
Submitted:
31 December 2023
Posted:
03 January 2024
Read the latest preprint version here
Abstract
I propose a new strategy to suppress human cancer completely with two entirely new drug compounds exploiting cancer's Warburg effect characterized by a defective mitochondrial aerobic respiration, substituted for by cytosolic aerobic fermentation/glycolysis of D-(+)-glucose into L-(+)-lactic acid. The two essentially new drugs compound 1 [P(op)T(est)162] and compound 3 (PT167) represent new highly symmetric, four-bladed propeller-shaped polyammonium cations. The in vitro antineoplastic highly efficacious drug compound 3 represents a covalent combination of compound 1 and compound 2 (PT166). The intermediate drug compound 2 is an entirely new colchic(in)oid derivative synthesized from colchicine. Compound 2's structure was determined by X-ray crystallography. Compound 1 and compound 3 were active in vitro versus 60 human cancer cell lines of the National Cancer Institute (NCI) Developmental Therapeutics Program (DTP) 60-cancer cell testing. Compound 1 and compound 3 both not only stop the growth of cancer cells to ±0% (cancerostatic effect), but completely kill nearly all 60 cancer cells to a level of almost −100% (tumoricidal effect). Compound 1 and compound 3 induce mitochondrial apoptosis (under cytochrome c release) in all cancer cells tested by (re)activating (in most cancers impaired) p53 function which results in a decrease of cancer's dysregulated cyclin D1 and an induction of the cell cycle-halting cyclin-dependent kinase inhibitor p21Waf1/p21Cip1.
Keywords:
cancer growth
; tumor metastasis
; cancer's Warburg effect
; p53
; p53 (re)activators
; mitochondrial apoptosis
; cytochrome c release
; tumoricidal drug effect
1. Introduction
In 1930 Otto Heinrich Warburg (1883−1970) published [1] his theory on the origin of cancer cells based on a series of
preceding investigations [2,3,4]. He summarized his theory on the origin of cancer cells in 1956 [5]. This theory was coined the Warburg
hypothesis [6]. The
essence of this hypothesis on the origin of cancer is that, under
aerobic conditions, malignant tissues metabolize approximately tenfold more
glucose to lactate in a constant time window than primary tissues, a phenomenon
known as the Warburg ’malignant’ effect. Warburg claimed that cancer cells heavily rely on aerobic glycolysis as an
energy source for malignant growth, rather than on the aerobic respiration,
thereby claiming a defect in cancer cell respiration. The Warburg
hypothesis was heavily debated for many years since its introduction [6], especially regarding the
respiratory defect misinterpreted by Warburg himself [6], and only in recent years many
details of the Warburg hypothesis were confirmed as correct [7–10]. Importantly, the Warburg
effect itself, which was proved to constitute a fact in vivo [11], has to be differentiated from
the complete Warburg hypothesis which was the cause of ongoing
scientific debate see [12–14].
In 2002 it was shown that human
cancer cells indeed suffer from defects in cellular respiration, either a
marked depletion in cellular mitochondrial content or a selective repression in
expression of the catalytic β-subunit of mitochondrial Complex V F-type ATPase (β-F1-ATPase)
concurrent with an increase in the expression of the glycolytic enzyme
glyceraldehyde 3-phosphate dehydrogenase [7]. Both mechanisms impair mitochondrial respiration and give support
for the Warburg hypothesis [7]. In rat rhabdomyosarcoma R1H cells mitochondrial function
was found to be deficient by a dysregulation of the mitochondrial
protein−to−cardiolipin ratio [10].
Mitochondrial respiration of R1H cells was significantly impaired, joined by
the incapacity of the rhabdomyosarcoma cells to differentiate into mature
striated skeletal muscle cells [10]. The
intriguing abnormalities in cardiolipin content were confirmed in vivo
utilizing brain tumors grown in mice [9]. The
compositional cardiolipin abnormalities involved an abundance of immature
molecular species and deficiencies of mature molecular species, suggesting
major defects in cardiolipin synthesis and remodeling in rodent brain tumor
tissue [9]. The tumor cardiolipin
abnormalities were also associated with significant reductions in both
individual and linked electron transport chain activities [9]. The acidic phospholipid cardiolipin [1,3-bis(sn-3′-phosphatidyl)-sn-glycerol,
1,3-diphosphatidyl-sn-glycerol] is known to anchor cytochrome c
together with cytochrome c oxidase at the inner mitochondrial membrane [15–18]. Abnormalities in cardiolipin content and
composition thus strongly impair cellular mitochondrial respiration by
impairment of cytochrome c/cytochrome c oxidase function(s) of Complexes
III (coenzyme Q – cytochrome c reductase) and IV (cytochrome c
oxidase) of the electron transport chain [9].
The intrinsic pathway of programmed cell death
(apoptosis) triggers cytochrome c release from the inner mitochondrial
membrane into the cytosol [19] where
cytochrome c associates with apoptotic protease activating factor 1
(Apaf-1, apoptotic peptidase activating factor 1) and procaspase 9 to form the
apoptosome [19]. The apoptosome in turn
activates the effector caspases 3, 6 and 7 to execute programmed cell death [19]. In cancer cells this mechanism is defective or
inefficient [20]. Therefore, in cancer therapy
one possibility to selectively and completely kill cancer cells is to induce
their apoptosis over the intrinsic pathway by pharmacological intervention [20]. Targeting mitochondria by antineoplastic drugs
was suggested as a novel strategy for cancer therapy [20].
Recently, I isolated a lipophilic polyammonium cation, compound 1, as a
by-product of the reaction of 1-aminoadamantane (amantadine) with
1,3-bis(chloromethyl)benzene (α,α′-dichloro-m-xylene) in refluxing
absolute ethanol. Amantadine is known as an antiparkinsonian (N-methyl-D-aspartate
ionotropic glutamate receptor antagonist) drug [21]
and chemotherapeutic antiviral drug inhibiting influenza A virus M2 protein
transmembrane proton channel [22]. This
synthesis was inspired by potential binding of a polyammonium cationic drug to
DNA and/or p53 tumor suppressor protein tetramerization domain [23]. It was found that compound 1 (PT162,
NSC 796018), a new compound never synthesized before [according to
Chemical Abstracts Service (CAS®) SciFinder® search], induced apoptosis in all
cell lines of the National Cancer Institute (NCI) Developmental Therapeutics
Program (DTP) 60-cancer cell 5-dose testing, excluding leukemia cell lines, in
the micromolar range of growth inhibition 50% (GI50). I decided to merge
compound 1 with the colchic(in)oid compound 2 (PT166, NSC
750423) which showed submicromolar GI50 in the NCI DTP 60-cancer
cell 5-dose testing, but did not induce cancer cell apoptosis. Compound 2
was synthesized from colchicine and thiosemicarbazide in an one-step procedure
and represents a new compound never synthesized before [according to Chemical
Abstracts Service (CAS®) SciFinder® search] just as compound 1. Compound
1 reacted with compound 2 under impact of sodium hydroxide (NaOH)
to give compound 3. Compound 3 (PT167, NSC 799315),
a new compound never synthesized before [according to Chemical Abstracts
Service (CAS®) SciFinder® search], showed submicromolar GI50 in the NCI
DTP 60-cancer cell 5-dose testing constantly in most cell lines including
leukemia cells. Importantly, compound 3 was able to induce apoptosis in
all investigated cancer cells, including leukemia cell lines, with a Mean of
Inhibition Data (MID) for total growth inhibition (TGI, growth
inhibition 100%) of 4.57 µM, and a MID for lethal concentration 50% (LC50)
of 15.85 µM. I report here the chemistry and NCI DTP 60-cancer cell 5-dose
testing data for compound 1, compound 2 and compound 3,
and demonstrate the apoptotic release of cytochrome c into the cytosol
and activation of effector caspases induced by compounds 1 and 3.
I propose that compound 1 and compound 3 induce apoptosis
according to the Warburg hypothesis of pre-damaged respiration as a
hallmark of cancer by exploiting the defect in mitochondrial
cardiolipin–cytochrome c association in cancer cells.
2. Chemistry
2.1. Compound 1
2.1.1. The Synthesis of Salt-Containing Compound 1 (PENTA)
To find a potential complexation
and/or stablization partner for retinazone
[24,25]
, an attempt was made to synthesize a polyammonium polycation from
the adamantane
[26]
-derived
influenza A virus inhibitor
[22]
and N-methyl-
D
-aspartate (NMDA) subtype glutamate receptor antagonist
[21]
amantadine × HCl. For that
purpose 1-aminoadamantane hydrochloride and an 1.5-fold molar excess of 1,3-bis(chloromethyl)benzene
were dissolved in aqueous ethanol. A solution of sodium hydroxide in water
(3-fold molar excess) was added and the mixture was refluxed for 3 h.
Successively acetone was added through the reflux condensor. After filtration,
dilution with water, acidification with HCl and volume reduction, the reaction
mixture was extracted with ethyl acetate to remove unreacted
1,3-bis(chloromethyl)benzene. Following additional volume reduction of the
aqueous phase a crude product could be isolated by freezing. The crude product
was dissolved in refluxing aqueous acetone and was hot filtrated. The filtrate
was evaporated from the acetone and was acidified with HCl. Instantly, a white
precipitate formed which represented the salt-containing compound 1 (PENTA).
2.1.2. The Synthesis of Pure Compound 1 [PT162 (NSC 796018)]
Pure compound 1 [PT162
(NSC 796018)] (Figure 1) was
synthesized from 1-aminoadamantane (amantadine) with
1,3-bis(chloromethyl)benzene (α,α′-dichloro-m-xylene) in refluxing
absolute ethanol. Compound 1 was a by-product of the synthesis obtained
in 12.4% yield. The lipophilic main product was not isolated and removed by
extraction with ethyl acetate. Crucial for successful synthesis of compound 1
was to utilize the free base 1-aminoadamantane instead of its commercial
hydrochloride. Preceding synthesis, the free base 1-aminoadamantane was
prepared from the hydrochloride by neutralization with NaOH, and this free base
was used in situ for synthesis of compound 1.
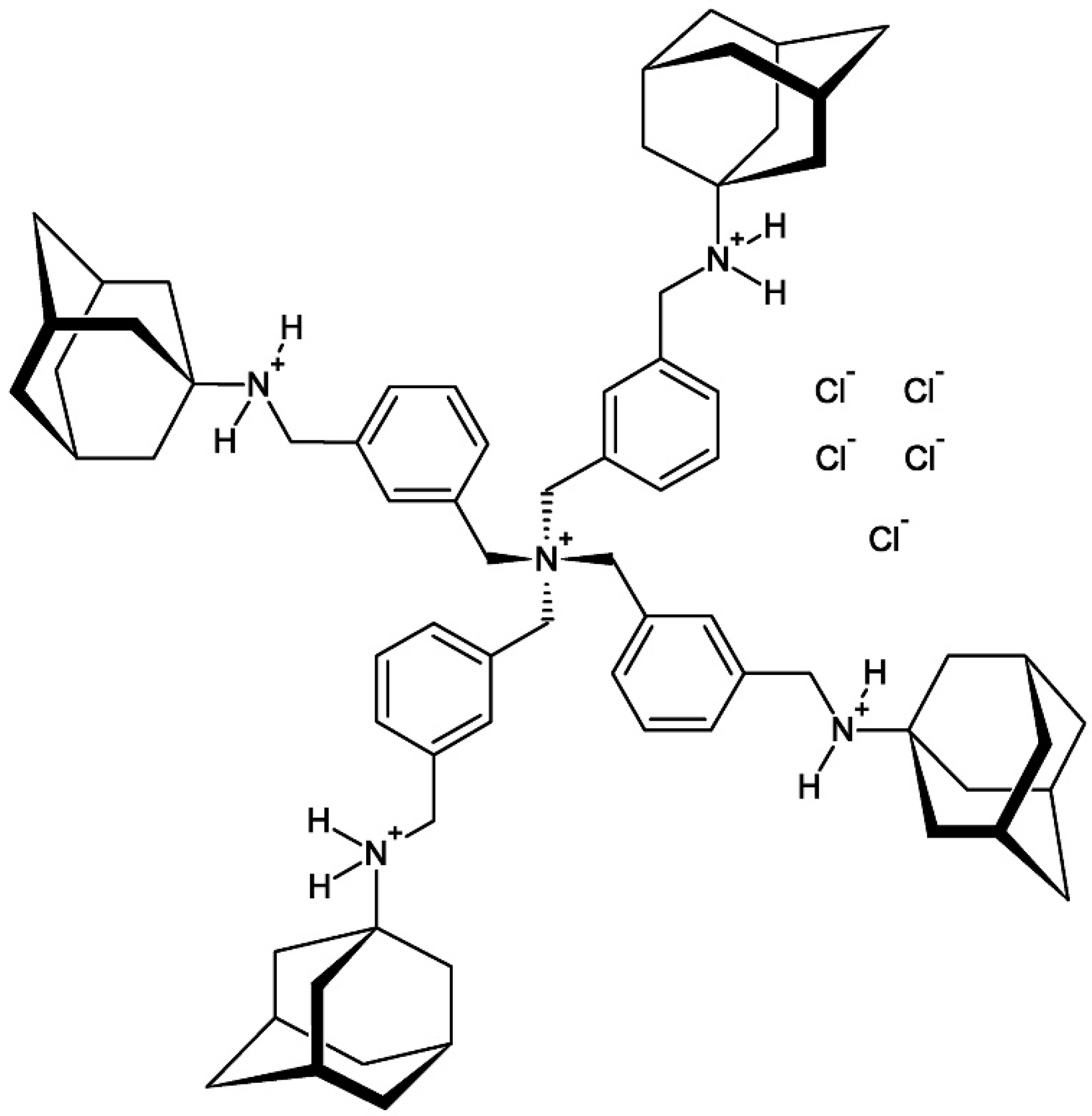
Figure 1.
The organic chemical structure formula of compound 1 [PT162 (NSC 796018)]. Four adamantan-1-ammonium cages are included within the four-bladed propeller-shaped polyammonium cation.
Figure 1.
The organic chemical structure formula of compound 1 [PT162 (NSC 796018)]. Four adamantan-1-ammonium cages are included within the four-bladed propeller-shaped polyammonium cation.
2.1.3. Structure Elucidation of Compound 1 [PT162 (NSC 796018)]
According to the structure
elucidation by nuclear magnetic resonance (NMR) spectroscopy compound 1
[PT162 (NSC 796018)] was formed from the
intermediate N-[3-(chloromethyl)benzyl]-1-adamantanamine (Scheme 1). The latter
intermediate quaternarized 1-aminoadamantane to give N,N,N-tris{3-[(tricyclo[3.3.1.13,7]decan-1-ylamino)methyl]benzyl}-1-adamantanammonium
hydroxide (Scheme 1),
which underwent Hofmann elimination [27] with 1-adamantanol being expelled. 1-Adamantanol is produced from
adamantene, a known reaction intermediate [28–30]. The resulting tris{3-[(tricyclo[3.3.1.13,7]decan-1-ylamino)methyl]benzyl}amine
in turn was quaternarized by another N-[3-(chloromethyl)benzyl]-1-adamantanamine
to yield, after acidification with HCl, tetrakis{3-[(tricyclo[3.3.1.13,7]decan-1-ammonio)methyl]benzyl}ammonium
pentachloride (compound 1) (Scheme 1).
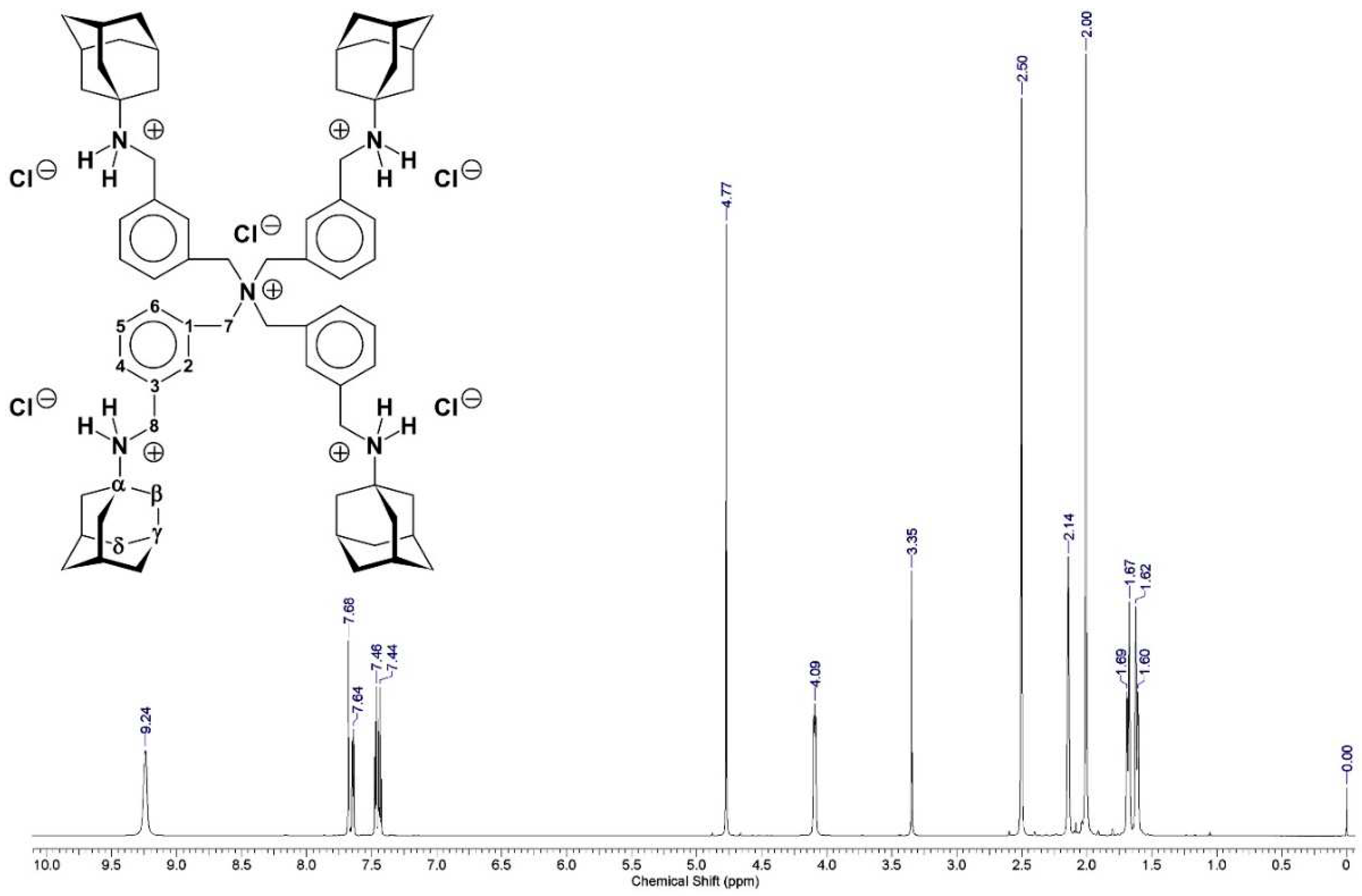
The structure of salt-containing
compound 1 (PENTA) was secured by analysis of its 1H-
(Figure S1) and 13C-NMR
(Figure S2) spectra
recorded in DMSO-d6. The proton NMR of the aliphatic part of salt-containing
compound 1 (PENTA) was analysed for (Figure S1): δ 1.62 (3 H, d; 2Jgem
= −12.1 Hz; δ-CHaxial), 1.69 (3 H, d; 2Jgem
= −12.4 Hz; δ-CHequatorial), 2.02 (6 H, s; β-CH2), 2.15 (3 H,
s; γ-CH),
4.10 (2 H, t; 3Jvicinal = 6.3 Hz; 8-CH2),
4.78 (2 H, s; 7-CH2). The proton NMR of the aromatic part of salt-containing
compound 1 (PENTA) was analysed for (Figure S1): δ 7.42‒7.49 (2 H,
m; H-4, H-6), 7.65 (1 H, d; 3Jortho = 7.1 Hz;
H-5), 7.69 (1 H, s; H-2). The three axial δ-protons of the adamantane
cage were split from the corresponding three equatorial δ-protons. A broad ammonium
resonance was found at δ 9.24 (2 H, br s; 8-NH2+ ammonium). The aliphatic
resonances contained an adamantane part, two methylene groups and an ammonium
resonance in the integrated proton ratio 15 : 2 : 2 : 2.
This pointed to a symmetric ammonium molecule with
two different m-xylylene methylene groups which were derived from
1,3-bis(chloromethyl)benzene (m-xylylene dichloride). Obviously, the
ammonium resonance originated from an (1-adamantyl-NH2-R)+
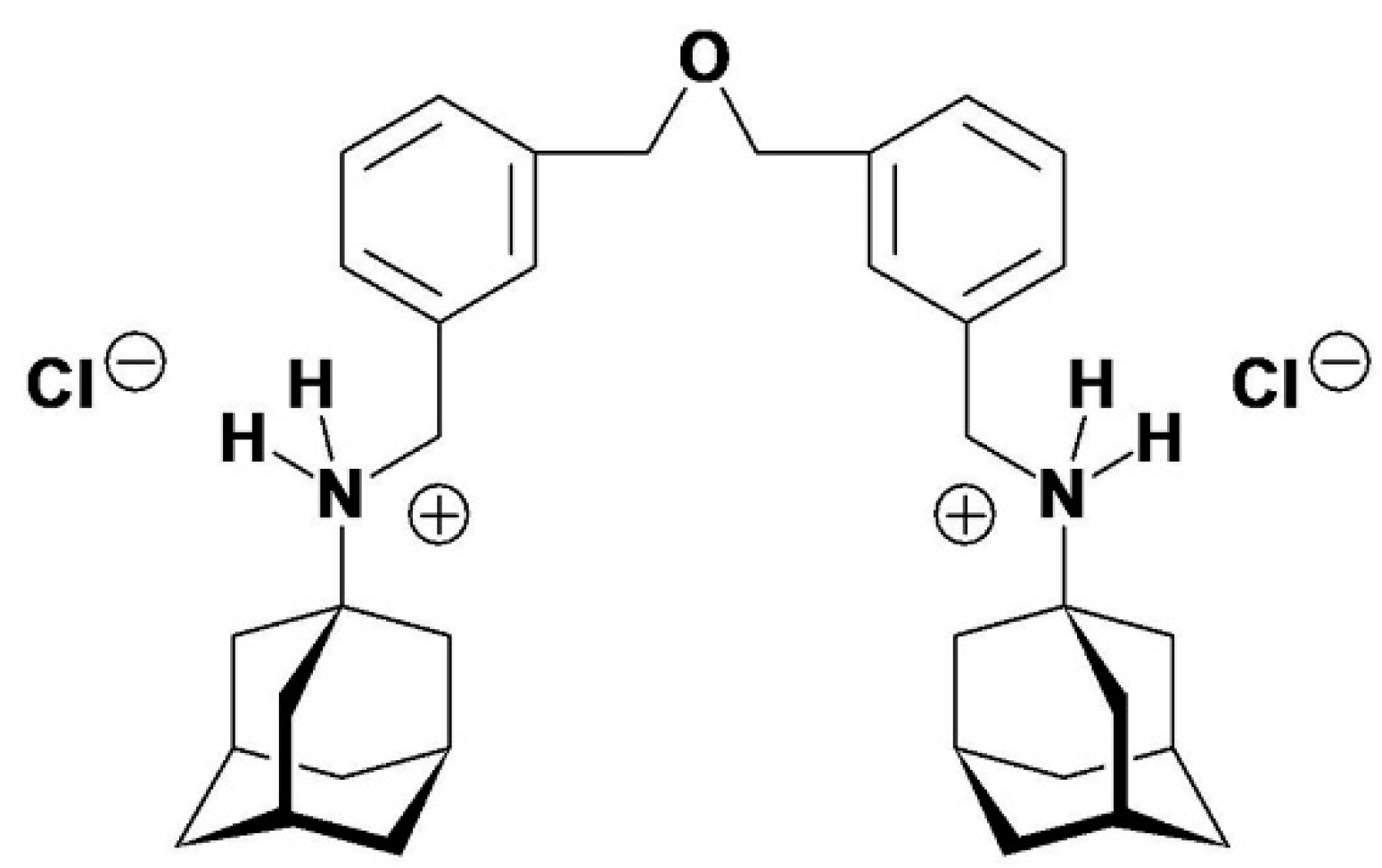
system. Therefore, only two reasonable possibilities for the structure of compound
1 (PT162) remained. The first would be di{3-[(tricyclo[3.3.1.13,7]decan-1-ammonio)methyl]benzyl}ether
dichloride (Figure 2), the second being
expressed by the already given formula (Figure 1).
The former structure could be ruled out for the following reasons: (i) the oxygen
elemental analysis of salt-containing compound 1 (PENTA) showed
only trace amounts of oxygen (w ≤ 0.82%) resulting from the ethanol and
acetone traces contained in salt-containing compound 1 (PENTA),
(ii) the absence of a commonly very strong dibenzyl ether band in the FT‒IR
spectrum (Figure S3) expected at wavenumber 1090 cm-1 for dibenzyl ether [31], (iii) the downfield methylene δ 4.78 (2 H, s; 7-CH2) did not
match the typical dibenzyl ether chemical shift found at δ 4.54 ppm (in CDCl3) [32], and (iv) the analysis of the carbon-13 NMR
spectrum of salt-containing compound 1 (PENTA) (Figure S2). The 13C-NMR of the
aliphatic part of salt-containing compound 1 (PENTA) was analysed
for (Figure S2): δ 28.50 (γ-CH),
35.25 (δ-CH2), 37.35 (β-CH2), 42.31 (8-CH2),
45.84 (7-CH2), 57.06 (α-C).
The 13C-NMR of the aromatic part of salt-containing compound 1
(PENTA) was analysed for (Figure S2):
δ 128.87 (C-4)*, 129.14 (C-6)*,
130.36 (C-2)*, 130.62 (C-5)*, 133.22 (C-3), 137.84 (C-1). The starred
assignments are tentative and interchangeable (they could not be assigned
unequivocally to the individual carbons). Clearly the structure proof could be
gained by looking at the downfield methylene δ
45.84 (7-CH2) which did not fit the typical dibenzyl ether α-CH2 chemical shift found at δ 72.10 ppm (in CDCl3) [32]. The 100.62 MHz 13C-Distortionless
Enhancement by Polarization Transfer Including Detection
of Quaternary Nuclei (DEPTQ) [33,34] (DEPTQ 13C-NMR) subspectrum (in DMSO-d6) of salt-containing compound 1
(PENTA) (Figure S4) secured the
given CH2 assignments, and proved that the resonance at δ 57.06 ppm originated from a quaternary
carbon. The assignments were further verified by analysis of the
gradient-selected Correlation Spectroscopy (gs-COSY)
two-dimensional 1H−1H-correlation spectrum [34] (Figure S5),
the gradient-selected Heteronuclear Multiple Quantum Coherence
(gs-HMQC) [34] (Figure S6), and the gradient-selected Heteronuclear Multiple Bond
Correlation (gs-HMBC) [34] (Figure S7) two-dimensional 1H‒13C-correlation spectra
of salt-containing compound 1 (PENTA).
Scheme 1.
The synthesis of the polyammonium polycation compound 1 from 1-aminoadamantane × HCl and 1,3-bis(chloromethyl)benzene. The intermediate N-[3-(chloromethyl)benzyl]-1-adamantanamine reacted with amantadine × HCl under influence of NaOH to N,N,N-tris{3-[(tricyclo[3.3.1.13,7]decan-1-ylamino)methyl]benzyl}-1-adamantanammonium hydroxide, which in turn underwent Hofmann elimination [27] on heating at reflux under elimination of 1-adamantanol (produced from adamantene [28−30]). Compound 1 resulted from quaternarization with another molecule N-[3-(chloromethyl)benzyl]-1-adamantanamine and acidification with hydrochloric acid (HCl).
Scheme 1.
The synthesis of the polyammonium polycation compound 1 from 1-aminoadamantane × HCl and 1,3-bis(chloromethyl)benzene. The intermediate N-[3-(chloromethyl)benzyl]-1-adamantanamine reacted with amantadine × HCl under influence of NaOH to N,N,N-tris{3-[(tricyclo[3.3.1.13,7]decan-1-ylamino)methyl]benzyl}-1-adamantanammonium hydroxide, which in turn underwent Hofmann elimination [27] on heating at reflux under elimination of 1-adamantanol (produced from adamantene [28−30]). Compound 1 resulted from quaternarization with another molecule N-[3-(chloromethyl)benzyl]-1-adamantanamine and acidification with hydrochloric acid (HCl).
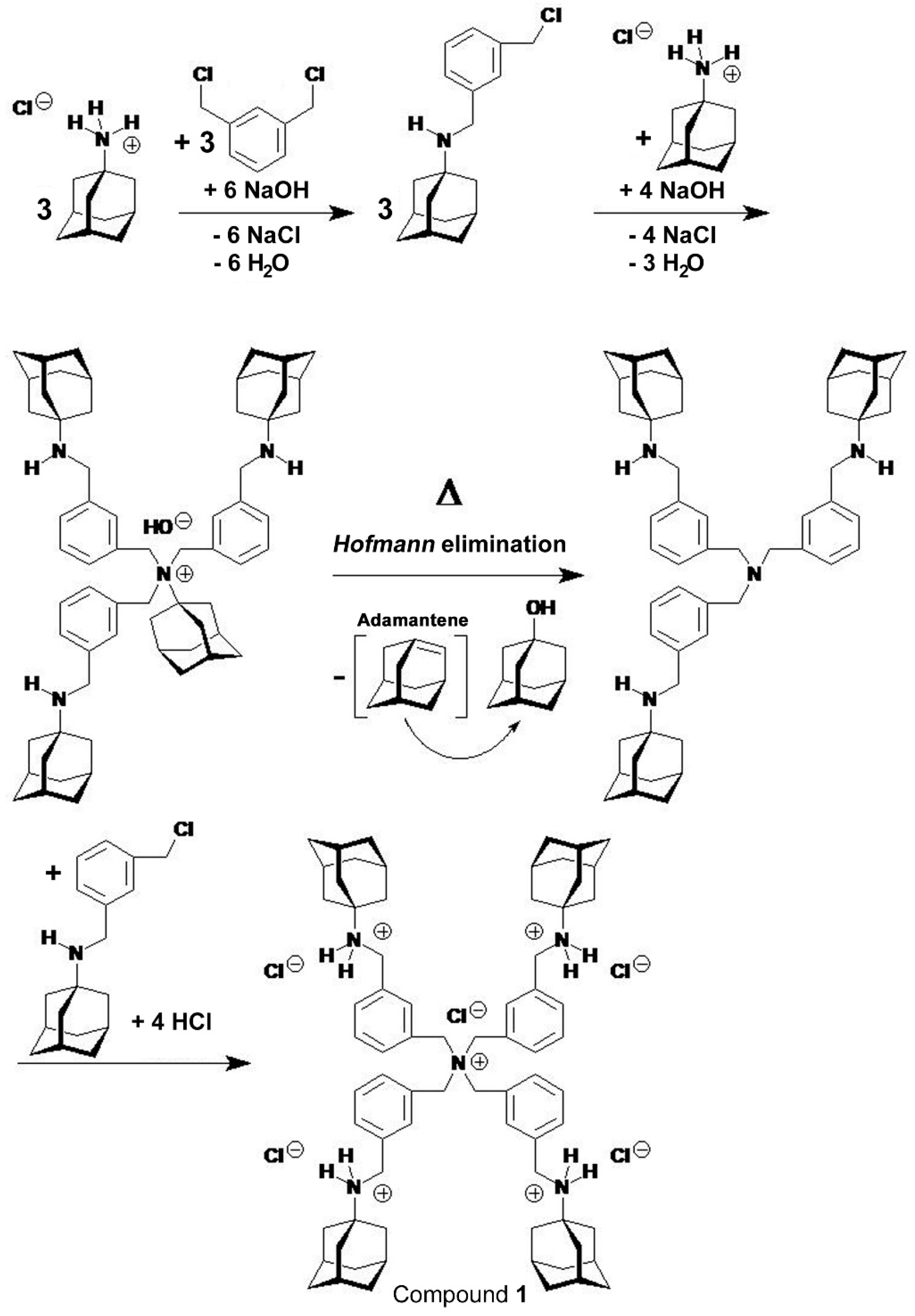
Figure 2.
The structure of the dibenzyl ether derivative di{3-[(tricyclo[3.3.1.13,7]decan-1-ammonio)methyl]benzyl}ether dichloride. This hypothetical structure was considered as alternative reaction product in the synthesis of compound 1.
Figure 2.
The structure of the dibenzyl ether derivative di{3-[(tricyclo[3.3.1.13,7]decan-1-ammonio)methyl]benzyl}ether dichloride. This hypothetical structure was considered as alternative reaction product in the synthesis of compound 1.
Conclusively, the second structure possibility, the true formula, must be depicted by compound 1 (Figure 1), because all other possibilities would not agree with the unequivocal NMR data. Finally, the FT‒IR spectrum of salt-containing compound 1 (PENTA) was compared to the corresponding spectrum of amantadine hydrochloride (Figure S3). Certain characteristics shared by both spectra could be recognized. The strong absorption band in salt-containing compound 1 (PENTA) at wavenumber 2925/2850 cm-1 was created by aliphatic ν(C‒H) stretching vibrations. In amantadine hydrochloride this absorption could be found at wavenumber 2914/2853 cm-1. Aliphatic progression bands associated to these peaks could be found in both spectra. These absorptions mainly could be ascribed to the adamantane cages present in both compounds. Typical bands were found at wavenumbers 1610/1585/1459/1074 cm-1 [salt-containing compound 1 (PENTA)], in amantadine hydrochloride at wavenumbers 1626/1594/1500/1086 cm-1. They probably originate from the 1-adamantanammonium structure shared by both compounds. In addition, salt-containing compound 1 (PENTA) clearly showed the m-xylylene aromatic linker resonances at wavenumbers 794/762/731/693 cm-1 which are not seen with amantadine hydrochloride (Figure S3).
The elemental analysis of salt-containing compound 1 (PENTA) revealed its sodium chloride content by calculating within ±0.3% for carbon and hydrogen. The NaCl obviously was co-precipitated in the final isolation step, which is not surprising when considering the polyammonium chloride character of compound 1. The nitrogen value was found to be 1.13% lower than calculated for C72H100N5Cl5 × 1.5 NaCl. The reason for this deficit could be the [N(CH2R)4]+ tetrasubstituted ammonium structure of the central ammonium nitrogen. Compounds of this type are known to cause combustion problems [35]. It could be speculated that the sodium content (NaCl) in salt-containing compound 1 (PENTA) led to formation of only partially combustible sodium nitrate (NaNO3). Mutually both effects could be responsible for the wrong nitrogen analysis of salt-containing compound 1 (PENTA). Since the other analytical data of salt-containing compound 1 (PENTA) are all very conclusive, the unsatisfactory nitrogen analysis should not be overrated. Taken together, the structure of salt-containing compound 1 (PENTA) could be demonstrated without doubt, especially when evaluating the unequivocal NMR data.
The structure of pure (salt-free) compound 1 [PT162 (NSC 796018)] was secured by 1H-NMR (Figure 3) and 13C-DEPTQ (data not shown) spectroscopy experiments, as well as elemental analysis and FT–IR spectroscopy. By 1H-NMR, through the integration of the proton resonance peaks, and by 13C-NMR, the highly symmetric structure of compound 1 was proved. Compound 1 represents a pentaammonium cation with a quaternary center and four secondary amine functions. The four functional bridges are m-xylylene linkers which connect the central quaternary ammonium cation chloride with four N-substituted 1-adamantanammonium chloride moieties. The resulting chemical structure of compound 1 is given (Figure 1). Compound 1 was registered by the National Cancer Institute (NCI) as NSC 796018.
Figure 3.
The 700.43 MHz 1H-NMR spectrum (in DMSO-d6) of the pure polyammonium polycation tetrakis{3-[(tricyclo[3.3.1.13,7] decan-1-ammonio)methyl]benzyl}ammonium pentachloride = pure (salt-free) compound 1 [PT162 (NSC 796018)] [DMSO-d5h1 quintet at δ 2.50 ppm, 2Jgem (2H−1H) = +1.9 Hz; water at δ 3.35 ppm].
Figure 3.
The 700.43 MHz 1H-NMR spectrum (in DMSO-d6) of the pure polyammonium polycation tetrakis{3-[(tricyclo[3.3.1.13,7] decan-1-ammonio)methyl]benzyl}ammonium pentachloride = pure (salt-free) compound 1 [PT162 (NSC 796018)] [DMSO-d5h1 quintet at δ 2.50 ppm, 2Jgem (2H−1H) = +1.9 Hz; water at δ 3.35 ppm].
2.2. Compound 2
2.2.1. The Synthesis of Colchic(in)oid Compound 2 [PT166 (NSC 750423)]
Compound 2 [PT166 (NSC 750423)] was synthesized from (–)-colchicine sesquihydrate (× 1½ H2O) and thiosemicarbazide under catalysis of sodium hydroxide (NaOH) in refluxing 90% (v/v) aqueous ethanol. The structure (Figure 4) of compound 2 was secured by X-ray crystallography (see Section 2.2.3.), 1H-NMR (Figure 5) and 13C-NMR spectroscopy (data not shown) experiments, as well as elemental analysis and FT–IR spectroscopy (Figure S8). The thiosemicarbazide moiety is attached at the former position of the 10-methoxy group in colchicine. The point of connection is the terminal nitrogen of the hydrazinyl moiety of thiosemicarbazide. Compound 2 was registered by National Cancer Institute (NCI) as NSC 750423.
Figure 4.
The organic chemical structure formula of compound 2 [PT166 (NSC 750423)]. It contains a water of crystallization and ⅔ (ethyl acetate).
Figure 4.
The organic chemical structure formula of compound 2 [PT166 (NSC 750423)]. It contains a water of crystallization and ⅔ (ethyl acetate).

Equimolar quantities of (–)-colchicine and thiosemicarbazide were dissolved in 90% (v/v) aqueous ethanol by refluxing for 5 min. After adding a slight excess of sodium hydroxide dissolved in water, the deep orange-red solution was refluxed for 5 min. The cold deep orange-red solution, after pre-cooling, was nearly neutralized by dropwise addition of hydrochloric acid. Afterwards, the volume of the solution was reduced in vacuo. The reddish-brown solution was then mixed with water, and was acidified with aqueous hydrochloric acid. The oily emulsion was extracted with ethyl acetate (EtOAc). The separated aqueous layer (pH 2) was additionally extracted with a second volume of EtOAc. After neutralization of this aqueous phase with sodium hydrogen carbonate, the aqueous phase (pH 7–8) was extracted twice with EtOAc each. The EtOAc phases were combined and washed twice with water. The washed EtOAc phase, which already precipitated, was mixed with acetone and frozen at –25 °C. If precipitation did not start spontaneously, the volume of the solution was reduced in vacuo until coagulation started. The evolved yellow crystalline precipitate of compound 2 was filtered and dried. From the combined aqueous phases by cooling a second crop of compound 2 could be obtained. The underlying molecular reactions for this synthesis are pictured (Scheme 2). Compound 2 was quite pure as judged by 1H-NMR spectroscopy. The representative 1H-NMR spectrum of compound 2 in DMSO-d6 is pictured (Figure 5).
Scheme 2.
The synthesis of the modified colchic(in)oid 10-(2-carbamothioylhydrazinyl)-10-demethoxycolchicine monohydrate × ⅔ (ethyl acetate) (compound 2, PT166, NSC 750423) by NaOH-catalysed amidation (amino-de-alkoxylation) of the vinylogous (tropolonic) carboxylic acid methyl ester (−)-colchicine. Methanol was split off by thiosemicarbazide (TSC) from (−)-colchicine during short refluxing. Subsequently, the product compound 2 was liberated from a monosodium salt intermediate by acidification with hydrochloric acid and extraction with ethyl acetate. The term colchic(in)oid is now well accepted and documented in literature [36].
Scheme 2.
The synthesis of the modified colchic(in)oid 10-(2-carbamothioylhydrazinyl)-10-demethoxycolchicine monohydrate × ⅔ (ethyl acetate) (compound 2, PT166, NSC 750423) by NaOH-catalysed amidation (amino-de-alkoxylation) of the vinylogous (tropolonic) carboxylic acid methyl ester (−)-colchicine. Methanol was split off by thiosemicarbazide (TSC) from (−)-colchicine during short refluxing. Subsequently, the product compound 2 was liberated from a monosodium salt intermediate by acidification with hydrochloric acid and extraction with ethyl acetate. The term colchic(in)oid is now well accepted and documented in literature [36].
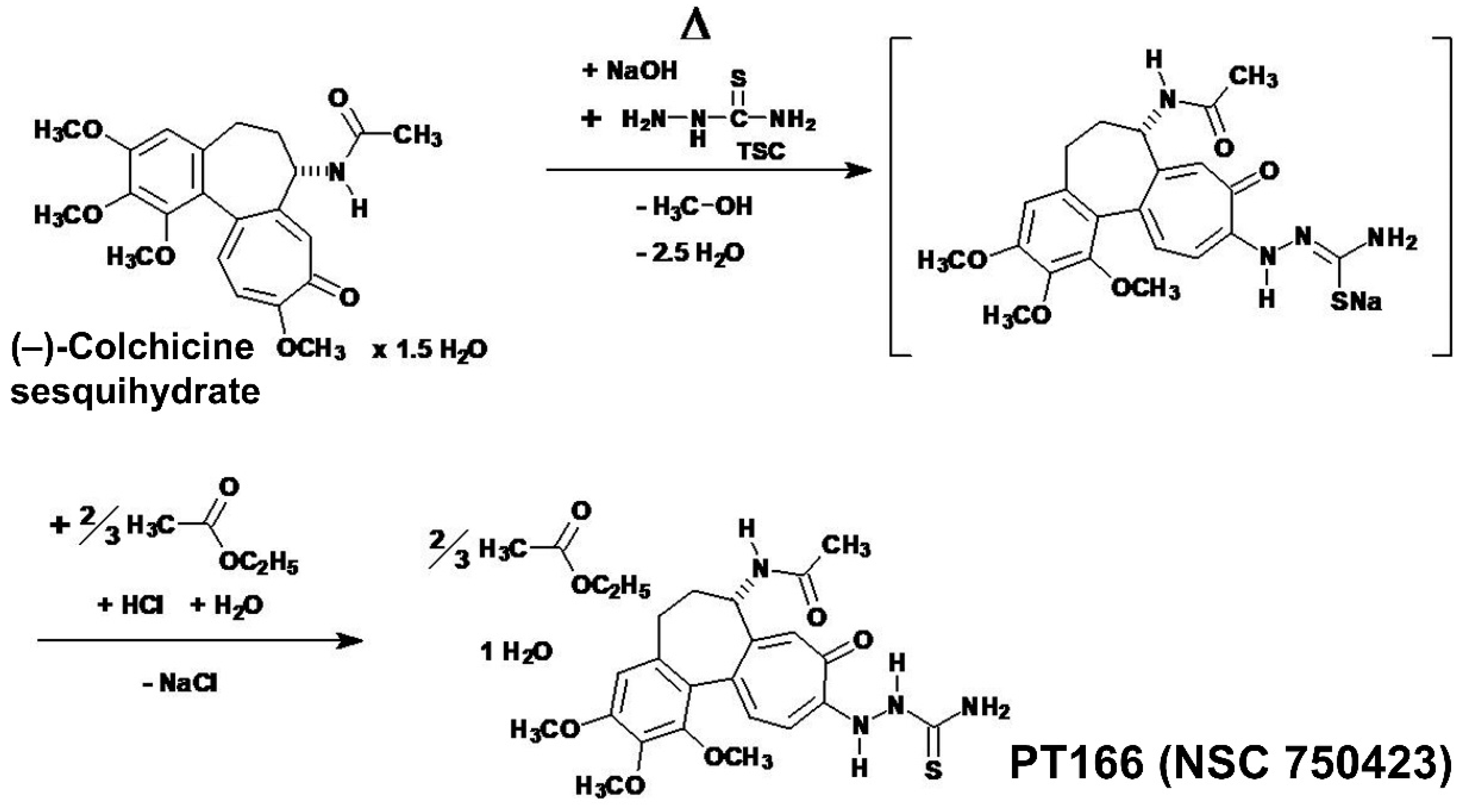
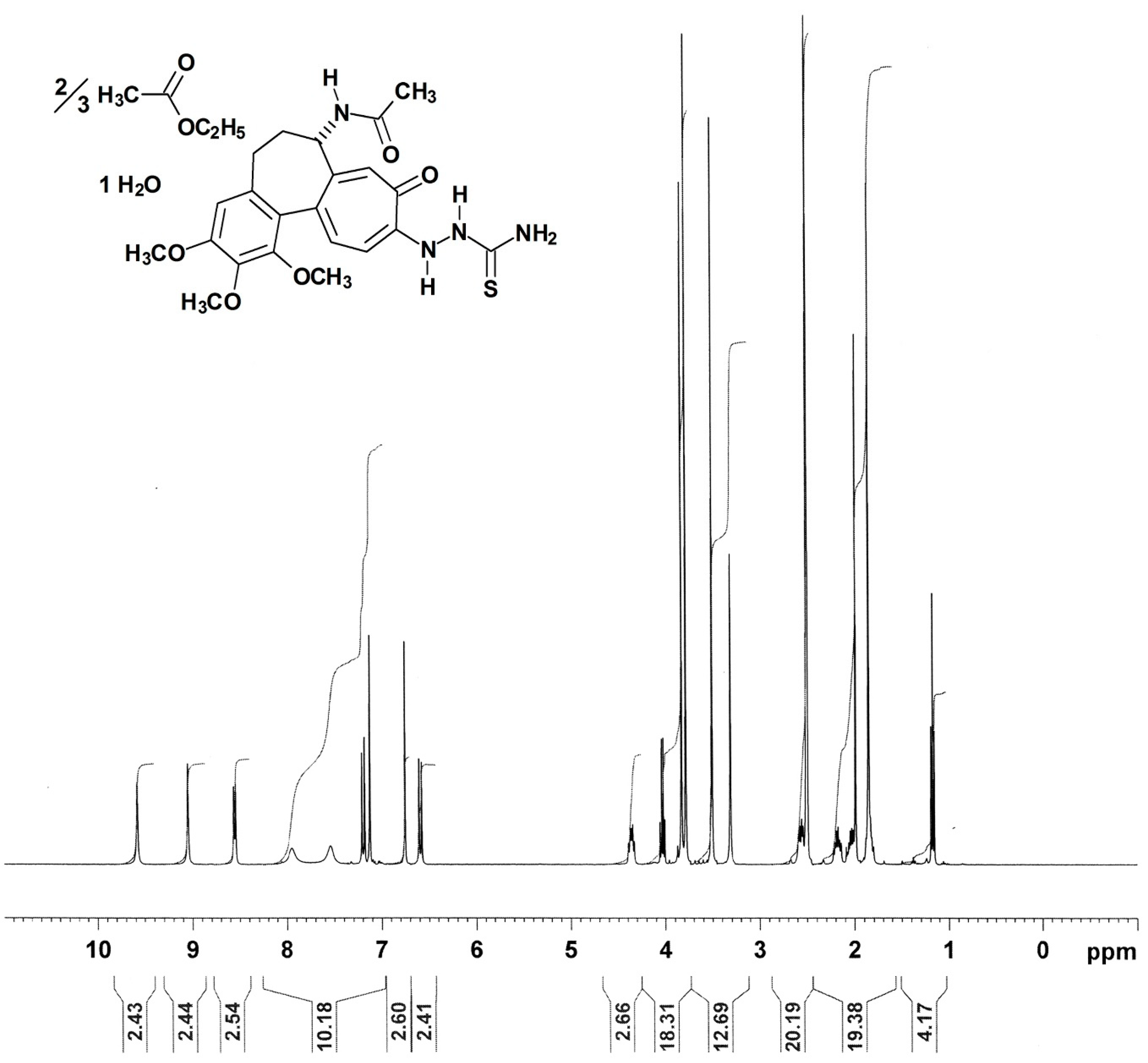
Figure 5.
The 400.13 MHz 1H-NMR spectrum (in DMSO-d6) [DMSO-d5h1 quintet at δ 2.50 ppm, 2Jgem (2H−1H) = +1.9 Hz; water at δ 3.32 ppm] of the colchic(in)oid 10-(2-carbamothioylhydrazinyl)-10-demethoxycolchicine monohydrate × ⅔ (ethyl acetate) = compound 2 [PT166 (NSC 750423)]. Downfield of δ 11 ppm no resonances were detected.
Figure 5.
The 400.13 MHz 1H-NMR spectrum (in DMSO-d6) [DMSO-d5h1 quintet at δ 2.50 ppm, 2Jgem (2H−1H) = +1.9 Hz; water at δ 3.32 ppm] of the colchic(in)oid 10-(2-carbamothioylhydrazinyl)-10-demethoxycolchicine monohydrate × ⅔ (ethyl acetate) = compound 2 [PT166 (NSC 750423)]. Downfield of δ 11 ppm no resonances were detected.
2.2.2. The Nuclear Magnetic Resonance Spectra of Compound 2
It is known that colchic(in)oids [36] have the tendency to retain solvents, like water [37] and/or ethyl acetate [38], very firmly. Natural (–)-colchicine itself retained chloroform [39,40,41], dibromomethane/diiodomethane [42], or water as dihydrate [42,43] or sesquihydrate [41,44]. Therefore, it could be understood that compound 2 was obtained as monohydrate × ⅔ (ethyl acetate) binary solvate, as judged by 1H-NMR and elemental analysis.
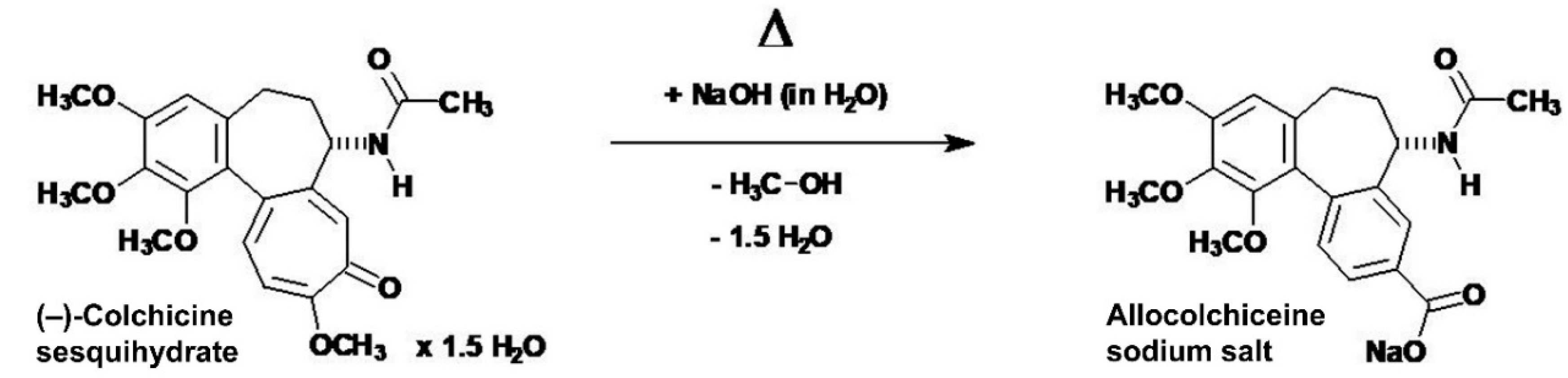
The 1H-NMR resonances of the colchic(in)oid compound 2 were assigned with help of literature [45,46,47,48], especially [46] which gave a complete assignment of the protons in the 1H-NMR spectrum of (–)-colchicine (in CDCl3). Compound 2 represents a completely new compound, never synthesized before [according to Chemical Abstracts Service (CAS) SciFinder®]. Therefore, the proton NMR spectrum (Figure 5) of compound 2 was interpreted by own effort to the point it was possible without doubt. Aliphatic proton resonances of compound 2 dissolved in DMSO-d6 could be differentiated as: δ 1.18 (1.5 H, t; 3J = 7.1 Hz; O–CH2–CH3 ethyl acetate), 1.85 (1 H, m; HA-6), 1.86 (3 H, s; 17-CH3), 1.99 (1.5 H, s; ROOC–CH3 ethyl acetate), 2.05 (1 H, m; HB-6), 2.19 (1 H, m; HA-5), 2.57 (1 H, m; HB-5), 3.51 (3 H, s; 13-OCH3)*, 3.79 (3 H, s; 15-OCH3)*, 3.83 (3 H, s; 14-OCH3)*, 4.03 (1 H, q; 3J = 7.1 Hz; O–CH2–CH3 ethyl acetate), 4.37 (1 H, m; H-7). The three starred assignments (*) are tentative and interchangeable [they could not be assigned unequivocally to the individual methoxyl protons, because their chemical shifts did nearly coincide (Figure 5)]. The gradient-selected Correlation Spectroscopy (gs-COSY) two-dimensional 1H−1H-correlation spectrum [34] proton–proton couplings (data not shown) in connection with the gradient-selected Heteronuclear Multiple Quantum Coherence (gs-HMQC) [34] 13C–1H couplings (data not shown) gave the required informations to assign the proton resonances of compound 2. Aromatic or troponic protons in compound 2 were identified as: δ 6.60 (1 H, d; 3J = 11.1 Hz; H-11), 6.76 (1 H, s; H-4), 7.14 (1 H, s; H-8), 7.20 (1 H, d; 3J = 10.9 Hz; H-12). The acetamide N–H, which coupled to H-7, could be recognized at δ 8.56 (1 H, d; 3J = 7.6 Hz; N–H acetamide). Exchangeable protons of the thiosemicarbazide moiety, detectable in DMSO-d6, could be unequivocally assigned as: δ 7.56 (1 H, br s; H2N–C=S amino, 4′-HA), 7.96 (1 H, br s; H2N–C=S amino, 4′-HB), 9.06 (1 H, s; 1′-N–H), 9.59 (1 H, s; 2′-N–H). In the gradient-selected Correlation Spectroscopy (gs-COSY) two-dimensional 1H−1H-correlation spectrum [34] (data not shown) of compound 2 (in DMSO-d6) no W-shaped long-range 4J (1H–1H) coupling, known as zig-zag (W) coupling, was found. This differentiates compound 2 from the thiosemicarbazones (E)-4-(dimethylamino)benzaldehyde thiosemicarbazone [24] and (E)-4-bromo-2-fluorobenzaldehyde thiosemicarbazone [24], where such a “W” coupling was observed [24]. This pointed to sterical fixation as prerequisite for observable W couplings in (E)-4-(dimethylamino)benzaldehyde thiosemicarbazone and (E)-4-bromo-2-fluorobenzaldehyde thiosemicarbazone, which obviously is not realized in compound 2. This proved that compound 2 is not a thiosemicarbazone. Furthermore, the protons of the tropone (ring C) could be unequivocally assigned, and their coupling constants secured that no benzilic-type rearrangement happened to the tropolone, a reaction seen with colchic(in)oids under certain (alkaline) conditions, leading occasionally to the rearrangement products allocolchicine (colchicic acid methyl ester) [49,50] or colchicic acid (allocolchiceine) [51], both being aromatic in ring C. Under the reaction conditions employed for the synthesis of compound 2, the allocolchiceine sodium salt (Scheme 3) could be expected as a side product, but was not observed. This benzilic-type rearrangement (Scheme 3) was elucidated by Šantavý [50] and Fernholz [51].
Scheme 3.
The possible side product in the synthesis of compound 2, the allocolchiceine (syn. colchinoic acid, colchicinoic acid, colchicic acid) monosodium salt, originating from hydrolytic ring C contraction by a benzilic-type rearrangement.
Scheme 3.
The possible side product in the synthesis of compound 2, the allocolchiceine (syn. colchinoic acid, colchicinoic acid, colchicic acid) monosodium salt, originating from hydrolytic ring C contraction by a benzilic-type rearrangement.
The 13C-NMR spectrum of compound 2 in DMSO-d6 was interpreted with help of literature data on (–)-colchicine [46] and (–)-colchiceine [52]. In addition, own experimental observations were applied in the following aliphatic carbon assignments: δ 14.05 (O–CH2–CH3 ethyl acetate), 20.72 (ROOC–CH3 ethyl acetate), 22.49 (C-17, CH3 acetamide), 29.33 (C-5), 36.34 (C-6), 51.38 (C-7), 55.84 (14-OCH3)**, 59.72 (O–CH2–CH3 ethyl acetate), 60.62 (13-OCH3, 15-OCH3)**. The two double-starred assignments (**) are tentative and interchangeable (they could not be assigned unequivocally to the individual carbons). The aromatic and troponic carbon resonances, the carbonyl and the thiocarbonyl resonances, were detected as: δ 107.61 (C-4), 108.27 (C-11), 126.23 (C-8), 131.57 (C-1a), 134.26 (C-4a), 137.21 (C-12), 140.71 (C-3)***, 150.34 (C-1)***, 150.40 (C-10), 150.46 (C-12a), 152.61 (C-2)***, 152.73 (C-7a), 168.39 (C-16, HN–C=O acetamide), 170.30 (C=O ester carbonyl, ethyl acetate), 174.81 (C-9, C=O carbonyl), 181.86 (C-3′, C=S thiocarbonyl). The three triple-starred resonances (***) could not being assigned unequivocally to their individual carbon.
By these analyses it was found that the amidation product of (–)-colchicine, the substituted thiosemicarbazide compound 2, was not cyclic with regard to the thiosemicarbazide unit at ring C of compound 2. This was quite surprising, since the reaction product of (–)-colchicine with thiourea was cyclic with respect to the thiourea substitution in ring C [53,54], which seemed surprising in turn, because the tropolonic C-9 carbonyl group in (–)-colchicine did not react with common carbonyl reagents like hydroxylamine or semicarbazide [55,56]. The reason for the latter irregularity could be the special tropylium oxide resonance type of tropones and tropolones [57,58,59,60,61]. Therefore, the synthesis of compound 2 clearly obeyed the common rules for chemical reactivity of tropolones, whereas the synthesis of the cyclic thiourea congener [53,54] of compound 2 did not follow the common chemical reactivity experience for tropolones.
The Fourier–transform infrared (FT–IR) absorption spectra of the colchic(in)oid 10-(2-carbamothioylhydrazinyl)-10-demethoxycolchicine monohydrate × ⅔ (ethyl acetate) = compound 2, and of the reference substance (−)-colchicine sesquihydrate [(−)-colchicine × 1½ H2O] are given in the Supplementary Information (Figure S8) for comparison.
Interestingly, the natural colchic(in)oids (–)-colchicine, and the partialsynthetic (–)-colchicine derivative N-acetylcolchinol methyl ether (Figure 6), occur in pure atropisomeric forms (Figure 6), as was elucidated by Brossi et al. [62,63]. The natural forms have the (aS,7S)-absolute configuration (Figure 6). The correct assignment of the absolute configuration of (–)-colchicine was given as (aS,7S) by Brossi et al. [63] according to the Cahn–Ingold–Prelog (CIP) rules [64,65]. The wrong (aR,7S)-absolute configuration was firstly postulated in 1981 during studies on tubulin binding by (–)-colchicine [66], and later in 1999 by Berg & Bladh [67]. The absolute configuration at C-7 was established earlier as (7S) [68] by chemical degradation of natural (–)-colchicine to N-acetyl-L-glutamic acid.
Figure 6.
The atropisomerism of natural (–)-colchicine and its degradation product N-acetylcolchinol methyl ether. Both exhibit the (aS,7S)-absolute configuration, as was proved by Brossi et al. [62,63].
Taken together, the structure of the modified colchic(in)oid compound 2 could be proved with considerable evidence, and biological effects, especially antineoplastic properties, are expected from biological testing of compound 2. Renewed interest in colchic(in)oid research is indicated by reports on conjugating (–)-colchicine to vitamin B12 (cobalamin) [69] or paclitaxel (taxol) [70]. These colchic(in)oid conjugates were suggested for the chemotherapy of various neoplastic conditions.
2.2.3. The X-Ray Crystallographic Crystal and Molecular Structure Determination of Compound 2
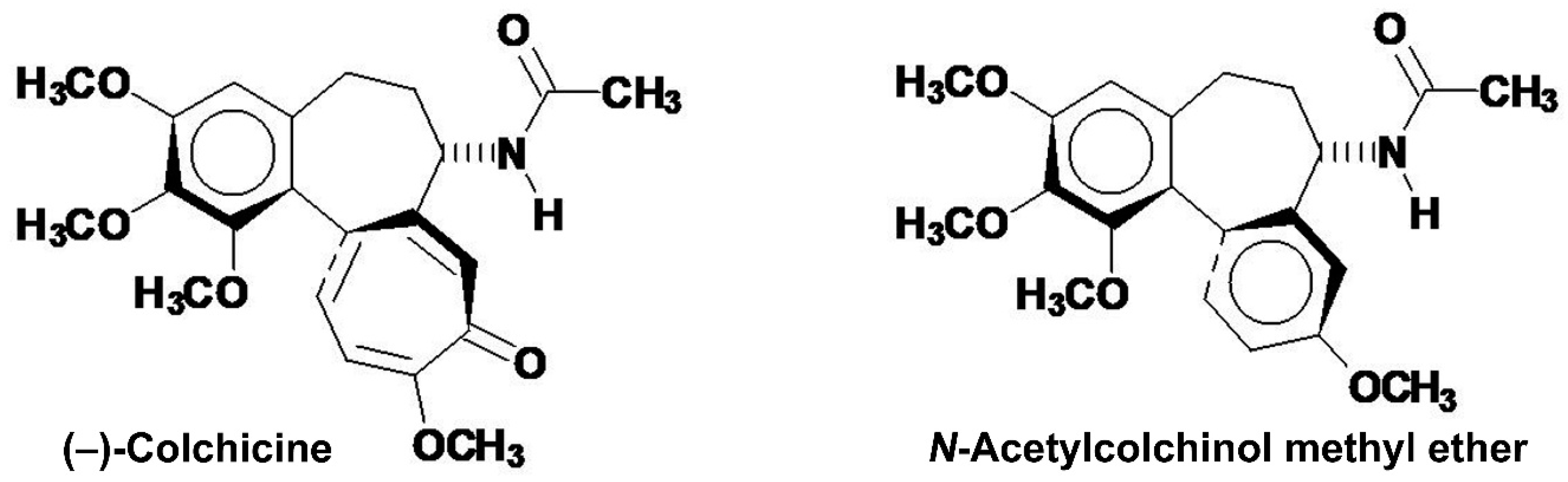
Compound 2 was crystallized from ethyl acetate and a single crystal was selected for X-ray crystallographic determination (at ϑ = 100 K) of the crystal and molecular structure of compound 2 (Figure 7, Figure 8, Figure 9, Figure S9, Figure S10). Compound 2 crystallized in the monoclinic space group P21 with ethyl acetate and water of crystallization [C22H26N4O5S × 1.5 H2O × 0.5 (C4H8O2)] (Z = 4) (Figure S9). The crystal packing (Figure 8) with indicated hydrogen bonds (Figure S10) in the unit cell (Z = 2) of compound 2 is depicted. It should be noted that the molecule is helical stereogenic and shows the [M(inus)]-helicity as N-[(aS,7S)-10-(2-carbamothioylhydrazinyl)-1,2,3-trimethoxy-9-oxo-5,6,7,9-tetrahydrobenzo[a]heptalen-7-yl]acetamide (Figure 9).
Figure 7.
The molecular view of one independent, isolated molecule of compound 2 as found in the single crystal. Compound 2 crystallized in the monoclinic space group P21 as sesquihydrate hemi(ethyl acetate) solvate (C24H33N4O7.50S) [C22H26N4O5S × 1.5 H2O × 0.5 (C4H8O2)] (Z = 4). Compound 2 is helical stereogenic and shows the (M)-helicity. The plane of the thiourea moiety in the 10-(thiosemicarbazide) substituent shows +119.1° torsion angle towards the 2-aminotropone plane.
Figure 7.
The molecular view of one independent, isolated molecule of compound 2 as found in the single crystal. Compound 2 crystallized in the monoclinic space group P21 as sesquihydrate hemi(ethyl acetate) solvate (C24H33N4O7.50S) [C22H26N4O5S × 1.5 H2O × 0.5 (C4H8O2)] (Z = 4). Compound 2 is helical stereogenic and shows the (M)-helicity. The plane of the thiourea moiety in the 10-(thiosemicarbazide) substituent shows +119.1° torsion angle towards the 2-aminotropone plane.
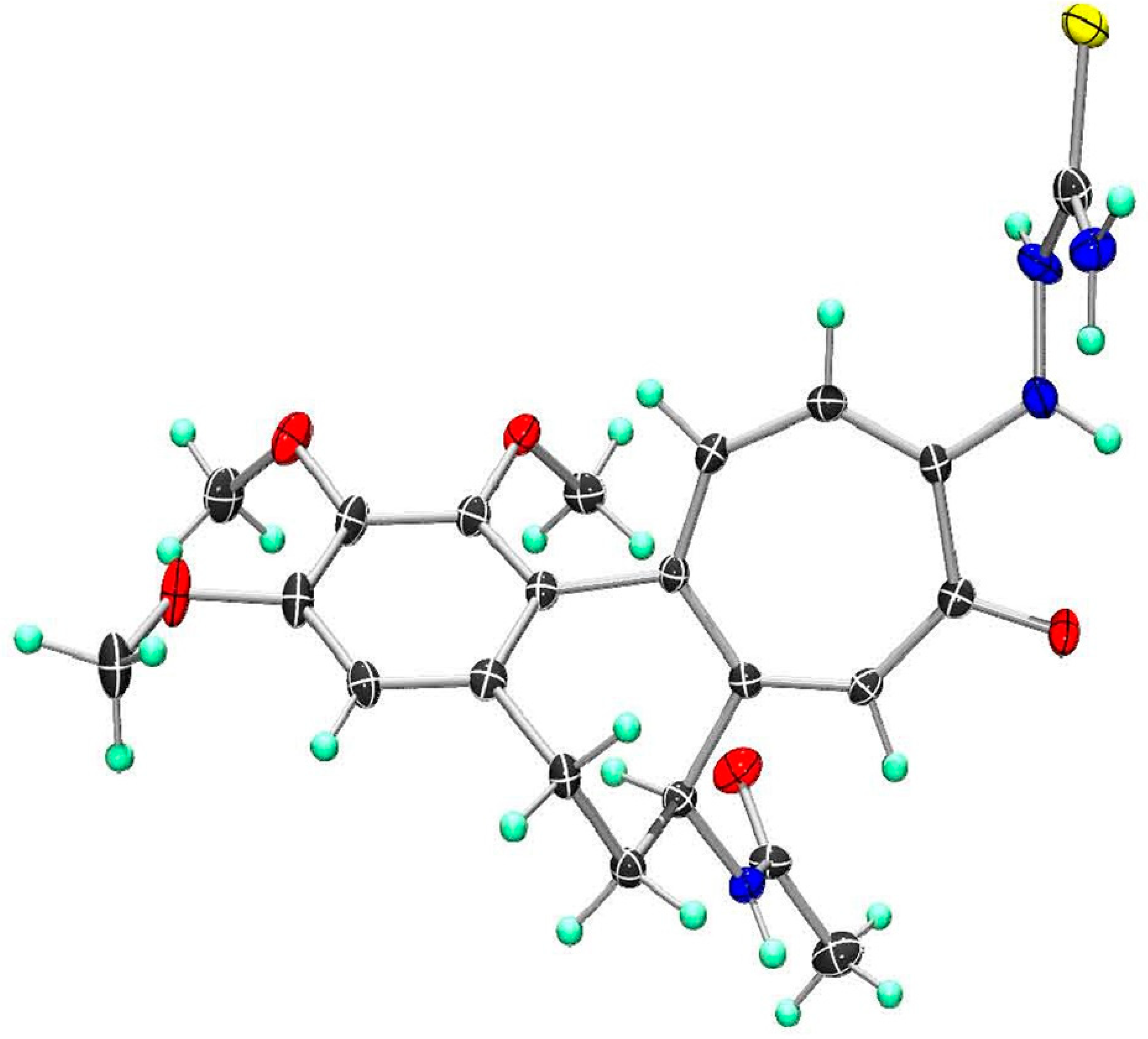
The helical axis atropisomerism view of one independent, isolated molecule of compound 2 as found in the single crystal is depicted (Figure 9). This stands in contradiction to a report that claimed the [P(lus)]-helicity (aR) for (–)-colchicine [67]. The classification of (M)-helicity for compound 2 followed the Cahn–Ingold–Prelog (CIP) rules for assignment of molecular helicity [64,65]. The (M)-helicity of (–)-colchicine was previously assigned correctly by Brossi et al. [63]. The X-ray crystallographic structure was deposited at The Cambridge Crystallographic Data Centre (CCDC) and assigned the deposition № CCDC 1839505 (ID: RIVGOW). The crystal data of the X-ray crystallographic determination of the crystal and molecular structure of compound 2 are tabulated (Table 1).
Figure 8.
Crystal packing in the monoclinic (space group P21) unit cell (Z = 2) of compound 2 crystallized as hydrate (ethyl acetate) solvate (2 C22H26N4O5S × 3 H2O × C4H8O2) C48H66N8O15S2 (M = 1059.21 g/mol). Unit cell dimensions (a, b, c are indicated from the origin o): a = 9.1886(5) Å, b = 20.9047(10) Å, c = 13.9841(7) Å, α = 90.00°, β = 106.153(2)°, γ = 90.00°, V = 2580.1(2) Å3.
Figure 8.
Crystal packing in the monoclinic (space group P21) unit cell (Z = 2) of compound 2 crystallized as hydrate (ethyl acetate) solvate (2 C22H26N4O5S × 3 H2O × C4H8O2) C48H66N8O15S2 (M = 1059.21 g/mol). Unit cell dimensions (a, b, c are indicated from the origin o): a = 9.1886(5) Å, b = 20.9047(10) Å, c = 13.9841(7) Å, α = 90.00°, β = 106.153(2)°, γ = 90.00°, V = 2580.1(2) Å3.
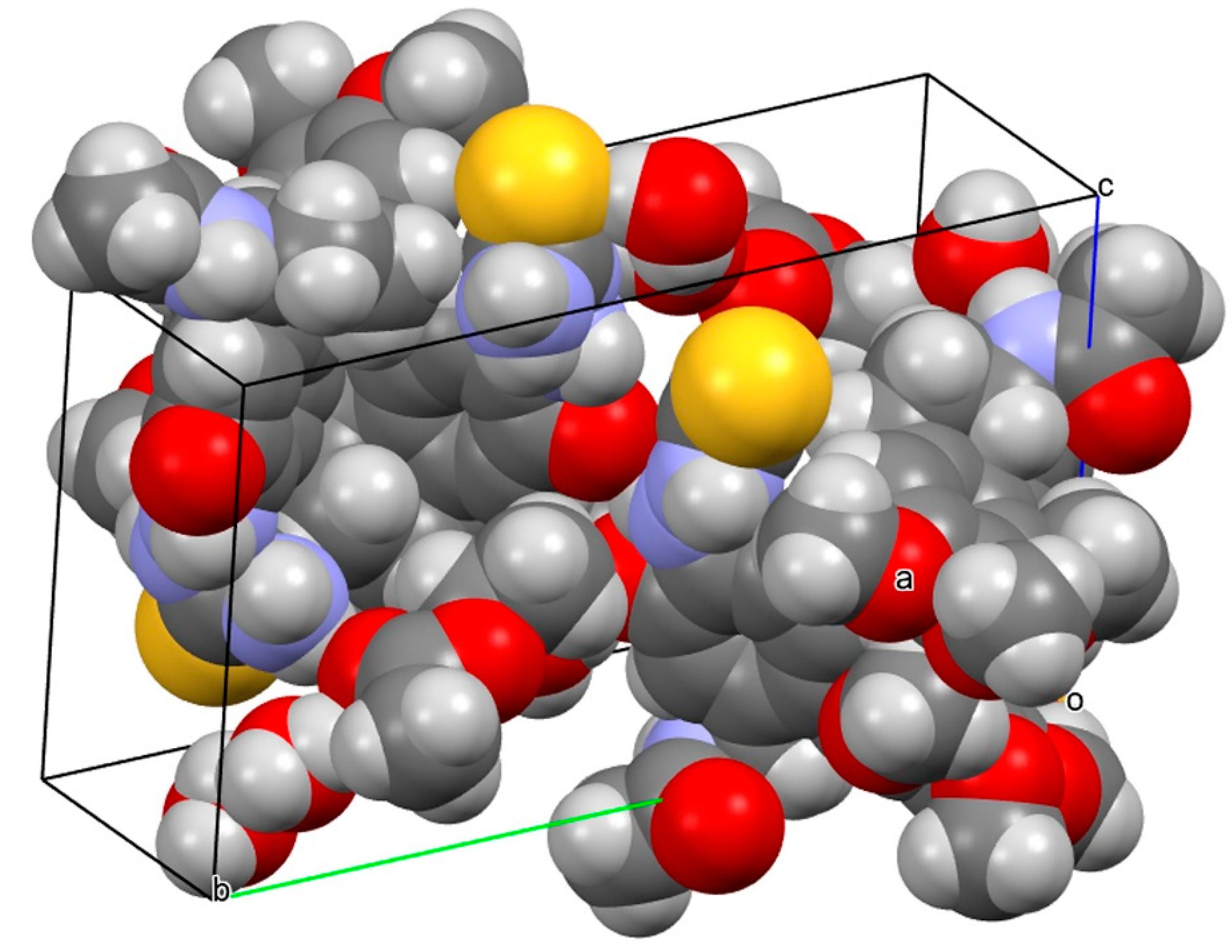
Figure 9.
The helical axis atropisomerism view of one independent, isolated molecule of compound 2 as found in the single crystal. Compound 2, the colchic(in)oid N-[(aS,7S)-10-(2-carbamothioylhydrazinyl)-1,2,3-trimethoxy-9-oxo-5,6,7,9-tetrahydrobenzo[a]heptalen-7-yl]acetamide, is helical stereogenic and shows the [M(inus)]-helicity. Priority rules [64,65] show the helical axis turn in (S).
Figure 9.
The helical axis atropisomerism view of one independent, isolated molecule of compound 2 as found in the single crystal. Compound 2, the colchic(in)oid N-[(aS,7S)-10-(2-carbamothioylhydrazinyl)-1,2,3-trimethoxy-9-oxo-5,6,7,9-tetrahydrobenzo[a]heptalen-7-yl]acetamide, is helical stereogenic and shows the [M(inus)]-helicity. Priority rules [64,65] show the helical axis turn in (S).

Table 1.
X-ray crystallographic data for compound 2, deposited and published as CSD Communication № CCDC 1839505 (ID: RIVGOW).
Table 1.
X-ray crystallographic data for compound 2, deposited and published as CSD Communication № CCDC 1839505 (ID: RIVGOW).
| Empirical formula | C48H66N8O15S2 |
| Formula weight, Mr (g·mol–1) | 1059.21 |
| Temperature, T (K) | 100(2) |
| Radiation, λ (Å) | MoK 0.71073 |
| Crystal system | Monoclinic |
| Space group | P21 |
| Unit cell dimensions | |
| a (Å) | 9.1886(5) |
| b (Å) | 20.9047(10) |
| c (Å) | 13.9841(7) |
| α (°) | 90.00 |
| β (°) | 106.153(2) |
| γ (°) | 90.00 |
| Unit cell volume, V (Å3) | 2580.1(2) |
| Formula units per unit cell, Z | 2 |
| Calculated density, ρcalc (Mg·m–3) | 1.363 |
| Absorption coefficient, μ (mm–1) | 0.178 |
| F(000) | 1124 |
| Theta (ϑ) range for collection | 1.80 to 26.05° |
| Reflections collected | 49598 |
| Independent reflections | 9686 |
| Minimum / maximum transmission, Tmin / Tmax | 0.9193 / 0.9929 |
| Absorption correction | Multi-scan, SADABS 2008/1 (G.M. Sheldrick, 2008) |
| Refinement method | Full-matrix least-squares on F2 |
| Data / parameters / restraints | 9686 / 693 / 5 |
| Goodness−of−fit on F2, S | 1.021 |
| Flack parameter, x | 0.09(6) |
| Final R indices [I > 2σ(I)] | R1 = 0.0506, wR2 = 0.1269 |
| R indices (all data) | R1 = 0.0594, wR2 = 0.1325 |
| Maximum / minimum residual electron density, Δρmax / Δρmin (e·Å–3) | 0.747 / –0.496 |
Summary of the crystal data for compound 2: C24H33N4O7.50S [C22H26N4O5S × 1½ H2O × ½ (C4H8O2)], Mr = 529.61 g/mol, colorless plate, 0.48 × 0.31 × 0.04 mm3, monoclinic space group P21, a = 9.1886(5) Å, b = 20.9047(10) Å, c = 13.9841(7) Å, β = 106.153(2)°, V = 2580.1(2) Å3, Z = 4, ρcalcd = 1.363 g·cm–3, μ = 0.178 mm–1, F(000) = 1124, T = 100(2) K, xFlack = 0.09(6), R1 = 0.0594, wR2 = 0.1325, 9686 independent reflections [2ϑ ≤ 52.1°] and 693 parameters. Computer programs utilized: APEX2 ver. 2008.3 (Bruker AXS, 2008), Saint+ ver. 7.53A (Bruker AXS, 2008), SHELXS97 (G.M. Sheldrick, 2008), SHELXL97 (G.M. Sheldrick, 2008), XP ver. 5.1 (Bruker AXS, 1998).
2.3. Compound 3
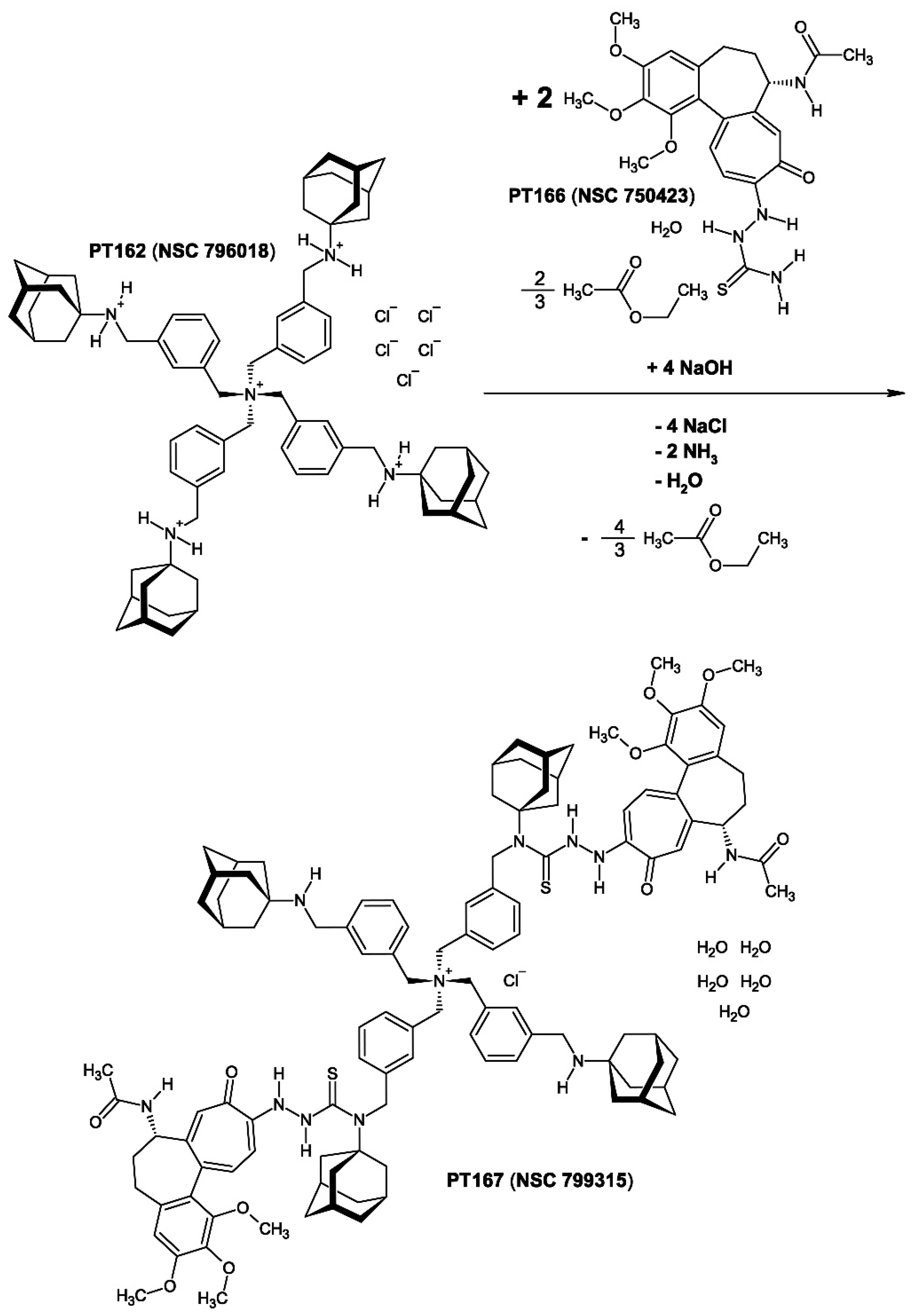
2.3.1. The Synthesis of Compound 3 [PT167 (NSC 799315)]
Compound 3 [PT167 (NSC 799315)] (Figure 10) was synthesized from compound 1 [PT162 (NSC 796018)] and compound 2 [PT166 (NSC 750423)] by trans-(thio)amidation catalysed by sodium hydroxide (NaOH) at room temperature. A similar trans-(thio)amidation originating from a thiosemicarbazide or thiosemicarbazone moiety was observed previously in the synthesis of retinazone, a retinoid thiosemicarbazone derivative [24,25]. As a result compound 2 was connected to compound 1 through a thioamide-bonding at an adamantanamine nitrogen (Scheme 4). The molecular stoichiometry of compound 3 was determined by 1H-NMR (Figure 11) spectroscopy experiments, as well as elemental analysis. Compound 1 recieved two molecules of compound 2 by trans-amidation to yield compound 3. The 1H-NMR spectrum of compound 3 (Figure 11) exhibits a peculiar resonance compression [the adamantane resonances 2.00 (24 H; β-CH2), 2.14 (12 H; γ-CH) could not being detected] induced by the large (macro)molecular structure of compound 3. This points to intramolecular (hydrophobic) interaction between the β-methylene and γ-methine structural elements of the compound 1-derived adamantanamine cages with the tropone ring (especially H-11) of the colchic(in)oid part of compound 3. An intramolecular interaction between the β-methylene and γ-methine structural elements of the adamantanamine cages with H-11 of the 10-(thiosemicarbazide)-substituted tropone ring in compound 3 could be demonstrated by molecular modeling (Figure 12). The resulting overall chemical structure of compound 3 is given (Figure 10). Compound 3 was registered by the National Cancer Institute (NCI) as NSC 799315.
Figure 10.
The organic chemical structure formula of compound 3 [PT167 (NSC 799315)] = C116H142N11O10S2+ × Cl− × 5 H2O = C116H152ClN11O15S2 (M = 2040.10 g/mol). The compound 3 contains one chloride anion (Cl−) and five water of crystallization (× 5 H2O).
Figure 10.
The organic chemical structure formula of compound 3 [PT167 (NSC 799315)] = C116H142N11O10S2+ × Cl− × 5 H2O = C116H152ClN11O15S2 (M = 2040.10 g/mol). The compound 3 contains one chloride anion (Cl−) and five water of crystallization (× 5 H2O).
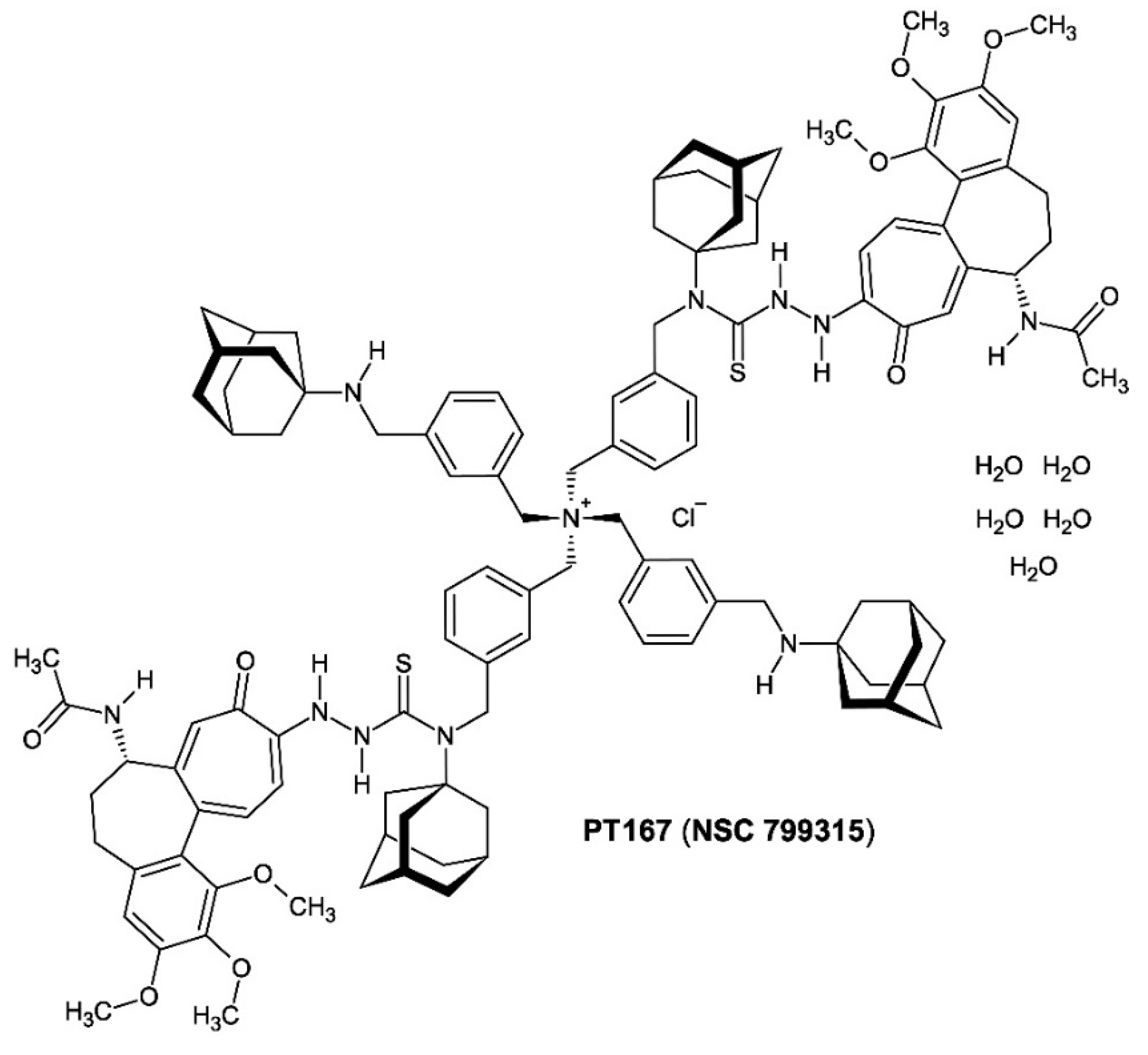
Figure 11.
The 700.43 MHz 1H-NMR spectrum of compound 3 = C116H142N11O10S2+ × Cl− × 5 H2O = C116H152ClN11O15S2 (M = 2040.10 g/mol) dissolved in DMSO-d6. Yellow marked is the solvent DMSO-d6 residual resonance [DMSO-d5h1 quintet at δ 2.50 ppm, 2Jgem (2H−1H) = +1.9 Hz] [DMSO]. The water broad singlet resonance is found at δ 3.32 ppm. Traces of the utilized synthesis solvents ethanol (triplet, δ 1.05 ppm; quartet, δ 3.44 ppm) and acetone (singlet, δ 2.09 ppm) can be detected.
Figure 11.
The 700.43 MHz 1H-NMR spectrum of compound 3 = C116H142N11O10S2+ × Cl− × 5 H2O = C116H152ClN11O15S2 (M = 2040.10 g/mol) dissolved in DMSO-d6. Yellow marked is the solvent DMSO-d6 residual resonance [DMSO-d5h1 quintet at δ 2.50 ppm, 2Jgem (2H−1H) = +1.9 Hz] [DMSO]. The water broad singlet resonance is found at δ 3.32 ppm. Traces of the utilized synthesis solvents ethanol (triplet, δ 1.05 ppm; quartet, δ 3.44 ppm) and acetone (singlet, δ 2.09 ppm) can be detected.
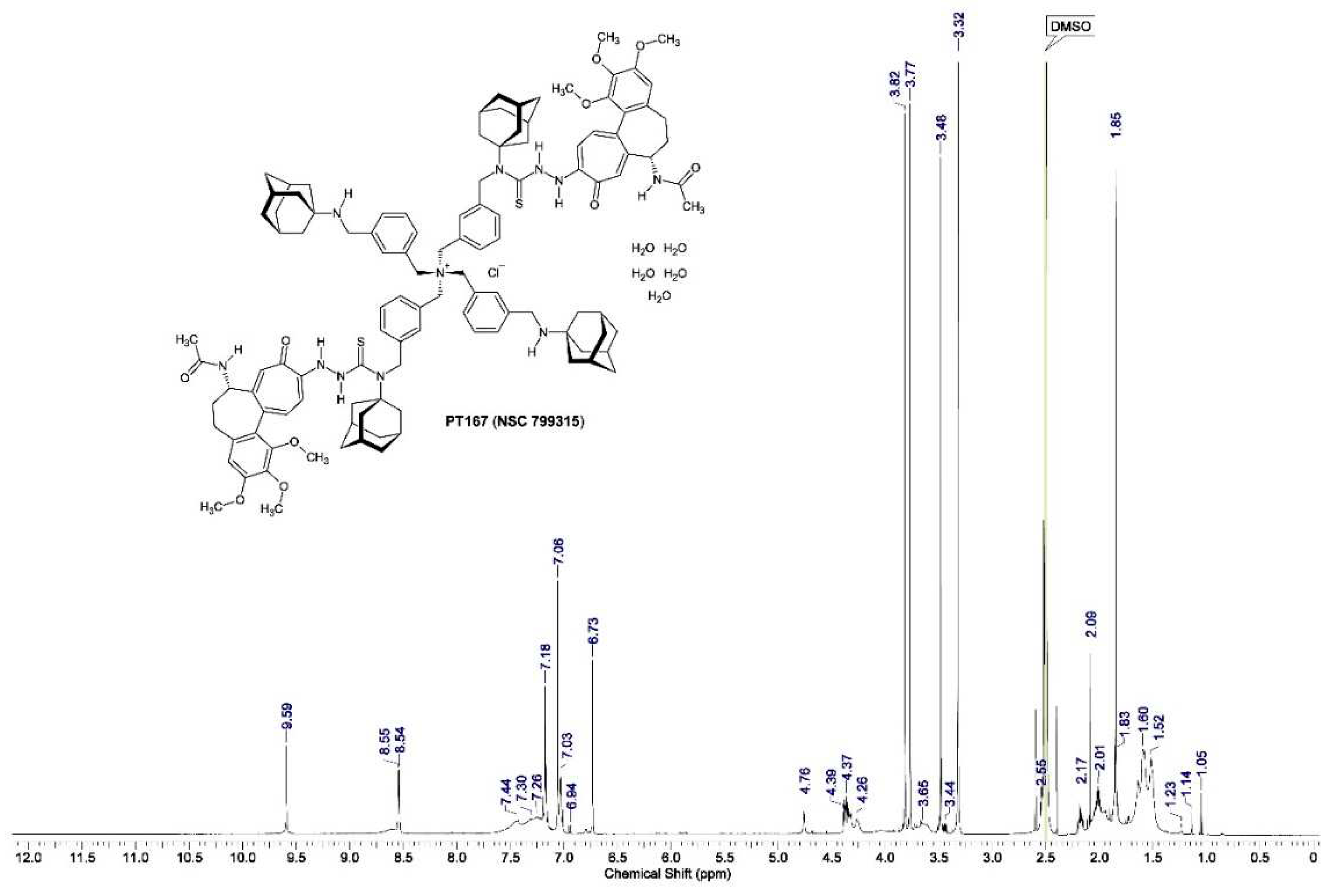
Figure 12.
Molecular modeling {ACD/Chem Sketch version 12.01 with integrated ACD/3D Viewer (Advanced Chemistry Development, Inc., Toronto, Ontario, Canada) and processed with Mercury 3.1 version 3.1.1 [The Cambridge Crystallographic Data Centre (CCDC), Cambridge, UK]} of compound 3 = C116H142N11O10S2+ × Cl− × 5 H2O = C116H152ClN11O15S2 (M = 2040.10 g/mol) without the chloride counteranion (Cl−) and the water of crystallization (pentahydrate). An intramolecular interaction between the β-methylene and γ-methine structural elements of the adamantanamine cages with H-11 of the 10-(thiosemicarbazide)-substituted tropone ring in compound 3 could be demonstrated.
Figure 12.
Molecular modeling {ACD/Chem Sketch version 12.01 with integrated ACD/3D Viewer (Advanced Chemistry Development, Inc., Toronto, Ontario, Canada) and processed with Mercury 3.1 version 3.1.1 [The Cambridge Crystallographic Data Centre (CCDC), Cambridge, UK]} of compound 3 = C116H142N11O10S2+ × Cl− × 5 H2O = C116H152ClN11O15S2 (M = 2040.10 g/mol) without the chloride counteranion (Cl−) and the water of crystallization (pentahydrate). An intramolecular interaction between the β-methylene and γ-methine structural elements of the adamantanamine cages with H-11 of the 10-(thiosemicarbazide)-substituted tropone ring in compound 3 could be demonstrated.
2.3.2. The Proton Nuclear Magnetic Resonance Spectrum (1H-NMR) of Freshly Synthesized Compound 3
Evidence for the formulation of compound 3 as the macromolecular entity [(bis{3-[(tricyclo[3.3.1.13,7]decan-1-ylamino)methyl]benzyl}ammonio)bis(methanediylbenzene-3,1-diylmethanediyl)]di-2-[(aS,7S)-7-(acetylamino)-1,2,3-trimethoxy-9-oxo-5,6,7,9-tetrahydrobenzo[a]heptalen-10-yl]-N-(tricyclo[3.3.1.13,7]decan-1-yl)hydrazinecarbothioamide chloride pentahydrate stems from the proton NMR (1H-NMR) spectrum of compound 3 [PT167 (NSC 799315)] dissolved in deuterated dimethyl sulfoxide (DMSO-d6) (Figure 11).
The proton resonances were measured as chemical shifts δ (ppm): δ 1.48–1.68 (24 H, br m; δ-CH2, adamantane), 1.83 (2 H, m; HA-6, colch), 1.85 (6 H, s; 17-CH3, colch), 2.01 (2 H, m; HB-6, colch), 2.17 (2 H, m; HA-5, colch), 2.52 (2 H, m; HB-5, colch), 3.48 (6 H, s; 13-OCH3, colch)*, 3.65 (2 H, br s; secondary amine N–H), 3.77 (6 H, s; 15-OCH3, colch)*, 3.82 (6 H, s; 14-OCH3, colch)*, 4.26 (br m; 8-CH2, m-xylylene), 4.32‒4.39 (2 H, br m; H-7, colch), 4.76 (s; 7-CH2, m-xylylene), 6.73 (2 H, s; H-4, colch), 7.03 (2 H, br m; H-11, colch), 7.06 (2 H, s; H-8, colch), 7.12‒7.72 (br m; H-4, H-6, H-5, H-2, m-xylylene), 7.17 (2 H, br m; H-12, colch), 8.54 (2 H, d; 3J = 7.7 Hz; N–H acetamide, colch), 9.59 (4 H, s; 1′-N–H, 2′-N–H, hydrazinecarbothioamide) [colch = the colchic(in)oid part of compound 3; * these assignments are tentative and interchangeable (they could not be assigned unequivocally to the individual methoxy groups); the adamantane resonances δ 2.00 ppm (β-CH2) and 2.14 ppm (γ-CH) were not detected due to paramagnetic resonance compression].
2.3.3. The Proton Nuclear Magnetic Resonance Spectrum (1H-NMR) of Compound 3 after Six Year Storage at +0−4 °C in the Refrigerator
The specimen of compound 3 which was synthesized at Saturday, May 27th, 2017, was investigated by proton NMR (Figure 13) at Wednesday, August 23rd, 2023, after over six years storage at +0−4 °C in the refrigerator. The specimen was newly dried over CaCl2 in vacuo for 19 h. The proton resonances were measured as chemical shifts δ (ppm): δ 1.14 (1 H, s; hydroxytropyl radical O–H), 1.45–1.81 (24 H, br m; δ-CH2, adamantane), 1.83 (2 H, m; HA-6, colch), 1.85 (6 H, s; 17-CH3, colch), 1.91 (2 H, m; HB-6, colch), 2.02–2.14 (12 H, br m; γ-CH), 2.18 (2 H, br m; HA-5, colch), 2.55 (2 H, br m; HB-5, colch), 2.86–3.08 [7 H, br s; 7-CH2, m-xylylene CH2 including ammonium ylide R−(CH−)N+(CH2R)3], 3.49 (6 H, s; 13-OCH3, colch)*, 3.67 (1 H, s; secondary amine N–H), 3.78 (6 H, s; 15-OCH3, colch)*, 3.82 (6 H, s; 14-OCH3, colch)*, 4.26‒4.39 (8 H, br m; H-7, colch; 3 × 8-CH2 m-xylylene), 4.76 (2 H, s; 1 × 8-CH2, m-xylylene at 8-NH+• radical cation), 6.72 (2 H, s; H-4, colch), 6.99 (2 H, br s; H-11, colch), 7.06 (2 H, s; H-8, colch), 7.17 (2 H, br s; H-12, colch), 7.30‒7.37 (12 H, br m; H-4, H-6, H-2, m-xylylene), 7.51 (4 H, br s; H-5, m-xylylene), 8.14 (1 H, s; 8-NH+• radical cation N–H), 8.48 (2 H, d; 3J = 7.9 Hz; N–H acetamide, colch), 9.56 (4 H, br s; 1′-N–H, 2′-N–H, hydrazinecarbothioamide) [colch = the colchic(in)oid part of compound 3; * these assignments are tentative and interchangeable (they could not be assigned unequivocally to the individual methoxy groups); the adamantane β-CH2 resonance δ 2.00 ppm was not detected due to paramagnetic resonance compression].
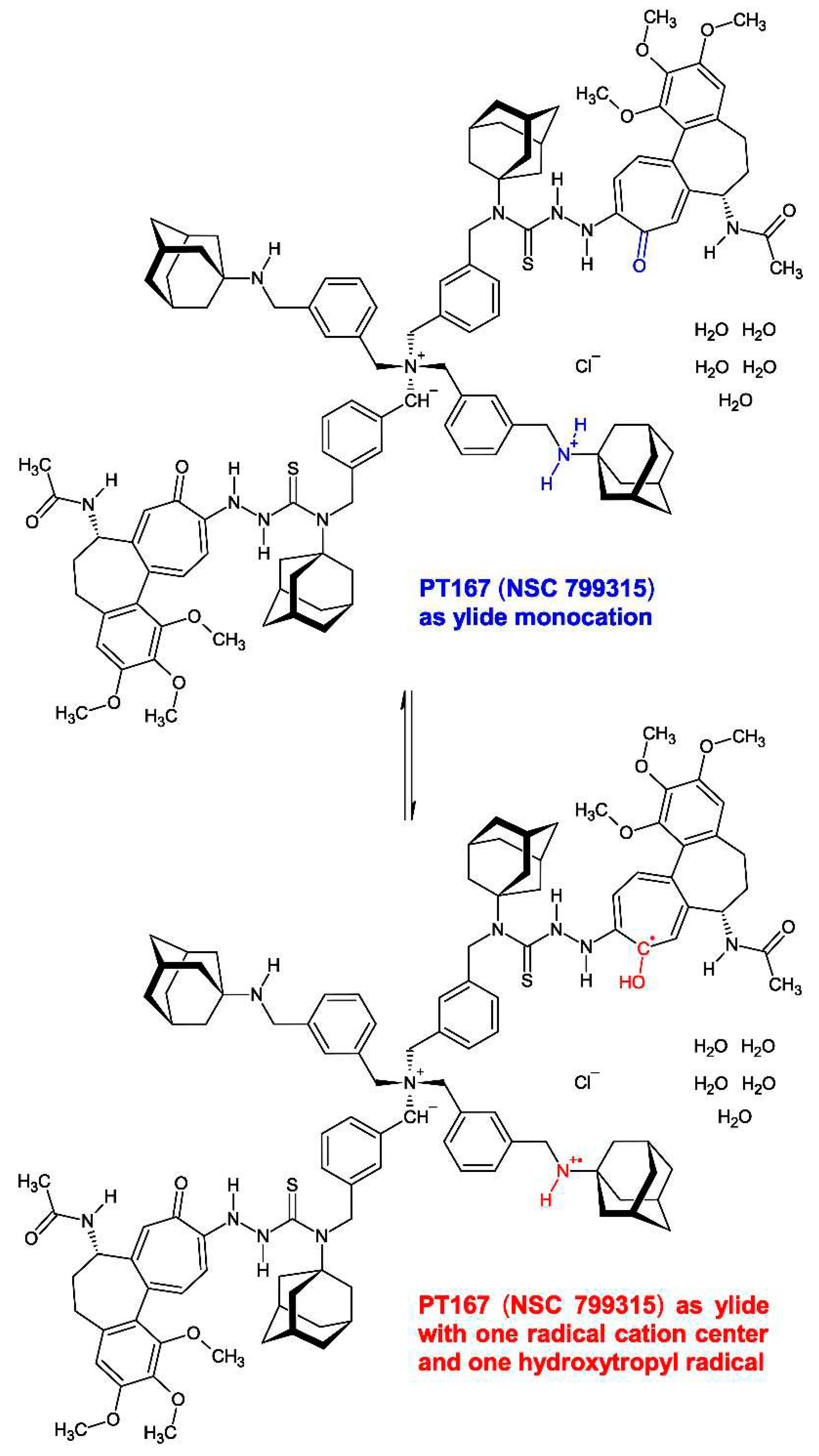
The 1H-NMR spectrum of newly dried (CaCl2, in vacuo, 19 h) compound 3 in DMSO-d6 after storing the substance over six years at +0−4 °C in the refrigerator (Figure 13) convincingly proves that (i) the substance is very pure even after six years storage, (ii) exhibits the chemical structure given by me, and (iii) points to proton transfer during storage yielding the ylide monocation (ylide monohydrochloride) with discernible resonances (7 H) for the ylide R−(CH−)N+(CH2R)3.
This substance bears a peculiar magnetic property. During NMR spectrometer shim it was detected that newly dried (CaCl2, in vacuo, 19 h) compound 3 is, in part, paramagnetic, because there were considerable difficulties in shimming the NMR spectrometer probe magnetic field (personal communication Robbin Schnieders). The operator told me that the DMSO-d6 solution of newly dried compound 3 had to be strongly diluted with DMSO-d6, and that the spectrum acquisition time had to be elongated considerably. This pointed to inclusion of a paramagnetic partial structure in newly dried compound 3. Indeed, the 1H-NMR spectrum (Figure 13) gives evidence for that interpretation which is depicted (Figure 14). The colchic(in)oid part of the molecule picks up one electron from the ylide monocation (ylide monohydrochloride) (Figure 14, in blue) yielding a resonance-stabilized 1-hydroxycyclohepta-2,4,6-trien-1-yl radical (hydroxytropyl radical) [71,72] producing in consequence the 8-NH+• radical cation (Figure 14, in red).
Figure 13.
The 499.86 MHz 1H-NMR spectrum of newly dried (CaCl2, in vacuo, 19 h) compound 3 in DMSO-d6 after storing the substance over six years at +0−4 °C in the refrigerator. The spectrum was measured at ϑ = 35 °C on a Varian Unity INOVA 500 instrument. Yellow marked are the solvent DMSO-d6 residual resonance [DMSO] and the water broad singlet resonance at δ 3.25 ppm [Water]. Only traces of the utilized synthesis solvent acetone (singlet, δ 2.08 ppm) are present.
Figure 13.
The 499.86 MHz 1H-NMR spectrum of newly dried (CaCl2, in vacuo, 19 h) compound 3 in DMSO-d6 after storing the substance over six years at +0−4 °C in the refrigerator. The spectrum was measured at ϑ = 35 °C on a Varian Unity INOVA 500 instrument. Yellow marked are the solvent DMSO-d6 residual resonance [DMSO] and the water broad singlet resonance at δ 3.25 ppm [Water]. Only traces of the utilized synthesis solvent acetone (singlet, δ 2.08 ppm) are present.
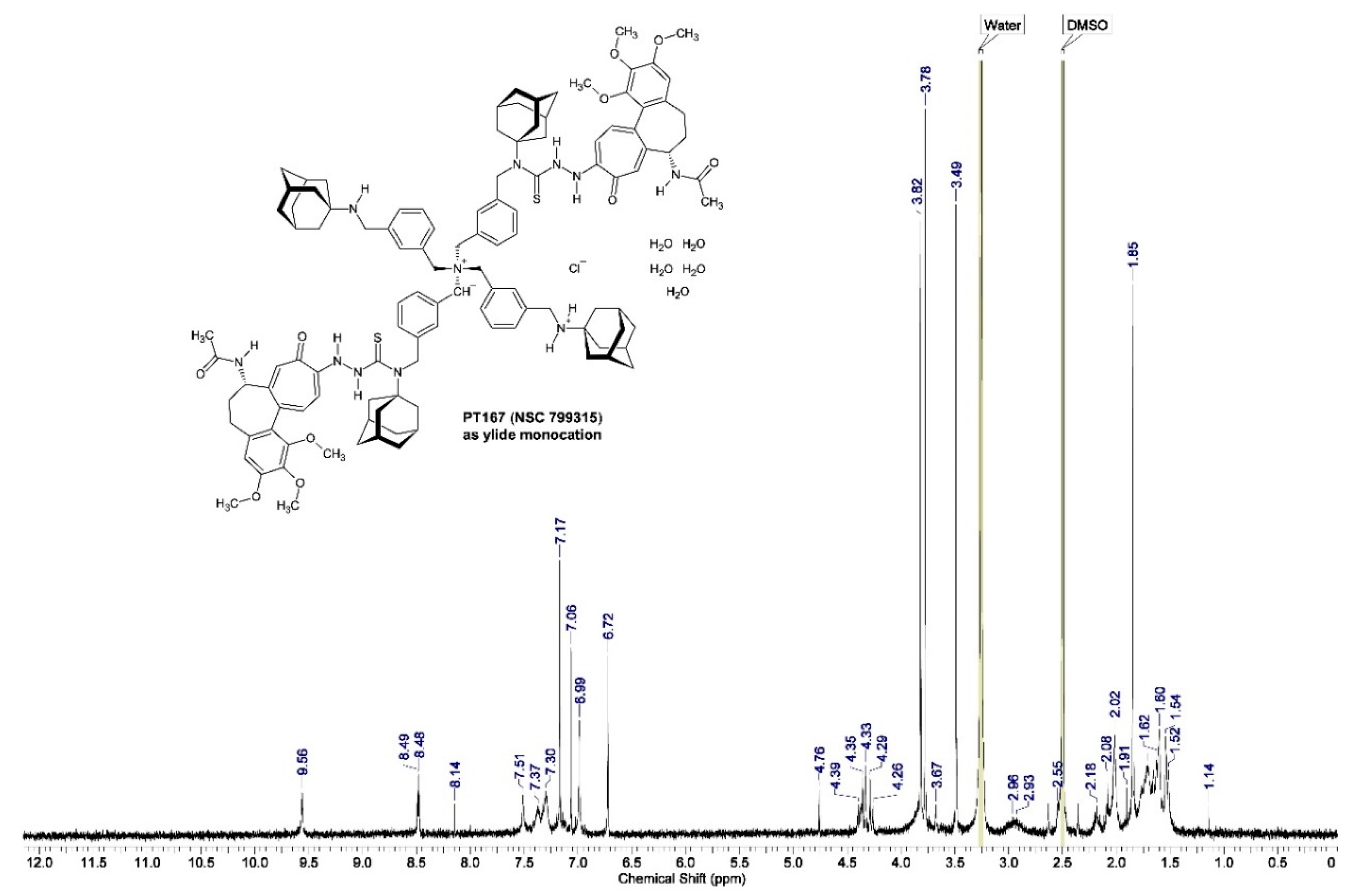
Figure 14.
The electron transfer in compound 3 yielding a partially paramagnetic molecular species. The colchic(in)oid part of the molecule picks up one electron from the ylide monocation yielding a resonance-stabilized 1-hydroxycyclohepta-2,4,6-trien-1-yl radical (hydroxytropyl radical) [71,72] producing the 8-NH+• radical cation.
Figure 14.
The electron transfer in compound 3 yielding a partially paramagnetic molecular species. The colchic(in)oid part of the molecule picks up one electron from the ylide monocation yielding a resonance-stabilized 1-hydroxycyclohepta-2,4,6-trien-1-yl radical (hydroxytropyl radical) [71,72] producing the 8-NH+• radical cation.
2.3.4. The Liquid Chromatographic (HPLC) Investigation of Compound 3
The specimen of compound 3 which was synthesized at Saturday, May 27th, 2017, was investigated at Friday, July 21st, 2023, after over six years storage at +0−4 °C in the refrigerator. The high-performance liquid chromatography (HPLC) (Figure 15A,B) was performed with a reversed phase C8 (RP8, n-octyl) column and gradient elution with eluent A = water/0.1% (v/v) formic acid (HCOOH) and eluent B = acetonitrile/0.1% (v/v) formic acid (HCOOH). The flowrate was 0.5 ml/min and the linear eluent gradient was t0min = 95% eluent A/5% eluent B to t13min = 5% eluent A/95% eluent B, t16min = stop. A 5 µl volume of a 50 µM compound 3 solution in acetonitrile (N≡C−CH3) was injected (0.25 nmol, 510.025 ng).
Figure 15.
(A) The total ion current (TIC) chromatogram of the liquid chromatographic (HPLC)/electrospray ionization (ESI) time−of−flight (ToF) mass spectrometric investigation of compound 3 after storing the substance over six years at +0−4 °C in the refrigerator. The high-performance liquid chromatography (HPLC) of compound 3 was performed on a reversed phase C8 (RP8, n-octyl) column with gradient elution. Indicated are the presumed multiple ionic species of compound 3 (see Table 2) which were separated due to the complicated ionization kinetics of the macromolecular substance compound 3 dissolved in acetonitrile with presence of 0.1% (v/v) HCOOH in the HPLC eluents. (B) The analogous liquid chromatographic (HPLC) investigation of compound 3 with UV detection at λ = 335 nm.
Figure 15.
(A) The total ion current (TIC) chromatogram of the liquid chromatographic (HPLC)/electrospray ionization (ESI) time−of−flight (ToF) mass spectrometric investigation of compound 3 after storing the substance over six years at +0−4 °C in the refrigerator. The high-performance liquid chromatography (HPLC) of compound 3 was performed on a reversed phase C8 (RP8, n-octyl) column with gradient elution. Indicated are the presumed multiple ionic species of compound 3 (see Table 2) which were separated due to the complicated ionization kinetics of the macromolecular substance compound 3 dissolved in acetonitrile with presence of 0.1% (v/v) HCOOH in the HPLC eluents. (B) The analogous liquid chromatographic (HPLC) investigation of compound 3 with UV detection at λ = 335 nm.
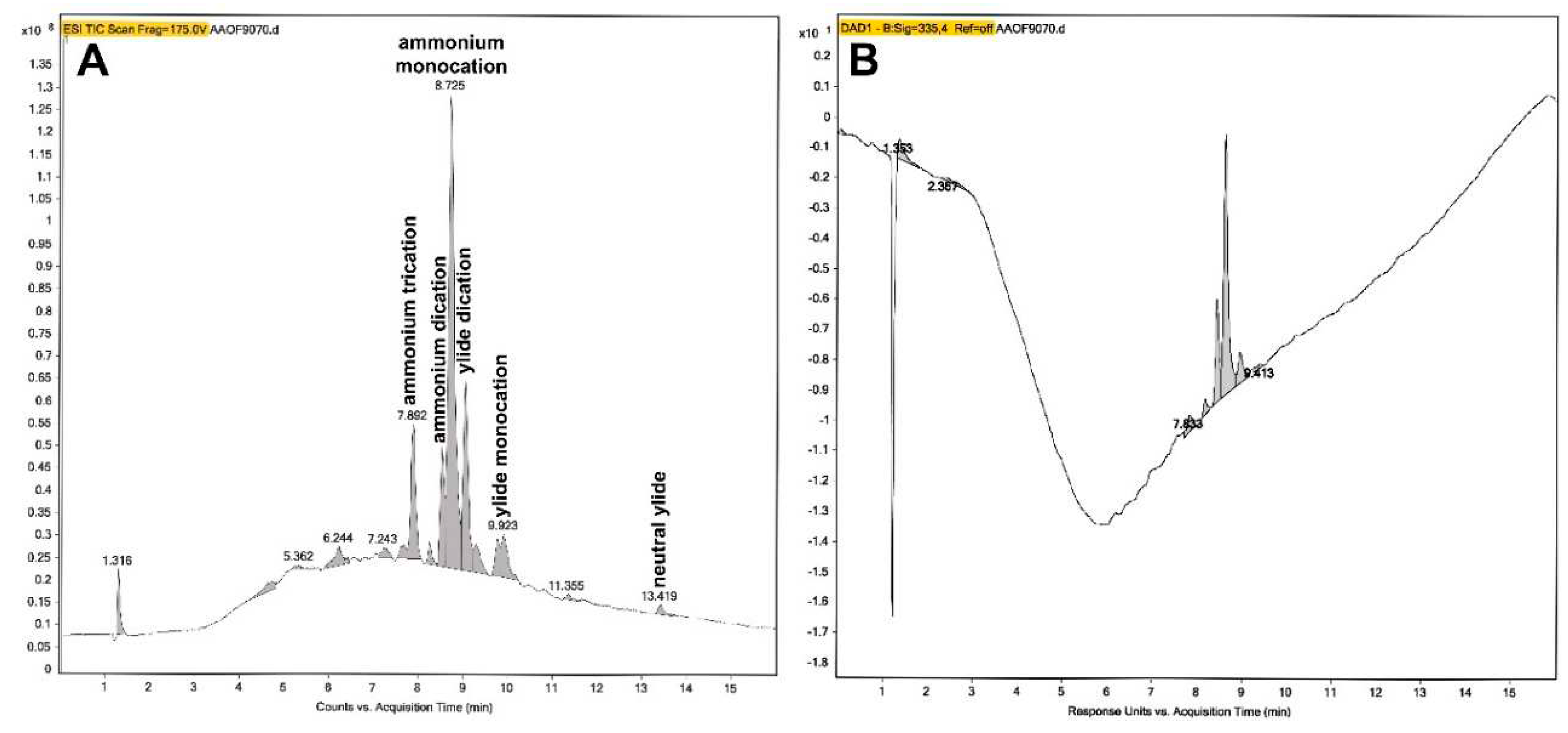
The total ion current (TIC) chromatogram is shown in Figure 15A, the chromatogram with UV detection at λ = 335 nm is depicted in Figure 15B. Multiple ionic species of compound 3 were separated (Table 2) due to the complicated ionization kinetics of compound 3 dissolved in acetonitrile (N≡C−CH3) with presence of 0.1% (v/v) HCOOH. The quaternary ammonium compound 3 can be protonated once or twice, or being not protonated like the in situ substance (Table 2). Moreover, compound 3 can exist in equilibrium as a neutral nitrogen ylide [73,74,75] at the central quaternary ammonium cation. This nitrogen ylide can be protonated once or twice, or being not protonated (neutral nitrogen ylide) (Table 2). The substance compound 3 was quite pure as judged from the chromatogram with UV detection at λ = 335 nm (Figure 15B).
Table 2.
The liquid chromatographic (HPLC)/electrospray ionization (ESI) time−of−flight (ToF) mass spectrometric investigation of compound 3 after storing the substance over six years at +0−4 °C in the refrigerator. Given are the HPLC peak regions in the total ion current (TIC) chromatogram (Figure 15A) which were scanned in the ESI mass spectrometer with the observed fragmentation peaks (in parentheses: relative peak intensity of the 100% base peak), and the presumed ionization identities of the ionized species of compound 3 dissolved in acetonitrile with presence of 0.1% (v/v) HCOOH in the HPLC eluents.
Table 2.
The liquid chromatographic (HPLC)/electrospray ionization (ESI) time−of−flight (ToF) mass spectrometric investigation of compound 3 after storing the substance over six years at +0−4 °C in the refrigerator. Given are the HPLC peak regions in the total ion current (TIC) chromatogram (Figure 15A) which were scanned in the ESI mass spectrometer with the observed fragmentation peaks (in parentheses: relative peak intensity of the 100% base peak), and the presumed ionization identities of the ionized species of compound 3 dissolved in acetonitrile with presence of 0.1% (v/v) HCOOH in the HPLC eluents.
| HPLC peak (min) | Peak identity | ESI−MS fragment peaks (m/z) (relative peak intensity of the 100% base peak) |
|---|---|---|
| 7.842−7.925 | ammonium trication | 290.1670 (100%), 678.3662 (12.5%), 135.1157 (9.6%) |
| 8.508−8.558 | ammonium dication | 712.3545 (100%), 123.0807 (16.5%), 356.6799 (2.1%), 483.2713 (1.3%) |
| 8.608−8.658 | ammonium monocation | 712.3550 (100%), 543.3511 (27.9%), 356.6804 (18.1%), 123.0806 (14.2%), 653.3701 (5.4%), 483.2714 (2.5%), 409.2404 (1.7%) |
| 8.691−8.741 | ammonium monocation | 712.3558 (100%), 356.6814 (19.1%), 578.2448 (6.3%), 135.1165 (5.8%) |
| 9.024−9.074 | ylide dication | 483.2727 (100%), 965.5391 (80.9%), 712.3534 (19.5%), 123.0798 (14.1%), 356.6793 (5.0%), 831.4260 (4.3%), 577.2110 (3.4%), 213.1784 (2.3%) |
| 9.740−9.973 | ylide monocation | 227.2000 (100%), 123.0804 (47.2%), 609.8644 (31.0%), 712.3522 (6.6%), 398.7700 (5.8%), 1218.7191 (5.0%), 796.5327 (4.0%), 483.2712 (2.5%) |
| 13.386−13.453 | neutral ylide | 123.0805 (100%), 637.3055 (45.6%), 227.1988 (7.1%), 439.2038 (5.8%), 712.3521 (3.2%) |
2.3.5. The Electrospray Ionization (ESI) Mass Spectrometric Investigation of Compound 3 after HPLC Separation
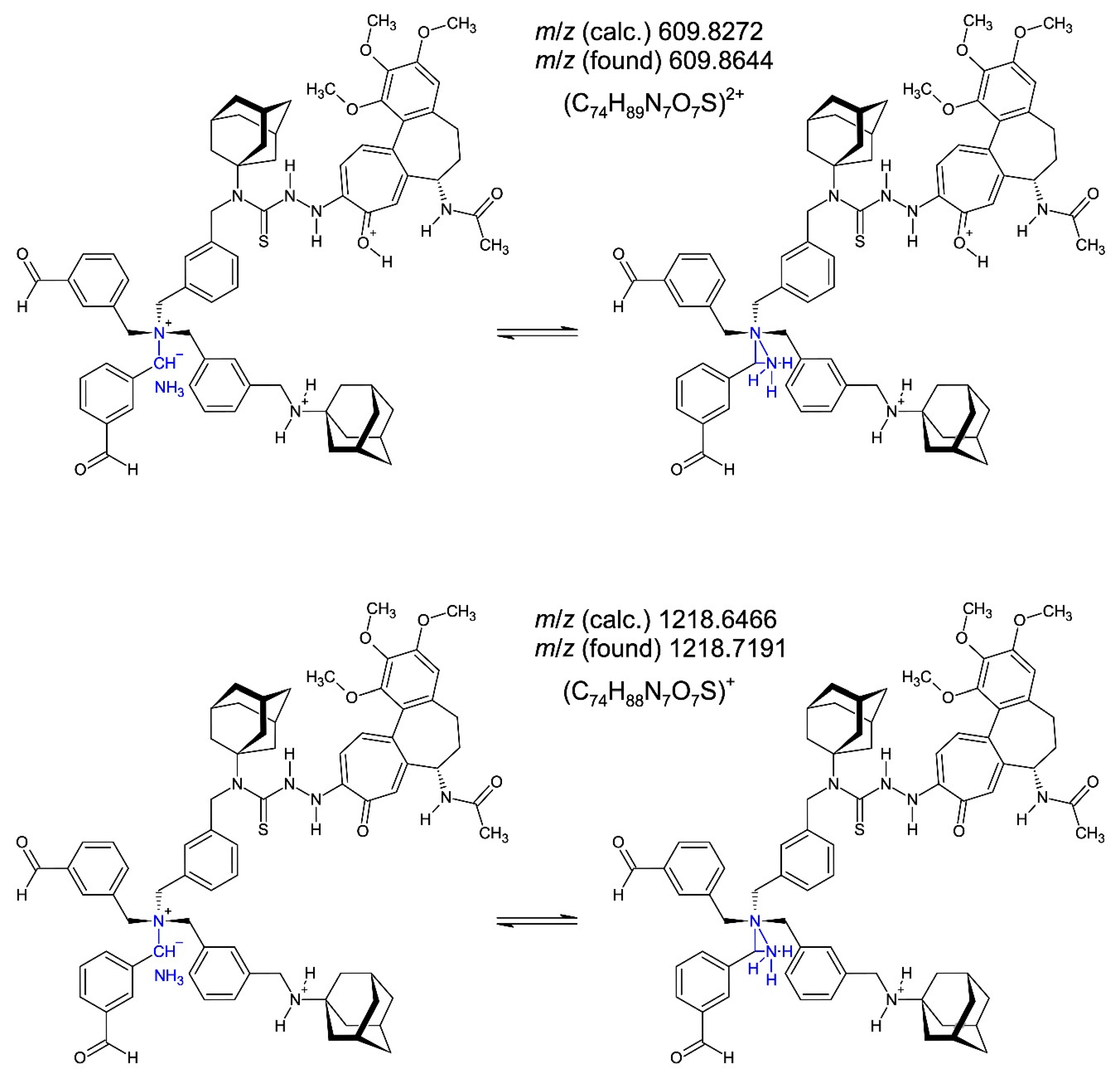
The six structure-proofing fragment cations in the electrospray ionization (ESI) time−of−flight (ToF) mass spectrometry of compound 3 (Figure S11−S20) following HPLC separation on a RP8 column (LC/MS coupling) are shown in Figure 16 (left, dications; right, monocations). The cations (C58H74N6O5S)2+ m/z 483.2727 (100%) (generated from ylide dication), (C58H73N6O5S)+ m/z 965.5391 (80.9%) (generated from ylide dication), (C74H89N7O7S)2+ m/z 609.8644 (31.0%) (generated from ylide monocation), (C74H88N7O7S)+ m/z 1218.7191 (5.0%) (generated from ylide monocation), (C40H49N4O6S)2+ m/z 356.6814 (19.1%) (generated from ammonium monocation), and (C40H50N5O5S)+ m/z 712.3550 (100%) (generated from ammonium monocation) are the major, structure-proofing fragments of compound 3 created in the ESI−ToF mass spectrometer under the fragmentor voltage Vf = 175 V. The molecule cation (C116H142N11O10S2)+ m/z 1913.0382 (or a higher protonated form) was not observed due to extensive molecule fragmentation.
The two peaks (C74H89N7O7S)2+ m/z 609.8644 (31.0%) (generated from ylide monocation) and (C74H88N7O7S)+ m/z 1218.7191 (5.0%) (generated from ylide monocation) (Figure 16) are to be formulated as ammonia (NH3) coordination-stabilized [73,74] nitrogen ylides [75] being in equilibrium with a mass spectrometric generated species exhibiting pentavalent nitrogen [76,77,78] according to Figure 17. The fragment cations (C18H21NNaO)+ m/z 290.17 [(3-{[(adamantan-1-yl)imino-κN]methyl}benzaldehyde-κO)sodium(1+)], (C8H7O2)+ m/z 135.12 [protonated isophthal(di)aldehyde], (C8H11O)+ m/z 123.08 [protonated m-xylyl alcohol (3-methylbenzyl alcohol)], and (C16H21N)+• m/z 227.20 [N-phenyladamantan-1-amine radical cation], accompany the greater fragments pointing to extensive fragmentation force under the fragmentor voltage Vf = 175 V. The N-phenyladamantan-1-amine radical cation results from 1-{[(adamantan-1-yl)amino]methyliumyl}cyclopenta-2,4-dien-1-ide (C16H21N, a zwitterionic fulvene) created from the benzyl species N-{[3-(aminomethyl)phenyl]methyl}adamantan-1-amine (C18H26N2) through loss of C2H5N and fulvene−to−benzene rearrangement [79,80].
In summary, the chemical structure of compound 3 could be substantiated as derivative of colchiceine hydrazide (10-hydrazinyl-10-demethoxycolchicine) [53,54] bridged over a thiocarbonyl to the compound 1 core, being quite pure. None compound 2 and compound 1, the synthesis starting substances, or colchiceine hydrazide, a possible degradation product, could be detected in the compound 3 preparation by highly sensitive detection methods.
Figure 16.
The six structure-proofing fragment cations (left, dications; right, monocations) in the electrospray ionization (ESI) time−of−flight (ToF) mass spectrometric investigation of compound 3 after storing the substance over six years at +0−4 °C in the refrigerator. The m/z 483.2727 (100%) (generated from ylide dication), m/z 965.5391 (80.9%) (generated from ylide dication), m/z 609.8644 (31.0%) (generated from ylide monocation), m/z 1218.7191 (5.0%) (generated from ylide monocation), m/z 356.6814 (19.1%) (generated from ammonium monocation), and m/z 712.3550 (100%) (generated from ammonium monocation) are the major, structure-proofing fragments of compound 3.
Figure 16.
The six structure-proofing fragment cations (left, dications; right, monocations) in the electrospray ionization (ESI) time−of−flight (ToF) mass spectrometric investigation of compound 3 after storing the substance over six years at +0−4 °C in the refrigerator. The m/z 483.2727 (100%) (generated from ylide dication), m/z 965.5391 (80.9%) (generated from ylide dication), m/z 609.8644 (31.0%) (generated from ylide monocation), m/z 1218.7191 (5.0%) (generated from ylide monocation), m/z 356.6814 (19.1%) (generated from ammonium monocation), and m/z 712.3550 (100%) (generated from ammonium monocation) are the major, structure-proofing fragments of compound 3.
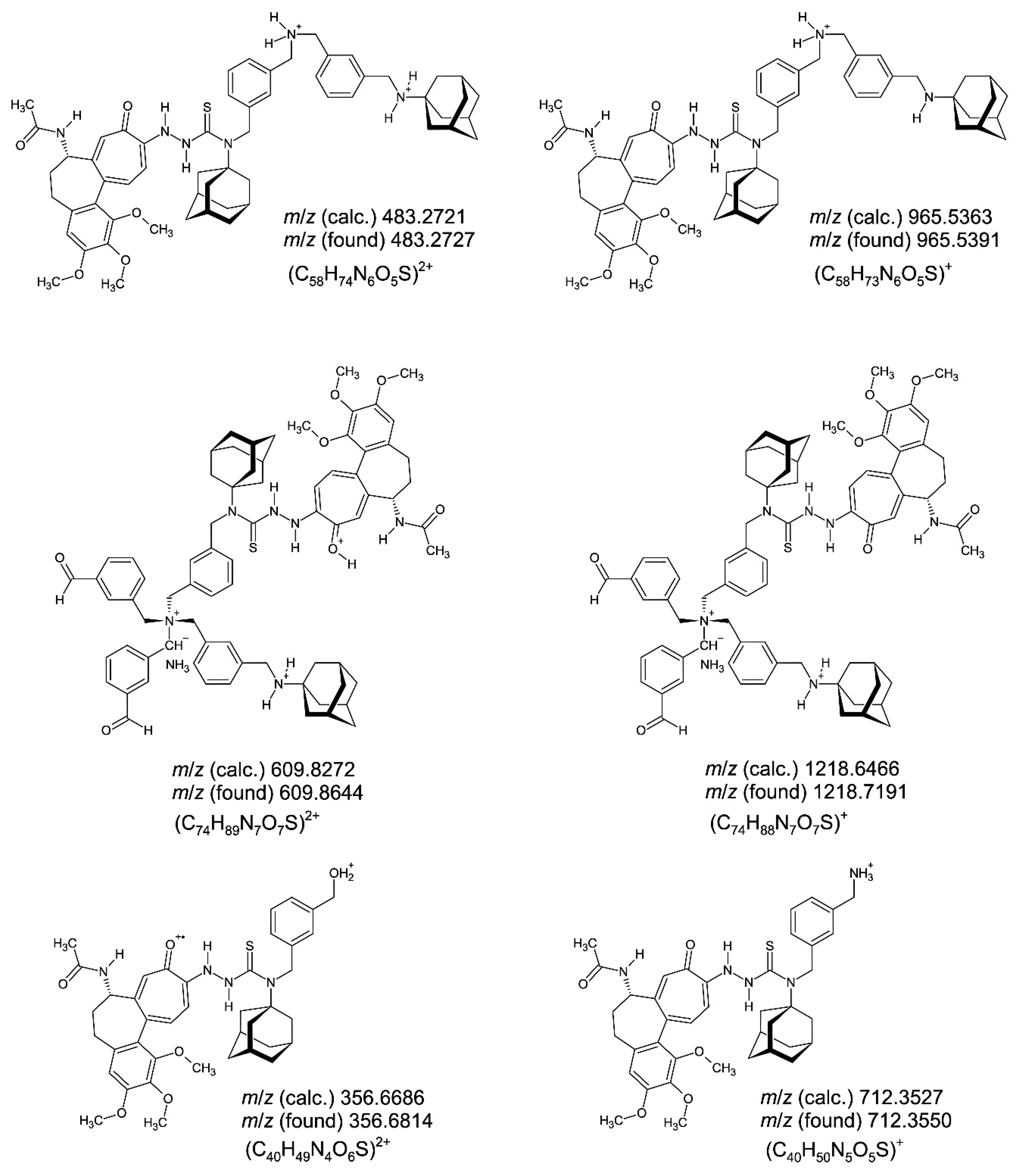
Figure 17.
The two cations (C74H89N7O7S)2+ m/z 609.8644 (31.0%) [generated from of compound 3 ylide monocation] and (C74H88N7O7S)+ m/z 1218.7191 (5.0%) [generated from of compound 3 ylide monocation] (see Figure 16) are to be formulated as ammonia (NH3)-stabilized [73,74] nitrogen ylides [75] (left, in blue) being in equilibrium with a mass spectrometric enabled species characterized by twice pentavalent nitrogen [76−78] (right, in blue).
Figure 17.
The two cations (C74H89N7O7S)2+ m/z 609.8644 (31.0%) [generated from of compound 3 ylide monocation] and (C74H88N7O7S)+ m/z 1218.7191 (5.0%) [generated from of compound 3 ylide monocation] (see Figure 16) are to be formulated as ammonia (NH3)-stabilized [73,74] nitrogen ylides [75] (left, in blue) being in equilibrium with a mass spectrometric enabled species characterized by twice pentavalent nitrogen [76−78] (right, in blue).
3. Biological Activities
3.1. NCI 60 Cell Five-Dose Screen with the Drugs Compound 1, Compound 2 and Compound 3
3.1.1. National Cancer Institute (NCI) Developmental Therapeutics Program (DTP) 60-Cancer Cell 5-Dose Testing
First page: graphic allover presentation of second page with All Cell Lines in one graphic and molar concentrations expressed in logarithmic log10 unit. The earlier the colored curves aspire to the bottom, the more potent is the drug. The more the curves strive from ±0% to –100%, the more the tumor is actually killed by the drug.
Second page: inhibition curves for tumor cell lines summarized in terms of tumor type. The Sample Concentrations are given at the x-axis in log10 units (−9 = 1 nM, −8 = 10 nM, −7 = 100 nM, −6 = 1 µM, −5 = 10 µM, −4 = 100 µM, −3 = 1,000 µM = 1 mM). The Percentage Growth are given at the y-axis in %, and spans from +100% to –100%. 0% Growth means that the tumor is still there but not growing anymore. –100% Growth means that the tumor cells are all dead and had died by apoptosis or necrosis, this is the ideal outcome. The smoother the curve, the more reliable is the tumor inhibition. Generally, it is not sufficient to reach only 0% Growth, since the tumor is still there. Ideal is –100% Growth, since the tumor cells were completely killed by the drug, but only few antineoplastic drugs in clinical use reach this. Cytostatics in clinical use generally reach only 0% Growth, this is called cytostasis (therefore the name cytostatics).
Third page: the actual used concentrations in log10 unit are given for five doses (Note: mostly these are not smooth values, but technically created values). Then the Mean Optical Densities of the used vital stain sulforhodamine B (SRB) retained in the cells are given. The higher the SRB optical density, the more is the number of living tumor cells. Then the Percent Growth is given again. The GI50 (Growth Inhibition 50%), TGI (Total Growth Inhibition = 0% Growth), and LC50 (Lethal Concentration 50% = –50% Growth) are given in linear concentration (E–6 = 10–6 M = µM, E–5 = 10–5 M...) units. A > 1.00E–X (for example: > 1.00E–4 = > 100 µM) means that the corresponding defined criterion is not reached by the drug = failure to reach the defind tumor inhibition criterion (GI50, TGI, or LC50). The NSC number of the test drug is given at the heading. The NSC Number is a standardized system of all anticancer compounds tested by NCI. The NSC number can be used for unequivocal identification and definition of all anticancer drugs, if registered and tested by NCI.
Fourth page: this is the summary of the results expressed in log10 units. The defined tumor inhibition criterion (GI50, TGI, or LC50) is recalculated in log10 expression, and the the colored bars which indicate then the sensitivity of the individual tumor cell line to the agent are given in log10 units. Bar to the left: less sensitive than mean; Bar to the right: more sensitive than mean. The most important feature is at the bottom of the page: the MID (Mean of Inhibition Data) indicates the mean concentration for all tested cell lines required for the drug to reach the defined tumor inhibition criterion (GI50, TGI, or LC50). It is given in Log10GI50, Log10TGI and Log10LC50. From that values the corresponding MID is calculated. The more negative the MID, the more potent is the drug. The MID can be transformed from logarithmic into linear concentrations by the formula: c = 10MID. The Delta and Range of the MID correspond to these definitions, expressed in logarithmic log10 unit:
The Delta is defined as:
Delta = Mean — Growth Percent of the drug’s most inhibited cell line
The Range is defined as:
Range = (Growth Percent of the drug’s least inhibited cell line — Mean) + Delta
3.1.2. Overall NCI 60 Cell Five-Dose Screen Results with the Drugs Compound 1, Compound 2 and Compound 3
Compound 1 (PT162, NSC 796018), compound 2 (PT166, NSC 750423) and compound 3 (PT167, NSC 799315) were screened in the NCI DTP 60-cancer cell 5-dose testing program. The results are summarized in the Table S1. Compound 1 and compound 3 generally started to inhibit cancer cell growth in the submicromolar range, whereby compound 3 was slightly more potent than compound 1. The responses of compound 1 and compound 3 regarding inhibition of cancer cell growth were remarkably smooth, regular and consistent. Nearly all cancer cell lines were inhibited by compound 1 and compound 3 in a very consistent fashion, including leukemia cell lines for compound 3. In contrast, the inhibiting effect of compound 2 on cancer cell growth was widely variable. Importantly, compound 1 and compound 3, including leukemia cell lines for compound 3, induced consistent cancer cell death in almost all cancer cell lines. In contrast, compound 2 failed to induce cancer cell death in nearly all cancer cell lines. The GI50 for compound 1 was 1.288 µM, for compound 2 0.933 µM, and for compound 3 1.349 µM (Table S1). The TGI for compound 1 was 4.677 µM, for compound 2 32.359 µM, and for compound 3 4.571 µM (Table S1). The LC50 for compound 1 was 16.596 µM, for compound 2 95.499 µM, and for compound 3 15.849 µM (Table S1). As can be clearly seen from these data compound 2 failed to induce cancer cell death and acts only cytostatic, whereas compound 1 and compound 3 successfully induced cancer cell death to nearly −100% cancer cell growth and can be classified as tumoricidal.
Compound 1 and compound 3 were consistently active versus wild-type p53-containing cancer cell lines and cancer cell lines with mutant or lost p53 protein (Table S1). The p53 status of the individual cancer cell lines in the NCI DTP 60-cancer cell 5-dose testing cell line panel was taken as published [81,82].
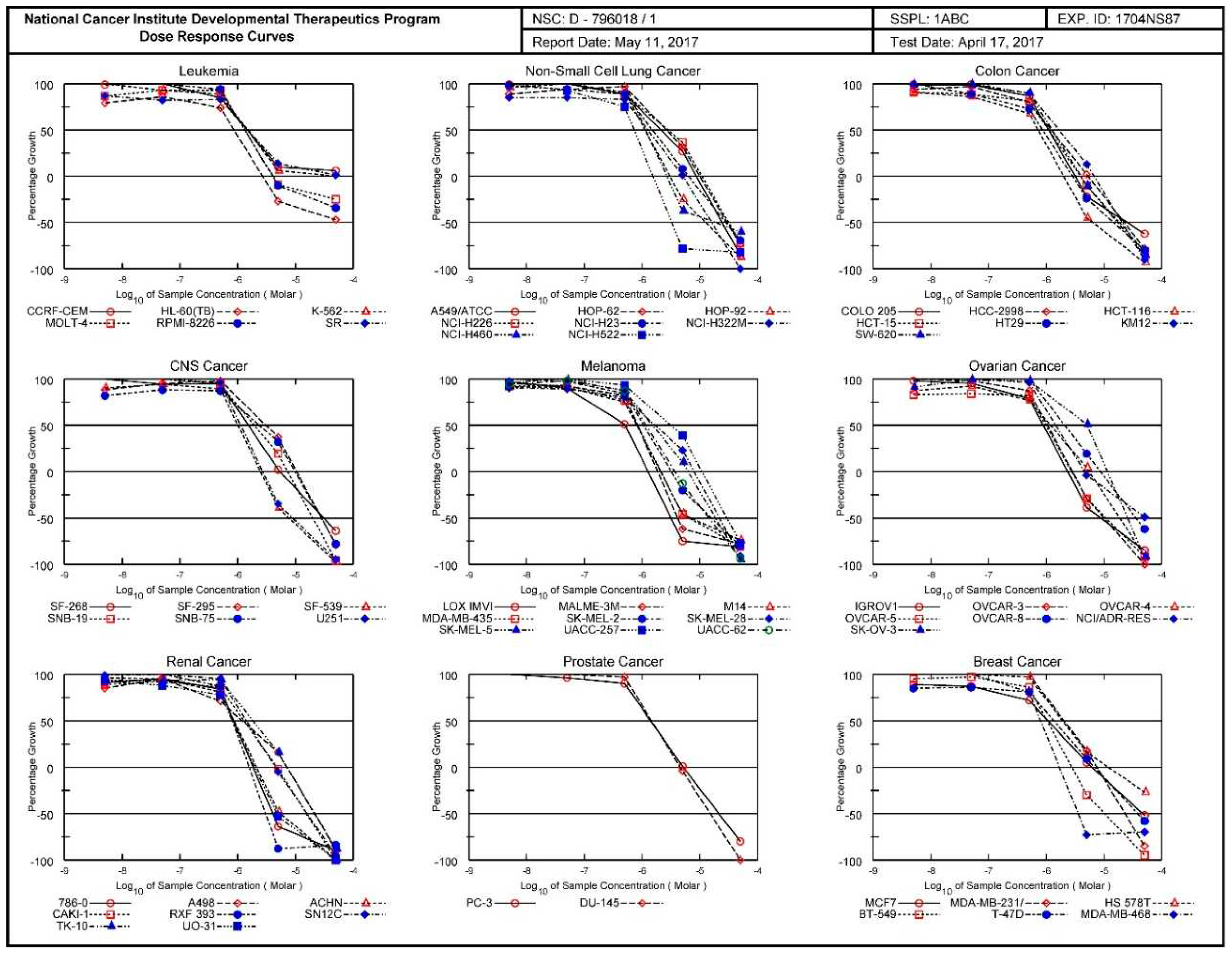
3.1.3. National Cancer Institute (NCI) Developmental Therapeutics Program (DTP) 60-Cancer Cell 5-Dose Testing Results with Compound 1
The four pages of the National Cancer Institute (NCI) Developmental Therapeutics Program (DTP) 60-cancer cell 5-dose testing results for compound 1 (PT162, NSC 796018) are given in succession. First page (Figure S21): graphic allover presentation of second page with All Cell Lines in one graphic. Second page (Figure 18): inhibition curves for tumor cell lines arranged/ordered for general tumor type. Third page (Figure S22): the Mean Optical Densities of the utilized vital stain sulforhodamine B (SRB) retained in the cells, the actual used concentrations in log10 unit for five doses, and the Percent Growth are given. The GI50 (Growth Inhibition 50%), TGI (Total Growth Inhibition = 0% Growth), and LC50 (Lethal Concentration 50% = –50% Growth) are given in linear concentration units. Fourth page (Figure S23): this is the summary of the results expressed in log10 units. The defined tumor inhibition criterion (GI50, TGI, or LC50) is recalculated in log10 expression, and the the colored bars which indicate then the sensitivity of the individual tumor cell line to the agent are given in log10 units.
Figure 18.
Graphic presentation of the antineoplastic activity exhibited by the pure drug compound 1 (PT162, NSC 796018) in the National Cancer Institute (NCI) Developmental Therapeutics Program (DTP) 60-cancer cell 5-dose testing with the inhibition curves for sixty human tumor cell lines arranged/ordered in terms of general tumor type.
Figure 18.
Graphic presentation of the antineoplastic activity exhibited by the pure drug compound 1 (PT162, NSC 796018) in the National Cancer Institute (NCI) Developmental Therapeutics Program (DTP) 60-cancer cell 5-dose testing with the inhibition curves for sixty human tumor cell lines arranged/ordered in terms of general tumor type.
3.1.4. National Cancer Institute (NCI) Developmental Therapeutics Program (DTP) 60-Cancer Cell 5-Dose Testing Results with Compound 2
The four pages of the National Cancer Institute (NCI) Developmental Therapeutics Program (DTP) 60-cancer cell 5-dose testing results for compound 2 (PT166, NSC 750423) are given in succession. First page (Figure S24): graphic allover presentation of second page with All Cell Lines in one graphic. Second page (Figure 19): inhibition curves for tumor cell lines arranged/ordered for general tumor type. Third page (Figure S25): the Mean Optical Densities of the utilized vital stain sulforhodamine B (SRB) retained in the cells, the actual used concentrations in log10 unit for five doses, and the Percent Growth are given. The GI50 (Growth Inhibition 50%), TGI (Total Growth Inhibition = 0% Growth), and LC50 (Lethal Concentration 50% = –50% Growth) are given in linear concentration units. Fourth page (Figure S26): this is the summary of the results expressed in log10 units. The defined tumor inhibition criterion (GI50, TGI, or LC50) is recalculated in log10 expression, and the the colored bars which indicate then the sensitivity of the individual tumor cell line to the agent are given in log10 units.
Figure 19.
Graphic presentation of the antineoplastic activity exhibited by the drug compound 2 (PT166, NSC 750423) in the National Cancer Institute (NCI) Developmental Therapeutics Program (DTP) 60-cancer cell 5-dose testing with the inhibition curves for sixty human tumor cell lines arranged/ordered in terms of general tumor type.
Figure 19.
Graphic presentation of the antineoplastic activity exhibited by the drug compound 2 (PT166, NSC 750423) in the National Cancer Institute (NCI) Developmental Therapeutics Program (DTP) 60-cancer cell 5-dose testing with the inhibition curves for sixty human tumor cell lines arranged/ordered in terms of general tumor type.

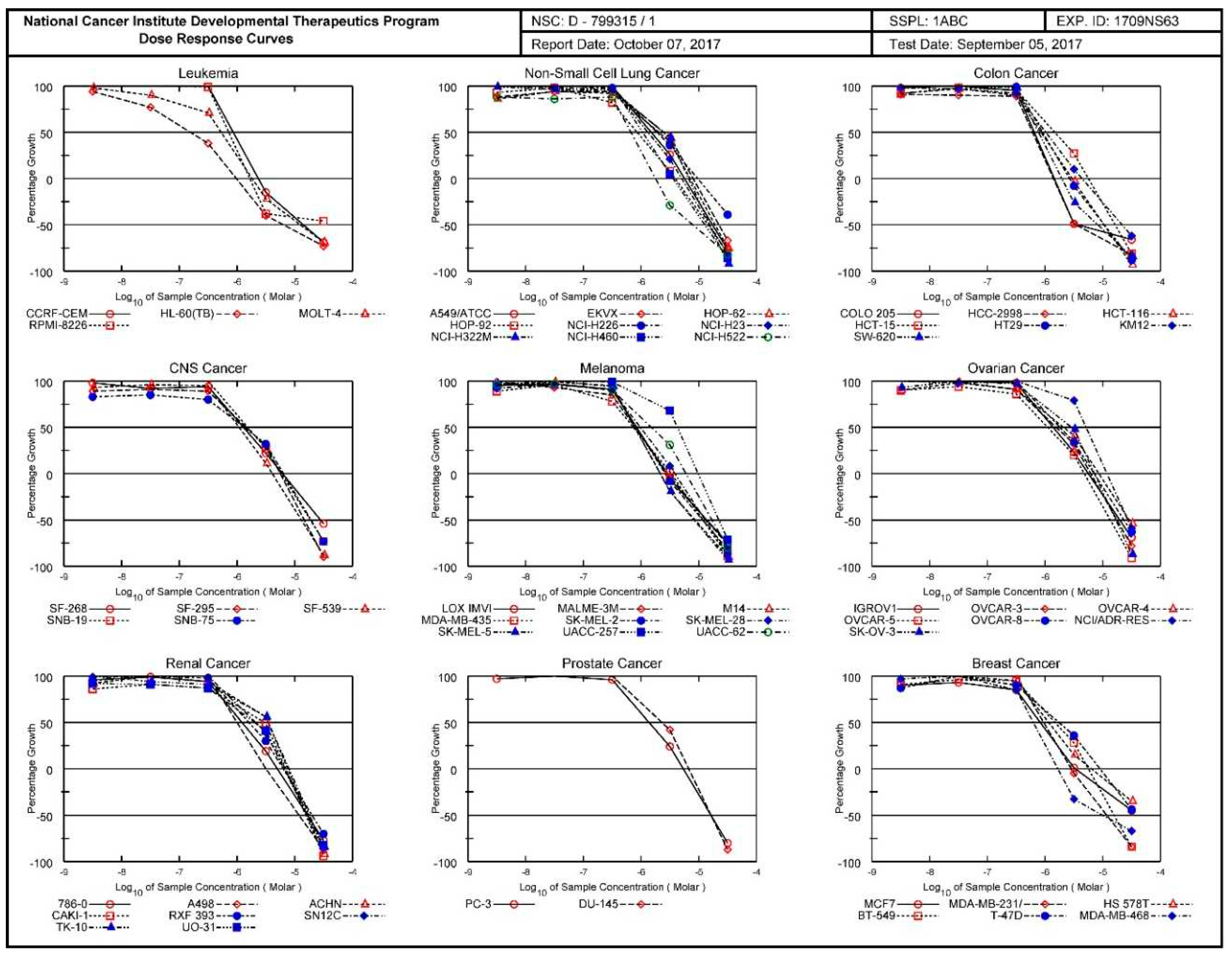
3.1.4. National Cancer Institute (NCI) Developmental Therapeutics Program (DTP) 60-Cancer Cell 5-Dose Testing Results with Compound 3
The four pages of the National Cancer Institute (NCI) Developmental Therapeutics Program (DTP) 60-cancer cell 5-dose testing results for compound 3 (PT167, NSC 799315) are given in succession. First page (Figure S27): graphic allover presentation of second page with All Cell Lines in one graphic. Second page (Figure 20): inhibition curves for tumor cell lines arranged/ordered for general tumor type. Third page (Figure S28): the Mean Optical Densities of the utilized vital stain sulforhodamine B (SRB) retained in the cells, the actual used concentrations in log10 unit for five doses, and the Percent Growth are given. The GI50 (Growth Inhibition 50%), TGI (Total Growth Inhibition = 0% Growth), and LC50 (Lethal Concentration 50% = –50% Growth) are given in linear concentration units. Fourth page (Figure S29): this is the summary of the results expressed in log10 units. The defined tumor inhibition criterion (GI50, TGI, or LC50) is recalculated in log10 expression, and the the colored bars which indicate then the sensitivity of the individual tumor cell line to the agent are given in log10 units.
Figure 20.
Graphic presentation of the antineoplastic activity exhibited by the drug compound 3 (PT167, NSC 799315) in the National Cancer Institute (NCI) Developmental Therapeutics Program (DTP) 60-cancer cell 5-dose testing with the inhibition curves for sixty human tumor cell lines arranged/ordered in terms of general tumor type.
Figure 20.
Graphic presentation of the antineoplastic activity exhibited by the drug compound 3 (PT167, NSC 799315) in the National Cancer Institute (NCI) Developmental Therapeutics Program (DTP) 60-cancer cell 5-dose testing with the inhibition curves for sixty human tumor cell lines arranged/ordered in terms of general tumor type.
3.1.5. Cytochrome c Assay (Mitochondrial and Cytosolic) with the Drugs Compound 1, Compound 2 and Compound 3 in the Human Prostate Cancer Cell Lines PC-3 and DU-145
To assess the nature of the cancer cell death-inducing effect of the drugs compound 1 (PT162, NSC 796018), compound 2 (PT166, NSC 750423), and compound 3 (PT167, NSC 799315), and to verify the failure to induce cancer cell death of compound 2, the cellular compartment of cytochrome c in the prostate cancer cell lines PC-3 and DU-145 under the action of compound 1, compound 2, and compound 3 was investigated (Figure 21). Compound 1 readily induced cytochrome c translocation from mitochondria into the cytosol at 25.0 µM concentration in PC-3 cells (Figure 21A) and 5.0 µM concentration in DU-145 cells (Figure 21B).
Figure 21.
Compound 1 (PT162, NSC 796018) and compound 3 (PT167, NSC 799315) induce cytochrome c translocation from mitochondria into the cytosol, a hallmark of the intrinsic pathway of apoptosis, whereas compound 2 (PT166, NSC 750423) fails to induce apoptotic cytochrome c translocation. The cytochrome c assays (mitochondrial and cytosolic) were performed with compound 1, 2 and 3 in the human prostate cancer cell lines PC-3 and DU-145. The protein content for each cellular fraction was calculated to determine the mass content of cytochrome c in each fraction (pg cytochrome c/mg total protein). The mass content of cytochrome c (pg cytochrome c/mg total protein) was normalized towards the corresponding blank DMSO control and is given in percent (%) of blank DMSO control. Each experiment was representative for the effects of compound 1, compound 2, and compound 3 in the human prostate cancer cell lines PC-3 and DU-145. (A) Experiment compound 1 in PC-3 cells. (B) Experiment compound 1 in DU-145 cells. (C) Experiment compound 2 in PC-3 cells. (D) Experiment compound 2 in DU-145 cells. (E) Experiment compound 3 in PC-3 cells. (F) Experiment compound 3 in DU-145 cells.
Figure 21.
Compound 1 (PT162, NSC 796018) and compound 3 (PT167, NSC 799315) induce cytochrome c translocation from mitochondria into the cytosol, a hallmark of the intrinsic pathway of apoptosis, whereas compound 2 (PT166, NSC 750423) fails to induce apoptotic cytochrome c translocation. The cytochrome c assays (mitochondrial and cytosolic) were performed with compound 1, 2 and 3 in the human prostate cancer cell lines PC-3 and DU-145. The protein content for each cellular fraction was calculated to determine the mass content of cytochrome c in each fraction (pg cytochrome c/mg total protein). The mass content of cytochrome c (pg cytochrome c/mg total protein) was normalized towards the corresponding blank DMSO control and is given in percent (%) of blank DMSO control. Each experiment was representative for the effects of compound 1, compound 2, and compound 3 in the human prostate cancer cell lines PC-3 and DU-145. (A) Experiment compound 1 in PC-3 cells. (B) Experiment compound 1 in DU-145 cells. (C) Experiment compound 2 in PC-3 cells. (D) Experiment compound 2 in DU-145 cells. (E) Experiment compound 3 in PC-3 cells. (F) Experiment compound 3 in DU-145 cells.
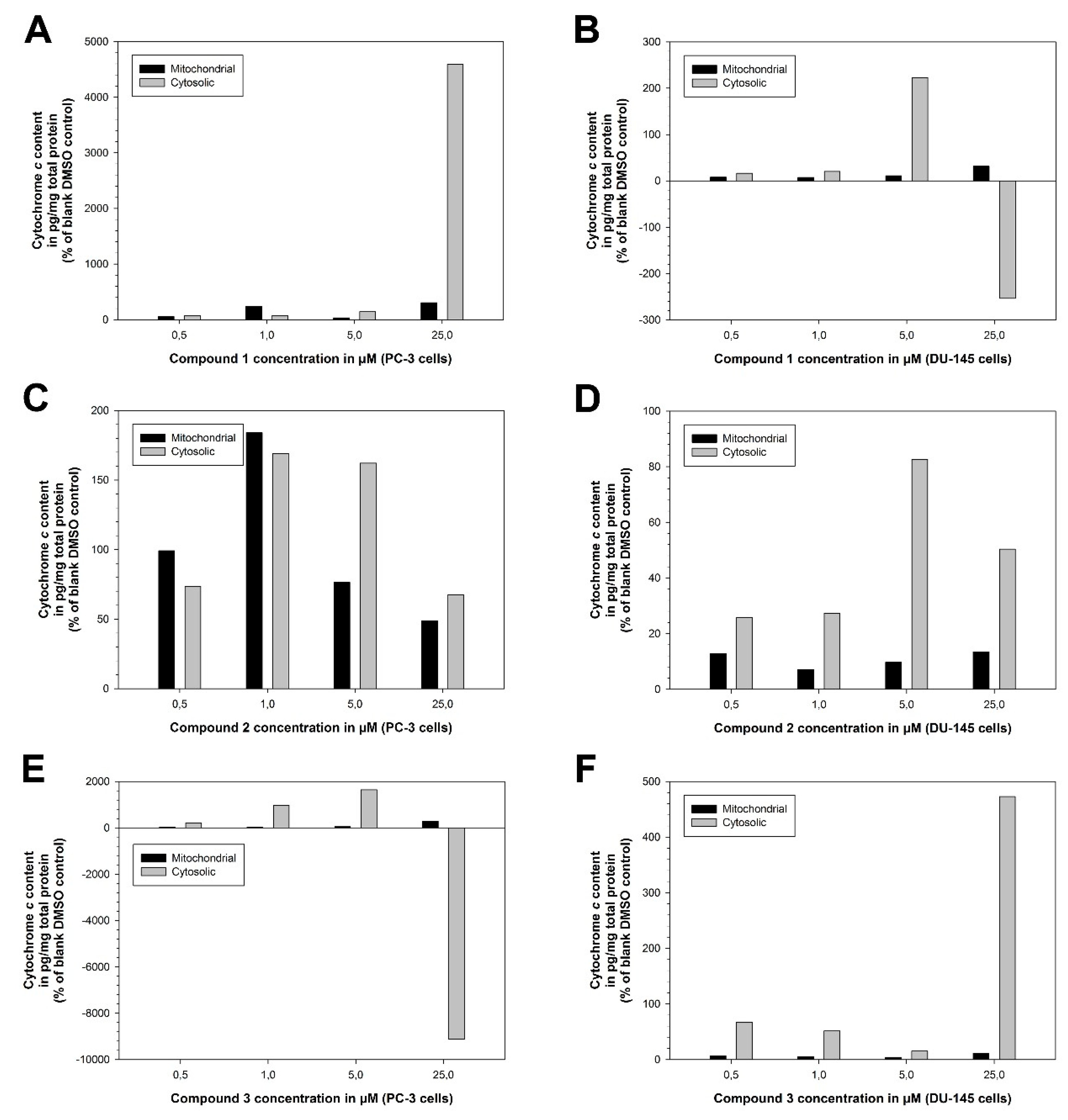
Compound 2 failed to induce a significant difference in the cytochrome c-residing cellular compartment in PC-3 cells (Figure 21C) and, less significant, in DU-145 cells (Figure 21D). Compound 3 readily induced cytochrome c translocation from mitochondria into the cytosol at 5.0 µM concentration in PC-3 cells (Figure 21E) and 25.0 µM concentration in DU-145 cells (Figure 21F). At 25.0 µM concentration compound 1 in DU-145 cells (Figure 21B) and compound 3 in PC-3 cells (Figure 21E) both induced complete cell death which resulted in massive depletion of cytochrome c in the cytosol, very probably by apoptotic degradation of cytochrome c protein by effector caspases. That means that cytochrome c apoprotein is digested by the cytosolic caspases executing apoptosis.
3.1.6. HIV-1LAI Replication Reverse Transcriptase Assay with the Drug Compound 1 in Primary PBL Cells
Compound 1 (PT162, NSC 796018) is inhibitory towards human immunodeficieny virus type 1 (HIV-1) strain LAI (HIV-1LAI) (= HIV-1BRU = LAV-1) replication in freshly explanted, primary human peripheral blood lymphocytes (PBL cells) (Table 3). The effective inhibitory concentration 50% (EC50) in PBL cells was 0.56 µM, the effective inhibitory concentration 90% (EC90) in PBL cells was 4.3 µM. The cyctotoxic concentration 50% (CC50) for the PBL cells was 2.2 µM, this yields a selectivity index 50% (SI50) = CC50/EC50 of 3.9. The r2 coefficient of determination (r2 measure of goodness–of‒fit) on EC50 and EC90 was 0.93. As a positive control served AZT (zidovudine, 3′-azido-3′-deoxythymidine). The given effective inhibitory concentrations (μM ± s.d.) for the positive control AZT were averaged and treated statistically from four (n = 4) independent determinations (Table 3). Furthermore, the cytotoxicity of compound 1 (PT162, NSC 796018) towards CCRF−CEM cells (ATCC® CCL-119™, acute lymphoblastic leukemia cells, tumorigenic CD4+ T lymphoblasts) [83] and Vero cells [African green monkey (grivet) Chlorocebus aethiops (syn. Cercopithecus aethiops) kidney epithelial cells] was determined (Table 3). The cyctotoxic concentration 50% (CC50) for the CCRF−CEM cells was < 1 µM with 60.0% growth inhibition of CCRF−CEM cells at 1 µM. The cytotoxic concentration 50% (CC50) for the Vero cells was 1.8 µM.
The inhibitory action of p53 on HIV-1 replication is well established [84,85,86]. Compound 1 (PT162, NSC 796018) clearly inhibits HIV-1LAI replication by inducing p53 and its associated cyclin-dependent kinase inhibitor p21Waf1/p21Cip1 [87]. The latter factor p21Waf1/p21Cip1 is induced by p53 activation [87] and inhibits HIV-1 replication [88,89]. In addition, tumor suppressor protein p53 interacts with HIV-1 trans-activator protein Tat (trans-activator of transcription) [90,91,92], HIV-1 Nef (negative regulatory factor) accessory protein [93], and HIV-1 Vpr (viral protein R, viral protein rapid) accessory protein [89,94]. The marked cytotoxic action of compound 1 (PT162, NSC 796018) on CCRF−CEM acute lymphoblastic leukemia cells clearly stems from its strong antileukemic/antineoplastic activity.
Table 3.
Cytotoxicity and antiviral activity of tetrakis{3-[(tricyclo[3.3.1.13,7]decan-1-ammonio)methyl]benzyl}ammonium pentachloride (compound 1, PT162, NSC 796018) versus human immunodeficiency virus type 1 strain LAI (HIV-1LAI) in mammalian cells.
Table 3.
Cytotoxicity and antiviral activity of tetrakis{3-[(tricyclo[3.3.1.13,7]decan-1-ammonio)methyl]benzyl}ammonium pentachloride (compound 1, PT162, NSC 796018) versus human immunodeficiency virus type 1 strain LAI (HIV-1LAI) in mammalian cells.
| Drug | Cytotoxicity CC50 (µM) and CCRF–CEM growth inhibition at fixed 1 µM concentration (%, in parentheses) | Anti-HIV-1LAI activity EC50 (µM)/EC90 (µM) in PBM cells | |||||
| PBM cells | CCRF −CEM |
Vero | EC50 | EC90 | SI50 | r2 | |
| Compound 1 (PT162, NSC 796018) |
2.2 | < 1 (60.0) |
1.8 | 0.56 | 4.3 | 3.9 | 0.93 |
| AZT* | > 100 | 14.3 | 56.0 | 0.0044 ± 0.0018 | 0.031 ± 0.015 | > 22,727 | 0.99 |
CC50, cytotoxic concentration 50%. EC50, effective inhibitory concentration 50%. EC90, effective inhibitory concentration 90%. SI50, selectivity index CC50/EC50. r2, coefficient of determination (r2 measure of goodness–of‒fit) on EC50 and EC90. AZT, zidovudine (3′-azido-3′-deoxythymidine). * The given effective inhibitory concentrations (µM ± s.d.) for the positive control AZT were averaged and treated statistically from four (n = 4) independent determinations.
4. Experimental Section
4.1. Materials and Methods
4.1.1. Chemicals
1-Adamantanammonium chloride (INN: amantadine hydrochloride) (1-aminoadamantane hydrochloride) [Lot: S4247215; w (m/m) = 100.0% (argentometric titration), mp > 360 °C (dec.)] and absolute ethanol pro analysi EMPLURA® [Lot: K48011060; w (n/n) = 99.9% (gas chromatography, area%), w (H2O) (m/m) = 0.06%, non-volatile matter < 0.0001%] were purchased from Merck KGaA–EMD Millipore Corp. (Darmstadt, Germany). Sodium hydroxide (NaOH) pearls pure (Ph.Eur., BP, Food Grade) [Lot: 2J002792; w (m/m) = 99.31% (titration), sodium carbonate < 0.5%, SiO2 < 0.001%, NaCl < 0.008%, Na2SO4 < 0.0025%, As < 0.00001 %, heavy metals (Cu, Fe, Mn, Ni, Pb, Hg) < 0.001%], 10.27 M [32% (m/m)] aqueous hydrochloric acid pro analysi [Lot: 3A001639; w (m/m) = 33.09% (titration), bromide < 0.005%, phosphate < 0.00005%, sulfate < 0.0001%, As < 0.000001 %, Fe < 0.00002%, heavy metals (Ni, Pb, Zn) < 0.000005%], sodium hydrogen carbonate (sodium bicarbonate) pro analysi NaHCO3 [Lot: 4W000829; w (m/m) = 100.42% (titration), pH (5% in H2O) 8.04 (20 °C), chloride ≤ 0.001%, sulfate ≤ 0.005%, phosphate ≤ 0.005%, cations (K, Mg, Ca) ≤ 0.005%, As ≤ 0.0001%, heavy metals (Cu, Fe, Pb) ≤ 0.0005%], and (–)-colchicine sesquihydrate (× 1½ H2O) = colchicine BioChemica [w (m/m) ≥ 98% (HPLC), specific rotation [20 °C, 589 nm (Na−D line)] –240° to –250° (c = 1 in EtOH), w (H2O) (m/m) ≤ 3% (Karl Fischer titration)] were purchased from AppliChem (Darmstadt, Germany). Acetone (USP, BP, Ph.Eur.) Pharma Quality [Lot: 0000869897; w (n/n) = 99.9% (gas chromatography, area%), w (H2O) (m/m) = 0.3%, non-volatile matter 0.0002%, methanol < 0.05%, propan-2-ol < 0.05%, benzene < 0.0002%, ethanol < 500 ppm, heavy metals (Fe, Zn) < 1,300 ppm, heavy metals (Cu, Mn) < 250 ppm], ethyl acetate pro analysi [Lot: 0000518022; w (n/n) = 99.9% (gas chromatography, area%), w (H2O) (m/m) = 0.01% (Karl Fischer titration), ethanol < 0.1%, methanol < 0.02%, methyl acetate < 0.02%, trace elements (Cr, Fe, Ni, Pb, Zn, P, S, K, Mg) < 0.00001%, Si < 0.00002%, Na < 0.0002%, non-volatile matter < 0.001%, acidity/alkalinity < 0.0005 meq/g] were purchased from PanReac AppliChem GmbH (Darmstadt, Germany). 1,3-Bis(chloromethyl)benzene (α,α′-dichloro-m-xylene) purum ≥ 98% (GC) [Lot: 385191/1; w (m/m) = 100.6% (argentometric titration after oxygen combustion), w (n/n) = 99.9% (gas chromatography, area %), mp 33.2–34.0 °C] was purchased from Fluka Chemie AG (Buchs, Switzerland). Thiosemicarbazide puriss. p.a. [Lot: 1167177V (Fluka); w (m/m) = 100.1% (iodometric titration), mp 181 °C (dec.), residue on ignition < 0.05%, metal trace analysis (inductively coupled plasma mass spectrometry) ≤ 50–5 mg/kg] was purchased from Sigma-Aldrich Corp. (St. Louis, MO, USA).
4.1.2. Cell Lines
The National Cancer Institute (NCI) Developmental Therapeutics Program (DTP) 60 cancer cell lines (see Figure 18, Figure 19, Figure 20, Table S1) were kindly provided by NCI [part of the National Institutes of Health (NIH), Bethesda, MD, USA]. The metastatic prostate cancer cell lines PC-3 (CRL-1435™) (bone metastasis, adenocarcinoma) and DU-145 (HTB-81™) (brain metastasis, adenocarcinoma) were retrieved from American Type Culture Collection (ATCC®, Manassas, VA, USA).
4.1.3. Analytical Methods
The Fourier-transform infrared (FT–IR) spectra were recorded with neat substance on JASCO FT/IR–4100 type A and FT/IR–6100 type A spectrometers (JASCO International Co. Ltd., Tokyo, Japan). Given FT–IR absorbance bands, expressed in wavenumbers (cm-1), are characterized in intensity as strong (str), middle (m), weak (w), and broad (br). The proton nuclear magnetic resonance (1H-NMR), carbon-13 nuclear magnetic resonance (13C-NMR) and 13C-Distortionless Enhancement by Polarization Transfer Including Detection of Quaternary Nuclei (DEPTQ 13C-NMR) [33,34] NMR spectroscopy experiments were recorded at a temperature of 25 °C using a Bruker Avance 700 III HD nuclear magnetic resonance spectrometer (Bruker BioSpin GmbH, Rheinstetten, Germany) [1H-NMR (700.43 MHz), 13C-NMR (176.12 MHz)]. The spectra were referenced to the center of the NMR solvent signal [1H-NMR: δ 2.51 (DMSO-d6); 13C-NMR: δ 39.41 (DMSO-d6)]. Additional 1H-NMR, 13C-NMR, DEPTQ 13C-NMR, gradient-selected Correlation Spectroscopy (gs-COSY) [34], gradient-selected Heteronuclear Multiple Quantum Coherence (gs-HMQC) [34] and gradient-selected Heteronuclear Multiple Bond Correlation (gs-HMBC) [34] spectra were recorded using a Bruker Avance III HD 400 nuclear magnetic resonance spectrometer (Bruker BioSpin GmbH, Rheinstetten, Germany) [1H-NMR (400.13 MHz), 13C-NMR (100.62 MHz)] at a temperature of 300.0 K. The spectra were referenced to the center of the NMR solvent signal [1H-NMR: δ 2.50 (DMSO-d6); 13C-NMR: δ 39.52 (DMSO-d6)]. Given chemical shifts δ [from tetramethylsilane (TMS): δ = 0] are specified as singlet (s), broad singlet (br s), doublet (d), triplet (t), quartet (q), multiplet (m), and broad multiplet (br m). Electrospray ionization time−of−flight mass spectrometry (ESI–ToF−MS) of compound 3 was conducted in positive ion mode on an Agilent LC−ESI−ToF instrument using an Agilent 1260 Infinity II liquid chromatography (LC) stack with an Agilent ZORBAX 300SB−C8 (4.6 mm ID × 50 mm) column (pore size 300 Å, particle size 5 µm) coupled to an Agilent 6230B time−of−flight LC/MS (LC/ToF) system (Agilent Technologies, Inc., Santa Clara, CA, USA). Source settings were ϑ (dry gas) = 350 °C, gas flow 10 l/min, nebulizer pressure pn = 35 psi (2.4132 bar). Capillary voltage was set to Vc = 3,500 V and fragmentor voltage was set to Vf = 175 V. The scan range was m/z 105−3000. Elemental analyses (C, H, N, S, O) were conducted on the EURO EA3000 CHNS–O elemental analyser (EuroVector SpA, Milan, Italy) by HEKAtech GmbH (Wegberg, Germany).
4.1.4. Software
Crystal data visualization and molecular modeling was performed with ACD/Chem Sketch version 2022.1.0 with integrated ACD/3D Viewer (Advanced Chemistry Development, Inc., Toronto, Ontario, Canada) and processed with Mercury 2022.3.0 [The Cambridge Crystallographic Data Centre (CCDC), Cambridge, UK]. Additional molecular modeling was performed with ACD/Chem Sketch version 12.01 with integrated ACD/3D Viewer (Advanced Chemistry Development, Inc., Toronto, Ontario, Canada) and processed with Mercury 3.1 version 3.1.1 [The Cambridge Crystallographic Data Centre (CCDC), Cambridge, UK]. The mass spectrometric m/z values were calculated with Scripps Research Core Service Molar Mass Calculator (The Scripps Research Institute, Center for Metabolomics and Mass Spectrometry, San Diego, CA, USA).
4.2. Biological Testing
4.2.1. National Cancer Institute (NCI) Developmental Therapeutics Program (DTP) 60-Cancer Cell 5-Dose Testing
Compounds which exhibit significant growth inhibition in the One-Dose Screen are evaluated against the 60-cell panel at five concentration levels [95]. The human tumor cell lines of the cancer screening panel are grown in RPMI-1640 medium containing 5% fetal bovine serum (FBS) and 2 mM L-glutamine. For a typical screening experiment, cells are inoculated into 96-well microtiter plates in 100 μl at plating densities ranging from 5,000 to 40,000 cells/well depending on the doubling time of individual cell lines. After cell inoculation, the microtiter plates are incubated at 37 °C, 5 % CO2, 95% air and 100% relative humidity for 24 h prior to addition of experimental drugs. After 24 h, two plates of each cell line are fixed in situ with trichloroacetic acid (TCA), to represent a measurement of the cell population for each cell line at the time of drug addition (Tz). Experimental drugs are solubilized in dimethyl sulfoxide (DMSO) at 400-fold the desired final maximum test concentration and stored frozen prior to use. At the time of drug addition, an aliquot of frozen concentrate is thawed and diluted to twice the desired final maximum test concentration with complete medium containing 50 μg/ml gentamicin. Additional four, 10-fold or ½ log serial dilutions are made to provide a total of five drug concentrations plus control. Aliquots of 100 μl of these different drug dilutions are added to the appropriate microtiter wells already containing 100 μl of medium, resulting in the required final drug concentrations.
Following drug addition, the plates are incubated for an additional 48 h at 37 °C, 5% CO2, 95% air, and 100% relative humidity. For adherent cells, the assay is terminated by the addition of cold TCA. Cells are fixed in situ by the gentle addition of 50 μl of cold 50% (m/v) TCA (final concentration, 10% TCA) and incubated for 60 min at 4 °C. The supernatant is discarded, and the plates are washed five times with tap water and air dried. Sulforhodamine B (SRB) solution (100 μl) at 0.4% (m/v) in 1% acetic acid is added to each well, and plates are incubated for 10 min at room temperature. After staining, unbound dye is removed by washing five times with 1% acetic acid and the plates are air dried. Bound stain is subsequently solubilized with 10 mM trizma® base [2-amino-2-(hydroxymethyl)-1,3-propandiol, tris base, trometamol], and the absorbance is read on an automated plate reader at a wavelength of λ = 515 nm. For suspension cells, the methodology is the same, except that the assay is terminated by fixing settled cells at the bottom of the wells by gently adding 50 μl of 80% TCA (final concentration, 16% TCA). Using the seven absorbance measurements [time zero, (Tz), control growth, (C), and test growth in the presence of drug at the five concentration levels (Ti)], the percentage growth is calculated at each of the drug concentrations levels. Percentage growth inhibition is calculated as:
[(Ti – Tz)/(C – Tz)] × 100 for concentrations for which Ti ≥ Tz
[(Ti – Tz)/Tz] × 100 for concentrations for which Ti < Tz.
Three dose response parameters are calculated for each experimental agent. Growth inhibition of 50% (GI50) is calculated from [(Ti – Tz)/(C – Tz)] × 100 = 50, which is the drug concentration resulting in a 50% reduction in the net protein increase (as measured by SRB staining) in control cells during the drug incubation. The drug concentration resulting in total growth inhibition (TGI) is calculated from Ti = Tz. The LC50 (concentration of drug resulting in a 50% reduction in the measured protein at the end of the drug treatment as compared to that at the beginning) indicating a net loss of cells following treatment is calculated from [(Ti – Tz)/Tz] × 100 = –50. Values are calculated for each of these three parameters if the level of activity is reached; however, if the effect is not reached or is exceeded, the value for that parameter is expressed as greater or less than the maximum or minimum concentration tested.
An outline for reading the NCI 60 Cell Five-Dose Screen data was given in Section 3.1.1. The numerical screening data are given in Supplementary Information (Table S1).
4.2.2. Cytochrome c Assay (Mitochondrial and Cytosolic)
Cell numbers of 2 × 106 PC-3 and DU-145 cells were seeded into a 100-well plate. At the next day cells were treated with either dimethyl sulfoxide (DMSO) or 6 concentrations of compound 1, 2 and 3 (500 nM, 1 µM, 5 µM, 25 µM, 50 µM, 75 µM) dissolved in DMSO for a time of 48 h. The concentrations of 25 µM, 50 µM and 75 µM of compounds 1, 2 and 3 were found to be highly toxic for both cell lines 48 h post-treatment. The enzyme-linked immunosorbent assay (ELISA) for cytochrome c was performed according to the manufacturer’s instructions [Cytochrome c (human), ELISA kit (Enzo Life Sciences, Inc., Farmingdale, NY, USA)] with two fractions (mitochondrial and cytosolic) of each cell line at the concentrations of compound 1, 2 and 3 of 500 nM, 1 µM, 5 µM, and 25 µM. The protein content for each fraction was calculated to determine the mass content of cytochrome c in each fraction (pg cytochrome c/mg total protein). The mass content of cytochrome c (pg cytochrome c/mg total protein) was normalized towards the corresponding blank DMSO control and is given in percent (%) of blank DMSO control.
4.2.3. Cytotoxicity and HIV-1LAI Replication Reverse Transcriptase Assays with Compound 1
The cytotoxicity and human immunodeficiency virus type 1 (HIV-1) strain LAI replication assays were performed in freshly explanted primary human peripheral blood mononuclear cells (PBM cells) according to published procedures [25]. The assays were conducted at least in triplicate and treated statistically (if possible). HIV-1LAI (= HIV-1BRU = LAV-1) was assayed in primary human peripheral blood lymphocyte (PBL) cells in the presence of a drug being evaluated. The parameter for antiviral activity was reduction of reverse transcriptase (RT) activity in the cell supernatant after Triton X‒100-mediated lysis of released virions, as measured by [5α-3H]dTTP (5α-tritiated thymidine 5′-triphosphate) incorporation into poly(rA)•poly(dT) directed by the primed RNA template poly(rA)•oligo(dT). It should be noted that the assay did not detect RT inhibition by potential RT inhibitors per se, but indirectly quantified the amount of released HIV-1 in the supernatant. The detailed assay methodology was reported by Schinazi et al. [96], as based on an older assay system of Spira et al. [97]. The experiments were conducted in triplicate and treated statistically by regression curve analysis (r2 coefficient of determination). The RT inhibitor AZT (zidovudine, 3′-azido-3′-deoxythymidine; RETROVIR™) served as a positive control. Cytotoxicity on PBL and the other cells (CCRF−CEM, Vero) exerted by the test compounds was determined as described by Stuyver et al. [98], by application of the CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega Corp., Madison, WI, USA). Briefly, the phenazine ethosulfate (PES)-coupled reduction of the tetrazolium salt 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) to a purple, water-soluble formazan by living, undamaged cells was measured.
4.3. X-Ray Crystallographic Determination of the Crystal and Molecular Structure of (M)-10-(2-Carbamothioylhydrazinyl)-10-demethoxycolchicine Sesquihydrate × ½ (Ethyl Acetate) = N-[(aS,7S)-10-(2-Carbamothioylhydrazinyl)-1,2,3-trimethoxy-9-oxo-5,6,7,9-tetrahydrobenzo[a]heptalen-7-yl]acetamide Sesquihydrate × ½ (Ethyl Acetate) (Crystalline Compound 2)
4.3.1. Crystallization of Compound 2 Single Crystals
Compound 2 was crystallized by atmospheric evaporation from ethyl acetate (EtOAc) overnight (time ~10 h) in an open Petri dish at room temperature (RT, ϑ = 14.0 °C). A suitable single crystal was selected and isolated under polarized light microscopic examination.
4.3.2. Crystal Data of Compound 2 Single Crystals
The crystal data of compound 2 were collected on a Bruker X8 APEX-II diffractometer with a CCD area detector and multi-layer mirror monochromated MoKα radiation. The structure was solved using direct methods, refined with the SHELX software package (G.M. Sheldrick, Acta Cryst., 2008, A64, 112–122) [99] and expanded using Fourier techniques. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were assigned to idealized positions and were included in structure factor calculations. The Flack parameter xFlack (H.D. Flack, Acta Cryst., 1983, A39, 876–881) [100] was near zero indicating that the right absolute configuration was solved.
4.3.3. Deposition of the X-Ray Crystallographic Structure Determination of Compound 2
Crystallographic data have been deposited with The Cambridge Crystallographic Data Centre (CCDC), 12 Union Road, Cambridge, CB2 1EZ, United Kingdom, as supplementary publication № CCDC 1839505 (ID: RIVGOW). These data can be obtained free of charge from CCDC via https://www.ccdc.cam.ac.uk/structures/.
4.4. Chemical Synthesis
4.4.1. Salt-Containing Tetrakis{3-[(tricyclo[3.3.1.13,7]decan-1-ammonio)methyl]benzyl}ammonium Pentachloride (Compound 1 × 1.5 NaCl, PT162 × 1.5 NaCl)
2.50 g 1-Aminoadamantane hydrochloride (13.32 mmol) and 3.50 g 1,3-bis(chloromethyl)benzene (α,α′-dichloro-m-xylene) (19.99 mmol) were dissolved in 30 ml of 90% (v/v) aqueous ethanol. A solution of 1.60 g sodium hydroxide (40.00 mmol) in 40 ml of water was added, and the mixture was refluxed for 3 h. After 20 min and 40 min reflux, 20 ml of acetone, each, were added through the reflux condensor. After 60 min, 80 min, and 100 min reflux, 40 ml of acetone, each, were added through the reflux condensor. After 120 min reflux, additional 20 ml of acetone were added through the reflux condensor. Then, after 10 min pre-cooling at +0–2 °C, the colorless solution with few suspended impurities was warm filtrated. The filtrate (pH 9) was mixed with 100 ml of water, and was evaporated in vacuo from the acetone to a volume of ca. 170 ml. Afterwards, 2 ml of 10.27 M [32% (m/v)] hydrochloric acid (20.54 mmol) were added, and the colorless solution (pH 2–3) was evaporated further in vacuo to a volume of ca. 120 ml.
Then the turbid suspension was mixed with 20 ml of water and was extracted with 100 ml of ethyl acetate (EtOAc) to remove unreacted 1,3-bis(chloromethyl)benzene (α,α′-dichloro-m-xylene). The aqueous phase was isolated and further evaporated in vacuo to a volume of ca. 50 ml. The aqueous phase was then frozen at –25 °C for 1½ h. The evolved white precipitate of crude compound 1 × 1.5 NaCl (PT162 × 1.5 NaCl) was filtered and dried over CaCl2 in vacuo (from the filtrate additional substance could be obtained which was treated as before). From the separated EtOAc phase by cooling at +0–2 °C for 3 h additional crude compound 1 × 1.5 NaCl (PT162 × 1.5 NaCl) could be obtained which was treated as before.
The combined crude product (730 mg) was dissolved in 50 ml of 80% (v/v) aqueous acetone by short (5 min) refluxing, and was hot filtrated to remove few impurities. The filtrate was transferred and mixed with 40 ml of 50% (v/v) aqueous acetone. Afterwards, the filtrate was evaporated in vacuo from the acetone. The resulting solution (pH 5) was acidified to pH 0–1 by addition of 0.6 ml of 10.27 M [32% (m/v)] hydrochloric acid (6.16 mmol). Instantly, a white precipitate formed. The suspension was supplemented with 10 ml of 50% (v/v) aqueous acetone and was frozen at –25 °C for 2½ h. The evolved white precipitate of compound 1 × 1.5 NaCl (PT162 × 1.5 NaCl) was filtered and dried over CaCl2 in vacuo.
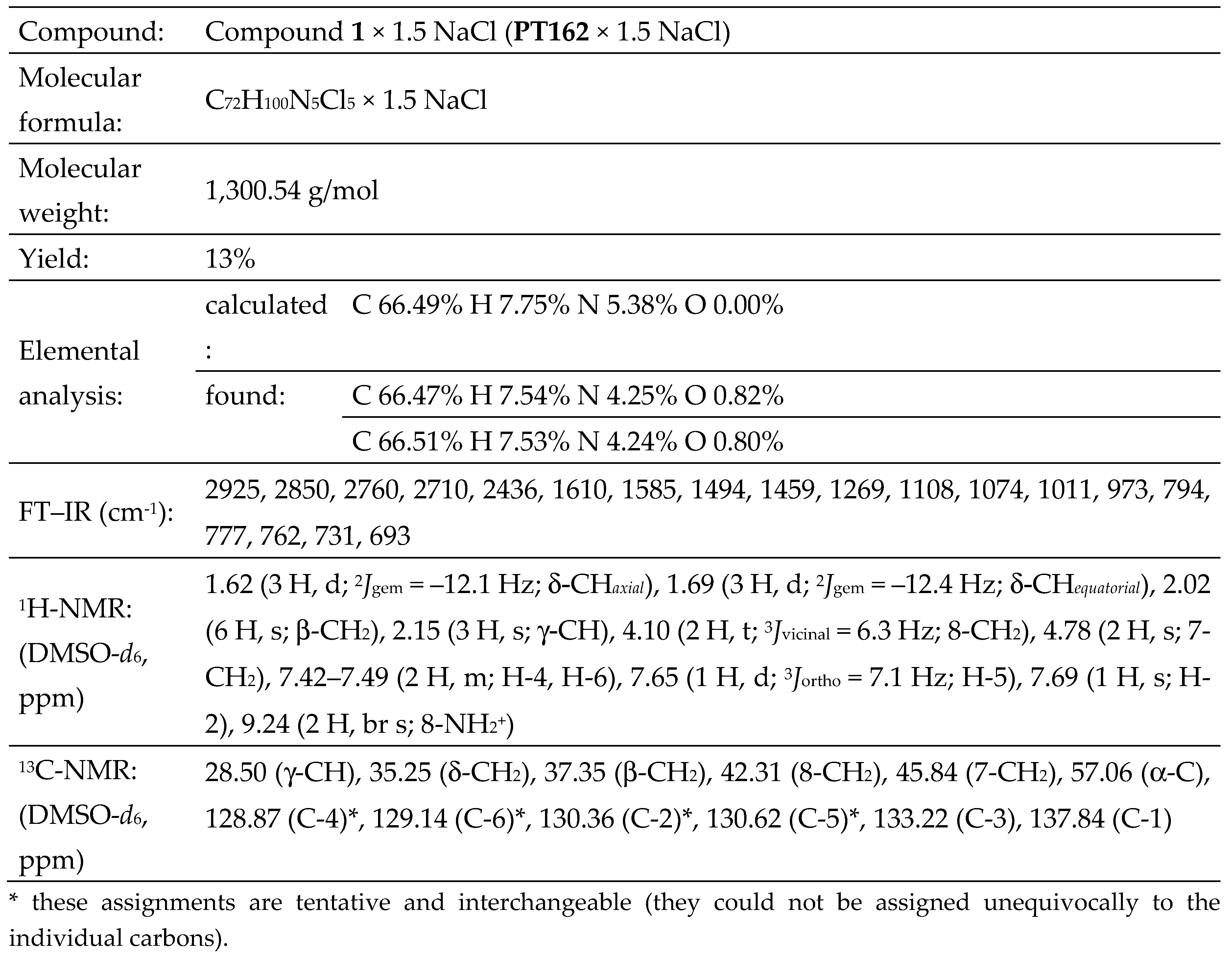
4.4.2. Pure (Salt-Free) Tetrakis{3-[(tricyclo[3.3.1.13,7]decan-1-ammonio)methyl]benzyl}ammonium Pentachloride (Compound 1, PT162, NSC 796018)
1-Aminoadamantane hydrochloride (M = 187.71 g/mol, 10.075 g, 53.6732 mmol) was dissolved in water (100 ml). A solution of sodium hydroxide NaOH (2.160 g, 54.0000 mmol) in water (20 ml) was added. Residues were transferred with water (20 ml). A heavy precipitate of the free base 1-aminoadamantane formed instantly. The suspension was frozen at –25 °C for 1 h. The evolved white precipitate of the yield of 1-aminoadamantane (free base) was filtered and vacuum-sucked dry (ca. 1 h).
The still wet 1-aminoadamantane (free base) and 1,3-bis(chloromethyl)benzene (α,α′-dichloro-m-xylene) (M = 175.06 g/mol, 7.498 g, 42.8310 mmol) were suspended in absolute ethanol (200 ml). The suspension was refluxed for 3 h. After 40 min reflux a clear colorless solution formed. After 5 min pre-cooling at +0–2 °C, the colorless solution with few suspended impurities was hot filtrated through one layer of filter paper. Residues were transferred and rinsed with absolute ethanol (10 ml) and acetone (30 ml). The filtrate was mixed with acetone (300 ml), 10.27 M [32% (m/v)] hydrochloric acid (3,200 µl, 32.8640 mmol), and ethyl acetate (EtOAc) (200 ml), and was frozen at –25 °C for 2.5 h. Afterwards, water (200 ml) and EtOAc (1,000 ml) were added, the mixture was shaken vigorously for 1 min, and was frozen at –25 °C for 2.5 h. Then 10.27 M [32% (m/v)] hydrochloric acid (3,000 µl, 30.8100 mmol) was added, the mixture was shaken vigorously for 1 min, and it was frozen at –25 °C for 30 min. The upper phase was then decanted and the lower aqueous phase was isolated. The isolated upper EtOAc phase was re-extracted with water (90 ml), the aqueous phase was isolated after phase separation, and was combined with the first aqueous phase. Finally, the isolated upper EtOAc phase was re-extracted with acidified {3,000 µl 10.27 M [32% (m/v)] hydrochloric acid, 30.8100 mmol} water (100 ml), the aqueous phase was isolated after phase separation, and was combined with the two prior aqueous phases. The combined aqueous phases (V = 500 ml) were then evaporated in vacuo at the lowest possible temperature to a volume of 200 ml until heavy crystallization started. The crystallizing suspension was then cooled at +0–2 °C for 6 h, and frozen at –25 °C for 20 min, to complete crystallization. The evolved first yield (1.543 g) of white crystals were filtered and dried over CaCl2 in vacuo. The filtrate was cooled at +0–2 °C for 50 h. The evolved second yield (62 mg) of white crystals was filtered and dried over CaCl2 in vacuo. Both yields were combined.

4.4.3. (M)-10-(2-Carbamothioylhydrazinyl)-10-demethoxycolchicine Monohydrate × ⅔ (Ethyl Acetate) = N-[(aS,7S)-10-(2-Carbamothioylhydrazinyl)-1,2,3-trimethoxy-9-oxo-5,6,7,9-tetrahydrobenzo[a]heptalen-7-yl]acetamide Monohydrate × ⅔ (Ethyl Acetate) (Compound 2, PT166, NSC 750423)
5.00 g (–)-Colchicine sesquihydrate (× 1½ H2O) (M = 426.46 g/mol, 11.72 mmol) and 1.08 g thiosemicarbazide (M = 91.13 g/mol, 11.85 mmol) were dissolved in 25 ml of 90% (v/v) aqueous ethanol by refluxing for 5 min. After adding a solution of 0.48 g sodium hydroxide (12.00 mmol) in 2 ml of water, the deep orange-red solution was refluxed for 5 min. The cold deep orange-red solution solution, after pre-cooling at –25 °C for 20 min, was titrated by dropwise addition of 1.1 ml of 10.27 M [32% (m/v)] hydrochloric acid (11.30 mmol) which was diluted with 2 ml of water. Afterwards, the volume of the solution was reduced in vacuo approximately by one half. The reddish-brown solution was then mixed with 100 ml of water, and was titrated with 1.1 ml of 10.27 M [32% (m/v)] hydrochloric acid (11.30 mmol) which was diluted with 2 ml of water. The oily emulsion was extracted with 50 ml of ethyl acetate (EtOAc). The separated aqueous layer (pH 2) was additionally extracted with 40 ml of EtOAc. After neutralization of this aqueous phase with sodium hydrogen carbonate NaHCO3, the aqueous phase (pH 7–8) was extracted twice with 40 ml of EtOAc each. The EtOAc phases were combined and washed twice with 100 ml of water each. The washed EtOAc phase, which already precipitated, was mixed with 50 ml of acetone and was frozen at –25 °C for 10 h. If precipitation did not start spontaneously, the volume of the solution was reduced in vacuo until coagulation started. The evolved yellow precipitate of compound 2 was filtered (1.01 g) and dried over CaCl2 in vacuo. From the combined aqueous phases by cooling two days at +0–2 °C a second crop of compound 2 could be obtained (1.75 g). It was treated as before and combined with the main yield.

4.4.4. [(Bis{3-[(tricyclo[3.3.1.13,7]decan-1-ylamino)methyl]benzyl}ammonio)bis(methanediylbenzene-3,1-diylmethanediyl)]di-2-[(aS,7S)-7-(acetylamino)-1,2,3-trimethoxy-9-oxo-5,6,7,9-tetrahydrobenzo[a]heptalen-10-yl]-N-(tricyclo[3.3.1.13,7]decan-1-yl)hydrazinecarbothioamide Chloride Pentahydrate (Compound 3, PT167, NSC 799315) (synthesized at Saturday, May 27th, 2017)
Materials:
- (Compound 1, PT162, NSC 796018) = tetrakis{3-[(tricyclo[3.3.1.13,7]decan-1-ammonio)methyl]benzyl}ammonium pentachloride (C72H100Cl5N5) (M = 1,212.86 g/mol) [w (n/n) ≥ 99% (1H-NMR and elemental analysis)], synthesized at Friday, December 30th, 2016.
- (Compound 2, PT166, NSC 750423) = N-[(7S)-10-(2-carbamothioylhydrazinyl)-1,2,3-trimethoxy-9-oxo-5,6,7,9-tetrahydrobenzo[a]heptalen-7-yl]acetamide monohydrate × ⅔ (ethyl acetate) [C22H26N4O5S × H2O × ⅔ (C4H8O2)] (M = 535.28 g/mol) [w (n/n) ≥ 98% (1H-NMR and elemental analysis)], synthesized at Thursday, January 29th, 2009.
Compound 1 [PT162 (NSC 796018)] (M = 1,212.86 g/mol, 300 mg, 247.3492 µmol) and compound 2 [PT166 (NSC 750423)] (M = 535.28 g/mol, 300 mg, 560.4543 µmol) were suspended in absolute ethanol (20 ml), and the yellow suspension was heated to 40–50 °C for 4 min (heatgun). Then water (1,000 µl) was added under stirring and all material dissolved to give a bright yellow solution. Afterwards, a solution of sodium hydroxide (37 mg, 925.0000 µmol) in water (2,000 µl) was added under stirring. The color of the solution changed to orange-yellow. After cooling at +0–2 °C for 12 min, the mixture was frozen at –25 °C for 10 min. After adding water (10 ml), the precipitating emulsion was frozen at –25 °C for 2 h. The evolved first yield (212 mg) of the bright yellow, amorphous substance compound 3 was filtered, carefully dried by vacuum suction for 30 min on the sintered glass filter filter, and dried over CaCl2 in vacuo. The filtrate was transferred with water (20 ml), and was frozen at –25 °C for 30 min. After cooling at +0–2 °C for 1 h, the evolved second yield (17 mg) of the bright yellow, amorphous substance compound 3 was filtered [the initial turbid filtrate was transferred with water (10 ml), and was re-filtered on the same sintered glass filter used before], carefully dried by vacuum suction for 30 min on the sintered glass filter filter, and dried over CaCl2 in vacuo. Both yields were combined.
5. Conclusions
Adenosine 5′-triphosphate (ATP) is required for normal cell proliferation and survival and comes primarily from two sources. The first is glycolysis, which comprises a series of reactions that metabolizes D-(+)-glucose to pyruvate in the cytoplasm to produce a net of 2 molecules ATP from each D-(+)-glucose. The other is the citric acid cycle (CAC), also known as the Krebs cycle, Szent Györgyi−Krebs cycle or the tricarboxylic acid cycle (TCA cycle), which uses pyruvate formed from glycolysis in a series of reactions that donate electrons via nicotinamide adenine dinucleotide (NADH/H+) and flavin adenine dinucleotide (FADH2) to the respiratory chain complexes in the mitochondria. With oxygen (O2) serving as the final electron acceptor, electron transfer across the mitochondrial inner membrane creates a proton gradient which forms 36 molecules ATP per one D-(+)-glucose molecule by catalysis of F1FO−ATP synthase. In conditions of oxygen limitation, such as within muscles under prolonged exercise, pyruvate is not utilized in the citric acid cycle (CAC), but is converted into L-(+)-lactic acid (’Fleischmilchsäure’) by lactate dehydrogenase (LDH) in a process termed anaerobic glycolysis.
Many cancer cells consume D-(+)-glucose heavily and produce L-(+)-lactic acid rather than catabolizing D-(+)-glucose via the citric acid cycle (CAC) which is normal for generating ATP in non-hypoxic healthy cells. The avid uptake of D-(+)-glucose by tumors is the prerequisite for the detection and monitoring of human tumors by 2-deoxy-2-[18F]fluoroglucose positron emission tomography (PET). More than 90 years ago, Otto Heinrich Warburg (1883−1970) observed that thin slices of human and animal tumors ex vivo displayed high levels of D-(+)-glucose uptake and L-(+)-lactate production. The shift toward L-(+)-lactate production in cancers, even in the presence of adequate oxygen, is termed the Warburg effect or aerobic glycolysis [1−8]. These observations have been confirmed, although the nuances of aerobic glycolysis and its molecular characteristics are still under investigation. Tumors display aerobic glycolysis partly through activation of proto-oncogenes or loss of tumor suppressors, which is then further intensified by stabilization of the evolutionary conserved hypoxia-inducible factor (HIF) via adaptive response to hypoxic microenvironment or through pathways that stabilize HIF under non-hypoxic conditions.
p53 was identified in 1979 by Sir David P. Lane & Lionel V. Crawford [101], Arnold J. Levine & Daniel I.H. Linzer [102], and Lloyd J. Old, Albert B. DeLeo & Colleagues [103]. p53 had been assumed before to exist as the target of the SV40 virus (Polyomaviridae, Betapolyomavirus; simian vacuolating virus 40, simian virus 40) which is tumorigenic. SV40 virus’ oncogenic potential involves suppression of the transcriptional properties of p53 [101,102] (and retinoblastoma) tumor suppressor protein(s) by the SV40 large T antigen [101,102]. The name p53 was coined in 1979 [103], describing the apparent molecular mass of the protein on sodium dodecyl sulfate−polyacrylamide gel electrophoresis (SDS−PAGE). p53 plays an important role in regulation of, or progression through, the cell cycle, in apoptosis and maintenance of genomic stability, by several mechanisms:
- p53 can activate DNA repair proteins when DNA has suffered sustained damage. Thus, it may be an important factor in aging.
- p53 can arrest growth by holding the cell cycle at the G1/S boundary on DNA damage recognition — if it stops the cell cycle here for long enough time, the DNA repair machinery will have time to fix the damage and the cells will be allowed to continue the cell cycle [104].
- p53 can initiate apoptosis (genetically programmed cell death) if DNA damage proves to be irreparable [104].
- p53 is essential for the senescence response to shortened telomeres.
- p53 acts as an inhibitor of angiogenesis generally mediated by vascular endothelial growth factor A (VEGF-A).
- p53 plays an important role in the differentiation and maintenance of human stem cells throughout embryonal development and adult human life.
Compound 1 induced the expression of the p53-induced cyclin-dependent kinase inhibitor Waf1/Cip1/Sdi1/CAP20/MDA-6/CDKN1A (p21Waf1/p21Cip1) [87], and downregulated the p53-suppressed cyclin D1 [105,106,107] protein factor in metastatic castration-resistant prostate cancer (mCRPC) cells, multiple myeloma U266 B lymphocyte lymphoblast cells and plasmacytoma RPMI-8226 B lymphocyte lymphoblast cells (data not shown). Moreover, compound 1 increased the p53-induced pro-apoptotic factor Bax expression in these cancer cells (data not shown). This is a clear indication of (re)activation of (mutant) p53 function by compound 1 in the tested cancer cells.
Compound 1 and compound 3 have, therefore, three antineoplastic mechanisms−of−action: (i) their binding to (mutant) p53 [101,102,103,104] core DNA-binding domain (DBD) [104], thereby restoring sequence-specific DNA-binding of p53 mutants, inhibiting mutant p53 prion-like amyloid aggregation, restoring Zn2+-binding to mutant p53 DBD, and reactivating wild-type p53 transcriptional activity, (ii) their binding to the tetramerization/oligomerization domain (OD) of mutant p53 [23,104] by ionic and hydrophobic interaction(s), thereby restoring the tetramerization of OD mutant p53, reinstalling the target promoter DNA-binding and transcriptional activation properties of OD mutant p53, and (iii) their binding to mitochondrial dianionic cardiolipin (Figure 22), thereby interrupting the anchoring of cytochrome c (and cytochrome c oxidase, Complex IV) at the inner mitochondrial membrane, inducing cytochrome c release into the cytosol with triggering of mitochondrial apoptosis (intrinsic pathway) [19].
Figure 22.
The organic chemical structural formula of cardiolipin [1,3-bis(sn-3′-phosphatidyl)-sn-glycerol, 1,3-diphosphatidyl-sn-glycerol]. The molecule is optically active, representing the L,L-form. Pictured is the predominant dianionic species in the inner mitochondrial membrane with C18:2 [linoleic acid, (9Z,12Z)-9,12-octadecadienoic acid] at each of the four acyl positions, as isolated from bovine heart, bovine heart cardiocyte mitochondria, and rat liver [108].
Figure 22.
The organic chemical structural formula of cardiolipin [1,3-bis(sn-3′-phosphatidyl)-sn-glycerol, 1,3-diphosphatidyl-sn-glycerol]. The molecule is optically active, representing the L,L-form. Pictured is the predominant dianionic species in the inner mitochondrial membrane with C18:2 [linoleic acid, (9Z,12Z)-9,12-octadecadienoic acid] at each of the four acyl positions, as isolated from bovine heart, bovine heart cardiocyte mitochondria, and rat liver [108].
In overall conclusion, compound 1 and compound 3 both trigger the mitochondrial pathway of apoptosis [19] after transcriptional (re)activation of p53 protein. Compound 2 is not able to trigger mitochondrial apoptosis, instead it represents a colchic(in)oid mitotic poison locking cells in anaphase of mitosis, probably binding to the well-known colchicine-binding site at β-tubulin [109,110,111,112,113]. There is no hint from my data that compound 3 can also occupy the colchic(in)oid-binding site at β-tubulin, probably the molecule is too large for that binding site. Therefore, compound 2 only stops tumor cell growth to ±0% growth index, but the cancer cells are all present still. In contrast, compound 1 and compound 3 both completely destroy the tumor cells to nearly −100% of their growth index by ’genetically programmed suicide’.
Importantly, compound 1 and compound 3 are tumoricidal versus both wild-type p53 protein-expressing tumor cell lines and mutant p53 protein-expressing or none p53-expressing cancer cell lines (Table S1). This pointed to an additional p53-independent induction of mitochondrial apoptosis by compound 1 and compound 3 through their binding to mitochondrial cardiolipin based on ionic plus−minus charge attraction and hydrophobic interaction(s), thereby disrupting the membrane-anchoring of cytochrome c and cytochrome c oxidase enzymes in the inner mitochondrial membrane leading to cytochrome c release into the cytosol. Compound 1 and compound 3 can be classified as essentially new antineoplastic drugs exploiting the cancer’s Warburg effect [114,115]. Cancer cell mitochondria exhibit abnormalities in their cardiolipin content, composition, and metabolism [9,10,116,117,118,119]. Therefore, cancer cell mitochondria bear a weakened interaction of cardiolipin (Figure 22) with cytochrome c and cytochrome c oxidase enzymes at the inner mitochondrial membrane. This fact is exploited by the polyammonium compounds 1 and 3 through binding to cancer cell’s dianionic cardiolipin, terminally interrupting its weakened interactions with respiratory chain enzymes preceding mitochondrial apoptosis [120,121,122,123,124].
6. Patents
The three compounds 1, 2 and 3 are proprietary to PopTest Oncology LLC/Palisades Therapeutics and are filed in: Altschul, R.L.; Theise, N.D.; Kesel, A.J.; Rapkin, M.; O’Brien, R.; Arment, A.R. Therapeutic Agents and Methods; PCT/WIPO Pat. Appl. WO/2018/067520 A2, 2018 (filed 3 October 2017); Eur. Pat. Appl. EP 3 534 910 A2, 2019 (filed 3 October 2017); U.S. Pat. Appl. 2019/0381038 A1, 2019 (filed 3 October 2017); U.S. Pat. Appl. US 11,040,037 B2, 2021 (filed 3 October 2017); U.S. Pat. Appl. 2021/0353623 A1, 2021 (filed 28 April 2021); U.S. Pat. Appl. US 11,224,599 B2, 2022 (filed 28 April 2021); U.S. Pat. Appl. 2022/0298203 A1, 2022 (filed 22 November 2021); U.S. Pat. Appl. 11,702,443 B2, 2023 (filed 22 November 2021); U.S. Pat. Appl. 2023/0382945 A1, 2023 (filed 4 May 2023).
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org: 1H-NMR, 13C-NMR, FT–IR, DEPTQ 13C-NMR, gs-COSY, gs-HMQC and gs-HMBC for 1, Figures S1−S7; FT–IR for 2, Figure S8; one formula unit and crystal packing showing the hydrogen bonds in the monoclinic unit cell of 2 (X-ray crystallography of 2), Figures S9 and S10; ESI mass spectra of the separated HPLC peak regions and enlargements of the ESI mass spectrum dication peaks of 3, Figures S11−S20; National Cancer Institute (NCI) Developmental Therapeutics Program (DTP) 60-cancer cell 5-dose testing of 1, 2 and 3 (with cellular p53 status), Table S1; Antineoplastic activity in the NCI DTP 60-cancer cell 5-dose testing of 1, Figures S21−S23; Antineoplastic activity in the NCI DTP 60-cancer cell 5-dose testing of 2, Figures S24−S26; Antineoplastic activity in the NCI DTP 60-cancer cell 5-dose testing of 3, Figures S27−S29.
Author Contributions
Conceptualization, A.J.K.; chemical synthesis, A.J.K.; analysis of data, A.J.K.; conclusions, A.J.K.; writing—original draft, A.J.K.; writing—review and editing, A.J.K.
Funding
No funding was available.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials. Small (~10−20 mg) specimens of the compounds 1, 2 and 3 can be obtained from the author upon request.
Acknowledgments
The author wants to heartily thank K. Gurova (Roswell Park Comprehensive Cancer Center, Buffalo, NY, USA) for the cytochrome c assays of compounds 1, 2 and 3. I heartily thank R.F. Schinazi [Center for AIDS Research (now: Center for ViroScience and Cure), Laboratory of Biochemical Pharmacology, Department of Pediatrics, Emory University School of Medicine, Atlanta, GA, USA, and (up to 2016) the Veterans Affairs Medical Center, Decatur, GA, USA, and Children’s Healthcare of Atlanta, Atlanta, GA, USA] for the HIV-1LAI testings of 1. In addition, the author wishes to heartily thank H. Braunschweig and his associated Senior Researchers T. Kupfer and K. Radacki (Institute for Anorganic Chemistry, Julius-Maximilians-University of Würzburg, Germany) for the X-ray crystallographic solving of the crystal and molecular structure of 2. I am grateful to E. Billings, M. Tobea, and, especially, B. Webb (The Scripps Research Institute, Center for Metabolomics and Mass Spectrometry, San Diego, CA, USA) for the LC/MS investigation service of the compound 3 specimen after more than six years storage in the refrigerator. I cordially thank C. Schnieders, G. Schnieders, R. Schnieders and T. Konrath (DEUTERO GmbH, Kastellaun, Germany) for measuring the 500 MHz spectrum of 3 after more than six years storage in the refrigerator. The author is deeply indepted to T. Westfeld, E.-M. May, D. Wiegel, W. Wübbolt, O. Meier, R. Sachs, A. Karbach, W. Bergmeier, J. Moldenhauer and H.-J. Hühn (Currenta GmbH & Co. OHG, Leverkusen, Germany) for analytical services. I am obliged to K. Hecker, K. Meuser and K. Hecker (HEKAtech GmbH, Wegberg, Germany) for expert elemental analyses.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Warburg, O. The Metabolism of Tumours (translated by Dickens, F.); Constable: London, 1930. [Google Scholar]
- Warburg, O.; Posener, K.; Negelein, E. Über den Stoffwechsel der Carcinomzelle. Biochem. Z. (Berl.) 1924, 152, 309–344. [Google Scholar] [CrossRef]
- Warburg, O. Über den Stoffwechsel der Carcinomzelle. Naturwissenschaften 1924, 12, 1131–1137. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Cuezva, J.M.; Krajewska, M.; López de Heredia, M.; Krajewski, S.; Santamaría, G.; Kim, H.; Zapata, J.M.; Marusawa, H.; Chamorro, M.; Reed, J.C. The bioenergetic signature of cancer: A marker of tumor progression. Cancer Res. 2002, 62, 6674–6681. [Google Scholar]
- Kim, J.-w.; Dang, C.V. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006, 66, 8927–8930. [Google Scholar] [CrossRef]
- Kiebish, M.A.; Han, X.; Cheng, H.; Chuang, J.H.; Seyfried, T.N. Cardiolipin and electron transport chain abnormalities in mouse brain tumor mitochondria: Lipidomic evidence supporting the Warburg theory of cancer. J. Lipid Res. 2008, 49, 2545–2556. [Google Scholar] [CrossRef]
- Jahnke, V.E.; Sabido, O.; Defour, A.; Castells, J.; Lefai, E.; Roussel, D.; Freyssenet, D. Evidence for mitochondrial respiratory deficiency in rat rhabdomyosarcoma cells. PLoS One 2010, 5, e8637. [Google Scholar] [CrossRef]
- Cori, C.F.; Cori, G.T. The carbohydrate metabolism of tumors. II. Changes in the sugar, lactic acid, and CO2-combining power of blood passing through a tumor. J. Biol. Chem. 1925, 65, 397–405. [Google Scholar] [CrossRef]
- Weinhouse, S. On respiratory impairment in cancer cells. Science 1956, 124, 267–269. [Google Scholar] [CrossRef]
- Warburg, O. Reply to the Editor. Science 1956, 124, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Burk, D.; Schade, A.L. Letter to the Editor. Science 1956, 124, 270–272. [Google Scholar] [CrossRef] [PubMed]
- Quinn, P.J.; Dawson, R.M.C. Interactions of cytochrome c and [14C]carboxymethylated cytochrome c with monolayers of phosphatidylcholine, phosphatidic acid and cardiolipin. Biochem. J. 1969, 115, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Vik, S.B.; Georgevich, G.; Capaldi, R.A. Diphosphatidylglycerol is required for optimal activity of beef heart cytochrome c oxidase. Proc. Natl. Acad. Sci. U. S. A. 1981, 78, 1456–1460. [Google Scholar] [CrossRef] [PubMed]
- Speck, S.H.; Neu, C.A.; Swanson, M.S.; Margoliash, E. Role of phospholipid in the low affinity reactions between cytochrome c and cytochrome oxidase. FEBS Lett. 1983, 164, 379–382. [Google Scholar] [CrossRef]
- Rytömaa, M.; Mustonen, P.; Kinnunen, P.K.J. Reversible, nonionic, and pH-dependent association of cytochrome c with cardiolipin-phosphatidylcholine liposomes. J. Biol. Chem. 1992, 267, 22243–22248. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Indran, I.R.; Tufo, G.; Pervaiz, S.; Brenner, C. Recent advances in apoptosis, mitochondria and drug resistance in cancer cells. Biochim. Biophys. Acta 2011, 1807, 735–745. [Google Scholar] [CrossRef]
- Danysz, W.; Parsons, C.G.; Kornhuber, J.; Schmidt, W.J.; Quack, G. Aminoadamantanes as NMDA receptor antagonists and antiparkinsonian agents – Preclinical studies. Neurosci. Biobehav. Rev. 1997, 21, 455–468. [Google Scholar] [CrossRef]
- Davies, W.L.; Grunert, R.R.; Haff, R.F.; McGahen, J.W.; Neumayer, E.M.; Paulshock, M.; Watts, J.C.; Wood, T.R.; Hermann, E.C.; Hoffmann, C.E. Antiviral activity of 1-adamantanamine (amantadine). Science 1964, 144, 862–863. [Google Scholar] [CrossRef] [PubMed]
- Kamada, R.; Nomura, T.; Anderson, C.W.; Sakaguchi, K. Cancer-associated p53 tetramerization domain mutants. Quantitative analysis reveals a low threshold for tumor suppressor inactivation. J. Biol. Chem. 2011, 286, 252–258. [Google Scholar] [CrossRef]
- Kesel, A.J. Broad-spectrum antiviral activity including human immunodeficiency and hepatitis C viruses mediated by a novel retinoid thiosemicarbazone derivative. Eur. J. Med. Chem. 2011, 46, 1656–1664. [Google Scholar] [CrossRef] [PubMed]
- Kesel, A.J.; Huang, Z.; Murray, M.G.; Prichard, M.N.; Caboni, L.; Nevin, D.K.; Fayne, D.; Lloyd, D.G.; Detorio, M.A.; Schinazi, R.F. Retinazone inhibits certain blood-borne human viruses including Ebola virus Zaire. Antivir. Chem. Chemother. 2014, 23, 197–215. [Google Scholar] [CrossRef]
- Landa, S.; Macháček, V. Sur l’adamantane, nouvel hydrocarbure extrait du naphte. Coll. Czech. Chem. Commun. 1933, 5, 1–5. [Google Scholar] [CrossRef]
- Hofmann, A.W. Einwirkung der Wärme auf die Ammoniumbasen. Ber. dtsch. chem. Ges. 1881, 14, 659–669. [Google Scholar] [CrossRef]
- Gano, J.E.; Eizenberg, L. Short-lived intermediates. IV. Adamantene. J. Am. Chem. Soc. 1973, 95, 972–974. [Google Scholar] [CrossRef]
- Bian, N.; Jones, M., Jr. More on adamantene. J. Am. Chem. Soc. 1995, 117, 8957–8961. [Google Scholar] [CrossRef]
- Tae, E.L.; Zhu, Z.; Platz, M.S. A matrix isolation, laser flash photolysis, and computational study of adamantene. J. Phys. Chem. A 2001, 105, 3803–3807. [Google Scholar] [CrossRef]
- Baek, J.Y.; Lee, S.J.; Han, B.H. Direct synthesis of symmetric ethers from carbonyl compounds using SbI3/PhSiH3. J. Korean Chem. Soc. 2004, 48, 220–224. [Google Scholar] [CrossRef]
- Nishiyama, T.; Kameyama, H.; Maekawa, H.; Watanuki, K. Ether synthesis using trifluoromethanesulfonic anhydride or triflates under mild reaction conditions. Can. J. Chem. 1999, 77, 258–262. [Google Scholar] [CrossRef]
- Burger, R.; Bigler, P. DEPTQ: Distorsionless enhancement by polarization transfer including the detection of quaternary nuclei. J. Magn. Res. 1998, 135, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.; Braun, S. 200 and More NMR Experiments. A Practical Course; 1st ed., Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2004. [Google Scholar]
- Kozłowski, E.; Biziuk, M. A new method for simultaneous determination of nitrogen, hydrogen and halogen in organic compounds. Mikrochim. Acta (Wien) 1979, 72, 1–17. [Google Scholar] [CrossRef]
- Sharifi, N.; Hamel, E.; Lill, M.A.; Risbood, P.; Kane, C.T., Jr.; Hossain, M.T.; Jones, A.; Dalton, J.T.; Farrar, W.L. A bifunctional colchicinoid that binds to the androgen receptor. Mol. Cancer Ther. 2007, 6, 2328–2336. [Google Scholar] [CrossRef] [PubMed]
- Mackay, M.F.; Morrison, J.D.; Gulbis, J.M. Crystal structure of triclinic colchiceine hemihydrate. Aust. J. Phys. 1985, 38, 413–420. [Google Scholar] [CrossRef]
- Silverton, J.V. The structure of colchiceine ethyl acetate–water solvate. Acta Crystallogr. B 1979, 35, 2800–2803. [Google Scholar] [CrossRef]
- Zeisel, S. Über Colchicin und Colchiceïn. Monatsh. Chem. 1883, 4, 162–164. [Google Scholar] [CrossRef]
- Zeisel, S. Über das Colchicin. I. Abhandlung. Monatsh. Chem. 1886, 7, 557–596. [Google Scholar] [CrossRef]
- Cook, J.W.; Loudon, J.D. Colchicine; In: Manske, R.H.F.; Holmes, H.L. (Eds.) The Alkaloids: Chemistry and Physiology; Academic Press Inc.: New York, 1952; Volume II, p. 265. [Google Scholar]
- King, M.V.; de Vries, J.L.; Pepinsky, R. An X-ray diffraction determination of the chemical structure of colchicine. Acta Crystallogr. 1952, 5, 437–440. [Google Scholar] [CrossRef]
- Lessinger, L.; Margulis, T.N. The crystal structure of colchicine. A new application of magic integers to multiple-solution direct methods. Acta Crystallogr. B 1978, 34, 578–584. [Google Scholar] [CrossRef]
- Clewer, H.W.B.; Green, S.J.; Tutin, F. The constituents of Gloriosa superba. J. Chem. Soc., Trans. 1915, 107, 835–846. [Google Scholar] [CrossRef]
- Clerici, F.; Mottadelli, S.; Rossi, L.M. 1H- and 13C-NMR spectra of thiocolchicine and derivatives: A complete analysis. J. Nat. Prod. 1995, 58, 259–263. [Google Scholar] [CrossRef]
- Meksuriyen, D.; Lin, L.-J.; Cordell, G.A.; Mukhopadhyay, S.; Banerjee, S.K. NMR studies of colchicine and its photoisomers, - and -lumicolchicines. J. Nat. Prod. 1988, 51, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.A.; Brossi, A.; Silverton, J.V. 7-Oxo-deacetamidocolchiceine and 7-benzylimino-deacetamido-colchiceine: Two novel products from the base catalysed reaction of (–)-N-benzylidene-deacetylcolchiceine. Helv. Chim. Acta 1978, 61, 1213–1220. [Google Scholar] [CrossRef]
- Hufford, C.D.; Capraro, H.-G.; Brossi, A. 13C- and 1H-NMR. Assignments for colchicine derivatives. Helv. Chim. Acta 1980, 63, 50–56. [Google Scholar] [CrossRef]
- Horowitz, R.M.; Ullyot, G.E. Colchicine. Some reactions of ring C. J. Am. Chem. Soc. 1952, 74, 587–592. [Google Scholar] [CrossRef]
- Šantavý, Fr. Préparation de l’acide colchicique à partir de la colchicine. Helv. Chim. Acta 1948, 31, 821–826. [Google Scholar] [CrossRef]
- Fernholz, H. Über die Umlagerung des Colchicins mit Natriumalkoholat und die Struktur des Ringes C. Justus Liebigs Ann. Chem. 1950, 568, 63–72. [Google Scholar] [CrossRef]
- Hufford, C.D.; Collins, C.C.; Clark, A.M. Microbial transformations and 13C-NMR analysis of colchicine. J. Pharm. Sci. 1979, 68, 1239–1243. [Google Scholar] [CrossRef]
- Nozoe, T.; Ikemi, T.; Itô, S. Some derivatives from colchicine. Proc. Japan Acad. (Nippon-gakushiin-kiyô, Tokyo) 1954, 30, 609–613. [Google Scholar] [CrossRef]
- Nozoe, T.; Ikemi, T.; Itô, S. Derivatives of colchicine. Sci. Rep. Tôhoku Univ. Ser. 1 (Tôhoku-daigaku-rika-hôkoku, dai 1-shû, kagaku, Sendai) 1954, 38, 117–129. [Google Scholar]
- Windaus, A.; Schiele, H. Die Konstitution des Colchicins. Nachr. Ges. Wiss. Göttingen Math.-Phys. 1924. [Google Scholar]
- Cook, J.W.; Loudon, J.D. Colchicine; In: Manske, R.H.F.; Holmes, H.L. (Eds.) The Alkaloids: Chemistry and Physiology; Academic Press Inc.: New York, 1952; Volume II, p. 273. [Google Scholar]
- Cook, J.W.; Loudon, J.D. The tropolones. Quart. Rev. Chem. Soc. (London) 1951, 5, 99–130. [Google Scholar] [CrossRef]
- Pauson, P.L. Tropones and tropolones. Chem. Rev. 1955, 55, 9–136. [Google Scholar] [CrossRef]
- Dauben, H.J., Jr.; Ringold, H.J. Synthesis of tropone. J. Am. Chem. Soc. 1951, 73, 876–876. [Google Scholar] [CrossRef]
- Doering, W. von E.; Detert, F.L. Cycloheptatrienylium oxide. J. Am. Chem. Soc. 1951, 73, 876–877. [Google Scholar] [CrossRef]
- Redington, R.L.; Bock, C.W. MO study of singlets, triplets, and tunneling in tropolone. 1. Geometries, tunneling, and vibrations in the ground electronic state. J. Phys. Chem. 1991, 95, 10284–10294. [Google Scholar] [CrossRef]
- Brossi, A.; Yeh, H.J.C.; Chrzanowska, M.; Wolff, J.; Hamel, E.; Lin, C.M.; Quin, F.; Suffness, M.; Silverton, J. Colchicine and its analogues: Recent findings. Med. Res. Rev. 1988, 8, 77–94. [Google Scholar] [CrossRef]
- Brossi, A.; Boyé, O.; Muzaffar, A.; Yeh, H.J.C.; Toome, V.; Wegrzynski, B.; George, C. aS,7S-absolute configuration of natural (–)-colchicine and allo-congeners. FEBS Lett. 1990, 262, 5–7. [Google Scholar] [CrossRef]
- Cahn, R.S.; Ingold, C.; Prelog, V. Spezifikation der molekularen Chiralität. Angew. Chem. 1966, 78, 413–447. [Google Scholar] [CrossRef]
- Cahn, R.S.; Ingold, C.; Prelog, V. Specification of Molecular Chirality. Angew. Chem. Int. Ed. Engl. 1966, 5, 385–415. [Google Scholar] [CrossRef]
- Detrich, H.W., III; Williams, R.C., Jr.; Macdonald, T.L.; Wilson, L.; Puett, D. Changes in the circular dichroic spectrum of colchicine associated with its binding to tubulin. Biochemistry 1981, 20, 5999–6005. [Google Scholar] [CrossRef] [PubMed]
- Berg, U.; Bladh, H. The absolute configuration of colchicine by correct application of the CIP rules. Helv. Chim. Acta 1999, 82, 323–325. [Google Scholar] [CrossRef]
- Corrodi, H.; Hardegger, E. Die Konfiguration des Colchicins und verwandter Verbindungen. Helv. Chim. Acta 1955, 38, 2030–2033. [Google Scholar] [CrossRef]
- Bagnato, J.D.; Eilers, A.L.; Horton, R.A.; Grissom, C.B. Synthesis and characterization of a cobalamin–colchicine conjugate as a novel tumor-targeted cytotoxin. J. Org. Chem. 2004, 69, 8987–8996. [Google Scholar] [CrossRef]
- Bombuwala, K.; Kinstle, T.; Popik, V.; Uppal, S.O.; Olesen, J.B.; Viña, J.; Heckman, C.A. Colchitaxel, a coupled compound made from microtubule inhibitors colchicine and paclitaxel. Beilstein J. Org. Chem. 2006, 2, 13. [Google Scholar] [CrossRef] [PubMed]
- Sustmann, R.; Brandes, D.; Lange, F.; Tashtoush, H.I. Substituent effects in cycloheptatrienyl radicals: An ESR spectroscopic study. Magn. Reson. Chem. 1989, 27, 340–343. [Google Scholar] [CrossRef]
- Fischer, K.H.; Hemberger, P.; Bodi, A.; Fischer, I. Photoionisation of the tropyl radical. Beilstein J. Org. Chem. 2013, 9, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.W. Organic Chemistry; Volume 7, Ylid Chemistry, Ed.; 1st ed., Academic Press: New York, London, 1966; p. 251283. [Google Scholar]
- Musker, W.K.; Stevens, R.R. Nitrogen ylides. V. Coordination chemistry of trimethylammonium methylide. Inorg. Chem. 1969, 8, 255–264. [Google Scholar] [CrossRef]
- Wittig, G.; Wetterling, M.-H. Darstellung und Eigenschaften des Trimethyl-ammonium-methylids. Justus Liebigs Ann. Chem. 1947, 557, 193–201. [Google Scholar] [CrossRef]
- Nekrasov, B.V. Pentavalent nitrogen. Bull. Acad. Sci. U. S. S. R., Div. Chem. Sci. (Russ. Chem. Bull.) 1979, 28, 1789–1790. [Google Scholar] [CrossRef]
- Kurzydłowski, D.; Zaleski-Ejgierd, P. Hexacoordinated nitrogen(V) stabilized by high pressure. Sci. Rep. 2016, 6, 36049. [Google Scholar] [CrossRef]
- Yan, C.; Takeshita, M.; Nakatsuji, J.-y.; Kurosaki, A.; Sato, K.; Shang, R.; Nakamoto, M.; Yamamoto, Y.; Adachi, Y.; Furukawa, K.; Kishi, R.; Nakano, M. Synthesis and properties of hypervalent electron-rich pentacoordinate nitrogen compounds. Chem. Sci. 2020, 11, 5082–5088. [Google Scholar] [CrossRef]
- Ikeda, E.; Tranter, R.S.; Kiefer, J.H.; Kern, R.D.; Singh, H.J.; Zhang, Q. The pyrolysis of methylcyclopentadiene: Isomerization and formation of aromatics. Proc. Combust. Inst. 2000, 28, 1725–1732. [Google Scholar] [CrossRef]
- Hanamirian, B.; Della Libera, A.; Pratali Maffei, L.; Cavallotti, C. Investigation of methylcyclopentadiene reactivity: Abstraction reactions and methylcyclopentadienyl radical unimolecular decomposition. J. Phys. Chem. A 2023, 127, 1314–1328. [Google Scholar] [CrossRef] [PubMed]
- Berglind, H.; Pawitan, Y.; Kato, S.; Ishioka, C.; Soussi, T. Analysis of p53 mutation status in human cancer cell lines. A paradigm for cell line cross-contamination. Cancer Biol. Ther. 2008, 7, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef]
- Foley, G.E.; Lazarus, H.; Farber, S.; Uzman, B.G.; Boone, B.A.; McCarthy, R.E. Continuous culture of human lymphoblasts from peripheral blood of a child with acute leukemia. Cancer 1965, 18, 522–529. [Google Scholar] [CrossRef]
- Subler, M.A.; Martin, D.W.; Deb, S. Inhibition of viral and cellular promoters by human wild-type p53. J. Virol. 1992, 66, 4757–4762. [Google Scholar] [CrossRef] [PubMed]
- Subler, M.A.; Martin, D.W.; Deb, S. Activation of the human immunodeficiency virus type 1 long terminal repeat by transforming mutants of human p53. J. Virol. 1994, 68, 103–110. [Google Scholar] [CrossRef]
- Duan, L.; Ozaki, I.; Oakes, J.W.; Taylor, J.P.; Khalili, K.; Pomerantz, R.J. The tumor suppressor protein p53 strongly alters human immunodeficiency virus type 1 replication. J. Virol. 1994, 68, 4302–4313. [Google Scholar] [CrossRef]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Sharifi, H.J.; DiGrigoli, S.; Kinnetz, M.; Mellon, K.; Hu, W.; de Noronha, C.M.C. Inhibition of HIV early replication by the p53 and its downstream gene p21. Virol. J. 2018, 15, 53. [Google Scholar] [CrossRef]
- Chowdhury, I.H.; Wang, X.-F.; Landau, N.R.; Robb, M.L.; Polonis, V.R.; Birx, D.L.; Kim, J.H. HIV-1 Vpr activates cell cycle inhibitor p21/Waf1/Cip1: A potential mechanism of G2/M cell cycle arrest. Virology 2003, 305, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Li, C.J.; Wang, C.; Friedman, D.J.; Pardee, A.B. Reciprocal modulations between p53 and Tat of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 5461–5464. [Google Scholar] [CrossRef] [PubMed]
- Longo, F.; Marchetti, M.A.; Castagnoli, L.; Battaglia, P.A.; Gigliani, F. A novel approach to protein‒protein interaction: Complex formation between the p53 tumor suppressor and the HIV Tat proteins. Biochem. Biophys. Res. Commun. 1995, 206, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Ariumi, Y.; Kaida, A.; Hatanaka, M.; Shimotohno, K. Functional cross-talk of HIV-1 Tat with p53 through its C-terminal domain. Biochem. Biophys. Res. Commun. 2001, 287, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Greenway, A.L.; McPhee, D.A.; Allen, K.; Johnstone, R.; Holloway, G.; Mills, J.; Azad, A.; Sankovich, S.; Lambert, P. Human immunodeficiency virus type 1 Nef binds to tumor suppressor p53 and protects cells against p53-mediated apoptosis. J. Virol. 2002, 76, 2692–2702. [Google Scholar] [CrossRef]
- Sawaya, B.E.; Khalili, K.; Mercer, W.E.; Denisova, L.; Amini, S. Cooperative actions of HIV-1 Vpr and p53 modulate viral gene transcription. J. Biol. Chem. 1998, 273, 20052–20057. [Google Scholar] [CrossRef] [PubMed]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef]
- Schinazi, R.F.; Sommadossi, J.-P.; Saalmann, V.; Cannon, D.L.; Xie, M.-Y.; Hart, G.C.; Smith, G.A.; Hahn, E.F. Activities of 3′-azido-3′-deoxythymidine nucleotide dimers in primary lymphocytes infected with human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 1990, 34, 1061–1067. [Google Scholar] [CrossRef]
- Spira, T.J.; Bozeman, L.H.; Holman, R.C.; Warfield, D.T.; Phillips, S.K.; Feorino, P.M. Micromethod for assaying reverse transcriptase of human T-cell lymphotropic virus type III/lymphadenopathy-associated virus. J. Clin. Microbiol. 1987, 25, 97–99. [Google Scholar] [CrossRef]
- Stuyver, L.J.; Lostia, S.; Adams, M.; Mathew, J.S.; Pai, B.S.; Grier, J.; Tharnish, P.M.; Choi, Y.; Chong, Y.; Choo, H.; Chu, C.K.; Otto, M.J.; Schinazi, R.F. Antiviral activities and cellular toxicities of modified 2′,3′-dideoxy-2′,3′-didehydrocytidine analogues. Antimicrob. Agents Chemother. 2002, 46, 3854–3860. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Flack, H.D. On enantiomorph-polarity estimation. Acta Crystallogr. A 1983, 39, 876–881. [Google Scholar] [CrossRef]
- Lane, D.P.; Crawford, L.V. T antigen is bound to a host protein in SV40-transformed cells. Nature 1979, 278, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Linzer, D.I.H.; Levine, A.J. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 1979, 17, 43–52. [Google Scholar] [CrossRef] [PubMed]
- DeLeo, A.B.; Jay, G.; Appella, E.; Dubois, G.C.; Law, L.W.; Old, L.J. Detection of a transformation-related antigen in chemically induced sarcomas and other transformed cells of the mouse. Proc. Natl. Acad. Sci. U. S. A. 1979, 76, 2420–2424. [Google Scholar] [CrossRef]
- Levine, A.J.; Lane, D.P. (Eds.) The p53 Family, Cold Spring Harbor Perspectives in Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2010. [Google Scholar]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef]
- Tashiro, E.; Tsuchiya, A.; Imoto, M. Functions of cyclin D1 as an oncogene and regulation of cyclin D1 expression. Cancer Sci. 2007, 98, 629–635. [Google Scholar] [CrossRef]
- Diehl, J.A. Cycling to cancer with cyclin D1. Cancer Biol. Ther. 2002, 1, 226–231. [Google Scholar] [CrossRef]
- Schlame, M.; Brody, S.; Hostetler, K.Y. Mitochondrial cardiolipin in diverse eukaryotes. Comparison of biosynthetic reactions and molecular acyl species. Eur. J. Biochem. 1993, 212, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Uppuluri, S.; Knipling, L.; Sackett, D.L.; Wolf, J. Localization of the colchicine-binding site of tubulin. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 11598–11602. [Google Scholar] [CrossRef] [PubMed]
- Andreu, J.M.; Perez-Ramirez, B.; Gorbunoff, M.J.; Ayala, D.; Timasheff, S.N. Role of the colchicine ring A and its methoxy groups in the binding to tubulin and microtubule inhibition. Biochemistry 1998, 37, 8356–8368. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Covell, D.G.; Pei, X.-F.; Ewell, J.B.; Nguyen, N.Y.; Brossi, A.; Hamel, E. Mapping the binding site of colchicinoids on -tubulin. 2-Chloroacetyl-2-demethylthiocolchicine covalently reacts predominantly with cysteine 239 and secondarily with cysteine 354. J. Biol. Chem. 2000, 275, 40443–40452. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, R.B.G.; Gigant, B.; Curmi, P.A.; Jourdain, I.; Lachkar, S.; Sobel, A.; Knossow, M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004, 428, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, H.; Gigant, B.; Yu, Y.; Wu, Y.; Chen, X.; Lai, Q.; Yang, Z.; Chen, Q.; Yang, J. Structures of a diverse set of colchicine binding site inhibitors in complex with tubulin provide a rationale for drug discovery. FEBS J. 2016, 283, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Pascale, R.M.; Calvisi, D.F.; Simile, M.M.; Feo, C.F.; Feo, F. The Warburg effect 97 years after its discovery. Cancers (Basel) 2020, 12, 2819. [Google Scholar] [CrossRef]
- Liu, C.; Jin, Y.; Fan, Z. Mechanism of Warburg effect-induced chemoresistance in cancer. Front. Oncol. 2021, 11, 698023. [Google Scholar] [CrossRef] [PubMed]
- Sapandowski, A.; Stope, M.; Evert, K.; Evert, M.; Zimmermann, U.; Peter, D.; Päge, I.; Burchardt, M.; Schild, L. Cardiolipin composition correlates with prostate cancer cell proliferation. Mol. Cell. Biochem. 2015, 410, 175–185. [Google Scholar] [CrossRef]
- Ahmadpour, S.T.; Mahéo, K.; Servais, S.; Brisson, L.; Dumas, J.-F. Cardiolipin, the mitochondrial signature lipid: Implication in cancer. Int. J. Mol. Sci. 2020, 21, 8031. [Google Scholar] [CrossRef]
- Di Carlo, C.; Sousa, B.C.; Manfredi, M.; Brandi, J.; Dalla Pozza, E.; Marengo, E.; Palmieri, M.; Dando, I.; Wakelam, M.J.O.; Lopez-Clavijo, A.F.; Cecconi, D. Integrated lipidomics and proteomics reveal cardiolipin alterations, upregulation of HADHA and long chain fatty acids in pancreatic cancer stem cells. Sci. Rep. 2021, 11, 13297. [Google Scholar] [CrossRef] [PubMed]
- Krieger, A.C.; Macias, L.A.; Goodman, J.C.; Brodbelt, J.S.; Eberlin, L.S. Mass spectrometry imaging reveals abnormalities in cardiolipin composition and distribution in astrocytoma tumor tissues. Cancers (Basel) 2023, 15, 2842. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z. The chemical biology of apoptosis: Exploring protein-protein interactions and the life and death of cells with small molecules. Chem. Biol. (Cell Chem. Biol.) 2002, 9, 1059–1072. [Google Scholar]
- Wong, R.S.Y. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Cavalcante, G.C.; Schaan, A.P.; Cabral, G.F.; Santana-da-Silva, M.N.; Pinto, P.; Vidal, A.F.; Ribeiro-Dos-Santos, Â. A cell’s fate: An overview of the molecular biology and genetics of apoptosis. Int. J. Mol. Sci. 2019, 20, 4133. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



