
Submitted:
27 December 2023
Posted:
29 December 2023
You are already at the latest version
Abstract
Targeted therapies are effective cancer treatments when accompanied by accurate diagnostic tests that can help identify patients that will respond to those therapies. The YAP/TAZ-TEAD axis is activated and plays a causal role in several cancer types, and TEAD inhibitors are currently in early phase clinical trials in cancer patients. However, mutations that predict YAP/TAZ-TEAD activation are not commonly found in most cancer types, making it hard to determine which tumors are de-pendent upon YAP/TAZ-TEAD. Here, we used a combination of RNA-seq and bioinformatics analysis of metastatic melanoma cells to develop a YAP/TAZ gene signature. We found that the genes in this signature are TEAD-dependent, and that their expression strongly correlates with YAP/TAZ activation in human melanomas. Using DepMap dependency data, we found that this YAP/TAZ signature was predictive of melanoma cell dependence upon YAP/TAZ or TEADs. Importantly, this was not limited to melanoma because this signature was also predictive when tested on a panel of over 1000 cancer cell lines representing numerous distinct cancer types. Our results suggest that YAP/TAZ gene signatures like ours may be an effective tool to predict tumor cell sensitivity to YAP/TAZ-TEAD inhibition, and thus provide a means to identify patients likely to benefit from TEAD inhibitors.
Keywords:
YAP
; TAZ
; WWTR1
; TEAD
; metastasis
; melanoma
; Hippo
; Gene Signature
1. Introduction
Despite decades of research and significant advances in our understanding of the molecular and cellular events that drive cancer development and progression, cancer remains a leading cause of death worldwide. Most cancer deaths result from metastatic disease, which requires systemic therapies because it cannot be effectively treated surgically. In melanoma specifically, cytotoxic chemotherapies are largely ineffective, so most patients are treated with immune therapies and targeted therapies. Immune therapies have proven successful, but not all patients have durable responses, and some patients are contraindicated due to other health issues. Targeted therapies, such as BRAF and MEK inhibitors, can also be effective, but patients typically develop resistance. Therefore, there is an urgent need for new targeted therapies and a way to predict which patients will benefit from them.
Yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ) are inappropriately active in many cancer types [4,5,6,7], including melanoma [1,8,9,10]. Hundreds of studies collectively demonstrate that increased YAP or TAZ activity can enhance tumor formation and growth, and also promote tumor progression and metastasis [4,6,7,11,12]. YAP and TAZ are negatively regulated by the Hippo Pathway, a serine/threonine kinase cascade that, when active, results in LATS-mediated phosphorylation of multiple serine residues in YAP and TAZ, promoting either cytoplasmic sequestration or proteasomal degradation [13,14,15,16]. In addition, numerous other cellular pathways can influence YAP and TAZ function either by regulating the Hippo Pathway or through Hippo Pathway-independent mechanisms [17,18,19]. As transcriptional co-activators, YAP and TAZ regulate gene expression programs to mediate their effects on cells. However, both lack DNA binding domains, so they must partner with other transcription factors to regulate target gene expression. Although YAP and TAZ can interact with numerous transcription factors, the TEAD family members play critical roles in YAP/TAZ-dependent gene expression [20,21].
Given the established roles for YAP, TAZ, and TEADs in cancer and other diseases, there has been substantial interest in developing compounds to target these proteins. Although YAP and TAZ have proven difficult to target directly, compounds that target the TEADs, and thus prevent YAP/TAZ-TEAD-mediated transcription, have great promise [21,22]. Indeed, numerous compounds that target YAP/TAZ-TEAD have been described (reviewed in [23]) and 3 TEAD inhibitors (VT3989, IK-930, IAG933) have recently entered early phase clinical trials in cancer patients (NCT04665206, NCT05228015, and NCT04857372). An antisense oligonucleotide inhibitor of YAP1 that proved effective in pre-clinical models has also entered clinical trials (NCT04659096). However, as is the case for most targeted therapies, the success of these treatments will depend upon whether tumors that are dependent upon YAP/TAZ-TEAD can be distinguished from those that are not. This will be straightforward for cancers driven by mutations known to cause YAP or TAZ activation, such as NF2-mutant mesothelioma [24], GNAQ mutant uveal melanoma [25], Epithelioid Hemangioendothelioma (EHE), which is driven by the oncogenic TAZ-CAMTA1 fusion protein [26,27,28], and other cancers driven by YAP fusions [29]. However, in most of the cancers where YAP and TAZ play causal roles, mutations or alterations in the Hippo Pathway or YAP and TAZ themselves are rare, so biomarkers that can predict sensitivity to YAP/TAZ-TEAD inhibition will be essential. Given the complexities of how YAP and TAZ are regulated, increased protein expression or nuclear localization do not necessarily indicate increased YAP/TAZ-TEAD activity. The expression of YAP/TAZ-TEAD target genes could provide a more direct readout for YAP/TAZ activation, but whether they can predict sensitivity to YAP/TAZ-TEAD inhibition remains unclear.
Here, we describe the development of a YAP/TAZ gene signature from metastatic human melanoma cells, which is also highly enriched in cell lines dependent upon YAP, TAZ, or TEADs. Despite being developed from melanoma cell lines, we show that the genes in this signature are YAP/TAZ-dependent in other cancer types and that this signature is predictive of cancer cell dependence upon YAP, TAZ, and TEAD. This work suggests that this YAP/TAZ-TEAD gene signature, and others like it, could have diagnostic value for identifying cancer patients that will benefit from TEAD inhibitors.
2. Materials and Methods
Cell lines, vectors, and cloning: Human melanoma cell lines (A375, A375-MA2, A2058) were cultured in growth media (DMEM+ 10% fetal bovine serum, 2 mM L-glutamine) at 37° C and 5% CO2 and maintained at low passage number. Cell lines were routinely tested for mycoplasma and other bacterial contaminants. A375 and A2058 cells were obtained from ATCC. A375-MA2 cells were derived from A375 by Richard Hynes [30]. All vectors used in this work are listed in Table S1. If vectors were previously described, received as gifts, or purchased from commercial vendors, the source of the vector is listed. New vectors were generated using standard cloning procedures, and the source constructs used for each insert and vector backbone are indicated.
Generation of retrovirus and lentivirus: Retrovirus and lentivirus were packaged as described previously [31]. Briefly, 293FT cells were plated on 6-well plates at roughly 50% confluence in growth media. After 16-24 hours, cells were transfected (according to the manufacturer’s protocol) with a transfection mixture containing 1 μg of viral vector, 0.5 μg of packaging vector (gag/pol), 0.5 μg of coat protein (VSVG), 5 μL of X-tremeGENE™ 9 (Sigma-Aldrich, Cat#6365779001), and 95 μL of Opti-MEM™ (Thermo Fisher Scientific, Cat#31985062). The transfection mixture was added to the cells for 24 hours, after which, the mixture was removed, and the cells were fed with fresh growth media. Culture supernatant was collected and filtered through a 0.45 μm filter 24 hours later. For stable transduction, cells at roughly 60-80% confluence were incubated with viral supernatant diluted 1:1 with fresh growth media and Polybrene (Sigma-Aldrich, Cat#45-H9268) (final concentration 8 µg/mL) for 24 hours and then viral supernatants were removed, and cells were fed with fresh growth media and stably selected with the appropriate antibiotic.
RNAi: siRNA experiments used Horizon Discovery SMARTPools and included a non-targeting control siRNA SMARTPool (Horizon Discovery ON-TARGETplus Non-targeting siRNA #1, Cat#D-001810-01-05) or SMARTPools targeting human YAP (Horizon Discovery ON-TARGETplus Human YAP1 (10413) siRNA, Cat#L-012200-00-0010) and human TAZ (Horizon Discovery ON-TARGETplus Human WWTR1 (25937) siRNA, Cat#L-016083-00-0010). Cells were plated at 4x105 cells per 6 cm well and cultured in growth media for 24 hours. A transfection mixture containing 9 μL (90 pmol) of siRNA, 27 μL of Lipofectamine™ RNAiMAX (Thermo Fisher Scientific Cat#13778075) and 900 μL Opti-MEMTM was setup according to the manufacturer’s protocol. For the combined knockdown of both YAP and TAZ, 4.5 μL (45pmol) of each siRNA SMARTPool was used so that the total volume of siRNA remained 9 μL (90 pmol). Twenty-four hours after the transfection, the cells were trypsinized and plated for Western blots or qPCR and cultured for an additional 48 hours in growth media before lysing as described below.
YAP/TAZ transcriptional activity: The YAP/TAZ-TEAD transcriptional reporter assays utilized a TEAD reporter construct (pGL3-5xMCAT(SV)-49 [31,32] that consists of 5 repeats of a TEAD binding element upstream of a minimal SV40 promoter that drives expression of the gene the encodes Firefly Luciferase. A control vector encoding a constitutively expressed Renilla luciferase (PRL-TK (Promega, Cat#E2231)) is co-transfected and used for normalization. Cells were plated on a 12-well in duplicate in 1 mL of growth media. Aer 24 hours cells were co-transfected with a transfection mixture containing 400 ng of a 20:1 mixture of pGL3-5xMCAT(SV)-49 and PRL-TK, 4 μL/well of Lipofectamine 3000, 2 μL/well of the P3000 reagent (Invitrogen, Cat#L3000001) and 94 μL of Opti-MEM. After 24 hours, luciferase activity was assayed using the Dual-Luciferase Reporter Assay System (Promega, Cat#E1910) and normalized luciferase levels were calculated as described previously [31]. For some experiments, cells were transfected with siRNAs or infected with viral constructs prior to assaying luciferase activity.
Western blotting and qPCR: For Western blots, cells were lysed in Cell Lysis Buffer (Cell Signaling Technology, Cat#9803) containing Pierce™ Protease Inhibitor Mini Tablets (Thermo Fisher Scientific, Cat#88665) and Pierce™ Phosphatase Inhibitor Mini Tablets (Thermo Fisher Scientific, Cat#88667). Protein concentration was determined by Pierce™ BCA protein assay kit (Thermo Fisher Scientific, Cat#23225) and equal protein (20-30 μg) was subjected to 10% SDS-PAGE, transferred to nitrocellulose membranes, and assayed by Western blot. The following primary antibodies were used at 1:1000 dilutions: total YAP (D8H1X) XP (Cell Signaling Technology, Cat#14074); total YAP (Cell Signaling Technology, Cat#4912,); total TAZ (V386) (Cell Signaling Technology, Cat#4883); and GAPDH Cell Signaling Technology, Cat#2118). The following horseradish-peroxidase-conjugated secondary antibodies were used at 1:5000 dilutions: goat anti-rabbit IgG (Thermo Fisher Scientific, Cat#31460); and goat anti-mouse IgG (Thermo Fisher Scientific, Cat#32430). Primary and secondary antibodies were diluted in 5% BSA. Western blotting images were captured with a Fujifilm LAS-3000 gel imager. For qPCR, cells were lysed with TRIzol (Thermo Fisher Scientific, Cat#15596018) and RNA was isolated following the manufacturer’s protocol. cDNA was made from 200 ng of the total RNA using qScript cDNA SuperMix at 42 °C (QuantaBio, Cat#95048) following the manufacturer’s protocol. qPCR reactions were carried out on 2 μL of cDNA, using 2 pmol of each primer (Table S1) and 10 µL of iTaq Universal SYBR Green (Bio-Rad, Cat#1725120). The reaction mixture was brought to a total of 20 µL with nuclease-free water. qPCR reactions were run using MyiQTM real-time PCR detection system according to the manufacturer’s instructions (Bio-Rad, Cat#1855201). PCR conditions were 95°C for 30 seconds, followed by 40 cycles of 95°C for 10 seconds, 60°C for 30 seconds, followed by a melt temperature analysis. For data processing, the Bio-Rad CFX Maestro software was used to calculate the fold change in mRNA for each indicated gene for each sample relative to a pre-determined control sample using the ΔΔCt method and GAPDH as a reference gene.
In vivo metastasis assays: The Albany Medical College Institutional Animal Care and Use Committee approved all mouse studies. Mice were housed in specific pathogen-free conditions in the Albany Medical College Animal Resources Facility, which is licensed by both the USDA and the NYS Department of Health, Division of Laboratories and Research, and accredited by the AAALAC. These studies used immunocompromised NOD/Scid mice (NOD/MrkBomTac-Prkdcscid, Taconic). To assay metastatic colonization, fluorescently labeled A375 cells expressing the indicated constructs were injected into the lateral tail veins of mice at 1x106 cells per mouse in 100 μL of PBS, and after 6 weeks, mice were euthanized, and lung metastases were counted using a fluorescent stereomicroscope.
RNA-seq: A375 cells stably expressing the indicated constructs were cultured in growth media, lysed with TRIzol (Thermo Fisher Scientific, Cat#15596018), and then RNA was isolated following the manufacturer’s protocol. cDNA was prepared with Illumina TruSeq chemistry and libraries were prepared using SPRIworks (Beckman Coulter) and sequenced using TruSeq SBS Kit v3 on the Illumina HiSeq2000. Sequence reads were aligned to the UCSC known genes version 2012 hg19 human assembly with using bowtie2 version 2.0.0-beta6 [33] and tophat version 2.0.4 [34]. Transcript assembly, gene summary, and differential expression was performed using cufflinks version 2.0.2 [35]. RNA-seq data is available in the NCBI Gene Expression Omnibus (GSE234083).
Bioinformatic Analyses: Publicly available gene expression datasets downloaded from the NCBI Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) are listed in Table S1, Tab 3. Analysis of datasets downloaded from The Cancer Genome Atlas (TCGA) (https://www.cancer.gov/tcga), The Broad Institute Cancer Dependency Map (DepMap) Portal (https://depmap.org/portal/), or The ENCODE Project Database (https://www.encodeproject.org/) [36] are described below. Metascape [2] was accessed via this link https://metascape.org. For GSEA, downloaded NCBI-GEO, TCGA, or DepMap datasets were used to generate .gct files, and .cls phenotype files were generated for the indicated comparisons. GSEA [37,38] was run to test for enrichment of genesets described in Table S1, Tab 4 using the appropriate CHIP platform and default settings. Probe/transcript Ids were collapsed to gene symbols, 1000 permutations were run, and the permutation type used was “phenotype” if the number of samples in each group was 8 or more, or “geneset” if the number of samples in each group was less than 8. Rank-ordered lists were generated by running GSEAPreranked with the same settings described above. Heatmaps were generated using MORPHEUS (https://software.broadinstitute.org/morpheus/). To calculate Z-Scored expression, TPM values were converted to log2(1+x) and Z-Scored in Morpheus. Similarity matrices were generated from the Z-Scored expression values by first calculating the Spearman’s Rank Correlation of each gene with either CTGF or CYR61 using the nearest neighbor function and 1000 permutations. Genes were then sorted by Spearman’s Rank Correlation values (highest to lowest) and the Similarity Matrix function was used to generate the Spearman’s Rank Correlation for each pairwise comparison of genes in the dataset.
ENCODE: Conservative IDR Threshold Peaks for the indicated ChIP-Seq datasets were downloaded from the ENCODE Portal [36] (https://www.encodeproject.org/) (Table S1, Tab 5). Using the ChIPseeker package in R [39], the datasets were annotated based on the GRCh38 genome assembly, with the transcriptional start sites being defined as +/- 1kb. ENTREZ IDs for annotated peaks were converted to gene symbols using the ENSEMBL Database Homo Sapiens V86 as a reference.
DepMap: The RNA-seq data file “OmicsExpressionProteinCodingGenesTPMLogp1.csv” was downloaded from “DepMap Public 23Q2 Primary Files” on The Broad Institute Cancer Dependency Map (DepMap) Portal (https://depmap.org/portal/). Dependency scores for TEADs 1-4, YAP1, and WWTR1 (TAZ) were downloaded from DepMap. The RNA-seq data from the 1019 cell lines that have reported YAP1, WWTR1, and TEADs dependency scores were analyzed for enrichment of genesets using GSEA analysis as described above. To generate ROC curves, the GSVA package in R [40] was used to calculate enrichment scores for the indicated genesets in the indicated cell lines, and the resulting GSVA enrichment scores were used to perform ROC curve analyses using MedCalc for Windows, version 22.013 (MedCalc Software, Ostend, Belgium). Cell lines were considered dependent upon a gene if the Chronos Dependency Score was < -0.65.
The Cancer Genome Atlas (TCGA): RNA-seq data from 303 human melanoma samples in the TCGA-Skin Cutaneous Melanoma (SKCM) project was downloaded from the NCI Genomic Data Commons Data Portal. Only datasets with an RNA integrity number between 7-10 were used. The transcripts per million (TPM) data for each sample was compiled and used for GSEA analysis, to generate heatmaps, and for similarity matrix calculations.
Statistical Analyses: Statistical analyses were performed in GraphPad Prism. The statistical test used to determine significance and the number of n’s is indicated in the legends. All scatter plots show mean + S.D unless noted otherwise in the legend.
3. Results
3.1. Identification of a YAP/TAZ gene signature in metastatic melanoma cells.
Figure 1.
YAP promotes melanoma metastasis in a TEAD and transactivation domain-dependent manner. GFP-expressing A375 cells were stably transduced with a control empty vector (MSCV-IRES-Hygro) or the indicated YAP constructs and then assayed by Western blot (A) or for TEAD transcriptional activity using a dual-luciferase reporter assay (B). (C) Cells from (A) were injected into NOD/SCID mice via the lateral tail vein and after 19 days, the numbers of GFP-positive metastases were counted in the lungs. (D) RNA-seq was performed on 3 independent RNA samples from A375 cells expressing an empty vector control (MSCV-IRES-Hygro) or the indicated YAP constructs, and the number of genes significantly up or down-regulated (fold change > 2, adjusted pValue < 0.05) by each YAP construct relative to control is shown. (E) The heatmap shows log2 fold change log2FC) for each indicated comparison for all 1,696 genes that were differentially expressed in the A375-YAP2SA vs. Control cells. Data used to generate this heatmap is found in Table S2. The plots in (B) and (C) show mean ± SD with each dot representing an n. n= 3 independent experiments in (B) and n=individual mice in (C). Statistical significance was determined using one-way ANOVA with Dunnett’s multiple comparisons test (B, left) and (C) or unpaired, two-tail t-test (B, right); ** p ≤ 0.01, **** p ≤ 0.0001, n.s. p>0.05.
Figure 1.
YAP promotes melanoma metastasis in a TEAD and transactivation domain-dependent manner. GFP-expressing A375 cells were stably transduced with a control empty vector (MSCV-IRES-Hygro) or the indicated YAP constructs and then assayed by Western blot (A) or for TEAD transcriptional activity using a dual-luciferase reporter assay (B). (C) Cells from (A) were injected into NOD/SCID mice via the lateral tail vein and after 19 days, the numbers of GFP-positive metastases were counted in the lungs. (D) RNA-seq was performed on 3 independent RNA samples from A375 cells expressing an empty vector control (MSCV-IRES-Hygro) or the indicated YAP constructs, and the number of genes significantly up or down-regulated (fold change > 2, adjusted pValue < 0.05) by each YAP construct relative to control is shown. (E) The heatmap shows log2 fold change log2FC) for each indicated comparison for all 1,696 genes that were differentially expressed in the A375-YAP2SA vs. Control cells. Data used to generate this heatmap is found in Table S2. The plots in (B) and (C) show mean ± SD with each dot representing an n. n= 3 independent experiments in (B) and n=individual mice in (C). Statistical significance was determined using one-way ANOVA with Dunnett’s multiple comparisons test (B, left) and (C) or unpaired, two-tail t-test (B, right); ** p ≤ 0.01, **** p ≤ 0.0001, n.s. p>0.05.
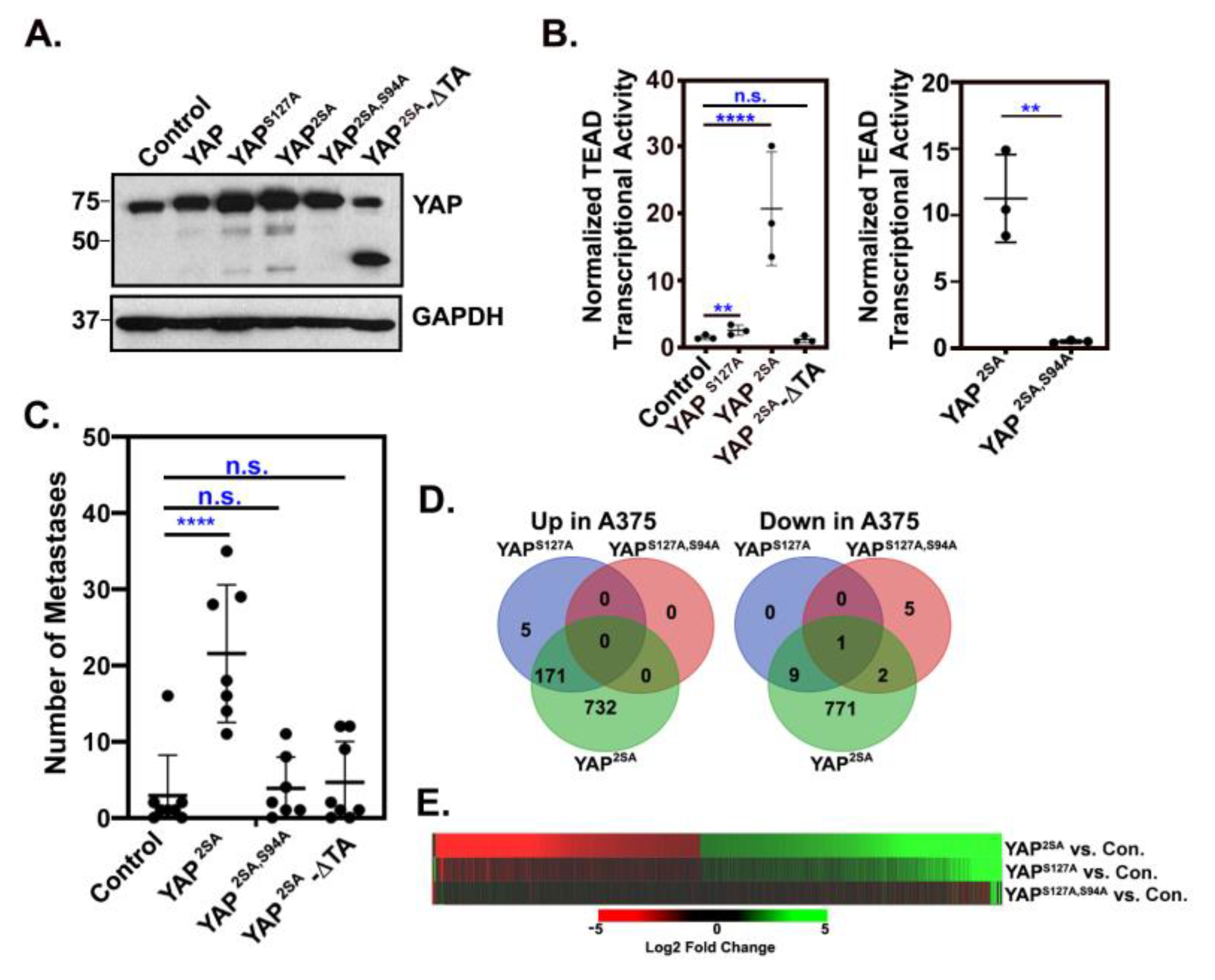
Our previous work in melanoma and breast cancer showed that abnormal activation of YAP promotes tumor growth and metastasis through TEAD interaction [41]. This suggests that YAP drives metastasis by enhancing TEAD-dependent gene expression. Consistently, we show here that expression of a LATS-insensitive form of YAP (YAPS127A, S381A, hence called YAP2SA) in A375 human melanoma cells promoted TEAD transcriptional activity and enhanced metastatic colonization of the lung (Figure 1A-C). However, mutant forms of YAP2SA unable bind to TEADs (YAP2SA,S94A) or that lack the transactivation domain (YAP2SA-∆TA) did not promote TEAD transcriptional activity or metastatic colonization (Figure 1A-C), suggesting that YAP-TEAD promotes metastasis by regulating the transcription of target genes. To elucidate changes in gene expression in cells rendered metastatic by YAP activation, we performed RNA sequencing on A375 cells stably expressing a control vector, LATS-insensitive forms of YAP (YAPS127A or YAP2SA) or a LATS-insensitive form of YAP that cannot bind TEADs (YAPS127A,S94A). Stable YAP2SA expression upregulated 903 genes and downregulated 783 genes, whereas YAPS127A upregulated 176 genes and downregulated 10 genes (Figure 1D and Table S2). The fact that TEAD transcriptional activity is more dramatically induced by YAP2SA than it is by YAPS127A (Figure 1B) likely explains why substantially more genes were differentially expressed in a statistically significant manner in the YAP2SA-expressing cells than in the YAPS127A-expressing cells. Indeed, most of the differentially regulated genes in the A375-YAP2SA cells were also upregulated or downregulated in the A375-YAPS127A cells, but to a lesser magnitude (Figure 1E and Table S2). Interestingly, only 8 genes were differentially expressed in the YAPS127A,S94A-expressing cells compared to control cells (Figure 1D&E and Table S2), suggesting that in A375 cells, most gene expression changes induced by YAP are mediated by TEADs.
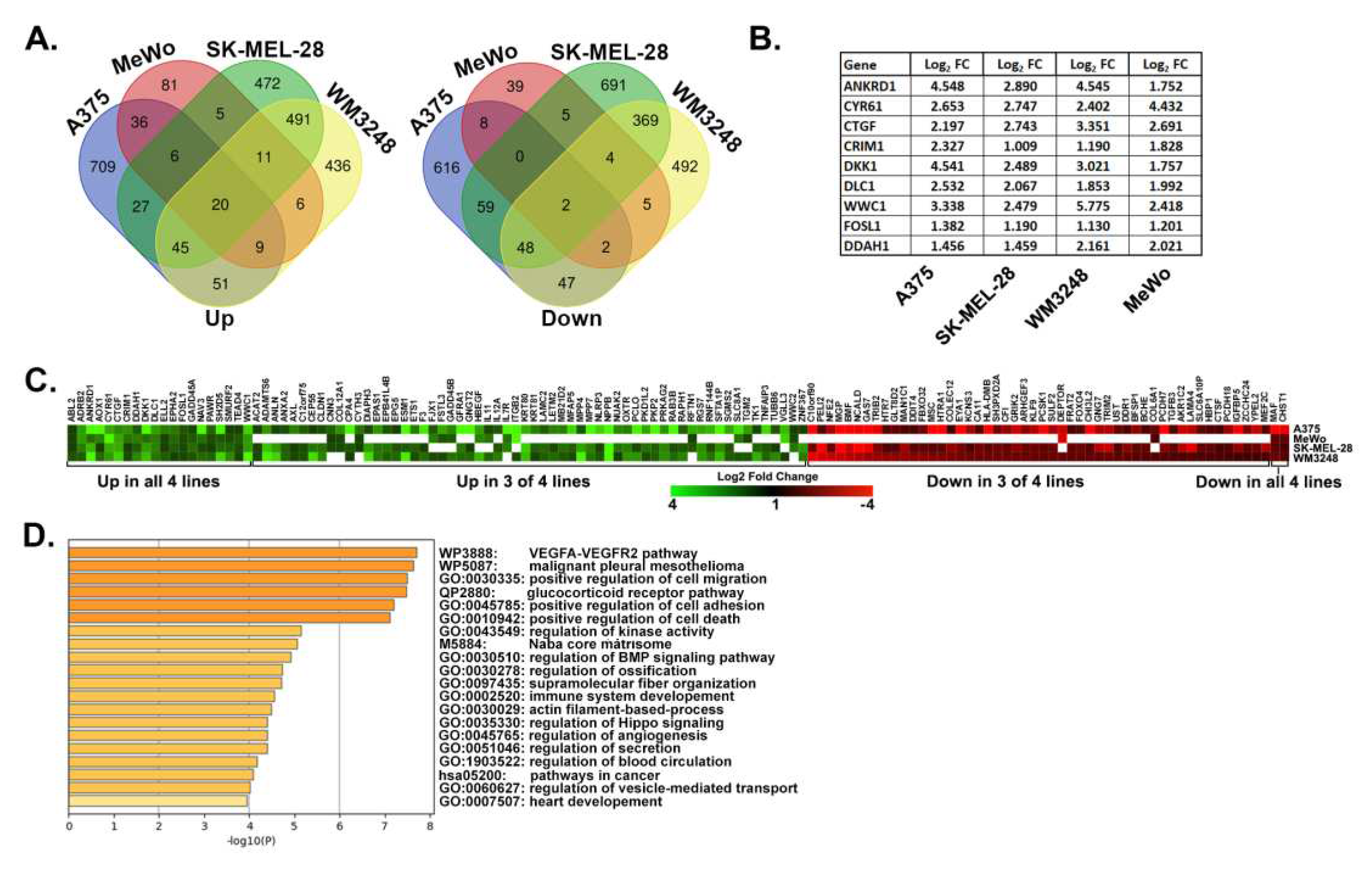
We next used publicly available gene expression datasets (Table S1, Tab 3) to test which YAP-dependent genes in A375 cells were also regulated by YAP or TAZ in other metastatic melanoma cell lines. This included MeWo cells rendered metastatic by the expression of LATS-insensitive YAP5SA [1] and two metastatic melanoma cell lines (SK-MEL-28 and WM3248) with both YAP and TAZ knocked down [42]. Genes were considered upregulated by YAP/TAZ if they were induced in A375-YAP2SA or MeWo-YAP5SA or if they were downregulated by YAP/TAZ siRNA in SK-MEL-28 or WM3248. Conversely, genes downregulated in A375-YAP2SA or MeWo-YAP5SA or upregulated by YAP/TAZ siRNA in SK-MEL-28 or WM3248 were considered downregulated by YAP/TAZ. Although many of the differentially expressed genes in each cell line were distinct, we identified a 132-gene YAP/TAZ signature consisting of genes that were upregulated (80 genes) or downregulated (52 genes) in A375-YAP2SA cells and at least 2 of the other metastatic melanoma cell lines (Figure 2A&C and Table S3). This signature included several known YAP/TAZ target genes (Figure 2B) and was enriched with genes involved in cell migration, cell adhesion, angiogenesis, core matrisome, and cancer pathways (Figure 2D).
Figure 2.
Identification of a YAP/TAZ gene signature in metastatic human melanoma cells. (A) Venn diagrams show overlap between genes upregulated (left) or downregulated (right) (>2 fold, pValue <0.05) in the following: A375-YAP2SA vs. Control cells (our data); MeWo YAP5SA vs. Control cells ([1]); Control siRNA vs. YAP/TAZ siRNA trans- fected SK- MEL-28 or WM3248 cells (GSE68599). (B) The table shows fold change for established YAP/TAZ target genes. All changes are statistically significant (adjusted pValue p< 0.001). (C) The heatmap shows fold change for the 132 YAP/TAZ dependent genes in our YAP/TAZ signature. White indicates the gene was not detected in A375, WM3248, and SK-MEL-28 or not differentially expressed in MeWo. Data used to generate this heatmap are found in Table S3, Tab 6. (D) Pathway analysis was performed with the YAP/TAZ Up geneset using Metascape [2]. The top 20 enriched clusters are shown.
Figure 2.
Identification of a YAP/TAZ gene signature in metastatic human melanoma cells. (A) Venn diagrams show overlap between genes upregulated (left) or downregulated (right) (>2 fold, pValue <0.05) in the following: A375-YAP2SA vs. Control cells (our data); MeWo YAP5SA vs. Control cells ([1]); Control siRNA vs. YAP/TAZ siRNA trans- fected SK- MEL-28 or WM3248 cells (GSE68599). (B) The table shows fold change for established YAP/TAZ target genes. All changes are statistically significant (adjusted pValue p< 0.001). (C) The heatmap shows fold change for the 132 YAP/TAZ dependent genes in our YAP/TAZ signature. White indicates the gene was not detected in A375, WM3248, and SK-MEL-28 or not differentially expressed in MeWo. Data used to generate this heatmap are found in Table S3, Tab 6. (D) Pathway analysis was performed with the YAP/TAZ Up geneset using Metascape [2]. The top 20 enriched clusters are shown.
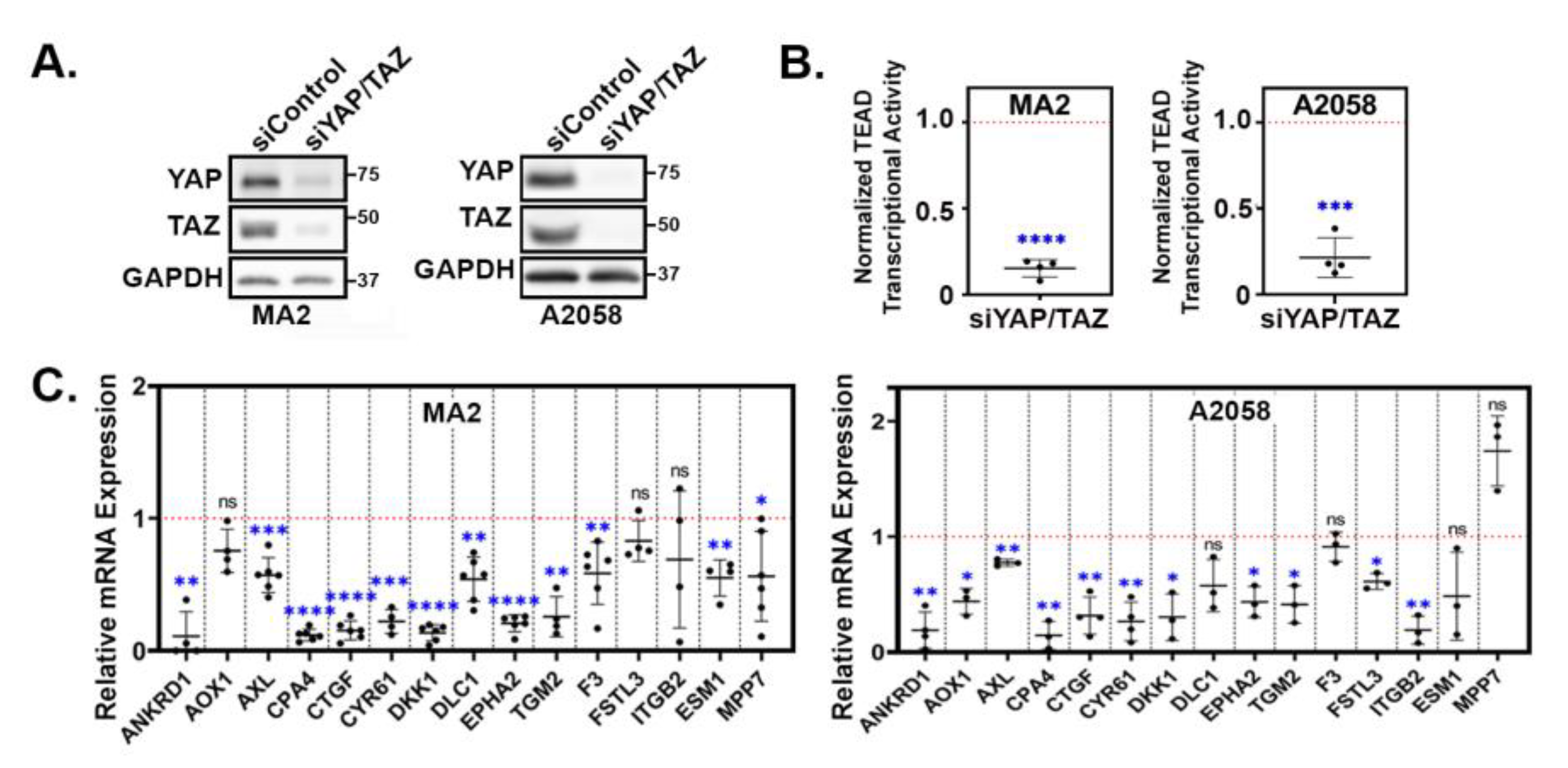
A parallel study from our lab found that the highly metastatic human melanoma cell lines A375-MA2 (hence called MA2) and A2058 require YAP and TAZ for metastatic colonization of the lungs (Warren et al. in preparation). To test if the YAP/TAZ target genes that we identified were also YAP/TAZ-dependent in these metastatic melanoma cell lines, we transfected MA2 and A2058 cells with either control siRNA pools or siRNA pools targeting both YAP and TAZ. Western blots confirmed that YAP and TAZ protein levels were dramatically reduced in the siYAP/TAZ transfected cells (Figure 3A). As expected, this led to a substantial inhibition in TEAD-mediated transcriptional activity (Figure 3B). We used qPCR to analyze the mRNA expression of several of the upregulated genes we identified in Figure 2 and found that YAP/TAZ knockdown reduced the expression of most of the genes in both cell lines (Figure 3C), indicating that these genes are YAP/TAZ dependent in these metastatic melanoma cell lines.
Figure 3.
YAP/TAZ signature genes are YAP/TAZ-dependent in metastatic melanoma cells. (A-C) A375-MA2 or A2058 cells were transfected with either a control siRNA SMARTpool or combined siRNA SMARTpools targeting YAP and TAZ for 24 hours. Cells were then then trypsinized and replated for an additional 24 hours and then assayed by (A) Western blot, (B) for TEAD transcriptional activity using a dual-luciferase reporter assay, or by (C) qPCR for the indicated genes. The plots (B&C) show the fold change in the siYAP/TAZ samples compared to the siControl samples, which were set to 1 (red dotted line) in each cell line. Each data point is an independent experiment and the mean ± SD is shown. Statistical significance was tested using a one-sample t-test; * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001, n.s. p>0.05.
Figure 3.
YAP/TAZ signature genes are YAP/TAZ-dependent in metastatic melanoma cells. (A-C) A375-MA2 or A2058 cells were transfected with either a control siRNA SMARTpool or combined siRNA SMARTpools targeting YAP and TAZ for 24 hours. Cells were then then trypsinized and replated for an additional 24 hours and then assayed by (A) Western blot, (B) for TEAD transcriptional activity using a dual-luciferase reporter assay, or by (C) qPCR for the indicated genes. The plots (B&C) show the fold change in the siYAP/TAZ samples compared to the siControl samples, which were set to 1 (red dotted line) in each cell line. Each data point is an independent experiment and the mean ± SD is shown. Statistical significance was tested using a one-sample t-test; * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001, n.s. p>0.05.
3.2. YAP/TAZ signature genes are TEAD-dependent.
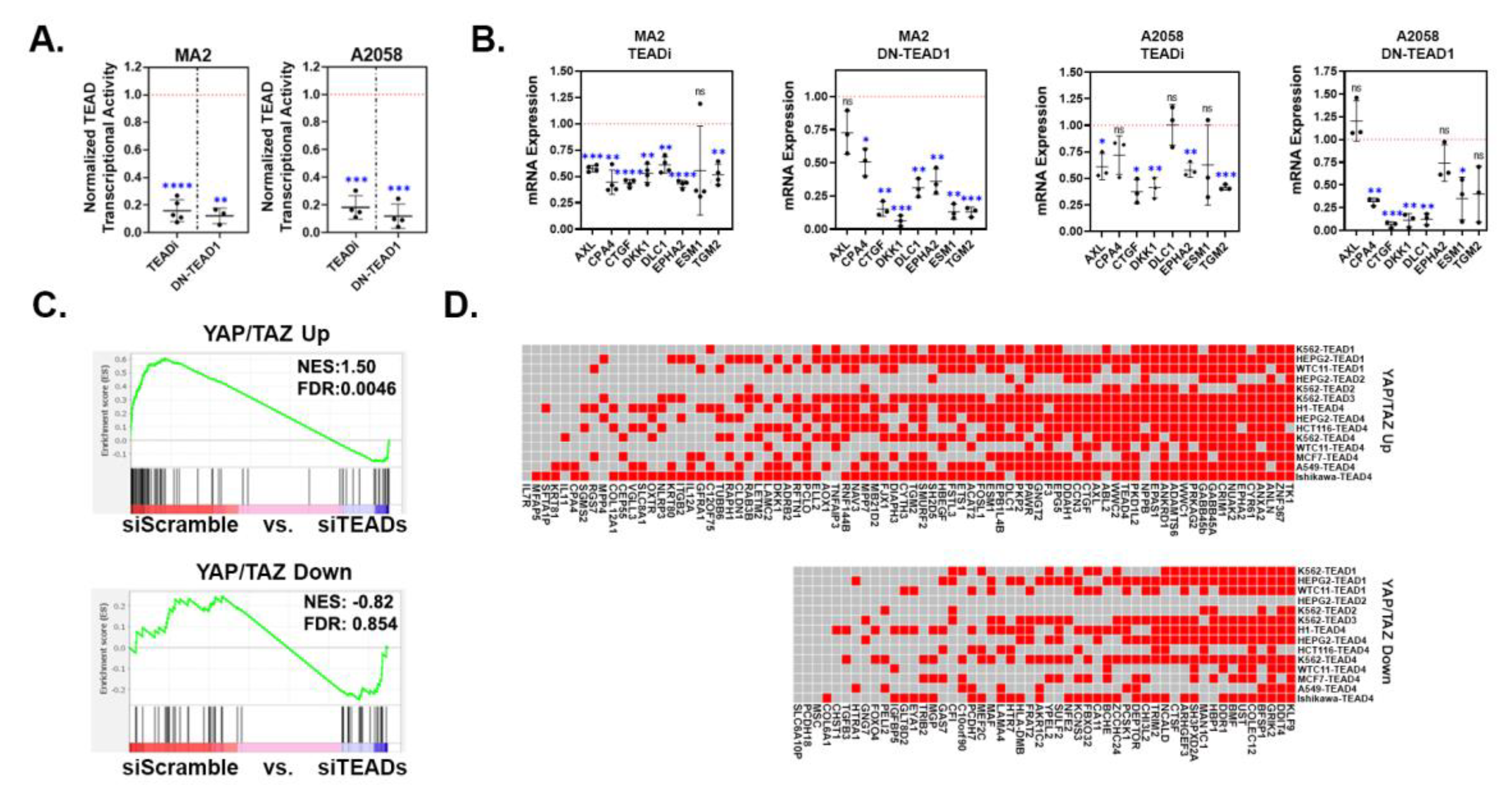
Many YAP/TAZ-dependent processes in cancer cells are mediated by the TEADs. Consistently, we found that the majority of the genes that were differentially expressed in A375-YAP2SA cells also changed in a similar manner in A375-YAPS127A cells, but did not change in A375 cells expressing the YAPS127A,S94A mutant that cannot bind TEADs (Figure 1E). Therefore, we next tested if genes in our YAP/TAZ signature were TEAD-dependent in additional melanoma cell lines. We blocked TEAD-mediated transcription in MA2 and A2058 cells by expressing either TEAD1Y421E, a dominant negative form of mouse TEAD1 (DN-TEAD1) [43] or a genetic TEAD inhibitor (TEADi) [44] and performed qPCR. As expected, both TEADi and DN-TEAD1 dramatically reduced the activity of the YAP/TAZ-TEAD reporter in both MA2 and A2058 cells (Figure 4A), indicating that these constructs effectively blocked TEAD-mediated transcription. Expression of either TEADi or DN-TEAD1 also reduced the expression of the genes from our YAP/TAZ signature that we tested (Figure 4B), confirming that these genes are dependent upon TEADs.
We next used GSEA to analyze available gene expression data from an invasive human melanoma cell line (MM047) in which all 4 TEADs were knocked down [3] to look for enrichment of our YAP/TAZ signature genes. Many of the 80 upregulated genes (YAP/TAZ Up) were strongly enriched (i.e. upregulated) in the control cells when compared to the TEAD knockdown cells (Figure 4C), indicating that these genes are TEAD-dependent. Surprisingly, only a few of the 52 downregulated genes (YAP/TAZ Down) were enriched in TEAD knockdown cells, suggesting that unlike the upregulated genes, most of the downregulated genes were not TEAD-dependent in MM047 cells (Figure 4C). Next, we analyzed several TEAD ChIP-seq datasets available in the ENCODE Database to determine which of our YAP/TAZ signature genes had associated TEAD peaks. Most of the 80 YAP/TAZ Up genes had TEAD peaks in several of the ChIP-seq datasets that we analyzed (Figure 4D). Although most of the YAP/TAZ Down genes also had TEAD peaks, fewer ChIP-seq datasets had peaks for the YAP/TAZ Down genes compared to the YAP/TAZ Up genes (Figure 4D), which could suggest that binding of TEADs to regulatory regions is more consistent for the genes upregulated by YAP and TAZ than it is for the genes downregulated by YAP and TAZ. Collectively, these results suggest that most of the genes upregulated by YAP/TAZ in our signature are TEAD-dependent.
Figure 4.
YAP/TAZ signature genes are TEAD-dependent. (A-B) A375-MA2 or A2058 cells stably transduced with control vector, TEADi, or TEAD1Y421E (DN-TEAD1) were assayed for (A) TEAD transcriptional activity using a dual-luciferase reporter assay or (B) by qPCR for the indicated genes. The plots (A&B) show the fold change in the TEADi or DN-TEAD samples compared to the control samples, which were set to 1 (red dotted line) in each cell line. Each data point is an independent experiment and the mean ± SD is shown. Statistical significance was tested using a one-sample t-test; * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001, n.s. p>0.05. (C) Gene Set Enrichment Analysis (GSEA) was performed using our YAP/TAZ Up and YAP/TAZ Down genesets and a publicly available dataset (GSE60664, [3]) in which all 4 TEADs were knocked down in human melanoma cells (MM047). Shown are the enrichment plots with Normalized Enrichment Score (NES) and False Discovery Rate (FDR) for each geneset. (D) ChIP-seq datasets with the indicated TEADs were downloaded from ENCODE and analyzed for the presence of peaks in the regulatory regions of each YAP/TAZ signature gene. The heatmap indicates which genes had a TEAD peak for each dataset. Red indicates at least 1 TEAD peak mapped to that gene, gray indicates no TEAD peaks mapped to that gene.
Figure 4.
YAP/TAZ signature genes are TEAD-dependent. (A-B) A375-MA2 or A2058 cells stably transduced with control vector, TEADi, or TEAD1Y421E (DN-TEAD1) were assayed for (A) TEAD transcriptional activity using a dual-luciferase reporter assay or (B) by qPCR for the indicated genes. The plots (A&B) show the fold change in the TEADi or DN-TEAD samples compared to the control samples, which were set to 1 (red dotted line) in each cell line. Each data point is an independent experiment and the mean ± SD is shown. Statistical significance was tested using a one-sample t-test; * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001, n.s. p>0.05. (C) Gene Set Enrichment Analysis (GSEA) was performed using our YAP/TAZ Up and YAP/TAZ Down genesets and a publicly available dataset (GSE60664, [3]) in which all 4 TEADs were knocked down in human melanoma cells (MM047). Shown are the enrichment plots with Normalized Enrichment Score (NES) and False Discovery Rate (FDR) for each geneset. (D) ChIP-seq datasets with the indicated TEADs were downloaded from ENCODE and analyzed for the presence of peaks in the regulatory regions of each YAP/TAZ signature gene. The heatmap indicates which genes had a TEAD peak for each dataset. Red indicates at least 1 TEAD peak mapped to that gene, gray indicates no TEAD peaks mapped to that gene.
3.3. Upregulated YAP/TAZ signature genes are predictive of cancer cell dependency upon YAP/TAZ-TEAD.
With TEAD inhibitors currently in clinical trials, an accurate way to predict sensitivity to YAP/TAZ-TEAD inhibition is necessary. To test if our YAP/TAZ signature has predictive value in melanoma, we used Chronos Dependency Scores from the DepMap Portal to identify melanoma cell lines that are dependent upon YAP, TAZ, or TEADs for viability, and then analyzed the expression of our YAP/TAZ signature genes in these cells. Of the 62 melanoma cell lines in the DepMap Portal, 17 are dependent upon TAZ (WWTR1), 2 are dependent upon YAP, 9 are dependent upon TEAD1, and 1 is dependent upon TEAD4 (Figure S1 and Table S4). None of the cell lines are dependent upon TEADs 2 or 3. We performed GSEA analysis to test for enrichment of our YAP/TAZ Up and YAP/TAZ Down genesets and other published YAP Up genesets [45,46,47] in the cell lines dependent upon either YAP or TAZ (YAP/TAZ) or upon any of the TEADs. The YAP/TAZ Up geneset was highly enriched (i.e. upregulated) in the TEAD-dependent and YAP/TAZ-dependent cell lines (Figure 5A). The Cordenonsi YAP Up and Wang YAP Up genesets also showed enrichment in the dependent cell lines; however, these genesets were not as strongly enriched as our YAP/TAZ Up geneset (lower normalized enrichment scores, higher false discovery rates, and lower % of genes in the leading edge). Our YAP/TAZ Down geneset showed only a slight negative enrichment in TEAD-dependent and YAP/TAZ-dependent cell lines, suggesting that upregulated genes may be better predictors of YAP/TAZ-TEAD dependence than downregulated genes.
Figure 5.
Upregulated YAP/TAZ signature genes are predictive of melanoma cell dependence upon YAP/TAZ-TEAD. RNA-seq data and dependency scores for 62 melanoma cell lines were downloaded from the DepMap Portal (Table S4). Cell lines were scored as dependent (Chronos Dependency Score of < -0.65) or independent (Chronos Dependency Score of > -0.65) for TEADs 1-4, YAP, or TAZ (WWTR1). (A) GSEA was performed on RNA-seq data to test for enrichment of the indicated genesets in cell lines dependent upon TEADs or either YAP or TAZ (YAP/TAZ). (B) The heatmap shows the relative expression (Z-Score of the log transformed TPM (log2(1+TPM))) of each of the 80 YAP/TAZ Up genes in the DepMap melanoma cell lines. Chronos Dependency Scores for TEADs, YAP, or TAZ are shown in the pink and white heatmap. Blue dots indicate cell lines with a Chronos Dependency Scores <-1.0, black dots indicate a score between -1.0 and -0.65. (C) GSVA was used to score enrichment of each indicated geneset in each of the 62 melanoma cell lines in (B) and then ROC curves were generated to test how well GSVA score could predict dependency upon YAP/TAZ or TEADs. The area under the curve (AUC), pValue, Youden Index J, Sensitivity, and Specificity values for each geneset are shown in the tables. Data used to generate this figure is found in Table S4.
Figure 5.
Upregulated YAP/TAZ signature genes are predictive of melanoma cell dependence upon YAP/TAZ-TEAD. RNA-seq data and dependency scores for 62 melanoma cell lines were downloaded from the DepMap Portal (Table S4). Cell lines were scored as dependent (Chronos Dependency Score of < -0.65) or independent (Chronos Dependency Score of > -0.65) for TEADs 1-4, YAP, or TAZ (WWTR1). (A) GSEA was performed on RNA-seq data to test for enrichment of the indicated genesets in cell lines dependent upon TEADs or either YAP or TAZ (YAP/TAZ). (B) The heatmap shows the relative expression (Z-Score of the log transformed TPM (log2(1+TPM))) of each of the 80 YAP/TAZ Up genes in the DepMap melanoma cell lines. Chronos Dependency Scores for TEADs, YAP, or TAZ are shown in the pink and white heatmap. Blue dots indicate cell lines with a Chronos Dependency Scores <-1.0, black dots indicate a score between -1.0 and -0.65. (C) GSVA was used to score enrichment of each indicated geneset in each of the 62 melanoma cell lines in (B) and then ROC curves were generated to test how well GSVA score could predict dependency upon YAP/TAZ or TEADs. The area under the curve (AUC), pValue, Youden Index J, Sensitivity, and Specificity values for each geneset are shown in the tables. Data used to generate this figure is found in Table S4.
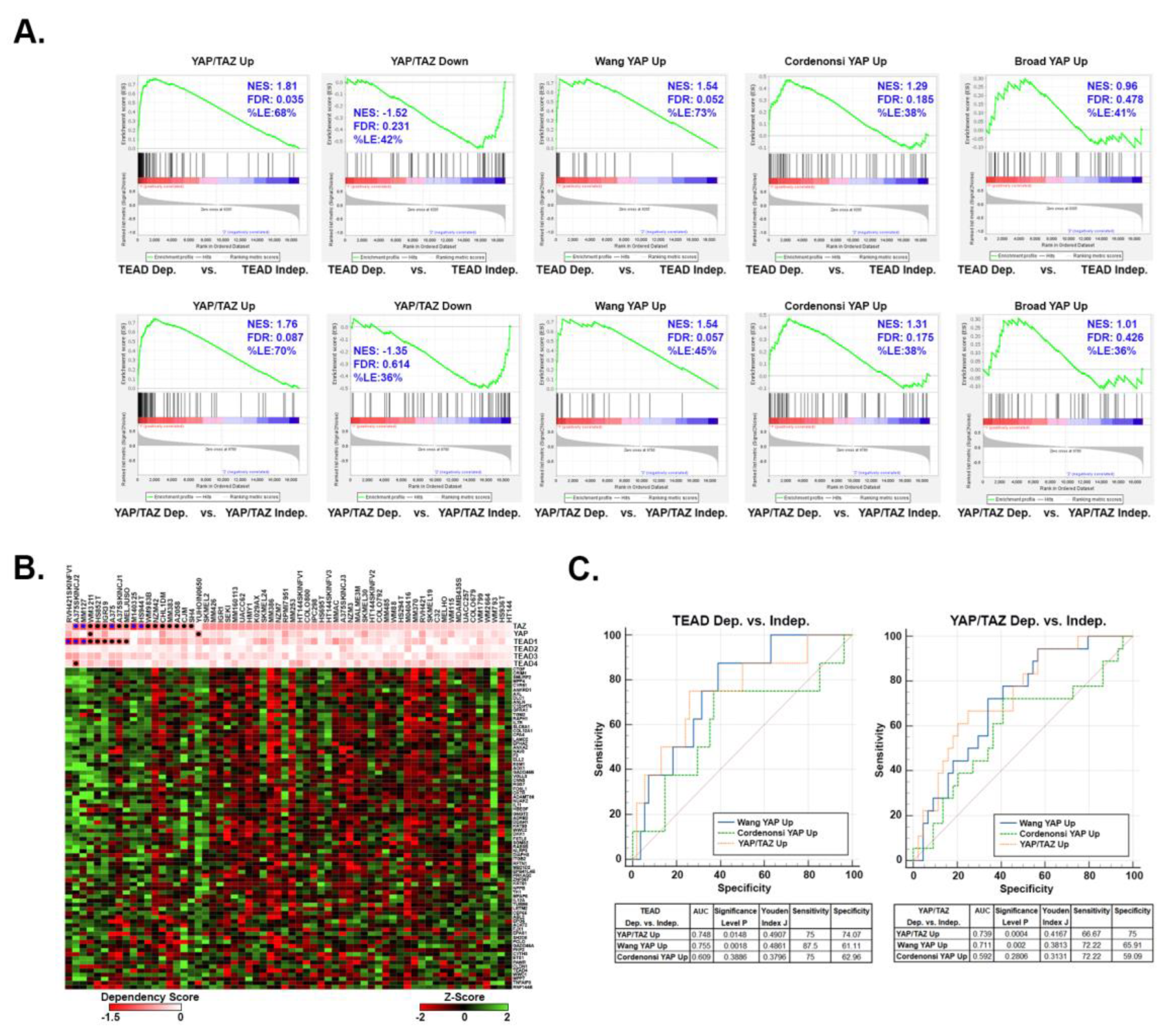
The above data suggests that cell lines dependent upon YAP/TAZ or TEADs have higher expression of the upregulated genes in our YAP/TAZ signature. Consistently, although there were a few exceptions, most of the YAP/TAZ Up genes were more highly expressed in the cell lines with the lowest dependency scores (i.e. the most dependent cell lines) (Figure 5B and Table S4). We next tested how well our YAP/TAZ Up geneset could predict dependency of these melanoma cell lines upon YAP/TAZ or TEADs relative to the other published YAP Up genesets. Gene Set Variance Analysis (GSVA) was used to score the relative enrichment of each geneset in each melanoma cell line, and then receiver operating characteristic (ROC) curves were generated to determine how well each geneset could predict dependency upon YAP/TAZ or TEADs. Although all 3 genesets were predictive of dependency upon TEADs or YAP/TAZ, our YAP/TAZ Up and the Wang YAP Up genesets were the most predictive (higher AUC and Youden Index values) (Figure 5C). Overall, these data show that enrichment of the upregulated genes in our YAP/TAZ signature can help determine if melanoma cell lines are dependent upon YAP/TAZ-TEAD, raising the intriguing possibility that this signature could be used to help identify melanoma patients that may respond to TEAD inhibition.
3.4. Upregulated YAP/TAZ signature genes strongly correlate with YAP/TAZ activation in human melanomas.
Our analysis so far has used gene expression data from cell lines cultured in vitro to identify YAP/TAZ-TEAD target genes and test their predictive value. We next sought to determine if our YAP/TAZ signature genes are also YAP/TAZ-dependent in human melanoma, so we analyzed RNA-seq data from The Cancer Genome Atlas (TCGA) Human Skin Cutaneous Melanoma (SKCM) project. Although increases in YAP1 or WWTR1 (TAZ) mRNA expression in a tumor could result in increased protein expression and thus, elevated YAP/TAZ activity, most of the regulation of YAP and TAZ occurs at the post-translational level, so YAP1 or WWTR1 mRNA expression is likely not the best readout for YAP/TAZ activity. Consistently, when we analyzed the TCGA-SKCM dataset, we found a poor correlation between YAP1 mRNA expression and the mRNA expression of several established YAP/TAZ target genes (CTGF/CCN2, CYR61/CCN1, ANKRD1, CRIM1, DDAH1, and F3) (Figure S2A). There was also not a significant difference in the expression of most of these genes in tumors with high YAP1 mRNA expression when compared to tumors with low YAP1 mRNA expression (Figure S2B). WWTR1 mRNA expression showed a stronger correlation with YAP/TAZ target genes, and the expression of these genes was generally elevated in tumors with high WWTR1 mRNA expression when compared to tumors with low expression (Figure S2A&B). However, these genes showed a much stronger correlation with the mRNA expression of the established YAP/TAZ target gene CTGF/CCN2 and their expression was significantly higher in CTGF high tumors compared to CTGF low tumors (Figure S2A&B). Consistently, genes in published YAP Up genesets [45,46] were more upregulated in CTGF high versus CTGF low tumors than they were in the YAP1 or WWTR1 high vs. low tumors (Figure S2C). Furthermore, DepMap Portal data showed that the expression of YAP1, WWTR1, or TEADs did not strongly correlate with dependence upon YAP, TAZ, or TEADs (Figure S1). Thus, the expression of YAP/TAZ target genes such as CTGF and CYR61 appears to be a more reliable indicator of increased YAP/TAZ activity than the mRNA expression of YAP1 and WWTR1 themselves.
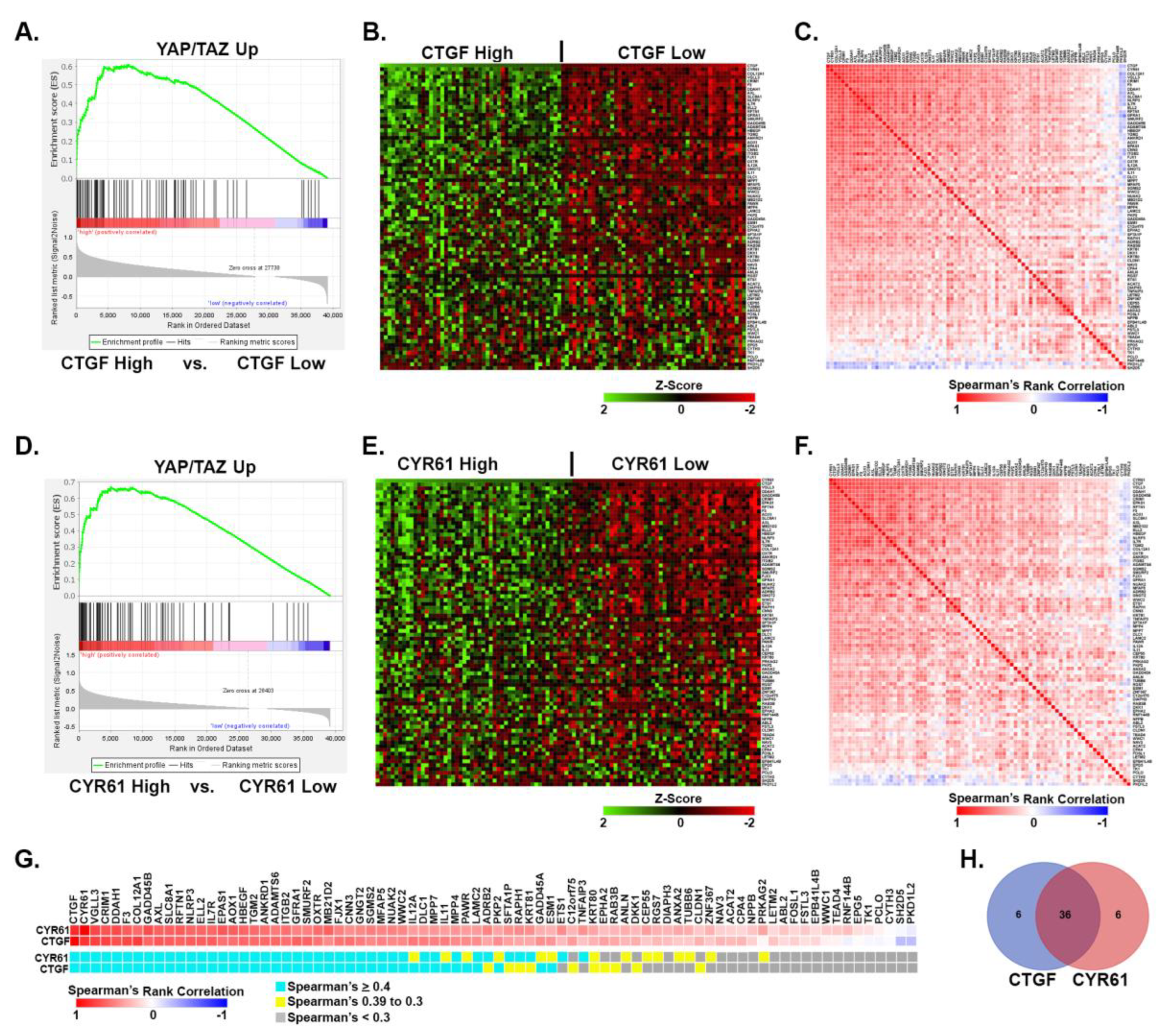
Given the above results, we next tested which of our YAP/TAZ signature genes were correlated with the expression of CTGF or CYR61 in the TCGA-SKCM tumors. GSEA analysis revealed that many of the YAP/TAZ Up genes were enriched in tumors with high CTGF or CYR61 mRNA expression compared to tumors with low expression (Figure 6A&D). Consistently, the expression of most of the YAP/TAZ Up genes was significantly higher in tumors with high CTGF or CYR61 expression compared to tumors with low expression (Figure 6B&E). To determine how strongly the expression of YAP/TAZ Up genes correlated with each other, we analyzed the TCGA-SKCM data using Spearman’s Rank Correlation similarity matrices. This revealed a large subset of the YAP/TAZ Up genes that showed a strong correlation with CTGF and CYR61 as well as with each other (Figure 6C&F). Of the 80 YAP/TAZ Up genes, 49 were strongly correlated with the mRNA expression of CTGF, CYR61, or both (Figure 6G&H and Table S5). In contrast, the YAP/TAZ Down genes did not show a strong negative correlation with CTGF or CYR61 mRNA expression. In fact, some of these genes were positively correlated with CTGF or CYR61 (Figure S3 and Table S5). This suggests that although these genes are downregulated by YAP and TAZ activation in metastatic melanoma cells in vitro, they are not consistently downregulated by YAP or TAZ in human melanomas in vivo. Similarly, data presented above (and below) demonstrate that, overall, YAP/TAZ Down genes are not as consistently regulated by YAP and TAZ in different cell lines as the YAP/TAZ Up genes are. Nevertheless, our data suggests that most of the YAP/TAZ Up genes in our signature are strongly correlated with YAP/TAZ activity in tumors isolated from melanoma patients.
Figure 6.
Upregulated YAP/TAZ signature genes are correlated with increased YAP/TAZ activity in human melanoma. RNA-seq data from the TCGA human skin cutaneous melanoma (SKCM) project was downloaded and tumors with high (> 1 standard deviation from the mean) or low (< -1 standard deviation from the mean) expression of CTGF (A-C) or CYR61 mRNA (D-F) were analyzed. (A&D) GSEA analysis was performed to test for enrichment of the YAP/TAZ Up geneset in CTGF or CYR61 high vs. low tumors (NES and FDR are indicated). (B&E) The heatmaps show the relative expression (Z-Score of the log transformed TPM (log2(1+TPM))) of each of the 80 YAP/TAZ Up genes in CTGF (B) or CYR61 (E) high vs. low tumors. Tumors are sorted by CTGF or CYR61 mRNA expression and genes are ranked from highest (top) to lowest (bottom) based on their Spearman Rank Correlation of the gene with either CTGF (B) or CYR61 (E). (C&F) Spearman similarity matrix analysis was performed on the expression data shown in (B) or (E) and the Spearman Rank Correlation value for each pairwise comparison across all tumors analyzed in each set is shown. Genes are ranked the same as in (B) and (E). (G) Shows the Spearman Rank Correlation values for each gene compared to CTGF or CYR61 (same data as 1st rows in C and F). Genes with Correlation values > 0.4 are indicated in blue, between 0.39 and 0.3 in yellow and < 0.3 in gray. (H) The Venn diagram shows the number of genes from each comparison in (G) with correlation values > 0.4. The data used to generate this figure is found in Table S5.
Figure 6.
Upregulated YAP/TAZ signature genes are correlated with increased YAP/TAZ activity in human melanoma. RNA-seq data from the TCGA human skin cutaneous melanoma (SKCM) project was downloaded and tumors with high (> 1 standard deviation from the mean) or low (< -1 standard deviation from the mean) expression of CTGF (A-C) or CYR61 mRNA (D-F) were analyzed. (A&D) GSEA analysis was performed to test for enrichment of the YAP/TAZ Up geneset in CTGF or CYR61 high vs. low tumors (NES and FDR are indicated). (B&E) The heatmaps show the relative expression (Z-Score of the log transformed TPM (log2(1+TPM))) of each of the 80 YAP/TAZ Up genes in CTGF (B) or CYR61 (E) high vs. low tumors. Tumors are sorted by CTGF or CYR61 mRNA expression and genes are ranked from highest (top) to lowest (bottom) based on their Spearman Rank Correlation of the gene with either CTGF (B) or CYR61 (E). (C&F) Spearman similarity matrix analysis was performed on the expression data shown in (B) or (E) and the Spearman Rank Correlation value for each pairwise comparison across all tumors analyzed in each set is shown. Genes are ranked the same as in (B) and (E). (G) Shows the Spearman Rank Correlation values for each gene compared to CTGF or CYR61 (same data as 1st rows in C and F). Genes with Correlation values > 0.4 are indicated in blue, between 0.39 and 0.3 in yellow and < 0.3 in gray. (H) The Venn diagram shows the number of genes from each comparison in (G) with correlation values > 0.4. The data used to generate this figure is found in Table S5.
3.5. YAP/TAZ signature genes are predictive of dependence upon YAP/TAZ-TEAD in other cancer types.
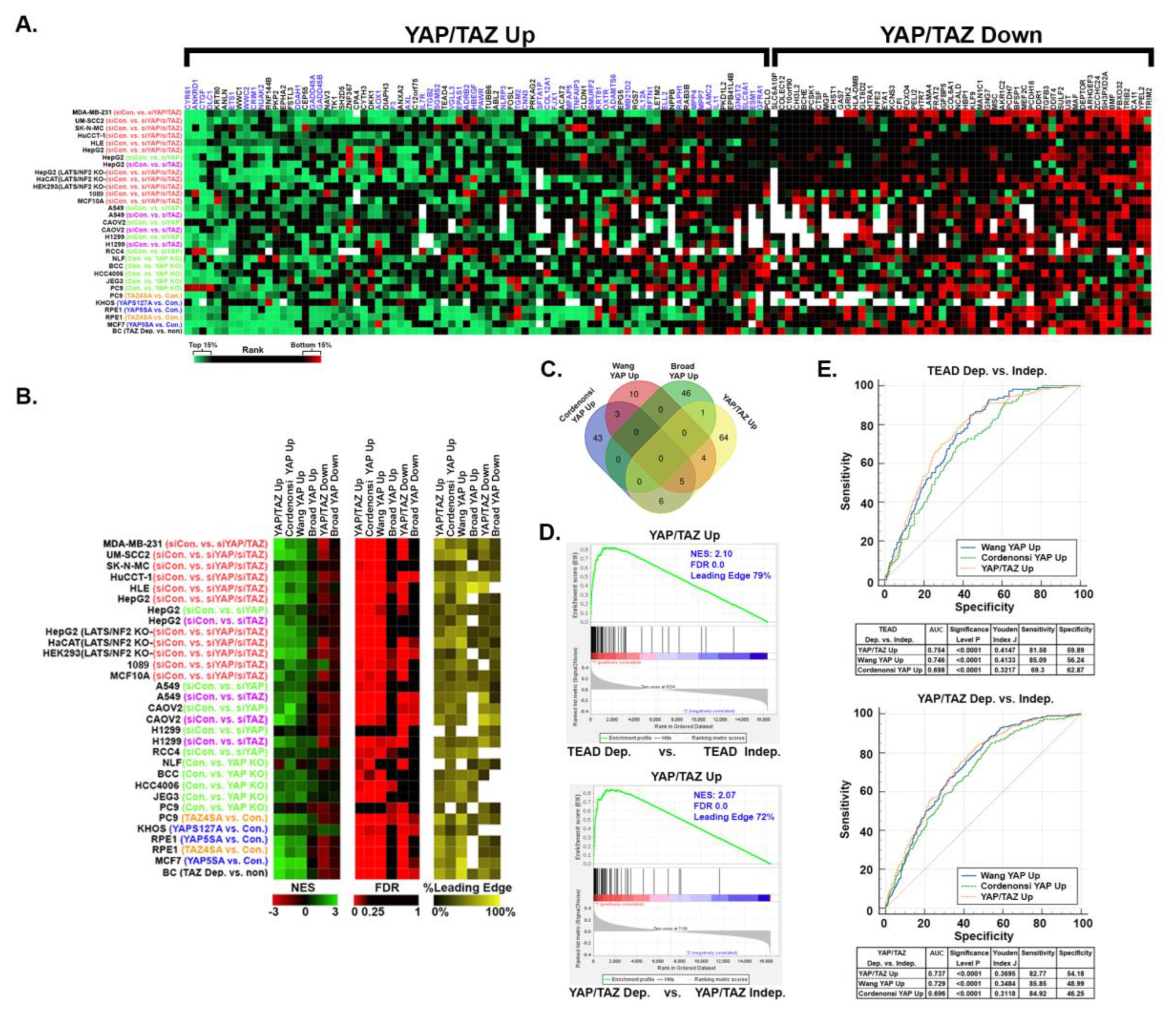
Although YAP and TAZ promote tumor growth and progression in multiple cancers (reviewed in [4,6,7,11], it is unknown to what degree YAP/TAZ-dependent gene expression is conserved across different cancer types. Since TEADs recruit YAP or TAZ to consensus motifs in the enhancer and promoter regions of target genes, YAP/TAZ-TEAD regulated genes in different cancers are likely to overlap to some degree. Indeed, several of the genes in our YAP/TAZ signature were established as YAP/TAZ targets in other cell types. However, tissue-specific transcription factors and epigenetic regulators could also influence which genes YAP and TAZ regulate in each cancer type. To determine which of our YAP/TAZ signature genes are also YAP/TAZ-responsive in other cancers, we analyzed 31 available gene expression datasets generated from cancer cell lines in which YAP and/or TAZ expression or function was altered (Table S1). Many of the 132 YAP/TAZ signature genes were also regulated by YAP/TAZ in several of the other cancer cell lines (Figure 7A and Table S6). The upregulated genes in our YAP/TAZ signature were significantly more conserved than the downregulated genes. Intriguingly, there was not a single gene that was YAP/TAZ-dependent in every cell line, and most of the genes were only YAP/TAZ-responsive in a subset of the cancer cell lines (Figure 7A and Table S6). This suggests that although most of the YAP/TAZ Up genes in our signature are regulated by YAP or TAZ in other cancers, which subsets of genes respond to changes in YAP/TAZ activity in each cancer cell line varies significantly.
YAP Up genesets, like those used above, are often used for geneset enrichment analysis to test for increased YAP/TAZ activity in cells or tissues. To test how well our YAP/TAZ signature could identify cancer cells with elevated YAP/TAZ activity, we performed GSEA analysis on each of the gene expression datasets analyzed in Figure 7A to compare enrichment of our YAP/TAZ Up and Down genesets with published YAP Up and Down genesets [45,46,47]. Interestingly, despite minimal overlap between these genesets, the Cordenonsi YAP Up, Wang YAP Up, and our YAP/TAZ Up gensets each showed significant enrichment in the cells with high YAP and/or TAZ when compared to cells with lower YAP and/or TAZ (Figure 7B&C and Table S6), indicating that the genes in these genesets are more highly expressed in cells with higher YAP/TAZ activity. Importantly, our YAP/TAZ Up geneset showed similar or greater enrichment (higher NES and lower FDR) than the other existing genesets (Figure 7B and Table S6). Although our YAP/TAZ Down geneset was negatively enriched in several of the datasets (Figure 7B and Table S6), the magnitude of this negative enrichment was weaker than the magnitude of the positive enrichment observed for the YAP/TAZ Up genes. This indicates that the downregulated genes are not as consistently YAP/TAZ dependent as the upregulated genes. The Broad YAP Up and Broad YAP Down genesets did not show strong positive or negative enrichment, respectively, potentially because they were generated from a single cell line (MCF10A), and likely contain genes whose YAP-dependence is unique to these cells.
Since our YAP/TAZ signature was enriched in cancer cells with elevated YAP/TAZ activity, we next tested whether it was predictive of YAP/TAZ-TEAD dependence in other cancer cell types like it was in melanoma. We used Chronos Dependency Scores to identify which of the 1019 cancer cell lines in DepMap Portal are dependent upon YAP, TAZ, or TEADs for viability. GSEA analysis showed that our YAP/TAZ Up geneset was highly enriched in cancer cell lines that are dependent on YAP/TAZ or TEADs compared to non-dependent cell lines (Figure 7D). ROC curve analysis showed that our YAP/TAZ Up geneset was similar or better than the other YAP Up genesets at predicting dependence upon either YAP/TAZ or TEADs (Figure 7E). Thus, despite being developed in melanoma cells, the upregulated genes in our YAP/TAZ signature are highly enriched in other cancer cell types with YAP/TAZ-TEAD activation and are predictive of cancer cell sensitivity to loss of YAP/TAZ or TEADs.
Figure 7.
YAP/TAZ signature genes are YAP/TAZ dependent and predictive of dependence upon YAP/TAZ-TEAD in other cancer types. Publicly available datasets (Table S1, Tab 3) were downloaded from NCBI-GEO and GSEA was used to generate rank-ordered lists for the indicated cell lines and comparisons. The heatmap shows the % Rank (the gene’s rank/total genes in rank-ordered list*100) of each of the 80 YAP/TAZ Up and 52 YAP/TAZ Down genes. Green indicates that the gene’s rank is between 0-15% (i.e. enriched in YAP and/or TAZ high cells), red between 85%-100% (i.e. enriched in the YAP and/or TAZ low cells), black between 15-85% (i.e. not enriched), and white indicates the gene was not included in the rank-ordered list for that dataset. (B) GSEA was performed on the same publicly available datasets using our YAP/TAZ Up and Down genesets and other published YAP Up genesets. The heatmaps show the Normalized Enrichment Score (NES), False Discovery Rate (FDR), and % of each gene set that was in the Leading Edge (% Leading Edge) for each comparison. (C) The Venn diagram shows overlap between our YAP/TAZ Up geneset and other published YAP Up genesets. Lists of each geneset are found in Table S1, Tab 4, GSEA results, and the % Rank values used to generate the heatmaps in this figure are found in Table S5. (D) RNA-seq data and dependency scores for all 1019 cell lines were downloaded from the DepMap Portal (see Table S4). Cell lines were scored as dependent (Chronos Dependency Score of < -0.65) or independent (Chronos Dependency Score of > -0.65) for TEADs 1-4, YAP, or TAZ. GSEA was performed on the RNA-seq data to test for enrichment of our YAP/TAZ Up genset. (E) GSVA was used to score enrichment of each indicated geneset in each of the 1019 cell lines in the DepMap Portal and then ROC curves were generated to test if GSVA score could predict dependency upon YAP/TAZ or TEADs. Area Under the Curve, pValue, Youden Index J, Sensitivity, and Specificity values for each geneset are shown in the tables.
Figure 7.
YAP/TAZ signature genes are YAP/TAZ dependent and predictive of dependence upon YAP/TAZ-TEAD in other cancer types. Publicly available datasets (Table S1, Tab 3) were downloaded from NCBI-GEO and GSEA was used to generate rank-ordered lists for the indicated cell lines and comparisons. The heatmap shows the % Rank (the gene’s rank/total genes in rank-ordered list*100) of each of the 80 YAP/TAZ Up and 52 YAP/TAZ Down genes. Green indicates that the gene’s rank is between 0-15% (i.e. enriched in YAP and/or TAZ high cells), red between 85%-100% (i.e. enriched in the YAP and/or TAZ low cells), black between 15-85% (i.e. not enriched), and white indicates the gene was not included in the rank-ordered list for that dataset. (B) GSEA was performed on the same publicly available datasets using our YAP/TAZ Up and Down genesets and other published YAP Up genesets. The heatmaps show the Normalized Enrichment Score (NES), False Discovery Rate (FDR), and % of each gene set that was in the Leading Edge (% Leading Edge) for each comparison. (C) The Venn diagram shows overlap between our YAP/TAZ Up geneset and other published YAP Up genesets. Lists of each geneset are found in Table S1, Tab 4, GSEA results, and the % Rank values used to generate the heatmaps in this figure are found in Table S5. (D) RNA-seq data and dependency scores for all 1019 cell lines were downloaded from the DepMap Portal (see Table S4). Cell lines were scored as dependent (Chronos Dependency Score of < -0.65) or independent (Chronos Dependency Score of > -0.65) for TEADs 1-4, YAP, or TAZ. GSEA was performed on the RNA-seq data to test for enrichment of our YAP/TAZ Up genset. (E) GSVA was used to score enrichment of each indicated geneset in each of the 1019 cell lines in the DepMap Portal and then ROC curves were generated to test if GSVA score could predict dependency upon YAP/TAZ or TEADs. Area Under the Curve, pValue, Youden Index J, Sensitivity, and Specificity values for each geneset are shown in the tables.
4. Discussion
4.1. A YAP/TAZ gene signature that predicts dependence upon YAP, TAZ, and TEADs
The experimental and clinical evidence linking YAP or TAZ activation to cancer development and progression suggests that TEAD inhibitors could be effective treatments for several different cancer types, and preclinical studies using TEAD inhibitors have yielded promising results [48,49,50,51]. However, YAP and TAZ are not the only drivers of tumor progression, so TEAD inhibition is only likely to be effective in the subset of patients whose tumors are reliant upon YAP/TAZ/TEADs. Furthermore, predicting sensitivity to TEAD inhibition will be particularly challenging in patients that lack mutations in Hippo Pathway genes. Our results suggest that YAP/TAZ gene signatures could be an effective way to identify cancers sensitive to TEAD inhibition. Consistently, our YAP/TAZ signature could identify cell lines dependent upon YAP/TAZ or TEADs when tested specifically in melanoma or when tested on over 1000 cell lines from different types of cancer. Our data also suggest that YAP/TAZ gene signatures are more effective at predicting dependence upon YAP/TAZ/TEADs than the mRNA levels of YAP1, WWTR1, or TEADs. Indeed, YAP and WWTR1 mRNA expression did not strongly correlate with the expression of known YAP/TAZ target genes (Figure S2) or with dependence upon YAP or TAZ (Figure S1). In contrast, our YAP/TAZ Up geneset could predict dependence upon YAP/TAZ, or TEADs (Figure 5 and Figure 7). Importantly, most of our YAP/TAZ Up genes were also enriched in human melanoma samples with high expression of established YAP/TAZ target genes, suggesting that these genes are also YAP/TAZ-dependent in human melanomas.
Despite these encouraging results, more work will be necessary to develop diagnostic YAP/TAZ signatures for clinical use. The DepMap dependency data used here is based on cell lines cultured in vitro and does not capture the complexity of a tumor. Furthermore, human cancers are heterogeneous, which is likely to influence the predictive power of any diagnostic signature. Testing using patient-derived xenograft models could help refine and improve our YAP/TAZ signature, but ultimately, data from clinical trials in patients treated with TEAD inhibitors will likely be necessary to further refine any signature developed using pre-clinical models. Despite the need for more work, our findings suggest that YAP/TAZ-TEAD gene signatures have potential diagnostic value that warrants further development.
The fact that our YAP/TAZ Up genset was predictive when tested on a diverse set of cancer cell lines (Figure 7D&E) suggests that it may not be necessary to develop cancer type-specific YAP/TAZ signatures. However, some of our findings suggest otherwise. We found that several of our YAP/TAZ signature genes that were highly conserved in melanoma cell lines and human melanomas were not YAP/TAZ-responsive in many other cancer types that we analyzed (Figure 7A). This suggests that these genes may be more reliable readouts for YAP/TAZ-TEAD activity in melanoma than in other cancers. Furthermore, our YAP/TAZ Up geneset, which was derived using melanoma cell lines, showed stronger enrichment in YAP/TAZ and TEAD-dependent melanoma cell lines than the other published YAP Up genesets that were derived from other cell types (Figure 5A). We also found that although some established YAP/TAZ target genes (CRIM1, F3, ANKRD1) correlated strongly with CTGF and CYR61 expression in human melanoma, others did not (WWC1, FOSL1, FSTL3) (Figure 6). Collectively, this suggests that while YAP/TAZ signatures may generally have predictive value across multiple cancer types, cancer type-specific YAP/TAZ signatures may provide an added level of accuracy for that cancer type.
Although our findings showed that increased expression of YAP/TAZ-TEAD target genes, which is likely due to elevated YAP/TAZ activity, was a reliable indicator of dependence upon YAP/TAZ-TEAD, there were some cell lines that did not appear to follow this pattern. For example, WM793 cells had high expression of our YAP/TAZ signature genes but were not sensitive to loss of YAP, TAZ, or TEADs, while NZM42 and CHL1DM cells, which were sensitive to loss of TAZ, had relatively low expression of YAP/TAZ signature genes (Figure 5). DepMap dependency data is based on CRISPR-mediated knockout of individual genes, so there could be compensation of YAP or TAZ for each other. TEADs could also compensate for each another. Although it is beyond the scope and feasibility of this study, testing a larger cohort of cell lines for sensitivity to pan-TEAD inhibitors could help refine our YAP/TAZ signature to enhance its predictive accuracy.
The development of therapeutic resistance is common in patients treated with targeted therapies and upregulation of YAP/TAZ-TEAD transcriptional activity has been found to be a resistance mechanism in cancer cells treated with targeted therapies [52,53]. For example, YAP/TAZ activation plays a causal role in resistance to KRAS, BRAF, MEK and EGFR inhibitors [42,51,54,55,56,57,58,59,60,61]. Collectively, this suggests that TEAD inhibitors could be effective second line therapies for some patients and that YAP/TAZ-TEAD signatures, like ours, might provide a means to identify which patients would benefit. Whether used as primary or secondary therapies, we should anticipate that some patients will develop resistance to TEAD inhibitors. Transcriptional profiling in a large cohort of cancer cell lines treated with pan-TEAD inhibitors could also help predict and prevent such resistance. Consistently, a recent study using a panel of cancer cell lines found that treatment with the TEAD inhibitor MGH-CP1 or YAP/TAZ silencing, each promoted VGLL3-mediated transcriptional activation of SOX4/PI3K/AKT signaling, which contributed to resistance to MGH-CP1 [62]. Importantly, this study further demonstrated that dual inhibition of AKT and TEAD could help overcome this resistance.
4.2. The complexity of YAP/TAZ-TEAD dependent gene expression in cancer cells
Several of our findings highlight the complexity of YAP/TAZ-TEAD-dependent gene expression programs. We found minimal overlap in the differentially expressed genes in each of the melanoma cell lines we analyzed (Figure 2). The genes that did overlap in melanoma cells were all also YAP/TAZ-dependent in some other cancer cell lines (Figure 7A), but the individual genes that responded to changes in YAP/TAZ activity in each cell line varied dramatically. Even well-established target genes like CTGF, CYR61, ANKRD1, and WWC1 appeared to be YAP/TAZ-independent in some cell lines. While some of the non-overlapping genes in the melanoma cell lines that we analyzed may be indirect targets, the majority of the genes in our YAP/TAZ signature had TEAD peaks in publicly available ChIP-Seq datasets (Figure 4D), indicating that these are direct YAP/TAZ-TEAD target genes. This suggests that contextual factors beyond the presence or absence of TEAD motifs in a gene’s regulatory elements influence YAP/TAZ-TEAD-dependent gene expression programs. Indeed, both YAP and TAZ can bind chromatin remodeling complexes and influence chromatin accessibility [63,64,65]. YAP and TAZ also bind a long and growing list of transcription factors and proteins that can dramatically influence the transcriptional landscapes of cells and tissues [20,63,66].
It is also important to note that the data we analyzed here does not consider temporal differences in YAP/TAZ-dependent gene expression. Indeed, a recent study nicely demonstrated that the regions of the genome occupied by TAZ change in a temporal manner following TAZ activation [67]. Our data also suggests that the magnitude of YAP or TAZ activation will influence which potential YAP/TAZ-TEAD target genes are changing in a substantial manner. For example, the same genes were influenced by YAP2SA and YAPS127A in A375 cells. However, in A375-YAPS127A cells, which have much lower YAP/TAZ-TEAD activity, only a fraction of those genes changed in a significant (greater than 2-fold) manner (Figure 1E). This suggests that as more YAP or TAZ enters the nucleus and binds to TEADs, a greater number of target genes will significantly change, which highlights the need to ensure that experimental manipulation of YAP/TAZ-TEAD activity mimics biologically relevant changes in activity that occur during disease processes.
Another interesting observation from our study is that although the number of genes upregulated and downregulated by YAP and TAZ were generally similar in the cell lines we analyzed, the upregulated genes were more highly conserved across different cancer types. Upregulated genes were also better predictors of YAP/TAZ and TEAD dependence (Figure 5 and Figure 7), and strongly correlated with other known YAP/TAZ target genes in human patient samples (Figure 6). In contrast, downregulated genes did not show the expected inverse correlation with known YAP/TAZ target genes in human patient samples (Figure S3). Most of the downregulated genes did have TEAD peaks in available ChIP-Seq datasets (Figure 4D), but the presence of these peaks was much less consistent across the datasets than it was for the upregulated genes. It is unclear why this is the case, but perhaps in the context of cancer, where YAP-TEAD and TAZ-TEAD tend to be more active, they are more likely to promote gene expression than to inhibit it.
While YAP/TAZ gene signatures offer potential diagnostic power for sensitivity to TEAD inhibition, individual YAP/TAZ target genes may also have therapeutic potential. Indeed, some of the genes in our signature have already been reported as drivers of YAP/TAZ-mediated tumor progression and metastasis. For example, YAP-mediated melanoma cell invasion was found to require YAP-dependent induction of, AXL, CRIM1, and CYR61 [1]. TAZ-mediated induction of AXL was also found to drive an AXL-ABL2-TAZ feed-forward loop that promoted lung adenocarcinoma brain metastasis [68]. Another study found that YAP promoted breast cancer metastasis through induction of the ITGB2 gene [69]. NUAK2 is a YAP/TAZ target gene that promotes tumor progression in liver and bladder cancer [70,71], and CTGF, a well-established YAP/TAZ target gene, has clear roles in cancer [72]. Several other genes in our signature have also been implicated in cancer, but their roles in YAP/TAZ-mediated tumor progression and metastasis have not yet been explored. Although determining which YAP/TAZ target genes are required for tumor progression and metastasis could reveal potential therapeutic targets, this is beyond the scope of this study, and could prove challenging if multiple genes with overlapping functions are involved.
5. Conclusions
With TEAD inhibitors already in clinical trials, the need for diagnostic tests that can identify patients who are likely to benefit from these drugs is essential. Since YAP and TAZ are extensively regulated at the posttranslational level through complex signaling networks, protein and mRNA expression levels of YAP or TAZ are not likely to be a reliable readout for YAP/TAZ-TEAD activity. Our study suggests that YAP/TAZ-TEAD gene signatures may provide a more reliable means to assess YAP/TAZ-TEAD activation and predict sensitivity to TEAD inhibition. The YAP/TAZ signature we developed here was predictive in melanoma cell lines and more broadly in a large set of cancer cell lines. The genes in this signature are TEAD dependent, and thus a direct readout for TEAD-dependent gene expression. Our work raises the intriguing possibility that with additional testing and refinement using pre-clinical models and patient data our YAP/TAZ signature could be used as a diagnostic test to help identify patients likely to respond favorably to TEAD inhibitors. Although we did not explore this in the current study, we hypothesize that since the genes in this signature are regulated by YAP/TAZ-TEAD, they may also have value as biomarkers to monitor the efficacy of TEAD inhibition during treatment with TEAD inhibitors or other drugs targeting the YAP/TAZ-TEAD axis.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org., Figure S1. DepMap dependency scores for melanoma cell lines.; Figure S2. YAP and WWTR1 mRNA expression do not strongly correlate with the mRNA expression of their target genes.; Figure S3. Downregulated YAP/TAZ signature genes are not negatively correlated with YAP/TAZ activity in human melanomas.; Table S1. Reagents and datasets used in this study.; Table S2. Processed A375 RNA-seq data.; Table S3. Lists of YAP/TAZ-regulated genes in metastatic melanoma cell lines.; Table S4. DepMap data.; Table S5. TCGA Data.; Table S6. YAP/TAZ signature genes are YAP/TAZ responsive in other cell lines. References [73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91] are cited in the Supplementary Materials.
Author Contributions
Conceptualization, J.M.L., R.O.H., E.N., R.K., P.S.; methodology, J.M.L., R.O.H., E.N., R.K., P.S.; validation, J.M.L., E.N., R.K., P.S.; formal analysis J.M.L., E.N., R.K., P.S.; investigation, J.M.L., E.N., R.K., P.S.; resources, J.M.L., R.O.H.; data curation, J.M.L., E.N., R.K., P.S.; writing J.M.L., E.N., R.K.; writing—review and editing, J.M.L., R.O.H., E.N., R.K., P.S.; visualization, J.M.L., E.N., R.K.; supervision, J.M.L., R.O.H.; project administration, J.M.L., R.O.H.; funding acquisition, J.M.L., R.O.H.; All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Institutes of Health (NIH) National Research Service Award CA1421442 (to J. M. L.); the Koch Institute Frontier Research Fund (to J. M. L.); The Albany Medical College Dean’s Office Bridge Grant (to J.M.L.); NCI, NIH, Integrative Center Biology Program Grant U54CA112967 (to R. O. H.); NIH Tumor Microenvironment Network Grant U54CA163109 (to R. O. H.); Department of Defense Breast Cancer Research Program Grant W81XWH-14-1-0240 (to R. O. H.); the MIT Ludwig Center for Molecular Oncology; and the Howard Hughes Medical Institute.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The RNA-seq data generated for this study are openly available in NCBI GEO accession number GSE234083. All 3rd party data used in these analyses are available in a the publicly accessible repositories listed in either the Supplemental Materials or Methods.
Acknowledgments
The results shown here are in whole or part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga, The Broad Institute Cancer Dependency Map (DepMap) Portal (https://depmap.org/portal/), and the ENCODE Project (https://www.encodeproject.org/). We thank Miss Sarah McMullan and Miss Chloe Purello for their critical reading of this manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Zhang, X.; Yang, L.; Szeto, P.; Abali, G.K.; Zhang, Y.; Kulkarni, A.; Amarasinghe, K.; Li, J.; Vergara, I.A.; Molania, R.; et al. The Hippo pathway oncoprotein YAP promotes melanoma cell invasion and spontaneous metastasis. Oncogene 2020, 39, 5267–5281. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, A.; Imrichova, H.; Atak, Z.K.; Dewaele, M.; Rambow, F.; Hulselmans, G.; Christiaens, V.; Svetlichnyy, D.; Luciani, F.; Van den Mooter, L.; et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat Commun 2015, 6, 6683. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Panciera, T.; Contessotto, P.; Cordenonsi, M. YAP/TAZ as master regulators in cancer: modulation, function and therapeutic approaches. Nat Cancer 2023, 4, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [PubMed]
- Janse van Rensburg, H.J.; Yang, X. The roles of the Hippo pathway in cancer metastasis. Cell Signal 2016, 28, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.J. YAP/TAZ: Drivers of Tumor Growth, Metastasis, and Resistance to Therapy. Bioessays 2020, 42, e1900162. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, J.Z.; Vergara, I.A.; Zhang, Y.; Szeto, P.; Yang, L.; Mintoff, C.; Colebatch, A.; McIntosh, L.; Mitchell, K.A.; et al. Somatic Hypermutation of the YAP Oncogene in a Human Cutaneous Melanoma. Mol Cancer Res 2019, 17, 1435–1449. [Google Scholar] [CrossRef]
- Nallet-Staub, F.; Marsaud, V.; Li, L.; Gilbert, C.; Dodier, S.; Bataille, V.; Sudol, M.; Herlyn, M.; Mauviel, A. Pro-invasive activity of the Hippo pathway effectors YAP and TAZ in cutaneous melanoma. J Invest Dermatol 2014, 134, 123–132. [Google Scholar] [CrossRef]
- Feng, Q.; Guo, P.; Kang, S.; Zhao, F. High expression of TAZ/YAP promotes the progression of malignant melanoma and affects the postoperative survival of patients. Pharmazie 2018, 73, 662–665. [Google Scholar] [CrossRef]
- Warren, J.S.A.; Xiao, Y.; Lamar, J.M. YAP/TAZ Activation as a Target for Treating Metastatic Cancer. Cancers 2018, 10, 115. [Google Scholar] [CrossRef] [PubMed]
- Calses, P.C.; Crawford, J.J.; Lill, J.R.; Dey, A. Hippo Pathway in Cancer: Aberrant Regulation and Therapeutic Opportunities. Trends Cancer 2019, 5, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 2007, 21, 2747–2761. [Google Scholar] [CrossRef] [PubMed]
- Lei, Q.Y.; Zhang, H.; Zhao, B.; Zha, Z.Y.; Bai, F.; Pei, X.H.; Zhao, S.; Xiong, Y.; Guan, K.L. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol Cell Biol 2008, 28, 2426–2436. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Zha, Z.Y.; Zhou, X.; Zhang, H.; Huang, W.; Zhao, D.; Li, T.; Chan, S.W.; Lim, C.J.; Hong, W.; et al. The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCFbeta-TrCP E3 ligase. J Biol Chem 2010, 285, 37159–37169. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Tumaneng, K.; Wang, C.Y.; Guan, K.L. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev 2010, 24, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Manning, S.A.; Kroeger, B.; Harvey, K.F. The regulation of Yorkie, YAP and TAZ: new insights into the Hippo pathway. Development 2020, 147. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Pan, D. The Hippo Signaling Pathway in Development and Disease. Dev Cell 2019, 50, 264–282. [Google Scholar] [CrossRef]
- Pocaterra, A.; Romani, P.; Dupont, S. YAP/TAZ functions and their regulation at a glance. J Cell Sci 2020, 133. [Google Scholar] [CrossRef]
- Kim, M.K.; Jang, J.W.; Bae, S.C. DNA binding partners of YAP/TAZ. BMB Rep 2018, 51, 126–133. [Google Scholar] [CrossRef]
- Holden, J.K.; Cunningham, C.N. Targeting the Hippo Pathway and Cancer through the TEAD Family of Transcription Factors. Cancers 2018, 10, 81. [Google Scholar] [CrossRef] [PubMed]
- Pobbati, A.V.; Kumar, R.; Rubin, B.P.; Hong, W. Therapeutic targeting of TEAD transcription factors in cancer. Trends Biochem Sci 2023, 48, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Barry, E.R.; Simov, V.; Valtingojer, I.; Venier, O. Recent Therapeutic Approaches to Modulate the Hippo Pathway in Oncology and Regenerative Medicine. Cells 2021, 10. [Google Scholar] [CrossRef]
- Sekido, Y.; Sato, T. NF2 alteration in mesothelioma. Front Toxicol 2023, 5, 1161995. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Degese, M.S.; Iglesias-Bartolome, R.; Vaque, J.P.; Molinolo, A.A.; Rodrigues, M.; Zaidi, M.R.; Ksander, B.R.; Merlino, G.; Sodhi, A.; et al. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer cell 2014, 25, 831–845. [Google Scholar] [CrossRef] [PubMed]
- Seavey, C.N.; Pobbati, A.V.; Rubin, B.P. Unraveling the Biology of Epithelioid Hemangioendothelioma, a TAZ-CAMTA1 Fusion Driven Sarcoma. Cancers (Basel) 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Seavey, C.N.; Pobbati, A.V.; Hallett, A.; Ma, S.; Reynolds, J.P.; Kanai, R.; Lamar, J.M.; Rubin, B.P. WWTR1(TAZ)-CAMTA1 gene fusion is sufficient to dysregulate YAP/TAZ signaling and drive epithelioid hemangioendothelioma tumorigenesis. Genes Dev 2021, 35, 512–527. [Google Scholar] [CrossRef]
- Lamar, J.M.; Motilal Nehru, V.; Weinberg, G. Epithelioid Hemangioendothelioma as a Model of YAP/TAZ-Driven Cancer: Insights from a Rare Fusion Sarcoma. Cancers (Basel) 2018, 10. [Google Scholar] [CrossRef]
- Szulzewsky, F.; Holland, E.C.; Vasioukhin, V. YAP1 and its fusion proteins in cancer initiation, progression and therapeutic resistance. Dev Biol 2021, 475, 205–221. [Google Scholar] [CrossRef]
- Xu, L.; Begum, S.; Hearn, J.D.; Hynes, R.O. GPR56, an atypical G protein-coupled receptor, binds tissue transglutaminase, TG2, and inhibits melanoma tumor growth and metastasis. Proc Natl Acad Sci U S A 2006, 103, 9023–9028. [Google Scholar] [CrossRef]
- Lamar, J.M.; Xiao, Y.; Norton, E.; Jiang, Z.G.; Gerhard, G.M.; Kooner, S.; Warren, J.S.A.; Hynes, R.O. SRC tyrosine kinase activates the YAP/TAZ axis and thereby drives tumor growth and metastasis. J Biol Chem 2019, 294, 2302–2317. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, W.M., Jr.; Hong, J.H.; Yaffe, M.B.; Farrance, I.K. The transcriptional co-activator TAZ interacts differentially with transcriptional enhancer factor-1 (TEF-1) family members. Biochem J 2005, 388, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Malladi, V.S.; Strattan, J.S.; Hitz, B.C.; Gabdank, I.; Narayanan, A.K.; Ho, M.; Lee, B.T.; et al. ENCODE data at the ENCODE portal. Nucleic Acids Res 2016, 44, D726–732. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; He, Q.Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 2013, 14, 7. [Google Scholar] [CrossRef]
- Lamar, J.M.; Stern, P.; Liu, H.; Schindler, J.W.; Jiang, Z.G.; Hynes, R.O. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc Natl Acad Sci U S A 2012, 109, E2441–2450. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Kim, J.; Hong, H.; Lee, S.H.; Lee, J.K.; Jung, E.; Kim, J. Actin remodeling confers BRAF inhibitor resistance to melanoma cells through YAP/TAZ activation. EMBO J 2016, 35, 462–478. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, M. A Sveinsson's chorioretinal atrophy-associated missense mutation in mouse Tead1 affects its interaction with the co-factors YAP and TAZ. Biochem Biophys Res Commun 2007, 361, 1022–1026. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Park, J.; Feng, A.; Awasthi, P.; Wang, Z.; Chen, Q.; Iglesias-Bartolome, R. YAP1/TAZ-TEAD transcriptional networks maintain skin homeostasis by regulating cell proliferation and limiting KLF4 activity. Nat Commun 2020, 11, 1472. [Google Scholar] [CrossRef] [PubMed]
- Cordenonsi, M.; Zanconato, F.; Azzolin, L.; Forcato, M.; Rosato, A.; Frasson, C.; Inui, M.; Montagner, M.; Parenti, A.R.; Poletti, A.; et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 2011, 147, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, X.; Maglic, D.; Dill, M.T.; Mojumdar, K.; Ng, P.K.; Jeong, K.J.; Tsang, Y.H.; Moreno, D.; Bhavana, V.H.; et al. Comprehensive Molecular Characterization of the Hippo Signaling Pathway in Cancer. Cell Rep 2018, 25, 1304–1317 e1305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Smolen, G.A.; Haber, D.A. Negative regulation of YAP by LATS1 underscores evolutionary conservation of the Drosophila Hippo pathway. Cancer Res 2008, 68, 2789–2794. [Google Scholar] [CrossRef]
- Tang, T.T.; Konradi, A.W.; Feng, Y.; Peng, X.; Ma, M.; Li, J.; Yu, F.X.; Guan, K.L.; Post, L. Small Molecule Inhibitors of TEAD Auto-palmitoylation Selectively Inhibit Proliferation and Tumor Growth of NF2-deficient Mesothelioma. Mol Cancer Ther 2021, 20, 986–998. [Google Scholar] [CrossRef]
- Kaneda, A.; Seike, T.; Danjo, T.; Nakajima, T.; Otsubo, N.; Yamaguchi, D.; Tsuji, Y.; Hamaguchi, K.; Yasunaga, M.; Nishiya, Y.; et al. The novel potent TEAD inhibitor, K-975, inhibits YAP1/TAZ-TEAD protein-protein interactions and exerts an anti-tumor effect on malignant pleural mesothelioma. Am J Cancer Res 2020, 10, 4399–4415. [Google Scholar]
- Fan, M.; Lu, W.; Che, J.; Kwiatkowski, N.P.; Gao, Y.; Seo, H.S.; Ficarro, S.B.; Gokhale, P.C.; Liu, Y.; Geffken, E.A.; et al. Covalent disruptor of YAP-TEAD association suppresses defective Hippo signaling. Elife 2022, 11. [Google Scholar] [CrossRef]
- Hagenbeek, T.J.; Zbieg, J.R.; Hafner, M.; Mroue, R.; Lacap, J.A.; Sodir, N.M.; Noland, C.L.; Afghani, S.; Kishore, A.; Bhat, K.P.; et al. An allosteric pan-TEAD inhibitor blocks oncogenic YAP/TAZ signaling and overcomes KRAS G12C inhibitor resistance. Nat Cancer 2023. [CrossRef] [PubMed]
- Reggiani, F.; Gobbi, G.; Ciarrocchi, A.; Ambrosetti, D.C.; Sancisi, V. Multiple roles and context-specific mechanisms underlying YAP and TAZ-mediated resistance to anti-cancer therapy. Biochim Biophys Acta Rev Cancer 2020, 1873, 188341. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.D.K.; Yi, C. YAP/TAZ Signaling and Resistance to Cancer Therapy. Trends Cancer 2019, 5, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Sabnis, A.J.; Chan, E.; Olivas, V.; Cade, L.; Pazarentzos, E.; Asthana, S.; Neel, D.; Yan, J.J.; Lu, X.; et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat Genet 2015, 47, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Honjo, S.; Jin, J.; Chang, S.S.; Scott, A.W.; Chen, Q.; Kalhor, N.; Correa, A.M.; Hofstetter, W.L.; Albarracin, C.T.; et al. The Hippo Coactivator YAP1 Mediates EGFR Overexpression and Confers Chemoresistance in Esophageal Cancer. Clin Cancer Res 2015, 21, 2580–2590. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Zheng, J.; Zhang, J.; Wang, H.; Shao, G.; Teng, L. Knockdown of TAZ modifies triple-negative breast cancer cell sensitivity to EGFR inhibitors by regulating YAP expression. Oncol Rep 2016, 36, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.C.; Stalnecker, C.A.; Jean Morales, A.; Taylor, K.E.; Klomp, J.E.; Klomp, J.A.; Waters, A.M.; Sudhakar, N.; Hallin, J.; Tang, T.T.; et al. TEAD Inhibition Overcomes YAP1/TAZ-Driven Primary and Acquired Resistance to KRASG12C Inhibitors. Cancer Res 2023, 83, 4112–4129. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Zhan, L.; Zhang, Y.; Zhang, J. Yes-activated protein promotes primary resistance of BRAF V600E mutant metastatic colorectal cancer cells to mitogen-activated protein kinase pathway inhibitors. J Gastrointest Oncol 2021, 12, 953–963. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, X.; Fang, L.; Lan, C.; Zheng, X.; Wang, Y.; Zhang, Y.; Han, X.; Liu, S.; Cheng, K.; et al. A combinatorial strategy using YAP and pan-RAF inhibitors for treating KRAS-mutant pancreatic cancer. Cancer Lett 2017, 402, 61–70. [Google Scholar] [CrossRef]
- Mudianto, T.; Campbell, K.M.; Webb, J.; Zolkind, P.; Skidmore, Z.L.; Riley, R.; Barnell, E.K.; Ozgenc, I.; Giri, T.; Dunn, G.P.; et al. Yap1 Mediates Trametinib Resistance in Head and Neck Squamous Cell Carcinomas. Clin Cancer Res 2021, 27, 2326–2339. [Google Scholar] [CrossRef]
- Fisher, M.L.; Grun, D.; Adhikary, G.; Xu, W.; Eckert, R.L. Inhibition of YAP function overcomes BRAF inhibitor resistance in melanoma cancer stem cells. Oncotarget 2017, 8, 110257–110272. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Hu, L.; Tao, Z.; Jarugumilli, G.K.; Erb, H.; Singh, A.; Li, Q.; Cotton, J.L.; Greninger, P.; Egan, R.K.; et al. Pharmacological blockade of TEAD-YAP reveals its therapeutic limitation in cancer cells. Nat Commun 2022, 13, 6744. [Google Scholar] [CrossRef]
- Battilana, G.; Zanconato, F.; Piccolo, S. Mechanisms of YAP/TAZ transcriptional control. Cell Stress 2021, 5, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Xie, S.; Yao, X.; Dey, A. Transcriptional Regulation of the Hippo Pathway: Current Understanding and Insights from Single-Cell Technologies. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Hillmer, R.E.; Link, B.A. The Roles of Hippo Signaling Transducers Yap and Taz in Chromatin Remodeling. Cells 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sheldon, M.; Sun, Y.; Ma, L. New Insights into YAP/TAZ-TEAD-Mediated Gene Regulation and Biological Processes in Cancer. Cancers (Basel) 2023, 15. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, Y.; Su, R.; Jia, Y.; Lai, X.; Su, H.; Fan, Y.; Wang, Y.; Xing, W.; Qin, J. Dimethyl Fumarate Combined With Vemurafenib Enhances Anti-Melanoma Efficacy via Inhibiting the Hippo/YAP, NRF2-ARE, and AKT/mTOR/ERK Pathways in A375 Melanoma Cells. Front Oncol 2022, 12, 794216. [Google Scholar] [CrossRef]
- Hoj, J.P.; Mayro, B.; Pendergast, A.M. A TAZ-AXL-ABL2 Feed-Forward Signaling Axis Promotes Lung Adenocarcinoma Brain Metastasis. Cell Rep 2019, 29, 3421–3434 e3428. [Google Scholar] [CrossRef]
- Liu, H.; Dai, X.; Cao, X.; Yan, H.; Ji, X.; Zhang, H.; Shen, S.; Si, Y.; Zhang, H.; Chen, J.; et al. PRDM4 mediates YAP-induced cell invasion by activating leukocyte-specific integrin beta2 expression. EMBO Rep 2018, 19. [Google Scholar] [CrossRef]
- Gill, M.K.; Christova, T.; Zhang, Y.Y.; Gregorieff, A.; Zhang, L.; Narimatsu, M.; Song, S.; Xiong, S.; Couzens, A.L.; Tong, J.; et al. A feed forward loop enforces YAP/TAZ signaling during tumorigenesis. Nat Commun 2018, 9, 3510. [Google Scholar] [CrossRef]
- Yuan, W.C.; Pepe-Mooney, B.; Galli, G.G.; Dill, M.T.; Huang, H.T.; Hao, M.; Wang, Y.; Liang, H.; Calogero, R.A.; Camargo, F.D. NUAK2 is a critical YAP target in liver cancer. Nat Commun 2018, 9, 4834. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.W.; Zhou, Y.D.; Chen, H.Z.; Luan, X.; Zhang, W.D. Targeting CTGF in Cancer: An Emerging Therapeutic Opportunity. Trends Cancer 2021, 7, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Duncan, A.W.; Ailles, L.; Domen, J.; Scherer, D.C.; Willert, K.; Hintz, L.; Nusse, R.; Weissman, I.L. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature 2003, 423, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, M.; Tan, T.Z.; Syed Sulaiman, N.B.; Lamar, J.M.; Bansal, P.; Cui, J.; Qiao, Y.; Ito, Y. RUNX1 and RUNX3 protect against YAP-mediated EMT, stem-ness and shorter survival outcomes in breast cancer. Oncotarget 2018, 9, 14175–14192. [Google Scholar] [CrossRef] [PubMed]
- Elster, D.; Tollot, M.; Schlegelmilch, K.; Ori, A.; Rosenwald, A.; Sahai, E.; von Eyss, B. TRPS1 shapes YAP/TEAD-dependent transcription in breast cancer cells. Nat Commun 2018, 9, 3115. [Google Scholar] [CrossRef] [PubMed]
- Enzo, E.; Santinon, G.; Pocaterra, A.; Aragona, M.; Bresolin, S.; Forcato, M.; Grifoni, D.; Pession, A.; Zanconato, F.; Guzzo, G.; et al. Aerobic glycolysis tunes YAP/TAZ transcriptional activity. EMBO J 2015, 34, 1349–1370. [Google Scholar] [CrossRef] [PubMed]
- Hiemer, S.E.; Zhang, L.; Kartha, V.K.; Packer, T.S.; Almershed, M.; Noonan, V.; Kukuruzinska, M.; Bais, M.V.; Monti, S.; Varelas, X. A YAP/TAZ-Regulated Molecular Signature Is Associated with Oral Squamous Cell Carcinoma. Mol Cancer Res 2015, 13, 957–968. [Google Scholar] [CrossRef]
- Toth, M.; Wehling, L.; Thiess, L.; Rose, F.; Schmitt, J.; Weiler, S.M.E.; Sticht, C.; De La Torre, C.; Rausch, M.; Albrecht, T.; et al. Co-expression of YAP and TAZ associates with chromosomal instability in human cholangiocarcinoma. BMC Cancer 2021, 21, 1079. [Google Scholar] [CrossRef]
- Gao, Y.; Xu, J.; Li, H.; Hu, Y.; Yu, G. Identification of Metastasis-Associated MicroRNAs in Metastatic Melanoma by miRNA Expression Profile and Experimental Validation. Front Genet 2021, 12, 663110. [Google Scholar] [CrossRef]
- Weiler, S.M.E.; Lutz, T.; Bissinger, M.; Sticht, C.; Knaub, M.; Gretz, N.; Schirmacher, P.; Breuhahn, K. TAZ target gene ITGAV regulates invasion and feeds back positively on YAP and TAZ in liver cancer cells. Cancer Lett 2020, 473, 164–175. [Google Scholar] [CrossRef]
- Mori, M.; Triboulet, R.; Mohseni, M.; Schlegelmilch, K.; Shrestha, K.; Camargo, F.D.; Gregory, R.I. Hippo signaling regulates microprocessor and links cell-density-dependent miRNA biogenesis to cancer. Cell 2014, 156, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Shreberk-Shaked, M.; Dassa, B.; Sinha, S.; Di Agostino, S.; Azuri, I.; Mukherjee, S.; Aylon, Y.; Blandino, G.; Ruppin, E.; Oren, M. A Division of Labor between YAP and TAZ in Non-Small Cell Lung Cancer. Cancer Res 2020, 80, 4145–4157. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.H.; Huang, Z.; Wu, J.; Ding, C.C.; Murphy, S.K.; Chi, J.T. A TAZ-ANGPTL4-NOX2 Axis Regulates Ferroptotic Cell Death and Chemoresistance in Epithelial Ovarian Cancer. Mol Cancer Res 2020, 18, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.H.; Lin, C.C.; Wu, J.; Chao, P.Y.; Chen, K.; Chen, P.H.; Chi, J.T. The Hippo Pathway Effector YAP Promotes Ferroptosis via the E3 Ligase SKP2. Mol Cancer Res 2021, 19, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Coggins, G.E.; Farrel, A.; Rathi, K.S.; Hayes, C.M.; Scolaro, L.; Rokita, J.L.; Maris, J.M. YAP1 Mediates Resistance to MEK1/2 Inhibition in Neuroblastomas with Hyperactivated RAS Signaling. Cancer Res 2019, 79, 6204–6214. [Google Scholar] [CrossRef] [PubMed]
- Maglic, D.; Schlegelmilch, K.; Dost, A.F.; Panero, R.; Dill, M.T.; Calogero, R.A.; Camargo, F.D. YAP-TEAD signaling promotes basal cell carcinoma development via a c-JUN/AP1 axis. EMBO J 2018, 37. [Google Scholar] [CrossRef]
- Kurppa, K.J.; Liu, Y.; To, C.; Zhang, T.; Fan, M.; Vajdi, A.; Knelson, E.H.; Xie, Y.; Lim, K.; Cejas, P.; et al. Treatment-Induced Tumor Dormancy through YAP-Mediated Transcriptional Reprogramming of the Apoptotic Pathway. Cancer cell 2020, 37, 104–122 e112. [Google Scholar] [CrossRef]
- Meinhardt, G.; Haider, S.; Kunihs, V.; Saleh, L.; Pollheimer, J.; Fiala, C.; Hetey, S.; Feher, Z.; Szilagyi, A.; Than, N.G.; et al. Pivotal role of the transcriptional co-activator YAP in trophoblast stemness of the developing human placenta. Proc Natl Acad Sci U S A 2020, 117, 13562–13570. [Google Scholar] [CrossRef]
- Morice, S.; Danieau, G.; Tesfaye, R.; Mullard, M.; Brion, R.; Dupuy, M.; Ory, B.; Brounais-Le Royer, B.; Corre, I.; Redini, F.; et al. Involvement of the TGF-beta Signaling Pathway in the Development of YAP-Driven Osteosarcoma Lung Metastasis. Front Oncol 2021, 11, 765711. [Google Scholar] [CrossRef]
- Lim, S.; Hermance, N.; Mudianto, T.; Mustaly, H.M.; Mauricio, I.P.M.; Vittoria, M.A.; Quinton, R.J.; Howell, B.W.; Cornils, H.; Manning, A.L.; et al. Identification of the kinase STK25 as an upstream activator of LATS signaling. Nat Commun 2019, 10, 1547. [Google Scholar] [CrossRef]
- Shen, H.; Chen, Y.; Wan, Y.; Liu, T.; Wang, J.; Zhang, Y.; Wei, L.; Hu, Q.; Xu, B.; Chernov, M.; et al. Identification of TAZ-Dependent Breast Cancer Vulnerabilities Using a Chemical Genomics Screening Approach. Front Cell Dev Biol 2021, 9, 673374. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



