
Submitted:
03 January 2024
Posted:
03 January 2024
You are already at the latest version
Abstract
Myotonic dystrophy is a hereditary disorder with systemic involvement. The Italian Neuro-Cardiology Network (INCN-RNC) is a unique collaborative experience involving neurology units combined with cardio-arrhythmology units. The INCN facilitates the creation of integrated neuro-cardiac teams in Neuromuscular Disease Centers for the management of cardiovascular involvement in the treatment of MD1.
Keywords:
Myotonic Dystrophy
; Cardiovascular Disease
; Neurological Disease
; Sudden Cardiac Death
; Arrythmogenic risk.
1. Introduction
The Italian Neuro-Cardiology Network (INCN-RNC) is a unique collaborative experience involving neurology units paired with cardio-arrhythmology unit. In January 2021 the 8th INCN-RNC annual meeting raised up the need of a coordinated and integrated model of care for patients with Myotonic Dystrophy type 1 (MD1). The board of neuromuscular disease experts discussed the current and emerging apparent gaps in the comprehensive care of cardiovascular involvement in MD1, including operational / logistical issues for health systems and integrated networks, to reach a consensus. Cardiovascular comorbidities in the MD1, despite substantial advances in research and clinical care, have relevant gaps in clinical evidence and cause uncertainty about best practices for the treatment and early diagnosis of arrhythmic and non-arrhythmic disorders. On January 2021 and May 2022 at the annual National meetings of INCN-RNC took place two symposia and round tables on "The practical management of cardiac involvement in patients with myotonic dystrophy: the need for interdisciplinary action". On February 2023, during the 10th annual meeting of the INCN-RNC, the council approved the first draft of this consensus document; on December 2023 the following definitive version was approved.
Myotonic Dystrophy
Myotonic Dystrophy (MD) is a dominantly inherited multisystem disorder caused by expanded CTG repeats in the 3’ UTR of the DMPK gene (MD1) or CCTG repeats in the first intron of the CNBP gene (Myotonic Dystrophy type 2 or MD2). The main pathogenic mechanism of MD is the toxic gain of function of RNAs transcribed from expanded alleles that fold into hairpin structures and accumulate in nuclear foci interfering with the activity of muscle-blind-like (MBNLs) and -CUGBP Elav-like Family Member 1 (CELF1) proteins[1]. These two classes of RNA binding proteins antagonistically regulate the alternative splicing of developmental genes and their alterations in MD tissues (skeletal muscle, heart, brain etc.) lead to an aberrant alternative splicing of multiple genes, with the preferential expression of immature protein isoforms[2]. Despite many clinical and molecular similarities, MD1 and MD2 manifest with different features, including lack of congenital or childhood forms, minimal facial signs and reduced risk of cardiac conduction defects in MD2 compared to MD1, together with different distribution of muscle involvement and pattern of muscle involvement at muscle biopsy and muscle MRI between the two forms [3,4]. This issue could be explained by the concurrence of other pathogenic mechanisms able to modulate the phenotype, including epigenetic modifications at the respective gene loci induced by the pathological expansions, the occurrence of antisense “RAN” translation and haploinsufficiency of the respective genes [5].
MD, as a whole, is the most common type of muscular dystrophy among adult Caucasians. However, the geographic and ethnic distribution of this disease is very uneven among different populations. Prevalence estimates of molecularly defined MD1 report values spanning over a very large range, between 0.43 and 158 cases per 100.000, depending on the population studied. This enormous variability reflects a very low prevalence of the disease in the Far East, as opposed to very high rates, depending on founder effects, observed among French Canadians, Basques and Afrikaners. On the other hand, myotonic dystrophies are virtually absent in native populations of the Americas, Africa and Oceania [6]. A recent study performed in the province of Rome, Italy, provided, for the first time, age-adjusted, sex- and age-specific prevalence estimate of MD1 and MD2 in the same area, with values of 8.35/100,000 for females and 11.07/100,000 for males in MD1. Values about tenfold lower were reported for MD2, with a slight female preponderance (6). MD1 and MD2 are progressive, multisystem disorders, characterized by muscle weakness, myotonia, cataracts, cardiac conduction defects and arrhythmia, respiratory and endocrine disturbances, excessive daytime sleepiness, cognitive and personality trait abnormalities, skin alterations etc. [7,8]. In addition, several features of the metabolic syndrome are common, including insulin resistance, increased waist circumference, dyslipidemia, and reduced levels of adiponectin. Surprisingly, despite the presence of all these metabolic risk factors, MD1 patients do not have higher chances of developing diabetes mellitus, coronary heart disease or stroke, compared to the general population. On the other hand, MD1 is associated with an increased risk of developing several types of benign or malignant tumors [9]. At present, there is no curative or disease-modifying treatment for MD1 or MD2, and management focuses on genetic counseling, preserving function and independence, preventing cardiopulmonary complications, and symptomatic treatment of myotonia, daytime sleepiness etc. [10,11]
1. Cardiovascular Involvement in MD1
Cardiac involvement occurs in 80% of MD1 patients and it often precedes the involvement of skeletal muscle [12]. Cardiac involvement in patients with MD1 occurs as a degenerative process, with progressive fibrosis and fatty replacement of the myocardium, which involves not only the specialized conduction system but also areas, initially unaffected, of the atrial and ventricular myocardium [13,14,15]. This anatomo-pathologic substrate may, on one hand, facilitate the development of cardiac conduction diseases, ventricular tachycardia and sudden cardiac death on the other hand, it may be responsible of ventricular dyssynchrony leading to cardiomyopathy with systolic dysfunction.
Conduction System Disease
Conduction system disease is the most prevalent cardiac abnormality in MD1 patients. The first-degree atrioventricular block (AVB) is reported in 28.2%- 34.1%and QRS complex> 120 ms in 18.4%- 19.9% [16,17]. These electrocardiographic conduction abnormalities are independent predictors for a prolonged His-ventricle (HV) interval ≥70 ms on electrophysiological study (EPS) [18], which early identifies a subgroup of MD1 patients in need of cardiac pacing [19].
Atrial Fibrillation
Atrial fibrillation (AF), often asymptomatic, frequently occurs in MD1 patients with a prevalence of 11%, about 70-fold higher than the general population [20,21]; however, it could be even higher, about 25%, if we consider cardiac implanted electronic device detected AF events [22]. MD1 patients affected by AF were more often males, had lower left ventricular ejection fraction (LVEF), electro-mechanical echocardiographic and electrocardiographic abnormalities [20,23,24]. AF has been associated with higher overall mortality in MD1 patients [20], however the association with sudden cardiac death is still controversial.
Ventricular Arrhythmias
The prevalence rates of non-sustained and sustained ventricular tachycardia (VT) were 2.2% and 0.8%, respectively[25]. The personal history of non-sustained VT has been recently identified as the only independent predictor of sustained VT [17] and it is considered a criterion to prefer implanted cardioverter defibrillator (ICD) over pacemaker (PM) for MD1 patients in need of permanent cardiac pacing [26]. For this reason, the early identification of a non-sustained VT in MD1 patients may be of pivotal importance to optimize clinical management and to choose the best device (PM vs ICD) in order to prevent sudden cardiac death [27]. Several studies have shown evidence of increased dispersion of ventricular repolarization (QTc dispersion, JTc dispersion, transmural dispersion of repolarization, QT variability index) and sympatho-vagal balance in patients with MD1 (Heart Rate Variability) suggesting a potential interest of these measures to predict ventricular arrhythmias [28,29,30,31].
Sudden Cardiac Death
MD1 patients have a three-fold higher risk of sudden cardiac death (SCD) than age-matched healthy controls. The annual incidence of SCD has been estimated at 0.53%–1.16% [32]. SCD accounts for up to 33% of all deaths in MD1. Even if the mechanisms leading to SCD remain controversial, the complete AVB, asystole and VT may represent the most prevalent cause of SCD in MD1 patients.
Independent predictors of SCD are (i) clinical diagnosis of atrial tachyarrhythmia and electrocardiogram (ECG) with one of the following features: any rhythm other than sinus rhythm, PR interval ≥ 240 ms, QRS duration ≥ 120 ms, and second- or third-degree atrioventricular block [32] and age, family history of SCD, and left bundle branch block [33].
Cardiomyopathy and Heart Failure
Differently from arrhythmias, little is still known about the epidemiology of left ventricular (LV) dysfunction and heart failure (HF) among MD1 patients [21,34]. The prevalence of LV systolic dysfunction (LVSD), assessed by trans-thoracic echocardiography (TTE), ranged from 0% to approximately 21% [34]. The causes of LVSD are not completely understood; however, they might include intra-ventricular (IV) and atrio-ventricular (AV) conduction time delay, atrial or ventricular arrhythmias and ventricular myocardial fibrosis. MD1 patients with prolonged PR or QRS intervals showed four-times higher risk to develop LVSD or HF [35]. Among MD1 patients with AF the prevalence of LVSD accounted up to 46% [34]. Contrast enhanced cardiac magnetic resonance imaging (MRI) studies have detected myocardial fibrosis in 13 to 40% of MD1 patients [36,37,38].
Data regarding LV diastolic dysfunction (LVDD) in MD1 patients are quite lacking. Up to date, mild diastolic dysfunction has been observed in 5% to 50% of MD1 patients [39]. The diastolic dysfunction in MD1 might be related to AF, fibrotic degenerative changes of the myocardium (likely affecting LV relaxation) and the impaired calcium metabolism in cardiomyocytes. No association between LVDD and AV or IV conduction defects has been observed[40]. Whether AV/ IV conduction defect may cause LV mechanical impairment or whether both electrical and mechanical impairments may be the common result of fibrosis of the myocardium and conduction system, still needs to be clarified.
The prevalence of symptomatic HF in MD1 subjects ranges from 0% to 7.1% [32], however HF symptoms should be underestimated due to the limited level of activity of MD1 patients. The early diagnosis of HF disease is of pivotal importance, since it increased four-times higher the risk of all-cause death, and a six-times higher the risk of cardiac death [35].
Therefore, although trials showing benefit from the treatment of HF in Steinert disease are lacking, it seems reasonable that treatment for HF should be started early. In particular, the administration of angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor antagonists (ARB) could be of particular benefit in MD1 due to anti-fibrotic properties [41] and is recommended when LVSD was< 50%. The use of beta-blockers should be reserved to patients without AV conduction defects or recipient of PM and/or ICD; the up-titrate drug dosage should be applied on the basis of individual response and toleration. Cardiac resynchronization therapy for patients with persistent symptomatic HF (New York Heart Association functional class III) due to LVSD (LVEF≤0.35%) with large QRS (> 150 ms) with left bundle branch block pattern and normal sinus rhythm while on optimal guideline-directed medical therapy [42,43].
Hypotension
It is generally recognized from clinicians, that MD1 subjects have low blood pressure (BP) values. However, only a few, non-systematic studies have showed that consecutive MD1 patients have significantly lower BP values than healthy control subjects [44]. It is still not clear whether low BP may be related to the pathophysiology of the disease or to autonomic cardiac dysfunction, predominantly parasympathetic, that is common in MD1 subjects, or if it may be a specific complication of the disease related to the genetic mutation. However, low BP values have recently demonstrated to be a marker of disease severity, and to contribute, when added to other clinical, electrocardiographic and respiratory parameters, to risk stratify MD1 patients at risk of death [45].
Stroke and Systemic Embolism
The prevalence of both symptomatic and asymptomatic ischemic strokes in MD1 patients was about 6.5%. The AF/flutter was found in 55% of MD1 patients with ischemic stroke. All patients with stroke had CHADS2 and CHA2DS2-VASc scores higher than 2 [46]. An expert consensus opinion of the AHA for the management of MD1 patients suggests use the CHA2DS2-VASc score to stratify the thromboembolic risk; however, it also outlines to carefully consider their increased fall risk of due to the underlying neuromuscular disease and muscle weakness. Since studies comparing vitamin K antagonists (VKAs) and direct oral anticoagulants (DOACs) in this clinical setting are lacking; a careful evaluation of renal function is warranted, eventually based on dosage of cystatin C, because serum creatinine may be low to non-detectable in the setting of low muscle mass (which is not uncommon in MD1 subjects) [42].
2. Non-Invasive Cardiac Evaluation
In order to optimize the clinical management of MD1 patients, neurologists should early identify referent cardiologists/electrophysiologists with expertise in neuromuscular disorders [42].
The cardiologic evaluation comprehensive of ECG, TTE and 24-hours Holter ECG monitoring are highly recommended at the time of disease diagnosis. The cardiologic clinical history investigation should focus on eventual warning symptoms, including heeling, dizziness, pre-syncope, syncope or breathlessness. Moreover, even for completely asymptomatic subjects, annual cardiologic visits with ECG are recommended, since MD1 is a progressive disease [43]. Given the increased prevalence of AF in MD1 patients and its association with the higher overall mortality, we suggest to perform careful electrocardiographic monitoring by 24-hours Holter ECG at least annually in the overall MD1 population and the daily remote monitoring of those with cardiac implantable electronic devices (CIEDs). For MD1 patients at increased risk of AF, according to electrocardiographic and echocardiographic risk parameters, an external loop recorder should be considered.
Twelve Lead ECG
Twelve lead ECG is an essential tool for the risk stratification of life-threatening arrhythmic disorders in MD1 patients. It’s indicated in all patients upon confirmation of MD1 diagnosis, and annually thereafter, due to the risk of disease progression.[42,43] A PR interval >200 ms and/or QRS duration >100 ms should be an indication to perform an EPS for detecting a prolonged HV interval (> 70 ms) in need of cardiac pacing [18,47,48]. However, it should be noted that up of 66.1% of MD1 patients with these electrocardiographic findings may have normal HV intervals [49]. Moreover, in MD1 patients with QRS >120 ms and PR >240 ms a pacemaker may be considered to reduce the risk of SCD [26,32].
24. -Hours Holter ECG Monitoring
Ambulatory electrocardiographic monitoring is a useful tool for identification of paroxysmal second or third degree AV blocks, or intermittent bundle branch block, that do not appear at rest ECG. It’s indicated at the time of MD1 diagnosis, and in case of occurrence of either ECG abnormalities (AV or intra-ventricular blocks) or symptoms including heeling, dizziness, pre-syncope, syncope [43]. Moreover, it may be useful to identify asymptomatic episode of non-sustained VT or paroxysmal AF, which may impact on the patients ‘prognosis and need to a careful management [17,50].
Transthoracic Echocardiogram
TTE is the most widely imaging tool used to obtain structural and functional information about the heart. MD1 patients should undergo cardiac imaging examination at baseline and every 1 to 5 years thereafter, if the initial imaging study is normal [43]. Particular attention should be given to subjects with baseline electrocardiographic abnormalities or arrhythmias, since the systolic dysfunction seems to be more common in these subgroups [40]. Moreover, new echocardiographic techniques, such as three-dimensional (3D) TTE or speckle tracking analysis, should be performed to empower bi-dimensional TTE diagnostic and prognostic ability[39,51].
Cardiac Magnetic Resonance
Contrast enhanced cardiac MRI is a highly sensitive non-invasive tool for the detection of functional and structural myocardial abnormalities. Besides parameters easily available with TTE examination, cardiac MRI may detect eventual myocardial damage suggestive of scar through late gadolinium enhancement (LGE). Moreover, it may quantify interstitial fibrosis through extracellular volume (ECV) fraction technique [52] and can detect even subtle myocardial deformation or contractility impairment (as per localized degenerated myocardial tissue) through the cardiac strain technique [53].
Several observational MRI studies showed cardiac structural abnormalities among MD1 subjects, including reduced LV [38,54] or right ventricle [37] systolic function, LV hypertrophy [38] and LV non-compaction [37]; moreover, reduced values of myocardial strain, both in the longitudinal, circumferential and strain area components, have been described among MD1 patients with preserved LVEF, as per an early detection of LV contractility impairment [54,55]. (Interestingly, a non-negligible prevalence of LV LGE with a non-ischemic distribution pattern (i.e. in the midwall or subepicardial myocardial layers), mostly located in the inter-ventricular septum or in the postero-lateral wall, has been detected in MD1 subjects, with a prevalence ranging from 12.5% [37,38] to 42% [56,57]. The prognostic role of LGE or interstitial fibrosis in MD1 patients is still debated and need of further studies.
3. Invasive Cardiac Evaluation and Treatment
Electrophysiological Study
EPS should increase the accuracy of the SCD risk stratification in MD1 patients with electrocardiographic abnormalities (PR interval > 200 ms or QRS>100 ms) through the evaluation of HV interval prolongation (> 70 ms), which identify those in need of prophylactic cardiac pacing. To date, little is still known about the timing and the role of programmed ventricular stimulation for arrhythmic risk stratification [27,58]. The ACADEMY 1, a prospective single-center study about the electrophysiological study guided ICD strategy in prevention of arrhythmic cardiac death in MD1 patients, suggest the inducibility of VT has a limited value in the arrhythmic risk stratification among MD1 patients [59].
Loop Recorder
The use of implanted loop recorder (ILR), a small well tolerated device for monitoring of the cardiac rhythm over several years, should be considered a feasible option for detecting the clinically asymptomatic progression of conduction disorders or spontaneous VT and for helping in the decision about the best device choice to prevent sudden cardiac death. It should be useful in MD1 patients with first-degree AV, fascicular, or bundle branch block and HV interval < 70 ms [60].
Pacemaker
The permanent cardiac pacing is indicated in patients with any second- and third-degree AVB or His-ventricle (HV) interval >70 ms, regardless the symptoms; and it may be considered in those with QRS >120 ms and PR >240 ms. Atrial pacing in the Bachmann bundle region was associated with a reduction of atrial electromechanical delay [61] and the risk of R-wave oversensing on the atrial lead [62], compared with right atrial stimulation, however, it showed no benefit for the prevention of AF onset [63]. The activation of right atrial preference pacing [64,65,66,67] and minimal ventricular pacing [68] algorithms seems to be an efficient strategy to reduce the risk of AF in MD1 patients implanted with a PM. An increase of the incidence AF has been shown in patients with a higher rate of right ventricular pacing and a lower rate of atrial stimulation [69].
Implantable Cardioverter-Defibrillator (ICD)
The ICD implantation may be considered for all MD1 in which permanent with permanent pacing indication and spontaneous or EPS inducible VT, even when asymptomatic or with preserved cardiac function. Because conduction system disease and the need for right ventricular pacing frequently accompany left ventricular systolic dysfunction in advanced neuromuscular disorders, the cardiac resynchronization therapy (CRT) may be an option for MD1 patients with bundle branch block (especially left bundle branch block), who need a permanent pacemaker implantation; however, there are currently only a few case reports about CRT therapy in MD1 patients [70,71,72].
CIED Remote Monitoring
Regardless of the type of cardiac implanted device (PM, ICD or ILR), a strategy of remote monitoring and interrogation, combined with at least annual in person evaluation, should be adopted to optimize the clinical management of the asymptomatic arrhythmias, to reduce the family-provided healthcare costs, and to overcome the logistic barriers for MD1 patients with motor disability [72,73,74,75].
4. Open issues/Improvement Areas in Organization of Services
Although there is a consensus in the general planning of cardiological assessments once a year, this remains arbitrary, as the lack of a reliable biological marker able to identify patients at higher cardiological risk do not allow to stratify patients on the basis of cardiological risk and therefore to establish a risk-based cardiological assessment follow-up. It is controversial, in this regard, the usefulness of genetic data as a predictor of cardiac complication in MD1. In fact, whereas CTG expansion length correlates with the age at onset of cardiac complications in those patients showing cardiac rhythm or conduction abnormalities, it seems to not predict the occurrence of cardiac complications in MD1 [76]. A possible explication of this leak correlation could be that CTG expansion is different among tissues and unstable over time. Possibly, CTG expansion detected on the leukocytes from blood sampling at the moment of diagnosis could not reflect the expansion in myocardiocytes at the moment of development of cardiac abnormalities, which could explain the weak correlation obtained in previous studies. Conversely, male gender seems to be associated to higher risk of cardiac complications in MD1, suggesting that in clinical assessment MD1 male patients should be monitored with higher attention in the clinical follow-up for the occurrence or progression of cardiac involvement [77]. Possibly, the routine use of cardiac MRI or electrophysiological studies could help to better stratify the risk among patients, even if these methods are expensive or invasive and timing of these studies is still controversial [48].
5. Disease Management Model: The “Neuro-Cardiac Team”
The comprehensive care of patients with neuromuscular diseases which of those affected by MD1 is an interdisciplinary challenge. The close collaboration of cardiologists and neurologists with expertise in neuromuscular diseases is essential to ensure optimal use of short- and long-term care and tests for the early diagnosis of cardiovascular involvement. This collaboration should be based on cooperative model to share a decision making tailored on clinical scenario (Neuro-Cardiac Team). The INCN facilitates the establishment of Neuro-Cardiac Teams integrated in the Centers for Neuromuscular Diseases for the management of cardiovascular involvement in the treatment of MD1.
6. Quality Improvement and Risk Management
The development of process, outcome, individual practitioner level and system level quality measures for MD1 patients are encouraged (Table 1). These measures are intended to be used to calculate performance or reporting at the practitioner level or system level. Measures have their greatest impact when they are used appropriately and are linked directly to operational steps that clinicians, patients, and health plans can apply in practice to improve care [10,78]. However, performance measurement may not achieve the desired goal of improving patient care by itself.
The function of clinical risk management is essentially to provide the Neuro-Cardiac Team with the information necessary to "learn from errors" or from preventable adverse events and from so-called "near-events" or near-miss. The Neuro-Cardiac Team, for this purpose, must at first prepare and implement tools aimed at the qualitative / quantitative identification of risks and specific critical issues using a proactive approach. The proactive analysis starts from the assumption that errors can be prevented by investigating the processes at all stages and aims to identify system criticalities and possible areas of human error, to prevent them from occurring. The Failure Mode Effects Analysis (FMEA) is a method that allows us to identify possible failure modes / errors, their effects and potential causes. The Failure Mode Effects and Criticality Analysis (FMECA) adds a quantitative analysis to the FMEA that allows to classify the Failure Modes / Errors based on an Index of Risk Priority (IPR). The numerical index (IPR) is constructed using scoring scales that consider the probability of the error occurring, the possibility of it being detected and the severity of its consequences. It is used in the application of the FMECA and defines the criticality level of a process. The value of the risk priority index helps to make decisions for the activation of prevention measures[79]. The application of the FMEA / FMECA consists in breaking down a process into individual tasks: Neuro-Cardiac Team starts the analysis from the review of existing processes and procedures, identifying, in the various phases, the critical points. This approach can also be used in the conception and design of new procedures, processes and technologies to create protective barriers that prevent human / active error in MD1 patients.
7. Integrated Interdisciplinary Comprehensive MD1 Pathway
A strategy based on the stratification of cardiovascular risk must be implemented by the neurologist already in the initial evaluation phase of the patient, in order to be able to decide the level of complexity of cardiological investigations according to practical clinical paths shared with the reference cardiologist (Figure 1).
8. Perspective
Noteworthy, forthcoming approaches to treat MD1 targets the toxic mRNA product aiming to reverse the pathophysiological mechanisms of the disease, possibly leading to multisystemic improvement. The first is the DYNE-101 (by DYNE Therapeutics), a molecule composed by an antigen-binding fragment antibody (Fab) conjugated to an antisense oligonucleotide (ASO). Preclinical data showed a reduction of nuclear foci and splicing restoring in patient cells, knockdown of toxic human mRNA and correction of splicing in a mouse model of MD1. DYNE-101 is able to improve myotonia after a single dose in mice. Finally, DYNE-101 showed a favourable safety profile and with significant reduction of wild-type DMPK RNA in non-human primates. The second product candidate is AOC 1001 (by AVIDITY Biosciences) a molecule composed by monoclonal antibody binding the transferrin receptor 1 (TfR1) conjugated with a small interfering RNA (siRNA). In preclinical studies, AOC 1001 successfully delivered siRNAs to muscle cells, resulting in durable, dose-dependent reductions of DMPK RNA across a broad range of muscles including skeletal, cardiac, and smooth muscle.
Both products are now completing the Phase 1/2 clinical trials (ACHIEVE Clinical Trial for DYNE-101 - NCT05481879, and MARINA Study for AOC 1001 - NCT05027269) and its open label extension study for MARINA study (MARINA-OLE - NCT05027269, http://www.clinicaltrials.gov).
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, V.R. and S.S.; validation, V.R; S.S.; formal analysis, A.M.; writing—review and editing, V.R.; G.A.; R.M, C.C., A.M., A.M.M., R.M,M. G., P.F., M.Z., C.G., A.D.A.; S.S.; visualization, A.M.; supervision, V.R.. All authors have read and agreed to the published version of the manuscript.” Please turn to the CRediT taxonomy for the term explanation. Authorship must be limited to those who have contributed substantially to the work reported.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- M. A. Hale, N. E. Johnson, and J. A. Berglund, “Repeat-associated RNA structure and aberrant splicing.,” Biochim Biophys Acta Gene Regul Mech, vol. 1862, no. 11–12, p. 194405, 2019. [CrossRef]
- A. López-Martínez, P. Soblechero-Martín, L. de-la-Puente-Ovejero, G. Nogales-Gadea, and V. Arechavala-Gomeza, “An Overview of Alternative Splicing Defects Implicated in Myotonic Dystrophy Type I.,” Genes (Basel), vol. 11, no. 9, Sep. 2020. [CrossRef]
- V. Pisani et al., “Preferential central nucleation of type 2 myofibers is an invariable feature of myotonic dystrophy type 2,” Muscle Nerve, vol. 38, no. 5, pp. 1405–1411, Nov. 2008. [CrossRef]
- M. Garibaldi et al., “Muscle magnetic resonance imaging in myotonic dystrophy type 1 (DM1): Refining muscle involvement and implications for clinical trials,” Eur J Neurol, vol. 29, no. 3, pp. 843–854, Mar. 2022. [CrossRef]
- C. A. Thornton, “Myotonic dystrophy.,” Neurol Clin, vol. 32, no. 3, pp. 705–19, viii, Aug. 2014. [CrossRef]
- V. N. Massa R., “Epidemiology of myotonic dystrophy in the molecular era: Implications for clinical trials and association studies. In: Myotonic Dystrophies: Epidemiology, Diagnosis and Therapeutic Challenges.,” 2015, pp. 1–12.
- M. Spaziani et al., “Hormonal and metabolic gender differences in a cohort of myotonic dystrophy type 1 subjects: a retrospective, case–control study,” J Endocrinol Invest, vol. 43, no. 5, pp. 663–675, May 2020. [CrossRef]
- S. Rossi et al., “Prevalence and predictor factors of respiratory impairment in a large cohort of patients with Myotonic Dystrophy type 1 (DM1): A retrospective, cross sectional study,” J Neurol Sci, vol. 399, pp. 118–124, Apr. 2019. [CrossRef]
- R. Alsaggaf et al., “Cancer Risk in Myotonic Dystrophy Type I: Evidence of a Role for Disease Severity,” JNCI Cancer Spectr, vol. 2, no. 4, Oct. 2018. [CrossRef]
- T. Ashizawa et al., “Consensus-based care recommendations for adults with myotonic dystrophy type 1,” Neurol Clin Pract, vol. 8, no. 6, pp. 507–520, Dec. 2018. [CrossRef]
- B. Schoser et al., “Consensus-based care recommendations for adults with myotonic dystrophy type 2,” Neurol Clin Pract, vol. 9, no. 4, pp. 343–353, Aug. 2019. [CrossRef]
- G. Pelargonio, “MYOTONIC DYSTROPHY AND THE HEART,” Heart, vol. 88, no. 6, pp. 665–670, Dec. 2002. [CrossRef]
- H. H. Nguyen, J. T. Wolfe, D. R. Holmes, and W. D. Edwards, “Pathology of the cardiac conduction system in myotonic dystrophy: A study of 12 cases,” J Am Coll Cardiol, vol. 11, no. 3, pp. 662–671, Mar. 1988. [CrossRef]
- O. Vignaux et al., “Right Ventricular MR Abnormalities in Myotonic Dystrophy and Relationship with Intracardiac Electrophysiologic Test Findings: Initial Results,” Radiology, vol. 224, no. 1, pp. 231–235, Jul. 2002. [CrossRef]
- D. Vinereanu, B. P. S. Bajaj, J. Fenton-May, M. T. Rogers, C. F. Mädler, and A. G. Fraser, “Subclinical cardiac involvement in myotonic dystrophy manifesting as decreased myocardial Doppler velocities,” Neuromuscular Disorders, vol. 14, no. 3, pp. 188–194, Mar. 2004. [CrossRef]
- H. Petri, J. Vissing, N. Witting, H. Bundgaard, and L. Køber, “Cardiac manifestations of myotonic dystrophy type 1,” Int J Cardiol, vol. 160, no. 2, pp. 82–88, Oct. 2012. [CrossRef]
- K. Wahbi et al., “Incidence and predictors of sudden death, major conduction defects and sustained ventricular tachyarrhythmias in 1388 patients with myotonic dystrophy type 1,” Eur Heart J, p. ehw569, Dec. 2016. [CrossRef]
- I. B. T. Joosten et al., “Electrocardiographic predictors of infrahissian conduction disturbances in myotonic dystrophy type 1,” EP Europace, vol. 23, no. 2, pp. 298–304, Feb. 2021. [CrossRef]
- A. Lazarus, J. Varin, D. Babuty, F. rédéric Anselme, J. Coste, and D. Duboc, “Long-term follow-up of arrhythmias in patients with myotonic dystrophy treated by pacing,” J Am Coll Cardiol, vol. 40, no. 9, pp. 1645–1652, Nov. 2002. [CrossRef]
- V. Russo et al., “Prevalence of atrial fibrillation in myotonic dystrophy type 1: A systematic review,” Neuromuscular Disorders, vol. 31, no. 4, pp. 281–290, Apr. 2021. [CrossRef]
- V. Russo, A. Rago, A. A. Papa, and G. Nigro, “Which is the true epidemiology of left ventricular dysfunction in patients with myotonic dystrophy type 1?,” J Chin Med Assoc, vol. 80, no. 11, pp. 740–741, Nov. 2017. [CrossRef]
- V. Russo et al., “Paroxysmal atrial fibrillation in myotonic dystrophy type 1 patients: P wave duration and dispersion analysis.,” Eur Rev Med Pharmacol Sci, vol. 19, no. 7, pp. 1241–8, Apr. 2015.
- V. Russo et al., “Interatrial block to predict atrial fibrillation in myotonic dystrophy type 1,” Neuromuscular Disorders, vol. 28, no. 4, pp. 327–333, Apr. 2018. [CrossRef]
- V. Russo et al., “The Role of the Atrial Electromechanical Delay in Predicting Atrial Fibrillation in Myotonic Dystrophy Type 1 Patients.,” J Cardiovasc Electrophysiol, vol. 27, no. 1, pp. 65–72, Jan. 2016. [CrossRef]
- K. Wahbi and, D. Furling, “Cardiovascular manifestations of myotonic dystrophy.,” Trends Cardiovasc Med, vol. 30, no. 4, pp. 232–238, May 2020. [CrossRef]
- S. G. Priori et al., “2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC).,” Eur Heart J, vol. 36, no. 41, pp. 2793–2867, Nov. 2015. [CrossRef]
- V. Russo, A. Rago, and G. Nigro, “Sudden cardiac death in neuromuscolar disorders: Time to establish shared protocols for cardiac pacing,” Int J Cardiol, vol. 207, pp. 284–285, Mar. 2016. [CrossRef]
- V. Russo et al., “Increased heterogeneity of ventricular repolarization in myotonic dystrophy type 1 population.,” Acta Myol, vol. 35, no. 2, pp. 100–106, Oct. 2016.
- V. Russo, A. A. Papa, A. Rago, and G. Nigro, “Arrhythmic risk evaluation in myotonic dystrophy: the importance of selection criteria and methodological approach.,” Clin Auton Res, vol. 27, no. 3, pp. 203–204, Jun. 2017. [CrossRef]
- D. Magrì et al., “Increased temporal dispersion of myocardial repolarization in myotonic dystrophy Type 1,” Int J Cardiol, vol. 156, no. 3, pp. 259–264, May 2012. [CrossRef]
- B. A. Hardin, M. R. Lowe, D. Bhakta, and W. J. Groh, “Heart Rate Variability Declines with Increasing Age and CTG Repeat Length in Patients with Myotonic Dystrophy Type 1,” Annals of Noninvasive Electrocardiology, vol. 8, no. 3, pp. 227–232, Jul. 2003. [CrossRef]
- W. J. Groh et al., “Electrocardiographic Abnormalities and Sudden Death in Myotonic Dystrophy Type 1,” New England Journal of Medicine, vol. 358, no. 25, pp. 2688–2697, Jun. 2008. [CrossRef]
- D. BHAKTA, C. SHEN, J. KRON, A. E. EPSTEIN, R. M. PASCUZZI, and W. J. GROH, “Pacemaker and Implantable Cardioverter-Defibrillator Use in a US Myotonic Dystrophy Type 1 Population,” J Cardiovasc Electrophysiol, vol. 22, no. 12, pp. 1369–1375, Dec. 2011. [CrossRef]
- V. Russo et al., “Prevalence of Left Ventricular Systolic Dysfunction in Myotonic Dystrophy Type 1: A Systematic Review,” J Card Fail, vol. 26, no. 10, pp. 849–856, Oct. 2020. [CrossRef]
- D. Bhakta, M. R. Groh, C. Shen, R. M. Pascuzzi, and W. J. Groh, “Increased mortality with left ventricular systolic dysfunction and heart failure in adults with myotonic dystrophy type 1,” Am Heart J, vol. 160, no. 6, pp. 1137-1141.e1, Dec. 2010. [CrossRef]
- H. Petri et al., “Myocardial fibrosis in patients with myotonic dystrophy type 1: a cardiovascular magnetic resonance study,” Journal of Cardiovascular Magnetic Resonance, vol. 16, no. 1, p. 59, Dec. 2014. [CrossRef]
- P. Choudhary et al., “Structural and electrical cardiac abnormalities are prevalent in asymptomatic adults with myotonic dystrophy,” Heart, vol. 102, no. 18, pp. 1472–1478, Sep. 2016. [CrossRef]
- M. C. Hermans et al., “Structural and functional cardiac changes in myotonic dystrophy type 1: a cardiovascular magnetic resonance study,” Journal of Cardiovascular Magnetic Resonance, vol. 14, no. 1, p. 48, Dec. 2012. [CrossRef]
- J. K. Lau, R. W. Sy, A. Corbett, and L. Kritharides, “Myotonic dystrophy and the heart: A systematic review of evaluation and management,” Int J Cardiol, vol. 184, pp. 600–608, Apr. 2015. [CrossRef]
- T. Paunic et al., “Routine echocardiography in patients with myotonic dystrophy type 1,” Journal of the Chinese Medical Association, vol. 80, no. 7, pp. 408–412, Jul. 2017. [CrossRef]
- V. Russo et al., “ACE inhibition to slow progression of myocardial fibrosis in muscular dystrophies,” Trends Cardiovasc Med, vol. 28, no. 5, pp. 330–337, Jul. 2018. [CrossRef]
- B. Feingold et al., “Management of Cardiac Involvement Associated With Neuromuscular Diseases: A Scientific Statement From the American Heart Association,” Circulation, vol. 136, no. 13, Sep. 2017. [CrossRef]
- E. M. McNally et al., “Clinical Care Recommendations for Cardiologists Treating Adults With Myotonic Dystrophy,” J Am Heart Assoc, vol. 9, no. 4, Feb. 2020. [CrossRef]
- T. O’Brien, P. S. Harper, and R. G. Newcombe, “Blood pressure and myotonic dystrophy,” Clin Genet, vol. 23, no. 6, pp. 422–426, Jun. 1983. [CrossRef]
- K. Wahbi et al., “Development and Validation of a New Scoring System to Predict Survival in Patients With Myotonic Dystrophy Type 1,” JAMA Neurol, vol. 75, no. 5, p. 573, May 2018. [CrossRef]
- K. Yoshida, Y. Aburakawa, Y. Suzuki, K. Kuroda, and T. Kimura, “The Frequency and Risk Factors for Ischemic Stroke in Myotonic Dystrophy Type 1 Patients,” Journal of Stroke and Cerebrovascular Diseases, vol. 27, no. 4, pp. 914–918, Apr. 2018. [CrossRef]
- K. Wahbi et al., “Electrophysiological Study With Prophylactic Pacing and Survival in Adults With Myotonic Dystrophy and Conduction System Disease,” JAMA, vol. 307, no. 12, p. 1292, Mar. 2012. [CrossRef]
- V. Russo and K. Wahbi, “Appropriate timing of electrophysiological study in myotonic dystrophy type 1: unsolved question.,” Europace, vol. 24, no. 6, p. 1036, Jul. 2022. [CrossRef]
- A. Creta et al., “A Normal Electrocardiogram Does Not Exclude Infra-Hisian Conduction Disease in Patients With Myotonic Dystrophy Type 1,” JACC Clin Electrophysiol, vol. 7, no. 8, pp. 1038–1048, Aug. 2021. [CrossRef]
- A. Gamet et al., “Twenty-four-hour ambulatory ECG monitoring relevancy in myotonic dystrophy type 1 follow-up: Prognostic value and heart rate variability evolution,” Annals of Noninvasive Electrocardiology, vol. 24, no. 1, Jan. 2019. [CrossRef]
- M. Galderisi et al., “Early changes of myocardial deformation properties in patients with dystrophia myotonica type 1: A three-dimensional Speckle Tracking echocardiographic study,” Int J Cardiol, vol. 176, no. 3, pp. 1094–1096, Oct. 2014. [CrossRef]
- P. Haaf, P. Garg, D. R. Messroghli, D. A. Broadbent, J. P. Greenwood, and S. Plein, “Cardiac T1 Mapping and Extracellular Volume (ECV) in clinical practice: a comprehensive review,” Journal of Cardiovascular Magnetic Resonance, vol. 18, no. 1, p. 89, Jan. 2017. [CrossRef]
- J.-U. Voigt and F. A. Flachskampf, “Strain and strain rate,” Zeitschrift f�r Kardiologie, vol. 93, no. 4, pp. 249–258, Apr. 2004. [CrossRef]
- J. A. Luetkens et al., “Comprehensive Cardiac Magnetic Resonance for Assessment of Cardiac Involvement in Myotonic Muscular Dystrophy Type 1 and 2 Without Known Cardiovascular Disease,” Circ Cardiovasc Imaging, vol. 12, no. 6, Jun. 2019. [CrossRef]
- M. Alì et al., “Rare Disease: Cardiac Risk Assessment With MRI in Patients With Myotonic Dystrophy Type 1,” Front Neurol, vol. 11, Mar. 2020. [CrossRef]
- L. Chmielewski et al., “Non-invasive evaluation of the relationship between electrical and structural cardiac abnormalities in patients with myotonic dystrophy type 1,” Clinical Research in Cardiology, vol. 108, no. 8, pp. 857–867, Aug. 2019. [CrossRef]
- A. Cardona, W. D. Arnold, J. T. Kissel, S. V. Raman, and K. M. Zareba, “Myocardial fibrosis by late gadolinium enhancement cardiovascular magnetic resonance in myotonic muscular dystrophy type 1: highly prevalent but not associated with surface conduction abnormality,” Journal of Cardiovascular Magnetic Resonance, vol. 21, no. 1, p. 26, Dec. 2019. [CrossRef]
- V. Russo, G. Nigro, and L. Politano, “Role of electrophysiological evaluation for the best device choice to prevent sudden cardiac death in patients with Myotonic Dystrophy Type 1 and Emery Dreifuss Muscular Dystrophy.,” Trends Cardiovasc Med, vol. 31, no. 1, pp. e1–e2, Jan. 2021. [CrossRef]
- V. Russo et al., “Arrhythmic CArdiac DEath in MYotonic dystrophy type 1 patients (ACADEMY 1) study: the predictive role of programmed ventricular stimulation,” EP Europace, vol. 24, no. 7, pp. 1148–1155, Jul. 2022. [CrossRef]
- V. Russo, “Editorial commentary: Myotonic Dystrophy: The ‘right weapons’ to fight the long battle against sudden cardiac death.,” Trends Cardiovasc Med, vol. 30, no. 4, pp. 239–240, May 2020. [CrossRef]
- V. Russo, A. Rago, A. A. Papa, G. Arena, L. Politano, and G. Nigro, “Bachmann bundle pacing reduces atrial electromechanical delay in type 1 myotonic dystrophy patients,” Journal of Interventional Cardiac Electrophysiology, vol. 51, no. 3, pp. 229–236, Apr. 2018. [CrossRef]
- V. Russo et al., “Far field R-wave sensing in Myotonic Dystrophy type 1: right atrial appendage versus Bachmann’s bundle region lead placement.,” Acta Myol, vol. 33, no. 2, pp. 94–9, Oct. 2014.
- G. Nigro et al., “Does Bachmann’s bundle pacing prevent atrial fibrillation in myotonic dystrophy type 1 patients? A 12 months follow-up study,” Europace, vol. 12, no. 9, pp. 1219–1223, Sep. 2010. [CrossRef]
- G. Nigro, V. Russo, A. Rago, A. Antonio Papa, A. Palladino, and L. Politano, “Right atrial preference pacing algorithm in the prevention of paroxysmal atrial fibrillation in myotonic dystrophy type 1 patients: a long term follow-up study.,” Acta Myol, vol. 31, no. 2, pp. 139–43, Oct. 2012.
- V. Russo et al., “Atrial fibrillation burden in Myotonic Dystrophy type 1 patients implanted with dual chamber pacemaker: the efficacy of the overdrive atrial algorithm at 2 year follow-up.,” Acta Myol, vol. 32, no. 3, pp. 142–7, Dec. 2013.
- V. Russo et al., “The effect of atrial preference pacing on atrial fibrillation electrophysiological substrate in Myotonic Dystrophy type 1 population.,” Acta Myol, vol. 33, no. 3, pp. 127–35, Dec. 2014.
- V. Russo et al., “The effect of atrial preference pacing on paroxysmal atrial fibrillation incidence in myotonic dystrophy type 1 patients: a prospective, randomized, single-bind cross-over study.,” Europace, vol. 14, no. 4, pp. 486–9, Apr. 2012. [CrossRef]
- V. Russo, A. A. Papa, A. Rago, C. Ciardiello, and G. Nigro, “Effect of dual-chamber minimal ventricular pacing on paroxysmal atrial fibrillation incidence in myotonic dystrophy type 1 patients: A prospective, randomized, single-blind, crossover study,” Heart Rhythm, vol. 15, no. 7, pp. 962–968, Jul. 2018. [CrossRef]
- V. Russo et al., “Does a high percentage of right ventricular pacing influence the incidence of paroxysmal atrial fibrillation in myotonic dystrophy type 1 patients?,” Kardiol Pol, vol. 71, no. 11, pp. 1147–1153, Nov. 2013. [CrossRef]
- T. KILIC et al., “Cardiac Resynchronization Therapy in a Case of Myotonic Dystrophy (Steinert’s Disease) and Dilated Cardiomyopathy,” Pacing and Clinical Electrophysiology, vol. 30, no. 7, pp. 916–920, Jul. 2007. [CrossRef]
- V. Russo, A. Rago, A. Antonio Papa, and G. Nigro, “Cardiac resynchronization improves heart failure in one patient with myotonic dystrophy type 1. A case report.,” Acta Myol, vol. 31, no. 2, pp. 154–5, Oct. 2012.
- V. Russo, A. Rago, A. D Andrea, L. Politano, and G. Nigro, “Early onset ‘electrical’ heart failure in myotonic dystrophy type 1 patient: the role of ICD biventricular pacing,” Anadolu Kardiyoloji Dergisi/The Anatolian Journal of Cardiology, Jul. 2012. [CrossRef]
- F. A. C. de Farias, C. M. Dagostini, Y. de A. Bicca, V. F. Falavigna, and A. Falavigna, “Remote Patient Monitoring: A Systematic Review,” Telemedicine and e-Health, vol. 26, no. 5, pp. 576–583, May 2020. [CrossRef]
- V. Russo et al., “Remote Monitoring of Atrial High Rate Episodes in Pacemaker Patients. The Rapid Study Design.,” J Atr Fibrillation, vol. 11, no. 2, p. 2075, Aug. 2018.
- V. Russo et al., “Seasonal trend of ventricular arrhythmias in a nationwide remote monitoring database of implantable defibrillators and cardiac resynchronization devices.,” Int J Cardiol, vol. 275, pp. 104–106, Jan. 2019. [CrossRef]
- E. Bucci et al., “A 34-year longitudinal study on long-term cardiac outcomes in DM1 patients with normal ECG at baseline at an Italian clinical centre,” J Neurol, vol. 265, no. 4, pp. 885–895, Apr. 2018. [CrossRef]
- M. Garibaldi et al., “Gender effect on cardiac involvement in myotonic dystrophy type 1,” Eur J Neurol, vol. 28, no. 4, pp. 1366–1374, Apr. 2021. [CrossRef]
- P. Narayanaswami et al., “Quality improvement in neurology,” Neurology, vol. 85, no. 10, pp. 905–909, Sep. 2015. [CrossRef]
- J. Reason, “Human error: models and management,” BMJ, vol. 320, no. 7237, pp. 768–770, Mar. 2000. [CrossRef]
Figure 1.
Flow chart of integrated interdisciplinary comprehensive MD1 pathway. ABPM: ambulatory blood pressure monitoring; HUTT: head up tilt test; ILR: implantable loop recoreder; ELR: external loop recorder; PM: pacemaker; ICD: implantable cardiovert defibrillator; AF: atrial fibrillation; VT: ventricular tachycardia; VF: ventricular fibrillation; BBR: bundle branch reentrant; CMR: cardiac magnetic resonance; CRT: cardiac resynchronization therapy nsVT: non-sustained ventricular tachycardia; LVEF: left ventricular ejection fraction.
Figure 1.
Flow chart of integrated interdisciplinary comprehensive MD1 pathway. ABPM: ambulatory blood pressure monitoring; HUTT: head up tilt test; ILR: implantable loop recoreder; ELR: external loop recorder; PM: pacemaker; ICD: implantable cardiovert defibrillator; AF: atrial fibrillation; VT: ventricular tachycardia; VF: ventricular fibrillation; BBR: bundle branch reentrant; CMR: cardiac magnetic resonance; CRT: cardiac resynchronization therapy nsVT: non-sustained ventricular tachycardia; LVEF: left ventricular ejection fraction.
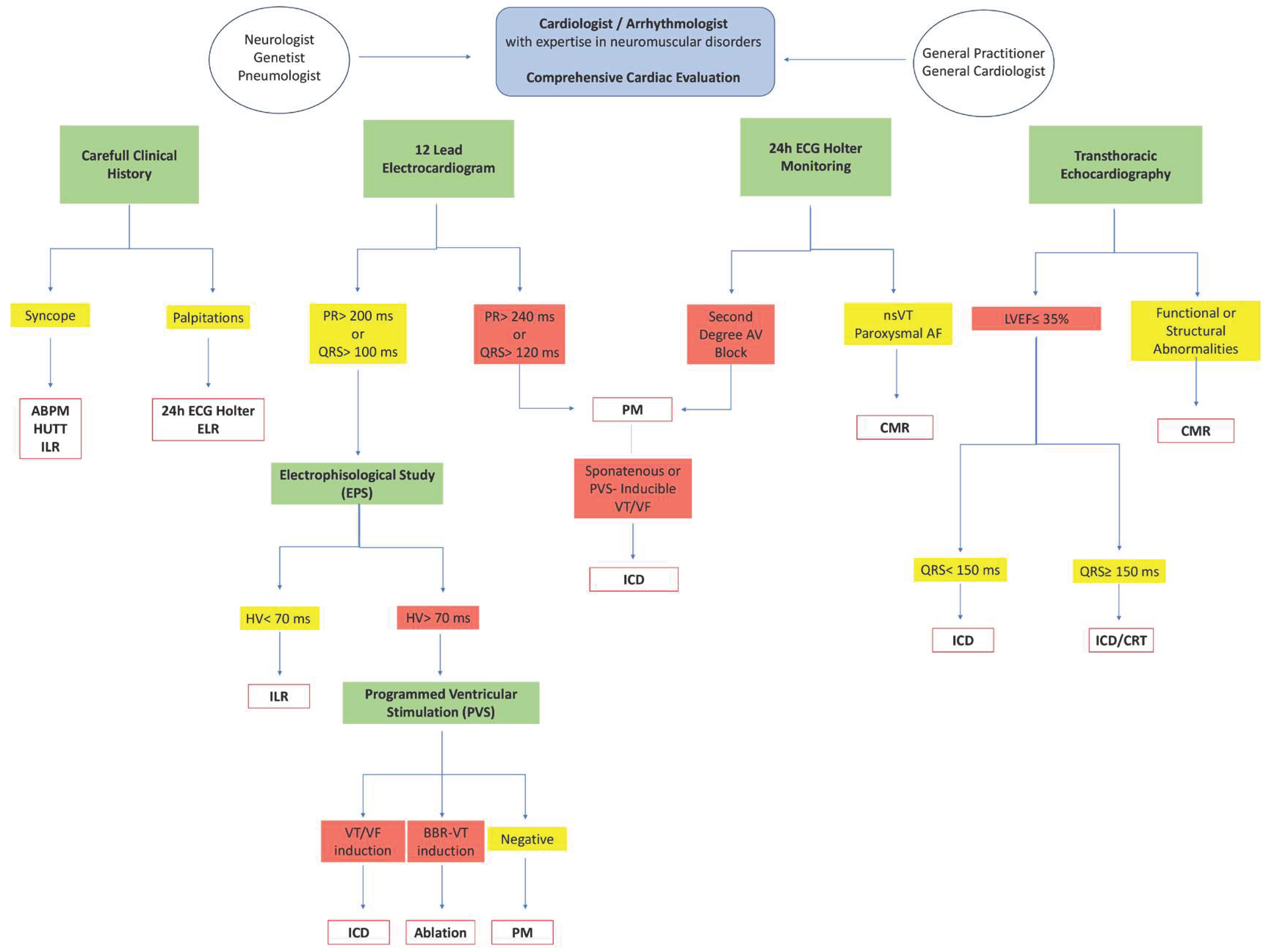

Table 1.
Quality set measures proposed for MD1 patients from the Italian Neuro-Cardiology Network (INCN)§.
Table 1.
Quality set measures proposed for MD1 patients from the Italian Neuro-Cardiology Network (INCN)§.
| Quality set measures proposed for MD1 patients from the Italian Neuro-Cardiology Network (INCN) |
|---|
| MD1 Pharmacological Treatment |
|
| MD1 Management |
|
MD1 Planning and Patient Engagement
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



