
Submitted:
27 December 2023
Posted:
28 December 2023
You are already at the latest version
Abstract
Solid tumors universally possess the ability to evade contact inhibition of proliferation (CIP), a mechanism halting cell proliferation when cell-cell contacts occur. Merlin, an ERM-like protein, crucially regulates CIP and is frequently deactivated in various cancers, indicating its signifi-cance as a tumor suppressor in cancer biology. Despite extensive investigations into Merlin's role in cancer, its lack of intrinsic catalytic activity and frequent conformation changes has made it notoriously challenging to study. To address this challenge, we harnessed innovative luciferase technologies to create and validate a split-luciferase biosensor system. This system enables precise quantification of Merlin's conformation and activity both in vitro and within living cells. This biosensor significantly enhances the study of Merlin's molecular functions, serving as a potent tool to explore its contributions to CIP and tumorigenesis.
Keywords:
Hippo Pathway
; Merlin
; tumor suppressor
; Biosensor
; NanoLuc
; NanoBiT
; Cell contact inhibition
1. Introduction
Merlin (Moesin, Ezrin, Radixin like protein) was initially discovered as the protein product of the NF2 gene in 1993[1,2]. In line with the two-hit model of tumour suppression, biallelic mutation of NF2 leads to the development of neurofibromatosis type II (NF2)[3], a tumorigenic, genetic disease characterized by bilateral schwannoma formation along the vestibulocochlear cranial nerve[4]. NF2 patients also display an elevated risk of developing schwannomas at other locations, or brain tumours of different varieties (i.e., meningiomas and ependymomas)[5]. The penetrance of the disease is nearly 100%, and most patients experience a shortened overall life expectancy and will progress to hearing loss[3,4]. Currently, management of NF2 is primarily focused on surgical intervention. An improved understanding of molecular Merlin function is needed in order to identify opportunities for targeted therapeutic intervention.
In spontaneous tumours, Merlin inactivation is ubiquitous or near-ubiquitous in schwannomas[5,6], and highly frequent in meningiomas and ependymomas[7,8,9,10]. Genetic NF2 mutation also occurs in roughly half of all mesotheliomas, and Merlin loss is thought to contribute greatly to disease progression[11,12]. Less frequently, Merlin is genetically inactivated in a wide variety of other cancers[13,14,15,16]. In mesotheliomas, breast cancers, and prostate cancers without genetic or transcriptional inactivation of NF2, there is evidence of functional Merlin inactivation at the level of post-translational modification[14,17,18]. Additionally, Merlin deficiency may contribute to tumour metastasis[19,20,21], drug resistance[22], and sensitivity to an iron-dependent form of cell death known as ferroptosis[23]. Overall, Merlin appears to be an influential tumour suppressor of general importance in cancer biology. However, the cellular signalling events underlying Merlin’s inhibition of tumour progression are incompletely understood.
In the context of tumour suppression, Merlin coordinates contact inhibition of proliferation (CIP), the mechanism by which intercellular contacts engage signalling to stop proliferative growth of cells. A hallmark feature of solid tumours is the ability to circumvent or overcome CIP[24,25,26]. As such, loss of CIP is regarded as a key event in tumorigenesis. Merlin-deficient cells lose the ability to be contact-inhibited in cell culture and form tumours in vivo, establishing a crucial role for Merlin in tumour suppression [14,21,27]. However, Merlin is a difficult protein to study because of its diverse subcellular localization, lack of intrinsic catalytic activity, and frequent conformation changes. As such, the molecular functions of Merlin in tumour suppressive cell signalling are not fully understood, despite nearly 3 decades of research efforts.
Merlin shares close sequential and structural similarity to the ERM (Ezrin, Radixin, Moesin) family of actin-cytoskeleton linker proteins[28,29,30,31]. Like other ERM proteins, Merlin activity is regulated by a change in conformation. Merlin adopts 2 distinct conformations: a ‘closed’ conformation characterized by an N-to-C terminal interaction, and a more linear ‘open’ conformation[28,32]. In its closed conformation, the intramolecular Merlin association masks binding sites targeting downstream effectors. As such, this conformation is thought to be inactive in tumour suppression, whereas open-form Merlin is hypothesized to be active as a tumour suppressor[8,28,32]. However, there are no crystallographic data of full-length Merlin in either conformation, and speculation regarding Merlin’s conformation-activity relationship has been a subject of intense study and controversy. Unique biochemical techniques must be developed to enable more accurate study of Merlin’s conformation-activity relationship. Merlin’s change in conformation is mediated by phosphorylation at serine-518 (S518). S518 is the best-studied site of post-translational Merlin modification and is phosphorylated by P21-activated kinases (PAK) and protein kinase A (PKA)[33,34,35]. Phosphorylation of S518 promotes closed-conformation, inactive Merlin, although this residue is unlikely to participate directly in the N-to-C terminal binding interface[36]. Instead, phosphorylation is thought to inhibit binding with other upstream regulators of Merlin activity, such as angiomotin, to prevent transition to the open conformation. Due to the well-established influence of phosphorylation at S518 on Merlin activity and purported conformation, phosphodeficient (S518A; A, alanine) and phosphomimetic (S518D; D, aspartic acid) mutants are commonly applied to study Merlin activity. However, there is currently no method to directly monitor the impact of phosphorylation on Merlin’s conformational state.
Furthermore, Merlin is a potent upstream regulator of the Hippo signalling pathway[36,37,38,39,40], an evolutionarily conserved kinase cascade with well-recognized and multifarious roles in protecting against tumorigenesis[41,42,43,44,45]. Merlin potentiates the activity of LATS, the core Hippo kinase[36,46], through a direction interaction that coordinates the colocalization of LATS alongside other Hippo components at the plasma membrane[40]. A 7 amino acid, evolutionarily conserved ‘blue-box’ region in Merlin’s F2 subdomain (Figure 1A) is heavily involved in this interaction[36,40,47,48], and deletion or alanine-substitution of the ‘blue-box’ residues abolish Merlin’s tumour suppressive capacity[49,50]. Moreover, the ‘blue-box’ residues are masked by the C-terminal tail of Merlin in its closed conformation[36,47], and mutations that lock Merlin into a closed conformation are unable to bind LATS and are deficient in tumour suppression[36,46]. Functionally, the Hippo pathway is also well-established as a key downstream signalling mechanism involved in coordination of CIP[51,52,53]. Together, these findings strongly support a crucial role for Merlin’s interaction with LATS as a key mediator of its function.
We have recently developed several bioluminescent biosensors monitoring the levels and activities of the Hippo pathway components (e.g. LATS, YAP/TAZ-TEAD PPI) in vitro and in vivo[54,55,56,57,58,59]. By using these biosensors, we have identified many upstream regulators and small molecule (SM) drugs modulating the Hippo pathway for cancer therapies. In this study, we propose to use similar biosensor system as an improved method to study Merlin activity. In brief, a split luciferase system allows for the development of complementation assays, wherein a functional luciferase is split into 2 non-functional components[59]. The constituents can be fused onto a pair of interacting proteins, and will reconstitute a functional luciferase and luminesce upon protein-protein interaction (PPI) between the interaction pair of interest. This provides a sensitive and accurate method to quantify a given PPI in real-time. Promega has recently engineered NanoBiT: a split luciferase system demonstrating improved sensitivity, thermal stability, and size when compared with previous split-luciferase technologies[60,61]. NanoBiT is comprised of the 18kDa LgBiT subunit and the 1.3 kDa, 11-amino acid SmBiT subunit.
In this work, we develop and validate a split-luciferase system aimed at studying Merlin’s intramolecular association. First, we clone the LgBiT and SmBiT luciferase constituents onto the N and C-terminus, respectively, of full-length Merlin in a unimolecular biosensor system. We hypothesize that, upon N-to-C-terminal interaction and transition to Merlin’s ‘closed’ conformation, this intramolecular biosensor (Mer-Intra-BS) will emit increased luminescent activity, indicative of LgBiT and SmBiT complementation (Figure 1C). In contrast, less luminescent activity will be observed when Merlin exists primarily in an open conformation. This system is validated using mutations to disrupt or promote the binding event of interest and thus change the expected luminescent activity of the biosensors. This biosensor serves as a powerful tool to study Merlin’s molecular function in real time, and can be applied in future work to offer unique insights into the elusive tumour suppressive functions of this protein.
2. Results
Development and Validation of Mer-Intra-BS
LgBiT and SmBiT are cloned onto the N and C-terminus, respectively, of full-length Merlin (Figure 1B). Upon conformation change from open form to an autoinhibited, closed form, we expect the luciferase constituents of this Mer-Intra-BS to reconstitute a functional luciferase and emit light (Figure 1C). In contrast, we anticipate less luminescent activity of the biosensor system when Merlin exists primarily in an open conformation (Figure 1C). As expected, the Mer-Intra-BS showed a dramatic increase in luminescent activity as compared to untransfected (mock) controls, or controls transfected with pBiT1.1-N vector expressing LgBiT alone (Figure 1D). Of note, LgBiT displays a small degree of luminescent activity without needing to complement with SmBiT. However, compared to LgBiT, about 149-fold increase in luminescent activity was observed in the Mer-Intra-BS, demonstrating that luciferase complementation is occurring as predicted. Expression of the Mer-Intra-BS was also verified by western blot (Figure 1E).
After confirming luciferase complementation and successful construction of the Mer-Intra-BS as designed, we next validated the hypothesis that luciferase complementation responds to Merlin’s change of conformational state. To do this, we first generated a mutant biosensor system that is constitutively locked into a closed conformation. Substitution of an alanine (A) at position 585 with a tryptophan (W) residue (A585W) was previously shown to promote interaction between Merlin’s N and C-terminus and stabilize the closed conformation of the protein[36,46]. Therefore, an A585W mutant biosensor system should show increased luminescent activity when compared to an identical wild-type (WT) Mer-Intra-BS under the same promoter. After site directed mutagenesis and cloning of the A585W mutant Mer-Intra-BS, we observe a significant increase in luminescent activity of the A585W biosensor when compared to the WT Mer-Intra-BS (Figure 2A). This increase in luminescent activity did not correlate with increased expression as detected by western blot (Figure 2B). Furthermore, a recent study pinpointed the crucial role of the last two amino acids, glutamic acid (E) and leucine (L), in forming the closed form of Merlin. The Merlin mutant lacking these residues, denoted as ΔEL, adopts an open conformation[46,62]. To explore this phenomenon, we engineered an intramolecular biosensor for the Merlin ΔEL mutant, referred to as Mer-ΔEL-intra-BS. In comparison to the wild-type (WT) control, we observed a significant decrease in luciferase activity for Mer-ΔEL-intra-BS (Figure 2C), despite similar expression levels (Figure 2D). These results provide compelling evidence that the Mer-Intra-BS accurately reflects a conformational shift within living cells.
Next, we validated the Mer-Intra-BS in a cotransfection experiment with PAK1, the best established upstream regulator of Merlin conformation and activity. PAK1 mediated phosphorylation of S518 is thought to promote the closed conformation of the protein, although there is some ambiguity remaining in the field. Thus, in the context of the current Mer-Intra-BS, cotransfection with PAK1 may increase Mer-Intra-BS luciferase activity by promoting LgBiT and SmBiT complementation. Upon cotransfection of the Mer-Intra-BS with a constitutively active (CA) PAK1, but not with a kinase-dead (KD) control, a statistically significant increase in luminescent activity was observed (Figure 3, upper panel). Notably, this increase in luminescent activity was not associated with increased expression of the Mer-Intra-BS as measured by western blot (Figure 3, lower panel), again demonstrating that increased luminescent activity of the Mer-Intra-BS was associated with change of conformation, rather than an alteration of protein stability. This provides strong support for the current model that S518 phosphorylation mediates transition to the closed conformation of Merlin. In combination with the data from the A585W and ΔEL mutant biosensor system, these findings strongly suggest that Mer-Intra-BS can accurately quantify Merlin’s conformational state.
After rigorous validation, we established stable cell lines expressing the Mer-Intra-BS under the control of a Dox-inducible promoter using lentiviral transgene introduction. Both HEK293T and HCT-116 cells were infected with lentivirus carrying the Mer-Intra-BS construct. Similar to the results obtained from transient transfection luciferase assays (Figure 2), the induction of Mer-Intra-BS protein expression by Dox led to a remarkable increase in luminescent activity in these cells (Figure 4), as evidenced by the elevated levels of Mer-Intra-BS protein observed in Figure 4. Live cell bioluminescent imaging.
(BLI) further substantiated these findings, demonstrating a positive correlation between the bioluminescent signal in living cells and cell numbers (Figure 5). Importantly, the establishment of these cell lines expressing Mer-Intra-BS in a stable manner represents a valuable resource for future investigations, facilitating the quantitative assessment of Merlin activity and its role as a tumor suppressor within living cells. By employing these firmly established cell lines, our investigation unveils a dynamic pattern in Mer-Intra-BS signaling. Notably, the signal intensifies during cell proliferation at lower densities (1x104 cells, 40-50% confluency) in HEK293T cells. Intriguingly, despite maintaining comparable expression levels at different cell densities, the signal experiences a significant reduction as cell density increases (2-8x104 cells, 80-100% confluency) (Figure 6).
This observation suggests that the Mer-Intra-BS signal has the capacity to monitor the contact inhibition effect of Merlin. The signal is elevated in its closed form at low cell density and reduced in its open form at high cell density. Furthermore, cell lines maintaining stable expression of Mer-Intra-BS present promising applications in in vivo xenograft mouse models. The integration of bioluminescent imaging and luciferase analysis in these models offers a robust platform for comprehensive assessments of Merlin's activity and its influence on tumor suppression.
In-depth characterization of the Mer-intra-BS was carried out in vitro, involving the purification of a His-tagged Mer-intra-BS fusion protein in bacterial cells, as illustrated in Figure 7A. Notably, luciferase activity exhibited a positive correlation with the quantities of purified His-Mer-intra-BS used in the assays (Figure 7B). This purified biosensor fusion protein stands as a valuable tool for precise quantification and validation of proteins, kinases, or drugs that influence Merlin's intramolecular interaction and activity. Its availability paves the way for comprehensive studies on the modulation of Merlin's function, enabling a deeper understanding of its regulatory mechanisms and potential therapeutic interventions.
3. Discussion
In summary, we have developed and validated a luciferase-based biosensor to study Merlin’s conformation changes and activity. Merlin’s conformation-activity relationship has long been a subject of controversy, and is yet unresolved despite intense biochemical investigation. The Mer-Intra-BS provides strong evidence in support of the current model of Merlin conformation-activity relationship, wherein S518 phosphorylation by PAK promotes the closed-form protein that is inactive in tumour suppression[33,34].
The development and validation of the Mer-Intra-BS present several significant contributions to our current understanding of Merlin biology. Firstly, the creation of a unimolecular Merlin-NanoBiT biosensor stands as an exciting proof of concept. This achievement is particularly noteworthy considering that many other proteins such as the FERM family (e.g. Ezrin and Moesin) also undergo similar N-to-C terminal autoinhibitory conformational changes. Secondly, the data obtained from cotransfection experiments with PAK1 (Figure 3) offer compelling and direct evidence supporting the prevailing consensus model of Merlin's conformation-activity relationship. This model posits that phosphorylation promotes a closed-form, rendering the protein inactive.
Furthermore, the A585W and ΔEL mutants of Mer-intra-BS provide unequivocal evidence confirming that these mutations, now widely employed, indeed represent closed and open forms, respectively, of Merlin conformations. These findings align with and support research results from other studies [36,46]. Thirdly, activation of Merlin by transition from closed to open form has been proposed as a mechanism by which Merlin exerts its tumor suppressor function in regulating CIP [14,21,27]. Our finding using the Mer-Intra-BS provides the first evidence that Merlin is indeed transformed from a closed into an open form during increased cell-cell contract (Figure 6). Lastly, the establishment of two stable cell lines expressing the Mer-Intra-BS provides a valuable toolkit for future investigations focused on characterizing Merlin's intramolecular conformation within living cells and xenograft mouse models. These cell lines offer a robust platform for delving deeper into the intricate dynamics of Merlin's conformational states, facilitating a more comprehensive understanding of its biological functions and opening avenues for potential therapeutic exploration.
Moving forward, there are several promising avenues for exploration utilizing the Merlin biosensor system we have developed. Firstly, functional characterization of cells stably expressing the Mer-Intra-BS is essential to confirm its retention of tumor-suppressive activity. Rigorous testing is warranted to establish the biosensor's efficacy in capturing Merlin's biological functions. Secondly, the creation of additional mutant biosensor systems, incorporating patient-derived substitutions, presents an exciting opportunity to investigate the molecular consequences of these variants on Merlin's intramolecular associations. For instance, certain NF2 patient-derived substitutions have been observed to impede the phosphorylation of YAP, a process normally facilitated by WT Merlin [36]. Employing point-mutant Mer-intra-BS constructs can directly assess whether these substitutions correlate with decreased luciferase activity, shedding light on the functional implications of these mutations.
Moreover, the Mer-intra-BS system holds potential for conducting gain-of-function screens, specifically focusing on kinases that regulate Merlin's intramolecular interaction and tumor suppressor activity. Additionally, the biosensor can be employed in screening endeavors aimed at identifying small molecule drugs capable of disrupting Merlin's intramolecular interactions and activating its tumor suppressor function, a strategy successfully implemented in our previous work [54,55,56,57]. By delving into these research directions, we can gain valuable insights into Merlin's intricate regulatory network and explore novel therapeutic interventions for cancer treatment.
4. Materials and Methods
4.1. Biosensor Design and Construction
Structurally, Merlin consists of a N-terminal FERM domain, a central helical domain, and a C-terminal domain (CTD) (Figure 1A). The Mer-Intra-BS construct consisted of an N-terminal LgBiT constituent, central full-length human Merlin component (accession number NM_000268.3), and C-terminal SmBiT constituent. Human Merlin was amplified by polymerase chain reaction (PCR). To fuse the SmBiT luciferase constituent onto Merlin, a 33-nucleotide sequence encoding SmBiT was included as an overhang on the reverse PCR primer, alongside a DNA segment encoding a flexible glycine-serine (G/S) region that is necessary for efficient luciferase complementation. As such, SmBiT and its G/S linker region were incorporated onto the C-terminus of the Merlin PCR product (Table S1). Following PCR, the Mer-SmBiT product was digested and ligated into the EcoR1/Nhe1 sites of pBiT1.1-N vector (Promega, Wisconsin, USA) containing an N-terminal sequence encoding LgBiT and a G/S linker in frame with the Merlin-SmBiT construct. Thus, a LgBiT-linker-Merlin-linker-SmBiT intramolecular Merlin biosensor was cloned (Figure 1B). This construct was later amplified and cloned into the pcDNA3.1 hygro(+) vector.
4.2. Site-Directed Mutagenesis
4.3. Cell Culture
HEK293T (human embryonic kidney) cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Sigma D6429, Oakville, Canada) containing 10% Fetal Bovine Serum (FBS) and 1% Penicillin/Streptomycin (P/S; Invitrogen, CA, USA). HCT-116 (human colon cancer) cells were cultured in McCoy’s 5A medium (Sigma M4892) supplemented with 10% Fetal Bovine Serum (FBS) and 1% Penicillin/Streptomycin (P/S; Invitrogen, CA, USA). All cells were cultured at 370C with 5% CO2.
4.4. Protein Extraction and Western Blot Analysis
Cells were lysed with 1×Passive Lysis Buffer (1×PLB; Promega) according to the manufacturer’s instructions. For western blotting, membranes were blocked with 5% skim milk for 1 hour at RT, then incubated with anti-Merlin (Cell Signaling #6995; diluted 1:1000), anti-β-actin (Sigma Aldrich #A5441; diluted 1:10,000), or anti-Myc (Cell Signaling #2278; diluted 1:1000) for 1 hour at RT or 12-16 hours at 40C. After washing, membranes were incubated with HRP-conjugated secondary antibodies diluted at 1:2500 for 15 minutes, followed by incubation with ClarityTM Western ECL Substrate (Bio-Rad) for 1 minute. Images were processed in an Amersham imager 600 series (GE Healthcare).
4.5. Lentivirus Production and Stable Cell Line Generation
Stable cell lines were generated by lentivirus-mediated transgene introduction into HEK293T and HCT-116 cells. The Mer-Intra-BS was cloned into the lentiviral pTRIPZ vector, which confers puromycin resistance and contains a Tet-on promoter to allow for Doxycycline (Dox)-inducible transgene expression. For lentivirus production, HEK293T cells were grown to 90-100% confluence on a 60mm plate, and then transfected with 1µg Mer-Intra-BS/pTRIPZ, 0.75µg psPAX (encoding lentiviral packaging components), and 0.25µg PMD2G (encoding components for viral envelope) using Polyjet Transient Transfection Reagent according to the manufacturer’s instructions (SignaGen). 24 hours after transfection, NaButryrate was added to the culture media to a final concentration of 10mM to increase lentivirus production. Cells were grown for another 24 hours, and media containing lentivirus was harvested, passed through a 0.45 μm filter, and concentrated using Lenti-X Concentrator (Clontech 631231). 100µL of concentrated virus was used to infect HEK293T and HCT116 cells cultured with 8µg/mL Polybrene. 2 days after infection, infected cells were selected in 2µg/mL puromycin.
4.6. Luciferase Assays
For luciferase assays, cells were transfected with Mer-Intra-BS alone or with other plasmids using Polyjet Transfection Reagent (SignaGen) and lysed with 1×PLB (Promega). Protein lysates or purified biosensor fusion proteins The Nano-Glo Live Cell Assay System (Promega) was used to measure luciferase activity with furimazine as the luciferin substrate according to manufacturer’s instructions as previously described[55]. The luminescent activity was measured using a Turner Biosystems 20/20 Luminometer (Promega), or GloMax Navigator Microplate Luminometer (Promega). All data for luciferase assays are presented as luminescence relative to no biosensor control (RTC).
4.7. Bioluminescent imaging (BLI) analysis
Increasing numbers (0.5-5×105 cells) of HEK293T or HCT116 cells stably expressing the Mer-Intra-BS were seeded into each well of 12 - well plate. Bioluminescent signals were measured using the Perkin Elmer IVIS Ilumina III after addition of Nano-Glo Live Cell substrate (Promega).
4.8. Monitoring cell density dependent activation of Mer-Intra-BS
Triplicates of HEK293T-Mer-Intra-BS cells at increasing densities (1-8x104) were plated in individual wells of a 24-well plate. Mer-Intra-BS induction was achieved by treating with Dox (1 μg/μl) for a duration of 2 days. Subsequently, protein extraction, concentration measurement, and luciferase analysis were performed. The relative light units (RLU) per microgram of protein lysate were calculated for each sample. The experiment was replicated twice to ensure reproducibility. The mean and standard deviation (S.D.) of RLU/μg were determined for each cell density. Statistical analysis, specifically a student t-test, was conducted to compare the RLU/μg values between the 1x104 cell density and the other cell densities.
4.9. Purification of His-tagged Mer-Intra-BS
LgBiT-Merlin-SmBiT is subcloned into pET28b vector. The construct is transformed into BL21 DE3 competent bacteria. A single colony is inoculated into 25ml of 2xYT medium containing ampicillin and incubated in an incubator at 37 ◦C with shaking at 250rpm overnight. The overnight grown bacteria were diluted to OD600 of 0.2 in 250ml 2xYT medium and incubated at 37 ◦C until the OD600 was between 0.6–0.8. Protein expression was induced with 0.3 mM IPTG (isopropyl β-D-1-thiogalactopyranoside) overnight at 25 ◦C. Bacterial cells were lysed by sonication, bacterial lysates were centrifuged to collect soluble fractions, and His-tagged proteins were isolated from the supernatant via Ni-NTA affinity purification. Proteins were concentrated using an Amicon Ultra-4 Centrifugal Filter Unit (Millipore-Sigma) in a standard buffer (30 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM MgCl2, and 3 mM DTT). Concentrated proteins were analyzed by SDS-PAGE, and stored at −80 ◦C.
5. Conclusions
Our newly established Merlin intramolecular biosensor will provide a very useful tool for studying the biochemical and biological function of tumor suppressor Merlin both in vitro and in vivo. It will have significant impact on our understanding of the roles of Merlin in cancer biology and therapy.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: List of Primers Used for PCR.
Author Contributions
Conceptualization, X.Y.; methodology, A.P., T.K., M.C, C.N., X.Y.; formal analysis, A.P., T.K., M.C.; investigation, A.P., T.K., M.C.; writing—original draft preparation, A.P.; writing—review and editing, X.Y.; supervision, X.Y.; project administration, X.Y.; funding acquisition, X.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Canadian Institute of Health Research (CIHR), grant number 186142 & 148629”.
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable.
Data Availability Statement
No extra data except the data presented in the manuscript is presented.
Acknowledgments
We will acknowledge excellent technical support from Yawei Hao.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rouleau, G. A.; Merel, P.; Lutchman, M.; Sanson, M.; Zucman, J.; Marineau, C.; Hoang-Xuan, K.; Demczuk, S.; Desmaze, C.; Plougastel, B., Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature 1993, 363, (6429), 515-521. [CrossRef]
- Trofatter, J. A.; MacCollin, M. M.; Rutter, J. L.; Murrell, J. R.; Duyao, M. P.; Parry, D. M.; Eldridge, R.; Kley, N.; Menon, A. G.; Pulaski, K.; et al., A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell 1993, 75, (4), 826. [CrossRef]
- Evans, D. G., Neurofibromatosis type 2 (NF2): a clinical and molecular review. Orphanet J Rare Dis 2009, 4, 16. [CrossRef]
- Asthagiri, A. R.; Parry, D. M.; Butman, J. A.; Kim, H. J.; Tsilou, E. T.; Zhuang, Z.; Lonser, R. R., Neurofibromatosis type 2. Lancet 2009, 373, (9679), 1974-86. [CrossRef]
- Blakeley, J. O.; Plotkin, S. R., Therapeutic advances for the tumors associated with neurofibromatosis type 1, type 2, and schwannomatosis. Neuro Oncol 2016, 18, (5), 624-38. [CrossRef]
- Subbiah, V.; Slopis, J.; Hong, D. S.; Ketonen, L. M.; Hamilton, J.; McCutcheon, I. E.; Kurzrock, R., Treatment of patients with advanced neurofibromatosis type 2 with novel molecularly targeted therapies: from bench to bedside. J Clin Oncol 2012, 30, (5), e64-8. [CrossRef]
- Plotkin, S. R.; Stemmer-Rachamimov, A. O.; Barker, F. G., 2nd; Halpin, C.; Padera, T. P.; Tyrrell, A.; Sorensen, A. G.; Jain, R. K.; di Tomaso, E., Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med 2009, 361, (4), 358-67. [CrossRef]
- Gusella, J. F.; Ramesh, V.; MacCollin, M.; Jacoby, L. B., Merlin: the neurofibromatosis 2 tumor suppressor. Biochim Biophys Acta 1999, 1423, (2), M29-36. [CrossRef]
- McClatchey, A. I.; Giovannini, M., Membrane organization and tumorigenesis--the NF2 tumor suppressor, Merlin. Genes & development 2005, 19, (19), 2265-2277.
- McClatchey, A. I., Merlin and ERM proteins: unappreciated roles in cancer development? Nat Rev Cancer 2003, 3, (11), 877-83. [CrossRef]
- Hanahan, D.; Weinberg, R. A., Hallmarks of cancer: the next generation. Cell 2011, 144, (5), 646-74. [CrossRef]
- Zirn, B.; Arning, L.; Bartels, I.; Shoukier, M.; Hoffjan, S.; Neubauer, B.; Hahn, A., Ring chromosome 22 and neurofibromatosis type II: proof of two-hit model for the loss of the NF2 gene in the development of meningioma. Clin Genet 2012, 81, (1), 82-7. [CrossRef]
- Knudson, A. G., Jr., Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A 1971, 68, (4), 820-3. [CrossRef]
- Petrilli, A. M.; Fernández-Valle, C., Role of Merlin/NF2 inactivation in tumor biology. Oncogene 2016, 35, (5), 537-48. [CrossRef]
- Brastianos, P. K.; Horowitz, P. M.; Santagata, S.; Jones, R. T.; McKenna, A.; Getz, G.; Ligon, K. L.; Palescandolo, E.; Van Hummelen, P.; Ducar, M. D.; Raza, A.; Sunkavalli, A.; Macconaill, L. E.; Stemmer-Rachamimov, A. O.; Louis, D. N.; Hahn, W. C.; Dunn, I. F.; Beroukhim, R., Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat Genet 2013, 45, (3), 285-9. [CrossRef]
- Lee, S.; Karas, P. J.; Hadley, C. C.; Bayley, V. J.; Khan, A. B.; Jalali, A.; Sweeney, A. D.; Klisch, T. J.; Patel, A. J., The Role of Merlin/NF2 Loss in Meningioma Biology. Cancers (Basel) 2019, 11, (11). [CrossRef]
- Gutmann, D. H.; Giordano, M. J.; Fishback, A. S.; Guha, A., Loss of merlin expression in sporadic meningiomas, ependymomas and schwannomas. Neurology 1997, 49, (1), 267-70. [CrossRef]
- Bianchi, A. B.; Mitsunaga, S. I.; Cheng, J. Q.; Klein, W. M.; Jhanwar, S. C.; Seizinger, B.; Kley, N.; Klein-Szanto, A. J.; Testa, J. R., High frequency of inactivating mutations in the neurofibromatosis type 2 gene (NF2) in primary malignant mesotheliomas. Proc Natl Acad Sci U S A 1995, 92, (24), 10854-8. [CrossRef]
- Sekido, Y.; Sato, T., NF2 alteration in mesothelioma. Front Toxicol 2023, 5, 1161995. [CrossRef]
- Sato, T.; Sekido, Y., NF2/Merlin Inactivation and Potential Therapeutic Targets in Mesothelioma. Int J Mol Sci 2018, 19, (4). [CrossRef]
- Bianchi, A. B.; Hara, T.; Ramesh, V.; Gao, J.; Klein-Szanto, A. J.; Morin, F.; Menon, A. G.; Trofatter, J. A.; Gusella, J. F.; Seizinger, B. R.; et al., Mutations in transcript isoforms of the neurofibromatosis 2 gene in multiple human tumour types. Nat Genet 1994, 6, (2), 185-92. [CrossRef]
- Morrow, K. A.; Das, S.; Metge, B. J.; Ye, K.; Mulekar, M. S.; Tucker, J. A.; Samant, R. S.; Shevde, L. A., Loss of tumor suppressor Merlin in advanced breast cancer is due to post-translational regulation. J Biol Chem 2011, 286, (46), 40376-85. [CrossRef]
- Cačev, T.; Aralica, G.; Lončar, B.; Kapitanović, S., Loss of NF2/Merlin expression in advanced sporadic colorectal cancer. Cell Oncol (Dordr) 2014, 37, (1), 69-77.
- Luo, Z. L.; Cheng, S. Q.; Shi, J.; Zhang, H. L.; Zhang, C. Z.; Chen, H. Y.; Qiu, B. J.; Tang, L.; Hu, C. L.; Wang, H. Y.; Li, Z., A splicing variant of Merlin promotes metastasis in hepatocellular carcinoma. Nat Commun 2015, 6, 8457. [CrossRef]
- Benhamouche, S.; Curto, M.; Saotome, I.; Gladden, A. B.; Liu, C. H.; Giovannini, M.; McClatchey, A. I., Nf2/Merlin controls progenitor homeostasis and tumorigenesis in the liver. Genes & development 2010, 24, (16), 1718-1730.
- Horiguchi, A.; Zheng, R.; Shen, R.; Nanus, D. M., Inactivation of the NF2 tumor suppressor protein merlin in DU145 prostate cancer cells. Prostate 2008, 68, (9), 975-84. [CrossRef]
- Thurneysen, C.; Opitz, I.; Kurtz, S.; Weder, W.; Stahel, R. A.; Felley-Bosco, E., Functional inactivation of NF2/merlin in human mesothelioma. Lung Cancer 2009, 64, (2), 140-7. [CrossRef]
- Chishti, A. H.; Kim, A. C.; Marfatia, S. M.; Lutchman, M.; Hanspal, M.; Jindal, H.; Liu, S. C.; Low, P. S.; Rouleau, G. A.; Mohandas, N.; Chasis, J. A.; Conboy, J. G.; Gascard, P.; Takakuwa, Y.; Huang, S. C.; Benz, E. J., Jr.; Bretscher, A.; Fehon, R. G.; Gusella, J. F.; Ramesh, V.; Solomon, F.; Marchesi, V. T.; Tsukita, S.; Tsukita, S.; Hoover, K. B.; et al., The FERM domain: a unique module involved in the linkage of cytoplasmic proteins to the membrane. Trends Biochem Sci 1998, 23, (8), 281-2. [CrossRef]
- Michie, K. A.; Bermeister, A.; Robertson, N. O.; Goodchild, S. C.; Curmi, P. M. G., Two Sides of the Coin: Ezrin/Radixin/Moesin and Merlin Control Membrane Structure and Contact Inhibition. Int J Mol Sci 2019, 20, (8). [CrossRef]
- Cooper, J.; Giancotti, F. G., Molecular insights into NF2/Merlin tumor suppressor function. FEBS letters 2014, 588, (16), 2743-2752. [CrossRef]
- Shimizu, T.; Seto, A.; Maita, N.; Hamada, K.; Tsukita, S.; Tsukita, S.; Hakoshima, T., Structural basis for neurofibromatosis type 2. Crystal structure of the merlin FERM domain. J Biol Chem 2002, 277, (12), 10332-6. [CrossRef]
- Bretscher, A.; Edwards, K.; Fehon, R. G., ERM proteins and merlin: integrators at the cell cortex. Nat Rev Mol Cell Biol 2002, 3, (8), 586-99. [CrossRef]
- Kissil, J. L.; Johnson, K. C.; Eckman, M. S.; Jacks, T., Merlin phosphorylation by p21-activated kinase 2 and effects of phosphorylation on merlin localization. J Biol Chem 2002, 277, (12), 10394-9. [CrossRef]
- Xiao, G. H.; Beeser, A.; Chernoff, J.; Testa, J. R., p21-activated kinase links Rac/Cdc42 signaling to merlin. J Biol Chem 2002, 277, (2), 883-6. [CrossRef]
- Alfthan, K.; Heiska, L.; Grönholm, M.; Renkema, G. H.; Carpén, O., Cyclic AMP-dependent protein kinase phosphorylates merlin at serine 518 independently of p21-activated kinase and promotes merlin-ezrin heterodimerization. J Biol Chem 2004, 279, (18), 18559-66. [CrossRef]
- Li, Y.; Zhou, H.; Li, F.; Chan, S. W.; Lin, Z.; Wei, Z.; Yang, Z.; Guo, F.; Lim, C. J.; Xing, W.; Shen, Y.; Hong, W.; Long, J.; Zhang, M., Angiomotin binding-induced activation of Merlin/NF2 in the Hippo pathway. Cell Res 2015, 25, (7), 801-17. [CrossRef]
- Hamaratoglu, F.; Willecke, M.; Kango-Singh, M.; Nolo, R.; Hyun, E.; Tao, C.; Jafar-Nejad, H.; Halder, G., The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nature cell biology 2006, 8, (1), 27-36. [CrossRef]
- Yu, J.; Zheng, Y.; Dong, J.; Klusza, S.; Deng, W. M.; Pan, D., Kibra functions as a tumor suppressor protein that regulates Hippo signaling in conjunction with Merlin and Expanded. Dev Cell 2010, 18, (2), 288-99. [CrossRef]
- Zhang, N.; Bai, H.; David, K. K.; Dong, J.; Zheng, Y.; Cai, J.; Giovannini, M.; Liu, P.; Anders, R. A.; Pan, D., The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Developmental cell 2010, 19, (1), 27-38. [CrossRef]
- Yin, F.; Yu, J.; Zheng, Y.; Chen, Q.; Zhang, N.; Pan, D., Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell 2013, 154, (6), 1342-55. [CrossRef]
- Wu, Z.; Guan, K. L., Hippo Signaling in Embryogenesis and Development. Trends Biochem Sci 2021, 46, (1), 51-63. [CrossRef]
- Sarmasti Emami, S.; Zhang, D.; Yang, X., Interaction of the Hippo Pathway and Phosphatases in Tumorigenesis. Cancers (Basel) 2020, 12, (9). [CrossRef]
- Azad, T., Ghahremani M., Yang, X., The role of YAP and TAZ in angiogenesis and vascular mimicry. Cells 2019, 8, 407. [CrossRef]
- Zhao, Y.; Yang, X., Chapter 8: Targeting the Hippo pathway to improve response to chemotherapy. Tarteting Cell Survival Pathways to Enhance Response to Chemotherapy. Elsevier Publishing Company: 2019; Vol. 3.
- Wu, L.; Yang, X., Targeting the Hippo pathway for breast cancer therapy. Cancers (Basel) 2018, 10, (11), E422. [CrossRef]
- Sher, I.; Hanemann, C. O.; Karplus, P. A.; Bretscher, A., The tumor suppressor merlin controls growth in its open state, and phosphorylation converts it to a less-active more-closed state. Dev Cell 2012, 22, (4), 703-5. [CrossRef]
- Primi, M. C.; Rangarajan, E. S.; Patil, D. N.; Izard, T., Conformational flexibility determines the Nf2/merlin tumor suppressor functions. Matrix Biol Plus 2021, 12, 100074. [CrossRef]
- Hong, A. W.; Meng, Z.; Plouffe, S. W.; Lin, Z.; Zhang, M.; Guan, K. L., Critical roles of phosphoinositides and NF2 in Hippo pathway regulation. Genes Dev 2020, 34, (7-8), 511-525. [CrossRef]
- LaJeunesse, D. R.; McCartney, B. M.; Fehon, R. G., Structural analysis of Drosophila merlin reveals functional domains important for growth control and subcellular localization. J Cell Biol 1998, 141, (7), 1589-99. [CrossRef]
- Johnson, K. C.; Kissil, J. L.; Fry, J. L.; Jacks, T., Cellular transformation by a FERM domain mutant of the Nf2 tumor suppressor gene. Oncogene 2002, 21, (39), 5990-7. [CrossRef]
- Zhao, B.; Wei, X.; Li, W.; Udan, R. S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; Zheng, P.; Ye, K.; Chinnaiyan, A.; Halder, G.; Lai, Z. C.; Guan, K. L., Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes & Development 2007, 21, (21), 2747-2761. [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; Elvassore, N.; Piccolo, S., Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, (7350), 179-183. [CrossRef]
- Ma, S.; Meng, Z.; Chen, R.; Guan, K. L., The Hippo Pathway: Biology and Pathophysiology. Annu Rev Biochem 2019, 88, 577-604. [CrossRef]
- Azad, T. N., K; Janse van Rensburg, HJ; Maritan, SM; Wu, L; Hao, Y; Montminy, T; Yu, J; Khanal, K; Mulligan, LM; Yang, X, A gain-of-functional screen identifies the Hippo pathway as a central mediator of receptor tyrosine kinases during tumorigenesis. Oncogene 2020, 39, 334-355. [CrossRef]
- Nouri, K. A., T; Lightbody, E; Khanal, P; Nicol, CJ; Yang, X, A kinome-wide screen using a NanoLuc LATS luminescent biosensor identifies ALK as a novel regulator of the Hippo pathway in tumorigenesis and immune evasion. FASEB Journal 2019, 33, 12487-12499.
- Nouri, K.; Azad, T.; Ling, M.; van Rensburg, H. J. J.; Pipchuk, A.; Shen, H.; Hao, Y.; Zhang, J.; Yang, X., Identification of celastrol as a novel YAP-TEAD inhibitor for cancer therapy by high throughput screening with ultrasensitive YAP/TAZ-TEAD biosensors. Cancers (Basel) 2019, 11, (10), 1596. [CrossRef]
- Azad, T.; Janse van Rensburg, H. J.; Lightbody, E. D.; Neveu, B.; Champagne, A.; Ghaffari, A.; Kay, V. R.; Hao, Y.; Shen, H.; Yeung, B.; Croy, B. A.; Guan, K. L.; Pouliot, F.; Zhang, J.; Nicol, C. J. B.; Yang, X., A LATS biosensor functional screen identifies VEGFR as a novel regulator of the Hippo pathway in angiogenesis. Nat.Commun. 2018, 9, 1061.
- Wu, L., Ge, A., Hao, Y., Yang, X, Development of a New HiBiT Biosensor Monitoring Stability of YAP/TAZ Proteins in Cells. Chemosensors 2023, 11, (9), 492. [CrossRef]
- Pipchuk, A.; Yang, X., Using Biosensors to Study Protein-Protein Interaction in the Hippo Pathway. Front Cell Dev Biol 2021, 9, 660137. [CrossRef]
- Dixon, A. S.; Schwinn, M. K.; Hall, M. P.; Zimmerman, K.; Otto, P.; Lubben, T. H.; Butler, B. L.; Binkowski, B. F.; Machleidt, T.; Kirkland, T. A.; Wood, M. G.; Eggers, C. T.; Encell, L. P.; Wood, K. V., NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chemical Biology 2016, 11, (2), 400-408. [CrossRef]
- Hall, M. P.; Unch, J.; Binkowski, B. F.; Valley, M. P.; Butler, B. L.; Wood, M. G.; Otto, P.; Zimmerman, K.; Vidugiris, G.; Machleidt, T.; Robers, M. B.; Benink, H. A.; Eggers, C. T.; Slater, M. R.; Meisenheimer, P. L.; Klaubert, D. H.; Fan, F.; Encell, L. P.; Wood, K. V., Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS chemical biology 2012, 7, (11), 1848-1857. [CrossRef]
- Nguyen, R.; Reczek, D.; Bretscher, A., Hierarchy of merlin and ezrin N- and C-terminal domain interactions in homo- and heterotypic associations and their relationship to binding of scaffolding proteins EBP50 and E3KARP. J Biol Chem 2001, 276, (10), 7621-9. [CrossRef]
- Hao, Y.; Chun, A.; Cheung, K.; Rashidi, B.; Yang, X., Tumor suppressor LATS1 is a negative regulator of oncogene YAP. The Journal of Biological Chemistry 2008, 283, (9), 5496-5509. [CrossRef]
Figure 1.
Establishment of the Mer-Intra-BS. (A) Domain structure of Merlin. The N-terminal FERM domain consists of 3 subdomains: F1, F2 and F3. The F2 subdomain contains a ‘Blue-Box’ region that is not found in ERM proteins and participates in binding with downstream Merlin effectors. (B) Construct design of the Mer-Intra-BS. The intramolecular Merlin-NanoBiT fusion protein was designed by fusing NanoBiT constituents LgBiT and SmBiT onto the N and C-terminus, respectively, of full-length Merlin. Each NanoBiT constituent is linked onto Merlin through a flexible glycine/serine linker region. (C) Hypothesized mechanism of action for the Mer-Intra-BS. In the context of the Mer-Intra-BS, luciferase complementation is expected when Merlin exists primarily its closed conformation, but not when Merlin exists in an ‘open’ conformation. Therefore, the closed conformation Mer-Intra-BS should display more luminescent activity. (D) Luciferase activity of the Mer-Intra-BS. The Mer-Intra-BS displays a dramatic (~150-fold, P <0.0005) increase in luminescent activity as compared to cells transfected with LgBiT alone, demonstrating that luciferase complementation occurs within the Mer-Intra-BS. Luminescence is presented (henceforth) as luminescence relative to control (Mock; RTC). Data are presented on a logarithmic scale. (E) The Mer-Intra-BS is detectable by western blot with anti-Merlin antibody. β-actin was used as internal control.
Figure 1.
Establishment of the Mer-Intra-BS. (A) Domain structure of Merlin. The N-terminal FERM domain consists of 3 subdomains: F1, F2 and F3. The F2 subdomain contains a ‘Blue-Box’ region that is not found in ERM proteins and participates in binding with downstream Merlin effectors. (B) Construct design of the Mer-Intra-BS. The intramolecular Merlin-NanoBiT fusion protein was designed by fusing NanoBiT constituents LgBiT and SmBiT onto the N and C-terminus, respectively, of full-length Merlin. Each NanoBiT constituent is linked onto Merlin through a flexible glycine/serine linker region. (C) Hypothesized mechanism of action for the Mer-Intra-BS. In the context of the Mer-Intra-BS, luciferase complementation is expected when Merlin exists primarily its closed conformation, but not when Merlin exists in an ‘open’ conformation. Therefore, the closed conformation Mer-Intra-BS should display more luminescent activity. (D) Luciferase activity of the Mer-Intra-BS. The Mer-Intra-BS displays a dramatic (~150-fold, P <0.0005) increase in luminescent activity as compared to cells transfected with LgBiT alone, demonstrating that luciferase complementation occurs within the Mer-Intra-BS. Luminescence is presented (henceforth) as luminescence relative to control (Mock; RTC). Data are presented on a logarithmic scale. (E) The Mer-Intra-BS is detectable by western blot with anti-Merlin antibody. β-actin was used as internal control.
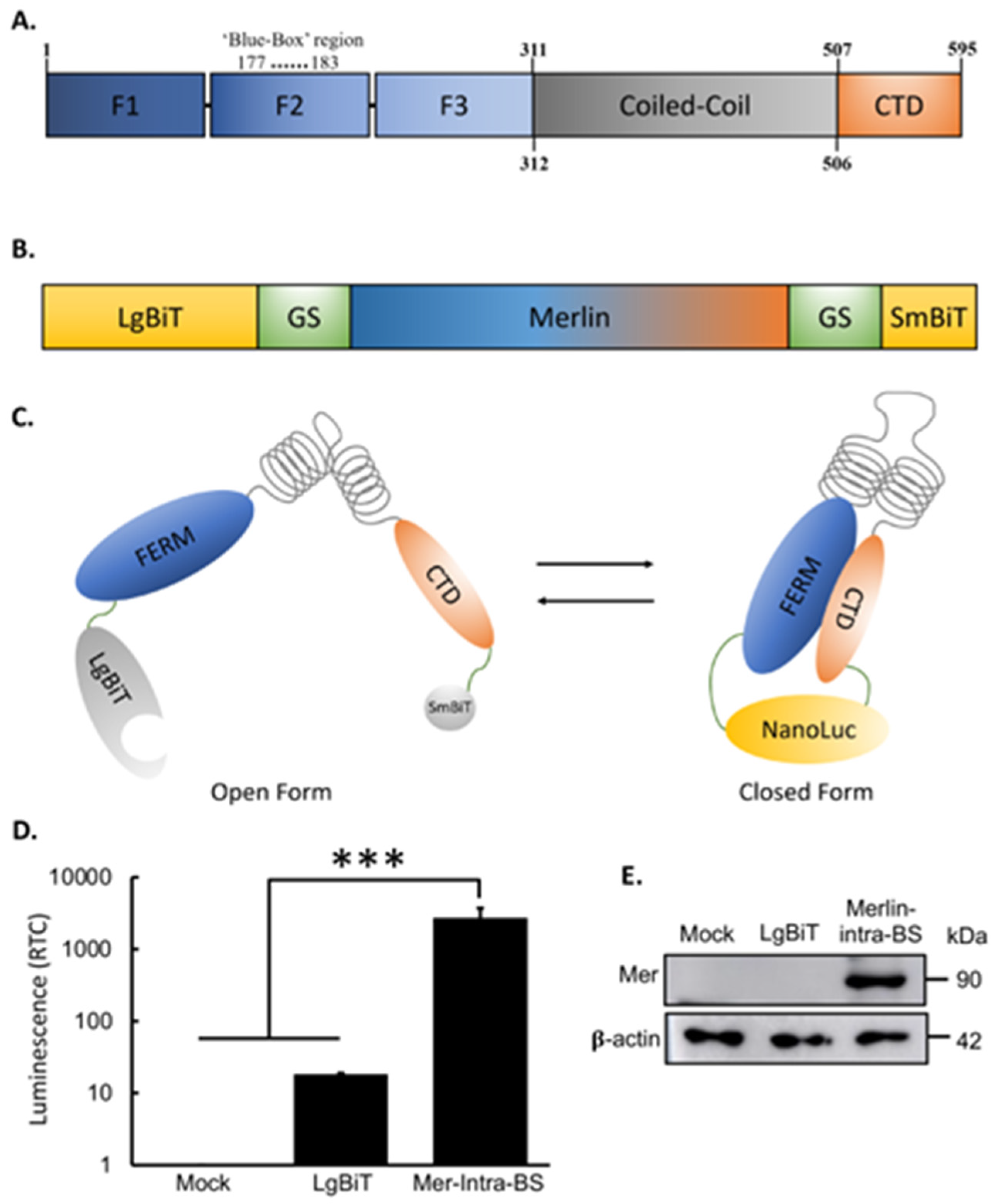
Figure 2.
Validation of Mer-Intra-BS. (A) Validation of the Mer-Intra-BS using an A585W mutant biosensor. The A585W mutant promotes the closed conformation Merlin by introducing a strong hydrophobic interaction between the N and C-terminus of Merlin. Wild-type (WT) or A585W mutant biosensor plasmid was transfected into HEK293T cells, followed by luciferase assay. The protein lysates were subjected to western blot analysis using anti-Merlin antibody (B). Protein expression as detected by western blot (B). (C) Validation of the Mer-Intra-BS using Mer-ΔEL-intra-BS. The Mer-ΔEL-intra-BS promotes the open conformation Merlin by disrupting the interaction between the N and C-terminus of Merlin. No plasmid (mock) or plasmids expressing wild-type Mer-Intra-BS or Mer-ΔEL-Intra-BS was transfected into HEK293T cells, followed by luciferase assays. The protein lysates were subjected to western blot analysis using anti-Merlin antibody (D). RTC, Relative to control (mock). ***, p<0.001; **, p<0.01, statistically significant.
Figure 2.
Validation of Mer-Intra-BS. (A) Validation of the Mer-Intra-BS using an A585W mutant biosensor. The A585W mutant promotes the closed conformation Merlin by introducing a strong hydrophobic interaction between the N and C-terminus of Merlin. Wild-type (WT) or A585W mutant biosensor plasmid was transfected into HEK293T cells, followed by luciferase assay. The protein lysates were subjected to western blot analysis using anti-Merlin antibody (B). Protein expression as detected by western blot (B). (C) Validation of the Mer-Intra-BS using Mer-ΔEL-intra-BS. The Mer-ΔEL-intra-BS promotes the open conformation Merlin by disrupting the interaction between the N and C-terminus of Merlin. No plasmid (mock) or plasmids expressing wild-type Mer-Intra-BS or Mer-ΔEL-Intra-BS was transfected into HEK293T cells, followed by luciferase assays. The protein lysates were subjected to western blot analysis using anti-Merlin antibody (D). RTC, Relative to control (mock). ***, p<0.001; **, p<0.01, statistically significant.
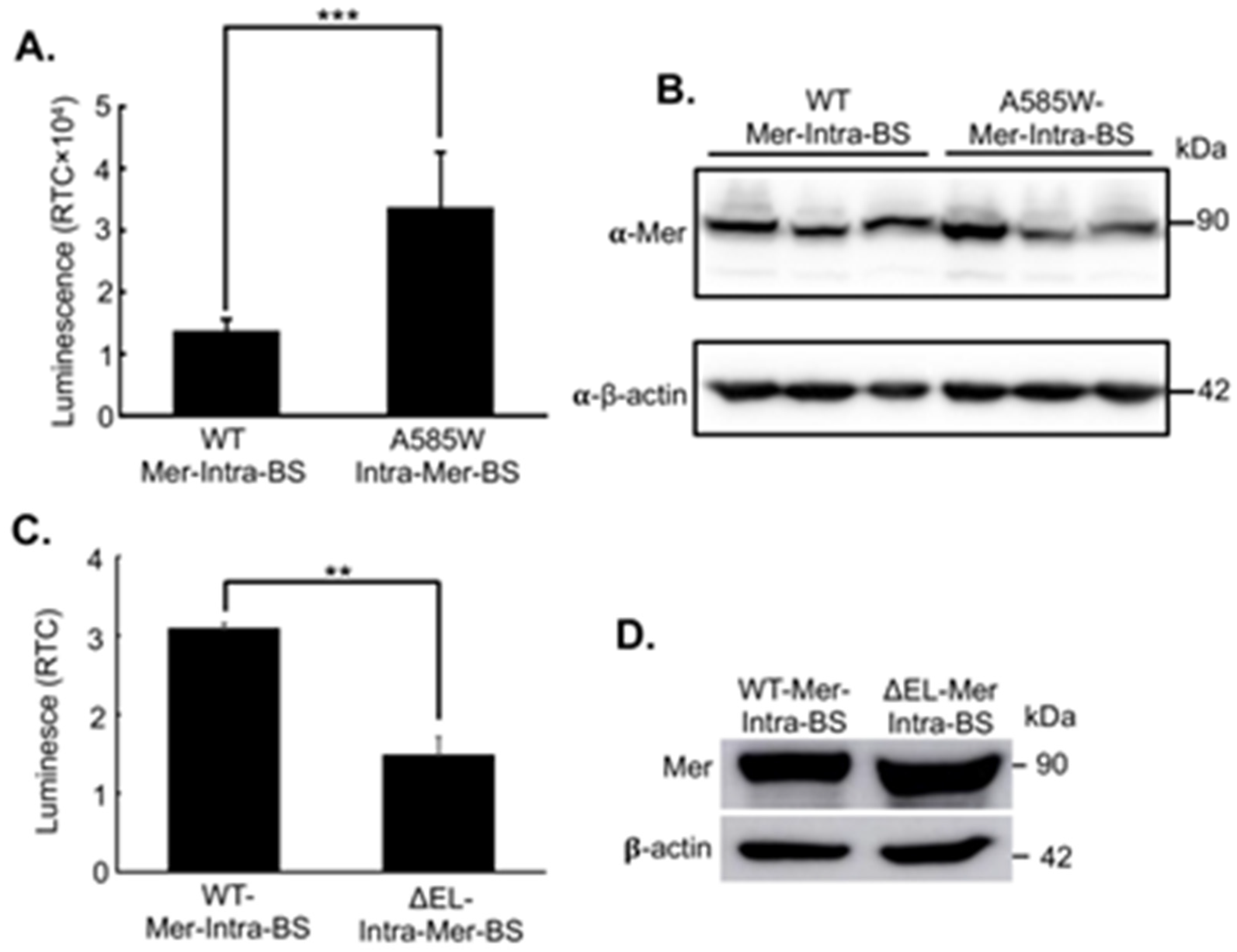
Figure 3.
Validation of the Mer-Intra-BS by cotransfection with PAK1. Luciferase activity (Upper panel) was measured for protein lysates extracted from HEK293T cells transfected with Mer-Intra-BS transfected alone or alongside kinase-dead (KD) and constitutively active (CA) PAK1. RTC, Relative to control (mock). **, p<0.01, statistically significant. The protein lysates were subjected to western blot analysis using anti-Merlin antibody. 𝛃-actin was used as internal loading control.
Figure 3.
Validation of the Mer-Intra-BS by cotransfection with PAK1. Luciferase activity (Upper panel) was measured for protein lysates extracted from HEK293T cells transfected with Mer-Intra-BS transfected alone or alongside kinase-dead (KD) and constitutively active (CA) PAK1. RTC, Relative to control (mock). **, p<0.01, statistically significant. The protein lysates were subjected to western blot analysis using anti-Merlin antibody. 𝛃-actin was used as internal loading control.
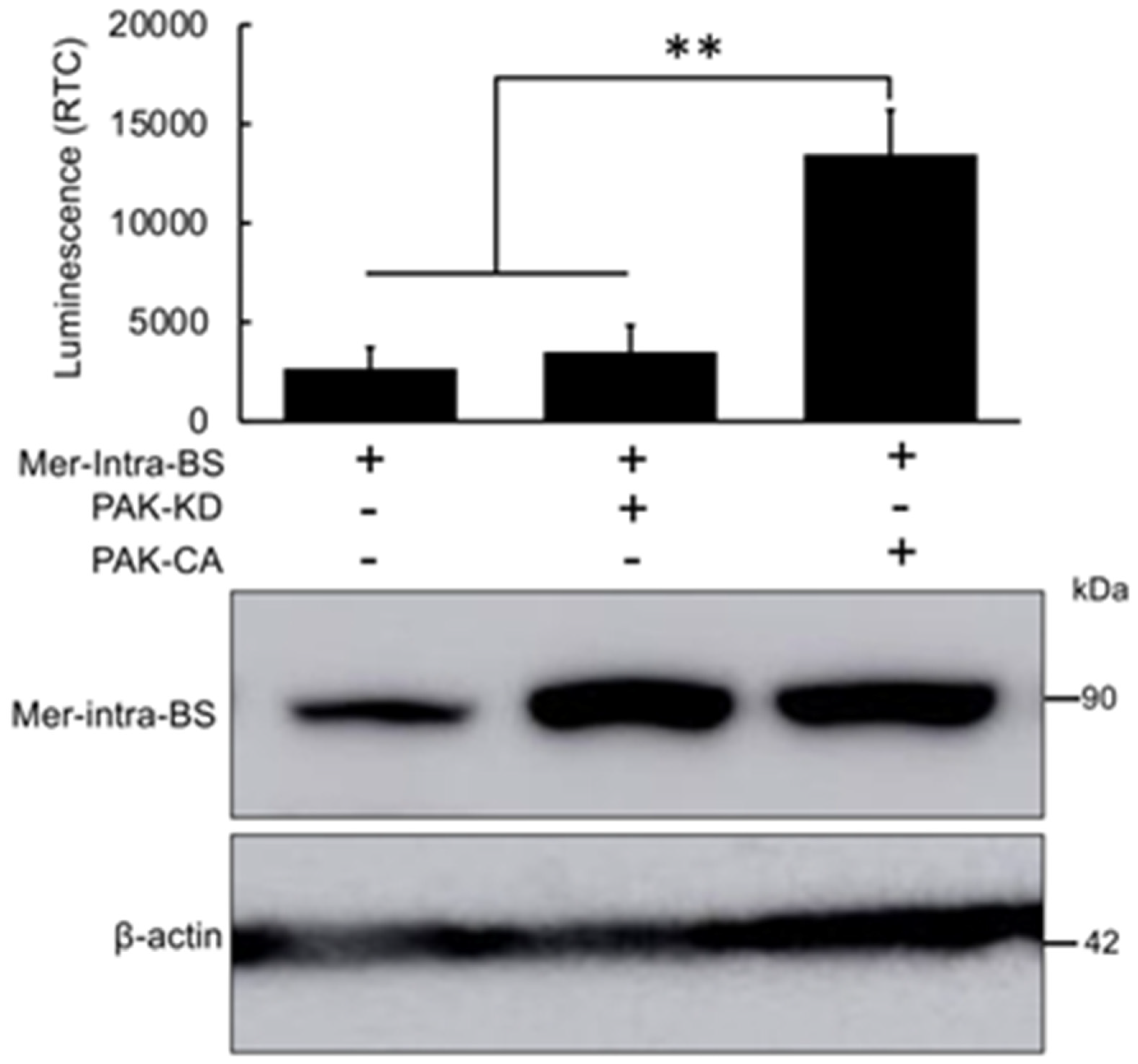
Figure 4.
Cells Lines with Dox-inducible Expression of the Mer-Intra-BS. Exposure to doxycycline dramatically increases expression (upper panel) and luminescent activity (Lower panel) of HEK293T and HCT16 cell lines stably transfected with the Mer-Intra-BS under a dox-inducible promoter. These stable cell lines present useful tools with which to study Merlin conformation under various conditions.
Figure 4.
Cells Lines with Dox-inducible Expression of the Mer-Intra-BS. Exposure to doxycycline dramatically increases expression (upper panel) and luminescent activity (Lower panel) of HEK293T and HCT16 cell lines stably transfected with the Mer-Intra-BS under a dox-inducible promoter. These stable cell lines present useful tools with which to study Merlin conformation under various conditions.
Figure 5.
Bioluminescent imaging analysis of Mer-Intra-BS. Duplicates of increasing number (1-8x104) of HEK293T or HCT116 stably expressing Dox-inducible Mer-Intra-BS were seeded into 24-well plate, followed by incubation in the presence of Dox (+Dox, 1 ug/ml) for 2 days. As a control, duplicate of 8x104 cells were also seeded and incubated in the absence of Dox (-Dox) for 2 days. BLI was analyzed using after addition of furimazine substrate. A heatmap of signal counts represent luminescence.
Figure 5.
Bioluminescent imaging analysis of Mer-Intra-BS. Duplicates of increasing number (1-8x104) of HEK293T or HCT116 stably expressing Dox-inducible Mer-Intra-BS were seeded into 24-well plate, followed by incubation in the presence of Dox (+Dox, 1 ug/ml) for 2 days. As a control, duplicate of 8x104 cells were also seeded and incubated in the absence of Dox (-Dox) for 2 days. BLI was analyzed using after addition of furimazine substrate. A heatmap of signal counts represent luminescence.
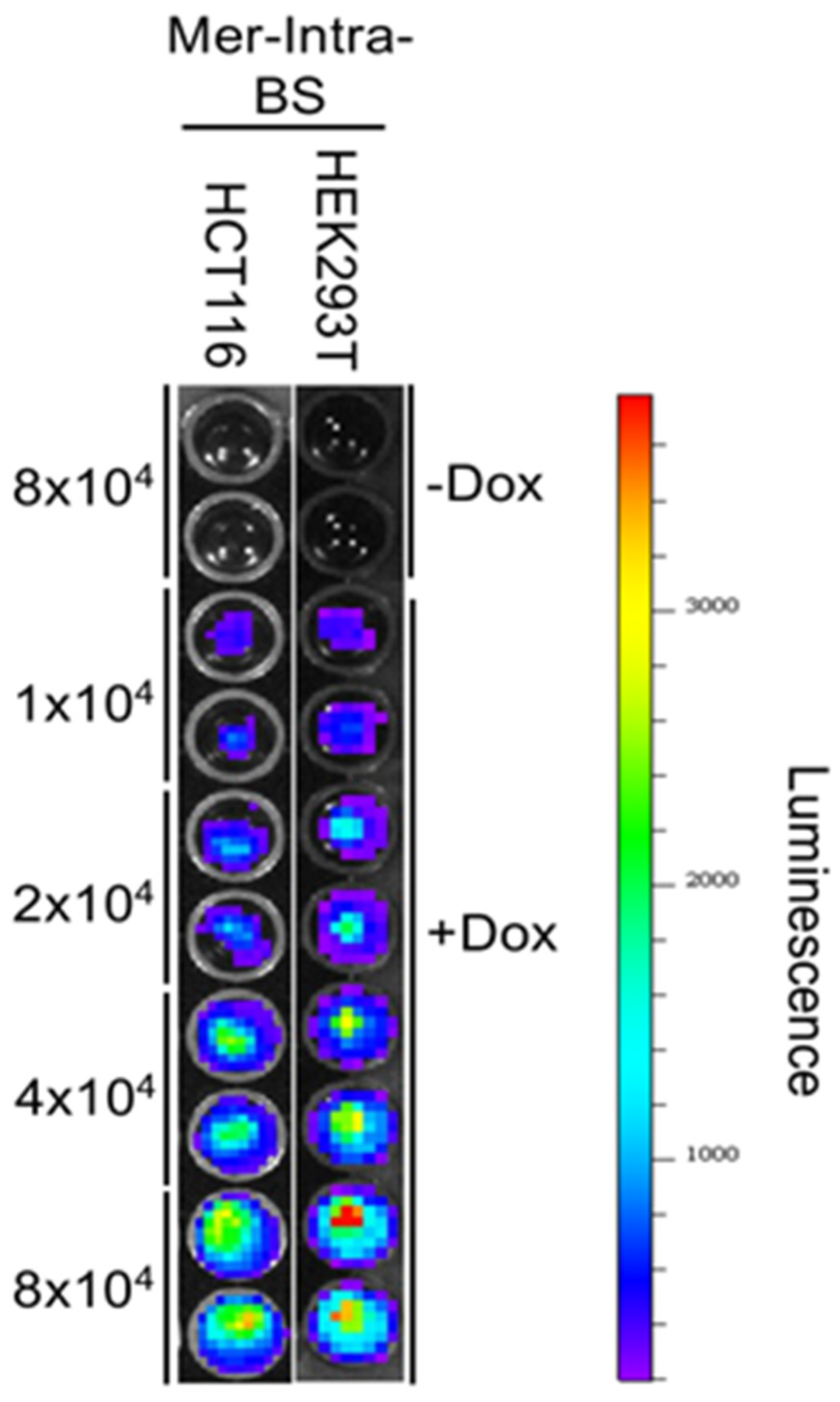
Figure 6.
Density-Dependent Luciferase Activity of the Mer-Intra-BS. Triplicates of HEK293T cells stably transfected with the Mer-Intra-BS under a Dox-inducible promoter were seeded into 24-well plate, following by induction of Mer-Intra-BS by Dox (1 μg/ml) for 2 days. Following lysis, protein lysates were quantified and the luciferase activity was measured. Upper panel: Luciferase assay. Protein concentration of protein lysates was measured. Relative luminescence (RLU) was normalized by protein concentration (RLU/μg). Lower Panel: Western blot analysis of Mer-Intra-BS expression. 10µg of lysates were subjected to western blot analysis using anti-Merlin antibody. Anti-𝛃-actin is used as internal loading control. *, p<0.05; **, p<0.01, statistically significant.
Figure 6.
Density-Dependent Luciferase Activity of the Mer-Intra-BS. Triplicates of HEK293T cells stably transfected with the Mer-Intra-BS under a Dox-inducible promoter were seeded into 24-well plate, following by induction of Mer-Intra-BS by Dox (1 μg/ml) for 2 days. Following lysis, protein lysates were quantified and the luciferase activity was measured. Upper panel: Luciferase assay. Protein concentration of protein lysates was measured. Relative luminescence (RLU) was normalized by protein concentration (RLU/μg). Lower Panel: Western blot analysis of Mer-Intra-BS expression. 10µg of lysates were subjected to western blot analysis using anti-Merlin antibody. Anti-𝛃-actin is used as internal loading control. *, p<0.05; **, p<0.01, statistically significant.
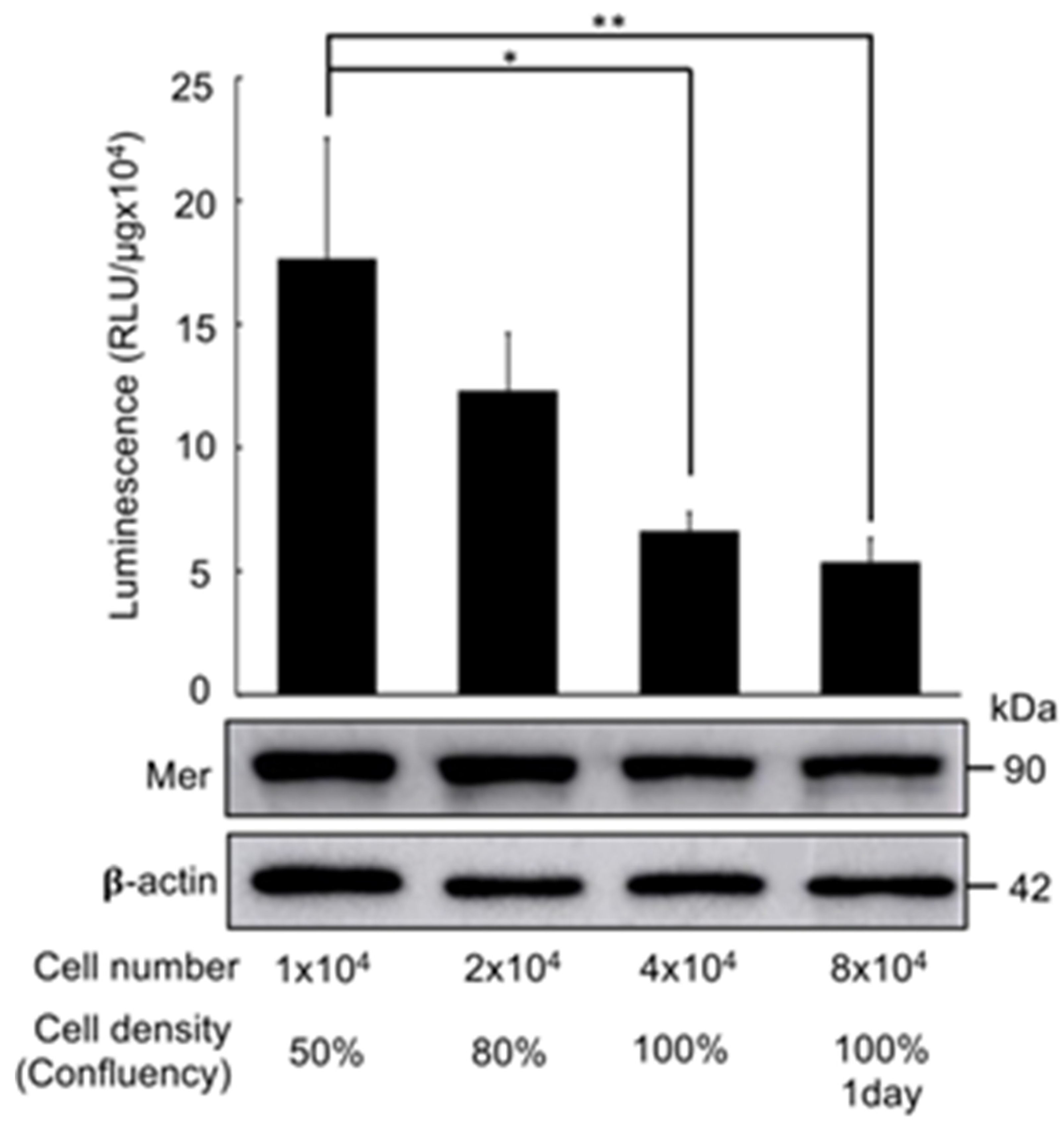
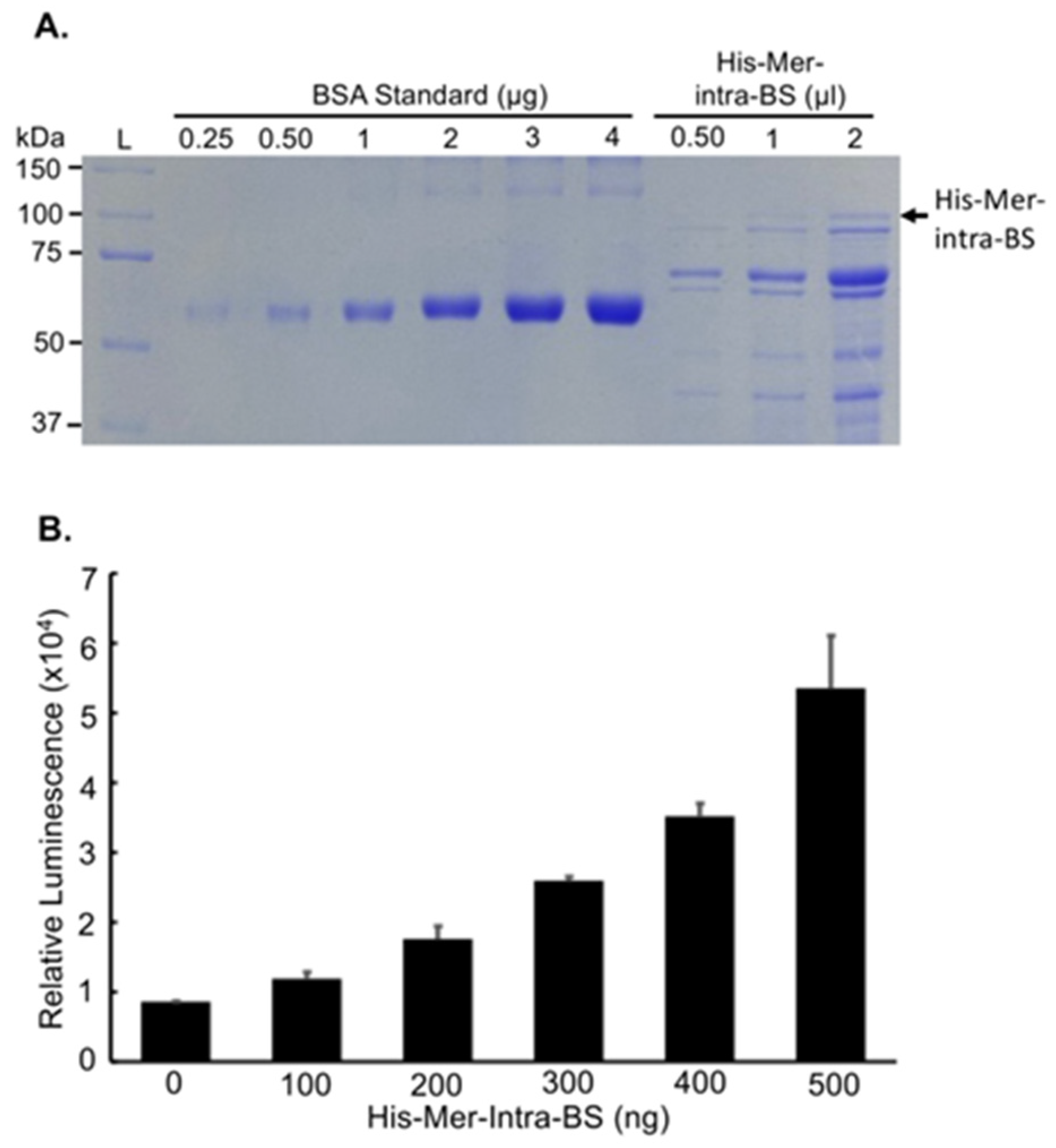
Figure 7.
In vitro characterization of Mer-Intra-BS in vitro. A. Purification of the Mer-Intra-BS. The Mer-Intra-BS was cloned in to the PET28B(+) vector at NDE1 and BAMH1 restriction enzyme cutting sites to append a 6xHistidine tag. The His-Mer-Intra-BS was purified by affinity chromatography and quantified alongside BSA standards (0.25-4 ug). B. Luciferase assays. Increasing amounts (0-500ng) of purified His-Mer-Intra-BS were subjected to luciferase assay in vitro.
Figure 7.
In vitro characterization of Mer-Intra-BS in vitro. A. Purification of the Mer-Intra-BS. The Mer-Intra-BS was cloned in to the PET28B(+) vector at NDE1 and BAMH1 restriction enzyme cutting sites to append a 6xHistidine tag. The His-Mer-Intra-BS was purified by affinity chromatography and quantified alongside BSA standards (0.25-4 ug). B. Luciferase assays. Increasing amounts (0-500ng) of purified His-Mer-Intra-BS were subjected to luciferase assay in vitro.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



