to roughly +0.3 |e|. It suggested less charge transfers from the ZnCu diatom of CunZn3O3 (n = 3,4) to the O-1 and O-3 atoms. As compensation, the newly added Cu2 and Cu3 units in CunZn3O3 (n = 3,4) transfer charges of +0.51 |e| and +0.56 |e| to the O-1 and O-3 atoms. To analyze the interaction of Cu aggregation (Cun-1) with CuZn3O3 cluster, the binding energies (Eb,2) of the isolated Cun-1 clusters with CuZn3O3 cluster were calculated by the following formula:
Eb,2 = E(CunZn3O3) – E(CuZn3O3) – E(Cun-1)
E(CunZn3O3), E(CuZn3O3) and E(Cun-1) represents the ground-state energy of CunZn3O3, CuZn3O3 and Cun-1 clusters, respectively. The Eb,2 of Cu2 in Cu3Zn3O3 cluster is calculated to be -1.60 eV, and that of Cu3 in Cu4Zn3O3 cluster is -3.21 eV. The more negative Eb,2 means the stronger interaction between Cu aggregation (Cun-1) and CuZn3O3 cluster, and higher stability of Cun-1 on the CuZn3O3 seven-membered ring. Here, the more negative binding energies (Eb,2) coincides with the more transferred charge from Cun-1 to CuZn3O3. For Cu/ZnO catalysts, the addition of ZnO is conducive to increasing the dispersion and reducibility of the active copper component.[42] From the perspective of Bader charge, the Cun-1 in CunZn3O3 (n = 3,4) is more reducible than the Cun in CunZn3O3 (n = 1,2). The synergistic interaction between Cu and Zn in CuZn3O3 may enhance the reducibility of Cu species in CunZn3O3 (n = 3,4).
4.3. Reactivity of CunZn3O3 (n = 1-4) Clusters
The model of d-band center was developed by Nørskov and co-workers[43] and was used as an important descriptor to determine the reactivity of surfaces and clusters.[44-48] The partial density of states (PDOS) for the d-orbitals of metal atoms in CunZn3O3 (n = 0-4) clusters are depicted in Figure 6, and the d-band centers (εd) are denoted by the red solid line. For the open-shell systems, the spin up (α) and spin down (β) d-band centers (εd) were calculated separately (Table S3), and the spin down ones were always higher in energy. So we uniformly use the spin down d-band centers (εd) for the subsequent comparison. The energy level of highest occupied molecular orbital (HOMO-β) are marked by the blue dashed line. For comparison, all HOMO energy levels in Figure 6 are shifted to zero. As shown in Figure 6f, the εd move toward HOMO-β as the Cu content (n) increases. It suggests the interaction between nucleophilic molecules and the metal atoms become stronger as the Cu content (n) increases.[44,47] It also infers the reactivity of CunZn3O3 (n = 0-4) clusters increase as the Cu content (n) increases.
The electrostatic potential (ESP) provides a way of identifying the active sites.[44,49] The surface ESP for CunZn3O3 (n = 1-4) clusters are shown in Figure 7. Obviously, the red-colored (positive ESP) regions are positioned at the metal atoms, and the ESP of CunZn3O3 clusters are less localized compared with the Zn3O3 clusters. Additionally, the cyan and yellow tiny spheres in Figure 7 point out the locations of the extreme points of the surface ESP, and the arrows indicate the extreme points with the maximum absolute values. The sites with the most positive values of molecular ESP are associated with the ideal adsorption positions for nucleophilic reagents, whereas the most negative ESP are related to that of electrophilic reagents. In this series of CunZn3O3 (n = 1-4) clusters, the most positive regions of ESP are always nearby the Zn-2 atom except for Cu2Zn3O3. Except for Cu2Zn3O3, the other CunZn3O3 clusters can be viewed as adding the Cu2 and Cu3 units to the CuZn3O3 cluster linked by two 3-fold coordinated oxygen atoms (O-1 and O-3). For Cu2Zn3O3, the newly added Cu atom (Cu-2) expands the seven-membered ring of CuZn3O3 to the eight-membered ring. The most positive region of ESP of Cu2Zn3O3 is nearby the newly added Cu-2 atom. In CunZn3O3 (n = 1-4) clusters, the ESP of 3-fold coordinated oxygen atoms are more negative than that of 2-fold coordinated oxygen atoms. For CuZn3O3 and Cu2Zn3O3, the most negative regions are located near the O-1 or O-3 atom. For Cu3Zn3O3 and Cu4Zn3O3, the most negative regions are located near the O-2 atoms. They infer the sensitivity of reactivity to the structures.
5. Conclusions
We report a systematic theoretical study of a series of copper-doped zinc oxide clusters: CunZn3O3 (n = 1-4). The geometric, electronic structures and chemical bonding of CunZn3O3 (n = 1-4) clusters are investigated by extensive density functional theory (DFT) and coupled cluster theory (CCSD(T)) calculations. The structural evolutions of CunZn3O3 (n = 1-4) clusters are found in our work. At the low Cu content (n = 1,2), Cu atoms prefer to insert into the Zn-O bond of Zn3O3 first, then aggregate to form the ZnCu2 units. The six-membered ring of Zn3O3 gradually expands to the eight-membered ring of Cu2Zn3O3. When the Cu content further increases (n = 3,4), the extra Cu atoms aggregate with each other to form Cun-1 units on the CuZn3O3 cluster. Additionally, relative stability of CunZn3O3 (n = 1-4) clusters is evaluated. The CunZn3O3 clusters become more stable as the Cu atoms doping (n = 1-4). Bader charge analysis suggests that as the Cu content (n) increases, the reducibility of Cu aggregation (Cun-1) on the CuZn3O3 cluster increase. The studies on d-band centers of CunZn3O3 (n = 0-4) clusters infer the reactivity also increase as the Cu content (n) increases. The information on the possible reaction site of CunZn3O3 (n = 1-4) clusters are predicted by the surface electrostatic potential (ESP) calculations.
Supplementary Materials
he following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: Calculated results at the B3LYP/BS level for the bond lengths, binding energies and other properties of ZnO and CuO along with the corresponding available experimental data. Table S2: Relative energies of CunZn3O3 (n = 1-4) clusters which were further refined by the CCSD(T) single-point calculations. Table S3: The calculated d-band centers for the spin up (α), spin down (β) and both spin modes of CunZn3O3 (n = 0-4) clusters. Figures S1-S4: Alternative optimized structures for CunZn3O3 (n = 1-4) clusters at the B3LYP/BS level. Table S4: Cartesian coordinates for the optimized CunZn3O3 (n = 0-4) clusters.
Author Contributions
Investigation, Z.W.T.; visualization, H.Y.Z. and H.H.L.; writing—original draft preparation, Z.W.T. and H.Y.Z.; writing—review and editing, B.W. and W.J.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (21301030 and 21603117).
Data Availability Statement
The data presented in this study are available from the corresponding authors upon reasonable request.
Acknowledgments
The authors gratefully acknowledge supports from the National Natural Science Foundation of China (21301030and 21603117).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Xiao, C.; Zhang, J. Architectural Design for Enhanced C2 Product Selectivity in Electrochemical CO2 Reduction Using Cu-Based Catalysts: A Review. ACS Nano 2021, 15, 7975–8000. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.; Xiao, S.; Lai, Q.; Wang, D.; Huang, Y.; Feng, G.; Zhang, R.; Wang, T. Advances in Enhancing the Stability of Cu-Based Catalysts for Methanol Reforming. Catalysts 2022, 12, 747. [Google Scholar] [CrossRef]
- Saw, S.K.; Datta, S.; Chavan, P.D.; Gupta, P.K.; Kumari, S.; Sahu, G.; Chauhan, V. Significance and influence of various promoters on Cu-based catalyst for synthesizing methanol from syngas: a critical review. Journal of Chemical Technology And Biotechnology 2023, 98, 1083–1102. [Google Scholar] [CrossRef]
- Teng, M.; Ye, J.; Wan, C.; He, G.; Chen, H. Research Progress on Cu-Based Catalysts for Electrochemical Nitrate Reduction Reaction to Ammonia. Industrial & Engineering Chemistry Research 2022, 61, 14731–14746. [Google Scholar] [CrossRef]
- Hou, R.; Qiu, R.; Sun, K. Progress in the Cu-based catalyst supports for methanol synthesis from CO2. Chemical Industry and Engineering Progress 2020, 39, 2639–2647. [Google Scholar]
- Velu, S.; Suzuki, K. Selective Production of Hydrogen for Fuel Cells Via Oxidative Steam Reforming of Methanol Over CuZnAl Oxide Catalysts: Effect of Substitution of Zirconium and Cerium on the Catalytic Performance. Topics in Catalysis 2003, 22, 235–244. [Google Scholar] [CrossRef]
- Ranjekar, A.M.; Yadav, G.D. Steam Reforming of Methanol for Hydrogen Production: A Critical Analysis of Catalysis, Processes, and Scope. Industrial & Engineering Chemistry Research 2021, 60, 89–113. [Google Scholar] [CrossRef]
- Hou, C.-C.; Wang, H.-F.; Li, C.; Xu, Q. From metal–organic frameworks to single/dual-atom and cluster metal catalysts for energy applications. Energy Environ. Sci. 2020, 13, 1658–1693. [Google Scholar] [CrossRef]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nature Chemistry 2011, 3, 634–641. [Google Scholar] [CrossRef]
- Qin, R.; Liu, P.; Fu, G.; Zheng, N. Strategies for Stabilizing Atomically Dispersed Metal Catalysts. Small Methods 2018, 2, 1700286. [Google Scholar] [CrossRef]
- Liu, Y.-Q.; Qiu, Z.-Y.; Zhao, X.; Wang, W.-W.; Dang, J.-S. Trapped copper in [6]cycloparaphenylene: a fully-exposed Cu7 single cluster for highly active and selective CO electro-reduction. Journal of Materials Chemistry A 2021, 9, 25922–25926. [Google Scholar] [CrossRef]
- Palagin, D.; Knorpp, A.J.; Pinar, A.B.; Ranocchiari, M.; van Bokhoven, J.A. Assessing the relative stability of copper oxide clusters as active sites of a CuMOR zeolite for methane to methanol conversion: size matters? Nanoscale 2017, 9, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Matxain, J.M.; Fowler, J.E.; Ugalde, J.M. Small clusters of II-VI materials: ZniOi, i=1 –9. Physical Review A 2000, 62, 053201. [Google Scholar] [CrossRef]
- Fernando, A.; Dimuthu, K.L.; Weerawardene, M.; Karimova, N.V.; Aikens, C.M. Quantum Mechanical Studies of Large Metal, Metal Oxide, and Metal Chalcogenide Nanoparticles and Clusters. Chemical Reviews 2015, 115, 6112–6216. [Google Scholar] [CrossRef] [PubMed]
- Yong, Y.; Wang, Z.; Liu, K.; Song, B.; He, P. Structures, stabilities, and magnetic properties of Cu-doped ZnnOn (n=3,9,12) clusters: A theoretical study. Computational and Theoretical Chemistry 2012, 989, 90–96. [Google Scholar] [CrossRef]
- Tayade, N.T.; Mane, S.M.; Shende, A.T.; Tirpude, M.P.; Shin, J.C. Dissociation of ZnO ring from Zn3O3 cluster by CASSCF. Chemical Physics 2021, 542, 111077. [Google Scholar] [CrossRef]
- Jin, W.; Chen, G.; Duan, X.; Yin, Y.; Ye, H.; Wang, D.; Yu, J.; Mei, X.; Wu, Y. Adsorption behavior of formaldehyde on ZnO (101¯0) surface: A first principles study. Applied Surface Science 2017, 423, 451–456. [Google Scholar] [CrossRef]
- Wang, B.; Xia, C.-J.; Fang, H.-L.; Chen, W.-J.; Zhang, Y.-F.; Huang, X. Mononuclear thorium halide clusters ThX4 (X = F, Cl): gas-phase hydrolysis reactions. Physical Chemistry Chemical Physics 2018, 20, 21184–21193. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Xie, L.; Liu, X.-J.; Chen, W.-J.; Zhang, Y.-F.; Huang, X. Structural Evolution and Chemical Bonding of Di-Niobium Boride Clusters Nb2Bx-/0 (x = 1-6): Hexagonal Bipyramid Nb2B6−/0 Species. European Journal Of Inorganic Chemistry 2018, 2018, 940–950. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, S.-Y.; Ye, L.-H.; Zhang, X.-F.; Zhang, Y.-F.; Chen, W.-J. Exploring the Reaction Mechanism of H2S Decomposition with MS3 (M = Mo, W) Clusters. ACS Omega 2020, 5, 13324–13332. [Google Scholar] [CrossRef]
- Wang, B.; Wu, N.; Zhang, X.-B.; Huang, X.; Zhang, Y.-F.; Chen, W.-K.; Ding, K.-N. Probing the Smallest Molecular Model of MoS2 Catalyst: S2 Units in the MoSn–/0 (n= 1–5) Clusters. Journal of Physical Chemistry A 2013, 117, 5632–5641. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Dolg, M. ABCluster: the artificial bee colony algorithm for cluster global optimization. Physical Chemistry Chemical Physics 2015, 17, 24173–24181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Dolg, M. Global optimization of clusters of rigid molecules using the artificial bee colony algorithm. Physical Chemistry Chemical Physics 2016, 18, 3003–3010. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. Journal of Chemical Physics 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Physical Review B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. Journal of Physical Chemistry 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01. Gaussian, Inc., Wallingford CT 2010. [Google Scholar]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theoretica Chimica Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Küchle, W.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted pseudopotentials for the actinides. Parameter sets and test calculations for thorium and thorium monoxide. Journal of Chemical Physics 1994, 100, 7535–7542. [Google Scholar] [CrossRef]
- Cao, X.; Dolg, M. Segmented contraction scheme for small-core actinide pseudopotential basis sets. Journal of Molecular Structure (THEOCHEM) 2004, 673, 203–209. [Google Scholar] [CrossRef]
- Cao, X.; Dolg, M.; Stoll, H. Valence basis sets for relativistic energy-consistent small-core actinide pseudopotentials. Journal of Chemical Physics 2003, 118, 487–496. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning Jr., T. H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. Journal of Chemical Physics 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Dunning Jr., T. H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. Journal of Chemical Physics 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Werner, H.J.; Knowles, P.J.; Manby, F.R.; Schütz, M.; Celani, P.; Knizia, G.; Korona, T.; Lindh, R.; Mitrushenkov, A.; Rauhut, G.; et al. MOLPRO, version 2010.1, a package of ab initio programs; see http://www.molpro.net.
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. Journal of Computational Chemistry 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Zhang, J.; Lu, T. Efficient evaluation of electrostatic potential with computerized optimized code. Physical Chemistry Chemical Physics 2021, 23, 20323–20328. [Google Scholar] [CrossRef] [PubMed]
- Lide, D.R. CRC Handbook of Chemistry and Physics, 89th edition; CRC Press/Taylor and Francis: Boca Raton, Florida, 2008. [Google Scholar]
- Chen, Z.W.; Yan, J.M.; Zheng, W.T.; Jiang, Q. Cu4 Cluster Doped Monolayer MoS2 for CO Oxidation. Scientific Reports 2015, 5, 11230. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.W.; Chen, L.X.; Yang, C.C.; Jiang, Q. Atomic (single, double, and triple atoms) catalysis: frontiers, opportunities, and challenges. Journal of Materials Chemistry A 2019, 7, 3492–3515. [Google Scholar] [CrossRef]
- Nicholls, D. Copper. In Complexes and First-Row Transition Elements; Macmillan Education UK: London, 1974; pp. 201–206. [Google Scholar]
- Thang, H.V.; Pacchioni, G. Spontaneous Formation of Gold Cluster Anions on ZnO/Cu(111) Bilayer Films. The Journal of Physical Chemistry C 2019, 123, 7644–7653. [Google Scholar] [CrossRef]
- Fierro, G.; Lo Jacono, M.; Inversi, M.; Porta, P.; Cioci, F.; Lavecchia, R. Study of the reducibility of copper in CuO-ZnO catalysts by temperature-programmed reduction. Applied Catalysis A: General 1996, 137, 327–348. [Google Scholar] [CrossRef]
- Hammer, B.; Nørskov, J.K. Electronic factors determining the reactivity of metal surfaces. Surface Science 1995, 343, 211–220. [Google Scholar] [CrossRef]
- Rodríguez-Kessler, P.L.; Rodríguez-Domínguez, A.R.; Muñoz-Castro, A. On the structure and reactivity of PtnCun (n = 1–7) alloy clusters. Physical Chemistry Chemical Physics 2021, 23, 7233–7239. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Waclawik, E.R.; Du, A. Computational screening of two-dimensional coordination polymers as efficient catalysts for oxygen evolution and reduction reaction. Journal of Catalysis 2017, 352, 579–585. [Google Scholar] [CrossRef]
- Takagi, N.; Ishimura, K.; Fukuda, R.; Ehara, M.; Sakaki, S. Reaction Behavior of the NO Molecule on the Surface of an Mn Particle (M = Ru, Rh, Pd, and Ag; n = 13 and 55): Theoretical Study of Its Dependence on Transition-Metal Element. The Journal of Physical Chemistry A 2019, 123, 7021–7033. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Guo, L. Mechanism of the Reverse Water–Gas Shift Reaction Catalyzed by Cu12TM Bimetallic Nanocluster: A Density Functional Theory Study. Journal of Cluster Science 2018, 29, 867–877. [Google Scholar] [CrossRef]
- Megha, *!!! REPLACE !!!*; Mondal, K.; Ghanty, T.K.; Banerjee, A. Adsorption and Activation of CO2 on Small-Sized Cu–Zr Bimetallic Clusters. The Journal of Physical Chemistry A 2021, 125, 2558–2572. [Google Scholar] [CrossRef]
- Arteca, G.A.; Hernández-Laguna, A.; Rández, J.J.; Smeyers, Y.G.; Mezey, P.G. A topological analysis of molecular electrostatic potential on van der Waals surfaces for histamine and 4-substituted derivatives as H2-receptor agonists. Journal of Computational Chemistry 1991, 12, 705–716. [Google Scholar] [CrossRef]
Figure 1.
Optimized structures (ΔE ≤ 0.50 eV) for Zn3O3 and CuZn3O3. The bond lengths are in angstroms and the relative energies (ΔE) in eV are in the parentheses.
Figure 1.
Optimized structures (ΔE ≤ 0.50 eV) for Zn3O3 and CuZn3O3. The bond lengths are in angstroms and the relative energies (ΔE) in eV are in the parentheses.
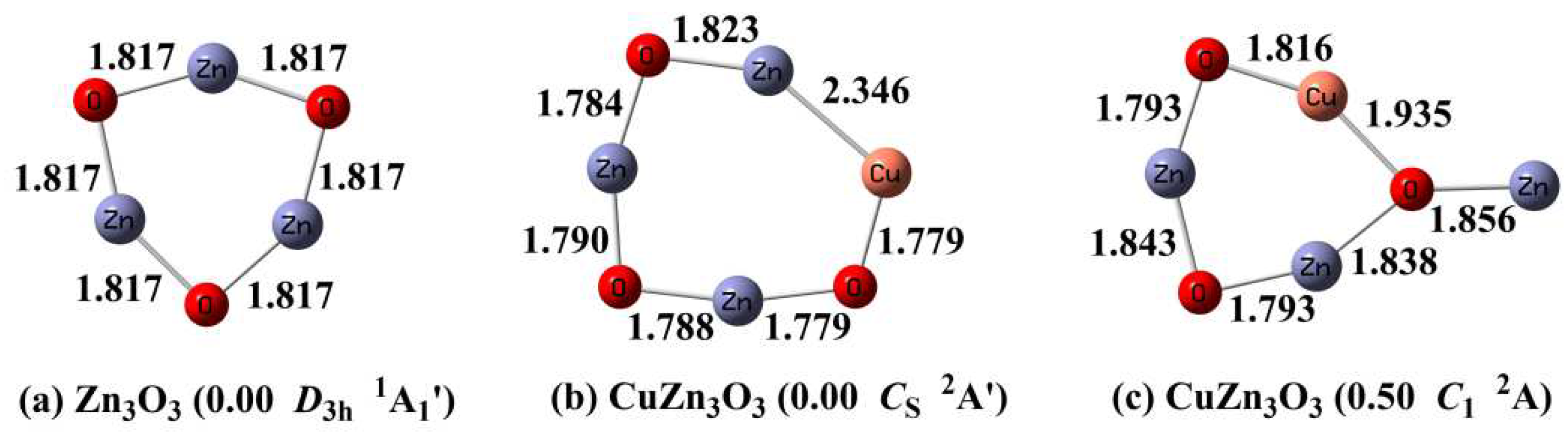
Figure 2.
Optimized structures (ΔE < 0.50 eV) for Cu2Zn3O3. The bond lengths are in angstroms and the relative energies (ΔE) in eV are in the parentheses.
Figure 2.
Optimized structures (ΔE < 0.50 eV) for Cu2Zn3O3. The bond lengths are in angstroms and the relative energies (ΔE) in eV are in the parentheses.
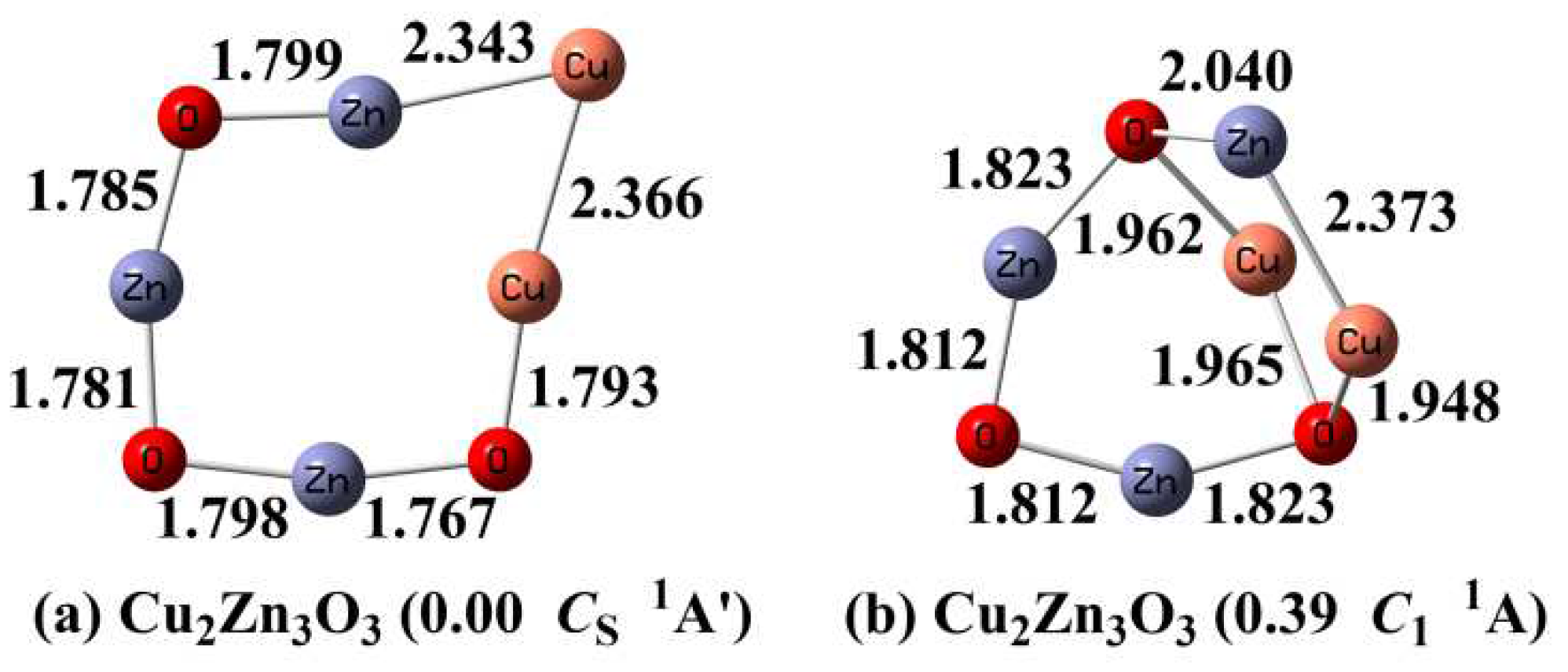
Figure 3.
Optimized structures (ΔE < 0.50 eV) for Cu3Zn3O3. The bond lengths are in angstroms and the relative energies (ΔE) in eV are in the parentheses.
Figure 3.
Optimized structures (ΔE < 0.50 eV) for Cu3Zn3O3. The bond lengths are in angstroms and the relative energies (ΔE) in eV are in the parentheses.

Figure 4.
Optimized structures (ΔE < 0.50 eV) for Cu2Zn3O3. The bond lengths are in angstroms and the relative energies (ΔE) in eV are in the parentheses.
Figure 4.
Optimized structures (ΔE < 0.50 eV) for Cu2Zn3O3. The bond lengths are in angstroms and the relative energies (ΔE) in eV are in the parentheses.
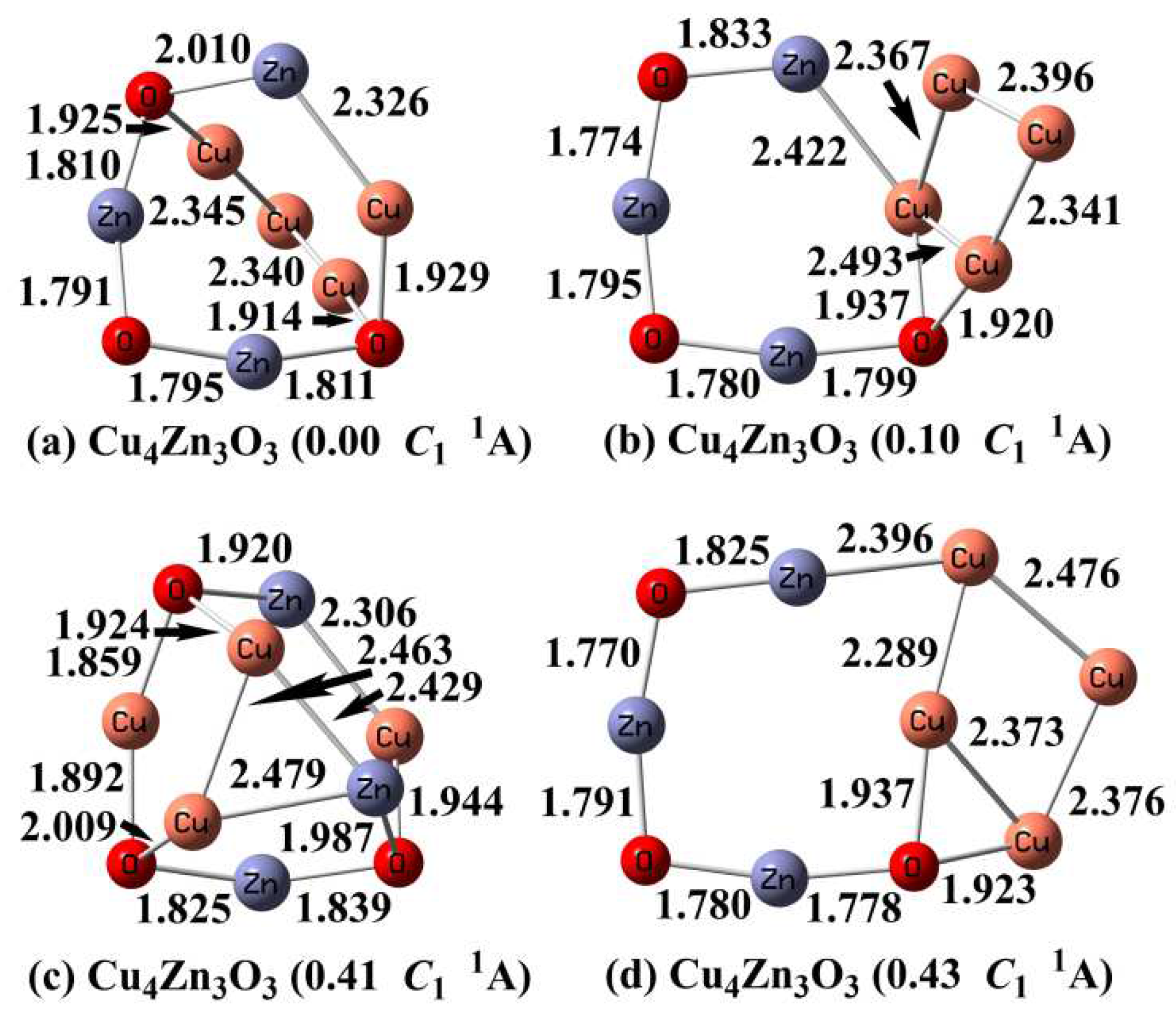
Figure 5.
Structural evolution of CunZn3O3 (n = 1-4) clusters.
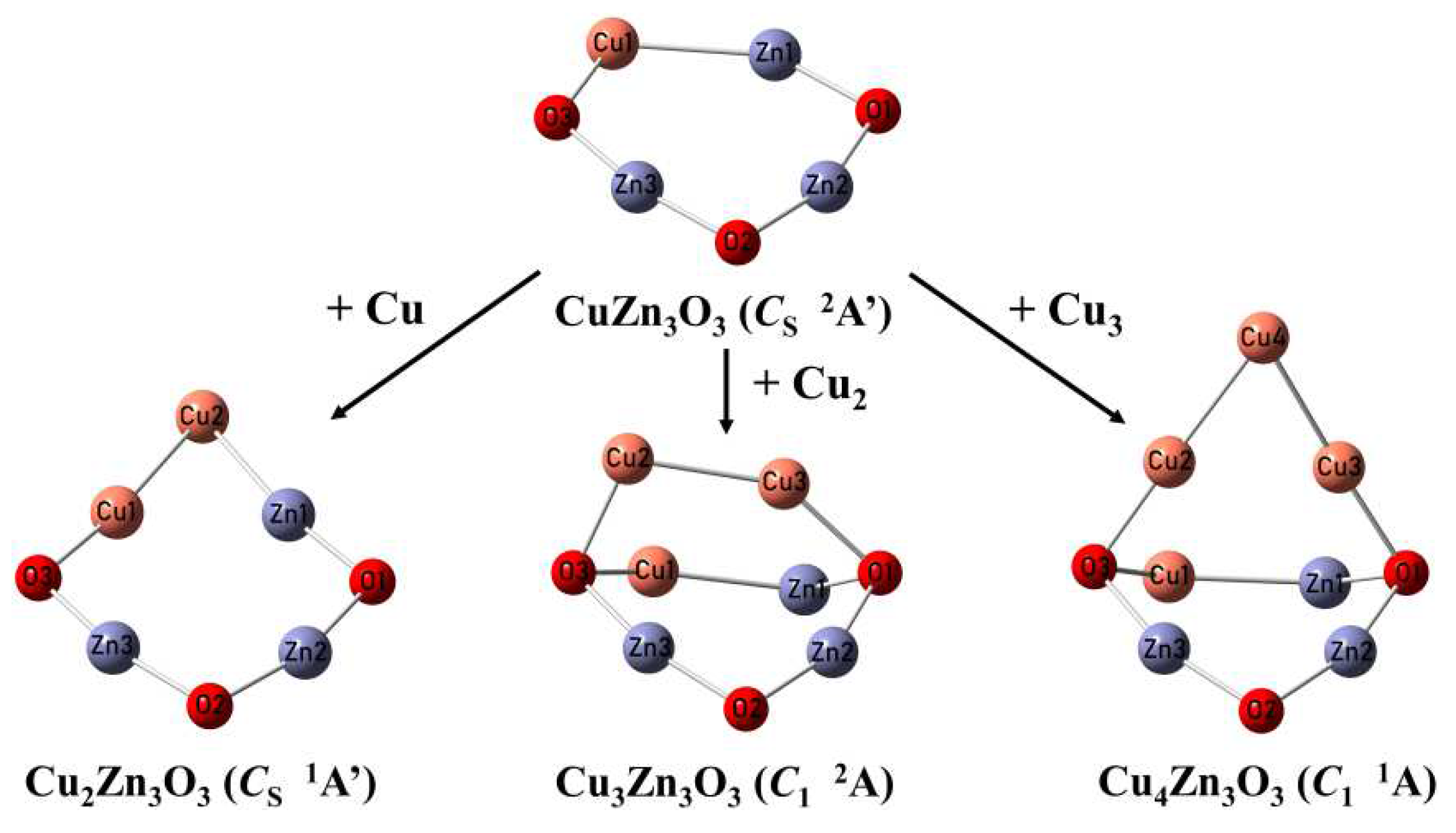
Figure 6.
(a)-(e) The d-band density of states for the lowest-energy CunZn3O3 (n = 0-4) clusters. The inset is the enlarged drawing of the d-band center. (f) The d-band center (εd) as a function of Cu content (n) in CunZn3O3 (n = 0-4) clusters.
Figure 6.
(a)-(e) The d-band density of states for the lowest-energy CunZn3O3 (n = 0-4) clusters. The inset is the enlarged drawing of the d-band center. (f) The d-band center (εd) as a function of Cu content (n) in CunZn3O3 (n = 0-4) clusters.
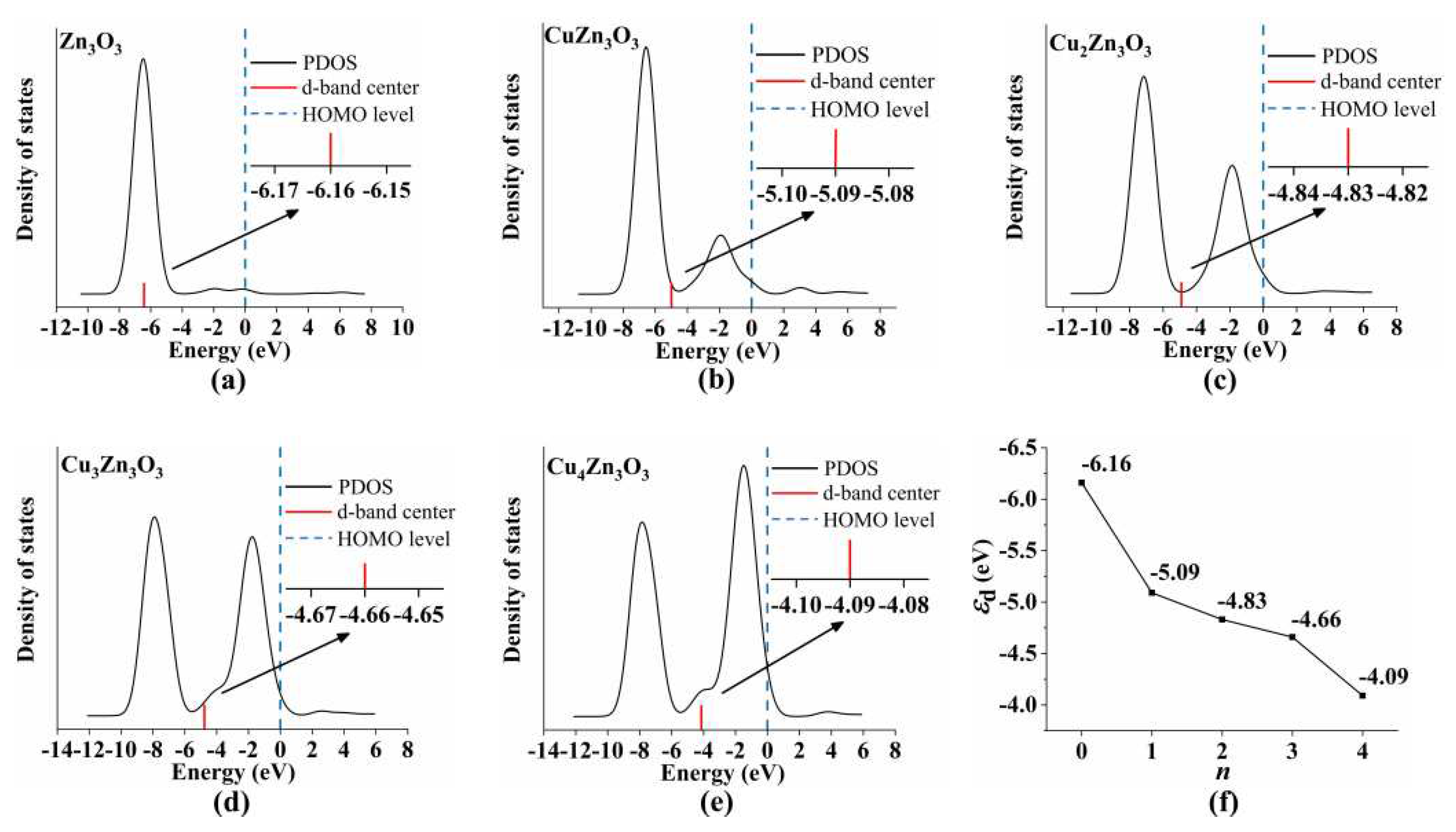
Figure 7.
The electrostatic potential (ESP) map on the van der Waals surface for the lowest-energy CunZn3O3 (n = 0-4) clusters.
Figure 7.
The electrostatic potential (ESP) map on the van der Waals surface for the lowest-energy CunZn3O3 (n = 0-4) clusters.
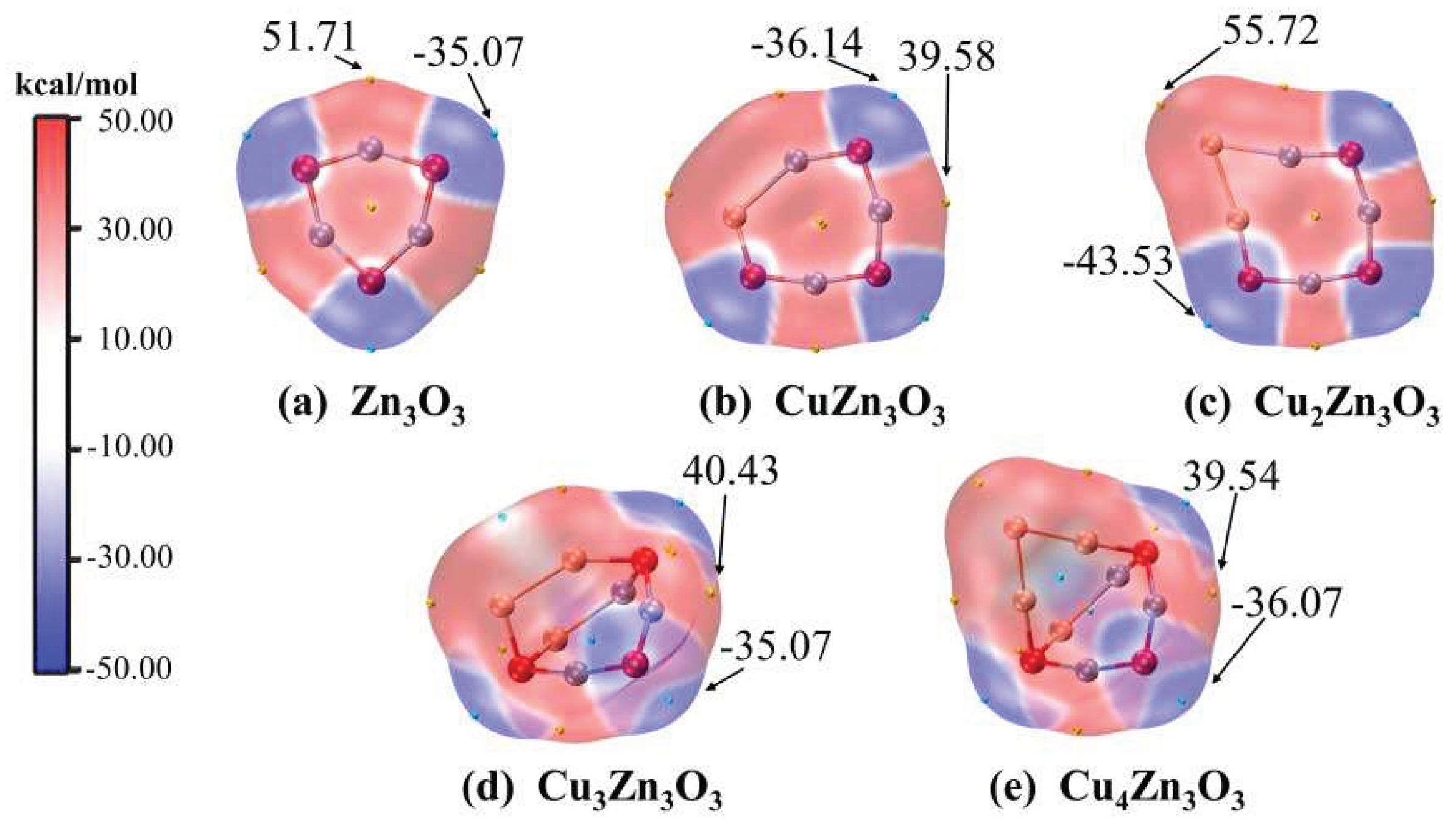

Table 1.
Atomization energy (Eb,1) of CunZn3O3 cluster. The energies are in eV.
| Cluster | Zn3O3 | CuZn3O3 | Cu2Zn3O3 | Cu3Zn3O3 | Cu4Zn3O3 |
| Eb,1 | 22.07 | 24.03 | 26.68 | 28.28 | 31.27 |
Table 2.
Bader charges (|e|) analysis of CunZn3O3 (n = 0-4).
| Cluster | Zn-1 | Zn-2 | Zn-3 | O-1 | O-2 | O-3 | Cu-1 | Cu-2 | Cu-3 | Cu-4 |
| Zn3O3 | 1.13 | 1.13 | 1.13 | -1.13 | -1.13 | -1.13 | ||||
| CuZn3O3 | 0.64 | 1.12 | 1.15 | -1.15 | -1.14 | -1.06 | 0.43 | |||
| Cu2Zn3O3 | 0.75 | 1.14 | 1.12 | -1.15 | -1.15 | -1.10 | 0.37 | 0.02 | ||
| Cu3Zn3O3 | 0.37 | 1.11 | 1.12 | -1.16 | -1.14 | -1.09 | 0.28 | 0.25 | 0.26 | |
| Cu4Zn3O3 | 0.37 | 1.12 | 1.11 | -1.17 | -1.14 | -1.11 | 0.26 | 0.31 | 0.31 | -0.06 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



