Submitted:
02 January 2024
Posted:
04 January 2024
You are already at the latest version
Abstract
Various attempts to amplify an AQP11 cDNA from tissues of the spiny dogfish (Squalus acanthias) were made. Two pairs of deoxy-inosine-containing degenerate primers were designed based on conserved amino acid sequences from an AQP11 alignment. These primers yielded some faint bands from gill cDNA that were sequenced. Blast searches with the sequences showed they were not AQP11. An elasmobranch AQP11 nucleotide sequence alignment was produced to identify conserved regions to make further degenerate primers. One primer pair produced a short 148bp fragment showing particularly strong expression in gill and intestine. It was sequenced and represented a piece of the AQP11 gene. However, as the fragment may have resulted from contaminating genomic DNA (in total RNA used to make cDNA), 5’ and 3’ RACE was performed to amplify the two ends of the putative cDNA. 5’ and 3’ RACE amplifications depend on the presence of a 5’ cap nucleotide and a poly A tail respectively on the putative AQP11 mRNA. Hence successful amplification was only possible from cDNA and not genomic DNA. Nested RACE amplifications were performed using gill and intestinal RACE cDNA but none of the DNA fragments were AQP11. Consequently, the spiny dogfish AQP11 gene may represent a pseudogene.
Keywords:
Aquaporin
; Spiny Dogfish
; Pseudogene
; Degenerate PCR
; Colony PCR
; 5’ RACE PCR
; 3’ RACE PCR
; Amino acid Alignment
; Nucleotide sequence alignment
; DNA cloning
1. Introduction
Aquaporins (AQPs) are ubiquitous family of membrane proteins found across the spectrum of life from bacteria to humans [1]. In mammals there are thirteen aquaporins numbered AQP0-12 [2,3,4,5], whereas in other animals, there are up to seventeen AQP genes [1]. Mammalian aquaporins fall into 3 main groups, orthodox or classical aquaporins (AQP0, AQP1, AQP2, AQP4, AQP5, AQP6, and AQP8), aquaglyceroporins (AQP3, AQP7, AQP9 and AQP10) and unorthodox or superaquaporins (AQP11 and AQP12) [6,7]. AQP11 and AQP12 were originally identified and described as AQPX1 and AQPX2 respectively [8]. AQP11 in mammals is widely expressed in a number of tissues including brain, heart, liver, spleen, GI tract, kidney, testes, ovary, muscle, and leukocytes [3]. Mammalian AQP11 exhibits water permeability in expression studies [3], although it is additionally thought to represent a glyceroporin and a peroxiporin transporting hydrogen peroxide and it localizes to the endoplasmic reticulum membrane internally within cells [9].
In teleost fish, there are duplicate copies of the AQP11 gene (AQP11a and AQP11b) [10]. In zebrafish (Danio rario), AQP11b is expressed in GI tract, liver and ovary [10]. In American eel (Anguilla rostrata) AQP11a is expressed at highest levels in liver, GI tract, and kidney, whereas AQP11b is found in the brain, gill, GI tract, kidney with lower levels in the eye, heart, swim bladder, spleen and liver [11].
Elasmobranchs, such as the spiny dogfish (Squalus acanthias), have a subset of the seventeen animal AQPs but with two individual gene duplications. Their AQP genomic complement includes AQP0, AQP1, AQP3C1, AQP3C2, AQP4, AQP8, AQP9, AQP10C1, AQP10C2, AQP11, AQP12, AQP14 and AQP15 [1,12,13,14,15,16,17]. All of these genes have now been cloned and sequenced from spiny dogfish cDNA except for AQP11 [18].
Up until very recently though there has been very little sequence data available in gene banks regarding AQPs from elasmobranch species. Sequences for AQP11 from a number of elasmobranch species began to be submitted to gene banks in 2021 and those sequences helped facilitate this study. As no AQP11 sequence was available for the spiny dogfish, the initial purpose of the study was to obtain a copy of the coding region of the AQP11 mRNA transcript using complementary DNA (cDNA) derived from various tissue total RNA samples and using degenerate PCR techniques successfully deployed many times [14,15,17,19,20,21,22,23,24,25]. With AQP11 sequences from closely related shark species available, it was initially thought that it would be a relatively easy and straight forward task to amplify a piece of the spiny dogfish AQP11 cDNA but that proved to be far from the reality of the situation.
2. Results
2.1. Amino acid-based degenerate PCR
Initial studies to amplify a piece of the spiny dogfish AQP11 transcript from cDNA utilized an established degenerate PCR technique. Amino acid sequences from various elasmobranch and coelacanth AQP11 genes were aligned and conserved stretches of sequence were identified to allow the design of inosine-containing primers (Figure 1.; see also methods).
Initially, one pair of primers were made (Elas AQP11 Sense and Elas AQP11 Anti 2) these should have resulted in an approximate 578bp fragment being generated from various spiny dogfish tissue cDNAs in degenerate PCR. An initial PCR run using an annealing step of 55˚C resulted in no bands around the correct size in any tissue. A second run was performed using an annealing step of 50˚C. This resulted in a couple of faint bands in the gill but when cloned and sequenced these transpired to be unrelated to AQP11. A further pair of degenerate primers were then made using amino acid sequences from other regions of the AQP11 gene (Elas AQP11 Sen 2 and Elas AQP11 Anti 3). These new primers allowed for them to be used in conjunction with the original primers in addition to being used on their own (possible combinations Sense-Anti 3, Sen 2–Anti 2, Sen 2-Anti 3). An example gel resulting from some the amplifications performed with these primers at 55˚C is seen in Figure 2. Although some bands were seen with the gill cDNA and cloned and sequenced (3 bands), none of these proved to be related to AQP11. It should be noted at this point that both of the sense primer sites were located within the expected first exon of the gene, whereas the antisense primers were mostly or entirely within the expected second exon of the gene and hence it was only possible to form AQP11 amplification products of the expected size from the cDNA and not from any genomic DNA contamination present in the total RNA/cDNA.
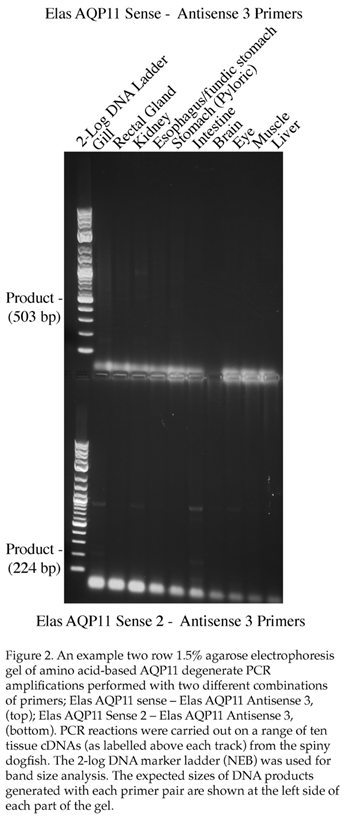
2.2. Nucleotide-based PCR
Although this initial series of amino-acid-sequence-based degenerate PCRs suggested that there was no expression of AQP11 in any of the spiny dogfish tissue cDNA samples, given the shark AQP11 sequences appeared fairly highly conserved, it was decided to attempt to obtain an AQP11 amplification product using nucleotide sequence-based degenerate PCR primers. The nucleotide sequences of various shark AQP11 genes were aligned and three conserved regions were identified (see Figure 3. and Table 2.). With three sites available, both sense and antisense primers were designed for the middle site such that three sets of PCR amplifications could be performed (AQP11 sense-AQP11 antisense.
Figure 3.
Partial Clustal Omega alignment of seven elasmobranch AQP11 cDNA sequences, showing only the relevant alignment blocks used to design nucleotide-based degenerate primers. Primer locations are indicated with underlined and bolded text in the catshark sequence. Names of the AQP11 primers produced from each region are indicated underneath alignment blocks. * indicate positions where the nucleotide sequence is identical in all species.
Figure 3.
Partial Clustal Omega alignment of seven elasmobranch AQP11 cDNA sequences, showing only the relevant alignment blocks used to design nucleotide-based degenerate primers. Primer locations are indicated with underlined and bolded text in the catshark sequence. Names of the AQP11 primers produced from each region are indicated underneath alignment blocks. * indicate positions where the nucleotide sequence is identical in all species.


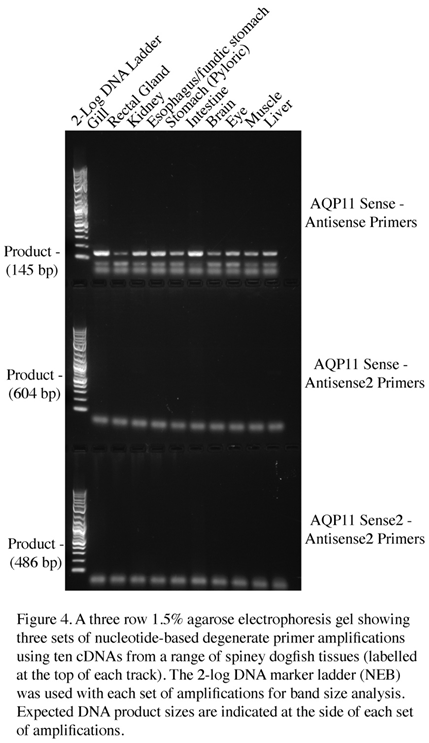
AQP11 sense-AQP11 antisense 2, AQP11 sense 2- AQP11 antisense 2; see Figure 4). With the set of amplifications performed with the AQP11 sense-AQP11 antisense primers, a band of approximately the expected size of 145bp (it was actually 148bp) was identified in every tissue cDNA. The highest level of amplification was seen in the gill and intestine, so these bands were subsequently cloned and sequenced. The fragments were essentially copies of a piece of the spiny dogfish AQP11 gene (see Figure 5.).
However, it was also evident that both of the primers used to make the successful fragment resided within the expected exon 1 of the AQP11 gene. Hence the fragment could have amplified off either cDNA or any contaminating genomic DNA within the cDNA samples.
2.3. RACE PCR
To attempt to determine whether the AQP11 fragment obtained was from a cDNA or genomic DNA origin, primers were designed from the obtained AQP11 sequence for RACE PCR in order to amplify the two ends of the cDNA sequence. The RACE PCR relies for its success on the presence of a poly A tail on the 3’ end of mRNAs and a 5’ cap on the other end of the mRNA. Without the presence of these nucleotide structures the cDNA would fail to be produced and the RACE PCR would then also subsequently fail to amplify AQP11 fragments. Of course, the poly A tail and 5’ cap nucleotide are features of the mRNA added enzymatically after transcription and are not present within the genomic copy of genes. Hence RACE PCR differentiates between sequences amplified from cDNA and genomic DNA and will not produce amplification products from the genomic copies of genes.
Nested RACE PCR was performed (see Figure 6. And Table 3.) and while some strong bands occurred particularly in the first round of 3’ RACE amplification, the banding in the second round was diminished rather than increased as would be expected. The three main intestinal 3’ RACE bands were cloned and sequenced. There were 2 sizes of clones present in sequences that resulted. Both had utilized the AQP11 gene specific primer sequences (3R1) from the first round of RACE amplification to amplify (i.e. they were first not second round amplification products) and Blast searches with the sequences showed both were angiotensin converting enzyme (ACE) splice variants most closely related to those from the bamboo shark and zebra shark respectively.
For 5’ RACE, the banding did increase in intensity in the second round of amplification. To amplify to the end of the transcript there was a minimum expected size of around 260bp, also as the primers for the second round of nested RACE PCR are internal to the initial round AQP11 fragment, the size of AQP11 should decrease by a known amount (73bp) in the second round of nested RACE PCR. So one band from the intestinal cDNA was chosen for cloning and sequencing as it was the strongest amplifying band on the gel and there was a somewhat larger band in the initial round of amplifications. When the cloning experiment was analyzed there were two sizes of clones present (417bp and 368bp) which were both sequenced and Blast searches with the sequences were most similar to genomic DNA on catshark chromosome 21 and great white shark dual specificity phosphatase 5 (dusp5) respectively.
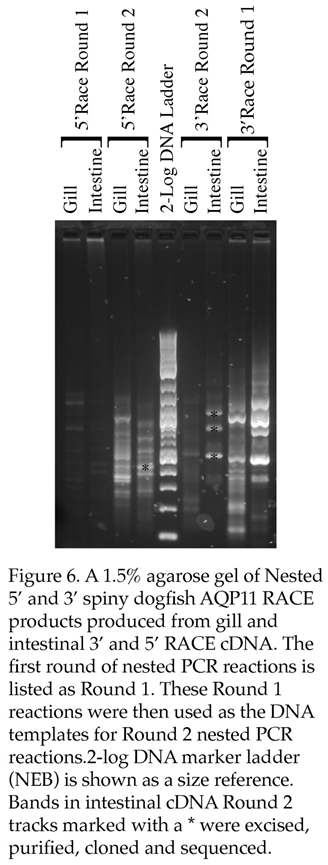
To summarize, none of the 5’ or 3’ RACE products sequenced represented AQP11 sequences.
3. Discussion
The fact that the amino acid-based degenerate PCR failed to generate a AQP11 PCR product was surprising as the AQP11 gene in elasmobranchs appears to be fairly highly conserved and therefore AQP11 should have represented a relatively easy target for this approach as the AQP8 gene [17] had done previously even though that study didn’t have any prior elasmobranch sequences to work with. Additionally, AQP11 expression was expected to be present in most of the tissue cDNAs utilized in this study such as brain, liver, gill, GI tract, and kidney, based on the AQP11 expression profile in mammals (such as in humans [3]) or in teleost fish (such as in the American eel [11]). So the lack of amplification in the amino acid-based degenerate PCRs was unexpected but would be understandable if AQP11 is a pseudogene because the primers used span across the expected exons of the gene and therefore potential products generated from genomic copies of the gene would be much larger (due to the presence of an intron). A second line of circumstantial evidence that is in line with this study is the fact that a transcriptomics study carried out on spiny dogfish cDNAs from brain, kidney, liver, and ovary, failed to identify an AQP11 transcript [26]. On the other hand, the transcriptomics study seems to have not been ultimately exhaustive in identifying every transcript present in these tissues as AQP8 which was subsequently shown to be expressed in spiny dogfish brain [17] was not identified either. The amplification of a small AQP11 fragment by one of the nucleotide-based degenerate PCR primers might have suggested that AQP11 expression was present in spiny dogfish tissue cDNAs, but it was known in advance that the total RNA samples used to make cDNAs were contaminated with a small amount of genomic DNA and the fragment generated (see Figure 5.) would be contained within the expected exon 1 of the genomic copy or the AQP11 gene and would amplify with primers used. The AQP11 antisense 2 primer would be expected to be located in exon 2 of the AQP11 gene and therefore this primer would only produce a product of the expected size from the cDNA version of AQP11. Again, the failure of the AQP11 antisense 2 primer to produce PCR products of the expected size in any tissue is consistent with a lack of AQP11 expression in these tissue which would be the case if AQP11 is a psuedogene. Finally the RACE amplification using primers designed from the spiny dogfish AQP11 fragment generated (see Figure 5) again would only likely amplify from cDNA copies of AQP11 because the cDNA synthesis reactions that incorporate a Smarter DNA sequence (see methods) rely on the binding of an oligo dT part of the cDNA synthesis primer to bind to the AQP11 mRNAs poly A tail and for 5’ RACE the incorporation of the Smarter oligo sequence at the 5’ end relies on the presence of a 5’ cap nucleotide on the mRNA. Both poly A tails and 5’ cap nucleotides are only present on mRNAs and are not found within the genomic sequences of genes. This is the reason why it would be unlikely that 5’ and 3’ RACE AQP11 fragments would be generated by the RACE reactions unless the AQP11 gene was transcribed into mRNA.
Ultimately, though of course this study is not proof that expression of the AQP11 gene in the spiny dogfish does not exist. In mammals the highest level of AQP11 expression is in the testes [3] (although in zebrafish no expression was found there; [10]) as this study did not have any access to any gonadal total RNA/cDNA samples, this is impossible to rule out here. Likewise in any case, AQP11 expression might be limited to other organs not utilized in this study such as pancreas or in tissue not part of major organ such as cartilage or in the integument. It is however interesting to note that in a recent article concerning the superaquaporin subfamily [9], the authors noted that due to the lack of significant AQP11 phenotypes in knock-out animals, “that AQP11 is dispensable similar to AQP10 which has turned to a pseudogene in some animals ”. The reference to AQP10 refers to its lack of expression in cows and their relatives [27]
4. Methods
4.1. cDNAs
The total RNA/cDNAs used in this study were the same as those in [28]. Complementary DNAs (cDNAs) were made by a modification of the manufacturer instructions. One μg of previously extracted total RNA samples from the tissues an adult spiny dogfish (when the animal was originally euthanized the work had IACUC approval from the Mount Desert Island Bio Lab) were incubated with 1μl of 100μM Oligo d(T)26 primer, 1μl of 10mM dNTPs (Lamda Biotech), 1μl of SUPERas.in RNase inhibitor (Thermofisher) and dH2O to a total of 6μl was mixed and incubated at 65˚C for 5 mins and then placed on ice for 1 min. Then 2μl of 5x RT buffer, and 1μl 100mM dithiothreitol (DTT) were added and the samples were mixed by pipetting. Finally, 1ul (200 units) of Superscript IV reverse transcriptase (Thermofisher) was added and the samples were mixed and incubated at 55˚C for 30 minutes. Samples were then incubated at 80˚C to inactivate the reverse transcriptase and then they were placed on ice and diluted to 200μl with dH2O.
4.2. Amino acid-based degenerate PCR
AQP11 degenerate PCR primers were designed based on reverse-translated conserved amino acid sequences as shown in figure 1. Gene Alignments were performed using Clustal Omega (at www. ebi.ac.uk) and the sequence of the spiny dogfish DNA fragment was translated using an online Translate tool (web.expasy.org/translate/). Deoxy-inosine was used at positions of uncertainty in the sequences (often the 3rd base of codons; see Table 1.). Primers were manufactured by Eurofins genomics. Degenerate PCR was performed using an Eppendorf Mastercycler pro S thermocycler. For each primer pair reactions were carried out initially with a cycle annealing temperature of 55˚C and then subsequently at 50˚C. Cycle extension times at 72˚C were approximately 1min/kbp of DNA fragment. Denaturation during each cycle was carried out at 94˚C for 30 seconds. PCR reactions contained 1μl of each 100μM degenerate primer and 1μl of cDNA as separate drops in a 200μl reaction tube. A master mix containing 2μl 10x Taq polymerase buffer (NEB), 0.4μl 10mM dNTP’s, 0.25μl Taq DNA polymerase (NEB) and dH2O to 17μl was created and added to PCR reaction tubes immediately before they were added to the thermocycler which was paused at 92˚C. Once all the tubes were added to the thermocycler, its cycling program was initiated for 40 cycles.
4.3. Agarose gel electrophoresis
A 135ml 1.5% (W/V) agarose gel was produced. 2.025g agarose (Lamda Biotech) was weighed into a conical flask and 132ml dH2O and 2.7ml 50x TAE buffer were added. Cling film covered the flask mouth with a hole poke in it to release steam. The flask was heated on full power for 45 sec, and the contents were swirled and microwaved for a further 1 min on 30% power. The flask was placed in a waterbath to cool at >45˚C. 13.5 μl of 10000x gel red (Biotium) electrophoresis dye was added and mixed by swirling. The gel was poured into a taped (masking taped ends) gel tray. Gel well combs were added to the gel and it was allowed to set. The gel was placed in a fridge for >20 minutes after setting before removal of the comb(s). PCR samples were added to the wells. A 2-log ladder DNA marker sample (NEB) was prepared with 1x TAE buffer and bromophenol blue loading dye. Samples were run initially for up to 2 min at 500 mAmps, then subsequently at a constant 265 mAmps. The gels were visualized and photographed using a Syngene G box gel documentation system.
4.4. Cloning and Sequencing
DNA bands were excised from agarose gels using a no.11 scalpel blade and DNA was purified using a Monarch DNA purification kit (NEB). Purified DNA was cloned into a pCR4 TOPO plasmid vector hosted in E.coli using a TOPO TA Cloning Kit for sequencing (Thermofisher). Bacterial colonies from LB agar plates were grown in 1ml terrific broth and then processed. 50μl of bacterial cultures was centrifuge and the culture medium removed. The bacteria were resuspended in 0.5ml dH2O. 0.5μl of each suspension was used for colony PCR. Colony PCR reactions were performed with 1μl each of 4μM M13 forward and reverse plasmid primers also contained 2μl 10x Taq polymerase buffer, 0.4μl 10mM dNTPs, and 0.1μl (0.5 units) Taq DNA polymerase. 4μl of each colony PCR sample were run on agarose gels and the best three were selected for sequencing. The remains of the colony PCR samples selected were purified using a Quickstep 2 PCR purification kit (Edge Biosystems). 1μl of the purified DNA was then run on another agarose gel with Logic DNA marker ladder (Lamda Biotech) and the amount of DNA in samples was quantified using the Syngene G box gel documentation system. 20ng/kb of each DNA sample was placed in a 200μl PCR tube, 1μl of 4μM T3 plasmid primer was added and the volume was made up to 8μl with dH2O. Samples were then sent for sequencing (Eton Biosciences).
4.5. Nucleotide-based degenerate PCR
Primers were designed (and manufactured; Eton Biosciences) using conserved regions of an alignment of the AQP11 nucleotide sequences from various elasmobranch species (see Figure 3. and Table 2.). Primers were made sufficiently long to allow the use of 2-step PCR using Phusion DNA polymerase (NEB). The cycle program was 30sec initial denaturation at 98˚C followed by 35 cycles of 72˚C for 30sec/kb (+8 sec) then 98˚C for 10sec per cycle. There was then a final extension step of 2minutes at 72˚C.
4.6. RACE PCR
Pre-existing spiny dogfish tissue 5’ and 3’ RACE cDNAs were used for the study which were manufactured using a SMARTer RACE cDNA synthesis kit (Takara) and using the manufacturer’s instructions, except the kits reverse transcriptase steps were replaced by those needed for Superscript IV reverse transcriptase and SUPERas.in RNase inhibitor. Some of the cDNAs generated had been used successfully previously for other genes (for example see [15,17]). The cDNAs for 5’RACE and 3’RACE are made separately and a Smarter oligo sequence is incorporated at the 5’cap in 5’ RACE cDNA and as the rear part of the oligo dT cDNA synthesis primer in 3’ RACE cDNA. PCR is conducted using a Universal Smarter RACE primer mix from the kit and a gene specific primer (5R1 for 5’ RACE or 3R1 for 3’ RACE; See Table 3.). As in the manufacturer’s instructions, initial (Round 1) PCR amplifications were conducted using touchdown PCR with an initial annealing temperature for 5 cycles of 72˚C, then at 70˚C for 5 further cycles, and then 68˚C for a further 25 cycles. The kits polymerase was replaced by Phusion DNA polymerase (NEB) and the extension time at 72˚C was 2 min per cycle. The PCR reactions generated were then used to seed a second set of nested PCR reactions (Round 2) using the kit’s nested primer (Universal primer short) and a second nested gene specific primer (5R2 for 5’ RACE or 3R2 for 3’ RACE; See Table 3.). These reactions were performed for 35 cycles using 98˚C denaturation for 10 sec and 2 minutes at 72˚C, 2-step PCR, followed by a final extension step of 5 min at 72˚C.
4.7. Sequence accession numbers
Coelacanth AQP11, XM_006006304; Catshark AQP11, XM_038818680; Great white shark AQP11, XM_041199754; Bamboo shark AQP11, XM_043692428; Zebra shark AQP11, XM_048532354; Dogfish AQP11, XXX; Whale shark AQP11 X1, XM_020529003; Whale shark AQP11 X2, XM_048597661; Whale shark AQP11 X3, XM_020529003; Thorny skate AQP11, XM_033023374; Sawfish AQP11, XM_052026755 Great white shark dusp5, XM_41209980; Catshark chromosome 21, LR744050.1; Bamboo shark ACE, XM_043674648.1; Zebra shark ACE, XM_059638649.1.
Author Contributions
C.P.C. was responsible for writing this article and was involved in laboratory experimental work. M.E.C. and E.O. carried out significant critical laboratory experimental work.
Funding
Funding was provided by Georgia Southern University and C.P.C.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AQP | Aquaporin |
| cDNA | Complementary DNA |
| RACE | Rapid Amplification of cDNA Ends |
| PCR | Polymerase Chain Reaction |
| Elas | Elasmobranch |
| ACE | Angiotensin Converting Enzyme |
| Dusp5 | dual specificity phosphatase 5 |
| RNase | Ribonuclease |
| DTT | dithiothreitol |
| dNTP | De-oxy nucleotide tri-phosphates |
| Taq | Thermus aquatus |
| IUPAC | International Union of Pure and Applied Chemistry |
| E.coli | Escherichia coli |
| NEB | New England Biolabs |
References
- Finn, R.N.; Chauvigné, F.; Hlidberg, J.B.; Cutler, C.P.; Cerdà, J. The lineage-specific evolution of aquaporin gene clusters facilitated tetrapod terrestrial adaptation. PLoS ONE 2014, 9, (11): e113686 p1-38. [CrossRef]
- Nejsum, L.N. The renal plumbing system: aquaporin water channels. Cell. Mol. Life Sci. 2005, 62, 1692-1706. [CrossRef] [PubMed]
- Ishibashi, K.; Hara, S.; Kondo, S. Aquaporin water channels in mammals. Clin. Exp. Nephrol. 2009, 13, 107-117. [CrossRef] [PubMed]
- Kitchen, P.; Day, R.E.; Salman M.M.; Conner, M.T.; Conner, A.C. Beyond water homeostasis: Diverse functional roles of mammalian aquaporins. Biochim. Biophys. Acta 2015, 1850, 2410-2421. [CrossRef] [PubMed]
- Login, F.H.; Nejsum, L.N. Aquaporin water channels: roles beyond renal water handling. 2023, Nature Rev. Nephrol. 2023, 19, 604-618. [CrossRef]
- Ishibashi, K.; Tanaka, Y.; Morishita, Y. The role of superaquaporins inside the cell. Biochim. Biophys. Acta 2014, 1840, 1507-1512. [CrossRef]
- Edamana, S.; Login, F.H.; Yamada, S.; Nejsum, L.N. Aquaporin water channels as regulators of cell-cell adhesion proteins. Am. J. Physiol. 2021, 320, C771-C777. [CrossRef]
- Ishibashi, K.; Kuwahara, M.; Kageyama, Y.; Sesaki, S.; Suzuki, M.;Imai, M. Molecular cloning of a new aquaporin superfamily in mammals. In Molecular Biology of water and solute transport. Hohman, S.; Neilsen, S. Eds. Springer, Boston, USA. 2000, pp. 123-126.
- Ishibashi, K.; Tanaka, Y.; Morishita, Y. The role of superaquaporins inside the cell: an update. Biochim. Biophys. Acta 2021, 1863, 183617. [CrossRef]
- Tingaud-Sequeira, A.; Calusinska, M.; Finn, R.N.; Chauvigné, F.; Lozano, J.; Cerdà, J. The zebrafish genome encodes the largest vertebrate repertoire of functional aquaporins with dual parology and substrate specificities similar to tetrapods. BMC Evol Biol. 2010, 10, 38. [CrossRef] [PubMed]
- Cutler, C.P.; Kurt, K. Georgia Southern University, Statesboro, Georgia, USA. 2019 (unpublished work).
- Yilmaz, O.; Chauvigné, F.; Ferré. A.; Nilsen, F.; Fjelldal, P.G.; Cerdà, J.; Finn, R.N. Unravelling the complex duplication history of deuterostome glycerol transporters. Cells 2020, 9, 1663. [CrossRef]
- Cutler, C.P.; Harmon, S.; Walsh.; Burch, K. Characterization of aquaporin 4 protein expression and localization in tissues of the dogfish (Squalus acanthias). Front. Physiol. 2012a, 3, 21, 1-13. [CrossRef]
- Cutler, C.P.; MacIver, B.; Cramb, G.; Zeidel, M.L. Aquaporin 4 is a ubiquitously expressed isoform in the dogfish (Squalus acanthias) shark. Front. Physiol. 2012b, 2, 107, 1-9. [CrossRef] [PubMed]
- Cutler, C.P.; Murray, D.; Ojo, T.; Harmon, S.; MacIver, B.; Cramb, G.; Zeidel, M.L. Aquaporin (AQP) channels in the spiny dogfish, Squalus acanthias I: Characterization of AQP3 and AQP15, and localization of their proteins in the gill and spiral valve intestine. Comp. Biochem. Physiol. 2022a, 258, 1-11. [CrossRef] [PubMed]
- Cutler, C.P.; Kurt, K.; Campbell, K.E.; Tolulope Ojo, T. Aquaporin (AQP) channels in the spiny dogfish, Squalus acanthias II: Localization of AQP3, AQP4 and AQP15 in the kidney. Comp. Biochem. Physiol. 2022b, 258, 1-11. [CrossRef] [PubMed]
- Cutler, C.P.; Mainer, S.; Ojo, T. The aquaporin 8 (AQP8) membrane channel gene is present in the elasmobranch dogfish (Squalus acanthias) genome and is expressed in brain but not in gill, kidney or intestine. Comp. Biochem. Physiol. 2022, 260, 1-8. [CrossRef] [PubMed]
- Cutler, C.P. Georgia Southern University, Statesboro, Georgia, USA. 2019 (includes some unpublished work).
- Cutler, C.P.; Sanders, I.L.; Hazon, N.; Cramb, G. Primary sequence, tissue specificity and expression of the Na+,K+-ATPase 1 subunit in the European eel (Anguilla anguilla). Fish Physiol. Biochem. 1995, 14, 423-429. [CrossRef] [PubMed]
- Cutler, C.P.; Sanders, I.L.; Cramb, G. Expression of Na+,K+-ATPase subunit isoforms in the European eel (Anguilla anguilla). Fish Physiol. Biochem. 1997, 17, 371-376. [CrossRef]
- Cutler, C.P.; Brezillon, S.; Bekir, S.; Sanders, I.L.; Hazon, N., Cramb, G. Expression of a duplicate Na+,K+-ATPase 1 isoform in the European eel (Anguilla anguilla). Am. J. Physiol. 2000, 279, R222-R229.
- Cutler, C.P.; Cramb, G. Branchial expression of an aquaporin 3 (AQP-3) homologue is downregulated in the European eel Anguilla anguilla following seawater acclimation. J. Exp. Biol. 2002, 205, 2643-2651. [CrossRef] [PubMed]
- Cutler, C.P.; Cramb, G. Two isoforms of the Na+/K+/2Cl- cotransporter (NKCC1) are expressed in the European eel (Anguilla anguilla). Biochimica. Biophysica. Acta. 2002, 1566, 92-103. [CrossRef] [PubMed]
- Martinez, A-S.; Cutler, C.P.; Wilson, G.; Phillips, C.; Hazon, N.; Cramb, G. Regulation of expression of two aquaporin homologues in the intestine of the European eel: Effects of seawater acclimation and cortisol treatment. Am. J. Physiol. 2005, 288, R1733-43.
- Cutler, C.P.; Cramb, G. Differential expression of absorptive cation-chloride cotransporters in the intestinal and renal tissues of the European eel (Anguilla anguilla). Comp. Biochem. Physiol. 2008, 149, 63-73. [CrossRef] [PubMed]
- Chana-Munoz, A.; Jendroszek, A.; Sønnichsen, M.; Kristiansen R.; Jensen, J.K.; Andreason, P.A.; Bendixen, C.; Panitz, F. Multi-tissue RNA-seq and transcriptome characterization of the spiny dogfish shark (Squalus acanthias) provides a molecular tool for biological research and reveals new genes involved in osmoregulation. PLOS ONE 2017, 12(8), e0182756. [CrossRef]
- Tanaka, Y.; Ishibashi, K.; Morishita, Y. Aquaporin 10 is a pseudogene in cattle and their relatives. Biochem. Biophys. Rep. 2015 16-21. [CrossRef]
- Madsen, S.S.; Engelund, M.; Cutler, C.P. Water transport and the functional dynamics of aquaporins in osmoregulatory organs of fishes. Biological Bulletin. 2015, 229, 1, 70-92. [CrossRef] [PubMed]
Figure 1.
Clustal omega alignment of the amino acid sequences of elasmobranch and coelacanth AQP11 amino acid sequences. The alignment includes a translation of the partial spiny dogfish AQP11 sequence resulting from this study. Underling in the whale shark X1 sequence represents locations of conserved amino acid sequences used to design amino acid-based degenerate PCR primers (see also Table 1.). * are locations in the alignment with the same amino acid in all species. : or . are positions in the alignment with chemically similar substitutions in some species.
Figure 1.
Clustal omega alignment of the amino acid sequences of elasmobranch and coelacanth AQP11 amino acid sequences. The alignment includes a translation of the partial spiny dogfish AQP11 sequence resulting from this study. Underling in the whale shark X1 sequence represents locations of conserved amino acid sequences used to design amino acid-based degenerate PCR primers (see also Table 1.). * are locations in the alignment with the same amino acid in all species. : or . are positions in the alignment with chemically similar substitutions in some species.


Figure 5.
Nucleotide sequence of the partial spiney dogfish AQP11 148bp fragment generated with the AQP11 sense and AQP11 antisense nucleotide-based degenerate PCR primers (see Table 2.). The primer sequences themselves have been removed as they are unreliable. The 5’ RACE primers produced from this sequence are bolded, the 3’ RACE primers are underlined.
Figure 5.
Nucleotide sequence of the partial spiney dogfish AQP11 148bp fragment generated with the AQP11 sense and AQP11 antisense nucleotide-based degenerate PCR primers (see Table 2.). The primer sequences themselves have been removed as they are unreliable. The 5’ RACE primers produced from this sequence are bolded, the 3’ RACE primers are underlined.
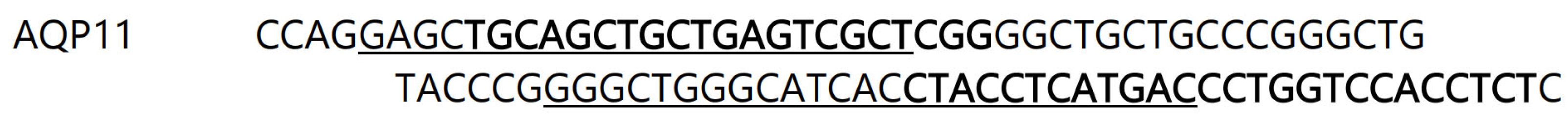

Table 1.
Amino acid-based degenerate PCR primers.
| Elas AQP11 Sense | AC[I]YT[I]CARYT[I]TGYATHTGYAC[I]CAYGARYT |
| Amino Acid sequence used | TLQLCICTHEL |
| Elas AQP11 Anti 2 | AR[I]GG[I]CC[I]ARCCARTA[I]ACRAARCARTAYTC |
| Amino Acid sequence used | EYCFVYWLGPL |
| Elas AQP11 Sen 2 | CAYTGGTAYCAYCAYCARCR[I]CARTAYAARTG |
| Amino Acid sequence used | HWYHHQR/HQYKC |
| Elas AQP11 Anti 3 | AR[I]RC[I]GGRTTRAA[I]AC[I]GC[I]CC[I]GT[I]ARRTC |
| Amino Acid sequence used | DLTGAVFNPV/AL |
| Expected product sizes:- | Sense - Anti 2 =578bp |
| Sense - Anti 3 = 503bp | |
| Sen 2 - Anti 2 = 299bp | |
| Sen 2 - Anti3 = 224bp | |
| [I] = Deoxyinosine. Other IUPAC codes used indicate wobbles e.g. R =A/G | |
Table 2.
Nucleotide-based degenerate PCR primers.
| AQP11 Sense | TCTCCACCCTGCAGCTGTGYRTCTGCAC |
| AQP11 Sense 2 | CACCTTYRGCDGYGCCASCTGCAAYCC |
| AQP11 Antisense | GGRTTGCAGSTGGCRCHGCYRAAGGTG |
| AQP11 Antisense 2 | GTCATYRCACCYADYARRGGTCCAAGCCAGTA |
| Expected product sizes:- | Sense - Antisense = 145bp |
| Sense – Antisense 2 = 604bp | |
| Sense 2 - Antisense 2 = 486bp | |
| IUPAC codes used indicate wobbles e.g. R =A/G | |
Table 3.
5’ and 3’ RACE PCR primers.
| 5R1 | AGAGGTGGAC CAGGGTCATG AGGTAG |
| 5R2 | CCGAGCGACT CAGCAGCTGC A |
| 3R1 | GAGCTGCAGC TGCTGAGTCG CT |
| 3R2 | GGGCTGGGCA TCACCTACCT CATGAC |
| Minimum expected product sizes:- | |
| 5’ RACE | = 260 bp |
| 3’ RACE | = 632 bp |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



