
Submitted:
28 December 2023
Posted:
03 January 2024
You are already at the latest version
Abstract
East Lake in Wuhan, China, harbors a high number of freshwater fish species of great conservation value, concurrently serving as vital resources for local livelihoods. However, the ecosystem is threatened by an array of anthropogenic activities, thus requiring consistent monitoring of the local fish community to enable more efficacious conservation management. In place of conventional surveying methods, we undertook the first analysis of the fish distribution within East Lake via metabarcoding of environmental DNA (eDNA). The accuracy and efficacy of eDNA metabarcoding relies heavily upon selecting an appropriate primer set for PCR amplification. Given the varying environmental conditions and taxonomic diversity across distinct study systems, it remains a challenge to propose an optimal genetic marker for universal use. Thus, it becomes necessary to select PCR primers suitable for the composition of fish in the East Lake. Here, we evaluated the performance of two primer sets, Mifish-U and Metafish, designed to amplify 12S rRNA barcoding genes in fishes. Our results detected a total of 116 taxonomic units and 51 fish species, with beta diversity analysis indicating significant differences in community structure diversity between the 6 sampling locations encompassing East Lake. While it was difficult to accurately compare the species-level discriminatory power and amplification bias of the two primers, Mifish outperformed Metafish in terms of taxonomic specificity for fish taxa and reproducibility. These findings will assist with primer selection for eDNA-based fish monitoring and biodiversity conservation in the East Lake and other freshwater ecosystems.
Keywords:
Environmental DNA
; Metabarcoding
; Fish Biodiversity
; East Lake
1. Introduction
East Lake, also known as Donghu, is a large, shallow freshwater lake located in Wuhan, China, that previously held the record as the largest urban lake in the country (Ge et al., 2014). The lake has been intensively utilized for fish production, with silver carp (Hypophthalmichthys molitrix) and bighead carp (H. nobilis) introduced as the main species for fish stocking (Liu & Chen, 1994). Yet, akin to other global freshwater and marine habitats, fish biodiversity and population in East Lake face a multitude of anthropogenic challenges, such as habitat disruption, overexploitation, climate change, pollution, infectious diseases, and foreign species invasion (Yao et al., 2022). Located in the heart of the Yangtze River Delta, one of China's most prosperous regions, East Lake previously encountered severe environmental issues. These included lake eutrophication triggered by substantial domestic sewage, as well as oil waste from the catering industry and the discharge of industrial and agricultural pollutants, over recent decades (Gan and Guo, 2004; Du et al., 2011; Yang et al., 2014). Nevertheless, since the early 2000s, the local government of Wuhan has vigorously promoted the sustainable development of the ecological environment. The establishment of the 3,367-hectare East Lake National Wetland Park has curtailed any potential lake-contaminating sewage intake and has concurrently planted contamination-absorbing aquatic vegetation, steadily restoring the previously ecologically compromised habitat. With the ongoing progress in East Lake’s wetland conservation and ecological restoration, it is crucial to frequently and accurately evaluate the local fish community to facilitate effective fish conservation management (Rees et al., 2014; Clements et al., 2018).
Nonetheless, conducting regular monitoring of vast aquatic ecosystems, such as lakes, rivers, and reservoirs, poses significant difficulties. This is primarily due to the labor-intensive nature of fieldwork, which becomes increasingly complex when managing multiple sites and equipment. Additionally, the process can be intrusive to the biological communities being studied. Furthermore, a considerable lack of taxonomic expertise is presently available, which is necessary for accurate identification and assessment (Hopkins and Freckleton, 2002; Hering et al., 2018; Zhang et al., 2020). Furthermore, these approaches are hampered by systematic sampling bias, limitations in morphological identification, and an increased risk of false-negative results, leading to underestimations of species diversity (Yamamoto et al., 2016; Wang et al., 2021; Burian et al., 2023). In contrast, environmental DNA (eDNA)-based approaches offer non-invasive, efficient, and economical alternatives for characterizing marine and freshwater biodiversity (Osathanunkul et Minamoto, 2020).
Environmental DNA (eDNA)-based approaches are emerging as a tool for characterizing marine and freshwater biodiversity that can complement traditional surveys. eDNA denotes a composite of DNA molecules shed into the environment by organisms, primarily via their skin, saliva, and secretions, which are widely distributed in various environmental media, such as water, soil, sediment, and air (Bohmann, et al., 2014). The eDNA metabarcoding refers to a rapidly emerging tool for biomonitoring that involves direct extraction of total DNA from environmental samples, followed by Polymerase Chain Reaction (PCR) amplification using primers designed to amplify a barcoding gene (i.e., COI, 16s, 18s) across a specific taxonomic group, and subsequent identification of target species sequences through sequencing and bioinformatics analyses (Wang et al., 2021). Several research endeavors have corroborated the efficacy of eDNA metabarcoding through High-Throughput Sequencing, demonstrating that it yields equivalent or superior species richness and uncovers biodiversity at a significantly lower cost compared to traditional surveys. Consequently, this technology promises immense potential as a complementary instrument for established monitoring methodologies in the realm of aquatic species ecology and conservation.(Port et al., 2016; Shaw et al. 2016, Valentini et al., 2016).
Given the relatively novel and rapidly evolving nature of the eDNA metabarcoding, numerous aspects of this technology remain to be validated and adapted for specific study systems, including the development of PCR primer pairs for DNA amplification, which is a crucial step in the process(Collins et al., 2019; Polanco et al., 2021). Universal primers target organisms that share close taxonomic relations and therefore possess conserved primer binding sequences, while amplified barcodes should encompass variable sites among distinct species for taxonomic classification (Zhang et al., 2020). Ideally, a well-designed universal PCR primer pair should fulfill the subsequent requirements: (1) exhibit high specificity and coverage for the target taxa (e.g., fishes); (2) ensure even amplification across species without PCR dropouts; and (3) demonstrate high discriminatory power for unambiguous taxonomic assignment (Clarke et al. 2014, Miya et al., 2020; Bylemans et al., 2018; Wilcox et al., 2013). Considering the trace amounts of highly degraded DNA from the study organisms in environmental samples, a small barcode size (usually < 200 bp) is recommended for higher PCR success rates (Freeland, 2017; Zhang et al., 2020). Furthermore, effective biodiversity analyses relies on comprehensive and accurate relevant reference databases to avoid limitations in taxonomic assignment for species without available information (Marques et al., 2020).
Generally, mitochondrial genes serve as standard markers for metabarcoding due to their taxonomic discriminatory power, abundant copies in cells shed by organisms, and slower degradation rates compared to nuclear genes (Jo et al., 2022). Previous eDNA metabarcoding studies focusing on fish in both freshwater and marine environments have targeted on mitochondrial cytochrome B (cytb), cytochrome oxidase subunit I (COI), 12S rRNA, and 16S rRNA genes (Shu et al., 2021). Several most used primer pairs, including 12S-V5 (ca. 106 bp) (Riaz et al., 2011; Kelly et al., 2014 ), MiFish-U (ca. 170 bp) (Miya et al., 2015), and Teleo (ca. 65 bp) (Valentini et al., 2016), target various regions of the 12S rRNA sequence. Nonetheless, although numerous research teams have designed versatile primers for fish community assessments, all of which effectively illustrate regional fish diversity, comparing the efficiency of these metabarcoding primers across various studies and geographical areas remains challenging. Although some research has assessed the effectiveness of multiple universal primer sets, the majority of these assessments rely solely on in silico PCR without subsequent in vitro validation (Valentini et al., 2016), which can lead to overly optimistic outcomes (Polanco et al., 2021). Furthermore, the scarce comparisons involving multiple primers often detect substantial discrepancies in the specificity of amplified taxa and the discrimination power of species, both in silico and in situ (Bylemans et al., 2018; Zhang et al., 2020). For instance, Zhang et al. (2020) demonstrated that among 22 primer sets, the two longest pairs in the 12S region, Ac12S (Evans et al., 2016) and AcMDB07 (Bylemans et al., 2018), exhibited the best performance in terms offish diversity amplified in China's freshwater river ecosystems. However, the efficiency of eDNA metabarcoding and primer pairs can significantly differ across diverse abiotic and biotic conditions of the studied ecosystems, as well as within species assemblages containing distinct lineage compositions or complexities (Abell et al., 2008; Bellemain et al., 2010; Clarke et al., 2014). Consequently, it is difficult to propose an optimal genetic marker or the most appropriate suite of primers for universal use, and for this reason, it remains necessary to select PCR primers for the composition of fish in the specific ecosystem to ensure an effective and accurate assessment of the community of interest (García-Machado et al. 2023).
As of now, the fish community of East Lake has yet to be analyzed via the technology of eDNA metabarcoding. It is therefore necessary to determine the suitable primers for the fish species in the local ecosystem. This study compares the performances of Mifish Universal Teleost Primers, the most frequently used primer pair so far (Xiong et al., 2022), and Metafish, a new 12S metabarcoding primer set designed by Nanjing University based on the mitochondrial genome of common Chinese fish found in the middle and lower reaches of the Yangtze River (Yang & Zhang, 2019). The outcomes of this study will contribute to a better understanding of primer selection for future eDNA-based fish community surveys within the East Lake and other related freshwater systems.
2. Methods
2.1. Sample collection
Water sampling was carried out in East Lake (30°32’ N, 114°23’ E) in June 2023. Sampling was conducted at 6 sites: Lingbo Gate (LBM), Liyuan (LY), Baima Road (BMXD), Luoyan Scenic Spot (LYJQ), Donghu Yangguang (DHYG), Ma’anshan Forest Park (SLGY). The location details of these samples are mapped out in Figure 1. At each site, 3 water samples, except for BMXD were collected from the surface using a Tri-Mode eDNA Sampler, an equipment developed by the Institute of Hydrobiology that includes a filter head extended for filtration. This device automatically filters water samples until it reaches the maximum loading capacity of the filter membrane, at which point the machine stops filtering automatically. Because BMXD is located within the river bay and is relatively close to the highway interchange, its surrounding environment is complex. In order to minimize the sampling randomness-associated errors, we collected 7 samples at this location. The mean value was used at all sampling points in the subsequent data analysis. The filtered membrane samples were stored on ice and transported to the laboratory at the Institute of Hydrobiology for DNA extraction within 12 h.
2.2. Metabarcoding of eDNA Samples
The analysis of our samples was conducted using two universal primer pairs, Mifish and Metafish, to amplify the V5 region of the mitochondrial 12S rRNA gene. eDNA metabarcoding employing the universal MiFish primer pairs has been demonstrated to generate short fragments of fish DNA from various taxa in environmental samples (Miya et al., 2020). Metafish was created by Yang et al. (2019), and it has been suggested to be included in group standard of China society of environmental sciences. DNA extraction from sample filters was performed using the DNeasy Blood and Tissue Kit (Thermo Scientific, Waltham, MA, USA) according to the method described in Zhang et al., 2022. PCRs were performed on eDNA extracts and negative controls (including filtration, extraction, and no-template PCR blanks), each utilizing uniquely tagged primers to facilitate the identification of individual PCR amplicons during the analysis of sequencing data (Zhang et al., 2020). The multiplex polymerase chain reaction (PCR) volume was 50 µL, including 20 µL of sterile distilled H2O, 25 µL of Taq 2× Master Mix (Vazyme, Nanjing, China), 1 µL of each primer (Mifish-U-F: 5′- GTCGGTAAAWCTCGTGCCAGC -3′; Mifish-U-R: 5′- CATAGTGGGGTATCTAATCCYAGTTTG -3′; Metafish-F: 5′- TCGTGCCAGCCACCGCGGTTA -3′; and Metafish-R: 5′- ATAGTGGGGTATCTAATCCCAG -3′) (Figure 2), and 1 µL of DNA solution. The thermal cycling PCR process consisted of an initial denaturation step of 2 minutes at 94 °C, followed by 30 cycles of denaturation at 98 °C for 5 seconds each. This was followed by an annealing step at 50 °C for 10 seconds, an extension step at 72 °C for 10 seconds, and a final extension at 72 °C for 5 minutes. Upon completion of the PCR, equal amounts of 1× loading buffer (containing SYBR green) and PCR products were mixed and loaded onto 1% agarose gels for electrophoresis. Samples with a bright main strip of 297 ± 25 bp were selected. The mixed PCR products were then purified using the Qiaquick Gel Extraction Kit (Thermo Scientific, Waltham, MA, USA). Library construction and sequencing were carried out by the Beijing Genomics Institute sequencing service in Wuhan, China, using 2 × 150 bp paired-end sequencing on a HiSeq 2500 System (Illumina Inc., San Diego, CA, USA).
2.3. Bioinformatics and statistical analyses
The quality assessment was carried out on paired-end reads in FASTQ format. To analyze the original double C-terminal sequencing data, a sliding window method was employed with a window size of 10 bp. The analysis revealed that the data started to shift at 1 bp from the 5′ end of the first base position. A quality score of 20 (Q20) was required to achieve 99% accuracy in the FASTQ data. The first value was below average quality due to a truncated sequence, which ended at 150 bp. No ambiguous bases (Ns) were allowed.
The sequence analysis was conducted using QIIME2 (Bolyen et al., 2019). Qualified raw sequences were combined and sorted based on index and barcode information, removing barcode sequences in the process. Subsequently, sequences underwent quality control, denoising, combination, and chimera removal using DADA2 (Callahan et al., 2017). Deduplicated sequences generated by DADA2 quality assurance were considered ASVs (amplicon sequence variants) (Katoh et al., 2002). ASVs are equivalent to OTUs with 100% similarity clustering (McDonald et al., 2012). ASVs with fewer than 20 reads were filtered out. Taxonomy was assigned using databases downloaded from NCBI (https://www.ncbi.nlm.nih.gov/ (28 February 2021)) and MitoFish (http://mitofish.aori.u-tokyo.ac.jp (02 August 2022)). The classification of ASVs was carried out using QIIME 2's q2-feature-classifier plugin, setting the sequence similarity threshold at 99% (Bokulich et al., 2018). The taxonomy dataset for each sample was utilized to calculate the observed species and Bray-Curtis indices, which were then employed to create non-metric multidimensional scaling (NMDS) plots using the vegan 2.3_5 package in the statistical software R (Oksanen et al., 2016). Data points were visualized in relation to the used primers and sampling locations. Functional regressions of the data points against each NMDS dimension were performed in Matlab to assess the significance of the observed patterns (Ricker, 1973). The Bray-Curtis distance ranges from 0 to 1, with a value of 0 indicating identical community compositions and a value of 1 indicating that the communities have no shared taxa. The correlation between fish communities and sample properties (sampling locations and amplification primers) was calculated using Mantel test (Goslee and Urban 2007). The significance of the difference between two sets of data was assessed using T-test
3. Results
3.1. Species Composition and Diversity
Figure 489. total sequence reads were retained for each library. A total of 72 taxonomic units was identified from 6 sampling sites (Table 1 and Table S1), with no difference in the total number of detected species between the two sets of primers for the same location (Figure S1). Detected taxa encompassed 51 fish species, 36 genera, and 16 families, with high percentages of Xenocyprididae, Cyprinidae, Oxudercidae, Gobionidae, Channidae, and Poeciliidae. An analysis by taxonomic order revealed that Cypriniformes consistently accounted for the largest portions of fish taxa for both primers (30.4-91.1%), and Gobiiformes was overall the second most abundant order. Other frequently detected orders included Anabantiformes, Cyprinodontiformes, and Perciformes. The 10 most common species detected in East Lake were Carassius auratus, H. nobilis, Rhinogobius similis, Cyprinus carpio, Hemiculter leucisculus, Chanodichthys dabryi, Pseudorasbora parva, Mugilogobius myxodermus, Channa argus, Culter alburnus. Certain invasive species, like Gambusia affinis, were also detected. The dominant species in the basin, alongside smaller-sized fish like Rhinogobius cliffordpopei, also comprise of economically valuable fish, such as C. carpio, H. nobilis, and C. auratus (Figure 3).
3.2. Community Diversity
The NMDS plots revealed that fish communities at the different sampling sites have different fish compositions and that communities revealed by both markers in most locations are more similar between them than between sampling sites (Figure 4). A regression of the Bray-Curtis data points against the NMDS axes revealed a significant relationship with dimension 1 (R2 = 0.563, p < 0.001) but less so with dimension 2 (R2 = 0.210, p = 0.028). To evaluate the impact of sampling location and amplification primers on the fish community, the Mantel test (i.e., Bray–Curtis distance) was performed. The results showed that the dissimilarity of fish communities was strongly correlated with sampling locations (r=0.653, p < 0.001) and amplification primers (r=0.167, p < 0.05). This result indicated that both sampling locations and amplification primers had significant influence on fish communities, with the former exhibiting a stronger correlation with community structure. The Bray-Curtis dissimilarity among samples from the same location (all six locations) was lower when amplified with Mifish primers than that amplified with Metafish primers, with three locations (BMXD, LBM, and SLGY) showing significant difference with p-value <0.05 (Figure 5).
3.3. Species Distribution by Primers
There were distinctions in taxonomic identity and relative abundance of taxa between primer pairs. Notably, for samples collected at LBM and LY, Metafish detected a considerably higher percentage of unclassified non-fish taxa (52.7% and 53.5%, respectively) compared to Mifish primers (5.44% and 4.38%, respectively). Several dominant fish species were detected with differing relative read proportions between the two primer sets, including C. auratus (17.5% for Mifish and 7.28% for Metafish in LY samples); H. nobilis (22.1% for Mifish and 8.13% for Metafish in LBM samples); R. similis (13.1% for Mifish and 4.68% for Metafish in LBM samples); C. dabryi (19.2% for Mifish and 8.69% for Metafish in LBM samples); and H. molitrix (0.04% for Mifish and 6.26% for Metafish in LYJQ samples). Some fish species were also detected by only one primer set at a low relative frequency (<1%) but were absent entirely from the other primer set in certain sampling sites, such as C. dabryi (detected only by Metafish for SLGY); G. affinis (detected only by Metafish for LBM); C. alburnus (detected only by Metafish for SLGY and by Mifish for LY and LYJQ); and Acheilognathus rhombeus (detected only by Mifish for LBM and BMXD and by Metafish for SLGY). In addition, certain fish species were exclusively detected by only one of the primer sets across all sampling sites. Fish species observed only by Mifish include Saurogobio xiangjiangensis (native), Saurogobio lissilabris (native), Paramisgurnus dabryanus (native), Misgurnus bipartitus (native), Pagellus bellottii (invasive), Coptodo zillii (invasive), and Gambusia holbrooki (invasive). On the other hand, fish species that were detected only by Metafish include Mystacoleucus marginatus (native), Misgurnus anguillicaudatus (native), Hyporhamphus intermedius (native), Hemibarbus barbus (native), Carassius carassius (native), and Acheilognathus tonkinensis (native) (Table 2). In total, Mifish and Metafish detected similar numbers of total fish species (45 vs. 44) (Table 1).
3.4. Species Distribution by Location
Scheme 36. 6-43.5% of relative read frequency for BMXD but less than 8% for LBM, SLGY, and LYJQ; R. similis represented 45% of detected fish for SLGY but less than 0.2% for BMXD; H. nobilis contributed to 38.7-48.2% for relative frequency for LYJQ but less than 5% for SLGY and LY. Moreover, fish species of the Oxudercidae family accounted for significant percentages for SLGY (54.9%) but less than 1.5% in BMXD; Conversely, fish species of the Cyprinidae family were found in higher relative frequency (52.3-60.1%) for BMXD but less than 7% for SLGY (Table 2). Overall, the highest number of total fish taxa detected by Metafish and Mifish was from samples collected at DHYG (43 and 46, respectively), while the fewest taxa units were found at SLGY and BMXD (32 and 28; 28 and 32, respectively) (Figure S1).
4. Discussion
In this research, we explored the possibility of utilizing eDNA-based monitoring methods to assess freshwater fish communities in East Lake. Repeated monitoring is essential for safeguarding these populations and addressing the anthropogenic pressures these habitats are subjected to. We employed two 12S primer sets, which have been proven to outperform other gene region-targeting assays in evaluating fish communities (Kelly et al., 2014; Miya et al., 2015; Polanco et al., 2021; Zhang et al, 2020). Although the two primer sets produced similar results in some aspects (such as taxonomic coverage and community characterization), the Mifish primer demonstrated superior taxonomic specificity and reproducibility compared to the Metafish primer.
High taxonomic specificity for the target taxonomic group is a crucial consideration when choosing metabarcoding primers. Insufficient specificity for the target group can lead to the excessive amplification of non-target sequences, causing the desired taxa to be overwhelmed and resulting in inefficient utilization of sequencing resources. Our research demonstrates that both primers amplified sequences of non-fish organisms at all sampling sites. However, for LBM and LY sampling sites, Metafish primers detected significantly higher relative proportions of unclassified taxa (52.7% and 53.5%, respectively) compared to Mifish primers (5.4% and 4.4%). This discrepancy suggests that the Mifish primer offers a higher degree of specificity for fish taxa, making it a more suitable choice for fish community assessments.
The high read abundances of unclassified taxa from water samples collected at LBM and LY could potentially be attributed to the increased level of human activity present at these sites: LBM is situated at Wuhan University, while LY encompasses the Liyuan Hospital. As a result, the extracted eDNA cannot avoid being contaminated with various human DNA-containing waste products. The amplification of human sequences using metabarcoding primers from environmental samples has been previously reported (Kelly et al., 2014; Miya et al. 2015). Efforts have been made to reduce human DNA amplification by employing general fish primers in combination with a blocking oligonucleotide. However, Zhang et al. (2020) demonstrated that blocking oligos might also prevent primers from binding to or amplifying certain desired fish sequences. Therefore, they suggested increasing sequencing depth as a preferred alternative to compensate for non-target amplification.
In addition to having a high taxonomic specificity for the target species, it is essential to have broad taxonomic coverage within the target group and a high ability to assign species levels for generating comprehensive and accurate biodiversity data using metabarcoding primers alone detected 4 native and 3 invasive fish species, while the Metafish primers uniquely detected 6 native fish species. Thus, both primers showed similar performance in our study system regarding taxonomic coverage and species-level assignment (45 for Mifish and 44 for Metafish). This consistency is probably due to the comparable barcode size and significant overlap in the barcode sequence of the two primers. As shown in Figure 2, Metafish is essentially a modified version of Mifish, with the reverse barcode sequence of the Metafish primer entirely encompassed by that of the Mifish primer set.
Regarding the amplification bias of the two primers, several dominant fish species displayed substantial variation in relative frequency of taxa between assays amplified by these primers. Prior studies have suggested a positive correlation between taxa relative abundance or biomass and taxa sequence counts (Evans et al., 2016; Klobucar et al., 2017; Ushio et al., 2018), which supports the potential of using metabarcoding sequencing data for multispecies quantitative estimations. However, Lu et al. (2021) found that not all species displayed such positive relationships in species-specific data analysis. Sequence read counts cannot be relied upon as an accurate measure of fish abundance or biomass, as they are prone to biases originating from multiple factors such as sampling methods, laboratory procedures, and analytical stages (including amplification, sequencing, and bioinformatics), apart from the source and fate of species eDNA in various environments (Shaw et al., 2016; Ushio et al., 2018; Evans & Lamberti, 2018; Lacoursiere-Roussel & Deiner, 2019). Moreover, the presence of unclassified taxa, particularly at high percentages in samples collected at LBM and LY, complicates the comparison of Mifish and Metafish primer performance in terms of even amplification across species. Given that most of the differences in relative abundance of dominant fish taxa were identified among the LBM and LY samples, it is even more difficult to ascertain which designed universal PCR primer has unbiasedly amplified 12S gene fragments across the target taxa without PCR dropouts. Consequently, it becomes challenging to accurately comparing the species distribution across sampling locations, particularly for fish species detected with very low relative frequency by both primers. Ushio et al. (2018) suggested incorporating an internal standard DNA (i.e., a known copy number of short DNA fragments from non-target species) into eDNA samples to detect potential biases within metabarcoding data for quantitative fish eDNA analysis. This method was effectively employed by Stoeckle et al. (2022), who discovered that Riaz 12S gene metabarcoding with an internal DNA standard can quantify marine bony fish eDNA across a range of approximately 10-5000 copies per reaction, indicating no significant PCR bias among teleost species.
Nevertheless, analysis by proportional reads-based Bray−Curtis indices indicated that eDNA amplified by both primer sets generated different profiles of fish diversity for samples collected from different locations. Although both primer choice for amplification and sampling location appeared to contribute to the significant relationship observed between the regression of Bray-Curtis data points and the NMDS axes, the Mantel test revealed that the latter had a stronger correlation with the dissimilarity of fish communities. More importantly, while it was difficult to determine which diversity profile detected by the two primers represented that of East Lake most accurately, the Bray-Curtis dissimilarity among samples from the same location (all six sites) was lower when amplified with Mifish primers compared to Metafish primers. This suggests that Mifish primers possess greater reproducibility than Metafish. Though, it is worth noting that unlike observed species, which is based on presence/absence of taxa, Bray-Curtis indices take into account read abundances, a factor influenced by PCR biases (Kelly et al., 2018) and eDNA dynamics (Allan et al., 2020).
In the future, to select a suitable primer set for eDNA-based monitoring in the East Lake, it is necessary to gain a more comprehensive and comparative evaluation of a wider selection of metabarcoding primers. Since different primers may have varying taxonomic ranges in amplification, employing multiple primer sets in combination can enhance taxonomic coverage and species discriminatory power (Miya et al., 2015). For instance, as proposed by Zhang et al. (2020), besides primers aimed at Actinopterygii species, those effective in amplifying Chondrichthyes species from environmental samples should also be included. Moreover, the success of metabarcoding applications in recovering biodiversity and assigning accurate taxonomy depends on the comprehensiveness and sequence quality of corresponding reference databases. Therefore, constructing high-quality reference databases for local biological communities should be a priority in DNA-based biodiversity monitoring. The combined use of both local and global databases can increase the detection probability of native, invasive, and rare species.
5. Conclusions
In this study, we examined the primer performance of Mifish-U and Metafish in assessing fish eDNA composition of samples collected from 6 different locations across East Lake. While it was difficult to compare the species-level discriminatory power and even amplification of the two primers, our results revealed Mifish outperformed Metafish in terms of taxonomic specificity and reproducibility. These findings will contribute to the usage of eDNA technology in future fish biodiversity assessments, particularly for other lakes in the Yangtze basin. However, it is crucial to remember that the community composition and complexity can vary significantly between geographical locations and different ecosystems, which may imply that the primer performance in this study might not be entirely applicable to other situations.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Barplots showing the number of total taxonomic units detected by both primers at the 6 sampling sites. Darker blue bars represent Metafish, and lighter blue bars represent Mifish.
Data Availability Statement
All the data required to assess the findings of the paper are provided in the main text and/or the Supplementary Materials. The raw sequencing data associated with this study can be accessed on the NCBI's SRA database under BioProject ID: PRJNA957488 (www.ncbi.nlm.nih.gov/bioproject/957488).
Acknowledgments
The experiments were conducted in compliance with the Ethics Committee of the Institute of Hydrobiology at the Chinese Academy of Sciences (CAS). The policies were implemented in accordance with the Chinese Association for Laboratory Animal Sciences and the Institutional Animal Care and Use Committee (IACUC) guidelines. This study was funded by a grant from the National Natural Science Foundation of China (32200367) and the National Key Research and Development Program of China (2022YFF0608200).
References
- Allan, E.A. , Zhang, W.G., Lavery, A.C., et al., 2020. Environmental DNA shedding and decay rates from diverse animal forms and thermal regimes. Environmental DNA. [CrossRef]
- Bellemain, E. , Carlsen, T., Brochmann, C., et al., 2010. ITS as an environmental DNA barcode for fungi: an in silico approach reveals potential PCR biases. BMC Microbiology. [CrossRef]
- Bohmann, K. , Evans, A., Gilbert, M.T.P., et al., 2014. Environmental DNA for wildlife biology and biodiversity monitoring. Trends in Ecology & Evolution. [CrossRef]
- Bokulich, N.A. , Kaehler, B.D., Rideout, J.R., et al., 2018. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome. [CrossRef]
- Bolyen, E. , Rideout, J.R., Dillon, M.R., et al., 2019. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Natural Biotechnology. [CrossRef]
- Bylemans, J. , Gleeson, D.M., Hardy, C.M., et al., 2018. Toward an ecoregion scale evaluation of eDNA metabarcoding primers: A case study for the freshwater fish biodiversity of the Murray-Darling Basin (Australia). Ecology and Evolution, 8697. [Google Scholar] [CrossRef]
- Burian, A. , Bruce, K., Tovela E., et al., 2023. Merging two eDNA metabarcoding approaches and citizen-science-based sampling to facilitate fish community monitoring along vast Sub-Saharan coastlines. Molecular Ecology Resources. [CrossRef]
- Callahan, B.J. , McMurdie, P.J., Holmes, S.P., 2017. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME Journal, 2643. [Google Scholar] [CrossRef]
- Clarke, L.J. , Soubrier, J., Weyrich, L.S., et al., 2014. Environmental metabarcodes for insects: in silico PCR reveals potential for taxonomic bias. Molecular Ecology Resources, 1170. [Google Scholar] [CrossRef]
- Clements, C.F. , Blanchard, J.L., Nash, K.L., et al., 2017. Body size shifts and early warning signals precede the historic collapse of whale stocks. Nature Ecology & Evolution. [CrossRef]
- Collins, R.A. , Bakker, J., Wangensteen, O.S., et al., 2019. Non-specific amplification compromises environmental DNA metabarcoding with COI. Methods in Ecology and Evolution, 1985. [Google Scholar] [CrossRef]
- Du, Y. , Xue, H.P., Wu, S.J., et al., 2011. Lake area changes in the middle Yangtze region of China over the 20th century. Journal of Environmental Management, 1248. [Google Scholar] [CrossRef]
- Evans, N.T. , Olds, B.P., Renshaw, M.A., et al., 2016. Quantification of mesocosm fish and amphibian species diversity via environmental DNA metabarcoding. Molecular Ecology Resources. [CrossRef]
- Evans, N.T. , Lamberti, G.A., 2018. Freshwater fisheries assessment using environmental DNA: A primer on the method, its potential, and shortcomings as a conservation tool. Fisheries Research. [CrossRef]
- Freeland, J.R. , 2017. The importance of molecular markers and primer design when characterizing biodiversity from environmental DNA. Genome. [CrossRef]
- Gan, Y. , Guo, Y., 2004. Evaluation analysis and remedy strategy for eutrophication in Wuhan lake Donghu. Resources and Environment in the Yangtze.
- García-Machado, E. , Normandeau, E., Côté, G., Bernatchez, L., 2023. How eDNA data filtration, sequence coverage, and primer selection influence assessment of fish communities in northern temperate lakes. Environmental DNA. [CrossRef]
- Ge, J. , Liu, M., Yun, X., et al., 2014. Occurrence, distribution and seasonal variations of polychlorinated biphenyls and polybrominated diphenyl ethers in surface waters of the East Lake, China. Chemosphere. [CrossRef]
- Goslee, S.C. , Urban, D.L., 2007. The ecodist package for dissimilarity-based analysis of ecological data. Journal of Statistical Software, 10.18637/jss.v022.i07.
- Hering, D. , Borja, A., Jones, J.I., et al., 2018. Implementation options for DNA-based identification into ecological status assessment under the European Water Framework Directive. Water Resources. [CrossRef]
- Hopkins, G. , Freckleton, R., 2002. Declines in the numbers of amateur and professional taxonomists: Implications for conservation. Animal Conservation 5 (3), 245–249. [CrossRef]
- Jo, T.S. , Tsuri, K., Yamanaka, H., 2022, Can nuclear aquatic environmental DNA be a genetic marker for the accurate estimation of a species abundance? The Science of Nature. [CrossRef]
- Lacoursiere-Roussel, A. , Deiner, K., 2019. Environmental DNA is not the tool by itself. Journal of Fish Biology. [CrossRef]
- Liu, J. , Chen, S., 1994. An assessment of the impact of fish stocking on lake eutrophication in China. SIL Communications. [CrossRef]
- Katoh, K. , Misawa, K., Kuma, K., et al., 2002. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Research, 3066. [Google Scholar] [CrossRef]
- Kelly, R.P. , Port, J.A., Yamahara, K.M., et al., 2014. Using environmental DNA to census marine fishes in a large mesocosm. PLoS One, 8617. [Google Scholar] [CrossRef]
- Kelly, R.P. , Gallego, R., Jacobs-Palmer, E., 2018. The effect of tides on nearshore environmental DNA. PeerJ. [CrossRef]
- Klobucar, S.L. , Rodgers, T.W., Budy, P., 2017. At the forefront: Evidence of the applicability of using environmental DNA to quantify the abundance of fish populations in natural lentic waters with additional sampling considerations. Canadian Journal of Fisheries and Aquatic Sciences, 2030. [Google Scholar] [CrossRef]
- Marques, V. , Guérin, P.-É., Rocle, M., et al., 2020. Blind assessment of vertebrate taxonomic diversity across spatial scales by clustering environmental DNA metabarcoding sequences. Ecography, 1779. [Google Scholar] [CrossRef]
- McDonald, D. , Price, M.N., Goodrich, J., et al., 2012. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME Journal. [CrossRef]
- Miya, M. , Gotoh, R.O., Sado, T., 2020. Mifish metabarcoding: A high-throughput approach for simultaneous detection of multiple fish species from environmental DNA and other samples. Fisheries Science. [CrossRef]
- Oksanen, J. , Blanchet, F.G., Friendly, M., et al., 2016. vegan: Community Ecology Package. R package version 2.4-3. Vienna: RFoundation for Statistical Computing.
- Osathanunkul, M. , Minamoto, T., 2020. A molecular survey based on eDNA to assess the presence of a clown featherback (Chitala ornata) in a confined environment. PeerJ, 0338. [Google Scholar] [CrossRef]
- Polanco, A. , Richards, E., Flück, B., et al., 2006. Comparing the performance of 12S mitochondrial primers for fish environmental DNA across ecosystems. Environmental DNA, 1113. [Google Scholar] [CrossRef]
- Port, J.A. , O’Donnell, J.L., Romero-Maraccini, O.C., et al., 2016. Assessing vertebrate biodiversity in a kelp forest ecosystem using environmental DNA. Molecular Ecology. [CrossRef]
- Shaw, J.L.A. , Clarke, L.J., Wedderburn, S.D., et al., 2016. Comparison of environmental DNA metabarcoding and conventional fish survey methods in a river system. Biological Conservation. [CrossRef]
- Shu, L. , Ludwig, A., Peng, Z., 2021. Environmental DNA metabarcoding primers for freshwater fish detection and quantification: In silico and in tanks. Ecological Evolution, 8281. [Google Scholar] [CrossRef]
- Stoeckle, M.Y. , Ausubel, J.H., Coogan, M., 2022. 12S gene metabarcoding with DNA standard quantifies marine bony fish environmental DNA, identifies threshold for reproducible detection, and overcomes distortion due to amplification of non-fish DNA. Environmental DNA. [CrossRef]
- Rees, H.C. , Maddison, B.C., Middleditch, D.J., et al., 2014. The detection of aquatic animal species using environmental DNA – A review of eDNA as a survey tool in ecology. Journal of Applied Ecology, 1450. [Google Scholar] [CrossRef]
- Riaz, T. , Shehzad, W., Viari, A., et al., 2011. ecoPrimers: inference of new DNA barcode markers from whole genome sequence analysis. Nucleic Acids Research. [CrossRef]
- Ricker, W.E. , 1973. Linear Regressions in Fishery Research. Journal of the Fisheries Research Board of Canada. [CrossRef]
- Ushio, M. , Murakami, H., Masuda, R., et al., 2018. Quantitative monitoring of multispecies fish environmental DNA using high-throughput sequencing. Metabarcoding and Metagenomics. [CrossRef]
- Valentini, A. , Taberlet, P., Miaud, C., et al., 2016. Next-generation monitoring of aquatic biodiversity using environmental DNA metabarcoding. Molecular Ecology 25. [CrossRef]
- Wang, S. , Yan, Z., Hänfling, B., et al., 2021. Methodology of fish eDNA and its applications in ecology and environment. Science of the Total Environment 755. [CrossRef]
- Wilcox, T.M. , McKelvey, K.S., Young, M.K., et al., 2013. Robust detection of rare species using environmental DNA: The importance of primer specificity. PLoS One, 5952. [Google Scholar] [CrossRef]
- Xiong, F. , Shu, L., Zeng, H., et al., 2022. Methodology for fish biodiversity monitoring with environmental DNA metabarcoding: the primers, databases and bioinformatic pipelines. Water Biology and Security, 1000. [Google Scholar] [CrossRef]
- Yamamoto, S. , Minami, K., Fukaya, K., et al., 2016. Environmental DNA as a ‘snapshot’ of fish distribution: A case study of Japanese Jack Mackerel in Maizuru Bay, sea of Japan. PLoS One, 0149. [Google Scholar] [CrossRef]
- Yang, J. , Zhang, X.W., 2019. Universal metabarcoding amplification primers for freshwater fish mitochondria 12S and application method thereof. China, CN109943645B, 04 2023. [Google Scholar]
- Yang, J. , Zhang, L., Mu, Y. et al., 2023. Small changes make big progress: A more efficient eDNA monitoring method for freshwater fish. Environmental DNA. [CrossRef]
- Yang, Y. , Yun, X., Liu, M., et al., 2014. Concentrations, distributions, sources, and risk assessment of organochlorine pesticides in surface water of the East Lake, China. Environmental Science and Pollution Research 21, 3050. [Google Scholar] [CrossRef]
- Yao, M. , Zhang, S., Lu, Q., et al., 2022. Fishing for Fish Environmental DNA: Ecological Applications, Methodological Considerations, Surveying Designs, and Ways Forward. Molecular Ecology, 5164. [Google Scholar] [CrossRef]
- Zhang, S. , Zheng, Y.T., Zhan, A.B., et al., 2022. Environmental DNA captures native and non-native fish community variations across the lentic and lotic systems of a megacity. Science Advances. [CrossRef]
- Zhang, S. , Zhao, J., Yao, M. 2020. A comprehensive and comparative evaluation of primers for metabarcoding eDNA from fish. Methods in Ecology and Evolution, 1609. [Google Scholar] [CrossRef]
Figure 1.
Map of East Lake showing the sampling sites (N=6), including Lingbo Gate (LBM), Liyuan (LY), Baima Road (BMXD), Luoyan Scenic Spot (LYJQ), Donghu Yangguang (DHYG), Ma’anshan Forest Park (SLGY).
Figure 1.
Map of East Lake showing the sampling sites (N=6), including Lingbo Gate (LBM), Liyuan (LY), Baima Road (BMXD), Luoyan Scenic Spot (LYJQ), Donghu Yangguang (DHYG), Ma’anshan Forest Park (SLGY).
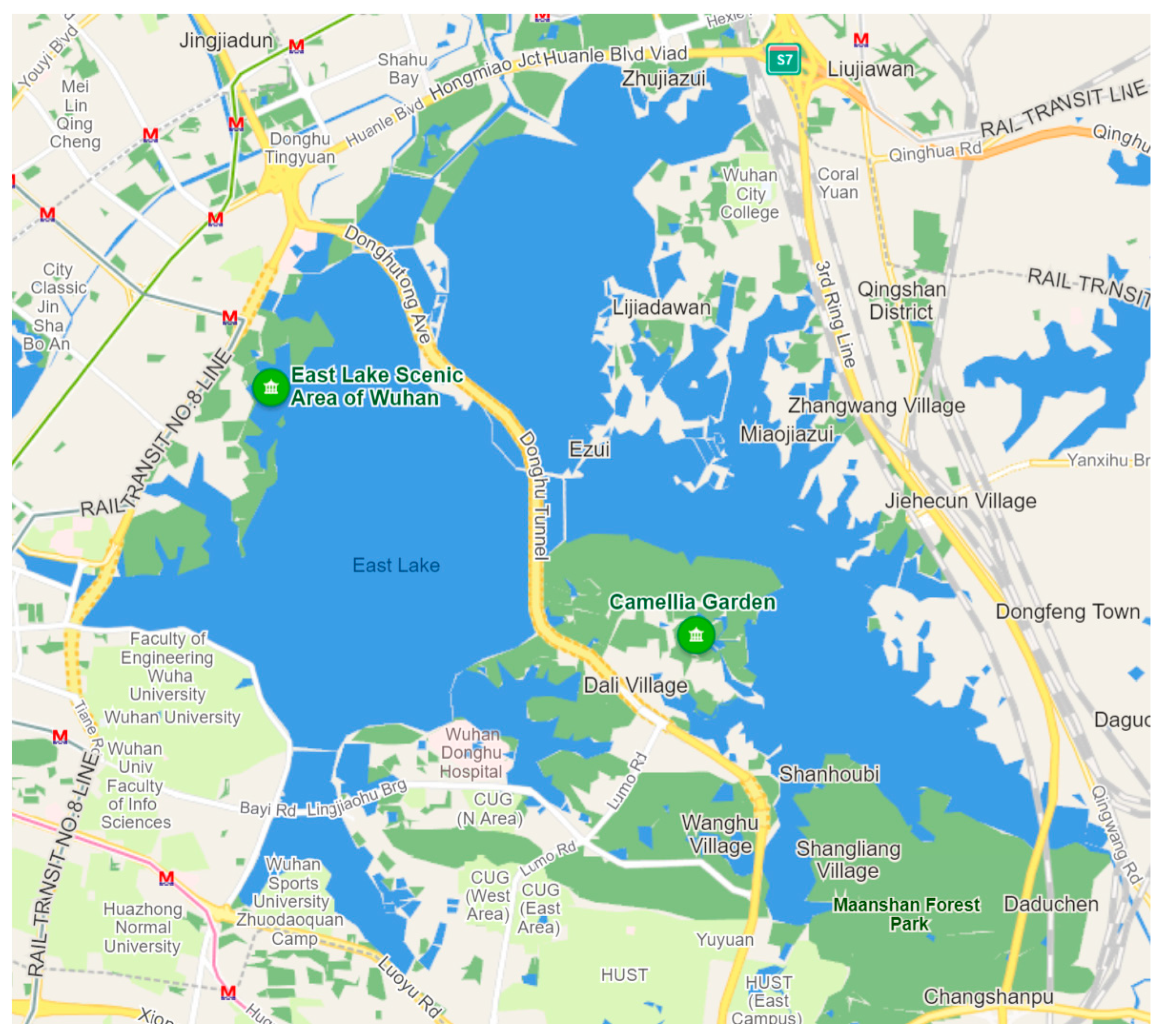
Figure 2.
Locations of the 2 fish metabarcoding primer pairs and amplicons on the 12S rRNA mitochondrial gene. Gene sequences of the grass carp (Ctenopharyngodon Idella; GenBank Acc. No. MG827396.1) were used as templates. Note amplicon sizes of the primer sets may vary depending on the fish species.
Figure 2.
Locations of the 2 fish metabarcoding primer pairs and amplicons on the 12S rRNA mitochondrial gene. Gene sequences of the grass carp (Ctenopharyngodon Idella; GenBank Acc. No. MG827396.1) were used as templates. Note amplicon sizes of the primer sets may vary depending on the fish species.
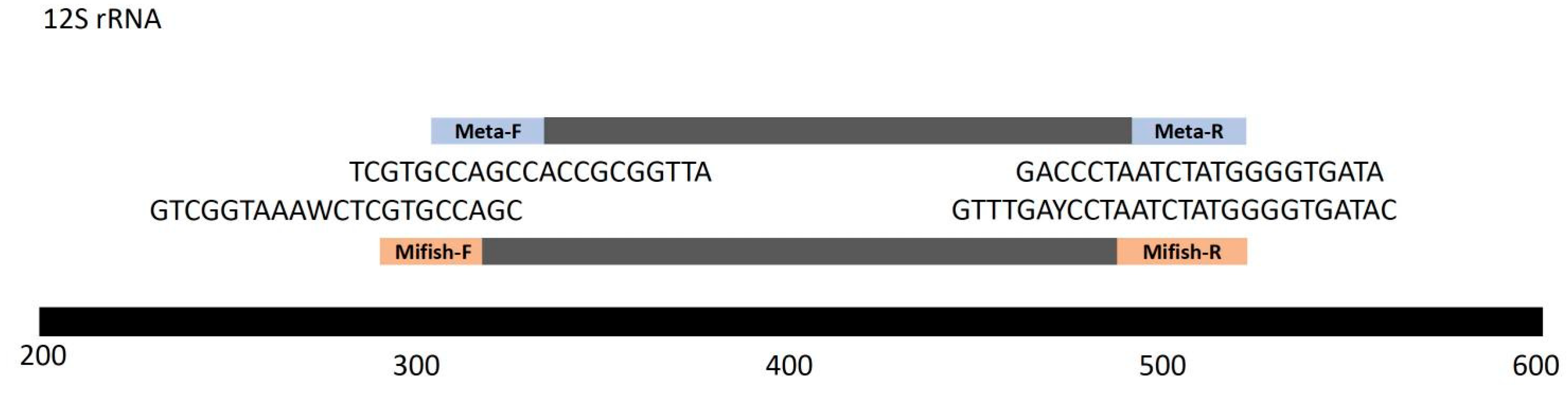
Figure 3.
Barplots showing average taxonomic distributions of amplified sequences for the six different locations and two metabarcoding primer sets.
Figure 3.
Barplots showing average taxonomic distributions of amplified sequences for the six different locations and two metabarcoding primer sets.
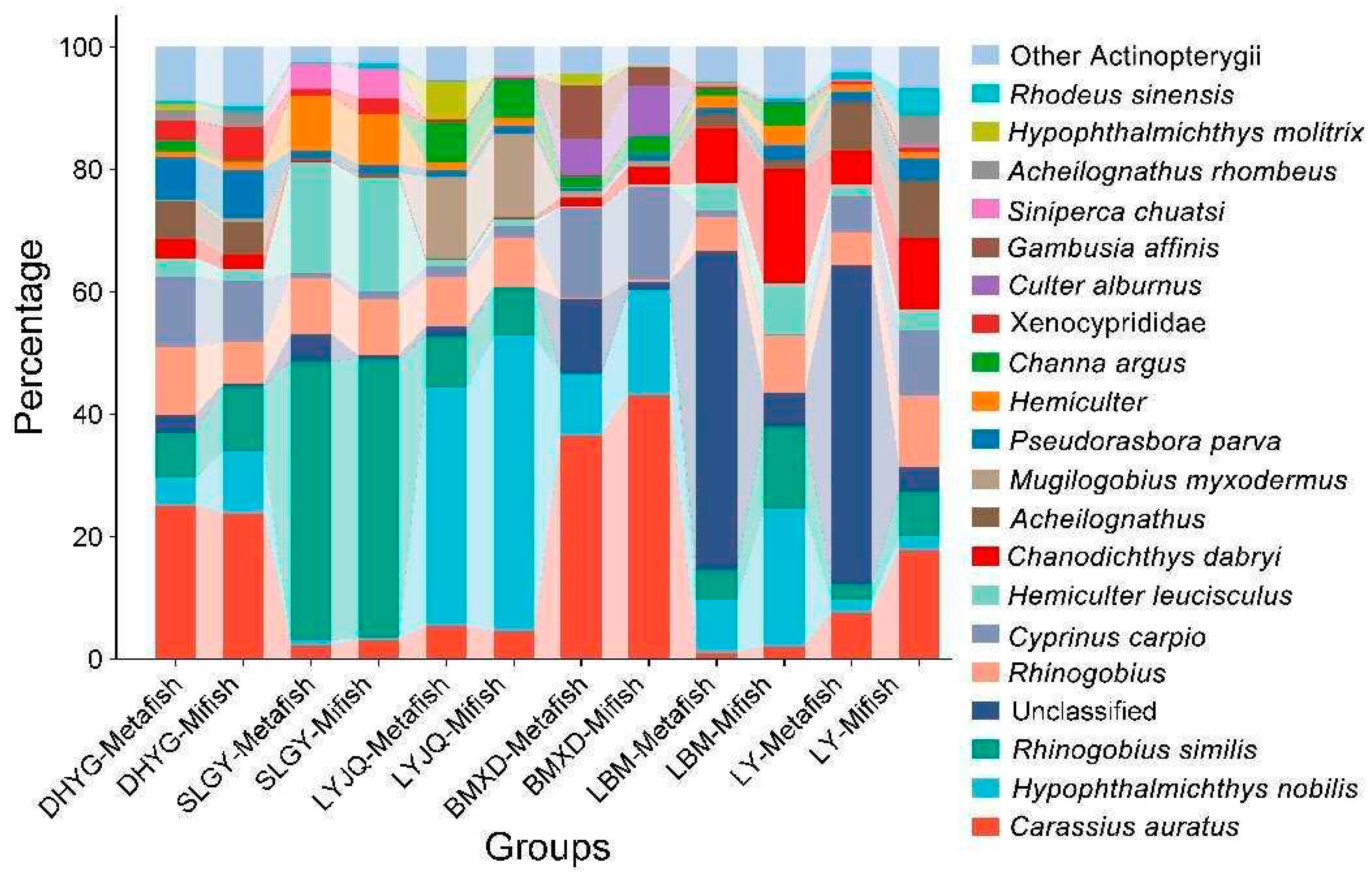
Figure 4.
NMDS plots of eDNA data based on Bray-Curtis indices (stress=0.181). Bray-Curtis indices are based on log-transformed data. The filled squares indicate samples amplified with “Mifish” primer, and the hollow squares indicate samples amplified with “Metafish” primer. The colors indicate sampling locations.
Figure 4.
NMDS plots of eDNA data based on Bray-Curtis indices (stress=0.181). Bray-Curtis indices are based on log-transformed data. The filled squares indicate samples amplified with “Mifish” primer, and the hollow squares indicate samples amplified with “Metafish” primer. The colors indicate sampling locations.
Figure 5.
The Bray-Curtis dissimilarity among sub-samples of a sampling location. The symbol “**” indicates a significant difference as p < 0.01, “*” indicates p < 0.05.
Figure 5.
The Bray-Curtis dissimilarity among sub-samples of a sampling location. The symbol “**” indicates a significant difference as p < 0.01, “*” indicates p < 0.05.
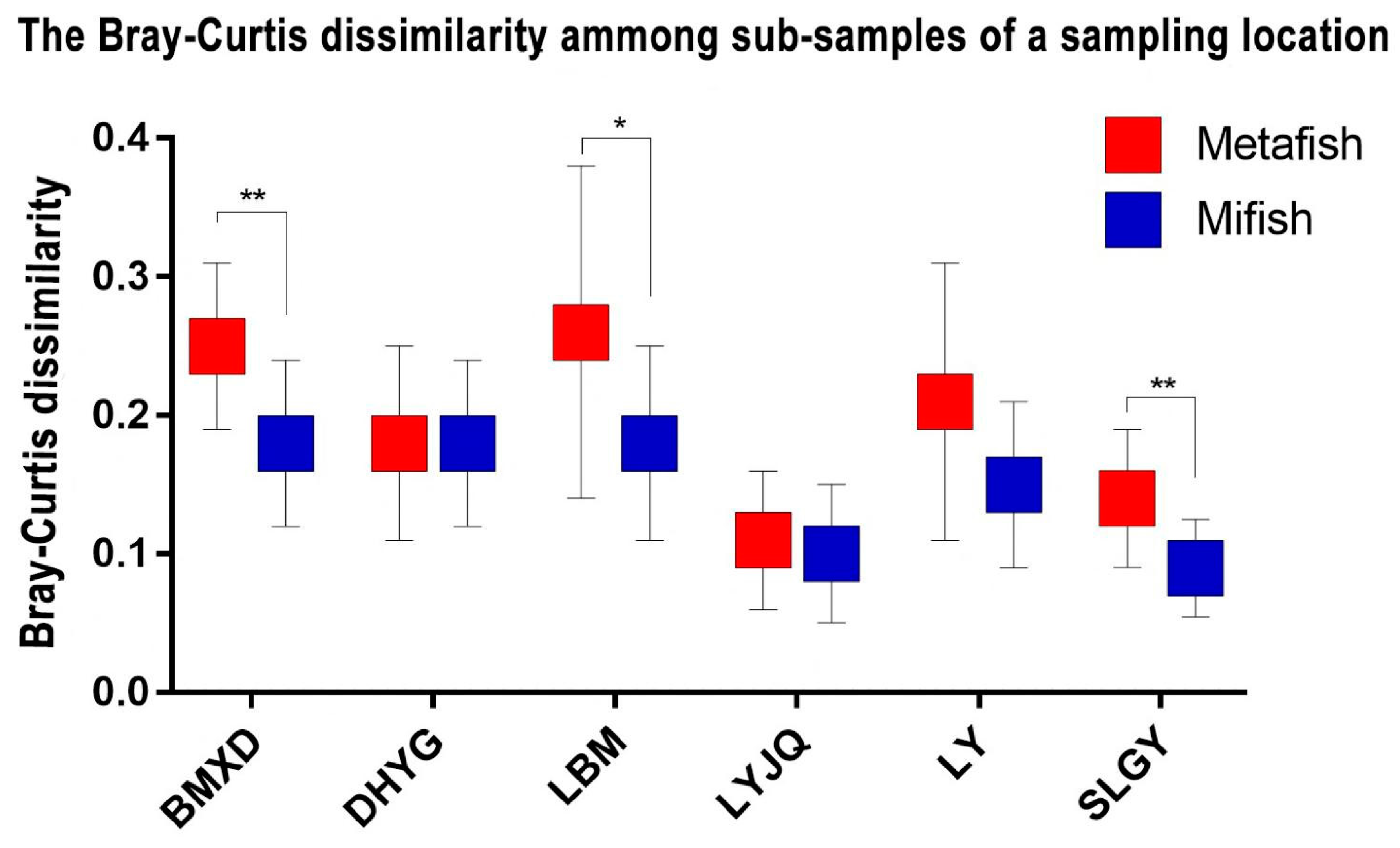

Table 1.
Table of the number of species, genus, family detected by Metafish and Mifish at each sampling site and in total.
Table 1.
Table of the number of species, genus, family detected by Metafish and Mifish at each sampling site and in total.
| Location-Primer | Species | Genus | Family |
|---|---|---|---|
| DHYG-Metafish | 29 | 8 | 4 |
| SLGY-Metafish | 22 | 6 | 2 |
| LYJQ-Metafish | 30 | 7 | 2 |
| BMXD-Metafish | 20 | 5 | 1 |
| LBM-Metafish | 23 | 9 | 3 |
| LY-Metafish | 25 | 10 | 1 |
| DHYG-Mifish | 30 | 11 | 2 |
| SLGY-Mifish | 18 | 7 | 2 |
| LYJQ-Mifish | 28 | 7 | 2 |
| BMXD-Mifish | 22 | 7 | 1 |
| LBM-Mifish | 21 | 10 | 2 |
| LY-Mifish | 24 | 10 | 2 |
| Total-Metafish | 44 | 32 | 14 |
| Total-Mifish | 45 | 34 | 15 |
| Total-Metafish & Mifish | 51 | 36 | 16 |
Table 2.
Table of the relative frequency of fish species that vary across sampling sites or by primer choice.
Table 2.
Table of the relative frequency of fish species that vary across sampling sites or by primer choice.
| Species | DHYG-Metafish | SLGY-Metafish | LYJQ-Metafish | BMXD-Metafish | LBM-Metafish | LY-Metafish | DHYG-Mifish | SLGY-Mifish | LYJQ-Mifish | BMXD-Mifish | LBM-Mifish | LY-Mifish |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Unclassified | 3.0 | 4.5 | 1.7 | 12.1 | 52.1 | 52.1 | 0.5 | 0.5 | <0.1 | 1.3 | 5.6 | 4.1 |
| Acheilognathus rhombeus | 1.7 | <0.1 | <0.1 | 0 | 0 | <0.1 | 2.7 | 0 | <0.1 | <0.1 | <0.1 | 5.1 |
| Acheilognathus tonkinensis | <0.1 | 0 | 0 | 0 | 0 | <0.1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Culter alburnus | 0 | <0.1 | 0 | 5.9 | 0 | 0 | 0 | 0 | <0.1 | 8.2 | 0 | <0.1 |
| Carassius auratus | 25.1 | 2.2 | 5.3 | 36.6 | 1.0 | 7.6 | 23.8 | 3.2 | 4.5 | 43.1 | 2.0 | 17.7 |
| Carassius carassius | 0 | 0 | 0 | 0 | <0.1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Channa argus | 1.6 | 0 | 6.6 | 1.8 | 1.2 | 0 | 0.2 | 0 | 6.4 | 1.8 | 3.6 | 0 |
| Chanodichthys dabryi | 3.1 | 0.2 | <0.1 | 1.5 | 9.1 | 5.7 | 2.4 | 0 | 0.13 | 2.8 | 18.8 | 11.7 |
| Coptodon zillii | 0 | 0 | 0 | 0 | 0 | 0 | <0.1 | <0.1 | 0 | <0.1 | <0.1 | <0.1 |
| Gambusia affinis | 0 | 0 | 0 | 9.0 | 0.3 | 0 | 0 | 0 | 0 | 3.0 | 0 | 0 |
| Gambusia holbrooki | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | <0.1 | 0 | 0 |
| Hemibarbus barbus | 0 | 0 | <0.1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Hypophthalmichthys molitrix | 1.1 | 0 | 6.3 | 1.9 | 0.4 | <0.1 | <0.1 | 0 | <0.1 | 0.2 | <0.1 | <0.1 |
| Hypophthalmichthys nobilis | 5.1 | 0.9 | 38.7 | 9.6 | 8.1 | 1.9 | 10.2 | 0.24 | 48.2 | 16.9 | 22.1 | 2.4 |
| Hyporhamphus intermedius | 0 | 0 | <0.1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Misgurnus anguillicaudatus | 0 | 0 | <0.1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Misgurnus bipartitus | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | <0.1 | 0 | 0 | 0 |
| Mystacoleucus marginatus | <0.1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Pagellus bellottii | 0 | 0 | 0 | 0 | 0 | 0 | <0.1 | 0 | 0 | 0 | 0 | 0 |
| Paramisgurnus dabryanus | 0 | 0 | 0 | 0 | 0 | 0 | <0.1 | 0 | <0.1 | 0 | 0 | 0 |
| Rhinogobius similis | 7.6 | 45.6 | 8.2 | <0.1 | 4.7 | 2.5 | 10.5 | 45.5 | 7.9 | <0.1 | 13.1 | 7.2 |
| Sarcocheilichthys sinensis | 0 | 0 | <0.1 | 0 | 0 | 0 | 0.3 | 0 | 0 | 0 | 0 | 0 |
| Saurogobio lissilabris | 0 | 0 | 0 | 0 | 0 | 0 | <0.1 | 0 | 0 | 0 | 0 | 0 |
| Saurogobio xiangjiangensis | 0 | 0 | 0 | 0 | 0 | 0 | <0.1 | 0 | 0 | 0 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



