
Submitted:
29 December 2023
Posted:
03 January 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
(1) Background: COVID-19 was responsible for the latest pandemic, shaking and reshaping healthcare systems worldwide. Its late clinical manifestations make it linger in medical memory as a debilitating illness over extended periods. (2) Methods: Recent literature was systematically analyzed to categorize and examine the symptomatology and pathophysiology of Long COVID across various bodily systems, including pulmonary, cardiovascular, gastrointestinal, neuropsychiatric, dermatological, renal, hematological, and endocrinological aspects. (3) Results: The review outlines the diverse clinical manifestations of Long COVID across multiple systems, emphasizing its complexity and challenges in diagnosis and treatment. Factors such as pre-existing conditions, initial COVID-19 severity, vaccination status, gender, and age were identified as influential in the manifestation and persistence of Long COVID symptoms. This condition is highlighted as a debilitating disease capable of enduring over an extended period and presenting new symptoms over time. (4) Conclusions: Long COVID emerges as a condition with intricate multi-systemic involvement, complicating its diagnosis and treatment. The findings underscore the necessity for a nuanced understanding of its diverse manifestations to effectively manage and address the evolving nature of this condition over time.
Keywords:
Long COVID
; COVID-19
; SARS-CoV-2
; hypercoagulability
; dysbiosis
1. Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) causes the disease we now call COVID-19. In early December 2019, the first cases of COVID-19 started to appear, igniting the beginning of a pandemic that has, by the time this article is written, taken 6.9 million lives worldwide. The report of the fifteenth meeting of the International Health Regulations Emergency Committee, which was held on the 4th of May 2023, concluded with the formal end of the public health emergency regarding the COVID-19 pandemic. With a decrease in deaths and hospitalizations due to acute COVID-19, the interest has now slowly shifted towards the long-term complications seen in both prolonged and recovered cases. Thus, the term Long COVID has emerged, encompassing multiple symptoms that manifest after a SARS-CoV-2 infection. Long COVID places additional pressure on organs that are already strained by the acute form of the disease, resulting in multi-systemic long-term symptoms. The description given for Long COVID by the World Health Organization (WHO) is based on its occurrence in patients with a known or suspected SARS-CoV-2 infection, its development within 3 months after the onset of symptoms, its persistence extending over 2 months or more and the inability to justify symptoms by any other differential diagnosis.
The symptomatology comprises a wide variety of symptoms, some more frequently encountered such as fatigue, exertional dyspnea, insomnia, malaise, cognitive impairment, myalgia, cough, anosmia, arthralgia, chest pain, fever, tachycardia, and palpitations [1,2,3,4,5], 7, [9,10,11,12,13,14], [16]. The establishment of accurate epidemiological risk factors is highly important, as they are extremely valuable both for diagnostic and research purposes. Most of the articles that were reviewed link a high incidence of manifestation or persistence of Long COVID symptoms in females [2,3,4,5,6,7,8,9,10,11,12,14,15], older patients [4,7,15], patients with previous pulmonary diseases [4,7,9,10,15], diabetic patients [15,16], obese patients [3,4,15], patients with previous cardiovascular pathology [15,16], patients with increased SARS-CoV-2 viral load [8], smokers [3,4], patients with depression [4,5,6,7,8,9,10,11] and patients that experienced severe COVID-19 [7,8,9,13,14].
This research aims to comprehensively detail and provide an understanding of the clinical manifestations of Long COVID, a newly emerged medical entity recognized in the aftermath of the pandemic. The research focuses on the involvement of specific systems or organs, with an emphasis on identifying and characterizing the unique features of these manifestations.
2. Materials and Methods
References were found in relevant databases such as the National Center for Biotechnology Information, the Centers for Disease Control and Prevention, the Scientific Electronic Library Online, and the Cochrane Database. For this review, utilizing searches within the publication interval of 2021/01/01 - 2023/12/01, along with linking the term “Long Covid” with one of the following: “cardiovascular diseases or renal diseases or respiratory diseases or gastrointestinal diseases or neuro-psychiatric diseases or endocrine diseases or skin diseases or pathology or dysbiosis or chronic fatigue syndrome or myalgic encephalomyelitis.” The obtained articles underwent examination. Their selection and rejection were carried out according to the flowchart (Figure 1).
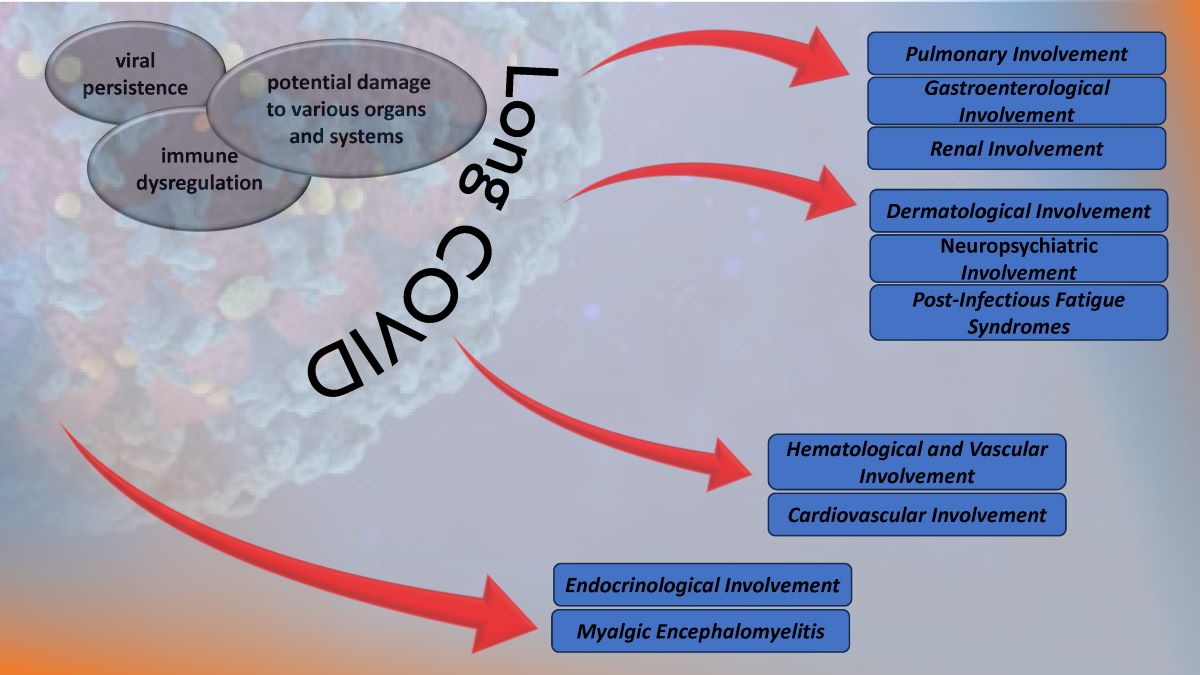
3. The Multisystem Impact of Long COVID
3.1. Pulmonary Involvement in Long COVID
The main impact of SARS-CoV-2 infection is on the respiratory system, the entrance of the virus into the epithelial lining of the lungs is facilitated by pneumonocytes that express the angiotensin-converting enzyme 2 (ACE2) receptors [17]. Upon infection by SARS-CoV-2, lung epithelial cells, which serve as the primary origin of inflammatory cytokines, engage in interactions with immune cells that have been recruited to the site of infection. This interaction plays a significant part in the development of inflammatory lung damage and subsequent respiratory failure [18]. The persistence and unregulated activation of cytokine storms can result in the impairment of the epithelial barrier. In the presence of such circumstances, the progression of lung fibrosis occurs due to the impairment of lung epithelial regeneration [19].
The injury inflicted by SARS-CoV-2 on a range of lung epithelial cells is attributed to the ACE2 receptors or transmembrane serine protease 2 (TMPRSS2) expression. ACE2 has been observed to be expressed in various types of cells in the airway epithelium, such as basal cells, ciliated cells, mucous cells, club cells, and intermediate cells [20]. According to a single-cell ribonucleic acid (RNA) sequencing analysis, it was observed that the average amount of ACE2 was comparatively higher in mucous cells as compared to other types of epithelial cells [21]. Alveolar type 1 (AT1) and alveolar type 2 (AT2) cells are susceptible to SARS-CoV-2 infection, with AT2 cells being the initial target [22]. Nonetheless, academic studies propose that a minor fraction of AT2 cells exhibit the expression of ACE2 while the expression of TMPRSS2 is minimal in the basal cells of the undifferentiated airway epithelium and more prominently expressed in the differentiated airway epithelium [23,24,25]. This proposed mechanism is accountable for the susceptibility of the lungs to COVID-19.
Dyspnea is the most common pulmonary symptom, because of Long COVID. Dyspnea is defined as an unpleasant and intolerable pattern of breathing, related to an inability to ventilate enough to provide the required amount of air for one normal breath. According to Hentsch L. et all, in research, published in 2021, three possible mechanisms were proposed for dyspnea induced by COVID-19 [26]. One of the mechanisms implies the interruption of afferent sensory signaling pathways by SARS-CoV-2 which may result in the inability of cortical structures responsible for processing the sensory aspects of dyspnea to receive afferent inputs from the brainstem. It is plausible that the virus may cause direct harm to the mechano- or irritant receptors located in the respiratory tract and/or chest wall, thereby impeding the transmission of afferent signals to the brainstem and higher brain structures. The second mechanism pertains to the possibility that SARS-CoV-2 may impede the capacity of cortical structures to identify or manage incoming sensory signals related to breathlessness originating from the brainstem. The occurrence of the conditions may arise either because of the direct impact of SARS-CoV-2 on nervous tissue or indirectly through the manifestation of inflammatory acute encephalopathy or cerebrovascular complications, including but not limited to ischemic or hemorrhagic stroke. The last mechanism indicates the cortical structures implicated in the perception of dyspnea may exhibit a facilitative influence on the perception of breathlessness, like that of pain, which may be disrupted by SARS-CoV-2. The induction of dyspnea-related panic attacks through experimental inhalation of 35% CO2 has been demonstrated in patients with bilateral amygdala damage [27]. The self-reported levels of panic and fear in this group were found to be remarkably higher than those acknowledged by the comparison group with intact neurological functioning. The findings of this study may indicate that the activation of extra-limbic brain structures by CO2 occurs directly, and subsequently, these structures are controlled in a downward manner by the amygdala [27].
Several mechanisms of pulmonary damage in COVID-19 have been identified, with viral and immune-mediated pathways being implicated. Pulmonary fibrosis may arise because of chronic inflammation or as an idiopathic, age-related fibroproliferative process that is influenced by genetic factors [28]. Pulmonary fibrosis is a recognized consequence of acute respiratory distress syndrome (ARDS). Nevertheless, the clinical relevance of persistent radiological abnormalities after ARDS is limited and has decreased with the implementation of protective lung ventilation techniques [29]. Research has indicated that a significant proportion of individuals diagnosed with COVID-19, specifically 40%, are prone to developing ARDS. Furthermore, it has been observed that 20% of ARDS cases are classified as severe [30]. The manifestation of post-COVID-19 fibrosis will require further observation, however, initial examination of patients with COVID-19 upon hospital release indicates that over 33% of recuperated individuals exhibit fibrotic irregularities. The defining characteristic of ARDS is the presence of diffuse alveolar damage (DAD). This is marked by an initial phase of acute inflammatory exudation, which is characterized by the presence of hyaline membranes. This is then followed by an organizing phase and a fibrotic phase [31]. Prior research has emphasized the significance of the duration of illness as a crucial factor in the development of pulmonary fibrosis after ARDS. The findings of this investigation indicate that a small proportion of patients (4%) who had a disease duration of less than one week, a notably larger proportion (24%) of patients with an illness lasting between 1-3 weeks, and a majority (61%) of patients who had a disease duration exceeding three weeks, experienced the development of fibrosis [32]. The development and progression of pulmonary fibrosis may be instigated and facilitated by a cytokine storm resulting from an atypical immune response. The release of matrix metalloproteinases during the inflammatory phase of ARDS leads to epithelial and endothelial injury. The process of fibrosis involves the participation of vascular endothelial growth factor and cytokines, including interleukin (IL) 6 and tumor necrosis factor (TNF) a. The etiology behind the differential outcomes of individuals who either recuperate from an insult or develop progressive pulmonary fibrosis characterized by the accumulation of fibroblasts and myofibroblasts, along with excessive collagen deposition, remains unclear [33]. While COVID-19-induced ARDS appears to be the primary indicator of pulmonary fibrosis, various studies have indicated that it differs from classical ARDS in terms of its high and low elastance types.
The computed tomography (CT) results of numerous cases of COVID-19 do not indicate classical ARDS. In addition, the presence of abnormal coagulopathy is a notable pathological characteristic of this ailment. The mechanism underlying pulmonary fibrosis in COVID-19 differs from that observed in other fibrotic lung diseases, such as idiopathic pulmonary fibrosis (IPF). Notably, pathological observations suggest that the site of injury in COVID-19-induced pulmonary fibrosis is primarily the alveolar epithelial cells, rather than the endothelial cells.
The act of coughing is a reflexive action that necessitates minimal conscious control. This reflex is initiated by the activation of peripheral sensory nerves that transmit signals to the vagus nerves. These nerves, in turn, provide sensory input to the brainstem at the solitary nucleus and the spinal trigeminal nucleus. The phenomenon of cough hypersensitivity has been established in the context of chronic cough, wherein the pathways responsible for coughing are believed to have undergone sensitization due to an increase in the magnitude of afferent signals transmitted to the brainstem. Coronaviruses, including SARS-CoV-2, gain access to host cells through specific receptors and proteases, namely ACE2, TMPRSS2, and furin [34]. SARS-CoV-2 may have the ability to directly interact with sensory neurons, as evidenced by the prevalence of sensory dysfunction such as coughing, as well as olfactory and taste impairments, among individuals who have been infected with the virus [35]. The expression of ACE2 or TMPRSS2 in human airway vagal sensory neurons and their susceptibility to SARS-CoV-2 infection remain unknown. The bronchopulmonary vagal sensory neurons in mice were subjected to single-cell sequencing, which revealed the absence of murine ACE2 expression [36].
The potential involvement of supplementary viral entry factors in the interplay between SARS-CoV-2 and neurons cannot be disregarded. One such factor is neuropilin-1, which is present in vagal and other sensory neurons [37]. The study by D. H. Brann et al, conducted a sequencing analysis on human olfactory mucosal cells, revealing the absence of ACE2 and TMPRSS2 in olfactory epithelial neurons [38]. However, a significant expression of these genes was observed in support cells of the olfactory epithelium and stem cells [38]. The veracity of the results was validated through the cellular histological localization of ACE2 in the specialized neuroepithelium of supporting cells surrounding neuronal dendritic projections. It is noteworthy that the neuroepithelium is comprised of odour-sensing cilia [39]. Hence, it is plausible that the onset of anosmia resulting from SARS-CoV-2 infection could be attributed to the impact of the infected epithelium on neuronal function.
The ACE2 gene has been identified in a specific group of sensory neurons located in the thoracic ganglia of humans. These neurons are also known to provide innervation to the lungs. It is worth noting that a particular group of nociceptive neurons, which express calcitonin-related polypeptide alpha (CALCA) or purinergic receptor P2X 3 (P2RX3), have been found to exhibit expression [40]. These neuronal subtypes are like those found in the vagal sensory ganglia, which play an important role in triggering coughing. The similarity in developmental lineage and molecular phenotype between certain vagal sensory neurons, particularly those implicated in cough, and dorsal root ganglion neurons suggests a potential correlation between ACE2 expression in human vagal sensory neurons. The etiology of persistent cough following SARS CoV-2 infection is presently unknown, despite the possibility that the involvement of dorsal root ganglion neurons that contain nociceptors could account for the joint and chest pain, headache, and dyspnea symptoms experienced Long COVID. The S1 spike protein of SARS CoV-2 can traverse the blood-brain barrier (BBB) in mice through absorptive transcytosis, indicating that a functional virus is not necessary for brain involvement [41]. Additional research is required to explore the potential direct interactions between the virus and the nervous system in the development of cough and other sensory symptoms in individuals with SARS-CoV-2 infection. The exclusion of pathological or structural causes is crucial in the clinical management of chronic cough following COVID-19. This includes assessing fibrosis damage of lung parenchyma or damage to the airways resulting from SARS-CoV-2 infection or critical care management [42]. The presence of lung parenchymal alterations is a frequent observation on computed tomography (CT) scans in adult individuals affected by COVID-19. Additionally, a proportion of 10-20% of patients may experience the development of lung fibrotic changes [42]. The presence of lung fibrosis has been found to potentially heighten the sensitivity of the cough reflex in reaction to mechanical stimuli applied on the chest wall, as evidenced in individuals diagnosed with idiopathic pulmonary fibrosis [43]. The presence of a persistent cough in individuals experiencing post-COVID symptoms may be attributed to neuroinflammation, resulting in a state of heightened laryngeal and cough hypersensitivity. This phenomenon serves as the underlying cause of chronic refractory or unexplained cough [44,45]. Neuromodulators such as gabapentin and pregabalin have demonstrated efficacy in managing chronic refractory cough. The aforementioned strategy could be deemed as a viable option for addressing the Long COVID, as these pharmaceutical agents may have utility in mitigating additional symptoms that coincide with coughing, such as discomfort, albeit with the possibility of exacerbating any cognitive impairment. The pulmonary manifestations of Long COVID are summarized in Figure 2.
3.2. Endocrinological Involvement in Long COVID
Endocrinologic sequelae of SARS-COV-2 are caused by physiopathological changes specific to the virus, inflammatory and immunological lesions, iatrogenic complications, deficiency of vitamin D and corticosteroid therapy. ACE2 receptor is the key component by which SARS-COV-2 obtains access to host cells. The spike glycoprotein of the virus is homotrimeric in nature and comprises S1 and S2 subunits. It extends outwards from the virus surface and plays a crucial role in binding to the ACE2 receptor [46,47]. Upon binding to ACE2, the S1 subunit is dissociated with the ACE2 receptor, in a process that requires the presence of TMPRSS2 [48]. Multiple endocrine tissues express abundant ACE2 and TMPRSS2, including the hypothalamus, pituitary, thyroid, adrenal, gonads, and pancreatic islets, which underlines the necessity to investigate further endocrine post-COVID manifestations related to these structures [49].
3.2.1. Pituitary Gland
Several case reports have indicated that there may be a high risk of pituitary apoplexy among patients with pituitary neoplasms and COVID-19 infection. While some of these studies included additional risk factors for apoplexy, such as pregnancy, and the majority involved patients with preexisting pituitary macroadenomas, others involved patients with microadenomas, which are typically associated with apoplexy less frequently [50,51,52].
3.2.2. Thyroid Gland
Thyroid tissue demonstrates a significant level of ACE2 messenger (m) RNA expression and regardless of gender, it expresses a significant level of TMPRSS2 [53,54]. Moreover, the thyroid’s location near the upper airway offers the virus an entry point to access the thyroid gland [55].
Reduced levels of thyroid-stimulating hormone (TSH) are accompanied by an increase in viral load in COVID-19 patients. IL-6 and TNF-a, which are pro-inflammatory cytokines, and increased ACE2 expression in vascular endothelium are the underlying mechanisms of post-COVID thyroid disturbances. COVID-19 thyroid involvement can manifest as sick euthyroid syndrome, subacute thyroiditis (SAT), Graves’ disease, postpartum thyroiditis, Hashimoto’s disease, and silent thyroiditis [55,56,57]. SARS-COV-2 may catalyze for SAT. In a study conducted by Fatourechi V. et al., among 38 patients who developed SAT following COVID, the onset of the complication was reported in days, weeks, or even months after the initial viral infection, exhibiting symptoms identical to typical cases of SAT, with most experiencing mild forms of COVID-19. Four cases of hypothyroidism were reported post-SAT, and there were no reported deaths [58].
3.2.3. Pancreas
The pancreas can be affected by COVID-19 through different mechanisms, including direct viral injury, systemic inflammation, lipotoxicity, and drug-induced pancreatitis [59]. Some cases of necrotizing pancreatitis and acute pancreatitis are considered long-term COVID complications [60]. Many patients experience glucose-impaired tolerance and insulin resistance, which may lead to type 2 diabetes (T2DM). Long-term COVID symptoms include various endocrine and metabolic conditions, such as newly diagnosed diabetes and severe metabolic complications like diabetic ketoacidosis [61]. The relationship between COVID-19 and diabetes appears to be bidirectional, with preexisting diabetes increasing the risk of severe COVID-19, and COVID-19 infection potentially causing new-onset diabetes during the long COVID course [62]. SARS-COV-2’s diabetogenic effects are significant, as shown by findings of hyperglycemia, severe insulin resistance, increased ketosis, and hyperosmolar hyperglycemic states in older individuals. These effects go beyond the usual stress response associated with serious illnesses [63]. The possible mechanism by which SARS-COV-2 triggers T2DM involves morphological changes in pancreatic islet remodelling, including the activation of the renin-angiotensin-aldosterone system (RAAS), islet redox, inflammation, amyloid formation, fibrosis, and beta cell dysfunction or loss, along with capillary rarefaction. The virus directly affects beta cells, leading to their malfunction, failure, and apoptosis, while the viral-virion storm and cytokine storm indirectly accelerate islet remodelling in the progression of T2DM [64].
Specifically, after the activation of the systemic and islet RAAS, various events occur before angiotensin II formation [65], a potent vasoconstrictor that, when present for a long time or in large quantities, could contribute to the development of T2DM. Similarly, SARS-COV-2 activates this system, increasing the potential for Long COVID T2DM. Additionally, the binding of SARS-CoV-2 to the ACE2 receptor may impact the pancreatic islet-exocrine interface through the involvement of exocrine pancreatic ductal cells [66]. Wang F. et al. assert in a study conducted on 52 COVID-19 patients with pneumonia that pancreatic injuries are present in approximately 17% of them [67]. Consequently, SARS-CoV-2 may affect both the endocrine and exocrine tissues of the pancreas during COVID-19 infections [67].
3.2.4. Adrenal Glands
Long COVID symptoms were reported to be like low cortisol symptoms [68,69]. Recent research indicates that astenia, myalgia, and arthralgia influence 65.5%, 50.6%, and 54.7% of Long COVID patients, respectively. This could result from the suppression of the hypothalamic-pituitary-adrenal axis caused by high doses of dexamethasone and direct free radical stress [70]. Furthermore, recent studies have shown a similarity in specific amino acid sequences between adrenocorticotropic hormone (ACTH) and SARS-CoV-2. This similarity has led researchers to theorize that COVID-19 infection might stimulate the creation of antibodies that react with both, potentially deactivating natural ACTH and eliminating cells that produce ACTH [71].
Hypocortisolism following COVID-19 infection was also associated with adrenal cortex impairment. ACE2 expression was detected in the zona fasciculata and zona reticularis of the adrenal cortex, suggesting that a COVID-19-induced adrenal tissue injury might affect the production of glucocorticoids [64]. Autopsies conducted on COVID-19 patients revealed necrosis of the adrenal cortical cells and the presence of the virus at this level [72].
3.2.5. Gonads
It is believed that SARS-COV-2 affects both testis cells and sperm due to increased expression of ACE2 receptors on both Sertoli and Leydig cells and in sperm leading to hypogonadism [70]. Postmortem histopathological examination of the testes of COVID-19 patients revealed Sertoli cell enlargement, vacuolization, and cytoplasmic thinning; tubule detachment from basement membranes; and a decreased number of Leydig cells [71]. In addition, SARS-CoV-2 infection associated with hypogonadism and impaired endothelial function contributes to erectile dysfunction. The prevalence of erectile dysfunction was found to be higher in individuals who have previously had COVID-19 [72].
Ovarian tissues, as well as the uterus, placenta, vagina, and breasts, express ACE2 receptors. However, SARS-CoV-2 does not appear to infect oocytes [73]. Infection with SARS-CoV-2 was associated with a change in menstrual volume and the duration of the menstrual cycle, regardless of the severity of the infection, according to the findings of a retrospective study [73]. In an international cohort study of women with long COVID syndrome, menstrual disturbances, particularly irregular menstruation, and unusually heavy periods/clots, were also observed [74]. The long-term effects of SARS-CoV-2 infection on the human endocrine system are summarized in Table 1.
3.3. Hematological and Vascular Mechanism of Long COVID
Long after the original infection, many hematological and vascular manifestations continue to persist [75]. Relapsing-remitting symptoms may be seen in long-term COVID patients [76]. Some of the most studied hematological and vascular SARS-CoV-2 mechanisms which can cause recurrent multi-organ damage are cell count alterations, viral persistence in the extracellular vesicles (EVs), the hypercoagulable state, active vascular inflammatory state, endothelial damage and microangiopathy [77].
3.3.1. Cell counts Alterations
The hematological consequences of COVID-19 are of serious concern, as they present significant lymphopenia and cluster of differentiation (CD) 8+ T-cell dysfunction, which leads eventually to a cytokine storm and a compromised immune system [78]. There is a considerable increase in mortality risk, associated with COVID-induced neutrophilia, which is most likely the context of bacterial superinfection with opportunistic pathogens, all these being particularly concerning [79]. Up to 75% of severely sick COVID-19 patients in the intensive care units (ICUs) have been found to have neutropenia and lymphopenia with the persistence of these blood counts for more than 6 weeks after discharge, and those who passed away had lymphocyte counts that were continuously declining by the time of death [79,80].
Low levels of thrombocytes are correlated with the intensity of the disease, and several analyses reveal that a low level of platelets is linked to a significantly greater chance of developing severe COVID-19, with an almost five-fold higher likelihood [78,80]. The primary mechanisms believed to contribute to the development of thrombocytopenia in COVID-19 include direct bone marrow toxicity resulting from the viral infection, endothelial damage induced by assisted ventilation, and aberrant platelet activation [78,80].
3.3.2. SARS-CoV-2 Persistence in Extracellular Vesicles
Various cell types release extracellular vesicles containing mRNA, microRNAs, deoxyribonucleic acid (DNA), lipids, and proteins. These EVs serve the purpose of facilitating the maintenance of the physiological state of neighbouring or distant cells. Recent investigations have suggested that SARS-CoV-2 has the potential to be transmitted to distant tissues and organs through EVs [93].
Additionally, numerous studies have demonstrated that EVs are crucial for the activation of coagulation. Chronic hypoxia brought on by Long COVID is frequently accompanied by pulmonary vascular abnormalities and impaired lung function. Additionally, hypoxia creates conditions for immune cells to release more pro-inflammatory cytokines [93,94]. Exosomes, microparticles (MPs), and apoptotic bodies are the three forms of EVs. Because of their small size, biogenesis technique, and cell entry mechanism, EVs and viruses share many structural similarities. Most enveloped RNA viruses break out from their host cells or from the plasma membrane by budding. The biosynthetic secretory pathway facilitates the release of identical SARS-CoV-2 particles from the endoplasmic reticulum (ER)-Golgi intermediate compartment (ERGIC) or Golgi apparatus into the external environment of the cells. According to some research, SARS-CoV-2 can exit cells as tiny secretory vesicles that later release the virus [95]. Most enveloped RNA viruses break out of their hosts’ cells or from the plasma membrane by budding. SARS-CoV-2 RNA was discovered in exosomal cargo in other experiments, raising the possibility that the virus might spread infection via the exocytic pathway [95]. This shows that one potential mechanism for the recurrence of COVID-19 infection may be the cellular transport pathway linked to the release of EVs containing SARS-CoV-2. EVs might act as a “trojan horse” for the viral RNA to resurface in COVID-19 patients who have recovered. SARS-CoV-2 has the potential to reside within particles outside the cells during cases of long-term COVID-19, subsequently re-engaging with various tissues and organs through the bloodstream [98]. In the end, persistent endothelial damage, widespread vascular endotheliitis, and thrombosis are caused by persistent viral presence, hypoxia, and inflammatory reactions [95].
3.3.3. Persistent Vascular Inflammatory State
The interaction between SARS-CoV-2 and the humoral innate immune system, more specifically the interaction between the coagulation, fibrinolytic, and complement systems is what primarily causes thromboinflammation. Endothelial cell activation and/or injury, leukocytes, and platelets all lead to thrombotic and inflammatory reactions [91]. Thromboembolic complications affecting both the venous and arterial circulation could result from the ongoing endothelial dysfunction that occurs during long Covid in combination with an impaired innate immune response. In addition to causing excessive thrombin production, fibrinolysis inhibition, and ongoing activation of the complement pathways, SARS-CoV-2-mediated endothelial dysfunction can further extend the microvascular system’s dysfunction and cause microthrombosis [92]. Acute COVID-19 can become more severe due to a cytokine storm. It was shown that those who went on to acquire long COVID typically had higher levels of cytokine biomarkers during the early stages of recovery, including TNF, interferon (IFN)-induced protein 10, and IL-6, associated with enhanced immune activation. According to some theories, chronic immune activation is caused by persistent viral RNA discharge. Elevated levels of IFN gamma and IL-2, pathological irregularities in CD4+ and CD8+ lymphocyte groups, as well as in monocyte CD14+ and CD16+ groups, along with deficiencies in B lymphocytes and monocytes, have been identified as indicative of immune system dysregulation in chronic COVID [98]. The advancement of COVID-19 is driven by heightened oxidative phosphorylation, inflammatory responses associated with reactive oxygen species, and the displacement of inflammatory responses mediated by TNF and IL-6. Therefore, long-term continuous chronic inflammation in COVID may activate endothelial cells (ECs), platelets, and other inflammatory cells, encourage the overexpression of procoagulant factors, and undermine the protective role of vascular endothelium, leading to aberrant coagulation. Due to these consequences, there is a feedback loop whereby thrombosis leads to inflammation and the ensuing blood clots may themselves cause further inflammation. An immediate connection between coagulation and inflammation is made possible by thrombin, which breaks down fibrinogen and stimulates the IL-1 [98].
3.3.4. Endothelial Damage and Dysfunction
Impaired endothelial function can serve as an independent risk factor for the development of long COVID syndrome. In lengthy COVID, vascular endothelial damage is also typical. In recovering COVID-19 patients, EC biomarkers such as von Willebrand factor (vWF) antigen, vWF propeptide (vWFpp), and factor VIII (FVIII: C) are markedly elevated [96]. The primary connection between the mechanisms that encourage thrombosis is vascular endothelial damage. The entire vascular system is covered by ECs, which also regulate the circulation of blood and coagulation, generate, and amplify inflammatory response, and keep vascular tension, shape, and homeostasis in balance [97]. Via low levels of antithrombin III, tissue factor pathway inhibitor, and protein C, vascular endothelial damage weakens the cell’s anticoagulant capabilities and at the same time increases permeability and leukocyte adhesion. By increasing the expression of tissue factor (TF), exposing phosphatidylserine (PS), and releasing vWF and factor VIII, injured ECs become procoagulant. Additionally, ECs can enhance the recruitment of neutrophils, up-regulate chemokine production on their surface, and take part in thrombosis [98]. ECs complications brought on by inflammation may result in a sharp rise in plasminogen activator, correlated with the elevated D-dimer levels in individuals with severe COVID-19. Additionally, capillary leakage can be accelerated, and the extracellular matrix can be modified because of plasmin’s effects on metalloproteinases. As a result, organ malfunction and post-acute symptoms may also be caused by endothelial injury and chronic dysfunction [98].
In COVID-19, endothelium injury brought on by the virus, inflammation, and hypoxia may result in decreased flow rate and wall shear stress, which can lead to platelet aggregation and thrombosis. In addition, one of the signs of endothelial dysfunction seen in many organs of deceased COVID-19 patients is intussusception angiogenesis (IA). This is a quick angiogenesis procedure that uses circulating angiogenic cells to divide the blood artery into two lumens. Important causes of IA include hypoxia, traditional angiogenic molecular factors, severe inflammation and cytokine storm, thrombosis, associated hemodynamic abnormalities, and dysfunction of RAAS products. Physiological laminar flow may also be hindered by the vascular control problems seen in focal vasoconstriction and progressively dilated artery segments [98].
3.3.5. Long Covid related Hypercoagulability
One of the mechanisms of the hypercoagulation state is the fact that endothelial damage is prone to evolve into endotheliitis, which increases the risk of hypercoagulability. The Long COVID patient is more likely to experience acute thrombotic events, which in turn trigger acute stroke, venous thromboembolism, disseminated intravascular coagulation (DIC) and acute ischemic events. Numerous clinical research and trials have demonstrated the necessity of routine blood counts and inflammatory marker tests for long COVID patients, namely for those who are older and weaker [77,99,100,101].
In addition to eliciting inflammatory immunological responses, the SARS-CoV-2 has the capability to cause direct harm to ECs through its attachment to ACE2 receptors during the acute phase of COVID-19. The consequences of endothelial cell damage include endothelial activation, impairment of barrier function, heightened permeability, and leakage in the microvasculature [81]. EC’s damage can lead to systemic and widespread endothelial dysfunction, as well as the activation of numerous immune-mediated inflammatory pathways and thrombus formation, all of which can cause serious multi-organ involvement and subsequently higher mortality [82]. SARS-CoV-2 has also been discovered in ECs, and patients hospitalized for COVID-19 have higher than average levels of circulating ECs, indicative of vascular damage. However, even after SARS-CoV-2 is not detected anymore, some pathogenic processes continue [83].
A large percentage of recovering patients had elevated markers of endothelial lesions and coagulation, which suggests infection may cause persistent coagulopathy, endotheliitis, and microvascular lesions, along with the creation of microthrombi [84]. Persistent endothelium lesions are frequent in the COVID-19 convalescent period, according to study findings overall. By producing antiplatelet and anticoagulant chemicals to prevent the development of fibrin clots or platelet aggregation, and inhibiting the binding of clotting proteins, the normal endothelium plays a critical role in avoiding thrombosis. It has been established in the past that endothelial dysfunction raises the risk of thrombosis. Therefore, endothelial dysfunction because of endothelial damage may be a secondary cause of long COVID thrombosis [85]. Anticoagulation has taken a prominent role in the complete care of COVID-19 infection, as hypercoagulability is becoming a more widely recognized consequence of COVID-19 infection [98,102].
The pathophysiology of both COVID-19 infection and its persistent phenotype, known as long COVID, is believed to be influenced to a large extent by the presence of abnormal microclot formation, specifically amyloid microclots, which are resistant to fibrinolysis. Additionally, there is an increase in 2-antiplasmin (2AP) levels and a surge of acute-phase inflammatory molecules. In a 2021 study conducted by Pretorius et al., an analysis of samples from COVID patients and those with Post-Acute Sequelae of COVID-19 (PASC), which is associated with persistent clotting issues in Long COVID, revealed notable increases in dysregulated molecules between acute COVID-19 cases and long-term COVID [89]. The prolonged inflammatory status in long COVID/PASC and persistent viral infection were considered possible factors. The study emphasizes the need for larger sample sets to validate these proteomics findings due to the limited statistical power of the current collection. Additionally, platelet aggregation assays, prothrombin time, and partial thromboplastin time analysis should be considered. The study concludes that circulating microclots and hyperactivated platelets, hypercoagulability driven by significant increases in inflammatory markers, and an aberrant fibrinolytic system result from dysregulated clotting proteins and lytic enzymes, with a substantial rise in 2AP playing a crucial role. Consequently, ongoing anticoagulant therapy could potentially benefit both acute COVID-19 infection and long-term COVID/PASC patients with clotting disorders.
3.3.6. Microangiopathy
Both initial COVID and long COVID, are characterized by coagulopathies and, the development of microclots in vivo [86]. Clinical research and studies have shown that these microclots are also amyloid in nature. The production of abnormal clots that take amyloid states and do not undergo fibrinolysis can be induced by adding pure, recombinant SARS-CoV-2 S1 spike protein to normal plasma [87]. It should be noted that exogenous thrombin was not used in these cases. It’s been demonstrated that these clots are identifiable within the bloodstream of individuals experiencing both acute and chronic COVID-19 [88,89]. These long COVID-associated microclots are resistant to fibrinolysis; they contain antitrypsin, making them resistant to lysis processes; they contain inflammatory molecules, which may cause inflammation upon lysis; and they are hypothesized to obstruct the microcirculation in different capillary beds, causing tissue dysfunction and eventually leading to organ dysregulation and dysfunction, which results in clinical presentations relating to an organ [90]. In a recent study provided by Pretorius E. et all, it was demonstrated that plasma from long COVID patients had microclots that were resistant to fibrinolysis [89]. Platelets have been reported to contain SARS-CoV-2 RNA, and severe acute infection is associated with platelet activation and degranulation. It is still unclear exactly how platelet activation caused by SARS-CoV-2 viral contacts works. However, several mechanisms might be able to account for this phenomenon. First, by directly interacting with the host platelet receptor, the viral spike protein of SARS-CoV-2 can connect with platelets and perhaps activate them. Indeed, the SARS-CoV-2 spike protein binds to the highly abundant ACE2 receptors on platelets with a strong affinity. When coated with antiviral antibodies capable of attaching to the human immunoglobulin receptor IIa (FcRIIa) CD 32a on platelets, SARS-CoV-2 may also bind to them [103]. The arginine-glycine-aspartic amino acid sequence found in the SARS-CoV-2 spike protein can interact with integrins and bind to CD 209 on platelets. The main receptor on the surface of platelets is glycoprotein IIb/IIIa, which can quickly bind a variety of ligands that contain the arginine-glycine-aspartic acid sequence. It has also been demonstrated that the SARS-CoV-2 envelope protein interacts with the Toll-like receptor (TLR) 2 present on platelets. Platelet aggregation and the creation of three-dimensional clots are caused by the release of thrombin (IIa) by activated platelets. This process is triggered by platelet adhesion receptor glycoprotein (GP) Ib-IX-V, which is found on the surface of platelets, binding to collagen and vWF that are exposed to injured artery walls [103].
3.3.7. Gastroenterological Involvement in Long COVID
Gastrointestinal (GI) manifestations are one of the main organ-specific complications of acute and long COVID [111] this is believed to be due to the ACE2-expressing cells on the brush border of the small intestinal mucosa [104,114]. The GI symptoms displayed during acute COVID–19 differ from those displayed in post-COVID – 19 syndromes. The acute symptoms are predominantly abdominal pain, nausea, vomiting, diarrhea, and abnormal liver function tests [110,113,115,116]. The post-COVID-19 syndrome manifestations present as loss of appetite, irritable bowel syndrome, weight loss, altered bowel motility and dysphagia [105,113]. The presence of GI symptoms during the acute stage of the infection is linked to a higher likelihood of long-term GI post-COVID- 19 problems [112]. As surmised in the systematic review by Choudhury et all, GI symptoms were reported in 14 out of the 50 studies conducted [107]. The 14 studies involved 296,487 patients and the frequency of which patients with COVID-19/long COVID experienced GI manifestations was 0.12/0.22 [107]. The relationship between Long COVID and inflammation has been evaluated in recent studies. Disruption of the gut-lung axis, an established indicator of illness severity in other respiratory conditions, may play a role [118]. Direct or indirect effects of SARS-CoV-2 on the GI tract can cause breakdowns in the integrity of the gut barrier, allowing gut bacteria and their products to translocate across the gut epithelium and escalating early systemic inflammation [117,120,126].
Acute COVID-19 has also been linked to elevated plasma levels of zonulin, a measure of tight junction permeability that promotes microbial translocation and inflammation [118]. There have also been reports of persistent shedding of SARS-CoV-2 in the faces and the existence of the virions in intestinal cells (enterocytes) [106,119,120]. This supports the hypothesis that the virus infects the GI tract through the fecal-oral route [120,123]. Gaebler C. et all demonstrated that the antigen persisted in the intestinal tract even after it had been cleared from the nasopharynx and the clinical illness resolved [106]. Of the biopsies obtained from the GI tracts of 14 former COVID-19-positive patients, 5 were found to have the SARS-CoV-2 protein in the enterocytes.
A study conducted in Texas aimed to evaluate the long-term sequelae of COVID-19 in patients with no associated pathology [108]. Of the 101 patients present at the COVID-19 recovery clinic, 25 patients had no comorbidities. As to be expected, almost all the patients reported respiratory symptoms (dyspnea and cough), as well as persistent GI manifestations e.g., abdominal pain, nausea, diarrhea, and vomiting in 84% of the patients. The study concluded that although comorbidities are a risk factor for severe COVID-19 symptoms, having no comorbidities does not protect against prolonged recovery [105].
A study conducted by Suárez-Fariñas M. et al, in 2021, set out to determine if COVID-19 influences inflammatory bowel disease (IBD) and IBD medication and if there is a shared pathological pathway between them [109]. The study found that genes associated with the inflammatory response in IBD overlap with genes that respond to COVID-19 infection. Through examining the blood of patients infected with COVID-19, it was observed that there was a higher expression of upregulated genes in the patients with IBD than there was with healthy control blood. The studies concluded that there is potential for a local, GI-associated replication of the SARS-CoV-2 because of the high expression of ACE2 and TMPRSS2.
3.4. Cardiovascular Involvement in Long COVID
The evaluation of prolonged cardiovascular issues is ongoing and understanding the actual impact of post-COVID-19 cardiovascular complications remains uncertain. COVID-19 appears to worsen preexisting cardiac pathology, but also causes new-onset cardiac diseases such as arrhythmias or hypertension, Figure 3.
While the causes behind enduring cardiac injury post-recovery are not yet fully grasped, a potential scenario points to a persistent inflammatory reaction reflected by the presence of circulating biomarkers for 3 to 8 months after acute infection: C-reactive protein, IL 6 and 8, ferritin, IFN beta and gamma1, procalcitonin (correlated with microvessel disease), chemokine ligand (CXCL) 9 (small cytokines that induce chemotaxis, promote differentiation and multiplication of leukocytes and cause tissue extravasation), CXCL10 (an ‘inflammatory’ chemokine), T-cell immunoglobulin mucin-3 (TIM-3) (involved in immune response), plasma ACE2 activity, pentraxin 3 (PTX3) (activates complement and facilitates pathogen recognition by macrophages). Some of these biomarkers were also found elevated in asymptomatic post-COVID patients during the lasting up to 8 months [127]. This chronic inflammatory status may be caused by persistent viral reservoirs such as intestines or other immune-privileged sites following the acute infection [106].
Immune exhaustion after prolonged antigen stimulation or immunosuppressive treatment may also facilitate the development of cardiac injury. Within the cardiac structure, SARS-CoV-2 primarily resided within interstitial cells and infiltrating macrophages within the myocardium. Viral entry depends upon the interaction between viral spike glycoprotein, the ACE2 receptor, a transmembrane aminopeptidase in the host cell membrane, and the host cell protease system such as TMPRSS2. ACE2 plays a crucial role in the neurohumoral regulation of the cardiovascular system and is primarily present in the vascular endothelium, cardiomyocytes, pericytes, cardiac fibroblasts, and epicardial adipose tissue. ACE2 naturally transforms angiotensin 1 and 2 into beneficial peptides like angiotensin 1-7 and angiotensin 1-9, which hold cardioprotective properties. SARS-CoV-2 causes downregulation of ACE2, thus preventing the synthesis of cardioprotective peptides and predisposes to cardiovascular damage.
Another proposed mechanism of myocardial damage is mediated by reactive oxygen species, which can induce the release of internal histones, damage-associated molecular patterns (DAMPs), and oxidized lipid-protein complexes. During the post-acute phase, these compounds trigger an inflammatory response that results in substantial myocardial tissue injury, leading to chronic myocardial scarring. This scarring may cause issues like reduced ventricular compliance, impaired blood flow within the heart muscle, decreased cardiac muscle contraction, and potential irregular heartbeats. Cytokine-induced damage can also manifest as blood clot formation, reduced oxygen delivery, destabilization of coronary plaques, the transition of chronic heart conditions into unstable states, increased metabolic requirements, diminished cardiac capacity, and inflammation in the heart valves [127].
A growing number of reports on cardiovascular autonomic dysfunction (CVAD) in PASC patients, namely patients who experience symptoms suggestive of postural orthostatic tachycardia syndrome (POTS), with exaggerated rise in heart rate and intolerance to standing, commonly affecting females and individuals aged between 15 and 45 years old.
Infections commonly trigger dysautonomia, and several potential ways in which SARS-CoV-2 might contribute to this condition have been suggested based on initial evidence: hypovolemia, brainstem involvement, and autoimmunity [129]. Hypovolemia is a recognized characteristic in patients with PASC, leading to a hyperadrenergic response. This reaction subsequently leads to cerebral hypoperfusion and disruption of central autonomic networks. Brainstem dysfunction encompasses various mechanisms, including direct invasion by the virus, neuroinflammation, brainstem compression, and vascular activation. Autoimmunity has an important role in the pathophysiology of post-viral POTS as supported by recent evidence [130]. A high prevalence of specific autoantibodies has been found in the sera of PASC patients presenting dysautonomia. They include G-protein coupled receptor (GPCR) antibodies which can activate adrenergic receptors and elicit a negative allosteric effect on muscarinic GPCRs. Autoantibodies can also activate cholinergic receptors and cause peripheral vasodilation. Patients experiencing POTS linked to PASC have exhibited the presence of serum anti-nuclear, anti-thyroid, anti-cardiac protein, anti-phospholipid, and Sjogren’s antibodies [131]. Alarmingly a possible shift in age distribution of Long Covid can be expected due to an increased number of young infected unvaccinated individuals in whom morbidity and mortality are difficult to predict according to the European Society of Cardiology [132].
3.5. Renal Involvement in Long COVID
Impairment of the kidneys in individuals who contracted COVID-19 have been widespread worldwide and manifests as hematuria, proteinuria, acute kidney injury (AKI), end-stage kidney disease (ESKD) and major adverse kidney events (MAKE) [133,7]. Even in the absence of coexisting elevation in serum creatinine concentration or a decline in estimated glomerular filtration rate (eGFR), the presence of low molecular weight proteinuria or hematuria indicates a subclinical AKI. The mechanisms behind these injuries are multiple and include direct injury to the renal cells, indirect injury through other organ destruction etc.
The ACE2 receptor is expressed in the epithelium of the kidneys and plays a major role in the entrance of the SARS-CoV-2. Previous studies have shown that ACE2 is the functional receptor for SARS-CoV in vivo and in vitro. Recent cryo-electron microscopy has shown that the spike protein of SARS-CoV-2 binds directly to ACE2 with an even greater affinity than SARS-CoV [138].
Several studies have revealed that SARS-CoV-2 relies on the widely expressed transmembrane glycoprotein, CD147, as a crucial element in infiltrating target cells [137,8]. CD147 is expressed in high amounts on the cell surface of the proximal tubular epithelial cells and is believed to have contributed to the pathogenesis of several renal diseases through its involvement in the immune-inflammatory response and dysregulated cell cycle [137]. It is established that the CD147 partners, cyclophilins, have a crucial role in the replication process of the SARS-CoV-2 and inhibiting these by cyclosporins has demonstrated effective suppression of intracellular viral propagation [136]. Su H. et all, in their autopsy results of patients with COVID-19, demonstrated by electron microscopic examination agglomerates of coronavirus-like particles having distinctive spikes within the tubular epithelium and podocytes [136]. They further described notable tubular impairment, especially within the early segment of the proximal tubule marked by an absence of microvilli, acute tubular necrosis, a notable decrease in megalin expression within the microvilli and the existence of intraluminal debris. All denoting the presence of AKI [136]. Further findings of the virus in urine only strengthen this discovery [139]. Diao B. et al, further cemented this discovery when they confirmed the findings of the SARS-CoV-2 in renal cells, further demonstrating that SARS-CoV-2 can replicate in vivo [140].
A study conducted by Chand S. et al., on 300 survivors of critical COVID-19, conducted in 2022 found that out of the patients who survived 120 days after covid resolution, 74.4% had AKI resolution while 25.6% did not have a resolution. Out of the 25.6% with AKI resolution 60% developed chronic kidney disease (CKD) and 40% required renal replacement therapy (RRT) [135].
According to Bowe B. et all in their cohort study conducted for 1 year on 1,726,683 US Veterans, and 89,216 patients who were 1-month survivors of SARS-CoV-2 infection, patients who successfully recovered from COVID-19 displayed kidney-related symptoms during the post-acute stage, such as eGFR decline, AKI, ESKD or MAKE. These symptoms increased in the post-acute period with the severity of the acute infection, and the risk for renal problems was higher in hospitalized patients, who developed AKI [134].
3.6. Dermatological Involvement in Long COVID
Dermatological manifestations and complications of COVID-19 are becoming increasingly acknowledged in the literature. There are many types of skin manifestations described in conjunction with COVID–19 such as maculopapular rash, urticaria, petechiae, purpura, vesicles, chilblains, livedo racemose and ischemia of the distal segments of the limbs [141,142,143]. These dermatological findings are significant as they can help the clinician to make a COVID-19 diagnosis. There are various hypothesized causes of rash in COVID-19 patients. Diffuse microvascular vasculitis, brought on by complement system activation, is the first. In one study carried out by Magro C. et al, in 2020, interstitial and perivascular neutrophilia with pronounced leukocytoclasia, considerable complement protein deposition in the dermal capillaries, and other findings point to vasculitis phenomena [141]. Other studies, performed by Sanchez A. et al, in 2020, suggest that the rash occurs as a direct effect of the virus. The justification is the presence of lymphocytosis (without eosinophils), papillary dermal oedema, epidermal spongiosis and lymphohistiocytic infiltrates [142]. Five clinical patterns were identified in a recent study conducted in Spain, that included 375 cases: maculopapular eruptions (47%), urticarial lesions (19%), various vesicular eruptions (9%) and livedo or necrosis (6%) [144]. Although the face is often spared, and the lesions are mostly restricted to the trunk and extremities (hands and feet).
The average lesions last a few days, although others have been reported to persist as short as 20 minutes or as long as four weeks. Nevertheless, in some individuals, lesions emerged 2 to 5 days before the beginning of COVID-19 symptomatology. In most cases, the mean latency that is, the amount of time it took for skin lesions to arise following the onset of the first typical COVID-19 symptoms was between 1 and 14 days [145]. There is still a lack of knowledge on the pathophysiological mechanisms of skin lesions in COVID-19 patients. The skin manifestations presented in COVID-19 may be divided into two categories regarding their pathological mechanisms [146]. The first is an immune response to the viral nucleotides of COVID-19, which clinically manifests in a similar way to viral exanthems. The second group is skin eruptions that result from the systemic effects of COVID-19, this is especially the case in vasculitis and thrombotic vasculopathy.
The possible actions of SARS-CoV-2 on the skin may be mediated by non-structural viral proteins (NSPs) that block the innate immune system (NSP3, NSP16), inhibit the effect of IFN (NSP5), or synthesize cytokines (NP3). The cytokine storm can stimulate dermal cells, leading to the appearance of various maculopapular or vesicular rashes. Secondary activation of complement by the surface viral antigen and microangiopathy may lead to the appearance of purpuric manifestations [145,147]. TMPRSS2 activation is crucial for the virus to attach to ACE2 through its spike protein. Androgen receptor elements near the TMPRSS2 gene on chromosome 21 suggest COVID-19 could affect men more due to androgen levels. The higher COVID-19 rates in Spanish men, possibly linked to androgenetic alopecia prevalence, hint at androgens playing a role in worsening the severity of the disease [148]. Another possible effect of SARS-CoV-2 is a direct impact through ACE2 in the epidermis, causing certain skin conditions (acantholysis and dyskeratosis) [149]. ACE2 appears in the skin’s basal epidermal layer, dermal blood vessel ECs, and eccrine tissue [145,150]. The reduced microcirculatory function across arterial beds in COVID-19 patients and its effects may be linked to COVID-19-related endotheliitis involving ACE2 [145,6].
3.7. Neuropsychiatric Mechanism of Long COVID
Up to one-third of individuals with long COVID, also known as PASC, may have ongoing neurological problems that manifest as anosmia, hypogeusia, lethargy, headaches, “brain fog,” dysautonomia, cognitive impairment, and peripheral neuropathy [151].
The neurological manifestations observed in cases of acute COVID-19 have been associated with various interconnected pathogenetic mechanisms. These mechanisms include viral invasion of the nervous system accompanied by abnormal immune responses, dysfunction of the blood-brain barrier due to epitheliopathy, coagulation disorders leading to neuronal injury caused by reduced oxygen supply, imbalances in metabolic processes, cascades of oxidative stress, and cellular apoptosis [152]. These factors are posited as potential causes for the enduring symptoms observed in individuals affected by COVID-19. SARS-CoV-2 can invade the stem and support cells located in the olfactory epithelium, which is situated outside the central nervous system. This invasion can lead to persistent alterations in the sense of smell. Anomalies in innate and adaptive immunity that may arise from SARS-CoV-2 infection include monocyte enlargement, T-cell exhaustion, and prolonged cytokine release [151]. These abnormalities may lead to neuroinflammatory reactions and microglia activation, abnormalities in the white matter, and microvascular alterations. Additionally, viral protease activity and complement activation can cause endotheliopathy, which can lead to hypoxic neuronal damage and BBB failure, and microvascular clot formation, which can obstruct capillaries [151].
3.7.1. SARS-CoV-2 Neurotropism
It is now believed that SARS-CoV-2’s direct neurotropic actions have a limited impact on the etiology of Long COVID, like the part that invasion of the nervous system played in acute COVID-19 [153]. A detailed study by Meinhardt J. et all proved the SARS-CoV-2 presence in the olfactory cells [153]. The serine protease TMPRSS2 is known to act on the cellular receptor ACE2 and to bind to the SARS-CoV, especially SARS-CoV-2, allowing the virus to enter human host cells. It has been demonstrated that non-neuronal cells in the human olfactory mucosa physiologically express ACE2 [153]. It was determined that the olfactory mucosa had the highest concentration of S protein [153]. Using immunohistochemistry, unique immunoreactivity for the S protein was discovered in cell types that differ morphologically and are suggestive of neuronal/neural origin [153]. This immunoreactivity included a distinctive granular, partially perinuclear pattern. The RNA of SARS-CoV-2 was identified in cells of the olfactory epithelium and nasal mucus [153].
Coronaviruses and other neurotropic viruses employ sensory and motor neural pathways to penetrate the central nervous system (CNS). The olfactory nerve is a type of neural route. The way olfactory nerves are structured differently in the nasal cavity and forebrain, as well as the olfactory bulb, mediates this. Inflammation and a demyelinating reaction may result from the virus getting into the brain and cerebrospinal fluid (CSF) in this way. In less than 7 days, once the infection is established, the viruses can infect the entire brain and CSF [155]. Hematogenous spread from severely infected airways and lungs is one way that SARS-CoV-2 may enter the CNS. This sort of spread would be made easier by systemic inflammation that raises BBB permeability. The brain’s circumventricular organs are fenestrated, highly permeable structures. This often enables circulating but non-BBB-crossing mediators to have an impact on brain activity directly. This permeability, however, can also allow for pathogen neuroinvasion during an acute infection. This can happen directly or by an indirect process where host immune cells actively carry intracellular infections into the central nervous system (CNS) [91].
SARS-CoV-2 showed choroid plexus basal (vascular side) epithelial tropism in an organoid model made from human pluripotent stem cells, despite more abundant ACE2 on the apical (CSF) side. Additionally, SARS-CoV-2 led to epithelial destruction and barrier leakage, which may have facilitated neuroinflammation by increasing the entry of immune cells, including those infected in a Trojan horse form, and circulating cytokines into the CNS [91].
Invading the stem cells, pericytes, columnar epithelial cells, and Bowman’s gland cells in the olfactory epithelium using the ACE2 receptor cause persistent filia thinning and volume loss in the olfactory bulb. Although there is limited proof of direct neuroinvasion in these regions, there is a correlation between the geographical distribution of ACE2 receptors and areas of hypometabolism in the brain. Instead, it is thought that these areas suffer from increased levels of oxidative stress, cytotoxic T lymphocyte infiltration, neurodegeneration, and demyelination because of neuroinvasion. These mechanisms probably continue because of persistent viral shedding, particularly in the GI tract where ACE2 co-regulates dopa-decarboxylase (DDC) and the dopamine metabolic pathway is active [151].
3.7.2. Brain-Gut Axis - Dysbiosis
The intestinal microbiome is a dynamic ecosystem composed of diverse communities of microorganisms including bacteria, archaea, and fungi, it begins its colonization early in life, undergoing dynamic changes in the first five years and achieving relative stability in adulthood. However, it remains responsive to various factors such as environmental changes, antibiotic use, diet, infections, or diseases. Changes in its composition are associated with multiple health conditions, although causality remains unclear in many cases. The association between microbiome changes and various health conditions like inflammatory bowel disease, obesity, and mental health disorders suggests a potential role of the microbiome in the pathogenesis of these conditions. However, establishing direct causation is challenging due to the complexity of microbiome-host interactions and numerous influencing factors affecting its composition and functionality.
During the COVID-19 pandemic, research has highlighted the impact of the infection on the intestinal microbiome. Cheng X et al., in a systematic analysis of 16 studies, observed a reduction in intestinal bacterial diversity in patients with acute COVID-19 and those who recovered from the illness [156]. Moreover, a decrease in bacteria producing anti-inflammatory effects mediated by butyrate production (Megasphaera, Dialister, Ruminococcus, Faecalibacterium, Roseburia, Lachnospira, and Prevotella) was noted, alongside an increase in microorganism’s species with pro-inflammatory properties (Streptococcus, Enterococcus, Corynebacterium, Blautia, and Dorea), leading to increased synthesis of proinflammatory cytokines [156,157]. These disruptions in the gut microbiome persisted into the recovery phase after the resolution of the initial infection, lasting for months, suggesting their potential contribution to PASC. Additionally, increases in fungi (Candida albicans) and Inoviruses [Pseudomonas phages (Pf1)], as well as a decrease in beneficial bacteria (Faecalibacterium prausnitzii, Eubacterium rectale, Bifidobacterium adolescentis), have been identified in patients with severe forms of COVID-19, leading to an increased risk of intestinal infections, heightened intestinal permeability, reduced intestinal immune response, and proinflammatory effects [158,159]. Furthermore, a significant increase in proinflammatory pathobionts (Prevotella species, Veillonella species, genus Actinomyces, Streptococcus anginosus, Gemella sanguinis) was observed in the oral microbiome of patients experiencing PASC [160]. The changes identified in the oral microbiome of these patients resembled those observed in individuals suffering from chronic fatigue syndrome, suggesting a potential link between the two conditions [160].
In COVID-19, several actions can lead to the onset of dysbiosis, including hypoxia, inflammation, antibiotic use, or mediated by decreased tryptophan absorption, Figure 4.
The absorption of dietary tryptophan, an essential amino acid absorbed in the small intestine, is mediated by ACE2, the enzyme involved in SARS-CoV-2 cell entry. SARS-CoV-2 leads to a decrease in tryptophan absorption through this pathway [157]. The majority (90%) of tryptophan binds to albumin and crosses the blood-brain barrier, entering the cells of the central nervous system (neurons, astrocytes, and microglia). Some tryptophan remains free in the blood, and the unabsorbed portion serves as a metabolic substrate for intestinal bacteria. Tryptophan is subsequently metabolized via three pathways: the kynurenine pathway, the serotonin pathway, and the Indole pathway [161]. Tryptophan is the sole precursor of serotonin (from which melatonin is later synthesized). Its synthesis primarily occurs in the intestine (enterochromaffin cells) and at neuronal levels (serotonergic neurons in the enteric nervous system and neurons in the central nervous system). A low level of serotonin can be responsible for brain fog, fatigue, depression, sleep disturbances, headaches, immunosuppression, and constipation. Constipation can, in turn, lead to alterations in the intestinal microbiome. The kynurenine pathway, primarily occurring hepato-renally, is the main metabolic route for tryptophan. It is stimulated by proinflammatory cytokines and corticosteroids, resulting in proinflammatory neurocatabolites (kynurenic acid and quinolinic acid). Almulla A.F. et all in a meta-analysis of 14 studies have shown that the kynurenine degradation pathway of tryptophan is highly stimulated in severe COVID-19 [162]. The resulting products are pro-inflammatory, leading to the activation of the aryl hydrocarbon receptor (AhRs), and the direct association with SARS-CoV-2 action on this receptor is responsible for the onset of systemic aryl hydrocarbon receptor activation syndrome (SAAS), responsible for fibrosis, chronic inflammation, impaired cognitive function, increased oxidative stress, thrombosis, and disorders in the functionality of various organs [162]. The third metabolic pathway is the indole pathway, producing indole derivatives, namely indole-3-propionic acid and indole-3-aldehyde, within the intestinal microbiome. The obtained metabolites have an immune-modulating effect, preventing the harmful effects of free radicals and oxidative stress. Additionally, they have a neuroprotective effect and prevent intestinal dysbiosis [161].
In cases of severe COVID-19, it has been observed that SARS-CoV-2 can continue to be present in the upper respiratory and gastrointestinal epithelium for around 1 month. This prolonged viral shedding is believed to contribute to an imbalance in the brain-gut axis, as the virus tends to deplete symbionts and disrupt the gut microbiome in the gastrointestinal tract. The disturbance of the microbiome is further perpetuated by the continued shedding of the virus. An analysis of human intestinal organoids infected with SARS-CoV-2 has shown that the ACE2 gene is co-regulated with DDC and genes involved in the dopaminergic pathway metabolism and the absorption of amino acid precursors for neurotransmitter synthesis. This finding provides additional evidence of an altered brain-gut axis [151].
3.7.3. Long COVID Neuroinflammation
The CNS has seen neuropathological alterations because of COVID-19. The production of pro-inflammatory mediators because of these modifications includes the induction of uncalled-for inflammatory reactions. Inflammation was induced in COVID-19 instances, according to recent clinical findings, and this induction was linked to an elevated level of cytokines, such as IL-1, IL-6, IL-10, and TNF. Previous research has shown that tight junction (TJ) proteins are degraded by cytokines, which change BBB integrity. Emerging data suggested that BBB permeability is increased by TJ degradation, specifically in claudin-5 and zonula occludens (ZO)-1. When the BBB’s integrity is altered, more viruses and cytokines have a chance to cross it and enter the CNS. This causes cerebral immune cells like microglia and astrocytes to become activated, which leads to cytokines-induced neuroinflammation [163].
It has been established that astrocytes and microglia have a very important role in neuroinflammation. Microglial cells have been demonstrated to activate in the CNS because of systemic infection, and they are more vulnerable to pathogens than astrocytes. Molecular signals like IL-1 and TNF stimulate astrocytes in response to the activation of microglia. In response to microglial activation, activated astrocytes can release a variety of inflammatory substances, such as TNF, reactive oxygen species (ROS), and nitric oxide (NO). This two-way interaction between astrocytes and microglia intensifies the ‘chain reaction pattern’ neuroinflammation [163].
TMPRSS2 was also highly expressed by astrocytes in the cerebral cortex of those patients. In general, ACE2 activates the AT1 receptor, which promotes neuroinflammation and oxidative stress. Additionally, it has been demonstrated that the high expression of inflammatory mediators and nuclear factor kappa-light-chain-enhancer (NF-kB) is linked to the elevated level of cathepsin L (CTSL) [163]. It is interesting to note that TMPRSS2 aids virus entrance into host cells, according to many studies. All the relevant studies on SARS-Cov-2 neuroinflammation suggest that the virus activated the CNS cells involved in the inflammation [163].
Additionally, COVID-19 encourages mast cell activation, neuroinflammation, and a high intracranial level of proinflammatory cytokines. Alcohol use and drug use disorders are two factors that can exacerbate the neuroinflammatory response caused by SARS-CoV-2, which differs between patients. To give one example, SARS-CoV-2 infection results in TJ loss, mast cell activation, and the production of inflammatory mediators, all of which have the potential to result in neuroinflammation, oedema, and bleeding, particularly in individuals with concurrent neurodegenerative disorders [163]. The impairment of the endothelial barrier and the subsequent increase in blood-brain barrier (BBB) permeability by the SARS-CoV-2 spike protein has been experimentally validated through the utilization of a three-dimensional tissue model of the BBB. The spike protein of SARS-CoV-2 has the potential to activate brain ECs, leading to an inflammatory reaction that could worsen the breakdown of the blood-brain barrier [163]. The in vitro studies demonstrated that the recombinant SARS-CoV-2 spike protein can decrease the expression of TJ proteins, such as ZO-1, ZO-2, Claudin-5, and junctional adhesion molecule (JAM-2), in human brain microvascular ECs [163]. Changes in junctional protein integrity could affect the BBB as a whole. It’s interesting to note that the level of cytokines, including TNF, IL-6, and IL-10, increased when the production of TJ proteins was downregulated. The anti-inflammatory enzyme indoleamine-2, 3-dioxygenase 1 (IDO1) is expressed by a variety of immune cells, including macrophages, monocytes, and microglia. Infection with SARS-CoV-2 leads to abnormal IDO1-mediated inflammation. Therefore, SARS-CoV-2-induced neurological problems may be caused by IDO1 [163].
3.8. Myalgic Encephalomyelitis and Chronic Fatigue Syndrome in Long COVID-19
Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) is clinically supported by the presence of a polymorphic, persistent clinical picture, Figure 5, [154,165,166,167].
The immunological response to COVID-19 resembles the traditional ME/CFS pattern in keyways. Important aspects of the immunological response to COVID-19 include the loss of basophils and plasmacytoid dendritic cells as well as the depletion of T cells, primarily CD8+ and T cells. The expression of cytokines, particularly IL-6, IL-10, and IFN-induced protein 10 (IP-10, formerly CXCL10), is elevated and closely linked with the course of the disease. IP-10 is of special significance because, like in ME/CFS patients, its concentrations usually remain high throughout the COVID-19 response. The immune system may be facing a bigger strain at higher levels, which could translate into more severe symptoms [165].
Patients diagnosed with COVID-19 exhibited indications of impaired mitochondrial function, alterations in metabolism characterized by heightened glycolysis, and elevated levels of cytokines in peripheral blood mononuclear cells. These observations align with the results of multiple autonomous investigations that have characterized ME/CFS as a state of reduced metabolic activity, involving dysfunction in various metabolic pathways. Additionally, there is a possibility that ME/CFS may be classified as a mitochondrial disorder due to heightened mitochondrial harm, diminished adenosine triphosphate (ATP) production, and impaired oxidative phosphorylation. Patients with COVID-19 infection have compromised mitochondrial activity, making it impossible for them to use this pathway to provide the energy they need. As a result, glycolysis is boosted to make up for high energy requirements. This is linked to an increase in the inflammatory response. Production of pro-inflammatory cytokines (IL-1 and IL-12) and a propensity for pyroptosis are brought on by inflammation activation. After pyroptosis, cell-free mitochondrial DNA is released, which exacerbates both local and systemic inflammation [165].
4. Conclusions
Long COVID is a complicated and multidimensional illness that affects a large proportion of those recovering from an acute COVID-19 infection. It has been linked to a variety of symptoms and problems, including chronic fatigue, cognitive impairment, respiratory troubles, cardiovascular irregularities, and psychological discomfort, according to research. The precise processes behind Long COVID are yet unknown. Pathology provides considerable issues for healthcare practitioners since it necessitates a multidisciplinary approach to diagnosis, treatment, and rehabilitation. Future research should concentrate on elucidating the pathogenesis of Long COVID, including the function of viral reservoirs, immune response dynamics, and potential long-term organ damage. To produce evidence-based guidelines for the diagnosis, treatment, and management of Long COVID, clinical trials are required. The research should also emphasize the development of biomarkers that can help in the condition’s early detection, prognosis, and monitoring. This COVID-19 complication should be the focus of public health campaigns, with a focus on increasing awareness, advocating preventative measures, and offering support and services to patients having long-term symptoms. Collaboration between researchers, healthcare professionals, and patient groups is critical for effectively addressing the issues posed by Long COVID and improving patient outcomes.
5. Patents
Author Contributions
Conceptualization, N.N. and P.M.; methodology, A.F.; software, M.G.; validation, N.N., P.M. and A.F.; formal analysis, M.G.; investigation, A.A.; resources, F.T.; data curation, A.A.; writing—original draft preparation, H.F., F.K., M.B., A.A., S.K., F.T.; writing—review and editing, N.N., F.K.; visualization, P.M.; supervision, N.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Carvalho-Schneider, C.; Laurent, E.; Lemaignen, A.; Beaufils, E.; Bourbao-Tournois, C.; Laribi, S.; Flament, T.; Ferreira-Maldent, N.; Bruyère, F.; Stefic, K.; Gaudy-Graffin, C.; Grammatico-Guillon, L.; Bernard, L. Follow-up of Adults with Non-Critical COVID-19 Two Months after Symptoms’ Onset. Clinical Microbiology and Infection 2020, 0 (0). [CrossRef]
- Sigfrid, L.; Drake, T. M.; Pauley, E.; Jesudason, E. C.; Olliaro, P.; Lim, W. S.; Gillesen, A.; Berry, C.; Lowe, D. J.; McPeake, J.; Lone, N.; Munblit, D.; Cevik, M.; Casey, A.; Bannister, P.; Russell, C. D.; Goodwin, L.; Ho, A.; Turtle, L.; O’Hara, M. E. Long Covid in Adults Discharged from UK Hospitals after Covid-19: A Prospective, Multicentre Cohort Study Using the ISARIC WHO Clinical Characterisation Protocol. The Lancet Regional Health - Europe 2021, 100186. [CrossRef]
- Desgranges, F.; Tadini, E.; Munting, A.; Regina, J.; Filippidis, P.; Viala, B.; Karachalias, E.; Suttels, V.; Haefliger, D.; Kampouri, E.; Van Singer, M.; Tschopp, J.; Rochat Stettler, L.; Schaad, S.; Brahier, T.; Hugli, O.; Mueller, Y.; Gouveia, A.; Opota, O.; Carron, P.-N. Post-COVID-19 Syndrome in Outpatients: A Cohort Study. Journal of General Internal Medicine 2022, 37 (8), 1943–1952. [CrossRef]
- Subramanian, A.; Nirantharakumar, K.; Hughes, S.; Myles, P.; Williams, T.; Gokhale, K. M.; Taverner, T.; Chandan, J. S.; Brown, K.; Simms-Williams, N.; Shah, A. D.; Singh, M.; Kidy, F.; Okoth, K.; Hotham, R.; Bashir, N.; Cockburn, N.; Lee, S. I.; Turner, G. M.; Gkoutos, G. V. Symptoms and Risk Factors for Long COVID in Non-Hospitalized Adults. Nature Medicine 2022, 28 (8), 1706–1714. [CrossRef]
- Pelà, G.; Goldoni, M.; Solinas, E.; Cavalli, C.; Tagliaferri, S.; Ranzieri, S.; Frizzelli, A.; Marchi, L.; Mori, P. A.; Majori, M.; Aiello, M.; Corradi, M.; Chetta, A. Sex-Related Differences in Long-COVID-19 Syndrome. Journal of Women’s Health (2002) 2022. [CrossRef]
- Gebhard, C. E.; Sütsch, C.; Bengs, S.; Todorov, A.; Deforth, M.; Buehler, K. P.; Meisel, A.; Schuepbach, R. A.; Zinkernagel, A. S.; Brugger, S. D.; Acevedo, C.; Patriki, D.; Wiggli, B.; Gysi, B.; Beer, J. H.; Friedl, A.; Twerenbold, R.; Kuster, G. M.; Pargger, H.; Tschudin-Sutter, S. Understanding the Impact of Sociocultural Gender on Post-Acute Sequelae of COVID-19: A Bayesian Approach. 2021. [CrossRef]
- Tleyjeh, I. M.; Saddik, B.; Ramakrishnan, R. K.; AlSwaidan, N.; AlAnazi, A.; Alhazmi, D.; Aloufi, A.; AlSumait, F.; Berbari, E. F.; Halwani, R. Long Term Predictors of Breathlessness, Exercise Intolerance, Chronic Fatigue and Well-Being in Hospitalized Patients with COVID-19: A Cohort Study with 4 Months Median Follow-Up. Journal of Infection and Public Health 2022, 15 (1), 21–28. [CrossRef]
- García-Abellán, J.; Padilla, S.; Fernández-González, M.; José Luis García; García, J. A.; María Andreo; Bibiana, S.; Galiana, A.; Félix Gutiérrez; Mar Masiá. Antibody Response to SARS-CoV-2 Is Associated with Long-Term Clinical Outcome in Patients with COVID-19: A Longitudinal Study. 2021, 41 (7), 1490–1501. [CrossRef]
- Asadi-Pooya, A. A.; Akbari, A.; Emami, A.; Lotfi, M.; Rostamihosseinkhani, M.; Nemati, H.; Barzegar, Z.; Kabiri, M.; Zeraatpisheh, Z.; Farjoud-Kouhanjani, M.; Jafari, A.; Sasannia, F.; Ashrafi, S.; Nazeri, M.; Nasiri, S.; Shahisavandi, M. Risk Factors Associated with Long COVID Syndrome: A Retrospective Study. Iranian Journal of Medical Sciences 2021, 46 (6), 428–436. [CrossRef]
- Munblit, D.; Bobkova, P.; Spiridonova, E.; Shikhaleva, A.; Gamirova, A.; Blyuss, O.; Nekliudov, N.; Bugaeva, P.; Andreeva, M.; DunnGalvin, A.; Comberiati, P.; Apfelbacher, C.; Genuneit, J.; Avdeev, S.; Kapustina, V.; Guekht, A.; Fomin, V.; Svistunov, A. A.; Timashev, P.; Subbot, V. S. Incidence and Risk Factors for Persistent Symptoms in Adults Previously Hospitalized for COVID-19. Clinical & Experimental Allergy 2021, 51 (9), 1107–1120. [CrossRef]
- Fernández-de-las-Peñas, C.; Martín-Guerrero, J. D.; Pellicer-Valero, Ó. J.; Navarro-Pardo, E.; Gómez-Mayordomo, V.; Cuadrado, M. L.; Arias-Navalón, J. A.; Cigarán-Méndez, M.; Hernández-Barrera, V.; Arendt-Nielsen, L. Female Sex Is a Risk Factor Associated with Long-Term Post-COVID Related-Symptoms but Not with COVID-19 Symptoms: The LONG-COVID-EXP-CM Multicenter Study. Journal of Clinical Medicine 2022, 11 (2), 413. [CrossRef]
- Bai, F.; Tomasoni, D.; Falcinella, C.; Barbanotti, D.; Castoldi, R.; Mulè, G.; Augello, M.; Mondatore, D.; Allegrini, M.; Cona, A.; Tesoro, D.; Tagliaferri, G.; Viganò, O.; Suardi, E.; Tincati, C.; Beringheli, T.; Varisco, B.; Battistini, C. L.; Piscopo, K.; Vegni, E. Female Gender Is Associated with “Long COVID” Syndrome: A Prospective Cohort Study. Clinical Microbiology and Infection 2021, 28 (4). [CrossRef]
- Chudzik, M.; Lewek, J.; Kapusta, J.; Banach, M.; Jankowski, P.; Bielecka-Dabrowa, A. Predictors of Long COVID in Patients without Comorbidities: Data from the Polish Long-COVID Cardiovascular (PoLoCOV-CVD) Study. Journal of Clinical Medicine 2022, 11 (17), 4980. [CrossRef]
- Chudzik, M.; Babicki, M.; Kapusta, J.; Kałuzińska-Kołat, Ż.; Kołat, D.; Jankowski, P.; Mastalerz-Migas, A. Long-COVID Clinical Features and Risk Factors: A Retrospective Analysis of Patients from the STOP-COVID Registry of the PoLoCOV Study. Viruses 2022, 14 (8), 1755. [CrossRef]
- Notarte, K. I.; de Oliveira, M. H. S.; Peligro, P. J.; Velasco, J. V.; Macaranas, I.; Ver, A. T.; Pangilinan, F. C.; Pastrana, A.; Goldrich, N.; Kavteladze, D.; Gellaco, M. M. L.; Liu, J.; Lippi, G.; Henry, B. M.; Fernández-de-las-Peñas, C. Age, Sex and Previous Comorbidities as Risk Factors Not Associated with SARS-CoV-2 Infection for Long COVID-19: A Systematic Review and Meta-Analysis. Journal of Clinical Medicine 2022, 11 (24), 7314. [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; Guan, L.; Wei, Y.; Li, H.; Wu, X.; Xu, J.; Tu, S.; Zhang, Y.; Chen, H.; Cao, B. Clinical Course and Risk Factors for Mortality of Adult Inpatients with COVID-19 in Wuhan, China: A Retrospective Cohort Study. The Lancet 2020, 395 (10229), 1054–1062. [CrossRef]
- Zhang, H.; Penninger, J. M.; Li, Y.; Zhong, N.; Slutsky, A. S. Angiotensin-Converting Enzyme 2 (ACE2) as a SARS-CoV-2 Receptor: Molecular Mechanisms and Potential Therapeutic Target. Intensive Care Medicine 2020, 46 (4), 586–590. [CrossRef]
- Thorne, L. G.; Reuschl, A.; Zuliani-Alvarez, L.; Whelan, M. V. X.; Turner, J.; Noursadeghi, M.; Jolly, C.; Towers, G. J. SARS-CoV-2 Sensing by RIG-I and MDA5 Links Epithelial Infection to Macrophage Inflammation. The EMBO Journal 2021, 40 (15). [CrossRef]
- Zhao, F.; Ma, Q.; Yue, Q.; Chen, H. SARS-CoV-2 Infection and Lung Regeneration. Clinical Microbiology Reviews 2022. [CrossRef]
- Zhang, H.; Rostami, M. R.; Leopold, P. L.; Mezey, J. G.; O’Beirne, S. L.; Strulovici-Barel, Y.; Crystal, R. G. Expression of the SARS-CoV-2 ACE2 Receptor in the Human Airway Epithelium. American Journal of Respiratory and Critical Care Medicine 2020. [CrossRef]
- Duclos, G. E.; Teixeira, V. H.; Autissier, P.; Gesthalter, Y. B.; Reinders-Luinge, M. A.; Terrano, R.; Dumas, Y. M.; Liu, G.; Mazzilli, S. A.; Brandsma, C.-A.; van den Berge, M.; Janes, S. M.; Timens, W.; Lenburg, M. E.; Spira, A.; Campbell, J. D.; Beane, J. Characterizing Smoking-Induced Transcriptional Heterogeneity in the Human Bronchial Epithelium at Single-Cell Resolution. Science Advances 2019, 5 (12). [CrossRef]
- Hou, Y. J.; Okuda, K.; Edwards, C. E.; Martinez, D. R.; Asakura, T.; Dinnon, K. H.; Kato, T.; Lee, R. E.; Yount, B. L.; Mascenik, T. M.; Chen, G.; Olivier, K. N.; Ghio, A.; Tse, L. V.; Leist, S. R.; Gralinski, L. E.; Schäfer, A.; Dang, H.; Gilmore, R.; Nakano, S. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell 2020, 182 (2). [CrossRef]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-Cell RNA-Seq Data Analysis on the Receptor ACE2 Expression Reveals the Potential Risk of Different Human Organs Vulnerable to 2019-NCoV Infection. Frontiers of Medicine 2020, 14 (2), 185–192. [CrossRef]
- Ziegler, C. G. K.; Allon, S. J.; Nyquist, S. K.; Mbano, I. M.; Miao, V. N.; Tzouanas, C. N.; Cao, Y.; Yousif, A. S.; Bals, J.; Hauser, B. M.; Feldman, J.; Muus, C.; Wadsworth, M. H.; Kazer, S. W.; Hughes, T. K.; Doran, B.; Gatter, G. J.; Vukovic, M.; Taliaferro, F.; Mead, B. E. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181 (5). [CrossRef]
- Lukassen, S.; Chua, R. L.; Trefzer, T.; Kahn, N. C.; Schneider, M. A.; Muley, T.; Winter, H.; Meister, M.; Veith, C.; Boots, A. W.; Hennig, B. P.; Kreuter, M.; Conrad, C.; Eils, R. SARS -CoV-2 Receptor ACE 2 and TMPRSS 2 Are Primarily Expressed in Bronchial Transient Secretory Cells. The EMBO Journal 2020, 39 (10). [CrossRef]
- Hentsch, L.; Cocetta, S.; Allali, G.; Santana, I.; Eason, R.; Adam, E.; Janssens, J.-P. Breathlessness and COVID-19: A Call for Research. Respiration 2021, 1–11. [CrossRef]
- Coccia, C. B. I.; Palkowski, G. H.; Schweitzer, B.; Motsohi, T.; Ntusi, N. Dyspnoea: Pathophysiology and a Clinical Approach. South African Medical Journal 2016, 106 (1), 32. [CrossRef]
- Liu, J.; Zheng, X.; Tong, Q.; Li, W.; Wang, B.; Sutter, K.; Trilling, M.; Lu, M.; Dittmer, U.; Yang, D. Overlapping and Discrete Aspects of the Pathology and Pathogenesis of the Emerging Human Pathogenic Coronaviruses SARS-CoV, MERS-CoV, and 2019-NCoV. Journal of Medical Virology 2020, 92 (5), 491–494. [CrossRef]
- Burnham, E. L.; Janssen, W. J.; Riches, D. W. H.; Moss, M.; Downey, G. P. The Fibroproliferative Response in Acute Respiratory Distress Syndrome: Mechanisms and Clinical Significance. European Respiratory Journal 2013, 43 (1), 276–285. [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; Zhang, Y.; Song, J.; Wang, S.; Chao, Y.; Yang, Z.; Xu, J.; Zhou, X.; Chen, D.; Xiong, W.; Xu, L. Risk Factors Associated with Acute Respiratory Distress Syndrome and Death in Patients with Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Internal Medicine 2020, 180 (7). [CrossRef]
- Liu, X.; Zhou, H.; Zhou, Y.; Wu, X.; Zhao, Y.; Lu, Y.; Tan, W.; Yuan, M.; Ding, X.; Zou, J.; Li, R.; Liu, H.; Ewing, R. M.; Hu, Y.; Nie, H.; Wang, Y. Risk Factors Associated with Disease Severity and Length of Hospital Stay in COVID-19 Patients. Journal of Infection 2020, 81 (1), e95–e97. [CrossRef]
- Rai, D. K.; Sharma, P.; Kumar, R. Post Covid 19 Pulmonary Fibrosis- Is It Reversible? The Indian Journal of Tuberculosis 2020. [CrossRef]
- George, P. M.; Wells, A. U.; Jenkins, R. G. Pulmonary Fibrosis and COVID-19: The Potential Role for Antifibrotic Therapy. The Lancet Respiratory Medicine 2020, 8 (8). [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell Entry Mechanisms of SARS-CoV-2. Proceedings of the National Academy of Sciences 2020, 117 (21). [CrossRef]
- Menni, C.; Valdes, A. M.; Freidin, M. B.; Sudre, C. H.; Nguyen, L. H.; Drew, D. A.; Ganesh, S.; Varsavsky, T.; Cardoso, M. J.; El-Sayed Moustafa, J. S.; Visconti, A.; Hysi, P.; Bowyer, R. C. E.; Mangino, M.; Falchi, M.; Wolf, J.; Ourselin, S.; Chan, A. T.; Steves, C. J.; Spector, T. D. Real-Time Tracking of Self-Reported Symptoms to Predict Potential COVID-19. Nature Medicine 2020, 1–4. [CrossRef]
- Mazzone, S. B.; Tian, L.; Moe, A. A. K.; Trewella, M. W.; Ritchie, M. E.; McGovern, A. E. Transcriptional Profiling of Individual Airway Projecting Vagal Sensory Neurons. Molecular Neurobiology 2020, 57 (2), 949–963. [CrossRef]
- Davies, J.; Randeva, H.; Chatha, K.; Hall, M.; Spandidos, D.; Karteris, E.; Kyrou, I. Neuropilin-1 as a New Potential SARS-CoV-2 Infection Mediator Implicated in the Neurologic Features and Central Nervous System Involvement of COVID-19. Molecular Medicine Reports 2020. [CrossRef]
- Brann, D. H.; Tsukahara, T.; Weinreb, C.; Lipovsek, M.; Berge, K. V. den; Gong, B.; Chance, R.; Macaulay, I. C.; Chou, H.-J.; Fletcher, R. B.; Das, D.; Street, K.; Bezieux, H. R. de; Choi, Y.-G.; Risso, D.; Dudoit, S.; Purdom, E.; Mill, J.; Hachem, R. A.; Matsunami, H. Non-Neuronal Expression of SARS-CoV-2 Entry Genes in the Olfactory System Suggests Mechanisms Underlying COVID-19-Associated Anosmia. Science Advances 2020, 6 (31), eabc5801. [CrossRef]
- Chen, M.; Shen, W.; Rowan, N. R.; Kulaga, H.; Hillel, A.; Ramanathan, M.; Lane, A. P. Elevated ACE-2 Expression in the Olfactory Neuroepithelium: Implications for Anosmia and Upper Respiratory SARS-CoV-2 Entry and Replication. European Respiratory Journal 2020, 56 (3), 2001948. [CrossRef]
- Shiers, S.; Ray, P. R.; Wangzhou, A.; Sankaranarayanan, I.; Tatsui, C. E.; Rhines, L. D.; Li, Y.; Uhelski, M. L.; Dougherty, P. M.; Price, T. J. ACE2 and SCARF Expression in Human Dorsal Root Ganglion Nociceptors: Implications for SARS-CoV-2 Virus Neurological Effects. Pain 2020, 161 (11), 2494–2501. [CrossRef]
- Rhea, E. M.; Logsdon, A. F.; Hansen, K. M.; Williams, L. M.; Reed, M. J.; Baumann, K. K.; Holden, S. J.; Raber, J.; Banks, W. A.; Erickson, M. A. The S1 Protein of SARS-CoV-2 Crosses the Blood–Brain Barrier in Mice. Nature Neuroscience 2020. [CrossRef]
- Ojha, V.; Mani, A.; Pandey, N. N.; Sharma, S.; Kumar, S. CT in Coronavirus Disease 2019 (COVID-19): A Systematic Review of Chest CT Findings in 4410 Adult Patients. European Radiology 2020. [CrossRef]
- Jones, R. M.; Hilldrup, S.; Hope-Gill, B. D.; Eccles, R.; Harrison, N. K. Mechanical Induction of Cough in Idiopathic Pulmonary Fibrosis. Cough 2011, 7 (1), 2. [CrossRef]
- Ryan, N. M.; Birring, S. S.; Gibson, P. G. Gabapentin for Refractory Chronic Cough: A Randomised, Double-Blind, Placebo-Controlled Trial. The Lancet 2012, 380 (9853), 1583–1589. [CrossRef]
- Vertigan, A. E.; Kapela, S. L.; Ryan, N. M.; Birring, S. S.; McElduff, P.; Gibson, P. G. Pregabalin and Speech Pathology Combination Therapy for Refractory Chronic Cough. Chest 2016, 149 (3), 639–648. [CrossRef]
- Walls, A. C.; Park, Y.-J.; Tortorici, M. A.; Wall, A.; McGuire, A. T.; Veesler, D. Structure, Function, and Antigenicity of the Sars-Cov-2 Spike Glycoprotein. Cell 2020, 181 (2), 281–292. [CrossRef]
- Gui, M.; Song, W.; Zhou, H.; Xu, J.; Chen, S.; Xiang, Y.; Wang, X. Cryo-Electron Microscopy Structures of the SARS-CoV Spike Glycoprotein Reveal a Prerequisite Conformational State for Receptor Binding. Cell Research 2016, 27 (1), 119–129. [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T. S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; Müller, M. A.; Drosten, C.; Pöhlmann, S. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181 (2), 271–280. [CrossRef]
- Lazartigues, E.; Qadir, M. M. F.; Mauvais-Jarvis, F. Endocrine Significance of SARS-CoV-2′S Reliance on ACE2. Endocrinology 2020, 161 (9). [CrossRef]
- Chan, J. L.; Gregory, K. D.; Smithson, S. S.; Naqvi, M.; Mamelak, A. N. Pituitary Apoplexy Associated with Acute COVID-19 Infection and Pregnancy. Pituitary 2020, 23 (6), 716–720. [CrossRef]
- Ghosh, R.; Roy, D.; Roy, D.; Mandal, A.; Dutta, A.; Naga, D.; Benito-León, J. A Rare Case of SARS-CoV-2 Infection Associated with Pituitary Apoplexy without Comorbidities. Journal of the Endocrine Society 2021, 5 (3). [CrossRef]
- Bordes, S. J.; Phang-Lyn, S.; Najera, E.; Borghei-Razavi, H.; Adada, B. Pituitary Apoplexy Attributed to COVID-19 Infection in the Absence of an Underlying Macroadenoma or Other Identifiable Cause. Cureus 2021, 13 (2). [CrossRef]
- Rotondi, M.; Coperchini, F.; Ricci, G.; Denegri, M.; Croce, L.; Ngnitejeu, S. T.; Villani, L.; Magri, F.; Latrofa, F.; Chiovato, L. Detection of SARS-COV-2 Receptor ACE-2 MRNA in Thyroid Cells: A Clue for COVID-19-Related Subacute Thyroiditis. Journal of Endocrinological Investigation 2020, 1–6. [CrossRef]
- Lazartigues, E.; Qadir, M. M. F.; Mauvais-Jarvis, F. Endocrine Significance of SARS-CoV-2′S Reliance on ACE2. Endocrinology 2020, 161 (9). [CrossRef]
- Croce, L.; Gangemi, D.; Ancona, G.; Liboà, F.; Bendotti, G.; Minelli, L.; Chiovato, L. The Cytokine Storm and Thyroid Hormone Changes in COVID-19. Journal of Endocrinological Investigation 2021, 44 (5), 891–904. [CrossRef]
- Kumari, K.; Chainy, G. B. N.; Subudhi, U. Prospective Role of Thyroid Disorders in Monitoring COVID-19 Pandemic. Heliyon 2020, 6 (12), e05712. [CrossRef]
- Popa, A.; Chereji, A.-I.; Dodu, M. A.; Chereji, I.; Fitero, A.; Daina, C. M.; Daina, L. G.; Badau, D.; Neculoiu, D. C.; Domnariu, C. The Impact of Changes Regarding Working Circumstances during COVID-19 Pandemic upon Patients Evaluated for Thyroid Dysfunction. International Journal of Environmental Research and Public Health 2022, 19 (16), 9856. [CrossRef]
- Fatourechi, V.; Aniszewski, J. P.; Fatourechi, G. Z. E.; Atkinson, E. J.; Jacobsen, S. J. Clinical Features and Outcome of Subacute Thyroiditis in an Incidence Cohort: Olmsted County, Minnesota, Study. The Journal of Clinical Endocrinology & Metabolism 2003, 88 (5), 2100–2105. [CrossRef]
- Correia de Sá, T.; Soares, C.; Rocha, M. Acute Pancreatitis and COVID-19: A Literature Review. World Journal of Gastrointestinal Surgery 2021, 13 (6), 574–584. [CrossRef]
- Amidifar, S.; Elahi, R.; Siahmansouri, A.; Maleki, A. J.; Moradi, A. Endocrine and Metabolic Complications of COVID-19: Lessons Learned and Future Prospects. Journal of Molecular Endocrinology 2022. [CrossRef]
- Rubino, F.; Amiel, S. A.; Zimmet, P.; Alberti, G.; Bornstein, S.; Eckel, R. H.; Mingrone, G.; Boehm, B.; Cooper, M. E.; Chai, Z.; Del Prato, S.; Ji, L.; Hopkins, D.; Herman, W. H.; Khunti, K.; Mbanya, J.-C.; Renard, E. New-Onset Diabetes in Covid-19. New England Journal of Medicine 2020. [CrossRef]
- Landstra, C. P.; de Koning, E. J. P. COVID-19 and Diabetes: Understanding the Interrelationship and Risks for a Severe Course. Frontiers in Endocrinology 2021, 12. [CrossRef]
- Rubino, F.; Amiel, S. A.; Zimmet, P.; Alberti, G.; Bornstein, S.; Eckel, R. H.; Mingrone, G.; Boehm, B.; Cooper, M. E.; Chai, Z.; Del Prato, S.; Ji, L.; Hopkins, D.; Herman, W. H.; Khunti, K.; Mbanya, J.-C.; Renard, E. New-Onset Diabetes in Covid-19. New England Journal of Medicine 2020. [CrossRef]
- Carlsson, P. O. The Renin-Angiotensin System in the Endocrine Pancreas. JOP: Journal of the pancreas 2001, 2 (1), 26–32.
- Goossens, G. H. The Renin-Angiotensin System in the Pathophysiology of Type 2 Diabetes. Obesity Facts 2012, 5 (4), 611–624. [CrossRef]
- Hayden, M. R.; Karuparthi, P. R.; Habibi, J.; Wasekar, C.; Lastra, G.; Manrique, C.; Stas, S.; Sowers, J. R. Ultrastructural Islet Study of Early Fibrosis in the Ren2 Rat Model of Hypertension. Emerging Role of the Islet Pancreatic Pericyte-Stellate Cell. JOP: Journal of the pancreas 2007, 8 (6), 725–738.
- Wang, F.; Wang, H.; Fan, J.; Zhang, Y.; Wang, H.; Zhao, Q. Pancreatic Injury Patterns in Patients with COVID-19 Pneumonia. Gastroenterology 2020. [CrossRef]
- Aloysius, M. M.; Thatti, A.; Gupta, A.; Sharma, N.; Bansal, P.; Goyal, H. COVID-19 Presenting as Acute Pancreatitis. Pancreatology 2020. [CrossRef]
- Lopez-Leon, S.; Wegman-Ostrosky, T.; Perelman, C.; Sepulveda, R.; Rebolledo, P. A.; Cuapio, A.; Villapol, S. More than 50 Long-Term Effects of COVID-19: A Systematic Review and Meta-Analysis. medRxiv: The Preprint Server for Health Sciences 2021. [CrossRef]
- Kamin, H. S.; Kertes, D. A. Cortisol and DHEA in Development and Psychopathology. Hormones and Behavior 2017, 89, 69–85. [CrossRef]
- Wheatland, R. Molecular Mimicry of ACTH in SARS – Implications for Corticosteroid Treatment and Prophylaxis. Medical Hypotheses 2004, 63 (5), 855–862. [CrossRef]
- Chu, K. Y.; Nackeeran, S.; Horodyski, L.; Masterson, T. A.; Ramasamy, R. COVID-19 Infection Is Associated with New Onset Erectile Dysfunction: Insights from a National Registry. Sexual Medicine 2022, 10 (1), 100478. [CrossRef]
- Barragan, M.; Guillén, J. J.; Martin-Palomino, N.; Rodriguez, A.; Vassena, R. Undetectable Viral RNA in Oocytes from SARS-CoV-2 Positive Women. Human Reproduction 2020, 36 (2), 390–394. [CrossRef]
- Li, K.; Chen, G.; Hou, H.; Liao, Q.; Chen, J.; Bai, H.; Lee, S.; Wang, C.; Li, H.; Cheng, L.; Ai, J. Analysis of Sex Hormones and Menstruation in COVID-19 Women of Child-Bearing Age. Reproductive BioMedicine Online 2021, 42 (1), 260–267. [CrossRef]
- Proal, A. D.; VanElzakker, M. B. Long COVID or Post-Acute Sequelae of COVID-19 (PASC): An Overview of Biological Factors That May Contribute to Persistent Symptoms. Frontiers in Microbiology 2021, 12. [CrossRef]
- Report: What Does COVID-19 Recovery Actually Look Like? Patient Led Research. https://patientresearchcovid19.com/research/report-1/.
- Erdinc, B.; Sahni, S.; Gotlieb, V. Hematological Manifestations and Complications of COVID-19. Advances in Clinical and Experimental Medicine 2021, 30 (1), 101–107. [CrossRef]
- Sava, C. N.; Bodog, T.-M.; Niulas, L. R.; Iuhas, A. R.; Marinau, C. P.; Negrut, N.; Balmos, A. B.; Pasca, B.; Roman, N. A.; Delia Nistor-Cseppento, C. Biomarker Changes in Pediatric Patients with COVID-19: A Retrospective Study from a Single Center Database. In Vivo (Athens, Greece) 2022, 36 (6), 2813–2822. [CrossRef]
- Amer Harky; Avesta Ala’Aldeen; None SundasButt; Duric, B.; Roy, S.; Zeinah, M. COVID-19 and Multiorgan Response: The Long-Term Impact. 2023, 101756–101756. [CrossRef]
- Barrett, T.J.; Bilaloglu, S.; Cornwell, M.; Burgess, H.M.; Virginio, V.W.; Drenkova, K.; Ibrahim, H.; Yuriditsky, E.; Aphinyanaphongs, Y.; Lifshitz, M.; et al. Platelets contribute to disease severity in COVID-19. J Thromb Haemost 2021, 19, 3139-3153. [CrossRef]
- Targosz-Korecka, M.; Kubisiak, A.; Kloska, D.; Kopacz, A.; Grochot-Przeczek, A.; Szymonski, M. Endothelial Glycocalyx Shields the Interaction of SARS-CoV-2 Spike Protein with ACE2 Receptors. Scientific Reports 2021, 11 (1), 12157. [CrossRef]
- Prasad, M.; Leon, M.; Lerman, L. O.; Lerman, A. Viral Endothelial Dysfunction: A Unifying Mechanism for COVID-19. Mayo Clinic Proceedings 2021. [CrossRef]
- Evangelos Oikonomou; Nektarios Souvaliotis; Stamatios Lampsas; Gerasimos Siasos; Garyphallia Poulakou; Panagiotis Theofilis; Papaioannou, T. G.; Anna-Bettina Haidich; Tsaousi, G.; Ntousopoulos Vasileios; Sakka Vissaria; Charalambous, G.; Rapti, V.; Raftopoulou, S.; Syrigos, K. N.; Costas Tsioufis; Dimitrios Tousoulis; Manolis Vavuranakis. Endothelial Dysfunction in Acute and Long Standing COVID−19: A Prospective Cohort Study. 2022, 144, 106975–106975. [CrossRef]
- Chioh, F. W.; Fong, S.-W.; Young, B. E.; Wu, K.-X.; Siau, A.; Krishnan, S.; Chan, Y.-H.; Carissimo, G.; Teo, L. L.; Gao, F.; Tan, R. S.; Zhong, L.; Koh, A. S.; Tan, S.-Y.; Tambyah, P. A.; Renia, L.; Ng, L. F.; Lye, D. C.; Cheung, C. Convalescent COVID-19 Patients Are Susceptible to Endothelial Dysfunction due to Persistent Immune Activation. eLife 2021, 10. [CrossRef]
- Fogarty, H.; Townsend, L.; Morrin, H.; Ahmad, A.; Comerford, C.; Karampini, E.; Englert, H.; Byrne, M.; Bergin, C.; O’Sullivan, J. M.; Martin-Loeches, I.; Nadarajan, P.; Bannan, C.; Mallon, P. W.; Curley, G. F.; Preston, R. J. S.; Rehill, A. M.; McGonagle, D.; Ni Cheallaigh, C.; Baker, R. I. Persistent Endotheliopathy in the Pathogenesis of Long COVID Syndrome. Journal of Thrombosis and Haemostasis 2021, 19 (10), 2546–2553. [CrossRef]
- Hernán Mejía-Rentería; Travieso, A.; A Sagir; Martínez-Gómez, E.; Carrascosa-Granada, A.; Toya, T.; Núñez-Gil, I. J.; Estrada, V.; Lerman, A.; Escaned, J. In-Vivo Evidence of Systemic Endothelial Vascular Dysfunction in COVID-19. 2021, 345, 153–155. [CrossRef]
- Manolis, A. S.; Manolis, T. A.; Manolis, A. A.; Papatheou, D.; Melita, H. COVID-19 Infection: Viral Macro- and Micro-Vascular Coagulopathy and Thromboembolism/Prophylactic and Therapeutic Management. Journal of Cardiovascular Pharmacology and Therapeutics 2020, 26 (1), 12–24. [CrossRef]
- Poor, H. D. Pulmonary Thrombosis and Thromboembolism in COVID-19. Chest 2021. [CrossRef]
- Pretorius, E.; Vlok, M.; Venter, C.; Bezuidenhout, J. A.; Laubscher, G. J.; Steenkamp, J.; Kell, D. B. Persistent Clotting Protein Pathology in Long COVID/Post-Acute Sequelae of COVID-19 (PASC) Is Accompanied by Increased Levels of Antiplasmin. Cardiovascular Diabetology 2021, 20 (1). [CrossRef]
- Pretorius, E.; Venter, C.; Laubscher, G. J.; Kotze, M. J.; Oladejo, S. O.; Watson, L. R.; Rajaratnam, K.; Watson, B. W.; Kell, D. B. Prevalence of Symptoms, Comorbidities, Fibrin Amyloid Microclots and Platelet Pathology in Individuals with Long COVID/Post-Acute Sequelae of COVID-19 (PASC). Cardiovascular Diabetology 2022, 21 (1). [CrossRef]
- Proal, A. D.; VanElzakker, M. B. Long COVID or Post-Acute Sequelae of COVID-19 (PASC): An Overview of Biological Factors That May Contribute to Persistent Symptoms. Frontiers in Microbiology 2021, 12. [CrossRef]
- Chen, W.; Pan, J. Y. Anatomical and Pathological Observation and Analysis of SARS and COVID-19: Microthrombosis Is the Main Cause of Death. Biological Procedures Online 2021, 23. [CrossRef]
- Guervilly, C.; Bonifay, A.; Burtey, S.; Sabatier, F.; Cauchois, R.; Abdili, E.; Arnaud, L.; Lano, G.; Pietri, L.; Robert, T.; Velier, M.; Papazian, L.; Albanese, J.; Kaplanski, G.; Dignat-George, F.; Lacroix, R. Dissemination of Extreme Levels of Extracellular Vesicles: Tissue Factor Activity in Patients with Severe COVID-19. Blood Advances 2021, 5 (3), 628–634. [CrossRef]
- Barberis, E.; Vanella, V. V.; Falasca, M.; Caneapero, V.; Cappellano, G.; Raineri, D.; Ghirimoldi, M.; De Giorgis, V.; Puricelli, C.; Vaschetto, R.; Sainaghi, P. P.; Bruno, S.; Sica, A.; Dianzani, U.; Rolla, R.; Chiocchetti, A.; Cantaluppi, V.; Baldanzi, G.; Marengo, E.; Manfredi, M. Circulating Exosomes Are Strongly Involved in SARS-CoV-2 Infection. Frontiers in Molecular Biosciences 2021, 8. [CrossRef]
- Eymieux, S.; Uzbekov, R.; Rouillé, Y.; Blanchard, E.; Hourioux, C.; Dubuisson, J.; Belouzard, S.; Roingeard, P. Secretory Vesicles Are the Principal Means of SARS-CoV-2 Egress. Cells 2021, 10 (8), 2047. [CrossRef]
- Fogarty, H.; Townsend, L.; Morrin, H.; Ahmad, A.; Comerford, C.; Karampini, E.; Englert, H.; Byrne, M.; Bergin, C.; O’Sullivan, J. M.; Martin-Loeches, I.; Nadarajan, P.; Bannan, C.; Mallon, P. W.; Curley, G. F.; Preston, R. J. S.; Rehill, A. M.; McGonagle, D.; Ni Cheallaigh, C.; Baker, R. I. Persistent Endotheliopathy in the Pathogenesis of Long COVID Syndrome. Journal of Thrombosis and Haemostasis 2021, 19 (10), 2546–2553. [CrossRef]
- Celestino Rodríguez; Luque, N.; Blanco, I.; Sebastian, L.; Joan Albert Barberà; Peinado, V. I.; Tura-Ceide, O. Pulmonary Endothelial Dysfunction and Thrombotic Complications in Patients with COVID-19. 2021, 64 (4), 407–415. [CrossRef]
- Wang, C.; Yu, C.; Jing, H.; Wu, X.; Novakovic, V. A.; Xie, R.; Shi, J. Long COVID: The Nature of Thrombotic Sequelae Determines the Necessity of Early Anticoagulation. Frontiers in Cellular and Infection Microbiology 2022, 12. [CrossRef]
- Sharma, P.; Behl, T.; Sharma, N.; Singh, S.; Grewal, A.S.; Albarrati, A.; Albratty, M.; Meraya, A.M.; Bungau, S. COVID-19 and diabetes: Association intensify risk factors for morbidity and mortality. Biomed Pharmacother 2022, 151, 113089. [CrossRef]
- Kabir, M.T.; Uddin, M.S.; Hossain, M.F.; Abdulhakim, J.A.; Alam, M.A.; Ashraf, G.M.; Bungau, S.G.; Bin-Jumah, M.N.; Abdel-Daim, M.M.; Aleya, L. nCOVID-19 Pandemic: From Molecular Pathogenesis to Potential Investigational Therapeutics. Front Cell Dev Biol 2020, 8, 616. [CrossRef]
- Gheorghe, G.; Ilie, M.; Bungau, S.; Stoian, A.M.P.; Bacalbasa, N.; Diaconu, C.C. Is There a Relationship between COVID-19 and Hyponatremia? Medicina (Kaunas) 2021, 57. [CrossRef]
- Tagde, P.; Tagde, S.; Tagde, P.; Bhattacharya, T.; Monzur, S.M.; Rahman, M.H.; Otrisal, P.; Behl, T.; ul Hassan, S.S.; Abdel-Daim, M.M.; et al. Nutraceuticals and Herbs in Reducing the Risk and Improving the Treatment of COVID-19 by Targeting SARS-CoV-2. Biomedicines 2021, 9, 1266. [CrossRef]
- Perumal, R.; Shunmugam, L.; Naidoo, K.; Wilkins, D.; Garzino-Demo, A.; Brechot, C.; Anders Vahlne; Janko Nikolich-Žugich. Biological Mechanisms Underpinning the Development of Long COVID. 2023, 26 (6), 106935–106935. [CrossRef]
- Mayte Suárez-Fariñas; Minami Tokuyama; Wei, G.; Huang, R.; Livanos, A. E.; Jha, D.; Anaïs Levescot; Haritz Irizar; Kosoy, R.; Cording, S.; Wang, W.-H.; Bojan Losic; Ungaro, R. C.; Di’Narzo, A.; Martinez-Delgado, G.; Suprun, M.; Corley, M. J.; Aleksandar Stojmirović; Houten, S. M.; Peters, L. A. Intestinal Inflammation Modulates the Expression of ACE2 and TMPRSS2 and Potentially Overlaps with the Pathogenesis of SARS-CoV-2–Related Disease. 2021, 160 (1), 287-301.e20. [CrossRef]
- Mehandru, S.; Merad, M. Pathological Sequelae of Long-Haul COVID. Nature Immunology 2022, 23 (2), 194–202. [CrossRef]
- Gaebler, C.; Wang, Z.; Lorenzi, J. C. C.; Muecksch, F.; Finkin, S.; Tokuyama, M.; Cho, A.; Jankovic, M.; Schaefer-Babajew, D.; Oliveira, T. Y.; Cipolla, M.; Viant, C.; Barnes, C. O.; Bram, Y.; Breton, G.; HägglöfT.; Mendoza, P.; Hurley, A.; Turroja, M.; Gordon, K. Evolution of Antibody Immunity to SARS-CoV-2. Nature 2021, 591. [CrossRef]
- Choudhury, A.; Tariq, R.; Jena, A.; Vesely, E. K.; Singh, S.; Khanna, S.; Sharma, V. Gastrointestinal Manifestations of Long COVID: A Systematic Review and Meta-Analysis. Therapeutic Advances in Gastroenterology 2022, 15, 175628482211184. [CrossRef]
- Adame, M. J.; Nada, K. M.; Seashore, J. Late Sequelae of COVID-19 Infection in Patients without Comorbidities. American Journal of Respiratory and Critical Care Medicine 2021.
- Suárez-Fariñas, M.; Tokuyama, M.; Wei, G.; Huang, R.; Livanos, A.; Jha, D.; Levescot, A.; Irizar, H.; Kosoy, R.; Cording, S.; et al. Intestinal Inflammation Modulates the Expression of ACE2 and TMPRSS2 and Potentially Overlaps With the Pathogenesis of SARS-CoV-2–related Disease. Gastroenterology 2021, 160, 287-301.e220. [CrossRef]
- Berna Akıncı Özyürek; Tuğçe Şahin Özdemirel; Esma Sevil Akkurt; Derya Yenibertiz; Zeynep Tilbe Saymaz; Sertaç Büyükyaylacı Özden; Zuhal Eroglu. What Are the Factors That Affect Post COVID 1st Month’s Continuing Symptoms? 2021, 75 (11). [CrossRef]
- Faycal, A.; Ndoadoumgue, A. L.; Sellem, B.; Blanc, C.; Dudoit, Y.; Schneider, L.; Tubiana, R.; Valantin, M.-A. .; Seang, S.; Palich, R.; Bleibtreu, A.; Monsel, G.; Godefroy, N.; Itani, O.; Paccoud, O.; Pourcher, V.; Caumes, E.; Ktorza, N.; Chermak, A.; Abdi, B. Prevalence and Factors Associated with Symptom Persistence: A Prospective Study of 429 Mild COVID-19 Outpatients. Infectious Diseases Now 2022, 52 (2), 75–81. [CrossRef]
- Fernández-de-las-Peñas, C.; Martín-Guerrero, J.; Navarro-Pardo, E.; Torres-Macho, J.; Canto-Diez, M. G.; Pellicer-Valero, O. Gastrointestinal Symptoms at the Acute COVID-19 Phase Are Risk Factors for Developing Gastrointestinal Post-COVID Symptoms: A Multicenter Study. Internal and Emergency Medicine 2021. [CrossRef]
- Kozak, R.; Armstrong, S. M.; Salvant, E.; Ritzker, C.; Feld, J.; Biondi, M. J.; Tsui, H. Recognition of Long-COVID-19 Patients in a Canadian Tertiary Hospital Setting: A Retrospective Analysis of Their Clinical and Laboratory Characteristics. Pathogens 2021, 10 (10), 1246. [CrossRef]
- Leth, S.; Gunst, J. D.; Mathiasen, V. D.; Hansen, K. S.; Søgaard, O. S.; Østergaard, L.; Jensen-Fangel, S.; Storgaard, M.; Agergaard, J. Persistent Symptoms in Hospitalized Patients Recovering from COVID-19 in Denmark. Open Forum Infectious Diseases 2021. [CrossRef]
- Lombardo, M. D. M.; Foppiani, A.; Peretti, G. M.; Mangiavini, L.; Battezzati, A.; Bertoli, S.; Martinelli Boneschi, F.; Zuccotti, G. V. Long-Term Coronavirus Disease 2019 Complications in Inpatients and Outpatients: A One-Year Follow-up Cohort Study. Open Forum Infectious Diseases 2021, 8 (8), ofab384. [CrossRef]
- Shang, Y. F.; Liu, T.; Yu, J. N.; Xu, X. R.; Zahid, K. R.; Wei, Y. C.; Wang, X. H.; Zhou, F. L. Half-Year Follow-up of Patients Recovering from Severe COVID-19: Analysis of Symptoms and Their Risk Factors. Journal of Internal Medicine 2021, 290 (2), 444–450. [CrossRef]
- Lamers, M. M.; Beumer, J.; Vaart, J. van der; Knoops, K.; Puschhof, J.; Breugem, T. I.; Ravelli, R. B. G.; Schayck, J. P. van; Mykytyn, A. Z.; Duimel, H. Q.; Donselaar, E. van; Riesebosch, S.; Kuijpers, H. J. H.; Schippers, D.; Wetering, W. J. van de; Graaf, M. de; Koopmans, M.; Cuppen, E.; Peters, P. J.; Haagmans, B. L. SARS-CoV-2 Productively Infects Human Gut Enterocytes. Science 2020, 369 (6499). [CrossRef]
- Mouchati, C.; Durieux, J. C.; Zisis, S. N.; Labbato, D.; Rodgers, M. A.; Ailstock, K.; Reinert, B. L.; Funderburg, N. T.; McComsey, G. A. Increase in Gut Permeability and Oxidized Ldl Is Associated with Post-Acute Sequelae of SARS-CoV-2. Frontiers in Immunology 2023, 14, 1182544. [CrossRef]
- Gupta, S.; Parker, J.; Smits, S.; Underwood, J.; Dolwani, S. Persistent viral shedding of SARS-CoV-2 in faeces - a rapid review. Colorectal Dis 2020, 22, 611-620. [CrossRef]
- Xing, Y.-H.; Ni, W.; Wu, Q.; Li, W.-J.; Li, G.-J.; Wang, W.-D.; Tong, J.-N.; Song, X.-F.; Wing-Kin Wong, G.; Xing, Q.-S. Prolonged Viral Shedding in Feces of Pediatric Patients with Coronavirus Disease 2019. Journal of Microbiology, Immunology and Infection 2020, 53 (3), 473–480. [CrossRef]
- Amirian, E. S. Potential Fecal Transmission of SARS-CoV-2: Current Evidence and Implications for Public Health. International Journal of Infectious Diseases 2020, 95, 363–370. [CrossRef]
- Chen, Y.; Chen, L.; Deng, Q.; Zhang, G.; Wu, K.; Ni, L.; Yang, Y.; Liu, B.; Wang, W.; Wei, C.; Yang, J.; Ye, G.; Cheng, Z. The Presence of SARS-CoV-2 RNA in the Feces of COVID-19 Patients. Journal of Medical Virology 2020, 92 (7), 833–840. [CrossRef]
- Jones, D. L.; Baluja, M. Q.; Graham, D. W.; Corbishley, A.; McDonald, J. E.; Malham, S. K.; Hillary, L. S.; Connor, T. R.; Gaze, W. H.; Moura, I. B.; Wilcox, M. H.; Farkas, K. Shedding of SARS-CoV-2 in Feces and Urine and Its Potential Role in Person-To-Person Transmission and the Environment-Based Spread of COVID-19. Science of The Total Environment 2020, 749, 141364. [CrossRef]
- Peng, L.; Liu, J.; Xu, W.; Luo, Q.; Chen, D.; Lei, Z.; Huang, Z.; Li, X.; Deng, K.; Lin, B.; Gao, Z. SARS-CoV-2 Can Be Detected in Urine, Blood, Anal Swabs, and Oropharyngeal Swabs Specimens. Journal of Medical Virology 2020. [CrossRef]
- Tian, Y.; Rong, L.; Nian, W.; He, Y. Review Article: Gastrointestinal Features in COVID-19 and the Possibility of Faecal Transmission. Alimentary Pharmacology & Therapeutics 2020, 51 (9), 843–851. [CrossRef]
- Khreefa, Z.; Barbier, M. T.; Koksal, A. R.; Love, G.; Del Valle, L. Pathogenesis and Mechanisms of SARS-CoV-2 Infection in the Intestine, Liver, and Pancreas. Cells 2023, 12 (2), 262. [CrossRef]
- Phetsouphanh, C.; Darley, D. R.; Wilson, D. B.; Howe, A.; Munier, C. M. L.; Patel, S. K.; Juno, J. A.; Burrell, L. M.; Kent, S. J.; Dore, G. J.; Kelleher, A. D.; Matthews, G. V. Immunological Dysfunction Persists for 8 Months Following Initial Mild-To-Moderate SARS-CoV-2 Infection. Nature Immunology 2022, 23 (2), 210–216. [CrossRef]
- Gaebler, C.; Wang, Z.; Lorenzi, J. C. C.; Muecksch, F.; Finkin, S.; Tokuyama, M.; Cho, A.; Jankovic, M.; Schaefer-Babajew, D.; Oliveira, T. Y.; Cipolla, M.; Viant, C.; Barnes, C. O.; Bram, Y.; Breton, G.; HägglöfT.; Mendoza, P.; Hurley, A.; Turroja, M.; Gordon, K. Evolution of Antibody Immunity to SARS-CoV-2. Nature 2021, 591. [CrossRef]
- Ståhlberg, M.; Reistam, U.; Fedorowski, A.; Villacorta, H.; Horiuchi, Y.; Bax, J.; Pitt, B.; Matskeplishvili, S.; Lüscher, T. F.; Weichert, I.; Thani, K. B.; Maisel, A. Post-Covid-19 Tachycardia Syndrome: A Distinct Phenotype of Post-Acute Covid-19 Syndrome. The American Journal of Medicine 2021. [CrossRef]
- Vernino, S.; Stiles, L. E. Autoimmunity in Postural Orthostatic Tachycardia Syndrome: Current Understanding. Autonomic Neuroscience 2018, 215, 78–82. [CrossRef]
- Bisaccia, G.; Ricci, F.; Recce, V.; Serio, A.; Iannetti, G.; Chahal, A.A.; Ståhlberg, M.; Khanji, M.Y.; Fedorowski, A.; Gallina, S. Post-Acute Sequelae of COVID-19 and Cardiovascular Autonomic Dysfunction: What Do We Know? J Cardiovasc Dev Dis 2021, 8. [CrossRef]
- Taquet, M.; Dercon, Q.; Luciano, S.; Geddes, J. R.; Husain, M.; Harrison, P. J. Incidence, Co-Occurrence, and Evolution of Long-COVID Features: A 6-Month Retrospective Cohort Study of 273,618 Survivors of COVID-19. PLOS Medicine 2021, 18 (9), e1003773. [CrossRef]
- Schiffl, H.; Lang, S. M. Long-Term Interplay between COVID-19 and Chronic Kidney Disease. International Urology and Nephrology 2023. [CrossRef]
- Bowe, B.; Xie, Y.; Xu, E.; Al-Aly, Z. Kidney Outcomes in Long COVID. Journal of the American Society of Nephrology 2021, 32 (11), 2851–2862. [CrossRef]
- Chand, S.; Kapoor, S.; Naqvi, A.; Thakkar, J.; Fazzari, M. J.; Orsi, D.; Dieiev, V.; Lewandowski, D. C.; Dicpinigaitis, P. V. Long-Term Follow up of Renal and Other Acute Organ Failure in Survivors of Critical Illness due to Covid-19. Journal of Intensive Care Medicine 2021, 37 (6), 736–742. [CrossRef]
- Su, H.; Yang, M.; Wan, C.; Yi, L.-X.; Tang, F.; Zhu, H.-Y.; Yi, F.; Yang, H.-C.; Fogo, A. B.; Nie, X.; Zhang, C. Renal Histopathological Analysis of 26 Postmortem Findings of Patients with COVID-19 in China. Kidney International 2020, 0 (0). [CrossRef]
- Wang, K.; Chen, W.; Zhang, Z.; Deng, Y.; Lian, J.-Q.; Du, P.; Wei, D.; Zhang, Y.; Sun, X.-X.; Gong, L.; Yang, X.; He, L.; Zhang, L.; Yang, Z.; Geng, J.-J.; Chen, R.; Zhang, H.; Wang, B.; Zhu, Y.-M.; Nan, G. CD147-Spike Protein Is a Novel Route for SARS-CoV-2 Infection to Host Cells. Signal Transduction and Targeted Therapy 2020, 5 (1). [CrossRef]
- Behl, T., Kaur, I., Aleya, L., Sehgal, A., Singh, S., Sharma, N., Bhatia, S., Al-Harrasi, A., Bungau, S.* CD147-spike protein interaction in COVID-19: Get the ball rolling with a novel receptor and therapeutic target. Sci. Tot. Environ. 2022, 808, 152072. [CrossRef]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Hurtado del Pozo, C.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905-913.e907. [CrossRef]
- Diao, B.; Wang, C.; Wang, R.; Feng, Z.; Zhang, J.; Yang, H.; Tan, Y.; Wang, H.; Wang, C.; Liu, L.; Liu, Y.; Liu, Y.; Wang, G.; Yuan, Z.; Hou, X.; Ren, L.; Wu, Y.; Chen, Y. Human Kidney Is a Target for Novel Severe Acute Respiratory Syndrome Coronavirus 2 Infection. Nature Communications 2021, 12 (1), 2506. [CrossRef]
- Magro, C.; Mulvey, J. J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement Associated Microvascular Injury and Thrombosis in the Pathogenesis of Severe COVID-19 Infection: A Report of Five Cases. Translational Research 2020. [CrossRef]
- Sanchez, A.; Sohier, P.; Benghanem, S.; L’Honneur, A.-S.; Rozenberg, F.; Dupin, N.; Garel, B. Digitate Papulosquamous Eruption Associated with Severe Acute Respiratory Syndrome Coronavirus 2 Infection. JAMA Dermatology 2020, 156 (7), 819. [CrossRef]
- Gottlieb, M.; Long, B. Dermatologic Manifestations and Complications of COVID-19. The American Journal of Emergency Medicine 2020. [CrossRef]
- Galván Casas, C.; Català, A.; Carretero Hernández, G.; Rodríguez-Jiménez, P.; Fernández Nieto, D.; Rodríguez-Villa Lario, A.; Navarro Fernández, I.; Ruiz-Villaverde, R.; Falkenhain, D.; Llamas Velasco, M.; García-Gavín, J.; Baniandrés, O.; González-Cruz, C.; Morillas-Lahuerta, V.; Cubiró, X.; Figueras Nart, I.; Selda-Enriquez, G.; Romaní, J.; Fustà-Novell, X.; Melian-Olivera, A. Classification of the Cutaneous Manifestations of COVID-19: A Rapid Prospective Nationwide Consensus Study in Spain with 375 Cases. British Journal of Dermatology 2020. [CrossRef]
- Kaya, G.; Kaya, A.; Saurat, J.-H. Clinical and Histopathological Features and Potential Pathological Mechanisms of Skin Lesions in COVID-19: Review of the Literature. Dermatopathology 2020, 7 (1), 3–16. [CrossRef]
- Suchonwanit, P.; Leerunyakul, K.; Kositkuljorn, C. Cutaneous Manifestations in COVID-19: Lessons Learned from Current Evidence. Journal of the American Academy of Dermatology 2020. [CrossRef]
- Criado, P. R.; Abdalla, B. M. Z.; de Assis, I. C.; van Blarcum de Graaff Mello, C.; Caputo, G. C.; Vieira, I. C. Are the Cutaneous Manifestations during or due to SARS-CoV-2 Infection/COVID-19 Frequent or Not? Revision of Possible Pathophysiologic Mechanisms. Inflammation Research 2020, 1–12. [CrossRef]
- Goren, A.; Vaño-Galván, S.; Wambier, C. G.; McCoy, J.; Gomez-Zubiaur, A.; Moreno-Arrones, O. M.; Shapiro, J.; Sinclair, R. D.; Gold, M. H.; Kovacevic, M.; Mesinkovska, N. A.; Goldust, M.; Washenik, K. A Preliminary Observation: Male Pattern Hair Loss among Hospitalized COVID-19 Patients in Spain – a Potential Clue to the Role of Androgens in COVID-19 Severity. Journal of Cosmetic Dermatology 2020, 19 (7), 1545–1547. [CrossRef]
- A. Mahé; E. Birckel; C. Merklen; Lefebvre, P.; Cédric Hannedouche; Jost, M.; L. Droy-Dupré. Histology of Skin Lesions Establishes That the Vesicular Rash Associated with COVID-19 Is Not “Varicella-Like.” 2020, 34 (10). [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.; Lely, A.; Navis, G.; van Goor, H. Tissue Distribution of ACE2 Protein, the Functional Receptor for SARS Coronavirus. A First Step in Understanding SARS Pathogenesis. The Journal of Pathology 2004, 203 (2), 631–637. [CrossRef]
- Leng, A.; Shah, M.; Ahmad, S. A.; Premraj, L.; Wildi, K.; Li Bassi, G.; Pardo, C. A.; Choi, A.; Cho, S.-M. Pathogenesis Underlying Neurological Manifestations of Long COVID Syndrome and Potential Therapeutics. Cells 2023, 12 (5), 816. [CrossRef]
- Stefanou, M.-I.; Palaiodimou, L.; Bakola, E.; Smyrnis, N.; Papadopoulou, M.; Paraskevas, G. P.; Rizos, E.; Boutati, E.; Grigoriadis, N.; Krogias, C.; Giannopoulos, S.; Tsiodras, S.; Gaga, M.; Tsivgoulis, G. Neurological Manifestations of Long-COVID Syndrome: A Narrative Review. Therapeutic Advances in Chronic Disease 2022, 13, 20406223221076890. [CrossRef]
- Balcom, E. F.; Nath, A.; Power, C. Acute and Chronic Neurological Disorders in COVID-19: Potential Mechanisms of Disease. Brain 2021. [CrossRef]
- Meinhardt, J.; Radke, J.; Dittmayer, C.; Franz, J.; Thomas, C.; Mothes, R.; Laue, M.; Schneider, J.; Brünink, S.; Greuel, S.; Lehmann, M.; Hassan, O.; Aschman, T.; Schumann, E.; Chua, R. L.; Conrad, C.; Eils, R.; Stenzel, W.; Windgassen, M.; Rößler, L. Olfactory Transmucosal SARS-CoV-2 Invasion as a Port of Central Nervous System Entry in Individuals with COVID-19. Nature Neuroscience 2020, 24 (2), 1–8. [CrossRef]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Hurtado del Pozo, C.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905-913.e907. [CrossRef]
- Cheng, X.; Zhang, Y.; Li, Y.; Wu, Q.; Wu, J.; Park, S.K.; Guo, C.; Lu, J. Meta-analysis of 16S rRNA microbial data identified alterations of the gut microbiota in COVID-19 patients during the acute and recovery phases. BMC Microbiol 2022, 22, 274. [CrossRef]
- Wang, M.; Zhang, Y.; Li, C.; Chang, W.; Zhang, L. The relationship between gut microbiota and COVID-19 progression: new insights into immunopathogenesis and treatment. Front Immunol 2023, 14, 1180336. [CrossRef]
- Liu, Q.; Su, Q.; Zhang, F.; Tun, H.M.; Mak, J.W.Y.; Lui, G.C.; Ng, S.S.S.; Ching, J.Y.L.; Li, A.; Lu, W.; et al. Multi-kingdom gut microbiota analyses define COVID-19 severity and post-acute COVID-19 syndrome. Nat Commun 2022, 13, 6806. [CrossRef]
- Yeoh, Y.K.; Zuo, T.; Lui, G.C.; Zhang, F.; Liu, Q.; Li, A.Y.; Chung, A.C.; Cheung, C.P.; Tso, E.Y.; Fung, K.S.; et al. Gut microbiota composition reflects disease severity and dysfunctional immune responses in patients with COVID-19. Gut 2021, 70, 698-706. [CrossRef]
- Haran, J.P.; Bradley, E.; Zeamer, A.L.; Cincotta, L.; Salive, M.C.; Dutta, P.; Mutaawe, S.; Anya, O.; Meza-Segura, M.; Moormann, A.M.; et al. Inflammation-type dysbiosis of the oral microbiome associates with the duration of COVID-19 symptoms and long COVID. JCI Insight 2021, 6. [CrossRef]
- Roth, W.; Zadeh, K.; Vekariya, R.; Ge, Y.; Mohamadzadeh, M. Tryptophan Metabolism and Gut-Brain Homeostasis. Int J Mol Sci 2021, 22. [CrossRef]
- Almulla, A.F.; Supasitthumrong, T.; Tunvirachaisakul, C.; Algon, A.A.A.; Al-Hakeim, H.K.; Maes, M. The tryptophan catabolite or kynurenine pathway in COVID-19 and critical COVID-19: a systematic review and meta-analysis. BMC Infect Dis 2022, 22, 615. [CrossRef]
- Almutairi, M. M.; Sivandzade, F.; Albekairi, T. H.; Alqahtani, F.; Cucullo, L. Neuroinflammation and Its Impact on the Pathogenesis of COVID-19. Frontiers in Medicine 2021, 8. [CrossRef]
- Fitero, A.; Bungau, S. G.; Tit, D. M.; Endres, L.; Khan, S. A.; Bungau, A. F.; Romanul, I.; Vesa, C. M.; Radu, A.-F.; Tarce, A. G.; Bogdan, M. A.; Nechifor, A. C.; Negrut, N. Comorbidities, Associated Diseases, and Risk Assessment in COVID-19—a Systematic Review. International Journal of Clinical Practice 2022, 2022, 1–24. [CrossRef]
- Moghimi, N.; Di Napoli, M.; Biller, J.; Siegler, J. E.; Shekhar, R.; McCullough, L. D.; Harkins, M. S.; Hong, E.; Alaouieh, D. A.; Mansueto, G.; Divani, A. A. The Neurological Manifestations of Post-Acute Sequelae of SARS-CoV-2 Infection. Current Neurology and Neuroscience Reports 2021, 21 (9). [CrossRef]
- Moga, T. D.; Nistor-Cseppento, C. D.; Bungau, S. G.; Tit, D. M.; Sabau, A. M.; Behl, T.; Nechifor, A. C.; Bungau, A. F.; Negrut, N. The Effects of the “Catabolic Crisis” on Patients’ Prolonged Immobility after COVID-19 Infection. Medicina 2022, 58 (6), 828. [CrossRef]
- Nistor-Cseppento, C. D.; Moga, T. D.; Bungau, A. F.; Tit, D. M.; Negrut, N.; Pasca, B.; Bochis, C. F.; Ghitea, T. C.; Jurcau, A.; Purza, A. L.; Uivarosan, D. The Contribution of Diet Therapy and Probiotics in the Treatment of Sarcopenia Induced by Prolonged Immobilization Caused by the COVID-19 Pandemic. Nutrients 2022, 14 (21), 4701. [CrossRef]
Figure 1.
The PRISMA flowchart for the selected studies.
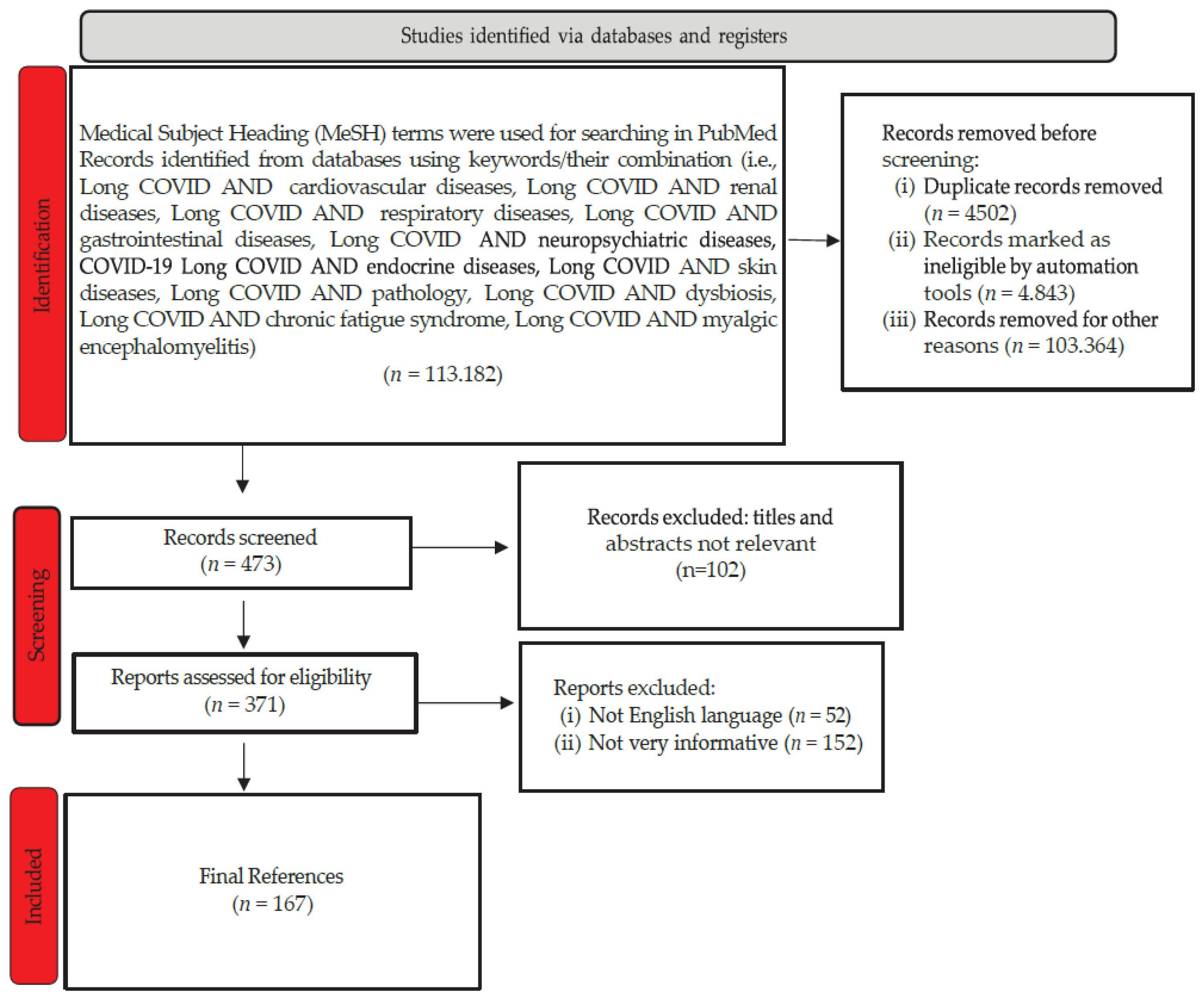
Figure 2.
The causes of the most common pulmonary manifestations in Long COVID.
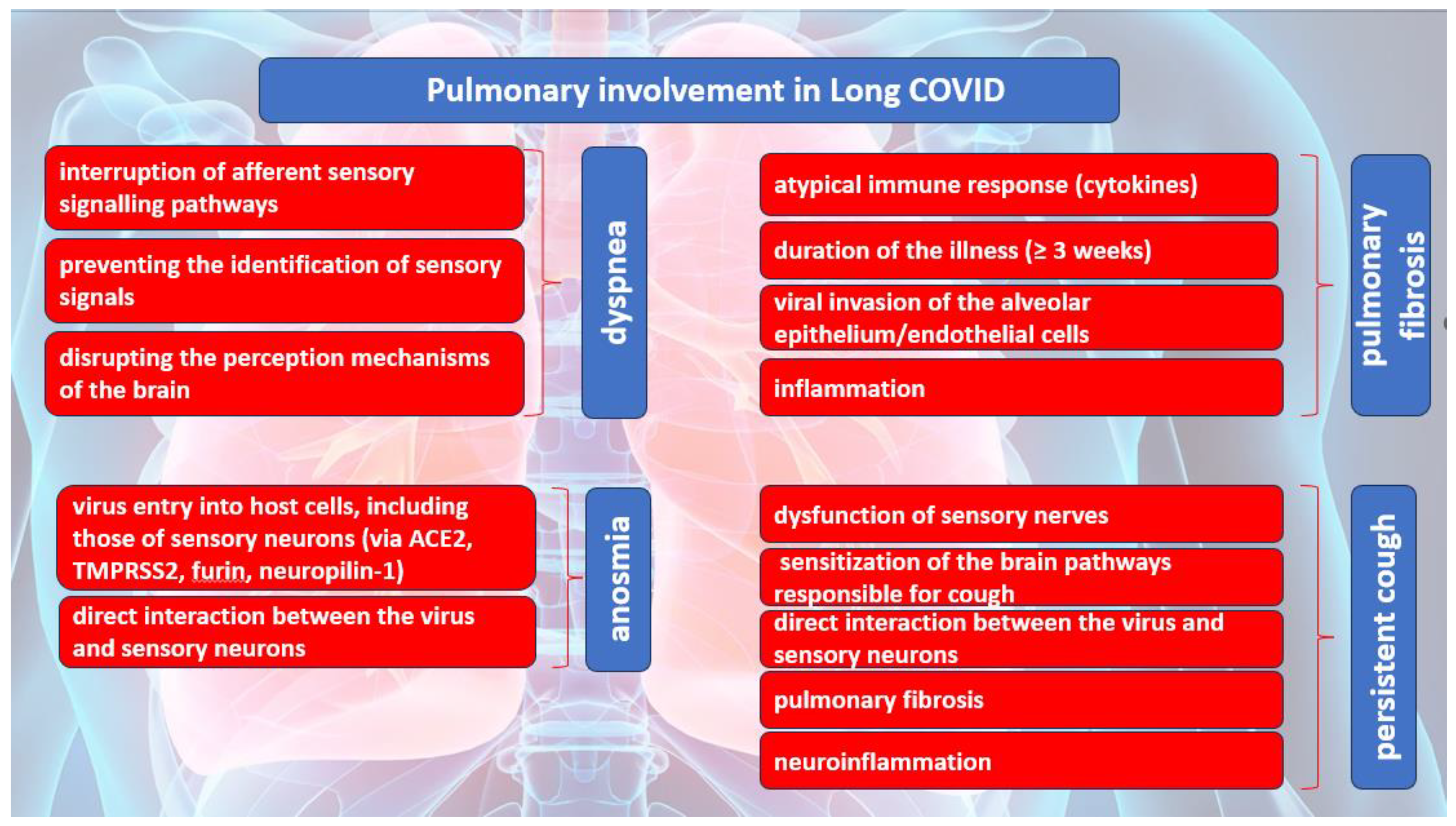
Figure 3.
Possible mechanisms of cardiac damage in COVID-19.
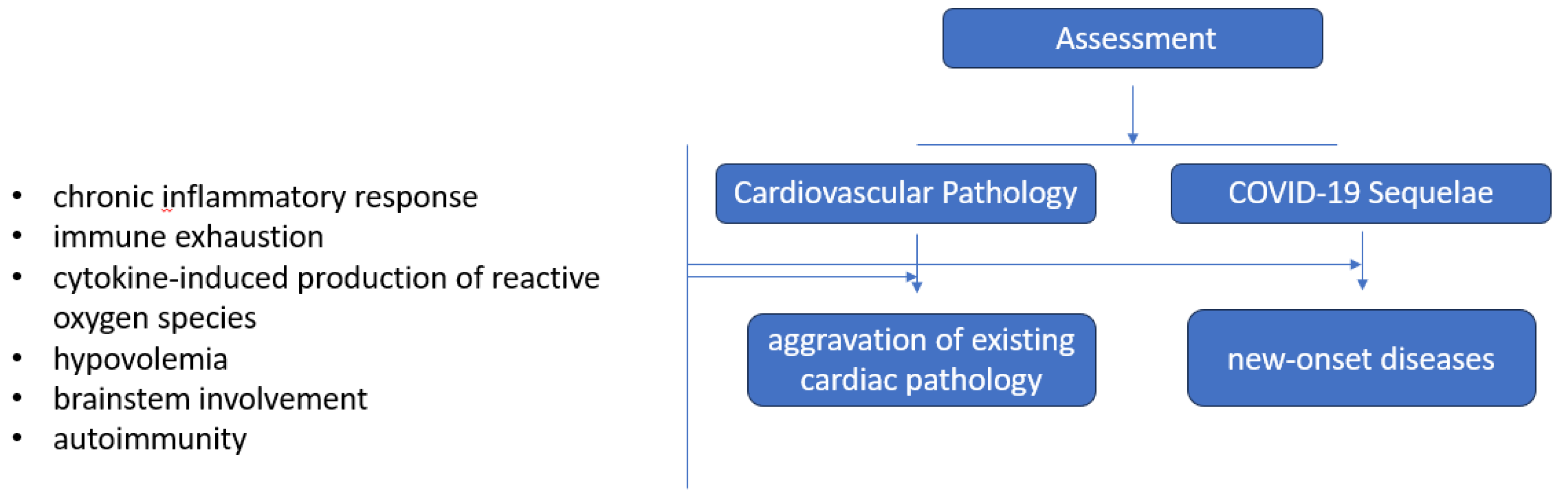
Figure 4.
Dysbiosis in COVID-19. PASC - Post-Acute Sequelae of COVID-19, Pf1 - Pseudomonas phages.
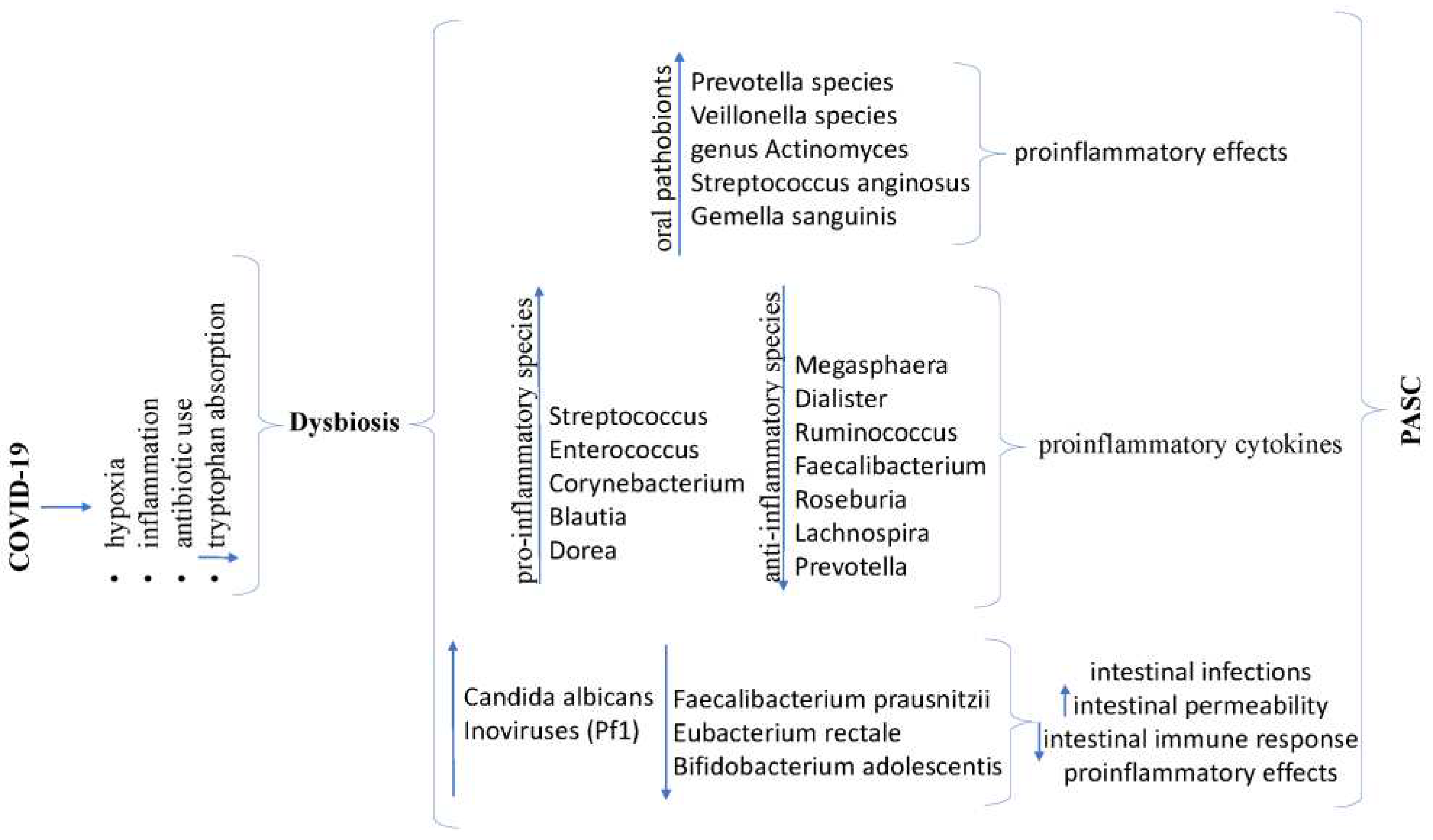
Figure 5.
ME/CFS clinical picture. ADL - activity of daily living, PEM - post-exertional malaise, IBS - irritable bowel syndrome, ME/CFS - Myalgic encephalomyelitis/chronic fatigue syndrome.
Figure 5.
ME/CFS clinical picture. ADL - activity of daily living, PEM - post-exertional malaise, IBS - irritable bowel syndrome, ME/CFS - Myalgic encephalomyelitis/chronic fatigue syndrome.
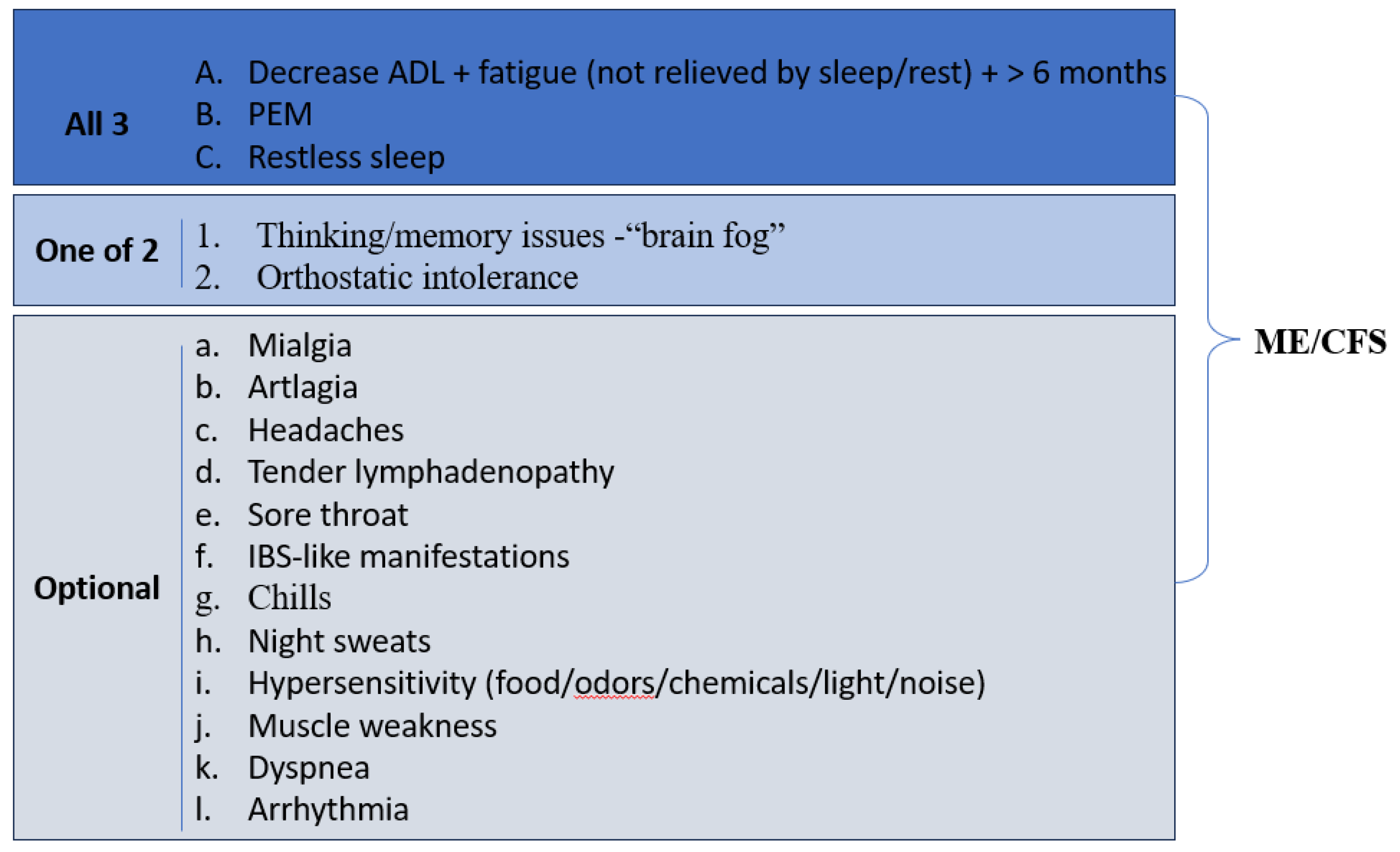

Table 1.
Long-term effects on the endocrine system.
| Gland | Clinical manifestation |
|---|---|
| 1. Pituitary gland | pituitary apoplexy |
| 2. Thyroid gland | euthyroid sick syndrome subacute thyroiditis Graves’ disease postpartum thyroiditis Hashimoto’s disease silent thyroiditis |
| 3. Pancreas | acute pancreatitis necrotizing pancreatitis impaired glucose tolerance insulin resistance diabetes mellitus severe metabolic complications (diabetic ketoacidosis) hyperglycemia abnormalities level of amylase/lipase |
| 4. Adrenal glands | necrosis hypocortisolism cross-reactive antibodies against endogenous ACTH |
| 5. Gonads | hypogonadism erectile dysfunction menstrual disturbances |
ACTH - adrenocorticotropic hormone.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



