
Submitted:
27 December 2023
Posted:
27 December 2023
You are already at the latest version
Abstract
Polycystic ovary syndrome (PCOS) is a multisystem disorder that presents with a variety of phenotypes involving metabolic, endocrine, reproductive, and psychological symptoms and signs. Women with PCOS are at increased risk of pregnancy complications including implantation failure, miscarriage, gestational diabetes, fetal growth restriction, preterm labour, and preeclampsia (PE). This may be attributed to the presence of specific susceptibility features associated with PCOS before and during pregnancy, such as chronic systemic inflammation, insulin resistance (IR), and hyperandrogenism, all of which have been associated with an increased risk of pregnancy complications. Many of the features of PCOS are reversible following lifestyle interventions such as diet and exercise, and pregnant women following a healthy lifestyle have been found to have a lower risk of complications, including PE. This review summarizes the evidence investigating the risk of PE and the role of nutritional factors in women with PCOS. The findings suggest that the beneficial aspects of lifestyle management of PCOS, as recommended in the evidenced-based international guidelines, extend to improved pregnancy outcomes. Identifying high-risk women with PCOS will allow targeted interventions, early pregnancy screening, and increased surveillance for PE. Women with PCOS should be included in risk assessment algorithms for PE.
Keywords:
PCOS
; pre-eclampsia
; pregnancy
; lifestyle
; nutrition
; placenta
; pathophysiology
; angiogenic ratio
; screening.
1. Introduction
Polycystic ovary syndrome is a systemic metabolic and endocrine disorder that results from a disturbance of adaptive interdependent homeostatic survival networks (metabolic, immune, and neuroendocrine) [1,2,3,4]. The pathophysiology is characterized by chronic systemic inflammation, IR, and hyperandrogenism [3,4,5,6,7,8,9]. This results in a range of phenotypic presentations, that when studied together, have been shown to confer an increased risk of pregnancy complications, including PE [10,11,12]. When considered separately, different PCOS phenotypes present with clinical and biochemical features that range from mild to severe [2,13]. Nevertheless, women with PCOS inherit individual susceptibility features, independent of other co-existing morbidities such as obesity, that increase their risk of pregnancy complications and justify their inclusion in PE risk assessment algorithms [10,13,14].
Recent evolutionary models characterize PCOS as an evolutionary mismatch disorder that arises from an interaction between genetic and environmental factors [1,2]. Rapid cultural changes in the contemporary environment have outpaced genetic adaptation and resulted in a mismatch between modern dietary and environmental exposures and behaviours, and selective metabolic and reproductive traits [1]. The influence of these modern exposures on developmentally programmed susceptibility genes, programs the embryo and fetus to express the phenotypic features of PCOS during childhood, adolescence, and adulthood [15,16,17,18,19]. As a result, adaptive physiological survival pathways that result in activation of inflammation, variation in insulin sensitivity, preferential abdominal fat accumulation, and downregulation of reproduction, become pathological following exposure to lifestyle and environmental factors [1,2]. The development of chronic systemic inflammation, IR, and hyperandrogenism, predispose women with PCOS to a range of chronic diseases and pregnancy complications [20,21,22,23,24].
Polycystic ovary syndrome affects 10-13% of reproductive age women and can present with a wide range of symptoms including menstrual irregularity, hirsutism, acne, alopecia, anxiety, depression, and subfertility, resulting in reduced quality of life [25]. PCOS can be a progressive metabolic condition that leads to obesity, hypertension, dyslipidemia, type 2 diabetes, metabolic syndrome, metabolic-associated hepatic steatosis, chronic kidney disease, cardiovascular disease, and cancer [1,22,26]. The population attributable risk of PCOS to type 2 diabetes is 19-28%, and the combined impact of PCOS makes a significant contribution to the chronic disease epidemic [22]. Multiple systematic reviews of large population-based studies over the past 40 years have reported a significantly increased risk of pregnancy complications, including PE, in women with PCOS [10,11,27,28,29,30].
Hypertensive disorders of pregnancy (HDP) (defined as chronic hypertension, gestational hypertension, preeclampsia-eclampsia and chronic hypertension with superimposed preeclampsia) are a leading cause of maternal-fetal morbidity and mortality worldwide [31,32]. Preeclampsia affects 3-5% of pregnancies and is responsible for 76,000 maternal and 500,000 fetal/neonatal deaths every year [31,33]. The definition of PE has evolved over time, in line with research developments into the underlying pathophysiology [34]. This has resulted in an increased awareness of factors involved in the prevention, prediction, diagnosis, treatment, and long-term consequences of PE. It has been estimated that over 300 million women are at risk of chronic health problems (neurodevelopmental, metabolic, cardiac) due to previous PE [35,36,37].
The International Society for the Study of Hypertension in Pregnancy (ISSHP) has defined PE as new-onset gestational hypertension at or after 20 weeks gestation accompanied by proteinuria, maternal organ involvement, or uteroplacental dysfunction [32]. The pathophysiology of PE is characterized by placental malperfusion that results in syncytiotrophoblast stress and release of soluble factors (pro-inflammatory cytokines, exosomes, extracellular vesicles, transcription factors, hormones, and anti-angiogenic factors), that cause maternal vascular endothelial injury resulting in hypertension and multi-organ involvement [38,39,40]. More recently, circulating angiogenic factors such as soluble fms-like tyrosine kinase (sFlt-1) and placental growth factor (PlGF), have been identified as markers of placental health and added to the diagnostic criteria in some countries [34]. In addition, advances in genetics, epigenetics, transcriptomics, metabolomics, artificial intelligence, organoid cultures of the endometrium and trophoblast, and stem cell research, have progressed our understanding of normal and pathological placentation [38,39].
It has long been appreciated that normal placental development requires a complex network of bidirectional communication signals (cytokines, metabolites, hormones, exosomes) between embryo-derived cells (trophoblast, macrophages) and maternal-derived cells (endometrial gland epithelium, stromal, macrophages, natural killer, dendritic, and T cells) [38,39]. Decades of epidemiological research has identified over 70 maternal risk factors that are associated with the development of PE [12,39]. This has resulted in a greater awareness of the possible role of pre-existing maternal pathophysiological features on the development of abnormal placentation and related complications, such as PE [38].
Although there may be differences in the pathogenesis of early (<34 weeks gestation) and late-onset PE, the pathophysiology and maternal/fetal consequences can be similar [38]. Pre-existing maternal pathological features such as chronic systemic inflammation [41], insulin resistance [42], and hyperandrogenemia [43], as occur in women with PCOS [3], may alter normal placental development, metabolism, and physiology, at all stages of pregnancy [44,45]. In addition, many of the metabolic, endocrine, reproductive, and neuroimmune disturbances that occur in women with PCOS, have been shown to be reversed following lifestyle interventions such as diet and exercise [1,25,46].
Comprehensive international guidelines recommend lifestyle interventions as the first line of management for all women with PCOS [25]. Healthy maternal dietary patterns have been associated with a lower risk of developing PE (odds ratio (OR): 0.78, 95% confidence interval (CI) 0.7-0.86) and increased consumption of ultra-processed foods confer a higher risk (OR: 1.28, CI 1.15-1.42) [47]. Evidenced-based reviews and prospective cohort studies have demonstrated the effectiveness of lifestyle interventions for reducing the risk of PE in women with PCOS [47,48]. In addition, over 75 randomized controlled trials have demonstrated the value of aspirin in the prevention of PE [49].
The following sections of this review will outline the evidence for the increased risk of PE in women with PCOS. The evidence supporting lifestyle and medical interventions will also be discussed and recommendations made for screening and medical therapy in pregnancy.
2. Evidence for Increased Risk of PE in Women with PCOS
Many observational studies, systematic reviews, and meta-analyses have reported an increased risk of pregnancy complications in women with PCOS over the past 40 years. These include miscarriage, gestational diabetes mellitus (GDM), intrauterine growth restriction, preterm birth, low birth weight, gestational hypertension, and PE [10,27,28,29,30]. In total, 8 systematic reviews published between 2006 and 2023, reported an increased relative risk of PE in women with PCOS of between 1.87 and 4.23 (Table 1) [10,27,28,29,30,50,51,52]. The most recent 2023 meta-analysis included 36 studies that compared rates of PE in women with and without PCOS [52]. Pooled meta-analysis of 34 studies in women not taking metformin, showed a significantly increased risk of PE in women with PCOS (OR: 2.35, 95% CI 1.93-2.86). Subgroup analysis of high-quality studies, after removal of low and medium quality studies, showed a significantly higher risk of PE in women with PCOS (OR: 3.05, 95% CI 1.20-7.8). Subgroup analysis in 7 body mass index (BMI) matched studies, showed that women with PCOS retained an increased risk of developing PE (OR: 2.39, 95% CI 1.14-4.99) [52]. These findings are in agreement with 2 previous meta-analyses that showed a higher prevalence of PE in BMI-matched women with and without PCOS [50,51]. Overall, these data suggest that women with PCOS have an increased risk of PE that is independent of BMI.
In addition, a large study from the United States National Inpatient Database of 71,436,308 weighted hospitalizations for deliveries, analyzed 195,675 women with PCOS for their risk of pregnancy complications [11]. Women with PCOS had a higher risk of PE, eclampsia, peripartum cardiomyopathy, and heart failure, during delivery hospitalizations. The risk of developing PE was significantly increased in women with PCOS after adjustment for age, race, demographic variables, and comorbidities, including BMI (OR: 1.56, 95% CI 1.54-1.59). In addition, delivery hospitalizations were associated with increased length and cost of hospitalization in women with PCOS [11]. Despite this large body of evidence, PCOS is not generally recognized as a risk factor for pregnancy complications and PE and is not included in national or international risk assessment models.
There is international consensus that women should be assessed for risk factors associated with PE in early pregnancy [12,32,53]. There is ongoing debate regarding which risk factors to include and the type of measures that should be part of risk assessment strategies [53]. Clinical practice guidelines (CPG) recommend a combination of predictive assessments that include clinical risk factors, biophysical markers such as mean arterial blood pressure and mean uterine artery pulsatility index (UtAPI), and biochemical markers such as pregnancy-associated plasma protein A (PAPP-A) and/or PlGF [54,55]. Various combinations of these measures are advised by different national bodies and professional societies [31,53,56]. Recent reviews and commentaries have highlighted the fact that PE risk factors in current CPG are poorly aligned with the evidence [12,57]. A recent review by an expert working group identified PCOS as a probable risk factor for the development of PE, having a similar relative risk and level of evidence to many other risks that are currently included in risk assessment models [12]. None of the current CPG include PCOS as a risk factor. More recent studies add further weight to this evidence and support the need for a review of strategies advocated by CPG [11].
The international evidenced-based guidelines for the assessment and management of PCOS recommend screening, monitoring, and management of risk profiles in women with PCOS, preconception, during pregnancy, and postpartum, in accordance with the recommendations for the general population [25,58]. These recommendations include assessment of blood glucose, body weight, blood pressure, smoking, alcohol consumption, diet, exercise, sleep, and emotional health. There are no specific recommendations for the identification, screening, and management of women with PCOS in pregnancy.
3. Evidence for the Role of Nutritional Factors in the Pathophysiology of PE
Maternal nutrition has been suspected to play a role in the pathogenesis of PE for over 100 years [59,60,61]. The “dietary” hypothesis was proposed by advocates in the United Kingdom (Theobald) and United States (Dieckmann) in the early 1900’s [60]. A 1926 review summarized the literature up to this time on the possible pathological effects of dietary macronutrients and their metabolites (protein and urea, fat and ketosis, carbohydrate restriction and ketosis) in patients with PE [61]. Although dietary management was common (milk, bread, rice, eggs, and fruit, with salt restriction) it was noted that there were “no series of experiments in which the effect of various diets in PE has been deliberately tested” [61]. Nevertheless, it was noted that the incidence of eclampsia fell significantly during the first and second world wars and increased in the post-war years in Germany and the Netherlands, respectively, and this was attributed to dietary restriction during the war years [60]. In addition, there were numerous publications between 1922 and 1957 that reported differences in the rates of PE and eclampsia in indigenous versus European and urban populations (Algeria 1922, India 1938, Ceylon 1946, South Africa 1947, New Guinea 1949, Fiji 1950, Indonesia 1952, Belgian Congo 1956) [59,60]. Indigenous diets were noted to consist of “wholefoods” containing starchy root vegetables, leafy greens, home-pounded grains, fruit, and small quantities of milk, fish, and meat, if they were available. This was contrasted with European and urban diets that were high in refined grains (white rice and flour), sugar, and salt, and low in meat, milk, vegetables, and fruit [60].
According to the Global Burden of Disease Study, poor quality diet is one of the leading risk factors for morbidity and mortality globally [62]. International guidelines for the assessment and management of PCOS have recommended that lifestyle interventions, such as diet and exercise, should be discussed with all women diagnosed with PCOS [63]. More recent observational and intervention studies provide contemporary evidence to support the role of nutritional factors in PE [64]. Evidence-based summaries from systematic reviews, meta-analyses, and representative PE expert groups (PRECISE), recommend healthy maternal dietary patterns to reduce the risk of PE [47,64,65,66].
A recent comprehensive evidenced-based expert review of nutritional factors that may protect or exacerbate the risk of PE by the PRECISE Conceptual Framework Working Group, highlighted the importance of focusing on research related to healthy diet patterns rather than single nutrients [47]. Nevertheless, they identified 25 nutritional factors in two umbrella reviews and twenty-two meta-analyses. Of these, 14 were found to be significantly associated with an increased incidence of PE. Healthy maternal diets containing fruits, vegetables, whole-grain foods, fish, and chicken, such as Mediterranean and New Nordic diets, were associated with 22% reduced odds of developing PE (OR: 0.78, 95% CI 0.70-0.86) [66]. In contrast, maternal diets high in ultra-processed foods and added sugars increased the odds of developing PE by 28% (OR: 1.28, 95% CI 1.15-1.42) [67]. Long-term longitudinal studies have shown that higher ultra-processed food intake in women is associated with increased cardiovascular risk and hypertension [68], as can occur in women with a history of PE [69]. These data support the recommendations of other expert reviews and the World Health Organization, on promoting healthy maternal diets [47,70,71].
Prospective cohort studies and observational research suggest that following a healthy lifestyle and diet prior to pregnancy is associated with reduced risk of PE. The multicenter prospective SCOPE study enrolled 5628 apparently healthy nulliparous women with singleton pregnancies, to examine the association of PCOS (354 women) with pregnancy complications, including PE [48]. The investigators reported that in this low-risk population the proportion of women with PE was similar in women with PCOS to those without PCOS (5.9% vs 6.7%; OR: 0.88, 95% CI 0.56-1.4). Pregnant women with PCOS were following a healthier lifestyle, including increased fruit and vegetable intake, more frequent vigorous exercise, lower alcohol consumption, and lower rates of smoking [48]. Analysis of data from the complete SCOPE cohort of 5628 women showed that lower intake of fruit and higher intake of fast food in the preconception period were associated with longer time to pregnancy [72]. A recent large meta-analysis of 21 cohort studies, showed that following a healthy lifestyle (diet and high physical activity) can also reduce the risk of developing GDM [73]. The authors highlighted the need for more randomized intervention trials. Taken together, these data suggest that nutritional factors may have an impact on fertilization, implantation, and placentation.
Intervention trials currently underway should help clarify the impact of lifestyle modification prior to pregnancy on in-utero metabolic factors and neonatal outcomes [74]. Recent developments in endometrial organoid research should provide insights into the molecular mechanisms involved in mediating the effects of metabolic and immune disturbances in women with PCOS with adverse pregnancy outcomes [75]. A recent endometrial organoid study compared cell-type-specific disease signatures and molecular pathways for PCOS-specific endometrial dysfunction in women with and without PCOS [76]. The investigators examined 248,694 nuclei from 6 endometrial cell subtypes. They reported a range of differentially expressed genes in cells and pathways related to processes involved in placentation. Women with PCOS were treated with either metformin or lifestyle management for 16 weeks, followed by repeat endometrial biopsy and establishment of a second endometrial organoid. Both treatments, either metformin or lifestyle intervention alone, restored multiple differentially expressed genes in each cellular subtype. This study provides new mechanistic insight into PCOS-specific endometrial dysfunction and the potential for reversibility with medical or lifestyle interventions.
Diet is a modifiable risk factor, and a balanced healthy diet has been shown to reduce the risk of PE [48]. Pregnant women should eat a diet rich in fruit, vegetables, and whole-grains, and healthy sources of fat and protein [65,71]. Women with PCOS should receive lifestyle-oriented counselling and advice, before, during, and after pregnancy, and be considered for inclusion in risk assessment algorithms for the prediction and assessment of PE as previously discussed.
4. Mechanisms of Action of Nutritional Factors in the Pathophysiology of PE
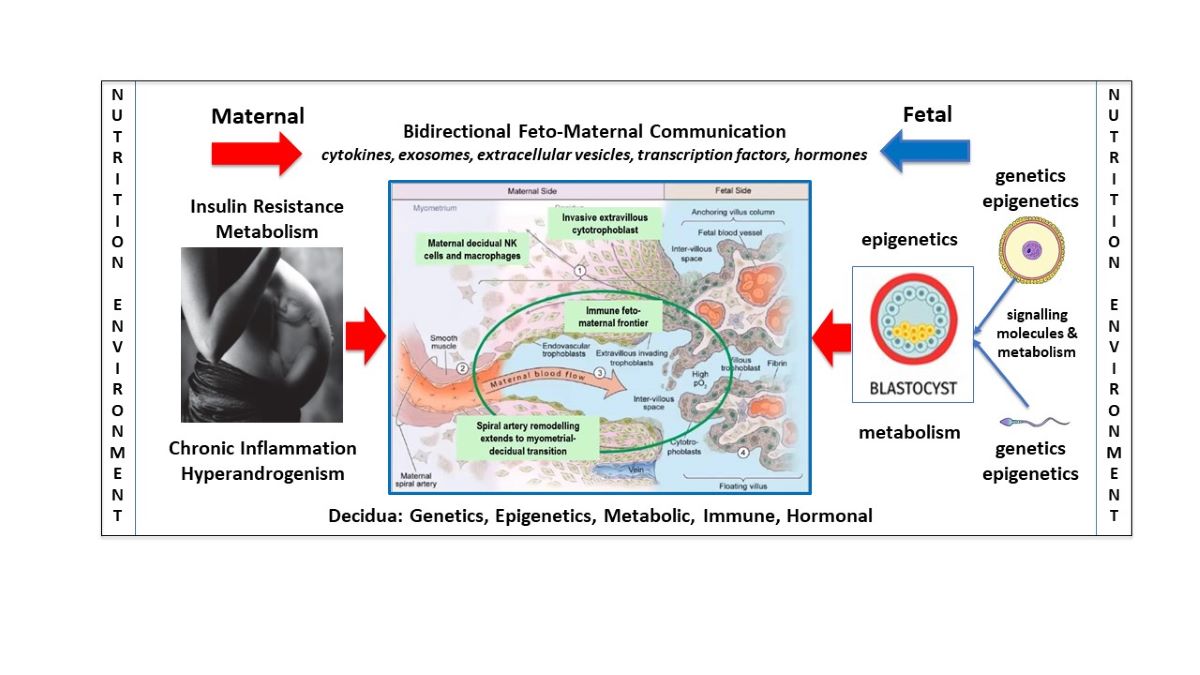
Advances in the understanding of normal placental development have paved the way for improved understanding of the pathogenesis and pathophysiology of abnormal placentation in PE [38]. The syndrome of PE may have multiple underlying causes that differ in early or late-onset presentations, all of which could be influenced by maternal nutritional disturbances [45,77]. Normal development of the placenta involves a complex network of communication signals between fetal-derived trophoblast and a broad range of maternal-derived endometrial cells [39]. The fetal-derived precursor cells of the placenta are affected by sperm-derived signals to endometrial cells prior to fertilization [78,79], paternal and maternal genetics [80], imprinted genes [81], epigenetic reprogramming following fertilization [82], nutritional components of oviduct fluid from cells lining the Fallopian tube prior to implantation [38,83], histotrophic nutrition during the first trimester [84], and haemotrophic nutrition following the establishment of significant blood flow into the placental intervillous space [85]. Disturbances of normal physiology in any of these components could lead to abnormal trophoblast-decidual dialogue and contribute to deficient trophoblast invasion into the uterus and impaired spiral artery remodeling, resulting in placental hypoperfusion and syncytiotrophoblast stress, as is known to occur in PE (Figure 1) [77].
It has been proposed that placental development, is independent of, and precedes, embryogenesis, because of a two-way feed-forward dialogue between trophoblast cells and endometrial glands [38]. Hormones from trophoblast (human chorionic gonadotrophin, human placental lactogen) and decidual cells (prolactin) stimulate glandular epithelial cells to upregulate production of nutrients (glucose, lipid droplets, glycoproteins) and growth factors (epidermal growth factor), that in turn feedback on trophoblast cells and promote further proliferation and growth of the placenta [38]. This new understanding has focused attention on the role of pre- and post-conception maternal pathophysiology, such as occurs in women with PCOS [3], altered nutrient supply, impaired bidirectional signalling, defective decidualization, and abnormal placentation in PE (Figure 1) [45,86,87,88,89].
Epigenetic processes also play an important role in the development and progression of PE [90] and a wide range of epigenetic changes involving methylation, histone deacetylation, and microRNA, have been identified in the placentas of women with PE [90,91]. Epigenetic processes are the mechanism by which environmental influences alter gene expression without changing the structure of DNA. In PE, reduced placental blood flow causes hypoxia and results in epigenetic changes that activate adaptive responses in the placenta and maternal circulation [92,93]. Nutrition, diet, and metabolism regulate epigenetic mechanisms and integrate environmental cues and exposures with cellular responses [94]. Cellular metabolites (acetyl coenzyme A, adenosine triphosphate, nicotinamide adenine dinucleotide) act as nutrient sensors and contribute to the regulation of gene expression via epigenetic mechanisms [95]. Reciprocal crosstalk between epigenetics and metabolism determines molecular programing and cellular function [94]. Maternal obesity can lead to placental dysfunction via chronic inflammation, dysregulation of metabolic pathways, and epigenetic changes in gene expression [96,97]. Unbalanced diets can alter normal metabolic and epigenetic processes and lead to disturbed cellular function and disease [98]. Recent research in nutritional epigenomics explains how lifestyle interventions (diet and exercise) can restore metabolic and epigenetic homeostasis [99,100]. The interaction between cellular metabolism and the epigenome is a fundamental process in placental development and PE that is influenced by maternal nutritional and environmental exposures [45,90,101,102,103,104,105,106,107,108].
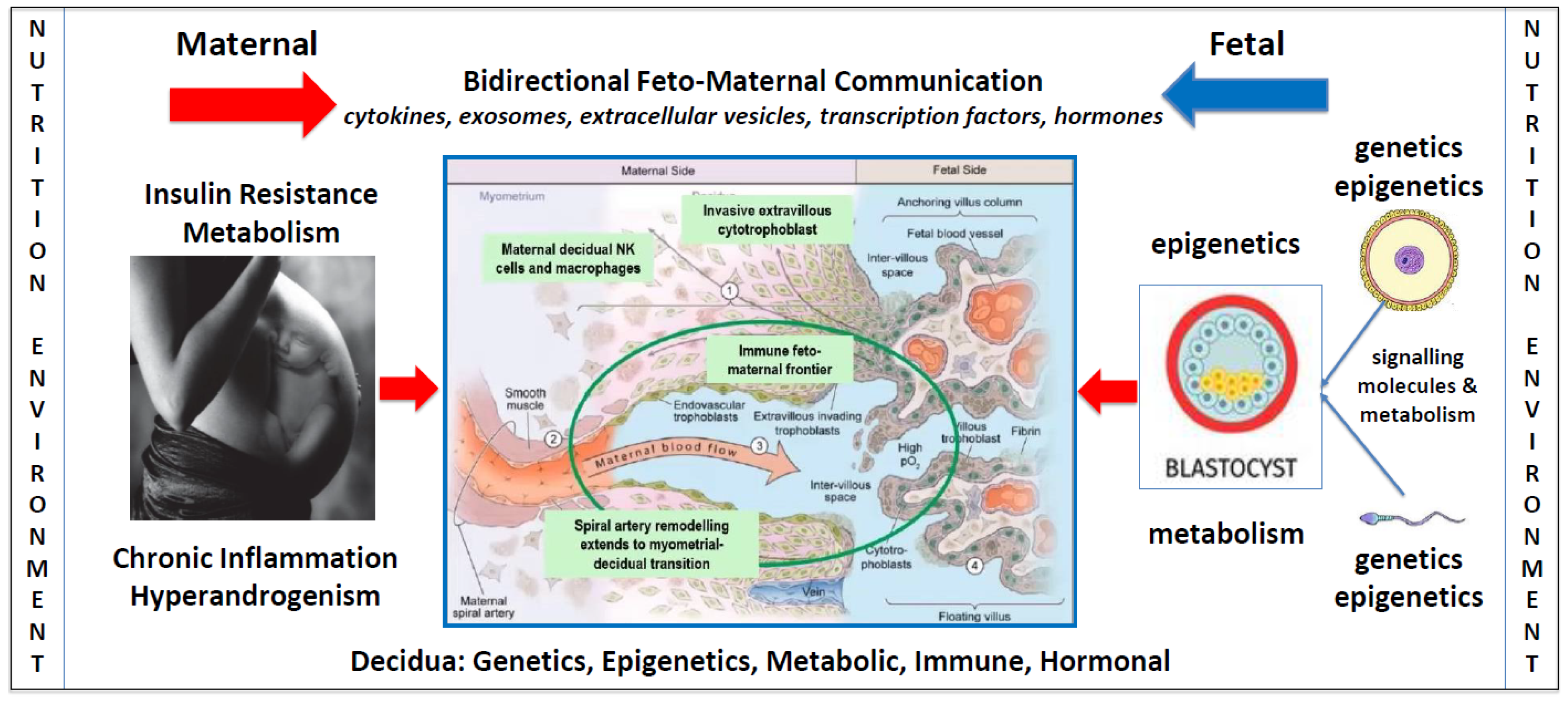
Figure 1.
Factors influencing bidirectional feto-maternal placentation. A schematic model showing the potential impact of nutritional and environmental factors at all stages of pregnancy, including gametogenesis, decidualization, implantation, and placental and fetal development. The blue arrow represents the paternal, maternal, and fetal components. Nutritional and environmental factors influence sperm maturation and development in males [109]. Once sperm enter the reproductive tract, they release signalling molecules that interact with decidual cells prior to fertilization [78]. Human oocytes develop in the mother during embryonic development and are subject to nutritional and environmental factors that influence epigenetic developmental programming [110]. Maternal and paternal nutritional and environmental factors can therefore influence sperm and oocytes prior to fertilization and have the potential to alter bidirectional communication signals during placentation. The red arrow represents the effect of nutritional and environmental factors in maternal pathophysiology and their impact on decidualization, placentation, and embryogenesis. Following fertilization, the zygote and morula receive nutrition from maternal secretions in the Fallopian tube [38]. During implantation and throughout the first trimester, both the placenta and embryo obtain nutrition from histotroph fluid that is derived from maternal endometrial gland secretions [38]. These secretions provide glucose, lipids, glycoproteins, and growth factors that stimulate rapid proliferation of villous trophoblast, extravillous trophoblast invasion, spiral artery remodeling, and normal development of the placenta [85,111]. At the start of the second trimester blood enters the intervillous space resulting in haemotrophic nutritional exchange between the maternal and fetal circulations [112]. Accumulating evidence suggests that pathophysiological changes in women with PCOS, such as insulin resistance, chronic inflammation, and hyperandrogenism, may influence the composition and quality of histotrophic and haemotrophic nutrition, alter bidirectional communication between decidual and placental cells, and effect normal placentation and fetal development. Central diagram in blue is adapted with permission from Kingdom and Drewlo 2011 [113].
Figure 1.
Factors influencing bidirectional feto-maternal placentation. A schematic model showing the potential impact of nutritional and environmental factors at all stages of pregnancy, including gametogenesis, decidualization, implantation, and placental and fetal development. The blue arrow represents the paternal, maternal, and fetal components. Nutritional and environmental factors influence sperm maturation and development in males [109]. Once sperm enter the reproductive tract, they release signalling molecules that interact with decidual cells prior to fertilization [78]. Human oocytes develop in the mother during embryonic development and are subject to nutritional and environmental factors that influence epigenetic developmental programming [110]. Maternal and paternal nutritional and environmental factors can therefore influence sperm and oocytes prior to fertilization and have the potential to alter bidirectional communication signals during placentation. The red arrow represents the effect of nutritional and environmental factors in maternal pathophysiology and their impact on decidualization, placentation, and embryogenesis. Following fertilization, the zygote and morula receive nutrition from maternal secretions in the Fallopian tube [38]. During implantation and throughout the first trimester, both the placenta and embryo obtain nutrition from histotroph fluid that is derived from maternal endometrial gland secretions [38]. These secretions provide glucose, lipids, glycoproteins, and growth factors that stimulate rapid proliferation of villous trophoblast, extravillous trophoblast invasion, spiral artery remodeling, and normal development of the placenta [85,111]. At the start of the second trimester blood enters the intervillous space resulting in haemotrophic nutritional exchange between the maternal and fetal circulations [112]. Accumulating evidence suggests that pathophysiological changes in women with PCOS, such as insulin resistance, chronic inflammation, and hyperandrogenism, may influence the composition and quality of histotrophic and haemotrophic nutrition, alter bidirectional communication between decidual and placental cells, and effect normal placentation and fetal development. Central diagram in blue is adapted with permission from Kingdom and Drewlo 2011 [113].
The accumulating molecular, endocrine, metabolic, and epigenetic evidence provides detailed mechanistic explanations for the role of nutritional factors in the pathophysiology of PE that support the evidenced-based research and recommendations previously discussed. The following sections provide a brief discussion of the pathophysiological components of PCOS that may be involved in the pathogenesis of altered placental development and function in PE.
4.1. Insulin Resistance
Insulin is a pleotropic hormone that has multiple cellular and tissue-specific actions such as regulation of glucose uptake in some cells (muscle, adipose, vascular endothelium) [114], increased production of endothelial nitric oxide resulting in vasodilation in systemic and cardiac blood vessels [115], reduced excretion of urate [116], enhanced sodium absorption in the kidney [117], and multiple metabolic effects [118]. Many of these physiological processes are known to be involved in the pathophysiology of both PCOS and PE [3,39].
Insulin resistance can be defined as an altered cellular response to insulin stimulation that occurs in selective cells and tissues throughout the body [118]. The development of physiological IR is a normal adaptation to pregnancy to ensure adequate placental growth and nutrient supply to the fetus [119]. All women develop progressive IR and hyperinsulinemia throughout pregnancy in response to various hormones (human placental growth hormone, human chorionic gonadotrophin, human placental lactogen) [120,121] and adipokines (adiponectin) produced by the placenta and maternal adipose tissue [122]. Pancreatic insulin secretion can increase by up to 250% during pregnancy to maintain euglycaemia [123]. Gestational diabetes is diagnosed if hyperglycaemia develops, as defined by national and international reference ranges, and is known to be associated with increased maternal and fetal complications [124,125]. Hyperinsulinaemia and/or hyperglycaemia related to GDM or PCOS may therefore be involved in pathological placental and fetal responses. Nutritional management is the cornerstone of treatment for both GDM and PCOS [25,126].
Most women diagnosed with PCOS prior to pregnancy have reduced insulin sensitivity, hyperinsulinaemia, and normoglycaemia [3,127]. Glucose metabolism has been shown to be important for preparation of the endometrium for embryo implantation. Pre-existing IR and hyperinsulinaemia have been associated with dysregulated decidualization and are thought to increase the risk of PE [128]. The expression of placental insulin receptors increases with gestational age, along with changes in the tissue distribution [129]. Although insulin sensitive glucose transporters are expressed in the placenta, the majority are not responsive to insulin stimulation [45]. Glucose transporters on the microvillous membrane of the syncytiotrophoblast move glucose into the cytoplasm by facilitated diffusion down the concentration gradient [85]. As a result, placental glucose transport to the fetus is mostly insulin independent. This may explain why maternal hyperglycaemia results in fetal hyperglycaemia and hyperinsulinaemia, which can be associated with adverse fetal (macrosomia) and placental effects [126]. Nevertheless, IR can interrupt glucose homeostasis and cause dysfunctional lipid metabolism and excessive inflammation, both of which are associated with PE [45].
Insulin resistance in early pregnancy has been shown to be predictive for the development of PE [130]. Pre-existing IR, coupled with the effects of chronic inflammation and hyperandrogenemia, is likely to have additive pathological effects on placental development and may be involved in the pathogenesis of PE [86,119]. Insulin has been shown to inhibit the activity of aromatase in human trophoblasts [131], which may provide a mechanism for connecting hyperinsulinaemia with placental androgen excess in women with PCOS. Elevated insulin levels were found to cause increased DNA damage, apoptosis, and decreased cell survival in cultured first trimester trophoblasts from healthy pregnancies [42]. Transcriptome signatures in placental trophoblasts exposed to insulin showed that the many biological processes (hormonal, cytokine, cell cycle, metabolic) were either up- or downregulated by insulin [132]. Trophoblast cells from the placenta of obese women were 30-times less sensitive to insulin than cells from normal-weight women. The investigators proposed that the enhancement of placental-specific genes supports the concept that insulin promotes both endocrine and growth functions in the placenta, and that IR and obesity can affect the structure and function of the placenta in early pregnancy [132]. Obese women have greater placental lipid accumulation (lipotoxicity and “fatty placenta”) than normal weight women [133]. Excess placental lipid may be due to changes in fatty acid uptake, decreased fatty acid oxidation, or increased esterification, and may contribute to increased lipid supply to the fetus and fetal adiposity [134]. These data support previous reports that show placental size and volume are strongly related to maternal insulin secretion in early pregnancy [135]. Nevertheless, obesity is often associated with hyperinsulinaemia, hyperglycaemia, and hypertriglyceridemia, which makes it difficult to separate the effects that may be driven by obesity from those caused by GDM or PCOS in women who have some or all these problems [134].
Maternal metabolism and cardiovascular physiology are altered in pregnancy in response to the increasing demands of placental and fetal growth [136]. Maternal vascular endothelial dysfunction is a classic feature of PE that is thought to be secondary to reduced placental blood flow and release of pro-inflammatory cytokines, reactive oxygen species, extracellular vesicles, and imbalance of angiogenic and anti-angiogenic factors [39,137]. Experimental studies have shown that women with PCOS have endothelial dysfunction (impaired endothelium-dependent vasodilation) and reduced response to the vasodilation effect of insulin [138,139,140,141]. The observed endothelial dysfunction may be related to both elevated androgen levels and IR [138,140]. In addition, PCOS is often associated with chronic hypertriglyceridemia [142], which is a known risk factor for endothelial dysfunction and may cause arteriolar vasoconstriction by altering the regulation of prostaglandins [143]. A systematic review and meta-analysis showed that women that develop PE had elevated serum lipids and triglycerides during all trimesters of pregnancy [88]. Both maternal and placental factors may therefore converge on the maternal endothelium to produce the observed pathological manifestations of PE [86]. Women with PCOS may be at increased risk of PE due to the combined effects of inflammation, hyperandrogenemia, hypertriglyceridemia, and IR, on vascular endothelial function.
In summary, accumulating evidence suggests that IR can affect normal placental development and may contribute to the pathophysiology of PE via a variety of mechanisms. Future research involving molecular techniques, computational advances, multiomics data, and endometrial organoids, should help our understanding of the effect of maternal IR on placental nutrient and energy utilization, growth pathways, and placental physiology. The investigation of maternal nutritional insults on placental development and function may open the way for interventions that mitigate the impact on adverse pregnancy-related outcomes, such as those that occur in PE.
4.2. Chronic Systemic Inflammation
Pregnancy is characterized by a state of chronic low-grade inflammation due to systemic release of a variety of placental cytokines that are required for normal placental development and function [132,144]. PCOS is also characterized by chronic inflammation that is thought to be secondary to poor-quality nutrition, nutritional excess and other environmental factors that affect metabolic and inflammatory signal transduction pathways [3,145,146]. Women with PCOS also have a chronic low-grade inflammatory state during pregnancy that has been found to be associated with a higher risk of adverse obstetric outcomes [147,148,149]. Abnormal maternal inflammation has been associated with altered uteroplacental development and function [150], although the precise mechanisms are largely unknown.
Women with PCOS appear to have a proinflammatory state that is intrinsic to the underlying pathophysiology [3]. This may contribute to altered endometrial immune cell (natural killer cells, macrophages, T cells) and cytokine profiles (interleukin 15 and 18, chemokine ligand 10), that compromise normal implantation [151,152]. Women with PE have a heightened inflammatory state with elevated proinflammatory cytokines and chemokines, both systemically and in the placenta [150,153]. Studies in rodents and non-human primates have provided evidence of mechanistic links between maternal inflammation and PE [150,154]. Lipopolysaccharide-induced inflammation in pregnant rats showed that inflammation was associated with deficient trophoblast invasion and spiral artery remodeling [150]. In addition, inflammation increased maternal mean arterial pressure and was associated with structural changes in the kidney and proteinuria, as is found in PE [150]. Wilson et al demonstrated increased syncytiotrophoblast inflammation in a testosterone-induced primate model of PCOS using a novel contrast-enhanced ultrasound technique [154].
Maternal diet during pregnancy can also influence systemic and placental inflammation and may provide a mechanistic link between PCOS and PE [155,156]. Diet quality has been found to influence insulin signaling and inflammatory pathways in rodents and humans [157]. Higher quality diet was shown to improve insulin signaling in the placenta using a mouse model of maternal obesity [157]. Francis et al examined the effect of consuming a healthy diet on a range of placental proteins involved in metabolic pathways and inflammation [156]. They assessed diet quality using the Healthy Eating Index, which is based on consumption of vegetables, fruit, dairy, protein, whole grains, and unsaturated fats, with lower intakes of red and processed meat, and added sugar [158]. They found that proteins of the p38MAPK inflammatory signaling pathway were lower in placental villi of pregnant women consuming a healthier diet. Placental p38MAPK is upregulated by proinflammatory stimuli and has been linked to placental angiogenesis and further production of proinflammatory cytokines [159]. Taken together, these data support the findings of observational studies linking healthy diets to improved pregnancy outcomes, and lower rates of PE, as previously discussed.
Pathological levels of maternal inflammation may result in altered decidual and placental inflammatory responses that affect placental development and function [41,150,160]. Inflammatory processes can contribute to, and exacerbate, the effects of insulin resistance and hyperandrogenism [1]. Previous reviews have discussed the role of chronic low-grade inflammation and altered immune function in PCOS [3] and PE [41].
4.3. Hyperandrogenism
The presence of hyperandrogenism in non-pregnant women with the PCOS is associated with increased metabolic and cardiovascular risk [161,162,163,164]. Although not all studies report increased adverse pregnancy outcomes in different PCOS phenotypes [165], the majority of published reports demonstrate an association between hyperandrogenism and complications such as gestational diabetes, preterm delivery, and PE [166,167,168]. PCOS is associated with altered histological structure of the placenta, including microscopic alterations in trophoblast invasion [169,170], that may be increased in women with hyperandrogenism [171]. Maternal hyperandrogenism has also been found to be an independent predictor of PE [168].
The relationship between androgens and maternal cardiovascular and placental function has been investigated in human and animal models [172]. Placental androgen receptor gene expression is increased and placental aromatase mRNA and protein expression are decreased, in the placenta of women with PE [173]. Serum testosterone levels of preeclamptic women are elevated (2-3 fold) and are correlated with vascular dysfunction [172]. Elevated androgens in pregnant rats are involved in gestational hypertension (reduced uterine arterial blood flow) [174], endothelial dysfunction (impaired nitric oxide-mediated relaxation in systemic and uterine vessels) [174,175], heightened vasoconstriction in response to angiotensin II [175], decreased spiral artery remodeling (inhibition of angiogenesis, reduced radial and spiral artery diameters, increased UtAPI) [176], placental hypoxia (increase in hypoxia-inducible factor) [176], and altered nutrient transport (reduced amino acid transport) [173]. All of these factors are known to be involved in the pathophysiology of PE.
Both IR and chronic inflammation can cause and exacerbate hyperandrogenism in women with PCOS [1,3]. Insulin stimulates androgen production in theca cells of normal ovaries and likely contributes to elevated maternal testosterone levels [172,177,178]. Insulin inhibits the aromatase enzyme in human trophoblasts which may result in a placental contribution to maternal hyperandrogenemia [131]. Chronic inflammation can cause hyperandrogenism via a number of mechanisms including disruption of signaling pathways, alteration to epigenetic processes, and posttranscriptional regulatory effects [9]. Hyperandrogenism in PCOS may be due to the synergistic actions of IR and chronic ovarian and systemic inflammation [3,179]. The degree of hyperandrogenism in women with PE varies depending on the sex of the fetus, with higher levels in pregnancies with a male fetus [173]. Therefore, women with PE may have elevated androgen levels due to a combination of fetal, maternal, and placental sources.
A detailed discussion of the effects of hyperandrogenism on maternal vascular and placental function and the implications for the pathogenesis of PE are beyond the scope of the present report and can be found in previous comprehensive reviews [172,180]. Taken together, these data strongly suggest that many androgen-mediated actions are important contributors to the pathophysiology of PE. The majority of women with PCOS have hyperandrogenemia [181] that may contribute to dysregulated androgen signaling in the placenta and increase the frequency of maternal-fetal complications associated with PE [172].
5. Identification, Assessment, and Management of Women with PCOS in Pregnancy
Early identification of women at increased risk of developing PE has been advocated to help reduce the associated maternal and perinatal morbidity and mortality [34,53]. There has been a general lack of awareness of the significant association between PCOS and adverse pregnancy outcomes, including PE. Healthcare professionals do not usually identify women with PCOS during antenatal care, delivery, or post-partum [182]. This is reflected in the absence of inclusion of PCOS in risk assessment algorithms for PE, and lack of specific recommendations for screening, monitoring, and treatment of women with PCOS during pregnancy [58]. The 2023 international guidelines include a meta-analysis of 109 studies that explored the relationship between PCOS and adverse pregnancy outcomes [63]. As previously discussed, women with PCOS had a significantly increased risk of developing PE on pooled analysis (OR: 2.28, 95% CI 1.88-2.77), that was even greater when only high-quality studies were assessed (OR: 3.05, 95% CI 1.20-7.8) [52].
Accumulated evidence from 8 systematic reviews over the past 40 years, therefore report a significantly increased risk of PE in women with PCOS [52]. These data, coupled with the increasing availability of early pregnancy screening for PE [34], suggest that women with PCOS should be included in risk assessment algorithms and be considered for screening and possible treatment to reduce the risk of developing PE.
Screening may include measurement of mean arterial blood pressure, ultrasound with mean UtAPI, and maternal serum biochemical markers (PAPP-A and/or PlGF) [54,183,184]. Several prospective studies have demonstrated the predictive value of measurements of the serum sFlt-1/PlGF ratio for diagnosis, monitoring, and management of women at high-risk of developing PE [34,53,185,186,187]. Low-dose aspirin treatment starting before 16 weeks and continued to 37 weeks, has been shown to significantly reduce the likelihood of preterm pre-eclampsia (62% reduction), preterm birth, and other associated complications [49]. Low-dose aspirin (<300mg per day) selectively inactivates endothelial cyclooxygenase and inhibits the biosynthesis of placental thromboxane A [33,188]. The mechanism by which aspirin prevents PE is unknown but may include improvements in placentation, inhibition of platelet aggregation, and anti-inflammatory effects [188,189,190,191]. Women with PCOS identified as high-risk on first trimester screening, could be offered low-dose aspirin prophylaxis followed by repeated angiogenic ratio measurements starting at 22 weeks gestation, as per established protocols (Figure 2) [53].
Low-dose aspirin has been co-administered with a variety of other medications for ovulation induction in women with PCOS-related infertility. These include clomid [192], letrozole [193], tamoxifen [194], and Chinese medicine [195]. When considered together, these studies report improved oocyte quality, higher pregnancy rates, and increased endometrial thickness. It is not possible to determine the impact of aspirin on placentation or pregnancy outcomes from these studies due to the combined administration with other medications. Nevertheless, aspirin is a non-steroidal anti-inflammatory medication with known hemodynamic and immunomodulatory effects and may have beneficial effects on placentation in high-risk women when initiated in the periconception period. A randomized trial sought to investigate this possibility by comparing low-dose aspirin monotherapy with placebo [196]. Preconception aspirin resulted in a non-significant increase in live birthrate among a large cohort of women with a history of pregnancy loss. A secondary analysis of women who were adherent to low-dose aspirin for at least 4 days per week showed improved reproductive outcomes [197]. A further secondary analysis in women with a history of low-grade inflammation, assessed by elevated high-sensitivity C-reactive protein (hsCRP), found that women in the lowest hsCRP tertile had increased live birth rates (RR: 1.35, 95% CI 1.08-1.67). Taken together, these studies raise some important issues for the management of women with PCOS as most women with PCOS have low-grade chronic inflammation and increased risk of pregnancy-related complications. Future studies are required to evaluate the impact of periconception aspirin in women with a history of adverse pregnancy outcomes and/or PCOS. In the meantime, the beneficial effects of lifestyle and postconception aspirin treatment are supported by a significant body of evidence, as previously discussed.
Recent studies have suggested that it may be possible to stop aspirin at 28 weeks gestation if the angiogenic ratio (sFlt-1:PlGF) and/or the UtAPI are normal [198,199]. Mendoza et al performed a multicentre, open label, randomized trial of 968 pregnant women who were at high-risk of preterm PE on first trimester screening (StopPRE trial) [198]. All women were commenced on aspirin 150mg per day before 16 weeks and 6 days gestation, followed by an sFlt-1:PlGF ratio at 24 to 28 weeks. Participants with a ratio less than 38 were randomized to either continue aspirin (control group) or discontinue aspirin treatment (intervention group). The incidence of preterm PE and delivery before 37 weeks gestation was 1.48% (7/473) in the intervention group and 1.73% (8/463) in the control group (absolute difference -0.25%; 95% CI: -1.86% to 1.36%) [198]. Aspirin commenced in high-risk women before 16 weeks and 6 days and discontinued at 24-28 weeks, was not inferior to aspirin continued to 37 weeks for preventing preterm PE in women who had a normal sFlt-1:PlGF ratio at 24-28 weeks. There were no significant differences in any other adverse pregnancy or neonatal outcomes.
Bonacina et al performed a post-hoc analysis of the StopPRE trial described above [199]. In the secondary analysis women with a UtAPI >90th percentile were excluded. A total of 836 women were randomized to continue (control group 416) or discontinue aspirin treatment (intervention group 420). Preterm PE occurred in 0.7% (3/409) in the intervention group and 1.3% (5/395) of the control group (absolute difference -0.53; 95% CI: -1.91 to 0.85). Discontinuation of aspirin at 24-28 weeks gestation in women with a UtAPI index <90th percentile was non-inferior to continuing aspirin treatment until 36 weeks for preventing preterm PE. Women in the intervention group had significantly less minor bleeding complications than women in the control group [199]. These findings are consistent with previous cohort studies showing that low-dose aspirin use during pregnancy may be associated with increased postpartum bleeding and haematoma [200].
The authors of the secondary analysis of the StopPRE trial concluded that high-risk women commenced on aspirin at <16 weeks gestation who had either a sFlt-1:PlGF ratio <38 or UtAPI <90th percentile, could discontinue aspirin at 24-28 weeks [199]. If further large trials confirm these findings, ceasing aspirin at 24-28 weeks could result in increased compliance and reduced bleeding risk, without loss of treatment efficacy.
First trimester triple test screening is based on references [54,184]. Use of aFlt-1:PIGF ratio is based on references [34,53]. Mean Arterial Pressure (MAP; Uterine Artery Pulsatility Index (UtAPI); Placental Growth Factor (PlGF); soluble fms-like tyrosine kinase-1 (sFlt-1).
In summary, the diagnosis of PCOS can reliably be made on history at first presentation in early pregnancy [201,202]. Women with PCOS should be offered nutritional advice from a dietitian and considered for prophylactic aspirin treatment as per Fetal Medicine Foundation or local guideline recommendations [183,203]. Women identified as high risk on screening should be offered low-dose aspirin (100-150mg/day, taken at night) starting before 16 weeks of pregnancy then followed by repeat angiogenic ratio measurement from 22 weeks gestation (Figure 2) [53,204]. In addition, health practitioners involved in antenatal care should be educated about the risk of pregnancy complications and PE in women with PCOS. Future research should be directed at investigating the underlying pathophysiology that predisposes women with PCOS to an increased risk of PE.
6. Conclusions
Polycystic ovary syndrome is a multisystem metabolic and endocrine disorder that is associated with an increased risk of pregnancy-related complications, including PE. International guidelines recommend lifestyle treatment, including diet and exercise, as the first line of management for all women with PCOS. A significant body of evidence supports the recommendations of expert advisory groups that healthy diet patterns reduce the risk of PE. PCOS is usually diagnosed in adolescence or early adulthood and presents an ideal opportunity for preventative lifestyle interventions that can be implemented prior to conception to reduce the risk of pregnancy complications. Women with a history of PCOS can also be assessed in early pregnancy and given lifestyle advice and support. In addition, women with PCOS can also be evaluated with first trimester triple test screening and advised about their eligibility for prophylactic medical therapy to reduce the risk of PE. Implementation of preventative intervention strategies have the potential to reduce pregnancy-related complications and future development of transgenerational chronic disease-related morbidity and mortality in women with PCOS and their offspring.
Author Contributions
Conceptualization, J.P., C.O.B., C.Y., F.L.G, S.B.; writing-original draft preparation, J.P.; writing-review and editing, J.P., C.O.B, C.Y., F.L.G., S.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
Nil.
Conflicts of Interest
No conflicts of interest to declare.
References
- Parker, J.; O’brien, C.; Hawrelak, J.; Gersh, F.L. Polycystic Ovary Syndrome: An Evolutionary Adaptation to Lifestyle and the Environment. Int J Environ Res Public Health. 2022, 19, 1336. [Google Scholar] [CrossRef]
- Dumesic, D.A.; Abbott, D.H.; Chazenbalk, G.D.; Scholar, G. An Evolutionary Model for the Ancient Origins of Polycystic Ovary Syndrome. J Clin Med. 2023, 12, 1–16. [Google Scholar]
- Parker, J. Pathophysiological Effects of Contemporary Lifestyle on Evolutionary-Conserved Survival Mechanisms in Polycystic Ovary Syndrome. Life. 2023, 13, 1056. [Google Scholar] [CrossRef]
- Dumesic, D.A.; Padmanabhan, V.; Chazenbalk, G.D.; Abbott, D.H. Polycystic ovary syndrome as a plausible evolutionary outcome of metabolic adaptation. Reprod Biol Endocrinol [Internet]. 2022, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dumesic, D.A.; Abbott, D.H.; Sanchita, S.; Chazenbalk, G.D. Endocrine–metabolic dysfunction in polycystic ovary syndrome: an evolutionary perspective. Curr Opin Endocr Metab Res [Internet]. 2020, 12, 41–48. [Google Scholar] [CrossRef]
- Parker, J.; O’Brien, C. Evolutionary and genetic antecedents to the pathogenesis of polycystic ovary syndrome (PCOS). J ACNEM. 2021, 40, 12–20. [Google Scholar]
- Stepto NK, Cassar S, Joham AE, Hutchison SK, Harrison CL, Goldstein RF, et al. Women with polycystic ovary syndrome have intrinsic insulin resistance on euglycaemic-hyperinsulaemic clamp. Hum Reprod. 2013, 28, 777–784. [CrossRef]
- Aboeldalyl, S.; James, C.; Seyam, E.; Ibrahim, E.M.; Shawki, H.E.D.; Amer, S. The role of chronic inflammation in polycystic ovarian syndrome—a systematic review and meta-analysis. Int J Mol Sci. 2021, 22, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Szukiewicz, D.; Trojanowski, S.; Kociszewska, A.; Szewczyk, G. Modulation of the Inflammatory Response in Polycystic Ovary Syndrome (PCOS)—Searching for Epigenetic Factors. Int J Mol Sci. 2022, 23. [Google Scholar] [CrossRef]
- Bahri Khomami M, Joham AE, Boyle JA, Piltonen T, Silagy M, Arora C, et al. Increased maternal pregnancy complications in polycystic ovary syndrome appear to be independent of obesity—A systematic review, meta-analysis, and meta-regression. Obes Rev. 2019, 20, 659–674. [CrossRef]
- Zahid S, Khan MZ, Gowda S, Faza NN, Honigberg MC, Vaught A, et al. Trends, Predictors, and Outcomes of Cardiovascular Complications Associated With Polycystic Ovary Syndrome During Delivery Hospitalizations: A National Inpatient Sample Analysis (2002–2019). J Am Heart Assoc. 2022, 11, 1–12.
- Elawad T, Scott G, Bone JN, Elwell H, Lopez CE, Filippi V, et al. Risk factors for pre-eclampsia in clinical practice guidelines: Comparison with the evidence. BJOG An Int J Obstet Gynaecol. 2022;(September):1–17.
- Dumesic DA, Phan JD, Leung KL, Grogan TR, Ding X, Li X, et al. Adipose insulin resistance in normal-weight women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2019, 104, 2171–2183.
- Brennan, K.M.; Kroener, L.L.; Chazenbalk, G.D.; Dumesic, D.A. Polycystic Ovary Syndrome: Impact of Lipotoxicity on Metabolic and Reproductive Health. Obstet Gynecol Surv. 2019, 74, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Abbott, D.H.; Dumesic, D.A.; Franks, S. Developmental origin of polycystic ovary syndrome - A hypothesis. J Endocrinol. 2002, 174, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Dumesic, D.A.; Hoyos, L.R.; Chazenbalk, G.D.; Naik, R.; Padmanabhan, V.; Abbott, D.H. Mechanisms of intergenerational transmission of polycystic ovary syndrome. Reproduction. 2019, 159, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Abbott, D.H.; Dumesic, D.A.; Abbott, D.H. Fetal androgen excess provides a developmental origin for polycystic ovary syndrome. Expert Rev Obs Gynecol. 2009, 4, 1–7. [Google Scholar] [CrossRef]
- Parker, J.; O’Brien, C.; Gersh, F.L. Developmental origins and transgenerational inheritance of polycystic ovary syndrome. Aust New Zeal J Obstet Gynaecol. 2021, 61, 922–926. [Google Scholar] [CrossRef] [PubMed]
- Meek, C.L. An unwelcome inheritance: childhood obesity after diabetes in pregnancy. Diabetologia [Internet]. 2023, 66, 1961–1970. [Google Scholar] [CrossRef] [PubMed]
- Zore, T.; Joshi, N.V.; Lizneva, D.; Azziz, R. Polycystic Ovarian Syndrome: Long-Term Health Consequences. Vol. 35, Seminars in Reproductive Medicine. 2017. p. 271–81.
- Reyes-Muñoz E, Castellanos-Barroso G, Ramírez-Eugenio BY, Ortega-González C, Parra A, Castillo-Mora A, et al. The risk of gestational diabetes mellitus among Mexican women with a history of infertility and polycystic ovary syndrome. Fertil Steril. 2012, 97, 1467–1471. [CrossRef]
- Rodgers RJ, Avery JC, Moore VM, Davies MJ, Azziz R, Stener-Victorin E, et al. Complex diseases and co-morbidities: Polycystic ovary syndrome and type 2 diabetes mellitus. Endocr Connect. 2019, 8, R71–5. [CrossRef]
- Wu, J.; Yao, X.Y.; Shi, R.X.; Liu, S.F.; Wang, X.Y. A potential link between polycystic ovary syndrome and non-alcoholic fatty liver disease: An update meta-analysis. Reprod Health. 2018, 15, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Palomba, S.; De Wilde, M.A.; Falbo, A.; Koster, M.P.H.; La Sala, G.B.; Fauser, B.C.J.M. Pregnancy complications in women with polycystic ovary syndrome. Hum Reprod Update. 2015, 21, 575–592. [Google Scholar] [CrossRef] [PubMed]
- Helena Teede, Chau Thien Tay JL, Anuja Dokras, Lisa Moran TP, Michael Costello JB, Leanne Redman JB, Robert Norman, Aya Mousa AJ. International evidence-based guideline for the assessment and management of polycystic ovary syndrome 2023. Natl Heal Med Res Counc. 2023;1–258.
- Du, Y.; Li, F.; Li, S.; Ding, L.; Liu, M. Causal relationship between polycystic ovary syndrome and chronic kidney disease : A Mendelian randomization study. Front Endocrinol (Lausanne). 2023, 14, 1120119. [Google Scholar] [CrossRef] [PubMed]
- Boomsma, C.M.; Eijkemans, M.J.C.; Hughes, E.G.; Visser, G.H.A.; Fauser, B.C.J.M.; Macklon, N.S. A meta-analysis of pregnancy outcomes in women with polycystic ovary syndrome. Hum Reprod Update. 2006, 12, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Kjerulff, L.E.; Sanchez-Ramos, L.; Duffy, D. Pregnancy outcomes in women with polycystic ovary syndrome: A metaanalysis. Am J Obstet Gynecol [Internet]. 2011, 204, 558–e1. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.Z.; Pang, L.H.; Li, M.J.; Fan, X.J.; Huang, R.D.; Chen, H.Y. Obstetric complications in women with polycystic ovary syndrome: A systematic review and meta-analysis. Reprod Biol Endocrinol. 2013, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.F.; Chen, H.S.; Rao, D.P.; Gong, J. Association between polycystic ovary syndrome and the risk of pregnancy complications A PRISMA-compliant systematic review and meta-analysis. Med (United States). 2016, 95, e4863. [Google Scholar]
- Magee LA, Brown MA, Hall DR, Gupte S, Hennessy A, Karumanchi SA, et al. The 2021 International Society for the Study of Hypertension in Pregnancy classification, diagnosis & management recommendations for international practice. Pregnancy Hypertens [Internet]. 2022;27(October 2021):148–69. Available from: https://doi.org/10.1016/j.preghy.2021.09.008. [CrossRef]
- Brown MA, Magee LA, Kenny LC, Karumanchi SA, McCarthy FP, Saito S, et al. Hypertensive disorders of pregnancy: ISSHP classification, diagnosis, and management recommendations for international practice. Hypertension. 2018, 72, 24–43. [CrossRef] [PubMed]
- Poon LC, Shennan A, Hyett JA, Kapur A, Hadar E, Divakar H, et al. The International Federation of Gynecology and Obstetrics (FIGO) initiative on pre-eclampsia: A pragmatic guide for first-trimester screening and prevention. Int J Gynecol Obstet. 2019;145(S1):1–33.
- Verlohren, S.; Dröge, L.A. The diagnostic value of angiogenic and antiangiogenic factors in differential diagnosis of preeclampsia. Am J Obstet Gynecol [Internet]. 2022, 226, S1048–58. [Google Scholar] [CrossRef]
- Fox, R.; Kitt, J.; Leeson, P.; Aye, C.Y.L.; Lewandowski, A.J. Preeclampsia: Risk factors, diagnosis, management, and the cardiovascular impact on the offspring. J Clin Med. 2019, 8, 1–22. [Google Scholar] [CrossRef]
- Davis EF, Lazdam M, Lewandowski AJ, Worton SA, Kelly B, Kenworthy Y, et al. Cardiovascular risk factors in children and young adults born to preeclamptic pregnancies: A systematic review. Pediatrics. 2012, 129, e1552–e1561. [CrossRef]
- Ahmed, R.; Dunford, J.; Mehran, R.; Robson, S.; Kunadian, V. Pre-eclampsia and future cardiovascular risk among women: A review. J Am Coll Cardiol [Internet]. 2014, 63, 1815–1822. [Google Scholar] [CrossRef]
- Burton, G.J.; Jauniaux, E. The human placenta: new perspectives on its formation and function during early pregnancy. Proc R Soc B Biol Sci. 2023, 290, 20230191. [Google Scholar] [CrossRef] [PubMed]
- Dimitriadis E, Rolnik DL, Zhou W, Estrada-Gutierrez G, Koga K, Francisco RPV, et al. Pre-eclampsia. Nat Rev Dis Prim. 2023, 9, 1–22.
- Chappell LC, Cluver CA, Kingdom J, Tong S. Pre-eclampsia. Lancet. 2021, 398, 341–354.
- Murthi, P.; Pinar, A.A.; Dimitriadis, E.; Samuel, C.S. Inflammasomes—a molecular link for altered immunoregulation and inflammation mediated vascular dysfunction in preeclampsia. Int J Mol Sci. 2020, 21, 1406. [Google Scholar] [CrossRef] [PubMed]
- Vega, M.; Mauro, M.; Williams, Z. Direct toxicity of insulin on the human placenta and protection by metformin. Fertil Steril [Internet]. 2019, 111, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Sun M, Sun B, Qiao S, Feng X, Li Y, Zhang S, et al. Elevated maternal androgen is associated with dysfunctional placenta and lipid disorder in newborns of mothers with polycystic ovary syndrome. Fertil Steril [Internet]. 2020, 113, 1275–1285. [CrossRef] [PubMed]
- Myers, J.E. What are the metabolic precursors which increase the risk of pre-eclampsia and how could these be investigated further. Placenta [Internet]. 2017, 60, 110–114. [Google Scholar] [CrossRef]
- Hu, M.; Li, J.; Baker, P.N.; Tong, C. Revisiting preeclampsia: a metabolic disorder of the placenta. FEBS J. 2022, 289, 336–354. [Google Scholar] [CrossRef]
- Parker, J.; Hawrelak, J.; Gersh, F.L. Nutritional role of polyphenols as a component of a wholefood diet in the management of polycystic ovary syndrome. J ACNEM. 2021, 40, 6–12. [Google Scholar]
- Kinshella MLW, Pickerill K, Bone JN, Prasad S, Campbell O, Vidler M, et al. An evidence review and nutritional conceptual framework for pre-eclampsia prevention. Br J Nutr. 2023, 130, 1065–1076. [CrossRef] [PubMed]
- Bahri Khomami M, Moran LJ, Kenny L, Grieger JA, Myers J, Poston L, et al. Lifestyle and pregnancy complications in polycystic ovary syndrome: The SCOPE cohort study. Clin Endocrinol (Oxf). 2019, 90, 814–821. [CrossRef] [PubMed]
- Rolnik DL, Wright D, Poon LC, O’Gorman N, Syngelaki A, de Paco Matallana C, et al. Aspirin versus Placebo in Pregnancies at High Risk for Preterm Preeclampsia. N Engl J Med. 2017, 377, 613–622. [CrossRef] [PubMed]
- Pan H, Xian P, Yang D, Zhang C, Tang H, He X, et al. Polycystic ovary syndrome is an independent risk factor for hypertensive disorders of pregnancy: A systematic review, meta-analysis, and meta-regression. Endocrine. 2021, 74, 518–529. [CrossRef] [PubMed]
- Riestenberg, C.; Jagasia, A.; Markovic, D.; Buyalos, R.P.; Azziz, R. Health Care-Related Economic Burden of Polycystic Ovary Syndrome in the United States: Pregnancy-Related and Long-Term Health Consequences. J Clin Endocrinol Metab. 2022, 107, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Mousa, A; Tay, CT; Teede H. Technical Report for the 2023 International Evidence-based Guideline for the Assessment and Management of Polycystic Ovary Syndrome. Monash University; 2023.
- Verlohren S, Brennecke SP, Galindo A, Karumanchi SA, Mirkovic LB, Schlembach D, et al. Clinical interpretation and implementation of the sFlt-1/PlGF ratio in the prediction, diagnosis and management of preeclampsia. Pregnancy Hypertens [Internet]. 2022;27(August 2021):42–50. Available from: https://doi.org/10.1016/j.preghy.2021.12.003. [CrossRef]
- Wright, D.; Wright, A.; Nicolaides, K.H. The competing risk approach for prediction of preeclampsia. Am J Obstet Gynecol [Internet]. 2020, 223, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Serrano B, Bonacina E, Rodo C, Garcia-Manau P, Sanchez-Duran MÁ, Pancorbo M, et al. First-trimester screening for pre-eclampsia and small for gestational age: A comparison of the gaussian and Fetal Medicine Foundation algorithms. Int J Gynecol Obstet. 2023, 160, 150–160. [CrossRef] [PubMed]
- ACOG Low-Dose Aspirin Use during Pregnancy. Am Coll Obstet Gynecol. 2018, 132, E44–52. [CrossRef]
- Kane, S.C.; Da Silva Costa, F. Risk factors for pre-eclampsia: Received wisdom versus reality. BJOG An Int J Obstet Gynaecol. 1732. [Google Scholar]
- Bahri Khomami, M.; Teede, H.J.; Joham, A.E.; Moran, L.J.; Piltonen, T.T.; Boyle, J.A. Clinical management of pregnancy in women with polycystic ovary syndrome: An expert opinion. Clin Endocrinol (Oxf). 2022, 97, 227–236. [Google Scholar] [CrossRef]
- Howeler, J.F. Hewson A. Dietary Fibre and Toxaemia of Pregnancy. Med J Aust. 1957;761–3.
- Hipsley, E. Dietary Fibre and Pregnancy Toxaemia. Br Med J. 1953;22 August:420–4222.
- Harding, V.J.; Van Wyck, H.B. Diet in the Treatment of Pre-Eclampsia. J Obstet Gynaecol. 1926, 33, 17–32. [Google Scholar] [CrossRef]
- Abbafati C, Abbas KM, Abbasi-Kangevari M, Abd-Allah F, Abdelalim A, Abdollahi M, et al. Global burden of 87 risk factors in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1223–1249. [CrossRef] [PubMed]
- Teede HJ, Misso ML, Costello MF, Dokras A, Laven J, Moran L, et al. Recommendations from the international evidence-based guideline for the assessment and management of polycystic ovary syndrome. Eur J Endocrinol [Internet]. 2023, 189, G43–64. [CrossRef]
- Traore SS, Bo Y, Amoah AN, Khatun P, Kou G, Hu Y, et al. A meta-analysis of maternal dietary patterns and preeclampsia. Clin Nutr Open Sci [Internet]. 2021, 40, 15–29. [CrossRef]
- Perry, A.; Stephanou, A.; Rayman, M.P. Dietary factors that affect the risk of pre-eclampsia. BMJ Nutr Prev Heal. 2022, 5, 118–133. [Google Scholar] [CrossRef] [PubMed]
- Kibret, K.T.; Chojenta, C.; Gresham, E.; Tegegne, T.K.; Loxton, D. Maternal dietary patterns and risk of adverse pregnancy (hypertensive disorders of pregnancy and gestational diabetes mellitus) and birth (preterm birth and low birth weight) outcomes: A systematic review and meta-analysis. Public Health Nutr. 2019, 22, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Paula, W.O.; Patriota, E.S.O.; Gonçalves, V.S.S.; Pizato, N. Maternal Consumption of Ultra-Processed Foods-Rich Diet and Perinatal Outcomes: A Systematic Review and Meta-Analysis. Nutrients. 2022, 14, 3242. [Google Scholar] [CrossRef] [PubMed]
- APant SGribbin PMachado AHodge LMoran SMarschner, S.Z. Association of ultra-processed foods with cardiovascular disease and hypertension in australian women. In: European Heart Journal [Internet]. Available from. [CrossRef]
- Wu P, Haththotuwa R, Kwok CS, Babu A, Kotronias RA, Rushton C, et al. Preeclampsia and future cardiovascular health. Circ Cardiovasc Qual Outcomes. 2017, 10, 1–9.
- Tuncalp Ö, Rogers LM, Lawrie TA, Barreix M, Peña-Rosas JP, Bucagu M, et al. WHO recommendations on antenatal nutrition: an update on multiple micronutrient supplements. BMJ Glob Heal. 2022, 5, 3–6.
- Marshall NE, Abrams B, Barbour LA, Catalano P, Christian P, Friedman JE, et al. The importance of nutrition in pregnancy and lactation: lifelong consequences. Am J Obstet Gynecol [Internet]. 2022, 226, 607–632. [CrossRef]
- Grieger JA, Grzeskowiak LE, Bianco-Miotto T, Jankovic-Karasoulos T, Moran LJ, Wilson RL, et al. Pre-pregnancy fast food and fruit intake is associated with time to pregnancy. Hum Reprod. 2018, 33, 1063–1070. [CrossRef] [PubMed]
- Sampathkumar, S.; Parkhi, D.; Ghebremichael-Weldeselassie, Y.; Sukumar, N.; Saravanan, P. Effectiveness of pre-pregnancy lifestyle in preventing gestational diabetes mellitus—a systematic review and meta-analysis of 257,876 pregnancies. Nutr Diabetes. 2023, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Erickson ML, Mey JT, Axelrod CL, Paul D, Gordesky L, Russell K, et al. Rationale and study design for lifestyle intervention in preparation for pregnancy (LIPP): A randomized controlled trial. Contemp Clin Trials [Internet]. 2020;94(May):106024. Available from: https://doi.org/10.1016/j.cct.2020.106024. [CrossRef]
- Gustaw Eriksson, Congru Li, Sanjiv Risal, Han-Pin Pui, Sara Torstensson, Angelica Linden Hirschberg, Sophie Petropoulos, Qiaolin Deng ES-V. Mapping Endometrial Cell-type-specific Disease Signatures A nd Endometrial Organoids In Polycystic Ovary Syndrome. J Endocr Soc. 2023;Oct 5;7(Suppl 1):bvad114.1587.
- Gustaw Eriksson, Congru Li, Sanjiv Risal, Han-Pin Pui ST, Angelica Linden Hirschberg, Sophie Petropoulos, Qiaolin Deng ES-V. Mapping Endometrial Cell-type-specific Disease Signatures And Endometrial Organoids In Polycystic Ovary Syndrome. J Endocr Soc. 2023;7(Supplement):A848.
- Redman, C.W.G.; Staff, A.C.; Roberts, J.M. Syncytiotrophoblast stress in preeclampsia: the convergence point for multiple pathways. Am J Obstet Gynecol [Internet]. 2022, 226, S907–27. [Google Scholar] [CrossRef] [PubMed]
- Schjenken JE, Sharkey DJ, Green ES, Chan HY, Matias RA, Moldenhauer LM, et al. Sperm modulate uterine immune parameters relevant to embryo implantation and reproductive success in mice. Commun Biol. 2021, 4, 1–14.
- Robertson, S.A.; Prins, J.R.; Sharkey, D.J.; Moldenhauer, L.M. Seminal Fluid and the Generation of Regulatory T Cells for Embryo Implantation. Am J Reprod Immunol. 2013, 69, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Ward, K.; Taylor, R.N. Genetic factors in the etiology of preeclampsia/eclampsia [Internet]. Fourth Edi. Chesley’s Hypertensive Disorders in Pregnancy, Fourth Edition. Elsevier Inc.; p. Available from. [CrossRef]
- Christians, J.K.; Leavey, K.; Cox, B.J. Associations between imprinted gene expression in the placenta, human fetal growth and preeclampsia. Biol Lett. 2017, 13, 20170643. [Google Scholar] [CrossRef]
- Ashraf, U.M.; Hall, D.L.; Rawls, A.Z.; Alexander, B.T. Epigenetic processes during preeclampsia and effects on fetal development and chronic health. Clin Sci. 2021, 135, 2307–2327. [Google Scholar] [CrossRef] [PubMed]
- Neubrand L, Pothmann H, Besenfelder U, Havlicek V, Gabler C, Dolezal M, et al. In vivo dynamics of pro - inflammatory factors , mucins , and polymorph nuclear neutrophils in the bovine oviduct during the follicular and luteal phase. Sci Rep [Internet]. 2023;1–14. Available from: https://doi.org/10.1038/s41598-023-49151-9. [CrossRef]
- Burton, G.J.; Cindrova-Davies, T.; Turco, M.Y. Review: Histotrophic nutrition and the placental-endometrial dialogue during human early pregnancy. Placenta [Internet]. 2020;102(February):21–6. Available from: https://doi.org/10.1016/j.placenta.2020.02.008. [CrossRef]
- O’Brien, K.; Wang, Y. The Placenta: A Maternofetal Interface. Annu Rev Nutr. 2023, 43, 301–325. [Google Scholar] [CrossRef]
- Conrad, K.P.; Rabaglino, M.B.; Post Uiterweer, E.D. Emerging role for dysregulated decidualization in the genesis of preeclampsia. Placenta [Internet]. 2017, 60, 119–129. [Google Scholar] [CrossRef]
- Rabaglino, M.B.; Conrad, K.P. Evidence for shared molecular pathways of dysregulated decidualization in preeclampsia and endometrial disorders revealed by microarray data integration. FASEB J. 2019, 33, 11682–11695. [Google Scholar] [CrossRef]
- Spracklen, C.N.; Smith, C.J.; Saftlas, A.F.; Robinson, J.G.; Ryckman, K.K. Maternal hyperlipidemia and the risk of preeclampsia: A meta-analysis. Am J Epidemiol. 2014, 180, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Tippetts, T.S.; Sieber, M.H.; Solmonson, A. Beyond energy and growth: the role of metabolism in developmental signaling, cell behavior and diapause. Development. 2023, 150, dev201610. [Google Scholar] [CrossRef] [PubMed]
- Kamrani A, Alipourfard I, Ahmadi-Khiavi H, Yousefi M, Rostamzadeh D, Izadi M, et al. The role of epigenetic changes in preeclampsia. BioFactors. 2019, 45, 712–724.
- Apicella, C.; Ruano, C.S.M.; Méhats, C.; Miralles, F.; Vaiman, D. The role of epigenetics in placental development and the etiology of preeclampsia. Int J Mol Sci. 2019, 20, 2837. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wong, R.J.; Stevenson, D.K. The impact of hypoxia in early pregnancy on placental cells. Int J Mol Sci.
- Chiarello DI, Abad C, Rojas D, Toledo F, Vázquez CM, Mate A, et al. Oxidative stress: Normal pregnancy versus preeclampsia. Biochim Biophys Acta - Mol Basis Dis [Internet]. 2020, 1866, 165354. [CrossRef]
- Gómez de Cedrón, M.; Moreno Palomares, R.; Ramírez de Molina, A. Metabolo-epigenetic interplay provides targeted nutritional interventions in chronic diseases and ageing. Front Oncol. 2023;13(June):1–20.
- Dai, Z.; Ramesh, V.; Locasale, J.W. The evolving metabolic landscape of chromatin biology and epigenetics. Nat Rev Genet [Internet]. 2020, 21, 782. [Google Scholar] [CrossRef] [PubMed]
- Doshani, A.; Konje, J.C. Placental dysfunction in obese women and antenatal surveillance. Best Pract Res Clin Obstet Gynaecol [Internet]. 2023, 91, 102407. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Young, S.L.; Grattan, D.R.; Jasoni, C.L. Obesity during pregnancy disrupts placental morphology, cell proliferation, and inflammation in a sex-specific manner across gestation in the mouse. Biol Reprod. 2014, 90, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tiffon, C. The impact of nutrition and environmental epigenetics on human health and disease. Int J Mol Sci. 2018, 19, 3425. [Google Scholar] [CrossRef]
- Barrón-Cabrera E, Ramos-Lopez O, González-Becerra K, Riezu-Boj JI, Milagro FI, Martínez-López E, et al. Epigenetic Modifications as Outcomes of Exercise Interventions Related to Specific Metabolic Alterations: A Systematic Review. Lifestyle Genomics. 2019;12(1–6):25–44.
- Berni Canani, R.; Di Costanzo, M.; Leone, L. The epigenetic effects of butyrate: Potential therapeutic implications for clinical practice. Clin Epigenetics. 2012, 4, 1–7. [Google Scholar] [CrossRef]
- Abraham E, Rousseaux S, Agier L, Giorgis-Allemand L, Tost J, Galineau J, et al. Pregnancy exposure to atmospheric pollution and meteorological conditions and placental DNA methylation. Environ Int. 2018;118(May):334–47.
- Peretz J, Vrooman L, Ricke WA, Hunt PA, Ehrlich S, Hauser R, et al. Bisphenol A and reproductive health: Update of experimental and human evidence, 2007-2013. Environ Health Perspect. 2014, 122, 775–786. [CrossRef]
- Ye, Y.; Tang, Y.; Xiong, Y.; Feng, L.; Li, X. Bisphenol A exposure alters placentation and causes preeclampsia-like features in pregnant mice involved in reprogramming of DNA methylation of WNT2. FASEB J. 2019, 33, 2732–2742. [Google Scholar] [CrossRef]
- Tait, S.; Tassinari, R.; Maranghi, F.; Mantovani, A. Bisphenol A affects placental layers morphology and angiogenesis during early pregnancy phase in mice. J Appl Toxicol. 2015, 35, 1278–1291. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.Q.; Qi, H.P.; Luo, Z.C.; Fraser, W.D. Maternal vitamin D status and adverse pregnancy outcomes: A systematic review and meta-analysis. J Matern Neonatal Med. 2013, 26, 889–899. [Google Scholar] [CrossRef]
- Bodnar, L.M.; Catov, J.M.; Simhan, H.N.; Holick, M.F.; Powers, R.W.; Roberts, J.M. Maternal vitamin D deficiency increases the risk of preeclampsia. J Clin Endocrinol Metab. 2007, 92, 3517–3522. [Google Scholar] [CrossRef] [PubMed]
- Indrio F, Martini S, Francavilla R, Corvaglia L, Cristofori F, Mastrolia SA, et al. Epigenetic matters: The link between early nutrition, microbiome, and long-term health development. Front Pediatr. 2017;5(August):1–14.
- Kinshella MLW, Omar S, Scherbinsky K, Vidler M, Magee LA, von Dadelszen P, et al. Maternal nutritional risk factors for pre-eclampsia incidence: findings from a narrative scoping review. Reprod Health. 2022, 19, 1–13.
- Trigg NA, Skerrett-Byrne DA, Xavier MJ, Zhou W, Anderson AL, Stanger SJ, et al. Acrylamide modulates the mouse epididymal proteome to drive alterations in the sperm small non-coding RNA profile and dysregulate embryo development. Cell Rep [Internet]. 2021, 37, 109787. [CrossRef]
- Perera, F.; Herbstman, J. Prenatal environmental exposures, epigenetics, and disease. Reprod Toxicol [Internet]. 2011, 31, 363–373. [Google Scholar] [CrossRef]
- Pollheimer, J.; Vondra, S.; Baltayeva, J.; Beristain, A.G.; Knöfler, M. Regulation of placental extravillous trophoblasts by the maternal uterine environment. Front Immunol.
- Foidart, J.M.; Hustin, J.; Dubois, M.; Schaaps, J.P. The human placenta becomes haemochorial at the 13th week of pregnancy. Int J Dev Biol. 1992, 36, 451–453. [Google Scholar]
- Kingdom JCP, Drewlo S. Is heparin a placental anticoagulant in high-risk pregnancies? Blood [Internet]. 2011, 118, 4780–4788. [CrossRef]
- Leto, D.; Saltiel, A.R. Regulation of glucose transport by insulin: Traffic control of GLUT4. Nat Rev Mol Cell Biol [Internet]. 2012, 13, 383–396. [Google Scholar] [CrossRef]
- Muniyappa, R. Iantorno, M. Quon M. An Integrated View of Insulin Resistance and Endothelial Dysfunction. Endocrinol Metab Clin North Am. 2008, 37, 685. [CrossRef] [PubMed]
- Mandal, A.K.; Leask, M.P.; Estiverne, C.; Choi, H.K.; Merriman, T.R.; Mount, D.B. Genetic and Physiological Effects of Insulin on Human Urate Homeostasis. Front Physiol.
- DeFronzo, R.A. The effect of insulin on renal sodium metabolism. Diabetologia. 1981, 21, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Shulman, G.I. Mechanisms of insulin action and insulin resistance. Physiol Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [PubMed]
- Kampmann, U.; Knorr, S.; Fuglsang, J.; Ovesen, P. Determinants of Maternal Insulin Resistance during Pregnancy: An Updated Overview. J Diabetes Res. 2019, 2019, 5320156. [Google Scholar] [CrossRef] [PubMed]
- Ma Q, Fan J, Wang J, Yang S, Cong Q, Wang R, et al. High levels of chorionic gonadotrophin attenuate insulin sensitivity and promote inflammation in adipocytes. J Mol Endocrinol. 2015, 54, 161–170. [CrossRef] [PubMed]
- Barbour, L.A.; McCurdy, C.E.; Hernandez, T.L.; Kirwan, J.P.; Catalano, P.M.; Friedman, J.E. Cellular mechanisms for insulin resistance in normal pregnancy and gestational diabetes. Diabetes Care. 2007;30(SUPPL. 2).
- Sonagra, A.D. Normal Pregnancy- A State of Insulin Resistance. J Clin Diagnostic Res. 2014, 8, CC01–CC03. [Google Scholar] [CrossRef] [PubMed]
- Catalano, P.M.; Huston, L.; Amini, S.B.; Kalhan, S.C. Longitudinal changes in glucose metabolism during pregnancy in obese women with normal glucose tolerance and gestational diabetes mellitus. Am J Obstet Gynecol. 1999, 180, 903–916. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, X.; Zuo, Y.; Gao, J.; Liu, Y.; Zheng, W. The risk factors of gestational diabetes mellitus in patients with polycystic ovary syndrome: What should we care. Med (United States). 2021, 100, E26521. [Google Scholar] [CrossRef]
- Yan, Q.; Qiu, D.; Liu, X.; Xing, Q.; Liu, R.; Hu, Y. The incidence of gestational diabetes mellitus among women with polycystic ovary syndrome: a meta-analysis of longitudinal studies. BMC Pregnancy Childbirth [Internet]. 2022, 22, 1–12. [Google Scholar] [CrossRef]
- Buchanan, T.A.; Xiang, A.H.; Page, K.A. Gestational diabetes mellitus: Risks and management during and after pregnancy. Nat Rev Endocrinol [Internet]. 2012, 8, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Cassar, S.; Misso, M.L.; Hopkins, W.G.; Shaw, C.S.; Teede, H.J.; Stepto, N.K. Insulin resistance in polycystic ovary syndrome: A systematic review and meta-analysis of euglycaemic-hyperinsulinaemic clamp studies. Hum Reprod. 2016, 31, 2619–2631. [Google Scholar] [CrossRef] [PubMed]
- Schulte, M.M.B.; Tsai, J.H.; Moley, K.H. Obesity and PCOS: The effect of metabolic derangements on endometrial receptivity at the time of implantation. Reprod Sci. 2015, 22, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Tumminia, A.; Scalisi, N.M.; Milluzzo, A.; Ettore, G.; Vigneri, R.; Sciacca, L. Maternal Diabetes Impairs Insulin and IGF-1 Receptor Expression and Signaling in Human Placenta. Front Endocrinol (Lausanne). 2021;12(March):1–10.
- Abhari, F.R.; Ghanbari Andarieh, M.; Farokhfar, A.; Ahmady, S. Estimating rate of insulin resistance in patients with preeclampsia using Homa-IR index and comparison with nonpreeclampsia pregnant women. Biomed Res Int. 2014, 2014, 140851. [Google Scholar] [CrossRef] [PubMed]
- Nestler, J.E. Regulation of the aromatase activity of human placental cytotrophoblasts by insulin, insulin-like growth factor-I, and -II. J Steroid Biochem Mol Biol. 1993;44(4–6):449–57.
- Lassance L, Haghiac M, Leahy P, Basu S, Minium J, Zhou J, et al. Identification of early transcriptome signatures in placenta exposed to insulin and obesity. Am J Obstet Gynecol [Internet]. 2015, 212, 647–e1. [CrossRef]
- Calabuig-Navarro V, Puchowicz M, Glazebrook P, Haghiac M, Minium J, Catalano P, et al. Effect of ω-3 supplementation on placental lipid metabolism in overweight and obese women. Am J Clin Nutr [Internet]. 2016, 103, 1064–1072. [CrossRef]
- Calabuig-Navarro V, Haghiac M, Minium J, Glazebrook P, Ranasinghe GC, Hoppel C, et al. Effect of maternal obesity on placental lipid metabolism. Endocrinology. 2017, 158, 2543–2555.
- O’Tierney-Ginn, P.; Presley, L.; Myers, S.; Catalano, P. Placental growth response to maternal insulin in early pregnancy. J Clin Endocrinol Metab. 2015, 100, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Leoni, M.; Padilla, N.; Fabbri, A.; Della-Morte, D.; Ricordi, C.I.M. Mechanisms of Insulin Resistance during Pregnancy [Internet]. Evolving Concepts in Insulin Resistance. In: Marco Infante. University of Rome. Italy, editor. Evolving Concepts in Insulin Resistance [Internet]. Intech Open; 2022. p. Downloaded 29 November 2023. Available from: https://www.intechopen.com/chapters/83989.
- Gou, R.; Zhang, X. Glycolysis: A fork in the path of normal and pathological pregnancy. FASEB J. 2023, 37, 1–20. [Google Scholar] [CrossRef]
- Paradisi G, Steinberg HO, Hempfling A, Cronin J, Hook G, Shepard MK, et al. Polycystic ovary syndrome is associated with endothelial dysfunction. Circulation. 2001, 103, 1410–1415. [CrossRef]
- Lambert EA, Teede H, Sari CI, Jona E, Shorakae S, Woodington K, et al. Sympathetic activation and endothelial dysfunction in polycystic ovary syndrome are not explained by either obesity or insulin resistance. Clin Endocrinol (Oxf). 2015, 83, 812–819. [CrossRef]
- Oncul M, Albayrak M, Sozer V, Karakus B, Gelisgen R, Karatas S, et al. Polycystic ovary syndrome and endothelial dysfunction: A potential role for soluble lectin-like oxidized low density lipoprotein receptor-1. Reprod Biol. 2020, 20, 396–401. [CrossRef] [PubMed]
- Chen, L.H.; Lin, C.P.; Wu, H.M.; Chu, P.H. Endothelial dysfunction in subfertile women with polycystic ovary syndrome. Reprod Biomed Online [Internet]. 2023, 46, 391–398. [Google Scholar] [CrossRef]
- Diamanti-Kandarakis, E.; Papavassiliou, A.G.; Kandarakis, S.A.; Chrousos, G.P. Pathophysiology and types of dyslipidemia in PCOS. Trends Endocrinol Metab. 2007, 18, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Avagliano L, Bulfamante G Pietro, Morabito A, Marconi AM. Abnormal spiral artery remodelling in the decidual segment during pregnancy: From histology to clinical correlation. J Clin Pathol. 2011, 64, 1064–1068. [CrossRef] [PubMed]
- Hauguel-de Mouzon, S.; Guerre-Millo, M. The Placenta Cytokine Network and Inflammatory Signals. Placenta. 2006, 27, 794–798. [Google Scholar] [CrossRef]
- Xiong, P.; Zhang, F.; Liu, F.; Zhao, J.; Huang, X. Metaflammation in glucolipid metabolic disorders : Pathogenesis and treatment. Biomed Pharmacother [Internet]. 2023;161(March):114545. Available from: https://doi.org/10.1016/j.biopha.2023.114545. [CrossRef]
- Tremellen, K.; Pearce, K. Dysbiosis of Gut Microbiota (DOGMA) - A novel theory for the development of Polycystic Ovarian Syndrome. Med Hypotheses [Internet]. 2012, 79, 104–112. [Google Scholar] [CrossRef]
- Wolf, M.; Sandler, L.; Hsu, K.; Vossen-Smirnakis, K.; Ecker, J.L.; Thadhani, R. First-trimester C-reactive protein and subsequent gestational diabetes. Diabetes Care. 2003, 26, 819–824. [Google Scholar] [CrossRef]
- Du M, Basu A, Fu D, Wu M, Centola M, Jenkins AJ, et al. Serum inflammatory markers and preeclampsia in type 1 diabetes: A prospective study. Diabetes Care. 2013, 36, 2054–2061. [CrossRef]
- Parchim NF, Wang W, Iriyama T, Ashimi OA, Siddiqui AH, Blackwell S, et al. Neurokinin 3 receptor and phosphocholine transferase: Missing factors for pathogenesis of C-reactive protein in preeclampsia. Hypertension. 2015, 65, 430–439. [CrossRef]
- Cotechini, T.; Komisarenko, M.; Sperou, A.; Macdonald-Goodfellow, S.; Adams, M.A.; Graham, C.H. Inflammation in rat pregnancy inhibits spiral artery remodeling leading to fetal growth restriction and features of preeclampsia. J Exp Med. 2014, 211, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Liu S, Hong L, Mo M, Xiao S, Chen C, Li Y, et al. Evaluation of endometrial immune status of polycystic ovary syndrome. J Reprod Immunol [Internet]. 2021;144(February):103282. Available from: https://doi.org/10.1016/j.jri.2021.103282. [CrossRef]
- Matteo M, Serviddio G, Massenzio F, Scillitani G, Castellana L, Picca G, et al. Reduced percentage of natural killer cells associated with impaired cytokine network in the secretory endometrium of infertile women with polycystic ovary syndrome. Fertil Steril [Internet]. 2010, 94, 2222-2227.e3. Available from: http://dx.doi.org/10.1016/j.fertnstert.2010.01.049. [CrossRef]
- Redman, C.W.G.; Sacks, G.P.; Sargent, I.L. Preeclampsia: An excessive maternal inflammatory response to pregnancy. Am J Obstet Gynecol. 4: I).
- Wilson, R.C.; Lo, J.O.; Romero Jimenez, G.; Lindner, J.R.; Slayden, O.D.; Roberts, V.H.J. Utilizing Contrast-Enhanced Ultrasonography with Phosphatidylserine Microbubbles to Detect Placental Inflammation in Rhesus Macaques. Molecules. 2023, 28, 2894. [Google Scholar] [CrossRef] [PubMed]
- Belkacemi, L.; Michael Nelson, D.; Desai, M.; Ross, M.G. Maternal undernutrition influences placental-fetal development. Biol Reprod. 2010, 83, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Francis, E.C.; Dabelea, D.; Boyle, K.E.; Jansson, T.; Perng, W. Maternal Diet Quality Is Associated with Placental Proteins in the Placental Insulin/Growth Factor, Environmental Stress, Inflammation, and MTOR Signaling Pathways: The Healthy Start ECHO Cohort. J Nutr. 2021, 152, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Rosario, F.J.; Powell, T.L.; Jansson, T. Activation of placental insulin and mTOR signaling in a mouse model of maternal obesity associated with fetal overgrowth. Am J Physiol - Regul Integr Comp Physiol. 2016, 310, R87–93. [Google Scholar] [CrossRef] [PubMed]
- Guenther PM, Kirkpatrick SI, Reedy J, Krebs-Smith SM, Buckman DW, Dodd KW, et al. The healthy eating Index-2010 is a valid and reliable measure of diet quality according to the 2010 dietary guidelines for Americans. J Nutr [Internet]. 2014, 144, 399–407. [CrossRef] [PubMed]
- Zarubin, T.; Han, J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005, 15, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Herrock, O.; Deer, E.; LaMarca, B. Setting a stage: Inflammation during preeclampsia and postpartum. Front Physiol. 2023;14(February):1–12.
- Daan NMP, Louwers Y V., Koster MPH, Eijkemans MJC, De Rijke YB, Lentjes EWG, et al. Cardiovascular and metabolic profiles amongst different polycystic ovary syndrome phenotypes: Who is really at risk? Fertil Steril [Internet] 2014, 102, 1444–1451. [CrossRef]
- Jeanes, Y.M.; Reeves, S. Metabolic consequences of obesity and insulin resistance in polycystic ovary syndrome: Diagnostic and methodological challenges. Nutr Res Rev. 2017, 30, 97–105. [Google Scholar] [CrossRef]
- Shroff, R.; Syrop, C.H.; Davis, W.; Van Voorhis, B.J.; Dokras, A. Risk of metabolic complications in the new PCOS phenotypes based on the Rotterdam criteria. Fertil Steril. 2007, 88, 1389–1395. [Google Scholar] [CrossRef] [PubMed]
- Persson, S.; Elenis, E.; Turkmen, S.; Kramer, M.S.; Yong, E.L.; Poromaa, I.S. Higher risk of type 2 diabetes in women with hyperandrogenic polycystic ovary syndrome. Fertil Steril. 2021, 116, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Mumm H, Jensen DM, Sørensen JA, Andersen LLT, Ravn P, Andersen M, et al. Hyperandrogenism and phenotypes of polycystic ovary syndrome are not associated with differences in obstetric outcomes. Acta Obstet Gynecol Scand. 2015, 94, 204–211. [CrossRef] [PubMed]
- de Wilde MA, Lamain-de Ruiter M, Veltman-Verhulst SM, Kwee A, Laven JS, Lambalk CB, et al. Increased rates of complications in singleton pregnancies of women previously diagnosed with polycystic ovary syndrome predominantly in the hyperandrogenic phenotype. Fertil Steril [Internet]. 2017, 108, 333–340. [CrossRef] [PubMed]
- Naver K V., Grinsted J, Larsen SO, Hedley PL, Jørgensen FS, Christiansen M, et al. Increased risk of preterm delivery and pre-eclampsia in women with polycystic ovary syndrome and hyperandrogenaemia. BJOG An Int J Obstet Gynaecol. 2014, 121, 575–581. [CrossRef]
- Jiang R, Yao Y, Wang T, Li B, Jiang P, Wang F, et al. Preeclampsia in pregnant women with polycystic ovary syndrome: risk factor analysis based on a retrospective cohort study. Ginekol Pol. 2023;Oct 20:Epub ahead of print. PMID: 37861221.
- Palomba S, Russo T, Falbo A, Di Cello A, Amendola G, Mazza R, et al. Decidual endovascular trophoblast invasion in women with polycystic ovary syndrome: An experimental case-control study. J Clin Endocrinol Metab. 2012, 97, 2441–2449. [CrossRef] [PubMed]
- Palomba S, Russo T, Falbo A, Di Cello A, Tolino A, Tucci L, et al. Macroscopic and microscopic findings of the placenta in women with polycystic ovary syndrome. Hum Reprod. 2013, 28, 2838–2847. [CrossRef]
- Palomba S, Falbo A, Chiossi G, Tolino A, Tucci L, La Sala GB, et al. Early trophoblast invasion and placentation in women with different PCOS phenotypes. Reprod Biomed Online [Internet]. 2014, 29, 370–381. [CrossRef] [PubMed]
- Sathish Kumar, Geoffrey H Gordon, David H Abbott, Mishra J. Androgens in maternal vascular and placental function: Implications for Preeclampsia Pathogenesis. Reproduction. 2018, 156, R155–67.
- Sathishkumar, K.; Balakrishnan, M.; Chinnathambi, V.; Chauhan, M.; Hankins, G.D.V.; Yallampalli, C. Fetal sex-related dysregulation in testosterone production and their receptor expression in the human placenta with preeclampsia. J Perinatol [Internet]. 2012, 32, 328–335. [Google Scholar] [CrossRef]
- Chinnathambi, V.; Balakrishnan, M.; Ramadoss, J.; Yallampalli, C.; Sathishkumar, K. Testosterone alters maternal vascular adaptations: Role of the endothelial no system. Hypertension. 2013, 61, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Chinnathambi V, Blesson CS, Vincent KL, Saade GR, Hankins GD, Yallampalli C, et al. Elevated testosterone levels during rat pregnancy cause hypersensitivity to angiotensin II and attenuation of endothelium-dependent vasodilation in uterine arteries. Hypertension. 2014, 64, 405–414. [CrossRef] [PubMed]
- Gopalakrishnan K, Mishra JS, Chinnathambi V, Vincent KL, Patrikeev I, Motamedi M, et al. Elevated Testosterone Reduces Uterine Blood Flow, Spiral Artery Elongation, and Placental Oxygenation in Pregnant Rats. Hypertension. 2016, 67, 630–639. [CrossRef] [PubMed]
- Diamond, M.P.; Grainger, D.; Diamond, M.C.; Sherwin, R.S.; Defronzo, R.A. Effects of methyltestosterone on insulin secretion and sensitivity in women. J Clin Endocrinol Metab. 1998, 83, 4420–4425. [Google Scholar] [CrossRef] [PubMed]
- Franks, S.; Gilling-Smith, C.; Watson, H.; Willis, D. Insulin action in the normal and polycystic ovary. Endocrinol Metab Clin North Am. 1999, 28, 361–378. [Google Scholar] [CrossRef] [PubMed]
- González, F. Inflammation in Polycystic Ovary Syndrome: Underpinning of insulin resistance and ovarian dysfunction. Steroids. 2012, 77, 300–305. [Google Scholar] [CrossRef]
- Kelley, A.S.; Smith, Y.R.; Padmanabhan, V. A Narrative Review of Placental Contribution to Adverse Pregnancy Outcomes in Women with Polycystic Ovary Syndrome. J Clin Endocrinol Metab. 2019, 104, 5299–5315. [Google Scholar] [CrossRef]
- Kanbour, S.A.; Dobs, A.S. Hyperandrogenism in Women with Polycystic Ovarian Syndrome: Pathophysiology and Controversies. Androgens. 2022, 3, 22–30. [Google Scholar] [CrossRef]
- Piltonen TT, Ruokojärvi M, Karro H, Kujanpää L, Morin-Papunen L, Tapanainen JS, et al. Awareness of polycystic ovary syndrome among obstetrician-gynecologists and endocrinologists in Northern Europe. PLoS One. 2019, 14, 1–12.
- Tan MY, Syngelaki A, Poon LC, Rolnik DL, O’Gorman N, Delgado JL, et al. Screening for pre-eclampsia by maternal factors and biomarkers at 11–13 weeks’ gestation. Ultrasound Obstet Gynecol. 2018, 52, 186–195. [CrossRef]
- O’Gorman N, Wright D, Syngelaki A, Akolekar R, Wright A, Poon LC, et al. Competing risks model in screening for preeclampsia by maternal factors and biomarkers at 11-13 weeks gestation. Am J Obstet Gynecol. 2016, 214, 103–e1.
- Zeisler H, Llurba E, Chantraine F, Vatish M, Staff AC, Sennström M, et al. Predictive Value of the sFlt-1:PlGF Ratio in Women with Suspected Preeclampsia. N Engl J Med. 2016, 374, 13–22. [CrossRef] [PubMed]
- Zeisler H, Llurba E, Chantraine FJ, Vatish M, Staff AC, Sennström M, et al. Soluble fms-like tyrosine kinase-1 to placental growth factor ratio: ruling out pre-eclampsia for up to 4 weeks and value of retesting. Ultrasound Obstet Gynecol. 2019, 53, 367–375. [CrossRef] [PubMed]
- Bian X, Biswas A, Huang X, Lee KJ, Li TKT, Masuyama H, et al. Short-Term Prediction of Adverse Outcomes Using the sFlt-1 (Soluble fms-Like Tyrosine Kinase 1)/PlGF (Placental Growth Factor) Ratio in Asian Women with Suspected Preeclampsia. Hypertension. 2019, 74, 164–172. [CrossRef] [PubMed]
- Rolnik, D.L.; Nicolaides, K.H.; Poon, L.C. Prevention of preeclampsia with aspirin. Am J Obstet Gynecol [Internet]. 2022, 226, S1108–19. [Google Scholar] [CrossRef] [PubMed]
- Panagodage S, Yong HEJ, Da Silva Costa F, Borg AJ, Kalionis B, Brennecke SP, et al. Low-Dose Acetylsalicylic Acid Treatment Modulates the Production of Cytokines and Improves Trophoblast Function in an in Vitro Model of Early-Onset Preeclampsia. Am J Pathol [Internet]. 2016, 186, 3217–3224. [CrossRef] [PubMed]
- Li, C.; Raikwar, N.S.; Santillan, M.K.; Santillan, D.A.; Thomas, C.P. Aspirin inhibits expression of sFLT1 from human cytotrophoblasts induced by hypoxia, via cyclo-oxygenase 1. Placenta. 2015, 36, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Khanabdali R, Shakouri-Motlagh A, Wilkinson S, Murthi P, Georgiou HM, Brennecke SP, et al. Low-dose aspirin treatment enhances the adhesion of preeclamptic decidual mesenchymal stem/stromal cells and reduces their production of pro-inflammatory cytokines. J Mol Med. 2018, 96, 1215–1225. [CrossRef]
- Tawfeek, M.; Hassan, M.; Ahmed, M.; Mohamed, N.; Ahmed, N. Low dose Aspirin with clomid in pco. Minia J Med Res. 2021, 32, 100–106. [Google Scholar] [CrossRef]
- Yu, Q.; Wang, Z.; Su, F.; Wang, M. Effectiveness and safety of aspirin combined with letrozole in the treatment of polycystic ovary syndrome: a systematic review and meta-analysis. Ann Palliat Med. 2021, 10, 4632–4641. [Google Scholar] [CrossRef]
- Aref, N.K.; Ahmed, W.A.S.; Ahmed, M.R.; Sedik, W.F. A new look at low-dose aspirin: Co-administration with tamoxifen in ovulation induction in anovulatory PCOS women. J Gynecol Obstet Hum Reprod [Internet]. 2019, 48, 673–675. [Google Scholar] [CrossRef]
- Zhao Y, Du B, Jiang X, Ma M, Shi L, Zhang Q, et al. Effects of combining lowdose aspirin with a Chinese patent medicine on follicular blood flow and pregnancy outcome. Mol Med Rep. 2014, 10, 2372–2376. [CrossRef] [PubMed]
- Schisterman EF, Silver RM, Perkins NJ, Mumford SL, Whitcomb BW, Stanford JB, et al. A randomised trial to evaluate the effects of low-dose aspirin in gestation and reproduction: Design and baseline characteristics. Paediatr Perinat Epidemiol. 2013, 27, 598–609. [CrossRef]
- Naimi AI, Perkins NJ, Sjaarda LA, Mumford SL, Platt RW, Silver RM, et al. The effect of preconception-initiated low-dose aspirin on human chorionic gonadotropin-detected pregnancy, pregnancy loss, and live birth: Per protocol analysis of a randomized trial. Ann Intern Med. 2021, 174, 595–601. [CrossRef] [PubMed]
- Mendoza M, Bonacina E, Garcia-Manau P, López M, Caamiña S, Vives À, et al. Aspirin Discontinuation at 24 to 28 Weeks’ Gestation in Pregnancies at High Risk of Preterm Preeclampsia: A Randomized Clinical Trial. Jama. 2023, 329, 542–550. [CrossRef] [PubMed]
- Bonacina E, Garcia-Manau P, López M, Caamiña S, Vives À, Lopez-Quesada E, et al. Mid-trimester uterine artery Doppler for aspirin discontinuation in pregnancies at high risk for preterm pre-eclampsia: Post-hoc analysis of StopPRE trial. BJOG An Int J Obstet Gynaecol. 2023, 00, 1–9.
- Hastie, R.; Tong, S.; Wikström, A.K.; Sandström, A.; Hesselman, S.; Bergman, L. Aspirin use during pregnancy and the risk of bleeding complications: a Swedish population-based cohort study. Am J Obstet Gynecol [Internet]. 2021, 224, 95–e1. [Google Scholar] [CrossRef]
- Bedrick, B.S.; Eskew, A.M.; Chavarro, J.E.; Jungheim, E.S. Self-Administered Questionnaire to Screen for Polycystic Ovarian Syndrome. Women’s Heal Reports. 2020, 1, 566–573. [Google Scholar] [CrossRef]
- Day FR, Hinds DA, Tung JY, Stolk L, Styrkarsdottir U, Saxena R, et al. Causal mechanisms and balancing selection inferred from genetic associations with polycystic ovary syndrome. Nat Commun. 2015, 6, 1–7.
- Lowe, S.A. Bowyer L, McMahon, LP. Morton, LP. North, RA. Paech, MJ. Said J. Guideline for the Management of Hypertensive Disorders of Pregnancy, 2014. Society of Obstetric Medicine of Australia and New Zealand. 2014.
- Ayala, D.E.; Ucieda, R.; Hermida, R.C. Chronotherapy with low-dose aspirin for prevention of complications in pregnancy. Chronobiol Int. 2013;30(1–2):260–79.
Figure 2.
Risk assessment and management of women with PCOS before and during pregnancy.
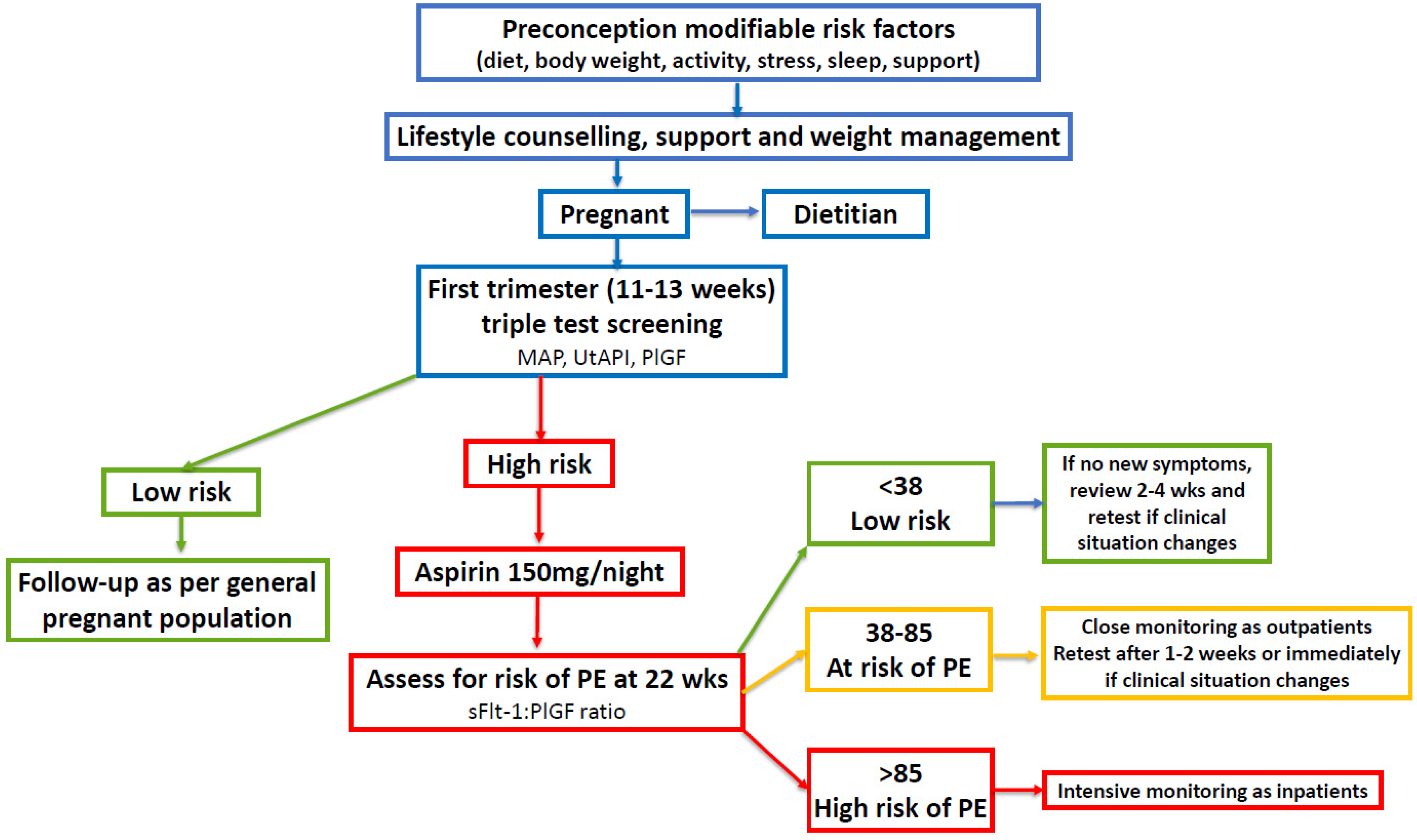

Table 1.
Systematic reviews of studies investigating the risk of pre-eclampsia in women with PCOS.
| Author (Year) | Odds Ratio (95% CI) 1 | Studies | Reference |
|---|---|---|---|
| Boomsma et al (2006) | 3.47 (1.95-6.17) | 8 | [27] |
| Kjerulff et al (2011) | 4.23 (2.77-6.46) | 12 | [28] |
| Qin et al (2013) | 3.28 (2.06-5.22) | 15 | [29] |
| Yu et al (2016) | 2.79 (2.29-3.38) | 25 | [30] |
| Khomami (2019) | 1.87 (1.55-2.25) | 26 | [10] |
| Pan (2021) Riestenberg (2022) Mousa (2023) |
2.07 (1.91-2.24) 2.03 (1.43-2.87) 2.28 (1.88-2.77) |
20 15 36 |
[50] [51] [52] |
1 CI = confidence interval.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions, or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



