
Submitted:
28 December 2023
Posted:
28 December 2023
You are already at the latest version
Abstract
Endothelial function is regulated by a myriad of complex pathways. In our previous study, the expression of miR-378a-3p was shown to inhibit endothelial autophagy and impair endothelial function. However, the mechanism underlying the effect of miR-378a-3p in endothelial cells remains to be elucidated. By employing an in silico approach, Protein Disulfide Isomerase 4 (PDIA-4) was identified as a target of miR-378-3p; however, its’ role in endothelial cells is not known. We here report that endothelial cell-specific loss of PDIA-4 leads to impaired autophagic flux accompanied by loss of endothelial function and apoptosis. Endothelial cell-specific loss of PDIA-4 also induced marked changes in endothelial cells’ architecture, accompanied with the loss of endothelial markers and gain of mesenchymal markers consistent with endothelial-to-mesenchymal transition (EndMT). Loss of PDIA-4 activates TGF-signaling, and inhibition of TGF-signaling inhibits EndMT in PDIA-4 silenced endothelial cells in vitro. Our findings help elucidate the role of PDIA-4 in endothelial autophagy and endothelial function and provide a potential target to modulate and/or limit autophagy and EndMT in (patho-)physiological conditions.
Keywords:
MicroRNA
; PDIA‐4
; Autophagy
; Apoptosis
; Endothelial‐to‐mesenchymal transition
; Endothelial function
; and Cardiovascular diseases
Introduction
Autophagy or “self-eating” is a highly conserved cellular process present in all eukaryotic cells. It clears damaged proteins or organelles and recycles cellular materials for reuse. Endothelial cells line the innermost layer of blood vessels and have critical roles in maintaining vascular homeostasis [1]. Imbalanced autophagy can lead to endothelial dysfunction and cardiovascular diseases [2,3,4,5,6]. Autophagy is a tightly regulated process in endothelial cells, and epigenetic regulation, particularly by microRNA (miRNA), is well established [3]. We have previously shown that genetic inhibition of autophagy by loss of autophagy-related gene 7 (ATG7) leads to impaired autophagy and induces TGF-β-mediated endothelial-to-mesenchymal transition (EndMT) in vitro and in vivo [7]. During EndMT, the endothelial cells acquire a migratory phenotype and begin to express different mesenchymal markers, including fibroblast-specific protein-1 (FSP-1), N-cadherin, α smooth muscle actin (α SMA), while downregulating endothelial cell markers including platelet endothelial cell adhesion molecule (CD31) and V-cadherin [8,9]. Finally, the cells lose endothelial phenotype and gain a mesenchymal phenotype with increased motility and contractile activities [10,11].
We identified miRNA-378-3p as the most up-regulated miRNA following the loss of ATG7 in endothelial cells, whether via genetic loss or pharmacologic inhibition of autophagy [12]. Using an in silico approach, we then identified Protein Disulfide Isomerase 4 (PDIA-4) as a target of miRNA-378-3p in endothelial cells [12,13]. Protein Disulfide Isomerases (PDIs) function as an oxidoreductase and as a chaperon that assists with the formation of disulfide bonds to help protein fold into its final conformation [14]. PDIA-4 belongs to the PDI family and has been shown to participate in platelet activation, endoplasmic reticulum stress response (ERS response), DNA repair machinery and tumor progression [15]. ERS response is believed to be a “self-protection” mechanism in response to stress, for example, the accumulation of misfolded protein [15]. This response can stimulate the formation of autophagosomes and trigger autophagy to clear damaged proteins [16]. Kyani et al. reported that inhibiting PDIA-4 in brain cancer could lead to autophagy-mediated ferroptosis [17]; however, there is currently no information about the molecular link between PDIA-4, endothelial autophagy and EndMT.
Methods
Cell Culture - Human umbilical vein endothelial cells [HUVECs [passage 5-7, (Lonza)], a common in vitro standard model to study endothelial cells [8,18,19], were cultured in endothelial growth medium (EGM-2, Lonza) supplemented with growth factors, serum and antibiotics at 37o C in humidified 5% CO2 incubator. HUVECs were transfected with 5nm PDIA-4 silencing RNA (siPDIA-4) (Santa Cruz: Cat # sc-44571) using a standard reverse transfection protocol (Lipofectamine ® 3000). RNAs or proteins were collected 24, 48 or 72-hrs post-transfection. TGFβ inhibitor experiments were conducted using the drug SB431542 (10uM). Cells were reverse transfected as previously described [18], incubated in complete media for 72hrs, and then treated with 10uM SB431542 for another 48hr.
cDNA Synthesis and Real-Time Quantitative PCR - To measure the transcript expression level of different genes, quantitative real-time PCR (RT-qPCR) was used. Total RNA was extracted from HUVECs in Trizol reagent (Invitrogen). RNA concentration was measured using the NanoDrop (Thermo Scientific). Complementary DNA (cDNA) was synthesized using the Quantitect Reverse Transcription Kit (Qiagen), followed by a polymerase chain reaction using Thermal Cycler Real-Time PCR machine (Eppendorf). RT-qPCR was conducted with the mixture of samples’ RNA, SYBR Select Master Mix (Applied Biosystems) and forward and reverse primers of PDIA-4, Cyclin-dependent Kinase Inhibitor 1a (P21) [18], Transforming Growth Factor-beta Receptor, Type 1 (TGFBR1), TGFBR2, SMAD3, SMAD4, SLUG, Transforming Growth Factor β (TGF β), Fibroblast Specific Protein 1 (FSP-1), Tek Tyrosine Kinase, Endothelial (Tie-2), N-Cadherin, CD31, Connective Tissue Growth Factor (CTGF) and GAPDH [9] (Table 1). RT-qPCR was run using the QuantStudio®3 Real-Time PCR Instrument (Applied Biosystems). The comparative Delta Delta CT method was employed for data analysis [20].
Western Blot - Cell lysates were first collected and extracted in RIPA buffer (Sigma) 24, 48, and 72-hrs post-transfection with either siPDIA-4 or its scrambled control. Equal amounts of protein were loaded on to SDS-polyacrylamide gels and transferred to the PVDF membranes (Thermo Fisher). Membranes were blocked for 1 hour in 1X TBS with 3% BSA and incubated overnight at 40 C with primary antibodies. The primary antibodies used in this study include p21 (Cell Signaling #2947S), Cleaved-Caspase-3 (Cell Signaling #9664S), LC3 I/II (Cell Signaling #4600IAP), GAPDH (Cell Signaling #5174S), p-SMAD3 (ABclonal #51451), SMAD3 (ABclonal #28379), Βeta Actin (Cell Signaling #4970S), Βeta Tubulin (Proteintech Cat#66240-1-Ig), BAX (Cell Signaling #2772S), BCL-2 (Cell Signaling #3498S), CD31 (ABclonal Cat#A4900), α smooth muscle actin (ABclonal Cat#A17910), N-Cadherin (ABclonal Cat#A19083), PDIA-4 (Cell Signaling #5033S), and P53 (Santa Cruz #sc-126). Proteins were then incubated with Goat anti-Rabbit secondary antibody (Enzo ADI-SAB-300-J) for 2 hrs at room temperature. Bands were visualized with the ECL substrates using a chemiluminescence channel and 700 channels in the LiCor Fc Odyssey system. The bands were quantified using LiCor Fc Odyssey inbuilt software.
Proliferation Assay - To analyze the proliferation of HUVECs after transfection with siPDIA-4 and respective scrambled controls, a colorimetric proliferation assay with WST-1 reagent was performed according to instructions from the manufacturer (Roche Applied Science, Rotkreuz, Switzerland, Cat. No. 11644807001). 1.5x105 cells/ml concentration per well were seeded in the 6-well assay plate and transfected with siPDIA-4 and scrambled control as described before. Cells were incubated with warm EGM-2 at 370C, 5% CO2 for 72 hrs and collected with trypsin. 100 μl cells were reseeded in triplicates for each biological replicate into 96 well plates, and 10 μl of WTS-1 reagent was added to each well. Cells were incubated with warm EGM-2 at 370C, 5% CO2 for 4 hrs and absorbance was measured at 440 nm. WST-1 is a red tetrazolium salt and is cleaved by metabolically active cells to formazan, which is dark red; by quantifying the amount of formazan formed, we can measure the amount of proliferating cells present [21].
Migration Assay - Briefly, 1.2-1.5x105 cells/ml concentration per well were seeded in the 6-well assay plate and transfected with siPDIA-4 or scrambled control as described previously. Transfected cells were incubated with warm EGM-2 media at 370C, 5% CO2 for 24 hrs or until 90% confluency was reached. A straight scratch was made in the middle of the cell monolayer using a p200 pipette tip, followed by washing with 1X PBS and incubation with low-serum (1%) media [22]. Cells were imaged immediately at T0 using phase contrast microscopy. Cells were then put back into the incubator and imaged every 4 hrs up to 20 hrs. The percent of open wound area at each time point was calculated using the ImageJ wound healing tool [23].
Angiogenesis Assay - In Vitro Angiogenesis Assay Kit (Millipore #ECM625) was used as instructed by the manufacturer [18]. Briefly, transfected cells were cultured on the 96-well plate coated with ECMatrix provided by the kit and subsequently evaluated for tube formation abilities as instructed by the manufacturer. Cells were imaged at multiple time points. Tubes were quantified using the Angiogenesis tool on ImageJ.
Immunofluorescence - Immunofluorescence experiments were carried out in 4-chamber microscopy slides performed as previously described [9]. Immunofluorescence signals from CD31 (Cell Signaling #3528S) and αSMA (Cell Signaling #19245S) staining were visualized using Alexa fluor-tagged anti-mouse (Fisher Scientific # A21202) and Alexa fluor-tagged anti-rabbit (Fisher Scientific # A11034) secondary antibody, respectively. Fluorescent microscopy images were captured using the Zeiss LSM700 confocal microscope and ZEN imaging software was utilized for image processing.
Statistical Analysis - Data are expressed as the mean ± SD. Student’s t-test was applied when the means of two groups were compared. An ANOVA with Tukey’s post-hoc correction was applied when the means of more than two groups were compared using GraphPad-Prism software. A p-value <0.05 was considered to indicate statistical significance.
Results
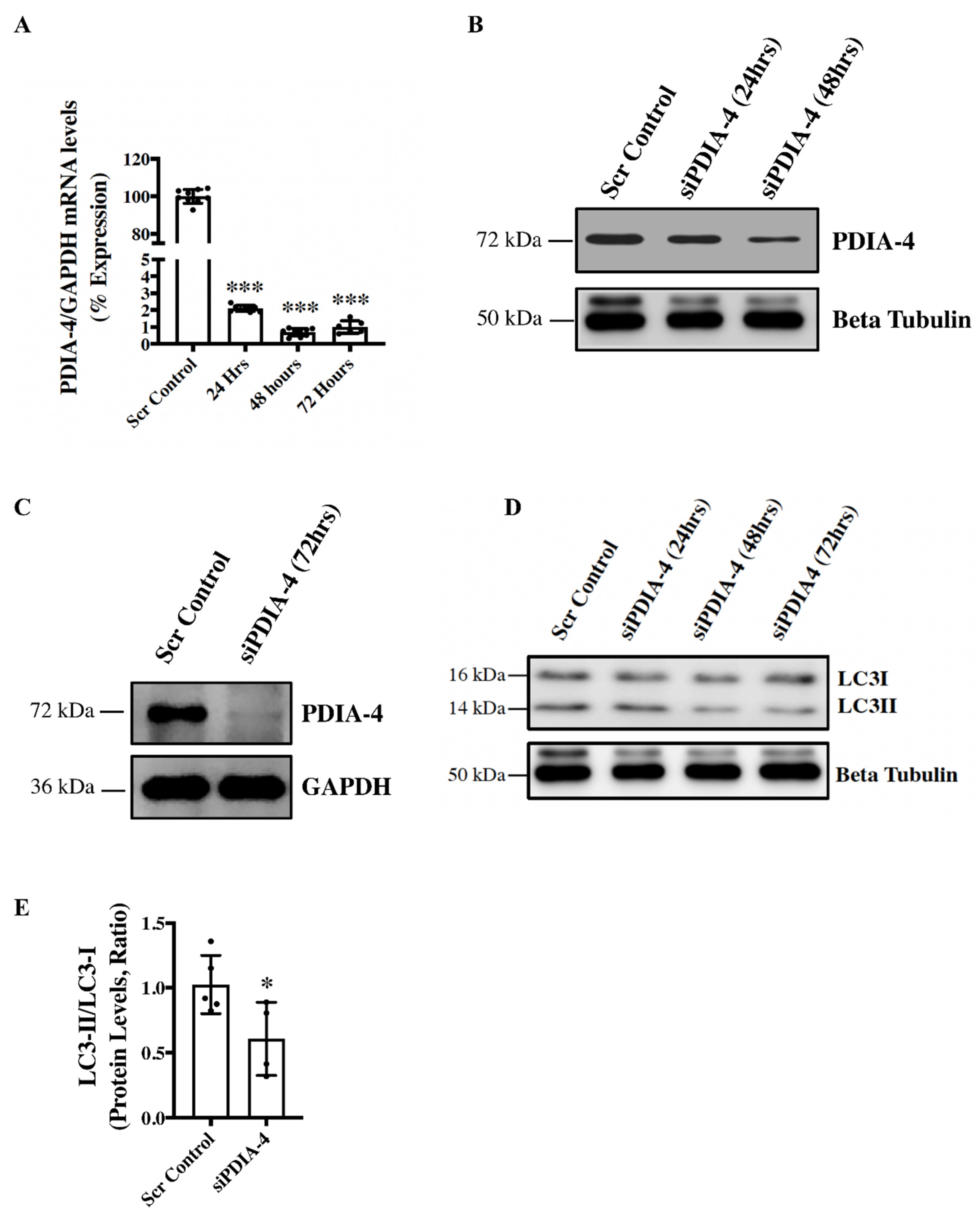
Loss of PDIA-4 inhibits autophagy in endothelial cells - To investigate the role of PDIA-4 in the endothelium, endothelial cells were transfected with either siPDIA-4 or scrambled control for 24, 48 or 72hrs, and cells were collected for RNA and protein extraction to perform qPCR and western blot, respectively. Our qPCR and western blot data on PDIA-4 confirmed successful silencing at both the transcript (Figure 1A) and the protein level, respectively (Figure 1B, C). Since overexpression of miRNA-378-3P impaired autophagy and miRNA-378-3P directly targets PDIA-4 [12], we next tested whether PDIA-4 affects autophagic activity in endothelial cells. Autophagic flux is commonly evaluated by measuring the ratio between LC3-II and LC3-I protein levels, and a lower LC3-II/LC3-I ratio indicates autophagic impairment [7]. Our western blot data showed a gradual decreased expression of LC3-II and increased expression of LC3-I, with an overall reduced LC3-II/LC3-I ratio indicating impaired autophagy in PDIA4-silenced in comparison to control endothelial cells (Figure 1D, E).
Figure 1.
Loss of PDIA-4 inhibits autophagy in endothelial cells. HUVECs were reverse transfected with either siPDIA-4 or scrambled control for 24, 48 or 72hrs. RNA and proteins were extracted, and (A) qPCR was conducted targeting PDIA-4 and GAPDH (n=3 in triplicate). (B, C) Western blot was performed for PDIA-4 and beta-tubulin or GAPDH as loading control. (D) Western blot was performed targeting LC3 and beta-tubulin (n=4-5). (E) Western blot data were quantified and then analyzed using the Student’s T-test, and data were expressed as mean ± SD. *represents p<0.05, ***represents p<0.001 versus respective Scr Cont.
Figure 1.
Loss of PDIA-4 inhibits autophagy in endothelial cells. HUVECs were reverse transfected with either siPDIA-4 or scrambled control for 24, 48 or 72hrs. RNA and proteins were extracted, and (A) qPCR was conducted targeting PDIA-4 and GAPDH (n=3 in triplicate). (B, C) Western blot was performed for PDIA-4 and beta-tubulin or GAPDH as loading control. (D) Western blot was performed targeting LC3 and beta-tubulin (n=4-5). (E) Western blot data were quantified and then analyzed using the Student’s T-test, and data were expressed as mean ± SD. *represents p<0.05, ***represents p<0.001 versus respective Scr Cont.
Loss of PDIA-4 impairs endothelial functions – Angiogenesis, migration, and proliferation are essential functions of endothelial cells that play an integral role in the maintenance of optimal vascular tone [24]. The in vitro angiogenesis assay is a surrogate for studying angiogenesis in cultured endothelial cells [25]. Loss of PDIA-4 resulted in a significant reduction in mesh area (Figure 2 A, B), number of nodes per field (Figure 2C) and tube length per field (Figure 2D), overall indicating reduced angiogenic potential in comparison to control endothelial cells. Migratory capacity is another essential function of endothelial cells, and our migration assay showed significantly reduced migration of PDIA-4-silenced endothelial cells in comparison to control endothelial cells (Figure 2E, F). A proliferation assay was performed to assess the proliferative ability of endothelial cells. Loss of PDIA-4 resulted in a significant reduction in cell proliferation compared to control cells (Figure 2G). In summary, loss of PDIA-4 led to reduced endothelial function in comparison to control endothelial cells.
Figure 2.
Loss of PDIA-4 impairs endothelial functions. HUVECs were transfected with either siPDIA-4 or scrambled control for 72hrs. (A-D) Cells were extracted and seeded on 96-well plates on Matrigel according to the manufacturer’s guidelines. Cells were imaged after 4hr incubation at 37o C in a humidified 5% CO2 incubator. Tubes were quantified using the ImageJ angiogenesis tool (n=3 in triplicates). (E, F) A scratch was made using a p200 pipette tip in the middle of the cell monolayer, and cells were incubated in low serum media (MCDB131 + 1% FBS). Cells were imaged immediately at T0 using phase contrast microscopy, then put back into the incubator and imaged every 4 hrs up to 20 hrs. The percent of open wound area at each time point was calculated using the ImageJ wound healing tool. Migration (the velocity of the cells to close the entire scratch area) was calculated (n=15-17). (G) HUVECS were transfected with siPDIA-4 or scrambled control into 96-well plates for 72 hours (n=9), and 10 μl of WST-1 reagent was added to each well. Cells were incubated with warm EGM2 at 370C, 5% CO2 for 4 hrs and absorbance was measured at 440 nm. HUVECs were reverse transfected with siPDIA-4 or scrambled control and incubated in complete EGM-2 medium for 72hrs. Protein was extracted, and (H) western blot was performed targeting p21 and beta-actin (loading control) (n=7-9). (I) The bands were quantified using LiCor Fc Odyssey inbuilt software. Data were analyzed using Student’s T-test and expressed as mean ± SD. *represents p<0.05, **represents p<0.01, and ***represents p<0.001 versus respective vehicle or Scr Cont.
Figure 2.
Loss of PDIA-4 impairs endothelial functions. HUVECs were transfected with either siPDIA-4 or scrambled control for 72hrs. (A-D) Cells were extracted and seeded on 96-well plates on Matrigel according to the manufacturer’s guidelines. Cells were imaged after 4hr incubation at 37o C in a humidified 5% CO2 incubator. Tubes were quantified using the ImageJ angiogenesis tool (n=3 in triplicates). (E, F) A scratch was made using a p200 pipette tip in the middle of the cell monolayer, and cells were incubated in low serum media (MCDB131 + 1% FBS). Cells were imaged immediately at T0 using phase contrast microscopy, then put back into the incubator and imaged every 4 hrs up to 20 hrs. The percent of open wound area at each time point was calculated using the ImageJ wound healing tool. Migration (the velocity of the cells to close the entire scratch area) was calculated (n=15-17). (G) HUVECS were transfected with siPDIA-4 or scrambled control into 96-well plates for 72 hours (n=9), and 10 μl of WST-1 reagent was added to each well. Cells were incubated with warm EGM2 at 370C, 5% CO2 for 4 hrs and absorbance was measured at 440 nm. HUVECs were reverse transfected with siPDIA-4 or scrambled control and incubated in complete EGM-2 medium for 72hrs. Protein was extracted, and (H) western blot was performed targeting p21 and beta-actin (loading control) (n=7-9). (I) The bands were quantified using LiCor Fc Odyssey inbuilt software. Data were analyzed using Student’s T-test and expressed as mean ± SD. *represents p<0.05, **represents p<0.01, and ***represents p<0.001 versus respective vehicle or Scr Cont.
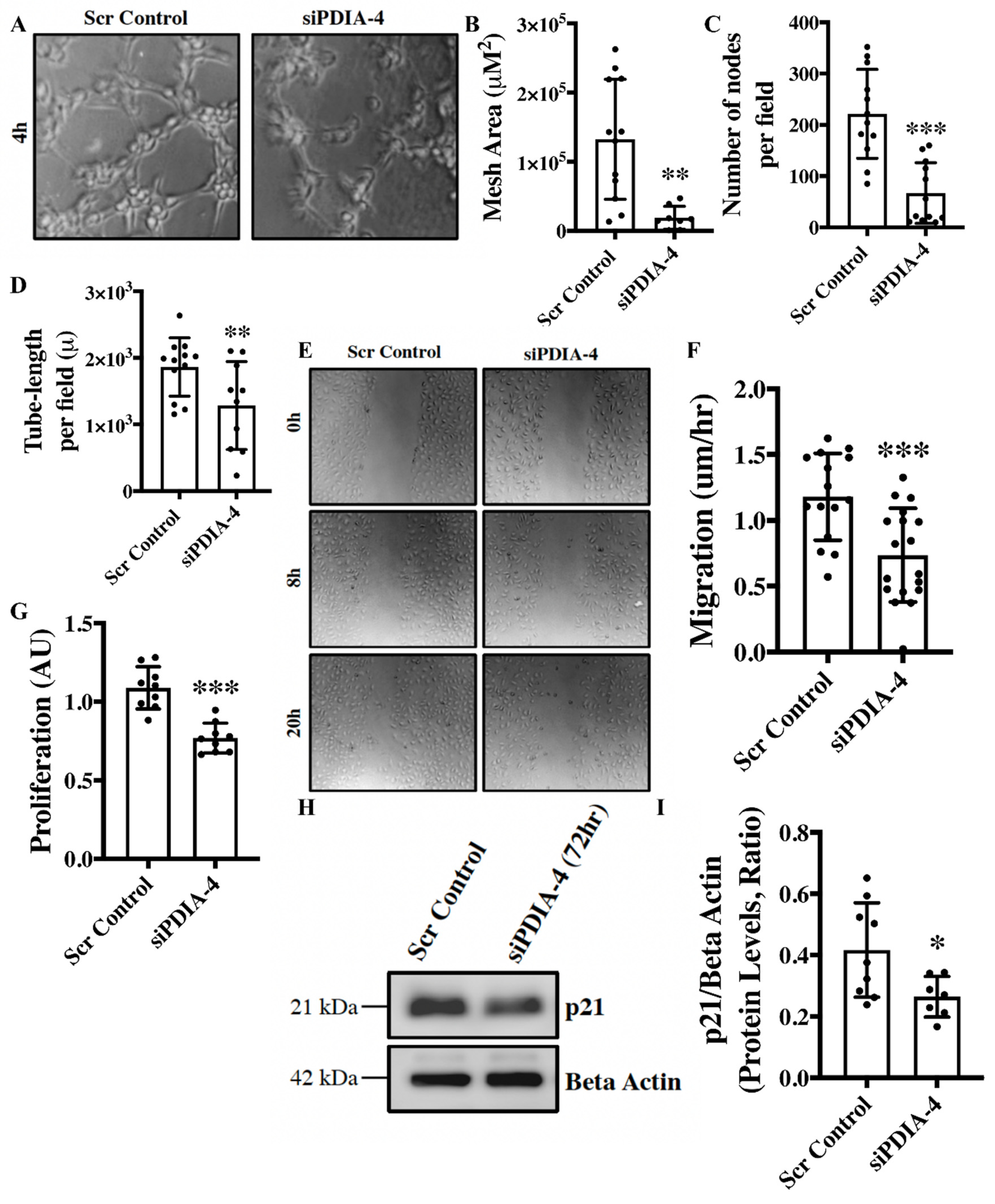
Effect of loss of PDIA-4 on regulators of endothelial functions – Nitric oxide (NO) produced by the activated endothelial NO synthase (eNOS, ser1177) is a fundamental determinant of endothelial function, and Akt mediates the activation of eNOS [26]. Endothelial cells constitutively express eNOS [26]. Accordingly, we next measured eNOS and AKT expression/activation. To our surprise, the loss of PDIA-4 did not show any significant difference in eNOS or AKT activation/expression in PDIA-4-silenced in compared to control endothelial cells (Supp. Figure 1A-E). To understand the mechanism behind the observed reduction of cellular proliferation in PDIA-4-silenced endothelial cells, we measured the expression level of the cell cycle inhibitor p21 in endothelial cells [27]. However, again, to our surprise, p21 was significantly down-regulated in PDIA-4-silenced endothelial cells in comparison to control endothelial cells (Figure 2H, I). Overall, these data indicate that loss of PDIA-4-associated impaired endothelial function was associated with reduced eNOS expression/activation.
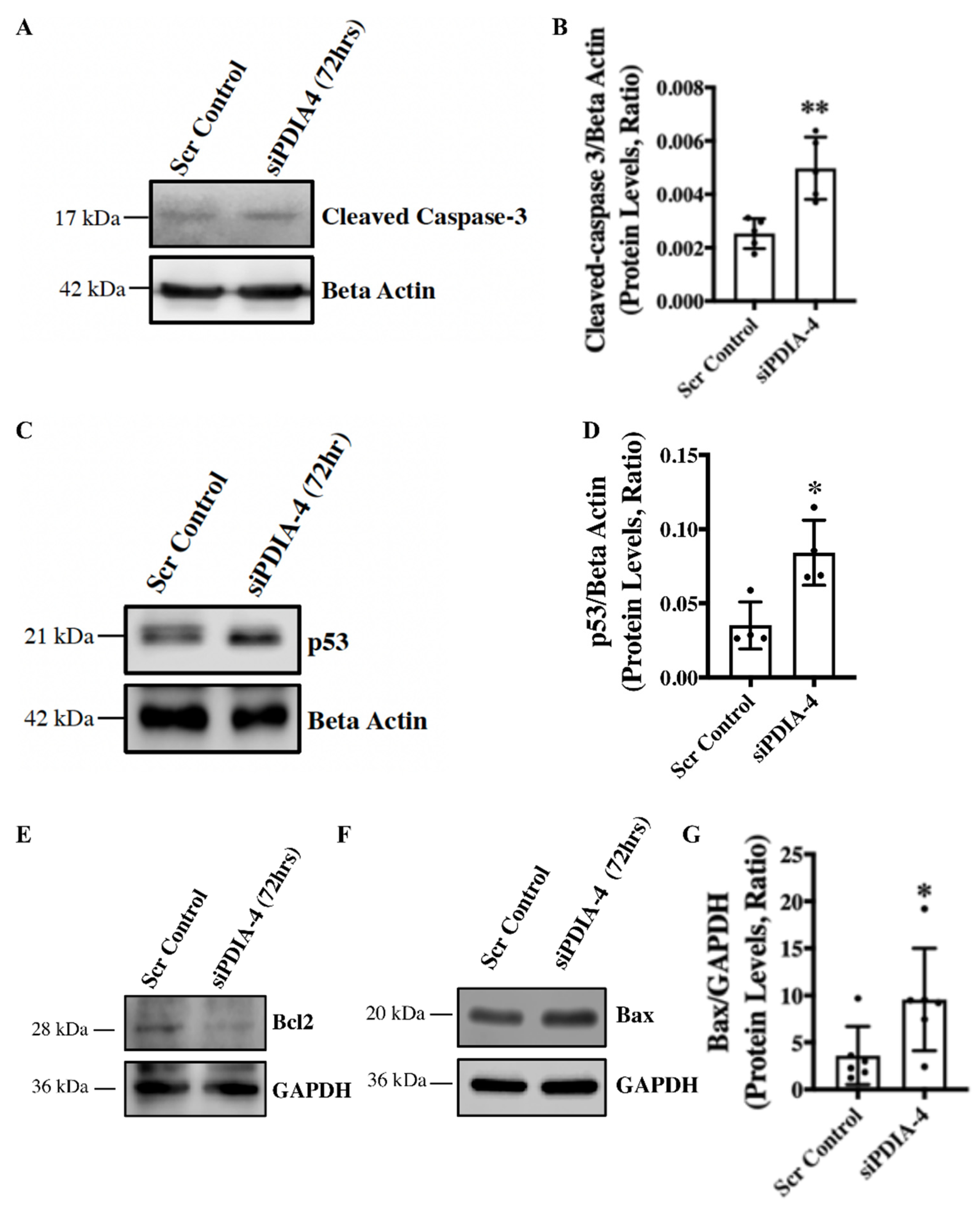
Loss of PDIA-4 induced p53-mediated apoptosis in endothelial cells - Apoptosis is an alternate cell death system to autophagy [28]. Instead of recycling selective organelles and cellular materials for future use, apoptosis destroys unwanted cells and results in cell necrosis or bursting of the cells. It is known that autophagy and apoptosis can be triggered by the same stress signals, and there is a crosstalk between the critical proteins involved in these two processes [29]. We observed impaired autophagy in PDIA-4-deficient endothelial cells, which was further associated with reduced cell proliferation. We next measured apoptosis in PDIA-4-silenced endothelial cells by measuring the cleaved caspase-3 levels. Our western blot data showed an increased level of cleaved caspase-3 protein expression, indicating increased apoptosis in PDIA-4-silenced endothelial cells in comparison to the controls (Figure 3A, B). P53 protein is an essential regulator of apoptosis [30], and to test whether the increased apoptosis in PDIA-4-silenced endothelial cells is mediated by p53, we measured p53 expression levels in PDIA-4-silenced and control endothelial cells. We observed a significant up-regulation of p53 expression in PDIA-4-silenced in comparison to control endothelial cells, indicating p53-mediated apoptosis in PDIA-4-silenced endothelial cells (Figure 3C, D). Furthermore, p53 is a transcription factor, and it induces apoptosis by regulating downstream targets like pro-apoptotic Bax and by shifting the ratio of the level of Bax to that of the pro-survival molecule Bcl-2 [31,32]. Accordingly, we measured the expression level of Bax and Bcl-2 in PDIA-4-silenced and control endothelial cells. In line, we did observe an increase in Bax expression and a reduced pro-survival Bcl-2 expression in PDIA-4-silenced endothelial cells (Figure 3E-G), shifting the overall balance from pro-survival to pro-apoptotic mode in PDIA-4-silenced endothelial cells.
Figure 3.
Loss of PDIA-4-induced p53-mediated apoptosis in endothelial cells. HUVECs were reverse transfected with siPDIA-4 or scrambled control for 72hrs. Proteins were extracted, and western blot was performed targeting (A) cleaved caspase-3 and beta-actin (loading controls), (C) p53 and beta-actin (loading controls), and (E) Bcl2, (G) Bax and GAPDH (loading controls). (B, D, G) The bands were quantified using LiCor Fc Odyssey inbuilt software (n=5-6). Data were analyzed using Student’s T-test. N = 3-4. Data are expressed as mean ± SD. *represents p<0.05 and **represents p<0.01 versus respective vehicle or Scr Cont. ns=non-significant.
Figure 3.
Loss of PDIA-4-induced p53-mediated apoptosis in endothelial cells. HUVECs were reverse transfected with siPDIA-4 or scrambled control for 72hrs. Proteins were extracted, and western blot was performed targeting (A) cleaved caspase-3 and beta-actin (loading controls), (C) p53 and beta-actin (loading controls), and (E) Bcl2, (G) Bax and GAPDH (loading controls). (B, D, G) The bands were quantified using LiCor Fc Odyssey inbuilt software (n=5-6). Data were analyzed using Student’s T-test. N = 3-4. Data are expressed as mean ± SD. *represents p<0.05 and **represents p<0.01 versus respective vehicle or Scr Cont. ns=non-significant.
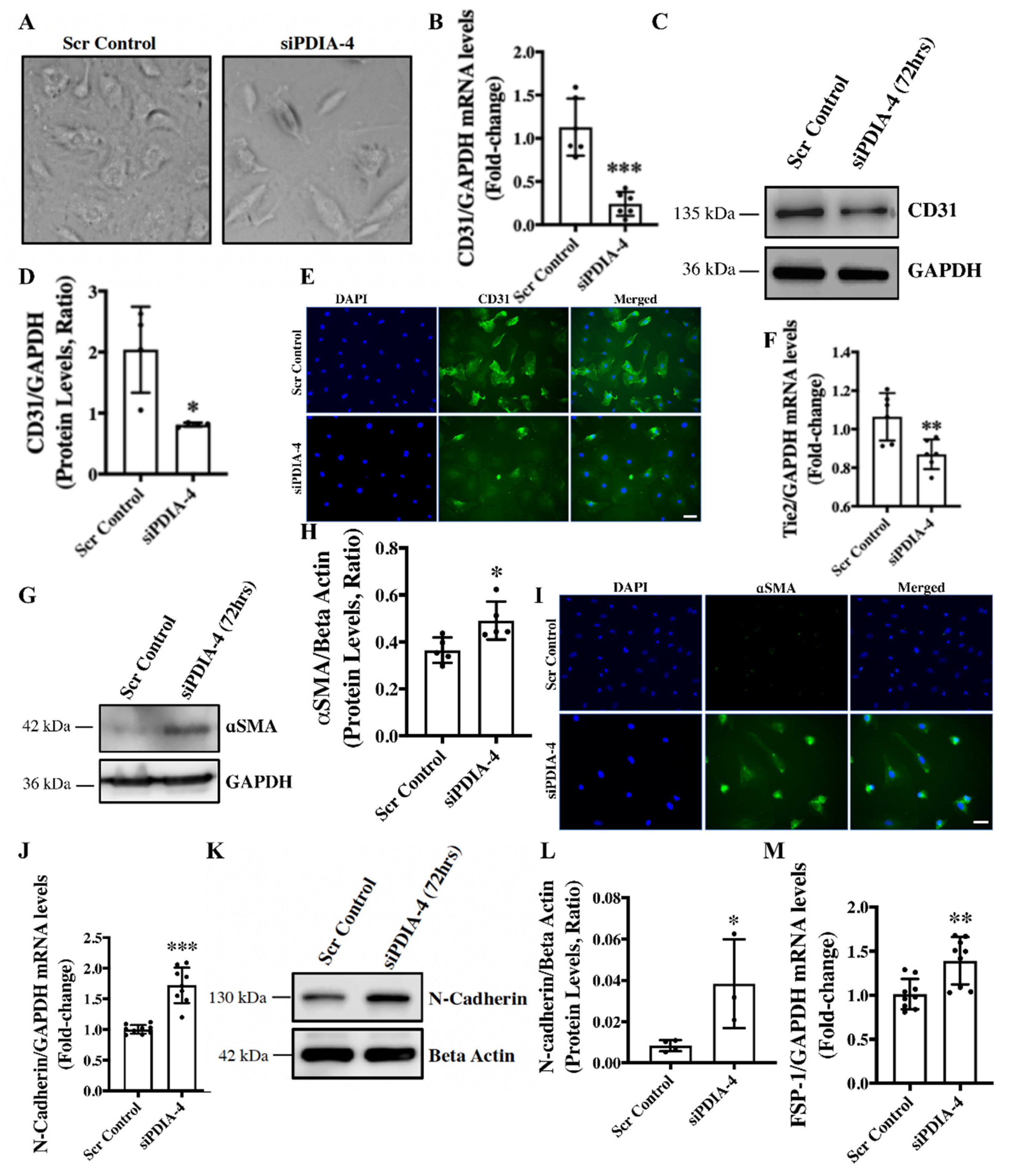
Loss of PDIA-4 induced End-MT in endothelial cells – We have previously reported that the loss of ATG7 in endothelial cells leads to impaired autophagy and End-MT in vitro and in vivo [7]. We next reported that loss of ATG7 in endothelial cells induced miR-378-3p expression that was inversely related to autophagy in endothelial cells and then confirmed PDIA-4 as a target of miR-378-3p in endothelial cells [12]. In this study, we have shown that the loss of PDIA-4 is associated with autophagy inhibition, which is in line with the loss of ATG7 and miR-378-3p upregulation-associated autophagic inhibition. Next, to clarify whether loss of PDIA-4 is also associated with loss of ATG-7 associated End-MT, we evaluated End-MT in PDIA-4-silenced and control endothelial cells. To our surprise, the loss of PDIA-4-deficient endothelial cells imaged under a phase contrast microscope displayed a conspicuous transition from the distinctive cobblestone-like appearance of endothelial cells to an enlarged spindle-shaped pattern that is consistent with fibroblast-like morphology of mesenchymal cells indicating End-MT in PDIA-4-silenced endothelial cells (Figure 4A) [7]. To confirm End-MT at the molecular level, next the expression levels of endothelial cell markers CD31 and Tie-2 and mesenchymal markers α smooth muscle actin (αSMA), N-cadherin and fibroblast-specific protein 1 (FSP-1) were measured in siPDIA-4- versus scrambled control-transfected endothelial cells. CD31 maintains endothelial cell junction integrity [33]; Tie-2 belongs to the receptor tyrosine kinase family [34]; αSMA is involved in the cytoskeletal reorganization during End-MT [35]; FSP-1 promotes cell differentiation and motility [36], and N-Cadherin serves as anchor points during cell-cell contact [37]. Expectedly, the loss of endothelial and gain of mesenchymal phenotype was mirrored as reduced expression of CD31 and Tie-2 (Figure 4B-F) and increased expression of mesenchymal marker αSMA, N-cadherin and FSP-1, and (Figure 4G-M) in PDIA-4-silenced in comparison to control endothelial cells. These data confirmed that loss of PDIA-4 in endothelial cells induced End-MT ultra-structurally and at the molecular level.
Figure 4.
Loss of PDIA-4 induced endothelial-to-mesenchymal transition in endothelial cells. (A) HUVECs were reverse transfected with siPDIA-4 or scrambled control for 72hrs. Images were taken under a phase contrast microscope. RNA was extracted, and qPCR was performed on (B) CD31 (F) Tie-2, (J) N-cadherin and (N) FSP-1 (n=2-3 in triplicates). Proteins were extracted, and western blot was performed targeting (C) CD31, (G) α SMA and (K) N-cadherin (n=3-5). (D, H, L) The bands were quantified using the LiCor Fc Odyssey inbuilt software. HUVECs were reverse transfected with siPDIA-4 or scrambled control for 72hrs on 4-chambers slides, and immunofluorescence was performed for (E) CD31 (green) and (I) αSMA (green). Bar = 100μm. Data were analyzed using Student’s T-test. Data are expressed as mean ± SD. * ** ***represent p<0.05, <0.01, <0.001, respectively versus Scr Cont.
Figure 4.
Loss of PDIA-4 induced endothelial-to-mesenchymal transition in endothelial cells. (A) HUVECs were reverse transfected with siPDIA-4 or scrambled control for 72hrs. Images were taken under a phase contrast microscope. RNA was extracted, and qPCR was performed on (B) CD31 (F) Tie-2, (J) N-cadherin and (N) FSP-1 (n=2-3 in triplicates). Proteins were extracted, and western blot was performed targeting (C) CD31, (G) α SMA and (K) N-cadherin (n=3-5). (D, H, L) The bands were quantified using the LiCor Fc Odyssey inbuilt software. HUVECs were reverse transfected with siPDIA-4 or scrambled control for 72hrs on 4-chambers slides, and immunofluorescence was performed for (E) CD31 (green) and (I) αSMA (green). Bar = 100μm. Data were analyzed using Student’s T-test. Data are expressed as mean ± SD. * ** ***represent p<0.05, <0.01, <0.001, respectively versus Scr Cont.
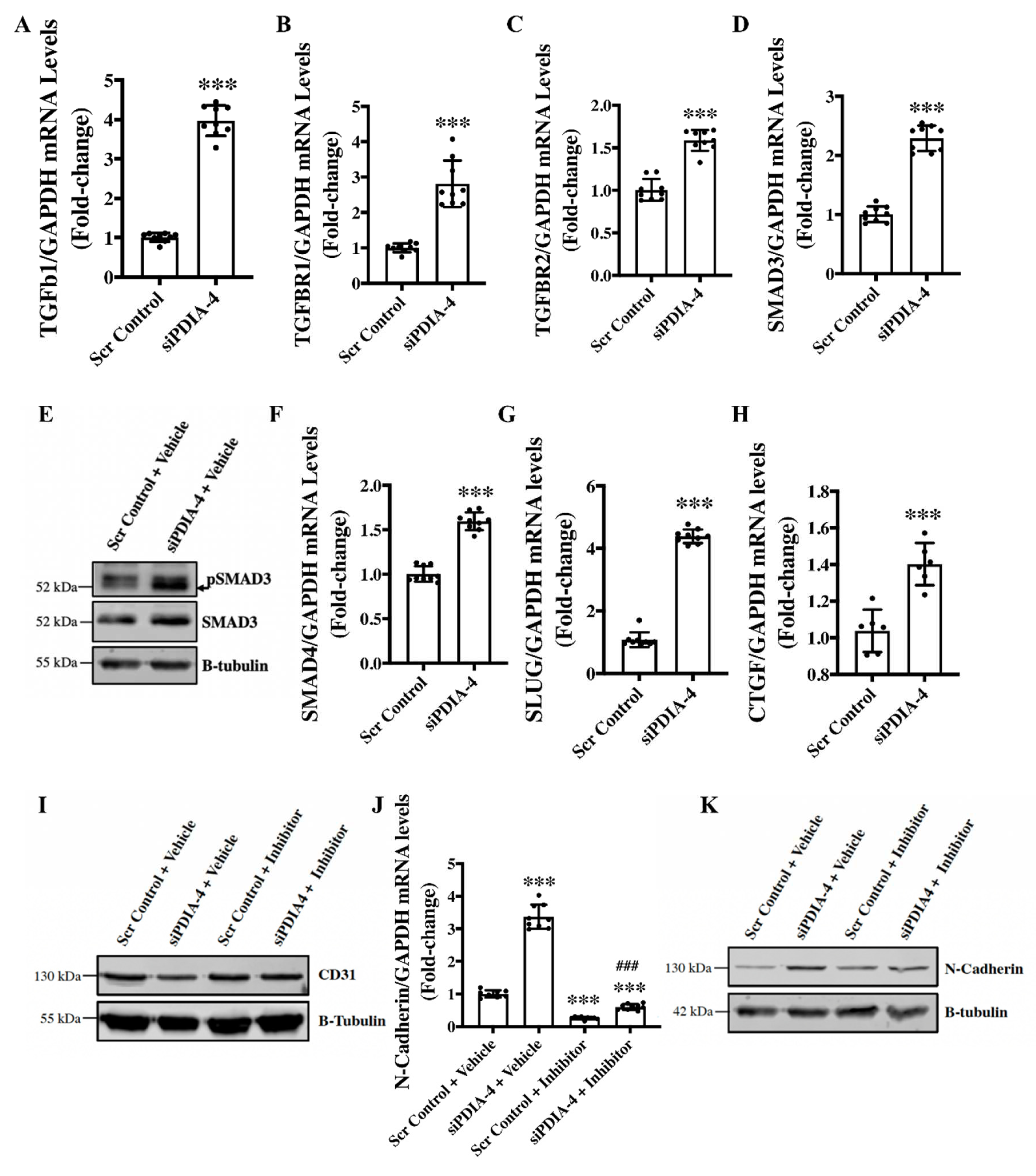
Loss of PDIA-4 induced End-MT is mediated by the TGFβ pathway - TGFβ signaling activation is the main stimulator of End-MT in endothelial cells [38]. Upon binding of TGFβ1 to TGFβ receptors, TGFBR1 and TGFBR2, SMAD2 gets phosphorylated and interacts with SMAD3 to translocate to the nucleus and activate transcription of genes involved in End-MT [39]. Next, to evaluate whether TGFβ mediates loss of PDIA-4 induced End-MT, expression level of the ligand TGFβ1 was measured, which demonstrated a significant upregulation of TGFβ1 expression in PDIA-4-silenced endothelial cells in comparison to control endothelial cells (Figure 5A). We then measured the expression level TGFβ1 receptors; TGFBR1 and TGFBR2 and observed a significantly increased expression of these receptors in PDIA-4-silenced endothelial cells in comparison to the control endothelial cells (Figure 5B, C). Next, to confirm whether increased expression of TGFβ1 and its receptors leads to increased activation of TGFβ signaling, we measured the activated level of SMADs in PDIA-4-silenced endothelial cells and observed an increased expression level of SMAD3 and SMAD4 and increased activation level of SMAD4 of the TGFβ signaling pathway (Figure 5D-F). We also measured TGFβ-dependent pro-fibrotic genes, such as CTGF, and, as expected, we observed upregulation in the PDIA-4-silenced endothelial cells in comparison to control endothelial cells (Figure 5G). We then measured transcription factor SLUG, which is a downstream effector in the TGFβ signaling pathway and observed a significant upregulation in PDIA-4-silenced endothelial cells compared to the controls (Figure 5H). To further confirm if loss of PDIA-4 induced End-MT is mediated by the TGFβ signaling, we inhibited TGFβ signaling and measured the expression level of End-MT markers in PDIA-4-silenced and control endothelial cells. TGFβ signaling inhibitor SB431542 is a potent and competitive inhibitor that inactivates the SMAD2/3 complex and blocks TGFβ signaling [40]. Inhibitor SB431542 treatment to the PDIA-4-silenced and control endothelial cells blocked End-MT in PDIA-4-silenced endothelial cells, which is evident by restored expression of endothelial marker CD31 (Figure 5I) and reduced expression of mesenchymal marker N-Cadherin (Figure 5J, K) in PDIA-4-silenced endothelial cells. Overall, these data confirm that TGFβ signaling mediates End-MT in PDIA-4-silenced endothelial cells.
Figure 5.
Loss of PDIA-4-induced End-MT is mediated by the TGFβ pathway in endothelial cells. HUVECS were reverse transfected with siPDIA-4 or scrambled control and incubated in complete EGM-2 medium for 72hr. RNA was extracted in Trizol, and qPCR was performed on targets (A) TGF β1, (B) TGFBR1, (C) TGFBR2, (D) SMAD3, (F) SMAD4, (G) SLUG, (H) CTGF and GAPDH (n=3 in triplicates). HUVECS were reverse transfected with siPDIA-4 or scrambled control and incubated in complete EGM-2 medium for 72hr and proteins were extracted to perform western blot for (E) SMAD3 and pSMAD3. After transfecting siPDIA-4 for 72hr, HUVECS were treated with either DMSO as vehicle control or TGF β inhibitor SB431542 (10uM). qPCR was performed targeting (J) N-cadherin (n=3 in triplicates). Western blot was performed targeting (I) N-cadherin and (K) CD31 (n=3). Data were analyzed using Student’s T-test. Data are expressed as mean ± SD. In Figure A-H, ***represents p<0.001 versus Scr Cont. In Figure J, ***represents p<0.001 versus Scr Cont + vehicle, and ### represents p<0.001 versus Scr Cont +inhibitor (n=3 in triplicates).
Figure 5.
Loss of PDIA-4-induced End-MT is mediated by the TGFβ pathway in endothelial cells. HUVECS were reverse transfected with siPDIA-4 or scrambled control and incubated in complete EGM-2 medium for 72hr. RNA was extracted in Trizol, and qPCR was performed on targets (A) TGF β1, (B) TGFBR1, (C) TGFBR2, (D) SMAD3, (F) SMAD4, (G) SLUG, (H) CTGF and GAPDH (n=3 in triplicates). HUVECS were reverse transfected with siPDIA-4 or scrambled control and incubated in complete EGM-2 medium for 72hr and proteins were extracted to perform western blot for (E) SMAD3 and pSMAD3. After transfecting siPDIA-4 for 72hr, HUVECS were treated with either DMSO as vehicle control or TGF β inhibitor SB431542 (10uM). qPCR was performed targeting (J) N-cadherin (n=3 in triplicates). Western blot was performed targeting (I) N-cadherin and (K) CD31 (n=3). Data were analyzed using Student’s T-test. Data are expressed as mean ± SD. In Figure A-H, ***represents p<0.001 versus Scr Cont. In Figure J, ***represents p<0.001 versus Scr Cont + vehicle, and ### represents p<0.001 versus Scr Cont +inhibitor (n=3 in triplicates).
Discussion
The main observation made in this study is that loss of endothelial PDIA-4 impairs autophagic flux and endothelial function and induces TGFβ-mediated End-MT. These data suggest that PDIA-4 may be a potentially novel and previously unrecognized target to modulate autophagy, endothelial function and End-MT in endothelial cells to limit impaired autophagy, endothelial dysfunction and/or End-MT associated disease.
We first reported that loss of ATG7 in endothelial cells is associated with the inhibition of autophagy and End-MT and the up-regulation of miR-378-3p in endothelial cells [12]. We then showed that miR-378-3p expression is inversely related to autophagy and endothelial function in endothelial cells and that PDIA-4 is a direct target of miR-378-3p in endothelial cells [12]. We continued this line of investigation, and now we report that loss of PDIA-4 inhibits autophagy and induces End-MT. The endoplasmic reticulum (ER) is known to be related to autophagy as it contributes to the formation of autophagosomes [41]. One similarity between ER and autophagy is that they are both involved in clearing waste products in the cells. Specifically for the ER, the accumulation of improperly folded protein serves as a stress signal and triggers ER stress response (ERS response) and autophagy to remove damaged proteins [42]. PDIA-4 functions as a protein chaperon and is upregulated during the ERS response [15]. There is no information linking PDIA-4 to endothelial autophagy, although Quan et al. found that autophagy-deficient β-cells were susceptible to ERS response and might contribute to diabetes [43]. Mice with autophagy-deficient β-cell interbred with obese mice, and the offspring were found to develop severe diabetes due to severe apoptosis in β cells [43]. Overall, this is the first report linking PDIA-4 with autophagy.
Similarly, we, for the first time, report that loss of PDIA-4 is associated with impaired endothelial function, with reduced angiogenic potential, cell migration, and cell proliferation. Xing et al. found that loss of PDIA-4 in cervical cancer cells significantly impaired cell proliferation and cell migration [44]. Additionally, it was found that the knockdown of PDIA-4 resulted in significantly lower cell proliferation in glioma cells [45]. Inhibition of PDIA-1 (an isoform of PDIA-4) in breast cancer cells and endothelial cells resulted in a significantly slower migration rate compared to the control [46]. Endothelial dysfunction is closely linked to atherosclerosis, mainly by making cells more susceptible to ERS response [47]. Civelek et al. found significant upregulation of genes involved in ERS response in athero-susceptible regions, linking endothelial dysfunction, ERS response and atherosclerosis [47]. Protein p21 is a cell cycle inhibitor [27], and we expected an increased p21 expression in PDIA-4-silenced endothelial cells. It is important to note that p21 also plays context-dependent roles, such as in apoptosis [48]. Here, we observed a reduced protein level of p21 in PDIA-4-silenced endothelial cells compared to control cells, indicating a p21-independent inhibition of the cell cycle in PDIA-4-silenced endothelial cells. It is possible that reduced cell proliferation due to the loss of PDIA-4 occurred through a p21-independent pathway.
Apoptosis often crosstalk with autophagy, and given the reduced angiogenic potential, migration and proliferation, which were not associated with regulators of endothelial function such as eNOS and AKT expression/activation, we next measured apoptosis. Interestingly, we did observe an increased apoptosis in PDIA-4-silenced endothelial cells. Pro-apoptotic p53 is an essential regulator of apoptosis [30]. We observed an increased expression of p53 following the loss of PDIA-4 endothelial cells. Bcl-2 is associated with cell survival, and Bax is associated with cell death, and both of them are downstream effectors of p53 [32]. We observed an increased Bax and reduced Bcl-2, indicating that increased apoptosis in PDIA-4-deficient endothelial cells was associated with the p53 apoptotic pathway. Overall, these results show that reduced endothelial function in PDIA-4-silenced endothelial cells is mainly attributed to increased apoptosis, indicating an important role played by PDIA-4 in maintaining endothelial cell function and survival.
We have previously reported that loss of ATG7 results in impaired autophagy, TGFβ-mediated End-MT and induction of miR-378-3p in endothelial cells [49]. We also showed that PDIA-4 is a target of miR-378-3p, and we next investigated whether loss of PDIA-4, which inhibits autophagy, would also induce End-MT. As expected, silencing PDIA-4 indeed induced End-MT, which was associated with the transition of endothelial cells from their characteristic cobblestone outlook to an elongated fibroblastic morphology. These findings were further confirmed by measuring the expression of endothelial and mesenchymal markers. Furthermore, TGFβ stimulates End-MT in autophagy-deficient ECs [49,50]. Accordingly, we observed an increased expression of ligand TGFβ1; its’ receptors TGFBR1, TGFBR2; effectors SMAD3, SMAD4; downstream transcription factor SLUG and TGFβ-responsive gene CTGF in PDIA-4-silenced compared to control endothelial cells, suggesting that loss of PDIA-4 induced End-MT through TGF β-dependent pathway. This was further confirmed by utilizing pharmacologic inhibition of the TGF β-pathway, which restored endothelial marker CD31 and mesenchymal marker N-Cadherin expression in PDIA-4-silenced ECs. These results are supported by the findings from Singh et al. that both genetic (via loss of ATG7) and pharmacologic (via bafilomycin) inhibition of autophagy led to TGFβ-mediated End-MT in endothelial cells. As mentioned previously, End-MT is associated with the pathology of many types of fibrosis, including pulmonary, cardiac, kidney and cancer fibrosis, mainly resulting from increased expression of extracellular proteins such as collagen [51,52,53]. The results from this study warrant further investigation into the therapeutic potential of PDIA-4 for treating impaired autophagy, impaired endothelial dysfunction or End-MT-associated diseases, such as atherosclerosis and organ fibrosis.
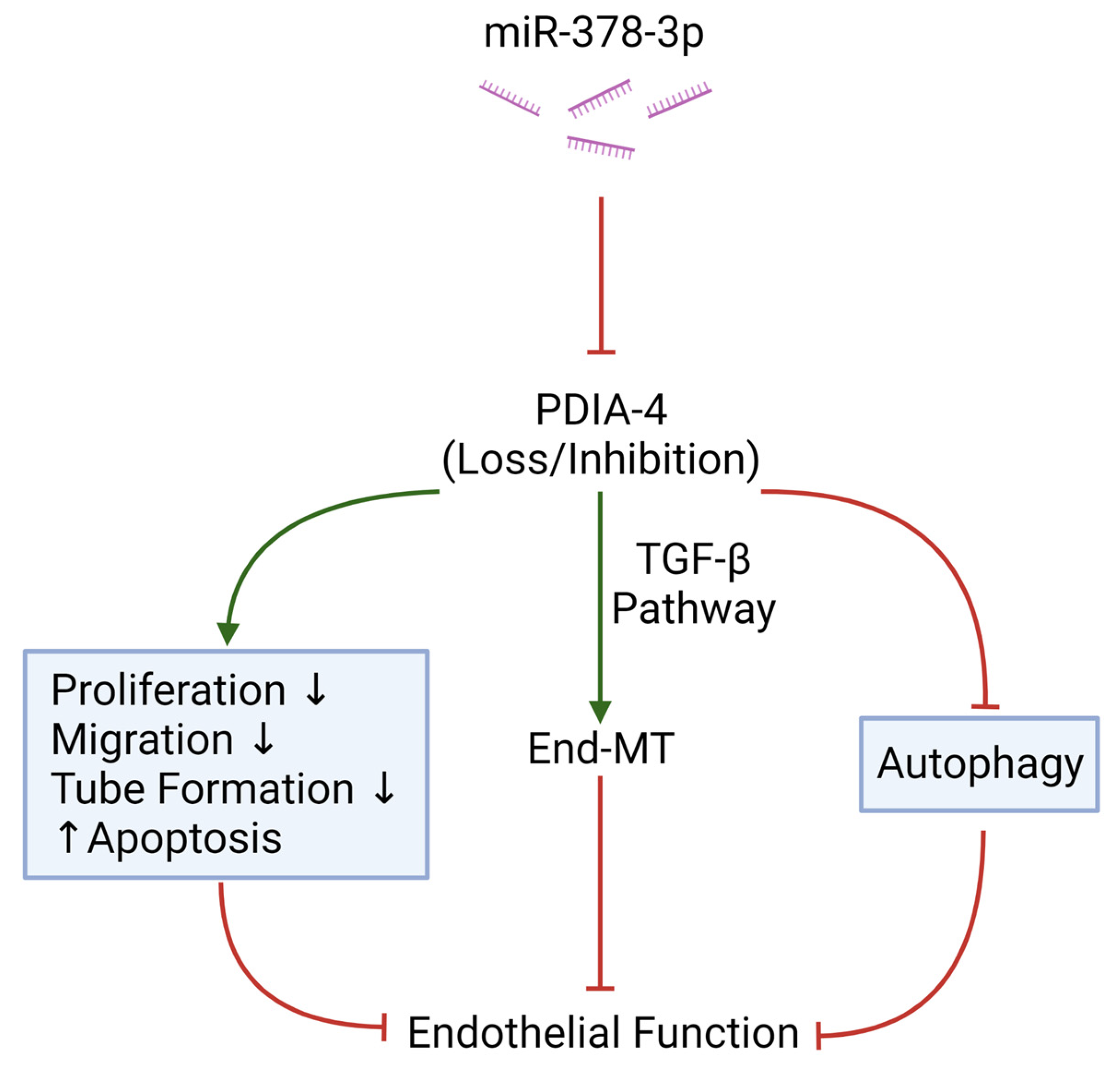
Overall, this is the first report linking PDIA-4 with endothelial autophagy, endothelial dysfunction, apoptosis and End-MT. There appears to be an interplay between ATG7, miR-378-3p and PDIA-4, resulting in the regulation of autophagy, endothelial function and End-MT (Figure 6). Results from our study add to the current knowledge of the relationship between miRNA, its regulatory targets and endothelial function, which may provide novel therapeutic targets to treat impaired endothelial autophagy, endothelial dysfunction or End-MT-associated diseases and warrant future investigations.
Figure 6.
Graphical Summary. MicroRNA miR-378-3p targets PDIA-4 to elicit its’effect on endothelial function, endothelial autophagy and endothelial survival. Red indicates “inhibition” and green arrows indicate “promotion”. Created with BioRender.com.
Figure 6.
Graphical Summary. MicroRNA miR-378-3p targets PDIA-4 to elicit its’effect on endothelial function, endothelial autophagy and endothelial survival. Red indicates “inhibition” and green arrows indicate “promotion”. Created with BioRender.com.
Author Contributions
KS conceived and designed the study. SB, AS, HN, BP, KB and NR carried out the experiments and analyzed the data. JF helped write and improve the discussion. KS, SB, AS, HN and JF wrote and assembled the manuscript with final figures and revision.
Funding
Funding for this project was provided by the Grant-in-Aid (#G-22-0032104), by Heart and Stroke Foundation of Canada, Canada to KS. KS is also the recipient of the 2018/19 National New Investigator Award- Salary Support from the Heart and Stroke Foundation of Canada, Canada.
Disclosure of Conflicts of Interest
The authors declare that they have no conflicts of interest with the contents of this article.
References
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Blood Vessels and Endothelial Cells. Molecular Biology of the Cell. 4th edition 2002. [Google Scholar]
- Nussenzweig, S.C.; Verma, S.; Finkel, T. The Role of Autophagy in Vascular Biology. Circ Res 2015, 116, 480–488. [Google Scholar] [CrossRef]
- Bu, S.; Singh, K.K. Epigenetic Regulation of Autophagy in Cardiovascular Pathobiology. International Journal of Molecular Sciences 2021, 22, 6544. [Google Scholar] [CrossRef]
- Singh, K.K.; Yanagawa, B.; Quan, A.; Wang, R.; Garg, A.; Khan, R.; Pan, Y.; Wheatcroft, M.D.; Lovren, F.; Teoh, H.; et al. Autophagy Gene Fingerprint in Human Ischemia and Reperfusion. J Thorac Cardiovasc Surg 2014, 147, 1065–1072.e1. [Google Scholar] [CrossRef]
- Torisu, K.; Singh, K.K.; Torisu, T.; Lovren, F.; Liu, J.; Pan, Y.; Quan, A.; Ramadan, A.; Al-Omran, M.; Pankova, N.; et al. Intact Endothelial Autophagy Is Required to Maintain Vascular Lipid Homeostasis. Aging Cell 2016, 15, 187–191. [Google Scholar] [CrossRef]
- Bu, S.; Nguyen, H.C.; Michels, D.C.R.; Rasheed, B.; Nikfarjam, S.; Singh, R.; Wang, L.; Patel, D.A.; Singh, S.; Qadura, M.; et al. Transcriptomics of Angiotensin II-Induced Long Noncoding and Coding RNAs in Endothelial Cells. J Hypertens 2022, 40, 1303–1313. [Google Scholar] [CrossRef]
- Singh, K.K.; Lovren, F.; Pan, Y.; Quan, A.; Ramadan, A.; Matkar, P.N.; Ehsan, M.; Sandhu, P.; Mantella, L.E.; Gupta, N.; et al. The Essential Autophagy Gene ATG7 Modulates Organ Fibrosis via Regulation of Endothelial-to-Mesenchymal Transition. J Biol Chem 2015, 290, 2547–2559. [Google Scholar] [CrossRef]
- Singh, S.; Adam, M.; Matkar, P.N.; Bugyei-Twum, A.; Desjardins, J.-F.; Chen, H.H.; Nguyen, H.; Bazinet, H.; Michels, D.; Liu, Z.; et al. Endothelial-Specific Loss of IFT88 Promotes Endothelial-to-Mesenchymal Transition and Exacerbates Bleomycin-Induced Pulmonary Fibrosis. Sci Rep 2020, 10, 4466. [Google Scholar] [CrossRef]
- Murugavel, S.; Bugyei-Twum, A.; Matkar, P.N.; Al-Mubarak, H.; Chen, H.H.; Adam, M.; Jain, S.; Narang, T.; Abdin, R.M.; Qadura, M.; et al. Valproic Acid Induces Endothelial-to-Mesenchymal Transition-Like Phenotypic Switching. Front Pharmacol 2018, 9, 737. [Google Scholar] [CrossRef]
- Bischoff, J. Endothelial to Mesenchymal Transition – Purposeful versus Maladaptive Differentiation. Circ Res 2019, 124, 1163–1165. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Jimenez, S.A. Endothelial to Mesenchymal Transition: Role in Physiology and in the Pathogenesis of Human Diseases. Physiol Rev 2019, 99, 1281–1324. [Google Scholar] [CrossRef]
- Bu, S.; Joseph, J.J.; Nguyen, H.C.; Ehsan, M.; Rasheed, B.; Singh, A.; Qadura, M.; Frisbee, J.C.; Singh, K.K. MicroRNA miR-378-3p Is a Novel Regulator of Endothelial Autophagy and Function. Journal of Molecular and Cellular Cardiology Plus 2023, 3, 100027. [Google Scholar] [CrossRef]
- Chanjiao, Y.; Chunyan, C.; Xiaoxin, Q.; Youjian, H. MicroRNA-378a-3p Contributes to Ovarian Cancer Progression through Downregulating PDIA4. Immun Inflamm Dis 2021, 9, 108–119. [Google Scholar] [CrossRef]
- Benham, A.M. The Protein Disulfide Isomerase Family: Key Players in Health and Disease. Antioxid Redox Signal 2012, 16, 781–789. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, H.; Cheng, Q. PDIA4: The Basic Characteristics, Functions and Its Potential Connection with Cancer. Biomed Pharmacother 2020, 122, 109688. [Google Scholar] [CrossRef]
- Yorimitsu, T.; Nair, U.; Yang, Z.; Klionsky, D.J. Endoplasmic Reticulum Stress Triggers Autophagy*. Journal of Biological Chemistry 2006, 281, 30299–30304. [Google Scholar] [CrossRef]
- Kyani, A.; Tamura, S.; Yang, S.; Shergalis, A.; Samanta, S.; Kuang, Y.; Ljungman, M.; Neamati, N. Discovery and Mechanistic Elucidation of a Class of Protein Disulfide Isomerase Inhibitors for the Treatment of Glioblastoma. ChemMedChem 2018, 13, 164–177. [Google Scholar] [CrossRef]
- Singh, S.; Nguyen, H.; Michels, D.; Bazinet, H.; Matkar, P.N.; Liu, Z.; Esene, L.; Adam, M.; Bugyei-Twum, A.; Mebrahtu, E.; et al. BReast CAncer Susceptibility Gene 2 Deficiency Exacerbates Oxidized LDL-Induced DNA Damage and Endothelial Apoptosis. Physiol Rep 2020, 8, e14481. [Google Scholar] [CrossRef]
- Singh, S.; Nguyen, H.C.; Ehsan, M.; Michels, D.C.R.; Singh, P.; Qadura, M.; Singh, K.K. Pravastatin-Induced Changes in Expression of Long Non-Coding and Coding RNAs in Endothelial Cells. Physiol Rep 2021, 9, e14661. [Google Scholar] [CrossRef]
- Yuan, J.S.; Reed, A.; Chen, F.; Stewart, C.N. Statistical Analysis of Real-Time PCR Data. BMC Bioinformatics 2006, 7, 85. [Google Scholar] [CrossRef]
- Protocol Guide: WST-1 Assay for Cell Proliferation and Viability. Available online: https://www.sigmaaldrich.com/CA/en/technical-documents/protocol/cell-culture-and-cell-culture-analysis/cell-counting-and-health-analysis/cell-proliferation-reagent-wst-1 (accessed on 24 January 2023).
- Jonkman, J.E.N.; Cathcart, J.A.; Xu, F.; Bartolini, M.E.; Amon, J.E.; Stevens, K.M.; Colarusso, P. An Introduction to the Wound Healing Assay Using Live-Cell Microscopy. Cell Adh Migr 2014, 8, 440–451. [Google Scholar] [CrossRef]
- Suarez-Arnedo, A.; Torres Figueroa, F.; Clavijo, C.; Arbeláez, P.; Cruz, J.C.; Muñoz-Camargo, C. An Image J Plugin for the High Throughput Image Analysis of in Vitro Scratch Wound Healing Assays. PLoS One 2020, 15. [Google Scholar] [CrossRef]
- Logsdon, E.A.; Finley, S.D.; Popel, A.S.; Gabhann, F.M. A Systems Biology View of Blood Vessel Growth and Remodelling. J Cell Mol Med 2014, 18, 1491–1508. [Google Scholar] [CrossRef]
- Nguyen, H.C.; Bu, S.; Nikfarjam, S.; Rasheed, B.; Michels, D.C.R.; Singh, A.; Singh, S.; Marszal, C.; McGuire, J.J.; Feng, Q.; et al. Loss of Fatty Acid Binding Protein 3 Ameliorates Lipopolysaccharide-Induced Inflammation and Endothelial Dysfunction. J Biol Chem 2023, 299, 102921. [Google Scholar] [CrossRef]
- Bu, S.; Nguyen, H.C.; Nikfarjam, S.; Michels, D.C.R.; Rasheed, B.; Maheshkumar, S.; Singh, S.; Singh, K.K. Endothelial Cell-Specific Loss of eNOS Differentially Affects Endothelial Function. PLoS One 2022, 17, e0274487. [Google Scholar] [CrossRef]
- Fujiwara, K.; Daido, S.; Yamamoto, A.; Kobayashi, R.; Yokoyama, T.; Aoki, H.; Iwado, E.; Shinojima, N.; Kondo, Y.; Kondo, S. Pivotal Role of the Cyclin-Dependent Kinase Inhibitor p21WAF1/CIP1 in Apoptosis and Autophagy *. Journal of Biological Chemistry 2008, 283, 388–397. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol Pathol 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Fan, Y.-J.; Zong, W.-X. The Cellular Decision between Apoptosis and Autophagy. Chin J Cancer 2013, 32, 121–129. [Google Scholar] [CrossRef]
- Lowe, S.W.; Ruley, H.E.; Jacks, T.; Housman, D.E. P53-Dependent Apoptosis Modulates the Cytotoxicity of Anticancer Agents. Cell 1993, 74, 957–967. [Google Scholar] [CrossRef]
- Pawlowski, J.; Kraft, A.S. Bax-Induced Apoptotic Cell Death. Proceedings of the National Academy of Sciences 2000, 97, 529–531. [Google Scholar] [CrossRef]
- Miyashita, T.; Krajewski, S.; Krajewska, M.; Wang, H.G.; Lin, H.K.; Liebermann, D.A.; Hoffman, B.; Reed, J.C. Tumor Suppressor P53 Is a Regulator of Bcl-2 and Bax Gene Expression in Vitro and in Vivo. Oncogene 1994, 9, 1799–1805. [Google Scholar]
- Lertkiatmongkol, P.; Liao, D.; Mei, H.; Hu, Y.; Newman, P.J. Endothelial Functions of PECAM-1 (CD31). Curr Opin Hematol 2016, 23, 253–259. [Google Scholar] [CrossRef]
- Wong, A.L.; Haroon, Z.A.; Werner, S.; Dewhirst, M.W.; Greenberg, C.S.; Peters, K.G. Tie2 Expression and Phosphorylation in Angiogenic and Quiescent Adult Tissues. Circ Res 1997, 81, 567–574. [Google Scholar] [CrossRef]
- Nakajima, Y.; Mironov, V.; Yamagishi, T.; Nakamura, H.; Markwald, R.R. Expression of Smooth Muscle Alpha-Actin in Mesenchymal Cells during Formation of Avian Endocardial Cushion Tissue: A Role for Transforming Growth Factor Beta3. Dev Dyn 1997, 209, 296–309. [Google Scholar] [CrossRef]
- Strutz, F.; Okada, H.; Lo, C.W.; Danoff, T.; Carone, R.L.; Tomaszewski, J.E.; Neilson, E.G. Identification and Characterization of a Fibroblast Marker: FSP1. J Cell Biol 1995, 130, 393–405. [Google Scholar] [CrossRef]
- Mrozik, K.M.; Blaschuk, O.W.; Cheong, C.M.; Zannettino, A.C.W.; Vandyke, K. N-Cadherin in Cancer Metastasis, Its Emerging Role in Haematological Malignancies and Potential as a Therapeutic Target in Cancer. BMC Cancer 2018, 18, 939. [Google Scholar] [CrossRef]
- Ma, J.; Sanchez-Duffhues, G.; Goumans, M.-J.; ten Dijke, P. TGF-β-Induced Endothelial to Mesenchymal Transition in Disease and Tissue Engineering. Frontiers in Cell and Developmental Biology 2020, 8. [Google Scholar] [CrossRef]
- Huang, F.; Chen, Y.-G. Regulation of TGF-β Receptor Activity. Cell & Bioscience 2012, 2, 9. [Google Scholar] [CrossRef]
- Inman, G.J.; Nicolás, F.J.; Callahan, J.F.; Harling, J.D.; Gaster, L.M.; Reith, A.D.; Laping, N.J.; Hill, C.S. SB-431542 Is a Potent and Specific Inhibitor of Transforming Growth Factor-β Superfamily Type I Activin Receptor-Like Kinase (ALK) Receptors ALK4, ALK5, and ALK7. Mol Pharmacol 2002, 62, 65–74. [Google Scholar] [CrossRef]
- Matsunaga, K.; Morita, E.; Saitoh, T.; Akira, S.; Ktistakis, N.T.; Izumi, T.; Noda, T.; Yoshimori, T. Autophagy Requires Endoplasmic Reticulum Targeting of the PI3-Kinase Complex via Atg14L. Journal of Cell Biology 2010, 190, 511–521. [Google Scholar] [CrossRef]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.J.; Kominami, E.; Momoi, T. ER Stress (PERK/eIF2α Phosphorylation) Mediates the Polyglutamine-Induced LC3 Conversion, an Essential Step for Autophagy Formation. Cell Death Differ 2007, 14, 230–239. [Google Scholar] [CrossRef]
- Quan, W.; Hur, K.Y.; Lim, Y.; Oh, S.H.; Lee, J.-C.; Kim, K.H.; Kim, G.H.; Kim, S.-W.; Kim, H.L.; Lee, M.-K.; et al. Autophagy Deficiency in Beta Cells Leads to Compromised Unfolded Protein Response and Progression from Obesity to Diabetes in Mice. Diabetologia 2012, 55, 392–403. [Google Scholar] [CrossRef]
- Xing, F.; Song, Z.; Cheng, Z. High Expression of PDIA4 Promotes Malignant Cell Behavior and Predicts Reduced Survival in Cervical Cancer. Oncol Rep 2022, 48, 184. [Google Scholar] [CrossRef]
- Li, H.; Liu, Q.; Xiao, K.; He, Z.; Wu, C.; Sun, J.; Chen, X.; Chen, S.; Yang, J.; Ma, Q.; et al. PDIA4 Correlates with Poor Prognosis and Is a Potential Biomarker in Glioma. Onco Targets Ther 2021, 14, 125–138. [Google Scholar] [CrossRef]
- Stojak, M.; Milczarek, M.; Kurpinska, A.; Suraj-Prazmowska, J.; Kaczara, P.; Wojnar-Lason, K.; Banach, J.; Stachowicz-Suhs, M.; Rossowska, J.; Kalviņš, I.; et al. Protein Disulphide Isomerase A1 Is Involved in the Regulation of Breast Cancer Cell Adhesion and Transmigration via Lung Microvascular Endothelial Cells. Cancers 2020, 12, 2850. [Google Scholar] [CrossRef]
- Civelek, M.; Manduchi, E.; Riley, R.J.; Stoeckert, C.J.; Davies, P.F. Chronic Endoplasmic Reticulum Stress Activates Unfolded Protein Response in Arterial Endothelium in Regions of Susceptibility to Atherosclerosis. Circulation Research 2009, 105, 453–461. [Google Scholar] [CrossRef]
- Coqueret, O. New Roles for P21 and P27 Cell-Cycle Inhibitors: A Function for Each Cell Compartment? Trends Cell Biol 2003, 13, 65–70. [Google Scholar] [CrossRef]
- Singh, K.K.; Lovren, F.; Pan, Y.; Quan, A.; Ramadan, A.; Matkar, P.N.; Ehsan, M.; Sandhu, P.; Mantella, L.E.; Gupta, N.; et al. The Essential Autophagy Gene ATG7 Modulates Organ Fibrosis via Regulation of Endothelial-to-Mesenchymal Transition *. Journal of Biological Chemistry 2015, 290, 2547–2559. [Google Scholar] [CrossRef]
- Goumans, M.-J.; Liu, Z.; ten Dijke, P. TGF-β Signaling in Vascular Biology and Dysfunction. Cell Res 2009, 19, 116–127. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-Mesenchymal Transition Contributes to Cardiac Fibrosis. Nat Med 2007, 13, 952–961. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Potenta, S.E.; Sugimoto, H.; Zeisberg, M.; Kalluri, R. Fibroblasts in Kidney Fibrosis Emerge via Endothelial-to-Mesenchymal Transition. J Am Soc Nephrol 2008, 19, 2282–2287. [Google Scholar] [CrossRef]
- Moonen, J.-R.A.J.; Lee, E.S.; Schmidt, M.; Maleszewska, M.; Koerts, J.A.; Brouwer, L.A.; van Kooten, T.G.; van Luyn, M.J.A.; Zeebregts, C.J.; Krenning, G.; et al. Endothelial-to-Mesenchymal Transition Contributes to Fibro-Proliferative Vascular Disease and Is Modulated by Fluid Shear Stress. Cardiovasc Res 2015, 108, 377–386. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



