Submitted:
26 December 2023
Posted:
27 December 2023
You are already at the latest version
Abstract
Hematopoietic stem cell (HSC) transplantation (HSCT) is used to treat various hematologic disorders. Use of genetically modified mouse models of hematopoietic cell transplantation have been critical in our fundamental understanding of HSC biology and developing approaches for human patients. Pre-clinical studies in animal models provide insight into the journey of transplanted HSCs from infusion to engraftment in bone marrow (BM) niches. Various signaling molecules and growth factors secreted by HSCs and the niche microenvironment play critical roles in homing and engraftment of the transplanted cells. The sustained equilibrium of these chemical and biologic factors ensures that engrafted HSCs generate healthy and durable hematopoiesis. Transplanted healthy HSCs compete with residual host cells to repopulate stem cell niches in the marrow. Stem cell niches, in particular, can be altered by the effects of previous treatments, aging, and paracrine effects of leukemic cells that create inhospitable bone marrow niches that are unfavorable for healthy hematopoiesis. More work to understand how stem cell niches can be restored to favour normal hematopoieis may be key to reducing leukemic relapses following transplant.
Keywords:
Hematopoietic stem cell transplantation
; xeno-transplantation
; homing
; engraftment
; clonal hematopoiesis
; clonal leukemogenesis
; competitive repopulation.
1. Introduction
Hematopoietic stem cell (HSC) transplantation (HCT) has been established as a powerful therapeutic and research tool for close to a century. HCT has been instrumental in understanding blood stem cell biology, both in homeostasis as well as disease conditions. A plethora of hematologic disorders including various forms of leukemia, sickle cell anemia, myelodysplastic syndrome, and bone marrow failure can be treated with HCT. While tremendous advancement has been made in the safety, efficacy, and application of HCT, challenges remain to improve this treatment method further. Preclinical animal models and especially xenotransplant models, where human cells can be transplanted into engineered mice that allow immune tolerance, have played a pivotal role in understanding human HSC biology. In this review we will highlight some key concepts such as HSC homing, engraftment, and competitive repopulation using the pre-clinical lens to allow greater fundamental understanding of the full potential of HCT.
2. Xenotransplantation Models in HCT Research
In recent years mice strains like NOD scid IL2RγNull (NSG) and NOD Rag1Null IL2RγNull (NRG) are being widely used for HCT research. Both strains are based on non-obese diabetic (NOD)/ severe combined immunodeficient (SCID) models. The NSG mice contain a mutation in the γ chain of interleukin 2 (IL-2) receptor rendering it non-functional, which deplete the immune system of these mice severely. These mice have been reported to be able to reconstitute a wider range of human hematopoietic cell types (T and B lymphocytes, myeloid cells, plasmacytoid dendritic cells, and NK cells) compared to the original NOD/SCID models following transplantation of human CD34+ cells. Capacity to support higher levels of human HSC engraftment is also greater in these mice [1,2]. The NSG mice are current gold-standard for HCT research and are widely used across the world. There are several variants of NSG mice that are used for specific research requirements. For example, mutation in Rag1 gene in NRG mice makes them more tolerant to the effects of irradiation (and genotoxic agents), but at the same time maintaining the similar level of human cell engraftment capacity. Therefore, these mice models can be used in cancer radiotherapy research where preservation of bone marrow niche is important for optimum HSC engraftment [3]. Thus, the use of different mice models is crucial in understanding different aspects of HCT process.
While the murine models are well-established and documented in HCT research, other model animals like zebrafish can also provide insight into HSC function and properties. Human HSCs have been xeno-transplanted in zebrafish to interrogate physiological as well as pathological processes involved. Transparency of the zebrafish embryos make them a suitable choice to be used for high resolution imaging techniques to visualize the growth and dynamics of the transplanted cells in-vivo, which is not an option when using murine models [4,5]. Thus, these animal model systems have become invaluable in understanding and studying HCT.
Research use of the animal models and translation of the findings to clinical practice present some unique challenges. The physiological, immunological, and genetic landscape are different between mice and humans, which can reduce the direct transferability of the experimental results [6,7]. For example, the evolving view of hematopoiesis, HSC, and progenitors from hierarchical model to continuum model demands more studies of the process in humans, as the current models are largely based on the studies in murine systems (extensively reviewed by Haas, Trumpp, and Milsom [8]). Moreover, NSG mice have a lifespan of about approximately 90 weeks [1], whereas global average human life expectancy is 73 years in 2023 [9]. Despite these differences, pre-clinical research using these animal models have enriched our understanding of the mechanistic aspects of HSC homing, engraftment, as well as patho-physiological changes in HSCs.
3. Mechanism of HSC Homing: from Tail Vein to Bone Marrow
The process of HSC translocation from the vein (usually tail vein in mice and one of the central veins in humans) to the bone marrow (BM) involves a series of intricate steps that are essential for the successful engraftment of transplanted cells. Upon infusion into the bloodstream, HSCs must navigate through the circulatory system and home to the BM niche, where they can proliferate and differentiate to reconstitute the hematopoietic system [10]. The fate of the infused HSCs is significantly influenced by the BM microenvironment, which consists of niches. Interactions between HSCs and non-stem cell neighbors are crucial for maintaining the quiescent state or promoting self-renewal and proliferation, but a precise understanding of the complex network of signals remains an active area of research. Following HSC transplantation, several critical events occur within the recipient’s body. These include homing and lodgment of transplanted HSCs within the BM niche, initiation of immune reconstitution and the process of inducing immune tolerance (incomplete immune tolerance can manifest as graft rejection or a graft-versus-host response), and early phases of hematopoietic recovery with the early emergence of donor-derived neutrophils and platelets [11] that arise primarily from short-term progenitors, and followed by waves of hematopoietic regeneration derived from HSCs. The BM niche is not a uniform environment, and HSCs are distributed within distinct anatomical locations. HSCs preferentially localize to specific regions within the BM, such as the endosteal surface, perivascular areas, and sinusoidal vessels [12]. The endosteal niche, which is in close proximity to bone surfaces, is characterized by interactions with mesenchymal stromal cells (MSCs) in relative hypoxia, osteoblastic cells and osteoblast-derived factors, such as CXCL12, stem cell factor (SCF), and angiopoietin-1. In contrast, the perivascular niche, surrounding sinusoidal vessels, is associated with interactions with endothelial cells, pericytes, and factors like CXCL12, SCF, and Notch ligands [13]. Notch signaling in HSCs enhances megakaryocyte production and platelet formation through Dll1 ligand, while Notch2 signaling through Jagged-1 generates short-term progenitor cells and long-term HSCs post-myeloablation, hindering myeloid differentiation [14,15]. Studies have shown that perivascular cells expressing Lepr and nestin+ reticular cells, as well as NG2+ pericytes, are associated with the regulation of HSC quiescence and proliferation [16,17]. Moreover, recent studies demonstrated that osteoblasts can expand hematopoietic progenitors in vitro, suggesting that genetic or pharmacologic manipulation of osteoblast numbers correlates with HSC counts in the BM [18]. Osteoblasts have been proposed to support HSC function by forming direct interactions via N-cadherin-mediated adhesion. Activated osteoblasts can produce osteopontin, angiopoietin-1, and thrombopoietin, which limit HSC expansion and contribute to HSC quiescence [19]. Furthermore, bone resorption and calcium release by osteoclasts also promote HSC maintenance and localization to the endosteal region. CXCL12-abundant reticular cells contact HSCs predominantly near sinusoids in endosteal and non-endosteal marrow [20]. Beside cellular components of the BM niche, innervation by the sympathetic nervous system regulates HSC mobilization through circadian release of noradrenaline, which modulates CXCL12 expression in the BM. The physical association between nerves and BM vasculature supports the importance of a vascular niche for HSC localization.
4. Role of Bone Marrow Niche in HSC Homing
Upon reaching the BM, the HSCs adhere to the endothelium of the BM vasculature, extravasate, and migrate to the BM stroma. The initial period of hematopoietic recovery is characterized by gradual reconstitution of donor-derived hematopoiesis with the emergence of progeny from the abundant short-term progenitors that have limited proliferative capacity. Hematopoiesis emerges in waves with a dynamic process that yields cells derived from increasingly rare progenitors with greater differentiation capacity, ending with everlasting multilineage hematopoiesis derived from HSCs with robust self-renewal capacity.
Homing of HSCs to the bone marrow stem cell niches is a crucial step towards successful engraftment after HSC transplantation and relies on intracellular signaling and interactions between chemokines, chemokine receptors, adhesion molecules, and proteases. E-endothelial and P-endothelial selectins are essential for cell movement in BM micro-vessels. Intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) play key roles in HSC homing [21,22], with α4β1/VLA-4 integrin and lectins playing primary roles in HSC attachment to marrow stromal cells [23,24]. Expression of the stromal-cell-derived factor-1 (SDF-1) ligand [25,26], also known as CXC chemokine ligand (CXCL)12, has been found to be increased in the BM microenvironment following the conditioning regimens for HSC transplantation. SDF-1 is a chemokine isolated from stromal fibroblasts and abundantly expressed by osteoblasts, endothelial cells, and a subset of reticular cells in the osteoblast and vascular niches of the BM. It interacts with the G-protein-coupled receptor CXCR4 to promote HSC quiescence and survival [27]. Activation of the CXCR4 receptor by SDF-1 has been one of the most studied transductional axes in recent years owing to its fundamental importance in regulating HSC trafficking to and from the BM. The interaction between SDF-1 and CXCR4 triggers chemotaxis via intracellular GTPase proteins, which are downregulated and ubiquitinated by E3 ubiquitin ligase [28]. Extracellular nucleotides (eNTPs) such as adenosine triphosphate (ATP), uridine triphosphate (UTP) [29], and sphingosine-1-phosphate (S1P) [30] act as potent chemotactic factors in modulating HSC migration in the presence of SDF-1. Pre-treatment with eUTP significantly increased the homing of HSCs to the BM, as demonstrated in immunodeficient mice. eNTPs act through P2 nucleotide receptors (P2Rs), particularly P2YRs, which activate their signal transduction pathways via phospholipase C or adenylate cyclase [29,31]. Although the influence of SDF-1 on HSC chemotactic responses is well established, its role in molecular pathways during the early stages of homing remains a contentious issue. Evidence suggests that HSCs can migrate to the BM independently of the SDF-1-CXCR4 axis. HSC homing in a murine model refractory to SDF-1 by incubation and co-injection with AMD3100 (now also called ‘plerixafor’: a CXCR4 receptor antagonist) showed normal or only slightly reduced BM cellularity. Studies have also shown CXCR4 knock down using an SDF-1 intracellular cytokine strategy were competent for engraftment [32,33].
Successfully engrafted cells can be of various potential in their ability of renewal, differentiation, and their preference for localization within specific BM niche compartments. Multiple studies using fluorescent-labeled monoclonal antibodies and fluorescence-activated cell sorting (FACS) have characterized the multilineage repopulation potential of different marrow cell populations. Long-term stem cells (LT-HSCs) are multipotent cells that self-renew to sustain the stem cell pool and differentiate into short-term HSCs (ST-HSCs) or lineage-restricted progenitors. These cells undergo extensive proliferation and differentiation to produce terminally differentiated hematopoietic cells [34]. Both ST-HSCs and LT-HSCs ’home’ to the bone-marrow microenvironment, where they self-renew and differentiate as needed [35]. Tracking the transplanted cells and their progenies with the murine system using bromo-deoxyuridine (BrdU) can provide valuable information on the homing capabilities of ST-HSC, LT-HSC, and their differentiation dynamics. BrdU can be incorporated into the DNA of replicating cells during the S phase of the cell cycle, and its detection provides a sensitive measure of cell division, even in rare cell populations like HSCs [36]. HSCs, defined as Ki-67− or BrdU label–retaining CD150+CD48−Lin−CD41− cells, are primarily found in small arterioles of the endosteal region, unsheathed by NG2 (CSPG4)+ pericytes. After activation, these cells move away from the NG2+ periarteriolar niche to the Lepr-expressing perisinusoidal niche [37]. Proliferating HSCs are associated with sinusoids, but their short distances are not statistically different from a random placement. Three-dimensional imaging has shown that HSCs are concentrated in a much larger endosteal fraction, with about 80% lying within 50% of the distance to the bone surface, and only 20% toward the central vein. It remains unclear how the bone influences this HSC distribution, as other confounding structures, such as arterioles, are also concentrated in this area [38,39].
5. Measurements of Engraftment in Mice Models
Measuring the engraftment status require assessment of multiple criteria, as during HSCT not only the HSCs but also the progenitors and mature cells are transfused. The progenitors and mature cells ensure rapid reconstitution of the recipient’s hematopoietic system as the HSCs home in the bone marrow niche, prior to going through the engraftment process of differentiated blood cell formation over the many weeks and months that follow. The more differentiated progenitors engraft quickly and provide initial short-term hematopoiesis limited to their respective lineage in the early days to weeks following myeloablation. ST-HSCs lack the ability to produce detectable GM progeny past 6-12 weeks, followed by other cells with more restricted differentiation potential such as multipotent progenitors (MPPs), common myeloid progenitors (CMPs), and common lymphoid progenitors (CLPs) [40]. Mice models have been pivotal in understanding and distinguishing between short- and long-term engraftment of HSCs, and the phenotype of the cells involved in each. In a recent study, transplantation of Lin-CD34+CD38loCD36- cells in NOD-SCID mice was shown to successfully engraft within 2 weeks and generate myelo-erythoid cells. This class of HSCs can maintain the hematopoietic system for short-term (usually 8 -12 weeks) [41]. Similarly, NSG mice have been used to define LT-HSCs, where CD34+CD38-CD45RA-Thy1+CD49f+ cells were found to maintain hematopoiesis for 16+ weeks [34,42]. Also, different methods have been developed to purify HSCs and isolate them into subpopulations that are easier to quantify based on their differentiation potentials. Purified adult mouse BM cells that yield >25% lymphomyeloid repopulating cells were defined as LT-HSCs, based on their ability to produce more than 1% of circulating WBCs for 16 weeks [43,44]. As observed different intrinsic mechanisms are involved which results in direct and indirect impacts on the engraftment potential. Gradual accumulation of different mutations in HSC clones is one such factors that impact the self-renewal and fate decision potentials of the HSCs.
6. Clonal Hematopoiesis
HSCs accumulate different somatic mutations with the aging of the individual. Once sufficient mutations have been accumulated, certain HSC clones acquire an advantage over others in producing progeny. Thus, the overrepresentation of mature blood cells from the same HSC clone leads to clonal hematopoiesis (CH) [45]. Prevalence of CH is low (~1%) in individuals under 50 years of age, however, it increases 10-fold after the age of 65 [46]. If the CH persist without any clinical consequences it is referred to as clonal hematopoiesis of indeterminate potential (CHIP) [47,48]. Transcription epigenetic regulatory genes such as DNA methyltransferase enzyme DNMT3A and DNA demethylase TET2, as well as the polycomb group protein transcriptional repressor ASXL1 are among the genes which have been found to be mutated in the individuals with CH [49,50,51]. Many factors play a role in the likelihood of accumulating these mutations, including the size of the clone, the number of mutations and the gene affected. These mutations have biases to subsets of HSCs, namely: lymphoid-biased, balanced, and myeloid-biased HSCs as categorized by Cho et al., where clonal composition analysis in the context of aging demonstrated loss of lymphoid-biased HSCs and hyper-prevalence of myeloid-biased HSCs [52]. This shift in HSC composition may be related to the impaired functions of the HSCs with aging. Thus, understanding the complex biology of normal HSCs and the progressive changes that give rise to clonal hematopoiesis and the various activity of HSPC pre- and post-transplantation could provide clarity on the biology of HSCs.
Developmental progression of hematopoietic cells depends on the functional contributions of key genes with an intrinsic regulation of short- and long-term renewal involving distinct processes. Studies have shown a consistency of the repopulation behaviour of clonally amplified cells both in-vivo and in-vitro clones. These clones exhibit a high degree of similarity suggesting a pre-set mechanism driving the changes which requires further exploration [53]. Barcoded vector libraries and retroviral integration sites can be utilized in animal models to better track the HSPC clones upon transplantation in animal models [54]. Recently, selective expansion of CRISPR-edited adult and umbilical cord blood derived CD34+ cell clones were demonstrated in murine models when ASXL1 function was lost [55]. These findings add to the complex regulation HSC biology that contributes to CH development.
CH has a wide range of physiological effects and has been identified to correlate with illnesses such as cardiovascular disease (CVD), due to correlation with an increase in inflammatory state and the susceptibility to coronary calcification, therefore, a higher rate of atherosclerosis in both human and mice [56]. This makes CH a useful biomarker for identifying various risk factors for diseases. Given these considerations regarding multitude of patho-physiological changes accumulated in HSCs with aging, younger donors could be preferable than aged donors in the context of HCT to maximize the engraftment potential, hematopoietic system reconstitution, and improvement of the BM microenvironment in a competitive manner in the recipient [57].
7. Competitive Repopulation
Healthy HSCs compete for space and resources and thus ensure their engraftment in the BM niche following transplantation (Figure 1) as evidenced by nonmyeloablated murine studies [58,59,60]. Although, a recent study using CD45.1 and CD45.2 mice provides evidence of empty niche locations which allow transplanted HSCs to engraft without directly competing with already established HSCs in the niche [61]. Regardless of niche space availability, CXCL12 and SCF secreted by CXCL12 abundant reticular cells and endothelial cells respectively help attract the transplanted HSCs to engraft in the niche in a competitive manner [62,63]. This physiological balance is disrupted in leukemia where leukemic stem cells (LSCs) start releasing increased amount of G-CSF, IL-1α, MIP-1β, and MIP-2 along with reduction in CXCL12 expression [64]. Moreover, leukemic myeloid cells can stimulate the MSCs to overproduce osteoblastic lineage cells (OBC) which have reduced expression of HSC retention factors like Lepr, CXCL12, N-cadherin, SCF, ANGPT1, and SLIT2. These OBCs simultaneously downregulate quiescence-enforcing TGFB1 while upregulating myeloid-promoting TGFB2 [65]. The combination of these changes in the LSCs and BM niche environment creates a hostile environment for normal HSCs, while providing the LSCs a competitive advantage. Therefore, successful treatment of leukemia and relapse prevention require improved engraftment of healthy HSCs as well as reversal of LSC-altered BM niche environment. Healthy HSCs rely on the optimum BM microenvironment condition for effective homing and engraftment. Therefore, different pre- and post-transplantation interventions can be considered to improve these aspects of HCT. For instance, co-infusion of MSCs, injection of G-CSF and GM-CSF as well as N-acetyl-L-cysteine (NAC) have been shown to restore the health of BM microenvironment [66,67,68]. These strategies may enhance the competitive repopulation capacity of the donor cells. Improving the competitive repopulation capacity of the transplanted HSCs can improve the patient outcome when using HCT to in the treatment of different diseases.
8. Long-Term Engraftment and its Influence on Leukemia Relapse
Despite recent advancement in leukemia treatment protocols, relapse of the disease remains a major cause of patient mortality to date [69,70,71]. The relapse is cause by a combination of factors that results from LSC properties as well as altered BM niche environment. Cellular signaling pathways like Wnt/β-catenin is activated by upregulation of FOXM1 that leads to β-catenin stabilization and therefore quiescence of LSCs [72]. On the other hand, activation of hedgehog pathway in leukemia can accelerate the disease pathogenesis [73]. Similarly, the BM niche also contributes to maintaining LSCs through CXCL12 by MSCs. CXCL12 has been shown to be responsible for LSC quiescence as well as resistance to tyrosine kinase inhibitor treatment [74]. Given the quiescent and treatment resistant nature of LSCs, one strategy to treat leukemia and minimize the chance of subsequent relapse would be to completely deplete the BM niche of LSCs and then perform a high-dose HSCT. Plerixafor’s ability of competitively binding to CXCR4 can be leveraged to displace the LSCs from BM niche in this strategy. Moreover, plerixafor treatment has shown to increase chemosensitivity of these LSCs [75,76]. This treatment in combination with established chemo- and radio-therapies has the potential to deplete the BM niche of the refractory/ quiescent LSCs. Subsequent high-dose HSCT then can repopulate the BM niche with healthy HSCs with long-term engraftment and reduce the chance of disease relapse. Strategies like these that aim to remodel pathological state of the BM niche could be worthwhile to explore in the future to develop robust treatments of hematological malignancies while improving engraftment.
9. Strategies to Improve Engraftment
Over the last two decades, the overall 3-year mortality of the recipients of HCT was about 50% due to various factors related to either the donor or the recipient [77]. Therefore, in the exploration to improve engraftment success, studies investigated various avenues including usage of better HSC sources, optimization of the stem cell numbers through mobilization procedures and the identification and enhancement of different signaling pathways that are involved in engraftment. Different sources of HSPCs for transplant have different engraftment potentials as well as advantages and disadvantages associated with them. For example, cells sourced from BM has a higher engraftment potential compared to the ones from umbilical cord blood (CB), as BM source contains higher HSPC dosage. However, finding rare types of human leukocyte antigen (HLA) matched donor can be a limiting factor for the BM source. Using CB as a source in these instances can be a good alternative with the added benefit of decreased incidence of GVHD [78,79]. Another source of HSCs is peripheral blood. HSC mobilizers like G-CSF is used to increase the HSC numbers in peripheral blood. Moreover, combining G-CSF with CXCR4 blockers like Plerixafor to has been proven to increase mobilization efficiency [80,81]. These strategies can mobilize sufficient dosage of cells for successful HCT. Cell sorting is another method to consider in this context to infuse HSC enriched population which may provide better engraftment in the long-term. Moreover, HSCs are rare populations and sufficient quantities are difficult to obtain. Therefore, ex-vivo platforms using a combination of small molecules in a synergistic manner has been shown to improve the HSC expansion and overcame these limitations [82]. In this context, route of delivery of the cells for HCT is of critical importance. Opting for direct inject of the HSPCs in the BM can bypass some of the elements of homing process and therby improve engraftment efficiency even where there is a low cell dosage from donor [83]. Another strategy to improve engraftment is the use of gene editing techniques like CRISPR-Cas9. For example, HBB, HBG1/2, and BCL11A genes for sickle cell disease and CCR5 gene for acquired immunodeficiency syndrome have been tested in this context. Gene edited hematopoietic stem and progenitor cells (HSPCs) have been found to engraft earlier than non-edited ones. However, persistence of the gene edited HSPCs in the recipient needs to be improved for this strategy to be used it as a standard treatment protocol in clinic [84].
Furthermore, the identification of different signaling pathways that are involved in engraftment provides an understanding of how it can be enhanced. The mechanism involving leukocyte migration and adhesion used by LT-HSC and ST-HSC are the ligand for E-selectin, sailyl Lewis-X (sLex), and the receptor for SDF-1, CXCR4 chemokine receptor [42]. SDF-1 belongs to α-chemokines which function as a chemoattractant for both committed and primitive hematopoietic progenitors, and a decrease in its level was shown to delay engraftment [85]. In addition, multiple studies have focused on factors that lead to stem cell aging to better understand factors that limit engraftment and may contribute to poor graft function after transplant. For instance, cyclin-dependent kinase inhibitor p16INK [4]a expression has been found to be increased with aging. p16Ink [4]a deficient aged HSC demonstrated rejuvenation in cell-cycle activity and engraftment capacity [86]. Other studies reported regulatory role of p19ARF on p53 stability through the inactivation of the ubiquitin ligase Mdm2. Moreover, mice overexpressing truncated isoforms of p53 (DNp53) or the short isoform of p53 (p44) exhibited accelerated aging and reduced HSC engraftment capabilities [87,88]. Furthermore, novel strategies like collecting the donor cells in hypoxic condition prior to transplant [89] as well as treatment of donor cells to short-term hyperthermia [90] have been shown to improve engraftment in murine studies. Knowledge of these intra- and extra-cellular factors gives a better understanding of how the HSC fate is regulated and thus can help optimize HCT strategies to improve engraftment. Table 1 provides a comprehensive list of different factors/ molecules, the study models, and the impact of the listed factor/ molecules on engraftment.
10. Conclusions
In this review we have explored different xeno-transplantation models in HCT research as well as HSC homing which allows HSCs to reach the bone marrow microenvironment, engraft, and proliferate. The anatomy and the various cellular components of BM niche plays a pivotal role to ensure efficient engraftment and maintenance of the transfused HSCs. In this context various animal models are used to ask and understand the important questions about HSC biology. While these xeno-transplantation models have advanced our understanding of HSC and HCT, there are fundamental differences with humans in terms of their genetic make-up and life spans. Therefore, interpreting these pre-clinical research data in clinical context requires considerations of these differences. Despite these short comings, the pre-clinical animal studies provide a better understanding of clinically relevant aspects of HCT and engraftment, such as CH and competitive repopulation. Finally, we highlighted current research and potential strategies to improve HCT success rate. These strategies include but are not limited to the use of various HSC sources with different engraftment potentials, ex-vivo expansion platforms, modifications of different chemokines that alter HSC engraftment capabilities, and the novel CRISPR-Cas9 gene editing technique. The knowledge from the pre-clinical studies continues to clarify the complex interplay of different factors affecting HCT and improves safety and efficacy of this widely used treatment modality.
References
- Shultz, L. D. et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol 174, 6477–6489 (2005). [CrossRef]
- Ishikawa, F. et al. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor {gamma} chain(null) mice. Blood 106, 1565–1573 (2005). [CrossRef]
- Pearson, T. et al. Non-obese diabetic-recombination activating gene-1 (NOD-Rag1 null) interleukin (IL)-2 receptor common gamma chain (IL2r gamma null) null mice: a radioresistant model for human lymphohaematopoietic engraftment. Clin Exp Immunol 154, 270–284 (2008). [CrossRef]
- Hamilton, N., Sabroe, I. & Renshaw, S. A. A method for transplantation of human HSCs into zebrafish, to replace humanised murine transplantation models. F1000Res 7, 594 (2018). [CrossRef]
- Konantz, M., Müller, J. S. & Lengerke, C. Zebrafish Xenografts for the In Vivo Analysis of Healthy and Malignant Human Hematopoietic Cells. Methods Mol Biol 2017, 205–217 (2019). [CrossRef]
- Hopper, S. E. et al. Comparative Study of Human and Murine Aortic Biomechanics and Hemodynamics in Vascular Aging. Front Physiol 12, 746796 (2021). [CrossRef]
- Bjornson-Hooper, Z. B. et al. A Comprehensive Atlas of Immunological Differences Between Humans, Mice, and Non-Human Primates. Front Immunol 13, 867015 (2022). [CrossRef]
- Haas, S., Trumpp, A. & Milsom, M. D. Causes and Consequences of Hematopoietic Stem Cell Heterogeneity. Cell Stem Cell 22, 627–638 (2018). [CrossRef]
- Life expectancy at birth (years). https://www.who.int/data/gho/data/indicators/indicator-details/GHO/life-expectancy-at-birth-(years).
- Calvi, L. M. & Link, D. C. The hematopoietic stem cell niche in homeostasis and disease. Blood 126, 2443–2451 (2015). [CrossRef]
- Ratajczak, M. Z. & Suszynska, M. Emerging Strategies to Enhance Homing and Engraftment of Hematopoietic Stem Cells. Stem Cell Rev Rep 12, 121–128 (2016). [CrossRef]
- Zhao, M. & Li, L. Dissecting the bone marrow HSC niches. Cell Research 2016 26:9 26, 975–976 (2016). [CrossRef]
- Bigas, A. & Espinosa, L. Hematopoietic stem cells: to be or Notch to be. Blood 119, 3226–3235 (2012). [CrossRef]
- Varnum-Finney, B. et al. Notch2 governs the rate of generation of mouse long-and short-term repopulating stem cells. J Clin Invest 121, (2011). [CrossRef]
- Maillard, I. et al. Canonical notch signaling is dispensable for the maintenance of adult hematopoietic stem cells. Cell Stem Cell 2, 356–366 (2008). [CrossRef]
- Xiao, Y. et al. Current insights into the bone marrow niche: From biology in vivo to bioengineering ex vivo. Biomaterials 286, 121568 (2022). [CrossRef]
- Boulais, P. E. & Frenette, P. S. Making sense of hematopoietic stem cell niches. Blood 125, 2621–2629 (2015). [CrossRef]
- Calvi, L. M. et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 425, 841–846 (2003). [CrossRef]
- Hosokawa, K. et al. Cadherin-based adhesion is a potential target for niche manipulation to protect hematopoietic stem cells in adult bone marrow. Cell Stem Cell 6, 194–198 (2010). [CrossRef]
- Zhang, J. et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 2003 425:6960 425, 836–841 (2003). [CrossRef]
- Frenette, P. S., Subbarao, S., Mazo, I. B., Von Andrian, U. H. & Wagner, D. D. Endothelial selectins and vascular cell adhesion molecule-1 promote hematopoietic progenitor homing to bone marrow. Proc Natl Acad Sci U S A 95, 14423–14428 (1998). [CrossRef]
- Mazo, I. B. et al. Hematopoietic Progenitor Cell Rolling in Bone Marrow Microvessels: Parallel Contributions by Endothelial Selectins and Vascular Cell Adhesion Molecule 1. Journal of Experimental Medicine 188, 465–474 (1998). [CrossRef]
- Ibbotson, G. C. et al. Functional α4-integrin: A newly identified pathway of neutrophil recruitment in critically ill septic patients. Nature Medicine 2001 7:4 7, 465–470 (2001). [CrossRef]
- Peled, A. et al. The chemokine SDF-1 activates the integrins LFA-1, VLA-4, and VLA-5 on immature human CD34+ cells: role in transendothelial/stromal migration and engraftment of NOD/SCID mice. Blood 95, 3289–3296 (2000).
- Lapidot, T., Dar, A. & Kollet, O. How do stem cells find their way home? Blood 106, 1901–1910 (2005). [CrossRef]
- Plett, P., Frankovitz, S. M., Wolber, F. M., Abonour, R. & Orschell-Traycoff, C. M. Treatment of circulating CD34+ cells with SDF-1α or anti-CXCR4 antibody enhances migration and NOD/SCID repopulating potential. Exp Hematol 30, 1061–1069 (2002). [CrossRef]
- Sugiyama, T., Kohara, H., Noda, M. & Nagasawa, T. Maintenance of the Hematopoietic Stem Cell Pool by CXCL12-CXCR4 Chemokine Signaling in Bone Marrow Stromal Cell Niches. Immunity 25, 977–988 (2006). [CrossRef]
- Marchese, A. et al. The E3 ubiquitin ligase AIP4 mediates ubiquitination and sorting of the G protein-coupled receptor CXCR4. Dev Cell 5, 709–722 (2003). [CrossRef]
- Rossi, L. et al. The extracellular nucleotide UTP is a potent inducer of hematopoietic stem cell migration. Blood 109, 533–542 (2007). [CrossRef]
- Adamiak, M. et al. Evidence for the involvement of sphingosine-1-phosphate in the homing and engraftment of hematopoietic stem cells to bone marrow. Oncotarget 6, 18819–18828 (2015). [CrossRef]
- Di Virgilio, F. et al. Nucleotide receptors: an emerging family of regulatory molecules in blood cells. Blood 97, 587–600 (2001). [CrossRef]
- Liles, W. C. et al. Mobilization of hematopoietic progenitor cells in healthy volunteers by AMD3100, a CXCR4 antagonist. Blood 102, 2728–2730 (2003). [CrossRef]
- Onai, N. et al. Impairment of lymphopoiesis and myelopoiesis in mice reconstituted with bone marrow–hematopoietic progenitor cells expressing SDF-1–intrakine. Blood 96, 2074–2080 (2000).
- NOTTA, F. et al. Isolation of Single Human Hematopoietic Stem Cells Capable of Long-Term Multilineage Engraftment. Science (American Association for the Advancement of Science) 333, 218–221 (2011). [CrossRef]
- Al-Amoodi, A. S. et al. Refining the migration and engraftment of short-term and long-term HSCs by enhancing homing-specific adhesion mechanisms. Blood Adv 6, 4373–4391 (2022). [CrossRef]
- Mathe et al 1965.
- Kunisaki, Y. et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 2013 502:7473 502, 637–643 (2013). [CrossRef]
- Chen, J., Hendriks, M., Chatzis, A., Ramasamy, S. K. & Kusumbe, A. P. Bone Vasculature and Bone Marrow Vascular Niches in Health and Disease. Journal of Bone and Mineral Research 35, 2103–2120 (2020). [CrossRef]
- Zhao, M. & Li, L. Dissecting the bone marrow HSC niches. Cell Research 2016 26:9 26, 975–976 (2016). [CrossRef]
- D, B., DJ, R. & IL, W. Hematopoietic stem cells: the paradigmatic tissue-specific stem cell. Am J Pathol 169, 439–443 (2006). [CrossRef]
- Mazurier, F., Doedens, M., Gan, O. I. & Dick, J. E. Rapid myeloerythroid repopulation after intrafemoral transplantation of NOD-SCID mice reveals a new class of human stem cells. Nat Med 9, 959–963 (2003). [CrossRef]
- Al-Amoodi, A. S. et al. Refining the migration and engraftment of short-term and long-term HSCs by enhancing homing-specific adhesion mechanisms. Blood Adv 6, 4373–4391 (2022). [CrossRef]
- Uchida, N., Dykstra, B., Lyons, K. J., Leung, F. Y. K. & Eaves, C. J. Different in vivo repopulating activities of purified hematopoietic stem cells before and after being stimulated to divide in vitro with the same kinetics. Exp Hematol 31, 1338–1347 (2003). [CrossRef]
- Dykstra, B. et al. High-resolution video monitoring of hematopoietic stem cells cultured in single-cell arrays identifies new features of self-renewal. Proc Natl Acad Sci U S A 103, 8185 (2006). [CrossRef]
- Jaiswal, S. & Ebert, B. L. Clonal hematopoiesis in human aging and disease. Science 366, (2019). [CrossRef]
- Genovese, G. et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 371, 2477–2487 (2014). [CrossRef]
- Skead, K. et al. Interacting evolutionary pressures drive mutation dynamics and health outcomes in aging blood. Nat Commun 12, (2021). [CrossRef]
- Silver, A. J. & Jaiswal, S. Clonal hematopoiesis: Pre-cancer PLUS. Adv Cancer Res 141, 85–128 (2019). [CrossRef]
- Busque, L. et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet 44, 1179–1181 (2012). [CrossRef]
- Steensma, D. P. et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 126, 9–16 (2015). [CrossRef]
- Xie, M. et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 20, 1472–1478 (2014). [CrossRef]
- Cho, R. H., Sieburg, H. B. & Muller-Sieburg, C. E. A new mechanism for the aging of hematopoietic stem cells: aging changes the clonal composition of the stem cell compartment but not individual stem cells. Blood 111, 5553–5561 (2008). [CrossRef]
- Dykstra, B. et al. Long-term propagation of distinct hematopoietic differentiation programs in vivo. Cell Stem Cell 1, 218–229 (2007). [CrossRef]
- Biasco, L. et al. In Vivo Tracking of Human Hematopoiesis Reveals Patterns of Clonal Dynamics during Early and Steady-State Reconstitution Phases. (2016). [CrossRef]
- Tothova, Z. et al. Multiplex CRISPR/Cas9-Based Genome Editing in Human Hematopoietic Stem Cells Models Clonal Hematopoiesis and Myeloid Neoplasia. Cell Stem Cell 21, 547-555.e8 (2017). [CrossRef]
- Jaiswal, S. et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med 377, 111–121 (2017). [CrossRef]
- Yuan, N. et al. Young donor hematopoietic stem cells revitalize aged or damaged bone marrow niche by transdifferentiating into functional niche cells. Aging Cell 22, (2023). [CrossRef]
- Stewart, F., Crittenden, R., Lowry, P. & Pearson-White, S. Long-term engraftment of normal and post-5-fluorouracil murine marrow into normal nonmyeloablated mice. (1993).
- Brecher, G., Ansell, J. D., Micklem, H. S., Tjio, J. H. & Cronkite, E. P. Special proliferative sites are not needed for seeding and proliferation of transfused bone marrow cells in normal syngeneic mice. Proc Natl Acad Sci U S A 79, 5085–5087 (1982). [CrossRef]
- Saxe, D., Boggs, S., hematology, D. B.-E. & 1984, undefined. Transplantation of chromosomally marked syngeneic marrow cells into mice not subjected to hematopoietic stem cell depletion. europepmc.orgDF Saxe, SS Boggs, DR BoggsExperimental hematology, 1984•europepmc.org.
- Shimoto, M., Sugiyama, T. & Nagasawa, T. Numerous niches for hematopoietic stem cells remain empty during homeostasis. Blood 129, 2124–2131 (2017). [CrossRef]
- Sugiyama, T., Kohara, H., Noda, M., Immunity, T. N.- & 2006, undefined. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. cell.comT Sugiyama, H Kohara, M Noda, T NagasawaImmunity, 2006•cell.com. [CrossRef]
- Ding, L., Saunders, T., Enikolopov, G., Nature, S. M.- & 2012, undefined. Endothelial and perivascular cells maintain haematopoietic stem cells. nature.comL Ding, TL Saunders, G Enikolopov, SJ MorrisonNature, 2012•nature.com. [CrossRef]
- Zhang, B. et al. Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell 21, 577–592 (2012). [CrossRef]
- Schepers, K. et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 13, 285–299 (2013). [CrossRef]
- Li, T. et al. Efficacy and safety of mesenchymal stem cells co-infusion in allogeneic hematopoietic stem cell transplantation: a systematic review and meta-analysis. Stem Cell Res Ther 12, (2021). [CrossRef]
- Arora, M. et al. Randomized comparison of granulocyte colony-stimulating factor versus granulocyte-macrophage colony-stimulating factor plus intensive chemotherapy for peripheral blood stem cell mobilization and autologous transplantation in multiple myeloma. Biology of Blood and Marrow Transplantation 10, 395–404 (2004). [CrossRef]
- Hu, L. et al. Antioxidant N-acetyl-L-cysteine increases engraftment of human hematopoietic stem cells in immune-deficient mice. Blood 124, e45–e48 (2014). [CrossRef]
- Maude, S. L. et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 371, 1507–1517 (2014). [CrossRef]
- Hu, Y. et al. CRISPR/Cas9-Engineered Universal CD19/CD22 Dual-Targeted CAR-T Cell Therapy for Relapsed/Refractory B-cell Acute Lymphoblastic Leukemia. Clin Cancer Res 27, 2764–2772 (2021). [CrossRef]
- Ravandi, F. et al. Updated results from phase I dose-escalation study of AMG 330, a bispecific T-cell engager molecule, in patients with relapsed/refractory acute myeloid leukemia (R/R AML). 10.1200/JCO.2020.38.15_suppl.7508 38, 7508-7508 (2020). [CrossRef]
- Sheng, Y. et al. FOXM1 regulates leukemia stem cell quiescence and survival in MLL-rearranged AML. Nat Commun 11, (2020). [CrossRef]
- Zhao, C. et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 458, 776–779 (2009). [CrossRef]
- Agarwal, P. et al. Mesenchymal niche-specific expression of Cxcl12 controls quiescence of treatment-resistant leukemia stem cells. Cell Stem Cell 24, 769 (2019). [CrossRef]
- Borthakur, G. et al. Phase 1 study of combinatorial sorafenib, G-CSF, and plerixafor treatment in relapsed/refractory, FLT3-ITD-mutated acute myelogenous leukemia patients. Am J Hematol 95, 1296–1303 (2020). [CrossRef]
- Mori, T. et al. Phase 1 study of plerixafor in combination with total body irradiation-based myeloablative conditioning for allogeneic hematopoietic stem cell transplantation. Int J Hematol 113, 877–883 (2021). [CrossRef]
- Du, J., Yu, D., Han, X., Zhu, L. & Huang, Z. Comparison of Allogeneic Stem Cell Transplant and Autologous Stem Cell Transplant in Refractory or Relapsed Peripheral T-Cell Lymphoma: A Systematic Review and Meta-analysis. JAMA Netw Open 4, (2021). [CrossRef]
- Pidala, J. et al. Race/ethnicity affects the probability of finding an HLA-A, -B, -C and -DRB1 allele-matched unrelated donor and likelihood of subsequent transplant utilization. Bone Marrow Transplant 48, 346–350 (2013). [CrossRef]
- Alotaibi, H. et al. Upfront Alternative Donor Transplant versus Immunosuppressive Therapy in Patients with Severe Aplastic Anemia Who Lack a Fully HLA-Matched Related Donor: Systematic Review and Meta-Analysis of Retrospective Studies, on Behalf of the Severe Aplastic Anemia Working Party of the European Group for Blood and Marrow Transplantation. Transplant Cell Ther 28, 105.e1-105.e7 (2022). [CrossRef]
- Cashen, A. et al. A phase II study of plerixafor (AMD3100) plus G-CSF for autologous hematopoietic progenitor cell mobilization in patients with Hodgkin lymphoma. Biol Blood Marrow Transplant 14, 1253–1261 (2008). [CrossRef]
- Li, C. et al. Single-dose MGTA-145/plerixafor leads to efficient mobilization and in vivo transduction of HSCs with thalassemia correction in mice. Blood Adv 5, 1239 (2021). [CrossRef]
- Manesia, J. K. et al. AA2P-mediated DNA demethylation synergizes with stem cell agonists to promote expansion of hematopoietic stem cells. Cell reports methods 3, 100663 (2023). [CrossRef]
- Gao, J. et al. Enhanced in vivo motility of human umbilical cord blood hematopoietic stem/progenitor cells introduced via intra-bone marrow injection into xenotransplanted NOD/SCID mouse. Exp Hematol 37, 990–997 (2009). [CrossRef]
- Maganti, H. B. et al. Persistence of CRISPR/Cas9 gene edited hematopoietic stem cells following transplantation: A systematic review and meta-analysis of preclinical studies. Stem Cells Transl Med 10, 996–1007 (2021). [CrossRef]
- Ratajczak, M. Z. & Suszynska, M. Emerging Strategies to Enhance Homing and Engraftment of Hematopoietic Stem Cells. Stem Cell Rev Rep 12, 121–128 (2016). [CrossRef]
- Janzen, V. et al. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature 443, 421–426 (2006). [CrossRef]
- Maier, B. et al. Modulation of mammalian life span by the short isoform of p53. Genes Dev 18, 306–319 (2004). [CrossRef]
- Tyner, S. D. et al. p53 mutant mice that display early ageing-associated phenotypes. Nature 415, 45–53 (2002). [CrossRef]
- Mantel, C. R. et al. Enhancing Hematopoietic Stem Cell Transplantation Efficacy by Mitigating Oxygen Shock. Cell 161, 1553–1565 (2015). [CrossRef]
- Capitano, M. L., Hangoc, G., Cooper, S. & Broxmeyer, H. E. Mild Heat Treatment Primes Human CD34(+) Cord Blood Cells for Migration Toward SDF-1α and Enhances Engraftment in an NSG Mouse Model. Stem Cells 33, 1975–1984 (2015). [CrossRef]
Figure 1.
Schematic representation of competitive repopulation assay to estimate the engraftment and hematopoietic system repopulation capacity of donor vs competitor HSCs. Donor and competitor BM cells are distinguished by unique genetic markers. Donor and competitor cells are then mixed and transplanted into lethally irradiated recipient mice. Following approximately 6 months of repopulation period, recipient BM is harvested to assess for cellular composition and contribution of donor vs competitor based on selected genetic markers. Figure created with BioRender.com.
Figure 1.
Schematic representation of competitive repopulation assay to estimate the engraftment and hematopoietic system repopulation capacity of donor vs competitor HSCs. Donor and competitor BM cells are distinguished by unique genetic markers. Donor and competitor cells are then mixed and transplanted into lethally irradiated recipient mice. Following approximately 6 months of repopulation period, recipient BM is harvested to assess for cellular composition and contribution of donor vs competitor based on selected genetic markers. Figure created with BioRender.com.
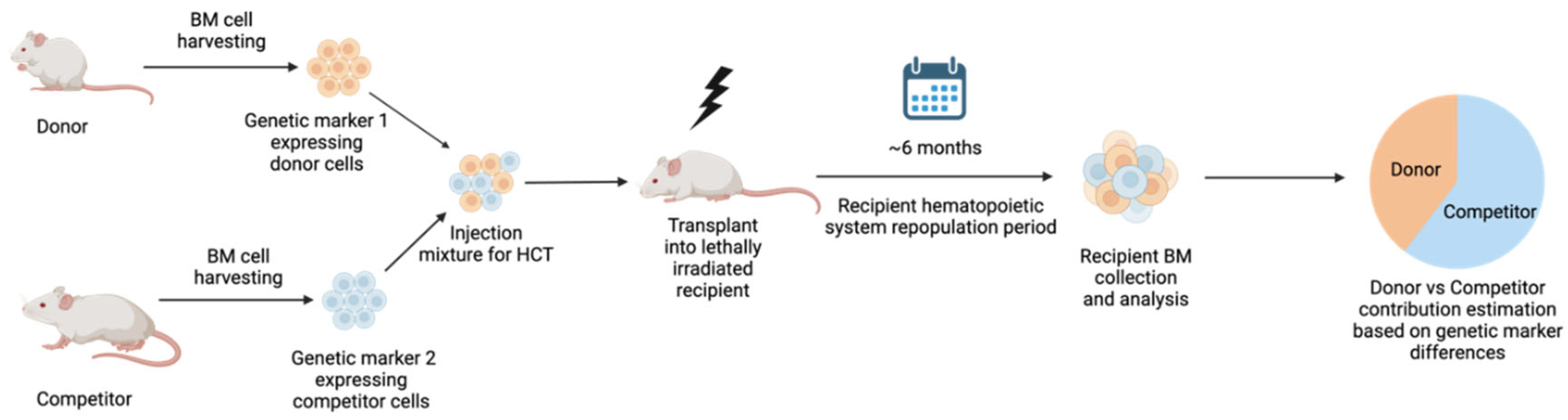

Table 1.
List of factors/ molecules that influence HSC engraftment and the animal models used to study these factors/ molecules.
Table 1.
List of factors/ molecules that influence HSC engraftment and the animal models used to study these factors/ molecules.
| Engraftment Factors | Function | Study model | Influence on engraftment | References |
| Stromal-cell-derived factor-1 (SDF-1) also known as CXCL12 | Chemokine isolated from stromal fibroblasts and abundantly expressed in BM. | NOD/LtSz-scid/scid (NOD/SCID) mice and MxCre-CXCR4f/null mice and C57BL/6 |
Actuate and promotes HSC maintenance and Improves engraftment | Lapidot, T. 2005 Plett, P. et al. 2002 Onai, N. et al. 2000 |
| Notch ligands | Signal through Jagged-1 generates short-term progenitor cells and long-term HSCs post-myeloablation, hindering myeloid differentiation | Transgenic Mice: Mx-Cre+ × ROSADNMAML/+ mice and C57BL/6 (B6, CD45.2+) and (B6-SJL, CD45.1+) |
Support HSC self-renewal and improves engraftment |
Varnum-Finney, B. et al. 2011 Maillard, I. et al. 2008 |
| Lepr and nestin+ reticular cells | Associated with the regulation of HSC quiescence and proliferation | Transgenic Mice: Tie2-cre and leptin receptor (LepR)-cre mice and Col1-caPPR mice |
Improves HSC frequency in the bone marrow | Xiao, Y. et al. 2022 Boulais, P. E. et al. 2015 |
| N-cadherin | Osteoblast direct interactions via N-cadherin-mediated adhesion support HSC function | Transgenic mice: Scl-tTA::TRE-BCR/ABL (BA) double-transgenic mouse - CML | Positively Regulates HSCs in BM niche | Hosokawa, K et al. 2010 Schepers, K et al 2013 |
| Osteopontin, angiopoietin-1, and thrombopoietin | Activated osteoblasts can produce osteopontin, angiopoietin-1, and thrombopoietin, which limit HSC expansion and contribute to HSC quiescence | Transgenic mice: Mx1-Cre+Bmpr1afx mutant mice |
Shown to positively impact HSC regulation | Hosokawa, K. et al. 2010 |
| Intercellular adhesion molecule-1 (ICAM-1) | Play a role in homing through mediating cellular adhesion interaction | Transgenic mice: C57BL/6 and 129S strains P/E-/- (C57/Bl6J×129S) Mice lacking the two selectins (P and E-) |
Positively Regulates HSCs in BM niche |
Frenette, P. S. et al. 1998 |
| Vascular cell adhesion molecule-1 (VCAM-1) | Play a role in homing through mediating rolling and firm adhesion of HPC in BM | Transgenic mice: C57/Bl6J×129S P/E-/- |
Positively Regulates HSCs in BM niche |
Mazo, I. B. et al. 1998 |
| α4β1/VLA-4 integrin and lectins | Primary roles in HSC attachment to marrow stromal cells | NOD/SCID | HSC homing by enabling attachment to the vascular endothelium | Peled et al 2000 |
| Adenosine triphosphate (ATP) and uridine triphosphate (UTP) | Extracellular nucleotide (eNTPs). act as potent chemotactic factors in modulating HSC migration in the presence of SDF-1 | NOD/SCID | UTP and ATP (to a lesser extent) modulate HSC motility and homing to BM niche | Rossi, L. et al. 2007 |
| Sphingosine-1-phosphate (S1P) | Extracellular nucleotide (eNTPs). act as potent chemotactic factors in modulating HSC migration in the presence of SDF-1 | Transgenic: B6.Cg-Tg(UBC-cre/ERT2)1Ejb/J | Homing of HSPC | Adamiak, M. et al. 2015 |
| N-acetyl-L-cysteine (NAC) | Shown to restore the health of BM microenvironment | NOD/SCID and NSG mice | Increase in human HSC engraftment and multilineage hematopoietic differentiation | Hu, L. et al 2014 |
| TGF-B1, TGF-B2, and SLIT2 | TGF-B2 promote myeloid differentiation and TGF-B1/SLIT2 are HSC retention factors, all Support HSC function | BCR/ABL (BA) mice | Regulate quiescence and self-renewal of HSCs | Schepers, K et al 2013 |
BM, Bone marrow; HSC, hematopoietic stem cell; CXC- chemokine ligand 12 (CXCL12)-abundant reticular cells; LepR + cells, leptin receptor (LepR)-positive cells; NG2+ cells, neural–glial antigen 2 (NG2)-positive cells; SCF, Stem Cell Factor; CXCL12, C-X-C Motif Chemokine Ligand 1; Angpt-1, Angiopoietin 1; Vcam1, Vascular cell adhesion protein 1; VLA-4, Very Late Antigen-4; TGF-β1/2, Transforming growth factor beta 1/ 2.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



