Submitted:
29 December 2023
Posted:
04 January 2024
You are already at the latest version
Abstract
Resistance to therapy and disease progression are the main causes of mortality in most cancers. In particular, the development of resistance is an important limitation affecting the efficacy of therapeutic alternatives for cancer, including chemotherapy, radiotherapy, and immunotherapy. Signaling pathways are largely responsible for the mechanisms of resistance to cancer treatment and progression, and multiple myeloma is no exception. p38 mitogen-activated protein kinase (p38) is downstream of several signaling pathways specific to treatment resistance and progression. Therefore, in recent years, developing therapeutic alternatives directed at p38 MAPK has been of great interest, in order to reverse chemotherapy resistance and prevent progression. In this review, we discuss recent findings on the role of p38, including recent advances in our understanding of its expression and activity as well as its isoforms, and its possible clinical role based on the mechanisms of resistance and progression in multiple myeloma.
Keywords:
multiple myeloma
; p38 mapk
; transcriptional factor
; cancer progression
; clinical implication
; gene expression
1. Introduction
Multiple myeloma (MM) is a hematologic malignancy that remains incurable, as most patients eventually relapse or become refractory to treatment [1]. Even though recent treatments have been improved, resistance to treatment persists in MM [2]. New therapeutic agents have recently been developed for the treatment of resistant or refractory MM, including immunomodulatory agents, proteasome inhibitors, monoclonal antibodies, and therapies directed at molecular targets in different signaling pathways. These therapeutic alternatives have shown relative antitumor activity in resistant and refractory MM, and recent combinations of these therapies have shown clinical effectiveness [3]. Recent studies have focused on therapies directed at molecular targets in refractory or relapsed MM, which are based on signaling pathways specifically activated in tumor cells, as is the case of p38 [3]. Mitogen-activated protein kinases (MAPK), allow cells to respond to a wide variety of stimuli and signals, such as DNA damage or inflammatory cytokine activity, and extracellular stimuli such as oxidative and osmotic stress and heat shock [4,5]. p38 has four well-characterized isoforms: p38α, β, γ, δ [6]. These four isoforms participate in the translation of signals that play a very important role in different cellular processes, such as cell proliferation, differentiation, glucose metabolism, and lipid secretion, senescence, stress response, apoptosis, autophagy, and cell migration [7,8,9]. Depending on the context, p38 can act as a tumor promoter or tumor suppressor [7,10]. p38 is constitutively activated in MM and has been implicated in osteoclast and osteoblast activity and bone destruction [11]. Recent studies have shown that treatment with bortezomib can induce p38 activation, and this activation may be related to chemoresistance [12,13]. However, other studies show that p38 activation is associated with the induction of apoptosis and autophagy in MM [14]. In this review, we discuss recent advances in our understanding of the regulation and expression of p38, as well as its activity in MM and its possible role as a therapeutic target, and the clinical implications. To construct the introduction of this review, general bibliographies of the molecular aspects of p38 were consulted, as well as review articles from the last 20 years. For references on the medullary part of p38 in MM, articles from the last 10 years were consulted with the keyword p38 in MM, inhibitors of p38 in MM and p38 regulation mechanism. In the PubMed and Google scholar databases in English. 80 publications found in the last 10 years with the words p38 in MM were analyzed and their content was analyzed and only 5 that did not highlight the importance of p38 in MM were discarded. 95 publications of p38 inhibitors in MM were found, and 45 that highlighted the importance of p38 and its inhibition in the response of MM were analyzed and considered.
2. p38 molecular signaling
The p38 family is a family of specific protein serine/threonine kinases characterized by a dual Thr-Gly-Tyr phosphorylation motif. Four isoforms have been identified, each encoded by four genes with high sequence homology: MAPK14, MAPK11, MAPK12, and MAPK13, which code for p38α, p38β, p38γ, and p38δ, respectively [15]. They share 75% homology at the amino acid level between p38α and p38β and between p38γ and p38δ [16]. On the other hand, p38γ shares 62% homology with p38α, whereas p38δ shares 61% homology with p39α [6,17]
p38α is one of the four isoforms that is expressed in all cells and tissues, so it is believed that it plays a very important role in the different cellular signaling cascades triggered by extracellular stimuli such as stress, proinflammatory cytokines or direct transcription activation. P38α act as integration points for multiple biochemical signals and are involved in a wide variety of cellular processes, including proliferation, differentiation, regulation of transcription, and development. This kinase is activated by various environmental stresses and proinflammatory cytokines. Its activation requires phosphorylation by MAP kinase kinases (MKKs), or autophosphorylation triggered by its interaction with the MAP3K7IP1/TAB1 protein. [18,19].
Phosphorylation of p38α can activate a wide range of stimuli, such as transcription factors, protein kinases, cytoplasmic substrates, and nuclear substrates [20]. Unlike p38α, p38β is mainly expressed in the brain [21]. Although p38α and p38β have been described as having shared functions, p38β is less expressed in different cell types. In general, p38α and p38β participate in cellular processes such as cell proliferation and differentiation, glucose and lipid metabolism, secretion, stress response, apoptosis, autophagy, and cell migration [9]. Under environmental stress, p38β is activated by phosphorylation mediated by MAP2K3/MKK3, MAP2K4/MKK4, and MAP2K6/MKK6. In addition, p38β selectively interacts with histone deacetylase HDAC3 in order to repress ATF2 activity, regulating TNF expression after LPS stimulation [22].
p38γ is mainly expressed in the skeleton and p38δ in the liver, pancreas, small intestine and testes [23,24]. Therefore, their expression is more restricted, which is why they are believed to have more specialized functions [5]. There is currently no evidence that the p38γ and p38δ isoforms perform the same functions as p38α; However, in certain cell lines, these isoforms carry out the same functions of tissue regeneration and immune response [16]. Although downregulation of p38α has been shown to induce the expression of p38γ and p38δ, the individual or combined function of the four p38 isoforms has not been described in detail [25]. p38γ signaling also positively regulates the expansion of transient amplifying myogenic precursor cells during muscle growth and regeneration [22,23,26]. p38δ is another of the four p38s that plays an important role in cascades of cellular responses elicited by extracellular stimuli such as proinflammatory cytokines or physical stress, leading to direct activation of transcription factors [27,28,29]. p38δ is one of the least studied isoforms of p38, its downstream targets include MAPKAPK2, which is phosphorylated and, in turn, phosphorylates subsequent targets. p38δ has been described to have a role in the regulation of protein translation through regulation of EEF2K protein, as well as in cytoskeletal remodeling through activation of MAPT and STMN1.
In the tumor context, the expression of the four isoforms varies and depends on the type of cancer. For example, significant levels of expression of p38α and p38δ have been reported in primary tumors of most cancers, while p38β and p38γ have significantly decreased expression in primary tumors of breast cancer, lung adenocarcinoma, and glioblastoma multiforme [9]. Other studies reported that non-Hodgkin lymphoma (NHL) patient samples and cell lines showed significant p38α expression correlated with malignancy [30].
Studies have shown that p38α induces the activation of pro-inflammatory and apoptosis-inhibiting signals, such as IL-6, IL-8 and IL12b, promoting cell survival and resistance to chemotherapy such as in MM, allowing post-treatment DNA repair [31]. Studies have reported that p38 also plays a role in cell invasion, by inducing epithelial mesenchymal trans differentiation [32], and inhibiting the matrix metalloproteins MMP-2 and MPP-9, which promotes metastasis and tumor invasion. Additionally, it has been shown that p38 promotes cell migration after induction of VEGF expression. [33,34].
2.1. MAPK p38 structure
MAPK14 (p38α): This gene is located on the short arm of chromosome 6 (6p21.31), (GRCh38/hg38by Entrez Gene) and consists of 96,433 base pairs in plus-strand orientation. It encodes a 360-amino-acid protein (MW = 41,293 Da) (NCBI Entrez Gene 1432). In its sequence it contains a protein kinase domain at residue 24-308 and a TXY phosphorylation motif at position 180-182 (Figure 1). There are two ATP binding sites at positions 30-38 and 53. Several post-translational modifications have been identified, including dual phosphorylation on Thr-180 and Tyr-182 by MAP2K3/MKK3, MAP2K4/MKK4, and MAP2K6/MKK6. Dual phosphorylation can also be mediated by TAB1-mediated autophosphorylation. TCR engagement in T-cells also leads to Tyr-323 phosphorylation by ZAP70. It is dephosphorylated and inactivated by DUPS1, DUSP10, DUSP16, and PPM1D [35]. It is acetylated at Lys-53 and Lys-152 by KAT2B and EP300. Acetylation at Lys-53 increases the affinity for ATP and enhances kinase activity. Lys-53 and Lys-152 are deacetylated by HDAC3. Ubiquitination occurs at Lys15, Lys45, Lys139, Lys152, Lys165, and Lys233 [36]. Five isoforms produced by alternative splicing have been reported: Q16539-1 (considered the canonical) to Q16539-5, with variant 4 being shorter (307 residues) [37,38,39,40]. Specific activation of isoform Q16539-3 is measured by mitogen stimulation and oxidative stress, while isoform Q16539-4 may play a role in the early initiation of apoptosis [41]. Its subcellular localization is in the cytosol and nucleus [42], with tissue expression predominantly in brain, heart, placenta, pancreas, and skeletal muscle, and less expression in lung, liver, and kidney (Expression Atlas [Q16539]).
MAPK11(p38β): This gene is located on the long arm of chromosome 22 (22q13.13) (GRCh37/hg19 by Entrez Gene) and consists of 7,055 base pairs in minus-strand orientation. It encodes a protein of 364 amino acids (MW = 41,357 Da) (NCBI Entrez Gene 5600). In its sequence it contains a protein kinase domain at residue 24-308 and a TXY phosphorylation motif at position 180-182 (Figure 1). There are two ATP binding sites at positions 30-38 and 53. Different post-translational modifications have been identified, such as dual phosphorylation on Thr-180 and Tyr-182 by MAP2K3/MKK3, MAP2K4/MKK4, and MAP2K6/MKK6 [43].
Two isoforms produced by alternative splicing have been reported: Q15759-1, considered the canonical, and Q15759-3, which is the shortest (213 residues) [44]. Its subcellular localization is in the cytosol and nucleus, with tissue expression predominantly in brain, heart, placenta, lung, liver, pancreas, kidney, and skeletal muscle (Q15759.
MAPK12 (p38γ): This gene is located on the long arm of chromosome 22 (22q13.33) (GRCh38/hg38 by Entrez Gene) and consists of 16,267 base pairs in minus-strand orientation. It encodes a protein of 367 amino acids (MW = 41,940 Da) (NCBI Entrez Gene P53778). In its sequence it contains a protein kinase domain at residue 27-311 and a TXY phosphorylation motif at position 183-185. There are two ATP-binding sites at positions 33-41 and 56 and one active site at residue 153 (Figure 1). Post-translational modifications have been identified, including dual phosphorylation on Thr-183 and Tyr-185 by the MAP2Ks MAP2K3/MKK3 and MAP2K6/MKK6 and ubiquitination for its degradation has been reported [23]. Two isoforms produced by alternative splicing have been reported: P53778-1, considered the canonical, and P53778-2, which is the shortest (357 residues, lacking residues 142-151) [45] (Figure 1). Its subcellular location is in the cytosol, nucleus, and mitochondria, with tissue expression predominantly in skeletal muscle and the heart [46,47].
MAPK13 (P38δ): This gene is located on the short arm of chromosome 6 (6p21.31) (GRCh38/hg38 by Entrez Gene) and consists of 16,716 base pairs in plus-strand orientation. It encodes a protein of 365 amino acids (MW = 42,090 Da) (NCBI Entrez Gene O15264). In its sequence it contains a protein kinase domain at residue 25-308 and a TXY phosphorylation motif at position 180-182. There are two ATP binding sites at position 31-39 and 54. Post-translational modifications have been identified, including dual phosphorylation on Thr-180 and Tyr-182 by MAP2K3/MKK3, MAP2K4/MKK4, MAP2K6/MKK6, and MAP2K7/MKK7. Two isoforms produced by alternative splicing have been reported: O15264-1, considered the canonical, and O15264-2, which is the shortest (257 residues, lacking residues 257-364) [48] (Figure 1). Its subcellular location is in the cytoplasm, cytosol, and nucleus, with tissue expression predominantly in testis, pancreas, small intestine, lung, and kidney.
2.2. P38 regulation
2.2.1. Transcription factors in the regulation of p38
As previously mentioned, the regulation of p38 can occur by various conditions, kinases, and transcription factors [15]. Zarubin et al. reported that regulation of p38 can occur by extracellular stimuli such as osmoregulation, heat, ultraviolet light, inflammatory cytokines such as TNF, growth factors such as CSF-1, or kinases such as MKK3 and MKK6, among other factors [10]. However, the participation of transcription factors is still a growing subject of study. Previously, a series of factors was suggested that might have a regulatory role, such as ATF-1 and 2, Elk-1, SRF, and CHOP-10, among others [49]. Downstream, the regulation of transcription factors such as YY1 and BCL6 by p38 has been reported [50], and several authors have referred to MEF2 as a p38 regulation target [51,52].
In this review, we analyzed the possible participation of transcription factors in the regulation of p38 using bioinformatic tools. Although they are not involved in the main mechanism of kinase regulation, they may be an important part of generating this at the transcriptional level. They may explain why the regulation of certain transcription factors affect the expression of p38, which becomes important for therapeutic purposes. We analyzed the promoter regions of the four isoforms of p38, namely, p38α (MAPK14), p38β (MAPK11), p38γ (MAPK12), and p38δ (MAPK13) [53], using the "search motif” tool of the EPD database (available at https://epd.epfl.ch/; accessed June 17, 2022). The region from -1000 to 100 relative to the transcription start site of promoters ID MAPK14_1, MAPK11_1, MAPK12_1, and MAPK13_1 was analyzed, which, according to Ensembl (https://www.ensembl.org), corresponds to p38 with a cut-off value of p= 0.001.
Transcription factors with at least one binding site in the mentioned region are listed in Table 1, including a selection of those that have been reported as relevant in hematological malignancies.
The assay allows us to assume that transcriptional regulation of p38 could be related to several malignant processes, given the nature of the transcription factors involved; however, it is necessary to experimentally validate the participation of each transcription factor. Recognizing that further research is required, here we describe some reported findings related to the participation of these transcription factors in the regulation of p38.
2.2.2. Non-coding RNAs in the regulation of p38
Interestingly, and contrary to what is observed with transcription factors, regulation of p38 by microRNAs has been studied in various pathologies.
Studies indicate that long non-coding RNAs (lncRNAs) play an important role in the pathophysiology of MM [54]. Recent studies have shown that prostate cancer-associated ncRNA transcript 1 (PCAT-1) plays an important role in the pathophysiology of MM and has a regulatory role in p38, where it induces an increase that correlates with proliferation and chemoresistance [55]. Therefore, regulating PCAT-1 could be an important therapeutic target in the context of regulating p38 in MM. Recent studies of glioblastoma have shown that the LncRNA small nucleolar RNA host gene 5 (SNHG5) promotes p38 protein and induces its activation through phosphorylation [56].
p53-induced noncoding transcript (PINT) is a long intergenic non-coding RNA (linc-RNA). A recent study reveals that nuclear PINT increases gene expression of the MAPKinase pathways [57]. Since p38 is related to treatment responses in MM, PINT likely increases p38 expression in MM cells and may activate a MKK6/p38 signaling kinase stress cascade in MM patients. Studies have shown that there is significant expression of miR-106b/25 cluster in MM cells [58]. It is a pro-oncogene, and interestingly, additional studies have shown that this cluster positively regulates p38 activation in MM. Therefore, inhibiting miR-106b/25 cluster and, as a result, p38, has been considered as a therapeutic alternative in MM [59].
In this review we used the miRtarbase (https://mirtarbase.cuhk.edu.cn/) to determine some of the miRNAs that probably regulate p38 (Table 2). microRNAs with a possible role in the regulation of MAPK14 were found: hsa-miR-124-3p, hsa-miR-24-3p, hsa-miR-199a-3p, hsa-miR-200a-3p, hsa-miR-141-3p, hsa-miR-125b-5p, hsa-miR-214-3p, hsa-miR-155-5p, hsa-miR-17-5p, and hsa-miR-106a-5p. The microRNAs found for MAPK11 were hsa-miR-122-5p, hsa-miR-124-3p, and hsa-let-7a-5p, and those for MAPK13 were hsa-miR-18a-5p, hsa-miR-150-5p, hsa-miR-18b-5p, hsa-miR-3134, hsa-miR-3691-5p, hsa-miR-4434, hsa-miR-4516, hsa-miR-4525, hsa-miR-4531, hsa-miR-4534, hsa-miR-4690-3p, hsa-miR-4731-5p, hsa-miR-4735-3p, hsa-miR-4761-5p, hsa-miR-4773, hsa-miR-5010-5p, hsa-miR-5187-5p, hsa-miR-5589-5p, hsa-miR-5685, hsa-miR-5703, hsa-miR-6778-3p, hsa-miR-6795-5p, hsa-miR-6798-5p, hsa-miR-6814-5p, hsa-miR-6887-5p, and hsa-miR-8082. No miRNAs were found for the isoform MAPK12.
To further confirm the correlation of the miRNAs/p38 axis, we used the Bio-predictive website (http://www.targetscan.org/vert_72/) (Table 2). TargetScan in conserved sites found possible binding involvement of the miRNAs hsa-miR-3681-3p, hsa-miR-128-3p, and has-miR-216-3p for the isoform MAPK14, while no conserved sites were found for MAPK13. According to TargetScan there are three conserved miRNA sites for MAPK12: hsa-miR-125a-5p, has-miR-125b-5p, and has-miR-4319. Finally, only one conserved site was found for MAPK11: hsa-miR-325-3p.
Studies of gastric adenocarcinoma showed that miR-141 could activate the p38 signaling pathway [60]. Additionally, studies have reported that miR-141 could modulate the response to oxidative stress and stimulate tumor growth in mouse models through direct regulation of p38α, which restricted tumorigenesis by blocking proliferation and promoting apoptosis [61,62]. Therefore, this regulatory capacity of miR-141 has been highlighted as a central factor in the response to chemotherapy [63]. It probably plays an important role in MM and chemoresistance, which is a field that offers opportunities to study it and its possible use as a therapeutic target based on its ability to regulate p38.
This analysis and many previous works allow us to establish that multiple miRNAs can regulate the expression of p38 and function in the growth, differentiation, apoptosis, and metastasis of tumors. Given the confirmed involvement of miRNAs in the regulation of p38, these miRNAs may serve as important regulators in various pathologies, including MM, and can be proposed as important therapeutic targets.
2.3. p38 chemicals inhibitors on cancer
Despite the evidence, the role and expression of p38 in tumorigenesis is controversial, since it is considered that it may have a dual oncogene or antitumor role. Therefore, different studies have proposed p38 as an important target for cancer therapy. In general, the expression level of p38 in some tumors is high; as in hematological neoplasms and others such as breast, ovarian, colorectal, and gastrointestinal cancer [64,65,66]]. In the search for specific compounds capable of inhibiting p38, many these have been developed and are in clinical studies, both in cancer and other diseases. For example, there are pyridinilimdazoles such as SB203580, which emerged as inhibitors of proinflammatory cytokines and their function was later attributed to inhibiting the catalytic activity of p38 through competitive binding in the ATP pocket [67]
-SB203580 is a specific inhibitor of p38α [68], although other pharmacological studies demonstrate that these inhibitors inhibit p38α/β but have no effect on p38γ/δ [69]. Studies have shown that this inhibitor can inhibit tumor metastasis and invasion in murine models, by inhibiting the expression of the E-cadherin protein, during gastrulation [70]. In the use of arsenic trioxide (ATO) for therapy in MM, high resistance to treatment has been reported, this attributed in part to the activation of p38 mediated by ATO in cell lines and primary cultures of MM. In vitro studies using the p38 inhibitor SB203580 increase the inhibition of growth and induction of apoptosis by treatment with ATO, as well as the inhibition of IL-6 secretion [71]. Therefore, the use of a combination of ATO and SB203580 has been proposed as an interesting therapeutic alternative in patients with MM. Additionally, as previously mentioned, the use of Syk inhibitors such as BAY61-3606, R406 or Piceatannol, have been used in the treatment of MM in vitro, showing inhibition of proliferation and induction of apoptosis mediated by the inhibition of MAPKs such as ERK and p38 [72]. Therefore, the use in combination of SB203580 with the Syk inhibitor significantly increases the induction of apoptosis in MM cells. Carfilzomib, a second-generation proteasome inhibitor approved for the treatment of multiple myeloma and shows an important effect in the treatment of osteosarcoma, however some resistance has developed. Studies show that Carfilzomib can induce p38 activation and this mediate resistance [73]. SB203580 is a small molecule originally designed as an anti-inflammatory and has been widely used in studies against various types of cancer alone or in combination with drugs [74,75,76], its toxicity is minimal and clinical studies continue as a neuroprotectant [77] and to prevent post-operative tissue adhesion [78]
-VX-745 is another p38 inhibitor, which was developed in 1998 [79], and it was |used in clinical studies in the treatment of rheumatoid arthritis. Studies have also shown that the VX-745 can inhibit the proliferation of MM cells, by inhibiting the secretion of IL-6 by bone marrow stromal cells (BMSCs) and by MM cells, preventing their adhesion implicated as a probable mechanism of chemo resistance [80]. VX745 was first described in 1998 and in 1999 a clinical trial was initiated for the treatment of rheumatoid arthritis [81]. Adverse effects were observed in the CNS, so another p38α inhibitor (VX702) was developed. that could not cross the blood-brain barrier. But its study continued in patients with dementia and its study is currently continuing in clinical phases for the treatment of Alzheimer's disease and dementia with Lewy bodies. Variants of VX745 have recently been developed and used in several diseases, but these variables are quite similar to VX745, but the pharmacokinetics improved [82]
-8-NH2-Ado is a nucleoside analogue that has been used in the treatment of hematological malignancies such as MM, but still not clear effectiveness in the clinic. However, in vitro it has been shown that 8-NH2-Ado has significant cytotoxic activity [83,84]. Recent studies have shown that the mechanism of action of 8-NH2-Ado, at least in MM, is the inhibition of the phosphorylation of many proteins such as ERK1/2, Akt and p38, and because of this inhibition of phosphorylation of these proteins is the induction of apoptosis in these MM cell lines treated with 8-NH2-Ado[84].
-SCIO-469, is a selective and active ATP-competitive p38α inhibitor that has been tested as monotherapy or in combination with bortezomib in relapsed MM, as well as in a murine model of MM, with excellent results [85,86], and even reduces the development of osteolytic bone lesions in this MM models [87]. Additional studies confirm that SCIO-469 treatment can suppress factors in the bone marrow microenvironment to inhibit MM cell proliferation and adhesion and alleviate osteolytic activation in MM [88]. Phase II clinical studies have shown that SCIO-469, as monotherapy or in combination with Bortezomib was well tolerated in patients with Relapsed Refractory Multiple Myeloma [89] Likewise, as in patients with myelodysplastic syndrome [89].
- BIRB-796 is a pan-p38 inhibitor with in vitro activity for p38α/β/γ/δ. It has been shown that treatment of MM with Bortezomib induces p38 activation and that inhibition of p38 reverses resistance to Bortezomib-dexamethasone or 17-AAG. Studies show that the use of BIRB-796 increases cytotoxicity and caspase activation, induced by the treatment [90]. Thus, also in BMSC, BIRB-796 inhibits the secretion of IL-6, VEGF, TNF-α and TGF-β1. BIRB-796 also inhibits IL-6 secretion induced in BMSCs by adherence to MM cells, thereby inhibiting tumor cell proliferation. Therefore, it is suggested that BIRB-796 reverses chemoresistance in the BM microenvironment, offering an important clinical alternative, alone and in combination with conventional chemotherapy, to improve patient outcome in MM [90]. Studies show that BIRB-796 has a greater affinity for p38δ and that BIRB-796 and its variables such as E1 decrease tumor volume and showed acceptable characteristics in vivo pharmacokinetic experiments and did not show any toxicity [82,91]
-LY2228820 a selective inhibitor of p38α and p38β, which was created for use in cancer [92], where it was shown to significantly inhibit tumor proliferation in in vitro and in vivo models of melanoma, non-small cell lung cancer, ovarian, glioma, myeloma, and breast cancer. However, recent studies have shown that it can inhibit the expression of EGFR, independent of p38 [93]. Recent studies have shown that DK7 (THZ1) inhibitors can inhibit the expression of p38 and induce tumor shrinkage; however, the use of the combination of THZ1 and LY2228820 has a synergistic effect in inhibiting the proliferation of cancer cells. [94]. In MM, as with other p38 inhibitors, the use of LY2228820 observed modest effect cytotoxic over tumor cells, but increased the tumor cytotoxicity of bortesomib and inhibited the secretion of IL-6 from BM stromal cells and BM mononuclear cells (BMMNC), derived from MM patients in remission and significantly inhibited osteoclastogenesis in vitro and in vivo in a xenographic model of human MM [95]. Suggesting that LY2228820 could be a new therapeutic alternative to improve outcome in patients with MM, both by improving the effect of bortezomib and by reducing osteoskeletal events. In clinical studies in advanced cancer patients, LY2228820 shown good results in tumor shrinkage with acceptable side effects at the minimum dose used. Treatment-related AEs were grade 1/2 with a treatment-related safety profile consisting primarily of rash, fatigue, constipation, and nausea. At the maximum dose explored, DLTs of grade 3 ataxia and grade 2 dizziness were reported. [96]
-SD-169 is an ATP-competitive inhibitor of p38α and also weakly inhibits p38β. Studies have shown that p38 is capable of activating osteoclasts and bone resorption and, on the other hand, inhibits osteoblasts and bone formation, both of which result in bone destruction in MM. In murine models of MM, it has been shown that inhibition of p38 with SD-169 decreases the ability of myeloma cells to cause bone destruction in vivo. Therefore, the use of SD-169 could be an effective treatment to treat tumor-induced osteolytic bone lesions in patients with myeloma [97].
It is clear that p38 expression, and especially the p38α/β isoforms plays an important role in the development and progression of multiple tumors including MM. Therefore, the use of chemical p38 inhibitors specifically designed around isoforms α/β, is of great relevance as therapeutic alternatives in patients with MM.
3. p38 expression in multiple myeloma
The participation of p38 in several cellular processes in the context of multiple myeloma has been reported. p38 is constitutively activated in myeloma and plays a fundamental role in bone destruction in this type of cancer, probably due to the regulatory effect on DKK-1 and MCP-1. Even attenuated p38 expression is sufficient to reduce lesions in bone in vivo, so it can be proposed as a potential therapeutic target for the treatment of bone lesions in myeloma [11]. This observation is consistent and as mentioned above by using a p38 inhibitor, VX-745 [80]. Additional studies using the p38 inhibitor 8-NH2-Ado, whose effects are described above, once again demonstrate the importance of the role of p38 in MM [98].
As already mentioned, p38 activation plays an important role in resistance to cytotoxic chemotherapeutic drugs in the treatment of MM. Studies on the roles played by the isoforms of p38 have revealed that the knocked down p38α isoform is more sensitive to bortezomib or arsenic trioxide treatment, while the knocked down p38β/γ isoform is more resistant to the same treatment, which suggests that p38α plays a more important role in resistance, while p38β/γ is not as relevant in this function [99]. Interestingly, knocking down p38δ shows a significant effect on proliferation without treatment, which is affected under treatment conditions. This suggests that the p38δ isoform plays an important role in the proliferation of MM cells. This is probably due to the phosphorylation capacity shown by p38δ on Erk1/2, while p38α has a greater activation effect on AKT and IKK, as well as the NFκB pathway. This would explain the differential roles of the MAPK p38 isoforms in MM cells.
In order to find studies reporting the clinical importance of p38 expression in MM, the public database Gene Expression Omnibus (GEO) NCBI. (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE146649), was interrogated using the words “multiple myeloma”, “healthy donors” and applying the filters for “homo sapiens” in the organism selected and Expression profiled by array in the study type finally, the results were manually curated to identify the information of the datasets showed. We select the GSE146649 dataset entitled: Expression data from bone marrow mesenchymal stromal cells obtained from healthy donors and myeloma patients and we performed an analysis of the p38 isoforms relative expression reported in the dataset. (Figure 2). Despite robust experimental evidence, when we compare the relative expression of isoforms MAPK11 and MAPK12, the expression at the diagnostic (DX) stage is decreased in comparison to the healthy donor (HD), complete response (CR), and early relapse (ER) stages (**p<0.01 ***p<0.001). On the other hand, isoform MAPK14 shows a decrease in relative expression in the DX stage but it is only significant vs. the CR stage (*p<0.05). MAPK13 shows no significative difference between stages. This analysis does not show specific results on the expression of each of the isoforms in MM and the different stages, probably due to the limitation in the sample size of each of the analyses. However, it allows us to suggest that at least for the p38α (MAPK 14) and p38β (MAPK11) isoforms, an increase in expression is observed after treatment and is maintained in the complete response and early relapse stages. This is consistent with what has been reported in vitro trials where the treatment induces an increase in the expression of p38 α/β, which can be favored clinically in combination with chemical inhibitors of p38 α/β.These results suggest a potential role of p38 in the development of MM; however, as mentioned above, there are few clinical studies. What suggests and as has been previously reported [80], treatment induces p38 expression in MM and this is maintained in patients who respond to treatment. Obviously, since it is a MAP kinase, rather than its expression, its activation or biological activity must be evaluated, which cannot be evaluated in these reviewed studies. Therefore, it would be very important to clinically evaluate the activation of the different p38 isoforms.
4. MAPK p38 as a molecular target in multiple myeloma therapy
Multiple studies have shown that interleukin-6 (IL-6) is an important growth factor involved in the physiopathogenesis of multiple myeloma [100], and through an autocrine/paracrine form promotes the survival and proliferation of myeloma cells. It has also been shown that growth factors such as granulocyte colony-stimulating factor (G-CSF), as well as IL-10, may also play a role in MM cell lines [101,102]. IL-6 can induce the activation of different signaling pathways that may be involved in the physiopathogenesis of this disease. As already mentioned, these include the Jak-Stat, Ras/Raf/Mek/Erk, and AKT pathways [101], which are essential for MM cell proliferation. Additionally, it has been shown that mutations in Ras (N-Ras and K-Ras) lead to constitutive activation that also influences a more malignant phenotype in patients with MM [103]. This Ras activation-dependent proliferation may be independent of the Mek/Erk pathway, which makes the use of inhibitors of this pathway unfeasible as a therapeutic alternative in cases of MM with malignant phenotypes [104]. Studies have shown that the expression of heme oxygenase-1 (HO-1), an enzyme that provides potent cytoprotection, cell proliferation, and drug resistance [105], is expressed and correlates with the expression of IL-6 in the bone marrow microenvironment of MM patients and autocrine IL-6 in MM patient cells, and both HO-1 and IL-6 have been associated with disease severity in MM [106]. Increased expression of HO-1 can induces increased expression of IL-6 through p38 and other MAPKs. Therefore, chemical inhibition of p38 significantly inhibits the expression of IL-6 mediated by HO-1, and this may have therapeutic importance in MM. Other growth factors, such as insulin-like growth factor 1 (IGF-1), have also been shown to induce activation of the Akt and Mek/Erk pathways in MM cell lines, where it appears that the PI-3/Akt pathway plays a major [101,102]
As already mentioned in the section on chemical inhibitors for p38, certain studies have associated the activity of the p38 pathway with the pathophysiology of MM, since inhibition of this pathway by the chemical inhibitor VX-745 induces inhibited IL-6 secretion in MM cells and inhibited cell proliferation. Thus, as well as the use of a combination of bortezomib with SCIO-469 or BIRB-796, as well LY2228820, which also inhibits osteoclastogenesis [12,90,95].
Therefore, it is believed that MAPK p38 indirectly participates in the physiopathogenesis of MM in an indirect way, and this inhibition may have therapeutic implications in the treatment of MM [12]. Studies have shown that treatment with bortezomib induces the induction of apoptosis by p38 inhibition, who is mediated by increased expression of p53 and decreased expression of Bcl-XL and Mcl-1 [86]. It has also been reported that p38 inhibition, decreases the expression of IL-6 and VGEF by BM stromal cells, resulting in the inhibition of MM cell proliferation and adhesion [88], thus decreasing the tumor burden and angiogenesis in murine models of MM [85,87].
The interaction between bone marrow stromal cells (BMSCs) and multiple myeloma cells is very important in the pathogenesis of MM by the secretion of growth factors, cytokines, and extracellular vesicles. Exosomes appear to play an important role in the communication between BMSCs and MM cells through the transfer of cytokines between other molecules. In an in vivo model, it was shown that BMSC and MM cells could exchange exosomes carrying certain cytokines, inducing increased growth of MM cells and resistance to bortezomib [107]. Thus, they also induce activation of several important pathways for survival, including p38. Therefore, these studies suggest that p38 inhibitors may have an important therapeutic role in preventing the effect of exosome-mediated BMSCs on the proliferation, migration, survival, and drug resistance of MM cells.
Recent studies have shown that TAK1 inhibitor induces inhibition of proliferation and apoptosis in MM cells through constitutive or melphalan-mediated inhibition of TAK1, NF-kB, and MAPK p38 [108]. Additional studies have shown that the selective TAK1 inhibitor 5Z-7-oxozeaene shows synergistic potential with bortezomib, inducing increased inhibition of proliferation and apoptosis [109]. This biological activity is related to the inhibition of TAK1, which induces inhibition of JNK, MAPK p38, and Erk, which are activated by TAK1 in MM cell lines. Therefore, various studies have reported the use of chemical inhibitors of growth signaling pathways in MM as a therapeutic alternative, such as inhibitors of the Stat3 and Erk2-dependent IL-6 activation pathways. Another candidate is the proteasome inhibitor PS-341, which has been shown to activate the JNK pathway and inhibit the Erk1/Erk2-dependent pathway [80]. The possibility that MAPK pathways participate in the growth and physiopathogenesis of MM in general has motivated the development of studies with the aim of using MAPK inhibitors as potential therapeutic agents alone or in combination with chemotherapy or other conventional or novel therapies.
MAPKAPK2 (MK2) is the direct substrate of MAPK p38. Recent studies have shown that MK2 plays an important role in the pathophysiology of MM [110]. These studies suggest that using p38 inhibitors that affect MK2 or direct MK2 inhibitors could be an important therapeutic alternative for MM. One study reported that rafoxanide, an antiparasitic, is capable of inducing mitochondria-dependent proliferation inhibition and apoptosis in MM cells [111]. This is a consequence of the inhibition of MAPK p38 and Stat-1. Therefore, non-specific inhibitors that impact the p38 pathway may also be therapeutic alternatives for MM.
It has recently been reported that p38 also regulates the expression of the NKG2D and DNAM-1 ligands in MM cells in a drug-dependent manner, sensitizing them to the induction of death by NK cells [112]. This has been determined to be mediated by p38 activation of transcription factor E2F1. These results suggest that p38 inhibition could be a therapeutic alternative in cases of immunoresistance in MM mediated by NK cells.
Spleen tyrosine kinase (Syk) is an intracellular enzyme that plays an important role in the activation of B cells or T cell receptors. Studies have shown that Syk inhibitors can inhibit proliferation and inducing apoptosis in these kinase cells in MM, where inhibition of the p38 pathway has been shown to be one of the consequences of such treatment [113]. The combination of Syk inhibitor and p38 inhibitor results in increased induction of apoptosis in MM cell lines.
Trifluoperazine is a drug used in psychosis, but recent studies have shown that it inhibits tumor growth in different types of cancer, including MM cells [114]. Additional studies found that this drug in combination with bortezomib had a cytotoxic effect in in vitro and in vivo models of MM by inducing proliferation inhibition and apoptosis mediated by p38 inhibition [114]
In vivo and in vitro studies have shown that celastrol, a pentacyclic nortriterpen quinone, has an antitumor effect in MM and other types of cancer [115]. Recent studies have shown that celastrol induces apoptosis in MM cell lines alone or in combination with bortezomib, and that the cytotoxic effect is also associated with inhibition of the IRAK4/ERK/p38 pathway [116].
Tris(dibenzylideneacetone)dipalladium (Tris DBA) is a small molecule of the palladium complex that induces inhibited cell growth and proliferation in multiple myeloma cells either alone or in combination with bortezomib. This effect is a consequence of the inhibition of downstream p38 signaling [117]. Interestingly, Tris DBA reverses hypoxia-mediated drug resistance by inhibiting p38 and Hif-1α [118].
The cell-derived protein kinase T-LAK/PDZ-binding kinase (TOPK/PBK) has been proposed as a potential therapeutic target due to its low expression in most normal tissues and high expression in various tumors, including MM. Recent studies have reported that OTS514, a TOPK/PBK inhibitor, had a cytotoxic effect on MM cell lines [119]. OTS514 induces apoptosis by inhibiting FOXM1, Akt, and p38.
Histone deacetylases are potential therapeutic targets in hematological malignancies. Recent studies have shown that a new histone deacetylase inhibitor induces cytotoxicity in MM cells via apoptosis and was able to induce cytotoxicity in myeloma cells co-cultured with bone mesenchymal stromal cells and osteoclasts previously treated with the inhibitor [120]. This inhibitor was found to suppress osteoclast differentiation and resorption in vitro by inhibiting ERK, p38, AKT, and JNK, which prevented MM-associated bone loss in an in vivo model. These results support the idea that inhibitors of MAPKs, and in particular p38, may have potential clinical use in multiple myeloma treatment in the near future.
Various groups have shown that p38 is constitutively active in myeloma cells and that this leads to osteolytic bone destruction [11,80,88]. Therefore, treatment with a p38 inhibitor, decreased tumor burden and bone lesions in a murine myeloma model, prolonging survival [87]. Additionally, inhibition of p38 has an important effect in reducing osteolytic bone lesions induced by MM, reducing osteoclastogenesis and improving osteoblastogenesis [11]. The important role of p38 in bone damage induced by MM was confirmed using shRNA specific for p38α in vitro, where lower bone resorption associated with the knockdown effect of p38 was observed. Therefore, p38 inhibition and in special p38α/β inhibitors is positioned as an important therapeutic alternative in the prevention of osteolytic bone lesions caused by MM [11,121].
The CXCR4 chemokine receptor is expressed in a wide variety of hematological malignancies, including MM [122], and in conjunction with its ligand SDF-1, it plays an important role in cancer progression. Studies in which researchers generated a humanized mAb, hz515H7, which binds to human CXCR4, have shown that this binding inhibits the signaling pathway induced by SDF-1, reducing the phosphorylation of Akt, Erk1/2, and p38, which strongly inhibits cell migration and proliferation [123]. Therefore, the inhibition of the p38 signaling pathway as part of the mechanism of action of humanized mAbs used therapeutically in MM may be important in their therapeutic efficacy. Additionally, studies using gambogic acid (GA), a xanthone that inhibits CXCR4 signaling, demonstrated that it is capable of suppressing osteoclastogenesis induced by MM cells [124], inhibiting activation of the factor NF-kB transcription pathway, which regulates CXCR4 expression. GA was found to suppress SDF-1α-induced chemotaxis of MM cells and signaling pathways downstream of CXCR4 and inhibit Akt, p38, and Erk1/2 activation. Interestingly, MM-mediated suppression of osteolytic bone damage is regulated by IL-6 inhibition and consequent p38 inhibition. These findings suggest that modulating chemotaxis factors and cytokines such as CXCR4 and IL-6, the common factors of which include activation of the p38 signaling pathway, may be an important therapeutic target, through bifunctional alternatives or directed toward inhibition of MAPK p38 in MM.
Studies have reported that the mTOR signaling pathway is involved in the pathophysiology of MM [125], and a small mTOR inhibitory molecule (SC06) was found to induce inhibition of tumor growth in an in vivo model of MM [126]. This inhibition did not affect other signaling pathways, such as AKT, ERK, c-Src, and JNK, but showed a significant effect on p38 in some of the analyzed MM cell lines. This suggests that the activation of the p38 signaling pathway in MM may be mediated in part by the mTOR pathway, and that the cytotoxic effect from the inhibition of the mTOR signaling pathway may be, among other pathways, the inhibition of the p38 pathway.
Studies have shown that MM cell lines can express high levels of TLR5, which, when activated with its specific ligand, flagellin, induces IL-6 expression through the activation of NF-κB, which is, in turn, activated by p38 and PI3K/AKT pathway signaling. This leads to greater resistance to chemotherapeutic agents [127]. In addition, MM cells have also been shown to express TLR7 and TLR9 [128], which also induce the expression of IL-6 and, by activating the p38 pathway, induces chemoresistance [129]. These results suggest that there are mechanisms of the innate immune system that may favor the development and chemoresistance of MM cells, which, by activating survival signaling pathways such as MAPK p38, may play a role in the pathophysiology of MM. Thus, once again, inhibition of the p38 pathway is emerging as an important therapeutic target.
All-trans retinoic acid (ATRA) has been used in the treatment of multiple myeloma, but ATRA-induced chemoresistance has been reported in myeloma patients. This is associated with the induction of apurinic endonuclease/redox factor-1 (Ape/Ref-1) expression, which leads to MDR transactivation [130,131]. Studies have revealed that ATRA activates the p38 pathway and can promote Ape/Ref-1 expression, and this was reversed by treatment with a chemical inhibitor of p38. These studies suggest that p38 has a role in chemoresistance to ATRA treatment, thus it may be an important target in ATRA-mediated chemoresistance.
Finally, as was previously discussed, the treatment with arsenic trioxide (ATO) promote p38 activation in MM cell lines, while treatment with ATO in combination with a p38 inhibitor, abolished resistance to ATO, so it was suggested that p38 could be involved in the promotion of ATO chemoresistance in MM cell lines [71].
All these data reinforce the idea that p38 activity, on specially p38 α/β, plays an important role in the pathophysiology of MM and that it directly or indirectly intervenes in the processes of chemoresistance, proliferation and growth of MM. As well as it plays a role in the physiopathogenesis of MM. Therefore, its therapeutic intervention in MM continues to be a valid strategy, whether its inhibition directly or by altering any of the mechanisms that regulate its expression and activation (Figure 3).
5. Conclusions
Many research papers have highlighted the importance of MAPKs in the physiopathogenesis of hematological malignancies and have shown that they have important roles in the regulation of growth and apoptosis of these hematological cells. In the specific case of MM, it has been described that the Raf/Mek/Erk pathway undoubtedly participates in its pathophysiology, contributing to its growth, while the JNK and p38 pathways seem to have an indirect role in growth or the mechanisms of resistance to current therapies.
Over the past two decades, the possibility of using chemical inhibitors of the MAPK pathways as therapeutic alternatives has been raised, in both in vitro assays and clinical trials. Currently, there are important advances in this area, with important limitations, probably due to the heterogeneity of gene regulation in MM; in addition, a flow cytometry analysis of phosphorylation profiles showed that the activation patterns of signaling pathways such as Stat-3 and MAPK p38 in cells from MM patients and MM cell lines are heterogeneous [132]. However, there are currently important clinical advances in MM treatment with the use of immunomodulators in combination with chemical inhibitors of the signaling pathways that could lead to therapeutic protocols with great potential.
In addition to studies that are currently under development to find new therapeutic alternatives for MM, Mek inhibitors alone or in combination have been used in the treatment of MM, as well as chemical inhibitors of p38α/β in MM patients.
The accumulated evidence on the role of MAPK in hematological malignancies has allowed the development of new therapeutic agents, some of which, as already mentioned, are in clinical trials. But without a doubt, greater knowledge regarding the participation of p38 and its isoforms in the pathophysiology of MM will allow us to significantly define its importance as a therapeutic target.
Studies have shown that the isoforms of p38 have different patterns of expression and biological functions, and efforts are currently focused on understanding the role of these isoforms in malignant neoplasms. In addition to the above, these isoforms present post-translational variants, and it is believed that they may also have a selective role in cancer, specifically in hematological malignancies.
Author Contributions
Conceptualization, M.M.-M. and M.I.V.; Methodology, M.M.-M. and M.I.V.; Software, M.M.-M.; Validation M.M.-M. and M.I.V.; Formal Analysis, M.M.-M. and M.I.V.; Investigation, M.M.-M. and M.I.V.; Resources, M.I.V.; Data Curation, M.M.-M. and M.I.V.; Writing—Original Draft Preparation, M.M.-M. and M.I.V.; Writing—Review and Editing, M.M.-M. and M.I.V.; Supervision, M.I.V.; Project Administration, M.I.V.; Funding Acquisition, M.I.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank Owen Vega for constructive criticism of the paper. .
Conflicts of Interest
No conflicts of interest, financial or otherwise, are declared by the authors.
Abbreviations
| ATP | Adenosine triphosphate |
| ATRA | All-trans retinoic acid |
| ATO | Arsenic trioxide |
| ATF2 | Activating Transcription Factor 2 |
| ATF1 | Activating Transcription Factor 1 |
| AP-1 | Jun Proto-Oncogene, AP-1 Transcription Factor Subunit |
| ASCL1 | Achaete-Scute Family BHLH Transcription Factor 1 |
| ATF4 | Activating Transcription Factor 4 |
| ATF7 | Activating Transcription Factor 7 |
| ARID3A | AT-Rich Interaction Domain 3A |
| ARID1A | AT-Rich Interaction Domain 1A |
| ASCL2 | Achaete-Scute Family BHLH Transcription Factor 2 |
| ARNT | Aryl Hydrocarbon Receptor Nuclear Translocator |
| ATRA | All-trans retinoic acid |
| ATO | Arsenic trioxide |
| AKT1 | AKT Serine/Threonine Kinase 1 |
| Ape/Ref-1 | apurinic endonuclease/redox factor-1 |
| BMSCs | Bone-marrow-derived mesenchymal stem cells |
| BCL6 | B-Cell Lymphoma 6 Protein |
| BACH2 | BTB Domain and CNC Homolog 2 |
| BM | Bone marrow |
| BMMNC | BM mononuclear cells. |
| BRAF | B-Raf Proto-Oncogene, Serine/Threonine Kinase |
| CML | Chronic Myeloid Leukemia |
| CRC | Colorectal cancer. |
| CDR1-AS | CDR1 Antisense RNA |
| CEBPA | CCAAT Enhancer Binding Protein Alpha |
| CTCF | CCCTC-Binding Factor |
| CTSC | cathepsin C |
| CHOP10 | DNA Damage Inducible Transcript 3 |
| CDC25 | Cell Division Cycle 25 |
| CXCL14 | C-X-C Motif Chemokine Ligand 14 |
| CXCR4 | C-X-C Motif Chemokine Receptor 4 |
| C-JUN | Jun Proto-Oncogene, AP-1 Transcription Factor Subunit |
| c-MYC | Myc Proto-Oncogene Protein |
| CREB1 | Cyclic AMP-Responsive Element-Binding Protein 1 |
| CDK | Cyclin-dependent kinase. |
| CDK7 | Cyclin Dependent Kinase 7 |
| DUSP1 | Dual Specificity Protein Phosphatase 1 |
| DUSP10 | Dual Specificity Protein Phosphatase 10 |
| DUSP16 | Dual Specificity Protein Phosphatase 16 |
| DBA | Dibenzylideneacetone |
| DCs | Dendritic cells |
| DKK-1 | Dickkopf WNT Signaling Pathway Inhibitor 1 |
| DNAM1 | CD226 Antigen |
| EPD | Eukaryotic Promotor Database |
| EMT | epithelial–mesenchymal transition |
| E2F1 | E2F Transcription Factor 1 |
| EGR1 | Early Growth Response 1 |
| EP300 | E1A Binding Protein P300 |
| ELK1 | ETS Transcription Factor ELK1 |
| ESCC | Esophageal squamous cell carcinoma |
| EPD | Eukaryotic Promotor Database |
| EEF2K | Eukaryotic Elongation Factor 2 Kinase |
| ESCC | Esophageal squamous cell carcinoma |
| ERK1/2 | Extracellular Signal-Regulated Kinase 2 (MAPK1) |
| FOXP3 | Forkhead Box P3 |
| FGFR1 | Fibroblast Growth Factor Receptor 1 |
| FOXM1 | Forkhead Box M1 |
| GA | Gambogic acid |
| GDF4 | Growth Differentiation Factor 4 |
| GADD45A | Growth Arrest and DNA Damage Inducible Alpha |
| G-CSF | granulocyte colony-stimulating factor |
| HEK293 | Human Embryonic Kidney cell line |
| HIF1A | Hypoxia Inducible Factor 1 Subunit Alpha |
| HDAC3 | Histone Deacetylase 3 |
| HDAC9 | Histone Deacetylase 9 |
| HCC | Hepatocellular carcinoma |
| HO-1 | heme oxygenase-1 |
| Imp 7/3 | Importin-7 |
| Imp 9/3 | Importin-9 |
| LncRNAs | Long noncoding RNA |
| IL-6 | Interleukin 6 |
| IL-8 | Interleukin 8 |
| IL-12B | Interleukin 12β |
| IAV | Influenza A virus |
| IKK | Inhibitor of Nuclear Factor Kappa-B Kinase |
| IRAK4 | Interleukin 1 Receptor Associated Kinase 4 |
| IGF-1 | insulin-like growth factor 1 |
| JUN | Jun Proto-Oncogene, AP-1 Transcription Factor Subunit |
| JAK | Janus Kinase |
| KLF4 | Kruppel-Like Factor 4 |
| KAT2B | Lysine Acetyltransferase 2B |
| KRAS | KRAS Proto-Oncogene, GTPase |
| LINCRNA | Long intergenic non-coding RNA |
| LPS | Lipopolysaccharide |
| MM | Multiple myeloma |
| MDR | Multidrug Resistance Protein |
| MAPK | Mitogen-activated protein kinase |
| MGUS | Monoclonal gammopathy of unknown significance |
| MKKs | MAP kinase kinases |
| MW | Molecular weight |
| MEF2 | Myocyte Enhancer Factor 2 |
| MSK1 | Nuclear Mitogen- And Stress-Activated Protein Kinase 1 |
| MSK2 | Nuclear Mitogen- And Stress-Activated Protein Kinase 2 |
| MAPKAPK2 | MAPK Activated Protein Kinase 2 |
| MART | Melanoma Antigen Recognized By T-Cells 1 |
| MAP3K71P1 | TGF-Beta Activated Kinase 1 (MAP3K7) Binding Protein 1 |
| MAP2K3 | Mitogen-Activated Protein Kinase Kinase 3 |
| MKK3 | MAP kinase kinases 3 |
| MAP2K4 | Mitogen-Activated Protein Kinase Kinase 4 |
| MKK4 | MAP kinase kinases 4 |
| MAP2K6 | Mitogen-Activated Protein Kinase Kinase 6 |
| MKK6 | MAP kinase kinases 6 |
| MYB | MYB Proto-Oncogene, Transcription Factor |
| MK2 | MAPK Activated Protein Kinase 2 |
| MK3 | MAPK Activated Protein Kinase 3 |
| mRNA | Messenger RNA |
| MMP-2 | Matrix Metallopeptidase 2 |
| MMP-9 | Matrix Metallopeptidase 9 |
| MYC | MYC Proto-Oncogene, BHLH Transcription Factor |
| MSCs | mesenchymal stem cells |
| MKK6 | Mitogen-Activated Protein Kinase Kinase 6 |
| MDS | Myelodysplastic syndromes. |
| MCP1 | C-C Motif Chemokine Ligand 2 |
| mTOR | Mechanistic Target Of Rapamycin Kinase |
| NHL | Non-Hodgkin lymphoma |
| NSPCs | Neural stem/progenitor cells |
| NFKB1 | Nuclear Factor Kappa B Subunit 1 |
| ND8/34 | Cell Line from mouse |
| NSCLC | Non-small cell lung cancer. |
| NOTCH3 | Notch Receptor 3 |
| NKG2D | NK Cell Receptor D |
| OSCs | Osteosarcoma cells |
| PINT | p53-induced noncoding transcript. |
| PCAT1 | Prostate Cancer Associated Transcript 1 |
| POU1F1 | Pituitary Transcript Factor 1 |
| PPMID | Protein Phosphatase Magnesium-Dependent 1 Delta |
| P53 | Tumor Protein P53 |
| P38 | Mitogen-activated protein kinase p38 |
| PRKD1 | Protein Kinase D1 |
| PRAK | MAPK Activated Protein Kinase 5 |
| rRNA | Ribosomal RNA |
| RAC3 | Rac Family Small GTPase 3 |
| RAF | Raf-1 Proto-Oncogene, Serine/Threonine Kinase |
| SRF | Serum Response Factor |
| STMN1 | Stathmin 1 |
| STAT1 | Signal Transducer and Activator of Transcription 1 |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| STAT4 | Signal Transducer and Activator of Transcription 4 |
| SMAD2 | SMAD Family Member 2 |
| SMAD3 | SMAD Family Member 3 |
| SMAD4 | SMAD Family Member 4 |
| SP1 | Specificity Protein 1 |
| SYK | Spleen Associated Tyrosine Kinase |
| SNGH5 | Small Nucleolar RNA Host Gene 5 |
| SDF1 | C-X-C Motif Chemokine Ligand 12 |
| TCR | T-cell receptor |
| TAB1 | TAK1-Binding Protein 1 |
| TNF-α | Tumor necrosis Factor alfa |
| TP53 | Tumor Protein P53 |
| TGF-β | Transforming Growth Factor Beta 1 |
| TNBC | Triple-negative breast cancer. |
| Tris DBA | Tris(dibenzylideneacetone)dipalladium. |
| TAK1 | Mitogen-Activated Protein Kinase Kinase Kinase 7 |
| TOPK/PBK | cell-derived protein kinase T-LAK/PDZ-binding kinase |
| TLR5 | Toll Like Receptor 5 |
| TLR7 | Toll Like Receptor 7 |
| TLR9 | Toll Like Receptor 9 |
| VEGF | Vascular endothelial growth factor |
| VDR | Vitamin D Receptor |
| XBP1 | X-Box-Binding Protein 1 |
| YY1 | YIN-YANG-1 |
| ZAP70 | 70 KDa Zeta-Chain Associated Protein |
| ZCCHC14 | Zinc Finger CCHC Domain-Containing Protein 14 |
References
- P. G. Richardson et al., “Bortezomib or High-Dose Dexamethasone for Relapsed Multiple Myeloma,” n engl j med, vol. 352, 2005, Accessed: Sep. 27, 2023. [Online]. Available: www.nejm.org.
- R. A. Kyle et al., “Review of 1027 patients with newly diagnosed multiple myeloma,” Mayo Clin Proc, vol. 78, no. 1, pp. 21–33, Jan. 2003. [CrossRef]
- P. de la Puente and A. K. Azab, “Contemporary drug therapies for multiple myeloma,” Drugs Today (Barc), vol. 49, no. 9, p. 563, 2013. [CrossRef]
- T. Gui, Y. Sun, A. Shimokado, and Y. Muragaki, “The Roles of Mitogen-Activated Protein Kinase Pathways in TGF-β-Induced Epithelial-Mesenchymal Transition,” J Signal Transduct, vol. 2012, pp. 1–10, Jan. 2012. [CrossRef]
- A. Martínez-Limón, M. Joaquin, M. Caballero, F. Posas, and E. de Nadal, “The p38 Pathway: From Biology to Cancer Therapy,” Int J Mol Sci, vol. 21, no. 6, Mar. 2020. [CrossRef]
- V. Sahu, A. Mohan, and S. Dey, “p38 MAP kinases: plausible diagnostic and prognostic serum protein marker of non small cell lung cancer,” Exp Mol Pathol, vol. 107, pp. 118–123, Apr. 2019. [CrossRef]
- P. P. Roux and J. Blenis, “ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions,” Microbiol Mol Biol Rev, vol. 68, no. 2, pp. 320–344, Jun. 2004. [CrossRef]
- J. Robidoux et al., “Selective activation of mitogen-activated protein (MAP) kinase kinase 3 and p38alpha MAP kinase is essential for cyclic AMP-dependent UCP1 expression in adipocytes,” Mol Cell Biol, vol. 25, no. 13, pp. 5466–5479, Jul. 2005. [CrossRef]
- S. Kudaravalli, P. den Hollander, and S. A. Mani, “Role of p38 MAP kinase in cancer stem cells and metastasis,” Oncogene 2022 41:23, vol. 41, no. 23, pp. 3177–3185, Apr. 2022. [CrossRef]
- T. Zarubin and J. Han, “Activation and signaling of the p38 MAP kinase pathway,” Cell Res, vol. 15, no. 1, pp. 11–18, 2005. [CrossRef]
- J. He et al., “p38 MAPK in myeloma cells regulates osteoclast and osteoblast activity and induces bone destruction,” Cancer Res, vol. 72, no. 24, pp. 6393–6402, Dec. 2012. [CrossRef]
- T. Hideshima et al., “p38 MAPK inhibition enhances PS-341 (bortezomib)-induced cytotoxicity against multiple myeloma cells,” Oncogene, vol. 23, no. 54, pp. 8766–8776, Nov. 2004. [CrossRef]
- Z. Jing, W. Yu, A. Li, X. Chen, Y. Chen, and J. Chen, “Trifluoperazine Synergistically Potentiates Bortezomib-Induced Anti-Cancer Effect in Multiple Myeloma via Inhibiting P38 MAPK/NUPR1,” Tohoku J Exp Med, vol. 257, no. 4, pp. 315–326, 2022. [CrossRef]
- X. Wu et al., “Dihydroartemisinin Modulates Apoptosis and Autophagy in Multiple Myeloma through the P38/MAPK and Wnt/ β-Catenin Signaling Pathways,” Oxid Med Cell Longev, vol. 2020, 2020. [CrossRef]
- J. Han, J. Wu, and J. Silke, “An overview of mammalian p38 mitogen-activated protein kinases, central regulators of cell stress and receptor signaling,” F1000Res, vol. 9, 2020. [CrossRef]
- B. Canovas and A. R. Nebreda, “Diversity and versatility of p38 kinase signalling in health and disease,” Nat Rev Mol Cell Biol, vol. 22, no. 5, pp. 346–366, May 2021. [CrossRef]
- S. Uddin, J. Ah-Kang, J. Ulaszek, D. Mahmud, and A. Wickrema, “Differentiation stage-specific activation of p38 mitogen-activated protein kinase isoforms in primary human erythroid cells,” Proc Natl Acad Sci U S A, vol. 101, no. 1, pp. 147–152, Jan. 2004. [CrossRef]
- M. Deak, A. D. Clifton, J. M. Lucocq, and D. R. Alessi, “Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB,” EMBO J, vol. 17, no. 15, pp. 4426–4441, Aug. 1998. [CrossRef]
- B. Pierrat, J. Da Silva Correia, J. L. Mary, M. Tomás-Zuber, and W. Lesslauer, “RSK-B, a novel ribosomal S6 kinase family member, is a CREB kinase under dominant control of p38alpha mitogen-activated protein kinase (p38alphaMAPK),” J Biol Chem, vol. 273, no. 45, pp. 29661–29671, Nov. 1998. [CrossRef]
- J. Han, J. Wu, J. Silke, J. D. Ashwell, and G. Sabio, “An overview of mammalian p38 mitogen-activated protein kinases, central regulators of cell stress and receptor signaling,” F1000Research 2020 9:653, vol. 9, p. 653, Jun. 2020. [CrossRef]
- S. H. Lee, J. Park, Y. Che, P.-L. Han, and J.-K. Lee, “Constitutive Activity and Differential Localization of p38 and p38 MAPKs in Adult Mouse Brain,” J. Neurosci. Res, vol. 60, pp. 623–631, 2000. [CrossRef]
- H. Enslen, J. Raingeaud, and R. J. Davis, “Selective activation of p38 mitogen-activated protein (MAP) kinase isoforms by the MAP kinase kinases MKK3 and MKK6,” J Biol Chem, vol. 273, no. 3, pp. 1741–1748, Jan. 1998. [CrossRef]
- X. Qi et al., “p38alpha antagonizes p38gamma activity through c-Jun-dependent ubiquitin-proteasome pathways in regulating Ras transformation and stress response,” J Biol Chem, vol. 282, no. 43, pp. 31398–31408, Oct. 2007. [CrossRef]
- I. Corre, F. Paris, and J. Huot, “The p38 pathway, a major pleiotropic cascade that transduces stress and metastatic signals in endothelial cells,” Oncotarget, vol. 8, no. 33, pp. 55684–55714, 2017. [CrossRef]
- N. Matesanz et al., “p38α blocks brown adipose tissue thermogenesis through p38δ inhibition,” PLoS Biol, vol. 16, no. 7, Jul. 2018. [CrossRef]
- X. Wang et al., “Involvement of the MKK6-p38gamma cascade in gamma-radiation-induced cell cycle arrest,” Mol Cell Biol, vol. 20, no. 13, pp. 4543–4552, Jul. 2000. [CrossRef]
- Y. Shi and M. Gaestel, “In the cellular garden of forking paths: how p38 MAPKs signal for downstream assistance,” Biol Chem, vol. 383, no. 10, pp. 1519–1536, Oct. 2002. [CrossRef]
- A. Cuadrado and A. R. Nebreda, “Mechanisms and functions of p38 MAPK signalling,” Biochem J, vol. 429, no. 3, pp. 403–417, Aug. 2010. [CrossRef]
- K. S. Robinson et al., “ZAKα-driven ribotoxic stress response activates the human NLRP1 inflammasome,” Science, vol. 377, no. 6603, pp. 328–335, Jul. 2022. [CrossRef]
- G. G. Vega et al., “P38 MAPK expression and activation predicts failure of response to CHOP in patients with Diffuse Large B-Cell Lymphoma,” BMC Cancer, vol. 15, no. 1, Oct. 2015. [CrossRef]
- Y. Feng, J. Wen, and C. C. Chang, “p38 Mitogen-activated protein kinase and hematologic malignancies,” Arch Pathol Lab Med, vol. 133, no. 11, pp. 1850–1856, Nov. 2009. [CrossRef]
- N. A. Bhowmick et al., “Transforming growth factor-β1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism,” Mol Biol Cell, vol. 12, no. 1, pp. 27–36, Oct. 2001. [CrossRef]
- L. Mao, L. Yuan, L. M. Slakey, F. E. Jones, M. E. Burow, and S. M. Hill, “Inhibition of breast cancer cell invasion by melatonin is mediated through regulation of the p38 mitogen-activated protein kinase signaling pathway,” Breast Cancer Research, vol. 12, no. 6, pp. 1–14, Dec. 2010. [CrossRef]
- T. T. T. Phan, N. V. Truong, W. G. Wu, Y. C. Su, T. S. Hsu, and L. Y. Lin, “Tumor suppressor p53 mediates interleukin-6 expression to enable cancer cell evasion of genotoxic stress,” Cell Death Discov, vol. 9, no. 1, Dec. 2023. [CrossRef]
- H. An, X. Lu, D. Liu, and W. G. Yarbrough, “LZAP inhibits p38 MAPK (p38) phosphorylation and activity by facilitating p38 association with the wild-type p53 induced phosphatase 1 (WIP1),” PLoS One, vol. 6, no. 1, 2011. [CrossRef]
- V. B. Pillai, N. R. Sundaresan, S. A. Samant, D. Wolfgeher, C. M. Trivedi, and M. P. Gupta, “Acetylation of a conserved lysine residue in the ATP binding pocket of p38 augments its kinase activity during hypertrophy of cardiomyocytes,” Mol Cell Biol, vol. 31, no. 11, pp. 2349–2363, Jun. 2011. [CrossRef]
- J. C. Lee et al., “A protein kinase involved in the regulation of inflammatory cytokine biosynthesis,” Nature, vol. 372, no. 6508, pp. 739–746, 1994. [CrossRef]
- A. S. Zervos, L. Faccio, J. P. Gatto, J. M. Kyriakis, and R. Brent, “Mxi2, a mitogen-activated protein kinase that recognizes and phosphorylates Max protein,” Proc Natl Acad Sci U S A, vol. 92, no. 23, pp. 10531–10534, Nov. 1995. [CrossRef]
- T. Sudo, Y. Yagasaki, H. Hama, N. Watanabe, and H. Osada, “Exip, a new alternative splicing variant of p38α, can induce an earlier onset of apoptosis in HeLa cells,” Biochem Biophys Res Commun, vol. 291, no. 4, pp. 838–843, 2002. [CrossRef]
- P. Wang, P. Yu, P. Gao, T. Shi, and D. Ma, “Discovery of novel human transcript variants by analysis of intronic single-block EST with polyadenylation site,” BMC Genomics, vol. 10, Nov. 2009. [CrossRef]
- G. Lominadze, M. J. Rane, M. Merchant, J. Cai, R. A. Ward, and K. R. McLeish, “Myeloid-related protein-14 is a p38 MAPK substrate in human neutrophils,” J Immunol, vol. 174, no. 11, pp. 7257–7267, Jun. 2005. [CrossRef]
- J. Raingeaud et al., “Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine,” J Biol Chem, vol. 270, no. 13, pp. 7420–7426, 1995. [CrossRef]
- U. Mahlknecht, J. Will, A. Varin, D. Hoelzer, and G. Herbein, “Histone deacetylase 3, a class I histone deacetylase, suppresses MAPK11-mediated activating transcription factor-2 activation and represses TNF gene expression,” J Immunol, vol. 173, no. 6, pp. 3979–3990, Sep. 2004. [CrossRef]
- T. Ota et al., “Complete sequencing and characterization of 21,243 full-length human cDNAs,” Nat Genet, vol. 36, no. 1, pp. 40–45, Jan. 2004. [CrossRef]
- J. E. Collins et al., “A genome annotation-driven approach to cloning the human ORFeome,” Genome Biol, vol. 5, no. 10, p. R84, 2004. [CrossRef]
- N. W. Court, C. G. Dos Remedios, J. Cordell, and M. A. Bogoyevitch, “Cardiac expression and subcellular localization of the p38 mitogen-activated protein kinase member, stress-activated protein kinase-3 (SAPK3),” J Mol Cell Cardiol, vol. 34, no. 4, pp. 413–426, Apr. 2002. [CrossRef]
- C. Lechner, M. A. Zahalka, J. F. Giot, N. P. H. Møller, and A. Ullrich, “ERK6, a mitogen-activated protein kinase involved in C2C12 myoblast differentiation,” Proc Natl Acad Sci U S A, vol. 93, no. 9, pp. 4355–4359, Apr. 1996. [CrossRef]
- D. S. Gerhard et al., “The status, quality, and expansion of the NIH full-length cDNA project: the Mammalian Gene Collection (MGC),” Genome Res, vol. 14, no. 10B, pp. 2121–2127, 2004. [CrossRef]
- L. New et al., “PRAK, a novel protein kinase regulated by the p38 MAP kinase,” EMBO J, vol. 17, no. 12, pp. 3372–3384, Jun. 1998. [CrossRef]
- G. Pantoja-Escobar, M. Morales-Martínez, G. G. Vega, G. Castro-Escarpulli, and M. I. Vega, “Cytotoxic effect caspase activation dependent of a genetically engineered fusion protein with a CD154 peptide mimetic (OmpC-CD154p) on B-NHL cell lines is mediated by the inhibition of bcl-6 and YY1 through MAPK p38 activation,” Leuk Lymphoma, vol. 60, no. 4, pp. 1062–1070, Mar. 2019. [CrossRef]
- M. Zhao et al., “Regulation of the MEF2 family of transcription factors by p38,” Mol Cell Biol, vol. 19, no. 1, pp. 21–30, Jan. 1999. [CrossRef]
- S.-H. Yang, A. Galanis, and A. D. Sharrocks, “Targeting of p38 mitogen-activated protein kinases to MEF2 transcription factors,” Mol Cell Biol, vol. 19, no. 6, pp. 4028–4038, Jun. 1999. [CrossRef]
- N. Umasuthan, S. D. N. K. Bathige, J. K. Noh, and J. Lee, “Gene structure, molecular characterization and transcriptional expression of two p38 isoforms (MAPK11 and MAPK14) from rock bream (Oplegnathus fasciatus),” Fish Shellfish Immunol, vol. 47, no. 1, pp. 331–343, Nov. 2015. [CrossRef]
- M. Zhou et al., “Prioritizing candidate disease-related long non-coding RNAs by walking on the heterogeneous lncRNA and disease network †,” 760 | Mol. BioSyst, vol. 11, p. 760, 2015. [CrossRef]
- Q. Yang, X. Shen, Z. Su, and S. Ju, “Emerging roles of noncoding RNAs in multiple myeloma: A review,” J Cell Physiol, vol. 234, no. 6, pp. 7957–7969, Jun. 2019. [CrossRef]
- L. Chen, X. Gong, and M. Huang, “YY1-Activated Long Noncoding RNA SNHG5 Promotes Glioblastoma Cell Proliferation Through p38/MAPK Signaling Pathway,” Cancer Biother Radiopharm, vol. 34, no. 9, pp. 589–596, Nov. 2019. [CrossRef]
- O. Marín-Béjar et al., “Pint lincRNA connects the p53 pathway with epigenetic silencing by the Polycomb repressive complex 2,” Genome Biol, vol. 14, no. 9, Sep. 2013. [CrossRef]
- A. Rocci, C. C. Hofmeister, and F. Pichiorri, “The potential of miRNAs as biomarkers for multiple myeloma,” Expert Rev Mol Diagn, vol. 14, no. 8, pp. 947–959, Nov. 2014. [CrossRef]
- C. Gu et al., “Integrative analysis of signaling pathways and diseases associated with the miR-106b/25 cluster and their function study in berberine-induced multiple myeloma cells,” Funct Integr Genomics, vol. 17, no. 2–3, pp. 253–262, May 2017. [CrossRef]
- S. Li, J. Zhu, J. Li, S. Li, and B. Li, “MicroRNA-141 inhibits proliferation of gastric cardia adenocarcinoma by targeting MACC1,” Arch Med Sci, vol. 14, no. 3, pp. 588–596, 2018. [CrossRef]
- L. Hui et al., “p38α suppresses normal and cancer cell proliferation by antagonizing the JNK–c-Jun pathway,” Nature Genetics 2007 39:6, vol. 39, no. 6, pp. 741–749, Apr. 2007. [CrossRef]
- E. K. Kim and E. J. Choi, “Pathological roles of MAPK signaling pathways in human diseases,” Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, vol. 1802, no. 4, pp. 396–405, Apr. 2010. [CrossRef]
- Y. Gao et al., “The Roles of MicroRNA-141 in Human Cancers: From Diagnosis to Treatment,” Cell Physiol Biochem, vol. 38, no. 2, pp. 427–448, 2016. [CrossRef]
- R. M. Schultz, “Potential of p38 MAP kinase inhibitors in the treatment of cancer,” Prog Drug Res, vol. 60, pp. 59–92, 2003. [CrossRef]
- C. Schultz, A. Link, and M. Leost, “Paullones, a series of cyclin-dependent kinase inhibitors: synthesis, evaluation of CDK1/cyclin B inhibition, and in vitro antitumor activity,” Journal of medicinal …, vol. 6, pp. 2909–2919, 1999. [CrossRef]
- M. I. Vega, S. Huerta-Yepaz, H. Garban, A. Jazirehi, C. Emmanouilides, and B. Bonavida, “Rituximab inhibits p38 MAPK activity in 2F7 B NHL and decreases IL-10 transcription: Pivotal role of p38 MAPK in drug resistance,” Oncogene, vol. 23, no. 20, pp. 3530–3540, Apr. 2004. [CrossRef]
- R. J. Gum et al., “Acquisition of sensitivity of stress-activated protein kinases to the p38 inhibitor, SB 203580, by alteration of one or more amino acids within the ATP binding pocket,” Journal of Biological Chemistry, vol. 273, no. 25, pp. 15605–15610, Jun. 1998. [CrossRef]
- A. Cuenda et al., “SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1,” FEBS Lett, vol. 364, no. 2, pp. 229–233, May 1995. [CrossRef]
- M. Goedert, A. Cuenda, M. Craxton, R. Jakes, and P. Cohen, “Activation of the novel stress-activated protein kinase SAPK4 by cytokines and cellular stresses is mediated by SKK3 (MKK6); comparison of its substrate specificity with that of other SAP kinases,” EMBO J, vol. 16, no. 12, pp. 3563–3571, Jun. 1997. [CrossRef]
- I. E. Zohn, Y. Li, E. Y. Skolnik, K. V. Anderson, J. Han, and L. Niswander, “p38 and a p38-Interacting Protein Are Critical for Downregulation of E-Cadherin during Mouse Gastrulation,” Cell, vol. 125, no. 5, pp. 957–969, Jun. 2006. [CrossRef]
- J. Wen et al., “P38 MAPK inhibition enhancing ATO-induced cytotoxicity against multiple myeloma cells,” Br J Haematol, vol. 140, no. 2, pp. 169–180, Jan. 2008. [CrossRef]
- R. M. Koerber et al., “Analysis of the anti-proliferative and the pro-apoptotic efficacy of Syk inhibition in multiple myeloma,” Exp Hematol Oncol, vol. 4, no. 1, p. 21, Aug. 2015. [CrossRef]
- L. Lei et al., “Resistance of osteosarcoma cells to the proapoptotic effects of carfilzomib involves activation of mitogen activated protein kinase pathways,” Exp Physiol, vol. 106, no. 2, pp. 438–449, Feb. 2021. [CrossRef]
- P. Dey, S. Biswas, R. Das, S. Chatterjee, and U. Ghosh, “p38 MAPK inhibitor SB203580 enhances anticancer activity of PARP inhibitor olaparib in a synergistic way on non-small cell lung carcinoma A549 cells,” Biochem Biophys Res Commun, vol. 670, pp. 55–62, Aug. 2023. [CrossRef]
- S. P. Davies, H. Reddy, M. Caivano, and P. Cohen, “Specificity and mechanism of action of some commonly used protein kinase inhibitors,” Biochem J, vol. 351, no. Pt 1, pp. 95–105, Oct. 2000. [CrossRef]
- J. Saklatvala, “The p38 MAP kinase pathway as a therapeutic target in inflammatory disease,” Curr Opin Pharmacol, vol. 4, no. 4, pp. 372–377, 2004. [CrossRef]
- H. S. Sharma et al., “Pathophysiology of blood-brain barrier in brain tumor. Novel therapeutic advances using nanomedicine,” Int Rev Neurobiol, vol. 151, pp. 1–66, Jan. 2020. [CrossRef]
- Y. Wu, X. Duan, Z. Gao, N. Yang, and F. Xue, “AICAR attenuates postoperative abdominal adhesion formation by inhibiting oxidative stress and promoting mesothelial cell repair,” PLoS One, vol. 17, no. 9, Sep. 2022. [CrossRef]
- J. P. Duffy et al., “The Discovery of VX-745: A Novel and Selective p38α Kinase Inhibitor,” ACS Med Chem Lett, vol. 2, no. 10, pp. 758–763, Oct. 2011. [CrossRef]
- T. Hideshima et al., “Targeting p38 MAPK inhibits multiple myeloma cell growth in the bone marrow milieu,” Blood, vol. 101, no. 2, pp. 703–705, Jan. 2003. [CrossRef]
- C. Ding, “Drug evaluation: VX-702, a MAP kinase inhibitor for rheumatoid arthritis and acute coronary syndrome,” Current Opinion in Investigational Drugs, vol. 7, no. 11, pp. 1020–1025, Jan. 2006, Accessed: Dec. 07, 2023. [Online]. Available: /articles/journal_contribution/Drug_evaluation_VX-702_a_MAP_kinase_inhibitor_for_rheumatoid_arthritis_and_acute_coronary_syndrome/23215691/1.
- V. R. Wydra, R. B. Ditzinger, N. J. Seidler, F. W. Hacker, and S. A. Laufer, “A patent review of MAPK inhibitors (2018 - present),” Expert Opin Ther Pat, vol. 33, no. 6, pp. 421–444, 2023. [CrossRef]
- V. Gandhi et al., “Compound GW506U78 in refractory hematologic malignancies: relationship between cellular pharmacokinetics and clinical response,” J Clin Oncol, vol. 16, no. 11, pp. 3607–3615, 1998. [CrossRef]
- K. Ghias, C. Ma, V. Gandhi, L. C. Platanias, N. L. Krett, and S. T. Rosen, “8-Amino-adenosine induces loss of phosphorylation of p38 mitogen-activated protein kinase, extracelluar signal-regulated kinase 1/2, and Akt kinase: Role in induction of apoptosis in multiple myeloma,” Mol Cancer Ther, vol. 4, no. 4, pp. 569–577, Apr. 2005. [CrossRef]
- S. Medicherla et al., “p38α-Selective MAP Kinase Inhibitor Reduces Tumor Growth in Mouse Xenograft Models of Multiple Myeloma,” Anticancer Res, vol. 28, no. 6A, pp. 3827–3833, Nov. 2008, Accessed: Sep. 27, 2023. [Online]. Available: https://ar.iiarjournals.org/content/28/6A/3827.
- T. A. Navas et al., “Inhibition of p38alpha MAPK enhances proteasome inhibitor-induced apoptosis of myeloma cells by modulating Hsp27, Bcl-X(L), Mcl-1 and p53 levels in vitro and inhibits tumor growth in vivo,” Leukemia, vol. 20, no. 6, pp. 1017–1027, 2006. [CrossRef]
- K. Vanderkerken et al., “Inhibition of p38α mitogen-activated protein kinase prevents the development of osteolytic bone disease, reduces tumor burden, and increases survival in murine models of multiple myeloma,” Cancer Res, vol. 67, no. 10, pp. 4572–4577, May 2007. [CrossRef]
- A. N. Nguyen et al., “Normalizing the bone marrow microenvironment with p38 inhibitor reduces multiple myeloma cell proliferation and adhesion and suppresses osteoclast formation,” Exp Cell Res, vol. 312, no. 10, pp. 1909–1923, Jun. 2006. [CrossRef]
- D. S. Siegel et al., “Phase II Trial of SCIO-469 as Monotherapy (M) or in Combination with Bortezomib (MB) in Relapsed Refractory Multiple Myeloma (MM).,” Blood, vol. 108, no. 11, p. 3580, Nov. 2006. [CrossRef]
- H. Yasui et al., “BIRB 796 enhances cytotoxicity triggered by bortezomib, heat shock protein (Hsp) 90 inhibitor, and dexamethasone via inhibition of p38 mitogen-activated protein kinase/Hsp27 pathway in multiple myeloma cell lines and inhibits paracrine tumour growth,” Br J Haematol, vol. 136, no. 3, pp. 414–423, Feb. 2007. [CrossRef]
- M. J. HOLTZMAN, A. G. ROMERO, B. J. GEROVAC, Z. HAN, S. P. KEELER, and K. WU, “MITOGEN-ACTIVATED PROTEIN KINASE INHIBITORS, METHODS OF MAKING, AND METHODS OF USE THEREOF,” Dec. 2019.
- R. M. Campbell et al., “Characterization of LY2228820 dimesylate, a potent and selective inhibitor of p38 MAPK with antitumor activity,” Mol Cancer Ther, vol. 13, no. 2, pp. 364–374, Feb. 2014. [CrossRef]
- D. Bhattacharjee et al., “Inhibition of a lower potency target drives the anticancer activity of a clinical p38 inhibitor,” Cell Chem Biol, vol. 30, no. 10, pp. 1211-1222.e5, Oct. 2023. [CrossRef]
- M. Lepore Signorile et al., “c-MYC Protein Stability Is Sustained by MAPKs in Colorectal Cancer,” Cancers (Basel), vol. 14, no. 19, Oct. 2022. [CrossRef]
- K. Ishitsuka et al., “p38 mitogen-activated protein kinase inhibitor LY2228820 enhances bortezomib-induced cytotoxicity and inhibits osteoclastogenesis in multiple myeloma; therapeutic implications,” Br J Haematol, vol. 141, no. 5, pp. 598–606, Jun. 2008. [CrossRef]
- A. L. T. A. P. K. B. M. R. D. W. G. S. J. C. M. F. M. E. S. M. J. L. M. T. F. C. E. J. M. P. C. X. C. R. S. G. I. R. R. Patnaik A, “First-in-human Phase I Study of Copanlisib (BAY 80-6946), an Intravenous Pan-Class I Phosphatidylinositol 3-kinase Inhibitor, in Patients With Advanced Solid Tumors and non-Hodgkin’s Lymphomas,” Annals of Oncology, vol. 27, no. 10, pp. 1928–1940, 2016. [CrossRef]
- J. Yang et al., “Constitutive activation of p38 MAPK in tumor cells contributes to osteolytic bone lesions in multiple myeloma,” Leukemia, vol. 26, no. 9, p. 2114, Sep. 2012. [CrossRef]
- K. Ghias, C. Ma, V. Gandhi, L. C. Platanias, N. L. Krett, and S. T. Rosen, “8-Amino-adenosine induces loss of phosphorylation of p38 mitogen-activated protein kinase, extracellular signal-regulated kinase 1/2, and Akt kinase: role in induction of apoptosis in multiple myeloma,” Mol Cancer Ther, vol. 4, no. 4, pp. 569–577, Apr. 2005. [CrossRef]
- H. Peng et al., “Characterization of p38 MAPK isoforms for drug resistance study using systems biology approach,” Bioinformatics, vol. 30, no. 13, pp. 1899–1907, Jul. 2014. [CrossRef]
- M. Hallek, P. L. Bergsagel, and K. C. Anderson, “Multiple Myeloma: Increasing Evidence for a Multistep Transformation Process,” Blood, vol. 91, no. 1, p. 3, Jan. 1998. [CrossRef]
- G. Pratt, “Molecular aspects of multiple myeloma,” Mol Pathol, vol. 55, no. 5, pp. 273–283, Oct. 2002. [CrossRef]
- L. C. Platanias, “Map kinase signaling pathways and hematologic malignancies,” Blood, vol. 101, no. 12, pp. 4667–4679, Jun. 2003. [CrossRef]
- P. Liu et al., “Activating Mutations of N- and K-ras in Multiple Myeloma Show Different Clinical Associations: Analysis of the Eastern Cooperative Oncology Group Phase III Trial,” Blood, vol. 88, no. 7, pp. 2699–2706, Oct. 1996. [CrossRef]
- B. Zhang and R. G. Fenton, “Proliferation of IL-6-independent multiple myeloma does not require the activity of extracellular signal-regulated kinases (ERK1/2),” J Cell Physiol, vol. 193, no. 1, pp. 42–54, Oct. 2002. [CrossRef]
- D. Ma et al., “Crucial role of heme oxygenase-1 in the sensitivity of acute myeloid leukemia cell line Kasumi-1 to ursolic acid,” Anticancer Drugs, vol. 25, no. 4, pp. 406–414, 2014. [CrossRef]
- W. Wu et al., “Potential crosstalk of the interleukin-6-heme oxygenase-1-dependent mechanism involved in resistance to lenalidomide in multiple myeloma cells,” FEBS J, vol. 283, no. 5, pp. 834–849, Mar. 2016. [CrossRef]
- J. Wang et al., “Bone marrow stromal cell-derived exosomes as communicators in drug resistance in multiple myeloma cells,” Blood, vol. 124, no. 4, pp. 555–566, Jul. 2014. [CrossRef]
- E. Håland et al., “TAK1-inhibitors are cytotoxic for multiple myeloma cells alone and in combination with melphalan,” Oncotarget, vol. 12, no. 21, pp. 2158–2168, Oct. 2021. [CrossRef]
- J. Zhang et al., “Synergistic action of 5Z-7-oxozeaenol and bortezomib in inducing apoptosis of Burkitt lymphoma cell line Daudi,” Tumour Biol, vol. 37, no. 1, pp. 531–539, Jan. 2016. [CrossRef]
- M. Guo et al., “Targeting MK2 Is a Novel Approach to Interfere in Multiple Myeloma,” Front Oncol, vol. 9, no. AUG, 2019. [CrossRef]
- W. Xiao et al., “Rafoxanide, an organohalogen drug, triggers apoptosis and cell cycle arrest in multiple myeloma by enhancing DNA damage responses and suppressing the p38 MAPK pathway,” Cancer Lett, vol. 444, pp. 45–59, Mar. 2019. [CrossRef]
- A. Soriani et al., “p38 MAPK differentially controls NK activating ligands at transcriptional and post-transcriptional level on multiple myeloma cells,” Oncoimmunology, vol. 6, no. 1, Jan. 2016. [CrossRef]
- R. M. Koerber et al., “Analysis of the anti-proliferative and the pro-apoptotic efficacy of Syk inhibition in multiple myeloma,” Exp Hematol Oncol, vol. 4, no. 1, Aug. 2015. [CrossRef]
- A. Li, X. Chen, Z. Jing, and J. Chen, “Trifluoperazine induces cellular apoptosis by inhibiting autophagy and targeting NUPR1 in multiple myeloma,” FEBS Open Bio, vol. 10, no. 10, pp. 2097–2106, Oct. 2020. [CrossRef]
- R. E. Tiedemann et al., “Identification of a potent natural triterpenoid inhibitor of proteosome chymotrypsin-like activity and NF-kappaB with antimyeloma activity in vitro and in vivo,” Blood, vol. 113, no. 17, pp. 4027–4037, 2009. [CrossRef]
- X. M. Xu et al., “[Effect of Celastrol Based on IRAK4/ERK/p38 Signaling Pathway on Proliferation and Apoptosis of Multiple Myeloma Cells],” Zhongguo Shi Yan Xue Ye Xue Za Zhi, vol. 30, no. 1, pp. 175–182, Feb. 2022. [CrossRef]
- S. S. Bhandarkar et al., “Tris (dibenzylideneacetone) dipalladium, a N-myristoyltransferase-1 inhibitor, is effective against melanoma growth in vitro and in vivo,” Clin Cancer Res, vol. 14, no. 18, pp. 5743–5748, Sep. 2008. [CrossRef]
- P. de la Puente, F. Azab, B. Muz, M. Luderer, J. Arbiser, and A. K. Azab, “Tris DBA palladium overcomes hypoxia-mediated drug resistance in multiple myeloma,” Leuk Lymphoma, vol. 57, no. 7, pp. 1677–1686, Jul. 2016. [CrossRef]
- A. T. Stefka, D. Johnson, S. Rosebeck, J. H. Park, Y. Nakamura, and A. J. Jakubowiak, “Potent anti-myeloma activity of the TOPK inhibitor OTS514 in pre-clinical models,” Cancer Med, vol. 9, no. 1, pp. 324–334, Jan. 2020. [CrossRef]
- J. He et al., “Therapeutic effects of the novel subtype-selective histone deacetylase inhibitor chidamide on myeloma-associated bone disease,” Haematologica, vol. 103, no. 8, pp. 1369–1379, Jul. 2018. [CrossRef]
- L. H, H. J, and Y. J, “Tumor cell p38 MAPK: A trigger of cancer bone osteolysis,” Cancer Cell Microenviron, vol. 2, no. 1, Jan. 2015. [CrossRef]
- L. Trentin et al., “Multiple myeloma plasma cells show different chemokine receptor profiles at sites of disease activity,” Br J Haematol, vol. 138, no. 5, pp. 594–602, Sep. 2007. [CrossRef]
- M. Broussas et al., “A New Anti-CXCR4 Antibody That Blocks the CXCR4/SDF-1 Axis and Mobilizes Effector Cells,” Mol Cancer Ther, vol. 15, no. 8, pp. 1890–1899, Aug. 2016. [CrossRef]
- M. K. Pandey et al., “Gambogic acid inhibits multiple myeloma mediated osteoclastogenesis through suppression of chemokine receptor CXCR4 signaling pathways,” Exp Hematol, vol. 42, no. 10, pp. 883–896, Oct. 2014. [CrossRef]
- J. Li, J. Zhu, B. Cao, and X. Mao, “The mTOR signaling pathway is an emerging therapeutic target in multiple myeloma,” Curr Pharm Des, vol. 20, no. 1, pp. 125–135, Jan. 2014. [CrossRef]
- K. Han et al., “SC06, a novel small molecule compound, displays preclinical activity against multiple myeloma by disrupting the mTOR signaling pathway,” Sci Rep, vol. 5, Sep. 2015. [CrossRef]
- H. Y. Cho and S. W. Lee, “TLR5 activation by flagellin induces doxorubicin resistance via interleukin-6 (IL-6) expression in two multiple myeloma cells,” Cell Immunol, vol. 289, no. 1–2, pp. 27–35, 2014. [CrossRef]
- J. Abdi, T. Mutis, J. Garssen, and F. Redegeld, “Characterization of the Toll-like receptor expression profile in human multiple myeloma cells,” PLoS One, vol. 8, no. 4, Apr. 2013. [CrossRef]
- G. Jego, D. Chiron, K. Berthenet, and C. Pellat-Deceunynck, “Modulation of normal and malignant plasma cells function by toll-like receptors,” Front Biosci (Elite Ed), vol. 4, no. 6, pp. 2289–2301, Jan. 2012. [CrossRef]
- Z. Liu, T. Li, K. Jiang, Q. Huang, Y. Chen, and F. Qian, “Induction of chemoresistance by all-trans retinoic acid via a noncanonical signaling in multiple myeloma cells,” PLoS One, vol. 9, no. 1, Jan. 2014. [CrossRef]
- T. Otsuki, H. Sakaguchi, T. Hatayama, P. Wu, A. Takata, and F. Hyodoh, “Effects of all-trans retinoic acid (ATRA) on human myeloma cells,” Leuk Lymphoma, vol. 44, no. 10, pp. 1651–1656, Oct. 2003. [CrossRef]
- C. Simard, M. Cloutier, and S. Néron, “Feasibility study: phosphospecific flow cytometry enabling rapid functional analysis of bone marrow samples from patients with multiple myeloma,” Cytometry B Clin Cytom, vol. 86, no. 2, pp. 139–144, Mar. 2014. [CrossRef]
Figure 1.
Illustration of the structure of the different p38 isoforms with their corresponding alternative splicing isoforms and their different domains. Solid blue line represents the gene sequence of the canonical isoform for each isoform. Blue line with segments in lighter blue represent the variants of each isoform where the light blue color represents segments with a different gene sequence than the canonical one. Yellow line indicates the protein kinase domain. Yellow circles with the letter P indicate the TXY phosphorylation motif (p38α reports an additional phosphorylation site by ZAP70 [T 323]). Orange circles indicate ATP binding sites. Pink line in the p38γ isoform variant P53778-2, represents lacking residues.
Figure 1.
Illustration of the structure of the different p38 isoforms with their corresponding alternative splicing isoforms and their different domains. Solid blue line represents the gene sequence of the canonical isoform for each isoform. Blue line with segments in lighter blue represent the variants of each isoform where the light blue color represents segments with a different gene sequence than the canonical one. Yellow line indicates the protein kinase domain. Yellow circles with the letter P indicate the TXY phosphorylation motif (p38α reports an additional phosphorylation site by ZAP70 [T 323]). Orange circles indicate ATP binding sites. Pink line in the p38γ isoform variant P53778-2, represents lacking residues.
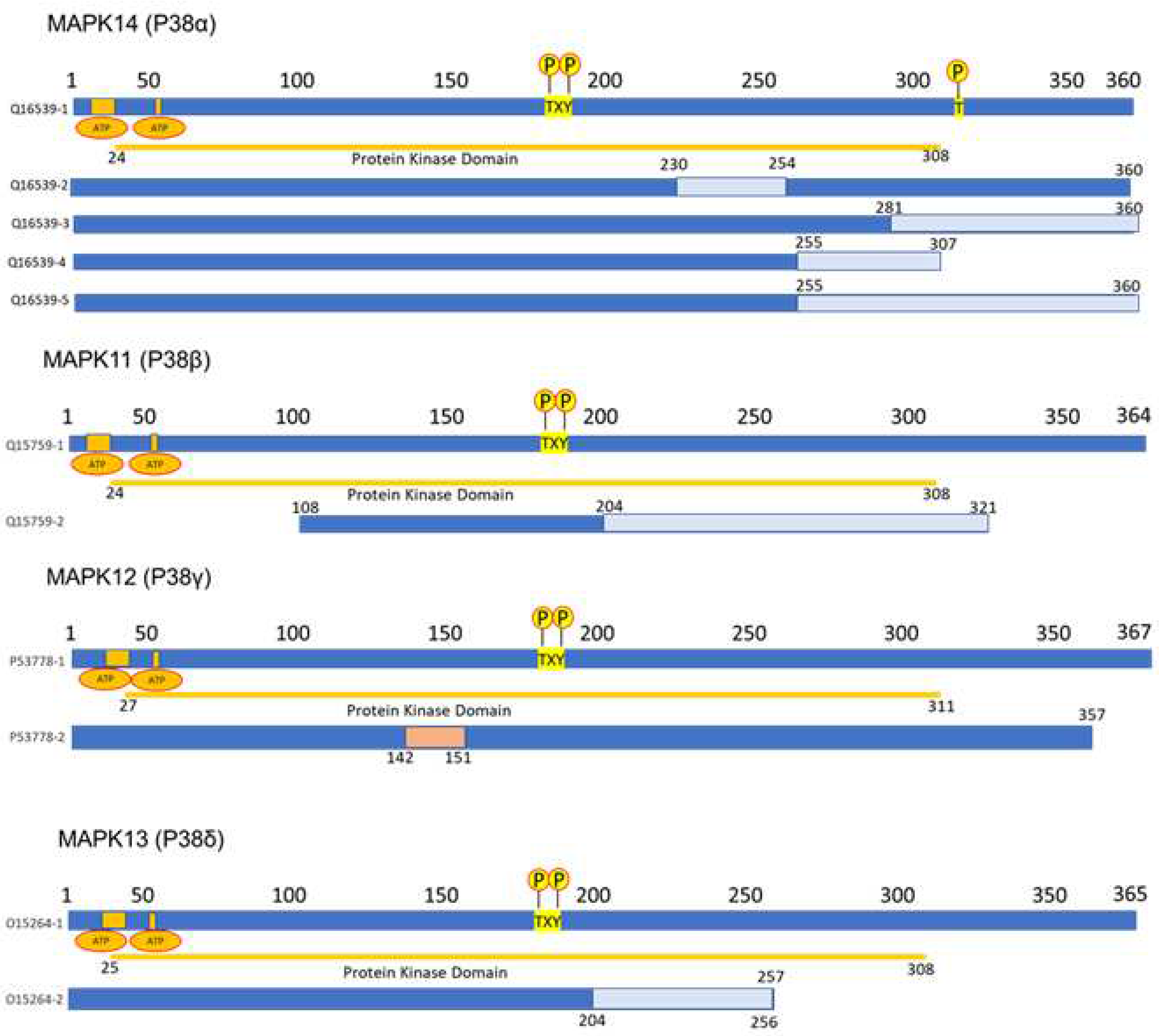
Figure 2.
Expression of four isoforms of p38 were evaluated using GEO Dataset: GSE146649 at different MM stages. HD, healthy donor; DX, diagnostic; CR, complete response; ER, early relapse. (n indicates the number of samples reported and used for this graph) (*p < 0.05, **p < 0.01, ***p < 0.001). ANOVA analysis was performed with a Bonferroni correction using GraphPad Prism 5.0.
Figure 2.
Expression of four isoforms of p38 were evaluated using GEO Dataset: GSE146649 at different MM stages. HD, healthy donor; DX, diagnostic; CR, complete response; ER, early relapse. (n indicates the number of samples reported and used for this graph) (*p < 0.05, **p < 0.01, ***p < 0.001). ANOVA analysis was performed with a Bonferroni correction using GraphPad Prism 5.0.
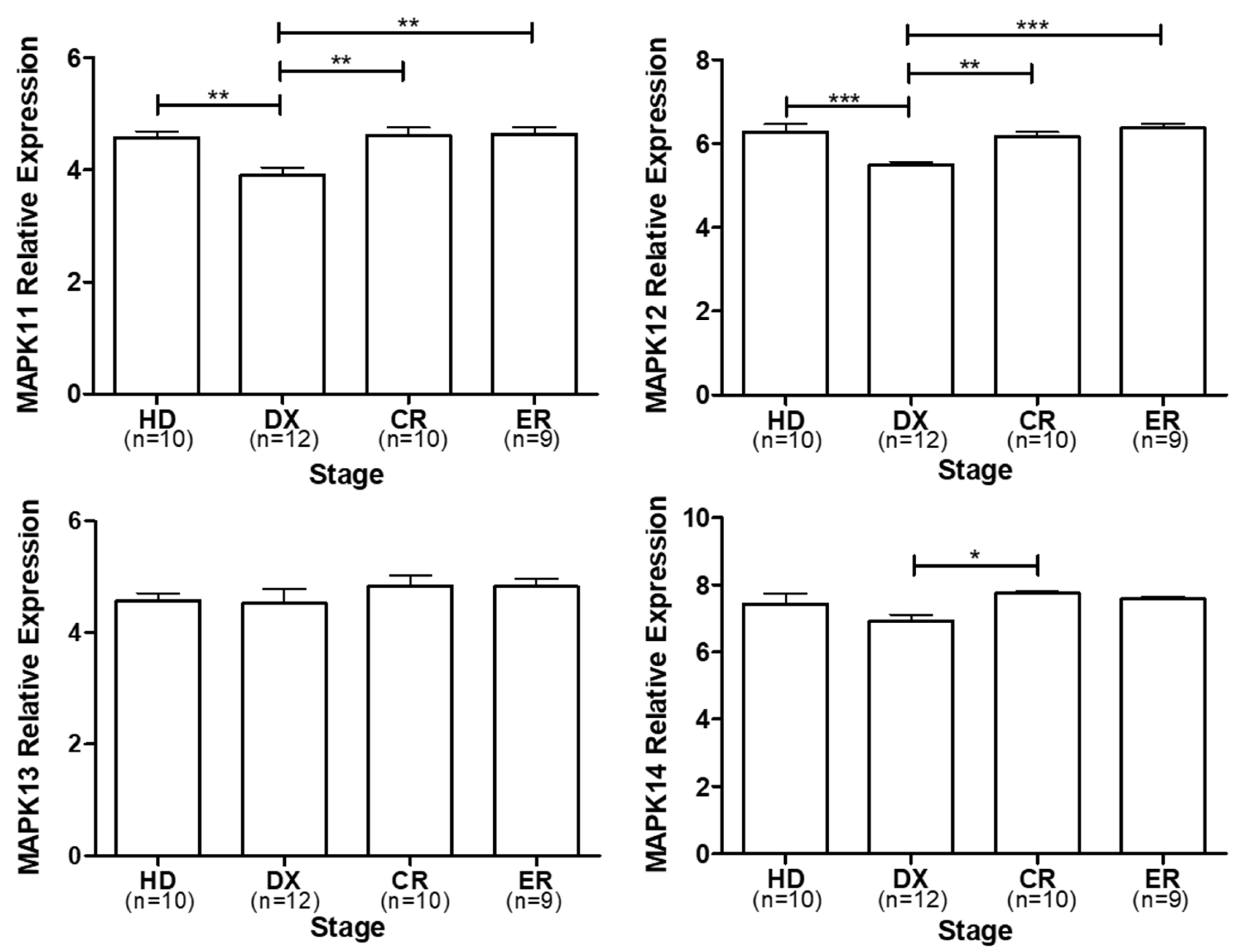
Figure 3.
Schematic representation of p38 role in multiple myeloma and its biological consequences of inhibition.
Figure 3.
Schematic representation of p38 role in multiple myeloma and its biological consequences of inhibition.
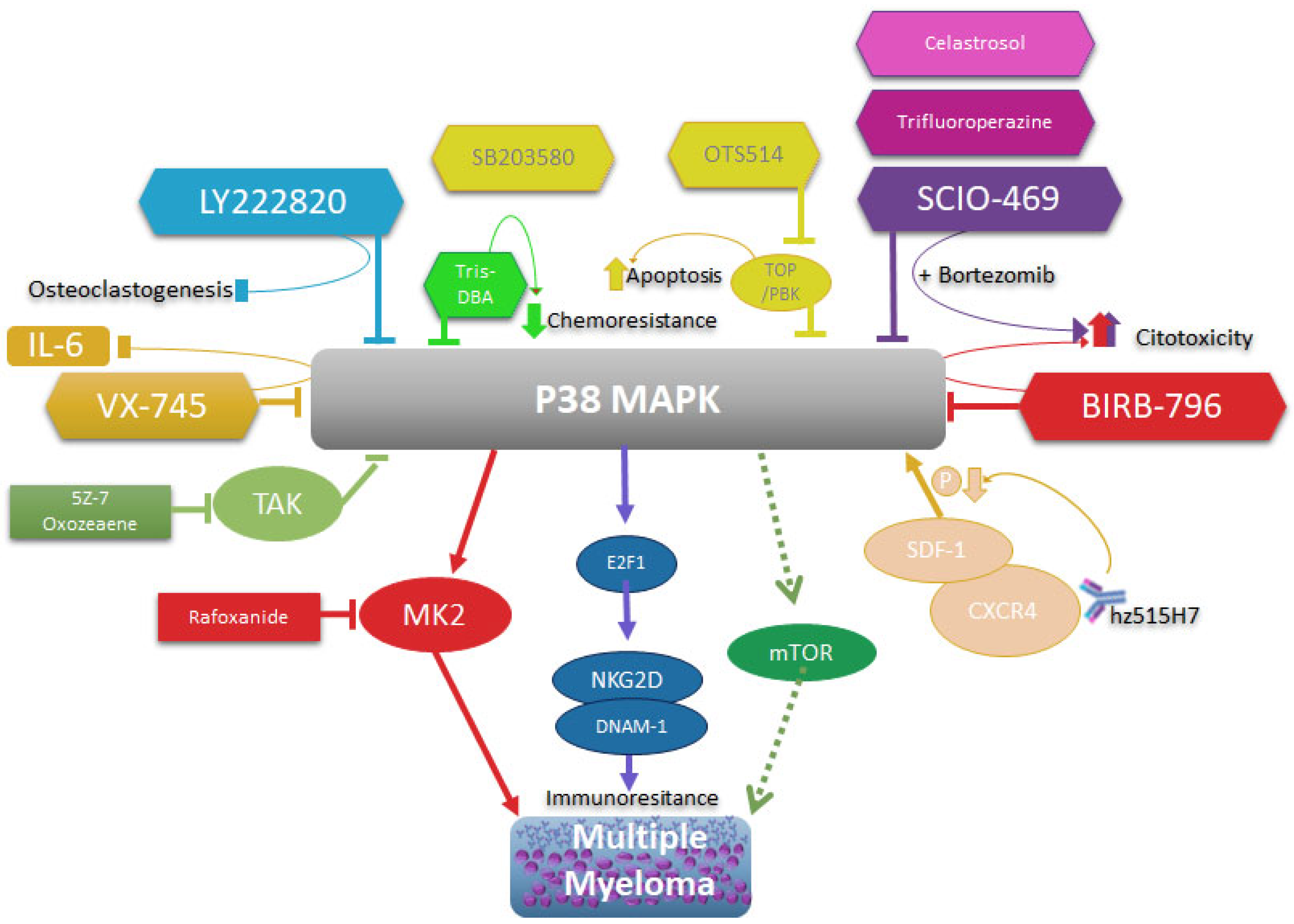

Table 1.
Putative binding sites for transcription factors in p38 promoter isoforms in EPD.
| Transcription factor | Putative binding site positions relative to TSS | |||
| MAPK14_1 | MAPK11_1 | MAPK12_1 | MAPK13_1 | |
| ARNT: HIF1A | -466, -455, -260 | -93, | -40, -88 | -120, 32 |
| ASCL1 | -933, -901, -880, -878, -785, -603, -602, -392, -391, -248, -59, 22, 2 | 74, 73, -459, -506, -550, -575, -609, -610, -755, -830, -937 | 44, 42, -2, -3, -190, -214, -215, -309, -310, -410, -411, -518, -523, -666, -688, -810, -812, -882, -883, -968, -982 | -993, -810, -809, -551, -278, -264, -254, -185, -184, -103, -102, -87, -40, -17, 25, 26 |
| ATF4 | -823, -777, -774 | |||
| ATF7 | -824, -823 | -92 | ||
| Ahr: Arnt | -466, -455, -334, -260 | -93 | -120, 32 | |
| Arid3a | -860 | -929, -490, -485 | ||
| Ascl2 | -901, -900, -785, -392, -391, -381, -380, -249, -248, -59, 24, 25 | -380, -829 | -2, -214, -309, -523, -524, -882, -883 | -809, -580, -278, -185, -184, -87 |
| Atf1 | -823 | -579 | -92 | -697 |
| BACH2 | -633, -629 | -392 | -508, -504 | |
| Bcl6 | -874 | -356, -497, -672, -732, -884 | -382, -927 | |
| CEBPA | -979, -823, -549 | -548 | -947 | |
| CTCF | -768, -513, -275, -209, 1 | -178, -219, -221, -253, -383, -584, -848 | 27, -52, -76, -146, -270, -336, -601, -672, -799, -891 | -870, -87, -54, -14 |
| E2F1 | -597, -596, -270, -269, -218, -217 | -183, -184, -290, -543, -544, -660 | 35, 34 | 15 |
| EGR1 | -465, -394, -28 | 34, -8, -252, -783, -903, -949 | 30, -256, -335, -602, -744, -917 | -866, -835, -604, -384, -278, -246, -147, -61, -39, -14 |
| ELF1 | -62, -28, 33 | 82, -126, -154, -489, -880 | 24, -120, -438 | -155, 73, 76 |
| JUN | -887, -822 | -609, -460 | ||
| KLF4 | -770, -587, -453, -334 | -91, -684 | -94, -290, -906 | -380, -122, -58 |
| MYC | -942, -785, -603, -602, -444, -392, -391, -266 | -230, -506 | -3, -87, -88, -518, -523, -643, -644 | -984, -606, -199, -185, -184, -103, -102, -87 |
| SMAD2:SMAD3: SMAD4 | -896, -464, -251, -204 | -147, -211, -926 | -260, -266, -563, -625, -829, -865, -910 | |
| YY1 | -977, -962, -547, -98 | -949, -814, -672 | ||
| XBP1 | -943 | -578 | -738 | |
| TP53 | -759, -758 | -578 | -754 | -959, -958, -437, -125, -124 |
| Stat4 | -419 | -672, -676 | -395 | |
| SP1 | -925, -881, -659, -633, -499, -225, -186, -141, -114, 72 | 63, -12, -33, -46, -87, -141, -165, -192, -200, -279, -294, -347, -508, -527, -543, -598, -650, -683, -759, -905 | 78, 28, 16, -19, -47, -103, -147, -164, -195, -206, -267, -289, -317, -592, -670, -740, -803, -822, -853, -905 | -868, -837, -666, -522, -329, -290, -257, -205, -59, -44, -25, -9 |
| POU1F1 | -575 | -407 | -919, -490, -395 | |
| NFKB1 | -982, -938, -552, -506, -32 | -69, -101, -102, -788, -945 | 34, 5, -62, -314, -513, -586, -899, -961 | -988, -944, -467, -466, -232, -211 |
Table 2.
Possible miRNAs regulating isoforms of p38.
| . | MAPK11 | MAPK12 | MAPK13 | MAPK14 |
| Predicted microRNAs (miRtarBase) | hsa-miR-122-5p, hsa-miR-124-3p, hsa-let-7a-5p | hsa-miR-18a-5p, hsa-miR-150-5p, hsa-miR-18b-5p, hsa-miR-3134, hsa-miR-3691-5p, hsa-miR-4434, hsa-miR-4516, hsa-miR-4525, hsa-miR-4531, hsa-miR-4534, hsa-miR-4690-3p, hsa-miR-4731-5p, hsa-miR-4735-3p, hsa-miR-4761-5p, hsa-miR-4773, hsa-miR-5010-5p, hsa-miR-5187-5p, hsa-miR-5589-5p, hsa-miR-5685, hsa-miR-5703, hsa-miR-6778-3p, hsa-miR-6795-5p, hsa-miR-6798-5p, hsa-miR-6814-5p, hsa-miR-6887-5p, hsa-miR-8082 | hsa-miR-124-3p, hsa-miR-24-3p, hsa-miR-199a-3p, hsa-miR-200a-3p, hsa-miR-141-3p, hsa-miR-125b-5p, hsa-miR-214-3p, hsa-miR-155-5p, hsa-miR-17-5p, hsa-miR-106a-5p | |
| Bio-Predicted(TargetScan) | hsa-miR-325-3p | hsa-miR-125a-5p, has-miR-125b-5p, has-miR-4319 | hsa-miR-3681-3p, hsa-miR-128-3p, has-miR-216-3p |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



