
Submitted:
29 December 2023
Posted:
03 January 2024
You are already at the latest version
Abstract
Mammalian fertilization initiates the reprogramming of oocytes and sperm, leading to the formation of a totipotent zygote. The zygotic genome remains transcriptionally silent throughout this intricate process, undergoing the maternal-to-zygotic transition (MZT) and subsequent zygotic genome activation (ZGA). Histone modifications are pivotal in shaping cellular identity and gene expression in many mammals. Recent advances in chromatin analysis enable a detailed exploration of histone modifications during ZGA. This review delves into conserved and unique regulatory strategies, providing essential insights into the dynamic changes in histone modifications and their variants during ZGA in mammals. The objective is to explore recent advancements in leading mechanisms related to histone modifications governing this embryonic development phase in depth. These considerations will be useful for informing future therapeutic approaches that target epigenetic regulation in diverse biological contexts, ranging from regenerative medicine to cancer research. It will also contribute to the extensive areas of evolutionary and developmental biology and possibly lay the foundation for future research and discussion on this seminal topic.
Keywords:
histone modifications
; chromatin landscape
; mammals
; embryonic development
; embryonic stem cells
1. Introduction
Mammalian fertilization commences with the fusion of an oocyte and a single sperm, a critical event in which these two terminally differentiated germ cells must undergo reprogramming to establish a totipotent zygote [1,2,3]. Notably, the zygotic genome remains transcriptionally silent during this reprogramming process [4]. As this intricate transformation unfolds, the reins of developmental control are handed over to the RNAs and proteins that have previously accumulated within the oocyte. This transition, during which maternal products are cleared, is commonly called the maternal-to-zygotic transition (MZT) [5,6]. This transition is meticulously coordinated with zygotic genome activation (ZGA), which signifies the initiation of transcriptional control and gene expression post-fertilization [7,8]. It becomes evident that epigenetic modifications are pivotal in orchestrating this fundamental transformation [9]. Subsequently, ZGA is succeeded by emerging distinct cell identities within embryonic cells, leading to their differentiation into the inner cell mass (ICM) and trophectoderm (TE) stages at the blastocyst stage [10].
Epigenetic modifications occurring in terminally differentiated gametes, including DNA methylation [11,12,13], histone modifications [14,15,16], chromatin accessibility [17,18,19], and 3D chromatin structure [20,21], can be reset to a foundational state following fertilization. This reset process is crucial for achieving totipotency and supporting the subsequent development of a new individual. The precise regulation of zygotic gene transcription is intricately linked to chromatin accessibility. It underscores epigenetic information’s pivotal role in upholding cellular identity and governing gene expression. The nucleosome, serving as the fundamental unit of chromatin, consists of octamers comprising two copies of the core histone proteins H2A, H2B, H3, and H4, collectively contributing to the formation of tightly packed heterochromatin [22,23]. The modulation of chromatin accessibility is mediated through the positioning and configuration of nucleosomes, factors influenced by histone variants, and the post-translational modification of histone N-terminal tails. Several studies have increasingly suggested that histone modifications and variants are pivotal in ensuring precise control over ZGA [24,25,26].
Recent advancements in low-input chromatin analysis technologies have introduced innovative methods to address the challenges associated with the inaccessibility of early-stage embryos [3]. These breakthrough approaches have enabled a comprehensive investigation of the epigenetic remodeling mechanisms at the whole-genome level. In this review, our primary goal is to provide an in-depth exploration of the recent advancements in our understanding of the dynamic changes in histone modifications during the activation of the zygotic genome in mammals. These considerations will be useful for informing future therapeutic approaches that target epigenetic regulation in diverse biological contexts, ranging from regenerative medicine to cancer research.
2. Methods
To compile this review, we conducted a comprehensive search of the Google Scholar and PubMed databases for relevant references from October up to December 2023 using the following terms: “epigenetics”, “histone modifications,” “chromatin landscape,” “mammals embryonic developments,” and “embryonic stem cells”. Also, the cited bibliography of each selected paper was revised to identify any further relevant publications that aligned with our search objectives. The full text of each article included in this review was examined. We finally selected only the articles written in English and Spanish.
3. Comprehensive Overview
ZGA is not a singular event but rather a period during which transcription gradually becomes activated, marked by two distinct transcriptional waves. The first, smaller wave occurs during the early cleavage divisions, while the second, more significant wave coincides with the pause in the first division cycle across diverse species [4,27]. Although the precise timing of these waves and the number of division cycles vary among species [28], the process within a given species is meticulously controlled, exhibiting highly reproducible temporal patterns. Species with rapid development, such as worms [29], frogs [30], fish [31,32], and flies [33,34], complete the MZT and enter gastrulation only hours after fertilization. In contrast, in mammals with a more prolonged development, such as mice [35,36] and humans [37,38], MZT takes one or more days. This disparity is believed to stem from the egg’s nature, suggesting that each egg’s unique requirements dictate different embryogenesis modes [39]. Despite these variations, fundamental processes are conserved, and in all animals, the precise onset of ZGA relies on intricately coordinated mechanisms [4]. In this context, it becomes evident that diverse regulatory mechanisms orchestrate gene expression to establish and define cellular identity and fate.
Among these regulatory mechanisms, the dynamic processes of promoting or removing methylation [24], acetylation [40], phosphorylation [41], SUMOylation [42], and ubiquitination [43,44] marks on histones actively participate in chromatin modification during ZGA. Histone modifications, particularly methylation and acetylation, are crucial in regulating transcription by altering chromatin structure and providing binding platforms for transcription factors and other regulators [45,46].
Histone methylation occurs on specific lysine and arginine residues of these proteins without altering their electrical charges [47]. Depending on the methylated residue, methylation can have diverse effects on gene transcription, either activating or repressing it [48]. In contrast, histone acetylation is closely associated with active gene transcription. This process, highly enriched at the transcription start site (TSS), involves changing the charge of lysine residues from basic to neutral. Histone acetylation has the effect of unpacking chromatin structure, increasing its accessibility for transcription processes [49].
The composition of a unique set of histones and their variants in the nucleosome is instrumental in loosening chromatin structure during ZGA. The subsequent sections delve into the specific roles of some of the most studied methylation and acetylation modifications in histone proteins, shedding light on their contributions during this critical developmental period.
4. H3K4me3 in Zygotic Genome Activation and Gene Expression Regulation
Histone H3 Lysine 4 trimethylation (H3K4me3) is an epigenetic modification associated with packaging DNA in eukaryotic cells, including human cells. It involves adding three methyl groups (trimethylation) to the fourth lysine residue on the histone H3 protein. This modification plays a crucial role in regulating gene expression by altering the accessibility of genes for transcription [50,51]. H3K4me3 is commonly linked to the activation of nearby gene transcription. It achieves this by facilitating chromatin remodeling through interactions with the Nucleosome remodeling factor (NURF) complex [52]. This process enhances the accessibility of DNA for transcription factors, enabling genes to be transcribed and expressed within the cell. Beyond its role in gene expression, H3K4me3 is a key player in genetic regulation related to stem cell potency and lineage determination [53]. It is predominantly found in DNA regions associated with development and the establishment of cell identity. This epigenetic modification contributes to the intricate control of gene expression, ultimately influencing cellular functions and fate.
4.1. H3K4me3 at Promoters and Across Species
H3K4me3 is a canonical activation mark typically present at gene TSSs [54], established at the 2-cell stage, and exhibiting variations among species [4]. In zebrafish, H3K4me3 is detected at many promoters before ZGA, priming genes for activation [31,54,55]. Similarly, H3K4me3 is observed in frogs before genome activation, increasing during gastrulation [56]. In contrast, in Drosophila, only a few promoters exhibit H3K4me3 before the major ZGA wave, implying that early transcription during the minor wave can proceed without this chromatin signature [57]. Nonetheless, a substantial upsurge in H3K4me3 accompanies the major wave of transcription in zebrafish, frogs, and Drosophila. In mice, early embryos exhibit broad domains of H3K4me3, which later become predominantly associated with conventional TSSs. Notably, depletion of the demethylases responsible for this shift leads to developmental arrest and downregulation of numerous ZGA genes [58].
In humans, signals of H3K4me3 can be identified at each stage of oocyte and embryonic development. The signal remains consistently uniform in all zygotes. The levels of H3K4me3 gradually decline from the germinal vesicle to the metaphase II stage and increase from the zygote stage to the four-cell stage, reaching their lowest point at the eight-cell stage [59]. A notable surge is then observed at the blastocyst stage. Notably, it’s important to highlight that H3K4me3 in human oocytes is linked to CpG-rich regions and significantly correlates with CpG density [60]. This association appears to be a species-specific feature, pivotal in marking genomic regions preferentially activated during the MZT.
4.2. Dynamic Reprogramming of H3K4me3
Recent advancements in low-input ChIP-seq methods have allowed researchers to investigate the genome-wide landscape of H3K4me3 during ZGA and the first cell fate decisions in early embryos [61,62]. After fertilization, the H3K4me3 peaks in the paternal genome undergo swift depletion in the zygote. Recent research has unveiled that both the promoter and global H3K4me3 patterns on the paternal allele of the zygote differ significantly from those observed in sperm, indicating extensive reprogramming upon fertilization [63]. Consequently, robust paternal H3K4me3 peaks are reinstated during the major ZGA, particularly from the late two-cell stage onward [64]. Conversely, the maternal genome contains a noncanonical form of H3K4me3 (ncH3K4me3), covering broad domains in both promoters and distal regions [65]. NcH3K4me3 is already established in mature oocytes (MII), unlike in growing oocytes, where H3K4me3 maintains a conventional pattern of narrow peaks at the gene promoters. The noncanonical form of H3K4me3 is deposited during oocyte maturation and remains unaltered by canonical until the major ZGA stage. The initiation and elimination of ncH3K4me3 represent a distinctive trait observed in the genomes of oocytes and early embryos [58,66], and its removal is a crucial process for normal ZGA (Figure 1A). The deposition of the ncH3K4me3 coincides with genome silencing from fully grown oocytes to the early 2-cell stage [53,67]. The underlying maternal factors within the oocyte cytoplasm are pivotal in orchestrating the rapid change in the H3K4me3 landscape. Identifying these key maternal factors holds the potential to shed light on the mechanisms of H3K4me3 remodeling.
Broad H3K4me3 domains in both promoters and distal regions are actively removed by lysine demethylases KDM5A and KDM5B [68,69]. These enzymes allow the removal of ncH3K4me marks and resetting canonical H3K4me3 peaks until the two-cell stage [70]. The absence of Kdm5b results in the extension of the broad ncH3K4me3 domains, disrupting precise lineage differentiation [71]. On the other hand, the overexpression of Kdm5b leads to transcriptome reactivation in mature oocytes, hinting at ncH3K4me3’s potential role in genome-wide silencing. Moreover, transposable elements such as B1/B2/B4 and ERVL exhibit significant overlap with ncH3K4me3 in distal regions [72], suggesting a correlation between ncH3K4me3 and repeat activities [65]. Nevertheless, the prevalence of repeats tends to be lower in MII oocytes and zygotes compared to late-stage embryos, indicating repeat activities alone cannot entirely account for the widespread distal H3K4me3 peaks observed in these cells.
During the human embryo pre-ZGA (4-cell stage), broader marks of H3K4me3 are already evident. 53% of these marks persist and become active in the 8-cell stage, while the remaining 47%, where H3K4me3 is lost, are in the promoters of development and differentiation genes [65]. These genes remain inactive during ZGA. Compared with promoter regions, weaker but widely distributed distal marks of H3K4me3 are evident in pre-ZGA embryos (Figure 1A), indicating de novo deposition of H3K4me3 [17]. These distal marks decrease in the 8-cell stage and are deposited in CpG-rich and hypomethylated regions. Many of these distal marks of H3K4me3 overlap with regulatory elements and exhibit high chromatin accessibility at the 4-cell stage [73].
The interplay between H3K4me3 and DNA methylation is pivotal for understanding genome-wide regulation. Demonstrably, DNA methylation in the maternal genome exhibits an inverse correlation with ncH3K4me3. The evidence indicates that oocytes featuring ncH3K4me3 display approximately 18% CpG methylation, starkly contrasting to the 57% observed in oocytes lacking ncH3K4me3 [12]. A significant correlation is also observed between H3K4me3 and CpG density in four-cell embryos, particularly for promoters with moderate CpG levels, showcasing transcriptional activity in post-ZGA stages [74]. H3K4me3 is recognized for its role in counteracting DNA methylation, along with repressive histone modifications such as Histone H3 Lysine 9 trimethylation (H3K9me3) and Histone H3 Lysine 27 trimethylation (H3K27me3) [53]. This interaction highlights the intricate regulatory mechanisms involved in gene expression. These bivalent domains play a crucial role in governing gene expression by maintaining a balance in the methylation levels of two histone proteins with opposing effects. This equilibrium enables them to remain suppressed, ready for activation in the absence of differentiation signals [75].
Another intriguing aspect of H3K4me3 dynamics involves cooperation with the histone variant H3.3, a crucial player after fertilization essential for embryo development. H3.3 turnover is associated with changes in histone modifications like H3K27me3 and H3K36me3 [76]. The specific correlation between H3.3 and ncH3K4me3 remains unclear, and further investigation is required to determine if H3.3 replacement is responsible for the removal of ncH3K4me3.
While the mechanism of H3K4me3 reprogramming remains an enigma, recent studies suggest that the Histone–lysine N-methyltransferase 2 (KMT2) complex may catalyze the establishment of broad H3K4me3 domains [5,77]. Identifying the transcription factors involved in this precisely controlled process is crucial for further exploration. These major reprogramming events of both promoter and distal H3K4me3 marks offer invaluable insights into the parental-to-zygotic transition in human pre-implantation embryos, providing a deeper understanding of early embryonic development and epigenetic regulation.
5. Unveiling H3K9me3: Orchestrating Epigenetic Landscapes in Development
H3K9me3 emerges as a sentinel, staunchly guarding the integrity of heterochromatin—a densely compacted chromatin state refractory to transcriptional activities [78,79]. This histone modification, characterized by adding three methyl groups to lysine 9 of histone H3, plays a pivotal role in orchestrating cellular processes by imposing a formidable barrier to cell fate transitions.
The functional significance of H3K9me3 lies in its ability to occlude DNA, rendering it inaccessible for transcription factors—a phenomenon intricately linked to the hindrance of the transcriptional machinery [80]. This repressive chromatin environment, governed by H3K9me3, serves as a regulatory mechanism controlling gene expression patterns critical for cellular identity and function.
Recent revelations from investigations into Somatic Cell Nuclear Transfer (SCNT) embryos add a layer of complexity to our understanding of H3K9me3 dynamics [81,82]. SCNT enables animal resurrection and the treatment of human diseases by reprogramming somatic cells into pluripotent states. In this context, a method fraught with challenges in achieving efficient reprogramming, the nuanced role of H3K9me3 comes to the fore [83,84,85]. Unlike fertilized embryos, SCNT embryos manifest a distinctive profile marked by gradual and incomplete demethylation of H3K9me2 and H3K9me3 [86,87]. This aberrant pattern significantly contributes to the failure of ZGA, highlighting the indispensable role of H3K9me3 in orchestrating the intricate balance between pluripotency and cellular differentiation.
5.1. Navigating Reprogramming Challenges and Zygotic Genome Activation
The dynamic features of H3K9me3 during early embryonic development are critical for natural reprogramming post-fertilization. It is remarkable that near-exclusive expression of the H3K9me3 demethylase named Lysine-specific demethylase 4A (KDM4D), is observed in MII oocytes and has been proven to be crucial for maintaining genomic stability and ZGA [88]. However, H3K9me3 emerges as a barrier in SCNT embryos, hindering proper ZGA and compromising development [89]. Studies indicate that H3K9me3 in donor cells obstructs somatic cell reprogramming, leading to abnormal ZGA in 2-cell SCNT embryos [90,91]. At this stage, regions resistant to reprogramming (RRRs) marked by H3K9me3 impede successful reprogramming, prompting strategies to overcome these challenges. For this reason, overexpressing Kdm4d in embryos or knocking down H3K9me3 methyltransferases (Suv39h1/h2) in donor cells proves effective in rescuing ZGA-related gene transcription and enhancing blastocyst development rates in SCNT embryos [82]. It should be noted that indiscriminate depletion of H3K9me3 can interfere with the lineage-specific deposition in SCNT blastocysts, highlighting the complexity of this process. Identifying new critical regulators involved in lineage-specific H3K9me3 deposition will add a new dimension to understanding the intricacies of epigenetic reprogramming challenges.
The regulatory influence of H3K9me3 extends to impact the expression of repeat elements and protein-coding genes in mouse pre-implantation embryos [79]. Early embryonic development triggers extensive demethylation of the mouse genome, leading to a significant fraction of long terminal repeats (LTRs) becoming hypomethylated and actively transcribed, a process crucial for normal development. Post-fertilization, H3K9me3 marks within LTR retrotransposons gradually increase during pre-implantation development (Figure 1B), with the Chromatin assembly factor 1 subunit A (CHAF1A) complex playing a pivotal role in blastocyst formation and cell fate decisions [86]. Properly regulating these LTRs is essential in later developmental stages to maintain genome stability [92]. Studies in mouse embryonic stem cells (mESCs) have suggested that multiple H3K9me3 modifiers, such as Suv39h1/h2 and ERG-associated protein with SET domain (ESET) complexes, are responsible for retrovirus silencing through the establishment of H3K9me3 modifications [93,94].
In the postimplantation stage, a dynamic interplay of H3K9me3 marks re-establishes in promoter regions, orchestrating the repression of lineage-specific genes [86]. Transcription factors such as Pou5f1, Sox12, and Zfp105 contribute to epiblast-specific H3K9me3, while Zbed6, Elf4, and Glis2 are involved in extraembryonic-specific H3K9me3 formation [65]. Mutant studies underscore the significance of precisely regulated H3K9me3 in ensuring proper embryonic development [80]. This multifaceted role of H3K9me3 emphasizes its complex involvement in reprogramming challenges and orchestrating crucial developmental events.
6. H3K27me3 and H3K27ac: Dual Epigenetic Players in Zygotic Genome Activation
In the intricate landscape of epigenetic regulation governing ZGA, two histone modifications stand out as key orchestrators—Histone 3 Lysine 27 trimethylation (H3K27me3) and acetylation (H3K27ac) [4,40]. These dual epigenetic players engage in a dynamic interplay, shaping the transcriptional destiny of critical genes during the pivotal events of early embryonic development.
6.1. Individual Roles of H3K27me3
H3K27me3 is a well-known repressive histone modification associated with gene silencing, primarily within facultative heterochromatin [95]. This modification involves adding three methyl groups to lysine 27 of histone H3 and is catalyzed by Polycomb Repressive Complex 2 (PRC2) [96,97] (Figure 2A). Playing a vital role in maintaining cellular identity by repressing genes associated with alternative cell fates, the dynamics of H3K27me3 during mammalian ZGA are crucial for unraveling the transition from a transcriptionally silent state to the activation of specific gene loci [4,98]. Recent studies have unveiled the intricate interplay between H3K27me3 and other histone modifications [75,99], shedding light on regulatory networks governing early embryonic development.
Before ZGA onset, observed in various organism models, including zebrafish, the genome undergoes intriguing pre-patterning, defined as establishing specific epigenetic marks and chromatin configurations [7,32,100]. In this context, H3K27me3, in conjunction with H3K4me3 and H3K9me3, delineates distinctive chromatin landscapes, suggesting the existence of a primed chromatin landscape before transcription onset [75]. Throughout the MZT, the dynamics of histone modifications unfold, with H3K27me3 gradually increasing and a pronounced elevation in H3K9me3 post-ZGA [31,101,102]. This temporal sequence paints a dynamic picture of chromatin reshaping during critical developmental stages, indicating an instructive role of histone modifications in shaping the developmental transcription program even before active transcription begins.
In mouse embryos, the large-scale loss of H3K27me3 in promoter regions occurs as early as pronuclei (PN)-5 zygotes, persisting through the morula to the ICM and TE stages [103]. This loss involves global erasure in the paternal genome and selective depletion in the maternal genome’s promoter regions [104,105]. Notably, a strong negative correlation between H3K27me3 enrichment and DNA methylation of promoter regions of genes in MII oocytes suggests a complex interplay between these epigenetic marks during early development [13]. On the other hand, the existence of non-canonical H3K27me3 patterns, pervasive and promiscuous in non-promoter regions [106], raises questions about their functional significance and the molecular mechanisms governing these patterns.
Examining H3K27 methylation dynamics during minor and major ZGA in mouse embryos reveals valuable insights [24]. The di-methylation and tri-methylation of H3K27 emerge as pivotal players in transcriptional silencing. H3K27me2, exclusively and robustly expressed in the female pronucleus during PN stages 2–3 and 4–5, significantly increases from 2- to 8-cell stage embryos, reaching its highest point in early blastocysts [107]. In contrast, H3K27me3 exhibits distinct staining patterns, which are prominent in the female pronucleus during PN stages 2–3 and detectable in both male and female pronuclei during PN stages 4–5 [108]. This expression weakens in 2-cell stage embryos, intensifies in 4-cell stage embryos, diminishes in 8-cell stage embryos, and becomes barely detectable in early blastocysts (Figure 2A). The dynamic fluctuations in H3K27me2 and H3K27me3 offer critical insights into their roles throughout the early stages of embryonic development [109], strongly suggesting their involvement in the intricate regulation of gene expression and chromatin accessibility.
SCNT embryos present a unique challenge regarding H3K27me3. While SCNT embryos exhibit strong H3K27me3 signals in all pseudopronuclei at the 1-cell stage, distinct from the asymmetric marks in normally fertilized embryos, the erasure of H3K27me3 from donor somatic cells doesn’t necessarily improve SCNT efficiency [110]. Studies indicate that overexpression of Lysine-specific demethylase 6A (KDM6A), an H3K27me3 demethylase, enhances ZGA-related gene expression in SCNT embryos [111,112], but this enhancement doesn’t translate into improved birth rates or Nuclear-transfer embryonic stem cells (ntESC) establishment efficiency [65]. Conversely, the knockdown of Lysine-specific demethylase 6B (KDM6B), another H3K27me3 demethylase, promotes both ZGA and SCNT efficiency [113]. The imprinting of H3K27me3-dependent genes is aberrant in SCNT embryos [114], emphasizing the crucial role of H3K27me3 in epigenome reprogramming. The complexity deepens with the discovery of maternal H3K27me3-mediated imprinting, transitioning to DNA methylation dependence in extra-embryonic cells after implantation [106,115].
In human embryonic development, H3K27me3 dynamics differ from those in mice. Notably, in human GV oocytes, H3K27me3 is selectively deposited in promoters of developmental genes and partially methylated domains, deviating from the pattern observed in mouse oocytes [116]. This initial divergence sets the stage for further discrepancies during human pre-implantation embryo development, where the resetting of H3K27me3 follows a unique trajectory compared to its counterpart in mice. Specifically, human embryos at ZGA (8-cell stage) manifest a striking absence of H3K27me3 signals [37], indicating a comprehensive erasure of this histone modification on both parental genomes. The correlation between the absence of core components of PRC2 in human embryos and the concurrent loss of H3K27me3 adds an intriguing layer to the understanding of epigenetic regulation [117]. This distinct scenario raises questions about regulatory mechanisms, such as the absence of imprinting regulation like X chromosome inactivation, which plays a pivotal role during early mouse embryogenesis.
Furthermore, the divergence continues as H3K27me3-mediated imprinted genes identified in mouse early embryos, and having human orthologs, seem to undergo de novo deposition of H3K27me3 in humans [95,115]. However, verifying the existence of H3K27me3-controlled imprinting in human early embryos necessitates further investigation. Further analysis reveals asymmetric H3K27me3 patterning between ICM- and TE-specific genes [118,119], hinting at potential preferential deposition between distinct cell types. These intricate dynamics of H3K27me3 provide a nuanced understanding of its roles in regulating gene expression, chromatin accessibility, and imprinting during the complex process of embryonic development.
6.2. Distinctive Functions of H3K27ac in ZGA
In stark contrast with H327m3, H3K27ac is generally associated with active transcription [54]. Catalyzed by histone acetyltransferases (HATs), adding acetyl groups to lysine 27 of histone H3 induces a more open chromatin structure, facilitating the binding of transcription factors and promoting gene expression (Figure 2B). As the zygotic genome undergoes activation, the presence of H3K27ac at specific genomic loci marks regions poised for transcriptional activity, a crucial indicator for the timely and precise expression of genes directing cell fate decisions and embryonic development [120].
Recent studies in mice have explored the existence of broad H3K27ac domains in embryos before the ZGA stage [40]. These investigations revealed that H3K27ac signals cover 17.6% of the mouse genome in zygotes, but this coverage drops below 10% in post-ZGA embryos [58]. Broad domains (>10 kb) of H3K27ac are widely detected in PN5 zygotes, some exceeding 20 kb. In contrast, broad domains > 20 kb become limited in mouse embryos at the 2-cell and 8-cell stages [40].
Comparing H3K27ac-enriched regions between mouse gametes and zygotes underscores a higher inheritance of regions from oocytes compared to those transmitted by spermatozoa. The data highlight the notable resemblance in the distribution of the H3K27ac signal across the entire genome of the mouse zygote to that of the mouse MII oocyte, in contrast to sperm and early post-ZGA embryos [58,63]. Additionally, a significant extension of H3K27ac domains, exceeding 20 kb, is observed in the zygote compared to oocytes and sperm, indicating a reprogramming of the H3K27ac pattern after fertilization.
In the mouse zygote, 68.3% of narrow H3K27ac peaks, with lengths less than 10 kb, originate de novo in regions without H3K27ac enrichment in mouse gametes. Most broad H3K27ac domains (> 10 kb) are established de novo by extending H3K27ac peaks in gametes post-fertilization. Of these domains, 8.0% are exclusively inherited from the oocyte, 4.8% are from the spermatozoa, and 0.5% are from shared broad domains between the oocyte and sperm. An additional comparison between H3K27ac patterns in spermatozoa and the human 2-cell embryo reveals that merely 0.4% of broad H3K27ac domains in the human 2-cell embryo are inherited from spermatozoa. Primarily, these broad H3K27ac domains in human 2-cell embryos do not originate from sperm [40].
Exploring the dynamics of histone H3K27ac in early human embryos unravels intriguing patterns. Utilizing an efficient ChIP-seq method, these studies demonstrate a genomic distribution of broad H3K27ac domains spanning over 10 kb in 2-cell and 4-cell embryos [54], with some domains exceeding 50 kb [40]. These broad domains markedly decreases in 8-cell embryos and subsequent stages, suggesting an association with zygotic genome activation in humans. Notably robust H3K27ac signal intensity is observed in zygotes, 2-cell, and 4-cell embryos before decreasing in 8-cell embryos (Figure 2B), supporting the observation of broad domains in the earliest stages of development.
Furthermore, enriched regions with H3K27ac in early embryos have been characterized, emphasizing their prevalence in intergenic and intronic regions. The H3K27ac signal spreads across the promoters of protein-coding genes at the 2- and 4-cell stages while being depleted at TSSs. However, after the 8-cell stage, the H3K27ac signal concentrates around the TSS [40]. Interestingly, most promoters with H3K27ac at the zygotic genome activation stage exhibit broad H3K27ac domains before this activation [8]. The association between broad H3K27ac domains and partially methylated domains (PMDs) suggests a dynamic interplay between H3K27ac marks and DNA methylation during the early stages of human embryonic development [121]. In particular, promoters covered by broad H3K27ac domains show higher CpG densities than those without a broad H3K27ac signal [122].
The dynamics of typical H3K27ac peaks during early embryogenesis reveal significant changes at the 8-cell stage. Broad H3K27ac domains transition to typical H3K27ac peaks in human embryos during this phase. A detailed analysis unveils that approximately 80% of H3K27ac peaks are in distal regions, i.e., non-promoter regions, at the 8-cell stage. Nevertheless, over 70% of human promoters exhibit H3K27ac enrichment at this stage [40]. Studies also confirm high expression in genes whose promoters are marked with H3K27ac and/or combined with the H3K4me3 modification [54,123]. Throughout early development, from the 8-cell stage to the 6-week stage, H3K27ac peaks in promoter regions exhibit greater stability than distal H3K27ac peaks, and the number of promoters marked with H3K4me3 undergoes minimal changes. Despite this stability, many stage-specific genes are expressed during the morula, blastocyst, and 6-week embryos, and this expression is correlated with stage-specific modifications of H3K27ac in promoters [40]. These findings reinforce the notion that establishing H3K27ac modification in promoters plays a crucial role in the temporal regulation of gene expression during human embryogenesis.
7. Dynamics of Other Histone Modifications in Early Embryonic Development
7.1. H3K36me3 Dynamics Unveiled: Allelic Reprogramming in Early Mouse Embryos
The methylation of H3K36 (H3K36me), a highly conserved process from yeast to humans, is intricately associated with transcribed regions, playing pivotal roles in transcription fidelity [124,125], RNA splicing [126,127], and DNA repair [128,129]. In mammals, SET domain containing 2 (SETD2) emerges as the primary methyltransferase responsible for catalyzing H3K36 trimethylation (H3K36me3) in vivo [130,131,132]. SETD2 facilitates the interaction with RNA polymerase II, orchestrating the coupling of H3K36me3 with transcription elongation [133]. Unlike H3K4me3, H3K36me3 exhibits a positive correlation with DNA methylation, recruiting DNA methyltransferase 3A and 3B (DNMT3A/B) and maintaining this association in various mammalian cells [134,135].
Several studies underscore the critical roles of SETD2 levels and H3K36me3 in establishing and safeguarding the maternal DNA methylome during oogenesis and early embryo development. In mice, SETD2-depleted oocytes experience a significant loss of H3K36me3, leading to invasions of H3K4me3 and H3K27me3 into regions formerly marked by H3K36me3 [65]. Additionally, SETD2-deficient oocytes result in an aberrant DNA methylome characterized by the loss of maternal imprints and anomalous deposition of H3K4me3 instead of DNA methylation, particularly at imprinted control regions (ICRs) [136].
Furthermore, the scarcity of SETD2 has been demonstrated to induce defects in oocyte maturation and embryonic lethality. Mice deficient in SETD2 do not survive beyond embryonic day (E) 10.5 - E11.5 [137]. Notably, the overexpression of H3.3K36M (lysine to methionine mutant) in mouse metaphase II (MII) oocytes results in reduced H3K36me3 levels and compromised embryo viability [138].
The study of allelic reprogramming of H3K36me3 post-fertilization in early mouse embryos has provided valuable insights. Given the transient inheritance of maternal marks H3K4me3 and H3K27me3, influencing processes like ZGA [53], imprinted X chromosome inactivation [139], and gene expression [140], the inquiry arises regarding the inheritance of H3K36me3 in early embryos and its potential interaction with other epigenetic marks. Recent immunofluorescence studies have uncovered the presence of H3K36me3 at all stages except in paternal pronuclei shortly after fertilization [136]. Analyses of H3K36me3 through ChIP-seq in sperm and discrimination of parental strains via single-nucleotide polymorphisms (SNPs) revealed a significant allelic imbalance in 1-cell embryos, with a notably higher number of maternal reads than paternal reads [141,142]. Surprisingly, H3K36me3 inherited from oocytes appears present in 1-cell embryos but diminishes considerably by the late 2-cell stage and is lost by the 8-cell stage [143]. Conversely, most, if not all, H3K36me3 peaks in sperm are lost in zygotes [73]. The temporal transition of H3K36me3 from parental to zygotic patterns aligns closely with ZGA, suggesting allelic reprogramming during early embryo development [144]. Notably, maternal H3K27me3 persists beyond ZGA in the blastocyst, potentially influencing the deposition of zygotic H3K36me3. Genes with paternal-specific H3K36me3, as opposed to maternal-specific H3K36me3, exhibit a preference for reciprocal allelic H3K27me3. This group includes a significant number of H3K27me3-controlled imprinted genes, indicating the occurrence of H3K36me3 in early embryos and its role in marking allele-specific gene expression [65].
7.2. Histone H3R26me2: Pivotal in Cell Fate Determination in Embryos
After ZGA, embryos undergo multiple cell divisions before the first segregation into cell lineages, giving rise to TE and ICM cells. TE lineage, marked by Cdx2 and Gata3 expression, plays a role in placental development [145,146,147]. In contrast, cells in the ICM, recognized by pluripotent factors, undergo differentiation into epiblast and primitive endoderm, giving rise to all embryonic tissues and certain extraembryonic membranes [148,149,150]. During differentiation, the HIPPO pathway regulates the TE lineage [151,152], and epigenetic modifications consolidate the ICM lineage [153].
Previously explored studies have highlighted the role of Histone 3 Arginine 26 dimethylation (H3R26me2) as a recently identified epigenetic mechanism [154]. This process, predominantly governed by Coactivator-associated arginine methyltransferase 1 (CARM1), has been reported to influence pluripotency in both mouse embryos and mESCs [155]. The overexpression of CARM1 in embryonic stem cells (ESC) and early mouse embryos’ blastomeres drives an increase in H3R26me2 at pluripotent gene promoters [156,157,158], such as Oct4/Pou5f1 [159], Nanog [160], and Sox2 [161,162]. This epigenetic mark linked to gene activation determines the elevated expression of these genes, closely related to cell fate determination and the pluripotent capacity of these cells.
The asymmetrical distribution of H3R26me2 in blastomeres is detected in embryos as early as the four-cell stage [157]. Blastomeres with higher levels of CARM1 and H3R26me2 contribute more significantly to the formation of the ICM [163]. Furthermore, higher levels of H3R26me2 enhance the expression of pluripotent genes and facilitate Sox2 binding to its targets, contributing to ICM specification. Additionally, CARM1 overexpression can increase the frequency of asymmetric cell divisions, leading cells to adopt a more internal position in the embryo [163]. Alongside CARM1, another chromatin regulator named PR domain-containing 14 (PRDM14) is also asymmetrically expressed in four-cell embryos, thus modulating the level of H3R26me2 to favor contribution to the ICM [164,165,166].
Early cell fate determination in mouse embryos has recently been suggested to commence at the late 2-cell stage [167]. At this point, a long non-coding RNA (lncRNA) known as LincGET is transiently and asymmetrically expressed in the nucleus, extending from the 2-cell to the 4-cell stage [159]. LincGET interacts with CARM1, accumulating it in nuclear granules that require the presence of the Nuclear Paraspeckle Assembly Transcript 1 (NEAT1) and its partner Nuclear RNA-binding protein 54 kDa (P54NRB) [167,168]. It results in a significant increase in the H3R26me2 level, activation of ICM-specific gene expression, positive regulation of transposons, and an increase in global chromatin accessibility. It is important to note that introducing LincGET into one of the blastomeres of 2-cell embryos can potentially redirect their differentiation toward the ICM [159].
The lncRNA named Neat1 is also required to mark H3R36me2, which is CARM1-dependent and crucial for ICM specification [169]. NEAT1 shows an asymmetrical expression among blastomeres in 4-cell embryos and recruits CARM1 in paraspeckle nuclear foci [144]. Disruption of NEAT1 results in a decrease in H3R26me2, an increase in Cdx2 expression, and a biased specification toward the TE lineage [157,170].
In summary, the asymmetry in the distribution of H3R26me2 emerges as one of the early signals guiding lineage specification. The variability in H3R26me2 is carefully regulated by the asymmetrical expression of CARM1, PRDM14, LincGET, and NEAT1 in the early blastomeres. Although the cause of the initial skewed expression of CARM1/PRDM14/LincGET/NEAT1 in 2-cell and 4-cell embryos is not fully understood, these findings provide a clearer understanding of the molecular events orchestrating cell fate determination in the early stages of embryonic development.
8. Functional Diversity of Histone Variants in the Activation of the Zygotic Genome
The histone variants exhibit distinct positioning and dynamics within cells, assembling into nucleosomes through different molecular chaperones. They interact with various chromatin remodeling complexes, replacing canonical histones, or undergoing substitution with other variants during cellular development and differentiation [171,172,173]. Structural differences introduced by a central histone variant can impact histone interactions, transforming nucleosome stability and chromatin opening or compaction [174]. Among these, histone H2A variants are recognized for coordinating early embryonic genome chromatin remodeling by replacing conventional H2A in a subset of nucleosomes [175,176].
The macroH2A histone variant is a central histone related to canonical H2A, possessing a long non-histone domain (NHD) at the C-terminal [177]. Previous studies have implicated macroH2A in epigenetic gene silencing events, including X chromosome inactivation [178,179]. However, macroH2A1 is expressed at similar levels in both male and female cells [180], suggesting its function extends beyond X chromosome inactivation. Further analyses have revealed that macroH2A can inhibit transcription by negatively regulating the binding of the NF-kappaB transcription factor and preventing SWI/SNF chromatin remodeling) [181,182,183]. MacroH2A is localized in the chromatin of germ vesicles in oocytes, associated with mature oocyte chromosomes, and abundant in the first polar body. After fertilization, a transient asymmetry is observed, with macroH2A preferentially associating with the female pronucleus. This maternal reserve of macroH2A is lost in late-stage 2 pronuclei (2PN), resulting in normal embryos at 2, 4, and 8-cell stages lacking macroH2A, except in residual polar bodies [181,184]. As macroH2A is a repressive H2A variant and should be progressively lost as the embryo becomes transcriptionally active, it is not detected in major ZGA [7,102]. MacroH2A protein expression reappears in embryos after the 8-cell stage and persists in morula and blastocysts, where nuclear macroH2A is present in both trophectodermal cells and the inner cell mass [181]. This finding suggests that embryos complete their initial three or four-cell cycles without macroH2A. These findings imply significant modifications in macroH2A variant content in the chromatin of developing embryos before implantation.
Another identified H2A variant is H2A.X, which plays a role in DNA repair [185,186]. In mammals, H2A.X shares up to 95% sequence similarities with canonical H2A and is highly conserved across species [187]. H2A.X contains a unique SQ motif at its C-terminus and is invariant in sequence and position relative to its C-terminus across species [188,189]. Recent studies have demonstrated that H2A.X regulates Cdx2 and its specific extraembryonic genes, determining the developmental potential of stem cells [190,191], and indicating regulatory functions of H2A.X in the transcriptional network related to cellular fate control.
H2A.X is the main H2A variant deposited on chromatin in cleavage-stage embryos in mice and humans with ZGA activity. This histone variant is specifically expressed in 1-2 cell stage mouse embryos [192] and shows an enrichment trend in human embryos at the 4-8 cell cleavage stage [174]. The proper amount of H2A.X ensures that genes involved in ZGA are at relatively normal expression levels. A recent study in ESCs identified that H2A.X inhibits the expression of genes mediated by Dux [193,194], a factor directly involved in ZGA stimulation, by binding to its locus, confirming that the dynamic incorporation of this histone variant finely modulates developmental progression.
The H2A.Z histone variant from yeast to mammals constitutes approximately 4 to 10% of total H2A histones [195]. Its multifaceted role includes crucial functions in transcriptional control [196], DNA repair [197], heterochromatin formation [198,199], and genetic stability [200,201]. Genomically, H2A.Z integrates into chromatin, playing an essential regulatory role in transcription. Despite its functional relevance, studies on H2A.Z in early developmental stages have been constrained by the lethality accompanying its mutation in various organisms [202,203,204]. However, multiple studies concur that it plays a crucial role as a regulator in the activation and transcription of genes during ZGA [195,205].
Two isoforms of H2A.Z have been identified, differing in only three amino acids. These variants, H2A.Z.1 and H2A.Z.2, are encoded by separate genes, H2AFZ and H2AFV, respectively [201]. Despite the subtle difference of three amino acids between these isoforms, they perform specialized functions related to their interactions. While H2A.Z.2 preferentially associates with H3K4me3, it has been confirmed that H2A.Z.1 interacts more efficiently with Bromodomain-containing protein 2 (BRD2) [196,206,207].
The deposition of H2A.Z on the TSS of the zygotic genome is facilitated by an ATPase chaperone known as Domino in Drosophila [208]. This deposition precedes ZGA and RNA polymerase II (Pol II) binding to chromatin, indicating its contribution to preparing genes for transcriptional activation [207]. Although the mammalian orthologs of Domino, Snf2 Related CREBBP Activator Protein (SRCAP) and E1A Binding Protein P400 (EP400) [209], have not been fully explored during early embryogenesis, previous studies have shown that EP400 is essential for the identity of ESCs [210], and EP400 mutant mice are lethal when homozygous [211]. This observation underscores the need for further research to better understand the dynamics of SRCAP and EP400 in the context of early developmental regulation.
During minor ZGA, H2A.Z is symmetrically expressed in male and female pronuclei in embryos at PN 2–3 and embryos at PN 4–5. However, variant expression slightly decreases in embryos at the 2-cell stage, reaching a higher level in embryos at the 4-cell stage. In embryos at the 8-cell stage and early blastocyst, the expression level of H2A.Z decreases, suggesting a temporal regulation of its function during later stages of embryonic development [212]. It has been verified that H2A.Z deposited by Domino in Drosophila and its mammalian orthologs, provided by the mother, are necessary for the transcriptional activation of thousands of genes at the onset of ZGA; the lack of expression of these, including regulators of this process, leads to embryonic death [195]. In mESCs, H2A.Z in chromatin is linked to H3K4me3, present in both active and bivalent promoters, but not in repressed genes [205]. This correlation pattern is maintained in human embryonic stem cells (hESCs) [213]. These findings expand our current understanding of ZGA regulation, emphasizing the importance of chromatin in this process. Given the evolutionary conservation of H2A.Z and the fundamental principles of ZGA, it is speculated that histone variants could play similar roles during mammalian embryogenesis. Future research in this direction will illuminate the complex process by which chromatin states and transcription factors jointly orchestrate zygotic genome activation.
Finally, we encounter the H3.3 variant, which has sparked considerable interest due to its distinctive role in remodeling the male and female genomes during fertilization and the early stages of embryonic development. This histone plays a vital function in maintaining genomic integrity in mammals [214]. Encoded by two different genes, H3f3a and H3f3b, H3.3 generates an identical protein product [215]. Its constitutive expression in cells and its incorporation into chromatin independently of DNA synthesis underscore its relevance in the biological context. In the case of mice, ZGA is associated with the extensive incorporation of the H3.3 variant into parental genomes [216]. Both in sperm and oocytes, H3.3 is enriched, with mature oocytes being particularly rich in H3 mRNA, leading to the formation of maternal H3.3 after activation. This histone also plays a crucial role in forming the male pronucleus during fertilization [217].
A detailed study using a mouse model marked with H3.3B-HA reveals the asynchronous activation of paternal and maternal genomes [76]. The early deposition of paternal H3.3 in the zygotic genome contrasts with the delay in maternal deposition until the four-cell stage. Maternally stored H3.3 in oocytes is essential for cleavage and the lesser ZGA [218]. Its global deposition in the paternal genome during the transition from protamine to histone [219], with a preferential enrichment in CpG-rich TSS, highlights its critical role. Depletion of maternal H3.3 can result in the loss of H3K27ac, leading to failure in the lesser ZGA and early embryo arrest [220]. Mechanically, the deposition of maternal H3.3 in the sperm genome removes repressive histone modifications, promotes the establishment of active modifications, and, in turn, enables the initiation of the lesser ZGA of the paternal genome. In summary, current findings emphasize that paternal chromatin remodeling mediated by H3.3 is essential for developing pre-implantation embryos and activating the paternal genome during embryogenesis, providing valuable insights into fundamental biological processes.
9. Future Perspectives and Applications of ZGA Histone Modifications in Stem Cell Research
In this review, we delved into the dynamics of histone modifications during the ZGA in mammals, unveiling an intricate network of molecular events that govern this pivotal process. The findings suggest potential therapeutic approaches for epigenetic regulation in biological contexts, particularly in regenerative medicine and cancer research.
The versatility of ESCs and their unique ability to regenerate any differentiated cell place them at the forefront as a pluripotent cell type of significant interest in research and medicine. This interest extends beyond conventional ethical and cultural considerations. The prospect of applying specific epigenetic marks to these cells generates considerable expectations for their use as revolutionary therapeutic alternatives, opening the possibility of addressing diverse diseases, including neurodegenerative conditions, osteoarticular disorders, and cancer.
In the field of neuroscience, disruptions in histone methylation have been implicated in various processes, including inflammation [221,222], quiescence of neural stem cells [223,224], and neurodegenerative and psychiatric disorders [225,226]. Recent research has established connections between epigenetic regulation and brain aging [227,228]. An increase in H3K4me3 has been associated with the re-entry of mature brain cells into the cell cycle [229], emphasizing the importance of maintaining a proper balance of this histone for neuronal function. The plasticity of histone methylation patterns provides a unique window for interventions with specific cellular therapies. While these findings are promising, they are not without ethical considerations, demanding a comprehensive understanding of the molecular complexities of the involved organ. This scenario continues to pave the way for deeper studies and critical reflections at the intersection of molecular biology and ethics.
Understanding the significance of histone modifications in modulating cellular changes has significantly advanced through reprogramming somatic cells into induced pluripotent stem cells (iPSCs) [230]. Adequate reprogramming of a somatic cell into an iPSC requires not only the introduction of stemness genes but also the reorganization of its chromatin, modifying specific epigenetic marks that enable pluripotency [231]. Histone modifications in neurodifferentiation with iPSCs have become a focus of various studies to improve therapy effectiveness [232,233]. In these pluripotent stem cells, genes associated with differentiation exhibit H3K27me3 as a repressive mark, while genes related to cell renewal display H3K4me3 as an activating mark. Recent developments in epigenetics have enabled the exploration of drugs that target these therapeutic pathways [234,235], facilitating the effective reprogramming of somatic cells into iPSCs.
The scientific community underscores the relevance of epigenetic regulations in bone development and repair within the realm of regenerative medicine [236]. A crucial regulatory sequence in the epigenetic control of the osteogenic commitment of Wharton’s jelly-derived mesenchymal stem cells (MSCs) has been identified [237]. These findings reveal that those repressive marks on the SP7 gene promoter result from a weak transcriptional response during osteoblast differentiation. Additionally, the enrichment of the H3K4me1 mark on the P1 promoter of the RUNX2 gene is associated with the repression of key regulatory genes. Conversely, the presence of activating histone marks in these regions is linked to the induction of osteogenic differentiation of Wharton’s jelly-derived MSCs. In this context, the epigenome emerges as a biomarker to assess the efficacy and safety of stem cell differentiation. The natural stimulation of the adult stem cell niche after bone tissue injury implies significant potential for clinical translation without requiring cell transplants. Pluripotency and cell differentiation can benefit from manipulating activating marks such as H3K27ac and H3K36me3, which could be essential for bone repair by stimulating osteogenic activity.
Histone modifications also provide an opportunity to address cancer through therapeutic strategies regulating the genetics of cancer cells [238,239]. Various studies have highlighted the central role of H3K9me3 in gene regulation, especially in hematopoietic disorders such as acute myeloid leukemia (AML) [240,241]. Other epigenetic changes, such as H3K27me3 and H3K4me3, are recognized as important markers in tumor progression [242,243] and cell differentiation [244], respectively, and are extensively studied in oncology. Additional research on post-translational histone modifications in cancer cell lines has identified specific patterns of modifications in histones H3 and H4 and their variants [245]. These studies describe how inhibiting Enhancer of zeste homolog 2 (EZH2), a key enzyme in H3K27 methylation, significantly reduces tumor burden in breast cancer cell lines in mice [246,247]. Similarly, the presence of H3K9me3 in double-strand breaks activates the acetyltransferase activity of Tip60 [248], which is essential for efficient DNA repair, suggesting that abnormal histone methylation patterns could influence DNA repair efficiency and contribute to cancer.
Our review provides a detailed insight into the dynamics of histone modifications during ZGA in mammals, highlighting various promising avenues as targets for application in the biological context of specific diseases, as outlined in this section. This knowledge could be crucial for developing protocols to differentiate ESCs and iPSCs with specific histone modifications, thus opening the possibility of applying these cellular therapies in humans to address medical conditions. Additionally, the mentioned perspectives have therapeutic implications and suggest intriguing paths for future research in the field of reproductive medicine. Another promising research direction is exploring specific transcription factors and cofactors orchestrating the reprogramming of other histones during ZGA. This approach would shed light on the molecular actors driving this critical process and could have applications in both fundamental understanding of biology and future therapeutic developments. Furthermore, conducting a comparative analysis of the dynamics of studied histones across different species could provide valuable evolutionary insights into the role of these modifications in early embryonic development, offering a broader understanding of comparative biology. Finally, understanding the specific mechanisms of histone reprogramming during early embryonic development in embryos produced by SCNT not only presents itself as a fundamental model for basic research but also paves the way for innovative approaches in animal cloning, regenerative medicine, and treating human diseases.
10. Conclusions
The in-depth analysis of ZGA accentuates the intricacies of this essential process in embryonic development. This review emphasizes that, regardless of variations in embryonic development, ZGA shares highly conserved control mechanisms in mammals. These finely orchestrated mechanisms establish gene expression, shaping cellular identity and fate. The pivotal role of histone modifications, including the methylation and acetylation of specific lysine and arginine residues on H3 and their variants, significantly impacts the activation or repression of gene transcription during ZGA. A comprehensive understanding of the dynamics of histone modifications during this period unveils novel perspectives for therapeutic applications. The potential to manipulate ESC and iPSC with specific histone modifications emerges as a promising approach for developing targeted cellular therapies.
Author Contributions
Conceptualization, F.S.L. and R.A.R.; writing—review and editing, F.S.L., R.A.R., N.I.B., Y.M.C.A., I.C.H., C.A.V.V. and A.A.B.H.; review and visualization, Y.V.C. All authors have read and approved the final version and accepted their responsibility in all aspects of the manuscript.
Funding
This research received no external financial.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Figures have been created with Biorender.com, accessed date 25 December 2023.
Conflicts of Interest
The authors express that there are no conflicts of interest.
Abbreviations
| AML | Acute myeloid leukemia |
| BRD2 | Bromodomain-containing protein 2 |
| CARM1 | Coactivator-associated arginine methyltransferase 1 |
| CHAF1A | Chromatin assembly factor 1 subunit A |
| ESCs | Embryonic stem cells |
| ESET | ERG-associated protein with SET domain |
| EZH2 | Enhancer of zeste homolog 2 |
| HATs | Histone acetyltransferases |
| hESCs | Human embryonic stem cells |
| ICM | Inner cell mass |
| ICRs | Imprinted control regions |
| iPSCs | Induced pluripotent stem cells |
| KDM4D | Lysine-specific demethylase 4A |
| KDM6A | Lysine-specific demethylase 6A |
| KDM6B | Lysine-specific demethylase 6B |
| KMT2 | Histone–lysine N-methyltransferase 2 |
| lncRNA | Non-coding RNA |
| LTRs | Long terminal repeats |
| mESCs | Mouse embryonic stem cells |
| MSCs | Mesenchymal stem cells |
| MZT | Maternal-to-zygotic transition |
| NEAT1 | Nuclear Paraspeckle Assembly Transcript 1 |
| NHD | Non-histone domain |
| ntESC | Nuclear-transfer embryonic stem cells |
| NURF | Nucleosome remodeling factor |
| P54NRB | Nuclear RNA-binding protein 54 kDa |
| PMDs | Partially methylated domains |
| PN | Pronuclei |
| PRC2 | Polycomb Repressive Complex 2 |
| PRDM14 | PR domain-containing 14 |
| RRRs | Regions resistant to reprogramming |
| SCNT | Somatic Cell Nuclear Transfer |
| SETD2 | SET domain containing 2 |
| SNPs | Single-nucleotide polymorphisms |
| TE | Trophectoderm |
| TSS | Transcription start site |
| ZGA | Zygotic genome activation |
References
- Siu, K. K.; Serrão, V. H. B.; Ziyyat, A.; Lee, J. E. The Cell Biology of Fertilization: Gamete Attachment and Fusion. Journal of Cell Biology 2021. [CrossRef]
- Bhakta, H. H.; Refai, F. H.; Avella, M. A. The Molecular Mechanisms Mediating Mammalian Fertilization. Development (Cambridge) 2019. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Xie, W. Epigenome in Early Mammalian Development: Inheritance, Reprogramming and Establishment. Trends in Cell Biology 2018. [CrossRef]
- Schulz, K. N.; Harrison, M. M. Mechanisms Regulating Zygotic Genome Activation. Nature Reviews Genetics 2019. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, L.; Guo, F.; Dai, X.; Zhang, X. Epigenetic Reprogramming during the Maternal-to-Zygotic Transition. MedComm 2023. [Google Scholar] [CrossRef]
- Vastenhouw, N. L.; Cao, W. X.; Lipshitz, H. D. The Maternal-to-Zygotic Transition Revisited. Development (Cambridge, England) 2019. [Google Scholar] [CrossRef] [PubMed]
- Lee, M. T.; Bonneau, A. R.; Giraldez, A. J. Zygotic Genome Activation during the Maternal-to-Zygotic Transition. Annual review of cell and developmental biology 2014. [CrossRef]
- Wu, E.; Vastenhouw, N. L. From Mother to Embryo: A Molecular Perspective on Zygotic Genome Activation. In Current Topics in Developmental Biology; 2020; Vol. 140. [CrossRef]
- Vallot, A.; Tachibana, K. The Emergence of Genome Architecture and Zygotic Genome Activation. Current Opinion in Cell Biology 2020. [CrossRef]
- Hackett, J. A.; Azim Surani, M. Regulatory Principles of Pluripotency: From the Ground State Up. Cell Stem Cell 2014. [Google Scholar] [CrossRef]
- Zhou, S.; Li, X.; Liu, Q.; Zhao, Y.; Jiang, W.; Wu, A.; Zhou, D. X. DNA Demethylases Remodel DNA Methylation in Rice Gametes and Zygote and Are Required for Reproduction. Mol Plant 2021, 14. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, J.; Duan, J.; Gao, X.; Zhu, W.; Lu, X.; Yang, L.; Zhang, J.; Li, G.; Ci, W.; et al. Programming and Inheritance of Parental DNA Methylomes in Mammals. Cell 2014, 157. [Google Scholar] [CrossRef]
- Guo, H.; Zhu, P.; Yan, L.; Li, R.; Hu, B.; Lian, Y.; Yan, J.; Ren, X.; Lin, S.; Li, J.; et al. The DNA Methylation Landscape of Human Early Embryos. Nature 2014, 511. [Google Scholar] [CrossRef]
- Robert, V. J. Histone Modifications in Germline Development and Maintenance. In Perinatal and Developmental Epigenetics: Volume 32 in Translational Epigenetics; 2022. [CrossRef]
- Hales, B. F.; Grenier, L.; Lalancette, C.; Robaire, B. Epigenetic Programming: From Gametes to Blastocyst. Birth Defects Research Part A - Clinical and Molecular Teratology, 2011. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Q.; Tang, F.; Yan, L.; Qiao, J. Epigenetic Regulation and Risk Factors during the Development of Human Gametes and Early Embryos. Annual Review of Genomics and Human Genetics 2019. [Google Scholar] [CrossRef]
- Wu, J.; Xu, J.; Liu, B.; Yao, G.; Wang, P.; Lin, Z.; Huang, B.; Wang, X.; Li, T.; Shi, S.; et al. Chromatin Analysis in Human Early Development Reveals Epigenetic Transition during ZGA. Nature 2018, 557. [Google Scholar] [CrossRef]
- Gorkin, D. U.; Barozzi, I.; Zhao, Y.; Zhang, Y.; Huang, H.; Lee, A. Y.; Li, B.; Chiou, J.; Wildberg, A.; Ding, B.; et al. An Atlas of Dynamic Chromatin Landscapes in Mouse Fetal Development. Nature 2020, 583. [Google Scholar] [CrossRef]
- Gao, L.; Wu, K.; Liu, Z.; Yao, X.; Yuan, S.; Tao, W.; Yi, L.; Yu, G.; Hou, Z.; Fan, D.; et al. Chromatin Accessibility Landscape in Human Early Embryos and Its Association with Evolution. Cell 2018, 173. [Google Scholar] [CrossRef] [PubMed]
- Bonev, B.; Cavalli, G. Organization and Function of the 3D Genome. Nature Reviews Genetics 2016. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Xu, Y.; Chen, X.; Feng, S.; Liu, Z.; Sun, Y.; Yao, X.; Li, F.; Zhu, W.; Gao, L.; et al. 3D Chromatin Structures of Mature Gametes and Structural Reprogramming during Mammalian Embryogenesis. Cell 2017, 170. [Google Scholar] [CrossRef] [PubMed]
- Koyama, M.; Kurumizaka, H. Structural Diversity of the Nucleosome. Journal of Biochemistry 2018. [CrossRef] [PubMed]
- Zhou, K.; Gaullier, G.; Luger, K. Nucleosome Structure and Dynamics Are Coming of Age. Nature Structural and Molecular Biology 2019. [CrossRef] [PubMed]
- Deng, M.; Chen, B.; Liu, Z.; Cai, Y.; Wan, Y.; Zhou, J.; Wang, F. Exchanges of Histone Methylation and Variants during Mouse Zygotic Genome Activation. Zygote 2020, 28. [Google Scholar] [CrossRef] [PubMed]
- Bu, G.; Zhu, W.; Liu, X.; Zhang, J.; Yu, L.; Zhou, K.; Wang, S.; Li, Z.; Fan, Z.; Wang, T.; et al. Coordination of Zygotic Genome Activation Entry and Exit by H3K4me3 and H3K27me3 in Porcine Early Embryos. Genome Res 2022, 32. [Google Scholar] [CrossRef] [PubMed]
- Shao, G. B.; Ding, H. M.; Gong, A. H. Role of Histone Methylation in Zygotic Genome Activation in the Preimplantation Mouse Embryo. In Vitro Cell Dev Biol Anim 2008, 44. [Google Scholar] [CrossRef] [PubMed]
- Darbo, E.; Herrmann, C.; Lecuit, T.; Thieffry, D.; van Helden, J. Transcriptional and Epigenetic Signatures of Zygotic Genome Activation during Early Drosophila Embryogenesis. BMC Genomics 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Pálfy, M.; Joseph, S. R.; Vastenhouw, N. L. The Timing of Zygotic Genome Activation. Current Opinion in Genetics and Development 2017. [Google Scholar] [CrossRef] [PubMed]
- Robertson, S.; Lin, R. The Maternal-to-Zygotic Transition in C. Elegans. In Current Topics in Developmental Biology; 2015; Vol. 113. [CrossRef]
- Blitz, I. L.; Cho, K. W. Y. Control of Zygotic Genome Activation in Xenopus. In Current Topics in Developmental Biology; 2021; Vol. 145. [CrossRef]
- Laue, K.; Rajshekar, S.; Courtney, A. J.; Lewis, Z. A.; Goll, M. G. The Maternal to Zygotic Transition Regulates Genome-Wide Heterochromatin Establishment in the Zebrafish Embryo. Nat Commun 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Jukam, D.; Shariati, S. A. M.; Skotheim, J. M. Zygotic Genome Activation in Vertebrates. Developmental Cell 2017. [Google Scholar] [CrossRef]
- Colonnetta, M. M.; Schedl, P.; Deshpande, G. Germline/Soma Distinction in Drosophila Embryos Requires Regulators of Zygotic Genome Activation. Elife 2023, 12. [Google Scholar] [CrossRef]
- Hamm, D. C.; Harrison, M. M. Regulatory Principles Governing the Maternal-to-Zygotic Transition: Insights from Drosophila Melanogaster. Open Biology 2018. [CrossRef]
- Li, L.; Lu, X.; Dean, J. The Maternal to Zygotic Transition in Mammals. Molecular Aspects of Medicine 2013. [Google Scholar] [CrossRef]
- Aoki, F. Zygotic Gene Activation in Mice: Profile and Regulation. Journal of Reproduction and Development 2022, 68. [Google Scholar] [CrossRef]
- Yuan, S.; Zhan, J.; Zhang, J.; Liu, Z.; Hou, Z.; Zhang, C.; Yi, L.; Gao, L.; Zhao, H.; Chen, Z. J.; et al. Human Zygotic Genome Activation Is Initiated from Paternal Genome. Cell Discov 2023, 9. [Google Scholar] [CrossRef] [PubMed]
- Tesarik, J. Control of Maternal-to-Zygotic Transition in Human Embryos and Other Animal Species (Especially Mouse): Similarities and Differences. International Journal of Molecular Sciences 2022. [CrossRef] [PubMed]
- Yuan, K.; Seller, C. A.; Shermoen, A. W.; O’Farrell, P. H. Timing the Drosophila Mid-Blastula Transition: A Cell Cycle-Centered View. Trends in Genetics 2016. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Fan, D.; Zhao, H.; Liu, Z.; Hou, Z.; Tao, W.; Yu, G.; Yuan, S.; Zhu, X.; Kang, M.; et al. Dynamics of Histone Acetylation during Human Early Embryogenesis. Cell Discov 2023, 9. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, D.; Avvakumov, N.; Côté, J. Histone Phosphorylation. Epigenetics 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Talamillo, A.; Barroso-Gomila, O.; Giordano, I.; Ajuria, L.; Grillo, M.; Mayor, U.; Barrio, R. The Role of SUMOylation during Development. Biochemical Society Transactions 2020. [CrossRef] [PubMed]
- Mattiroli, F.; Penengo, L. Histone Ubiquitination: An Integrative Signaling Platform in Genome Stability. Trends in Genetics 2021. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, Q.; Ding, J.; Yin, T.; Ye, P.; Zhang, Y. The Conceivable Functions of Protein Ubiquitination and Deubiquitination in Reproduction. Frontiers in Physiology 2022. [Google Scholar] [CrossRef]
- Millán-Zambrano, G.; Burton, A.; Bannister, A. J.; Schneider, R. Histone Post-Translational Modifications — Cause and Consequence of Genome Function. Nature Reviews Genetics 2022. [Google Scholar] [CrossRef]
- Liu, R.; Wu, J.; Guo, H.; Yao, W.; Li, S.; Lu, Y.; Jia, Y.; Liang, X.; Tang, J.; Zhang, H. Post-translational Modifications of Histones: Mechanisms, Biological Functions, and Therapeutic Targets. MedComm (Beijing) 2023, 4. [Google Scholar] [CrossRef]
- Smith, B. C.; Denu, J. M. Chemical Mechanisms of Histone Lysine and Arginine Modifications. Biochimica et Biophysica Acta - Gene Regulatory Mechanisms 2009. [Google Scholar] [CrossRef] [PubMed]
- Jambhekar, A.; Dhall, A.; Shi, Y. Roles and Regulation of Histone Methylation in Animal Development. Nature Reviews Molecular Cell Biology 2019. [CrossRef] [PubMed]
- Eberharter, A.; Becker, P. B. Histone Acetylation: A Switch between Repressive and Permissive Chromatin. EMBO Rep 2002, 3. [Google Scholar] [CrossRef]
- Wang, H.; Fan, Z.; Shliaha, P. V.; Miele, M.; Hendrickson, R. C.; Jiang, X.; Helin, K. H3K4me3 Regulates RNA Polymerase II Promoter-Proximal Pause-Release. Nature 2023, 615. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kim, G. W.; Kwon, S. H.; Lee, J. S. Broad Domains of Histone H3 Lysine 4 Trimethylation in Transcriptional Regulation and Disease. FEBS Journal 2020. [Google Scholar] [CrossRef]
- Wysocka, J.; Swigut, T.; Xiao, H.; Milne, T. A.; Kwon, S. Y.; Landry, J.; Kauer, M.; Tackett, A. J.; Chait, B. T.; Badenhorst, P.; et al. A PHD Finger of NURF Couples Histone H3 Lysine 4 Trimethylation with Chromatin Remodelling. Nature 2006, 442. [Google Scholar] [CrossRef]
- Liu, X.; Wang, C.; Liu, W.; Li, J.; Li, C.; Kou, X.; Chen, J.; Zhao, Y.; Gao, H.; Wang, H.; et al. Distinct Features of H3K4me3 and H3K27me3 Chromatin Domains in Pre-Implantation Embryos. Nature 2016, 537. [Google Scholar] [CrossRef]
- Beacon, T. H.; Delcuve, G. P.; López, C.; Nardocci, G.; Kovalchuk, I.; van Wijnen, A. J.; Davie, J. R. The Dynamic Broad Epigenetic (H3K4me3, H3K27ac) Domain as a Mark of Essential Genes. Clinical Epigenetics 2021. [Google Scholar] [CrossRef]
- Lindeman, L. C.; Andersen, I. S.; Reiner, A. H.; Li, N.; Aanes, H.; Østrup, O.; Winata, C.; Mathavan, S.; Müller, F.; Aleström, P.; et al. Prepatterning of Developmental Gene Expression by Modified Histones before Zygotic Genome Activation. Dev Cell 2011, 21. [Google Scholar] [CrossRef]
- Van Heeringen, S. J.; Akhtar, W.; Jacobi, U. G.; Akkers, R. C.; Suzuki, Y.; Veenstra, G. J. C. Nucleotide Composition-Linked Divergence of Vertebrate Core Promoter Architecture. Genome Res 2011, 21. [Google Scholar] [CrossRef] [PubMed]
- Ardehali, M. B.; Mei, A.; Zobeck, K. L.; Caron, M.; Lis, J. T.; Kusch, T. Drosophila Set1 Is the Major Histone H3 Lysine 4 Trimethyltransferase with Role in Transcription. EMBO Journal 2011, 30. [Google Scholar] [CrossRef] [PubMed]
- Dahl, J. A.; Jung, I.; Aanes, H.; Greggains, G. D.; Manaf, A.; Lerdrup, M.; Li, G.; Kuan, S.; Li, B.; Lee, A. Y.; et al. Broad Histone H3K4me3 Domains in Mouse Oocytes Modulate Maternal-to-Zygotic Transition. Nature 2016, 537. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Xu, B.; Sun, Y.; Lu, X.; Gu, R.; Wu, L.; Feng, Y.; Xu, C. Dynamic Changes of Histone H3 Trimethylated at Positions K4 and K27 in Human Oocytes and Preimplantation Embryos. Fertil Steril 2012, 98. [Google Scholar] [CrossRef] [PubMed]
- Sendžikaitė, G.; Kelsey, G. The Role and Mechanisms of DNA Methylation in the Oocyte. Essays in Biochemistry 2019. [CrossRef]
- Huang, X.; Gao, X.; Li, W.; Jiang, S.; Li, R.; Hong, H.; Zhao, C.; Zhou, P.; Chen, H.; Bo, X.; et al. Stable H3K4me3 Is Associated with Transcription Initiation during Early Embryo Development. Bioinformatics 2019, 35. [Google Scholar] [CrossRef]
- Brind’Amour, J.; Lorincz, M. C. Profiling Histone Methylation in Low Numbers of Cells. In Methods in Molecular Biology; 2022; Vol 2529. [CrossRef]
- Zhang, B.; Zheng, H.; Huang, B.; Li, W.; Xiang, Y.; Peng, X.; Ming, J.; Wu, X.; Zhang, Y.; Xu, Q.; et al. Allelic Reprogramming of the Histone Modification H3K4me3 in Early Mammalian Development. Nature 2016, 537. [Google Scholar] [CrossRef]
- Ishihara, T.; Griffith, O. W.; Suzuki, S.; Renfree, M. B. Presence of H3K4me3 on Paternally Expressed Genes of the Paternal Genome From Sperm to Implantation. Front Cell Dev Biol 2022, 10. [Google Scholar] [CrossRef]
- Xu, R.; Li, C.; Liu, X.; Gao, S. Insights into Epigenetic Patterns in Mammalian Early Embryos. Protein and Cell 2021. [Google Scholar] [CrossRef]
- Albert, T. K.; Kerl, K. A Histone Tale That EnCOMPASSes Pausing: New Insights into the Functional Repertoire of H3K4me3. Signal Transduction and Targeted Therapy 2023. [Google Scholar] [CrossRef]
- Sha, Q. Q.; Zhang, J.; Fan, H. Y. Function and Regulation of Histone H3 Lysine-4 Methylation During Oocyte Meiosis and Maternal-to-Zygotic Transition. Frontiers in Cell and Developmental Biology 2020. [CrossRef] [PubMed]
- Yoo, J.; Kim, G. W.; Jeon, Y. H.; Kim, J. Y.; Lee, S. W.; Kwon, S. H. Drawing a Line between Histone Demethylase KDM5A and KDM5B: Their Roles in Development and Tumorigenesis. Experimental and Molecular Medicine 2022. [Google Scholar] [CrossRef] [PubMed]
- Xhabija, B.; Kidder, B. L. KDM5B Is a Master Regulator of the H3K4-Methylome in Stem Cells, Development and Cancer. Seminars in Cancer Biology 2019. [CrossRef]
- Wang, Z.; Zhong, C.; Li, H. Histone Demethylase KDM5B Catalyzed H3K4me3 Demethylation to Promote Differentiation of Bone Marrow Mesenchymal Stem Cells into Cardiomyocytes. Mol Biol Rep 2022, 49. [Google Scholar] [CrossRef] [PubMed]
- Kidder, B. L.; Hu, G.; Yu, Z.-X.; Liu, C.; Zhao, K. Extended Self-Renewal and Accelerated Reprogramming in the Absence of Kdm5b. Mol Cell Biol 2013, 33. [Google Scholar] [CrossRef]
- Peaston, A. E.; Evsikov, A. V.; Graber, J. H.; de Vries, W. N.; Holbrook, A. E.; Solter, D.; Knowles, B. B. Retrotransposons Regulate Host Genes in Mouse Oocytes and Preimplantation Embryos. Dev Cell 2004, 7. [Google Scholar] [CrossRef]
- Xia, W.; Xu, J.; Yu, G.; Yao, G.; Xu, K.; Ma, X.; Zhang, N.; Liu, B.; Li, T.; Lin, Z.; et al. Resetting Histone Modifications during Human Parental-to-Zygotic Transition. Science (1979) 2019, 365. [Google Scholar] [CrossRef]
- Reshetnikov, V. V.; Kisaretova, P. E.; Ershov, N. I.; Merkulova, T. I.; Bondar, N. P. Data of Correlation Analysis between the Density of H3K4me3 in Promoters of Genes and Gene Expression: Data from RNA-Seq and ChIP-Seq Analyses of the Murine Prefrontal Cortex. Data Brief 2020, 33. [Google Scholar] [CrossRef]
- Sun, H.; Wang, Y.; Wang, Y.; Ji, F.; Wang, A.; Yang, M.; He, X.; Li, L. Bivalent Regulation and Related Mechanisms of H3K4/27/9me3 in Stem Cells. Stem Cell Reviews and Reports 2022. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.; Cui, H.; Xiao, S.; Song, E.; Zong, M.; Ling, S.; Rosenwaks, Z.; Gao, S.; Liu, X.; et al. Maternal H3.3-Mediated Paternal Genome Reprogramming Contributes to Minor Zygotic Genome Activation. bioRxiv 2023, 2023.11.07.566007. [Google Scholar] [CrossRef]
- Park, K.; Kim, J. A.; Kim, J. Transcriptional Regulation by the KMT2 Histone H3K4 Methyltransferases. Biochimica et Biophysica Acta - Gene Regulatory Mechanisms 2020. [Google Scholar] [CrossRef] [PubMed]
- Nicetto, D.; Zaret, K. S. Role of H3K9me3 Heterochromatin in Cell Identity Establishment and Maintenance. Current Opinion in Genetics and Development 2019. [Google Scholar] [CrossRef] [PubMed]
- Ninova, M.; Tóth, K. F.; Aravin, A. A. The Control of Gene Expression and Cell Identity by H3K9 Trimethylation. Development (Cambridge) 2019. [Google Scholar] [CrossRef] [PubMed]
- Nicetto, D.; Donahue, G.; Jain, T.; Peng, T.; Sidoli, S.; Sheng, L.; Montavon, T.; Becker, J. S.; Grindheim, J. M.; Blahnik, K.; et al. H3K9me3-Heterochromatin Loss at Protein-Coding Genes Enables Developmental Lineage Specification. Science (1979) 2019, 363. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Zhu, Q.; Zhao, Y.; Chen, M.; Yang, L.; Shen, S.; Yang, G.; Shi, Z.; Zhang, X.; Shi, Q.; et al. Unreprogrammed H3K9me3 Prevents Minor Zygotic Genome Activation and Lineage Commitment in SCNT Embryos. Nat Commun 2023, 14. [Google Scholar] [CrossRef]
- Matoba, S.; Liu, Y.; Lu, F.; Iwabuchi, K. A.; Shen, L.; Inoue, A.; Zhang, Y. Embryonic Development Following Somatic Cell Nuclear Transfer Impeded by Persisting Histone Methylation. Cell 2014, 159. [Google Scholar] [CrossRef]
- Matoba, S.; Zhang, Y. Somatic Cell Nuclear Transfer Reprogramming: Mechanisms and Applications. Cell Stem Cell 2018. [Google Scholar] [CrossRef]
- Srirattana, K.; Kaneda, M.; Parnpai, R. Strategies to Improve the Efficiency of Somatic Cell Nuclear Transfer. International Journal of Molecular Sciences 2022. [CrossRef]
- Moura, M. T. Cloning by SCNT: Integrating Technical and Biology-Driven Advances. In Methods in Molecular Biology; 2023; Vol 2647. [CrossRef]
- Wang, C.; Liu, X.; Gao, Y.; Yang, L.; Li, C.; Liu, W.; Chen, C.; Kou, X.; Zhao, Y.; Chen, J.; et al. Reprogramming of H3K9me3-Dependent Heterochromatin during Mammalian Embryo Development. Nat Cell Biol 2018, 20. [Google Scholar] [CrossRef]
- Sampaio, R. V.; Sangalli, J. R.; De Bem, T. H. C.; Ambrizi, D. R.; del Collado, M.; Bridi, A.; de Ávila, A. C. F. C. M.; Macabelli, C. H.; de Jesus Oliveira, L.; da Silveira, J. C.; et al. Catalytic Inhibition of H3K9me2 Writers Disturbs Epigenetic Marks during Bovine Nuclear Reprogramming. Sci Rep 2020, 10. [Google Scholar] [CrossRef]
- Sankar, A.; Lerdrup, M.; Manaf, A.; Johansen, J. V.; Gonzalez, J. M.; Borup, R.; Blanshard, R.; Klungland, A.; Hansen, K.; Andersen, C. Y.; et al. KDM4A Regulates the Maternal-to-Zygotic Transition by Protecting Broad H3K4me3 Domains from H3K9me3 Invasion in Oocytes. Nat Cell Biol 2020, 22. [Google Scholar] [CrossRef]
- Becker, J. S.; Nicetto, D.; Zaret, K. S. H3K9me3-Dependent Heterochromatin: Barrier to Cell Fate Changes. Trends in Genetics 2016. [Google Scholar] [CrossRef]
- Wu, D. Y.; Li, X.; Sun, Q. R.; Dou, C. L.; Xu, T.; He, H.; Luo, H.; Fu, H.; Bu, G. W.; Luo, B.; et al. Defective Chromatin Architectures in Embryonic Stem Cells Derived from Somatic Cell Nuclear Transfer Impair Their Differentiation Potentials. Cell Death Dis 2021, 12. [Google Scholar] [CrossRef]
- Chung, Y. G.; Matoba, S.; Liu, Y.; Eum, J. H.; Lu, F.; Jiang, W.; Lee, J. E.; Sepilian, V.; Cha, K. Y.; Lee, D. R.; et al. Histone Demethylase Expression Enhances Human Somatic Cell Nuclear Transfer Efficiency and Promotes Derivation of Pluripotent Stem Cells. Cell Stem Cell 2015, 17. [Google Scholar] [CrossRef]
- Geis, F. K.; Goff, S. P. Silencing and Transcriptional Regulation of Endogenous Retroviruses: An Overview. Viruses 2020. [CrossRef]
- Bulut-Karslioglu, A.; DeLaRosa-Velázquez, I. A.; Ramirez, F.; Barenboim, M.; Onishi-Seebacher, M.; Arand, J.; Galán, C.; Winter, G. E.; Engist, B.; Gerle, B.; et al. Suv39h-Dependent H3K9me3 Marks Intact Retrotransposons and Silences LINE Elements in Mouse Embryonic Stem Cells. Mol Cell 2014, 55. [Google Scholar] [CrossRef] [PubMed]
- Yang, B. X.; El Farran, C. A.; Guo, H. C.; Yu, T.; Fang, H. T.; Wang, H. F.; Schlesinger, S.; Seah, Y. F. S.; Goh, G. Y. L.; Neo, S. P.; et al. Systematic Identification of Factors for Provirus Silencing in Embryonic Stem Cells. Cell 2015, 163. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, Y.; Loh, Y. P.; Tng, J. Q.; Lim, M. C.; Cao, Z.; Raju, A.; Lieberman Aiden, E.; Li, S.; Manikandan, L.; et al. H3K27me3-Rich Genomic Regions Can Function as Silencers to Repress Gene Expression via Chromatin Interactions. Nat Commun 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- van Mierlo, G.; Veenstra, G. J. C.; Vermeulen, M.; Marks, H. The Complexity of PRC2 Subcomplexes. Trends in Cell Biology 2019. [CrossRef] [PubMed]
- Boros, J.; Arnoult, N.; Stroobant, V.; Collet, J.-F.; Decottignies, A. Polycomb Repressive Complex 2 and H3K27me3 Cooperate with H3K9 Methylation To Maintain Heterochromatin Protein 1α at Chromatin. Mol Cell Biol 2014, 34. [Google Scholar] [CrossRef] [PubMed]
- Brumbaugh, J.; Stefano, B. Di; Hochedlinger, K. Reprogramming: Identifying the Mechanisms That Safeguard Cell Identity. Development (Cambridge) 2019. [Google Scholar] [CrossRef] [PubMed]
- Charlet, J.; Duymich, C. E.; Lay, F. D.; Mundbjerg, K.; Dalsgaard Sørensen, K.; Liang, G.; Jones, P. A. Bivalent Regions of Cytosine Methylation and H3K27 Acetylation Suggest an Active Role for DNA Methylation at Enhancers. Mol Cell 2016, 62. [Google Scholar] [CrossRef] [PubMed]
- Andersen, I. S.; Reiner, A. H.; Aanes, H.; Aleström, P.; Collas, P. Developmental Features of DNA Methylation during Activation of the Embryonic Zebrafish Genome. Genome Biol 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Eckersley-Maslin, M. A.; Alda-Catalinas, C.; Reik, W. Dynamics of the Epigenetic Landscape during the Maternal-to-Zygotic Transition. Nature Reviews Molecular Cell Biology 2018. [CrossRef] [PubMed]
- Liu, C.; Ma, Y.; Shang, Y.; Huo, R.; Li, W. Post-Translational Regulation of the Maternal-to-Zygotic Transition. Cellular and Molecular Life Sciences 2018. [Google Scholar] [CrossRef]
- Zheng, H.; Huang, B.; Zhang, B.; Xiang, Y.; Du, Z.; Xu, Q.; Li, Y.; Wang, Q.; Ma, J.; Peng, X.; et al. Resetting Epigenetic Memory by Reprogramming of Histone Modifications in Mammals. Mol Cell 2016, 63. [Google Scholar] [CrossRef] [PubMed]
- Fraser, R.; Lin, C. J. Epigenetic Reprogramming of the Zygote in Mice and Men: On Your Marks, Get Set, Go! Reproduction 2016. [Google Scholar] [CrossRef]
- Rousseaux, S.; Reynoird, N.; Escoffier, E.; Thevenon, J.; Caron, C.; Khochbin, S. Epigenetic Reprogramming of the Male Genome during Gametogenesis and in the Zygote. Reprod Biomed Online 2008, 16. [Google Scholar] [CrossRef]
- Inoue, A.; Jiang, L.; Lu, F.; Suzuki, T.; Zhang, Y. Maternal H3K27me3 Controls DNA Methylation-Independent Imprinting. Nature 2017, 547. [Google Scholar] [CrossRef]
- Meng, T. G.; Zhou, Q.; Ma, X. S.; Liu, X. Y.; Meng, Q. R.; Huang, X. J.; Liu, H. L.; Lei, W. L.; Zhao, Z. H.; Ouyang, Y. C.; et al. PRC2 and EHMT1 Regulate H3K27me2 and H3K27me3 Establishment across the Zygote Genome. Nat Commun 2020, 11. [Google Scholar] [CrossRef]
- Pailles, M.; Hirlemann, M.; Brochard, V.; Chebrout, M.; Oudin, J. F.; Marks, H.; Jouneau, A.; Bonnet-Garnier, A. H3K27me3 at Pericentromeric Heterochromatin Is a Defining Feature of the Early Mouse Blastocyst. Sci Rep 2022, 12. [Google Scholar] [CrossRef]
- Juan, A. H.; Wang, S.; Ko, K. D.; Zare, H.; Tsai, P. F.; Feng, X.; Vivanco, K. O.; Ascoli, A. M.; Gutierrez-Cruz, G.; Krebs, J.; et al. Roles of H3K27me2 and H3K27me3 Examined during Fate Specification of Embryonic Stem Cells. Cell Rep 2016, 17. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, F.; Kou, Z.; Zhang, Y.; Gao, S. Defective Chromatin Structure in Somatic Cell Cloned Mouse Embryos. Journal of Biological Chemistry 2009, 284. [Google Scholar] [CrossRef]
- Zhou, C.; Wang, Y.; Zhang, J.; Su, J.; An, Q.; Liu, X.; Zhang, M.; Wang, Y.; Liu, J.; Zhang, Y. H3K27me3 Is an Epigenetic Barrier While KDM6A Overexpression Improves Nuclear Reprogramming Efficiency. FASEB Journal 2019, 33. [Google Scholar] [CrossRef]
- Bai, G. Y.; Song, S. H.; Zhang, Y. W.; Huang, X.; Huang, X. W.; Sun, R. Z.; Lei, L. Kdm6a Overexpression Improves the Development of Cloned Mouse Embryos. Zygote 2018, 26. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Song, L.; Liu, X.; Bai, L.; Li, G. KDM 6A and KDM 6B Play Contrasting Roles in Nuclear Transfer Embryos Revealed by MERVL Reporter System. EMBO Rep 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Matoba, S.; Wang, H.; Jiang, L.; Lu, F.; Iwabuchi, K. A.; Wu, X.; Inoue, K.; Yang, L.; Press, W.; Lee, J. T.; et al. Loss of H3K27me3 Imprinting in Somatic Cell Nuclear Transfer Embryos Disrupts Post-Implantation Development. Cell Stem Cell 2018, 23. [Google Scholar] [CrossRef] [PubMed]
- Raas, M. W. D.; Zijlmans, D. W.; Vermeulen, M.; Marks, H. There Is Another: H3K27me3-Mediated Genomic Imprinting. Trends in Genetics 2022. [Google Scholar] [CrossRef] [PubMed]
- Macrae, T. A.; Fothergill-Robinson, J.; Ramalho-Santos, M. Regulation, Functions and Transmission of Bivalent Chromatin during Mammalian Development. Nature Reviews Molecular Cell Biology 2023. [CrossRef] [PubMed]
- Saha, B.; Home, P.; Ray, S.; Larson, M.; Paul, A.; Rajendran, G.; Behr, B.; Paul, S. EED and KDM6B Coordinate the First Mammalian Cell Lineage Commitment To Ensure Embryo Implantation. Mol Cell Biol 2013, 33. [Google Scholar] [CrossRef] [PubMed]
- Dahl, J. A.; Reiner, A. H.; Klungland, A.; Wakayama, T.; Collas, P. Histone H3 Lysine 27 Methylation Asymmetry on Developmentally-Regulated Promoters Distinguish the First Two Lineages in Mouse Preimplantation Embryos. PLoS One 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y. H.; Yu, J. Epigenetic Disruptions of Histone Signatures for the Trophectoderm and Inner Cell Mass in Mouse Parthenogenetic Embryos. Stem Cells Dev 2015, 24. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Hilbert, L.; Oda, H.; Wan, Y.; Heddleston, J. M.; Chew, T. L.; Zaburdaev, V.; Keller, P.; Lionnet, T.; Vastenhouw, N.; et al. Histone H3K27 Acetylation Precedes Active Transcription during Zebrafish Zygotic Genome Activation as Revealed by Live-Cell Analysis. Development (Cambridge) 2019, 146. [Google Scholar] [CrossRef] [PubMed]
- Lavarone, E.; Barbieri, C. M.; Pasini, D. Dissecting the Role of H3K27 Acetylation and Methylation in PRC2 Mediated Control of Cellular Identity. Nat Commun 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Vavouri, T.; Lehner, B. Human Genes with CpG Island Promoters Have a Distinct Transcription-Associated Chromatin Organization. Genome Biol 2012, 13. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, Z.; Dong, Q.; Xiong, J.; Zhu, B. Histone H3K27 Acetylation Is Dispensable for Enhancer Activity in Mouse Embryonic Stem Cells. Genome Biol 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- DiFiore, J. V.; Ptacek, T. S.; Wang, Y.; Li, B.; Simon, J. M.; Strahl, B. D. Unique and Shared Roles for Histone H3K36 Methylation States in Transcription Regulation Functions. Cell Rep 2020, 31. [Google Scholar] [CrossRef]
- Sen, P.; Dang, W.; Donahue, G.; Dai, J.; Dorsey, J.; Cao, X.; Liu, W.; Cao, K.; Perry, R.; Lee, J. Y.; et al. H3K36 Methylation Promotes Longevity by Enhancing Transcriptional Fidelity. Genes Dev 2015, 29. [Google Scholar] [CrossRef]
- Leung, C. S.; Douglass, S. M.; Morselli, M.; Obusan, M. B.; Pavlyukov, M. S.; Pellegrini, M.; Johnson, T. L. H3K36 Methylation and the Chromodomain Protein Eaf3 Are Required for Proper Cotranscriptional Spliceosome Assembly. Cell Rep 2019, 27. [Google Scholar] [CrossRef]
- Kim, S.; Kim, H.; Fong, N.; Erickson, B.; Bentley, D. L. Pre-MRNA Splicing Is a Determinant of Histone H3K36 Methylation. Proc Natl Acad Sci U S A 2011, 108. [Google Scholar] [CrossRef]
- Li, F.; Mao, G.; Tong, D.; Huang, J.; Gu, L.; Yang, W.; Li, G. M. The Histone Mark H3K36me3 Regulates Human DNA Mismatch Repair through Its Interaction with MutSα. Cell 2013, 153. [Google Scholar] [CrossRef]
- Sun, Z.; Zhang, Y.; Jia, J.; Fang, Y.; Tang, Y.; Wu, H.; Fang, D. H3K36me3, Message from Chromatin to DNA Damage Repair. Cell and Bioscience 2020. [CrossRef]
- Wang, L.; Niu, N.; Li, L.; Shao, R.; Ouyang, H.; Zou, W. H3K36 Trimethylation Mediated by SETD2 Regulates the Fate of Bone Marrow Mesenchymal Stem Cells. PLoS Biol 2018, 16. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Workman, J. L. Regulation of SETD2 Stability Is Important for the Fidelity of H3K36me3 Deposition. Epigenetics Chromatin 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Zhu, B. Roles of H3K36-Specific Histone Methyltransferases in Transcription: Antagonizing Silencing and Safeguarding Transcription Fidelity. Biophys Rep 2018, 4. [Google Scholar] [CrossRef]
- Jha, D. K.; Strahl, B. D. An RNA Polymerase II-Coupled Function for Histone H3K36 Methylation in Checkpoint Activation and DSB Repair. Nat Commun 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, D. N.; Papillon-Cavanagh, S.; Chen, H.; Yue, Y.; Chen, X.; Rajagopalan, K. N.; Horth, C.; McGuire, J. T.; Xu, X.; Nikbakht, H.; et al. The Histone Mark H3K36me2 Recruits DNMT3A and Shapes the Intergenic DNA Methylation Landscape. Nature 2019, 573. [Google Scholar] [CrossRef]
- Yano, S.; Ishiuchi, T.; Abe, S.; Namekawa, S. H.; Huang, G.; Ogawa, Y.; Sasaki, H. Histone H3K36me2 and H3K36me3 Form a Chromatin Platform Essential for DNMT3A-Dependent DNA Methylation in Mouse Oocytes. Nat Commun 2022, 13. [Google Scholar] [CrossRef]
- Xu, Q.; Xiang, Y.; Wang, Q.; Wang, L.; Brind’Amour, J.; Bogutz, A. B.; Zhang, Y.; Zhang, B.; Yu, G.; Xia, W.; et al. SETD2 Regulates the Maternal Epigenome, Genomic Imprinting and Embryonic Development. Nat Genet 2019, 51. [Google Scholar] [CrossRef]
- Hu, M.; Sun, X. J.; Zhang, Y. L.; Kuang, Y.; Hu, C. Q.; Wu, W. L.; Shen, S. H.; Du, T. T.; Li, H.; He, F.; et al. Histone H3 Lysine 36 Methyltransferase Hypb/Setd2 Is Required for Embryonic Vascular Remodeling. Proc Natl Acad Sci U S A 2010, 107. [Google Scholar] [CrossRef]
- Aoshima, K.; Inoue, E.; Sawa, H.; Okada, Y. Paternal H3K4 Methylation Is Required for Minor Zygotic Gene Activation and Early Mouse Embryonic Development. EMBO Rep 2015, 16. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, Y. Maternal H3K27me3-Dependent Autosomal and X Chromosome Imprinting. Nature Reviews Genetics 2020. [Google Scholar] [CrossRef]
- Igolkina, A. A.; Zinkevich, A.; Karandasheva, K. O.; Popov, A. A.; Selifanova, M. V.; Nikolaeva, D.; Tkachev, V.; Penzar, D.; Nikitin, D. M.; Buzdin, A. H3K4me3, H3K9ac, H3K27ac, H3K27me3 and H3K9me3 Histone Tags Suggest Distinct Regulatory Evolution of Open and Condensed Chromatin Landmarks. Cells 2019, 8. [Google Scholar] [CrossRef]
- Lismer, A.; Lambrot, R.; Lafleur, C.; Dumeaux, V.; Kimmins, S. ChIP-Seq Protocol for Sperm Cells and Embryos to Assess Environmental Impacts and Epigenetic Inheritance. STAR Protoc 2021, 2. [Google Scholar] [CrossRef]
- Lismer, A.; Kimmins, S. Emerging Evidence That the Mammalian Sperm Epigenome Serves as a Template for Embryo Development. Nat Commun 2023, 14. [Google Scholar] [CrossRef]
- Cockrum, C. S.; Strome, S. Maternal H3K36 and H3K27 HMTs Protect Germline Development via Regulation of the Transcription Factor LIN-15B. Elife 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Zhang, C.; Zhang, Y. Epigenetic Regulation of Mouse Preimplantation Embryo Development. Current Opinion in Genetics and Development 2020. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Hu, B.; Wang, Z.; Wu, X.; Luo, L.; Li, S.; Wang, S.; Zhang, K.; Wang, H. Functional Role of GATA3 and CDX2 in Lineage Specification during Bovine Early Embryonic Development. Reproduction 2023, 165. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Guo, G.; Yuan, P.; Ralston, A.; Sun, L.; Huss, M.; Mistri, T.; Pinello, L.; Ng, H. H.; Yuan, G.; et al. The Role of Cdx2 as a Lineage Specific Transcriptional Repressor for Pluripotent Network during the First Developmental Cell Lineage Segregation. Sci Rep 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Home, P.; Ray, S.; Dutta, D.; Bronshteyn, I.; Larson, M.; Paul, S. GATA3 Is Selectively Expressed in the Trophectoderm of Peri-Implantation Embryo and Directly Regulates Cdx2 Gene Expression. Journal of Biological Chemistry 2009, 284. [Google Scholar] [CrossRef] [PubMed]
- Rebuzzini, P.; Zuccotti, M.; Garagna, S. Building Pluripotency Identity in the Early Embryo and Derived Stem Cells. Cells 2021. [CrossRef]
- Niwa, H. How Is Pluripotency Determined and Maintained? Development 2007. [Google Scholar] [CrossRef] [PubMed]
- Allègre, N.; Chauveau, S.; Dennis, C.; Renaud, Y.; Meistermann, D.; Estrella, L. V.; Pouchin, P.; Cohen-Tannoudji, M.; David, L.; Chazaud, C. NANOG Initiates Epiblast Fate through the Coordination of Pluripotency Genes Expression. Nat Commun 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Jho, E. H. The History and Regulatory Mechanism of the Hippo Pathway. BMB Reports 2018. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Guan, K. L. Hippo Signaling in Embryogenesis and Development. Trends in Biochemical Sciences 2021. [CrossRef] [PubMed]
- Yang, J.; Jiang, W. Dynamic Changes in Epigenetic Modifications During Mammalian Early Embryo Development. In Handbook of Epigenetics: The New Molecular and Medical Genetics, Third Edition; 2022. [CrossRef]
- Torres-Padilla, M. E.; Parfitt, D. E.; Kouzarides, T.; Zernicka-Goetz, M. Histone Arginine Methylation Regulates Pluripotency in the Early Mouse Embryo. Nature 2007, 445. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Bruce, A. W.; Jedrusik, A.; Ellis, P. D.; Andrews, R. M.; Langford, C. F.; Glover, D. M.; Zernicka-Goetz, M. CARM1 Is Required in Embryonic Stem Cells to Maintain Pluripotency and Resist Differentiation. Stem Cells 2009, 27. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Lyu, X.; Qin, N.; Liu, H.; Zhang, M.; Lai, Y.; Dong, B.; Lu, P. Coactivator-Associated Arginine Methyltransferase 1: A Versatile Player in Cell Differentiation and Development. Genes and Diseases 2023. [Google Scholar] [CrossRef]
- Hupalowska, A.; Jedrusik, A.; Zhu, M.; Bedford, M. T.; Glover, D. M.; Zernicka-Goetz, M. CARM1 and Paraspeckles Regulate Pre-Implantation Mouse Embryo Development. Cell 2018, 175. [Google Scholar] [CrossRef]
- Franek, M.; Legartová, S.; Suchánková, J.; Milite, C.; Castellano, S.; Sbardella, G.; Kozubek, S.; Bártová, E. CARM1 Modulators Affect Epigenome of Stem Cells and Change Morphology of Nucleoli. Physiol Res 2015, 64. [Google Scholar] [CrossRef]
- Wang, J.; Wang, L.; Feng, G.; Wang, Y.; Li, Y.; Li, X.; Liu, C.; Jiao, G.; Huang, C.; Shi, J.; et al. Asymmetric Expression of LincGET Biases Cell Fate in Two-Cell Mouse Embryos. Cell 2018, 175. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Jiang, J.; Xu, C.; Wang, Y.; Sun, L.; Guo, X.; Liu, H. MicroRNA-181 Regulates CARM1 and Histone Aginine Methylation to Promote Differentiation of Human Embryonic Stem Cells. PLoS One 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Goolam, M.; Scialdone, A.; Graham, S. J. L.; MacAulay, I. C.; Jedrusik, A.; Hupalowska, A.; Voet, T.; Marioni, J. C.; Zernicka-Goetz, M. Heterogeneity in Oct4 and Sox2 Targets Biases Cell Fate in 4-Cell Mouse Embryos. Cell 2016, 165. [Google Scholar] [CrossRef]
- Zhao, H. yong; Zhang, Y. jun; Dai, H.; Zhang, Y.; Shen, Y. fei. CARM1 Mediates Modulation of Sox2. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Cao, Z.; Tong, X.; Yin, H.; Zhou, N.; Zhang, X.; Zhang, M.; Wang, X.; Liu, Q.; Yan, Y.; Ma, Y.; et al. Histone Arginine Methyltransferase CARM1-Mediated H3R26me2 Is Essential for Morula-to-Blastocyst Transition in Pigs. Front Cell Dev Biol 2021, 9. [Google Scholar] [CrossRef]
- Burton, A.; Muller, J.; Tu, S.; Padilla-Longoria, P.; Guccione, E.; Torres-Padilla, M. E. Single-Cell Profiling of Epigenetic Modifiers Identifies PRDM14 as an Inducer of Cell Fate in the Mammalian Embryo. Cell Rep 2013, 5. [Google Scholar] [CrossRef]
- Wu, J.; Belmonte, J. C. I. The Molecular Harbingers of Early Mammalian Embryo Patterning. Cell 2016. [Google Scholar] [CrossRef] [PubMed]
- Nakaki, F.; Saitou, M. PRDM14: A Unique Regulator for Pluripotency and Epigenetic Reprogramming. Trends in Biochemical Sciences 2014. [CrossRef]
- Yao, C.; Zhang, W.; Shuai, L. The First Cell Fate Decision in Pre-Implantation Mouse Embryos. Cell Regeneration 2019. [Google Scholar] [CrossRef]
- Zhang, Y.; Duan, E. LncRNAs and Paraspeckles Predict Cell Fate in Early Mouse Embryo. Biol Reprod 2019, 100. [Google Scholar] [CrossRef]
- Grosch, M.; Ittermann, S.; Shaposhnikov, D.; Drukker, M. Chromatin-Associated Membraneless Organelles in Regulation of Cellular Differentiation. Stem Cell Reports 2020. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Wang, T.; Zhao, Z.; Wei, W.; Xin, W.; Yang, X.; Wang, X. Novel Insights into the Emerging Role of Neat1 and Its Effects Downstream in the Regulation of Inflammation. Journal of Inflammation Research 2022. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, S.; Smith, M. M. Histone Variants and Epigenetics. Cold Spring Harb Perspect Biol 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Talbert, P. B.; Henikoff, S. Histone Variants at a Glance. J Cell Sci 2021, 134. [Google Scholar] [CrossRef] [PubMed]
- Martire, S.; Banaszynski, L. A. The Roles of Histone Variants in Fine-Tuning Chromatin Organization and Function. Nature Reviews Molecular Cell Biology 2020. [CrossRef]
- Buschbeck, M.; Hake, S. B. Variants of Core Histones and Their Roles in Cell Fate Decisions, Development and Cancer. Nature Reviews Molecular Cell Biology 2017. [CrossRef]
- Herchenröther, A.; Wunderlich, T. M.; Lan, J.; Hake, S. B. Spotlight on Histone H2A Variants: From B to X to Z. Seminars in Cell and Developmental Biology 2023. [CrossRef]
- Oberdoerffer, P.; Miller, K. M. Histone H2A Variants: Diversifying Chromatin to Ensure Genome Integrity. Seminars in Cell and Developmental Biology 2023. [CrossRef]
- Chakravarthy, S.; Gundimella, S. K. Y.; Caron, C.; Perche, P.-Y.; Pehrson, J. R.; Khochbin, S.; Luger, K. Structural Characterization of the Histone Variant MacroH2A. Mol Cell Biol 2005, 25. [Google Scholar] [CrossRef]
- Buschbeck, M.; Uribesalgo, I.; Wibowo, I.; Rué, P.; Martin, D.; Gutierrez, A.; Morey, L.; Guigó, R.; López-Schier, H.; Di Croce, L. The Histone Variant MacroH2A Is an Epigenetic Regulator of Key Developmental Genes. Nat Struct Mol Biol 2009, 16. [Google Scholar] [CrossRef]
- Duthie, S. M. Mechanisms of X-inactivation. In eLS; 2001. [CrossRef]
- Rasmussen, T. P.; Mastrangelo, M. A.; Eden, A.; Pehrson, J. R.; Jaenisch, R. Dynamic Relocalization of Histone MacroH2A1 from Centrosomes to Inactive X Chromosomes during X Inactivation. Journal of Cell Biology 2000, 150. [Google Scholar] [CrossRef]
- Chang, C. C.; Ma, Y.; Jacobs, S.; Tian, X. C.; Yang, X.; Rasmussen, T. P. A Maternal Store of MacroH2A Is Removed from Pronuclei Prior to Onset of Somatic MacroH2A Expression in Preimplantation Embryos. Dev Biol 2005, 278. [Google Scholar] [CrossRef]
- Angelov, D.; Molla, A.; Perche, P. Y.; Hans, F.; Côté, J.; Khochbin, S.; Bouvet, P.; Dimitrov, S. The Histone Variant MacroH2A Interferes with Transcription Factor Binding and SWI/SNF Nucleosome Remodeling. Mol Cell 2003, 11. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C. J.; Meers, O.; Buschbeck, M.; Heidel, F. H. The Role of Macroh2a Histone Variants in Cancer. Cancers 2021. [CrossRef] [PubMed]
- Mermoud, J. E.; Tassin, A. M.; Pehrson, J. R.; Brockdorff, N. Centrosomal Association of Histone MacroH2A1.2 in Embryonic Stem Cells and Somatic Cells. Exp Cell Res 2001, 268. [Google Scholar] [CrossRef] [PubMed]
- Paull, T. T.; Rogakou, E. P.; Yamazaki, V.; Kirchgessner, C. U.; Gellert, M.; Bonner, W. M. A Critical Role for Histone H2AX in Recruitment of Repair Factors to Nuclear Foci after DNA Damage. Current Biology 2000, 10. [Google Scholar] [CrossRef]
- Collins, P. L.; Purman, C.; Porter, S. I.; Nganga, V.; Saini, A.; Hayer, K. E.; Gurewitz, G. L.; Sleckman, B. P.; Bednarski, J. J.; Bassing, C. H.; et al. DNA Double-Strand Breaks Induce H2Ax Phosphorylation Domains in a Contact-Dependent Manner. Nat Commun 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Sun, K. Y.; Guo, S. M.; Cheng, G. P.; Yin, Y.; He, X.; Zhou, L. Q. Cleavage-Embryo Genes and Transposable Elements Are Regulated by Histone Variant H2a.x. Journal of Reproduction and Development 2021, 67. [Google Scholar] [CrossRef] [PubMed]
- Schmücker, A.; Lei, B.; Lorković, Z. J.; Capella, M.; Braun, S.; Bourguet, P.; Mathieu, O.; Mechtler, K.; Berger, F. Crosstalk between H2A Variant-Specific Modifications Impacts Vital Cell Functions. PLoS Genet 2021, 17. [Google Scholar] [CrossRef] [PubMed]
- Kinner, A.; Wu, W.; Staudt, C.; Iliakis, G. Gamma-H2AX in Recognition and Signaling of DNA Double-Strand Breaks in the Context of Chromatin. Nucleic acids research 2008. [Google Scholar] [CrossRef]
- Nishiyama, A.; Xin, L.; Sharov, A. A.; Thomas, M.; Mowrer, G.; Meyers, E.; Piao, Y.; Mehta, S.; Yee, S.; Nakatake, Y.; et al. Uncovering Early Response of Gene Regulatory Networks in ESCs by Systematic Induction of Transcription Factors. Cell Stem Cell 2009, 5. [Google Scholar] [CrossRef]
- Wu, T.; Liu, Y.; Wen, D.; Tseng, Z.; Tahmasian, M.; Zhong, M.; Rafii, S.; Stadtfeld, M.; Hochedlinger, K.; Xiao, A. Histone Variant H2A.X Deposition Pattern Serves as a Functional Epigenetic Mark for Distinguishing the Developmental Potentials of IPSCs. Cell Stem Cell 2014, 15. [Google Scholar] [CrossRef]
- Wu, B. J.; Dong, F. L.; Ma, X. S.; Wang, X. G.; Lin, F.; Liu, H. L. Localization and Expression of Histone H2A Variants during Mouse Oogenesis and Preimplantation Embryo Development. Genetics and Molecular Research 2014, 13. [Google Scholar] [CrossRef]
- Sugie, K.; Funaya, S.; Kawamura, M.; Nakamura, T.; Suzuki, M. G.; Aoki, F. Expression of Dux Family Genes in Early Preimplantation Embryos. Sci Rep 2020, 10. [Google Scholar] [CrossRef]
- Ren, W.; Gao, L.; Mou, Y.; Deng, W.; Hua, J.; Yang, F. DUX: One Transcription Factor Controls 2-Cell-like Fate. International Journal of Molecular Sciences 2022. [CrossRef]
- Ibarra-Morales, D.; Rauer, M.; Quarato, P.; Rabbani, L.; Zenk, F.; Schulte-Sasse, M.; Cardamone, F.; Gomez-Auli, A.; Cecere, G.; Iovino, N. Histone Variant H2A.Z Regulates Zygotic Genome Activation. Nat Commun 2021, 12. [Google Scholar] [CrossRef]
- Giaimo, B. D.; Ferrante, F.; Herchenröther, A.; Hake, S. B.; Borggrefe, T. The Histone Variant H2A.Z in Gene Regulation. Epigenetics and Chromatin 2019. [Google Scholar] [CrossRef]
- Semer, M.; Bidon, B.; Larnicol, A.; Caliskan, G.; Catez, P.; Egly, J. M.; Coin, F.; Le May, N. DNA Repair Complex Licenses Acetylation of H2A.Z.1 by KAT2A during Transcription. Nat Chem Biol 2019, 15. [Google Scholar] [CrossRef]
- Tsukii, K.; Takahata, S.; Murakami, Y. Histone Variant H2A.Z Plays Multiple Roles in the Maintenance of Heterochromatin Integrity. Genes to Cells 2022, 27. [Google Scholar] [CrossRef]
- Cole, L.; Kurscheid, S.; Nekrasov, M.; Domaschenz, R.; Vera, D. L.; Dennis, J. H.; Tremethick, D. J. Multiple Roles of H2A.Z in Regulating Promoter Chromatin Architecture in Human Cells. Nat Commun 2021, 12. [Google Scholar] [CrossRef]
- Rudnizky, S.; Bavly, A.; Malik, O.; Pnueli, L.; Melamed, P.; Kaplan, A. H2A.Z Controls the Stability and Mobility of Nucleosomes to Regulate Expression of the LH Genes. Nat Commun 2016, 7. [Google Scholar] [CrossRef]
- Sales-Gil, R.; Kommer, D. C.; de Castro, I. J.; Amin, H. A.; Vinciotti, V.; Sisu, C.; Vagnarelli, P. Non-redundant Functions of H2A.Z.1 and H2A.Z.2 in Chromosome Segregation and Cell Cycle Progression. EMBO Rep 2021, 22. [Google Scholar] [CrossRef]
- Iouzalen, N.; Moreau, J.; Méchali, M. H2A.ZI, a New Variant Histone Expressed during Xenopus Early Development Exhibits Several Distinct Features from the Core Histone H2A. Nucleic Acids Res 1996, 24. [Google Scholar] [CrossRef]
- Clarkson, M. J.; Wells, J. R. E.; Gibson, F.; Saint, R.; Tremethick, D. J. Regions of Variant Histone His2AvD Required for Drosophila Development. Nature 1999, 399. [Google Scholar] [CrossRef]
- Shen, T.; Ji, F.; Wang, Y.; Lei, X.; Zhang, D.; Jiao, J. Brain-Specific Deletion of Histone Variant H2A.z Results in Cortical Neurogenesis Defects and Neurodevelopmental Disorder. Nucleic Acids Res 2018, 46. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, J.; Zhou, J.; Bu, G.; Zhu, W.; He, H.; Sun, Q.; Yu, Z.; Xiong, W.; Wang, L.; et al. Hierarchical Accumulation of Histone Variant H2A.Z Regulates Transcriptional States and Histone Modifications in Early Mammalian Embryos. Advanced Science 2022, 9. [Google Scholar] [CrossRef]
- Dryhurst, D.; Ishibashi, T.; Rose, K. L.; Eirín-López, J. M.; McDonald, D.; Silva-Moreno, B.; Veldhoen, N.; Helbing, C. C.; Hendzel, M. J.; Shabanowitz, J.; et al. Characterization of the Histone H2A.Z-1 and H2A.Z-2 Isoforms in Vertebrates. BMC Biol 2009, 7. [Google Scholar] [CrossRef]
- Mylonas, C.; Lee, C.; Auld, A. L.; Cisse, I. I.; Boyer, L. A. A Dual Role for H2A.Z.1 in Modulating the Dynamics of RNA Polymerase II Initiation and Elongation. Nat Struct Mol Biol 2021, 28. [Google Scholar] [CrossRef]
- Scacchetti, A.; Schauer, T.; Reim, A.; Apostolou, Z.; Sparr, A. C.; Krause, S.; Heun, P.; Wierer, M.; Becker, P. B. Drosophila SWR1 and NuA4 Complexes Are Defined by DOMINO Isoforms. Elife 2020, 9. [Google Scholar] [CrossRef]
- Scacchetti, A.; Becker, P. B. Variation on a Theme: Evolutionary Strategies for H2A.Z Exchange by SWR1-Type Remodelers. Current Opinion in Cell Biology 2021. [Google Scholar] [CrossRef]
- Fujii, T.; Ueda, T.; Nagata, S.; Fukunaga, R. Essential Role of P400/MDomino Chromatin-Remodeling ATPase in Bone Marrow Hematopoiesis and Cell-Cycle Progression. Journal of Biological Chemistry 2010, 285. [Google Scholar] [CrossRef]
- Dickinson, M. E.; Flenniken, A. M.; Ji, X.; Teboul, L.; Wong, M. D.; White, J. K.; Meehan, T. F.; Weninger, W. J.; Westerberg, H.; Adissu, H.; et al. High-Throughput Discovery of Novel Developmental Phenotypes. Nature 2016, 537. [Google Scholar] [CrossRef]
- Faast, R.; Thonglairoam, V.; Schulz, T. C.; Beall, J.; Wells, J. R. E.; Taylor, H.; Matthaei, K.; Rathjen, P. D.; Tremethick, D. J.; Lyons, I. Histone Variant H2A.Z Is Required for Early Mammalian Development. Current Biology 2001, 11. [Google Scholar] [CrossRef]
- McHaourab, Z. F.; Perreault, A. A.; Venters, B. J. ChIP-Seq and ChIP-Exo Profiling of Pol II, H2A.Z, and H3K4me3 in Human K562 Cells. Sci Data 2018, 5. [Google Scholar] [CrossRef]
- Loppin, B.; Berger, F. Histone Variants: The Nexus of Developmental Decisions and Epigenetic Memory. Annual Review of Genetics 2020. [Google Scholar] [CrossRef]
- Kumar, V. C.; Pai, R. Genes of the Month: H3.3 Histone Genes: H3F3A and H3F3B. Journal of clinical pathology 2021. [Google Scholar] [CrossRef]
- Ishiuchi, T.; Abe, S.; Inoue, K.; Yeung, W. K. A.; Miki, Y.; Ogura, A.; Sasaki, H. Reprogramming of the Histone H3.3 Landscape in the Early Mouse Embryo. Nat Struct Mol Biol 2021, 28. [Google Scholar] [CrossRef]
- Kong, Q.; Banaszynski, L. A.; Geng, F.; Zhang, X.; Zhang, J.; Zhang, H.; O’Neill, C. L.; Yan, P.; Liu, Z.; Shido, K.; et al. Histone Variant H3.3–Mediated Chromatin Remodeling Is Essential for Paternal Genome Activation in Mouse Preimplantation Embryos. Journal of Biological Chemistry 2018, 293. [Google Scholar] [CrossRef]
- Wen, D.; Rosenwaks, Z. Activation of the Maternal Genome Through Asymmetric Distribution of Oocyte-Genome-Associated Histone H3.3. bioRxiv 2023, 2023.11.01.565208. [Google Scholar] [CrossRef]
- Bao, J.; Bedford, M. T. Epigenetic Regulation of the Histone-to-Protamine Transition during Spermiogenesis. Reproduction 2016. [Google Scholar] [CrossRef]
- Wen, D.; Banaszynski, L. A.; Liu, Y.; Geng, F.; Noh, K. M.; Xiang, J.; Elemento, O.; Rosenwaks, Z.; David Allis, C.; Rafii, S. Histone Variant H3.3 Is an Essential Maternal Factor for Oocyte Reprogramming. Proc Natl Acad Sci U S A 2014, 111. [Google Scholar] [CrossRef]
- Lin, Y.; Qiu, T.; Wei, G.; Que, Y.; Wang, W.; Kong, Y.; Xie, T.; Chen, X. Role of Histone Post-Translational Modifications in Inflammatory Diseases. Frontiers in Immunology 2022. [Google Scholar] [CrossRef]
- Park, J.; Lee, K.; Kim, K.; Yi, S. J. The Role of Histone Modifications: From Neurodevelopment to Neurodiseases. Signal Transduction and Targeted Therapy 2022. [Google Scholar] [CrossRef]
- Fitzsimons, C. P.; Van Bodegraven, E.; Schouten, M.; Lardenoije, R.; Kompotis, K.; Kenis, G.; Van Den Hurk, M.; Boks, M. P.; Biojone, C.; Joca, S.; et al. Epigenetic Regulation of Adult Neural Stem Cells: Implications for Alzheimer’s Disease. Molecular Neurodegeneration 2014. [Google Scholar] [CrossRef]
- Audesse, A. J.; Webb, A. E. Mechanisms of Enhanced Quiescence in Neural Stem Cell Aging. Mech Ageing Dev 2020, 191. [Google Scholar] [CrossRef]
- Basavarajappa, B. S.; Subbanna, S. Histone Methylation Regulation in Neurodegenerative Disorders. International Journal of Molecular Sciences 2021. [CrossRef]
- Ritchie, F. D.; Lizarraga, S. B. The Role of Histone Methyltransferases in Neurocognitive Disorders Associated with Brain Size Abnormalities. Frontiers in Neuroscience 2023. [Google Scholar] [CrossRef]
- Vitorakis, N.; Piperi, C. Insights into the Role of Histone Methylation in Brain Aging and Potential Therapeutic Interventions. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Ding, Y.; Liu, C.; Zhang, Y. Aging-Related Histone Modification Changes in Brain Function. Ibrain 2023. [Google Scholar] [CrossRef]
- Berson, A.; Nativio, R.; Berger, S. L.; Bonini, N. M. Epigenetic Regulation in Neurodegenerative Diseases. Trends in Neurosciences 2018. [Google Scholar] [CrossRef]
- Omole, A. E.; Fakoya, A. O. J. Ten Years of Progress and Promise of Induced Pluripotent Stem Cells: Historical Origins, Characteristics, Mechanisms, Limitations, and Potential Applications. PeerJ, 2018, 2018 (MAY). [CrossRef]
- Papp, B.; Plath, K. Epigenetics of Reprogramming to Induced Pluripotency. Cell 2013. [Google Scholar] [CrossRef]
- Huang, M.; Xiao, X.; Ji, G.; Wu, Q. Histone Modifications in Neurodifferentiation of Embryonic Stem Cells. Heliyon 2022. [Google Scholar] [CrossRef]
- Castillo Bautista, C. M.; Sterneckert, J. Progress and Challenges in Directing the Differentiation of Human IPSCs into Spinal Motor Neurons. Frontiers in Cell and Developmental Biology 2023. [CrossRef]
- Ganesan, A.; Arimondo, P. B.; Rots, M. G.; Jeronimo, C.; Berdasco, M. The Timeline of Epigenetic Drug Discovery: From Reality to Dreams. Clinical Epigenetics 2019. [Google Scholar] [CrossRef]
- Holdgate, G. A.; Bardelle, C.; Lanne, A.; Read, J.; O’Donovan, D. H.; Smith, J. M.; Selmi, N.; Sheppard, R. Drug Discovery for Epigenetics Targets. Drug Discovery Today 2022. [Google Scholar] [CrossRef]
- Montecino, M.; Carrasco, M. E.; Nardocci, G. Epigenetic Control of Osteogenic Lineage Commitment. Frontiers in Cell and Developmental Biology. Frontiers Media S.A. January 8, 2021. [CrossRef]
- Sepulveda, H.; Aguilar, R.; Prieto, C. P.; Bustos, F.; Aedo, S.; Lattus, J.; van Zundert, B.; Palma, V.; Montecino, M. Epigenetic Signatures at the RUNX2-P1 and Sp7 Gene Promoters Control Osteogenic Lineage Commitment of Umbilical Cord-Derived Mesenchymal Stem Cells. J Cell Physiol 2017, 232. [Google Scholar] [CrossRef]
- Jin, M. L.; Jeong, K. W. Histone Modifications in Drug-Resistant Cancers: From a Cancer Stem Cell and Immune Evasion Perspective. Experimental and Molecular Medicine 2023. [Google Scholar] [CrossRef]
- Bajbouj, K.; Al-ali, A.; Ramakrishnan, R. K.; Saber-ayad, M.; Hamid, Q. Histone Modification in Nsclc: Molecular Mechanisms and Therapeutic Targets. International Journal of Molecular Sciences 2021. [CrossRef]
- Markouli, M.; Strepkos, D.; Piperi, C. Impact of Histone Modifications and Their Therapeutic Targeting in Hematological Malignancies. International Journal of Molecular Sciences 2022. [CrossRef]
- Monaghan, L.; Massett, M. E.; Bunschoten, R. P.; Hoose, A.; Pirvan, P. A.; Liskamp, R. M. J.; Jørgensen, H. G.; Huang, X. The Emerging Role of H3K9me3 as a Potential Therapeutic Target in Acute Myeloid Leukemia. Frontiers in Oncology 2019. [Google Scholar] [CrossRef]
- He, L. R.; Liu, M. Z.; Li, B. K.; Rao, H. L.; Liao, Y. J.; Guan, X. Y.; Zeng, Y. X.; Xie, D. Prognostic Impact of H3K27me3 Expression on Locoregional Progression after Chemoradiotherapy in Esophageal Squamous Cell Carcinoma. BMC Cancer 2009, 9. [Google Scholar] [CrossRef]
- Ngollo, M.; Lebert, A.; Daures, M.; Judes, G.; Rifai, K.; Dubois, L.; Kemeny, J. L.; Penault-Llorca, F.; Bignon, Y. J.; Guy, L.; et al. Global Analysis of H3K27me3 as an Epigenetic Marker in Prostate Cancer Progression. BMC Cancer 2017, 17. [Google Scholar] [CrossRef]
- Belhocine, M.; Simonin, M.; Abad Flores, J. D.; Cieslak, A.; Manosalva, I.; Pradel, L.; Smith, C.; Mathieu, E. L.; Charbonnier, G.; Martens, J. H. A.; et al. Dynamics of Broad H3K4me3 Domains Uncover an Epigenetic Switch between Cell Identity and Cancer-Related Genes. Genome Res 2022, 32. [Google Scholar] [CrossRef]
- Dutta, H.; Jain, N. Post-Translational Modifications and Their Implications in Cancer. Frontiers in Oncology 2023. [Google Scholar] [CrossRef]
- Guan, X.; Deng, H.; Choi, U. L.; Li, Z.; Yang, Y.; Zeng, J.; Liu, Y.; Zhang, X.; Li, G. EZH2 Overexpression Dampens Tumor-Suppressive Signals via an EGR1 Silencer to Drive Breast Tumorigenesis. Oncogene 2020, 39. [Google Scholar] [CrossRef]
- Hirukawa, A.; Smith, H. W.; Zuo, D.; Dufour, C. R.; Savage, P.; Bertos, N.; Johnson, R. M.; Bui, T.; Bourque, G.; Basik, M.; et al. Targeting EZH2 Reactivates a Breast Cancer Subtype-Specific Anti-Metastatic Transcriptional Program. Nat Commun 2018, 9. [Google Scholar] [CrossRef]
- Sun, Y.; Jiang, X.; Xu, Y.; Ayrapetov, M. K.; Moreau, L. A.; Whetstine, J. R.; Price, B. D. Histone H3 Methylation Links DNA Damage Detection to Activation of the Tumour Suppressor Tip60. Nat Cell Biol 2009, 11. [Google Scholar] [CrossRef]
Figure 1.
Epigenetic dynamics of H3K4me3 and H3K9me3 in early embryogenesis. (A) H3K4me3 experiences swift depletion in the paternal genome post-fertilization, only to be reinstated during ZGA. Conversely, the deposition of ncH3K4me3 coincides with the silencing of the genome originating from fully grown oocytes and is subsequently substituted by canonical H3K4me3 at the 2-cell stage. Throughout ZGA, chromatin accessibility becomes evident at the H3K4me3-marked TSS sites of active genes. Simultaneously, transposable elements exhibit accessibility and enrichment in H3K4me3. Progressing through development, there is a gradual escalation of H3K4me3 marks, culminating in forming a bivalent state with H3K27me3, priming the activation of developmental genes during the blastocyst stage. (B) Maternal genome-specific regions with H3K9me3 marks are more prevalent than paternal genome regions during early embryogenesis. The onset of early embryonic development triggers extensive genome-wide demethylation, resulting in the hypomethylation of actively transcribed regions, including LTRs. After fertilization, H3K9me3 marks within LTRs gradually increase during pre-implantation development. The majority of parental H3K9me3 regions are established de novo after fertilization. H3K9me3-enriched LTRs progressively emerge after the 4-cell stage and play a role in LTR silencing. Additionally, H3K9me3 marks in the promoters of developmental genes are erased after fertilization and subsequently restored after implantation. Abbreviations: LTRs, long terminal repeats; MZT, Maternal-to-zygotic transition; TSS, transcriptional start sites; ZGA, zygotic genome activation. Created with BioRender.com .
Figure 1.
Epigenetic dynamics of H3K4me3 and H3K9me3 in early embryogenesis. (A) H3K4me3 experiences swift depletion in the paternal genome post-fertilization, only to be reinstated during ZGA. Conversely, the deposition of ncH3K4me3 coincides with the silencing of the genome originating from fully grown oocytes and is subsequently substituted by canonical H3K4me3 at the 2-cell stage. Throughout ZGA, chromatin accessibility becomes evident at the H3K4me3-marked TSS sites of active genes. Simultaneously, transposable elements exhibit accessibility and enrichment in H3K4me3. Progressing through development, there is a gradual escalation of H3K4me3 marks, culminating in forming a bivalent state with H3K27me3, priming the activation of developmental genes during the blastocyst stage. (B) Maternal genome-specific regions with H3K9me3 marks are more prevalent than paternal genome regions during early embryogenesis. The onset of early embryonic development triggers extensive genome-wide demethylation, resulting in the hypomethylation of actively transcribed regions, including LTRs. After fertilization, H3K9me3 marks within LTRs gradually increase during pre-implantation development. The majority of parental H3K9me3 regions are established de novo after fertilization. H3K9me3-enriched LTRs progressively emerge after the 4-cell stage and play a role in LTR silencing. Additionally, H3K9me3 marks in the promoters of developmental genes are erased after fertilization and subsequently restored after implantation. Abbreviations: LTRs, long terminal repeats; MZT, Maternal-to-zygotic transition; TSS, transcriptional start sites; ZGA, zygotic genome activation. Created with BioRender.com .
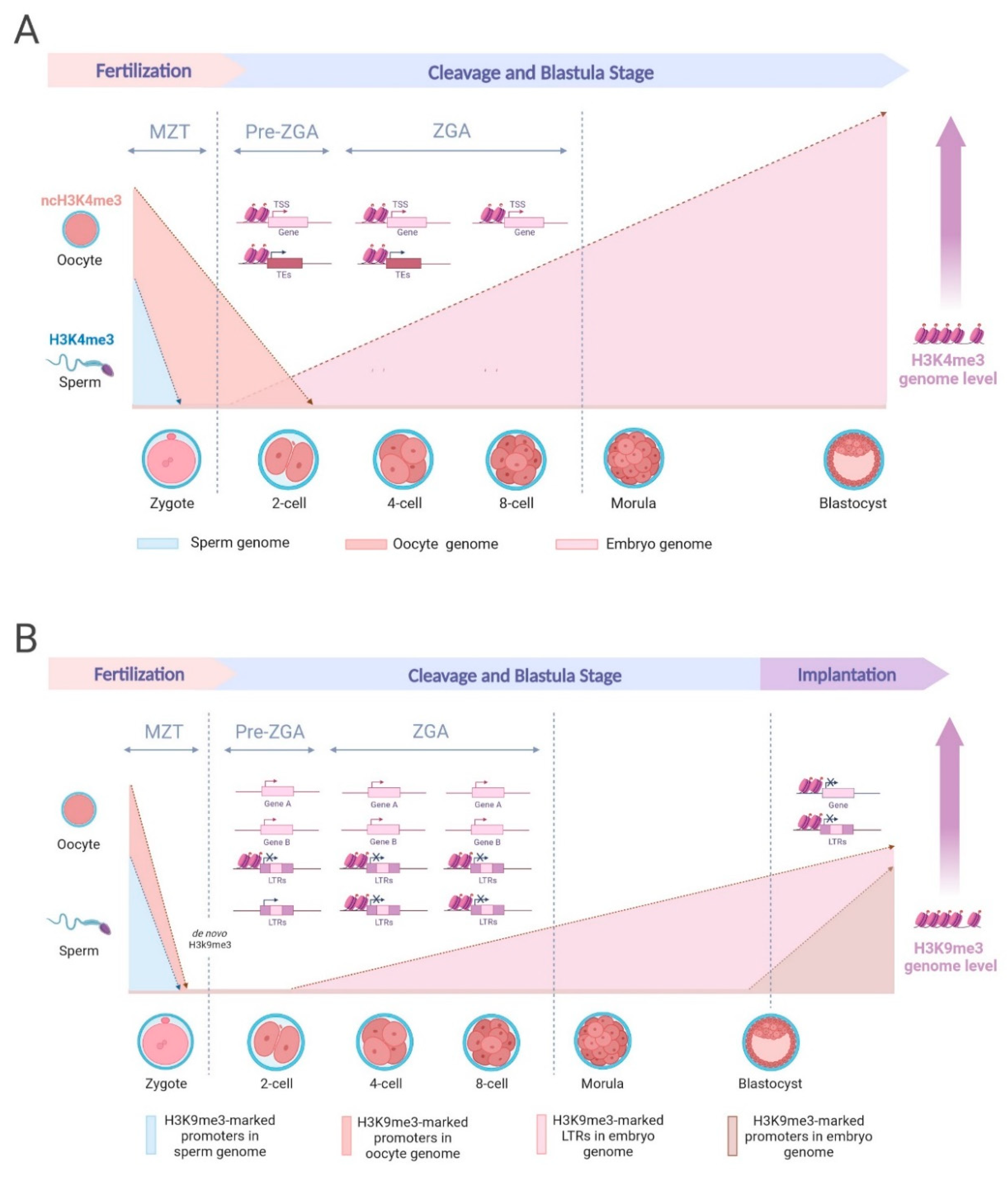
Figure 2.
Antagonistic activity between H3K27me3 and H3K27ac in ZGA gene expression. (A) A polycomb repressive complex (PRC2) orchestrates the trimethylation of histone 3 on lysine 27 through histone methyltransferase activity. The SAM cofactor in Enhancer of zeste homolog 2 (EZH2), one of the PCR2 subunits, is essential to recognize the H3K27 tail selectively. The introduction of the trimethylation mark on lysine 27 results in the downregulation of nearby genes by forming heterochromatic regions. H3K27me3 in promoter regions experiences a widespread loss at the 2-cell stage. Additionally, H3K27me3 manifests in non-promoter regions in a highly pervasive and promiscuous manner. (B) A HAT facilitates the acetylation of histone 3 on lysine 27 by transferring an acetyl group from acetyl CoA. H3K27ac is an active enhancer mark, promoting a more accessible chromatin structure. It is present in both distal and proximal regions of genes, with enrichment observed at TSS. The H3K27ac marks exhibit discernible intensity in zygotes, 2-cell, and 4-cell embryos but gradually decline in 8-cell embryos. Abbreviations: HAT, histone acetyltransferase; PRC2, Polycomb Repressive Complex 2; RNAPII, RNA polymerase II; SAM, S-adenosylmethionine; TSS, transcriptional start sites. Created with BioRender.com.
Figure 2.
Antagonistic activity between H3K27me3 and H3K27ac in ZGA gene expression. (A) A polycomb repressive complex (PRC2) orchestrates the trimethylation of histone 3 on lysine 27 through histone methyltransferase activity. The SAM cofactor in Enhancer of zeste homolog 2 (EZH2), one of the PCR2 subunits, is essential to recognize the H3K27 tail selectively. The introduction of the trimethylation mark on lysine 27 results in the downregulation of nearby genes by forming heterochromatic regions. H3K27me3 in promoter regions experiences a widespread loss at the 2-cell stage. Additionally, H3K27me3 manifests in non-promoter regions in a highly pervasive and promiscuous manner. (B) A HAT facilitates the acetylation of histone 3 on lysine 27 by transferring an acetyl group from acetyl CoA. H3K27ac is an active enhancer mark, promoting a more accessible chromatin structure. It is present in both distal and proximal regions of genes, with enrichment observed at TSS. The H3K27ac marks exhibit discernible intensity in zygotes, 2-cell, and 4-cell embryos but gradually decline in 8-cell embryos. Abbreviations: HAT, histone acetyltransferase; PRC2, Polycomb Repressive Complex 2; RNAPII, RNA polymerase II; SAM, S-adenosylmethionine; TSS, transcriptional start sites. Created with BioRender.com.
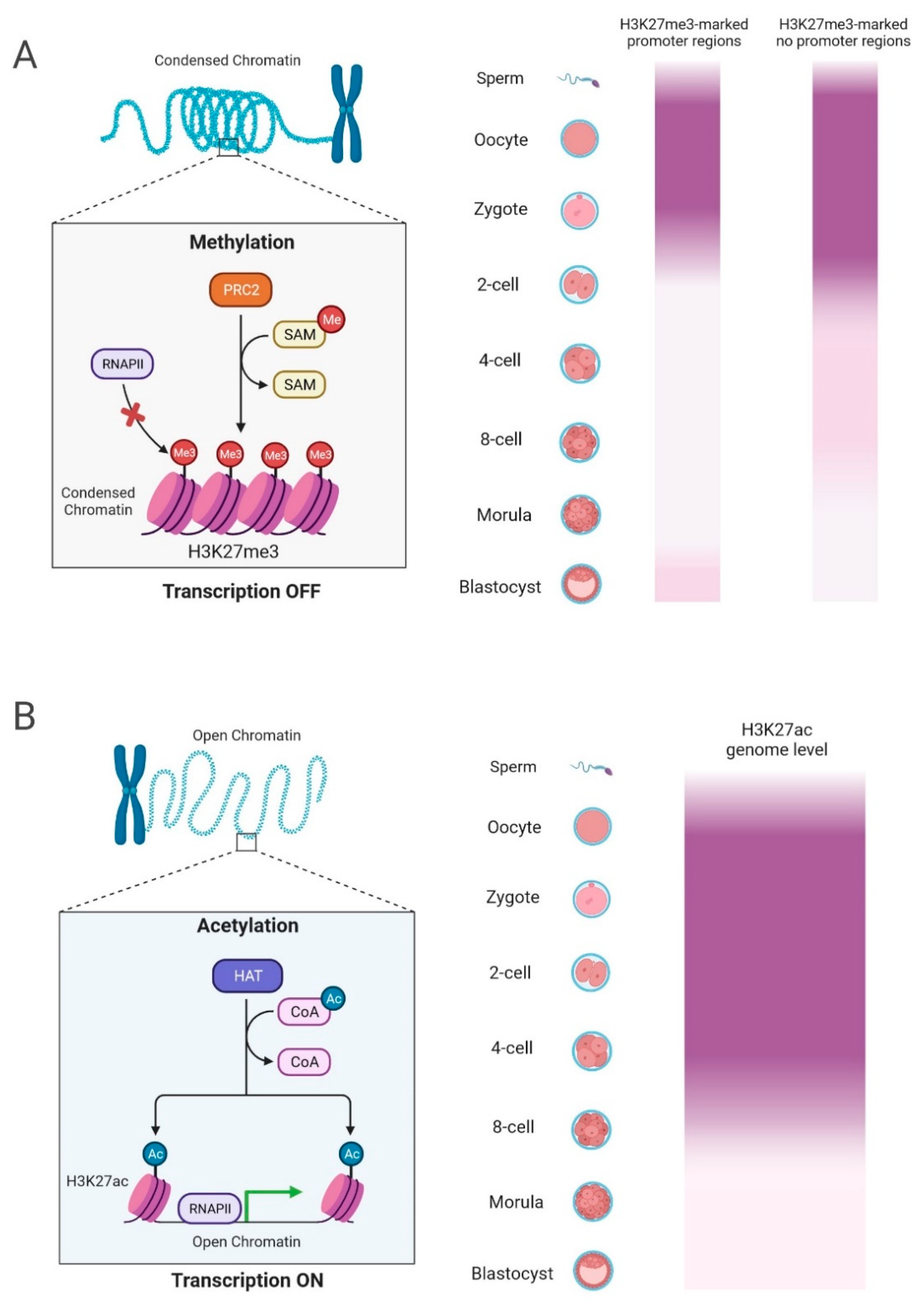
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



