
Submitted:
27 December 2023
Posted:
29 December 2023
You are already at the latest version
Abstract
The Fas associated Death Domain (FADD) is an adaptor protein that predominantly transduces apoptosis signal from death receptor (DR) to activate caspases, leading to the initiation of apoptotic signaling and the coordinated removal of damaged, infected, or unwanted cells. In addition to its apoptotic functions, FADD is involved in signaling pathways related to autophagy, cell proliferation, necroptosis, and cellular senescence, indicating its versatile role in cell survival and proliferation. The subcellular localization and intracellular expression of FADD play a crucial role in determining its functional outcomes, thereby highlighting the importance of spatiotemporal mechanisms and regulation. Furthermore, FADD has emerged as a key regulator in inflammatory signaling, contributing to immune responses and cellular homeostasis. This review provides a comprehensive summary and analysis of cellular dynamics of FADD in regulating of programmed cell death and inflammation through distinct molecular mechanisms associated with various signaling pathways.
Keywords:
Cancer
; FADD
; Apoptosis
; RIP Kinases
; Autophagy
; NF-κB
; Inflammation
; Therapy
1. Introduction
The Fas Associated Death Domain (FADD), also known as MORT-1, is an adaptor protein that facilitates molecular interactions between death receptors (DRs) and apical procaspase-8 and -10 to form a multimeric death inducing signaling complex (DISC) for the initiation of apoptosis signaling [1,2]. In addition to this well-characterized role, FADD has also been associated with diverse cellular events including; cell proliferation, autophagy, necroptosis, Inflammation and embryogenesis [1,3]. Expression of FADD is essential for maintaining the cell death regulatory mechanism during physiological and pathological perturbations [4]. Furthermore, the structural orientation and cellular expression/localization of FADD largely govern the functional diversity and its association with different cellular pathways [5,6,7]. Although, the dominant role of FADD in DR-induced apoptosis mostly describes in relation to its localization in the cytosol, some interesting studies have revealed a nuclear expression and role of FADD [8,9]. Importantly, the mechanistic significance of FADD in cytoplasmic to nuclear trafficking is mostly associated with distinct cell and cancer types [10]. Constitutive expression of FADD aids cellular homeostasis, however defective or low expression of FADD has been implicated in pathological manifestations in different type of cancers (Figure 1) [11]. Notably, dysregulated expression of FADD has been implicated in resistance to apoptosis in various types of human cancer cells and mice model systems [1]. An interesting study demonstrated a non-canonical role of FADD in the regulation of embryonic development [12]. Previous studies demonstrated the importance of FADD protein expression in cell cycle regulation [13], antioxidant and redox signaling [14], and antimicrobial immune responses [15]. Of note, the death domain (DD) of FADD interacts with the DD of autophagic protein Atg5 and promotes type II programmed cell death (autophagic cell death) [16]. Moreover, FADD deficiency in T cells provokes autophagic signaling and leads to caspase-independent cell death [17]. The versatility of FADD has also been reported in RIP1- and RIP3-dependent necroptosis signaling [18,19]. In the canonical pathway, DR stimulation in the presence of FADD proteolytically activates caspase-8 to cleave RIP1 and RIP3, thereby preventing necroptosis [20,21]. Interestingly, FADD proteins play an important role in inflammatory signaling and related disorders. Previous studies have demonstrated the importance of FADD in the regulation of NF-κB and TLR signaling in modulating interferon response against infectious exposures [22,23]. Post-translational modifications of FADD such as phosphorylation and ubiquitination have been implicated in the regulation of the cell cycle [24,25] and proapoptotic activity, respectively [26]. TNFα stimulation induces linear ubiquitination of endogenous FADD in Jurkat cells to induce pro-survival mechanisms [27]. In contrast, the E3 ligase Makorin Ring Finger Protein 1 (MKRN1) mediates ubiquitination and proteasomal degradation of FADD, thereby abrogating the activation of cell death. It has been shown that MKRN1 knockdown results in FADD protein stabilization and the formation of the DISC, which causes hypersensitivity to extrinsic apoptosis [26]. We have previously reported that FADD induces JNK1-dependent ubiquitination of the cFLIP protein to instigate death receptor mediated apoptosis [28,29]. Importantly, further in-depth investigation into FADD-mediated ubiquitin signaling is still needed and mapping novel regulators of FADD would be more promising for designing targeted therapy. Overall, FADD has multiple regulatory functions and serves as a unique molecule to regulate both apoptotic and non-apoptotic signaling. This review article comprehensively discusses the importance of FADD as a cellular intrinsic switch that helps regulate signaling dynamics to maintain cellular homeostasis.
Thus, comprehending the cellular dynamics of FADD in the regulation of programmed cell death signaling and inflammation may elucidate the potential of this protein to provide therapeutic intervention for cancer and inflammation.
2. Structure and cellular localization of FADD
2.1. The structural organization of FADD
The human FADD gene is located on chromosome 11q13.3 and consist of two coding exons that encode a 22kDa protein [31]. FADD contains two functional domains: an N-terminal Death Domain (DD) and a C-terminal Death Effector Domain (DED). The DD of FADD (FADD-DD) consists of 80 amino acids arranged in six antiparallel amphipathic -helices, which structurally resembles the CD95 DD. The FADD-DD is crucial for homophilic interaction with the DD of death receptors [6,7,32]. FADD-DD interacts with multiple receptors, including TRADD (TNFR-1 signaling), DR3, TRAIL receptors 1 (DR4) and TRAIL receptors 2 (DR5), to activate extrinsic apoptosis signaling [33,34]. The FADD-DED interacts with DEDs of procaspase-8 and/or -10 forming a death inducing signaling complex (DISC), which facilitate downstream apoptosis signaling [35,36]. The three-dimensional structural analysis of FADD has revealed that the DD and DED domains of FADD are arranged in an orthogonal tail-to-tail manner, with each domain having conserved backbone of six α-helices [31,37]. Further, studies have shown that the DD of FADD is enriched in positively charged residues (K110, R113, R114, R117, R127) near the α11/α12 interhelical loop, allowing for interaction with the DD of CD95/Fas receptor [37,38]. Moreover, the interfaces of the first and sixth α-helices of FADD-DD heteromerize with DD-containing proteins involved in cell death and inflammatory signaling [7,39]. Previous in vitro studies have demonstrated that purified DD of FADD can interact with independently with the DD of receptor-interacting protein kinase 1 (RIPK1 or RIP1) and plays a role in regulating the necrotic activities of RIPK1 [40]. Notably, NMR structural analysis has shown that each α-helix of FADD-DED is rich in conserved hydrophobic and negatively charged residues, such as Glu/Asp or Asn (at position 19) and the RXDL motif (at position 78-81). This provides a platform for DED-containing proteins to assemble DISC [41,42]. Previous research, including our own, has shown that overexpression of full-length FADD can induce apoptosis either independently or in response to death receptor activation [28,29,32,43]. Mutagenesis and biochemical studies have further revealed that FADD-DD alone cannot initiate downstream apoptosis signaling without the activation of death receptor and requires functional DEDs [37]. Interestingly, over-expression of DED is sufficient to induce cell death without the need of death receptor. In contrast, overexpression of FADD-DD inhibits downstream signaling and activation of Fas/CD95 and DR5 receptors mediated apoptosis [44,45]. Mutational analysis has shown that deletion or mutation in the DED of the FADD (FADD-DN) acts as a dominant negative in death receptor signaling and loss the ability to recruit executioner caspase-8 [46]. Another study using a DED mutant of FADD (deletion of 80-208) has shown impaired functionality of FADD in response to canonical death inducers and defects in T-cell proliferation [47]. Importantly, the functional outcomes of FADD-mediated signaling are partially dependent on its phosphorylation state. It has been demonstrated phosphorylation of serine 194, located at the C-terminus of FADD, regulates cell cycle progression [48]. Additionally, the G2/M stage has been identified as the most favorable phase for FADD phosphorylation during the cell cycle process, although the molecular mechanism behind this remains unclear [49]. It is worth noting that, the orthologs of human FADD have been characterized in the mice and xenopus. The mouse FADD (mFADD) and xenopus FADD (xFADD) proteins are 80% and 62% structural resemblance with human FADD (hFADD), respectively [44,50]. Induced expression of xFADD in mammalian cells leads to apoptosis, while a truncated (dominant negative) DED of xFADD abolishes apoptosis in response to Fas ligand stimulation [50]. In conclusion, the structural analysis of FADD emphasizes the critical role of the individual DD and DED domains in transmitting cell death signaling, while the phosphorylation state of FADD governs its functional outcomes.
2.2. Expression regulation and localization of FADD
There are numerous transcription factors (TFs) recognized for their role in regulating the expression of FADD in cancer cells (Figure 2; detailed analysis in Supplementary Table S1). The TF, hypoxia-inducible factor-1α (HIF-1α) inhibited the transcriptional activity of FADD gene in colon cancer cells [52]. Moreover, previous studies have demonstrated that the overexpression of BRCA1 in breast cancer cells which lack BRCA1 results in a significant upregulation of FADD expression. This increase can be attributed to the direct interaction between BRCA1 and the promoter region of FADD. Conversely, the depletion of BRCA1 has been shown to cause a marked decrease in FADD expression, both at the protein and messenger RNA (mRNA) levels [53]. The cytosolic localization of FADD is crucial for DR-induced DISC formation and apoptosis signaling [1]. Previous study demonstrated that the nuclear localization signal (NLS) and nuclear export signals (NES) in the DED facilitate the nucleo-cytoplasmic shuttling of FADD [8]. Furthermore, a mutation in a phenylalanine residue at position 25th of the DED region abolishes the nuclear translocation of FADD [10]. Some reports suggest that CK1α (casein kinase) and CK2β are key mediators of FADD phosphorylation, directing its translocation across the nucleus. CK1α induces the phosphorylation and nuclear translocation of FADD, while CK2β retains phosphorylated FADD inside the nucleus to inhibit death receptor signaling [10,54]. Importantly, the nucleo-cytoplasmic shuttling protein exportin-5 interacts with FADD to facilitate the import of phosphorylated FADD (pFADD) into the nucleus, and a mutation at Serine 194 (Ser194) disrupts FADD-exportin 5 interactions [10,55]. The nuclear translocation of pFADD strengthens the anti-apoptotic activity of NF-κB, promoting cell proliferation [25]. A previous report highlights that FADD predominantly translocates to the high non-condensed transcriptionally active region of chromatin, but in the absence of DNA binding motifs, FADD may not directly influence the transcriptional machinery [56]. Nevertheless, the DED of FADD interacts with the methyl-CpG binding domain protein 4 (MBD4) in the nucleus; however, the downstream signaling of FADD-MBD4 is not well defined [10]. Cytosolic FADD interacts with death receptor and autophagy intermediates [17], while nuclear localization of FADD strengthens its anti-apoptotic activity and promotes cell proliferation [25]. Importantly, further exploration of the unidentified function of nuclear FADD will contribute to our understanding of its nuclear significance apart from its well-established role in of apoptosis signaling transduction.
3. FADD: cell intrinsic molecular intraction
3.1. FADD and cFLIP interactions
Cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein (cFLIP) plays a major role in blocking death receptor (DR) mediated apoptosis, it also plays a fundamental role in apoptosis, immune receptor signaling, inflammation, autophagy, and necroptosis [57,58]. Similar to procasppase-8/ -10, the anti-apoptotic protein cFLIP also contains two death effector domains (DEDs) at the N-terminal, but it is an inactive enzymatic homologue of procaspase-8/ -10 [59]. The gene encoding regions of cFLIP is located near procaspase-8 and procaspase-10 on chromosome 2q33, suggesting a genetic link between these apoptosis regulatory genes [60]. The cFLIP protein has three isoforms: a long form cFLIPL (55 kDa), a short variant cFLIPS (27 kDa), and regulator cFLIPR (25 kDa). All isoforms of cFLIP contain two death effector domains (DEDs), but cFLIPL has an additional inactive caspase-like DED, while cFLIPS and cFLIPR lack the entire caspase-like DEDs [57,61]. Importantly, all three isoforms of cFLIP are believed to competitively inhibit procaspase-8 recruitment to the DISC through their DEDs [62,63]. In cancer cells, cFLIP occupies the majority of FADD, which serves as a common docking site for both procaspase-8 and cFLIP through DED interactions [28,29,64]. Computational analysis suggests that the DEDs of FADD, cFLIP, and procaspase-8 contain an ‘charged triad’ E/D-RxDL motif that is imcrucial for their downstream signaling [39]. Molecular docking studies have shown that FADD DED preferentially engages FLIP through its α1/α4 surface and procaspase-8 using its α2/α5 surface. These relative orientations contribute to FLIP being recruited to the DISC at comparable levels to procaspase-8, despite lower cellular expression [65]. Additionally, cFLIP has a higher binding affinity to FADD compared to procaspase-8 at the DISC [66]. The heterogeneous expression of FADD and cFLIP across different tumor types confers resistance to death receptor-induced apoptosis (Figure 3). Mechanistically, in response to TNF-α, TNF receptor (TNFR1) oligomerize with adaptor proteins TRADD, ubiquitin ligases TRAF2 and cIAP1/2, and RIP1 to form ‘complex I’ for the activation of NF-κB signaling [67]. At the transcriptional level, activated NF-κB induces the expression of anti-apoptotic proteins such as cFLIP, cIAPs and Bcl-2 family members to block apoptotic signaling [68,69]. Importantly, elevated expression of cFLIPL strengthens the complex I for constitutive NF-κB activation [70,71]. Notably, cFLIP protein undergoes post-translational modifications [72,73]. The phosphorylation of cFLIPL and cFLIPS has been observed in various cell lines, inhibiting their interaction with the adaptor molecule FADD and sensitizing cancer cells to apoptosis [74]. Furthermore, ubiquitin-mediated protein modification of cFLIP isoforms is essential for cellular homeostasis and proliferation [75,76]. We have previously shown that TNF-α stimulation of FADD-overexpressing cells induces JNK and E3 ligase ITCH-dependent ubiquitination of cFLIPL [28]. Panner et al. demonstrated PTEN-Akt-AIP4-mediated ubiquitination of FLIPS and sensitivity to TRAIL [77]. Moreover, lysine 167 (K167) residue of cFLIPL serves as a novel ubiquitination site for ROS-dependent degradation [78], and the DNA repair protein Ku70 interacts with cFLIP and protects it from polyubiquitination and proteasomal degradation [79]. However, despite significant advances in understanding the regulation of apoptosis and cell survival, the involvement of FADD and cFLIP in these processes remains elusive.
3.2. FADD and RIP1 interaction
RIP1 is an adaptor protein containing a death domain (DD) and is a crucial component of TNF-R1 and TRAIL-R1/R2 signaling [67,81]. The importance of the Receptor interacting protein kinase 1 (RIPK1) in cellular homeostasis was investigated in RIP1-deficient mice, which exhibited lethality due to extensive apoptosis in both lymphoid and adipose tissue [82]. It was observed that an interplay between FADD and RIP1 is critical for the regulation of apoptosis and necrosis during embryogenesis and lymphocyte function in a mouse model [83]. The heterogeneous expression of FADD and RIPK1 exacerbates the restriction of interaction and subsequent signaling in diverse tumor types. (Figure 3). Additionally, Rip1 kinase inactive mutations have distinct impacts on the embryogenesis of Fadd-deficient mice [84]. Knockdown of FADD or FADD-/- leukemia Jurkat T-cell failed to induce NF-κB signaling upon TRAIL stimulation [85]. Upon ligand-dependent receptor activation, intracellular FADD oligomerizes and recruits RIP1 and caspase-8, forming a complex called the RIPoptosome, which initiates programmed necrosis (necroptosis) [86]. Jang et al. demonstrated that, the RIP1 DD and FADD DD form a stable complex with a structure similar to that of the Fas DD/FADD DD complex [87]. Earlier structure-based mutagenesis studies revealed that, RIP1 DD point mutations K604E, E614K, G623K, E626K, M637K, K642D, and S657K disrupt the stability of the complex with FADD DD [87]. The pleiotropic nature of TNFR1 signaling plays an essential role in regulating apoptotic and non-apoptotic signaling pathways. Activation of TNFR1 leads to the formation of ‘complex I’, which includes TNF receptor-associated protein with a death domain (TRADD), TNF receptor-associated protein 2 (TRAF2), RIP1, and cellular inhibitor of apoptosis protein 1 and 2 (cIAP1 and cIAP2), and primarily regulates NF-κB signaling [67]. Moreover, during the regulatory inhibition of NF-κB signaling, TNFR1 signaling recruits FADD, caspase-8 and RIP1 to form a cell death-inducing complex known as ‘complex II’ [86,88]. Interestingly, a previous report demonstrated that RIP1-deficient cells failed to induce NF-κB activation, even in the absence of TNF-α stimulation [82]. Notably, NF-κB activation can be blocked by TNF-α-mediated signaling through caspsase-8 mediated cleavage of RIP1 [86,89]. These studies highlight the pleiotropic role of RIP1 in maintaining cellular homeostasis by regulating apoptosis machinery and cell survival pathways. Stimulation of TNFα leads to the autophosphorylation and polyubiquitination of RIP1. Polyubiquitination through Lysine 48 (K48) linkage leads to degradation, while Lysine 63 (K63) linkages result in the activation of IκB kinase and NF-κB activation [88]. Furthermore, a point mutation of RIP1 at Lysine 377 (K377R) blocks K63-linked polyubiquitination, preventing the recruitment of IκB kinase, IKKβ, and TAK1 complex to the TNF receptor and thereby inhibiting NF-κB activation [90]. Therefore, polyubiquitination of RIP1 is crucial for the activation of IKKβ, which phosphorylates IκB, an inhibitor of NF-κB, leading to its degradation via the proteasomal pathway [91]. As a result, NF-κB is released from the inhibitory complex, translocated to the nucleus, and activates the transcription of target genes involved in immunity, inflammation, and survival [92]. Downregulation of RIPK1 in HepG2 cells significantly reduces NF-kB transcriptional activity and promotes caspase-8 and caspase-3-mediated apoptosis [93]. In summary, in the absence or downregulation of complex I-mediated NF-κB activation, the RIP1-FADD-caspase-8 complex II reinforces apoptosis. In the following sections, we will discuss the details of RIPoptosome signaling.
4. Role of FADD in cell death and Inflammatory signaling
4.1. FADD in the regulation of the TNFα-NF-κB signaling axis
The TNF-α induced NF-κB signaling and downstream activation of anti-apoptotic genes have negative impact on apoptosis signaling. In the majority of cancer types, abnormal activation of NF-κB signaling promotes tumor development [67,94]. In addition to cancer cell signaling, dysregulation of TNF receptor (TNFR) signaling is associated with inflammatory disorders, such as arthritis and inflammatory bowel disease [95,96,97], making it a promising therapeutic target The role of FADD in regulating TNF-α induced NF-κB signaling activation has been the subject of ongoing debate, with several groups currently working to determine the underlying mechanisms. TNF-α is a multifunctional cytokine belonging to the tumor necrosis factor superfamily, with important roles in cellular immunity, cell differentiation, proliferation, inflammation, and cell death [68]. Dysregulation of NF-κB signaling is closely associated with various human diseases, including cancer [98]. Activation of NF-κB-associated signaling for evading tumor cell death is a major factor contributing to tumor cell proliferation [69]. We and other have previously demonstrated that the TNFα-NF-κB signaling axis promotes prolonged survival in various tumor cell types [28,29], but these cells remain susceptible to apoptosis induction by chemotherapeutic drugs and radiation [99,100]. TNF-α exerts its biological effects through cell surface TNF receptors (TNFRs), which consist of a cytoplasmic death domain (DD) approximately 80 amino acids in length. This domain is responsible for recruiting downstream components of the death machinery [68]. Activation of TNFR-1 leads to a conformational change in its cytoplasmic DD tail, allowing it to interact with the DD-containing adaptor protein TRADD (TNFR-associated death domain). TRADD can form both a pro-inflammatory/survival “complex I”, which recruits RIP1, TNFR-associated factor (TRAF)-2 and -5, and cIAP 1/2, as well as a pro-apoptotic signaling “complex II”, which recruits FADD and RIP1 [81,86]. While the DD of RIP1 can directly interact with the DD of ligand bound TNFR1, it generally prefers TRADD-mediated recruitment in complex I, possibly due to its high affinity for TRADD [101]. Formation of complex I leads to robust activation of NF-κB and AP-1 and upregulates several anti-apoptotic genes such as Bcl-xL, A1/Bfl-1, (c-IAP) 1/2, X-chromosome-linked IAP (XIAP; also known as hILP), and cFLIP [67,69]. Moreover, the binding of transforming growth factor-β-activated kinase (TAK-1) binding protein (TAB)-2/TAB-3 to ubiquitinated RIP1 stabilizes complex I, further activating NF-κB [102,103]. In contrast, the deubiquitinylation of RIP1 by the enzymes cylindromatosis (CYLD) or cIAPs subsequently dissociates RIP1 from complex I and allows I to interact with FADD and procaspase-8 forming complex II and triggering cell death [73,88]. Importantly, the oligomerization of the RIP1-FADD-procaspase-8 complex II is tightly regulated by the anti-apoptotic protein cFLIP [88]. In this context, tumor cell survival signaling pathways, including NF-κB, MAPK/ERK, and Akt, are known to transcriptionally upregulate cFLIP expression in a feedback mechanism [70]. We have demonstrated that the expression of FADD is critical for maintaining complex II-mediated apoptotic cell death by regulating the expression of cFLIP and the assembly of complex I [28]. Zhou et al. showed that pharmacological targeting of IAPs suppressed NF-κB activation and induced FADD-dependent apoptosis in multiple myeloma (MM) cells, highlighting the significant functional contribution of FADD [104]. Chaudhary et al. previously demonstrated that low FADD concentration induces the activation of NF-κB signaling in a time- and dose-dependent manner [105]. Furthermore, the bifurcated TNF-α signaling in the form of “complex I” and “complex II” represents independent mechanisms that may be specific to certain cell type [86,106]. The existence of these two opposing signaling complexes may explain the lack of response to TNF-α observed in many cells expressing TNF receptors. Subsequent studies have revealed a previously unappreciated role for the FADD protein as a molecular switch regulating the TNF-α- NF-κB signaling axis, thereby influencing both apoptosis and cell proliferation (Figure 4).
4.2. Role of FADD in Necroptosis
Necroptosis is a form of caspase-independent cell death that is mediated by the RIPK3 protein. RIPK3 phosphorylates and activates the pseudokinase Mixed Lineage Kinase-Like (MLKL), which then executes cell death in the absence of apoptotic pressure [107]. However, some apoptotic components have been found to be involved in the formation of the necroptotic complex and dependent cell death [108,109,110]. The signaling of necroptosis signaling regulates various cellular and pathological processes, ranging from development to the regulation of immune regulatory cells [19]. Necroptotic cell death is initiated by cytokines TNF-α, Fas, or TRAIL, which leads to dysregulation of mitochondrial reactive oxygen species (ROS) production and eventual collapse of cellular energy production [111,112]. Additionally, genotoxic agents and related cellular stress can also induce necroptosis [88,113].
The C-terminal DD of RIPK3 facilitates its interaction with the DD of RIPK1 through a RIP homotypic interaction motif (RHIM). The interaction leads to auto-phosphorylation and activation of RIPK3 as well as the assembly of “complex IIb”. At the molecular level, the RIPK1-RIPK3 complex IIb associates with FADD and caspase 8 to form the necroptotic complex [19,110,114]. It is worth noting that, FADD and caspase-8 are common components in complex II and complex IIb, and they constantly regulate RIPK3-mediated necrotic cell death [114,115]. Previous studies have shown that deletion of RIP3 completely restores cell proliferation in FADD mutated T cells [116,117]. Conversely, FADD deficient primary T cells fail to assemble the RIP1-RIP3 complex [118]. Furthermore, mouse embryonic fibroblasts (MEFs) lacking FADD show resistance to TNF-α-induced necrosis, and restoration of FADD expression restores both apoptotic and necrotic sensitivity to TNF-α [119]. Irrinki et al. demonstrate that the FADD-RIP1-RIP3-NEMO (NF-κB essential modulator) complex induces disintegration of mitochondrial bioenergetics to promote TNF-α-driven necroptosis [120]. An incoherent expression of FADD and RIPK2 is consistently observed across various cancer types, resulting in improper interaction and downstream signaling (Figure 3).
Ripk1-deficient mice can survive for a few days [82], and co-deletion fadd or caspase-8 does not prevent the perinatal lethality of ripk1−/− mice [121]. However, the combined deletion of FADD-caspase-8-mediated apoptosis and RIPK3-MLKL-mediated necroptosis provides protection from lethality and increased survival [122]. In MEFs, the ablation of Rip1 challenges TNF-α-induced expression of cFLIP, suggesting that TNF-induced caspase-8-mediated apoptosis partially contributes to the lethality of newborn ripk1−/− mice [123]. Furthermore, FADD has been reported to suppress RIP3-mediated chronic intestinal inflammation and necrosis in the epithelial cells [18]. Conversely, Fas signaling mediated suppression of RIP3 by caspase-8 or FADD facilitates T cell clonal expansion [17,117]. FADD has also been shown to neutralize virus-induced interferon production by enhancing the ubiquitin activity of E3 ligase TRIM21 [124]. Importantly, the E3 ubiquitin ligase MKRN1 regulates the ubiquitination of FADD and RIP1-RIP3 complex formation [26]. Thus, FADD-mediated regulation of necroptosis signaling could provide an opportunity to define the fate of cells in pathological consequences (Figure 4).
4.3. Role of FADD in Inflammation
FADD has emerged as an important regulator of innate immunity and inflammation [125,126]. The formation of the multimolecular complex known as the FADDosome, which consist of caspase-8-FADD-RIPK1 has been previously associated with the production of cytokine induced by TRAIL (TNF-related apoptosis-inducing ligand) [127]. In A549 cells, the removal of FADD or caspase-8 failed to activate NF-κB and production of pro-inflammatory cytokines in response to TRAIL. Additionally, the injection of FADD knockout A549 cells in mice resulted in the development of lung tumors, highlighting the role of the TRAIL-FADD-NF-κB signaling axis in cytokine and tumor regulation [128]. Essentially, the binding of TLR4/IL-1R triggers the interaction of the adaptor protein MyD88 (myeloid differentiation primary response 88) and downstream IL-1-receptor-associated kinase (IRAK) through DD interactions, leading to the activation of NF-κB signaling and the expression of pro-inflammatory cytokines( IL-6, IL-1β, and TNF) [129,130]. However, FADD can also compete with IRAK for DD interactions and interacts with MyD88, thus impairing NF-κB activation and downstream pro-inflammatory signaling [126,131]. Moreover, the loss of FADD enhances MyD88-IRAK1 interaction, suggesting that FADD balances the IRAK1 binding to MyD88 in response to TLR4 activation [132,133]. Depletion of FADD in myeloid cells induces RIP3- and MyD88-dependent systemic inflammation [125]. Another interesting finding suggest that FADD may differentially regulate Fas signaling of apoptosis and inflammation depending on the cell type and stimulation [134]. The NLRP3 (NOD-like receptor family, pyrin domain containing 3) inflammasome, an important component of innate immunity, is critical for the host’s immune defenses against pathogens. The NLRP3 inflammasome assembly consisting of NLRP family receptor, the adaptor protein ASC, and inflammatory caspase-1, is responsible for the processing and activation of the cytokines IL-1β and IL-18 [135]. In macrophages, the NF-κB-TRIF-MyD88 signaling axis stimulated by LPS primes the assembly of NLRP3 inflammasome and the expression of pro-IL-1β and pro-IL-18, leading to the maturation of theses cytokines [136]. Additionally, a caspase-8–FADD–RIPK1 has been reported to activate the NLRP3 inflammasome in human monocytic cell lines in response to LPS stimulation, independent of their apoptotic functions [137]. The genetic ablation of caspase-8 or Fadd in murine macrophages impairs both the transcriptional priming and activation of the NLRP3 inflammasome [138]. The activation of NLRP3 inflammasome leads to the cleavage of gasdermin D (GSDMD) into N-terminus GSDMD (N-GSDMD), releasesing large amounts of inflammatory cytokines and inducing inflammatory cell death known as pyroptosis [139]. Notably an investigation shown that the activation of NLRP3 inflammasome in human monocytes/macrophages induces the secretion of FADD through microvesicle shedding, without increased IL-1β release and pyroptosis [23]. Furthermore, NLRP3 inflammasome-mediated pyroptosis acts as a protective mechanism against viral infections, such as SARS-CoV-2, preventing a productive viral cycle [140]. Another study demonstrated that the co-treatment of TNF-α and IFN-γ induces the JAK/STAT1/IRF1 axis, leading to caspase-8/FADD-mediated PANoptosis (combination of Pyroptosis, Apoptosis and Necroptois) in murine bone marrow derived macrophages (BMDM. Blocking TNF-α and IFN-γ protected mice from mortality during SARS-CoV-2 infection [141], highlighting the significance of these finding in developing therapies targeting cytokine storm-induced mortality in COVID-19 [142,143,144]. Additionally, the proper antimicrobial responses of the innate immune cells and intestinal epithelial cells (IECs), such as macrophages and Paneth cells, plays crucial roles in regulating gut immune homeostasis [145,146], when these response fails, to maintain gut homeostasis chronic inflammations develops illness such as inflammatory bowel disease (IBD) [147,148]. In mice models with IEC-specific deficiencies in caspase-8 deficiency (Casp8fl/fl × Vil1-cre, Casp8IEC-KO) have been reported to develop ileitis [149], as well as impaired mucosal barrier function and bacterial clearance at the epithelial interface leads to colitis [150]. Moreover, mice with IEC-specific FADD deficiency (FADDIEC-KO), spontaneously developed epithelial cell necrosis with loss of Paneth cells and erosive colitis [18]. Collectively, these findings reveal the extensive expression and regulatory functionalities of FADD and caspase-8 in inflammatory pathways (Figure 4).
4.4. Role of FADD in Autophagy
The process of autophagy commonly referred to as the “self-eating” process, aims to degrade unwanted cytosolic constituents by transporting them to the lysosome to protect against stress-induced cell death [151]. While basal levels of autophagy help maintain cellular homeostasis, autophagy can induce cell death during pathological or physiological stress [152]. The pathways of apoptosis and autophagy are interconnected through key regulatory proteins that govern cell death and survival [153]. The DD of FADD interacts with autophagy related-protein 5 (ATG5), thereby triggering autophagic cell death in response to IFN-gamma stimulation [16]. Previous studies have reported that Atg5 has dual role in the regulation of autophagy, but under cell death stress, it may induce cell death [16]. Pua et al. observed significantly increased cell death in Atg5-/- CD8+ T lymphocytes and proposed that the co-regulation of Atg5 and FADD, either in the autophagic process or independent of autophagy, may transmit signals crucial for T cell proliferation [154]. Pyo et al. demonstrated that a Lysine residue in the middle and C-terminal region of Atg5 is conjugated with Atg12 and binds to FADD to induce cell death [16]. Additionally, Pyo et al. used immunoprecipitation analysis to detect the association of Atg5 and Atg12 with FADD in a complex [16]. Earlier reports have indicated that FADD and caspase-8 jointly regulate autophagic signaling for proper T cell proliferation [154,155]. Mitogenically activated T cells triggers the interaction between FADD and the Atg5:Atg12 complex leading to caspase-8 activation and autophagic cell death [154]. Depletion of FADD forces T cells to undergo hyperautophagy and activate RIP1-dependent necroptotic cell death, independent of caspase-8 activation [156]. Re-expression of full-length FADD in FADD-/- MEF restores basal level autophagy induced by serum deprivation [17]. Expression of FADD and concurrent activation of caspase-8 may inhibits hyperautophagy and necroptotic death and favor apoptotic death [157]. Although the crosstalk between apoptosis and autophagy may vary depending on the cell types and stimulus, a better understanding of FADD mediated regulation of both pathways could be valuable for designing a common strategy to regulate both processes (Figure 4).
5. FADD in cancer therapeutic
Cytosolic expression of the adaptor protein FADD is crucial for death receptor-mediated pathways and may serve as a promising therapeutic target in various disease conditions, such as malignancy, autoimmunity, and inflammation. Previous studies have demonstrated that altering FADD expression in T cells impairs resistance against Fas ligand (FasL)-induced apoptosis and promotes cell proliferation [158]. Additionally, FADD deficient mice (FADD-/-) develope thymic lymphoma as they age [159]. Dysregulated expression of FADD could serve as a prominent tumor biomarker and prognostic factor for developing appropriate treatment strategies [1]. We have previously demonstrated that FADD can effectively target the anti-apoptotic protein cFLIP and the pro-inflammatory NF-κB pathway in various tumor cell types [28,29,100,160,161], highlighting the potential of FADD as a therapeutic candidate. Previous approaches have successfully delivered a fusion of the FADD gene with human telomerase reverse transcriptase (hTERT) promoter, resulting in significant apoptosis induction in glioma cells [162]. Advancements in cancer therapy provide opportunities to manipulate the expression of apoptotic genes which can be utilized to regulate a wide spectrum of pathways [163]. Previous studies have demonstrated that adenovirus or retrovirus-mediated transfer of the FADD gene induces apoptosis in glioma cells [164]. Shinoura et al. showed that, adenoviral delivery of FADD adenovirus (Adeno-FADD) potentially induced apoptosis in Fas ligand resistant U251 glioma cells, suggesting that FADD could be a therapeutic modality for treating gliomas [165]. Adenoviral-mediated delivery of the FADD gene to rheumatoid arthritis (RA) synoviocyte cells induces apoptosis, and local injection of FADD adenovirus (Ad-FADD) eliminates human rheumatoid synoviocytes engrafted in severe combined immunodeficiency mice. This suggest that FADD gene transfer might be effective in the treatment of RA [166]. Ho et al. demonstrated that viral vector-mediated delivery of FasL and FADD effectively induced cell death in human glioma cells cultured from biopsy samples. Combined therapies of both genes, in the presence of temozolomide significantly improved the survival of mice bearing high-grade gliomas [167]. Furthermore, novel approaches involving the design of cell penetrating peptides (CPPs) for direct delivery of proteins into the cytoplasm of cells improve the prospects of developing cures for several incurable diseases [168,169]. Our previous research has shown that TAT peptide conjugated FADD protein successfully delivered to cancer cells through the caveolar pathway of endocytosis [170] and induces apoptosis signaling, simultaneously targeting pro-tumorigenic and pro-inflammatory NF-κB signaling [171]. Collectively, targeted delivery of genes or proteins could be effective in combination with conventional chemo- or radiotherapy for cancer treatment.
6. Future perspective
In this review, we have provided a comprehensive description of the dynamic role of FADD in the regulation of cell death and inflammatory pathways. We have also elucidated the molecular mechanisms through which FADD modulates downstream signaling, including NF-κB activation, RIPoptosome assembly, and NLRP3 inflammasome signaling. Further advancements in this field will necessitate a more profound mechanistic understanding of how FADD mediates the regulation of apoptotic and inflammatory pathways. Such understanding will be instrumental in the development of innovative treatment strategies.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
“Conceptualization, K.R. and C.P.; validation, K.R. and C.P.; writing—original draft preparation, review and editing.; supervision, C.P.; project administration, C.P. All authors have read and agreed to the published version of the manuscript.”
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tourneur, L.; Chiocchia, G. FADD: a regulator of life and death. Trends in immunology 2010, 31, 260-269. [CrossRef]
- Scaffidi, C.; Fulda, S.; Srinivasan, A.; Friesen, C.; Li, F.; Tomaselli, K.J.; Debatin, K.M.; Krammer, P.H.; Peter, M.E. Two CD95 (APO-1/Fas) signaling pathways. EMBO J 1998, 17, 1675-1687. [CrossRef]
- Imtiyaz, H.Z.; Zhou, X.; Zhang, H.; Chen, D.; Hu, T.; Zhang, J. The death domain of FADD is essential for embryogenesis, lymphocyte development, and proliferation. The Journal of biological chemistry 2009, 284, 9917-9926. [CrossRef]
- Marin-Rubio, J.L.; Vela-Martin, L.; Fernandez-Piqueras, J.; Villa-Morales, M. FADD in Cancer: Mechanisms of Altered Expression and Function, and Clinical Implications. Cancers (Basel) 2019, 11. [CrossRef]
- Algeciras-Schimnich, A.; Shen, L.; Barnhart, B.C.; Murmann, A.E.; Burkhardt, J.K.; Peter, M.E. Molecular ordering of the initial signaling events of CD95. Molecular and cellular biology 2002, 22, 207-220.
- Jeong, E.J.; Bang, S.; Lee, T.H.; Park, Y.I.; Sim, W.S.; Kim, K.S. The solution structure of FADD death domain. Structural basis of death domain interactions of Fas and FADD. The Journal of biological chemistry 1999, 274, 16337-16342.
- Scott, F.L.; Stec, B.; Pop, C.; Dobaczewska, M.K.; Lee, J.J.; Monosov, E.; Robinson, H.; Salvesen, G.S.; Schwarzenbacher, R.; Riedl, S.J. The Fas-FADD death domain complex structure unravels signalling by receptor clustering. Nature 2009, 457, 1019-1022. [CrossRef]
- Gomez-Angelats, M.; Cidlowski, J.A. Molecular evidence for the nuclear localization of FADD. Cell death and differentiation 2003, 10, 791-797. [CrossRef]
- O'Reilly, L.A.; Divisekera, U.; Newton, K.; Scalzo, K.; Kataoka, T.; Puthalakath, H.; Ito, M.; Huang, D.C.; Strasser, A. Modifications and intracellular trafficking of FADD/MORT1 and caspase-8 after stimulation of T lymphocytes. Cell death and differentiation 2004, 11, 724-736. [CrossRef]
- Screaton, R.A.; Kiessling, S.; Sansom, O.J.; Millar, C.B.; Maddison, K.; Bird, A.; Clarke, A.R.; Frisch, S.M. Fas-associated death domain protein interacts with methyl-CpG binding domain protein 4: a potential link between genome surveillance and apoptosis. Proceedings of the National Academy of Sciences of the United States of America 2003, 100, 5211-5216. [CrossRef]
- Tourneur, L.; Mistou, S.; Michiels, F.M.; Devauchelle, V.; Renia, L.; Feunteun, J.; Chiocchia, G. Loss of FADD protein expression results in a biased Fas-signaling pathway and correlates with the development of tumoral status in thyroid follicular cells. Oncogene 2003, 22, 2795-2804. [CrossRef]
- Park, S.M.; Schickel, R.; Peter, M.E. Nonapoptotic functions of FADD-binding death receptors and their signaling molecules. Current opinion in cell biology 2005, 17, 610-616. [CrossRef]
- Werner, M.H.; Wu, C.; Walsh, C.M. Emerging roles for the death adaptor FADD in death receptor avidity and cell cycle regulation. Cell cycle 2006, 5, 2332-2338.
- Cheng, W.; Zhang, R.; Yao, C.; He, L.; Jia, K.; Yang, B.; Du, P.; Zhuang, H.; Chen, J.; Liu, Z.; et al. A critical role of Fas-associated protein with death domain phosphorylation in intracellular reactive oxygen species homeostasis and aging. Antioxidants & redox signaling 2014, 21, 33-45. [CrossRef]
- Zhou, K.; Bai, L.; Nan, X.; Zhao, K.; Song, Y.; Li, W.; Wang, Q. FADD regulates antibacterial immune responses via the immune deficiency signaling pathway in the Chinese mitten crab. Dev Comp Immunol 2022, 128, 104326. [CrossRef]
- Pyo, J.O.; Jang, M.H.; Kwon, Y.K.; Lee, H.J.; Jun, J.I.; Woo, H.N.; Cho, D.H.; Choi, B.; Lee, H.; Kim, J.H.; et al. Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. The Journal of biological chemistry 2005, 280, 20722-20729. [CrossRef]
- Bell, B.D.; Leverrier, S.; Weist, B.M.; Newton, R.H.; Arechiga, A.F.; Luhrs, K.A.; Morrissette, N.S.; Walsh, C.M. FADD and caspase-8 control the outcome of autophagic signaling in proliferating T cells. Proceedings of the National Academy of Sciences of the United States of America 2008, 105, 16677-16682. [CrossRef]
- Welz, P.S.; Wullaert, A.; Vlantis, K.; Kondylis, V.; Fernandez-Majada, V.; Ermolaeva, M.; Kirsch, P.; Sterner-Kock, A.; van Loo, G.; Pasparakis, M. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature 2011, 477, 330-334. [CrossRef]
- Han, J.; Zhong, C.Q.; Zhang, D.W. Programmed necrosis: backup to and competitor with apoptosis in the immune system. Nature immunology 2011, 12, 1143-1149. [CrossRef]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nature immunology 2000, 1, 489-495. [CrossRef]
- Feng, S.; Yang, Y.; Mei, Y.; Ma, L.; Zhu, D.E.; Hoti, N.; Castanares, M.; Wu, M. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cellular signalling 2007, 19, 2056-2067. [CrossRef]
- Salaun, B.; Romero, P.; Lebecque, S. Toll-like receptors' two-edged sword: when immunity meets apoptosis. European journal of immunology 2007, 37, 3311-3318. [CrossRef]
- Mouasni, S.; Tourneur, L. FADD at the Crossroads between Cancer and Inflammation. Trends Immunol 2018, 39, 1036-1053. [CrossRef]
- Alappat, E.C.; Feig, C.; Boyerinas, B.; Volkland, J.; Samuels, M.; Murmann, A.E.; Thorburn, A.; Kidd, V.J.; Slaughter, C.A.; Osborn, S.L.; et al. Phosphorylation of FADD at serine 194 by CKIalpha regulates its nonapoptotic activities. Molecular cell 2005, 19, 321-332. [CrossRef]
- Bhojani, M.S.; Chen, G.; Ross, B.D.; Beer, D.G.; Rehemtulla, A. Nuclear localized phosphorylated FADD induces cell proliferation and is associated with aggressive lung cancer. Cell cycle 2005, 4, 1478-1481.
- Lee, E.W.; Kim, J.H.; Ahn, Y.H.; Seo, J.; Ko, A.; Jeong, M.; Kim, S.J.; Ro, J.Y.; Park, K.M.; Lee, H.W.; et al. Ubiquitination and degradation of the FADD adaptor protein regulate death receptor-mediated apoptosis and necroptosis. Nature communications 2012, 3, 978. [CrossRef]
- Goto, E.; Tokunaga, F. Decreased linear ubiquitination of NEMO and FADD on apoptosis with caspase-mediated cleavage of HOIP. Biochemical and biophysical research communications 2017, 485, 152-159. [CrossRef]
- Ranjan, K.; Pathak, C. FADD regulates NF-kappaB activation and promotes ubiquitination of cFLIPL to induce apoptosis. Sci Rep 2016, 6, 22787. [CrossRef]
- Ranjan, K.; Pathak, C. Expression of FADD and cFLIP(L) balances mitochondrial integrity and redox signaling to substantiate apoptotic cell death. Mol Cell Biochem 2016, 422, 135-150. [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res 2017, 45, W98-W102. [CrossRef]
- Kim, P.K.; Dutra, A.S.; Chandrasekharappa, S.C.; Puck, J.M. Genomic structure and mapping of human FADD, an intracellular mediator of lymphocyte apoptosis. Journal of immunology 1996, 157, 5461-5466.
- Chinnaiyan, A.M.; O'Rourke, K.; Tewari, M.; Dixit, V.M. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 1995, 81, 505-512.
- Hsu, H.; Shu, H.B.; Pan, M.G.; Goeddel, D.V. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell 1996, 84, 299-308.
- Barnhart, B.C.; Lee, J.C.; Alappat, E.C.; Peter, M.E. The death effector domain protein family. Oncogene 2003, 22, 8634-8644. [CrossRef]
- Boldin, M.P.; Goncharov, T.M.; Goltsev, Y.V.; Wallach, D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell 1996, 85, 803-815.
- Muzio, M.; Chinnaiyan, A.M.; Kischkel, F.C.; O'Rourke, K.; Shevchenko, A.; Ni, J.; Scaffidi, C.; Bretz, J.D.; Zhang, M.; Gentz, R.; et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death--inducing signaling complex. Cell 1996, 85, 817-827.
- Carrington, P.E.; Sandu, C.; Wei, Y.; Hill, J.M.; Morisawa, G.; Huang, T.; Gavathiotis, E.; Wei, Y.; Werner, M.H. The structure of FADD and its mode of interaction with procaspase-8. Molecular cell 2006, 22, 599-610. [CrossRef]
- Hill, J.M.; Morisawa, G.; Kim, T.; Huang, T.; Wei, Y.; Wei, Y.; Werner, M.H. Identification of an expanded binding surface on the FADD death domain responsible for interaction with CD95/Fas. The Journal of biological chemistry 2004, 279, 1474-1481. [CrossRef]
- Yang, J.K. Death effecter domain for the assembly of death-inducing signaling complex. Apoptosis : an international journal on programmed cell death 2015, 20, 235-239. [CrossRef]
- Park, Y.H.; Jeong, M.S.; Park, H.H.; Jang, S.B. Formation of the death domain complex between FADD and RIP1 proteins in vitro. Biochimica et biophysica acta 2013, 1834, 292-300. [CrossRef]
- Eberstadt, M.; Huang, B.; Chen, Z.; Meadows, R.P.; Ng, S.C.; Zheng, L.; Lenardo, M.J.; Fesik, S.W. NMR structure and mutagenesis of the FADD (Mort1) death-effector domain. Nature 1998, 392, 941-945. [CrossRef]
- Tibbetts, M.D.; Zheng, L.; Lenardo, M.J. The death effector domain protein family: regulators of cellular homeostasis. Nature immunology 2003, 4, 404-409. [CrossRef]
- Siegel, R.M.; Martin, D.A.; Zheng, L.; Ng, S.Y.; Bertin, J.; Cohen, J.; Lenardo, M.J. Death-effector filaments: novel cytoplasmic structures that recruit caspases and trigger apoptosis. The Journal of cell biology 1998, 141, 1243-1253.
- Zhang, J.; Winoto, A. A mouse Fas-associated protein with homology to the human Mort1/FADD protein is essential for Fas-induced apoptosis. Molecular and cellular biology 1996, 16, 2756-2763.
- Sandu, C.; Morisawa, G.; Wegorzewska, I.; Huang, T.; Arechiga, A.F.; Hill, J.M.; Kim, T.; Walsh, C.M.; Werner, M.H. FADD self-association is required for stable interaction with an activated death receptor. Cell death and differentiation 2006, 13, 2052-2061. [CrossRef]
- Chinnaiyan, A.M.; Tepper, C.G.; Seldin, M.F.; O'Rourke, K.; Kischkel, F.C.; Hellbardt, S.; Krammer, P.H.; Peter, M.E.; Dixit, V.M. FADD/MORT1 is a common mediator of CD95 (Fas/APO-1) and tumor necrosis factor receptor-induced apoptosis. The Journal of biological chemistry 1996, 271, 4961-4965.
- Zornig, M.; Hueber, A.O.; Evan, G. p53-dependent impairment of T-cell proliferation in FADD dominant-negative transgenic mice. Current biology : CB 1998, 8, 467-470.
- Matsuyoshi, S.; Shimada, K.; Nakamura, M.; Ishida, E.; Konishi, N. FADD phosphorylation is critical for cell cycle regulation in breast cancer cells. British journal of cancer 2006, 94, 532-539. [CrossRef]
- Scaffidi, C.; Volkland, J.; Blomberg, I.; Hoffmann, I.; Krammer, P.H.; Peter, M.E. Phosphorylation of FADD/ MORT1 at serine 194 and association with a 70-kDa cell cycle-regulated protein kinase. Journal of immunology 2000, 164, 1236-1242.
- Sakamaki, K.; Takagi, C.; Kominami, K.; Sakata, S.; Yaoita, Y.; Kubota, H.Y.; Nozaki, M.; Yonehara, S.; Ueno, N. The adaptor molecule FADD from Xenopus laevis demonstrates evolutionary conservation of its pro-apoptotic activity. Genes to cells : devoted to molecular & cellular mechanisms 2004, 9, 1249-1264. [CrossRef]
- Liska, O.; Bohar, B.; Hidas, A.; Korcsmaros, T.; Papp, B.; Fazekas, D.; Ari, E. TFLink: an integrated gateway to access transcription factor-target gene interactions for multiple species. Database (Oxford) 2022, 2022. [CrossRef]
- Hindryckx, P.; De Vos, M.; Jacques, P.; Ferdinande, L.; Peeters, H.; Olievier, K.; Bogaert, S.; Brinkman, B.; Vandenabeele, P.; Elewaut, D.; et al. Hydroxylase inhibition abrogates TNF-alpha-induced intestinal epithelial damage by hypoxia-inducible factor-1-dependent repression of FADD. Journal of immunology 2010, 185, 6306-6316. [CrossRef]
- Nguyen, D.D.; Lee, D.G.; Kim, S.; Kang, K.; Rhee, J.K.; Chang, S. Integrative Bioinformatics and Functional Analyses of GEO, ENCODE, and TCGA Reveal FADD as a Direct Target of the Tumor Suppressor BRCA1. International journal of molecular sciences 2018, 19. [CrossRef]
- Vilmont, V.; Filhol, O.; Hesse, A.M.; Coute, Y.; Hue, C.; Remy-Tourneur, L.; Mistou, S.; Cochet, C.; Chiocchia, G. Modulatory role of the anti-apoptotic protein kinase CK2 in the sub-cellular localization of Fas associated death domain protein (FADD). Biochimica et biophysica acta 2015, 1853, 2885-2896. [CrossRef]
- Zhang, J.; Zhang, D.; Hua, Z. FADD and its phosphorylation. IUBMB life 2004, 56, 395-401. [CrossRef]
- Cimino, Y.; Costes, A.; Damotte, D.; Validire, P.; Mistou, S.; Cagnard, N.; Alifano, M.; Regnard, J.F.; Chiocchia, G.; Sautes-Fridman, C.; et al. FADD protein release mirrors the development and aggressiveness of human non-small cell lung cancer. British journal of cancer 2012, 106, 1989-1996. [CrossRef]
- Ozturk, S.; Schleich, K.; Lavrik, I.N. Cellular FLICE-like inhibitory proteins (c-FLIPs): fine-tuners of life and death decisions. Experimental cell research 2012, 318, 1324-1331. [CrossRef]
- Safa, A.R. c-FLIP, a master anti-apoptotic regulator. Exp Oncol 2012, 34, 176-184.
- Tsuchiya, Y.; Nakabayashi, O.; Nakano, H. FLIP the Switch: Regulation of Apoptosis and Necroptosis by cFLIP. International journal of molecular sciences 2015, 16, 30321-30341. [CrossRef]
- Irmler, M.; Thome, M.; Hahne, M.; Schneider, P.; Hofmann, K.; Steiner, V.; Bodmer, J.L.; Schroter, M.; Burns, K.; Mattmann, C.; et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997, 388, 190-195. [CrossRef]
- Longley, D.B.; Wilson, T.R.; McEwan, M.; Allen, W.L.; McDermott, U.; Galligan, L.; Johnston, P.G. c-FLIP inhibits chemotherapy-induced colorectal cancer cell death. Oncogene 2006, 25, 838-848. [CrossRef]
- Bagnoli, M.; Ambrogi, F.; Pilotti, S.; Alberti, P.; Ditto, A.; Barbareschi, M.; Galligioni, E.; Biganzoli, E.; Canevari, S.; Mezzanzanica, D. c-FLIPL expression defines two ovarian cancer patient subsets and is a prognostic factor of adverse outcome. Endocrine-related cancer 2009, 16, 443-453. [CrossRef]
- Yu, J.W.; Jeffrey, P.D.; Shi, Y. Mechanism of procaspase-8 activation by c-FLIPL. Proceedings of the National Academy of Sciences of the United States of America 2009, 106, 8169-8174. [CrossRef]
- Panaitiu, A.E.; Basiashvili, T.; Mierke, D.F.; Pellegrini, M. An engineered construct of cFLIP provides insight into DED1 structure and interactions. Structure 2022, 30, 229-239 e225. [CrossRef]
- Majkut, J.; Sgobba, M.; Holohan, C.; Crawford, N.; Logan, A.E.; Kerr, E.; Higgins, C.A.; Redmond, K.L.; Riley, J.S.; Stasik, I.; et al. Differential affinity of FLIP and procaspase 8 for FADD's DED binding surfaces regulates DISC assembly. Nature communications 2014, 5, 3350. [CrossRef]
- Chang, D.W.; Xing, Z.; Pan, Y.; Algeciras-Schimnich, A.; Barnhart, B.C.; Yaish-Ohad, S.; Peter, M.E.; Yang, X. c-FLIP(L) is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. The EMBO journal 2002, 21, 3704-3714. [CrossRef]
- Dempsey, P.W.; Doyle, S.E.; He, J.Q.; Cheng, G. The signaling adaptors and pathways activated by TNF superfamily. Cytokine & growth factor reviews 2003, 14, 193-209.
- Waters, J.P.; Pober, J.S.; Bradley, J.R. Tumour necrosis factor and cancer. The Journal of pathology 2013, 230, 241-248. [CrossRef]
- Karin, M.; Lin, A. NF-kappaB at the crossroads of life and death. Nature immunology 2002, 3, 221-227. [CrossRef]
- Marques-Fernandez, F.; Planells-Ferrer, L.; Gozzelino, R.; Galenkamp, K.M.; Reix, S.; Llecha-Cano, N.; Lopez-Soriano, J.; Yuste, V.J.; Moubarak, R.S.; Comella, J.X. TNFalpha induces survival through the FLIP-L-dependent activation of the MAPK/ERK pathway. Cell death & disease 2013, 4, e493. [CrossRef]
- Kreuz, S.; Siegmund, D.; Rumpf, J.J.; Samel, D.; Leverkus, M.; Janssen, O.; Hacker, G.; Dittrich-Breiholz, O.; Kracht, M.; Scheurich, P.; et al. NFkappaB activation by Fas is mediated through FADD, caspase-8, and RIP and is inhibited by FLIP. The Journal of cell biology 2004, 166, 369-380. [CrossRef]
- Fulda, S.; Kufer, M.U.; Meyer, E.; van Valen, F.; Dockhorn-Dworniczak, B.; Debatin, K.M. Sensitization for death receptor- or drug-induced apoptosis by re-expression of caspase-8 through demethylation or gene transfer. Oncogene 2001, 20, 5865-5877. [CrossRef]
- Micheau, O.; Lens, S.; Gaide, O.; Alevizopoulos, K.; Tschopp, J. NF-kappaB signals induce the expression of c-FLIP. Molecular and cellular biology 2001, 21, 5299-5305. [CrossRef]
- Higuchi, H.; Yoon, J.H.; Grambihler, A.; Werneburg, N.; Bronk, S.F.; Gores, G.J. Bile acids stimulate cFLIP phosphorylation enhancing TRAIL-mediated apoptosis. The Journal of biological chemistry 2003, 278, 454-461. [CrossRef]
- Fukazawa, T.; Fujiwara, T.; Uno, F.; Teraishi, F.; Kadowaki, Y.; Itoshima, T.; Takata, Y.; Kagawa, S.; Roth, J.A.; Tschopp, J.; et al. Accelerated degradation of cellular FLIP protein through the ubiquitin-proteasome pathway in p53-mediated apoptosis of human cancer cells. Oncogene 2001, 20, 5225-5231. [CrossRef]
- Roberts, J.Z.; Crawford, N.; Longley, D.B. The role of Ubiquitination in Apoptosis and Necroptosis. Cell death and differentiation 2022, 29, 272-284. [CrossRef]
- Panner, A.; Crane, C.A.; Weng, C.; Feletti, A.; Parsa, A.T.; Pieper, R.O. A novel PTEN-dependent link to ubiquitination controls FLIPS stability and TRAIL sensitivity in glioblastoma multiforme. Cancer Res 2009, 69, 7911-7916. [CrossRef]
- Wilkie-Grantham, R.P.; Matsuzawa, S.; Reed, J.C. Novel phosphorylation and ubiquitination sites regulate reactive oxygen species-dependent degradation of anti-apoptotic c-FLIP protein. The Journal of biological chemistry 2013, 288, 12777-12790. [CrossRef]
- Kerr, E.; Holohan, C.; McLaughlin, K.M.; Majkut, J.; Dolan, S.; Redmond, K.; Riley, J.; McLaughlin, K.; Stasik, I.; Crudden, M.; et al. Identification of an acetylation-dependant Ku70/FLIP complex that regulates FLIP expression and HDAC inhibitor-induced apoptosis. Cell death and differentiation 2012, 19, 1317-1327. [CrossRef]
- Tang, G.; Cho, M.; Wang, X. OncoDB: an interactive online database for analysis of gene expression and viral infection in cancer. Nucleic Acids Res 2022, 50, D1334-D1339. [CrossRef]
- Sakurai, H.; Suzuki, S.; Kawasaki, N.; Nakano, H.; Okazaki, T.; Chino, A.; Doi, T.; Saiki, I. Tumor necrosis factor-alpha-induced IKK phosphorylation of NF-kappaB p65 on serine 536 is mediated through the TRAF2, TRAF5, and TAK1 signaling pathway. The Journal of biological chemistry 2003, 278, 36916-36923. [CrossRef]
- Kelliher, M.A.; Grimm, S.; Ishida, Y.; Kuo, F.; Stanger, B.Z.; Leder, P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity 1998, 8, 297-303.
- Zhang, H.; Zhou, X.; McQuade, T.; Li, J.; Chan, F.K.; Zhang, J. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature 2011, 471, 373-376. [CrossRef]
- Liu, Y.; Fan, C.; Zhang, Y.; Yu, X.; Wu, X.; Zhang, X.; Zhao, Q.; Zhang, H.; Xie, Q.; Li, M.; et al. RIP1 kinase activity-dependent roles in embryonic development of Fadd-deficient mice. Cell death and differentiation 2017, 24, 1459-1469. [CrossRef]
- Grunert, M.; Gottschalk, K.; Kapahnke, J.; Gundisch, S.; Kieser, A.; Jeremias, I. The adaptor protein FADD and the initiator caspase-8 mediate activation of NF-kappaB by TRAIL. Cell death & disease 2012, 3, e414. [CrossRef]
- Wang, L.; Du, F.; Wang, X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008, 133, 693-703. [CrossRef]
- Jang, T.H.; Zheng, C.; Li, J.; Richards, C.; Hsiao, Y.S.; Walz, T.; Wu, H.; Park, H.H. Structural study of the RIPoptosome core reveals a helical assembly for kinase recruitment. Biochemistry 2014, 53, 5424-5431. [CrossRef]
- Tenev, T.; Bianchi, K.; Darding, M.; Broemer, M.; Langlais, C.; Wallberg, F.; Zachariou, A.; Lopez, J.; MacFarlane, M.; Cain, K.; et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Molecular cell 2011, 43, 432-448. [CrossRef]
- Lin, Y.; Devin, A.; Rodriguez, Y.; Liu, Z.G. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes & development 1999, 13, 2514-2526.
- Ea, C.K.; Deng, L.; Xia, Z.P.; Pineda, G.; Chen, Z.J. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Molecular cell 2006, 22, 245-257. [CrossRef]
- Wertz, I.E.; Dixit, V.M. Regulation of death receptor signaling by the ubiquitin system. Cell death and differentiation 2010, 17, 14-24. [CrossRef]
- Chen, Z.J. Ubiquitin signalling in the NF-kappaB pathway. Nature cell biology 2005, 7, 758-765. [CrossRef]
- Sun, J.; Yu, X.; Wang, C.; Yu, C.; Li, Z.; Nie, W.; Xu, X.; Miao, X.; Jin, X. RIP-1/c-FLIPL Induce Hepatic Cancer Cell Apoptosis Through Regulating Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL). Med Sci Monit 2017, 23, 1190-1199. [CrossRef]
- Duckett, C.S. Apoptosis and NF-kappa B: the FADD connection. The Journal of clinical investigation 2002, 109, 579-580. [CrossRef]
- Kang, J.W.; Yan, J.; Ranjan, K.; Zhang, X.; Turner, J.R.; Abraham, C. Myeloid Cell Expression of LACC1 Is Required for Bacterial Clearance and Control of Intestinal Inflammation. Gastroenterology 2020, 159, 1051-1067. [CrossRef]
- Ranjan, K. Intestinal Immune Homeostasis and Inflammatory Bowel Disease: A Perspective on Intracellular Response Mechanisms. Gastrointestinal Disorders 2020, 2, 246-266.
- Kalliolias, G.D.; Ivashkiv, L.B. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol 2016, 12, 49-62. [CrossRef]
- Liu, D.; Zhong, Z.; Karin, M. NF-kappaB: A Double-Edged Sword Controlling Inflammation. Biomedicines 2022, 10. [CrossRef]
- Baldwin, A.S. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-kappaB. The Journal of clinical investigation 2001, 107, 241-246. [CrossRef]
- Ranjan, K.; Sharma, A.; Surolia, A.; Pathak, C. Regulation of HA14-1 mediated oxidative stress, toxic response, and autophagy by curcumin to enhance apoptotic activity in human embryonic kidney cells. Biofactors 2014, 40, 157-169. [CrossRef]
- Hayden, M.S.; Ghosh, S. Regulation of NF-kappaB by TNF family cytokines. Seminars in immunology 2014, 26, 253-266. [CrossRef]
- Blonska, M.; Shambharkar, P.B.; Kobayashi, M.; Zhang, D.; Sakurai, H.; Su, B.; Lin, X. TAK1 is recruited to the tumor necrosis factor-alpha (TNF-alpha) receptor 1 complex in a receptor-interacting protein (RIP)-dependent manner and cooperates with MEKK3 leading to NF-kappaB activation. The Journal of biological chemistry 2005, 280, 43056-43063. [CrossRef]
- Broglie, P.; Matsumoto, K.; Akira, S.; Brautigan, D.L.; Ninomiya-Tsuji, J. Transforming growth factor beta-activated kinase 1 (TAK1) kinase adaptor, TAK1-binding protein 2, plays dual roles in TAK1 signaling by recruiting both an activator and an inhibitor of TAK1 kinase in tumor necrosis factor signaling pathway. The Journal of biological chemistry 2010, 285, 2333-2339. [CrossRef]
- Zhou, L.; Zhang, Y.; Meads, M.B.; Dai, Y.; Ning, Y.; Hu, X.; Li, L.; Sharma, K.; Nkwocha, J.; Parker, R.; et al. IAP and HDAC inhibitors interact synergistically in myeloma cells through noncanonical NF-kappaB- and caspase-8-dependent mechanisms. Blood Adv 2021, 5, 3776-3788. [CrossRef]
- Chaudhary, P.M.; Eby, M.T.; Jasmin, A.; Kumar, A.; Liu, L.; Hood, L. Activation of the NF-kappaB pathway by caspase 8 and its homologs. Oncogene 2000, 19, 4451-4460. [CrossRef]
- Budd, R.C. Death receptors couple to both cell proliferation and apoptosis. The Journal of clinical investigation 2002, 109, 437-441. [CrossRef]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nature reviews. Molecular cell biology 2014, 15, 135-147. [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nature reviews. Molecular cell biology 2010, 11, 700-714. [CrossRef]
- Christofferson, D.E.; Yuan, J. Necroptosis as an alternative form of programmed cell death. Current opinion in cell biology 2010, 22, 263-268. [CrossRef]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nature chemical biology 2005, 1, 112-119. [CrossRef]
- Kalai, M.; Van Loo, G.; Vanden Berghe, T.; Meeus, A.; Burm, W.; Saelens, X.; Vandenabeele, P. Tipping the balance between necrosis and apoptosis in human and murine cells treated with interferon and dsRNA. Cell death and differentiation 2002, 9, 981-994. [CrossRef]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. Virus inhibition of RIP3-dependent necrosis. Cell host & microbe 2010, 7, 302-313. [CrossRef]
- Davis, C.W.; Hawkins, B.J.; Ramasamy, S.; Irrinki, K.M.; Cameron, B.A.; Islam, K.; Daswani, V.P.; Doonan, P.J.; Manevich, Y.; Madesh, M. Nitration of the mitochondrial complex I subunit NDUFB8 elicits RIP1- and RIP3-mediated necrosis. Free radical biology & medicine 2010, 48, 306-317. [CrossRef]
- Vandenabeele, P.; Declercq, W.; Van Herreweghe, F.; Vanden Berghe, T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Science signaling 2010, 3, re4. [CrossRef]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112-1123. [CrossRef]
- Ch'en, I.L.; Tsau, J.S.; Molkentin, J.D.; Komatsu, M.; Hedrick, S.M. Mechanisms of necroptosis in T cells. The Journal of experimental medicine 2011, 208, 633-641. [CrossRef]
- Lu, J.V.; Weist, B.M.; van Raam, B.J.; Marro, B.S.; Nguyen, L.V.; Srinivas, P.; Bell, B.D.; Luhrs, K.A.; Lane, T.E.; Salvesen, G.S.; et al. Complementary roles of Fas-associated death domain (FADD) and receptor interacting protein kinase-3 (RIPK3) in T-cell homeostasis and antiviral immunity. Proceedings of the National Academy of Sciences of the United States of America 2011, 108, 15312-15317. [CrossRef]
- Osborn, S.L.; Diehl, G.; Han, S.J.; Xue, L.; Kurd, N.; Hsieh, K.; Cado, D.; Robey, E.A.; Winoto, A. Fas-associated death domain (FADD) is a negative regulator of T-cell receptor-mediated necroptosis. Proceedings of the National Academy of Sciences of the United States of America 2010, 107, 13034-13039. [CrossRef]
- Lin, Y.; Choksi, S.; Shen, H.M.; Yang, Q.F.; Hur, G.M.; Kim, Y.S.; Tran, J.H.; Nedospasov, S.A.; Liu, Z.G. Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. The Journal of biological chemistry 2004, 279, 10822-10828. [CrossRef]
- Irrinki, K.M.; Mallilankaraman, K.; Thapa, R.J.; Chandramoorthy, H.C.; Smith, F.J.; Jog, N.R.; Gandhirajan, R.K.; Kelsen, S.G.; Houser, S.R.; May, M.J.; et al. Requirement of FADD, NEMO, and BAX/BAK for aberrant mitochondrial function in tumor necrosis factor alpha-induced necrosis. Molecular and cellular biology 2011, 31, 3745-3758. [CrossRef]
- Alvarez-Diaz, S.; Dillon, C.P.; Lalaoui, N.; Tanzer, M.C.; Rodriguez, D.A.; Lin, A.; Lebois, M.; Hakem, R.; Josefsson, E.C.; O'Reilly, L.A.; et al. The Pseudokinase MLKL and the Kinase RIPK3 Have Distinct Roles in Autoimmune Disease Caused by Loss of Death-Receptor-Induced Apoptosis. Immunity 2016, 45, 513-526. [CrossRef]
- Dillon, C.P.; Weinlich, R.; Rodriguez, D.A.; Cripps, J.G.; Quarato, G.; Gurung, P.; Verbist, K.C.; Brewer, T.L.; Llambi, F.; Gong, Y.N.; et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 2014, 157, 1189-1202. [CrossRef]
- Newton, K.; Wickliffe, K.E.; Maltzman, A.; Dugger, D.L.; Strasser, A.; Pham, V.C.; Lill, J.R.; Roose-Girma, M.; Warming, S.; Solon, M.; et al. RIPK1 inhibits ZBP1-driven necroptosis during development. Nature 2016, 540, 129-133. [CrossRef]
- Young, J.A.; Sermwittayawong, D.; Kim, H.J.; Nandu, S.; An, N.; Erdjument-Bromage, H.; Tempst, P.; Coscoy, L.; Winoto, A. Fas-associated death domain (FADD) and the E3 ubiquitin-protein ligase TRIM21 interact to negatively regulate virus-induced interferon production. The Journal of biological chemistry 2011, 286, 6521-6531. [CrossRef]
- Schock, S.N.; Young, J.A.; He, T.H.; Sun, Y.; Winoto, A. Deletion of FADD in macrophages and granulocytes results in RIP3- and MyD88-dependent systemic inflammation. PloS one 2015, 10, e0124391. [CrossRef]
- Ma, Y.; Liu, H.; Tu-Rapp, H.; Thiesen, H.J.; Ibrahim, S.M.; Cole, S.M.; Pope, R.M. Fas ligation on macrophages enhances IL-1R1-Toll-like receptor 4 signaling and promotes chronic inflammation. Nature immunology 2004, 5, 380-387. [CrossRef]
- Henry, C.M.; Martin, S.J. Caspase-8 Acts in a Non-enzymatic Role as a Scaffold for Assembly of a Pro-inflammatory "FADDosome" Complex upon TRAIL Stimulation. Molecular cell 2017, 65, 715-729 e715. [CrossRef]
- Hartwig, T.; Montinaro, A.; von Karstedt, S.; Sevko, A.; Surinova, S.; Chakravarthy, A.; Taraborrelli, L.; Draber, P.; Lafont, E.; Arce Vargas, F.; et al. The TRAIL-Induced Cancer Secretome Promotes a Tumor-Supportive Immune Microenvironment via CCR2. Molecular cell 2017, 65, 730-742 e735. [CrossRef]
- Kawai, T.; Akira, S. TLR signaling. Seminars in immunology 2007, 19, 24-32. [CrossRef]
- Waghela, B.N.; Vaidya, F.U.; Ranjan, K.; Chhipa, A.S.; Tiwari, B.S.; Pathak, C. AGE-RAGE synergy influences programmed cell death signaling to promote cancer. Mol Cell Biochem 2021, 476, 585-598. [CrossRef]
- Bannerman, D.D.; Tupper, J.C.; Kelly, J.D.; Winn, R.K.; Harlan, J.M. The Fas-associated death domain protein suppresses activation of NF-kappa B by LPS and IL-1 beta. The Journal of clinical investigation 2002, 109, 419-425. [CrossRef]
- Aliprantis, A.O.; Yang, R.B.; Weiss, D.S.; Godowski, P.; Zychlinsky, A. The apoptotic signaling pathway activated by Toll-like receptor-2. The EMBO journal 2000, 19, 3325-3336. [CrossRef]
- Zhande, R.; Dauphinee, S.M.; Thomas, J.A.; Yamamoto, M.; Akira, S.; Karsan, A. FADD negatively regulates lipopolysaccharide signaling by impairing interleukin-1 receptor-associated kinase 1-MyD88 interaction. Molecular and cellular biology 2007, 27, 7394-7404. [CrossRef]
- Bossaller, L.; Chiang, P.I.; Schmidt-Lauber, C.; Ganesan, S.; Kaiser, W.J.; Rathinam, V.A.; Mocarski, E.S.; Subramanian, D.; Green, D.R.; Silverman, N.; et al. Cutting edge: FAS (CD95) mediates noncanonical IL-1beta and IL-18 maturation via caspase-8 in an RIP3-independent manner. Journal of immunology 2012, 189, 5508-5512. [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. International journal of molecular sciences 2019, 20. [CrossRef]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nature reviews. Immunology 2013, 13, 397-411. [CrossRef]
- Moriwaki, K.; Bertin, J.; Gough, P.J.; Chan, F.K. A RIPK3-caspase 8 complex mediates atypical pro-IL-1beta processing. Journal of immunology 2015, 194, 1938-1944. [CrossRef]
- Gurung, P.; Anand, P.K.; Malireddi, R.K.; Vande Walle, L.; Van Opdenbosch, N.; Dillon, C.P.; Weinlich, R.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.D. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. Journal of immunology 2014, 192, 1835-1846. [CrossRef]
- Xue, Y.; Enosi Tuipulotu, D.; Tan, W.H.; Kay, C.; Man, S.M. Emerging Activators and Regulators of Inflammasomes and Pyroptosis. Trends Immunol 2019, 40, 1035-1052. [CrossRef]
- Sefik, E.; Qu, R.; Junqueira, C.; Kaffe, E.; Mirza, H.; Zhao, J.; Brewer, J.R.; Han, A.; Steach, H.R.; Israelow, B.; et al. Inflammasome activation in infected macrophages drives COVID-19 pathology. Nature 2022, 606, 585-593. [CrossRef]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-alpha and IFN-gamma Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149-168 e117. [CrossRef]
- Raman, R.; Patel, K.J.; Ranjan, K. COVID-19: Unmasking Emerging SARS-CoV-2 Variants, Vaccines and Therapeutic Strategies. Biomolecules 2021, 11. [CrossRef]
- Raman, R.; Patel, K.J.; Ranjan, K. SARS-CoV-2 Variants: Impact of Spike Mutations on Vaccine and Therapeutic Strategies. In Frontiers of COVID-19: Scientific and Clinical Aspects of the Novel Coronavirus 2019, Adibi, S., Griffin, P., Sanicas, M., Rashidi, M., Lanfranchi, F., Eds.; Springer International Publishing: Cham, 2022; pp. 143-160.
- Singh, S.; Kishore, D.; Singh, R.K.; Pathak, C.; Ranjan, K. Higher BCG-induced trained immunity prevalence predicts protection from COVID-19: Implications for ongoing BCG trials. Clin Transl Discov 2022, 2, e60. [CrossRef]
- Kaser, A.; Zeissig, S.; Blumberg, R.S. Inflammatory bowel disease. Annu Rev Immunol 2010, 28, 573-621. [CrossRef]
- Ranjan, K.; Hedl, M.; Abraham, C. The E3 ubiquitin ligase RNF186 and RNF186 risk variants regulate innate receptor-induced outcomes. Proceedings of the National Academy of Sciences of the United States of America 2021, 118. [CrossRef]
- Huang, C.; Hedl, M.; Ranjan, K.; Abraham, C. LACC1 Required for NOD2-Induced, ER Stress-Mediated Innate Immune Outcomes in Human Macrophages and LACC1 Risk Variants Modulate These Outcomes. Cell Rep 2019, 29, 4525-4539 e4524. [CrossRef]
- Ranjan, K.; Hedl, M.; Sinha, S.; Zhang, X.; Abraham, C. Ubiquitination of ATF6 by disease-associated RNF186 promotes the innate receptor-induced unfolded protein response. The Journal of clinical investigation 2021, 131. [CrossRef]
- Gunther, C.; Martini, E.; Wittkopf, N.; Amann, K.; Weigmann, B.; Neumann, H.; Waldner, M.J.; Hedrick, S.M.; Tenzer, S.; Neurath, M.F.; et al. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature 2011, 477, 335-339. [CrossRef]
- Stolzer, I.; Kaden-Volynets, V.; Ruder, B.; Letizia, M.; Bittel, M.; Rausch, P.; Basic, M.; Bleich, A.; Baines, J.F.; Neurath, M.F.; et al. Environmental Microbial Factors Determine the Pattern of Inflammatory Lesions in a Murine Model of Crohn's Disease-Like Inflammation. Inflamm Bowel Dis 2020, 26, 66-79. [CrossRef]
- Levine, B.; Yuan, J. Autophagy in cell death: an innocent convict? The Journal of clinical investigation 2005, 115, 2679-2688. [CrossRef]
- Klionsky, D.J. The molecular machinery of autophagy: unanswered questions. Journal of cell science 2005, 118, 7-18. [CrossRef]
- Mukhopadhyay, S.; Panda, P.K.; Sinha, N.; Das, D.N.; Bhutia, S.K. Autophagy and apoptosis: where do they meet? Apoptosis : an international journal on programmed cell death 2014, 19, 555-566. [CrossRef]
- Pua, H.H.; Dzhagalov, I.; Chuck, M.; Mizushima, N.; He, Y.W. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. The Journal of experimental medicine 2007, 204, 25-31. [CrossRef]
- Li, C.; Capan, E.; Zhao, Y.; Zhao, J.; Stolz, D.; Watkins, S.C.; Jin, S.; Lu, B. Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death. Journal of immunology 2006, 177, 5163-5168.
- Walsh, C.M.; Edinger, A.L. The complex interplay between autophagy, apoptosis, and necrotic signals promotes T-cell homeostasis. Immunological reviews 2010, 236, 95-109. [CrossRef]
- Tait, S.W.; Ichim, G.; Green, D.R. Die another way--non-apoptotic mechanisms of cell death. Journal of cell science 2014, 127, 2135-2144. [CrossRef]
- Newton, K.; Harris, A.W.; Bath, M.L.; Smith, K.G.; Strasser, A. A dominant interfering mutant of FADD/MORT1 enhances deletion of autoreactive thymocytes and inhibits proliferation of mature T lymphocytes. The EMBO journal 1998, 17, 706-718. [CrossRef]
- Newton, K.; Harris, A.W.; Strasser, A. FADD/MORT1 regulates the pre-TCR checkpoint and can function as a tumour suppressor. The EMBO journal 2000, 19, 931-941. [CrossRef]
- Ranjan, K.; Pathak, C. Expression of cFLIPL Determines the Basal Interaction of Bcl-2 With Beclin-1 and Regulates p53 Dependent Ubiquitination of Beclin-1 During Autophagic Stress. J Cell Biochem 2016, 117, 1757-1768. [CrossRef]
- Ranjan, K.; Surolia, A.; Pathak, C. Apoptotic potential of Fas-associated death domain on regulation of cell death regulatory protein cFLIP and death receptor mediated apoptosis in HEK 293T cells. J Cell Commun Signal 2012, 6, 155-168. [CrossRef]
- Komata, T.; Koga, S.; Hirohata, S.; Takakura, M.; Germano, I.M.; Inoue, M.; Kyo, S.; Kondo, S.; Kondo, Y. A novel treatment of human malignant gliomas in vitro and in vivo: FADD gene transfer under the control of the human telomerase reverse transcriptase gene promoter. International journal of oncology 2001, 19, 1015-1020.
- Pope, R.M. Apoptosis as a therapeutic tool in rheumatoid arthritis. Nature reviews. Immunology 2002, 2, 527-535. [CrossRef]
- Kondo, S.; Ishizaka, Y.; Okada, T.; Kondo, Y.; Hitomi, M.; Tanaka, Y.; Haqqi, T.; Barnett, G.H.; Barna, B.P. FADD gene therapy for malignant gliomas in vitro and in vivo. Human gene therapy 1998, 9, 1599-1608. [CrossRef]
- Shinoura, N.; Yoshida, Y.; Sadata, A.; Hanada, K.I.; Yamamoto, S.; Kirino, T.; Asai, A.; Hamada, H. Apoptosis by retrovirus- and adenovirus-mediated gene transfer of Fas ligand to glioma cells: implications for gene therapy. Human gene therapy 1998, 9, 1983-1993. [CrossRef]
- Kobayashi, T.; Okamoto, K.; Kobata, T.; Hasunuma, T.; Kato, T.; Hamada, H.; Nishioka, K. Novel gene therapy for rheumatoid arthritis by FADD gene transfer: induction of apoptosis of rheumatoid synoviocytes but not chondrocytes. Gene therapy 2000, 7, 527-533. [CrossRef]
- Ho, I.A.; Ng, W.H.; Lam, P.Y. FasL and FADD delivery by a glioma-specific and cell cycle-dependent HSV-1 amplicon virus enhanced apoptosis in primary human brain tumors. Mol Cancer 2010, 9, 270. [CrossRef]
- Zhang, Y.; Roise, J.J.; Lee, K.; Li, J.; Murthy, N. Recent developments in intracellular protein delivery. Curr Opin Biotechnol 2018, 52, 25-31. [CrossRef]
- Kumar, M.; Ranjan, K.; Singh, V.; Pathak, C.; Pappachan, A.; Singh, D.D. Hydrophilic Acylated Surface Protein A (HASPA) of Leishmania donovani: Expression, Purification and Biophysico-Chemical Characterization. Protein J 2017, 36, 343-351. [CrossRef]
- Pathak, C.; Vaidya, F.U.; Waghela, B.N.; Jaiswara, P.K.; Gupta, V.K.; Kumar, A.; Rajendran, B.K.; Ranjan, K. Insights of Endocytosis Signaling in Health and Disease. International journal of molecular sciences 2023, 24. [CrossRef]
- Ranjan, K.; Waghela, B.N.; Vaidya, F.U.; Pathak, C. Cell-Penetrable Peptide-Conjugated FADD Induces Apoptosis and Regulates Inflammatory Signaling in Cancer Cells. International journal of molecular sciences 2020, 21. [CrossRef]
Figure 1.
Expression of FADD in across different tumor types. mRNA expression levels of FADD were examined in tumor (red) and matched normal (green) tissues. Notably, downregulation of FADD was observed in colon, lung, and prostate tumor tissues when compared to their respective normal tissues. The dataset used in this analysis includes a varying number of cases for each tumor type, as follows: breast invasive carcinoma (BRCA), cholangiocarcinoma (CHOL), colon adenocarcinoma (COAD), glioblastoma (GBM), low-grade gliomas (LGG), lung adenocarcinoma (LUAD), lung squamous carcinoma (LUSC), ovarian tumor (OV), and prostate adenocarcinoma (PRAD). The normalized datasets were obtained from GEPIA. [30].
Figure 1.
Expression of FADD in across different tumor types. mRNA expression levels of FADD were examined in tumor (red) and matched normal (green) tissues. Notably, downregulation of FADD was observed in colon, lung, and prostate tumor tissues when compared to their respective normal tissues. The dataset used in this analysis includes a varying number of cases for each tumor type, as follows: breast invasive carcinoma (BRCA), cholangiocarcinoma (CHOL), colon adenocarcinoma (COAD), glioblastoma (GBM), low-grade gliomas (LGG), lung adenocarcinoma (LUAD), lung squamous carcinoma (LUSC), ovarian tumor (OV), and prostate adenocarcinoma (PRAD). The normalized datasets were obtained from GEPIA. [30].
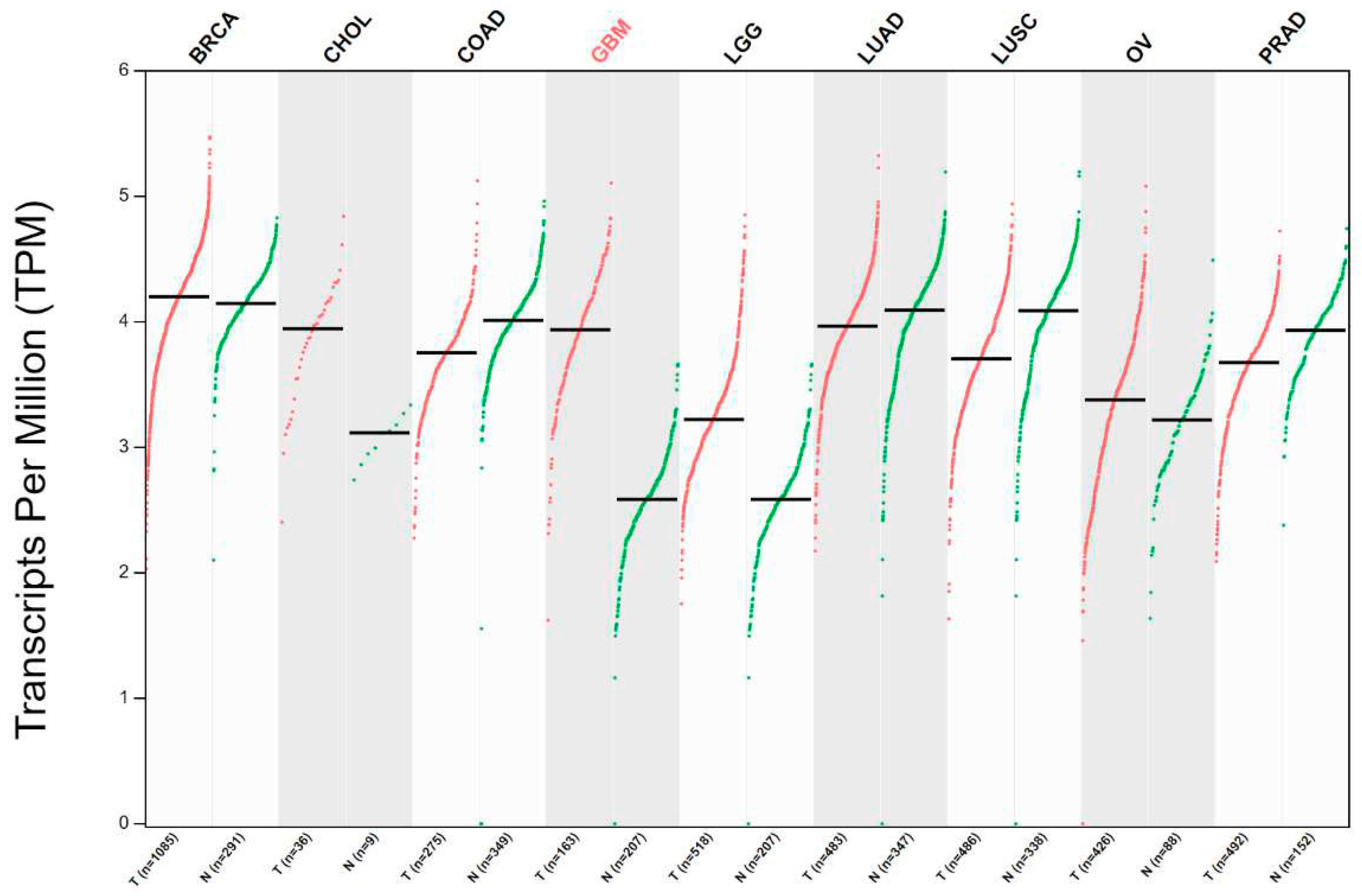
Figure 2.
Interacting network of transcription factors (TFs) regulating FADD. Interactive network visualization of FADD and regulatory TFs. Total 411 TFs experiemntally determined that regulates FADD promoter region. Datasets obtained from TFlink. Please refer Supplementary Table S1 for in-depth analysis [51].
Figure 2.
Interacting network of transcription factors (TFs) regulating FADD. Interactive network visualization of FADD and regulatory TFs. Total 411 TFs experiemntally determined that regulates FADD promoter region. Datasets obtained from TFlink. Please refer Supplementary Table S1 for in-depth analysis [51].

Figure 3.
mRNA expression correlation analysis of FADD with cFLIP, RIPK1, and RIPK2 across different cancer types. The analysis focuses on mRNA expression correlation in various cancer types, including Breast Invasive Carcinoma (BRCA, n=1063), Colon Adenocarcinoma (COAD, n=302), Brain Lower Grade Carcinoma (LGG, n=359), Lung Adenocarcinoma (LUAD, n=530), and Pancreatic Adenocarcinoma (PRAD, n=317). TPM, transcripts per million. The datasets used for this study were obtained from oncoDB [80].
Figure 3.
mRNA expression correlation analysis of FADD with cFLIP, RIPK1, and RIPK2 across different cancer types. The analysis focuses on mRNA expression correlation in various cancer types, including Breast Invasive Carcinoma (BRCA, n=1063), Colon Adenocarcinoma (COAD, n=302), Brain Lower Grade Carcinoma (LGG, n=359), Lung Adenocarcinoma (LUAD, n=530), and Pancreatic Adenocarcinoma (PRAD, n=317). TPM, transcripts per million. The datasets used for this study were obtained from oncoDB [80].
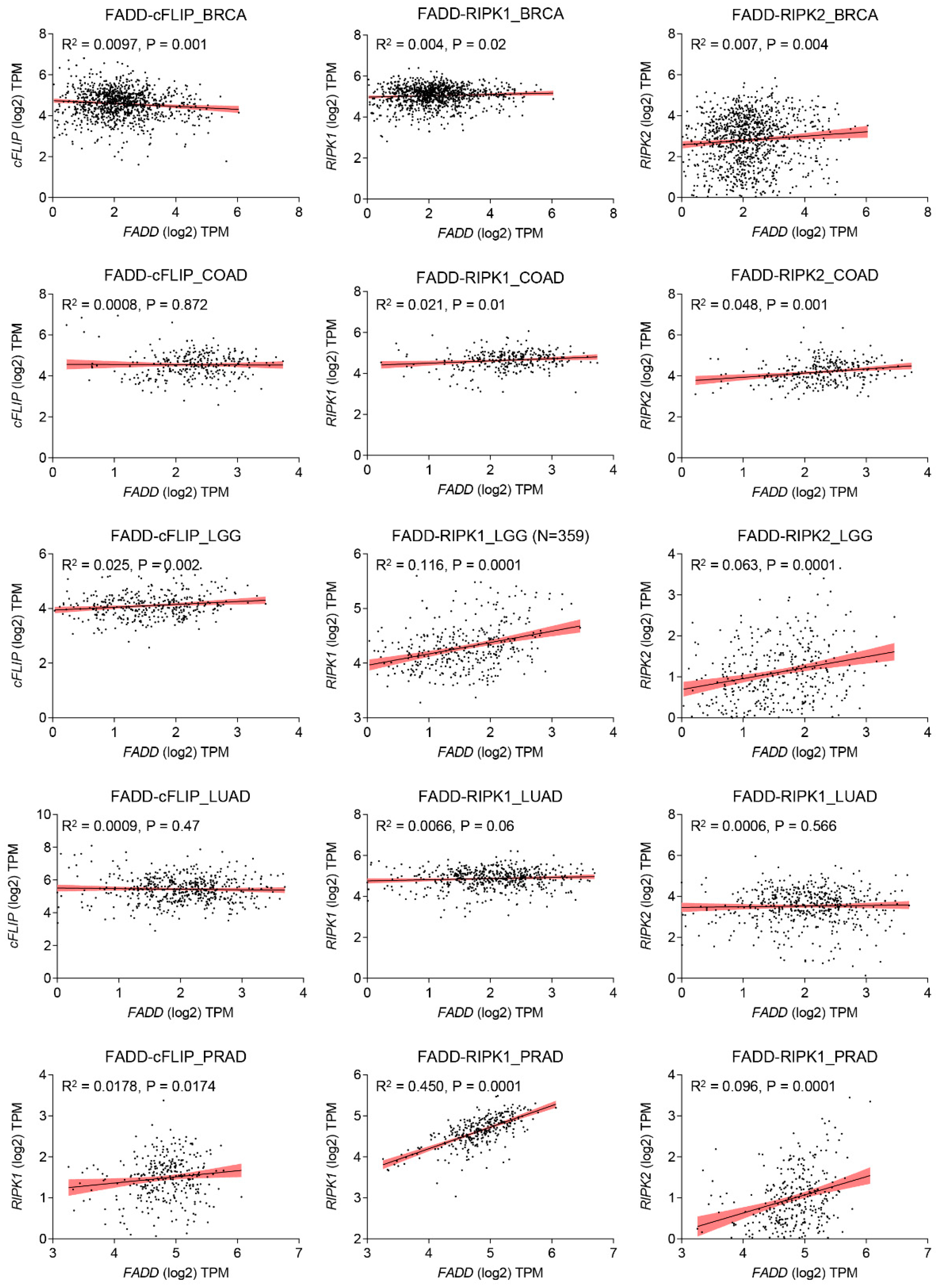
Figure 4.
FADD as a master regulator of cell death and inflammatory pathways. Cytosolic expression of FADD is crucial for the regulation of various intracellular pathways. The FADD protein either directly interacts with key pathway regulators (through DD interaction) or induces some pathways through post-translational modifications.
Figure 4.
FADD as a master regulator of cell death and inflammatory pathways. Cytosolic expression of FADD is crucial for the regulation of various intracellular pathways. The FADD protein either directly interacts with key pathway regulators (through DD interaction) or induces some pathways through post-translational modifications.
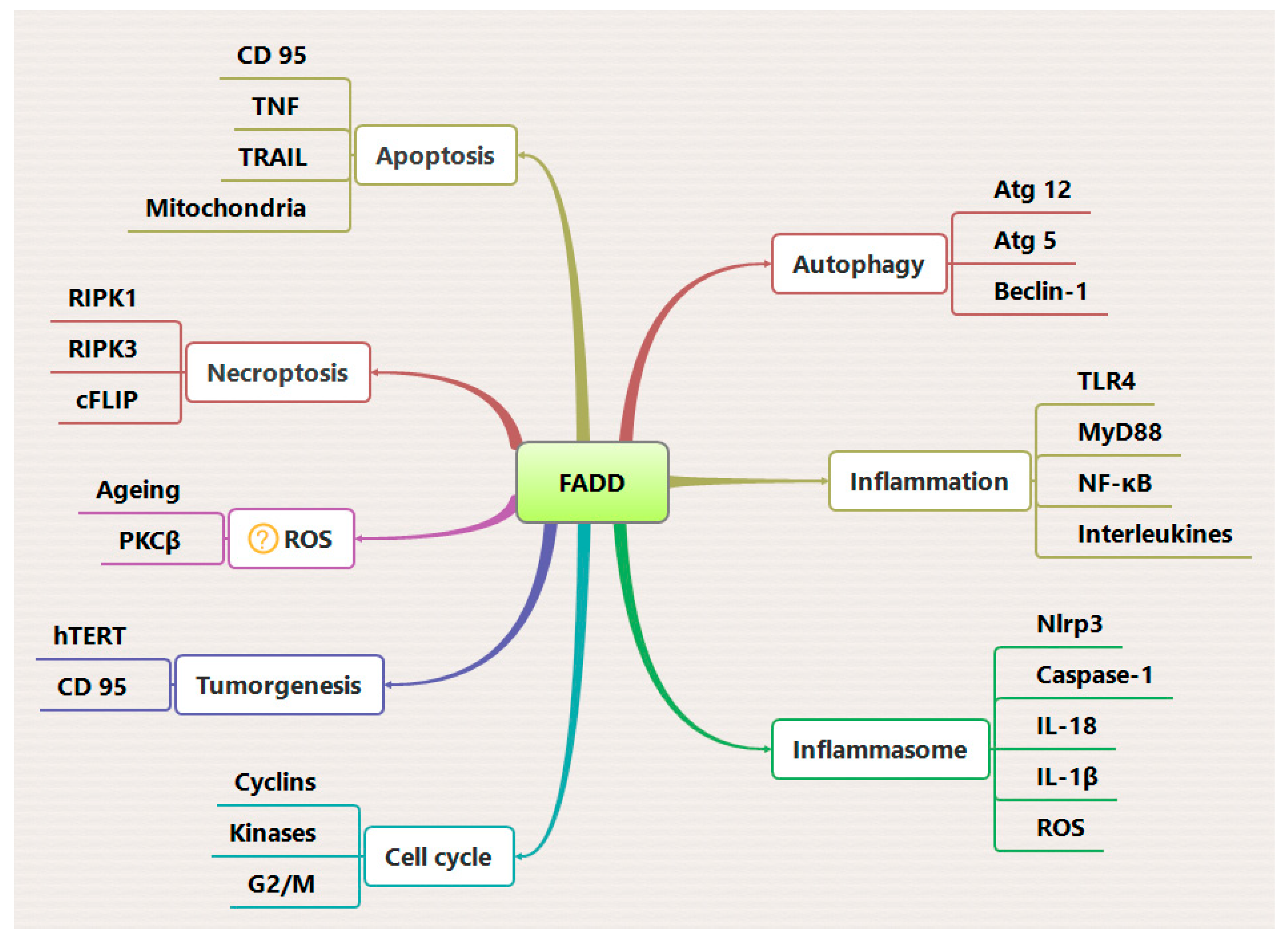
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



