
Submitted:
02 January 2024
Posted:
03 January 2024
You are already at the latest version
Abstract
New insights into the causes and mechanisms of congenital heart disease (CHD) have been gained through the use of next-generation sequencing. Examination of the whole exome sequence detects detrimental gene variations modifying single or contiguous nucleotides, that are characterised as pathogenicity based on statistical assessments of families. and correlates with congenital heart disease, elevated expression during heart development, and reduction of harmful protein-coding mutations in the general population. Patients with CHD and extracardiac abnormalities enriched for gene classes meeting these criteria. CHDs and extracardiac defects, supporting a common set of pathways in the organogenesis of CHDs. Single-cell transcriptomics data reveal the expression of genes associated with CHD in specific cell lineages, and emerging evidence suggests that genetic variants disrupt multicellular genes essential for cardiogenesis. Metrics and units are being tracked in whole genome sequence studies.
Keywords:
congenital heart disease
; first heart field
; second heart field
; whole exome sequencing
; whole genome sequencing
; loss-of-function variant
; copy number variants
; gene variants
; deletion
; de novo mutations
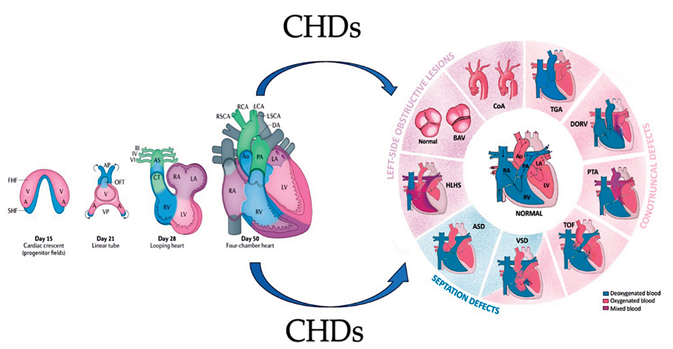
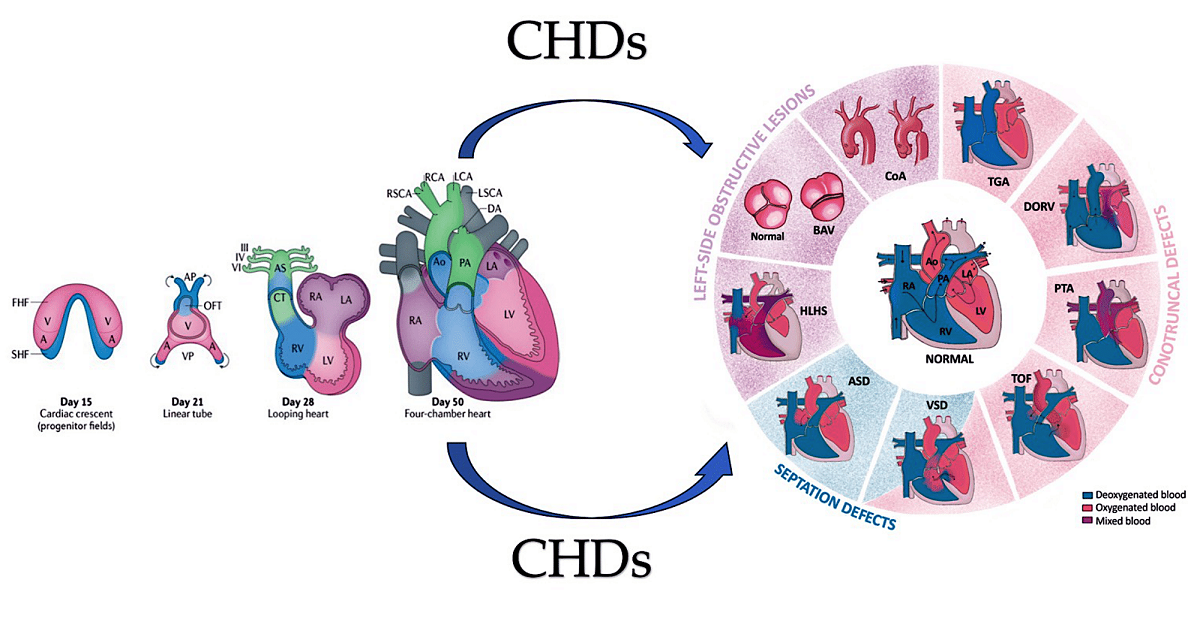
Graphical abstract. The image on the left illustrates the development of the human heart, beginning with the formation of the first heart field (FHF) or cardiac crescent at approximately embryonic day 15. Chamber-specific cardiac progenitors are arranged in a specific spatial distribution. Simultaneously, another group of cardiac progenitors, the second heart field (SHF), develops behind the FHF. By day 21 of embryonic development, the fusion of the FHF midline forms a linear cardiac tube. Cells from the SHF migrate into the arterial and venous poles of the tube, as indicated by the arrows. By day 28, the linear heart tube loops to form early ventricular chambers. Cardiac neural crest cells populate the pharyngeal arches III, IV and VI, the aortic sac (AS) and the conotruncal ridges (CT). At embryonic day 50, the heart structures resemble those of the mature heart, with four main chambers and two outflow tracts (OFTs). The diagram on the right displays the typical four-chambered heart of mammals and the structural abnormalities present in the most prevalent forms of congenital heart disease (CHDs). The black arrows indicate the direction of blood flow. Abbreviations; A, atrium, Ao, aorta, ASD. Atrial Septal Defect; AV, atrioventricular; BAV: Bicuspid Aortic Valve; CoA, coarctation of the aorta; DA, ductus arteriosus; DORV for Double-Outlet Right Ventricle; HLHS, hypoplasic left heart syndrome; LA, left atrium; LCA, left carotid artery; LSCA, left subclavian artery; LV, left ventricle; PA, pulmonary artery; , PTA, Persistent Truncus Arteriosus RA, right atrium; RCA, right carotid artery; RSCA, right subclavian artery; RV, right ventricle; TGA for Transposition of the Great Arteries; TOF for Tetralogy of Fallot; V, ventricle; VSD for Ventricular Septal Defect From Ibrahim et al Int. J. Mol. Sci. 2023, 24(22), 16258; https://doi.org/10.3390/ijms242216258
1. Current knowledge
Congenital heart disease (CHD) is widely acknowledged as the most frequent and frequently serious abnormality present at birth, with a prevalence of 6-13 per 1,000 newborn infants aged. [1,2,3,4,5,6] CHD includes a wide range of heart deformities that vary from a solitary irregularity such as an atrial or ventricular septal defect or an isolated dysplastic valve to complex conditions. Cardiac conditions characterized by multiple defects, namely tetralogy of Fallot or hypoplastic left heart syndrome. (Figure 1 and Figure 2) Improved surgical and medical techniques to palliate and definitively treat critical and corrective measures for critical malformations, which require early intervention for survival, have led to a significant reduction in the mortality rate from CHD. [7] As a result, more than 90% of patients with CHD are able to survive into adulthood, contributing to the increased prevalence of CHD in the general population. In addition, the extended lifespan of patients with CHD has prompted greater acknowledgement of the related comorbidities. Roughly 13% of neonates with CHD demonstrate extracardiac structural or functional abnormalities and may encounter neurodevelopmental delays [3,8] during childhood. CHD is classed as syndromic if other diagnoses are likely to have resulted from the same aetiology. Syndromic CHD commonly causes intricate heart malformations, which may lead to lifelong health issues affecting various bodily systems.
Therefore, comprehending the aetiologies and mechanisms of CHD can provide valuable insights into both normal and abnormal developmental processes of multiple organs.
1.1. Genetic
As a result of significant technical advances in human genome research, genetics plays a key role in the development of CHD. Initially, evidence emerged from examining patients with syndromic CHD through karyotyping, identifying aneuploidies such as trisomy 13, 18 or 21 (Down syndrome) and monosomy X (Turner syndrome) in approximately 12% of CHD patients. [9] Cytogenetic analysis and genomic arrays have helped to understand subchromosomal structural rearrangements, highlighting frequent 3-Mb deletions at
22q11.2 in DiGeorge (velocardiofacial) syndrome. [10,11,12,13] Deletions are present in approximately 2% of all cases of CHD and in 13% of individuals with specific cardiac malformations, as determined by fluorescence in situ hybridization [14,15] and targeted amplification. [16] About 25% of sporadic CHD cases are caused by the coexistence of karyotype or microarray-detected abnormalities. [17,18] The preliminary detection of monogenic causes of familial CHD has been facilitated by genome-wide linkage causes of familial CHD, encompassing variations in genes encoding transcription factors and transcriptional regulators of genes involved in heart growth. [19,20] Enhanced comprehension of the human genome at the base pair level, along with advancements in sequencing technologies, has facilitated the identification of genes linked to CHD. Advanced whole exome sequencing (WES) and whole genome sequencing (WGS) platforms, combined with bioinformatics tools and large, compiled sequencing datasets from population-based studies enable the detection of deleterious variants, including missense mutations, loss-of-function (LOF) variants, minor insertions or deletions, copy number variants (CNVs), and structural variants. The implementation of these technologies has exposed harmful variants that are present in family cases of CHD, show de novo mutations in CHD probands with unaffected parents, and appear at notably higher frequencies in around 20% of CHD patients in large cohort studies. [18,19,20,21,22,23,24,25]
Taken together, in 45% of patients with CHD, these methods can detect deleterious coding variants in definitive and candidate genes for CHD. [18,19,20,21,22,23,24,25] Genes firmly linked to CHD manifest mutations that show significant co-segregation within families impacted by CHD, are significantly more prevalent in unrelated patients faced with CHD compared to control groups, or account for heart defects in individuals with CHD linked to syndromes. CHD candidate genes exhibit shared features, but their properties lack statistical significance for definitive categorisation. Various types of variants, including aneuploidies, CNVs, structural variants, and LOF variants, alter the dosage of CHD-related genes. Conversely, deleterious missense variants can maintain the physiological gene dosage whilst impairing the function of the encoded protein. [21,24] Figure 3
Genes associated with CHD offer fresh insights into the essential molecules and pathways implicated in cardiogenesis, adding to and elaborating on the extensive information garnered from exploring heart development in experimental models. Studying spontaneous gene variants in sporadic CHD can reveal novel genes implicated in cardiogenesis, while providing detailed information. [17,21,24] Clinical evaluations enable the identification of a wider range of cardiac and extracardiac phenotypes compared to studies conducted on experimental models. As the majority of human variants have a heterozygous dosage, while experimental models typically examine homozygous variants, CHD data obtained from human studies can reveal information regarding less evident impacts on heart formation. Furthermore, patients with critical and complex malformations display the greatest frequency of de novo damaging variants in genes linked to CHD, which provides convincing evidence of severe adverse effects on reproductive health; and the evolutionary restriction on numerous genes connected with CHD. [8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25]
The question of why CHD is the most prevalent congenital abnormality is intriguing. One hypothesis suggests that the intricate developmental processes involved in heart formation are highly responsive to alterations in gene dosage for many essential genes and pathways. Such changes can cause developmental errors, many of which the 18-week gestational age screening ultrasonography can readily detect. [26,27] To showcase the intricate molecular orchestration of heart formation, this review provides a brief summary of cardiac development. This is followed by a discussion of the latest genomic discoveries and strategies at the forefront of current CHD research over the past decade. Key signals that govern cardiac morphogenesis have been examined extensively in several informative reviews which are recommended for further details, including publications. [28,29,30,31,32,33,34,35]. Throughout the review, we identify in bold those genes with harmful variants detected in patients with CHD. Figure 4 [22,23,36,37,38,39,40]
2. Development of heart
The heart develops during the early stages of embryogenesis. The cardiac precursor fields differentiate and coalesce to create the cardiac crescent. During human gestation, the heart tube grows, undergoes regional transformation, and emerges as a nearly fully formed organ by 8 weeks. The process involves a linear and then looped structure. During heart development, differentiating precursor cell clusters interact to generate specialised heart cells with well-defined three-dimensional architecture within restricted regions.
2.1. A look at the lines of progenitor cells and their morphogenesis.
The first heart field (FHF), derived from the lateral plate mesoderm, the second heart field (SHF), derived from the lateral plate splanchnic mesoderm, and the migrating cardiac neural crest, which populates the III, IV and VI pharyngeal arches, are three major cell lineages involved in the formation of heart structures [35,41,42,43] (Figure 5). The FHF and SHF, which are symmetrical, merge anteriorly to form the heart crescent, which then coalesces into a linear cardiac tube with ventricular and atrial precursors aligned along the anterior-posterior axis. The heart cells originating from the FHF are involved in the development of the atria and the left ventricle, while the cells from the SHF are involved in the development of the right ventricle and the outflow tract and also contribute to both atria. Asymmetric growth causes ballooning of the outer curvature of the heart tube, which in turn increases the size of the ventricular chamber [44,45]. The separation of the subsequent outflow tract and the differentiation of smooth muscle occur with the contribution of the cardiac neural crest.
2.2. Heart morphogenesis under genetic control
Several important biomolecules play a role in differentiating progenitor cells into cardiac cell lineages and directing cardiac morphogenesis. Genes associated with CHD have identified some of these molecules. FHF arises from cardiac mesoderm through signalling including bone morphogenetic protein (BMP) and fibroblast growth factor (FGF), which are induced by the adjacent endoderm [46,47,48,49]. The expression of key cardiac transcripts required for cardiomyocyte lineage commitment is reduced in experimental models by deletion of genes encoding BMPs [50]. The expression of genes such as GATA4, GATA6, NKX2-5 and TBX5 control sarcomere formation and contraction. [28,29,30,31,32,33,51] Ventricular cardiomyocyte specification is promoted by the expression of IRX4 in both the FHF and SHF of the cardiac crescent. [52] Factors such as retinoic acid within the mesodermal progenitor fields initiate commitment to an atrial lineage. [53,54]. Further specification leads to cardiomyocytes that will populate the left ventricle (IRX4, HAND1 and MSX1), [55,56,57] the right ventricle (IRX4 and GATA6) [55,58] and the outflow tract (GATA6, HAND2, ARID3B and TEAD45). [56,58,59,60] FGF signalling is required for SHF contributions to the outflow tract, allowing neural crest migration and induction of tbx1 expression in the SHF in zebrafish. [61] Although there are stereotyped differentiation pathways, plasticity is maintained in the progenitor fields during cardiac development. For example, ablation of FHF cells in zebrafish results in SHF compensation that regenerates ventricular cardiomyocytes. [62] Additional pathways and gene networks involved in cardiac neural crest differentiation have been discovered. In mice, normal differentiation of cardiac neural crest cells is prevented in the absence of retinoic acid signalling, [63] which can be ameliorated by maternal vitamin A supplementation. [64] This highlights the genotype-environment interactions in CHD. In contrast, a missense variant of GATA6, which is frequently found in patients with CHD and impairs outflow track formation, significantly enhances retinoic acid signalling. [58] FOXC2 expression is essential for cardiac neural crest migration, as it mediates the activation of Sema3c expression in the SHF. TBX1 expression, on the other hand, results in counterbalancing inhibitory signals. [65,66]
In the atrioventricular canal and outflow tract, the heart contains non-chambered cardiomyocytes, as well as valvular endothelial and smooth muscle cells. Regulatory proteins, such as TBX2, TBX3, [67], BMP2, and BMP4, [68] repress chamber-specific genes to specify non-chamber myocardium. Some valvular endothelial cells undergo a process called endothelial-to-mesenchymal transition to populate the endocardial cushions. This process is guided by signalling cascades that include vascular endothelial growth factor (VEGF) [69] and transforming growth factor-β (TGFβ). [70] Investigations into the differentiation of the outflow tract in mouse models have also shown that histone deacetylase 3 (encoded by Hdac3) is necessary for the differentiation of smooth muscle, whereas FGF and sonic hedgehog signalling are needed for the pulmonary arteries and veins. [71,72,73] Through transdifferentiation, cardiomyocytes can also change into cells that are not cardiac chambers. During the development of the mouse heart, a population of cardiomyocytes was found to transdifferentiate into vascular smooth muscle cells of the great arteries. [74]
The left-right signalling axis is established in the embryo within the embryonic node. This occurs through signals mediated by motile and sensory cilia, leading to the asymmetric expression of genes that encode downstream regulatory signals.
The genes encoding downstream regulatory signals include those coding for the TGFβ family members Nodal, Lefty1, Lefty2 and Zic3 [75,76,77]. These cues are critical for proper rightward looping of the linear heart tube, which properly positions the atria, ventricles and outflow tract and gives the sinoatrial node in the right atria. Disruption of Sonic hedgehog or activin signalling in the chick embryo [78] or knockout of mutations in Lefty2, which encodes left-right axis signalling proteins and transcriptional regulators, can cause defects in cardiac looping and result in congenital heart defects in mice. [79]
3. Interfering with Human Cardiac Development
Studying the anatomy of the human embryo has helped to understand how the human heart develops and the causes of cardiac malformations resulting from disrupted spatiotemporal dynamics. CHD is often divided into anatomical subgroups that are the result of abnormalities in common embryonic processes. Therefore, insights into the downstream developmental and physiological consequences will be gained from the discovery of genes with pathogenic variants that cause specific malformations.
3.1. Atrial and ventricular septal defects.
Valve and atrioventricular canal abnormalities, including defects in septum and valve function, may result from abnormal formation of endocardial cushions and/or dorsal mesenchymal protrusion. Only about 5% of individuals with CHD [80] have complete atrioventricular canal defects. However, almost half of the approximately 40% of individuals with trisomy 21 who have CHD have this defect. [81,82] This association suggests that genes located within a 0.96-Mb region of chromosome 21 play a crucial role in endocardial cushion development. [83] However, the responsible gene(s) are still unknown. As with other syndromes, not all people with trisomy 21 will go on to develop CHD, and up to 60% will have a structurally normal heart. [81,82]. Single atrial or ventricular septal defects are the most frequent congenital heart malformation; however, the causal etiology is rarely defined in individuals who have these defects sporadically in the absence of a genetic syndrome. Deleterious variants in NKX2-5 and GATA4 were first detected in patients with rare autosomal dominant familial septal malformation. In families with septal defects and associated limb defects, TBX5 variants were identified as the cause of Holt-Oram syndrome. In model systems, the proteins of these genes physically interact with each other to promote the correct septation of the heart chambers during the separation of the atria and ventricles. [84,85,86,87,88,89]. Figure 6
3.2. Left-sided obstructive lesions.
A number of narrowing or obstructive abnormalities of the left ventricle and valves are included in this group of CHDs. These may occur in isolation, such as mitral stenosis and bicommissural aortic valve, [90,91] (Figure 7) aortic stenosis and coarctation of the aorta, (Figure 8) or in combination. [90] This is a condition known as Shone complex, which is characterised by an annulo-leaflet mitral ring, parachute mitral valve, subaortic stenosis, coarctation of the aorta and hypoplastic left heart syndrome with mitral and aortic stenosis or atresia and hypoplastic left ventricle. (Figure 9) [90] Compared to other congenital heart defects, left-sided obstructive lesions have a higher heritability. Families with a range of malformations show a higher incidence of CHDs, suggesting a shared genetic pathway in the development of these structures. [90,91,92]
Bicommissural aortic valve (15-30%), aortic coarctation (7-20%) and, less commonly, hypoplastic left heart syndrome [93,94] are common in patients with Turner syndrome (45, XO karyotype). Patients with Turner syndrome have an increased risk of left-sided obstruction lesions due to altered gene dosages on chromosome X and copy number polymorphisms at the 12q13.31 locus. [95,96,97] However, it is currently unknown how these genotypes affect the development of left-sided structures. Jacobsen syndrome is a genetic disorder caused by a deletion in chromosome 11q23. [98] This syndrome can cause severe left-sided obstructive lesions in 33% of patients. The development of these lesions is partly due to a requirement for the protein C-ets-1, which is encoded by ETS1. This protein is necessary for supporting cardiomyocyte proliferation and extracellular matrix organization in the developing endocardium. [99,100,101] Bicuspid aortic valve is caused by monogenic damaging variants in GATA5, [102,103] while hypoplastic left heart syndrome is caused by such variants in RBFOX2. [104] Recently, in a family with autosomal dominant BAV, a novel heterozygous GATA6 mutation, p.E386X, was identified. [105]
Mutations in MYH6, which encodes for α-myosin heavy chain, can lead to Shone complex [22] and hypoplastic left heart syndrome [106,107]. MYH6 is a contractile protein highly expressed during cardiogenesis and is one of the few CHD-associated genes not associated to transcription or translation. Figure 10
3.3. Right-sided obstructive lesions.
These conditions reflect a wide range of embryological abnormalities that hinder the blood flow from the right side of the heart to the pulmonary capillary beds. Obstruction to the flow of systemic venous blood can occur in various conditions, such as tricuspid atresia, pulmonary atresia with intact ventricular septum, isolated pulmonary valve stenosis, supravalvular and branch pulmonary artery stenosis. Tricuspid atresia is associated with chromosomal trisomy in humans [108] and Zfpm2 LOF in mice [109] and is caused by the absence of the right-sided atrioventricular valve. Noonan syndrome is associated with pulmonary valve stenosis in more than 50% of children, particularly in those with variants in the PTPN11 gene. [110] PTPN11 variants cause pulmonary valve stenosis (PVS) by activating mitogen-activated protein kinases (MAPKs) in endocardial cells. This leads to excess endothelial-to-mesenchymal transition during pulmonary valve development. [111,112,113]
Other syndromes such as Costello and cardiofaciocutaneous syndromes, which are caused by excessive activation of the RAS-MAPK pathway, often result in PVS. [113,114,115]. Recently, the molecular determinants of Costello syndrome (CS) have been investigated, with the activation of mitochondrial bioenergetics and quality control resulting in restored organelle function in HRAS p.G12A and p.G12S cell models. In addition, reduced left ventricular hypertrophy was observed in CS mice and the incidence of developmental defects was reduced in the CS zebrafish model. Together, these findings underscore the importance of mitochondrial proteostasis and bioenergetics in the pathophysiology of RASopathies and indicate that patients with CS may benefit from the use of mitochondrial modulators. [115] Figure 11
Robinow syndrome can cause isolated pulmonary valvular stenosis and conotruncal lesions, such as tetralogy of Fallot [116,117,118], in both autosomal dominant and recessive forms. The pathogenic variants in genes encoding receptors and ligands controlling planar cell polarity (WNT5A, ROR2, DVL1 and DVL3) are responsible for the syndrome. [116,117,118,119] These findings demonstrate the importance of planar cell polarity. The development of the right ventricular infundibulum and the outflow tract cushions in the SHF is influenced by the cell polarity pathway. Tetralogy of Fallot, PVS and/or stenosis of the peripheral pulmonary arterial branches can occur in individuals with autosomal dominant Alagille syndrome (caused by mutations in the JAG1 and NOTCH2 genes). This demonstrates the sensitivity of Notch signalling in the development of the right ventricular outflow tract, pulmonary valve formation, and vasculogenesis, which contributes to the pulmonary arterial tree. [120,121,122] Figure 12
3.4. Defects in the left-right pattern.
Heterotaxy syndrome is caused by defects in left-right axis patterning, resulting in abnormal positioning of the viscera and congenital heart defects, including structural malformations and aberrant arterial and venous connections. Critical to left-right axis patterning is normal ciliary function. Some patients with primary ciliary dyskinesia have heterotaxy syndrome with CHD. Transposition of the great arteries is typical in patients with non-syndromic congenital heart disease and deleterious variants in cilia-related genes. [123,124]. Patients with cardiac left-right patterning defects also frequently have deleterious variants in genes involved in laterality, such as NODAL, FOXH1, ZIC3, ACVR2B and SMAD2. [123,124,125] Defects can cause CHD through autosomal dominant or autosomal recessive mechanisms. Variants in ciliary genes involved in laterality defects can result in CHDs in either autosomal dominant or autosomal recessive inheritance. Variants associated with CHD in genes encoding Nodal and TGFβ signalling proteins are dominant, whilst ZIC3 variants are associated with X-linked heterotaxy syndrome in hemizygous males. [126,127,128,129,130]
3.5. Conotruncal defects.
Conotruncal defects, including tetralogy of Fallot, persistent truncus arteriosus, and transposition of the great arteries (as shown in Figure 13), result from underdevelopment or misalignment of the ventricular septum, outflow tract, and/or great arteries.
Autosomal dominant 22q11.2 deletion syndrome often leads to conotruncal defects, which are commonly associated with the deletion of the CHD-associated gene TBX1. [131,132,133,134] Similarly, CHARGE syndrome, which is often caused by deletions or variants in the CHD-associated gene CHD7, [135,136,137,138] can also result in conotruncal defects. These disorders are associated with cardiac, facial and head malformations, which originate in neural crest cells, and reveal effects of harmful gene variants on cell lineages involved in extra-cardiac malformations. Both of these congenital heart defects display variable penetrance and expressivity [138,139,140], suggesting that additional factors influence the the clinical phenotype. A genetic modifier that influences conotruncal defects in 22q11.2 deletion syndrome has been reported, although these additional factors are largely unknown. [131,141] Recurring duplications and deletions at 1q21.1 involving the CHD candidate genes GJA5 encoding connexin 40 and CHD1L linked to CHD7 gene are often associated with tetralogy of Fallot, sometimes (but not invariably) accompanied by extracardiac abnormalities such as mental retardation and dysmorphic features of the face [142,143,144]. The deletion in the 1q21 proximal region cannot evolve toward thrombocytopenia-absent radius syndrome critical region or the RBM8A gene. [144]
Nonsyndromic conotruncal defects are also primarily linked with monogenic loss of function variants in genes involved in two important signalling pathways that govern development of processes in the second heart field (SHF) and cardiac neural crest cells. These pathways are the Notch signaling (NOTCH1) and the VEGF signal trasduction (KDR, FLT4, VEGFA, FGD5, BCAR1, IQGAP1, FOXO1 and PRDM1). [145,146,147,148] The reason why these progenitor fields and developing tissues are particularly susceptible to alterations in the VEGF pathway is still unknown. Additionally, conotruncal defects are frequently caused by autosomal dominant damaging variants in ZFPM2, which encodes the transcription factor ZFPM2, and in the genes encoding its binding partners GATA4 and GATA6. [66,149,150,151,152,153] The GATA genes are transcriptional regulators that act as "pioneer workers" for transcription. They bind to and modulate closed regions of chromatin, allowing the later binding of transcription signals. This demonstrates the importance of the transcriptional regulatory network in conotruncal formation. [151]
4. Cohort studies reveal new CHD genes.
Analyses of CHD cohorts recruited from single centres or through national and international collaborations are increasingly identifying genes associated with CHD. [22,25,154] The majority of studies include patients with severe or critical CHD. There is an under-representation of patients with prevalent simple CHD findings and an absence of patients with confirmed genetic syndromes. [155] WES and case-control analyses are commonly used to detect variants. These investigations indicate that individuals with CHD have the same proportion of total de novo variants but substantially more uncommon (allele frequency ≤ 1 × 10-5 in the general population) deleterious variants relative to the general population. These comprise a 9% excess of de novo deleterious variants, a 7% excess of dominant inherited variants and a 1% excess of recessively heritable variants. [22,23,24,25]
In patients with undetermined sporadic CHD, deleterious variants are detected in an estimated 8-10% of patients. [22,25] The highest rate of de novo harmful in patients with syndromic CHD, while the highest rate of heritable LOF variants is found among patients with isolated CHD. [25] Cohort studies have supported the relevance of previously defined biological signalling pathways in CHD pathogenesis. These include genes involved in the Notch [156] and VEGF [156] signalling cascades. Chromatin remodelling is, however, the most striking functional class of newly identified CHD-associated genes. [23,36] Patients with CHD who had extracardiac anomalies and neurodevelopmental delay most often had deleterious variants in these genes encoding chromatin remodelling proteins. The broad expression pattern of the genes suggests that organ development uses common epigenetic and transcriptional regulatory pathways.
The importance of ciliary function in cardiogenesis is supported by WES data from patients with CHD [157]. It is often the case that deleterious variants in cilia-related genes are inherited from an unaffected parent, as has been shown in other recessive diseases. In contrast, harmful variations in genes associated with chromatin alteration are more likely to occur de novo, which indicates that the damage caused by even a single mutant allele in this class of genes is significant.
4.1. CHD definitive genes.
WES data were analysed from two cohorts of patients with congenital heart defects: 2056 patients with isolated congenital heart defects, 2994 with syndromic congenital heart defects and 808 with congenital heart defects and unknown extracardiac status. The analysis had sufficient statistical power to detect a significant excess of harmful variants in patients with CHD compared to matched controls, with correction for analyses of all protein-coding genes (P ≤ 2 × 10-6) [22,25,158]. From these cohorts, the genes that are identified have a definitive direct link to CHD, as with the genes related to the rare familial forms of CHD. However, the genes discovered in large study cohorts have a greater contribution to the overall population prevalence of CHD. In these two WES cohorts, a total of 18 genes were significantly enriched. The genes are ADNP, ANKRD11, CDK13, CHD4, CHD7, DDX3X, DYRK1A, FLT4, KMT2A, KMT2D, NOTCH1, NSD1, PACS1, PRKD1, PTPN11, RBFOX2, SMAD6 and TAB2. [22,25,146,147] Although patients with syndromic CHD were excluded at enrolment, the analysis identified variants in CHD7 (CHARGE syndrome), KMT2D (Kabuki syndrome), NSD1 (Sotos syndrome) and PTPN11 (Noonan syndrome). This suggests that formes frustes of syndromic phenotypes, which escape clinical recognition, may arise from some of the variants associated with syndromic CHD. Mutations in CDK13, CHD4 and PRKD1 are more common in syndromic CHD, [22] whereas SMAD6 mutations are more common in isolated CHDs. [159] Previous studies have emphasised the biological significance of certain genes associated with CHD. In mice, heart malformations have been observed as a result of null mutations in the murine homologues of SMAD6 and NOTCH1. [160,161] RBFOX2 transcript levels are reduced in some CHD patient tissues compared to controls. [162] Additionally, the protein encoded by DYRK1A is a therapeutic target for neurodevelopmental phenotypes in individuals with trisomy 2. [163,164] New phenotype correlations have been discovered through recurrent variants in other genes. NOTCH1 and FLT4 mutations are causative factors in tetralogy of Fallot, accounting for 7% of patients in one study. [22,145,146,147] Additionally, genes previously linked to intellectual disability, such as ADNP, [165] ANKRD11 (KBG syndrome), [166] CDK13, [167,168] and DDX3X, [169] are now accepted to be implicated in CHD. However, many definitive roles of CHD in heart development remain unknown.
4.2. Genes linked with CHD that have deleterious variants in humans.
According to WES data, around 400 genes are believed to contribute to CHD, which is significantly more than the number currently known. [36] Although some of these genes may contain harmful variants in CHD patients, they cannot be classified as definitive CHD-associated genes due to a lack of statistical significance. [22,25,146,158] Morton et al conducted meta-analyses of WES data from two large cohorts of patients with congenital heart disease to overcome limitations in statistical power caused by small sample sizes. Patients with aneuploidies or 22q11 deletions were excluded. The WES data were compared with data from a cohort of 128 individuals without CHD, as well as with the Genome Aggregation Database. The authors identified 132 candidate genes for CHD. 66 of these variants were enriched for LOF and 78 were enriched for deleterious missense. Patients with aneuploidies or 22q11 deletions were excluded from the analysis. [158]
This meta-analysis supports the involvement of CHD-associated genes in transcriptional regulation of transcription. Several of the genes identified as candidates for CHD with loss-of-function variants have functional annotations related to chromatin modification, transcription factors, or RNA processing, and some have been previously studied. Implicated in CHD or in organogenesis, accounting for associated extracardiac phenotypes. Patients with loss-of-function variants in the group of definitive and candidate genes for congenital heart disease regulation of gene expression exhibited a higher incidence of extracardiac phenotypes than those with damaging missense variants. [22,25] The analyses confirmed that de novo LOF variants were more common in patients with CHD and extracardiac abnormalities in genes related to chromatin modification. In patients with CHD and extracardiac abnormalities, there was an enrichment of inherited missense variants, while in patients with isolated CHD, there was an enrichment of transmitted LOF variants. These observations suggest a potential genetic basis for the presence or absence of extracardiac abnormalities in CHD patients. Some chromatin-modifying proteins may have cardiac-specific functions.
New technologies have enabled the comparison of results from studies on people of different ethnic and racial backgrounds [22,25,170]. The functional assessment of deleterious missense variants is expected to reclassify some candidate variant genes for CHD as definitive, improving genotype-phenotype correlations and increasing our knowledge of their precise roles in cardiac development.
5. Whole-genome sequencing.
Modern whole genome sequencing offers technical advantages over whole exome sequencing. (Figure 1) It eliminates the need for PCR amplification, provides more consistent genome coverage, and has superior validation rates for single nucleotide polymorphisms and copy number variations [171]. WGS can also identify rare or common mitochondrial sequences, microRNAs, non-coding RNAs, and promoter and regulatory genes, in addition to detecting exomes. WGS has successfully identified MYH6 variants associated with aortic coarctation in patients. Additionally, adults with tetralogy of Fallot and a missing pulmonary valve have higher rates of deleterious variants in VEGF. A comparison was made between adults with tetralogy of Fallot and a missing pulmonary valve and those with a present pulmonary valve. [172,173,174]
5.1. Variation in non-coding sequences through whole genome sequencing
Only about 1% of the genome encodes protein, while the remaining 99% consists of non-coding sequences. These sequences include promoters, enhancers, and factors that influence chromatin topology or the accessibility of binding sites for RNA or protein factors and other unknown factors. [175,176,177,178,179,180] Variants in non-coding sequences can be harmful and may cause or influence the expression of CHD-associated genes. Topological association underlies this hypothesis. During cardiomyocyte differentiation in vitro, there is evidence of a relationship between non-coding regions and gene promoters [181,182,183]. Additionally, assessing non-coding sequences within CNVs can aid in defining them. The regions involved in CHD are likely to be those that are most involved in the development of the CHD. [184] In fact, targeted analyses of non-coding sequences TBX5 flanking regions were analysed and three potential enhancers were identified. One of these enhancers contained a homozygous variant in a patient with CHD. [185] The homozygous non-coding variant prevented cardiac expression in model organisms when compared to the normal enhancer sequence. [185]
A study was conducted to analyze de novo variants in non-coding sequences in CHD. The focus was on variants in non-coding elements that are likely to regulate gene expression. Two related techniques were used [186,187]. Variants were identified using WGS based on scores derived from machine learning analysis of various genomic annotations or by their location within regions of active transcription during differentiation of human induced pluripotent stem cells (hiPSCs) into cardiomyocytes (hiPSC-CMs). Variants were identified using WGS based on scores obtained from machine learning assessment of different genomic annotations or by their position within domains of active transcription during human induced pluripotent stem cell (hiPSC) differentiation into cardiomyocytes (hiPSC-CMs). Functional assays were used to validate the non-coding variants prioritised by both strategies. Further analysis showed that in patients with CHD, compared to controls, there was an increase in non-coding variants within the binding sites of RNA-binding proteins. These findings suggest potential implications. Non-coding variants, especially those found in enhancers and RNA-binding protein sites, may account for 4-12% of CHD cases. While this is a promising start, the number of enhancer elements and RNA-binding protein sites throughout the genome is vast. Further analysis of larger cohorts will be necessary to confirm and expand upon these initial findings. [186,187]
5.2. Using whole genome sequencing to identify structural variation.
WGS allows for the detection of structural mutations with higher resolution than existing methods and increases the repertoire of LOF mutations identified in human disease. [188,189,190] Earlier work has suggested that small structural mutations can lead to CHD. These include an 8-kb deletion in FLT4 linked to tetralogy of Fallot [147] and 10 kb deletion in CHD7 associated with CHARGE syndrome caused by insertion of an Alu element. Genomic inversions and complex rearrangements can be identified by WGS. [18,19,20,21,22,23,24,25] (Figure 1) These structural mutations have been reported in patients with unexplained Alagille syndrome, affecting the expression of the JAG1 and NOTCH2 genes. It is expected that the number of deletions, insertions, translocations and other genomic rearrangements identified in CHD will increase significantly with advances in structural variant algorithms for interrogating whole genome sequencing. [145,146,147,191,192]
5.3. Future uses of genomics.
Undiscovered genomic features offer ongoing exciting possibilities to increase our knowledge of CHD genetics and explain the 55% of unexplained cases of CHD. WGS allows for more complete genotyping of an individual's SNPs and structural variants, allowing for the investigation of oligogenic causes of CHD, including both rare and common variants. Expanded analyses of genetic variants across the genome may provide insights into the penetrance and phenotypic diversity of congenital heart disease (CHD), both in familial cases and among unrelated patients with shared CHD genotypes. Additional research is required to comprehend the impact of environmental exposures on the consequences of a CHD genotype. This is due to the significance of the following factors: CHD-associated genes and chromatin remodelling have a strong correlation, and environmental factors can also affect chromatin structure [193,194]. Some puzzling features of CHD may be elucidated by exploring these complexities. For instance, epidemiological evidence indicates that the recurrence rate of CHD in families ranges from 3% to 7%, which is lower than what is predicted by current knowledge of CHD genotypes. Recurrence rates of 25% and 50% would be expected for autosomal recessive and dominant conditions, respectively. [195,196,197]
6. Deriving causality by integrating biology
An important source of evidence for causality in the clinical setting is the identification of genes and variants with a potential causal relationship to CHD, in combination with investigations of gene regulation during development and functional studies in model systems.
6.1. Insights gained from single cell transcriptomics.
The emergence of single-nuclear RNA sequencing and single-cell RNA sequencing (scRNA-seq) has allowed for a thorough analysis of the numerous cells present in both the developing mouse heart [198] and the mature human heart [199,200].
These studies have revealed differences in gene expression between various cell lineages and unique cell states within each lineage that were previously unknown when analyzing bulk RNA sequencing data. For instance, the healthy adult human heart is composed of four subsets of ventricular cardiomyocytes and five subsets of atrial cardiomyocytes. [199] New questions are being raised by these findings. Studies on the developmental origins, plasticity, and physiological functions of distinct cell subsets are ongoing. These findings provide valuable insights into the cellular composition of the heart during fetal development. Transcriptional signatures of cardiomyocytes (TNNI3 and TNNT2), fibroblast-like cells (COL1A1, COL1A2 and POSTN), endothelial cells (PECAM1 and KDR) and valve cells (SOX9) have been detected in recent scRNA-seq investigations of human fetal hearts. [201] During gestational week 7, genes related to the Notch signalling pathway are significantly more abundant in endocardial cells, which coincides with the compaction of the myocardium. Meanwhile, genes associated with the BMP pathway are expressed in both endocardial and fibroblast-like cells from gestational week 5 to week 25. This reflects periods of endocardial-to-mesenchymal transition. [201]
To complement and extend the limited data sets from human tissues, analyses of scRNA-seq data from the developing mouse embryo are presented. In the text, the complexity and transcriptional profiles of lineages derived from three germ layers are described: the early lateral plate mesoderm, the paraxial mesoderm and the neural mesoderm [198]. The expression of Mesp1 in mouse cells during embryonic day (E) 6.75 to E7.5 indicates a loss of pluripotency and the acquisition of a commitment to the cardiac lineage. [202] Hand2 expression initially specifies the second heart field, although it thereafter specifies cells of the outflow tract. [41] At E9.5 and later stages, cardiomyocytes cease dividing and start exhibiting gene expression patterns specific to each chamber. [201,203] The significant crosstalk between cell lineages driving cardiac differentiation and morphogenesis is also revealed by these data sets. Using these resources to investigate the temporal and lineage-specific expression of genes that contain harmful variants linked to CHD offers more granular detail than is possible from transcriptional analyses of bulk cardiac tissues. Pijuan-Sala al. reported significant findings in the presentation of scRNA-seq data from embryonic germ layer derivatives that contribute to heart development in mice. [204] These genes reveal the temporal emergence of cardiomyocytes, endothelial cells, and neural crest cells. They highlight the role of definitive and candidate CHD genes previously discovered in the meta-analysis. They reveal diverse lineage-specific and temporal expression in embryonic cells that are critical for normal cardiovascular development. As anticipated, genes with harmful variants found in patients with tetralogy of Fallot-associated CHD), such as Kdr, Flt1, and Flt4, exhibit high expression in E8.25-E8.5 endothelial cells, in line with the known functions of the encoded proteins in the VEGF pathway. [145,147] In contrast, three genes, Rpl19, Rps24 and Rps26, which have been implicated in Diamond-Blackfan anaemia and CHD, have different cellular and temporal patterns in the developing mouse heart. At E7.5-E7.75, Rpl19 and Rps26 are enriched in the mesenchyme, while Rps24 is expressed in cardiomyocytes at E8.5. [205] It is possible that these genes have different roles in cardiac development due to their enrichment in cardiomyocytes at E8.5. Lastly, genes encoding chromatin-modifying proteins that have an integral function in the development of the heart and that have been shown to be affected by deleterious mutations in patients with CHD (Chd4, Chd7, Kat6a, Kat6b, Kmt2 and Kmt2d) exhibit diverse cellular and temporal expression profiles in the developing mouse heart. [182] This implies that altered transcriptional dynamics in many cell types probably contribute to CHD. Morton et al. [158,187] predicted that datasets will aid in understanding the role of poorly studied genes enriched for damaging patients and identifying disrupted pathways. These insights will improve our understanding of the morphological consequences of deleterious variants and aid in resolving CHD. Cellular deficits can contribute to lifelong adverse events in patients with CHD. (182,183,206)
6.2. Findings from pluripotent stem cell models of congenital heart disease.
HipSCs are sourced either from patients or from isogenic cell lines. They are edited using CRISPR-Cas9, an experimental approach for the functional analysis of damaging variants found in patients with CHD. The process of differentiating hiPSC-CMs exhibits characteristics similar to those of FHF [207,208] or SHF, cardiac neural crest or pre-valvular endocardial cells. [209,210,211] This allows for analysis of early developmental stages, such as the differentiation of mesoderm progenitor cells into early cardiomyocytes [207,208]. This allows for analysis of early developmental stages, such as the differentiation of mesoderm progenitor cells into early cardiomyocytes. [178] hiPSC-CMs offer a window into the mechanism of early cardiogenesis that is difficult to achieve with model organisms when combined with epigenetic and transcriptional profiling. [58,209,210,211]
Mutant hiPSCs can be characterised molecularly. This will enable the assessment of the functional impact of CHD-associated variants on gene expression, chromatin status, and differentiation states. The hiPSC-CMs have provided valuable insights into the pathogenesis of CHD, although these in vitro assays do not take into account the effects of in vivo morphology and haemodynamics. Patients with GATA6 LOF variants exhibit disruption of crucial downstream regulators linked to outflow tract development, specifically KDR and HAND2. This disruption explains why GATA6 LOF variants are associated with outflow tract malformations. The study used hiPSCs. [58] In contrast, hiPSCs carrying a recurrent GATA6 missense variant found in patients with CHD and pancreatic agenesis exhibit a significantly altered epigenetic landscape and increased retinoic acid signalling. [58] Transcriptomic analyses of TBX5-haploinsufficient hiPSCs revealed that gene regulatory networks controlling cardiomyocyte differentiation are sensitive to TBX5 dosage. Additionally, novel genetic tools that may affect CHD phenotypes have been identified. [212] HipSCs carrying a missense variant in GATA4, which is associated with septal defects and pulmonary stenosis, exhibit a loss of GATA4-TBX5 interactions and inappropriate expression of endothelial cell genes in iPSC-CMs. [84]
The use of hiPSCs holds potential for evaluating non-coding variants in CHD. This overcomes the challenge of limited conservation of non-coding sequences between species that hinders analysis in experimental models. Using the large-scale parallel assays designed for hiPSCs can differentiate likely contributory non-coding variants from numerous benign polymorphisms can help identify the associated genes. [213] Additionally, profiling the epigenetics and transcription of hiPSCs during cellular differentiation offers insight. It is challenging to achieve early cardiogenesis in model organisms. [182]
7. Moving genomics into the clinical setting
The field of clinical genetic testing for people with CHD is constantly in evolution as a result of improvements in testing methods and the identification of associations between genotypes and clinical phenotypes over time. Traditional genetic testing strategies have focused mainly on syndromic CHD or selected CHD phenotypes that are strongly associated with a particular genotype, such as the 22q11 deletion. [214] Genetic testing for congenital heart disease has traditionally targeted syndromic CHD or selected CHD phenotypes that are closely linked to a particular genotype (such as the 22q11 deletion). [214] However, the availability and cost-effectiveness of clinical WES is increasing, making gene-based diagnosis of both syndromic and non-syndromic CHD more common. WES provides comprehensive analysis that can identify the causes of CHD and detect incidental findings. If the results of genetic testing are clinically actionable or unclear, it is essential to obtain informed consent and seek guidance from genetic counsellors or experienced clinicians. However, clinical whole exome sequencing (WES) is becoming more widely available and cost-effective, enabling gene-based diagnosis of both syndromic and non-syndromic CHD. Informed consent and guidance from genetic counsellors or experienced clinicians is essential, since WES provides comprehensive analysis to identify the causes of CHD as well as incidental findings that are clinically actionable and/or of unclear relevance.
Furthermore, as new findings emerge, it is necessary to establish criteria for determining the pathogenicity of CHD requires criteria that are similar to those proposed by ClinGen and the American College of Medical Genetics and Genomics. [215,216,217,218]
Patient care can be significantly impacted by the identification of definitive causal variants for congenital heart disease (CHD). Genotyping to inform the risk of recurrence, prenatal genetic counselling and pre-implantation genotyping will benefit both the family of a newborn with CHD and adults with repaired or mitigated CHD. [219] The genotype can increasingly indicate the risk for adverse extracardiac conditions or comorbidities, such as neurodevelopmental delay. [220,221] This emphasises the importance of monitoring negative postoperative results [220,221] and the potential development of cancers. [222,223,224]
7.1. CHD in association with extracardiac abnormalities or neurodevelopmental disabilities.
The likelihood of detecting a pathogenic CHD genotype increases from 8% to 22% when there is an extracardiac anomaly or neurodevelopmental delay, especially in genes that modify chromatin. [22] Studies of cohorts with neurocognitive disorders have shown an enrichment of damaging variants in chromatin-modifying genes [23,225,226,227], indicating a significant genetic contribution to the association. It is important to note the specific genetic factors that contribute to this association. between CHD and neurodevelopmental delay. Identifying chromatin-related damaging genotypes in newborns with CHD can inform pre-emptive interventions to improve their learning and social skills. [225,226,227]
De novo copy number variations have an impact on perioperative outcomes.
CHD genotypes can help identify high-risk patients for cardiac surgery. Patients with CHD and 22q11.2 deletions experience longer cardiopulmonary bypass durations and frequent repeat operations. Patients with CHD who have other large CNVs (>300 kb) also have a higher risk of death or requiring transplantation and prolonged admission time in intensive care compared to those without the deletion. [228,229,230,231,232] WES is continuing to extend these findings. Patients with congenital heart disease (CHD) who have de novo copy number variations (CNVs) or damaging single-nucleotide variants experience longer time to extubation and reduced transplantation-free survival. [220] These negative outcomes are particularly significant for CHD patients without extracardiac anomalies. Some genomic regions associated with risk are linked to cardiac trabeculation and myocardial performance, which may underlie the risk of heart failure and transplantation in some patients with CHD. [233] The link between genotype and morphological structure is important to consider.
7.2. Cancer risk in patients with congenital heart disease.
The incidence of cancer is 1.4 to 2 times higher in adults with CAD than in the general population. [222,223,224] Certain CHD genotypes may also increase the risk of cancer [152] in combination with increased radiation exposure associated with therapeutic interventions. [234,235] This is because many genes with harmful variants associated with CHD are also recognised as genes associated with cancer risk. Since patients with CHD with harmful variants in genes associated with cancer risk often have extracardiac manifestations, this association may be related to the underlying and widespread developmental role of some CHD-associated genes.
7.3. Forthcoming applications in the clinic.
Investigating the relationship between causal variants associated with CHD and clinical outcomes presents a significant opportunity. Some of the questions that need to be answered include: How do variants in specific genes or defects in signalling pathways relate to the observed variability in CHD phenotypes? Additionally, do genetic variants linked to CHD or other genetic variants impact survival rates following surgical procedures? How do structural heart disease-causing variants relate to long-term cardiovascular function measures in patients with CHD? To answer these questions, we need large genome-wide sequencing datasets. These datasets need to be harmonised with comprehensive, high-quality clinical data. Multicentre collaborative efforts are likely to be crucial given the expense and magnitude of the datasets necessary to answer these questions.
8. Conclusions
The genetic architecture of CHD has been discovered through technological advances. This includes damaging germline and mosaic variants, copy number and structural variants that affect multiple genes, and variants in non-coding sequences. These discoveries have implications beyond the understanding of the molecular basis for this common birth defect. These insights define important genes and pathways involved in the development and function of the heart and other organs. Variants associated with CHD also identify genetic variants that increase the risk of neurocognitive dysfunction and cancer, and confer specific risks on patients requiring surgical intervention for CHD. Although progress has been made in understanding the genetic architecture of CHD, there is still much to learn. The causes of CHD remain unknown in over 50% of patients. To address this challenge, we need to explore new directions. One potential avenue is identifying abnormal gene expression. To increase productivity, direct analysis of human CHD tissues, comprehensive assessment of non-coding sequences and oligogenic variants, and examination of the effects of environmental exposures may be useful. Results from these and other studies offer great promises not only to improve the diagnosis, classification and clinical care of patients with CHD, but also to enrich the basic biological and genetic insights in developmental and genomic biology.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ibrahim S, Gaborit B, Lenoir M, Collod-Beroud G, Stefanovic S. Maternal Pre-Existing Diabetes: A Non-Inherited Risk Factor for Congenital Cardiopathies. Int J Mol Sci. 2023 Nov 13;24(22):16258. [CrossRef]
- Dilli D, Akduman H, Zenciroğlu A, Çetinkaya M, Okur N, Turan Ö, Özlü F, Çalkavur Ş, Demirel G, Koksal N, Çolak R, Örün UA, Öztürk E, Gül Ö, Tokel NK, Erdem S, Meşe T, Erdem A, Bostan ÖM, Polat TB, Taşar M, Hatemi AC, Doyurgan O, Özkan M, Avşar MK, Sarıosmanoğlu ON, Uğurlucan M, Sığnak IŞ, Başaran M. Neonatal Outcomes of Critical Congenital Heart Defects: A Multicenter Epidemiological Study of Turkish Neonatal Society: Neonatal Outcomes of CCHD. Pediatr Cardiol. 2023 Dec 28. [CrossRef]
- Hossin MZ, de la Cruz LF, McKay KA, Oberlander TF, Sandström A, Razaz N. Association of pre-existing maternal cardiovascular diseases with neurodevelopmental disorders in offspring: a cohort study in Sweden and British Columbia, Canada. Int J Epidemiol. 2023 Dec 27: dyad184. [CrossRef]
- Bakker MK, Bergman JEH, Krikov S, Amar E, Cocchi G, Cragan J, de Walle HEK, Gatt M, Groisman B, Liu S, Nembhard WN, Pierini A, Rissmann A, Chidambarathanu S, Sipek A Jr, Szabova E, Tagliabue G, Tucker D, Mastroiacovo P, Botto LD. Prenatal diagnosis and prevalence of critical congenital heart defects: an international retrospective cohort study. BMJ Open. 2019 Jul 2;9(7):e028139. [CrossRef]
- Hoffman JIE, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002 Jun 19;39(12):1890-900. [CrossRef]
- Reller MD, Strickland MJ, Riehle-Colarusso T, Mahle WT, Correa A Prevalence of congenital heart defects in metropolitan Atlanta. J Pediatr. 2008 Dec;153(6):807-13. [CrossRef]
- Spector LG, Menk JS, Knight JH, McCracken C, Thomas AS, Vinocur JM, Oster ME, St Louis JD, Moller JH, Kochilas L. Trends in Long-Term Mortality After Congenital Heart Surgery. J Am Coll Cardiol. 2018 May 29;71(21):2434-2446. [CrossRef]
- Egbe A, Lee S, Ho D, Uppu S, Srivastava S. Prevalence of congenital anomalies in newborns with congenital heart disease diagnosis. Ann Pediatr Cardiol. 2014 May;7(2):86-91. [CrossRef]
- Wang H, Lin X, Lyu G, He S, Dong B, Yang Y. Chromosomal abnormalities in fetuses with congenital heart disease: a meta-analysis. Arch Gynecol Obstet. 2023 Sep;308(3):797-811. [CrossRef]
- Diniz BL, Deconte D, Gadelha KA, Glaeser AB, Guaraná BB, de Moura AÁ, Rosa RFM, Zen PRG. Congenital Heart Defects and 22q11.2 Deletion Syndrome: A 20-Year Update and New Insights to Aid Clinical Diagnosis.
- J Pediatr Genet. 2023 Feb 17;12(2):113-122. [CrossRef]
- Thienpont B, Mertens L, de Ravel T, Eyskens B, Boshoff D, Maas N, Fryns JP, Gewillig M, Vermeesch JR, Devriendt K. Submicroscopic chromosomal imbalances detected by array-CGH are a frequent cause of congenital heart defects in selected patients. Eur Heart J. 2007 Nov;28(22):2778-84. [CrossRef]
- Wilson DI, Cross IE, Goodship JA, Brown J, Scambler PJ, Bain HH, Taylor JF, Walsh K, Bankier A, Burn J, et al. A prospective cytogenetic study of 36 cases of DiGeorge syndrome. Am J Hum Genet. 1992 Nov;51(5):957-63.
- Sgardioli IC, Vieira TP, Simioni M, Monteiro FP, Gil-da-Silva-Lopes VL 22q11.2 Deletion Syndrome: Laboratory Diagnosis and TBX1 and FGF8 Mutation Screening. J Pediatr Genet. 2015 Mar;4(1):17-22. [CrossRef]
- Agergaard P, Olesen C, Østergaard JR, Christiansen M, Sørensen KM The prevalence of chromosome 22q11.2 deletions in 2,478 children with cardiovascular malformations. A population-based study. Am J Med Genet A. 2012 Mar;158A(3):498-508. [CrossRef]
- Agergaard P, Hebert A, Sørensen KM, Østergaard JR, Olesen C. Can clinical assessment detect 22q11.2 deletions in patients with cardiac malformations? A review. Eur J Med Genet. 2011 Jan-Feb;54(1):3-8. [CrossRef]
- Peyvandi S, Lupo PJ, Garbarini J, Woyciechowski S, Edman S, Emanuel BS, Mitchell LE, Goldmuntz E.. 22q11.2 deletions in patients with conotruncal defects: data from 1,610 consecutive cases. Pediatr Cardiol. 2013 Oct;34(7):1687-94. [CrossRef]
- Dehghan B, Sabri MR, Ahmadi A, Ghaderian M, Mahdavi C, Ramezani Nejad D, Sattari M. Identifying the Factors Affecting the Incidence of Congenital Heart Disease Using Support Vector Machine and Particle Swarm Optimization. Adv Biomed Res. 2023 May 19;12:130. [CrossRef]
- Pierpont ME, Basson CT, Benson DW Jr, Gelb BD, Giglia TM, Goldmuntz E, McGee G, Sable CA, Srivastava D, Webb CL; American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young. Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation. 2007 Jun 12;115(23):3015-38. [CrossRef]
- Goldmuntz, E. 22q11.2 deletion syndrome and congenital heart disease. Am J Med Genet C Semin Med Genet. 2020 Mar;184(1):64-72. [CrossRef]
- Fahed AC, Gelb BD, Seidman JG, Seidman CE Genetics of congenital heart disease: the glass half empty. Circ Res. 2013 Feb 15;112(4):707-20. [CrossRef]
- LaHaye S, Corsmeier D, Basu M, Bowman JL, Fitzgerald-Butt S, Zender G, Bosse K, McBride KL, White P, Garg V. Utilization of Whole Exome Sequencing to Identify Causative Mutations in Familial Congenital Heart Disease. Circ Cardiovasc Genet. 2016 Aug;9(4):320-9. [CrossRef]
- Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, DePalma SR, Zeng X, Qi H, Chang W, Sierant MC, Hung WC, Haider S, Zhang J, Knight J, Bjornson RD, Castaldi C, Tikhonoa IR, Bilguvar K, Mane SM, Sanders SJ, Mital S, Russell MW, Gaynor JW, Deanfield J, Giardini A, Porter GA Jr, Srivastava D, Lo, et al.. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet. 2017 Nov;49(11):1593-1601. [CrossRef]
- Homsy J, Zaidi S, Shen Y, Ware JS, Samocha KE, Karczewski KJ, DePalma SR, McKean D, Wakimoto H, Gorham J, Jin SC, Deanfield J, Giardini A, Porter GA Jr, Kim R, Bilguvar K, López-Giráldez F, Tikhonova I, Mane S, Romano-Adesman A, Qi H, Vardarajan B, Ma L, Daly M, Roberts AE, et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science. 2015 Dec 4;350(6265):1262-6. [CrossRef]
- Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017 Feb 23;542(7642):433-438. [CrossRef]
- Sifrim A, Hitz MP, Wilsdon A, Breckpot J, Turki SH, Thienpont B, McRae J, Fitzgerald TW, Singh T, Swaminathan GJ, Prigmore E, Rajan D, Abdul-Khaliq H, Banka S, Bauer UM, Bentham J, Berger F, Bhattacharya S, Bu'Lock F, Canham N, Colgiu IG, Cosgrove C, Cox H, Daehnert I, Daly A, Danesh J, Fryer A, Gewillig M, Hobson E, Hoff K, Homfray T; INTERVAL Study; Kahlert AK, Ketley A, Kramer HH, Lachlan K, Lampe AK, Louw JJ, Manickara AK, Manase D, McCarthy KP, Metcalfe K, Moore C, Newbury-Ecob R, Omer SO, Ouwehand WH, Park SM, Parker MJ, Pickardt T, Pollard MO, Robert L, Roberts DJ, Sambrook J, Setchfield K, Stiller B, Thornborough C, Toka O, Watkins H, Williams D, Wright M, Mital S, Daubeney PE, Keavney B, Goodship J; UK10K Consortium; Abu-Sulaiman RM, Klaassen S, Wright CF, Firth HV, Barrett JC, Devriendt K, FitzPatrick DR, Brook JD; Deciphering Developmental Disorders Study; Hurles ME. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat Genet. 2016 Sep;48(9):1060-5. [CrossRef]
- Shan W, Yuanqing X, Jing Z, Xi W, Huifeng G, Yi W. Risk factor analysis for adverse prognosis of the fetal ventricular septal defect (VSD). BMC Pregnancy Childbirth. 2023 Sep 21;23(1):683. [CrossRef]
- International Society of Ultrasound in Obstetrics & Gynecology. Cardiac screening examination of the fetus: guidelines for performing the ‘basic’ and ‘extended basic’ cardiac scan. Ultrasound Obstet Gynecol2006 Jan;27(1):107-113.
- Meilhac SM, Buckingham ME The deployment of cell lineages that form the mammalian heart. Nat Rev Cardiol. 2018 Nov;15(11):705-724. [CrossRef]
- Kathiriya IS, Nora EP, Bruneau BG Investigating the transcriptional control of cardiovascular development. Circ Res. 2015 Feb 13;116(4):700-14. [CrossRef]
- Maas RGC, van den Dolder FW, Yuan Q, van der Velden J, Wu SM, Sluijter JPG, Buikema JW. Harnessing developmental cues for cardiomyocyte production. Development. 2023 Aug 1;150(15):dev201483. [CrossRef]
- Günthel M, Barnett P, Christoffels VM Development, proliferation, and growth of the mammalian heart. Mol Ther. 2018 Jul 5;26(7):1599-1609. [CrossRef]
- Cui M, Wang Z, Bassel-Duby R & Olson EN Genetic and epigenetic regulation of cardiomyocytes in development, regeneration and disease. Development 145, dev171983 (2018).
- Chien KR, Domian IJ, Parker KK. Cardiogenesis and the complex biology of regenerative cardiovascular medicine.Science. 2008 Dec 5;322(5907):1494-7. [CrossRef]
- Jain R, Epstein JA Competent for commitment: you’ve got to have heart! Genes Dev. 2018 Jan 1;32(1):4-13. [CrossRef]
- Zaffran S, Kelly RG, Meilhac SM, Buckingham ME, Brown NA. Right ventricular myocardium derives from the anterior heart field. Circ Res. 2004 Aug 6;95(3):261-8. [CrossRef]
- Zaidi, S.; Choi, M.; Wakimoto, H.; Ma, L.; Jiang, J.; Overton, J.D.; Romano-Adesman, A.; Bjornson, R.D.; Breitbart, R.E.; Brown, K.K.; et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature 2013, 498, 220–223. [Google Scholar] [CrossRef]
- Hedermann, G.; Hedley, P.L.; Thagaard, I.N.; Krebs, L.; Ekelund, C.K.; Sørensen, T.I.A.; Christiansen, M. Maternal obesity and metabolic disorders associate with congenital heart defects in the offspring: A systematic review. PLoS ONE 2021, 16, e0252343. [Google Scholar] [CrossRef] [PubMed]
- Cavadino, A.; Sandberg, L.; Öhman, I.; Bergvall, T.; Star, K.; Dolk, H.; Loane, M.; Addor, M.C.; Barisic, I.; Cavero-Carbonell, C.; et al. Signal Detection in EUROmediCAT: Identification and Evaluation of Medication-Congenital Anomaly Associations and Use of VigiBase as a Complementary Source of Reference. Drug Saf. 2021, 44, 765–785. [Google Scholar] [CrossRef] [PubMed]
- Kalisch-Smith, J.I.; Ved, N.; Sparrow, D.B. Environmental Risk Factors for Congenital Heart Disease. Cold Spring Harb. Perspect. Biol. 2020, 12, a037234. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, K.J.; Correa, A.; Feinstein, J.A.; Botto, L.; Britt, A.E.; Daniels, S.R.; Elixson, M.; Warnes, C.A.; Webb, C.L. American Heart Association Council on Cardiovascular Disease in the Y: Noninherited risk factors and congenital cardiovascular defects: Current knowledge: A scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young: Endorsed by the American Academy of Pediatrics. Circulation 2007, 115, 2995–3014. [Google Scholar] [PubMed]
- Hutson MR, Kirby ML Model systems for the study of heart development and disease. Semin Cell Dev Biol. 2007 Feb;18(1):101-10. [CrossRef]
- de la Pompa JL, Timmerman LA, Takimoto H, Yoshida H, Elia AJ, Samper E, Potter J, Wakeham A, Marengere L, Langille BL, Crabtree GR, Mak TW. Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum.Nature. 1998 Mar 12;392(6672):182-6. [CrossRef]
- Lin C-J, Lin C-Y, Chen C-H, Zhou B, Chang C-P Partitioning the heart: mechanisms of cardiac septation and valve development. Development. 2012 Sep;139(18):3277-99. [CrossRef]
- Wu B, Wu B, Benkaci S, Shi L, Lu P, Park T, Morrow BE, Wang Y, Zhou B. Crk and Crkl Are Required in the Endocardial Lineage for Heart Valve Development. J Am Heart Assoc. 2023 Sep 19;12(18):e029683. [CrossRef]
- Christoffels VM, Habets PE, Franco D, Campione M, de Jong F, Lamers WH, Bao ZZ, Palmer S, Biben C, Harvey RP, Moorman AF. Chamber formation and morphogenesis in the developing mammalian heart. Dev Biol. 2000 Jul 15;223(2):266-78. [CrossRef]
- Alsan BH, Schultheiss TM Regulation of avian cardiogenesis by Fgf8 signaling. Development. 2002 Apr;129(8): 1935-43. [CrossRef]
- Astrof S, Arriagada C, Saijoh Y, Francou A, Kelly RG, Moon A. Aberrant differentiation of second heart field mesoderm prefigures cellular defects in the outflow tract in response to loss of FGF8. Dev Biol. 2023 Jul;499:10-21. [CrossRef]
- Ivanovitch K, Soro-Barrio P, Chakravarty P, Jones RA, Bell DM, Mousavy Gharavy SN, Stamataki D, Delile J, Smith JC, Briscoe J. Ventricular, atrial, and outflow tract heart progenitors arise from spatially and molecularly distinct regions of the primitive streak. PLoS Biol. 2021 May 17;19(5):e3001200. [CrossRef]
- Itoh N, Ohta H, Nakayama Y & Konishi M Roles of FGF signals in heart development, health, and disease. Front. Cell Dev. Biol. 4, 110 (2016).
- de Pater E, Ciampricotti M, Priller F, Veerkamp J, Strate I, Smith K, Lagendijk AK, Schilling TF, Herzog W, Abdelilah-Seyfried S, Hammerschmidt M, Bakkers J. Bmp signaling exerts opposite effects on cardiac differentiation. Circ Res. 2012 Feb 17;110(4):578-87. [CrossRef]
- Targoff KL, Colombo S, George V, Schell T, Kim SH, Solnica-Krezel L, Yelon D. Nkx genes are essential for maintenance of ventricular identity.Development. 2013 Oct;140(20):4203-13. [CrossRef]
- Nelson DO, Lalit PA, Biermann M, Markandeya YS, Capes DL, Addesso L, Patel G, Han T, John MC, Powers PA, Downs KM, Kamp TJ, Lyons GE. Irx4 Marks a Multipotent, Ventricular-Specific Progenitor Cell. Stem Cells. 2016 Dec;34(12):2875-2888. [CrossRef]
- Goldfracht I, Protze S, Shiti A, Setter N, Gruber A, Shaheen N, Nartiss Y, Keller G, Gepstein L. Generating ring-shaped engineered heart tissues from ventricular and atrial human pluripotent stem cell-derived cardiomyocytes.
- Nat Commun. 2020 Jan 7;11(1):75. [CrossRef]
- Giacomelli E, Bellin M, Orlova VV, Mummery CL. Co-Differentiation of Human Pluripotent Stem Cells-Derived Cardiomyocytes and Endothelial Cells from Cardiac Mesoderm Provides a Three-Dimensional Model of Cardiac Microtissue. Curr Protoc Hum Genet. 2017 Oct 18; 95:21.9.1-21.9.22. [CrossRef]
- Cheng Z et al. Two novel mutations of the IRX4 gene in patients with congenital heart disease. Hum. Genet. 130, 657–662 (2011).
- de Soysa TY et al. Single-cell analysis of cardiogenesis reveals basis for organ-level developmental defects. Nature 572, 120–124 (2019).
- Chen YH, Ishii M, Sun J, Sucov HM & Maxson RE Msx1 and Msx2 regulate survival of secondary heart field precursors and post-migratory proliferation of cardiac neural crest in the outflow tract. Dev. Biol. 308, 421–437 (2007).
- Sharma A et al. GATA6 mutations in hiPSCs inform mechanisms for maldevelopment of the heart, pancreas, and diaphragm. eLife 9, e53278 (2020).
- Uribe V et al. Arid3b is essential for second heart field cell deployment and heart patterning. Development 141,4168–4181 (2014).
- Wang Z, Wang DZ, Hockemeyer D, McAnally J, Nordheim A, Olson EN. Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature. 2004 Mar 11;428(6979):185-9. [CrossRef]
- Felker A, Prummel KD, Merks AM, Mickoleit M, Brombacher EC, Huisken J, Panáková D, Mosimann C. Continuous addition of progenitors forms the cardiac ventricle in zebrafish. Nat Commun. 2018 May 21;9(1):2001. [CrossRef]
- Sánchez-Iranzo H, Galardi-Castilla M, Minguillón C, Sanz-Morejón A, González-Rosa JM, Felker A, Ernst A, Guzmán-Martínez G, Mosimann C, Mercader N. Tbx5a lineage tracing shows cardiomyocyte plasticity during zebrafish heart regeneration. Nat Commun. 2018 Jan 30;9(1):428. [CrossRef]
- Jiang X, Choudhary B, Merki E, Chien KR, Maxson RE, Sucov HM.Normal fate and altered function of the cardiac neural crest cell lineage in retinoic acid receptor mutant embryos. Mech Dev. 2002 Sep;117(1–2):115-22. [CrossRef]
- Zhou C, Häneke T, Rohner E, Sohlmér J, Kameneva P, Artemov A, Adameyko I, Sahara M. STRA6 is essential for induction of vascular smooth muscle lineages in human embryonic cardiac outflow tract development. Cardiovasc Res. 2023 May 22;119(5):1202-1217. [CrossRef]
- Sanchez J, Miyake R, Cheng A, Liu T, Iseki S, Kume T. Conditional inactivation of Foxc1 and Foxc2 in neural crest cells leads to cardiac abnormalities.Genesis. 2020 Jul;58(7):e23364. [CrossRef]
- Kodo K, Shibata S, Miyagawa-Tomita S, Ong SG, Takahashi H, Kume T, Okano H, Matsuoka R, Yamagishi H. Regulation of Sema3c and the interaction between cardiac neural crest and second heart field during outflow tract development. Sci Rep. 2017 Jul 28;7(1):6771. [CrossRef]
- Greulich F, Rudat C, Kispert A. Mechanisms of T-box gene function in the developing heart. Cardiovasc Res. 2011 Jul 15;91(2):212-22. [CrossRef]
- MacGrogan D, Nus M, de la Pompa JL Notch signaling in cardiac development and disease.Curr Top Dev Biol. 2010;92:333-65. [CrossRef]
- Stankunas K, Ma GK, Kuhnert FJ, Kuo CJ, Chang CP. VEGF signaling has distinct spatiotemporal roles during heart valve development.Dev Biol. 2010 Nov 15;347(2):325-36. [CrossRef]
- Alvandi Z, Bischoff J. Endothelial-Mesenchymal Transition in Cardiovascular Disease.Arterioscler Thromb Vasc Biol. 2021 Sep;41(9):2357-2369. [CrossRef]
- Singh N, Trivedi CM, Lu M, Mullican SE, Lazar MA, Epstein JA. Histone deacetylase 3 regulates smooth muscle differentiation in neural crest cells and development of the cardiac outflow tract. Circ Res. 2011 Nov 11;109(11):1240-9. [CrossRef]
- Kelly RG, Papaioannou VE. Visualization of outflow tract development in the absence of Tbx1 using an FgF10 enhancer trap transgene.Dev Dyn. 2007 Mar;236(3):821-8. [CrossRef]
- Peng T, Tian Y, Boogerd CJ, Lu MM, Kadzik RS, Stewart KM, Evans SM, Morrisey E. Coordination of heart and lung co-development by a multipotent cardiopulmonary progenitor. Nature. 2013 Aug 29;500(7464):589-92. [CrossRef]
- Liu X, Chen W, Li W, Li Y, Priest JR, Zhou B, Wang J, Zhou Z. Single-Cell RNA-seq of the developing cardiac outflow tract reveals convergent development of the vascular smooth muscle cells. Cell Rep. 2019 Jul 30;28(5):1346-1361.e4. [CrossRef]
- Forrest K, Barricella AC, Pohar SA, Hinman AM, Amack JD. Understanding laterality disorders and the left-right organizer: Insights from zebrafish. Front Cell Dev Biol. 2022 Dec 23;10:1035513. [CrossRef]
- Lopes Floro K, Artap ST, Preis JI, Fatkin D, Chapman G, Furtado MB, Harvey RP, Hamada H, Sparrow DB, Dunwoodie S. Loss of Cited2 causes congenital heart disease by perturbing left-right patterning of the body axis. Hum Mol Genet. 2011 Mar 15;20(6):1097-110. [CrossRef]
- Bellchambers HM, Ware SM. ZIC3 in Heterotaxy. Adv Exp Med Biol. 2018;1046:301-327. [CrossRef]
- Levin, M. Left-right asymmetry in vertebrate embryogenesis. Bioessays. 1997 Apr;19(4):287-96. [CrossRef]
- Chang H, Zwijsen A, Vogel H, Huylebroeck D, Matzuk MM. Smad5 is essential for left-right asymmetry in mice Dev Biol. 2000 Mar 1;219(1):71-8. [CrossRef]
- Kumar S, Donofrio M, Frank L, He D, Jonas R. Complete atrioventricular canal with guarded primum septal defect.Pediatr Cardiol. 2011 Apr;32(4):503-5. [CrossRef]
- Paladini D, Tartaglione A, Agangi A, Teodoro A, Forleo F, Borghese A, Martinelli P. The association between congenital heart disease and Down syndrome in prenatal life. Ultrasound Obstet Gynecol. 2000 Feb;15(2):104-8. [CrossRef]
- Santoro M, Coi A, Spadoni I, Bianchi F, Pierini A. Sex differences for major congenital heart defects in Down Syndrome: A population based study. Eur J Med Genet. 2018 Sep;61(9):546-550. [CrossRef]
- Pelleri MC, Locatelli C, Mattina T, Bonaglia MC, Piazza F, Magini P, Antonaros F, Ramacieri G, Vione B, Vitale L, Seri M, Strippoli P, Cocchi G, Piovesan A, Caracausi M. Partial trisomy 21 with or without highly restricted Down syndrome critical region (HR-DSCR): report of two new cases and reanalysis of the genotype-phenotype association. BMC Med Genomics. 2022 Dec 21;15(1):266. [CrossRef]
- Ang Y-S, Rivas RN, Ribeiro AJS, Srivas R, Rivera J, Stone NR, Pratt K, Mohamed TMA, Fu JD, Spencer CI, Tippens ND, Li M, Narasimha A, Radzinsky E, Moon-Grady AJ, Yu H, Pruitt BL, Snyder MP, Srivastava D. Disease model of GATA4 mutation reveals transcription factor cooperativity in human cardiogenesis. Cell. 2016 Dec 15;167(7):1734-1749.e22. [CrossRef]
- Misra C, Sachan N, McNally CR, Koenig SN, Nichols HA, Guggilam A, Lucchesi PA, Pu WT, Srivastava D, Garg V. Congenital heart disease-causing Gata4 mutation displays functional deficits in vivo. PLoS Genet. 2012;8(5):e1002690. [CrossRef]
- Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K, Matsuoka R, Cohen JC, Srivastava D. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003 Jul 24;424(6947):443-7. [CrossRef]
- Brown CO 3rd, Chi X, Garcia-Gras E, Shirai M, Feng XH, Schwartz RJ. The cardiac determination factor, Nkx2-5, is activated by mutual cofactors GATA-4 and Smad1/4 via a novel upstream enhancer. J Biol Chem. 2004 Mar 12;279(11):10659-69. [CrossRef]
- Vecoli C, Pulignani S, Foffa I, Andreassi MG. Congenital heart disease: the crossroads of genetics, epigenetics and environment. Curr Genomics. 2014 Oct;15(5):390-9. [CrossRef]
- Kirklin/Barratt-Boyes Cardiac Surgery 4th Edition by James K Kirklin MD (Author), Eugene H. Blackstone MD.
- McBride KL Pignatelli R, Lewin M, Ho T, Fernbach S, Menesses A, Lam W, Leal SM, Kaplan N, Schliekelman P, Towbin JA, Belmont JW. Inheritance analysis of congenital left ventricular outflow tract obstruction malformations: segregation, multiplex relative risk, and heritability. Am J Med Genet A. 2005 Apr 15;134A(2):180-6. [CrossRef]
- Nappi F, Giacinto O, Lusini M, Garo M, Caponio C, Nenna A, Nappi P, Rousseau J, Spadaccio C, Chello M. Patients with Bicuspid Aortopathy and Aortic Dilatation. J Clin Med. 2022 Oct 11;11(20):6002. [CrossRef]
- Agasthi P, Pujari SH, Tseng A, Graziano JN, Marcotte F, Majdalany D, Mookadam F, Hagler DJ, Arsanjani R. Management of adults with coarctation of aorta. World J Cardiol. 2020 May 26;12(5):167-191. [CrossRef]
- Silberbach M et al. Cardiovascular health in Turner syndrome: a scientific statement from the American Heart Association. Circulation. Genomic Precis. Med. 11, e000048 (2018).
- Lara DA, Ethen MK, Canfield MA, Nembhard WN & Morris SA A population-based analysis of mortality in patients with Turner syndrome and hypoplastic left heart syndrome using the Texas Birth Defects Registry. Congenit. Heart Dis. 12, 105–112 (2017).
- Bouayed Abdelmoula N, Abdelmoula B, Smaoui W, Trabelsi I, Louati R, Aloulou S, Aloulou W, Abid F, Kammoun S, Trigui K, Bedoui O, Denguir H, Mallek S, Ben Aziza M, Dammak J, Kaabi O, Abdellaoui N, Turki F, Kaabi A, Kamoun W, Jabeur J, Ltaif W, Chaker K, Fourati H, M'rabet S, Ben Ameur H, Gouia N, Mhiri MN, Rebai T. Left-sided congenital heart lesions in mosaic Turner syndrome. Mol Genet Genomics. 2018 Apr;293(2):495-501. [CrossRef]
- Ribé L, Shihadeh FD, Afifi RO, Estrera AL, Prakash SK. Outcomes of cardiothoracic surgery in women with Turner syndrome. Ann Cardiothorac Surg. 2023 Nov 27;12(6):569-576. [CrossRef]
- Prakash SK, Bondy CA, Maslen CL, Silberbach M, Lin AE, Perrone L, Limongelli G, Michelena HI, Bossone E, Citro R; BAVCon Investigators, GenTAC Registry Investigators; Lemaire SA, Body SC, Milewicz DM. Autosomal and X chromosome structural variants are associated with congenital heart defects in Turner syndrome: The NHLBI GenTAC registry. Am J Med Genet A. 2016 Dec;170(12):3157-3164. [CrossRef]
- Wenger SL, Grossfeld PD, Siu BL, Coad JE, Keller FG, Hummel M. Molecular characterization of an 11q interstitial deletion in a patient with the clinical features of Jacobsen syndrome. Am J Med Genet A. 2006 Apr 1;140(7):704-8. [CrossRef]
- Crucean A, Alqahtani A, Barron DJ, Brawn WJ, Richardson RV, O'Sullivan J, Anderson RH, Henderson DJ, Chaudhry B. Re-evaluation of hypoplastic left heart syndrome from a developmental and morphological perspective. Orphanet J Rare Dis. 2017 Aug 10;12(1):138. [CrossRef]
- Miao Y, Tian L, Martin M, Paige SL, Galdos FX, Li J, Klein A, Zhang H, Ma N, Wei Y, Stewart M, Lee S, Moonen JR, Zhang B, Grossfeld P, Mital S, Chitayat D, Wu JC, Rabinovitch M, Nelson TJ, Nie S, Wu SM, Gu M.. Intrinsic endocardial defects contribute to hypoplastic left heart syndrome. Cell Stem Cell. 2020 Oct 1;27(4):574-589.e8. [CrossRef]
- Jiang Y, Habibollah S, Tilgner K, Collin J, Barta T, Al-Aama JY, Tesarov L, Hussain R, Trafford AW, Kirkwood G, Sernagor E, Eleftheriou CG, Przyborski S, Stojković M, Lako M, Keavney B, Armstrong LAn induced pluripotent stem cell model of hypoplastic left heart syndrome (HLHS) reveals multiple expression and functional differences in HLHS-derived cardiac myocytes. Stem Cells Transl Med. 2014 Apr;3(4):416-23. [CrossRef]
- Shi LM, Tao JW, Qiu XB, Wang J, Yuan F, Xu L, Liu H, Li RG, Xu YJ, Wang Q, Zheng HZ, Li X, Wang XZ, Zhang M, Qu XK, Yang YQ.. GATA5 loss-of-function mutations associated with congenital bicuspid aortic valve. Int J Mol Med. 2014 May;33(5):1219-26. [CrossRef]
- Bonachea EM et al. Rare GATA5 sequence variants identified in individuals with bicuspid aortic valve. Pediatr. Res. 76, 211–216 (2014).
- Huang M, Akerberg AA, Zhang X, Yoon H, Joshi S, Hallinan C, Nguyen C, Pu WT, Haigis MC, Burns CG, Burns CE. Intrinsic myocardial defects underlie an Rbfox-deficient zebrafish model of hypoplastic left heart syndrome. Nat Commun. 2022 Oct 5;13(1):5877. [CrossRef]
- Xu YJ, Di RM, Qiao Q, Li XM, Huang RT, Xue S, Liu XY, Wang J, Yang YQ. GATA6 loss-of-function mutation contributes to congenital bicuspid aortic valve. Gene. 2018 Jul 15; 663:115-120. [CrossRef]
- Theis JL, Hu JJ, Sundsbak RS, Evans JM, Bamlet WR, Qureshi MY, O'Leary PW, Olson TM. Genetic Association Between Hypoplastic Left Heart Syndrome and Cardiomyopathies. Circ Genom Precis Med. 2021 Feb;14(1):e003126. [CrossRef]
- Theis JL, Zimmermann MT, Evans JM, Eckloff BW, Wieben ED, Qureshi MY, O'Leary PW, Olson TM. Recessive MYH6 mutations in hypoplastic left heart with reduced ejection fraction. Circ Cardiovasc Genet. 2015 Aug;8(4):564-71. [CrossRef]
- Berg C, Lachmann R, Kaiser C, Kozlowski P, Stressig R, Schneider M, Asfour B, Herberg U, Breuer J, Gembruch U, Geipel A. Prenatal diagnosis of tricuspid atresia: intrauterine course and outcome. Ultrasound Obstet Gynecol. 2010 Feb;35(2):183-90. [CrossRef]
- Sarkozy A, Conti E, D'Agostino R, Digilio MC, Formigari R, Picchio F, Marino B, Pizzuti A, Dallapiccola B. ZFPM2/FOG2 and HEY2 genes analysis in nonsyndromic tricuspid atresia. Am J Med Genet A. 2005 Feb 15;133A(1):68-70. [CrossRef]
- Pierpont ME, Digilio MC. Cardiovascular disease in Noonan syndrome. Curr Opin Pediatr. 2018 Oct;30(5):601-608. [CrossRef]
- Lee ST, Ki CS, Lee HJ. Mutation analysis of the genes involved in the Ras-mitogen-activated protein kinase (MAPK) pathway in Korean patients with Noonan syndrome. Clin Genet. 2007 Aug;72(2):150-5. [CrossRef]
- Pandit B, Sarkozy A, Pennacchio LA, Carta C, Oishi K, Martinelli S, Pogna EA, Schackwitz W, Ustaszewska A, Landstrom A, Bos JM, Ommen SR, Esposito G, Lepri F, Faul C, Mundel P, López Siguero JP, Tenconi R, Selicorni A, Rossi C, Mazzanti L, Torrente I, Marino B, Digilio MC, Zampino G, Ackerman MJ, Dallapiccola B, Tartaglia M, Gelb BD. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet. 2007 Aug;39(8):1007-12. [CrossRef]
- Roberts A, Allanson J, Jadico SK, Kavamura MI, Noonan J, Opitz JM, Young T, Neri G. The cardiofaciocutaneous syndrome. J Med Genet. 2006 Nov;43(11):833-42. [CrossRef]
- Tidyman WE, Rauen KA. Noonan, Costello and cardio-facio-cutaneous syndromes: dysregulation of the Ras-MAPK pathway.Expert Rev Mol Med. 2008 Dec 9;10: e37. [CrossRef]
- Dard L, Hubert C, Esteves P, Blanchard W, Bou About G, Baldasseroni L, Dumon E, Angelini C, Delourme M, Guyonnet-Dupérat V, Claverol S, Fontenille L, Kissa K, Séguéla PE, Thambo JB, Nicolas L, Herault Y, Bellance N, Dias Amoedo N, Magdinier F, Sorg T, Lacombe D, Rossignol R. HRAS germline mutations impair LKB1/AMPK signaling and mitochondrial homeostasis in Costello syndrome models. J Clin Invest. 2022 Apr 15;132(8):e131053. [CrossRef]
- White JJ, Mazzeu JF, Hoischen A, Bayram Y, Withers M, Gezdirici A, Kimonis V, Steehouwer M, Jhangiani SN, Muzny DM, Gibbs RA; Baylor-Hopkins Center for Mendelian Genomics; van Bon BWM, Sutton VR, Lupski JR, Brunner HG, Carvalho CMB. DVL3 Alleles Resulting in a -1 Frameshift of the Last Exon Mediate Autosomal-Dominant Robinow Syndrome. Am J Hum Genet. 2016 Mar 3;98(3):553-561. [CrossRef]
- Rai A, Patil SJ, Srivastava P, Gaurishankar K, Phadke SR. Clinical and molecular characterization of four patients with Robinow syndrome from different families. Am J Med Genet A. 2021 Apr;185(4):1105-1112. [CrossRef]
- Hu R, Qiu Y, Li Y, Li J. A novel frameshift mutation of DVL1-induced Robinow syndrome: A case report and literature review. Mol Genet Genomic Med. 2022 Mar;10(3):e1886. [CrossRef]
- Danyel M, Kortüm F, Dathe K, Kutsche K, Horn D. Autosomal dominant Robinow syndrome associated with a novel DVL3 splice mutation. Am J Med Genet A. 2018 Apr;176(4):992-996. [CrossRef]
- Mašek J, Andersson ER. The developmental biology of genetic Notch disorders. Development. 2017 May 15;144(10):1743-1763. [CrossRef]
- Hofmann JJ, Briot A, Enciso J, Zovein AC, Ren S, Zhang ZW, Radtke F, Simons M, Wang Y, Iruela-Arispe ML Endothelial deletion of murine Jag1 leads to valve calcification and congenital heart defects associated with Alagille syndrome. Development. 2012 Dec 1;139(23):4449-60. [CrossRef]
- McCright B, Lozier J, Gridley T. A mouse model of Alagille syndrome: Notch2 as a genetic modifier of Jag1 haploinsufficiency. Development. 2002 Feb;129(4):1075-82. [CrossRef]
- Liu X, Chen W, Li W, Priest JR, Fu Y, Pang K, Ma B, Han B, Liu X, Hu S, Zhou Z.. Exome-based case-control analysis highlights the pathogenic role of ciliary genes in transposition of the great arteries. Circ Res. 2020 Mar 27;126(7):811-821. [CrossRef]
- Blue GM, Mekel M, Das D, Troup M, Rath E, Ip E, Gudkov M, Perumal G, Harvey RP, Sholler GF, Gecz J, Kirk EP, Liu J, Giannoulatou E, Hong H, Dunwoodie SL, Winlaw DS. Whole genome sequencing in transposition of the great arteries and associations with clinically relevant heart, brain and laterality genes. Am Heart J. 2022 Feb;244:1-13. [CrossRef]
- Li AH, Hanchard NA, Azamian M, D'Alessandro LCA, Coban-Akdemir Z, Lopez KN, Hall NJ, Dickerson H, Nicosia A, Fernbach S, Boone PM, Gambin T, Karaca E, Gu S, Yuan B,. Genetic architecture of laterality defects revealed by whole exome sequencing. Eur J Hum Genet. 2019 Apr;27(4):563-573. [CrossRef]
- Yi T, Sun H, Fu Y, Hao X, Sun L, Zhang Y, Han J, Gu X, Liu X, Guo Y, Wang X, Zhou X, Zhang S, Yang Q, Fan J, He Y. Genetic and Clinical Features of Heterotaxy in a Prenatal Cohort. Front Genet. 2022 Apr 19; 13:818241. [CrossRef]
- Papanayotou C, Collignon J.Philos Trans R Soc Lond B Activin/Nodal signalling before implantation: setting the stage for embryo patterning. Biol Sci. 2014 Dec 5;369(1657):20130539. [CrossRef]
- Mohapatra B, Casey B, Li H, Ho-Dawson T, Smith L, Fernbach SD, Molinari L, Niesh SR, Jefferies JL, Craigen WJ, Towbin JA, Belmont JW, Ware SM.. Identification and functional characterization of NODAL rare variants in heterotaxy and isolated cardiovascular malformations. Hum Mol Genet. 2009 Mar 1;18(5):861-71. [CrossRef]
- Bedard JE, Haaning AM, Ware SM. Identification of a novel ZIC3 isoform and mutation screening in patients with heterotaxy and congenital heart disease. PLoS One. 2011;6(8):e23755. [CrossRef]
- D'Alessandro LC, Latney BC, Paluru PC, Goldmuntz E. The phenotypic spectrum of ZIC3 mutations includes isolated d-transposition of the great arteries and double outlet right ventricle. Am J Med Genet A. 2013 Apr;161A(4):792-802. [CrossRef]
- Zhao Y, Wang Y, Shi L, McDonald-McGinn DM, Crowley TB, McGinn DE, Tran OT, Miller D, Lin JR, Zackai E, Johnston HR, Chow EWC, Vorstman JAS, Vingerhoets C, van Amelsvoort T, Gothelf D, Swillen A, Breckpot J, Vermeesch JR, Eliez S, Schneider M, van den Bree MBM, Owen MJ, Kates WR, Repetto GM,et al. Chromatin regulators in the TBX1 network confer risk for conotruncal heart defects in 22q11.2DS. NPJ Genom Med. 2023 Jul 18;8(1):17. [CrossRef]
- Khositseth A, Tocharoentanaphol C, Khowsathit P, Ruangdaraganon N. Chromosome 22q11 deletions in patients with conotruncal heart defects. Pediatr Cardiol. 2005 Sep-Oct;26(5):570-3.
- Ziolkowska L, Kawalec W, Turska-Kmiec A, Krajewska-Walasek M, Brzezinska-Rajszys G, Daszkowska J, Maruszewski B, Burczynski P. Chromosome 22q11.2 microdeletion in children with conotruncal heart defects: frequency, associated cardiovascular anomalies, and outcome following cardiac surgery. Eur J Pediatr. 2008 Oct;167(10):1135-40. [CrossRef]
- Carotti A, Digilio MC, Piacentini G, Saffirio C, Di Donato RM, Marino B. Cardiac defects and results of cardiac surgery in 22q11.2 deletion syndrome. Dev Disabil Res Rev. 2008;14(1):35-42. [CrossRef]
- Corsten-Janssen N, du Marchie Sarvaas GJ, Kerstjens-Frederikse WS, Hoefsloot LH, van Beynum IM, Kapusta L, van Ravenswaaij-Arts CM. CHD7 mutations are not a major cause of atrioventricular septal and conotruncal heart defects. Am J Med Genet A. 2014 Dec;164A(12):3003-9. [CrossRef]
- Corsten-Janssen N, Scambler PJ. Clinical and molecular effects of CHD7 in the heart. Am J Med Genet C Semin Med Genet. 2017 Dec;175(4):487-495. [CrossRef]
- Jongmans MC, Admiraal RJ, van der Donk KP, Vissers LE, Baas AF, Kapusta L, van Hagen JM, Donnai D, de Ravel TJ, Veltman JA, Geurts van Kessel A, De Vries BB, Brunner HG, Hoefsloot LH, van Ravenswaaij CM. CHARGE syndrome: the phenotypic spectrum of mutations in the CHD7 gene. J Med Genet. 2006 Apr;43(4):306-14. [CrossRef]
- Meisner JK, Martin DM. Congenital heart defects in CHARGE: The molecular role of CHD7 and effects on cardiac phenotype and clinical outcomes. Am J Med Genet C Semin Med Genet. 2020 Mar;184(1):81-89. [CrossRef]
- Zentner GE, Layman WS, Martin DM, Scacheri PC. Molecular and phenotypic aspects of CHD7 mutation in CHARGE syndrome. Am J Med Genet A. 2010 Mar;152A (3):674-86. [CrossRef]
- Rozas MF, Benavides F, León L, Repetto GM Association between phenotype and deletion size in 22q11.2 microdeletion syndrome: systematic review and meta-analysis. Orphanet J Rare Dis. 2019 Aug 9;14(1):195. [CrossRef]
- Zhao Y, Diacou A, Johnston HR, Musfee FI, McDonald-McGinn DM, McGinn D, Crowley TB, Repetto GM, Swillen A, Breckpot J, Vermeesch JR, Kates WR, Digilio MC, Unolt M, Marino B, Pontillo M, Armando M, Di Fabio F, Vicari S, van den Bree M, Moss H, Owen MJ, Murphy KC, Murphy CM, Murphy D, Schoch K, Shashi V, Tassone F, Simon TJ, Shprintzen RJ, Campbell L. Complete sequence of the 22q11.2 allele in 1,053 subjects with 22q11.2 deletion syndrome reveals modifiers of conotruncal heart defects. Am J Hum Genet. 2020 Jan 2;106(1):26-40. [CrossRef]
- He GW, Maslen CL, Chen HX, Hou HT, Bai XY, Wang XL, Liu XC, Lu WL, Chen XX, Chen WD, Xing QS, Wu Q, Wang J, Yang Q. Identification of novel rare copy number variants associated with sporadic tetralogy of Fallot and clinical implications.Clin Genet. 2022 Nov;102(5):391-403. [CrossRef]
- Mefford HC, Sharp AJ, Baker C, Itsara A, Jiang Z, Buysse K, Huang S, Maloney VK, Crolla JA, Baralle D, Collins A, Mercer C, Norga K, de Ravel T, Devriendt K, Bongers EM, de Leeuw N, Reardon W, Gimelli S, Bena F, Hennekam RC, Male A, Gaunt L, Clayton-Smith J, Simonic I, Park SM, Mehta SG, Nik-Zainal S, Woods CG, Firth HV, Parkin G, Fichera M, Reitano S, Lo Giudice M, Li KE, Casuga I, Broomer A, Conrad B, Schwerzmann M, Räber L, Gallati S, Striano P, Coppola A, et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N Engl J Med. 2008 Oct 16;359(16):1685-99. [CrossRef]
- Ceylan AC, Sahin I, Erdem HB, Kayhan G, Simsek-Kiper PO, Utine GE, Percin F, Boduroglu K, Alikasifoglu M.J An eight-case 1q21 region series: novel aberrations and clinical variability with new features. Intellect Disabil Res. 2019 Jun;63(6):548-557. [CrossRef]
- Reuter MS, Chaturvedi RR, Jobling RK, Pellecchia G, Hamdan O, Sung WWL, Nalpathamkalam T, Attaluri P, Silversides CK, Wald RM, Marshall CR, Williams SG, Keavney BD, Thiruvahindrapuram B, Scherer SW, Bassett AS. Clinical Genetic Risk Variants Inform a Functional Protein Interaction Network for Tetralogy of Fallot. Circ Genom Precis Med. 2021 Aug;14(4):e003410. [CrossRef]
- Page DJ, Miossec MJ, Williams SG, Monaghan RM, Fotiou E, Cordell HJ, Sutcliffe L, Topf A, Bourgey M, Bourque G, Eveleigh R, Dunwoodie SL, Winlaw DS, Bhattacharya S, Breckpot J, Devriendt K, Gewillig M, Brook JD, Setchfield KJ, Bu'Lock FA, O'Sullivan J, Stuart G,. Whole exome sequencing reveals the major genetic contributors to nonsyndromic tetralogy of Fallot. Circ Res. 2019 Feb 15;124(4):553-563. [CrossRef]
- Reuter MS et al. Haploinsufficiency of vascular endothelial growth factor related signaling genes is.
- associated with tetralogy of Fallot. Genet. Med. 21, 1001–1007 (2019).
- Škorić-Milosavljević D, Lahrouchi N, Bosada FM, Dombrowsky G, Williams SG, Lesurf R, Tjong FVY, Walsh R, El Bouchikhi I, Breckpot J, Audain E, Ilgun A, Beekman L, Ratbi I, Strong A, Muenke M, Heide S, Muir AM, Hababa M, Cross L, Zhou D, Pastinen T; et al. Rare variants in KDR, encoding VEGF Receptor 2, are associated with tetralogy of Fallot. Genet Med. 2021 Oct;23(10):1952-1960. [CrossRef]
- Tan ZP, Huang C, Xu ZB, Yang JF, Yang YF. Novel ZFPM2/FOG2 variants in patients with double outlet right ventricle. Clin Genet. 2012 Nov;82(5):466-71. [CrossRef]
- Huang X, Niu W, Zhang Z, Zhou C, Xu Z, Liu J, Su Z, Ding W, Zhang H. Identification of novel significant variants of ZFPM2/FOG2 in non-syndromic Tetralogy of Fallot and double outlet right ventricle in a Chinese Han population. Mol Biol Rep. 2014;41(4):2671-7. [CrossRef]
- Su W, Zhu P, Wang R, Wu Q, Wang M, Zhang X, Mei L, Tang J, Kumar M, Wang X, Su L, Dong N. Congenital heart diseases and their association with the variant distribution features on susceptibility genes. Clin Genet. 2017 Mar;91(3):349-354. [CrossRef]
- Yang YQ, Gharibeh L, Li RG, Xin YF, Wang J, Liu ZM, Qiu XB, Xu YJ, Xu L, Qu XK, Liu X, Fang WY, Huang RT, Xue S, Nemer G. GATA4 loss-of-function mutations underlie familial tetralogy of fallot. Hum Mutat. 2013 Dec ;34(12):1662-71. [CrossRef]
- Kodo K, Nishizawa T, Furutani M, Arai S, Yamamura E, Joo K, Takahashi T, Matsuoka R, Yamagishi H. GATA6 mutations cause human cardiac outflow tract defects by disrupting semaphorinplexin signaling. Proc Natl Acad Sci U S A. 2009 Aug 18;106(33):13933-8. [CrossRef]
- Lahaye S, Corsmeier D, Basu M, Bowman JL, Fitzgerald-Butt S, Zender G, Bosse K, McBride KL, White P, Garg V. Utilization of whole exome sequencing to identify causative mutations in familial congenital heart disease. Circ Cardiovasc Genet. 2016 Aug;9(4):320-9. [CrossRef]
- Hoang TT, Goldmuntz E, Roberts AE, Chung WK, Kline JK, Deanfield JE, Giardini A, Aleman A, Gelb BD, Mac Neal M, Porter GA Jr, Kim R, Brueckner M, Lifton RP, Edman S, Woyciechowski S, Mitchell LE, Agopian AJ. The congenital heart disease genetic network study: cohort description. PLoS One. 2018 Jan 19;13(1):e0191319. [CrossRef]
- Preuss C, Capredon M, Wünnemann F, Chetaille P, Prince A, Godard B, Leclerc S, Sobreira N, Ling H, Awadalla P, Thibeault M, Khairy P; MIBAVA Leducq consortium; Samuels ME, Andelfinger G Family based whole exome sequencing reveals the multifaceted role of Notch signaling in congenital heart disease. PLoS Genet. 2016 Oct 19;12(10):e1006335. [CrossRef]
- Sevim Bayrak C, Zhang P, Tristani-Firouzi M, Gelb BD, Itan Y. De novo variants in exomes of congenital heart disease patients identify risk genes and pathways. Genome Med. 2020 Jan 15;12(1):9.
- Morton SU, Shimamura A, Newburger PE, Opotowsky AR, Quiat D, Pereira AC, Jin SC, Gurvitz M, Brueckner M, Chung WK, Shen Y, Bernstein D, Gelb BD, Giardini A, Goldmuntz E, Kim RW, Lifton RP, Porter GA Jr, Srivastava D, Tristani-Firouzi M, Newburger JW, Seidman JG, Seidman CE. Association of damaging variants in genes with increased cancer risk among patients with congenital heart disease. JAMA Cardiol. 2021 Apr 1;6(4):457-462. [CrossRef]
- Park JE, Park JS, Jang SY, Park SH, Kim JW, Ki CS, Kim DK. A novel SMAD6 variant in a patient with severely calcified bicuspid aortic valve and thoracic aortic aneurysm. Mol Genet Genomic Med. 2019 May;7(5):e620. doi10.1002/mgg3.620.
- Raya A, Kawakami Y, Rodriguez-Esteban C, Buscher D, Koth CM, Itoh T, Morita M, Raya RM, Dubova I, Bessa JG, de la Pompa JL, Izpisua Belmonte JC. Notch activity induces Nodal expression and mediates the establishment of left-right asymmetry in vertebrate embryos.Genes Dev. 2003 May 15;17(10):1213-8. [CrossRef]
- Galvin KM, Donovan MJ, Lynch CA, Meyer RI, Paul RJ, Lorenz JN, Fairchild-Huntress V, Dixon KL, Dunmore JH, Gimbrone MA Jr, Falb D, Huszar D. A role for Smad6 in development and homeostasis of the cardiovascular system. Nat. Genet. Nat Genet. 2000 Feb;24(2):171-4. [CrossRef]
- McKean DM, Homsy J, Wakimoto H, Patel N, Gorham J, DePalma SR, Ware JS, Zaidi S, Ma W, Patel N, Lifton RP, Chung WK, Kim R, Shen Y, Brueckner M, Goldmuntz E, Sharp AJ, Seidman CE, Gelb BD, Seidman JG. Loss of RNA expression and allele-specific expression associated with congenital heart disease. Nat Commun. 2016 Sep 27; 7:12824. [CrossRef]
- Brault V, Nguyen TL, Flores-Gutiérrez J, Iacono G, Birling MC, Lalanne V, Meziane H, Manousopoulou A, Pavlovic G, Lindner L, Selloum M, Sorg T, Yu E, Garbis SD, Hérault Y. Dyrk1a gene dosage in glutamatergic neurons has key effects in cognitive deficits observed in mouse models of MRD7 and Down syndrome. PLoS Genet. 2021 Sep 29;17(9):e1009777. [CrossRef]
- Feki A, Hibaoui Y. DYRK1A Protein, A Promising Therapeutic Target to Improve Cognitive Deficits in Down Syndrome.
- Brain Sci. 2018 Oct 16;8(10):187. [CrossRef]
- Vandeweyer G, Helsmoortel C, Van Dijck A, Vulto-van Silfhout AT, Coe BP, Bernier R, Gerdts J, Rooms L, van den Ende J, Bakshi M, Wilson M, Nordgren A, Hendon LG, Abdulrahman OA, Romano C, de Vries BB, Kleefstra T, Eichler EE, Van der Aa N, Kooy RF. The transcriptional regulator ADNP links the BAF (SWI/SNF) complexes with autism. Am J Med Genet C Semin Med Genet. 2014 Sep ;166C (3):315-26. [CrossRef]
- Ockeloen CW, Willemsen MH, de Munnik S, van Bon BW, de Leeuw N, Verrips A, Kant SG, Jones EA, Brunner HG, van Loon RL, Smeets EE, van Haelst MM, van Haaften G, Nordgren A, Malmgren H, Grigelioniene G, Vermeer S, Louro P, Ramos L, Maal TJ, van Heumen CC, Yntema HG, Carels CE, Kleefstra T. Further delineation of the KBG syndrome phenotype caused by ANKRD11 aberrations. Eur J Hum Genet. 2015 Sep ;23(9):1176-85. [CrossRef]
- Hamilton MJ, Caswell RC, Canham N, Cole T, Firth HV, Foulds N, Heimdal K, Hobson E, Houge G, Joss S, Kumar D, Lampe AK, Maystadt I, McKay V, Metcalfe K, Newbury-Ecob R, Park SM, Robert L, Rustad CF, Wakeling E, Wilkie AOM, Study TDDD, Twigg SRF, Suri M. Heterozygous mutations affecting the protein kinase domain of CDK13 cause a syndromic form of developmental delay and intellectual disability. J Med Genet. 2018 Jan;55(1):28-38. [CrossRef]
- van den Akker WMR, Brummelman I, Martis LM, Timmermans RN, Pfundt R, Kleefstra T, Willemsen MH, Gerkes EH, Herkert JC, van Essen AJ, Rump P, Vansenne F, Terhal PA, van Haelst MM, Cristian I, Turner CE, Cho MT, Begtrup A, Willaert R, Fassi E, van Gassen KLI, Stegmann APA, de Vries BBA, Schuurs-Hoeijmakers JHM. De novo variants in CDK13 associated with syndromic ID/DD: Molecular and clinical delineation of 15 individuals and a further review. Clin Genet. 2018 May;93(5):1000-1007. [CrossRef]
- Beal B, Hayes I, McGaughran J, Amor DJ, Miteff C, Jackson V, van Reyk O, Subramanian G, Hildebrand MS, Morgan AT, Goel H. Expansion of phenotype of DDX3X syndrome: six new cases. Clin Dysmorphol. 2019 Oct;28(4):169-174. [CrossRef]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M,. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016 Aug 18;536(7616):285-91. [CrossRef]
- Belkadi A, Bolze A, Itan Y, Cobat A, Vincent QB, Antipenko A, Shang L, Boisson B, Casanova JL, Abel L Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants.
- Proc Natl Acad Sci U S, A. 2015 Apr 28;112(17):5473-8. [CrossRef]
- Hsieh A, Morton SU, Willcox JAL, Gorham JM, Tai AC, Qi H, DePalma S, McKean D, Griffin E, Manheimer KB, Bernstein D, Kim RW, Newburger JW, Porter GA Jr, Srivastava D, Tristani-Firouzi M, Brueckner M, Lifton RP, Goldmuntz E, Gelb BD, Chung WK, Seidman CE, Seidman JG, Shen Y. EM-mosaic detects mosaic point mutations that contribute to congenital heart disease. Genome Med. 2020 Apr 29;12(1):42. [CrossRef]
- Manheimer KB, Richter F, Edelmann LJ, D'Souza SL, Shi L, Shen Y, Homsy J, Boskovski MT, Tai AC, Gorham J, Yasso C, Goldmuntz E, Brueckner M, Lifton RP, Chung WK, Seidman CE, Seidman JG, Gelb BD. Robust identification of mosaic variants in congenital heart disease. Hum Genet. 2018 Feb;137(2):183-193. [CrossRef]
- Wei W, Keogh MJ, Aryaman J, Golder Z, Kullar PJ, Wilson I, Talbot K, Turner MR, McKenzie CA, Troakes C, Attems J, Smith C, Sarraj SA, Morris CM, Ansorge O, Jones NS, Ironside JW, Chinnery PF. Frequency and signature of somatic variants in 1461 human brain exomes. Genet Med. 2019 Apr;21(4):904-912. [CrossRef]
- .Zech M, Jech R, Boesch S, Škorvánek M, Weber S, Wagner M, Zhao C, Jochim A, Necpál J, Dincer Y, Vill K, Distelmaier F, Stoklosa M, Krenn M, Grunwald S, Bock-Bierbaum T, Fečíková A, Havránková P, Roth J, Příhodová I, Adamovičová M, Ulmanová O, Bechyně K, Danhofer P, Veselý B, Haň V, Pavelekova P, Gdovinová Z, Mantel T, Meindl T, Sitzberger A, Schröder S, Blaschek A, Roser T, Bonfert MV, et al. Monogenic variants in dystonia: an exome-wide sequencing study Lancet Neurol. 2020 Nov;19(11):908-918. [CrossRef]
- Gardner EJ, Sifrim A, Lindsay SJ, Prigmore E, Rajan D, Danecek P, Gallone G, Eberhardt RY, Martin HC, Wright CF, FitzPatrick DR, Firth HV, Hurles ME. Detecting cryptic clinically relevant structural variation in exome-sequencing data increases diagnostic yield for developmental disorders. Am J Hum Genet. 2021 Nov 4;108(11):2186-2194. [CrossRef]
- Wright CF, Fitzgerald TW, Jones WD, Clayton S, McRae JF, van Kogelenberg M, King DA, Ambridge K, Barrett DM, Bayzetinova T, Bevan AP, Bragin E, Chatzimichali EA, Gribble S, Jones P, Krishnappa N, Mason LE, Miller R, Morley KI, Parthiban V, Prigmore E, Rajan D, Sifrim A, Swaminathan GJ, Tivey AR, Middleton A, Parker M, Carter NP, Barrett JC, Hurles ME, FitzPatrick DR, Firth HV; DDD study. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet. 2015 Apr 4;385(9975):1305-14. [CrossRef]
- Padhi EM, Hayeck TJ, Cheng Z, Chatterjee S, Mannion BJ, Byrska-Bishop M, Willems M, Pinson L, Redon S, Benech C, Uguen K, Audebert-Bellanger S, Le Marechal C, Férec C, Efthymiou S, Rahman F, Maqbool S, Maroofian R, Houlden H, Musunuri R, Narzisi G, Abhyankar A, Hunter RD, Akiyama J, Fries LE, Ng JK, Mehinovic E, Stong N, Allen AS, Dickel DE, Bernier RA, Gorkin DU, Pennacchio LA, Zody MC, Turner TN. Coding and noncoding variants in EBF3 are involved in HADDS and simplex autism. Hum Genomics. 2021 Jul 13;15(1):44. [CrossRef]
- Choy MK, Javierre BM, Williams SG, Baross SL, Liu Y, Wingett SW, Akbarov A, Wallace C, Freire-Pritchett P, Rugg-Gunn PJ, Spivakov M, Fraser P, Keavney BD. Promoter interactome of human embryonic stem cell-derived cardiomyocytes connects GWAS regions to cardiac gene networks. Nat Commun. 2018 Jun 28;9(1):2526. [CrossRef]
- Hoelscher SC, Stich T, Diehm A, Lahm H, Dreßen M, Zhang Z, Neb I, Aherrahrou Z, Erdmann J, Schunkert H, Santamaria G, Cuda G, Gilsbach R, Hein L, Lange R, Hassel D, Krane M, Doppler SA. miR-128a Acts as a Regulator in Cardiac Development by Modulating Differentiation of Cardiac Progenitor Cell Populations. Int J Mol Sci. 2020 Feb 10;21(3):1158. [CrossRef]
- Kheradpour P, Ernst J, Melnikov A, Rogov P, Wang L, Zhang X, Alston J, Mikkelsen TS, Kellis M. Systematic dissection of regulatory motifs in 2000 predicted human enhancers using a massively parallel reporter assay. Genome Res. 2013 May;23(5):800-11. [CrossRef]
- Akerberg BN, Gu F, VanDusen NJ, Zhang X, Dong R, Li K, Zhang B, Zhou B, Sethi I, Ma Q, Wasson L, Wen T, Liu J, Dong K, Conlon FL, Zhou J, Yuan GC, Zhou P, Pu WT. A reference map of murine cardiac transcription factor chromatin occupancy identifies dynamic and conserved enhancers. Nat Commun. 2019 Oct 28;10(1):4907. [CrossRef]
- Vanoudenhove J, Yankee TN, Wilderman, Cotney J Epigenomic and transcriptomic dynamics during human heart organogenesis. Circ Res. 2020 Oct 9;127(9):e184-e209. [CrossRef]
- Hussein IR, Bader RS, Chaudhary AG, Bassiouni R, Alquaiti M, Ashgan F, Schulten HJ, Al Qahtani MH. dentification of De Novo and Rare Inherited Copy Number Variants in Children with Syndromic Congenital Heart Defects Pediatr Cardiol. 2018 Jun;39(5):924-940. [CrossRef]
- van Ouwerkerk AF, Bosada FM, van Duijvenboden K, Houweling AC, Scholman KT, Wakker V, Allaart CP, Uhm JS, Mathijssen IB, Baartscheer T, Postma AV, Barnett P, Verkerk AO, Boukens BJ, Christoffels VM. Patient-Specific TBX5-G125R Variant Induces Profound Transcriptional Deregulation and Atrial Dysfunction. Circulation. 2022 Feb 22;145(8):606-619. [CrossRef]
- Richter F, Morton SU, Kim SW, Kitaygorodsky A, Wasson LK, Chen KM, Zhou J, Qi H, Patel N, DePalma SR, Parfenov M, Homsy J, Gorham JM, Manheimer KB, Velinder M, Farrell A, Marth G, Schadt EE, Kaltman JR, Newburger JW, Giardini A, Goldmuntz E, Brueckner M, et al. Genomic analyses implicate noncoding denovo variants in congenital heart disease. Nat Genet. 2020 Aug;52(8):769-777. [CrossRef]
- Morton SU, Pereira AC, Quiat D, Richter F, Kitaygorodsky A, Hagen J, Bernstein D, Brueckner M, Goldmuntz E, Kim RW, Lifton RP, Porter GA Jr, Tristani-Firouzi M, Chung WK, Roberts A, Gelb BD, Shen Y, Newburger JW, Seidman JG, Seidman CE. Genome-Wide De Novo Variants in Congenital Heart Disease Are Not Associated With Maternal Diabetes or Obesity. Circ Genom Precis Med. 2022 Apr;15(2):e003500. [CrossRef]
- Wang T, Zhao PA, Eichler EE. Rare variants and the oligogenic architecture of autism. Trends Genet. 2022 Sep;38(9):895-903. [CrossRef]
- Karczewski KJ et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581,434–443 (2020).
- Collins RL, Brand H, Karczewski KJ, Zhao X, Alföldi J, Francioli LC, Khera AV, Lowther C, Gauthier LD, Wang H, Watts NA, Solomonson M, O'Donnell-Luria A, Baumann A, Munshi R, Walker M, Whelan CW, Huang Y, Brookings T, Sharpe T, Stone MR, Valkanas E, Fu J, Tiao G, Laricchia KM, Ruano-Rubio V, Stevens C, Gupta N, Cusick C, Margolin L; Genome Aggregation Database Production Team; Genome Aggregation Database Consortium; Taylor KD, Lin HJ, Rich SS, Post WS, Chen YI, Rotter JI, Nusbaum C, Philippakis A, Lander E, Gabriel S, Neale BM, Kathiresan S, Daly MJ, Banks E, MacArthur DG, Talkowski ME.A structural variation reference for medical and population genetics. Nature. 2020 May;581(7809):444-451. [CrossRef]
- Hureaux M, Guterman S, Hervé B, Till M, Jaillard S, Redon S, Valduga M, Coutton C, Missirian C, Prieur F, Simon-Bouy B, Beneteau C, Kuentz P, Rooryck C, Gruchy N, Marle N, Plutino M, Tosca L, Dupont C, Puechberty J, Schluth-Bolard C, Salomon L, Sanlaville D, Malan V, Vialard F. Chromosomal microarray analysis in fetuses with an isolated congenital heart defect: A retrospective, nationwide, multicenter study in France. Prenat Diagn. 2019 May;39(6):464-470. [CrossRef]
- Hatim O, Pavlinov I, Xu M, Linask K, Beers J, Liu C, Baumgärtel K, Gilbert M, Spinner N, Chen C, Zou J, Zheng W. Generation of an Alagille syndrome (ALGS) patient-derived induced pluripotent stem cell line (TRNDi032-A) carrying a heterozygous mutation (p.Cys682Leufs*7) in the JAG1 gene. Stem Cell Res. 2023 Dec; 73:103231. [CrossRef]
- Legoff L, D’Cruz SC, Tevosian S, Primig M, Smagulova F Transgenerational inheritance of environmentally induced epigenetic alterations during mammalian development. Cells. 2019 Dec 3;8(12):1559. [CrossRef]
- Barua S, Junaid MA Lifestyle, pregnancy and epigenetic effects. Epigenomics. 2015;7(1):85-102. [CrossRef]
- Yokouchi-Konishi T, Yoshimatsu J, Sawada M, Shionoiri T, Nakanishi A, Horiuchi C, Tsuritani M, Iwanaga N, Kamiya CA, Neki R, Miyake A, Kurosaki K, Shiraishi I.. Recurrent congenital heart diseases among neonates born to mothers with congenital heart diseases. Pediatr Cardiol. 2019 Apr;40(4):865-870. [CrossRef]
- Ellesøe SG, Workman CT, Bouvagnet P, Loffredo CA, McBride KL, Hinton RB, van Engelen K, Gertsen EC, Mulder BJM, Postma AV, Anderson RH, Hjortdal VE, Brunak S, Larsen LA. Familial co-occurrence of congenital heart defects follows distinct patterns. Eur Heart J. 2018 Mar 21;39(12):1015-1022. [CrossRef]
- Øyen N, Poulsen G, Boyd HA, Wohlfahrt J, Jensen PK, Melbye M. Recurrence of congenital heart defects in families. Circulation. 2009 Jul 28;120(4):295-301. [CrossRef]
- Argelaguet R, Clark SJ, Mohammed H, Stapel LC, Krueger C, Kapourani CA, Imaz-Rosshandler I, Lohoff T, Xiang Y, Hanna CW, Smallwood S, Ibarra-Soria X, Buettner F, Sanguinetti G, Xie W, Krueger F, Göttgens B, Rugg-Gunn PJ, Kelsey G, Dean W, Nichols J, Stegle O, Marioni JC, Reik W. Multi-omics profiling of mouse gastrulation at single-cell resolution. Nature. 2019 Dec;576(7787):487-491. [CrossRef]
- Tucker NR, Chaffin M, Fleming SJ, Hall AW, Parsons VA, Bedi KC Jr, Akkad AD, Herndon CN, Arduini A, Papangeli I, Roselli C, Aguet F, Choi SH, Ardlie KG, Babadi M, Margulies KB, Stegmann CM, Ellinor PT. Transcriptional and Cellular Diversity of the Human Heart. Circulation. 2020 Aug 4;142(5):466-482. [CrossRef]
- Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, Zhang F, Mundlos S, Christiansen L, Steemers FJ, Trapnell C, Shendure J. The single-cell transcriptional landscape of mammalian organogenesis. Nature. 2019 Feb;566(7745):496-502. [CrossRef]
- Cui Y, Zheng Y, Liu X, Yan L, Fan X, Yong J, Hu Y, Dong J, Li Q, Wu X, Gao S, Li J, Wen L, Qiao J, Tang F.. Single-cell transcriptome analysis maps the developmental track of the human heart. Cell Rep. 2019 Feb 12;26(7):1934-1950.e5. [CrossRef]
- Lescroart F, Wang X, Lin X, Swedlund B, Gargouri S, Sànchez-Dànes A, Moignard V, Dubois C, Paulissen C, Kinston S, Göttgens B, Blanpain C.. Defining the earliest step of cardiovascular lineage segregation by single‐cell.
- RNA-seq. Science. 2018 Mar 9;359(6380):1177-1181. [CrossRef]
- DeLaughter DM, Bick AG, Wakimoto H, McKean D, Gorham JM, Kathiriya IS, Hinson JT, Homsy J, Gray J, Pu W, Bruneau BG, Seidman JG, Seidman CE. Single-cell resolution of temporal gene expression during heart development. Dev Cell. 2016 Nov 21;39(4):480-490. [CrossRef]
- Pijuan-Sala B, Griffiths JA, Guibentif C, Hiscock TW, Jawaid W, Calero-Nieto FJ, Mulas C, Ibarra-Soria X, Tyser RCV, Ho DLL, Reik W, Srinivas S, Simons BD, Nichols J, Marioni JC, Göttgens B. A single-cell molecular map of mouse gastrulation and early organogenesis. Nature. 2019 Feb;566(7745):490-495. [CrossRef]
- Ulirsch JC, Verboon JM, Kazerounian S, Guo MH, Yuan D, Ludwig LS, Handsaker RE, Abdulhay NJ, Fiorini C, Genovese G, Lim ET, Cheng A, Cummings BB, Chao KR, Beggs AH, Genetti CA, Sieff CA, Newburger PE, Niewiadomska E, Matysiak M, Vlachos A, Lipton JM, Atsidaftos E. The genetic landscape of Diamond-Blackfan anemia. Am J Hum Genet. 2018 Dec 6;103(6):930-947. [CrossRef]
- Bramel EE, Creamer TJ, Saqib M, Camejo Nunez WA, Bagirzadeh R, Roker LA, Goff LA, MacFarlane EG. Postnatal Smad3 Inactivation in Murine Smooth Muscle Cells Elicits a Temporally and Regionally Distinct Transcriptional Response. Front Cardiovasc Med. 2022 Apr 8;9:826495. [CrossRef]
- Bissoli I, D'Adamo S, Pignatti C, Agnetti G, Flamigni F, Cetrullo S. Induced pluripotent stem cell-based models: Are we ready for that heart in a dish? Front Cell Dev Biol. 2023 Jan 19; 11:1129263. [CrossRef]
- Baumann, K. Achieving pluripotency. Nat Rev Mol Cell Biol. 2010 Oct;11(10):677. [CrossRef]
- Zhang J, Tao R, Campbell KF, Carvalho JL, Ruiz EC, Kim GC, Schmuck EG, Raval AN, da Rocha AM, Herron TJ, Jalife J, Thomson JA, Kamp TJ.Nat Commun. 2019 May 20;10(1):2238. [CrossRef]
- Liu JA, Cheung M. Neural crest stem cells and their potential therapeutic applications. Dev Biol. 2016 Nov 15;419(2):199-216. [CrossRef]
- Neri T, Hiriart E, van Vliet PP, Faure E, Norris RA, Farhat B, Jagla B, Lefrancois J, Sugi Y, Moore-Morris T, Zaffran S, Faustino RS, Zambon AC, Desvignes JP, Salgado D, Levine RA, de la Pompa JL, Terzic A, Evans SM, Markwald R, Pucéat M.Nat Commun. 2019 Apr 26;10(1):1929. [CrossRef]
- Kathiriya IS, Rao KS, Iacono G, Devine WP, Blair AP, Hota SK, Lai MH, Garay BI, Thomas R, Gong HZ, Wasson LK, Goyal P, Sukonnik T, Hu KM, Akgun GA, Bernard LD, Akerberg BN, Gu F, Li K, Speir ML, Haeussler M, Pu WT, Stuart JM, Seidman CE, Seidman JG, Heyn H, Bruneau BG. Dev Cell. 2021 Feb 8;56(3):292-309.e9. [CrossRef]
- Rydzanicz M, Zwoliński P, Gasperowicz P, Pollak A, Kostrzewa G, Walczak A, Konarzewska M, Płoski R. A recurrent de novo variant supports KCNC2 involvement in the pathogenesis of developmental and epileptic encephalopathy. Am J Med Genet A. 2021 Nov;185(11):3384-3389. [CrossRef]
- Pierpont ME, Brueckner M, Chung WK, Garg V, Lacro RV, McGuire AL, Mital S, Priest JR, Pu WT, Roberts A, Ware SM, Gelb BD, Russell MW; American Heart Association Council on Cardiovascular Disease in the Young; Council on Cardiovascular and Stroke Nursing; and Council on Genomic and Precision Medicine. Genetic basis for congenital heart disease: revisited: a scientific statement from the American Heart Association. Circulation. 2018 Nov 20;138(21):e653-e711. [CrossRef]
- DiStefano MT, Hemphill SE, Oza AM, Siegert RK, Grant AR, Hughes MY, Cushman BJ, Azaiez H, Booth KT, Chapin A, Duzkale H, Matsunaga T, Shen J, Zhang W, Kenna M, Schimmenti LA, Tekin M, Rehm HL, Tayoun ANA, Amr SS; ClinGen Hearing Loss Clinical Domain Working Group. ClinGen expert clinical validity curation of 164 hearing loss gene-disease pairs. Genet Med. 2019 Oct;21(10):2239-2247. [CrossRef]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015 May;17(5):405-24. [CrossRef]
- Boycott KM, Azzariti DR, Hamosh A, Rehm HL. Seven years since the launch of the Matchmaker Exchange: The evolution of genomic matchmaking. Hum Mutat. 2022 Jun;43(6):659-667. [CrossRef]
- Patel RY, Shah N, Jackson AR, Ghosh R, Pawliczek P, Paithankar S, Baker A, Riehle K, Chen H, Milosavljevic S, Bizon C, Rynearson S, Nelson T, Jarvik GP, Rehm HL, Harrison SM, Azzariti D, Powell B, Babb L, Plon SE, Milosavljevic A; ClinGen Resource. ClinGen Pathogenicity Calculator: a configurable system for assessing pathogenicity of genetic variants. Genome Med. 2017 Jan 12;9(1):3. [CrossRef]
- Yu Y, Lei W, Yang J, Wei YC, Zhao ZL, Zhao ZA, Hu S. Functional mutant GATA4 identification and potential application in preimplantation diagnosis of congenital heart. Gene. 2018 Jan 30; 641:349-354. [CrossRef]
- Boskovski MT, Homsy J, Nathan M, Sleeper LA, Morton S, Manheimer KB, Tai A, Gorham J, Lewis M, Swartz M, Alfieris GM, Bacha EA, Karimi M, Meyer D, Nguyen K, Bernstein D, Romano-Adesman A, Porter GA Jr, Goldmuntz E, Chung WK, Srivastava D, et al. De novo damaging variants, clinical phenotypes and post-operative outcomes in congenital heart disease. Circ Genom Precis Med. 2020 Aug;13(4):e002836. [CrossRef]
- Zomer AC, Ionescu-Ittu R, Vaartjes I, Pilote L, Mackie AS, Therrien J, Langemeijer MM, Grobbee DE, Mulder BJ, Marelli AJ. Sex differences in hospital mortality in adults with congenital heart disease: the impact of reproductive health.J Am Coll Cardiol. 2013 Jul 2;62(1):58-67. [CrossRef]
- Gurvitz M et al. Prevalence of cancer in adults with congenital heart disease compared with the general population. Am. J. Cardiol. 118, 1742–1750 (2016).
- Mandalenakis Z, Karazisi C, Skoglund K, Rosengren A, Lappas G, Eriksson P, Dellborg M. Risk of cancer among children and young adults with congenital heart disease compared with healthy controls. JAMA Netw Open. 2019 Jul 3;2(7):e196762. [CrossRef]
- Cohen S, Gurvitz MZ, Beauséjour-Ladouceur V, Lawler PR, Therrien J, Marelli AJ. Cancer Risk in Congenital Heart Disease-What Is the Evidence? Can J Cardiol. 2019 Dec;35(12):1750-1761. [CrossRef]
- Lim ET, Uddin M, De Rubeis S, Chan Y, Kamumbu AS, Zhang X, D'Gama AM, Kim SN, Hill RS, Goldberg AP, Poultney C, Minshew NJ, Kushima I, Aleksic B, Ozaki N, Parellada M, Arango C, Penzol MJ, Carracedo A, Kolevzon A, Hultman CM, Weiss LA, Fromer M, Chiocchetti AG, Freitag CM; Autism Sequencing Consortium; Church GM, Scherer SW, Buxbaum JD, Walsh CA. Rates, distribution and implications of postzygotic mosaic mutations in autism spectrum disorder. Nat Neurosci. 2017 Sep ;20(9):1217-1224. [CrossRef]
- Freed D, Pevsner J. The Contribution of Mosaic Variants to Autism Spectrum Disorder. PLoS Genet. 2016 Sep 15;12(9):e1006245. [CrossRef]
- Dou Y, Yang X, Li Z, Wang S, Zhang Z, Ye AY, Yan L, Yang C, Wu Q, Li J, Zhao B, Huang AY, Wei L. Postzygotic single-nucleotide mosaicisms contribute to the etiology of autism spectrum disorder and autistic traits and the origin of mutations. Hum Mutat. 2017 Aug;38(8):1002-1013. [CrossRef]
- Yu X, Tao Y, Liu X, Yu F, Jiang C, Xiao Y, Zhang H, He Y, Ye L, Wang Y, Zhou C, Wang J, Jiang Z, Hong H. The implication of chromosomal abnormalities in the surgical outcomes of Chinese pediatric patients with congenital heart disease. Front Cardiovasc Med. 2023 May 24;10:1164577. [CrossRef]
- Putotto C, Pugnaloni F, Unolt M, Maiolo S, Trezzi M, Digilio MC, Cirillo A, Limongelli G, Marino B, Calcagni G, Versacci P. 22q11.2 Deletion Syndrome: Impact of Genetics in the Treatment of Conotruncal Heart Defects. Children (Basel). 2022 May 25;9(6):772. [CrossRef]
- Mercer-Rosa L, Pinto N, Yang W, Tanel R, Goldmuntz E 22q11.2 deletion syndrome is associated with perioperative outcome in tetralogy of Fallot. J Thorac Cardiovasc Surg. 2013 Oct;146(4):868-73. [CrossRef]
- O’Byrne ML, O'Byrne ML, Yang W, Mercer-Rosa L, Parnell AS, Oster ME, Levenbrown Y, Tanel RE, Goldmuntz E.22q11.2 deletion syndrome is associated with increased perioperative events and more complicated postoperative course in infants undergoing infant operative correction of truncus arteriosus communis or interrupted aortic arch. J Thorac Cardiovasc Surg. 2014 Oct;148(4):1597-605. [CrossRef]
- Kim DS, Kim JH, Burt AA, Crosslin DR, Burnham N, Kim CE, McDonald-McGinn DM, Zackai EH, Nicolson SC, Spray TL, Stanaway IB, Nickerson DA, Heagerty PJ, Hakonarson H, Gaynor JW, Jarvik GP.. Burden of potentially pathologic copy number variants is higher in children with isolated congenital heart disease and significantly impairs covariate-adjusted transplant-free survival. J Thorac Cardiovasc Surg. 2016 Apr;151(4):1147-51.e4. [CrossRef]
- Bai W, Suzuki H, Huang J, Francis C, Wang S, Tarroni G, Guitton F, Aung N, Fung K, Petersen SE, Piechnik SK, Neubauer S, Evangelou E, Dehghan A, O'Regan DP, Wilkins MR, Guo Y, Matthews PM, Rueckert D. A population-based phenome-wide association study of cardiac and aortic structure and function. Nat Med. 2020 Oct;26(10):1654-1662. [CrossRef]
- Cohen S, Liu A, Gurvitz M, Guo L, Therrien J, Laprise C, Kaufman JS, Abrahamowicz M, Marelli AJ. Exposure to Low-Dose Ionizing Radiation From Cardiac Procedures and Malignancy Risk in Adults With Congenital Heart Disease. Circulation. 2018 Mar 27;137(13):1334-1345. [CrossRef]
- Danieli C, Cohen S, Liu A, Pilote L, Guo L, Beauchamp ME, Marelli AJ, Abrahamowicz M. Flexible Modeling of the Association Between Cumulative Exposure to Low-Dose Ionizing Radiation From Cardiac Procedures and Risk of Cancer in Adults With Congenital Heart Disease. Am J Epidemiol. 2019 Aug 1;188(8):1552-1562. [CrossRef]
Figure 1.
shows the common types of CHD from the Paediatric Cardiac Genomics Consortium, with percentages denoting the prevalence of malformation in the cohort of patients. It is important to note that the percentages exceed 100 due to coexisting structural cardiac abnormalities in the participants. Abbreviation; AVCD, atrioventricular canal defect; CHD, congenital heart disease; CTM; conotruncal malformation; LVOTO, left ventricular outflow tract obstruction; SD; septal defect. Ref [17,18].
Figure 1.
shows the common types of CHD from the Paediatric Cardiac Genomics Consortium, with percentages denoting the prevalence of malformation in the cohort of patients. It is important to note that the percentages exceed 100 due to coexisting structural cardiac abnormalities in the participants. Abbreviation; AVCD, atrioventricular canal defect; CHD, congenital heart disease; CTM; conotruncal malformation; LVOTO, left ventricular outflow tract obstruction; SD; septal defect. Ref [17,18].
Figure 2.
shows a simplified scheme of CHD defects divided according to anatomical subtype and their corresponding oxygen saturation levels. Anomalies in the connections between the left and right ventricles, such as atrial or ventricular septum defects or atrioventricular canal defects, usually lead to acyanotic states due to the left-right shunt of oxygenated blood into the pulmonary circulation. LVOTOs can range from isolated aortic stenosis to a combination of anomalies, such as hypoplastic left heart syndrome with mitral and aortic stenosis, which can cause cyanosis. Conotruncal malformations, such as tetralogy of Fallot and transposition of the great arteries, are often associated with cyanosis due to shunting of deoxygenated blood into the systemic circulation. This information is from the Paediatric Cardiac Genomics Consortium. Abbreviations; Ao, aorta, LA, left atrium; LV, left ventricle; PA, pulmonary artery; RA, right atrium; RV; right ventricle. Ref. [17,18].
Figure 2.
shows a simplified scheme of CHD defects divided according to anatomical subtype and their corresponding oxygen saturation levels. Anomalies in the connections between the left and right ventricles, such as atrial or ventricular septum defects or atrioventricular canal defects, usually lead to acyanotic states due to the left-right shunt of oxygenated blood into the pulmonary circulation. LVOTOs can range from isolated aortic stenosis to a combination of anomalies, such as hypoplastic left heart syndrome with mitral and aortic stenosis, which can cause cyanosis. Conotruncal malformations, such as tetralogy of Fallot and transposition of the great arteries, are often associated with cyanosis due to shunting of deoxygenated blood into the systemic circulation. This information is from the Paediatric Cardiac Genomics Consortium. Abbreviations; Ao, aorta, LA, left atrium; LV, left ventricle; PA, pulmonary artery; RA, right atrium; RV; right ventricle. Ref. [17,18].
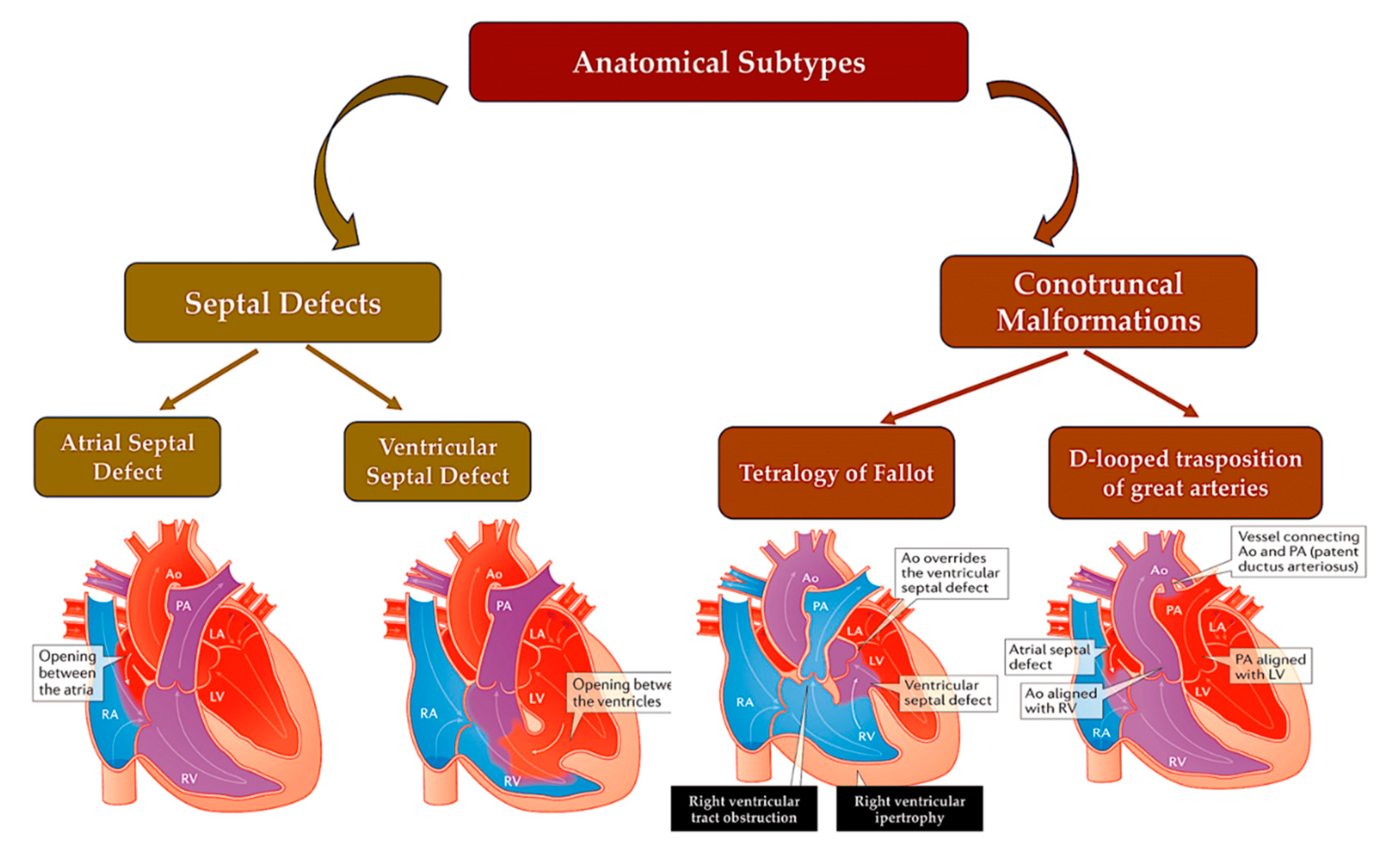
Figure 3.
Detection of genes associated with CHD in human genome. Bioinformatic analytical tools such as WES and WGS platforms can be used to identify deleterious missense and LOF variants, small insertions or deletions, CNVs and structural variants in large, aggregated, population-based sequencing datasets. Abbreviations; CHD, congenital heart disease; CNV, copy number variant; LOF, loss-of-function; WES, whole-exome sequencing; WGS, whole-genome sequencing. Ref [18,19,20,21,22,23,24,25].
Figure 3.
Detection of genes associated with CHD in human genome. Bioinformatic analytical tools such as WES and WGS platforms can be used to identify deleterious missense and LOF variants, small insertions or deletions, CNVs and structural variants in large, aggregated, population-based sequencing datasets. Abbreviations; CHD, congenital heart disease; CNV, copy number variant; LOF, loss-of-function; WES, whole-exome sequencing; WGS, whole-genome sequencing. Ref [18,19,20,21,22,23,24,25].
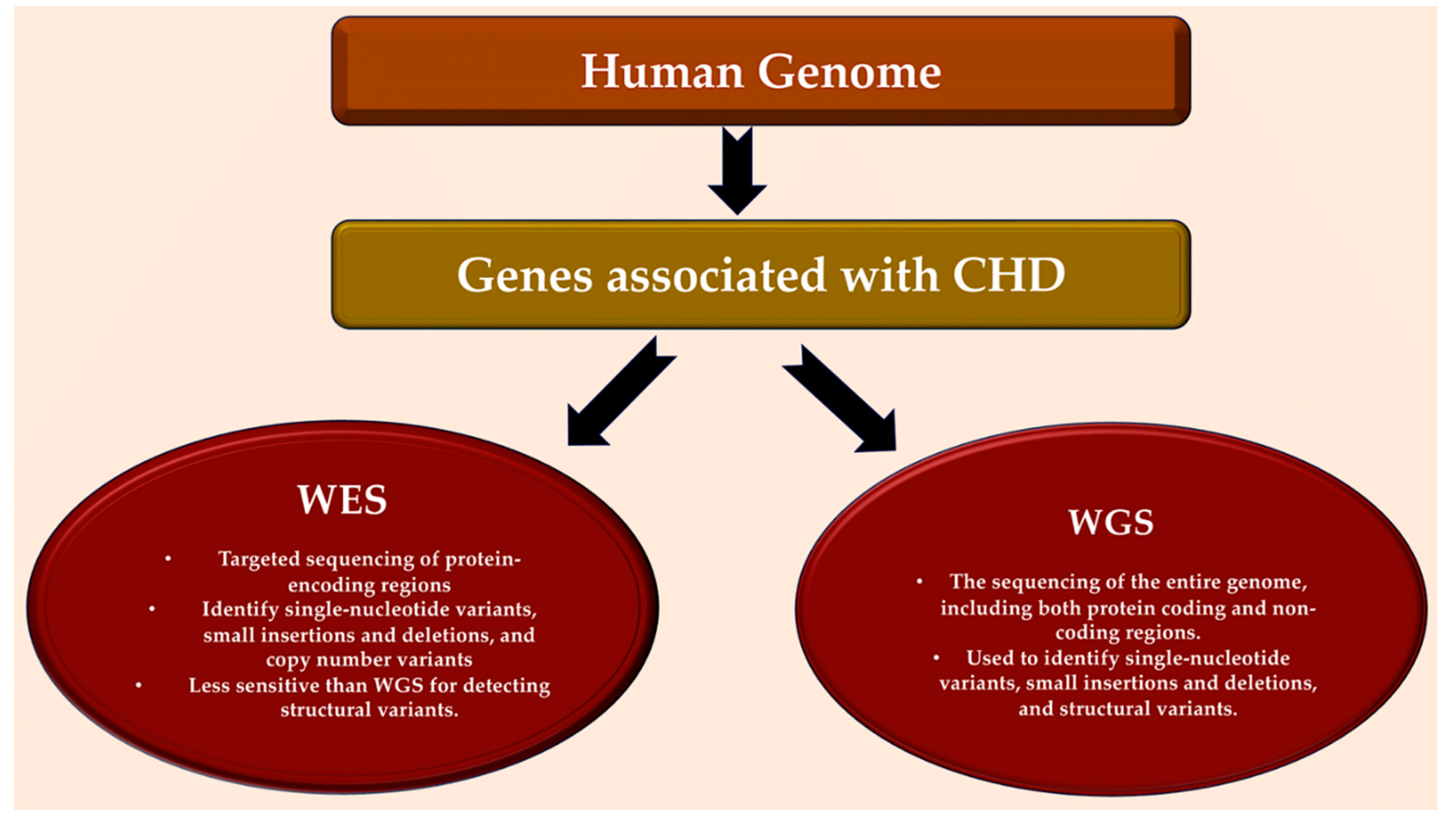
Figure 4.
The pie chart illustrates the various causes of CHD and their relative proportions in contributing to the disease. Zaidi et al [36] were the first to estimate the contribution of de novo mutations to CHD, while Jin et al [22] presented the first model to systematically investigate inherited CHD mutations. Recessive transmitted causes involve variants in cilia and known CHD genes. Dominant causes of TOF include LOFs associated with FLT4, KDR, TBX1, and NOTCH1. DNMs may involve chromatin modifiers and dominant CHD genes. Aneuploidy etiology is related to trisomy 13, 18, or 21 and monosomy X. Non-genetic causes of CHD include teratogenic exposures such as alcohol, antiseizure and antiretroviral medication, and illnesses and infectious agents such as diabetes, hypercholesterolemia, and rubella. The causes of CHD are investigated through gene-gene interaction, gene-environment interaction, polygenic inheritance, and epigenetics. CNVs, including Del22q11.2, Del20p12, Del17q11, Del11q24-25, Del8p23.1, Del1q21.1, Dup12q24, and Dup7q11.23, are also considered. Non-genetic causes of CHD are discussed in references 23,37-40. Abbreviations; CHD, congenital heart disease; CNVs, copy number variations; Del, deletion, DMS, de novo mutations; Dup, duplication; LOF, loss of function.
Figure 4.
The pie chart illustrates the various causes of CHD and their relative proportions in contributing to the disease. Zaidi et al [36] were the first to estimate the contribution of de novo mutations to CHD, while Jin et al [22] presented the first model to systematically investigate inherited CHD mutations. Recessive transmitted causes involve variants in cilia and known CHD genes. Dominant causes of TOF include LOFs associated with FLT4, KDR, TBX1, and NOTCH1. DNMs may involve chromatin modifiers and dominant CHD genes. Aneuploidy etiology is related to trisomy 13, 18, or 21 and monosomy X. Non-genetic causes of CHD include teratogenic exposures such as alcohol, antiseizure and antiretroviral medication, and illnesses and infectious agents such as diabetes, hypercholesterolemia, and rubella. The causes of CHD are investigated through gene-gene interaction, gene-environment interaction, polygenic inheritance, and epigenetics. CNVs, including Del22q11.2, Del20p12, Del17q11, Del11q24-25, Del8p23.1, Del1q21.1, Dup12q24, and Dup7q11.23, are also considered. Non-genetic causes of CHD are discussed in references 23,37-40. Abbreviations; CHD, congenital heart disease; CNVs, copy number variations; Del, deletion, DMS, de novo mutations; Dup, duplication; LOF, loss of function.
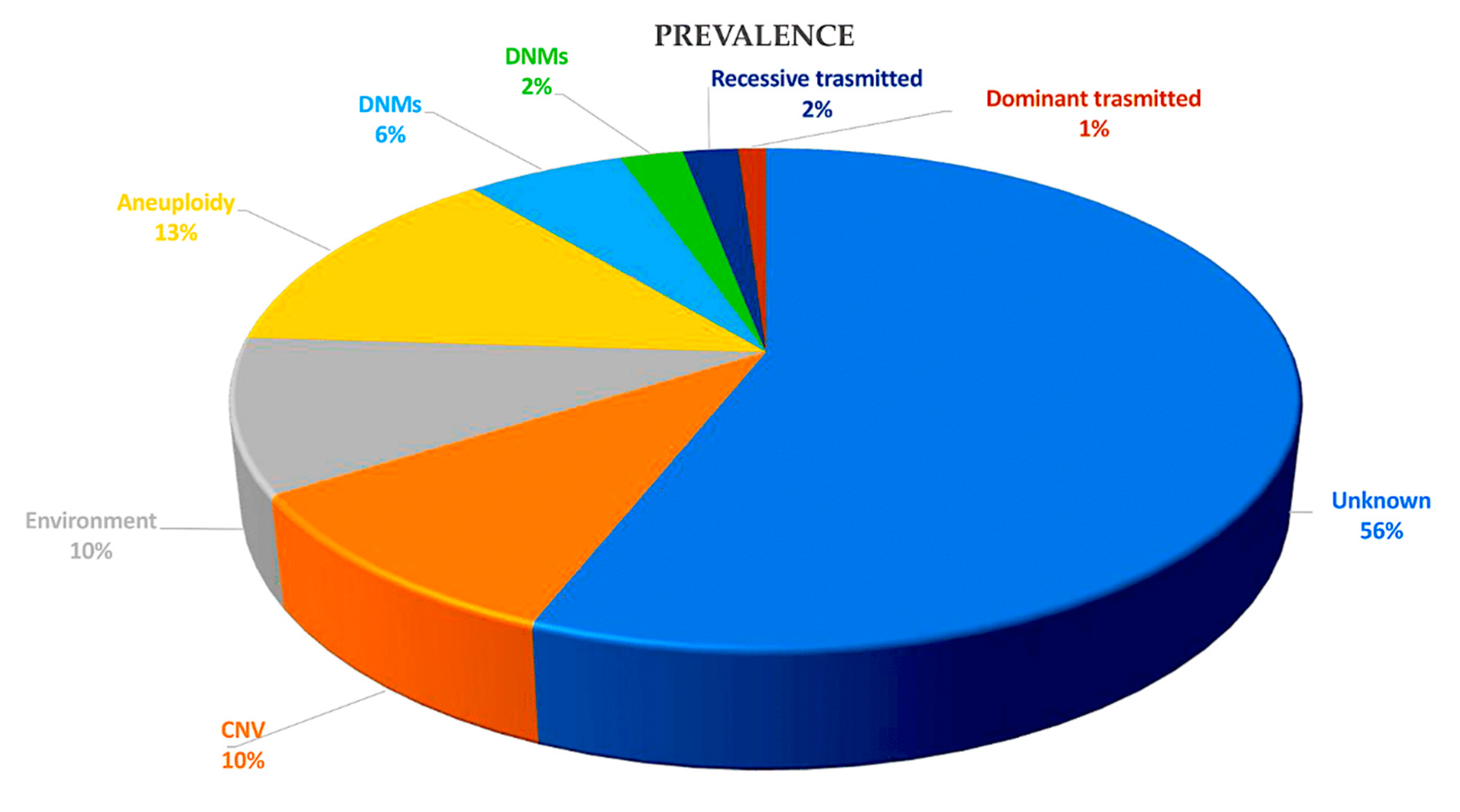
Figure 5.
This schematic illustrates the embryonic development of the human heart, including first and second heart field formation, heart tube formation and pumping, looping, neural crest migration, and septation. The process results in a fully developed heart at the end of gestation. Abbreviation: HF, heart field. Ref [41,42,43,44,45].
Figure 5.
This schematic illustrates the embryonic development of the human heart, including first and second heart field formation, heart tube formation and pumping, looping, neural crest migration, and septation. The process results in a fully developed heart at the end of gestation. Abbreviation: HF, heart field. Ref [41,42,43,44,45].
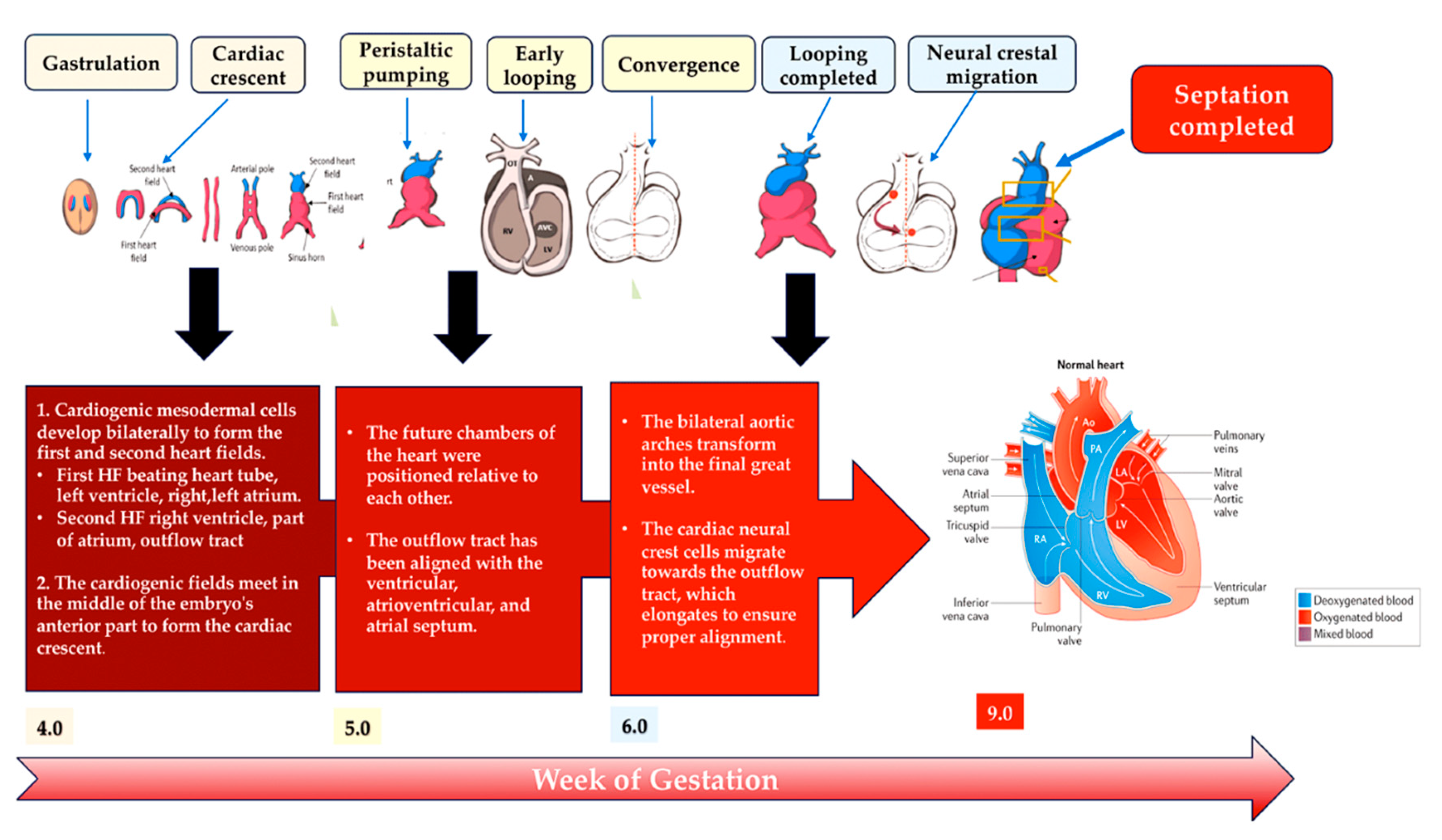
Figure 6.
The left diagram shows that two types of congenital heart defects (ASD and VSD) are linked to gene mutations in transcription factors and signaling molecules. The right diagram illustrates abnormal connections between the left and right chambers of the heart, including atrial or ventricular septal defects or atrioventricular canal defects. These defects typically do not result in cyanosis because oxygenated blood moves from the left to the right side of the heart and into the pulmonary circulation. Abbreviations; ASD, atrial septal defect; VSD, ventricular septal defect. The image was created using modified images licensed under a Creative Commons Attribution 3.0 Unported License. Ref [88,89].
Figure 6.
The left diagram shows that two types of congenital heart defects (ASD and VSD) are linked to gene mutations in transcription factors and signaling molecules. The right diagram illustrates abnormal connections between the left and right chambers of the heart, including atrial or ventricular septal defects or atrioventricular canal defects. These defects typically do not result in cyanosis because oxygenated blood moves from the left to the right side of the heart and into the pulmonary circulation. Abbreviations; ASD, atrial septal defect; VSD, ventricular septal defect. The image was created using modified images licensed under a Creative Commons Attribution 3.0 Unported License. Ref [88,89].
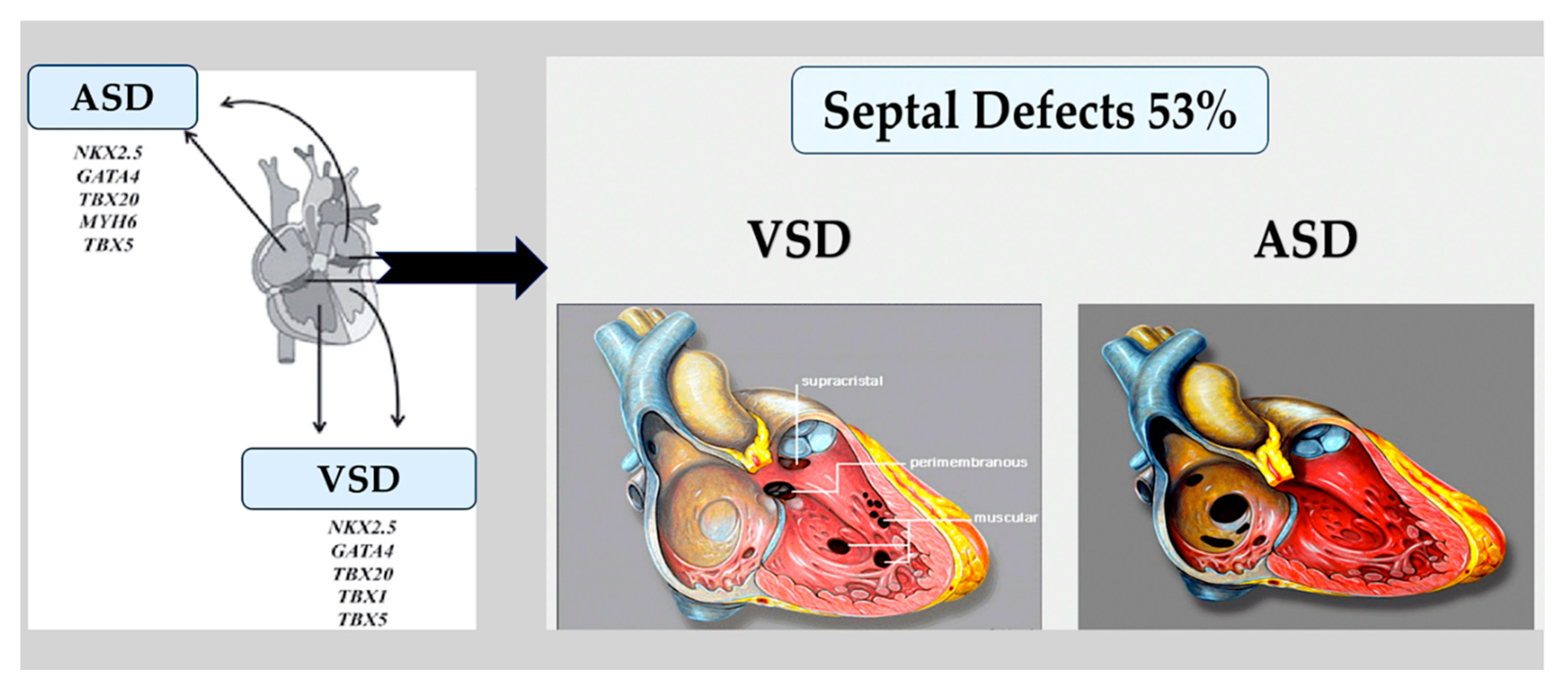
Figure 7.
Fused bicuspid aortic valve. Panel A represents a short-axis normal tricuspid aortic pattern with anatomical proximities. Panel B C and D represent three cusps fusion patterns seen in the short heart axis. All BAVs have three sinuses. Raphe structure is between the fused cusps. Non fused cusp is prominent in respect to the fused ones. B) left-noncoronary fusion pattern; C) Right-noncoronary fusion pattern; D) right-left fusion pattern. The commissure angle of the non-fused cusp has a degree<180°. L, left coronary sinus; LA, left atrium; LC, left cusp; LCA, left coronary artery; MV, mitral valve; N, non-coronary sinus; NC, non-coronary cusp; PA, pulmonary artery; R, right coronary sinus; RC, right cusp; RCA, right coronary artery; RV, right ventricle. Licenses Centre Cardiologique du Nord (with permission). License Number 5644110549132 License date Oct 08, 2023; publication NEJM; Title: Mitral valve Repair for Mitral valve prolapse. Ref [91].
Figure 7.
Fused bicuspid aortic valve. Panel A represents a short-axis normal tricuspid aortic pattern with anatomical proximities. Panel B C and D represent three cusps fusion patterns seen in the short heart axis. All BAVs have three sinuses. Raphe structure is between the fused cusps. Non fused cusp is prominent in respect to the fused ones. B) left-noncoronary fusion pattern; C) Right-noncoronary fusion pattern; D) right-left fusion pattern. The commissure angle of the non-fused cusp has a degree<180°. L, left coronary sinus; LA, left atrium; LC, left cusp; LCA, left coronary artery; MV, mitral valve; N, non-coronary sinus; NC, non-coronary cusp; PA, pulmonary artery; R, right coronary sinus; RC, right cusp; RCA, right coronary artery; RV, right ventricle. Licenses Centre Cardiologique du Nord (with permission). License Number 5644110549132 License date Oct 08, 2023; publication NEJM; Title: Mitral valve Repair for Mitral valve prolapse. Ref [91].
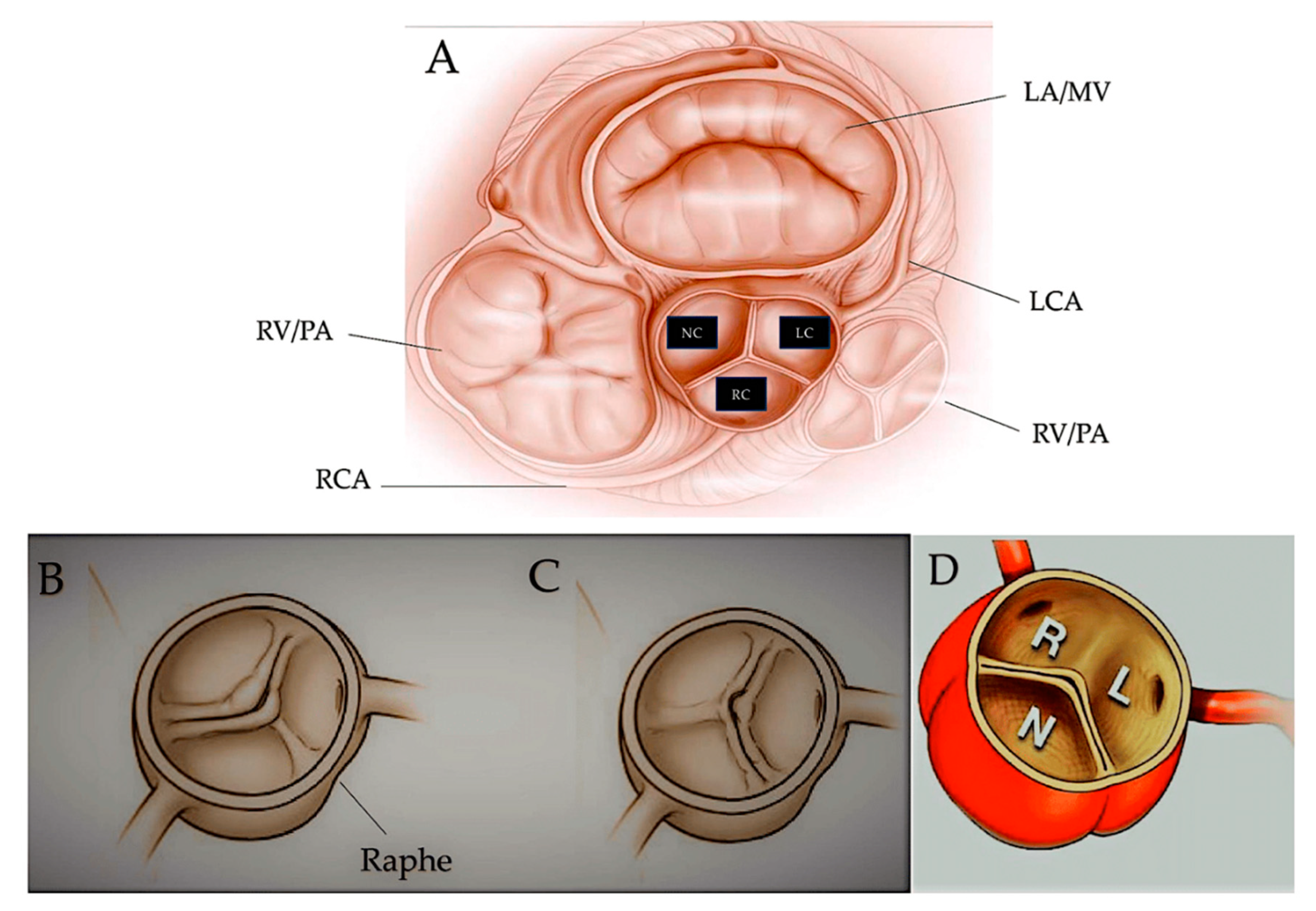
Figure 8.
Two examples of left-sided obstructive lesions, represented by a normal and a pathological aortic valve with the corresponding surgical treatment, and an aortic coarctation with the corresponding surgical approach. Left up: Fetal aortic valve stenosis is a heart condition where the aortic valve, located between the left ventricle and aorta, cannot fully open. This causes blood to flow out of the left ventricle and enlarges the right ventricle, which can lead to poor fetal growth due to changes in blood flow. Left down: The procedure performed is an extended 3-patch supravalvular aortic stenosis repair. In this procedure, the ascending aorta is transected at its narrowest point, and three incisions are made into the sinuses of Valsalva. The sinuses are then enlarged using three pericardial patches, and the patch from the noncoronary sinus is extended into the ascending aorta to ensure symmetrical enlargement of the narrow segment. Right up and down: The diagram illustrates the surgical procedure employed to treat severe aortic coarctation. Extended resection is performed, followed by end-to-end anastomosis. Abbreviations; PDA, Patent ductus Arteriosus; L.C.C, left common carotid artery; L.S.A, Left subclavian artery. The image was created using modified images licensed under a Creative Commons Attribution 3.0 Unported License. Ref [89,92].
Figure 8.
Two examples of left-sided obstructive lesions, represented by a normal and a pathological aortic valve with the corresponding surgical treatment, and an aortic coarctation with the corresponding surgical approach. Left up: Fetal aortic valve stenosis is a heart condition where the aortic valve, located between the left ventricle and aorta, cannot fully open. This causes blood to flow out of the left ventricle and enlarges the right ventricle, which can lead to poor fetal growth due to changes in blood flow. Left down: The procedure performed is an extended 3-patch supravalvular aortic stenosis repair. In this procedure, the ascending aorta is transected at its narrowest point, and three incisions are made into the sinuses of Valsalva. The sinuses are then enlarged using three pericardial patches, and the patch from the noncoronary sinus is extended into the ascending aorta to ensure symmetrical enlargement of the narrow segment. Right up and down: The diagram illustrates the surgical procedure employed to treat severe aortic coarctation. Extended resection is performed, followed by end-to-end anastomosis. Abbreviations; PDA, Patent ductus Arteriosus; L.C.C, left common carotid artery; L.S.A, Left subclavian artery. The image was created using modified images licensed under a Creative Commons Attribution 3.0 Unported License. Ref [89,92].
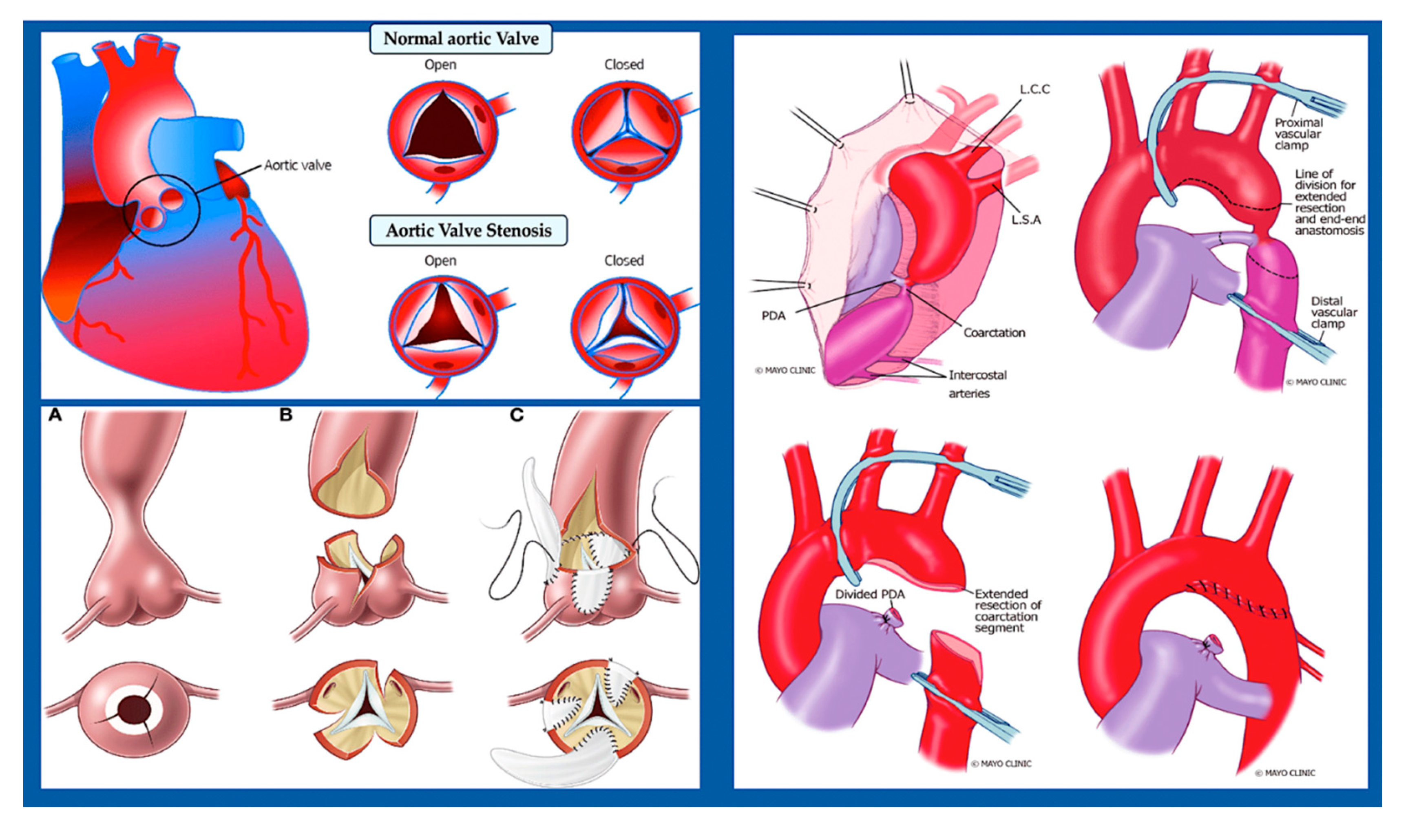
Figure 9.
The image shows a comparison between a normal heart (down) and a heart (up) affected by hypoplastic left heart syndrome. The different treatment strategies are represented. On the left: Diagram of hypoplastic left heart syndrome. The mitral and aortic valves, left ventricular cavity, and aorta are severely underdeveloped, resulting in systemic blood flow being supplied by the patent ductus arteriosus. On the right: Staged reconstruction for hypoplastic left heart syndrome. Stage I of the Norwood reconstruction involves using a modified Blalock-Taussig (BT) shunt. The second stage is the Norwood reconstruction or the Norwood reconstruction using the Sano modification. The hybrid procedure is also an option. The first stage is the Norwood reconstruction, which can be performed using a modified Blalock-Taussig (BT) shunt or the Sano modification. The final stage is the Fontan procedure. Asterisk denotes Native ascending aorta. LA stands for left atrium. LPA represents left pulmonary artery. LV stands for left ventricle. RA denotes right atrium. RPA represents right pulmonary artery. RV stands for right ventricle. SVC denotes superior vena cava. The image was created using modified images licensed under a Creative Commons Attribution 3.0 Unported License. Ref [89].
Figure 9.
The image shows a comparison between a normal heart (down) and a heart (up) affected by hypoplastic left heart syndrome. The different treatment strategies are represented. On the left: Diagram of hypoplastic left heart syndrome. The mitral and aortic valves, left ventricular cavity, and aorta are severely underdeveloped, resulting in systemic blood flow being supplied by the patent ductus arteriosus. On the right: Staged reconstruction for hypoplastic left heart syndrome. Stage I of the Norwood reconstruction involves using a modified Blalock-Taussig (BT) shunt. The second stage is the Norwood reconstruction or the Norwood reconstruction using the Sano modification. The hybrid procedure is also an option. The first stage is the Norwood reconstruction, which can be performed using a modified Blalock-Taussig (BT) shunt or the Sano modification. The final stage is the Fontan procedure. Asterisk denotes Native ascending aorta. LA stands for left atrium. LPA represents left pulmonary artery. LV stands for left ventricle. RA denotes right atrium. RPA represents right pulmonary artery. RV stands for right ventricle. SVC denotes superior vena cava. The image was created using modified images licensed under a Creative Commons Attribution 3.0 Unported License. Ref [89].
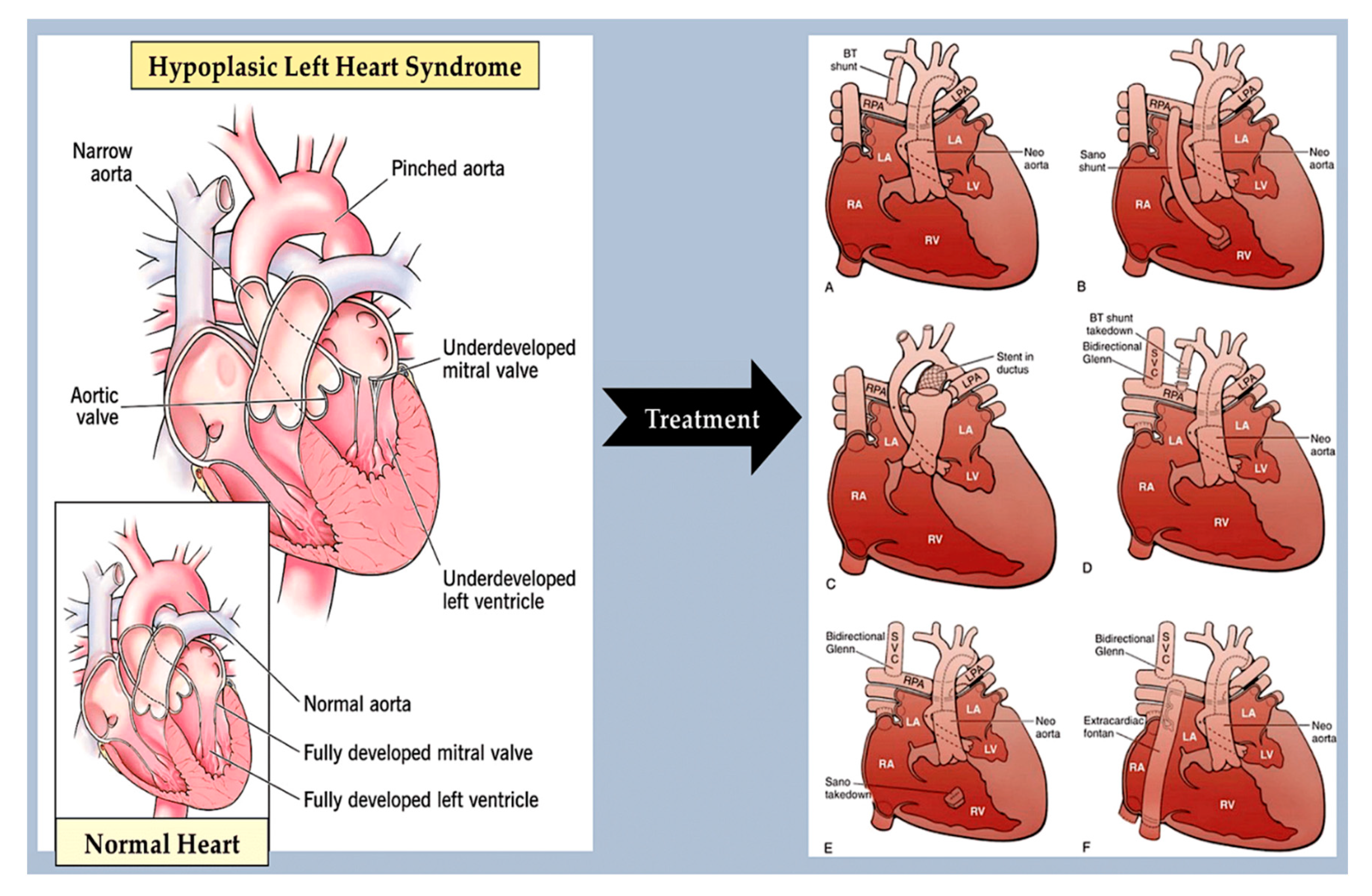
Figure 10.
A: The figure displays a Venn diagram that shows the overlap of genes associated with LSOLs based on disease subtype. Genes without robust evidence of association are marked with an asterisk. B: shows the genetic landscape of left-sided obstructive lesions. The genes marked with an asterisk have no robust evidence of association. The genes code for transcription factors, structural or contractile proteins, and cell signaling components. Abbreviations; AS, aortic stenosis; BAV, bicuspid aortic valve; CoA, coarctation of the aorta; HLHS, hyperplastic left heart syndrome; IAA, interrupted aortic arch; LSOL; left-sided obstructive lesion. The image was created using modified images licensed under a Creative Commons Attribution 3.0 Unported License.
Figure 10.
A: The figure displays a Venn diagram that shows the overlap of genes associated with LSOLs based on disease subtype. Genes without robust evidence of association are marked with an asterisk. B: shows the genetic landscape of left-sided obstructive lesions. The genes marked with an asterisk have no robust evidence of association. The genes code for transcription factors, structural or contractile proteins, and cell signaling components. Abbreviations; AS, aortic stenosis; BAV, bicuspid aortic valve; CoA, coarctation of the aorta; HLHS, hyperplastic left heart syndrome; IAA, interrupted aortic arch; LSOL; left-sided obstructive lesion. The image was created using modified images licensed under a Creative Commons Attribution 3.0 Unported License.
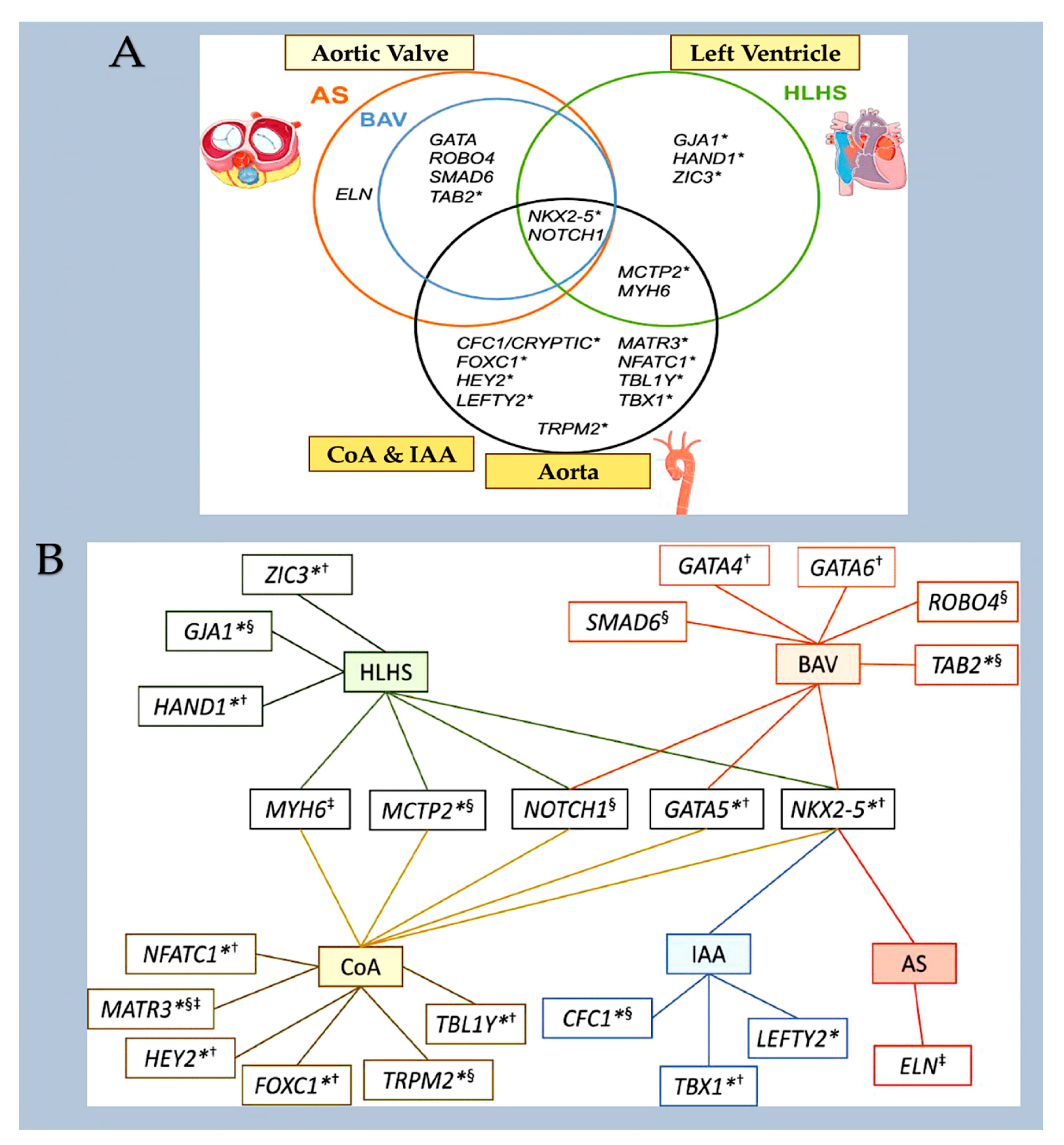
Figure 11.
Germline mutations that activate RAS/MAPK signalling cause the "RASopathies". These are a group of rare human developmental disorders affecting over 400,000 individuals in the United States alone. Costello syndrome (CS) is a syndrome of multiple congenital anomalies. It is caused by heterozygous activating germline mutations in HRAS [1]. Most individuals with CS have a mutation in HRAS at position G12, with over 80% having a p.G12S substitution. Children with CS typically present with increased birth weight, dysmorphic craniofacial features, failure to thrive, and gastroesophageal reflux with oral aversion, especially in the neonatal period. CS can also affect the skin, causing excessive wrinkling and redundancy over the dorsum of the hands and feet, and deep plantar and palmar folds. In addition, individuals with CS have an increased risk of developing benign or malignant tumours. From Dard et al. J Clin Invest. 2022 Apr 15;132(8):e131053.
Figure 11.
Germline mutations that activate RAS/MAPK signalling cause the "RASopathies". These are a group of rare human developmental disorders affecting over 400,000 individuals in the United States alone. Costello syndrome (CS) is a syndrome of multiple congenital anomalies. It is caused by heterozygous activating germline mutations in HRAS [1]. Most individuals with CS have a mutation in HRAS at position G12, with over 80% having a p.G12S substitution. Children with CS typically present with increased birth weight, dysmorphic craniofacial features, failure to thrive, and gastroesophageal reflux with oral aversion, especially in the neonatal period. CS can also affect the skin, causing excessive wrinkling and redundancy over the dorsum of the hands and feet, and deep plantar and palmar folds. In addition, individuals with CS have an increased risk of developing benign or malignant tumours. From Dard et al. J Clin Invest. 2022 Apr 15;132(8):e131053.
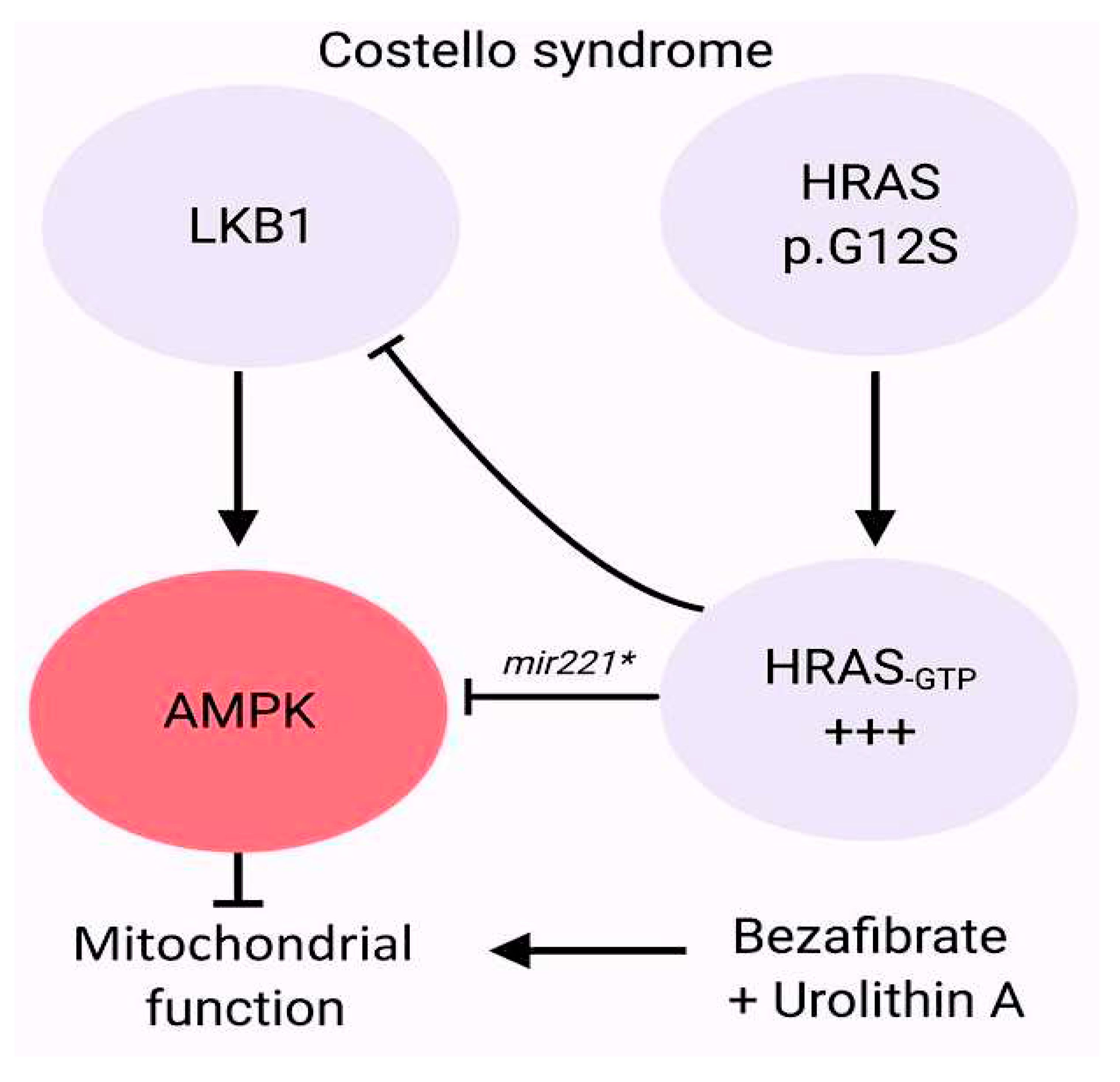
Figure 12.
The diagram illustrates a type of obstructive lesion on the right side. PA/IVS is an abnormality in which there is complete closure of the communication from the RV to the PA and no ventricular septal defect. PA/IVS is a disorder with varying morphology. The RV is often small or hypoplastic, or it can be significantly dilated, as seen in association with Ebstein’s anomaly or TV dysplasia and severe TV regurgitation. Although extracardiac anomalies are unusual in PA/IVS, many different cardiac anomalies can be observed. PA/IVS is typically associated with abnormalities of the right side of the heart, including dysplastic TV leaflets and thickened chordae that may cause severe regurgitation in utero. Atrial septal defects are also common. PA/IVS is typically associated with abnormalities of the right side of the heart, including dysplastic TV leaflets and thickened chordae that may cause severe regurgitation in utero. It is worth noting that the TV annulus is often small in many cases. In some cases, the tricuspid valve may be fully sealed off, causing tricuspid atresia or, more commonly, it may be moderately to severely underdeveloped, resulting in tricuspid stenosis. Especially in the setting of a hypertrophied, hypertensive RV with little or no tricuspid regurgitation, coronary abnormalities are common in PA/IVS. An inverse relationship exists between RV size and coronary anomalies - the smaller the RV and tricuspid annulus, the greater the likelihood of coronary anomalies. Abbreviations; PAPA/IVS, Pulmonary atresia with intact ventricular septum; PA, pulmonary artery; RV, right ventricle; TV, tricuspid valve. The image was created using modified images licensed under a Creative Commons Attribution 3.0 Unported License. Ref [89].
Figure 12.
The diagram illustrates a type of obstructive lesion on the right side. PA/IVS is an abnormality in which there is complete closure of the communication from the RV to the PA and no ventricular septal defect. PA/IVS is a disorder with varying morphology. The RV is often small or hypoplastic, or it can be significantly dilated, as seen in association with Ebstein’s anomaly or TV dysplasia and severe TV regurgitation. Although extracardiac anomalies are unusual in PA/IVS, many different cardiac anomalies can be observed. PA/IVS is typically associated with abnormalities of the right side of the heart, including dysplastic TV leaflets and thickened chordae that may cause severe regurgitation in utero. Atrial septal defects are also common. PA/IVS is typically associated with abnormalities of the right side of the heart, including dysplastic TV leaflets and thickened chordae that may cause severe regurgitation in utero. It is worth noting that the TV annulus is often small in many cases. In some cases, the tricuspid valve may be fully sealed off, causing tricuspid atresia or, more commonly, it may be moderately to severely underdeveloped, resulting in tricuspid stenosis. Especially in the setting of a hypertrophied, hypertensive RV with little or no tricuspid regurgitation, coronary abnormalities are common in PA/IVS. An inverse relationship exists between RV size and coronary anomalies - the smaller the RV and tricuspid annulus, the greater the likelihood of coronary anomalies. Abbreviations; PAPA/IVS, Pulmonary atresia with intact ventricular septum; PA, pulmonary artery; RV, right ventricle; TV, tricuspid valve. The image was created using modified images licensed under a Creative Commons Attribution 3.0 Unported License. Ref [89].
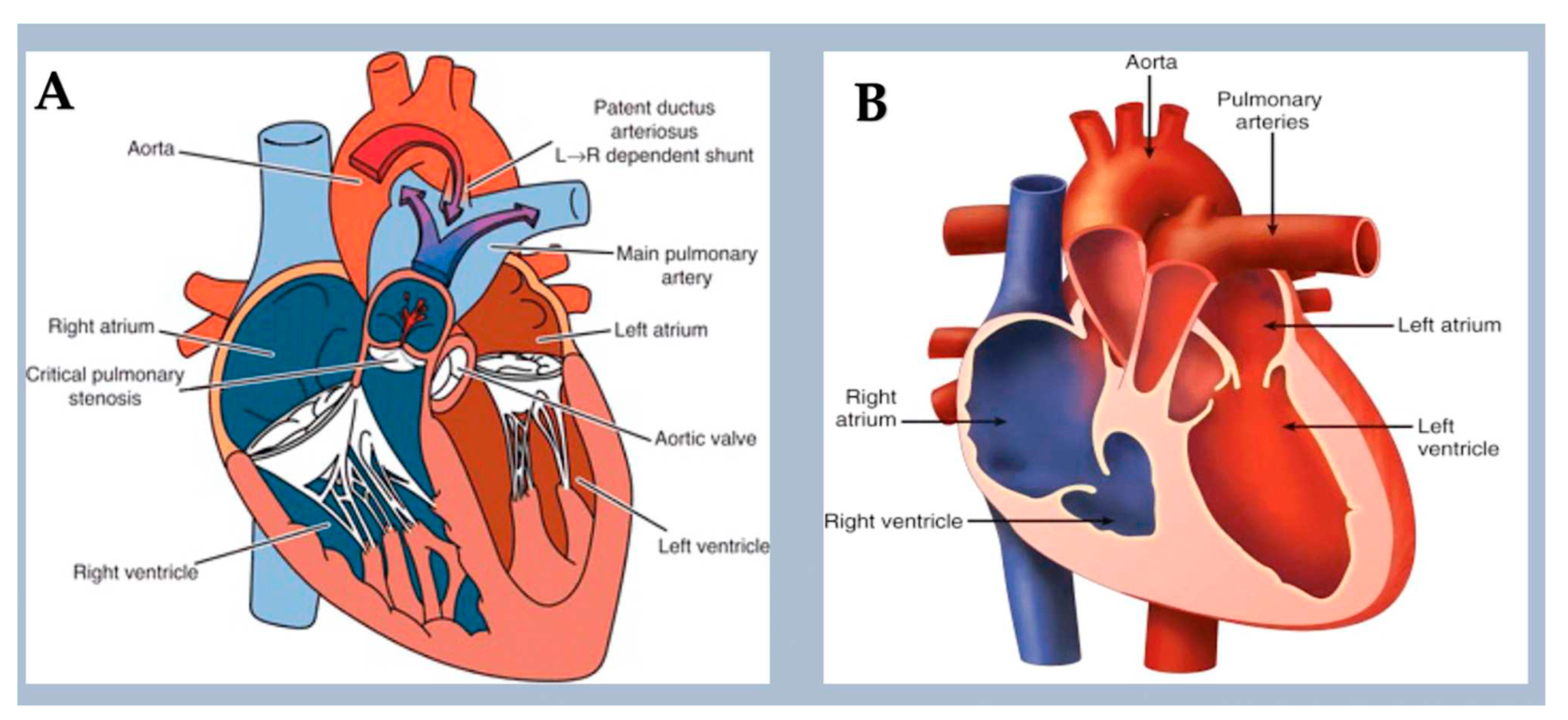
Figure 13.
Conotruncal defects include conditions such as tetralogy of Fallot, persistent truncus arteriosus and transposition of the great arteries, which are caused by underdevelopment or malposition of the ventricular septum, outflow tract and/or great arteries. Conotruncal malformations often result in cyanosis due to the shunting of deoxygenated blood into the systemic circulation. The image was created using modified images licensed under a Creative Commons Attribution 3.0 Unported License. Ref [89].
Figure 13.
Conotruncal defects include conditions such as tetralogy of Fallot, persistent truncus arteriosus and transposition of the great arteries, which are caused by underdevelopment or malposition of the ventricular septum, outflow tract and/or great arteries. Conotruncal malformations often result in cyanosis due to the shunting of deoxygenated blood into the systemic circulation. The image was created using modified images licensed under a Creative Commons Attribution 3.0 Unported License. Ref [89].
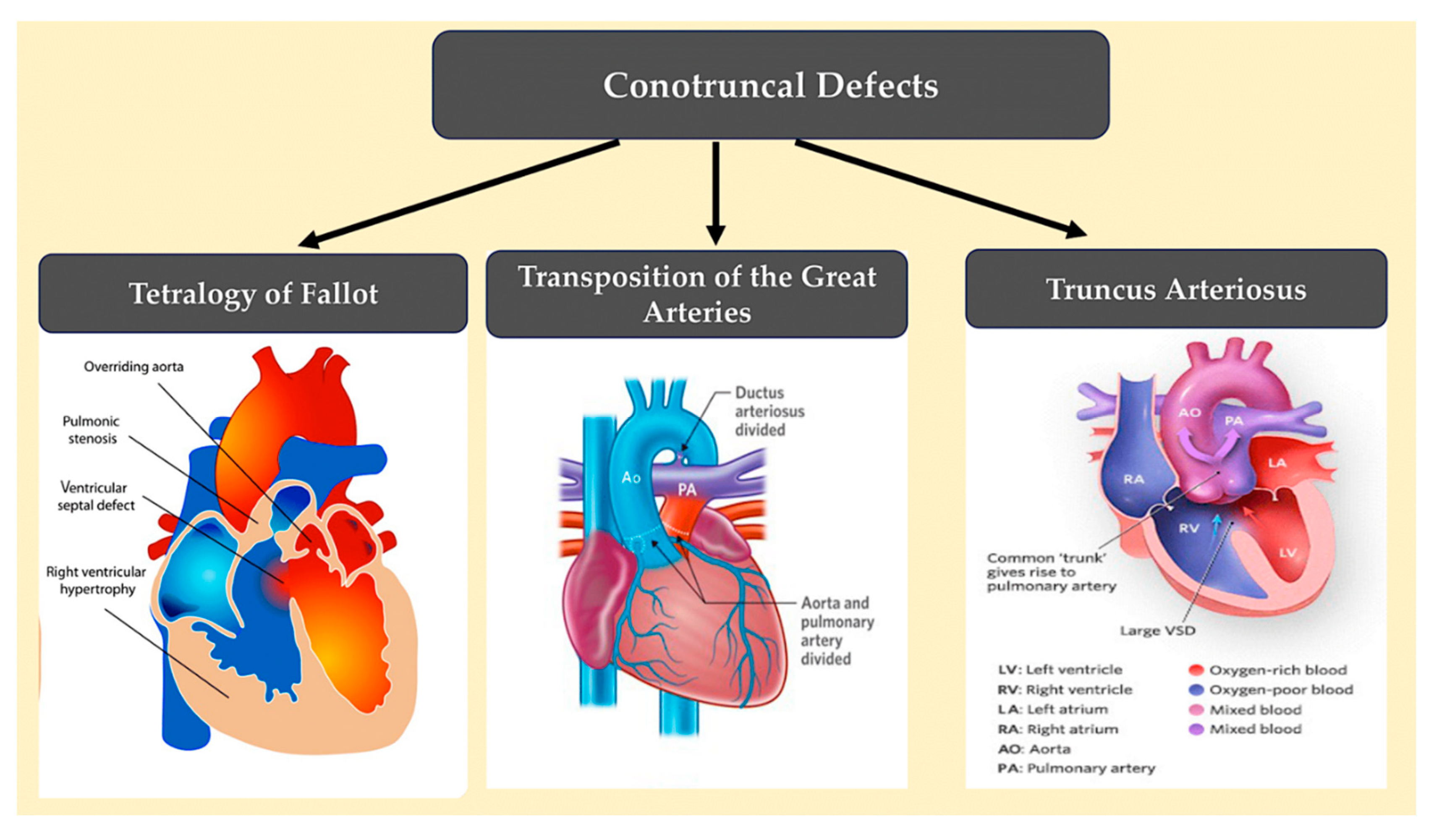
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



