
Submitted:
27 December 2023
Posted:
29 December 2023
You are already at the latest version
Abstract
Synaptic transmission is essential for nervous system function and the loss of synapses is a known major contributor to dementia. Alzheimer’s disease dementia (ADD) is characterized by synaptic loss in the mesial temporal lobe and cerebral neocortex, brain areas associated with memory and cognition. The association of synaptic loss and ADD was established in the late 1980s and it has been estimated that 30-50% of neocortical synaptic protein is lost in ADD, but there has not yet been a quantitative profiling of different synaptic proteins in different brain regions in ADD from the same individuals. Very recently, PET imaging of synapses is being developed, accelerating the focus on the role of synaptic loss in ADD and other conditions. In this study, we quantified densities of two synaptic proteins, the presynaptic protein SNAP25 and the postsynaptic protein PSD95 in human brain using enzyme-linked immunosorbent assays (ELISA). Protein was extracted from the cingulate, hippocampus, frontal, visual, and entorhinal cortex from cognitively unimpaired controls, subjects with mild cognitive impairment (MCI) and demented subjects with different levels of Alzheimer’s pathology. SNAP25 is significantly reduced in ADD when compared to controls in frontal cortex, visual cortex and cingulate; while hippocampus showed a smaller, non-significant reduction and entorhinal cortex concentrations were not different. In contrast, all brain areas showed lower PSD95 concentrations in ADD when compared to non-demented controls, although in hippocampus this failed to reach significance. Interestingly, cognitively unimpaired cases with high levels of AD pathology had higher levels of both synaptic proteins in all brain regions. SNAP25 and PSD95 concentrations significantly correlated with densities of neurofibrillary tangles, amyloid plaques and Mini Mental State Examination scores. Our results suggest that synaptic transmission is affected by ADD in multiple brain regions. Differences were less marked in entorhinal cortex and hippocampus, most likely due to a ceiling effect imposed by the very early development of neurofibrillary tangles in older people in these brain regions.
Keywords:
aging
; SNAP25
; PSD95
; neurofibrillary tangles
; amyloid plaques
; Braak stage
; cerebral cortex
; PET imaging
1. Introduction
Neurodegenerative diseases such as Alzheimer’s disease dementia (ADD) are characterized by brain weight loss probably due to progressive degeneration and death of nerve cells that cause loss of brain matter[1,2,3]. This includes brain matter loss in areas that are associated with cognition and thus is a potentially useful biomarker. While brain weight loss could be explained by neuronal loss, axonal loss, or synapse loss, previous stereological studies have failed to show any significant neocortical neuronal loss in ADD [4,5,6,7,8,9]. Therefore, the greater brain weight loss that we and others have observed in ADD [10] is most likely due to axonal loss and/or synaptic loss. Synaptic integrity is affected in multiple neurodegenerative diseases, and highly correlates with cognitive decline in both human and animal models. A common hypothesis has proposed that the progression of ADD is accompanied by synapse loss due to the accumulation of pathologic hyperphosphorylated tau and amyloid-β [11,12,13]. Beyond actual loss, some have suggested that synaptic dysfunction might precede late-stage features of many neurological conditions such as ADD [11,14,15,16,17,18,19]. The correlation between synaptic loss and Alzheimer’s disease dementia (ADD) was established in the late 1980s using electron microscopy (EM) techniques [3]. These methods are precise but are limited by the laborious tissue processing required and by their practical restriction to extremely small tissue samples. In the 1990s, immunochemical quantification became possible and confirmed 30-50% neocortical synaptic protein losses in ADD [19]. Very recently, PET imaging of synapses is being developed, accelerating the focus on the role of synaptic loss in ADD and other conditions [6,20,21]. However, there has not yet been an extensive profiling of different synaptic proteins in different brain regions in ADD. In this study we have done immunochemical assays to estimate the relative expression of presynaptic protein Synaptosome Associated Protein 25 (SNAP25) and Postsynaptic Density Protein 95 (PSD95) in different brain regions in cognitively unimpaired subjects (CU), ADD and cases with moderate levels of AD pathology that were either cognitively unimpaired (CU-HP) or had mild cognitive impairment (MCI).
2. Methods
Subjects were all volunteers in the Arizona Study of Aging and Neurodegenerative Disorders (AZSAND), a longitudinal clinicopathological study of normal aging, cognition, and movement in the elderly since 1996 in Sun City, Arizona [22,23]. Autopsies are performed by the Banner Sun Health Research Institute Brain and Body Donation Program (BBDP; www.brainandbodydonation program.org). All subjects sign Institutional Review Board-approved informed consents allowing both clinical assessments during life and several options for brain and/or bodily organ donation after death. Most subjects are clinically characterized with annual standardized test batteries consisting of general neurological, cognitive and movement disorders components, including the Mini Mental State Examination (MMSE) [22,23]. Subjects for the current study (Table 1; n=101) were chosen by searching the BBDP database for cases that had a clinicopathological diagnosis of ADD (n=35), a final clinical diagnosis of cognitively unimpaired (CU; n=33), defined as those lacking dementia or MCI and having low levels of AD pathology according to the National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease (NIA-AA) [24], and a final clinical diagnosis of mild cognitive impairment (MCI) with moderate levels of AD pathology or who were cognitively unimpaired individuals with high levels of AD pathology (CU-HP). Brain weights were determined at autopsy, after removal of 10-30 cc of ventricular cerebrospinal fluid but prior to fixation. The complete neuropathological examination was performed using standard AZSAND methods [22,23]. Thick (40-80 um), large-format (up to half of each cerebral hemisphere) sections of un-embedded fixed brain were taken using a sliding freezing microtome while standard-sized paraffin-embedded blocks were sectioned (5-6 um) using a rotary microtome. Microscopic observations included assessment of frontal, parietal, temporal and occipital lobes, all major diencephalic nuclei and major subdivisions of the brainstem, cerebellum and spinal cord (limited to cervical cord for brain-only autopsies). Both slide sets were stained with hematoxylin and eosin and the large-format set was also stained for senile plaques, neurofibrillary changes and other neuronal and glial tauopathies using thioflavin S, Gallyas and Campbell-Switzer methods [22,23,25,26,27]. In all cases, an additional set of paraffin sections was immunohistochemically stained for phosphorylated α-synuclein (p-syn), while staining for phosphorylated TDP-43 (p-TDP43) was done for a subset of subjects [28,29,30]. Neuritic plaque and neurofibrillary tangle (NFT) densities were graded blindly as recommended by CERAD with separate semi-quantitative density estimates of none, sparse, moderate, or frequent [31]. All scores were converted to a 0–3 scale for statistical purposes. Regions scored included cortical gray matter from frontal (F), temporal (T), parietal (P), hippocampal CA1 (H), and entorhinal (E) regions. Summation of all scores from all areas were used for statistical correlations, with a maximum score of 15. Neurofibrillary degeneration was staged on the thick frozen sections by the original method of Braak [25,27], and neuropathological ADD diagnoses were made when neuritic plaque densities and Braak stage met “intermediate” or “high” criteria according to NIA-AA criteria [24,32,33,34]. Non-ADD conditions were diagnosed using standard clinicopathological criteria, with international consensus criteria for those disorders where these were available.
2.1. Synaptic densities
Frozen samples consisted of 30 cryostat sections (approximately equivalent to 100 mg of tissue) from frontal cortex, cingulate, visual cortex area 17, hippocampus and entorhinal cortex. Tissue was homogenized for protein extraction in 1mL of RIPA buffer plus Protein Inhibitor Cocktail (PIC) using an OmniTH tissue grinder. Homogenates were then centrifuged at 40,000 x g for 30 min at 4 C and the supernatant was collected and stored at -80 C. Total protein was quantified using Micro BCA Protein Assay (Pierce 23235). Laboratory-developed sandwich enzyme-linked immunosorbent assays (ELISA) were used to measure the concentration of presynaptic protein, SNAP25, and post-synaptic protein, PSD95. The protocol is a modification from Gottschall et al, 2010 [4]. For the SNAP25 assay, mouse monoclonal anti-SNAP25, 1:200 (clone SP14, MAB331, Millipore) was used as the capture antibody and polyclonal rabbit anti-SNAP25, 1:1,000 (IgG fraction, S9684, Sigma, St. Louis, MO) was used as the detection antibody. For PSD95, mouse anti-PSD95 antibody at 1:100 (clone 7E3-1B8, EMD Millipore, MAB1598) was the capture antibody while rabbit anti-PSD95, 1:400 (Abcam ab18258) was used as the detection antibody. Absorbance was measured at 450 nm on a Bio-Rad iMark absorbance microplate reader (Bio-Rad, Hercules, CA). Standard curves were created by dilution of mixtures of cortical brain derived from two male and female control subjects.
Briefly, capture antibody was incubated in the ELISA plates overnight at room temperature. Then the plates were washed with buffer B (10 mM phosphate buffered saline, pH 7.5, 0.05% Tween 20), and all wells were blocked with blocking/dilution buffer (PBS 0.05% Tween 20 +1% BSA + 50mM glycine + PIC) for one hour with shaking. Standard curve and study protein samples were diluted in blocking/dilution buffer and then incubated in the coated plates for two hours at room temperature with shaking. After removing the samples and washing again, the plates were incubated with the detection antibody for an additional 2 hrs at room temperature with shaking. Plates were washed. Developing antibody consisted of (horse radish peroxidase conjugated) AffiniPure goat anti-rabbit IgG (1:10,000) (Jackson Laboratory, 111-035-144) diluted in blocking/dilution buffer and added as 100 µl. The plates were shaken at room temperature for 45 min. Wells were washed five times with buffer B and 100 µl of tetramethylbenzidine substrate (Sigma T8665) was added, the plate incubated in the dark until the zero substrate blank becomes the most faint, pale blue (usually 20 to 30 min depending on the assay). To stop development, 50 µl of 1 M H2SO4 was added and absorbance measured immediately at 450 nm on a Bio-Rad iMark absorbance microplate reader.
2.2. Statistical Methods
Univariate analyses were used as an initial screen to indicate which variables might have significant relationships with diagnostic groups, synaptic protein concentrations and/or MMSE scores. For comparing group measures, the Mann-Whitney U-Test, one way analysis of variance (ANOVA), Kruskal-Wallis ANOVA and contrast analyses were used as appropriate. The chi-squared test was used to compare proportions and Spearman’s method was used to test univariate correlations. Variables that were significantly affected on this initial screen were included in multivariable logistic regression models.
3. Results
The group age means differed significantly (p <0.0001). The youngest group (ADD) had a mean age of 82, while the oldest group (CU-HP) mean age was 92. The CU mean age was 83 and the MCI group averaged 90 years (Table 1). As expected, AD pathology was significantly different between groups, with CU having the lowest densities of amyloid plaques and neurofibrillary tangles, ADD having the highest densities of such pathologies and MCI and CU-HP showing almost identical moderate levels of AD pathology (Table 1 and Figure 1). MMSE scores were significantly lower in ADD when compared to the other groups, while CU-HP and MCI had lower scores than the CU group, but these did not meet statistical significance. PMI and sex distribution were not significantly different between groups and brain weights were only significantly lower in ADD.
Presynaptic protein SNAP25 expression was significantly reduced in ADD frontal cortex, Brodmann area 17, cingulate cortex, and hippocampus, to 44%, 57%, 38% and 62%, of CU levels, while entorhinal cortex showed similar protein expression in both groups. MCI cases, relative to CU cases, expressed significantly? higher concentrations of SNAP25 in almost all brain regions, while CU-HD groups showed higher concentrations in all brain regions, but these differences did not reach statistical significance. PSD95 protein levels were significantly reduced in almost all brain regions of the ADD group, to 46%, 48%, 56%, and 47%, in frontal, cingulate, visual, and entorhinal areas, respectively, when compared to CU; hippocampus showed a 46% reduction but did not reach the significance level. Expression of PSD95 in MCI and CU-HD groups, as for SNAP25, was greater than in the CU group but this did not reach the significance level. In addition, we observed a greater synaptic protein reduction, for both SNAP25 and PSD95, in females with ADD than in males, relative to CU. These differences reached significance in the frontal, cingulate and visual cortices, while no sex differences were observed in entorhinal cortex and hippocampus, except for PSD95 expression, that was also reduced to a greater level in these regions in females with ADD (Figure2).
Univariable correlations were significant, for both SNAP25 and PSD95, with plaque and NFT densities as well as with MMSE and age (Table 2) but not with brain weight. Logistic regression models confirmed that the MMSE and NFT correlations with synaptic protein concentrations were significant even after correcting for sex and age.
4. Discussion
It has been accepted for decades that synapses are lost and dysregulated in ADD [5,11,14,15,16,18], yet to our knowledge this is the first time that synaptic densities from multiple areas in the brain have been evaluated in a large set of pathologically confirmed cases. SNAP25 is a component of the SNARE complex, which is central to synaptic vesicle exocytosis, fusion and eventually neurotransmitter release. It is suggested that chronic reductions of SNAP25 levels are associated with intellectual impairment, psychiatric disorders and ADD [35,36,37]. In addition, some studies suggest that reduction of this protein may impair the structure and/or function of postsynaptic proteins such as PSD95. PSD95 is suggested to be one of the most abundant postsynaptic proteins. The protein is located at excitatory synapses, an essential component involved in the regulation of synaptic strength and plasticity by regulating glutamatergic receptor trafficking, which is impaired in AD. Therefore, multiple studies suggest that PSD95 disruption is associated with cognitive and learning deficits. Reduced expression has been also observed in the brains of subjects who die with ADD and in AD animal models [37,38,39,40]. Our results support the previous understanding of synaptic loss in ADD as measured by synaptic protein concentrations. We observed considerable variability within each group that could be attributed to multiple factors, such as sex differences, and variable levels of AD pathology but in addition we observed a possible ceiling effect on reduction in areas affected by ADD early in life such as the entorhinal cortex and the hippocampus. The entorhinal area is the main interface between the hippocampus and neocortex, making this area crucial for memory formation and consolidation. Our data suggest that synaptic loss in these regions might happen often in normal aging [2,3]. The mean age at death of individuals in our program and at many other centers is in the 80s, suggesting that the loss of synapses might be occurring a decade or more before this. Therefore, preventing or reversing synaptic loss might best be accomplished long before the first signs of cognitive decline. Another puzzling result from this study is the apparent upregulation of synaptic proteins in multiple brain regions in the MCI and CU-HP group that suggest a compensatory effect at early stages of the disease. This phenomenon has been previously observed by others and could explain resilience of a group of individuals that potentially could “tolerate” greater amounts of pathology resulting in a delay of disease progression. However, some studies hypothesize that the upregulation of these synaptic proteins trigger detrimental events in the synaptic terminals that result in synaptic loss. [18,41,42,43]. In addition, our study suggests important sex differences that we would like to explore in future studies that would include a larger number of subjects per sex in each group. Our data suggests that cognitively normal females have higher densities of presynaptic protein in cortical regions and therefore in ADD experience a proportionately greater loss than males [44,45,46]. Studying such differences will not only help us understand better the mechanisms through which synapses might be lost in ADD and aging but will also be an important consideration in future treatment trails [42].
Future studies that could allow us to visually quantify synapses in multiple brain regions of the same individuals are still needed to determine whether our results are due to loss of synapses or loss of protein concentrations per synapse. This will also allow us to understand better the increased synaptic protein expression in the CU-HP group. Are these subjects expressing more synaptic protein or do they have more synapses? It will be important to understand if these subjects’ cognitive abilities are still preserved, even with moderate levels of AD pathology, because they had greater pre-existing synaptic densities or synaptic protein expression, or whether these were developed as a compensatory response to progressive AD pathology. If the latter, this suggests that therapeutic agents might be developed to initiate these responses.
Figure 2.
Diagnostic group and sex as predictors of brain regional SNAP25 and PSD95 protein. Odds ratios for each comparison and p values were calculated using non-parametric correlations. P<0.05 *; SFG: Superior Frontal Gyrus; ENT: Entorhinal Cortex; HIP: Hippocampus; CING: Cingulate; A17: Visual cortex area 17.
Figure 2.
Diagnostic group and sex as predictors of brain regional SNAP25 and PSD95 protein. Odds ratios for each comparison and p values were calculated using non-parametric correlations. P<0.05 *; SFG: Superior Frontal Gyrus; ENT: Entorhinal Cortex; HIP: Hippocampus; CING: Cingulate; A17: Visual cortex area 17.
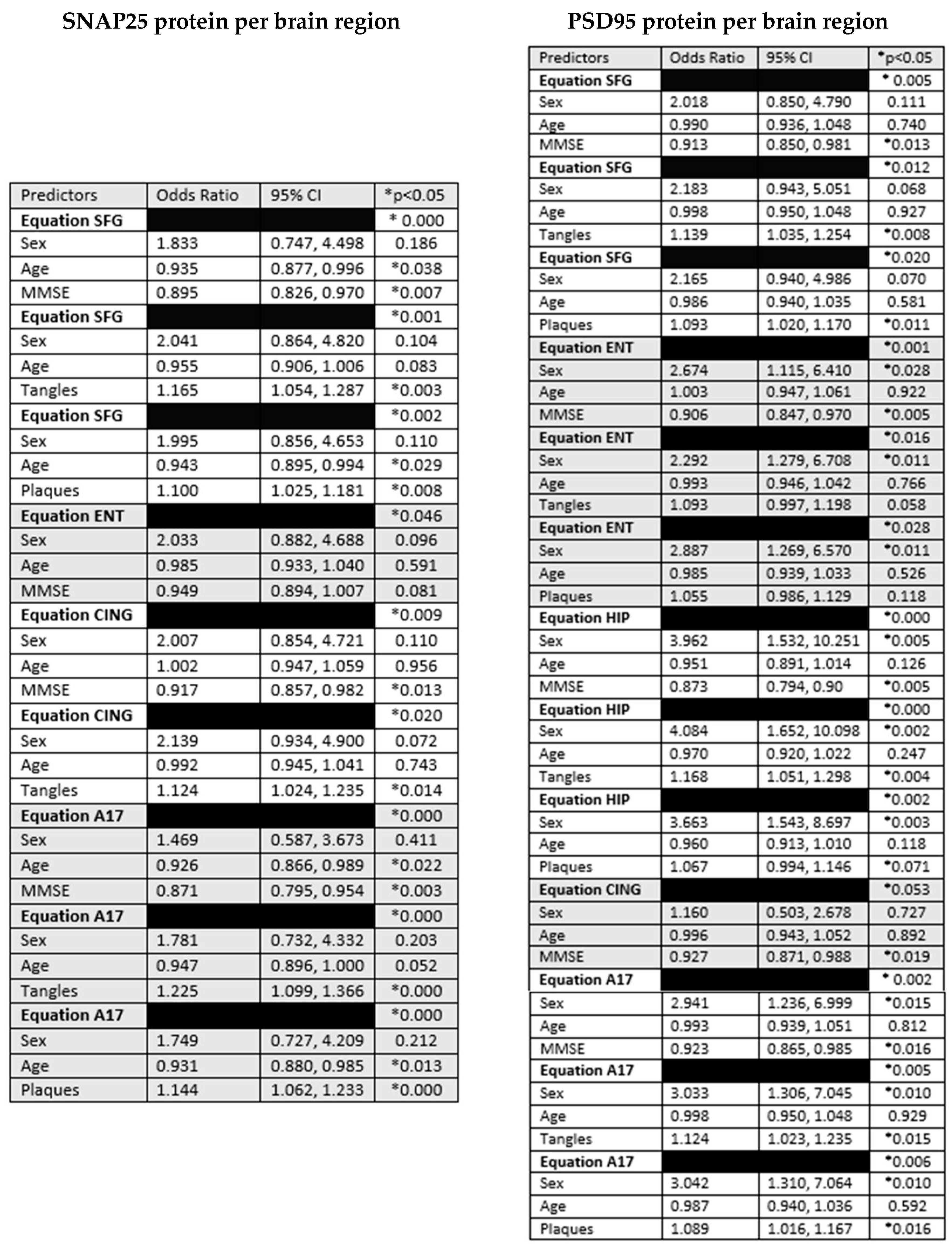
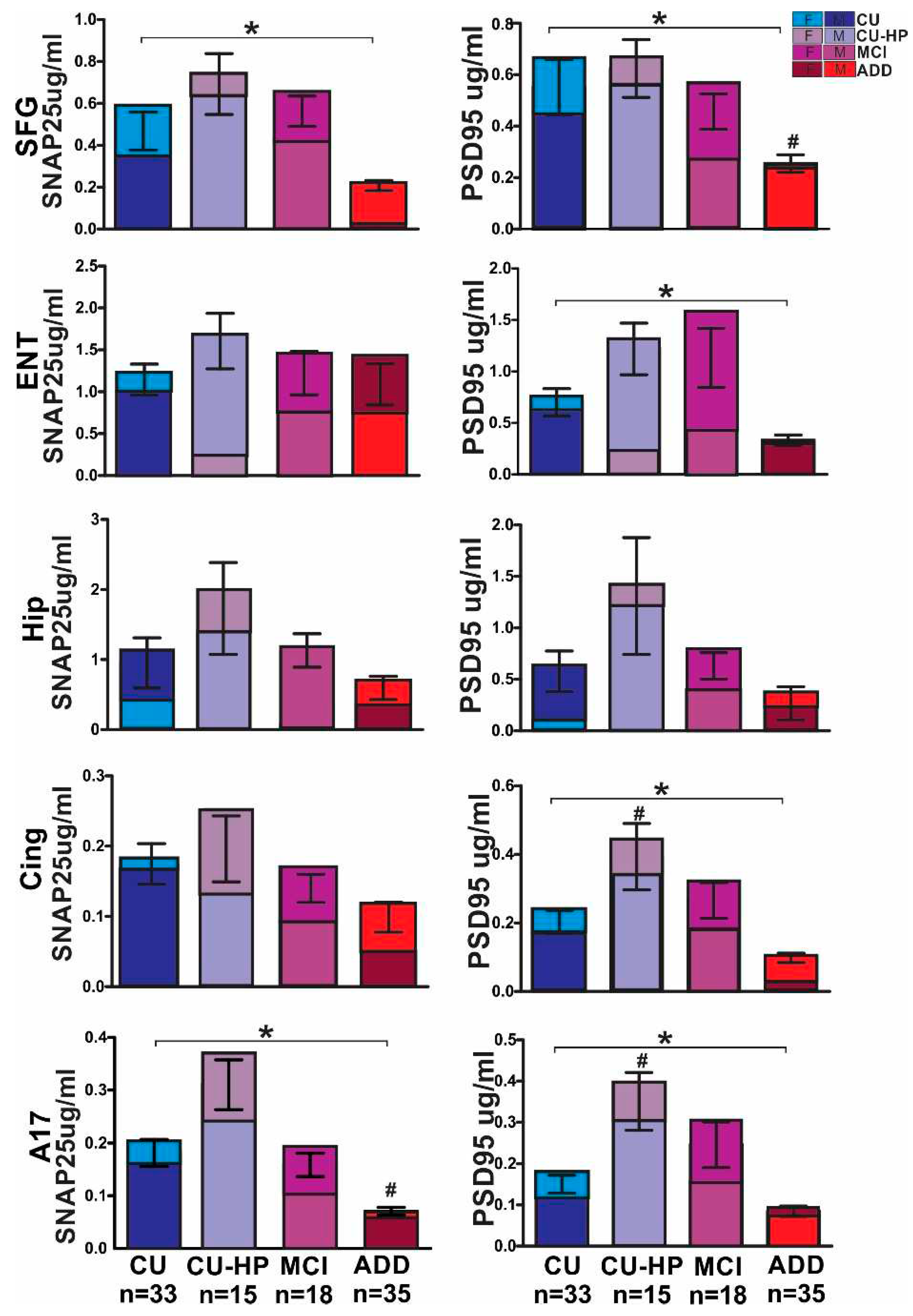
Figure 3.
Protein expression of presynaptic protein SNAP25 and the postsynaptic protein PSD95 in human brain measured by ELISA. SNAP25 is significantly reduced in ADD when compared to CU controls in frontal and visual cortex and cingulate, while hippocampus showed non-statistically significant reduction and entorhinal cortex showed similar protein concentration. PSD95 concentration was significantly lower in ADD when compared to non-demented controls, in all brain regions except hippocampus (* ANOVA p<0.05; # Dunnett’s multiple comparison test against CU p>0.05; f= females; m= males).
Figure 3.
Protein expression of presynaptic protein SNAP25 and the postsynaptic protein PSD95 in human brain measured by ELISA. SNAP25 is significantly reduced in ADD when compared to CU controls in frontal and visual cortex and cingulate, while hippocampus showed non-statistically significant reduction and entorhinal cortex showed similar protein concentration. PSD95 concentration was significantly lower in ADD when compared to non-demented controls, in all brain regions except hippocampus (* ANOVA p<0.05; # Dunnett’s multiple comparison test against CU p>0.05; f= females; m= males).
References
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, B.M.; Anderson, J.M. A quantitative study of cerebral atrophy in old age and senile dementia. J. Neurol. Sci. 1981, 50, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Scheff, S.W.; Neltner, J.H.; Nelson, P.T. Is synaptic loss a unique hallmark of Alzheimer’s disease? Biochem. Pharmacol. 2014, 88, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Gottschall, P.E.; Ajmo, J.M.; Eakin, A.K.; Howell, M.D.; Mehta, H.; Bailey, L.A. Panel of synaptic protein ELISAs for evaluating neurological phenotype. Exp. Brain Res. 2010, 201, 885–893. [Google Scholar] [CrossRef]
- Clare, R.; King, V.G.; Wirenfeldt, M.; Vinters, H.V. Synapse loss in dementias. J. Neurosci. Res. 2010, 88, 2083–2090. [Google Scholar] [CrossRef] [PubMed]
- Mecca, A.P.; O'Dell, R.S.; Sharp, E.S.; Banks, E.R.; Bartlett, H.H.; Zhao, W.; Lipior, S.; Diepenbrock, N.G.; Chen, M.K.; Naganawa, M.; Toyonaga, T. Synaptic density and cognitive performance in Alzheimer’s disease: A PET imaging study with [(11) C]UCB-J. Alzheimers Dement. 2022, 18, 2527–2536. [Google Scholar] [CrossRef] [PubMed]
- Pakkenberg, B.; Pelvig, D.; Marner, L.; Bundgaard, M.J.; Gundersen, H.J.G.; Nyengaard, J.R.; Regeur, L. Aging and the human neocortex. Exp. Gerontol. 2003, 38, 95–99. [Google Scholar] [CrossRef]
- Pelvig, D.P.; Pakkenberg, H.; Regeur, L.; Oster, S.; Pakkenberg, B. Neocortical glial cell numbers in Alzheimer’s disease. A stereological study. Dement. Geriatr. Cogn. Disord. 2003, 16, 212–219. [Google Scholar] [CrossRef]
- Regeur, L.; Jensen, G.B.; Pakkenberg, H.; Evans, S.; Pakkenberg, B. No global neocortical nerve cell loss in brains from patients with senile dementia of Alzheimer’s type. Neurobiol. Aging 1994, 15, 347–352. [Google Scholar] [CrossRef]
- Filon, J.R.; Intorcia, A.J.; Sue, L.I.; Arreola, E.V.; Wilson, J.; Davis, K.J.; Sabbagh, M.N.; Belden, C.M.; Caselli, R.J.; Adler, C.H.; et al. Gender Differences in Alzheimer Disease: Brain Atrophy, Histopathology Burden, and Cognition. Journal of Neuropathology & Experimental Neurology 2016, 75, 748–754. [Google Scholar]
- Wu, M.; Zhang, M.; Yin, X.; Chen, K.; Hu, Z.; Zhou, Q.; Cao, X.; Chen, Z.; Liu, D. The role of pathological tau in synaptic dysfunction in Alzheimer’s diseases. Transl. Neurodegener. 2021, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Kent, S.A.; Spires-Jones, T.L.; Durrant, C.S. The physiological roles of tau and Abeta: implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol. 2020, 140, 417–447. [Google Scholar] [CrossRef] [PubMed]
- Spires-Jones, T.L.; Hyman, B.T. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Serra, R.; Alonso-Nanclares, L.; Cho, K.; Giese, K.P. Emerging insights into synapse dysregulation in Alzheimer’s disease. Brain Commun. 2022, 4, fcac083. [Google Scholar] [CrossRef] [PubMed]
- Tzioras, M.; McGeachan, R.I.; Durrant, C.S.; Spires-Jones, T.L. Synaptic degeneration in Alzheimer disease. Nat. Rev. Neurol. 2023, 19, 19–38. [Google Scholar] [CrossRef]
- Wang, X.; Christian, K.M.; Song, H.; Ming, G.-L. Synaptic dysfunction in complex psychiatric disorders: from genetics to mechanisms. Genome Med. 2018, 10, 9. [Google Scholar] [CrossRef]
- Henstridge, C.M.; Pickett, E.; Spires-Jones, T.L. Synaptic pathology: A shared mechanism in neurological disease. Ageing Res. Rev. 2016, 28, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.; Jambrina, E.; Li, J.; Marston, H.; Menzies, F.; Phillips, K.; Gilmour, G. Targeting the Synapse in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 735. [Google Scholar] [CrossRef] [PubMed]
- Piccioni, G.; Mango, D.; Saidi, A.; Corbo, M.; Nisticò, R. Targeting Microglia-Synapse Interactions in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef]
- Carson, R.E.; Naganawa, M.; Toyonaga, T.; Koohsari, S.; Yang, Y.; Chen, M.-K.; Matuskey, D.; Finnema, S.J. Imaging of Synaptic Density in Neurodegenerative Disorders. J. Nucl. Med. 2022, 63 (Suppl. 1), 60S–67S. [Google Scholar] [CrossRef]
- Chen, M.-K.; Mecca, A.P.; Naganawa, M.; Finnema, S.J.; Toyonaga, T.; Lin, S.-F.; Najafzadeh, S.; Ropchan, J.; Lu, Y.; McDonald, J.W.; et al. Assessing Synaptic Density in Alzheimer Disease With Synaptic Vesicle Glycoprotein 2A Positron Emission Tomographic Imaging. JAMA Neurol. 2018, 75, 1215–1224. [Google Scholar] [CrossRef]
- Beach, T.G.; Sue, L.I.; Walker, D.G.; Roher, A.E.; Lue, L.; Vedders, L.; Connor, D.J.; Sabbagh, M.N.; Rogers, J. The Sun Health Research Institute Brain Donation Program: description and experience, 1987-2007. Cell Tissue Bank 2008, 9, 229–245. [Google Scholar] [CrossRef]
- Beach, T.G.; Adler, C.H.; Sue, L.I.; Serrano, G.; Shill, H.A.; Walker, D.G.; Lue, L.; Roher, A.E.; Dugger, B.N.; Maarouf, C.; et al. Arizona Study of Aging and Neurodegenerative Disorders and Brain and Body Donation Program. Neuropathology 2015, 34, 354–389. [Google Scholar] [CrossRef] [PubMed]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 2012, 123, 1–11. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Gallyas, F. An argyrophil III method for the demonstration of fibrous neuroglia. Acta Morphol. Acad. Sci. Hung. 1981, 29, 185–193. [Google Scholar]
- Braak, H.; Braak, E. Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol. 1991, 1, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Arai, T.; Nonaka, T.; Kametani, F.; Yoshida, M.; Hashizume, Y.; Beach, T.G.; Buratti, E.; Baralle, F.; Morita, M.; et al. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 2008, 64, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Beach, T.G.; White, C.L.; Hamilton, R.L.; Duda, J.E.; Iwatsubo, T.; Dickson, D.W.; Leverenz, J.B.; Roncaroli, F.; Buttini, M.; Hladik, C.L.; et al. Evaluation of alpha-synuclein immunohistochemical methods used by invited experts. Acta Neuropathol. 2008, 116, 277–288. [Google Scholar] [CrossRef]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef]
- Mirra, S.S.; Heyman, A.; McKeel, D.; Sumi, S.M.; Crain, B.J.; Brownlee, L.M.; Vogel, F.S.; Hughes, J.P.; Belle, G.V.; Berg, L.; Participating CERAD Neuropathologists The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991, 41, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Aging, N.I.O. Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Neurobiol. Aging 1997, 18 (Suppl. 4), S1–S2. [Google Scholar]
- Hyman, B.T.; Trojanowski, J.Q. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J. Neuropathol. Exp. Neurol. 1997, 56, 1095–1097. [Google Scholar] [CrossRef] [PubMed]
- Mirra, S.S. The CERAD neuropathology protocol and consensus recommendations for the postmortem diagnosis of Alzheimer’s disease: a commentary. Neurobiol. Aging 1997, 18 (Suppl. 4), S91–S94. [Google Scholar] [CrossRef]
- Antonucci, F.; Corradini, I.; Morini, R.; Fossati, G.; Menna, E.; Pozzi, D.; Pacioni, S.; Verderio, C.; Bacci, A.; Matteoli, M. Reduced SNAP-25 alters short-term plasticity at developing glutamatergic synapses. EMBO Rep. 2013, 14, 645–651. [Google Scholar] [CrossRef]
- Fossati, G.; Morini, R.; Corradini, I.; Antonucci, F.; Trepte, P.; Edry, E.; Sharma, V.; Papale, A.; Pozzi, D.; Defilippi, P.; et al. Reduced SNAP-25 increases PSD-95 mobility and impairs spine morphogenesis. Cell Death Differ. 2015, 22, 1425–1436. [Google Scholar] [CrossRef] [PubMed]
- Kivisäkk, P.; Carlyle, B.C.; Sweeney, T.; Quinn, J.P.; Ramirez, C.E.; Trombetta, B.A.; Mendes, M.; Brock, M.; Rubel, C.; Czerkowicz, J.; Graham, D. Increased levels of the synaptic proteins PSD-95, SNAP-25, and neurogranin in the cerebrospinal fluid of patients with Alzheimer’s disease. Alzheimers Res. Ther. 2022, 14, 58. [Google Scholar] [CrossRef]
- Coley, A.A.; Gao, W.J. PSD95: A synaptic protein implicated in schizophrenia or autism? Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 82, 187–194. [Google Scholar] [CrossRef]
- Chen, X.; Nelson, C.D.; Li, X.; Winters, C.A.; Azzam, R.; Sousa, A.A.; Leapman, R.D.; Gainer, H.; Sheng, M.; Reese, T.S. PSD-95 is required to sustain the molecular organization of the postsynaptic density. J. Neurosci. 2011, 31, 6329–6338. [Google Scholar] [CrossRef]
- Keith, D.; El-Husseini, A. Excitation Control: Balancing PSD-95 Function at the Synapse. Front. Mol. Neurosci. 2008, 1, 4. [Google Scholar] [CrossRef]
- Honer, W.G. Pathology of presynaptic proteins in Alzheimer’s disease: more than simple loss of terminals. Neurobiol. Aging 2003, 24, 1047–1062. [Google Scholar] [CrossRef] [PubMed]
- Merlo, S.; Spampinato, S.F.; Sortino, M.A. Early compensatory responses against neuronal injury: A new therapeutic window of opportunity for Alzheimer’s Disease? CNS Neurosci. Ther. 2019, 25, 5–13. [Google Scholar] [CrossRef] [PubMed]
- DeKosky, S.T.; Ikonomovic, M.D.; Styren, S.D.; Beckett, L.; Wisniewski, S.; Bennett, D.A.; Cochran, E.J.; Kordower, J.H.; Mufson, E.J. Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann. Neurol. 2002, 51, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Nanclares, L.; Gonzalez-Soriano, J.; Rodriguez, J.R.; DeFelipe, J. Gender differences in human cortical synaptic density. Proc. Natl. Acad. Sci. U S A 2008, 105, 14615–14619. [Google Scholar] [CrossRef] [PubMed]
- Hyer, M.M.; Phillips, L.L.; Neigh, G.N. Sex Differences in Synaptic Plasticity: Hormones and Beyond. Front. Mol. Neurosci. 2018, 11, 266. [Google Scholar] [CrossRef]
- Martínez-Pinilla, E.; Ordóñez, C.; del Valle, E.; Navarro, A.; Tolivia, J. Regional and Gender Study of Neuronal Density in Brain during Aging and in Alzheimer’s Disease. Front. Aging Neurosci. 2016, 8, 213. [Google Scholar] [CrossRef]
Figure 1.
Semiquantitative scores for AD pathology. Neuritic plaque and neurofibrillary tangle (NFT) densities were graded blindly as recommended by CERAD with separate semi-quantitative density estimates of none, sparse, moderate, or frequent in each region of interest which included cortical gray matter from frontal (F), temporal (T), parietal (P), hippocampal CA1 (H), and entorhinal (E) regions. In this study the summation of plaque and NFT scores ranged from 0 in some cognitively unimpaired controls to 15 in most ADD (* ANOVA p<0.05).
Figure 1.
Semiquantitative scores for AD pathology. Neuritic plaque and neurofibrillary tangle (NFT) densities were graded blindly as recommended by CERAD with separate semi-quantitative density estimates of none, sparse, moderate, or frequent in each region of interest which included cortical gray matter from frontal (F), temporal (T), parietal (P), hippocampal CA1 (H), and entorhinal (E) regions. In this study the summation of plaque and NFT scores ranged from 0 in some cognitively unimpaired controls to 15 in most ADD (* ANOVA p<0.05).
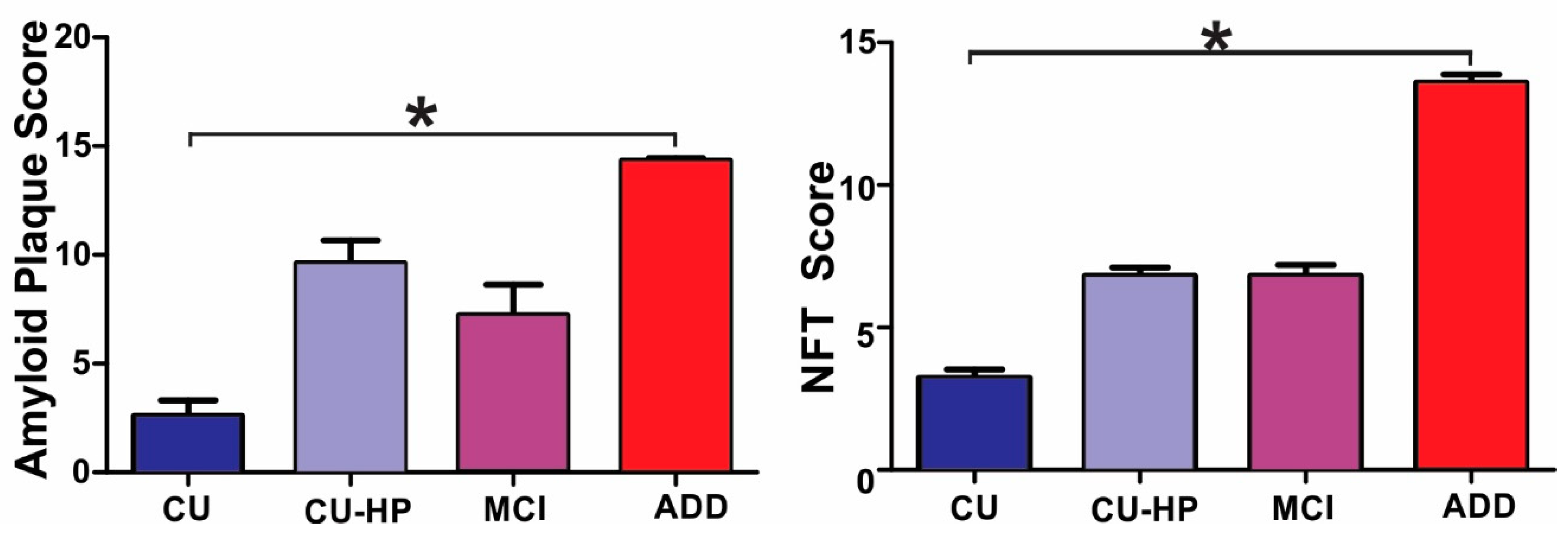

Table 1.
General and pathological characteristics of study subjects.
| DX (n) | Age (SD) | Gender (M:F) | PMI (SD) | Brain weight (SD) |
MMSE (SD) |
Total plaques | Thal stage | Total tangle | Braak stage | |
|---|---|---|---|---|---|---|---|---|---|---|
| CU (33) | 83 (7)* | 17:16 | 3 (1) | 1211 (123)* | 29 (1) | 3 (4)* | 1 (2)* | 3 (2)* | 2 (.8)* | |
| CU-HP (15) | 92 (6)# | 7:8 | 4 (3) | 1203 (128) | 28 (2) | 10 (4)# | 3 (1)# | 7 (1)# | 4 (0)# | |
| MCI (18) | 90 (7)# | 7:11 | 4 (2) | 1149 (87) | 26 (3) | 7 (6) | 2 (2)# | 7 (2)# | 4 (.4)# | |
| ADD (35) | 82 (8) | 20:15 | 3 (1) | 1089 (125)# | 15 (8) | 14 (1)# | 5 (.6)# | 14 (2)# | 6 (.5)# | |
* p < 0.05 for group comparisons; # p < 0.05 for comparison with controls. N: number of cases; SD: standard deviations; M: males; F: females; MMSE: Mini Mental State Examination; PMI, postmortem interval; CU: cognitively unimpaired low pathology controls; CU-HP cognitively unimpaired high pathology; MCI: mild cognitive impairment; ADD: Alzheimer’s disease dementia.
Table 2.
Univariable comparison screening of factors for effects on synaptic protein loss. Spearman rho (ρ) for each comparison and p values were calculated using non-parametric correlations. P<0.05 *; . P<0.01 **; . P<0.001 ***; MMSE: Mini Mental State Examination; A17: Visual cortex area 17.
Table 2.
Univariable comparison screening of factors for effects on synaptic protein loss. Spearman rho (ρ) for each comparison and p values were calculated using non-parametric correlations. P<0.05 *; . P<0.01 **; . P<0.001 ***; MMSE: Mini Mental State Examination; A17: Visual cortex area 17.
| SNAP25 | MMSE | Age | Total tangles | Total plaques | PSD95 |
|---|---|---|---|---|---|
| Frontal | 0.29 ** | 0.18 (NS) | -0.213 ** | -0.292 ** | 0.655 *** |
| Entorhinal | 0.26 * | 0.22 * | -0.050 | -0.081 | 0.737 *** |
| Hippocampus | 0.16 (NS) | 0.12 (NS) | -0.085 | -0.062 | 0.698 *** |
| Cingulate | 0.39 *** | 0.18 (NS) | -0.294 ** | -.286 ** | 0.629 *** |
| A17 | 0.49 *** | 0.32 ** | -0.430 *** | -0.445 *** | 0.674 *** |
| PSD95 | |||||
| Frontal | 0.31 ** | 0.06 (NS) | -.0267 * | -0.213 * | |
| Entorhinal | 0.41 *** | 0.20 * | -0.191 * | -0.233 * | |
| Hippocampus | 0.43 *** | 0.20 * | -0.324 ** | -0.307 ** | |
| Cingulate | 0.32 ** | 0.19 (NS) | -0.257 ** | -0.260 ** | |
| A17 | 0.33 ** | 0.22 * | -0.231 ** | -0.291 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



