
Submitted:
30 December 2023
Posted:
03 January 2024
You are already at the latest version
Abstract
There is an urgent need to develop new therapies for cancer treatment due to high rates of resistance and tumor relapse. Moreover, chemotherapy and radiation weaken the immune system and leave patients susceptible to sudden death from infections and organ failure since antineoplastic drugs lack specificity. Hence, scientists worldwide continue the search for natural alternatives with anticancer activities and fewer side effects. In this regard, traditional medicine offers strategies for preventing and treating numerous diseases and nowadays scientific studies worldwide highlight some of the mechanisms underlying their potential as an effective alternative for the management of cancer. In Mexico, Mayan healers have been prescribed mainly medicinal plants for more than 2,000 years. Furthermore, their ethnomedical knowledge has led to drug discovery or the development of herbal remedies. Medicinal plants from the genus of Cissus remain an important element of all indigenous medical systems in Mexico including the Mayan tribes in Yucatan, particularly for the management of gastrointestinal illnesses, sores, cutaneous diseases, and tumors. In this review, we present the reported bioactivities against cancer cells of the extract and compounds from the species of Cissus with a particular focus on recent studies on the phytochemistry of C. trifoliata which is widely distributed in Mexican territory and used for tumor management in traditional medicine.
Keywords:
Cissus sp.
; Phytochemicals
; Apoptosis
; Proliferation
; EMT
; Anticancer
Introduction
Cancer is the second leading cause of morbidity and mortality worldwide, with approximately 19 million new cases per year, which are expected to rise by about 70% over the next two decades. Moreover, cancer is responsible for 10 million deaths annually, representing 1 of 6 deaths 1. In Mexico, cancer is the third leading cause of death, and 45% of deceases occur in the economically active population. Malignant tumors from the lung, liver, prostate, breast, and cervix account for approximately half of all cancer deaths 2. In this respect, neoplasias that arise from epithelial tissues are classified as carcinomas and account for 90% of human cancers 3. Hence, developing successful therapies for them may result in dramatic changes in the mortality associated with malignant neoplasias 3. Cancer is a disease characterized by the uncontrolled growth of abnormal cells that impair the biological process of tissues. In addition, malignant cells spread by invasion of nearby tissues and metastasize to distant organs, compromising their function and ultimately resulting in death. Importantly, metastasis is the leading cause of cancer-associated deceases since they usually display increased resistance to radio and chemotherapy 3. The malignant neoplasias from the epithelia possess an enhanced tolerance to cell death along with the sustained proliferative signaling and the evasion of growth suppression signals that lead to tumorigenesis 4, whereas the process of epithelial to mesenchymal transition (EMT) promotes the generation of metastasis and the emergence of drug resistance 5. Therefore, compounds with the ability to trigger apoptosis, cell cycle arrest, or prevent the process of EMT may represent promising therapeutic strategies against carcinomas 4, 5.
Cancer management includes surgery, radiation, and systemic drugs such as the conventional cytotoxic and endocrine agents. More recently, the knowledge of the molecular biology of tumors has given rise to molecules designed to target specific oncogenes hoping to reduce the side effects of common drugs 6. However, most employed therapeutic regimens for cancer include at least one type of cytotoxic drug; whose mechanisms of action may include the generation of DNA damage, the inhibition of essential enzymes, microtubules, and topoisomerases, which ultimately generates unspecific cell death 6. Hence, antineoplasic treatment causes toxicity in normal tissues leading to side effects that include vomit, alopecia, diarrhea, constipation, and life-threatening consequences such as myelosuppression, cystitis, gastric ulcers, lung fibrosis, cardiotoxicity, hepatotoxicity, mucositis, and nephrotoxicity. Additionally, second malignant neoplasms can develop as a result of chemotherapy regimens or radiation within the first year after treatment, being acute leukemias the more common 7. Moreover, treatment is expensive and reduces the quality of life during and after their administration 8. Importantly, over half of patients are diagnosed in advanced stages in which therapies are mostly ineffective 9. Therefore, despite advances in cancer care tumor resistance and toxicity derived from therapy remain the big challenges limiting the survival of patients 10.
Plants as a potential source of new anticancer drugs
Drug discovery from medicinal plants has played an important role in the treatment of cancer 11. Antineoplastic agents from plants in clinical use include the vinca alkaloids (vinblastine, vincristine and vinorelbine), epipodophyllotoxin lignans (etoposide and teniposide), taxane diterpenoids (paclitaxel and docetaxel) and camptothecin alkaloids (topotecan and irinotecan) 12. According to the National Cancer Institute (NCI), the initial screenings for plants with potential anticancer activity should be performed in established cell lines, in which the toxic effects of extracts or isolated compounds are measured. Following that guideline, a plant is considered anticancer if its extracts or isolated compounds show a half maximal inhibitory concentration (IC50) value of ≤30 µg/ml or ≤4 µg/ml, respectively 13. In general, the MTT method is selected to evaluate the cytotoxic activity of both in different concentrations. The assay relies on mitochondrial metabolism since living cells reduce the tetrazolium salts to formazan products. Then, this reaction is quantified by optical density in an ELISA reader providing information on the amount of living cells. Hence, the MTT assay uses the IC50 value as a parameter to measure cytotoxic activity, indicating the amount of extract or compound concentration needed to inhibit half of the proliferation of cancer cells 14. In this contribution is reviewed the reported phytochemical composition of C. trifoliata and the bioactivity of their extracts or compounds against cancer cells along with the underlying mechanism of action. Emphasis was made on recovering the IC50 against the cell models of lung cancer (A549), liver cancer (HepG2), breast cancer (MCF7), cervix cancer (HeLa), and prostate cancer (PC3) since they represent the most studied cultures of the most common types of carcinomas diagnosed in Mexican population 15. In addition, evidence from those phytochemicals against the process of proliferation and EMT was also discussed due to their importance in tumorigenesis and for the emergence of drug resistance and metastasis in carcinomas.
Anticancer effects of C. trifoliata
Cissus plants are 350 species of lianas widely distributed in the tropical and subtropical regions in Asia, the Americas, Africa, and Australia 16. They belong to the family Vitaceae and have been used in traditional medicine for the management of several diseases, such as diabetes, infections, arthritis, menopause, obesity, pain, and cancer 17. Cissus trifoliata is widely distributed in tropical America and Mexican territory, being native to Baja California, Chihuahua, Coahuila, Durango, Nuevo León, San Luis Potosí, Sinaloa, Sonora, Tamaulipas, Puebla, Michoacán, Veracruz, Oaxaca, Quintana Roo and Yucatán. Is also present in the United States, Venezuela, Colombia, and Ecuador 18. Moreover is also dispersed among some islands in the Caribbean such as Aruba, Bahamas, Cuba, Haiti, Jamaica, and Puerto Rico 19. Cissus trifoliata mexican specimens have also been referred to as C. incisa (Nutt.) Des MouL 19, but recent genetic studies confirm the lack of differences between both species 20. Although C. trifoliata (L.) is the accepted botanical name; is also referred to as C. acida, C. carnifolia, Vitis incisa, V. trifoliata, possum-grape, sorrelvine, vine-sorrel, Hierba del buey in spanish and called Xbolontibi in Maya 19. Cissus trifoliata has been an important medicinal plant for the Mayan tribes in Yucatan, particularly for the management of gastrointestinal illnesses 21, sores 22, cutaneous diseases, and tumors 23, 24. Likewise, in Northeastern Mexico, is used by the native population to treat skin infections, inflammation, abscesses, and tumors 25. In other parts of America ethnobotanical uses for C. trifoliata include burns, sore, and tumors 19. Modern studies of its biological activities against cancer include the evaluation of the cytotoxic activity of the stem extracts against the human cancer cell lines A549, Hep3B, MCF7, HeLa, and PC3 obtained from the ATCC. Notably, the in vitro evaluation was consistent with the antitumor properties suggested by traditional medicine since the hexane extract showed exceptional activity against the cancer cell lines Hep3B with an IC50 value of 26 µg/ml and MCF7 with an IC50 value of 30 µg/ml whereas the aqueous extract also displayed antiproliferative properties on MCF7 cells with an IC50 value of 30 µg/ml. Although the CHCl3-MeOH extract lacks activity at the desired range of IC50 ≤30 µg/ml, all tested cells were sensitive to all extracts at doses below 100 µg/ml 15 (Table 1). In this regard, based on other authors, plant extracts in the range of IC50 from 20-100 μg/ml are classified as moderately active against cancer cells 26.
Although scarce, similar studies have been performed for other species of the genus Cissus. Namely, the hexane extract of C. quadrangularis stems displayed low activity against KB (keratin-forming tumor cell line HeLa) and A431 (epidermoid carcinoma) with an estimated IC50 value of 200 μg/ml for both 27. In addition, the acetonic extract was more active against HepG2 with an estimated IC50 value of 43 µg/ml and IC50 value of 48 μg/ml for KB cells 28. Likewise, the ethyl acetate extract from the stems of C. sicyoides showed an IC50 value of 43 μg/ml against the HepG2 29 while the methanolic extract from the stems of C. debilis inhibits the colon carcinoma cell line CaCo-2 with an estimated IC50 value of 50 µg/ml 30. Finally, the ethyl acetate and ethanolic extracts of C. verticillata exhibit an IC50 value of 43 µg/ml and 50 µg/ml against the HepG2 and NCI-H292 cells respectively 30. Therefore, most Cissus plant extracts can be considered moderately active against all carcinoma cells tested according to their calculated IC50 value. The presence of active extracts suggests that tested plants possess bioactive compounds with potential anticancer activities. Therefore, following the NCI guideline further studies are desirable in those specimens 13. In the next sections, current work on the phytochemistry of C. trifoliata is provided, along with the role of compounds in plant physiology and their cytotoxic, antiproliferative, and anti-EMT activities against the carcinoma cell lines of interest.
Cissus trifoliata compounds and their role in plant physiology
Metabolic profiling has been previously useful to understand the chemical diversity of a medicinal plant. Chromatography coupled with mass spectrometry is the most widely applied technology used for the analysis of samples in very complex matrices such as those of plant extracts 15. Accordingly, the metabolic profile of the hexane, CHCl3-MeOH, and aqueous extracts of C. trifoliata stems was assessed by Gas chromatography-mass spectrometry (GC-MS) and Ultraperformance Liquid Chromatography-Quadrupole Time of Fly-Mass Spectrometry (UPLC-QTOF-MS) analysis. Broadly, the analysis showed a metabolic profile composed of flavonoids, stilbenes, phenolics, fatty acids, sterols, alkanes, alcohols, and triterpenes 15. Furthermore, results were consistent with chemical classes reported in previous studies in some other species of Cissus plants 31 and well-characterized specimens from Vitaceae 32. The most abundant lipid compounds corresponded to alkanes, fatty acids, fatty alcohols, sterols, and terpenes. The alkanes comprise the hentriacontane, nonacosane, and octacosane, and the alcohol triacontanediol. The fatty acids include palmitic acid, stearic acid, and arachidic acid. Finally, identified sterols consist of campesterol, stigmasterol, and sitosterol, whereas the triterpenes involve squalene, betulinic acid, ursolic acid, and lupeol. Overall, the lipid composition seems consistent with typical plant waxes 33, 34, since major constituents are usually unsaturated linear hydrocarbons, long-chain saturated fatty acids 35, long-chain saturated esters 36, terpenes, and sterols 37. Furthermore, the alkanes identified octacosane, nonacosane, and hentriacontane are predominant cuticular wax components in plants 38. In the case of triacontanediol, is a common fatty alcohol present in the plan cuticular wax 38 that also plays a role as a growth regulator 39. Palmitic and steric acids are the most abundant plant membrane fatty acids that play structural roles 40 and influence the fluidity of cellular membranes of higher plants 41. In the case of eicosanoic acid in general is a minor constituent of plant cell membranes 42. Likewise, sterols regulate the fluidity of membranes, and cellular developmental processes in plants, acting as precursors of plant hormones 43. While a variety of sterols exist, campesterol, stigmasterol, and β-sitosterol are the most abundant in plants 43 playing structural roles in plant membrane fluidity 44. However, when plants are infestated by insects changes in plant sterol structure confer them an antifeedant role 45. The ursolic acid also exhibits antifeedant properties against insect larvae 46 whereas the biosynthesis of lupeol is induced by pathogens and exerts antimicrobial activities 47. Squalene increases the rigidity and the size of the cell membrane, enhancing the polarity and hydrophobic interactions, contributing to membrane reconstitution, functional regulation of proteins, and movement of ions. Therefore, squalene plays an important role in electrochemical cell gradient 48. Finally, betulinic acid has been detected in different plant species, and its common occurrence in exposed organs such as leaves, bark, or fruits, suggests a defensive role for this triterpenoid 49.
Most lipid compounds present in C. trifoliata have been previously identified as constituents in other Cissus plants (Table 2). For example, the alkane hentriacontane was reported in C. quadrangularis stems 50, while the nonacosane in C. cornifolia 51. Similarly, the analysis of the hexane extracts of C. quadrangularis stems identified as the main components the hexadecanoic acid ethyl ester, the octadecanoic acid ethyl ester, and phytol 52. The palmitic acid is also the principal component of the hexane extract of the stems of C. quadrangularis 52 and their aerial parts (aqueous alcoholic extract) 53. Palmitic acid is also present in C. vitiginea 50, and its content in plant extracts is high in low-polarity solvents. The steric acid was found as a major constituent of the hexane extract of stems of C. quadrangularis, and in the methanolic extract of the entire plant 52, 54. In the case of eicosanoic acid, was also a major constituent of hexane extract of roots in C. quadrangularis 52. Regarding sterols, β-sitosterol and stigmasterol were previously isolated from the hexane extract of stems of C. quadrangularis 52 and the methanolic and ethyl acetate extracts of the aerial parts and roots of C. assamica 55, C. polyantha 56, C. rheoifolia 57, 58 and C. pteroclada 58. Campesterol was isolated from hexane 52 and ethanolic extract of C. quadrangularis stems 59 and is also found in the methanolic extract of the roots of C. rheifolia 57. Concerning terpenes, squalene was previously isolated from Cissus quadrangularis 60, lupeol was found as a major constituent of hexane extract of stems and roots in C. quadrangularis 52, 60, ursolic acid was previously isolated from C. assamica 55 and C. repens 61 and the ursolic and betulic acids in C. assamica 62. Overall, the lipid composition reported for C. trifoliata is widely distributed in the plant kingdom 37 and corresponds mainly to constituents of plant cuticular waxes and membranes 63. In contrast, the polar composition presented the chemical classes of phenolic acids, flavonoids, and stilbenes, with these last phytocompounds characterized by a narrow distribution within plants 15. The phenolic compounds detected include isoferulic acid, protochatechuic acid, trans-p-coumaric acid, and trigallic acid. Flavonoids represent the most abundant class with glucosides of apigenin, chrysoeroil, cyanidin, delphinidin, dihydrokaempferol, kampferol, myricetin, naringenin, quercetin, and syringetin. Finally, the stilbenes identified comprise resveratrol, piceatannol, pallidol, piceid and viniferin.
Phenolic compounds are of considerable physiological and morphological importance in plants, contributing to growth and reproduction, protection against pathogens or predators, and to the color and sensory characteristics of fruits and vegetables. For example, gallic acid derivatives like the protocatechuic acid, and trigallic acid are secondary metabolites widely distributed in the plant kingdom that play a regulatory role in the induction of abiotic stress tolerance or to enhance the direct defense against insects 64. Isoferulic acid is a structural component in the plant cell wall and serves to increase its rigidity and strength 65. Trans-p-coumaric acid biosynthesis and storage in the plant cell play a vital role in response to pathogenic infections 66. Concerning flavonoids, they have multiple functions including the response to environmental injuries, the regulation of cell growth, and the attraction of pollinators 67. For example, flavonoids yield stress protection by acting as ROS scavengers or inducers of antioxidant enzymes. In this regard, apigenin and other flavones protect photosynthetic tissues from oxidative damage and confer tolerance to salinity 68. Chrysoeriol provides strong antifeedant activity since it is toxic for insects 69. In the case of the anthocyanins cyanidin and delphinidin, both are involved in responses to oxidative stress induced by heat conditions, water or nutrient deficit, and mechanical damage due to herbivore attack, insect infestation, or fungal infection 70. Kaempferol and quercetin are the two main flavonoid species abundantly present in plants. Both were found to be associated with auxin-regulated cell division acting as signaling molecules. In addition, environmental and pathogenic stress induces kaempferol accumulation, whereas quercetin and anthocyanins, alter an interaction between jasmonic acid and gibberellic acid which results in the regulation of the defense system to cope with stress. Likewise, in some stressful conditions, plants convert kaempferol and quercetin glycosides into anthocyanins, which promote the plant defense system. Flavonoids accumulate in mesophyll cells, vacuole, and chloroplasts reducing ROS generation. For example, quercetin generated in the plant epidermal tissue protects from oxidative stress induced by an intense light 71. Moreover, the concomitant increase in flavonoids with the concentration of heavy metals in plant tissue suggests a role in alleviating stress. Indeed, myricetin in conjunction with kaempferol was observed to increase the phytoremediation capacity of some plants due to the high accumulation of heavy metals 72. Likewise, dihydrokaempferol and quercetin increased salt tolerance 73 and the naringenin suppressed the growth of annual plant species, acting as an allelochemical 74. This inhibitory effect of naringenin was attributed at least to some extent, to impaired auxin transport. In addition, recent studies find that naringenin alleviates osmotic and salinity stresses by regulating photosynthetic machinery and chloroplastic antioxidant metabolism 74. Hence, the induction of flavonoids correlates with mechanisms against different types of stress such as ozone, light, heat, and salinity but also with exposure to biotic aggressors. For instance, bacterial or fungal-mediated infection is inhibited by flavonols such as myricetin and anthocyanins like delphinidin 74. Transcriptional upregulation of flavonols, anthocyanin, and proanthocyanidins biosynthesis was found to promote the accumulation of quercetin, kaempferol, and anthocyanins and enhance resistance to infections. Additionally, higher levels of flavonols such as kaempferol, isorhamnetin, and syringetin, reduce oxidative damage and susceptibility to fungal infections 75. Finally, in contrast to phenolics and flavonoids, stilbenes show a narrow distribution in the plant kingdom. They have been identified in 72 plant species belonging to Pinaceae, Gnetaceae, Fabaceae, Polygonaceae, Moraceae, and Vitaceae. Stilbene compounds mostly derive from resveratrol although different structures can be found in specific plant families. In C. trifoliata, resveratrol was identified along to piceatannol, pallidol, piceid and viniferin. Stilbenes are mainly involved in constitutive and inducible protection of the plant against phytopathogens, hence displaying antibacterial, antifungal, nematocidal, and insecticidal properties. In addition, their levels increase in response to drought, heat, radiation, heavy metals, salts, air pollutants, and mechanical stress 76, 77. Broadly, the presence of phenolics in C. trifoliata includes gallic acid derivatives such as protocatechuic acid, trigallic acid, and methyl digallate which are secondary metabolites widely distributed in the plant kingdom 64. Apigenin was previously isolated from C. adnata 78, C. ibuensis 79, C. digitata 80, C. repens 81, C. verticillata 82 and C. quadrangularis 83. Kaempferol presence has been identified in C. quadrangularis 84, C. ibuensis 79, C. repens 81, and C. sicyoides 85 whereas dihydrokaempferol and myricetin in C. quadrangularis 86. Quercetin has been reported on alcoholic extracts from C. digitate 80, C. quadrangularis 83, and C. repens 81. The anthocyanidins, cyanidin and delphinidin were identified in the methanolic extract of C. sicyoides 87 and petroleum ether extract of the stems of C. quadrangularis 86. Chrysoeriol was found in C. aralioides, C. lageniflora and C. petiolata 88 and naringenin in C. rotundifolia 89. Syringetin is not previously reported in Cissus plants, however, is a common flavonol identified in the close relative Vitis vinifera, which is also rich in several glycosides with quercetin, myricetin, and kaempferol 90. Finally, resveratrol, piceatannol, and pallidol were isolated and characterized in ethanolic extracts from the stems of C. quadrangularis 31, whereas pallidol in C. pallida 91. Additionally, the glucosides of the stilbenes piceatanol and ε-viniferin were previously identified in C. quadrangularis 31 and C. repens 81 (Table 3).
Mechanisms implicated in the cytotoxicity of C. trifoliata compounds
The process of apoptosis is characterized by several morphological changes including cell shrinkage, membrane blebbing, and nuclear DNA fragmentation. Broadly speaking, this form of cell death is the result of two mechanisms, one is the cytoplasmic pathway triggered by extracellular signals through death receptors located on the cell membrane of the target cell that belong to the TNF superfamily. On the other hand, the intrinsic pathway is activated by intracellular signals that induce the release of cytochrome-C to the cytoplasm from the intermembrane area of the mitochondria and leads to the activation of the apoptosome complex composed of cytosolic factor Apaf-1, ATP and active caspase 9. Caspases are inactive cysteine proteases until their proteolytic cleavage, caspases 2,8,9 and 10 act as initiators while 3,6 and 7 as effectors. On the other hand, members of the Bcl-2 family include proteins that prevent apoptosis such as Bcl-2, and Bcl-xL, and those that promote it such as Bax, Bad, Bid, Bak, and Bcl-xS 92. Given that the apoptosis of cancer cells continues to be one of the main objectives in preliminary screenings in the search for antineoplastic drugs 93, the following section provides the in vitro experimental evidence of the compounds present in C. trifoliata that have a calculated IC50 against all the carcinoma cell lines of interest and the apoptosis-inducing mechanisms that have been described to date (Table 4).
Terpenes are derived from five-carbon isoprene units and classified based on the number of them. In nature, triterpenoids are biosynthesized from five isoprene units using the mevalonate pathway and are usually found as tetracyclic or pentacyclic structures 94. Plant sterols belong to this subclass of terpenes and are composed of three 6-carbon rings and a 5-carbon ring with a double bond between carbons 5 and 6, a hydroxyl group on carbon 3, and a hydrocarbon side chain at the 17C position 94. The more abundant sterols are the β-sitosterol, campesterol and stigmasterol. In vitro studies have revealed potential anticancer effects of phytosterols, particularly from β-sitosterol and stigmasterol 94. In this regard, β-sitosterol shows cytotoxic activity against the cell lines A549 (231 µM) 95, Hep3B (60 μM) 96, MCF7 (603 μM) 95, PC3 (178 μM) 97, and HeLa (410 μM) 98. Treatment of MCF7 cells with β-sitosterol result in increased caspase-8 activity 99 and defects in sphingolipid metabolism, causing apoptosis and cell growth inhibition in a dose-dependent manner 100. Stigmasterol also display cytotoxic activity against A549 (51 μM), MCF7 (22 μM), PC3 (18 μM) 95, HeLa (412 μM) 98 and Hep3B (30 μM) cells 101. Evidence from HepG2 cultures indicates that apoptosis is triggered by the upregulation of Bax and p53 expression and the downregulation of Bcl-2 102. The pentacyclic triterpenes also have anticancer activity, they are generally present in higher plants and contain the ursane, oleanane, lupane, and friedelane skeletons 103. Lupeol is the form of lupan in which hydrogen at the 3β position is replaced by a hydroxy group. It shows cytotoxic activity against A549 (49 μM) 95, HepG2 (112 μM) 104, MCF7 (75 μM), PC3 (70 μM) 95, and HeLa (88 μM) cell lines 105. In PC3 cells, lupeol induces apoptosis through the mitochondrial cell death pathway by downregulation of Bcl-2 expression and cell cycle arrest 106. In addition, the exposition of liver cancer cells to lupeol suppresses STAT3 activation along with cyclin D1, Bcl-2, Bcl-xL, and survivin expression 107. Betulinic acid is a lupane-type pentacyclic triterpenoid having a double bond at position 20, a 3 beta-hydroxy and 28-carboxy substituents. It has been shown to induce apoptotic cell death in A549 cells (33 μM) through the mitochondrial intrinsic pathway 108. Betulinic acid produces cell rounding, chromatin condensation, nuclear fragmentation, membrane blebbing, and formation of apoptotic bodies in HepG2 cells (23 μM) 108. The cytotoxic activity in vitro against MCF7 (20 μM) cells resulted in a dose-dependent inhibition of cell proliferation and apoptosis independent of the p53 pathway 109. Betulinic acid also induces apoptosis on HeLa cells (23 μM) by the sequential activation of caspases 9, 3, and 7 and the cleavage of poly (ADP-ribose) polymerase (PARP), a nuclear enzyme fragmented during the programmed cell death 108. Its study on PC3 cells (22 μM) reveals that apoptosis also resulted from NF-κB inhibition, associated with a decrease in the activity of IKKβ, the serine/threonine protein kinase that phosphorylates IκBα, which negatively regulates the activation of the transcription factor 110. Ursolic acid derives from the ursane skeleton, hence is an alkene at C12-C13, but is also substituted by a beta-hydroxy group at position 3 and a carboxylic moiety at carbon 28. It triggers apoptosis of human lung cancer cell line A549 (40 μM) by upregulation of Fas/APO-1, a member of the TNF receptor superfamily, and downregulation of NF-κB, Bcl-2, and Bcl-xL 111. Against Hep3B (50 μM), it inhibits cell viability by negative modulation of the Janus kinase 2/signal transducer and activator of transcription 3 (JAK2/STAT3) pathway and downregulation of Bcl-xL 112. MCF7 cells exposed to ursolic acid (53 μM) undergo apoptosis through the intrinsic mitochondrial pathway by modulating the glucocorticoid receptor, Activator Protein-1 (AP-1), and decreasing Bcl-2 protein and PARP cleavage 113. Ursolic acid also inhibited the cell viability of PC3 (32 μM) and HeLa (10 μM) cells by activation of the mitochondrial pathway of apoptosis 114.
Flavonoids are a large group of polyphenolic compounds characterized by a core structure of 15 carbons arranged as two benzene rings connected by a heterocyclic pyran ring 115. Naringenin is a flavanon, a subclass of flavonoid that is characterized by a benzopyran bearing a ketone at the carbon C4. In MCF7 cells (1719 μM) it showed a dose-dependent response, reducing cell viability and inducing apoptosis 116. Naringenin also induces apoptosis in HepG2 (200 μM) cells as evidenced by the exposure of phosphatidylserine in the outer layer of the cell membrane and the activities of caspases 9, 8, and 3. Furthermore, the protein expression levels of Bax and Bak were increased whereas the level of Bcl-xL was decreased 117. Antiproliferative activities on naringenin treatment of PC3 (50 μM) were evidenced by the low abundance of PCNA, a marker of mitosis. In addition, naringenin induced apoptosis by DNA fragmentation and downregulation of Bcl-2 in a dose-dependent manner 118. In A549 cells (800 μM) naringenin induces ROS production and Bax-mediated mitochondrial apoptotic cell death by formation of the apoptosome complex and activation of caspase-3. In addition, it enhances the expression of death receptor 5 and upregulates TRAIL-induced apoptosis 119. The cytotoxic effect and apoptosis induction of naringenin in HeLa (195 μM) cells showed a similar effect with an increment in the expression of Bax and decreased expression of Bcl-2 120. The enzyme flavone synthase catalyzes the conversion of naringenin to apigenin by forming a single double bond between the C2-C3 atoms of the pyran ring. The treatment of MCF7 cells (30 μM) with this flavonoid increased apoptosis evidenced by p53 expression, PARP cleavage, and augmenting the release of cytochrome c into the cytosol. In the case of PC3 cells (40 μM), apigenin induced a significant decrease in Akt phosphorylation at Serine 473; inhibiting its kinase activity, which was confirmed by reduced phosphorylation of the proapoptotic proteins Bad and glycogen synthase kinase-3, their essential downstream targets. Exposure to apigenin induced caspase-9 activity and decreased the survival of PC3 cells in a dose-dependent manner 121. The molecular mechanism and signaling pathway of apigenin in induced cytotoxicity in A549 (72 μM) was accompanied by morphological changes, DNA damage, reduction of cell viability, and apoptosis. In addition, it induces protein production of p53, Bid, and Bax while decreasing the levels of Bcl-2 122. Treatment of HepG2 cells (81 μM) with this flavone resulted in the induction of DNA fragmentation triggering apoptosis as evident from the morphology of cells 123. Apigenin also exerted concentration-dependent cytotoxic effects on HeLa cells (10 μM) inducing pronounced morphological changes, retraction of cytoplasm, and detachment from the plate associated with apoptosis 124. Concerning flavonols, they are flavonoids that have an unsaturated pyran ring at the C2-C3 position, oxidized at C4, and hydroxylated at C3. Quercetin, kaempferol, and myricetin are the main flavonols and exhibit a myriad of anticancer properties 115. They differ in the number of hydroxyl groups at the B-ring, in which kaempferol is hydroxylated at C4´, quercetin at C3´and C4´, and myricetin at C3´, C4´, and C5´ positions. Quercetin exerts cytotoxic effects against many different types of cancer cells. In MCF7 (165 μM) cells exposed to quercetin, their nuclei exhibit chromatin condensation and changes in the expression of the proapoptotic proteins Bax, and caspase-3 125. Regarding PC3 cells (46 μM), their exposure to quercetin results in cell death via downregulation of NF-κB, mTOR and Bcl-2 while increasing the activity of caspase-3 126, 127. Quercetin in HepG2 cells (24 μM) induced cell death via caspase 3 and 9 activation, regulation of Bcl-2, and inhibition of PI-3-Kinase/Akt and ERK pathways 128. Likewise, in HeLa cells (185 μM), it modulates the PI3K/Akt pathway and induces apoptosis by caspase-3 activation 129, mitochondrial dysfunction, and the generation of ROS 130. Quercetin is also able to induce apoptosis through the regulation of Bcl-2 and Bax in A549 cells (74 μM) 131. Kaempferol triggers apoptosis in a dose-dependent manner in MCF7 cells (168 μM) by increasing the generation of ROS, cell shrinkage, and loss of adhesion 132. A study on A549 cells (35 μM) found that kaempferol leads to cell death via upregulation of caspase-7 and downregulation of Bcl-2 and Bcl-xL 133. Similarly, the cytotoxicity against HeLa cells (48 μM) was accompanied by increased expressions of p21, p53, caspase-3, and decreased expression of Bcl-2 causing apoptosis 134. Consistent with those results, the exposition of HepG2 cells (40 μM) with kaempferol induces apoptosis by the activation of caspase-3, and caspase-4 135. In the prostate cancer cells PC3 (58 μM), kaempferol also promotes apoptosis in a dose-dependent manner although the mechanisms behind this effect remain to be clarified 136. Myricetin has been found to suppress cell viability of MCF7 cells (80 μM) by apoptosis through inhibition of the protein p21-activated kinase 1 and phosphorylated extracellular mitogen-activated protein kinase (ERK1/2) with negative modulation of β-catenin pathway, survivin, and activation of caspase-3 137. The treatment of PC3 (48 μM) with myricetin also induced apoptosis with upregulation of the expression levels of caspase-3 and caspase-9 and inhibition of the phosphorylation of ERK1/2 and AKT 138. It has also been reported that myricetin exposition to Hela (60 μM) cells results in apoptosis via caspase-3 activation with loss of mitochondrial membrane potential 139. Likewise, it triggers apoptosis in A549 cells (50 μM) by changes in the mitochondria, ROS generation, and p53 expression 140. Myricetin treatment also leads to apoptosis in HepG2 cells (95 μM) through the intrinsic pathway increasing Bad expression 141, 142. Finally, resveratrol is a non-flavonoid polyphenol of the stilbene class that shares a structure characterized by a 14-carbon skeleton composed of two benzene rings linked by an ethylene bridge. This phytoalexin displays a wide array of capabilities against numerous malignancies 143. The exposition of HepG2 cells (52 μM) resulted in the induction of apoptosis via activation of caspase-9, and caspase-3, upregulation of p53 expression, and downregulation of Bcl-2 144. Resveratrol also induces apoptosis of MCF7 cells (196 μM) through a caspase-independent mechanism with downregulation of Bcl-2 and NF-κB 145. Cell death by resveratrol against A549 cells (8 μM) was found to be mediated by apoptosis with upregulation of caspase-3, Bax and downregulation of the Bcl-2 146, 147. Studies on HeLa cells (90 μM) revealed that resveratrol also induces apoptosis by ROS overload and mitochondrial function impairment 148. Similarly, PC3 cells (202 μM) exposed to resveratrol experience growth inhibition by interfering with glucose metabolism 149 and undergo p53-independent apoptosis by cytochrome c release and caspase activation 150. Naturally-derived compounds are considered to have less toxic side effects when compared to cancer drugs 151. For example, the IC50 value of paclitaxel against PC3 cells is 5 nM 152 whereas the lowest concentration reviewed against the same cancer cell line was for resveratrol, with an IC50 value of 8 μM. In other words compounds present in C. trifoliata harbor low cytotoxicity since the concentration to induce cell death by apoptosis needs to be one thousand times higher than molecules used in antineoplasic therapy. Thankfully, terpenes and phenolics exert anticancer activities through a variety of mechanisms different from apoptosis including the inhibition of proliferation 153 and metastasis 154. Furthermore, cancer management has been progressing from the use of general cytotoxic agents to molecules able to attenuate signaling pathways that control other important aspects involved in the tumoral progression 151. In this regard, since cancer cells develop a degree of autonomy from growth signals, they undergo uncontrolled growth and proliferation leading to tumorigenesis. Therefore compounds that block proliferation may hinder tumor mass growth and result in overall anticancer effects 155. On the other hand, effectively targeting the process of EMT has the potential to improve carcinoma therapy due to its relevance for invasion, metastasis, and drug resistance 5. Hence, in an attempt to further understand the potential anticancer effects of C. trifoliata, the antiproliferative and anti-EMT activities of compounds in Table 4 were also reviewed in the next section. In addition, when possible, the potential mechanisms underlying those activities were retrieved from findings in the carcinoma cell lines of interest.
Antitumoral and antimetastatic activities of C. trifoliata compounds
Approved drugs for the treatment of cancer are highly toxic and induce resistance, which ultimately results in tumor recurrence and metastasis. In addition, the non-specific toxicities towards normal cells also limit their anticancer activities 156. In contrast, natural products have been observed to influence multiple oncogenic signaling pathways simultaneously by modulating the activity or expression of their molecular targets. Moreover, phytocompounds have chemical diversity, low toxicity, safety, and availability, which make them an attractive and affordable alternative to synthetic products 151. The relationship between cancer and the cell cycle seems obvious since neoplasms are considered to be a disease largely caused by the lack of regulation in cell proliferation. Furthermore, for many years it has been considered that such conditions lead to an increase in errors during DNA replication, thereby increasing the chances of acquiring favorable mutations for tumor progression 4. The cell cycle has four sequential stages where the S phase produces DNA replication, and the M phase divides the cell into two daughter cells. Among them, there are two separation phases called G1 and G2. During G1 the cell is sensitive to positive and negative signals from growth signaling networks. In G2, which occurs after the S phase, the cell prepares to enter mitosis. On the other hand, when cells have reversibly withdrawn from the cell division cycle in response to high cell density or mitogen deprivation, they are considered to be in the G0 phase. The progression through the cell cycle is controlled by the family of cyclin-dependent serine/threonine kinases (CDKs) and their regulators, the cyclins. Specifically, cyclin D-CDK4, cyclin D-CDK6, and cyclin E-CDK2 drive G1 progression while S phase is initiated by cyclin A-CDK2 and cyclin B-CDK1 regulates progression through G2 and entry into mitosis. If sensing mechanisms detect aberrant events during such processes, an arrest in the cell cycle can be triggered until the problem is resolved. Those functions are driven by members of the Ink4 family, such as p16, p15, p18, and p19, which suppresses CDK4 and CDK6, and the Cip/Kip family of inhibitors, which block the activity of CDK2, such as the proteins p21, p27, and p57 that can reversibly arrest cell cycle progression 157. Given that one of the main characteristics of tumor cells is their uncontrolled proliferation, it is not surprising that many of the compounds with anticancer potential have been evaluated in the context of cell cycle effectors 158.
The biological activities of phytocompounds against cancer cells other than apoptosis are usually observable when cells are exposed at sublethal doses and for longer periods. For example, the exposition of MCF7 cells to β-Sitosterol at low concentrations (19 μM) for 3 days decreased almost 90% the proliferation rate 100. Similarly, stigmasterol (20 μM) impairs proliferation by G1 cell cycle arrest diminishing the activity of cyclin D-CDK6 in Ishikawa endometrial cancer cells 159. Regarding the pentacyclic triterpenes, HeLa cells exposed to lupeol (25 μM) undergo S-phase cell cycle arrest after 24 hours of incubation along with decreased protein levels of Cyclin E, Cyclin A, and CDK2 160. Treatment of MCF7 cells with betulinic acid (20 μM) halts cell proliferation and induces G0/G1 phase cell cycle arrest in HepG2 cells at a relatively low concentration (11 μM) 108. Ursolic acid (40 µM) inhibits the 85% proliferation of A549 cells by G1 phase cell arrest associated with a marked decrease in the protein expression of cyclins D1, D2, and E and CDK2, 4, and 6 with induction of p21 111. Furthermore even at a lower concentration (10 μM) can decrease the 80% of proliferation in MCF7 cells 161. Regarding the phenolic compounds, naringenin (100 μM) inhibits HepG2 proliferation inducing cell cycle arrest at G2/M phase 162. In the same model, treatment for 48 hours with apigenin (30 μM) induces the expression of p53, p21, and the accumulation of cells arrested in G2/M phase 123. Kaempferol (40 µM) also inhibits cell proliferation through G1 cell cycle arrest by activation of p53 gene expression and downregulation of cyclin D1, CDK4, and CDK6 in HepG2 cells 163. The exposition of PC3 to quercetin (100 μM) for 24 hours led to cell cycle arrest and 80% diminution of cell proliferation by downregulation NF-κB and mTOR protein expression 126, 127. Treatment for 24 h of HepG2 cells with myricetin (66 µM) also resulted in G2/M phase arrest and inhibition of proliferation mediated by decreased protein levels of CDK2 and cyclin B-CDK1 142. On the other hand, the stilbene resveratrol (25 μM) was shown to induce cell cycle arrest in the G0/G1 phase on A549 cells through downregulation in the expression levels of cyclin D1, CDK4, and CDK6, and upregulation of p21 and p27 164.
The process of metastasis involves the migration of cancer cells from the primary neoplasia to different locations in the body and the generation of new tumor colonies. One of the most important events in gaining metastatic qualities in cells derived from carcinomas is the induction of EMT. This process of transdifferentiation is characterized by the loss of adhesion to neighboring cells and the cellular matrix as well as the adoption of a mesenchymal phenotype. Broadly speaking, cells that undergo EMT exhibit increased resistance to anticancer therapy and tend to produce more aggressive lesions with an overall worse prognosis. Markers linked to this event include N-cadherin, vimentin, and fibronectin, while the expression of epithelial junctional proteins such as E-cadherins, claudins, and occludins are downregulated. Among the transcription factors capable of inducing this process are the proteins Snail, Slug, Zeb, and Twist, which in turn can be activated by the increase in the production of cytokines such as IL-6, TNF-α and TGF-β in the tumoral microenvironment 5.
β-sitosterol (32 µM) prevents EMT, migration, and invasion of HepG2 cells by downregulation of N-cadherin, Twist, vimentin, and Snail expression while inducing upregulation of E-cadherin 154. In the Ishikawa endometrial carcinoma cells, stigmasterol (20 µM) was found to suppress Zeb, Slug, and N-cadherin while increasing the expression of E-cadherin 159. Lupeol (50 µM) also exhibited anti-EMT properties against A549 cells as shown by the downregulation of N-cadherin and vimentin with repression of migration 165. The potential of betulinic acid in preventing EMT was assessed by the exposition (20 µM) of the human gastric carcinoma cell line SNU-16 imparing migration and invasion through the downregulation of N-cadherin 166. Similar results were found with ursolic acid (25 μM) treatment against the migration capabilities of the human gastric carcinoma cells BGC823 associated with a negative modulation of the NF-κB pathway and vimentin, Snail, and Twist gene expression 167. Concerning polyphenols, naringenin (50 µM) prevents TGF-β induction of EMT in the pancreatic carcinoma cells panc-1 by inhibiting the Smad3 pathway, impairing migration, invasion, and the mesenchymal phenotype as noted by the repression of vimentin and N-cadherin 168. Interestingly, apigenin (20 μM) reverses EMT in the human hepatocarcinoma cells Bel-7402 as indicated by changes in cell morphology and reexpression of E-cadherin and claudin-3 linked to repression of the NF-κB and Snail axis 169. Kaempferol (25 µM) was also able to suppress EMT induced by TGF-β in A549 cells along with the prevention of their invasive potential through downmodulation of Akt1 and Smad pathways and inactivation of MMP-2 170. In PC3 cells, quercetin (25 µM) treatment prevented the invasiveness via down-regulation of N-cadherin and vimentin and reexpression of E-cadherin 171. Dual effects were shown on MCF7 exposed to quercetin (37 μM) since cells undergo G1 phase arrest with cyclinD1, p21, and Twist gene expression suppression suggesting anti-proliferative and anti-EMT activities 172. The flavonol myricetin (37 μM) also inhibited migration, invasion, and EMT in PC3 cells by downregulation of vimentin 138. The effects of resveratrol (37 μM) in preventing EMT on HeLa cells were shown to suppress migration and invasion by downregulation of IL-6 and STAT signaling, along with a reduction in the protein levels of N-cadherin and vimentin 173.
Conclusion and perspectives
The development of antineoplastic therapy based on medicinal plants begins with the evaluation of total extracts against a diversity of cell lines for a possible anticancer biological activity followed by the purification of active phytochemicals based on the fractionation guided by in vitro bioassays. Then, candidate molecules can be tested in vivo using preclinical models of cancer to establish the preliminary efficacy, toxicity, pharmacokinetic, and safety profile and decide whether a compound should be taken further for clinical trials 174. The bioassay-guided study of C. trifoliata allowed us to find the most active fraction against PC3 cells, composed of ursolic acid, betulinic acid, naringenin, apigenin, kaempferol, and resveratrol 175. That is, triterpenes, flavonoids, and stylbenes, this resembles results from the bioassay-guided study performed in Vitis vinifera extracts in which the fraction that reduces most of the proliferation in MCF7 cells was composed of β-sitosterol, oleanolic acid, betulinic acid, resveratrol and ε-viniferin 176. Furthermore, the bioassay-guided study of C. quadrangularis performed on MCF7 cells showed a highly antiproliferative fraction characterized by the presence of the flavonoids quercetin and rutin 83. Furthermore, ursolic acid 177, betulinic acid 178, naringenin 179, apigenin 121, kaempferol 180, and resveratrol 181 have been shown antitumoral activities in different preclinical models of carcinomas. However the medicinal use of natural products is now considering mixtures rather than purified compounds as a more effective strategy due to potential synergies 182. In addition, cancer is a disease regulated by multiple pathways and numerous studies suggest that resistance is less likely to occur when a combination of compounds is provided instead of single active constituents. In addition, plants evolved to fight adversity through the combined action of structurally and functionally diverse constituents 182. As such, the evaluation of complex mixtures might be valuable in improving the therapy against cancer, but few studies have been performed to test this possibility. In this regard, the extracts from leaves of C. sicyoides suppressed the Ehrlich carcinoma in mice and the bioactivity was related to the content of β-sitosterol and resveratrol 183. Hence, seems plausible that C. trifoliata extracts may exhibit antitumoral activities in vivo as suggested by their chemical composition and ethnomedical uses. Furthermore, the study of the molecular mechanisms implicated in the synergies resulting from fractions, extracts, or from the entire plant may help to clarify their potential uses against carcinomas.
References
- Sung, H.; Ferlay, J.; Siegel, R. L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. , Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians, 2021, 71 (3), 209-249.
- Mohar-Betancourt, A.; Reynoso-Noverón, N.; Armas-Texta, D.; Gutiérrez-Delgado, C.; Torres-Domínguez, J. A. , Cancer trends in Mexico: essential data for the creation and follow-up of public policies. Journal of Global Oncology 2017, 3(6), 740–748. [Google Scholar] [CrossRef] [PubMed]
- Méndez-López, L. F. , Revisiting epithelial carcinogenesis. International Journal of Molecular Sciences 2022, 23(13), 7437. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R. A. , Hallmarks of cancer: the next generation. Cell 2011, 144(5), 646–674. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Settleman, J. , EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 2010, 29(34), 4741–4751. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. , The different mechanisms of cancer drug resistance: a brief review. Advanced Pharmaceutical Bulletin 2017, 7(3), 339. [Google Scholar] [CrossRef] [PubMed]
- Boffetta, P.; Kaldor, J. M. , Secondary malignancies following cancer chemotherapy. Acta Oncologica 1994, 33(6), 591–598. [Google Scholar] [CrossRef]
- Cutler, D. M. , Are we finally winning the war on cancer? The Journal of Economic Perspectives 2008, 22(4), 3–26. [Google Scholar] [CrossRef]
- Manders, D. B.; Kehoe, S. M.; Miller, D. S.; Lea, J. S.; Richardson, D. L. , Third-line salvage chemotherapy for recurrent carcinoma of the cervix is associated with minimal response rate and high toxicity. American Journal of Clinical Oncology 2017. [CrossRef] [PubMed]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D. B.; Johnston, P. G. , Cancer drug resistance: an evolving paradigm. Nature Reviews Cancer 2013, 13(10), 714–726. [Google Scholar] [CrossRef]
- Siddiqui, A. J.; Jahan, S.; Singh, R.; Saxena, J.; Ashraf, S. A.; Khan, A.; Choudhary, R. K.; Balakrishnan, S.; Badraoui, R.; Bardakci, F. , Plants in anticancer drug discovery: from molecular mechanism to chemoprevention. BioMed Research International 2022, 2022. [Google Scholar] [CrossRef]
- Oberlies, N. H.; Kroll, D. J. , Camptothecin and taxol: historic achievements in natural products research. Journal of Natural Products 2004, 67(2), 129–135. [Google Scholar] [CrossRef]
- Grever, M. R.; Schepartz, S. A.; Chabner, B. A. In The National Cancer Institute: cancer drug discovery and development program, Seminars in Oncology, Elsevier: 1992; pp 622-638.
- Suffness, M.; Pezzuto, J. , Assays related to cancer drug discovery. Academic Press: London, 1990; Vol. 6, p 360.
- Méndez-López, L. F.; Garza-González, E.; Ríos, M. Y.; Ramírez-Cisneros, M. Á.; Alvarez, L.; González-Maya, L.; Sánchez-Carranza, J. N.; Camacho-Corona, M. d. R. , Metabolic profile and evaluation of biological activities of extracts from the stems of Cissus trifoliata. International Journal of Molecular Sciences 2020, 21(3), 930. [Google Scholar] [CrossRef]
- Wen, J.; Lu, L. M.; Nie, Z. L.; Liu, X. Q.; Zhang, N.; Ickert-Bond, S.; Gerrath, J.; Manchester, S. R.; Boggan, J.; Chen, Z. , A new phylogenetic tribal classification of the grape family (Vitaceae). Journal of Systematics and Evolution 2018. [CrossRef]
- Banu, J. , Medicinal properties of plants from the genus Cissus: A review. Journal of Medicinal Plants Research 2012, 6(16), 3080–3086. [Google Scholar]
- McCartney, P. , SEINet: metadata-mediated access to distributed ecological data. LTER. DataBits Spring 2003, 1. [Google Scholar]
- Standley, P. C. , Trees and shrubs of Mexico. Smithsonian Institution: 1967; Vol. 1.
- Adams, N. F.; Collinson, M. E.; Smith, S. Y.; Bamford, M. K.; Forest, F.; Malakasi, P.; Marone, F.; Sykes, D. , X-rays and virtual taphonomy resolve the first Cissus (Vitaceae) macrofossils from Africa as early-diverging members of the genus. American Journal of Botany 2016, 103(9), 1657–1677. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.; Ankli, A.; Frei, B.; Weimann, C.; Sticher, O. , Medicinal plants in Mexico: Healers' consensus and cultural importance. Social Science and Medicine 1998, 47(11), 1859–1871. [Google Scholar] [CrossRef] [PubMed]
- de las Mercedes Rodríguez, L. , Etnobotánica maya: Algunas plantas de uso medicinal en estomatología. Revista ADM, 2015, 72 (1).
- Alejandro, M.; Alberto, M.; Gama Campillo, L. M.; Mariaca Méndez, R. , El uso de las plantas medicinales en las comunidades Maya-Chontales de Nacajuca, Tabasco, México. Polibotánica, 2010, (29), 213-262.
- Mendieta, R. M.; Rodríguez, A. Plantas medicinales del estado de Yucatán. Boletín de la Sociedad Botánica de México, (43), 94-95.
- Estrada-Castillón, E.; Soto-Mata, B. E.; Garza-López, M.; Villarreal-Quintanilla, J. Á.; Jiménez-Pérez, J.; Pando-Moreno, M.; Sánchez-Salas, J.; Scott-Morales, L.; Cotera-Correa, M. , Medicinal plants in the southern region of the State of Nuevo León, México. Journal of Ethnobiology and Ethnomedicine 2012, 8(1), 45. [Google Scholar] [CrossRef] [PubMed]
- Atjanasuppat, K.; Wongkham, W.; Meepowpan, P.; Kittakoop, P.; Sobhon, P.; Bartlett, A.; Whitfield, P. J. , In vitro screening for anthelmintic and antitumour activity of ethnomedicinal plants from Thailand. Journal of Ethnopharmacology 2009, 123(3), 475–482. [Google Scholar] [CrossRef]
- Bhujade, A.; Gupta, G.; Talmale, S.; Das, S.; Patil, M. , Induction of apoptosis in A431 skin cancer cells by Cissus quadrangularis Linn stem extract by altering Bax-Bcl2 ratio, release of cytochrome c from mitochondria and PARP cleavage. Food and Function 2013, 4(2), 338–346. [Google Scholar] [CrossRef]
- Opoku, A.; Geheeb-Keller, M.; Lin, J.; Terblanche, S.; Hutchings, A.; Chuturgoon, A.; Pillay, D. , Preliminary screening of some traditional Zulu medicinal plants for antineoplastic activities versus the HepG2 cell line. Phytotherapy Research 2000, 14(7), 534–537. [Google Scholar] [CrossRef] [PubMed]
- Sáenz, M.; Garcia, M.; Quilez, A.; Ahumada, M. , Cytotoxic activity of Agave intermixta L.(Agavaceae) and Cissus sicyoides L.(Vitaceae). Phytotherapy Research, 2000, 14 (7), 552-554.
- Line-Edwige, M.; Raymond, F. G.; François, E.; Edouard, N. , Antiproliferative effect of alcoholic extracts of some Gabonese medicinal plants on human colonic cancer cells. African Journal of Traditional, Complementary and Alternative Medicines, 2009, 6 (2).
- Adesanya, S. A.; Nia, R.; Martin, M.-T.; Boukamcha, N.; Montagnac, A.; Païs, M. , Stilbene derivatives from Cissus quadrangularis. Journal of Natural Products 1999, 62(12), 1694–1695. [Google Scholar] [CrossRef]
- Billet, K.; Houillé, B.; de Bernonville, T. D.; Besseau, S.; Oudin, A.; Courdavault, V.; Delanoue, G.; Guérin, L.; Clastre, M.; Giglioli-Guivarc'h, N. , Field-based metabolomics of vitis vinifera l. Stems provides new insights for genotype discrimination and polyphenol metabolism structuring. Frontiers in Plant Science 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Gniwotta, F.; Vogg, G.; Gartmann, V.; Carver, T. L.; Riederer, M.; Jetter, R. J. , What do microbes encounter at the plant surface? Chemical composition of pea leaf cuticular waxes. Plant Physiology 2005, 139(1), 519–530. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.; Ali, M.; Singh, V.; Singla, R. K. , Isolation and characterization of phytoconstituents from the stems of Ichnocarpus frutescens. Chinese Journal of Natural Medicines 2010, 8(6), 401–404. [Google Scholar] [CrossRef]
- Hannoufa, A.; McNevin, J.; Lemieux, B. , Epicuticular waxes of eceriferum mutants of Arabidopsis thaliana. Phytochemistry 1993, 33(4), 851–855. [Google Scholar] [CrossRef]
- Nyemb, J. N.; Magnibou, L. M.; Talla, E.; Tchinda, A. T.; Tchuenguem, R. T.; Henoumont, C.; Laurent, S.; Mbafor, J. , Lipids constituents from gardenia aqualla stapf & hutch. Open Chemistry 2018, 16(1), 371–376. [Google Scholar]
- Kunst, L.; Samuels, A. , Biosynthesis and secretion of plant cuticular wax. Progress in Lipid Research 2003, 42(1), 51–80. [Google Scholar] [CrossRef] [PubMed]
- Lara, I.; Belge, B.; Goulao, L. F. , A focus on the biosynthesis and composition of cuticle in fruits. Journal of Agricultural and Food Chemistry 2015, 63(16), 4005–4019. [Google Scholar] [CrossRef]
- Koonce, S. D.; Brown, J. , A study of the alcohols of carnauba wax. Journal of the American Oil Chemists' Society, 1944, 21 (8), 231-234.
- Heldt, H. , Plant biochemistry. Elsevier Academic Press. XXIV: 2005.
- Zhukov, A. , Palmitic acid and its role in the structure and functions of plant cell membranes. Russian Journal of Plant Physiology 2015, 62(5), 706–713. [Google Scholar] [CrossRef]
- Gunstone, F. D.; Harwood, J. L.; Dijkstra, A. J. , The lipid handbook with CD-ROM. CRC press: 2007.
- Piironen, V.; Lindsay, D. G.; Miettinen, T. A.; Toivo, J.; Lampi, A. M. , Plant sterols: biosynthesis, biological function and their importance to human nutrition. Journal of the Science of Food and Agriculture 2000, 80(7), 939–966. [Google Scholar] [CrossRef]
- Griebel, T.; Zeier, J. , A role for β-sitosterol to stigmasterol conversion in plant-pathogen interactions. The Plant Journal 2010, 63(2), 254–268. [Google Scholar] [CrossRef]
- Jing, X.; Grebenok, R. J.; Behmer, S. T. , Plant sterols and host plant suitability for generalist and specialist caterpillars. Journal of Insect Physiology 2012, 58(2), 235–244. [Google Scholar] [CrossRef]
- Santos Júnior, H. M.; Lopes, K. C.; Alves, D. S.; Carvalho, G. A.; Oliveira, D. F. , Ursolic acid and cis-tilroside produced by Merremia tomentosa affect oviposition of Leucoptera coffeella on coffee plants. Química Nova, 2018, 41 (3), 302-309.
- Wal, P.; Wal, A.; Sharma, G.; Rai, A. , Biological activities of lupeol. Systematic Reviews in Pharmacy 2011, 2(2), 96. [Google Scholar] [CrossRef]
- Lozano-Grande, M. A.; Gorinstein, S.; Espitia-Rangel, E.; Dávila-Ortiz, G.; Martínez-Ayala, A. L. , Plant sources, extraction methods, and uses of squalene. International Journal of Agronomy 2018. [CrossRef]
- Hiebert-Giesbrecht, M. R.; Escalante-Erosa, F.; García-Sosa, K.; Dzib, G. R.; Calvo-Irabien, L. M.; Peña-Rodríguez, L. M. , Spatio-temporal variation of terpenoids in wild plants of Pentalinon andrieuxii. Chemistry and Biodiversity 2016, 13(11), 1521–1526. [Google Scholar] [CrossRef]
- Rosy, B. A.; Rosakutty, P. , GC-MS analysis of methanol wild plant and callus extracts from three Cissus species, Family Vitaceae. Journal of Chemical and Pharmaceutical Research 2012, 4(7), 3420–3426. [Google Scholar]
- Chipiti, T.; Ibrahim, M. A.; Koorbanally, N. A.; Islam, M. S. , In vitro antioxidant activity and GC-MS analysis of the ethanol and aqueous extracts of Cissus cornifolia (Baker) Splanch (Vitaceae) parts. ActaPoloniae Pharmaceutica Drug Research 2015, 72(1), 119–127. [Google Scholar]
- Vishnuthari., N.; Sripathi., S. K. 52. Vishnuthari., N.; Sripathi., S. K., GC-MS Analysis of hexane extract of stems and roots of the ethnomedicinal plant Cissus quadrangularis Linn. Journal of Chemical, Biological and Physical Sciences, 2015, 5 (4), 3954-3963.
- Kumar, S.; Anandan, A.; Jegadeesan, M. , Identification of chemical compounds in Cissus quadrangularis L. Variant I of different samples using GC-MS analysis. Archives of Applied Science Research, 2012, 4 (4), 1782-1787.
- Eswaran, R.; Anandan, A.; Doss, A.; Sangeetha, G.; Anand, S. , Analysis of chemical composition of Cissus quadrangularis linn by GC-MS. Asian Journal of Pharmaceutical and Clinical Research 2012, 2, 139–40. [Google Scholar]
- Xie, Y.; Deng, P.; Zhang, Y.; Yu, W. , Studies on the chemical constituents from Cissus assamica. Journal of Chinese Medicinal Materials 2009, 32(2), 210–213. [Google Scholar]
- Sani, Y.; Musa, A.; Tajuddeen, N.; Abdullahi, S.; Abdullahi, M.; Pateh, U.; Idris, A. , Isoliquiritigenin and sitosterol from Cissus polyantha Tuber Glig and Brandt. Journal of Medicinal Plants Research 2015, 9(35), 918–921. [Google Scholar]
- Saifah, E.; Vaisiriroj, V.; Kelley, C. J.; Higuchi, Y. , Constituents of the roots of Cissus rheifolia. Journal of Natural Products 1987, 50(2), 328–328. [Google Scholar] [CrossRef]
- Pan, G.; Li, W.; Luo, P.; Qin, J.; Su, G. , Study on steroidal and triterpenoid constituents from Cissus pteroclada. Journal of Chinese Medicinal Materials 2013, 36(8), 1274–1277. [Google Scholar] [PubMed]
- Chanda, S.; Baravalia, Y.; Nagani, K. , Spectral analysis of methanol extract of Cissus quadrangularis L. stem and its fractions. Journal of Pharmacognosy and Phytochemistry, 2013, 2 (4).
- Pathomwichaiwat, T.; Ochareon, P.; Soonthornchareonnon, N.; Ali, Z.; Khan, I. A.; Prathanturarug, S. , Alkaline phosphatase activity-guided isolation of active compounds and new dammarane-type triterpenes from Cissus quadrangularis hexane extract. Journal of Ethnopharmacology 2015, 160, 52–60. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Z.; He, H.; Gao, S.; Kong, N.; Ding, M.; Hao, X. , Lignans and triterpenoids from Cissus repens (Vitaceae). Acta Botanica Yunnanica 2006, 28(4), 433–437. [Google Scholar]
- Chan, Y. Y.; Wang, C. Y.; Hwang, T. L.; Juang, S. H.; Hung, H. Y.; Kuo, P. C.; Chen, P. J.; Wu, T. S. , The constituents of the stems of cissus assamica and their bioactivities. Molecules 2018, 23(11), 2799. [Google Scholar] [CrossRef] [PubMed]
- Heredia-Guerrero, J. A.; Benítez, J. J.; Domínguez, E.; Bayer, I. S.; Cingolani, R.; Athanassiou, A.; Heredia, A. , Infrared and Raman spectroscopic features of plant cuticles: a review. Frontiers in Plant Science 2014, 5, 305. [Google Scholar] [CrossRef]
- Choubey, S.; Varughese, L. R.; Kumar, V.; Beniwal, V. J. , Medicinal importance of gallic acid and its ester derivatives: a patent review. Pharmaceutical Patent Analyst 2015, 4(4), 305–315. [Google Scholar] [CrossRef] [PubMed]
- Mathew, S.; Abraham, T. E. , Ferulic acid: an antioxidant found naturally in plant cell walls and feruloyl esterases involved in its release and their applications. Critical Reviews in Biotechnology, 2004, 24 (2-3), 59-83.
- Liu, S.; Jiang, J.; Ma, Z.; Xiao, M.; Yang, L.; Tian, B.; Yu, Y.; Bi, C.; Fang, A.; Yang, Y. , The role of hydroxycinnamic acid amide pathway in plant immunity. Frontiers in Plant Science 2022, 13, 922119. [Google Scholar] [CrossRef] [PubMed]
- Batra, P.; Sharma, A. K. , Anti-cancer potential of flavonoids: recent trends and future perspectives. 3 Biotech 2013, 3(6), 439–459. [Google Scholar] [CrossRef]
- Mekawy, A. M. M.; Abdelaziz, M. N.; Ueda, A. , Apigenin pretreatment enhances growth and salinity tolerance of rice seedlings. Plant Physiology Biochemistry 2018, 130, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Ruttanaphan, T.; Thitathan, W.; Piyasaengthong, N.; Nobsathian, S.; Bullangpoti, V. , Chrysoeriol isolated from Melientha suavis Pierre with activity against the agricultural pest Spodoptera litura. Chemical Biological Technologies in Agriculture 2022, 9(1), 1–7. [Google Scholar] [CrossRef]
- Mannino, G.; Gentile, C.; Ertani, A.; Serio, G.; Bertea, C. M. , Anthocyanins: Biosynthesis, distribution, ecological role, and use of biostimulants to increase their content in plant foods A review. Agriculture 2021, 11(3), 212. [Google Scholar] [CrossRef]
- Jan, R.; Khan, M.; Asaf, S.; Lubna; Asif, S. ; Kim, K. M., Bioactivity and therapeutic potential of kaempferol and quercetin: New insights for plant and human health. Plants 2022, 11(19), 2623. [Google Scholar] [CrossRef] [PubMed]
- Manan, F. A.; Mamat, D. D.; Samad, A. A.; Ong, Y. S.; Ooh, K. F.; Chai, T. T. , Heavy metal accumulation and antioxidant properties of Nephrolepis biserrata growing in heavy metal-contaminated soil. Global NEST 2015.
- Shomali, A.; Das, S.; Arif, N.; Sarraf, M.; Zahra, N.; Yadav, V.; Aliniaeifard, S.; Chauhan, D. K.; Hasanuzzaman, M. , Diverse physiological roles of flavonoids in plant environmental stress responses and tolerance. Plants 2022, 11(22), 3158. [Google Scholar] [CrossRef] [PubMed]
- Yildiztugay, E.; Ozfidan-Konakci, C.; Kucukoduk, M.; Turkan, I. , Flavonoid naringenin alleviates short-term osmotic and salinity stresses through regulating photosynthetic machinery and chloroplastic antioxidant metabolism in Phaseolus vulgaris. Frontiers in Plant Science 2020, 11, 682. [Google Scholar] [CrossRef] [PubMed]
- Samkumar, A.; Karppinen, K.; McGhie, T. K.; Espley, R. V.; Martinussen, I.; Jaakola, L. , Flavonoid biosynthesis is differentially altered in detached and attached ripening bilberries in response to spectral light quality. Frontiers in Plant Science 2022, 13, 969934. [Google Scholar] [CrossRef] [PubMed]
- Valletta, A.; Iozia, L. M.; Leonelli, F. , Impact of environmental factors on stilbene biosynthesis. Plants 2021, 10(1), 90. [Google Scholar] [CrossRef]
- Keller, M.; Viret, O.; Cole, F. M. , Botrytis cinerea infection in grape flowers: defense reaction, latency, and disease expression. Phytopathology 2003, 93(3), 316–322. [Google Scholar] [CrossRef]
- Laitonjam, W. S.; Yumnam, R. S.; Kongbrailatpam, B. D. , Study on isolation and comparison of the chemical compositions of Cissus adnata Roxb. leaves and Smilax lanceaefolia Roxb. roots and their free radical scavenging activities. International Research Journal of Pure and Applied Chemistry 2011, 1(1), 1. [Google Scholar] [CrossRef]
- Ahmadu, A.; Onanuga, A.; Aquino, R. , Flavonoid glycosides from the leaves of Cissus ibuensis hook (vitaceae). African Journal of Traditional, Complementary and Alternative Medicines, 2010, 7 (3).
- Al-Said, M.; Khalifa, A.; Al-Azizi, M. , Flavonoids from Cissus digitata. International Journal of Pharmacognosy 1991, 29(4), 281–283. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Zhang, Z.-K.; He, H.-P.; Wang, J.-S.; Zhou, H.; Ding, M.; Hao, X.-J. , Stilbene C-glucosides from Cissus repens. Journal of Asian Natural Products Research 2007, 9(7), 631–636. [Google Scholar] [CrossRef]
- Barbosa, W.; Vincieri, F.; Gallori, S.; Pinto, L.; Silva, A.; Anunciacao, J.; di Scienze Farmaceutiche, D.; Barbosa, W. L. R. , Characterisation of flavonoid glycosides in pharmacopoeial preparation of Cissus verticillata (l) nicolson & ce jarvis) using HPLC-DAD and HPLC-MS. International Journal of Pharmaceutical Sciences and Research 2013, 4(10), 3871. [Google Scholar]
- Vijayalakshmi, A.; Kumar, P.; Sakthi Priyadarsini, S.; Meenaxshi, C. , In vitro antioxidant and anticancer activity of flavonoid fraction from the aerial parts of Cissus quadrangularis linn against human breast carcinoma cell lines. Journal of Chemistry 2013, 2013. [Google Scholar]
- Thakur, A.; Jain, V.; Hingorani, L.; Laddha, K. , Improved high-performance liquid chromatography-DAD method for the simultaneous analysis of quercetin and kaempferol in the stems of Cissus quadrangularis Linn. Acta Chromatographica 2009, 21(1), 95–103. [Google Scholar] [CrossRef]
- Beltrame, F. L.; Sartoretto, J. L.; Bazotte, R. B.; Cuman, R. N.; Cortez, D. A. G.; Fernandes, L. C.; Tchaikovski, O. , Phytochemical study and evaluation of the antidiabetic potential of Cissus sicyoides L.(Vitaceae). Química Nova, 2001, 24 (6), 783-785.
- Kaur, J.; Dhiman, V.; Bhadada, S.; Katare, O.; Ghoshal, G. , LC/MS guided identification of metabolites of different extracts of Cissus quadrangularis. Food Chemistry Advances 2022, 1, 100084. [Google Scholar] [CrossRef]
- Toledo, M.; Reyes, F.; Iaderoza, F.; Francis, F.; Draettao, S. , Anthocyanins from anil trepador Cissus sicyoides. Journal of Food Science 1983, 48. [Google Scholar] [CrossRef]
- Onyeweaku, G.; Nyananyo, B.; Ozimede, C. , Taxonomic studies on the genus Cissus L.(Vitaceae) present in Obio/Akpor local government area of Rivers State, Nigeria. Journal of Applied Sciences Environmental Management, 2020, 24 (1), 139-145.
- Mohan, S.; Prabhakaran, V.-S.; Narayanaswamy, R. , In silico analysis of cissus rotundifolia constituents as human neutrophil elastase (HNE), matrix metalloproteinases (MMP 2 and MMP 9), and tyrosinase inhibitors. Applied Biochemistry Biotechnology 2022, 1–14. [Google Scholar]
- De Rosso, M.; Tonidandel, L.; Larcher, R.; Nicolini, G.; Dalla Vedova, A.; De Marchi, F.; Gardiman, M.; Giust, M.; Flamini, R. , Identification of new flavonols in hybrid grapes by combined liquid chromatography–mass spectrometry approaches. Food Chemistry 2014, 163, 244–251. [Google Scholar] [CrossRef]
- Khan, M. A.; Nabi, S. G.; Prakash, S.; Zaman, A. , Pallidol, a resveratrol dimer from Cissus pallida. Phytochemistry 1986, 25(8), 1945–1948. [Google Scholar] [CrossRef]
- Strasser, A.; O'Connor, L.; Dixit, V. M. , Apoptosis signaling. Annual Review of Biochemistry 2000, 69(1), 217–245. [Google Scholar] [CrossRef] [PubMed]
- Hickman, J. A. , Apoptosis induced by anticancer drugs. Cancer Metastasis Reviews 1992, 11, 121–139. [Google Scholar] [CrossRef]
- Grattan, B. , Plant sterols as anticancer nutrients: evidence for their role in breast cancer. Nutrients 2013, 5(2), 359–387. [Google Scholar] [CrossRef]
- Tahsin, T.; Wansi, J. D.; Al-Groshi, A.; Evans, A.; Nahar, L.; Martin, C.; Sarker, S. D. , Cytotoxic properties of the stem bark of citrus reticulata blanco (Rutaceae). Phytotherapy Research 2017, 31(8), 1215–1219. [Google Scholar] [CrossRef]
- Samita, F.; Ochieng, C. O.; Owuor, P. O.; Manguro, L. O. A.; Midiwo, J. O. , Isolation of a new β-carboline alkaloid from aerial parts of Triclisia sacleuxii and its antibacterial and cytotoxicity effects. Natural Product Research 2017, 31(5), 529–536. [Google Scholar] [CrossRef]
- Luo, H.; Cai, Y.; Peng, Z.; Liu, T.; Yang, S. , Chemical composition and in vitroevaluation of the cytotoxic and antioxidant activities of supercritical carbon dioxide extracts of pitaya (dragon fruit) peel. Chemistry Central Journal 2014, 8(1), 1. [Google Scholar] [CrossRef]
- Ayaz, M.; Sadiq, A.; Wadood, A.; Junaid, M.; Ullah, F.; Khan, N. Z. , Cytotoxicity and molecular docking studies on phytosterols isolated from Polygonum hydropiper L. Steroids 2019, 141, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.; Chinnam, M.; Fink, C.; Bradford, P. , β-Sitosterol activates Fas signaling in human breast cancer cells. Phytomedicine 2007, 14(11), 747–754. [Google Scholar] [CrossRef]
- Awad, A. B.; Barta, S. L.; Fink, C. S.; Bradford, P. G. , β-Sitosterol enhances tamoxifen effectiveness on breast cancer cells by affecting ceramide metabolism. Molecular Nutrition and Food Research 2008, 52(4), 419–426. [Google Scholar] [CrossRef]
- Chen, Y. C.; Lee, H. Z.; Chen, H. C.; Wen, C. L.; Kuo, Y. H.; Wang, G. J. , Anti-inflammatory components from the root of Solanum erianthum. International Journal of Molecular Sciences 2013, 14(6), 12581–12592. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y. S.; Li, X. F.; Kang, K. H.; Ryu, B.; Kim, S. K. , Stigmasterol isolated from marine microalgae Navicula incerta induces apoptosis in human hepatoma HepG2 cells. BMB Reports 2014, 47(8), 433. [Google Scholar] [CrossRef] [PubMed]
- Gill, B. S.; Kumar, S. ; Navgeet, Triterpenes in cancer: significance and their influence. Molecular Biology Reports 2016, 43, 881–896. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Liu, F.; Zhang, L.; Wu, Y.; Hu, B.; Zhang, Y.; Li, Y.; Liu, H. , Growth inhibition and apoptosis induced by lupeol, a dietary triterpene, in human hepatocellular carcinoma cells. Biological and Pharmaceutical Bulletin 2011, 34(4), 517–522. [Google Scholar] [CrossRef] [PubMed]
- Kang, S. C.; Lim, S. Y.; Song, Y. J. , Lupeol is one of active components in the extract of Chrysanthemum indicum Linne that inhibits LMP1-induced NF-κB activation. PLoS One 2013, 8(11), e82688. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Nigam, N.; Kalra, N.; Shukla, Y. , Regulation of signaling pathways involved in lupeol induced inhibition of proliferation and induction of apoptosis in human prostate cancer cells. Molecular Carcinogenesis 2008, 47(12), 916–924. [Google Scholar] [PubMed]
- Siveen, K.; Nguyen, A.; Lee, J.; Li, F.; Singh, S.; Kumar, A. P.; Low, G.; Jha, S.; Tergaonkar, V.; Ahn, K. , Negative regulation of signal transducer and activator of transcription-3 signalling cascade by lupeol inhibits growth and induces apoptosis in hepatocellular carcinoma cells. British Journal of Cancer 2014, 111(7), 1327. [Google Scholar] [CrossRef] [PubMed]
- Król, S. K.; Kiełbus, M.; Rivero-Müller, A.; Stepulak, A. , Comprehensive review on betulin as a potent anticancer agent. Biomed Research International 2015, 2015. [Google Scholar] [CrossRef]
- Damle, A. A.; Pawar, Y. P.; Narkar, A. A. , Anticancer activity of betulinic acid on MCF-7 tumors in nude mice. Indian Journal of Experimental Biology 2013.
- Zhang, X.; Hu, J.; Chen, Y. , Betulinic acid and the pharmacological effects of tumor suppression. Molecular Medicine Reports 2016, 14(5), 4489–4495. [Google Scholar] [CrossRef]
- Hsu, Y. L.; Kuo, P. L.; Lin, C. C. , Proliferative inhibition, cell-cycle dysregulation, and induction of apoptosis by ursolic acid in human non-small cell lung cancer A549 cells. Life Sciences 2004, 75(19), 2303–2316. [Google Scholar] [CrossRef]
- Liu, T.; Ma, H.; Shi, W.; Duan, J.; Wang, Y.; Zhang, C.; Li, C.; Lin, J.; Li, S.; Lv, J. , Inhibition of STAT3 signaling pathway by ursolic acid suppresses growth of hepatocellular carcinoma. International Journal of Oncology 2017, 51(2), 555–562. [Google Scholar] [CrossRef]
- Kassi, E.; Sourlingas, T.; Spiliotaki, M.; Papoutsi, Z.; Pratsinis, H.; Aligiannis, N.; Moutsatsou, P. , Ursolic acid triggers apoptosis and Bcl-2 downregulation in MCF-7 breast cancer cells. Cancer Investigation 2009, 27(7), 723–733. [Google Scholar] [CrossRef]
- Kassi, E.; Papoutsi, Z.; Pratsinis, H.; Aligiannis, N.; Manoussakis, M.; Moutsatsou, P. , Ursolic acid, a naturally occurring triterpenoid, demonstrates anticancer activity on human prostate cancer cells. Journal of Cancer Research and Clinical Oncology 2007, 133(7), 493–500. [Google Scholar] [CrossRef]
- Kubina, R.; Iriti, M.; Kabała-Dzik, A. , Anticancer potential of selected flavonols: Fisetin, kaempferol, and quercetin on head and neck cancers. Nutrients 2021, 13(3), 845. [Google Scholar] [CrossRef]
- Rhman, M. A.; Devnarain, N.; Khan, R.; Owira, P. M. , Synergism potentiates oxidative antiproliferative effects of naringenin and quercetin in MCF-7 breast cancer cells. Nutrients 2022, 14(16), 3437. [Google Scholar] [CrossRef]
- Banjerdpongchai, R.; Wudtiwai, B.; Khawon, P. , Induction of human hepatocellular carcinoma HepG2 cell apoptosis by naringin. Asian Pacific Journal of Cancer Prevention 2016, 17(7), 3289–3294. [Google Scholar]
- Lim, W.; Park, S.; Bazer, F. W.; Song, G. , Naringenin-induced apoptotic cell death in prostate cancer cells is mediated via the PI3K/AKT and MAPK signaling pathways. Journal of Cellular Biochemistry 2017, 118(5), 1118–1131. [Google Scholar] [CrossRef]
- Lu, W. L.; Yu, C. T. R.; Lien, H. M.; Sheu, G. T.; Cherng, S. H. , Cytotoxicity of naringenin induces Bax-mediated mitochondrial apoptosis in human lung adenocarcinoma A549 cells. Environmental toxicology 2020, 35(12), 1386–1394. [Google Scholar] [CrossRef]
- Larasati, L.; Kusharyanti, I.; Hermawan, A.; Susidarti, R. A.; Meiyanto, E. , Naringenin enhances the anti-tumor effect of doxorubicin on HeLa cervical cancer cells through cytotoxic activity and apoptosis induction. Indonesian Journal of Cancer Chemoprevention 2011, 2(3), 325–333. [Google Scholar] [CrossRef]
- Kaur, P.; Shukla, S.; Gupta, S. , Plant flavonoid apigenin inactivates Akt to trigger apoptosis in human prostate cancer: an in vitro and in vivo study. Carcinogenesis 2008, 29(11), 2210–2217. [Google Scholar] [CrossRef] [PubMed]
- Lu, H. F.; Chie, Y. J.; Yang, M. S.; Lee, C. S.; Fu, J. J.; Yang, J. S.; Tan, T. W.; Wu, S. H.; Ma, Y. S. ; Apigenin induces caspase-dependent apoptosis in human lung cancer A549 cells through Bax-and Bcl-2-triggered mitochondrial pathway. International Journal of Oncology 2010, 36(6), 1477–1484. [Google Scholar] [PubMed]
- Chiang, L. C.; Ng, L. T.; Lin, I. C.; Kuo, P. L.; Lin, C. C. , Anti-proliferative effect of apigenin and its apoptotic induction in human Hep G2 cells. Cancer Letters 2006, 237(2), 207–214. [Google Scholar] [CrossRef] [PubMed]
- Souza, R. P.; Bonfim-Mendonça, P. d. S.; Gimenes, F.; Ratti, B. A.; Kaplum, V.; Bruschi, M. L.; Nakamura, C. V.; Silva, S. O.; Maria-Engler, S. S.; Consolaro, M. E. , Oxidative stress triggered by Apigenin induces apoptosis in a comprehensive panel of human cervical cancer-derived cell lines. Oxidative Medicine and Cellular Longevity 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Khorsandi, L.; Orazizadeh, M.; Niazvand, F.; Abbaspour, M.; Mansouri, E.; Khodadadi, A. , Quercetin induces apoptosis and necroptosis in MCF-7 breast cancer cells. Bratislavske Lekarske Listy 2017, 118(2), 123–128. [Google Scholar] [CrossRef] [PubMed]
- Bhat, F. A.; Sharmila, G.; Balakrishnan, S.; Singh, P. R.; Srinivasan, N.; Arunakaran, J. , Epidermal growth factor-induced prostate cancer (PC3) cell survival and proliferation is inhibited by quercetin, a plant flavonoid through apoptotic machinery. Biomedicine & Preventive Nutrition, 2014, 4 (4), 459-468.
- Haddad, A.; Venkateswaran, V.; Viswanathan, L.; Teahan, S.; Fleshner, N.; Klotz, L. , Novel antiproliferative flavonoids induce cell cycle arrest in human prostate cancer cell lines. Prostate Cancer and Prostatic Diseases 2006, 9(1), 68. [Google Scholar] [CrossRef]
- Granado-Serrano, A. B.; Martín, M. A.; Bravo, L.; Goya, L.; Ramos, S. , Quercetin induces apoptosis via caspase activation, regulation of Bcl-2, and inhibition of PI-3-kinase/Akt and ERK pathways in a human hepatoma cell line (HepG2). The Journal of nutrition 2006, 136(11), 2715–2721. [Google Scholar] [CrossRef] [PubMed]
- Li, X. M.; Luo, X. G.; He, J. F.; Wang, N.; Zhou, H.; Yang, P. L.; Zhang, T. C. , Induction of apoptosis in human cervical carcinoma HeLa cells by active compounds from Hypericumáascyron L. Oncology Letters 2018, 15(3), 3944–3950. [Google Scholar]
- Li, X. M.; Liu, J.; Pan, F. F.; Shi, D. D.; Wen, Z. G.; Yang, P. L. , Quercetin and aconitine synergistically induces the human cervical carcinoma HeLa cell apoptosis via endoplasmic reticulum (ER) stress pathway. PloS one 2018, 13(1), e0191062. [Google Scholar] [CrossRef]
- Klimaszewska-Wiśniewska, A.; Hałas-Wiśniewska, M.; Izdebska, M.; Gagat, M.; Grzanka, A.; Grzanka, D. , Antiproliferative and antimetastatic action of quercetin on A549 non-small cell lung cancer cells through its effect on the cytoskeleton. Acta Histochemica 2017, 119(2), 99–112. [Google Scholar] [CrossRef]
- Harrath, A. H.; Jalouli, M.; Oueslati, M. H.; Farah, M. A.; Feriani, A.; Aldahmash, W.; Aldawood, N.; Al-Anazi, K.; Falodah, F.; Swelum, A. , The flavonoid, kaempferol-3-O-apiofuranosyl-7-O-rhamnopyranosyl, as a potential therapeutic agent for breast cancer with a promoting effect on ovarian function. Phytotherapy Research 2021, 35(11), 6170–6180. [Google Scholar] [CrossRef]
- Nguyen, T.; Tran, E.; Ong, C.; Lee, S.; Do, P.; Huynh, T.; Nguyen, T.; Lee, J.; Tan, Y.; Ong, C. , Kaempferol-induced growth inhibition and apoptosis in A549 lung cancer cells is mediated by activation of MEK-MAPK. Journal of Cellular Physiology 2003, 197(1), 110–121. [Google Scholar] [CrossRef] [PubMed]
- Kashafi, E.; Moradzadeh, M.; Mohamadkhani, A.; Erfanian, S. , Kaempferol increases apoptosis in human cervical cancer HeLa cells via PI3K/AKT and telomerase pathways. Biomedicine and Pharmacotherapy 2017, 89, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ren, F.; Zhang, L.; Zhang, X.; Yang, R.; Xie, B.; Li, Z.; Hu, Z.; Duan, Z.; Zhang, J. , Kaempferol induces apoptosis in HepG2 cells via activation of the endoplasmic reticulum stress pathway. Molecular Medicine Reports 2016, 13(3), 2791–2800. [Google Scholar] [CrossRef]
- Da, J.; Xu, M.; Wang, Y.; Li, W.; Lu, M.; Wang, Z. , Kaempferol promotes apoptosis while inhibiting cell proliferation via androgen-dependent pathway and suppressing vasculogenic mimicry and invasion in prostate cancer. Analytical Cellular Pathology 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Jiao, D.; Zhang, X. D. , Myricetin suppresses p21-activated kinase 1 in human breast cancer MCF-7 cells through downstream signaling of the β-catenin pathway. Oncology Reports 2016, 36(1), 342–348. [Google Scholar] [CrossRef]
- Ye, C.; Zhang, C.; Huang, H.; Yang, B.; Xiao, G.; Kong, D.; Tian, Q.; Song, Q.; Song, Y.; Tan, H. , The natural compound myricetin effectively represses the malignant progression of prostate cancer by inhibiting Pim1 and disrupting the PIM1/CXCR4 interaction. Cellular Physiology and Biochemistry 2018, 48(3), 1230–1244. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.-L.; Shi, S.; Shen, Y.-L.; Wang, L.; Chen, H.-Y.; Zhu, J.; Ding, Y. , Myricetin and methyl eugenol combination enhances the anticancer activity, cell cycle arrest and apoptosis induction of cis-platin against HeLa cervical cancer cell lines. International Journal of Clinical and Experimental Pathology 2015, 8(2), 1116. [Google Scholar] [PubMed]
- Shih, Y.-W.; Wu, P.-F.; Lee, Y.-C.; Shi, M.-D.; Chiang, T.-A. , Myricetin suppresses invasion and migration of human lung adenocarcinoma A549 cells: possible mediation by blocking the ERK signaling pathway. Journal of Agricultural and Food Chemistry 2009, 57(9), 3490–3499. [Google Scholar] [CrossRef]
- Zhang, X. H.; Chen, S. Y.; Tang, L.; Shen, Y. Z.; Luo, L.; Xu, C. W.; Liu, Q.; Li, D. , Myricetin induces apoptosis in HepG2 cells through Akt/p70S6K/bad signaling and mitochondrial apoptotic pathway. Anti-Cancer Agents in Medicinal Chemistry, 2013, 13 (10), 1575-1581.
- Zhang, X. H.; Zou, Z. Q.; Xu, C. W.; Shen, Y. Z.; Li, D. , Myricetin induces G2/M phase arrest in HepG2 cells by inhibiting the activity of the cyclin B/Cdc2 complex. Molecular Medicine Reports 2011, 4(2), 273–277. [Google Scholar]
- Xue, Y. Q.; Di, J. M.; Luo, Y.; Cheng, K. J.; Wei, X.; Shi, Z. , Resveratrol oligomers for the prevention and treatment of cancers. Oxidative Medicine Cellular Longevity 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Chen, Y.; Cheng, X.; Zhang, X.; He, Q. , Potentiation of resveratrol-induced apoptosis by matrine in human hepatoma HepG2 cells. Oncology Reports 2014, 32(6), 2803–2809. [Google Scholar] [CrossRef] [PubMed]
- Pozo-Guisado, E.; Merino, J. M.; Mulero-Navarro, S.; Lorenzo-Benayas, M. J.; Centeno, F.; Alvarez-Barrientos, A.; Salguero, P. M. F. , Resveratrol-induced apoptosis in MCF-7 human breast cancer cells involves a caspase-independent mechanism with downregulation of Bcl-2 and NF-κB. International Journal of Cancer 2005, 115(1), 74–84. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Li, J.; Yang, Y.; Zhao, X.; Liu, Y.; Jiang, Y.; Zhou, L.; Feng, Y.; Yu, Y.; Cheng, Y. , Resveratrol modulates the apoptosis and autophagic death of human lung adenocarcinoma A549 cells via a p53-dependent pathway: Integrated bioinformatics analysis and experimental validation. International Journal of Oncology 2020, 57(4), 925–938. [Google Scholar] [CrossRef]
- Hu, S.; Li, X.; Xu, R.; Ye, L.; Kong, H.; Zeng, X.; Wang, H.; Xie, W. , The synergistic effect of resveratrol in combination with cisplatin on apoptosis via modulating autophagy in A549 cells. Acta Biochimica et Biophysica Sinica 2016, 48(6), 528–535. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Enríquez, S.; Pacheco-Velázquez, S. C.; Marín-Hernández, Á.; Gallardo-Pérez, J. C.; Robledo-Cadena, D. X.; Hernández-Reséndiz, I.; García-García, J. D.; Belmont-Díaz, J.; López-Marure, R.; Hernández-Esquivel, L. , Resveratrol inhibits cancer cell proliferation by impairing oxidative phosphorylation and inducing oxidative stress. Toxicology Applied Pharmacology 2019, 370, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, J.; Moradi, F.; Maddalena, L. A.; Ferreira-Tollstadius, B.; Selim, S.; Stuart, J. A. , Resveratrol integrates metabolic and growth effects in PC3 prostate cancer cells-involvement of prolyl hydroxylase and hypoxia inducible factor-1. Oncology Letters 2019, 17(1), 697–705. [Google Scholar] [CrossRef]
- Gogada, R.; Prabhu, V.; Amadori, M.; Scott, R.; Hashmi, S.; Chandra, D. , Resveratrol induces p53-independent, X-linked inhibitor of apoptosis protein (XIAP)-mediated Bax protein oligomerization on mitochondria to initiate cytochrome c release and caspase activation. Journal of Biological Chemistry 2011, 286(33), 28749–28760. [Google Scholar] [CrossRef]
- Naeem, A.; Hu, P.; Yang, M.; Zhang, J.; Liu, Y.; Zhu, W.; Zheng, Q. , Natural products as anticancer agents: Current status and future perspectives. Molecules 2022, 27(23), 8367. [Google Scholar] [CrossRef]
- Li, Y.; Zeng, Y.; Mooney, S. M.; Yin, B.; Mizokami, A.; Namiki, M.; Getzenberg, R. H. , Resistance to paclitaxel increases the sensitivity to other microenvironmental stresses in prostate cancer cells. Journal of Cellular Biochemistry 2011, 112(8), 2125–2137. [Google Scholar] [CrossRef]
- Jeong, J. H.; An, J. Y.; Kwon, Y. T.; Rhee, J. G.; Lee, Y. J. , Effects of low dose quercetin: Cancer cell-specific inhibition of cell cycle progression. Journal of Cellular Biochemistry 2009, 106(1), 73–82. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, Y.; Wang, N.; Liu, R.; Wu, Q.; Pei, H.; Li, W. , β-sitosterol suppresses hepatocellular carcinoma growth and metastasis via FOXM1-regulated Wnt/β-catenin pathway. Authorea 2023. [CrossRef]
- Topcul, M.; Cetin, I. , Endpoint of cancer treatment: targeted therapies. Asian Pacific Journal of Cancer Prevention 2014, 15(11), 4395–4403. [Google Scholar] [CrossRef]
- Bordoloi, D.; Roy, N.; Monisha, J.; Kunnumakkara, A. , Multi-targeted agents in cancer cell chemosensitization: What we learnt from curcumin thus far. Recent Patents on Anti-Cancer Drug Discovery 2016, 11 (1); 67-97,
- Williams, G. H.; Stoeber, K. , The cell cycle and cancer. The Journal of Pathology 2012, 226(2), 352–364. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Moralli, S.; Tarrado-Castellarnau, M.; Miranda, A.; Cascante, M. , Targeting cell cycle regulation in cancer therapy. Pharmacology Therapeutics 2013, 138(2), 255–271. [Google Scholar] [CrossRef]
- Wang, W.-L.; Chen, S.-M.; Lee, Y.-C.; Chang, W.-W. , Stigmasterol inhibits cancer stem cell activity in endometrial cancer by repressing IGF1R/mTOR/AKT pathway. Journal of Functional Foods 2022, 99, 105338. [Google Scholar] [CrossRef]
- Prasad, N.; Sabarwal, A.; Yadav, U. C.; Singh, R. P. , Lupeol induces S-phase arrest and mitochondria-mediated apoptosis in cervical cancer cells. Journal of Biosciences 2018, 43, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Mishra, T.; Arya, R. K.; Meena, S.; Joshi, P.; Pal, M.; Meena, B.; Upreti, D.; Rana, T.; Datta, D. , Isolation, characterization and anticancer potential of cytotoxic triterpenes from Betula utilis bark. PloS one 2016, 11(7), e0159430. [Google Scholar] [CrossRef]
- Arul, D.; Subramanian, P. , Naringenin (citrus flavonone) induces growth inhibition, cell cycle arrest and apoptosis in human hepatocellular carcinoma cells. Pathology Oncology Research 2013, 19, 763–770. [Google Scholar] [CrossRef]
- Luo, H.; Rankin, G. O.; Li, Z.; DePriest, L.; Chen, Y. C. , Kaempferol induces apoptosis in ovarian cancer cells through activating p53 in the intrinsic pathway. Food Chemistry 2011, 128(2), 513–519. [Google Scholar] [CrossRef]
- Yuan, L.; Zhang, Y.; Xia, J.; Liu, B.; Zhang, Q.; Liu, J.; Luo, L.; Peng, Z.; Song, Z.; Zhu, R. , Resveratrol induces cell cycle arrest via a p53-independent pathway in A549 cells. Molecular Medicine Reports 2015, 11(4), 2459–2464. [Google Scholar] [CrossRef]
- Bhatt, M.; Patel, M.; Adnan, M.; Reddy, M. N. , Anti-metastatic effects of lupeol via the inhibition of MAPK/ERK pathway in lung cancer. Anti-Cancer Agents in Medicinal Chemistry, 2021, 21 (2), 201-206.
- Chen, Y.; Wu, X.; Liu, C.; Zhou, Y. , Betulinic acid triggers apoptosis and inhibits migration and invasion of gastric cancer cells by impairing EMT progress. Cell Biochemistry Function 2020, 38(6), 702–709. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Dai, C.; Shen, L. , Ursolic acid inhibits epithelial-mesenchymal transition through the Axl/NF-B pathway in gastric cancer cells. Evidence-Based Complementary Alternative Medicine, 2019, 2019.
- Lou, C.; Zhang, F.; Yang, M.; Zhao, J.; Zeng, W.; Fang, X.; Zhang, Y.; Zhang, C.; Liang, W. , Naringenin decreases invasiveness and metastasis by inhibiting TGF-β-induced epithelial to mesenchymal transition in pancreatic cancer cells. PloS one 2012, 7(12), e50956. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Zhao, D.; Zhou, H.; Wang, X.; Zhong, W. l.; Chen, S.; Gu, W.; Wang, W.; Zhang, C.; Liu, Y. , Apigenin inhibits NF-κB and snail signaling, EMT and metastasis in human hepatocellular carcinoma. Oncotarget 2016, 7(27), 41421. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.; Park, S. J.; Choi, Y. S.; Jeon, W.-K.; Kim, B.-C. , Kaempferol suppresses transforming growth factor-β1–induced epithelial-to-mesenchymal transition and migration of A549 lung cancer cells by inhibiting Akt1-mediated phosphorylation of Smad3 at threonine-179. Neoplasia 2015, 17(7), 525–537. [Google Scholar] [CrossRef] [PubMed]
- Bhat, F. A.; Sharmila, G.; Balakrishnan, S.; Arunkumar, R.; Elumalai, P.; Suganya, S.; Singh, P. R.; Srinivasan, N.; Arunakaran, J. , Quercetin reverses EGF-induced epithelial to mesenchymal transition and invasiveness in prostate cancer (PC-3) cell line via EGFR/PI3K/Akt pathway. The Journal of Nutritional Biochemistry 2014, 25(11), 1132–1139. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, S.; Halagowder, D.; Sivasithambaram, N. D. , Quercetin suppresses twist to induce apoptosis in MCF-7 breast cancer cells. PloS one 2015, 10(10), e0141370. [Google Scholar] [CrossRef]
- Sun, X.; Xu, Q.; Zeng, L.; Xie, L.; Zhao, Q.; Xu, H.; Wang, X.; Jiang, N.; Fu, P.; Sang, M. , Resveratrol suppresses the growth and metastatic potential of cervical cancer by inhibiting STAT3Tyr705 phosphorylation. Cancer Medicine 2020, 9(22), 8685–8700. [Google Scholar] [CrossRef] [PubMed]
- Choudhari, A. S.; Mandave, P. C.; Deshpande, M.; Ranjekar, P.; Prakash, O. , Phytochemicals in cancer treatment: From preclinical studies to clinical practice. Frontiers in Pharmacology 2020, 10, 1614. [Google Scholar] [CrossRef]
- Méndez-López, L. F.; Caboni, P.; Arredondo-Espinoza, E.; Carrizales-Castillo, J. J.; Balderas-Rentería, I.; Camacho-Corona, M. d. R. , Bioassay-guided identification of the antiproliferative compounds of Cissus trifoliata and the transcriptomic effect of resveratrol in prostate cancer PC3 cells. Molecules 2021, 26(8), 2200. [Google Scholar] [CrossRef]
- Amico, V.; Barresi, V.; Chillemi, R.; Tringali, C. , Bioassay-guided isolation of antiproliferative compounds from grape (Vitis vinifera) stems. Natural Product Communications 2008, 4(1), 27–34. [Google Scholar] [CrossRef]
- Shanmugam, M. K.; Rajendran, P.; Li, F.; Nema, T.; Vali, S.; Abbasi, T.; Kapoor, S.; Sharma, A.; Kumar, A. P.; Ho, P. C. , Ursolic acid inhibits multiple cell survival pathways leading to suppression of growth of prostate cancer xenograft in nude mice. Journal of Molecular Medicine 2011, 89(7), 713. [Google Scholar] [CrossRef] [PubMed]
- Damle, A. A.; Pawar, Y. P.; Narkar, A. A. , Anticancer activity of betulinic acid on MCF-7 tumors in nude mice. Indian Journal of Experimental Biology 2013, 51, 485–491. [Google Scholar] [PubMed]
- Qin, L.; Jin, L.; Lu, L.; Lu, X.; Zhang, C.; Zhang, F.; Liang, W. , Naringenin reduces lung metastasis in a breast cancer resection model. Protein Cell 2011, 2(6), 507–516. [Google Scholar] [CrossRef] [PubMed]
- Kim, S. H.; Hwang, K. A.; Choi, K. C. , Treatment with kaempferol suppresses breast cancer cell growth caused by estrogen and triclosan in cellular and xenograft breast cancer models. The Journal of Nutritional Biochemistry 2016, 28, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Harper, C. E.; Patel, B. B.; Wang, J.; Arabshahi, A.; Eltoum, I. A.; Lamartiniere, C. A. , Resveratrol suppresses prostate cancer progression in transgenic mice. Carcinogenesis 2007, 28(9), 1946–1953. [Google Scholar] [CrossRef]
- Caesar, L. K.; Cech, N. B. , Synergy and antagonism in natural product extracts: when 1+ 1 does not equal 2. Natural Product Reports 2019, 36(6), 869–888. [Google Scholar] [CrossRef]
- Lucena, F. R.; Almeida, E. R.; Aguiar, J. S.; Silva, T. G.; Souza, V. M.; Nascimento, S. C. , Cytotoxic, antitumor and leukocyte migration activities of resveratrol and sitosterol present in the hidroalcoholic extract of Cissus sicyoides L., Vitaceae, leaves. Revista Brasileira de Farmacognosia, 2010, 20 (5), 729-733.

Table 1.
Activity of C. trifoliata stem extracts against cancer cell lines (IC50 µg/ml).
| Cell line | Hexane | CHCl3-MeOH | Aqueous |
|---|---|---|---|
| Lung cancer A549 | 51 | 85 | 94 |
| Liver cancer Hep3B | 24 | 81 | 81 |
| Breast cancer MCF7 | 30 | 78 | 30 |
| Cervical cancer HeLa | 35 | 82 | 90 |
| Prostate cancer PC3 | 62 | 61 | 58 |
Table 2.
Lipid constituents in the stems of Cissus trifoliata.
| Function in plants | Compound | Presence in Cissus plants |
|---|---|---|
| Components of cuticular wax | Triacontanediol | C. trifoliata |
| Hentriacontane | C. quadrangularis | |
| Nonacosane | C. cornifolia | |
| Octacosane | C. trifoliata | |
| Structural role in cellular membranes | Arachidic acid | C. quadrangularis |
| Stearic acid | C. quadrangularis | |
| Palmitic acid | C. quadrangularis, C. vitiginea | |
| β-sitosterol | C. quadrangularis, C. assamica, C. polyantha, C. rheoifolia, C. pteroclada | |
| Campesterol | C. quadrangularis, C. rheifolia | |
| Stigmasterol | C. quadrangularis, C. assamica, C. polyantha, C. rheoifolia, C. pteroclada | |
| Squalene | C. quadrangularis | |
| Defensive role, acting as antifeedant, and antimicrobial | Ursolic acid | C. assamica, C. repens |
| Betulinic acid | C. assamica | |
| Lupeol | C. quadrangularis, C. assamica, C. repens. |
Table 3.
Polar constituents in the stems of Cissus trifoliata.
| Function in plants | Compound | Presence in Cissus plants |
|---|---|---|
| Structural component in the cell wall, and serves to enhance its rigidity and strength | Isoferulic acid | C. trifoliata |
| Regulatory role in inducing abiotic stress tolerance and enhancing the direct defense against insects | Protocatechuic acid | C. trifoliata |
| Antimicrobial properties and protection against oxidative stress | Trigallic acid | C. trifoliata |
| Vital role in response to pathogenic infections | Trans-p-coumaric acid | C. trifoliata |
| Protect photosynthetic tissues from oxidative damage, and confer enhanced salinity tolerance and growth | Apigenin | C. adnata, C. digitata, C. verticillata and C.quadrangularis |
| Protect from oxidative stress induced by heat, water, nutrients, or mechanical damage due to insects, or fungal infections | Cyanidin | C. sicyoides, C. quadrangularis |
| Promote plant defense systems to environmental and pathogenic stress | Delphinidin | C. sicyoides, C. quadrangularis |
| Inhibits bacterial or fungal infections | Myricetin | C. quadrangularis |
| Involved in auxin-regulated cell division as a signaling molecule. Increases grown in heavy metal-contaminated soils | Kaempferol | C. ibuensis, C. sicyoides, C. quadrangularis |
| Regulate response to salt stress tolerance | Dihydrokaempferol | C. quadrangularis |
| Allelochemical, impairing auxin transport and alleviates short-term osmotic and salinity stresses | Naringenin | C. rotundifolia |
| Antifeedant activity and toxicity against insects | Chrysoeriol | C. aralioides, C. lageniflora and C. petiolata |
| Protect epidermal tissue from oxidative stress in response to high light intensity. Regulates the antioxidant enzymes at the transcriptional level | Quercetin | C. ibuensis, C. digitata and C.quadrangularis |
| Reduce the oxidative damage and susceptibility to fungal infections | Syringetin | C. trifoliata |
| Antimicrobial, nematocidal, and insecticidal activities, increase resistance to drought, thermal stress, ultraviolet radiation, mechanical stress, heavy metals, salts, and air pollutants | Resveratrol | C. ibuensis, C. sicyoides, C. quadrangularis |
| Contribute to resistance against fungal infection | ε-viniferin | C. repens, C. sicyoides |
| Display antifungal activities | Pallidol | C. quadrangularis, C. pallida |
| Produced in response to infection, has antimicrobial activity and increases the tolerance to stress | Piceatannol | C. quadrangularis |
Table 4.
Cytotoxic activity (IC50 µg/ml)† of compounds present in Cissus trifoliata against carcinoma cells.
Table 4.
Cytotoxic activity (IC50 µg/ml)† of compounds present in Cissus trifoliata against carcinoma cells.
| Compound | A549 | MCF7 | HeLa | PC3 | Hep* | Mechanisms underlying apoptosis |
 |
96 | 250 | 170 | 74 | 25 | Increases caspase-8 activity and impairs sphingolipid metabolism |
 |
21 | 9 | 170 | 8 | 12 | Upregulation of Bax and p53 expression and downregulation of Bcl-2 |
 |
21 | 32 | 38 | 30 | 48 | Triggers mitochondrial cell death pathway by downregulation of Bcl-2, Bcl-xL, and survivin expression with a negative modulation of STAT3 activities |
 |
15 | 9 | 10 | 10 | 10 | Cell death through intrinsic pathway promoting the activities of caspases 9, 3, and 7 and the cleavage PARP and inhibition of NF-κB pathway |
 |
18 | 24 | 5 | 15 | 22 | Upregulation of Fas/APO-1 and downregulation of JAK2/STAT3 and NF-κB pathways, Bcl-2, and Bcl-xL expression |
 |
218 | 468 | 53 | 13 | 54 | Enhances TRAIL death receptor expression, the formation of the apoptosome complex, caspase-3 activation, and decreases Bcl-2 expression |
 |
20 | 8 | 3 | 11 | 22 | Induces protein production of p53, Bid, and Bax while decreasing the levels of Bcl-2. It also causes the release of cytochrome c, caspase activation, and suppresses the phosphorylation of Akt, Bad, and glycogen synthase kinase-3 |
 |
10 | 48 | 14 | 17 | 11 | Upregulates p21, p53, caspases 3, 4, 7, and downregulates of Bcl-2 and Bcl-xL |
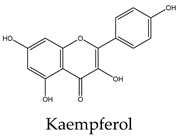 |
22 | 50 | 56 | 14 | 24 | Induces the expression of Bax, and caspase-3 with downregulation of NF-κB, mTOR, Bcl-2, and the PI3k/Akt and ERK pathways |
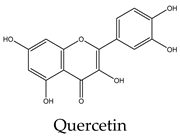 |
16 | 26 | 19 | 15 | 30 | Inhibits survivin, protein p21-activated kinase 1, PI3k/Akt, ERK, and the β-catenin pathway, with activation of caspases 3, 9 and increasing the expression of Bad and p53 |
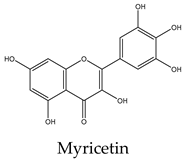 |
2 | 45 | 20 | 46 | 12 | Induces cytochrome c release and activation of caspases 3 and 9, while upregulates p53, Bax and downregulates Bcl-2 and NF-κB |
† All IC50 values were subjected to unit conversion since according to NCI active compounds must show inhibitory activity at concentration ≤4 µg/ml. * Column groups data from HepG2 or Hep3B cells, both derived from liver carcinoma.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



