
Submitted:
27 December 2023
Posted:
28 December 2023
You are already at the latest version
Abstract
The Golgi apparatus, long known for protein processing and vesicular trafficking, has recently emerged as an essential player in innate immune signaling pathways. This review article discusses our growing understanding of Golgi cells' roles in initiating and activating innate immunity pathways. We elaborate on the relevance of membrane connections between Golgi organelles and other organelles such as endoplasmic reticulum, mitochondria, endosomes, and autophagosomes that facilitate efficient access to innate immune signal transduction and subsequent effector responses. Additionally, we discuss microbial strategies that exploit the Golgi while diminishing its associated innate immune responses. By providing greater illumination of its multiple functions and mechanisms of operation, this article offers invaluable insight into how protein processing impacts innate immunity and vice versa.
Keywords:
Immunology
; Innate-Immunity
; Golgi-Apparatus
; Intracellular-Signaling
; NLRP3-Inflammasome
1. Introduction
Camillo Golgi's 1898 discovery of the Golgi apparatus using silver nitrate staining marked a pivotal moment. Initially called the internal reticular apparatus, it was later recognized as a crucial cellular component rather than an artifact [1].
The Golgi apparatus is vital for modifying and sorting newly synthesized proteins and lipids. Its dynamic structure, composed of stacked cisternae layers, undergoes regulated disassembly and reassembly during cell cycles. Positioned near nuclei in mammalian cells, it responds to stressors and mediates the transport of proteins and lipids from the endoplasmic reticulum to the trans-Golgi network (TGN) [2,3].
Eukaryotic cells rely on the secretory pathway for protein synthesis, processing, and delivery. Proteins originating in the endoplasmic reticulum progress to the Golgi complex for further processing and sorting. The Golgi's role in cargo movement is debated between the cisternal maturation and vesicular transport models. The Cisternal Maturation Model envisions cisternae maturation from the ER to the trans-Golgi network (TGN) cisterna, while the Vesicular Transport Model emphasizes compartmental stability and vesicular trafficking [4].
The controlled release of cytokines and inflammatory mediators in response to stimuli involves transcription, translation, post-translational modifications, and secretion via the ER, Golgi complex, or cell surface. Cytokines such as IL-2, IL-3, IL-6, IL-10, IL-12, and TNF are directed to the ER for folding via signal peptides. Properly folded cytokines are packaged into vesicles coated with coat protein complex II (COPII) for transport to the ER-Golgi intermediate compartment (ERGIC) and further to the medial-Golgi network (medial Golgi network/TGN) for sorting and modifications. Additionally, the Golgi apparatus plays a crucial role in cargo sorting, separating transmembrane and soluble cytokines, and contributing to protein trafficking [5,6].
The innate immune system orchestrates inflammatory responses to infections and tissue damage. Pattern recognition receptors (PRRs), including Toll-like receptors (TLRs) and RIG-I-like receptors (RLRs), detect microbial and damage-related molecules. Non-professional immune cells, such as epithelial and endothelial cells, also participate in innate immune responses [7].
Toll-like receptor (TLR) transport to endosomes is meticulously regulated. TLR7's interaction with Unc93B1 in the Golgi complex facilitates direct transport to endosomes, bypassing intermediate cell surface steps [8,9].
Inflammasomes are protein complexes that activate pro-inflammatory cytokines, such as IL-1b, through caspase-1-dependent pathways. The NLRP3 inflammasome's distinct responsiveness to various stimuli, including antibiotics and ATP, has implications for various disorders. Recent insights highlight trans-Golgi network (TGN) disruption during NLRP3 inflammasome activation, facilitating inflammasome assembly [10,11]. STING, an ER-associated membrane protein, is crucial for pathogen recognition and activates type I interferon (IFN-I) pathways via the TBK1/IRF3 signaling axis [12,13].
This review integrates the Golgi apparatus's role within innate immunity pathways, exploring its interactions with inflammasomes, cGAS-STING pathways, and TLR/RLR signaling. By shedding light on its involvement, we unveil how the Golgi apparatus shapes immune responses to infections and cellular damage.
2. Golgi Apparatus and NLRP3 Inflammasome
The Golgi apparatus has long been known for its role in protein modification and trafficking. However, recent studies have unveiled the Golgi apparatus's influence on the activation of inflammasomes. Inflammasomes, complex molecular structures that emerge as a response to pathogens or danger signals, are critical components of the body's defense mechanism. Their precise control is essential for effective host defense in complex organisms.
Central to this intricate interplay is the Golgi apparatus's involvement in the assembly and activation of the NLR family pyrin domain containing 3 protein (NLRP3) inflammasome (see Figure 1). The NLRP3 inflammasome comprises three major components: the sensor protein NLRP3, the adaptor protein ASC (also known as PYCARD), and the effector protein caspase 1. NLRP3 belongs to the NOD-like receptor (NLR) protein family, consisting of proteins with an NOD-like domain (NACHT), an N-terminal effector domain such as Pyrin domains (PYD), caspase recruitment domains (CARD) or baculovirus inhibitor of apoptosis protein repeat (BIR), and a C-terminal leucine-rich repeats (LRR). An ATPase activity within their central NACHT domain facilitates receptor oligomerization while their N-terminal PYD or CARD interact with ASC/caspase-1 domains via interaction ATPase activity [14]. While the roles of these components have been extensively documented, recent research has unveiled the Golgi apparatus's pivotal role in this process.
Activation of the NLRP3 inflammasome triggers the maturation and release of proinflammatory cytokines such as IL-1β and IL-18 [15,16], crucial for initiating early inflammation responses and a form of rapid cell death called pyroptosis [17]. The Golgi apparatus's participation in these processes has emerged as a groundbreaking revelation. This dynamic organelle, responsible for protein modification and trafficking, contributes significantly to the assembly and activation of the NLRP3 inflammasome [18]. Signals like potassium (K+) or chloride ions (Cl-), calcium ion influx, disruption of lysosomes, mitochondrial dysfunction, metabolic changes, and metabolic disruption [18] may also be involved in NLRP3 activation.
One of the key findings is the Golgi apparatus's role in positioning and localizing NLRP3-an aspect that plays a pivotal role in inflammasome activation. Upon activation, NLRP3 directly interacts with mitochondria-associated endoplasmic reticulum membranes (MAMs), and substances capable of activating NLRP3 facilitate the proximity of MAMs to Golgi membranes [17]. Furthermore, Zhang et al recently proved an increased level of diacylglycerol (DAG) in Golgi organelles [17]. When activated with inflammasome activators, MAMs localize near Golgi membranes; an increase in diacylglycerol DAG levels at Golgi promotes recruitment and activation of protein kinase D (PKD). Once is activated, PKD phosphorylates NLRP3, leading to its release from MAMs and full maturation within the cytosol [17,19]. This spatial relationship between the Golgi apparatus and NLRP3 hints at the Golgi's regulatory function in the assembly and activation of the NLRP3 inflammasome.
The activation of the NLRP3 inflammasome involves two distinct signals: priming signals and activation signals. Priming signals are typically initiated by microbes or endogenous cytokines, while activation signals are triggered by pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs). These signals set off a cascade of events, culminating in the assembly of the NLRP3 inflammasome [20]. Inflammasome activation plays an essential role in protecting against invading pathogens. Still, excessive activation and gain-of-function mutations of these proteins have been implicated in numerous inflammatory diseases, including gout, type 2 diabetes, Alzheimer's disease, atherosclerosis, and arthritis - in addition to autoinflammatory conditions like CAPS/FMF/MAS syndromes and cryopyrin-associated periodic syndromes (CAPS/FMF/MAS) [21,22]. NLRP3 inflammasome has also been implicated in an inflammatory response triggered by dying cells without evidence of infection, which may contribute to tissue damage and worsen diseases such as acute kidney injury [23,24].
The Golgi apparatus's involvement in inflammasome activation goes beyond its conventional roles. As a central hub for cellular trafficking and modification processes, the Golgi apparatus assumes a regulatory role in coordinating precise component positioning and molecular interactions. This orchestration optimizes the assembly and activation of the NLRP3 inflammasome, thereby contributing to efficient and controlled responses against viral infections. In collective alignment, these discoveries underscore the Golgi apparatus's profound role as a central orchestrator of immune signaling events. Its remarkable ability to regulate inflammasome activation, enhance immune responses, and intersect with intricate cellular processes, including cholesterol metabolism, highlights its significance.
The comprehensive utilization of the Golgi apparatus's multifaceted contributions holds exceptional promise in shaping innovative therapeutic strategies. These strategies, aiming to intricately modulate immune responses, bear the potential to alleviate inflammatory disorders and usher in a new era of targeted interventions.
In summary, the Golgi apparatus's multifaceted contributions to inflammasome activation are increasingly recognized as essential for immune responses. Its role in modulating NLRP3 assembly, optimizing spatial relationships, and enhancing immune signaling sheds light on new dimensions of innate immunity regulation. Understanding the Golgi apparatus's involvement holds the potential to shape novel therapeutic strategies, targeting immune responses and offering insights into the treatment of inflammatory disorders.
3. Golgi Apparatus and cGAS–STING Signaling
This section undertakes a comprehensive exploration of STING (Stimulator of Interferon Genes), a pivotal player in immune signaling. While STING predominantly activates transcription pathways NF-kB and IRF3, culminating in the production of type I interferons as a fundamental antiviral defense mechanism, it is crucial to emphasize the contribution of the Golgi apparatus in these processes.
Mammalian cells intricately employ DNA as a signaling trigger to initiate immune defense responses, driving type-I interferon production through the cGAMP-STING pathway. This pathway originates from DNA molecules producing cyclic-GMP-AMP, subsequently activating STING protein. The ensuing downstream activation of transcription factors IRF3 and IRF7 amplifies this process. This collaborative effort of the Golgi apparatus and STING accentuates the significance of Golgi-mediated trafficking in immune response orchestration.
The cGAS-STING pathway stands as a cornerstone in DNA sensing within mammalian cells. Here, the interactions between DNA sensing enzyme cGAS, cyclic GMP-AMP (its product), and STING acquire paramount importance in combatting infections, leading to the production of type I interferons and proinflammatory cytokines. Through meticulous exploration, we showcase how the Golgi apparatus intricately modulates this process, highlighting the Golgi's role in fine-tuning immune responses [25].
3.1. cGAMP-STING Pathway: Golgi-Mediated Regulation of DNA-Triggered Immune Response
DNA, a vital trigger for immune defense mechanisms within mammalian cells, catalyzes type-I interferon production via the cGAMP-STING pathway. The detection of cytosolic DNA serves as a critical alert, initiating defense mechanisms. For instance, the formation of cyclic-GMP-AMP from DNA molecules, followed by binding and activation of STING protein, triggers a cascade of events culminating in the activation of transcription factors IRF3 and IRF7, ultimately fostering IFNb production. This multi-faceted process benefits from the Golgi apparatus's participation in orchestrating timely and efficient protein trafficking, thereby enhancing the immune response [26].
3.2. cGAS-STING pathway: Golgi's Intricate Participation in Pathway Initiation
After STING binds to cGAMP, it exits the endoplasmic reticulum (ER). It translocates to the Golgi apparatus, where it triggers type I interferon and proinflammatory responses through the activation of interferon regulatory factor 3 (IRF3) and nuclear factor-kappa B (NF-κB) [27]. Recent studies have shown that Golgi apparatus-synthesized sulfated glycosaminoglycans (sGAGs) interact with STING to initiate its oligomerization at the Golgi apparatus, thus identifying sGAGs as necessary coligands for STING activation and downstream IFN signaling [28]. Palmitoylation of STING at the Golgi is essential for the activation of STING. Treatment of cells with palmitoylation inhibitor 2-bromopalmitate (2-BP) inhibits type I interferon response without affecting the trafficking of STING from the ER. After reaching the Golgi, STING undergoes palmitoylation and activates TBK1 at the trans-Golgi network [28].
3.3. cGas activation: Golgi's Intricate Participation in Pathway Initiation
Direct activation of the DNA sensor cGAS by DNA binding triggers conformational changes that activate its enzyme and subsequent enzymatic activity [29,30,31]. The production of cGAMP, an endogenous second messenger with specific mixed phosphodiester linkages, takes place. The liquid-liquid phase separation resulting from DNA binding creates microreactors where active cGAS can thrive, augmenting cGAMP production. In the context of these intricate processes, the Golgi apparatus's strategic role in facilitating these reactions comes to the fore, underscoring its contribution to immune response regulation.
3.4. Downstream Signaling Events: Golgi-Mediated Regulation of STING Activation
Direct binding of STING to cyclic dinucleotides produced by bacteria holds the key to initiating downstream signaling events crucial for immune responses. The conformational change STING undergoes upon binding to cGAMP takes center stage. While in its quiescent state, STING resides within the endoplasmic reticulum under the guidance of its calcium sensor, Stromal Interaction Molecule 1 (STIM1). Upon binding to cGAMP, a sequence of events including Golgi apparatus-mediated translocation from the ER to the ER-Golgi intermediate compartment (ERGIC) and Golgi apparatus ensues. This concerted effort involving the Golgi apparatus sheds light on its role in shaping STING's activation dynamics [32].
3.5. STING interaction with TBK1: Golgi-Mediated Antiviral Response Modulation
Translocation of STING to the Golgi sets the stage for its interaction with TANK-binding kinase 1 (TBK1), a pivotal kinase in the activation of interferon regulatory factor 3 (IRF3) transcription factor. Here, the Golgi apparatus's role is pivotal in facilitating this interaction, ensuring proper phosphorylation of STING's C-terminal tail region. This action provides an access port for IRF3, marking the Golgi's influence in directing immune responses. The subsequent activation of IRF3 and its downstream effects, including transcription of type I interferon-beta (IFNb) and induction of interferon-stimulated genes (ISGs), reinforces the Golgi apparatus's profound impact on the immune landscape [33,34].
4. Golgi Apparatus and TLR/RLR Signaling
Viral infections have evolved strategies to exploit host factors for their replication within target cells, distinguishing them from bacteria, fungi, and helminths. The distinct molecular patterns present in viral genomes and replication intermediates render them discernible as pathogens, allowing the immune system to detect viral RNA while tolerating self-RNA. This delicate balance forms the basis of an effective antiviral response without inadvertently activating host systems. To achieve this, various families of RNA sensors have been identified. Among them, RIG-I-like receptors (RLRs) function in the cytosol to detect viral RNA, while Toll-like receptors (TLRs) recognize it within endolysosomes.
The Golgi apparatus, a central hub for cellular trafficking and processing, emerges as a pivotal player in the orchestration of these immune pathways. While the focus remains on RLR and TLR signaling, we will elucidate the integral role of the Golgi apparatus in enhancing these processes.
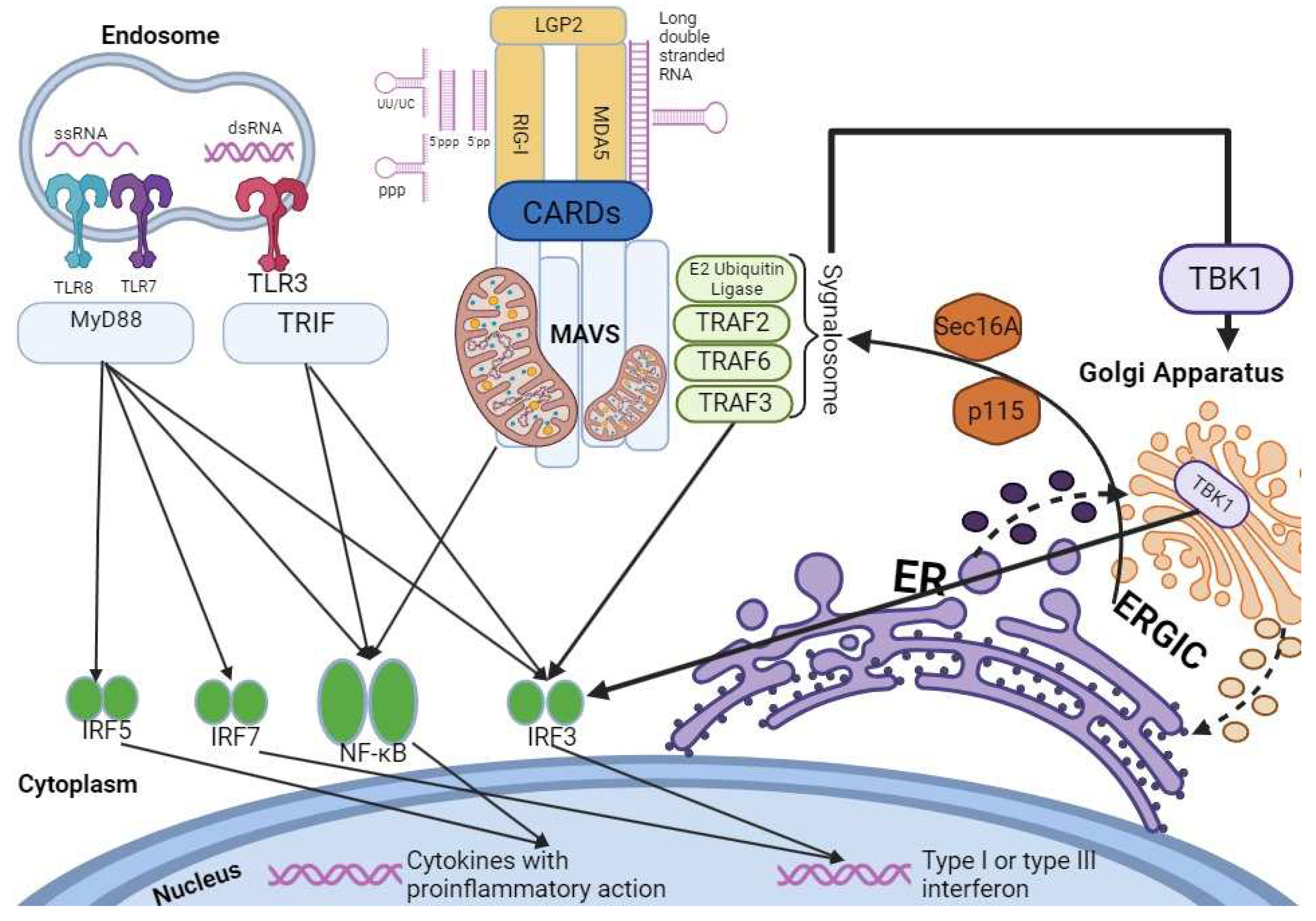
Upon recognizing pathogen-associated molecular pattern (PAMP) RNA, RLRs such as RIG-I and MDA5 undergo conformational changes, releasing their caspase activation and recruitment domains (CARDs) from autorepression. These CARDs then engage an adaptor molecule known as mitochondrial antiviral signaling protein (MAVS). Notably, the Golgi apparatus plays a significant role in these interactions, acting as a platform where MAVS forms prion-like aggregates. These aggregates attract E3 ubiquitin ligases and downstream effector proteins like TNF receptor-associated factor 2 (TRAF2), TRAF3, and TRAF6, forming an active signaling complex termed the "signalosome." This intricate cascade leads to phosphorylation, nuclear translocation of key innate immune transcription factors including interferon regulatory factor 3 (IRF3) and IRF7, and activation of NF-kB. Ultimately, this cascade results in the induction of type I and III interferons, as well as genes encoding proinflammatory cytokines and chemokines, collectively working to restrict viral infections [35].
The Golgi apparatus further comes into focus in the context of TBK1, a central component of this pathway (see Figure 2). TBK1's critical role in type I interferon (IFN) production becomes evident, as its absence significantly diminishes the induction of type I IFNs, which are crucial for antiviral immune responses. Notably, recent studies highlight that upon stimulation of RLR or TLR3 pathways, TBK1 can be targeted to the Golgi apparatus through its interaction with optineurin (OPTN), an adaptor protein binding to ubiquitin [36]. This interaction initiates the assembly of complexes between TBK1 and OPTN, leading to trans-autophosphorylation and activation of TBK1. Subsequently, TBK1 phosphorylates interferon regulatory factor 3 (IRF3), a key step in promoting type I IFN production and triggering an effective antiviral response.
Furthermore, the Golgi apparatus's impact extends to the regulation of interferon responses through TLR activation and intracellular cytoplasmic receptors like protein kinase R (PKR). The intricate network of TNF receptor-associated factors (TRAFs) plays a pivotal role in connecting upstream receptor signals with downstream gene activation. Of special note is TRAF3, which facilitates IRF3 activation through TNF receptor-associated factors, leading to IFN-beta (IFN-b) production [37].
As we delve into the role of the Golgi apparatus in these immune pathways, it becomes evident that the Golgi's dynamic and multifaceted functions extend beyond protein trafficking. Recent investigations have unveiled novel interactors of TRAF3, such as Sec16A and p115, which are integral to the Endoplasmic Reticulum-to-Golgi Vesicular Transport system [38,39,40]. These proteins modulate vesicle assembly and trafficking at the ER-Golgi interface, accentuating the Golgi apparatus's role in immune signaling pathways. The fragmentation of the Golgi apparatus upon activation of the RLH and cytoplasmic DNA sensor pathways further highlights its involvement, as it creates punctate structures that enable TRAF3 colocalization with MAVS, reinforcing its significance in these signaling cascades [36,37,38].
A study conducted by Beachboard DC et al. illustrates RAB1B's vital role in positively regulating the RIG-I pathway, which induces type I interferon (IFN) [41]. Notably, RAB1B's interaction with TRAF3 as an integral component of this pathway underscores its importance in facilitating interactions between TRAF3 and MAVS. This discovery evidence the role of RAB1B as a cellular trafficking protein, simultaneously controlling host responses to viral infections [41].
5. Conclusion
After many years of studying and debating its existence, the Golgi apparatus seems to function not only as a central organelle in vesicular trafficking and protein and lipid transport but also as a platform for innate immunity signaling and subsequent effectors. This review summarizes the new discoveries on how Golgi connects intracellular compartments such as ER, and mitochondria and therefore plays a vital role in cell defense. Although many new studies are revealing the crucial part that this dynamic organelle plays in innate immunity, we need to better understand the detailed mechanisms that regulate the cellular response to pathogens. The NLRP3 Inflammasome formation still needs to be better investigated, and perhaps other roles of Golgi in innate immune pathways will be brought to light by further inquiring the ways that the cellular compartments such as ER and mitochondria communicate with each other to orchestrate cellular defense. Also, the type I Interferon response initiated by activation of Toll-like receptors is vital in limiting viral infections and, as many emerging studies show, is regulated at the Golgi by TBK1, situating Golgi in the first line of cell defense. By better understanding the signaling pathways that stand at the molecular base of innate immunity, we may be able to reveal how exactly can we intervene to potentiate the innate immune system to better fight microbial infection and to also refresh the way we view the immune response as a complex and very thorough regulated molecular process that happens due to collaboration of many intracellular compartments in every infected cell. Finally, continuous research is much needed to reveal ideas and new therapeutical approaches in treating both infectious and inflammatory diseases and, why not, even improve the evolution of autoimmune diseases, a heavy burden for millions of patients around the world.
Author Contributions
Conceptualization, I.M., and I.C.; Data curation, I.M., and I.C.; Methodology, I.M., I.C., A.E.C and R.A.C.B.; Analyzation, A.E.C., R.A.C.B., I.M., I.C.; Supervision, I.C., and I.M.; Validation, I.C.; Writing original draft, I.M., and A.E.C.; Writing review and editing, I.M., A.E.C., R.A.C.B. and I.C. All authors have read and agreed to the published version of the manuscript.
References
- Saceleanu, V.M.; Covache-Busuioc, R.A.; Costin, H.P.; Glavan, L.A.; Ciurea, A.V. An Important Step in Neuroscience: Camillo Golgi and His Discoveries. Cells 2022, 11, 4112. [Google Scholar] [CrossRef]
- Shorter, J.; Warren, G. Golgi Architecture and Inheritance. Annu. Rev. Cell Dev. Biol. 2002, 18, 379–420. [Google Scholar] [CrossRef]
- Tang, D.; Wang, Y. Cell cycle regulation of Golgi membrane dynamics. Trends Cell Biol. 2013, 23, 296–304. [Google Scholar] [CrossRef]
- Boncompain, G.; Weigel, A.V. Transport and sorting in the Golgi complex: multiple mechanisms sort diverse cargo. Curr. Opin. Cell Biol. 2018, 50, 94–101. [Google Scholar] [CrossRef]
- Lieu, Z.Z.; Lock, J.G.; Hammond, L.A.; La Gruta, N.L.; Stow, J.L.; Gleeson, P.A. A trans -Golgi network golgin is required for the regulated secretion of TNF in activated macrophages in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 3351–3356. [Google Scholar] [CrossRef]
- Micaroni, M.; Stanley, A.C.; Khromykh, T.; Venturato, J.; Wong, C.X.F.; Lim, J.P.; et al. Rab6a/a’ Are Important Golgi Regulators of Pro-Inflammatory TNF Secretion in Macrophages. Johannes L, editor. PLoS ONE 2013, 8, e57034. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell. 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, M.M.; Spooner, E.; Hoebe, K.; Beutler, B.; Ploegh, H.L.; Kim, Y.M. The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. J. Cell Biol. 2007, 177, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Brinkmann, M.M.; Paquet, M.E.; Ploegh, H.L. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature. 2008, 452, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, Z.J. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature. 2018, 564, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef]
- Dobbs, N.; Burnaevskiy, N.; Chen, D.; Gonugunta, V.K.; Alto, N.M.; Yan, N. STING Activation by Translocation from the ER Is Associated with Infection and Autoinflammatory Disease. Cell Host Microbe. 2015, 18, 157–168. [Google Scholar] [CrossRef]
- Mukai, K.; Konno, H.; Akiba, T.; Uemura, T.; Waguri, S.; Kobayashi, T. Activation of STING requires palmitoylation at the Golgi. Nat. Commun. 2016, 7, 11932. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.Y. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; et al. Inflammatory caspases are innate immune receptors for intracellular, L. P.S. Nature. 2014, 514, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.K.; Kim, J.K.; Shin, D.M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef]
- Zhang, Z.; Meszaros, G.; He W ting Xu, Y.; De Fatima Magliarelli, H.; Mailly, L. Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J. Exp. Med. 2017, 214, 2671–2693. [Google Scholar] [CrossRef] [PubMed]
- Di, A.; Xiong, S.; Ye, Z.; Malireddi, R.K.S.; Kometani, S.; Zhong, M.; et al. The TWIK2 Potassium Efflux Channel in Macrophages Mediates NLRP3 Inflammasome-Induced Inflammation. Immunity. 2018, 49, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Chi, Z.; Jiang, D.; Xu, T.; Yu, W.; Wang, Z.; et al. Cholesterol Homeostatic Regulator SCAP-SREBP2 Integrates NLRP3 Inflammasome Activation and Cholesterol Biosynthetic Signaling in Macrophages. Immunity. 2018, 49, 842–856. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; et al. Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, V.; Campelo, F. PKD Regulates Membrane Fission to Generate TGN to Cell Surface Transport Carriers. Cold Spring Harb. Perspect. Biol. 2011, 3, a005280. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, N.; Natarajan, K.; Clatworthy, M.R.; Wang, Z.; Germain, R.N. The Adaptor MAVS Promotes NLRP3 Mitochondrial Localization and Inflammasome Activation. Cell 2013, 153, 348–361. [Google Scholar] [CrossRef]
- Park, S.; Juliana, C.; Hong, S.; Datta, P.; Hwang, I.; Fernandes-Alnemri, T.; et al. The Mitochondrial Antiviral Protein MAVS Associates with NLRP3 and Regulates Its Inflammasome Activity. J. Immunol. 2013, 191, 4358–4366. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef]
- Taguchi, T.; Mukai, K.; Takaya, E.; Shindo, R. STING Operation at the ER/Golgi Interface. Front. Immunol. 2021, 12, 646304. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.; Jiang, Q.; Guan, Y.; et al. Golgi apparatus-synthesized sulfated glycosaminoglycans mediate polymerization activation of the cGAMP sensor, S. T.I.N.G. Immunity. 2021, 54, 962–975. [Google Scholar] [CrossRef]
- Gao, P.; Ascano, M.; Wu, Y.; Barchet, W.; Gaffney, B.L.; Zillinger, T.; et al. Cyclic [G(2′,5′)pA(3′,5′)p] Is the Metazoan Second Messenger Produced by DNA-Activated Cyclic GMP-AMP Synthase. Cell 2013, 153, 1094–1107. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, J.; Du, F.; Xu, H.; Sun, L.; Chen, Z.; et al. The Cytosolic DNA Sensor cGAS Forms an Oligomeric Complex with DNA and Undergoes Switch-like Conformational Changes in the Activation Loop. Cell Rep. 2014, 6, 421–430. [Google Scholar] [CrossRef]
- Shang, G.; Zhu, D.; Li, N.; Zhang, J.; Zhu, C.; Lu, D.; et al. Crystal structures of STING protein reveal basis for recognition of cyclic, d. i.-G.M.P. Nat Struct Mol Biol. 2012, 19, 725–727. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, E.S.; Marinho, F.V.; de Queiroz, N.M.G.P.; Antunes, M.M.; Oliveira, S.C. Impact of STING Inflammatory Signaling during Intracellular Bacterial Infections. Cells 2021, 11, 74. [Google Scholar] [CrossRef] [PubMed]
- Agalioti, T.; Lomvardas, S.; Parekh, B.; Yie, J.; Maniatis, T.; Thanos, D. Ordered Recruitment of Chromatin Modifying and General Transcription Factors to the IFN-β Promoter. Cell 2000, 103, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef]
- Chow, K.T.; Gale, M.; Loo, Y.M. RIG-I and Other RNA Sensors in Antiviral Immunity. Annu. Rev. Immunol. 2018, 36, 667–694. [Google Scholar] [CrossRef]
- Pourcelot, M.; Zemirli, N.; Silva Da Costa, L.; Loyant, R.; Garcin, D.; Vitour, D.; et al. The Golgi apparatus acts as a platform for TBK1 activation after viral RNA sensing. BMC Biol. 2016, 14, 69. [Google Scholar] [CrossRef] [PubMed]
- Oganesyan, G.; Saha, S.K.; Guo, B.; He, J.Q.; Shahangian, A.; Zarnegar, B.; et al. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature 2006, 439, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.; VanScoy, S.; Cheng, T.F.; Gomez, D.; Reich, N.C. IRF-3-dependent and augmented target genes during viral infection. Genes. Immun. 2008, 9, 168–175. [Google Scholar] [CrossRef]
- van Zuylen, W.J.; Doyon, P.; Clément, J.F.; Khan, K.A.; D’Ambrosio, L.M.; Dô, F.; et al. Proteomic profiling of the TRAF3 interactome network reveals a new role for the ER-to-Golgi transport compartments in innate immunity. PLoS Pathog. 2012, 8, e1002747. [Google Scholar] [CrossRef]
- Iinuma, T.; Shiga, A.; Nakamoto, K.; O’Brien, M.B.; Aridor, M.; Arimitsu, N.; et al. Mammalian Sec16/p250 Plays a Role in Membrane Traffic from the Endoplasmic Reticulum. J. Biol. Chem. 2007, 282, 17632–17639. [Google Scholar] [CrossRef]
- Beachboard, D.C.; Park, M.; Vijayan, M.; Snider, D.L.; Fernando, D.J.; Williams, G.D.; et al. The small GTPase RAB1B promotes antiviral innate immunity by interacting with TNF receptor–associated factor 3 (TRAF3). J. Biol. Chem. 2019, 294, 14231–14240. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
PLC releases Ca2+ from the ER through inositol-1, 4, 5-trisphosphate (InsP3) Receptor.Ca 2+ accumulates in the mitochondria which may lead to mitochondrial damage.(1) Damaged mitochondria release several factors that trigger activation of the NLRP3 inflammasome.(2) NLRP3 was shown to directly bind to mitochondria-associated ER membranes (MAMs).(black arrow) The other product of PLC activation, diacylglycerol (DAG), accumulates in the Golgi (3) and increases the activity of PKD (protein kinase D) which then phosphorylates NLRP3-Inflammasome and activates it (3*), releasing it from MAMs.(4).
Figure 1.
PLC releases Ca2+ from the ER through inositol-1, 4, 5-trisphosphate (InsP3) Receptor.Ca 2+ accumulates in the mitochondria which may lead to mitochondrial damage.(1) Damaged mitochondria release several factors that trigger activation of the NLRP3 inflammasome.(2) NLRP3 was shown to directly bind to mitochondria-associated ER membranes (MAMs).(black arrow) The other product of PLC activation, diacylglycerol (DAG), accumulates in the Golgi (3) and increases the activity of PKD (protein kinase D) which then phosphorylates NLRP3-Inflammasome and activates it (3*), releasing it from MAMs.(4).
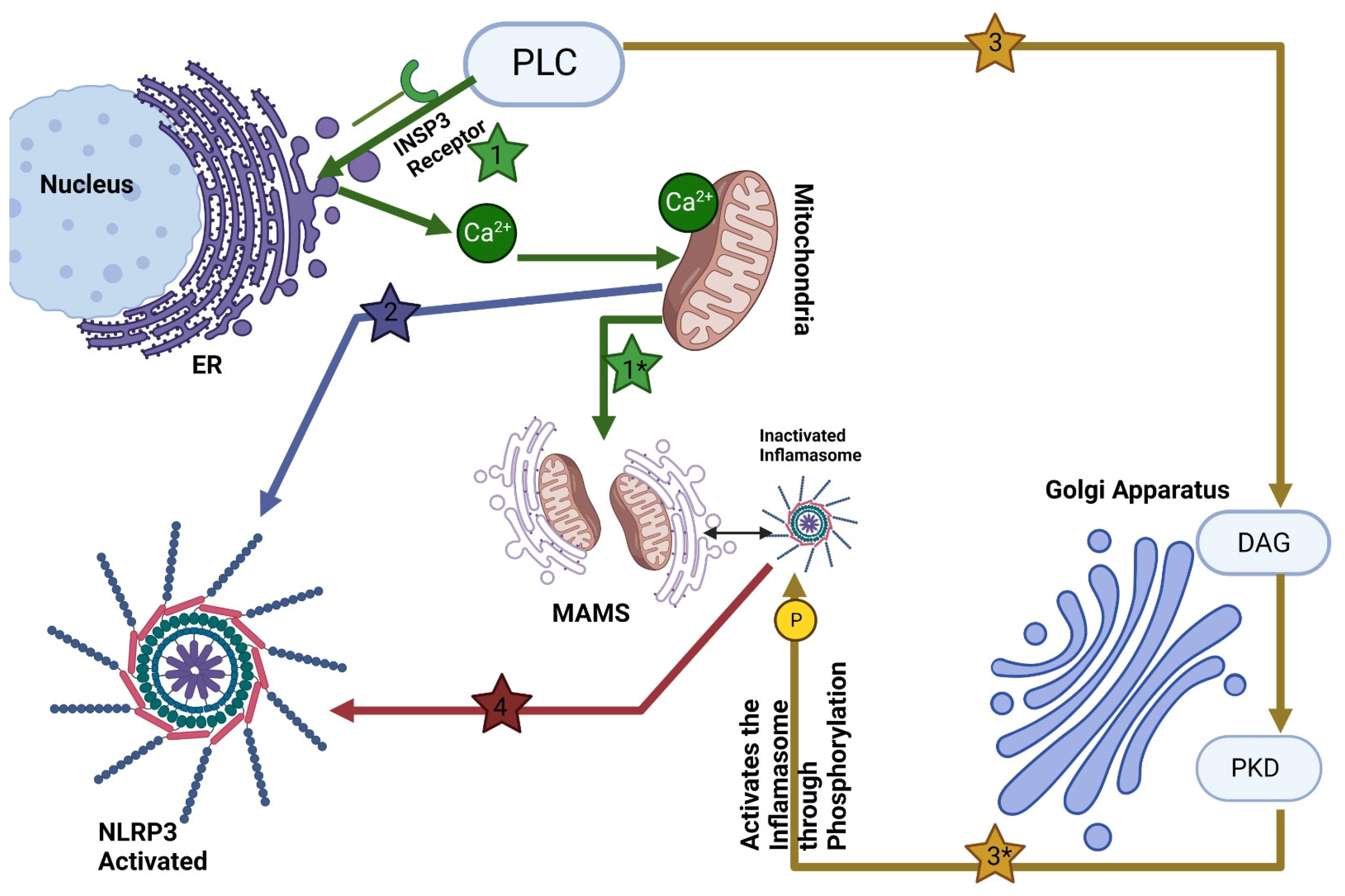
Figure 2.
TLR3, TLR7, and TLR8, which are Toll-like receptors (TLRs), are located predominantly within endosomes. TLR3 specifically detects double-stranded RNA (dsRNA), while TLR7 and TLR8 can be activated by single-stranded RNA (ssRNA) as well as imidazoquinoline compounds. TLR3 uses the adapter protein TRIF to activate transcription factors IRF3 and NF-kB; MyD88 can signal activation of transcription factors IRF3, IRF5, IRF7 as well as NF-kB depending on cell type. RLRs (RIG-I-like receptors) can be found within the cytosol. RIG-I can identify short dsRNA and ssRNA motifs with specific sequences such as poly-U/UC (containing sequences of uracil interspersed with occasional cytosines) or adenine/uracil (AU) rich sequences with 5' triphosphate (5'ppp) or possibly monophosphate (5'pp) termination sites. MDA5 RLR recognizes long and structured dsRNA. When binding their ligands, RLRs change their conformation and release caspase activators and recruitment domains (CARDs). CARDs then engage the adaptor protein MAVS (mitochondrial antiviral signaling protein). MAVS then form prion-like aggregates at the Golgi which attract E3 ubiquitin ligases and TNF receptor-associated factor 2 (TRAF2), TRAF3, and TRAF6, forming the "signalosome" and initiate downstream signaling pathways involving IRF3 and NF-kB which lead to phosphorylation and nuclear translocation of IRF 3, IRF 7 and NF-kB activation and also activation of TBK-I. TBK1 phosphorylates interferon regulatory factor 3 (IRF3), a key step in promoting type I IFN production. TRAF 3 also activates IRF 3 leading to INF beta production. This cascade results in the induction of type I and III interferons, as well as genes encoding proinflammatory cytokines and chemokines.
Figure 2.
TLR3, TLR7, and TLR8, which are Toll-like receptors (TLRs), are located predominantly within endosomes. TLR3 specifically detects double-stranded RNA (dsRNA), while TLR7 and TLR8 can be activated by single-stranded RNA (ssRNA) as well as imidazoquinoline compounds. TLR3 uses the adapter protein TRIF to activate transcription factors IRF3 and NF-kB; MyD88 can signal activation of transcription factors IRF3, IRF5, IRF7 as well as NF-kB depending on cell type. RLRs (RIG-I-like receptors) can be found within the cytosol. RIG-I can identify short dsRNA and ssRNA motifs with specific sequences such as poly-U/UC (containing sequences of uracil interspersed with occasional cytosines) or adenine/uracil (AU) rich sequences with 5' triphosphate (5'ppp) or possibly monophosphate (5'pp) termination sites. MDA5 RLR recognizes long and structured dsRNA. When binding their ligands, RLRs change their conformation and release caspase activators and recruitment domains (CARDs). CARDs then engage the adaptor protein MAVS (mitochondrial antiviral signaling protein). MAVS then form prion-like aggregates at the Golgi which attract E3 ubiquitin ligases and TNF receptor-associated factor 2 (TRAF2), TRAF3, and TRAF6, forming the "signalosome" and initiate downstream signaling pathways involving IRF3 and NF-kB which lead to phosphorylation and nuclear translocation of IRF 3, IRF 7 and NF-kB activation and also activation of TBK-I. TBK1 phosphorylates interferon regulatory factor 3 (IRF3), a key step in promoting type I IFN production. TRAF 3 also activates IRF 3 leading to INF beta production. This cascade results in the induction of type I and III interferons, as well as genes encoding proinflammatory cytokines and chemokines.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



