
Submitted:
03 January 2024
Posted:
04 January 2024
You are already at the latest version
Abstract
Pulmonary Hypertension (PH) is a terminal disease characterized by severe pulmonary vascular remodeling. Unfortunately, targeted therapy to prevent disease progression is limited. Here, the vascular cell populations that contribute to the molecular and morphological changes of PH in conjunction with current animal models for studying vascular remodeling in PH will be examined. The status quo of epigenetic targeting for treating vascular remodeling in different PH subtypes will be dissected, while parallel epigenetic threads between pulmonary hypertension and pathogenic cancer provide insight into future therapeutic PH opportunities.
Keywords:
vascular remodeling
; Pulmonary Hypertension
; epigenetic
; lung cancer
Introduction
Pulmonary hypertension (PH) is a progressive and incurable cardiopulmonary vascular disease that is heterogeneous and multifactorial in nature. In PH, delayed diagnosis is associated with diagnostic challenges due to the late onset of symptoms, epidemiological, sex-related, and pathophysiological differences1. Recently, the diagnostic mean pulmonary artery pressure (mPAP) threshold was lowered from >25 mm Hg to >20 mm Hg as studies suggest that lowering pulmonary vascular resistance standards from ≥3.0 Wood units (WU) to >2.0 WU is warranted to promote early identification of PH patients, with the goal of reducing long term disease burden2. Discovering therapeutic approaches to manage PH vascular remodeling is critical to meeting this target and enhancing PH patients’ quality of life.
As the evolution of PH is multifactorial, encompassing exposure to pulmonary vascular insults such as genetic mutations, hypoxia, drugs, pathogens, and environmental pollutants etc, the challenge is to identify specific treatable targets. Therefore, it is vital that we understand not only key triggering factors that initiate vascular remodeling, but also the growing list of cellular phenotypes that contribute to this aberrant process. Here, we discuss the molecular and morphological reprogramming of different vascular cell populations, the pathogenic pathways that resemble cancer, and their epigenetic impact on pulmonary vascular remodeling in PH with a particular emphasis on group 1 PH, also named as pulmonary arterial hypertension (PAH). Vascular cell populations in this review include endothelial cells (ECs), smooth muscle cells (SMCs), pericytes, fibroblasts and myofibroblasts in different experimental PH animal models.
Epigenetics
Epigenetic modifications encompass any process that results in a change in the gene expression pattern without external factors altering the genetic code. Highly organized, eukaryotic DNA is tightly packaged in nucleosomes with histones in the nucleus. Epigenetic chromatin remodeling modifications occur mainly via three mechanisms including 1) DNA/ RNA methylation, 2) histone post-translational modifications and 3) RNA interference through non-coding RNAs (ncRNAs), including small (microRNAs (miRNAs), small interfering RNAs (siRNAs) and Piwi-interacting RNAs (piRNAs) and long noncoding RNAs (lncRNAs), resulting in activation or silencing of genes. DNA methylation, defined by the addition of a methyl group (CH3) to cytosine residues to form 5-methylcytosine by DNA methyltransferases (DNMTs), usually turns off gene expression by facilitating chromatin condensation. Importantly, the histone octamer that DNA is wound around consists of dimers of core histones H2A, H2B, H3, and H4. Within the histone core, the N- and C-terminal amino acid tails of the histone core can undergo post-translational modifications including methylation, acetylation, phosphorylation, sumoylation, ubiquitylation, ribosylation, resulting in the modulation of DNA accessibility by providing binding platforms for transcription factors (TFs) and DNA/chromatin-modifying or remodeling enzymes. An altered epigenetic landscape may drive PH disease pathogenesis3.
PH disease models
One of the major challenges in the PH research field is the availability of a simple and severe PH animal model. Multiple animal models have been designed and utilized to investigate the pathobiology, molecular, and mechanistic insights of PH. Commonly used experimental PH models include chronic hypoxia, Sugen hypoxia, Monocrotaline, gene editing, and pneumonectomy models4.
Chronic hypoxic modeling causes PH-associated pulmonary vascular remodeling with decent predictability and repeatability. However, most of these vascular changes can be restored by returning to a normoxic environment. Combination of the chronic hypoxia model with the vascular endothelial growth factor receptor (VEGFR)-2 antagonist Sugen 5416 (SU5416; semaxanib) (SuHx) results in more severe remodeling and development of plexiform lesions that are more resistant to reversal under normoxic environment. These hypoxic models, however, can have gender, strain, and age differences5.
Monocrotaline (MCT), an 11-membered macrocyclic pyrrolizidine alkaloid that is metabolized into the toxic metabolite MCT pyrrole (MCTP) by liver cytochrome P450 3A4 (CYP3A4), results in vascular EC damage and inflammation-inducing PH, associated RV hypertrophy and an increase in the medial thickness of pulmonary arteries in rats. Although the MCT model has low cost, simplicity, and reproducibility, its broad toxicity causes this model to be less in line with human PAH6.
More severe phenotypes in PH can be achieved by imposing second vascular insults such as chronic hypoxia or an MCT challenge to gene editing models such as BMPR2 mutants and IL6 knockouts. While a left/right pneumonectomy model with MCT or hypoxia has been utilized to model flow-induced PH. Moreover, a recent study suggests that an extended pneumonectomy, achieved by simultaneous removal of the left lung and right caval lobe in mice, is a promising animal model to study the cellular response and molecular mechanisms contributing to compensatory lung growth and flow-induced PH7. In addition to these models, bleomycin and Schistosome treatment can induce PH in rodents4.
Cellular Epigenetic changes
There are multiple vascular cell types known to be involved in the pathogenesis of PAH including endothelial cells, smooth muscle cells, fibroblasts, and pericytes.
Endothelial Cells
The most inner wall of the blood vessel is lined by a monolayer of endothelial cells (ECs). This EC monolayer is responsible for the flux and exchange of various substances across the circulation into the parenchymal tissue, preservation of the vascular barrier, maintenance of vascular tone, thrombosis prevention, inflammation, and modulation of neighboring mural cells including smooth muscle cells and pericytes. Early during the onset of PH, following vascular insults damaged ECs are triggered to undergo apoptosis. With PH progression, apoptosis-resistant ECs emerge1 and eventually in later stages, ECs become senescent resulting in a non-reversible dysfunctional endothelium that contributes to neointima formation and endothelial to mesenchymal transition (EndoMT). Together this contributes to the progression of PAH as there is less protection against thrombosis, leakage, and inflammation.
Multiple epigenetic mechanisms have been implicated in PH endothelial dysfunction. Extensive remodeling of the active enhancer landscape with acetylated histone H3K27 mark was reported in pulmonary arterial endothelial cells (PAECs) derived from PH patients8. Moreover, H3K27 acetylation required for EC regeneration was absent in PAH. In other studies, histone H3K9 acetylation-mediated BOLA3 deficiency, increased endothelial proliferation, survival, and vasoconstriction while leading to decreased angiogenic potential in multiple PH models including hypoxic mice. In contrast, the mimic of bromodomain and extra-terminal (BET) proteins that bind to acetylated histones was found to decrease inflammation and cell cycle progression of pulmonary ECs in PH9. Epigenetic enzymes, such as SIRT3, can modulate EC metabolism via the acetylation of non-histone proteins10. DNA methylation in EC was previously shown to modulate endothelial NO synthase (eNOS) activity via the methylation of a proximal promoter of eNOS resulting in interrupted vasodilation and endothelial homeostasis. Moreover, a previous DNA methylation analysis in pulmonary ECs of PH patients identified a set of genes mainly involved in the lipid transport pathway that could be relevant to PAH pathophysiology11. Other studies found that N6-methyladenosine (m6A)-modified transcripts of lncRNAs have a role in Pyroptosis (a form of programmed cell death) in ECs in hypoxic mediated PH via DNA methylation. Thus, demonstrating a collaboration of multiple epigenetic modifications12. Lastly, a recent study showed long non-coding RNA growth arrest-specific transcript 5 (GAS5) promoted spermidine (SP)-induced autophagy in pulmonary ECs in PH and in a hypoxia rat model by targeting miRNA-31-5p13. The therapeutic potential of microRNAs has been assessed in multiple studies including miR-150 supplementation that demonstrated an attenuation in pulmonary endothelial damage induced by vascular stresses14. Further studies are needed to better understand the epigenetic influence and influencers of endothelial dysregulation.
Smooth Muscle Cells
Vascular smooth muscle cells (SMCs), the medial layer of arteries, are significant contributors to maintaining the integrity of structure and function in the pulmonary vessels. Extensive pulmonary vascular remodeling in PH lung is mainly seen in the small to mid-size arteries, < 500 μm in diameter, that possess a significant thickening of media. Importantly, SMC can reversibly undergo phenotypic switching where it can go from a quiescent to a contractile or synthetic phenotype while acquiring or losing proliferative and migratory potential. Moreover, during phenotypic switching, the capacity to synthesize the extracellular matrix (ECM) is altered, while single-cell transcriptomic studies show the significance of heterogeneity of SMCs in remodeled PAs15. Thus a conceptual relationship between PH vascular remodeling and pathologic cancer theory, supported by pulmonary SMC hyperproliferation, resistance to apoptosis, and increased migration1, continues to gain traction.
Accumulating studies show the role of epigenetics in SMC phenotypic switching or transition from contractile to synthetic, with increased proliferative and migratory capacities. An epigenetic modifier, Switch-independent 3a (SIN3a) overexpression decreased the HDAC activity and methylation level of the BMPR2 promoter attenuating SMCs hyperproliferation and migration in PH in both patients’ tissues and MCT and SuHx PH models16. Furthermore, recent studies define an important role for histone acetylation in PH-SMCs vascular remodeling. For example, increased levels of aldehyde dehydrogenase, ALDH1A3 (aldehyde dehydrogenase family 1 member 3) promote acetyl coenzyme A to acetylate histone H3K27, while inducing highly proliferative and glycolytic pulmonary artery SMC (PASMC) in PH17. Histone H3K9 acetylation was significantly increased in the lungs of PH patients and promoted PASMC proliferation via Sphingosine Kinase 218. CircRNA, Hsa_circ_0001402 acts as an miR-183-5p sponge to inhibit SMC proliferation and migration while activating VSMC autophagy to alleviate neointimal hyperplasia19. Another SMC epigenetic target is SETD2, the main enzyme that catalyzes the trimethylation of H3K36 (H3K36me3) as SMCs-specific SETD2 deficiency ameliorated the pathological pulmonary vascular remodeling in a hypoxia-induced mouse model of PAH20. Moreover, emerging studies suggest that pulmonary ECs can undergo EndoMT transition, resulting in mesenchymal/smooth muscle-like cells in PH. Although these studies support a broader role for epigenetics in SMC phenotypic switching, factors regulating this process and their downstream molecular targets require further exploration.
Adventitial Fibroblasts
Fibroblasts are the most common cell type in the adventitial layer. The activated fibroblast, also known as a myofibroblast, is hyperproliferative, synthesizes increased ECM, and secrete inflammatory cytokines.
Recent studies determined that fibroblasts from PH patients treated with combined therapy of sildenafil (vasodilator) and HDAC inhibitor exhibited synergistic inhibitory effects on PH-fibroblast proliferation and induced metabolic reprogramming21. A recent study in multiple PH models suggests that pharmacological inhibition of the P300/CREB-binding transcriptional co-activators and histone acetyl transferase (HAT) complex rescues distal pulmonary vascular remodeling and hemodynamics in multiple PH models as well as the vascular remodeling in precision-cut tissue slices from human PAH lungs ex vivo 22. Other studies demonstrated via Integrative analysis of RNA-seq and ChIP-seq data of PH-fibroblasts, that the altered epigenetic landscape signatures of fibroblasts in PH were similar to those found in lung morphogenesis 22 highlighting a reactivation of developmental pathways in PH progression. While a role of miR-124 in PH fibroblasts has been demonstrated to modulate proliferative, migratory, and inflammatory phenotype of fibroblasts in PH23. These findings support the importance of expanding our understanding of the epigenetic factors that reawaken latent lung development tendencies and their contribution to the development of PH.
Pericytes
Pericytes provide the structural support for the endothelial tube and participate in vascular tone maintenance24,25. Accumulation of pericytes in the distal pulmonary arteries is seen in PH patients26,27, with single-cell transcriptomics findings supportive of the recruitment of pericytes to the inflamed pulmonary arteries in PH and the potential of pericytes to differentiate into smooth muscle-like cells 26,27. Emerging studies implicate a role for pericytes in PH, where tyrosine kinase receptor–inducing lncRNA and TUG1 lncRNA induce a pro-proliferative phenotype in PH26,28,29. Unfortunately, broadening our understanding of epigenetic factors on pericyte contribution to PH vascular remodeling is challenged by a lack of consistent molecular markers and the subsequent difficulties associated with distinguishing pericytes from other cell types. However, recent advancements in multiomics, single cell and spatial transcriptomics, CUT&RUN, and CUT&TAG technologies will likely improve pericyte research in PH.
Cell – Cell interactions:
Single-cell transcriptomic data demonstrate that the cell-cell communication in PH is significantly altered and transforms more towards SMC- fibroblasts contacts30. Recent studies highlight the importance of SMC–EC contact for EC regeneration. Importantly, this process is mediated by epigenetic histone acetylation31. Other studies determined that endothelial-secreted factors promote the hyperproliferation of SMCs by epigenetic mechanisms such as histone acetylation and microRNA32,33. Future exploration is needed to further determine the epigenetic contribution that cell-cell interaction has in PH vascular remodeling and to assess the reversibility of epigenetic targeting in PH (Figure 1).
Vascular Remodeling: Unveiling Common Epigenetic Threads in Pulmonary Hypertension and Associated Proliferative Diseases such as Lung Cancer
Parallels between numerous respiratory disorders, particularly lung cancer (LC), chronic obstructive pulmonary disease (COPD), and idiopathic pulmonary fibrosis (IPF), have long piqued the interest of researchers. Notably, in 1998 PH was linked to malignant disorders due to its pathogenic pathways resembling some elements of cancer34, with recent studies revealing significant genetic and epigenetic overlap between different types of LC and PH35–37.
The similarity between cancer behaviors and some disease characteristics of PH was a startling discovery with PH PASMCs and ECs exhibiting cellular traits typical of cancer including hyper-proliferation and resistance to apoptosis35–37. In contrast to tumor cell hyperproliferation, aberrant proliferation in PH causes vascular remodeling and contributes considerably to the unique symptoms of PH of pulmonary artery constriction and increased pulmonary vascular resistance38. Several studies have found a strong link between PH and LC39–42 as a considerable number of LC patients also develop PH39,40,43. In LC, tumor epithelial cells affect the tumor microenvironment, causing vascular remodeling and contributing to the establishment of PH. This association has been corroborated across various mouse models, underscoring the intricate interplay between LC epithelial cells, SMC, and ECs.
The development of PH in LC patients creates complications, possibly affecting surgery outcomes and, in extreme situations, hindering resection feasibility43. Despite recent advances in understanding the frequency of both LC and PH, the complicated processes underlying their relationship remain unknown. However, the presence of PH in LC patients provides an opportunity to use surrogate PH indicators to predict LC prognosis. Consequently, an in-depth investigation of the development of PH in LC patients is required, with the goal of using it as diagnostic and prognostic indicators. Aligned with the central theme of this review focusing on PH and epigenetics, this segment will focus on the epigenetic factors influencing the concurrent occurrence of PH in LC patients. Additionally, it will contemplate the prospect of employing these epigenetic markers for novel diagnostic and prognostic applications for PH management in LC patients.
SMCs and ECs play critical roles in PH35,37,38. To investigate epigenetic abnormalities in LC patients that may contribute to the development of PH, a targeted analysis of epigenetic variables impacting these particular cell types within the lung environment is required. However, the relationship between LC epithelial cells and their cellular counterparts is complex. As epigenetic modifications in LC epithelial cells may cause changes in the chemicals produced by tumor cells, potentially training SMCs and ECs to be hyperproliferative and apoptosis-resistant, leading to PH. Epigenetic changes inside SMCs and ECs, mediated by interactions with tumor epithelial cells, may, on the other hand, induce such traits in these cell types. Understanding the epigenetic remodeling that leads to PH in LC patients requires a holistic approach to both illnesses.
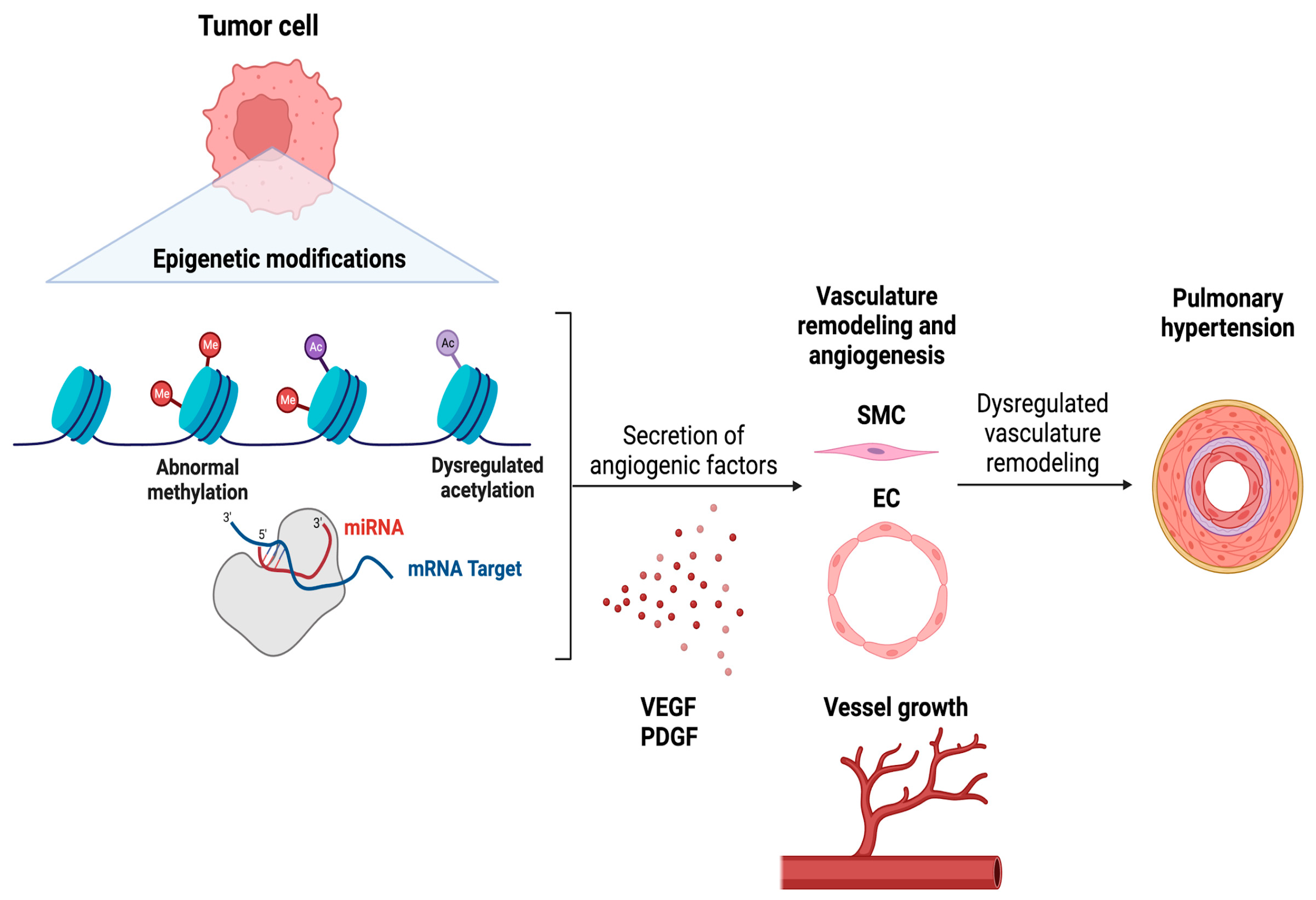
Tumor epithelial cells release cytokines, chemokines, and other growth factors to alter the tumor microenvironment and promote tumor proliferation and migration44,45. Driven in part by the tumor microenvironment that contains a diverse range of stromal and immune cell types39,46–48. By hijacking the immune system, tumors create an immunosuppressive environment to avoid immune cell-mediated tumor cell death, and more crucially, modify the vasculature to maintain the input of oxygen and nutrients to continue growing49–51. To induce angiogenesis, tumor epithelial cells release angiogenic factors such as Vascular endothelial growth factor (VEGF) and Platelet-derived growth factor (PDGF). Given their function in vascular remodeling, plasma levels of these factors are understandably higher in patients with severe PH52,53. As a result, focusing on the control of the angiogenic factors by lung tumor cells, as well as how it is altered by epigenetic alterations, may help us understand the common incidence of PH in LC patients. This section will attempt to unravel how lung tumor epithelial cells alter the lung vasculature to increase PH via epigenetic modifications while addressing whether these epigenetic markers can be utilized to predict the incidence of PH in LC patients and be targeted to treat PH in LC patients (Figure 2).
Posttranslational histone modifications, DNA changes, and microRNAs (miRNAs) are the most prevalent epigenetic alterations used by lung tumor cells to induce angiogenesis and modify the vasculature54,55. Histone acetylation and methylation are the two most frequent epigenetic changes that influence angiogenesis in LC54 with Histone deacetylases (HDACs), particularly HDAC1, playing a key role in vascular integrity56. Although research on HDACs and angiogenesis in LC is scarce, their impact on angiogenesis in other cancers57,58 suggests a potential role for HDACs in LC-mediated vascular modifications. Another epigenetic alteration that controls LC angiogenesis is DNA methylation. Studies indicate that the methylation state of a promoter regulates genes critical to vascular remodeling such as VEGF expression59. MicroRNAs are another epigenetic regulator that affects angiogenesis in LC. MiRNAs, short single-stranded non-coding RNAs with total lengths ranging from 19 to 25 nucleotides, exert control over gene expression by triggering translational repression via fast mRNA degradation by deadenylation. MicroRNAs that target angiogenic factors such as VEGF-A (miR-128, miR-200b) are downregulated in60–62, but MicroRNAs that target anti-angiogenic factors (miR-221/222 cluster, miR-210) are elevated. These discoveries indicate that targeting these epigenetic alterations that PH linked to lung cancer.
Implications on LC Associated PH Management
The relationship between angiogenesis in LC and probable PH raises an essential question: can we use these common epigenetic modifications in LC patients as diagnostic tools for PH? The key to unraveling this conundrum is determining how these modifications that occur in tumor epithelial cells, SMCs, and ECs, may be targeted for PH treatment in LC. Despite scant research on specific epigenetic alterations in LC-related PH, investigating shared changes in both disorders provides insights into possible indicators and therapeutic methods for this condition.
Several studies suggest that blood VEGF levels can be used to determine the severity of LC. Interestingly, VEGF plasma levels are also increased in patients with severe PH63,64, with VEGF and VEGF receptor 2 (VEGFR2) being strongly expressed in complicated vascular lesions in the lungs of PH patients65. Taken together, this might be considered a realistic alternative for predicting LC-linked PH, while targeting epigenetic markers, in addition to anti-angiogenic medications, could be a viable strategy for preventing/treating PH in these patients. Histone acetylation, as previously established, is one of the major epigenetic alterations that govern angiogenesis in LC. Surprisingly, it is a modification that occurs often in PH as well33,65,66. Unfortunately, medications that inhibit HDAC (HDACi) such as Vorinostat (Zolinza), Romidepsin (Istodax), and Belinostat (Beleodaq), already approved by the FDA as safe for use in certain lymphoma cases, lack conclusive data regarding their efficacy in treating solid tumors or whether tumors with specific genetic changes may respond better to these drugs66–68. Nonetheless, considering the importance of HDACs and HATs in reprogramming vasculature in LC, they may have substantial promise as a therapy not just for LC, but also for LC-linked PH. As mentioned previously, DNA methylation is another significant epigenetic alteration in LC that affects vascular remodeling. Like histone acetylation, DNA methylation is also seen extensively in PH69,70. DNA methylation can be inhibited using DNA methyltransferase inhibitors (DNMTis)71. Unfortunately, there are no FDA-approved DNMTis for the treatment of LC. Azacitidine and decitabine, both DNA methyltransferase inhibitors, have undergone clinical studies in several malignancies, including LC, to determine their efficacy and safety72. Their approvals include myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML)73, but not have been approved for the treatment of any solid tumors. Based on the function of DNA methylation and vascular remodeling, these inhibitors may influence the epigenetic pathways involved in PH development, thereby opening a novel treatment route in LC-associated PH. To alleviate PH, they might act on both tumors and cells responsible for vascular remodeling. Finally, like HDACis and DNMTis, no miRNA mimics or inhibitors are available for the therapy of solid malignancies. Furthermore, miRNA-related medicines are still in the early stages of research for a variety of disorders. Of note, four novel RNAi-based therapies have been authorized by the FDA: patisiran, givosiran, lumasiran, inclisiran, and Vutrisiran74–77. Each of these siRNA medicines targets distinct mRNA transcripts to treat conditions other than cancer. As a result, numerous advances in the use of miRNA-related medicines for the treatment of LC-related PH still need to be made. The epigenetic mechanisms leading to PH development in LC patients are summarized in Figure 2.
Taken as a whole, the holistic review of the existing literature reveals that, while addressing epigenetic changes has the potential for treating LC-related PH, significant research in this area is absent. Notably, using epigenetic targeting in conjunction with standard anti-angiogenic medications to target angiogenesis in LC may be promising. Because LC and PH share epigenetic changes, focusing on these similarities has a double advantage. Targeting these changes might effectively address the complex interaction between lung tumor epithelial cells, ECs, and SMCs, thereby preventing the development of pulmonary hypertension in LC patients.
Conclusion
Epigenetic mediators impact PH vascular remodeling by molecular and morphological reprogramming of ECs, SMCs, pericytes, fibroblasts, and myofibroblasts vascular cell populations. With activated pathogenic pathways not only resembling cancer, but they also afford valuable insight into future therapeutic opportunities. Taken together, these studies support the importance of the identification of PH epigenetic mediators and their targets.
Authors Contribution
ADCUR conceived the project, prepared figures, drafted and wrote the manuscript, PT conceived the project, prepared figures, drafted and wrote the manuscript, and MAS conceived the project, wrote, revised and critically reviewed the manuscript. All authors read and approved the manuscript.
Sources of funding
This publication was made possible in part by 1R56HL163583 (MAS) from the NIH and by an award from the Indiana University School of Medicine (MAS). The content is solely the responsibility of the authors and does not necessarily represent the official views of the Indiana University School of Medicine.
Acknowledgments
Schematic diagrams are created using BioRender.com.
References
- Rabinovitch, M. Molecular Pathogenesis of Pulmonary Arterial Hypertension. J Clin Invest 2012, 122, 4306–4313. [Google Scholar] [CrossRef]
- Maron, B. A. Revised Definition of Pulmonary Hypertension and Approach to Management: A Clinical Primer. J Am Heart Assoc 2023, 12, 29024. [Google Scholar] [CrossRef]
- Dushani, C.U. Ranasinghe, A.; Schwarz, M. A. Integrating Epigenetics and Metabolomics to Advance Treatments for Pulmonary Arterial Hypertension. Biochem Pharmacol 2022, 115245. [Google Scholar] [CrossRef]
- Wu, X. H.; Ma, J. L.; Ding, D.; Ma, Y. J.; Wei, Y. P.; Jing, Z. C. Experimental Animal Models of Pulmonary Hypertension: Development and Challenges. Animal Model Exp Med 2022, 5, 207–216. [Google Scholar] [CrossRef]
- Zhao, L. Chronic Hypoxia-Induced Pulmonary Hypertension in Rat: The Best Animal Model for Studying Pulmonary Vasoconstriction and Vascular Medial Hypertrophy. Drug Discov Today Dis Models 2010, 7, 83–88. [Google Scholar] [CrossRef]
- Tang, C.; Luo, Y.; Li, S.; Huang, B.; Xu, S.; Li, L. Characteristics of Inflammation Process in Monocrotaline-Induced Pulmonary Arterial Hypertension in Rats. Biomedicine & Pharmacotherapy 2021, 133, 111081. [Google Scholar] [CrossRef]
- Klouda, T.; Tsikis, S. T.; Kim, H.; Liu, T.; Visner, G.; Fernandez-Gonzalez, A.; Kourembanas, S.; Puder, M.; Raby, B.; Yuan, K. Pericytes Contribute to Flow-Induced Pulmonary Hypertension. Am J Respir Cell Mol Biol 2023, 68, 705–708. [Google Scholar] [CrossRef]
- Reyes-Palomares, A.; Gu, M.; Grubert, F.; Berest, I.; Sa, S.; Kasowski, M.; Arnold, C.; Shuai, M.; Srivas, R.; Miao, S.; Li, D.; Snyder, M. P.; Rabinovitch, M.; Zaugg, J. B. Remodeling of Active Endothelial Enhancers Is Associated with Aberrant Gene-Regulatory Networks in Pulmonary Arterial Hypertension. Nature Communications 2020 11:1 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mumby, S.; Gambaryan, N.; Meng, C.; Perros, F.; Humbert, M.; Wort, S. J.; Adcock, I. M. Bromodomain and Extra-Terminal Protein Mimic JQ1 Decreases Inflammation in Human Vascular Endothelial Cells: Implications for Pulmonary Arterial Hypertension. Respirology 2017, 22, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Egnatchik, R. A.; Brittain, E. L.; Shah, A. T.; Fares, W. H.; Ford, H. J.; Monahan, K.; Kang, C. J.; Kocurek, E. G.; Zhu, S.; Luong, T.; Nguyen, T. T.; Hysinger, E.; Austin, E. D.; Skala, M. C.; Young, J. D.; Roberts, L. J.; Hemnes, A. R.; West, J.; Fessel, J. P. Dysfunctional BMPR2 Signaling Drives an Abnormal Endothelial Requirement for Glutamine in Pulmonary Arterial Hypertension. Pulm Circ 2017, 7, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Hautefort, A.; Chesné, J.; Preussner, J.; Pullamsetti, S. S.; Tost, J.; Looso, M.; Antigny, F.; Girerd, B.; Riou, M.; Eddahibi, S.; Deleuze, J. F.; Seeger, W.; Fadel, E.; Simonneau, G.; Montani, D.; Humbert, M.; Perros, F. Pulmonary Endothelial Cell DNA Methylation Signature in Pulmonary Arterial Hypertension. Oncotarget 2017, 8, 52995–53016. [Google Scholar] [CrossRef]
- Wang, X.; Li, Q.; He, S.; Bai, J.; Ma, C.; Zhang, L.; Guan, X.; Yuan, H.; Li, Y.; Zhu, X.; Mei, J.; Gao, F.; Zhu, D. LncRNA FENDRR with M6A RNA Methylation Regulates Hypoxia-Induced Pulmonary Artery Endothelial Cell Pyroptosis by Mediating DRP1 DNA Methylation. Mol Med 2022, 28. [Google Scholar] [CrossRef]
- Wu, Q.; Zhou, X.; Wang, Y.; Hu, Y. LncRNA GAS5 Promotes Spermidine-induced Autophagy through the MiRNA-31-5p/NAT8L Axis in Pulmonary Artery Endothelial Cells of Patients with CTEPH. Mol Med Rep 2022, 26. [Google Scholar] [CrossRef] [PubMed]
- Russomanno, G.; Jo, K. B.; Abdul-Salam, V. B.; Morgan, C.; Endruschat, J.; Schaeper, U.; Osman, A. H.; Alzaydi, M. M.; Wilkins, M. R.; Wojciak-Stothard, B. MiR-150-PTPMT1-Cardiolipin Signaling in Pulmonary Arterial Hypertension. Mol Ther Nucleic Acids 2021, 23, 142–153. [Google Scholar] [CrossRef]
- Crnkovic, S.; Valzano, F.; Fließer, E.; Gindlhuber, J.; Puthenparampil, H. T.; Basil, M.; Morley, M. P.; Katzen, J.; Gschwandtner, E.; Klepetko, W.; Cantu, E.; Wolinski, H.; Olschewski, H.; Lindenmann, J.; Zhao, Y. Y.; Morrisey, E. E.; Marsh, L. M.; Kwapiszewska, G. Single-Cell Transcriptomics Reveals Skewed Cellular Communication and Phenotypic Shift in Pulmonary Artery Remodeling. JCI Insight 2022, 7. [Google Scholar] [CrossRef] [PubMed]
- Bisserier, M.; Mathiyalagan, P.; Zhang, S.; Elmastour, F.; Dorfmüller, P.; Humbert, M.; David, G.; Tarzami, S.; Weber, T.; Perros, F.; Sassi, Y.; Sahoo, S.; Hadri, L. Regulation of the Methylation and Expression Levels of the BMPR2 Gene by SIN3a as a Novel Therapeutic Mechanism in Pulmonary Arterial Hypertension. Circulation 2021, 144, 52–73. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Shao, N. Y.; Moonen, J. R. M. D.; Zhao, Z.; Shi, M.; Otsuki, S.; Wang, L.; Elaine Yan, T. N.; Marciano, D. P.; Contrepois, K.; Li, C. G.; Wu, J. C.; Snyder, M. P.; Rabinovitch, M. ALDH1A3 Coordinates Metabolism with Gene Regulation in Pulmonary Arterial Hypertension. Circulation 2021, 143, 2074–2090. [Google Scholar] [CrossRef] [PubMed]
- Ranasinghe, A. D. C. U.; Holohan, M.; Borger, K. M.; Donahue, D. L.; Kuc, R. D.; Gerig, M.; Kim, A.; Ploplis, V. A.; Castellino, F. J.; Schwarz, M. A. Altered Smooth Muscle Cell Histone Acetylome by the SPHK2/S1P Axis Promotes Pulmonary Hypertension. Circ Res 2023, 133, 704–719. [Google Scholar] [CrossRef] [PubMed]
- Lin, J. J.; Chen, R.; Yang, L. Y.; Gong, M.; Du, M. Y.; Mu, S. Q.; Jiang, Z. A.; Li, H. H.; Yang, Y.; Wang, X. H.; Wang, S. F.; Liu, K. X.; Cao, S. H.; Wang, Z. Y.; Zhao, A. Q.; Yang, S. Y.; Li, C.; Sun, S. G. Hsa_circ_0001402 Alleviates Vascular Neointimal Hyperplasia through a MiR-183-5p-Dependent Regulation of Vascular Smooth Muscle Cell Proliferation, Migration, and Autophagy. J Adv Res 2023. [Google Scholar] [CrossRef]
- Zhou, X. L.; Huang, F. J.; Li, Y.; Huang, H.; Wu, Q. C. SEDT2/METTL14-Mediated M6A Methylation Awakening Contributes to Hypoxia-Induced Pulmonary Arterial Hypertension in Mice. Aging 2021, 13, 7538–7548. [Google Scholar] [CrossRef]
- Zhang, H.; D’Alessandro, A.; Li, M.; Reisz, J. A.; Riddle, S.; Muralidhar, A.; Bull, T.; Zhao, L.; Gerasimovskaya, E.; Stenmark, K. R. Histone Deacetylase Inhibitors Synergize with Sildenafil to Suppress Purine Metabolism and Proliferation in Pulmonary Hypertension. Vascul Pharmacol 2023, 149. [Google Scholar] [CrossRef] [PubMed]
- Chelladurai, P.; Kuenne, C.; Bourgeois, A.; Günther, S.; Valasarajan, C.; Cherian, A. V.; Rottier, R. J.; Romanet, C.; Weigert, A.; Boucherat, O.; Eichstaedt, C. A.; Ruppert, C.; Guenther, A.; Braun, T.; Looso, M.; Savai, R.; Seeger, W.; Bauer, U. M.; Bonnet, S.; Pullamsetti, S. S. Epigenetic Reactivation of Transcriptional Programs Orchestrating Fetal Lung Development in Human Pulmonary Hypertension. Sci Transl Med 2022, 14, 5407. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, H.; Li, M.; Frid, M. G.; Flockton, A. R.; McKeon, B. A.; Yeager, M. E.; Fini, M. A.; Morrell, N. W.; Pullamsetti, S. S.; Velegala, S.; Seeger, W.; McKinsey, T. A.; Sucharov, C. C.; Stenmark, K. R. MicroRNA-124 Controls the Proliferative, Migratory, and Inflammatory Phenotype of Pulmonary Vascular Fibroblasts. Circ Res 2014, 114, 67–78. [Google Scholar] [CrossRef]
- Garrison, A. T.; Bignold, R. E.; Wu, X.; Johnson, J. R. Pericytes: The Lung-Forgotten Cell Type. Front Physiol 2023, 14. [Google Scholar] [CrossRef]
- Guo, L.; Yang, Q.; Wei, R.; Zhang, W.; Yin, N.; Chen, Y.; Xu, C.; Li, C.; Carney, R. P.; Li, Y.; Feng, M. Enhanced Pericyte-Endothelial Interactions through NO-Boosted Extracellular Vesicles Drive Revascularization in a Mouse Model of Ischemic Injury. Nature Communications 2023 14:1 2023, 14, 1–18. [Google Scholar] [CrossRef]
- Bordenave, J.; Tu, L.; Berrebeh, N.; Thuillet, R.; Cumont, A.; Le Vely, B.; Fadel, E.; Nadaud, S.; Savale, L.; Humbert, M.; Huertas, A.; Guignabert, C. Lineage Tracing Reveals the Dynamic Contribution of Pericytes to the Blood Vessel Remodeling in Pulmonary Hypertension. Arterioscler Thromb Vasc Biol 2020, 40, 766–782. [Google Scholar] [CrossRef]
- Ricard, N.; Tu, L.; Le Hiress, M.; Huertas, A.; Phan, C.; Thuillet, R.; Sattler, C.; Fadel, E.; Seferian, A.; Montani, D.; Dorfmüller, P.; Humbert, M.; Guignabert, C. Increased Pericyte Coverage Mediated by Endothelial-Derived Fibroblast Growth Factor-2 and Interleukin-6 Is a Source of Smooth Muscle-like Cells in Pulmonary Hypertension. Circulation 2014, 129, 1586–1597. [Google Scholar] [CrossRef]
- Dave, J.; Jagana, V.; Janostiak, R.; Bisserier, M. Unraveling the Epigenetic Landscape of Pulmonary Arterial Hypertension: Implications for Personalized Medicine Development. Journal of Translational Medicine 2023 21:1 2023, 21, 1–18. [Google Scholar] [CrossRef]
- Zehendner, C. M.; Valasarajan, C.; Werner, A.; Boeckel, J. N.; Bischoff, F. C.; John, D.; Weirick, T.; Glaser, S. F.; Rossbach, O.; Jae, N.; Demolli, S.; Khassafi, F.; Yuan, K.; De Jesus Perez, V. A.; Michalik, K. M.; Chen, W.; Seeger, W.; Guenther, A.; Wasnick, R. M.; Uchida, S.; Zeiher, A. M.; Dimmeler, S.; Pullamsetti, S. S. Long Noncoding RNA TYKRIL Plays a Role in Pulmonary Hypertension via the P53-Mediated Regulation of PDGFRb. Am J Respir Crit Care Med 2020, 202, 1445–1457. [Google Scholar] [CrossRef]
- Crnkovic, S.; Valzano, F.; Fließer, E.; Gindlhuber, J.; Puthenparampil, H. T.; Basil, M.; Morley, M. P.; Katzen, J.; Gschwandtner, E.; Klepetko, W.; Cantu, E.; Wolinski, H.; Olschewski, H.; Lindenmann, J.; Zhao, Y. Y.; Morrisey, E. E.; Marsh, L. M.; Kwapiszewska, G. Single-Cell Transcriptomics Reveals Skewed Cellular Communication and Phenotypic Shift in Pulmonary Artery Remodeling. JCI Insight 2022, 7. [Google Scholar] [CrossRef]
- Miyagawa, K.; Shi, M.; Chen, P. I.; Hennigs, J. K.; Zhao, Z.; Wang, M.; Li, C. G.; Saito, T.; Taylor, S.; Sa, S.; Cao, A.; Wang, L.; Snyder, M. P.; Rabinovitch, M. Smooth Muscle Contact Drives Endothelial Regeneration by BMPR2-Notch1-Mediated Metabolic and Epigenetic Changes. Circ Res 2019, 124, 211–224. [Google Scholar] [CrossRef]
- Su, Y.; Tan, R.; Sun, M.; Yuan, L.; Ruiz, M.; Dupuis, J.; Hu, Q.; Zhu, L. MiR-1249 on Endothelial Extracellular Vesicles Mediates Cigarette Smoke-Induced Pulmonary Hypertension by Inhibiting HDAC10 (Histone Deacetylase 10)-NFκB (Nuclear Factor ΚB)-CaSR (Calcium-Sensing Receptor) Cascade. Hypertension 2022, 79, 2721–2732. [Google Scholar] [CrossRef] [PubMed]
- Ranasinghe, A. D. C. U.; Holohan, M.; Borger, K. M.; Donahue, D. L.; Kuc, R. D.; Gerig, M.; Kim, A.; Ploplis, V. A.; Castellino, F. J.; Schwarz, M. A. Altered Smooth Muscle Cell Histone Acetylome by the SPHK2/S1P Axis Promotes Pulmonary Hypertension. Circ Res 2023, 133, 704–719. [Google Scholar] [CrossRef] [PubMed]
- Voelkel, N. F.; Cool, C.; Lee, S. D.; Wright, L.; Geraci, M. W.; Tuder, R. M. Primary Pulmonary Hypertension between Inflammation and Cancer. Chest 1998, 114 (3 Suppl), 225S–230S. [Google Scholar] [CrossRef]
- Lévy, M.; Maurey, C.; Celermajer, D. S.; Vouhé, P. R.; Danel, C.; Bonnet, D.; Israël-Biet, D. Impaired Apoptosis of Pulmonary Endothelial Cells Is Associated with Intimal Proliferation and Irreversibility of Pulmonary Hypertension in Congenital Heart Disease. J Am Coll Cardiol 2007, 49, 803–810. [Google Scholar] [CrossRef]
- Ameshima, S.; Golpon, H.; Cool, C. D.; Chan, D.; Vandivier, R. W.; Gardai, S. J.; Wick, M.; Nemenoff, R. A.; Geraci, M. W.; Voelkel, N. F. Peroxisome Proliferator-Activated Receptor Gamma (PPARgamma) Expression Is Decreased in Pulmonary Hypertension and Affects Endothelial Cell Growth. Circ Res 2003, 92, 1162–1169. [Google Scholar] [CrossRef]
- Tuder, R. M.; Cool, C. D.; Yeager, M.; Taraseviciene-Stewart, L.; Bull, T. M.; Voelkel, N. F. The Pathobiology of Pulmonary Hypertension. Endothelium. Clin Chest Med 2001, 22, 405–418. [Google Scholar] [CrossRef]
- Lan, N. S. H.; Massam, B. D.; Kulkarni, S. S.; Lang, C. C. Pulmonary Arterial Hypertension: Pathophysiology and Treatment. Diseases 2018, 6. [Google Scholar] [CrossRef]
- Pullamsetti, S. S.; Kojonazarov, B.; Storn, S.; Gall, H.; Salazar, Y.; Wolf, J.; Weigert, A.; El-Nikhely, N.; Ghofrani, H. A.; Krombach, G. A.; Fink, L.; Gattenlöhner, S.; Rapp, U. R.; Schermuly, R. T.; Grimminger, F.; Seeger, W.; Savai, R. Lung Cancer-Associated Pulmonary Hypertension: Role of Microenvironmental Inflammation Based on Tumor Cell-Immune Cell Cross-Talk. Sci Transl Med 2017, 9. [Google Scholar] [CrossRef]
- McHugh, S.; Vanchiere, C.; Oliveros, E.; Islam, S.; Luceno, S.; Vaidya, A.; Forfia, P. Malignancy-Related Pulmonary Hypertension Presenting as a Pulmonary Veno-Occlusive–Like Syndrome: A Single-Center Case Series. JACC Case Rep 2021, 3, 1044. [Google Scholar] [CrossRef]
- Wieshammer, S.; Dreyhaupt, J.; Müller, D.; Momm, F.; Jakob, A. Venous Thromboembolism and Persistent Pulmonary Hypertension in Cancer Patients: A Cross-Sectional Study. Thromb J 2016, 14. [Google Scholar] [CrossRef]
- he, X. wu; Tang, yi H.; Luo, Z. Q.; Gong, L. di; Cheng, T. O. Subacute Cor Pulmonale Due to Tumor Embolization to the Lungs. Angiology 1989, 40, 11–17. [CrossRef]
- Tello, K.; Wilhelm, J.; Gattenlöhner, S.; Sibelius, U.; Grimminger, F.; Seeger, W.; Savai, R. Noninvasive Surrogate Markers of Pulmonary Hypertension Are Associated with Poor Survival in Patients with Lung Cancer. Am J Respir Crit Care Med 2021, 203, 1316–1319. [Google Scholar] [CrossRef]
- Esquivel-Velázquez, M.; Ostoa-Saloma, P.; Palacios-Arreola, M. I.; Nava-Castro, K. E.; Castro, J. I.; Morales-Montor, J. The Role of Cytokines in Breast Cancer Development and Progression. Journal of Interferon & Cytokine Research 2015, 35, 1. [Google Scholar] [CrossRef]
- Kartikasari, A. E. R.; Huertas, C. S.; Mitchell, A.; Plebanski, M. Tumor-Induced Inflammatory Cytokines and the Emerging Diagnostic Devices for Cancer Detection and Prognosis. Front Oncol 2021, 11, 2641. [Google Scholar] [CrossRef]
- Yang, L.; Achreja, A.; Yeung, T. L.; Mangala, L. S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J. C.; Ni, J.; Nakahara, R.; Wahlig, S.; Chiba, L.; Kim, S. H.; Morse, J.; Pradeep, S.; Nagaraja, A. S.; Haemmerle, M.; Kyunghee, N.; Derichsweiler, M.; Plackemeier, T.; Mercado-Uribe, I.; Lopez-Berestein, G.; Moss, T.; Ram, P. T.; Liu, J.; Lu, X.; Mok, S. C.; Sood, A. K.; Nagrath, D. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab 2016, 24, 685. [Google Scholar] [CrossRef] [PubMed]
- Diaz Bessone, M. I.; Gattas, M. J.; Laporte, T.; Tanaka, M.; Simian, M. The Tumor Microenvironment as a Regulator of Endocrine Resistance in Breast Cancer. Front Endocrinol (Lausanne) 2019, 10, 547. [Google Scholar] [CrossRef]
- Ni, Y.; Zhou, X.; Yang, J.; Shi, H.; Li, H.; Zhao, X.; Ma, X. The Role of Tumor-Stroma Interactions in Drug Resistance Within Tumor Microenvironment. Front Cell Dev Biol 2021, 9. [Google Scholar] [CrossRef]
- Bates, J. P.; Derakhshandeh, R.; Jones, L.; Webb, T. J. Mechanisms of Immune Evasion in Breast Cancer. BMC Cancer 2018, 18, 1–14. [Google Scholar] [CrossRef]
- Lee, W. S.; Yang, H.; Chon, H. J.; Kim, C. Combination of Anti-Angiogenic Therapy and Immune Checkpoint Blockade Normalizes Vascular-Immune Crosstalk to Potentiate Cancer Immunity. Experimental & Molecular Medicine 2020 52:9 2020, 52, 1475–1485. [Google Scholar] [CrossRef]
- Thomas, R.; Al-Khadairi, G.; Decock, J. Immune Checkpoint Inhibitors in Triple Negative Breast Cancer Treatment: Promising Future Prospects. Front Oncol 2021, 10, 3464. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, A. I.; Zakynthinos, E.; Kostikas, K.; Kiropoulos, T.; Koutsokera, A.; Ziogas, A.; Koutroumpas, A.; Sakkas, L.; Gourgoulianis, K. I.; Daniil, Z. D. Serum VEGF Levels Are Related to the Presence of Pulmonary Arterial Hypertension in Systemic Sclerosis. BMC Pulm Med 2009, 9, 18. [Google Scholar] [CrossRef]
- Wu, K.; Tang, H.; Lin, R.; Carr, S. G.; Wang, Z.; Babicheva, A.; Ayon, R. J.; Jain, P. P.; Xiong, M.; Rodriguez, M.; Rahimi, S.; Balistrieri, F.; Rahimi, S.; Valdez-Jasso, D.; Simonson, T. S.; Desai, A. A.; Garcia, J. G. N.; Shyy, J. Y. J.; Thistlethwaite, P. A.; Wang, J.; Makino, A.; Yuan, J. X. J. Endothelial Platelet-Derived Growth Factor-Mediated Activation of Smooth Muscle Platelet-Derived Growth Factor Receptors in Pulmonary Arterial Hypertension. Pulm Circ 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Tan, H. W.; Xu, Y. M.; Qin, S. H.; Chen, G. F.; Lau, A. T. Y. Epigenetic Regulation of Angiogenesis in Lung Cancer. J Cell Physiol 2021, 236, 3194–3206. [Google Scholar] [CrossRef]
- Aspriţoiu, V. M.; Stoica, I.; Bleotu, C.; Diaconu, C. C. Epigenetic Regulation of Angiogenesis in Development and Tumors Progression: Potential Implications for Cancer Treatment. Front Cell Dev Biol 2021, 9. [Google Scholar] [CrossRef]
- Chen, C.; Wei, M.; Wang, C.; Sun, D.; Liu, P.; Zhong, X.; He, Q.; Yu, W. The Histone Deacetylase HDAC1 Activates HIF1α/VEGFA Signal Pathway in Colorectal Cancer. Gene 2020, 754, 144851. [Google Scholar] [CrossRef]
- Kaluza, D.; Kroll, J.; Gesierich, S.; Manavski, Y.; Boeckel, J. N.; Doebele, C.; Zelent, A.; Rössig, L.; Zeiher, A. M.; Augustin, H. G.; Urbich, C.; Dimmeler, S. Histone Deacetylase 9 Promotes Angiogenesis by Targeting the Antiangiogenic MicroRNA-17-92 Cluster in Endothelial Cells. Arterioscler Thromb Vasc Biol 2013, 33, 533–543. [Google Scholar] [CrossRef]
- Turtoi, A.; Peixoto, P.; Castronovo, V.; Bellahcène, A. Histone Deacetylases and Cancer-Associated Angiogenesis: Current Understanding of the Biology and Clinical Perspectives. Crit Rev Oncog 2015, 20, 119–137. [Google Scholar] [CrossRef]
- Cooper, M. P.; Keaney, J. F. Epigenetic Control of Angiogenesis via DNA Methylation. Circulation 2011, 123, 2916–2918. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y. C.; Yoon, S.; Jeong, Y.; Yoon, J.; Baek, K. Regulation of Vascular Endothelial Growth Factor Signaling by MiR-200b. Mol Cells 2011, 32, 77. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y. L.; Guo, F.; Liu, F.; Gao, F. L.; Zhang, P. Q.; Niu, X.; Guo, S. C.; Yin, J. H.; Wang, Y.; Deng, Z. F. MiR-210 Activates Notch Signaling Pathway in Angiogenesis Induced by Cerebral Ischemia. Mol Cell Biochem 2012, 370, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhang, Y.; Li, M.; Liu, X.; Darvishi, M. The Various Role of MicroRNAs in Breast Cancer Angiogenesis, with a Special Focus on Novel MiRNA-Based Delivery Strategies. Cancer Cell International 2023 23:1 2023, 23, 1–22. [Google Scholar] [CrossRef]
- Eddahibi, S.; Humbert, M.; Sediame, S.; Chouaid, C.; Partovian, C.; Maitre, B.; Teiger, E.; Rideau, D.; Simonneau, G.; Sitbon, O.; Adnot, S. Imbalance between Platelet Vascular Endothelial Growth Factor and Platelet-Derived Growth Factor in Pulmonary Hypertension. 2012, 162 (4 I), 1493–1499. [CrossRef]
- Selimovic, N.; Bergh, C. H.; Andersson, B.; Sakiniene, E.; Carlsten, H.; Rundqvist, B. Growth Factors and Interleukin-6 across the Lung Circulation in Pulmonary Hypertension. European Respiratory Journal 2009, 34, 662–668. [Google Scholar] [CrossRef]
- Tuder, R. M.; Chacon, M.; Alger, L.; Wang, J.; Taraseviciene-Stewart, L.; Kasahara, Y.; Cool, C. D.; Bishop, A. E.; Geraci, M.; Semenza, G. L.; Yacoub, M.; Polak, J. M.; Voelkel, N. F. Expression of Angiogenesis-Related Molecules in Plexiform Lesions in Severe Pulmonary Hypertension: Evidence for a Process of Disordered Angiogenesis. J Pathol 2001, 195, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Libby, E. N.; Becker, P. S.; Burwick, N.; Green, D. J.; Holmberg, L.; Bensinger, W. I. Panobinostat: A Review of Trial Results and Future Prospects in Multiple Myeloma. Expert Rev Hematol 2015, 8, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Slingerland, M.; Guchelaar, H. J.; Gelderblom, H. Histone Deacetylase Inhibitors: An Overview of the Clinical Studies in Solid Tumors. Anticancer Drugs 2014, 25, 140–149. [Google Scholar] [CrossRef]
- Laubach, J. P.; Moreau, P.; San-Miguel, J. F.; Richardson, P. G. Panobinostat for the Treatment of Multiple Myeloma. Clinical Cancer Research 2015, 21, 4767–4773. [Google Scholar] [CrossRef]
- Archer, S. L.; Marsboom, G.; Kim, G. H.; Zhang, H. J.; Toth, P. T.; Svensson, E. C.; Dyck, J. R. B.; Gomberg-Maitland, M.; Thébaud, B.; Husain, A. N.; Cipriani, N.; Rehman, J. Epigenetic Attenuation of Mitochondrial Superoxide Dismutase 2 (SOD2) in Pulmonary Arterial Hypertension: A Basis for Excessive Cell Proliferation and a New Therapeutic Target. Circulation 2010, 121, 2661. [Google Scholar] [CrossRef]
- Liu, D.; Yan, Y.; Chen, J. W.; Yuan, P.; Wang, X. J.; Jiang, R.; Wang, L.; Zhao, Q. H.; Wu, W. H.; Simonneau, G.; Qu, J. M.; Jing, Z. C. Hypermethylation of BMPR2 Promoter Occurs in Patients with Heritable Pulmonary Arterial Hypertension and Inhibits BMPR2 Expression. Am J Respir Crit Care Med 2017, 196, 925–928. [Google Scholar] [CrossRef]
- Derissen, E. J. B.; Beijnen, J. H.; Schellens, J. H. M. Concise Drug Review: Azacitidine and Decitabine. Oncologist 2013, 18, 619. [Google Scholar] [CrossRef]
- Juergens, R. A.; Wrangle, J.; Vendetti, F. P.; Murphy, S. C.; Zhao, M.; Coleman, B.; Sebree, R.; Rodgers, K.; Hooker, C. M.; Franco, N.; Lee, B.; Tsai, S.; Delgado, I. E.; Rudek, M. A.; Belinsky, S. A.; Herman, J. G.; Baylin, S. B.; Brock, M. V.; Rudin, C. M. Combination Epigenetic Therapy Has Efficacy in Patients with Refractory Advanced Non Small Cell Lung Cancer. Cancer Discov 2011, 1, 598. [Google Scholar] [CrossRef] [PubMed]
- Gore, S. D. New Ways to Use DNA Methyltransferase Inhibitors for the Treatment of Myelodysplastic Syndrome. Hematology / the Education Program of the American Society of Hematology. American Society of Hematology. Education Program 2011, 2011, 550. [Google Scholar] [CrossRef] [PubMed]
- Ray, K. K.; Wright, R. S.; Kallend, D.; Koenig, W.; Leiter, L. A.; Raal, F. J.; Bisch, J. A.; Richardson, T.; Jaros, M.; Wijngaard, P. L. J.; Kastelein, J. J. P. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N Engl J Med 2020, 382, 1507–1519. [Google Scholar] [CrossRef] [PubMed]
- Scott, L. J.; Keam, S. J. Lumasiran: First Approval. Drugs 2021, 81, 277–282. [Google Scholar] [CrossRef]
- Scott, L. J. Givosiran: First Approval. Drugs 2020, 80, 335–339. [Google Scholar] [CrossRef]
- Kristen, A. V.; Ajroud-Driss, S.; Conceição, I.; Gorevic, P.; Kyriakides, T.; Obici, L. Patisiran, an RNAi Therapeutic for the Treatment of Hereditary Transthyretin-Mediated Amyloidosis. Neurodegener Dis Manag 2019, 9, 5–23. [Google Scholar] [CrossRef]
Figure 1.
The progression of pulmonary hypertension via epigenetic reprogramming.
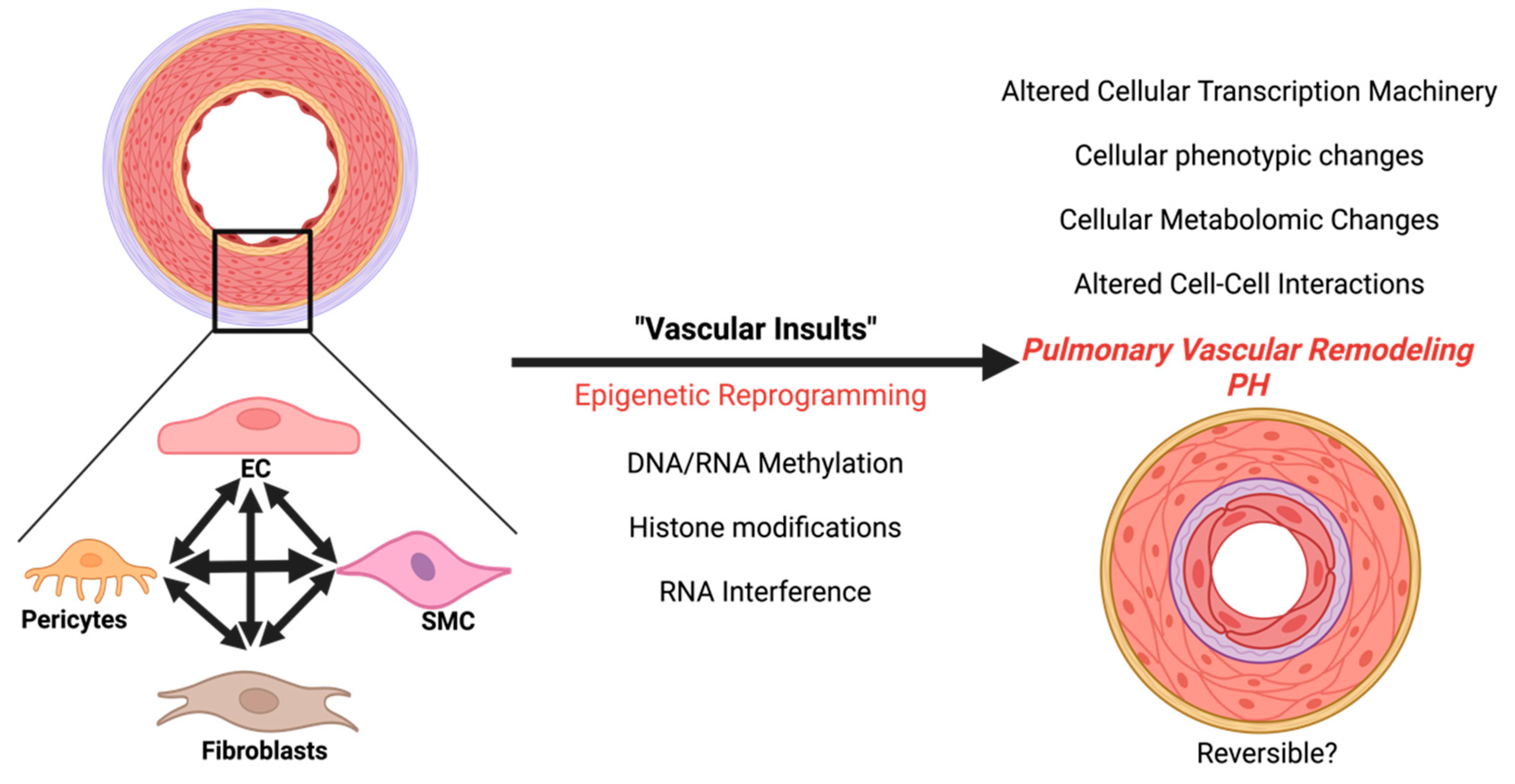
Figure 2.
Role of epigenetic reprogramming in contemporary pulmonary hypertension in lung cancer.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



