
Submitted:
03 January 2024
Posted:
04 January 2024
You are already at the latest version
Abstract
Progress in understanding the pathogenesis and treatment of Alzheimer's disease (AD) is based on the recognition of the primary causes of the disease, which can be deduced from the knowledge of risk factors and biomarkers measurable in the early stages of the disease. Insights into risk factors and the time course of biomarker abnormalities point to a role for mitochondrial dysfunction and oxidative stress in the onset and development of AD. Coenzyme Q10 (CoQ10) is a lipid antioxidant and electron transporter in the mitochondrial electron transport system. The availability and activity of CoQ10 is crucial for proper mitochondrial function and cellular bioenergetics. Based on the mitochondrial hypothesis of AD and the hypothesis of oxidative stress, the regulation of the efficiency of the oxidative phosphorylation system by means of CoQ10 can be considered promising in restoring the mitochondrial function impaired in AD, or in preventing the onset of mitochondrial dysfunction and the development of amyloid and tau pathology in AD.
Keywords:
Alzheimer’s disease
; Coenzyme Q10
; Mitochondrial dysfunction
; Oxidative stress
; Drug
1. Introduction
Neurodegeneration and aging of the brain are influenced by genetic and epigenetic factors, external and internal environment, lifestyle, trauma and diseases. Impaired synaptic and structural neuroplasticity in aging is associated with pathophysiological, functional, and morphological changes in the brain that may serve as biomarkers of brain aging and neurodegeneration. Pathophysiological biomarkers of brain aging include factors that are linked to the mitochondrial dysfunction [1]. Due to the key role of mitochondria in bioenergetics, oxidative stress, metabolism, neuroinflammation, neuroplasticity, and apoptosis [2,3,4], attention has long been paid to mitochondrial dysfunction in aging and age-related neurodegenerative diseases such as Alzheimer's disease (AD) [5,6,7].
In general, mitochondrial dysfunction is associated with reduced ATP production, increased production of reactive oxygen species (ROS), release of proapoptotic factors, and disturbed calcium homeostasis. The reduced production of ATP is mainly caused by the impaired function of the oxidative phosphorylation system (OXPHOS), which includes a series of redox reactions ending in oxygen, during which the mitochondrial electron transport system (ETS) transfers electrons between the complexes of the respiratory chain with the formation of the proton motive force, but also of superoxide [8]. The essential carrier of electrons from complex I or from complex II to complex III is coenzyme Q (CoQ), especially CoQ10 in humans. The availability and activity of CoQ10 and the Q-cycle play a significant role in the effectiveness of the OXPHOS system. In addition, CoQ10 functions as a lipid antioxidant and a necessary factor for controlling protein uncoupling and opening mitochondrial permeability transition pores (mPTPs).
CoQ10 is endogenously synthesized in every cell; it is also partially absorbed through food, which can affect its availability and activity. Supplementation of CoQ10 and its analogs is tested in the treatment of diseases related to oxidative stress and disruption of cellular energy, including neurodegenerative diseases [9]. This review summarizes findings on the role of oxidative stress and mitochondrial dysfunction in the pathophysiology of AD, with a focus on the role of CoQ10.
2. Alzheimer’s disease
AD is a degenerative disease characterized by amyloid beta (Aβ) and tau protein pathology, where increased formation of neurotoxic Aβ oligomers and plaques and tau hyperphosphorylation (leading to microtubule disruption and the formation of neurotoxic tau oligomers and neurofibrillary tangles (NFTs)) are associated with progressive neuronal loss, a decline in cognitive functions and the development of dementia [10]. Mitochondrial dysfunction, neuroinflammation, oxidative stress, disruption of neurotransmission, metabolic disorders, accumulation of transition metals, vascular damage, and chronic hypoperfusion of brain tissue are also involved in the pathogenesis of AD, which is captured in various interconnected biological hypotheses of AD (Figure 1) [11].
Aβ pathology and tauopathy are mainly involved in the etiology of AD [12,13]. The direct cause of brain cell damage and neuroplasticity in AD is primarily the effects of free radicals and apoptotic processes. The targets of new potential AD drugs are mainly processes related to Aβ and P-tau neurotoxicity, mitochondrial dysfunction, oxidative stress, metabolic disorders, and neuroinflammation.
The main pathophysiological feature of AD is impaired proteostasis of pathways involved in the synthesis, folding, post-translational modifications, aggregation, targeting, and degradation of Aβ and tau protein in the brain. Mitochondria, mitochondria-associated membranes (MAM), and endoplasmic reticulum (ER), which are connected to the ubiquitin proteasome system, autophagy system, ROS production, regulation of free cytosolic calcium, and apoptosis, are significantly involved in these processes.
2.1. Risk factors
The main risk factor for the sporadic form of AD is age. Aging is a complex event, for the explanation of which suitable biomarkers are sought at the molecular, cellular and physiological levels, which cause or accompany aging [3]. In general, age-related neurodegeneration is influenced by environmental factors (age, diet, exercise, lifestyle, and cognitive reserve), metabolic and oxidative stress, mitochondrial dysfunction, genetics and epigenetics, cerebrovascular dysfunction, blood-brain barrier dysfunction, neurotoxicity, and neuroinflammation [14]. The essence of the aging hypothesis, as well as the hypothesis of neurodegenerative diseases, is a gradual increase in cellular dysfunctions caused by the accumulation of protein, lipid, and nucleic acid dysfunctions.
The biology of aging and neurodegeneration is associated with metabolic and oxidative stress, inflammation, DNA mutations and related processes [15]. Biomarkers of aging include genomic instability, telomere attrition, epigenetic changes, loss of proteostasis, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, altered intercellular communication, chronic inflammation, and dysbiosis [16].
The exact cause of AD onset remains controversial. Progress is expected from longitudinal studies allowing the identification of risk factors and early biomarkers of AD detectable in peripheral blood long before the onset of clinical symptoms of the disease [17]. Occurrence of the APOE ɛ4 allele is a major genetic risk factor for late-onset sporadic AD [18,19], as ApoE4 increases the neurotoxicity of Aβ and tau, which have a role in AD pathology [20]. Epigenetic changes, including changes in mtDNA, have been shown to be important in the pathogenesis of AD [21]. The autosomal dominant (familial) form of AD, which is defined as pathologically confirmed dominantly inherited AD, occurs in less than 1% of all cases [22]. However, all forms of AD are thought to share similar pathophysiological processes.
The main risk factor for the most common sporadic form of AD is age. Therefore, progress in understanding the pathophysiology of AD is largely linked to progress in understanding the mechanisms of aging-related neurodegeneration. Other risk factors for AD are female gender [23,24], other genetic and epigenetic variations [18,25], brain injury [26] and internal and external environmental factors and stressors [27,28], including low levels of education [29], lifestyle [30], infection [31], cardiovascular disease [32] and metabolic dysregulation [33] such as type 2 diabetes mellitus (T2DM) [34]. The most significant environmental risk factors for the development of AD are late-life depression and T2DM [28].
2.2. Biomarkers
Validated biochemical biomarkers of AD are low concentrations of Aβ42 in CSF, which reflect Aβ deposition in the brain, and increased tau in CSF, which is a marker of neuronal degeneration or damage. Tau biomarkers include both total tau (T-tau) and phosphorylated tau (P-tau) [35]. Currently, Alzheimer's disease biomarkers are mainly sought and studied (i) neuroimaging (focused on structural and functional changes, decreased connectivity, hypometabolism, and pathological aggregates of Aβ and tau [36,37,38,39], (ii) proteomic and metabolomic [40], and (iii) oxidative stress [41], mitochondrial [42,43], and neuroinflammatory [44].
The time course of measurable pathophysiological biomarkers in relation to the clinical course of AD can be used as a basis for the development of new drugs in AD targeting pathophysiological processes in the early stages of the disease. A hypothetical time course of biomarker abnormalities and pathological changes in AD is proposed based on longitudinal measurement of Aβ and P-tau in CSF or in brain by PET, measurement of neurodegeneration by FDG-PET (hypometabolism) and MRI (hippocampal atrophy), and synaptic dysfunction by FDG -PET and fMRI, neuroinflammatory changes, and mitochondrial dysfunction [17,22,37,45,46]. According to this model, the onset of AD (even decades before the recognition of clinical symptoms of the disease) is associated with age-related mitochondrial dysfunction and the neurotoxicity of Aβ oligomers, while neurodegeneration and progression of AD is more associated with the neurotoxicity of P-tau oligomers and NFTs.
2.3. Mitochondrial hypothesis
The mitochondrial cascade hypothesis of AD posits that mitochondrial dysfunction determines the initiation and development of this disease. According to this hypothesis, primary changes in mitochondrial function may induce a cascade of processes that lead to AD-specific neuropathological changes. Aβ and tau pathology/neurotoxicity may also be potentiated by interaction with mitochondrial proteins and membranes. According to the original mitochondrial cascade hypothesis [47], the basic level of mitochondrial function is genetically determined, and the decline of mitochondrial function is determined by aging processes, genetic factors, and environmental influences. If the decline in mitochondrial function exceeds a certain threshold, then the histological and pathophysiological changes specific to AD are triggered.
According to the mitochondrial cascade hypothesis, AD neuropathology arises secondary to mitochondrial dysfunction when the age-associated decline in mitochondrial function reaches a point where compensatory mechanisms are no longer effective [17,48,49]. The primary cause of the disease may not only be mitochondrial dysfunction, amyloidopathy or tauopathy, but also changes in the activity of factors that can cause them and which are localized in mitochondria, such as ApoE4 [50], glycogen synthase kinase 3 [51,52], and monoamine oxidase [53]. It appears that AD may have multiple initiating pathological factors that interact with each other.
Amyloid and tau pathology are considered to be specific for AD, but due to the mutual interactions and feedback effects of the aforementioned processes, it is not yet clear what triggers AD. According to the integrative amyloid-tau-mitochondrial hypothesis [17], the interaction of risk factors and biomarkers and their mutual synergy rather than the primary effects of one particular factor are decisive for the development of AD.
Mitochondrial-associated endoplasmic reticulum (ER) membranes (MAMs) play a key role in maintaining calcium homeostasis, phospholipid and cholesterol metabolism, import of lipids from the ER into mitochondria, initiation of autophagy, mitochondrial fission, and apoptosis. Thus, MAMs have a role in the development of neurodegenerative diseases such as AD [54,55,56,57]. According to the MAM hypothesis, AD is mainly a communication disorder between ER and mitochondria [58].
The mitochondrial hypothesis of AD is supported by the observation that aging (as the most important risk factor for AD) affects the function of mitochondria in brain cells, and thus the generation of ATP, calcium homeostasis, and the regulation of gene expression [1,6]. Changes with age occur in the gene expression of mitochondrial proteins, the morphology of mitochondria and their fission and fusion [59], oxidative damage of mtDNA [60], the function of the OXPHOS system [61], depolarization of the inner membrane and opening of mPTPs [62]; cellular NAD+ levels and the NAD:NADH ratio also decrease [63].
Mitochondrial dysfunction in AD includes bioenergetic impairment, increased ROS production, impaired mitochondrial dynamics and trafficking, and DNA mutations [64,65]. Mitochondrial abnormalities, including impaired function of mitochondrial ETS complexes and ATP production, have been described in AD [66]. One possibility to regulate the processes associated with neurodegeneration in AD is the regulation of the OXPHOS system through the availability and activity of CoQ10 using metabolic modulators, drugs, diet, and exercise [67,68].
Disruption of synaptic plasticity is one of the first steps in the neurodegeneration process associated with aging and the development of neurodegenerative diseases such as AD. Early deficits in synaptic mitochondria in AD include increased Aβ accumulation, mitochondrial dysfunction, increased mPTP, decreased mitochondrial respiration, and decreased complex IV activity [69]. At the same time, Aβ and tau pathologies are in a reciprocal relationship with mitochondrial dysfunction and oxidative stress in AD [17,70,71,72]. In a mouse model of AD, disruption of mitochondrial bioenergetics has been shown to precede the development of AD pathology [73]. Aβ accumulates in mitochondria and reduces the enzymatic activity of complex II and IV, reduces mitochondrial respiration, and impairs mitochondrial dynamics [74,75,76]. Damage to mitochondrial bioenergetics in AD was demonstrated both by measurements in the brains of AD transgenic mice and by PET neuroimaging in human AD brains [65]. Aβ and tau appear to act synergistically to damage the OXPHOS system, with tau damaging complex I and Aβ damaging complex IV [77].
2.4. Oxidative stress hypothesis
According to the free radical theory of aging [78,79,80], aging and age-related diseases are associated with overproduction of ROS, primarily from mitochondria, and subsequent damage to cellular proteins, lipids, and nucleic acids. Although oxidative stress is accepted as a key modulator of the biological processes of aging and neurodegeneration [81], useful endogenous mechanisms regulated by ROS [82] must also be taken into account in therapeutic interventions and attention must also be paid to the role of other manifestations of mitochondrial dysfunction, such as impaired bioenergetics, inflammation, mtDNA mutation, impaired mitophagy and retrograde signaling from mitochondria to the nucleus [83,84,85,86].
According to the oxidative stress hypothesis, the cause of the development of AD is oxidative stress, where damage to brain cells by ROS contributes to neurodegeneration and cognitive decline. At the same time, mitochondrial dysfunction, but also Aβ and P-tau can contribute to the increased production of RONS [87]. ROS can trigger an inflammatory response and, conversely, inflammation induces oxidative stress [88]. Damage to synapses and brain cells due to oxidative stress may be both a consequence and a cause of Aβ and tau neurotoxicity in AD [89]. Mitochondrial dysfunction is also involved in the development of oxidative stress, so administration of CoQ10 as an antioxidant and/or regulators of CoQ10 activity in oxidative phosphorylation could be effective in AD therapy.
The oxidative stress hypothesis is based on the observation that oxidative stress, i.e. an imbalance between the production and elimination of ROS, occurs in neurodegeneration associated with AD and with aging. Increased ROS production and/or reduced antioxidant defenses lead to the accumulation of dysfunctional proteins, lipids and nucleic acids (including mtDNA) and impaired mitophagy. In neurons, superoxide is mainly produced in the mitochondrial matrix during electron transfer in ETS (mainly generated by the mitochondrial complexes I and III). Superoxide is converted directly in the matrix by superoxide dismutase to less reactive hydrogen peroxide, which, however, passes through membranes and can be converted to a very reactive hydroxyl radical in the cytosol [8]. Mitochondrial dysfunction can lead to increased oxidative and nitrosative stress, as impaired electron transfer in the mitochondrial ETS leads to increased production of ROS and the formation of reactive hydroxyl radicals, and impaired transport of calcium ions into the mitochondrial matrix induces increased production of nitric oxide (NO) and the formation of reactive peroxynitrite. These radicals can initiate increased peroxidation of membrane lipids in AD [90]. Lipophilic antioxidants such as CoQ10 have a protective effect on the peroxidation of membrane lipids.
Damage to synapses and brain cells due to oxidative stress may be both a consequence and a cause of Aβ and tau neurotoxicity in AD [89]. Mitochondrial dysfunction is also involved in the development of oxidative stress, so administration of CoQ10 and/or regulation of CoQ10 activity in the ETS is being tested in AD therapy. Administration of CoQ10 as an antioxidant had no significant effect on biomarkers associated with amyloid and tau pathology in AD measured in CSF (Aβ42, tau, and P-tau), nor on cognitive function [91]. Also, administration of CoQ10 did not have a significant therapeutic effect in other neurodegenerative diseases such as Parkinson's disease and Huntington's disease [92].
3. Coenzyme Q10
Coenzyme Q10 (CoQ10, ubiquinone, 2,3 dimethoxy-5-methyl-6-decaprenyl-1,4-benzoquinone) is the most common form of coenzyme Q in humans [93]; it is found in all cell membranes, where it acts as an electron carrier and antioxidant. CoQ10 occurs in three redox forms, as oxidized form (ubiquinone, C59H90O4), reduced form (ubiquinol, C59H92O4), and semi-oxidized form (semiquinone, C59H91O4; under physiological conditions it occurs as an ion C59H90O4–) (Figure 2).
The main cellular functions of CoQ10 include (i) electron transport in the mitochondrial electron transfer system (ETS) with a key role in the formation of ATP, (ii) antioxidant action including protection against lipid peroxidation, participation in the reduction/recycling of other antioxidant molecules (α-tocopherol and ascorbate), and stabilization of the plasma membrane and cell redox balance (iii) apoptosis and modulation of mPTPs, (iv) signaling modulation of gene expression, including anti-inflammatory effects, (v) maintenance of proton gradient on the lysosomal membrane, and (vi) activation of mitochondrial uncoupling proteins [94,95,96,97].
Coenzyme Q10 (ubiquinone, CoQ10) is present in mitochondria as part of the electron transport chain. CoQ10 is a lipophilic molecule located in cell membranes, mainly in the inner mitochondrial membrane (IMM); is crucial for electron transfer between complex I or II and complex III of the respiratory chain [98] and also from other dehydrogenases [9,99,100] localized on the outer or inner surface of the IMM. Furthermore, CoQ10 is an important factor in the activation of protein uncoupling, controls the mPTP, participates in the transport of electrons in plasma membranes and lysosomes, affects the structure and fluidity of membranes and acts as an endogenous lipid antioxidant [9,101]. Moreover, CoQ10 and its redox state could indirectly modulate a number of mitochondrial and non-mitochondrial metabolic pathways, such as sulfide metabolism, one-carbon metabolism, glutathione, and ferroptosis; it can therefore be included in the pathophysiology of some metabolic diseases [100].
In addition to the main role in electron transport in the mitochondrial ETS, CoQ10 has other mitochondrial functions, mainly as a cofactor in the activation of uncoupling [102] and in the control of the mPTP [103]. CoQ10 is also involved in the function of redox systems in various membranes, the structure and fluidity of membranes [104], gene expression, cell growth, differentiation, and apoptosis [105]. The reduced form of CoQ10(H2) (ubiquinol) acts as an antioxidant and scavenger of free radicals, thereby preventing oxidative damage to lipids, proteins and nucleic acids. On the other hand, CoQ10 can also have a pro-oxidant role, as it can participate in the formation of superoxide and hydrogen peroxide [106].
The internal synthesis of CoQ10 starts from the amino acid tyrosine (benzoquinone ring) and from the mevalonate pathway (isoprenoid side chain) (Figure 2) [107,108]. Current knowledge about CoQ10 biosynthesis is described in detail in a series of reviews [109,110,111]. A collection of enzymes that produces CoQ10 (termed as complex Q) is localized in the inner mitochondrial membrane and in the endoplasmic reticulum [94,95].
3.1. CoQ10 in OXPHOS system
High-energy electrons enter the mitochondrial ETS via complex I (from the reduced coenzyme nicotinamide adenine dinucleotide, NADH) or via complex II (from the reduced coenzyme flavin adenine dinucleotide, FADH2). The product of redox reactions in the complex I is the transfer of 4 protons per 1 NADH molecule into the intermembrane space. Electrons are transferred from complex I or complex II to complex III (coenzyme Q : cytochrome c – oxidoreductase, sometimes called the cytochrome bc1 complex) by ubiquinol (reduced CoQ10(H2)) and enter protonmotive Q-cycle of the complex III. In the Q-cycle mechanism, protons are translocated across the inner mitochondrial membrane (IMM) as a result of reoxidation of ubiquinol at Qo-site at the outer side of the IMM and reduction of ubiquinone (CoQ10) at Qi-site at the matrix side of the IMM (Figure 3). Respiration subunits in the Q-cycle are cytochrome c1, cytochrome b (with low (bL) and a higher (bH) potential hemes), and Rieske protein. The substrates of redox reactions catalyzed by complex III are ubiquinol and two molecules of ferricytochrome c, the products are ubiquinone, two molecules of ferrocytochrome c, and four protons (released into the intermembrane space and used by ATP synthase during ATP biosynthesis) [97,112,113,114]. Total mitochondrial ETS activity is most easily measured as the kinetics of mitochondrial oxygen consumption [115,116]. Note, that the Qo-site of the complex III is an important site for generation of superoxide and thus has a role in oxidative damage during aging and neurodegeneration.
The efficiency of the OXPHOS system, especially the transfer of electrons by means of CoQ10 and the Q-cycle, is influenced by the existence and organization of respiratory supercomplexes [117,118]. The most common supercomplexes (respirasomes) observed are complex I/III, complex I/III/IV, and complex III/IV. Supercomplex assembly is dynamic and their formation and stabilization depends on the lipid composition of the IMM, especially on the presence of cardiolipin [119]; the initiation of their formation can be associated with the membrane potential. The formation of supercomplexes can significantly increase the efficiency of the OXPHOS system by optimizing the utilization of ETS substrates, stabilizing complex I, and reducing the formation of ROS [120,121]. A constitutive part of respirasomes is CoQ10, which exists in different CoQ10 pools in the IMM [122].
Dependence between respiratory complexes, supercomplex assembly dynamics, and the existence of CoQ10 pools in the effectiveness of the ETS system is intensively studied [123]. According to the classic fluid model, respiratory complexes I-IV are randomly distributed in the IMM and electron transfer between them is realized by electron carriers, CoQ10 located inside the IMM and cyt c located on the external surface of the IMM (Figure 3). The random collision model [124] assumed that mitochondrial electron transport is a process of random collisions based on the diffusion of individual components in the fluid IMM. The discovery of the existence of supercomplexes in the respiratory chain [125] and the recognition of their function [126] led to the modification of the fluid model to the plasticity model, which adds to the fluid model a new view of the structural and functional complexity in the transfer of electrons in the mitochondrial ETS as well as the function of the CoQ10 pool in the IMM [122,127,128].
The plasticity model assumes that respiratory complexes can function either individually or as components of supercomplexes. It has long been assumed that free CoQ10 resides in the IMM in a homogeneous pool [129]. With the discovery of respirasomes and their function, it is shown that there is a segmentation of CoQ10 molecules, into a pool attached by supercomplex I/III (CoQNADH pool) and into a pool available for complex II and other enzymes (CoQFADH pool) [120], while the two pools interact with each other. The majority of CoQ that receives electrons from FADH2 and the majority of cyt c apparently remain unbound to supercomplexes.
3.2. CoQ10 in AD pathophysiology
CoQ10 is mainly formed endogenously, so the cause of a deficiency in CoQ10 can mainly be its impaired biosynthesis, increased degradation, or increased usage. In mitochondrial diseases, CoQ10 deficiency can be caused by mutations in genes responsible for CoQ10 biosynthesis, or secondarily by defects in other genes [130]. Endogenous metabolites, which are also involved in cholesterol production, may be involved in the regulation of CoQ10 biosynthesis [95]. By upregulating the synthesis of CoQ10, not only its concentration but also its appropriate mitochondrial localization can be achieved. However, dietary intake can also contribute to CoQ10 availability, especially when its endogenous production is reduced with aging or with genetic mutations primarily or secondarily involved in CoQ10 biosynthesis.
Protective effects of CoQ10 against Aβ a neurotoxicity [131] and the observation that serum CoQ10 levels may be inversely associated with dementia risk [132] suggests that determination of serum CoQ10 levels could be useful for predicting the development of AD. Other studies, however, did not find significant differences in plasma CoQ10 concentrations in AD and controls. Although the results of some studies show that serum CoQ10 could be a predictor of dementia [132], CoQ10 deficiency has not been sufficiently demonstrated in peripheral blood [133]. But an association between clinical assessment of cognitive decline and plasma CoQ10 concentration was found, which may mean that even a small variation in CoQ10 availability or activity can trigger mitochondrial dysfunction.
In permeabilized platelets from AD patients, reduced capacity of the electron transport system, altered activity of respiratory chain complexes (increased complex I activity and decreased complex IV activity) [43,134], and reduced plasma CoQ10 concentration were found, with complex IV activity being negatively correlated and respiratory capacity being positively correlated with cognitive MMSE early [134,135,136]. The exact role of CoQ10 in the pathogenesis of AD and the treatment of neurodegenerative diseases is yet to be clarified [96].
Oxidative stress is also thought to contribute to AD progression by inducing Aβ overexpression and accumulation, while it is unclear what is the primary cause of disease development. Oxidative stress modulates proteostasis, which is strongly impaired in AD [137]. Therefore, antioxidants are among the new clinically tested potential drugs in AD [138]. The reduced form of CoQ10(H2) is a known antioxidant that also affects Aβ pathology [139]. Protective effects of CoQ10 and other antioxidants against Aβ accumulation in the brain and against neuroinflammation and hypoxia have been reported [140].
A study with a mouse model of AD showed that there is a relationship between changes in the hippocampus and cerebral cortex and oxidative stress, proteostasis, and bioenergetics and that CoQ10 may act preventively [141]. Evaluation of the therapeutic response to CoQ10 administration to AD patients showed efficacy in mitochondrial activation [142], but the change in CoQ10 levels in AD patients and its effects on improving neuropsychological assessments are not clearly confirmed [133]. Thus, the role of CoQ10 in the pathogenesis of AD is not fully understood.
A role for CoQ10 in AD pathophysiology is supported by the observation that CoQ induces tau aggregation and that CoQ is present in paired helical filaments (PHF) [143]. Cell culture studies have shown a protective effect of CoQ10 on Aβ neurotoxicity [131,139,144]. In a mouse model of AD, it was shown that early intervention with ubiquinol can act preventively against the deregulation of proteostasis (disorders caused by an imbalance in the protein homeostasis network - synthesis, folding and transport of proteins; by post-translational modification and degradation or clearance of misfolded proteins) in AD.
4. AD treatment
4.1. Targets of novel AD drugs
Drugs currently approved or recommended for the treatment of AD belong to the category of agents targeting neurotransmitter receptors (cholinesterase inhibitors and NMDA receptor antagonists) and Aβ (monoclonal antibodies directed against Aβ plaques, protofibrils, and oligomers). In clinical use, there are drugs aimed at suppressing or alleviating the symptoms of AD (donepezil, rivastigmine, galantamine, memantine and the memantine/donepezil combination). Two disease-modifying drugs (DMDs) targeting Aβ pathology (aducanumab and lecanemab) have recently received accelerated approval from the United States Food and Drug Administration (FDA). Attention is mainly devoted to the development of new effective and specific DMDs. However, there is also ongoing testing of symptomatic substances aimed at improving cognitive functions and neuropsychiatric symptoms in AD, and these are often drugs already approved for the treatment of other diseases or substances used in alternative and complementary medicine [145].
Oxidative stress and categories of biological processes and AD drug targets that are closely related to mitochondrial function (metabolism and bioenergetics, synaptic plasticity/neuroprotection, and cell death) are included in the CADRO classification system (“Common Alzheimer's and Related Dementias Research Ontology”; https://iadrp.nia.nih.gov/about/cadro). According to the periodic annual review of drug development for AD [138] based on an analysis of data from ClinicalTrials.gov, a total of 141 agents were tested for the treatment of AD and mild cognitive impairment (MCI) in phase 1, 2, or 3 clinical trials at the beginning of 2023 with AD. Their most common targets are inflammation (17.0%), Aβ (15.6%), synaptic plasticity/neuroprotection (12.9%), tau (9.2%), metabolism and bioenergetics (7.1%), and oxidative stress (5.0%).
In addition to new DMD drugs targeting the primary causes of AD onset and progression, appropriate combinations of approved drugs with adjuvant agents are also being sought and tested. These supplements, such as CoQ10, ω-3 fatty acids, soy, ginkgo biloba, B vitamins, vitamin D plus calcium, vitamin C, or β-carotene have not yet been shown to prevent cognitive dysfunction in AD [133,146]. It can be expected that if adjuvants have anti-amyloid, anti-tau, neurochemical, mitochondrial, antioxidant or anti-inflammatory effects [131,147], they may also have therapeutic potential to moderate the progression of cognitive impairment in AD. Drugs in phase 2 or 3 clinical trials targeted to oxidative stress include hydralazine, icosapent ethyl, PUFA, omega-3, edavarone, and Flos gossypii flavonoids. Metabolism and bioenergetics are targeted by metformin, insulin intranasal (+ empagliflozin), dapagliflozin, empagliflozin, semaglutide, T3D-959, Chinese traditional medicine, and obicetrapib [138].
Antioxidants are only taken as adjuncts in the administration of approved AD drugs. The lack of effectiveness of antioxidants in AD therapy can be explained by their non-specific intervention in the balance between ROS activity and the activity of the antioxidant system, which can suppress the useful and necessary role of free radicals in certain areas of the brain. Nevertheless, the testing of CoQ10 and its analogues as a supportive therapy for AD continues to be important, mainly associated with the suppression of excessive peroxidation of membrane lipids.
A suitable target for substances that restore or increase mitochondrial function is the stimulation of mitochondrial biogenesis. Mitochondrial biogenesis is associated not only with cell division, but also with the response to oxidative stress (that is, with the demand for increased cellular energy consumption), exercise, hormones, electrical stimulation, etc. Mitochondrial biogenesis has become the target of new drugs for diseases associated with mitochondrial dysfunction, including neurodegenerative disorders such as AD. Pathways associated with mitochondrial biogenesis and activated in response to energy deficit include activation of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) axis, activation of AMP-activated protein kinase (AMPK), and Sirtuin 1 activation [65].
In summary, the main approach in the search for new AD disease-modifying drugs (DMD) is targeting the pathophysiological processes causing the onset of the disease, which, according to current AD hypotheses, are mainly Aβ and tau pathology, mitochondrial dysfunction, oxidative stress, neuroinflammation, disturbed neurotransmission, and disorders of brain metabolism. If we assume that age is the main risk factor for AD, then we can focus on the regulation of processes and biomarkers discussed in the oxidative stress hypothesis and in the mitochondrial hypothesis.
4.2. CoQ10 in AD treatment
Substances that act as antioxidants or increase mitochondrial bioenergetics have potential for the treatment of neurodegenerative diseases such as AD. From the point of view of the mitochondrial hypothesis of AD and the possibility of pharmacologically influencing mitochondrial dysfunction, synaptic plasticity and metabolism of brain cells, CoQ10 and mitochondrial proteins and lipids interacting with it also appear to be possible targets of new AD drugs. These are substances capable of regulating the availability and activity of CoQ10 and thus the function of the OXPHOS system.
CoQ10 and other antioxidants or bioenergetics stimulators may be potentially effective in the treatment of neurodegenerative diseases [148,149]. CoQ10 is well characterized as a neuroprotective antioxidant in animal models and in human studies of neurodegenerative disorders. CoQ10 has been shown to reduce oxidative stress and amyloid pathology in a mouse model of AD [150]. Age-related decline in mitochondrial function has been shown in a mouse model to be accompanied by decreased levels of mitochondrial CoQ, and exogenous administration of water-soluble CoQ can lead to restoration of mitochondrial function [151]. A number of studies have confirmed significant neuroprotective effects of CoQ10 in experimental biological models, but the suitability of CoQ10 as a biomarker or drug has not been confirmed in AD patients [133].
Given the role of CoQ10 in bioenergetics and antioxidant activity and the observation that CoQ10 has protective effects against Aβ-induced cell toxicity and impaired synaptic plasticity [144,152], CoQ10 and the processes regulated by it have strong potential as new AD drug targets [153]. Although studies in animal models of AD show significant improvements in cognition, clinical trials have not been very successful. Therefore, upregulation of brain CoQ10 biosynthesis appears to be more suitable for treatment of neurodegenerative disorders. An increase in CoQ10 biosynthesis is possible in a physiological way (cold adaptation and exercise) [107] or by targeting of isoprenoid regulation within mevalonate pathway [154], e.g. by administration of substances, such as epoxidized all-trans polyisoprenoids [95].
Reduced availability of CoQ10 can be eliminated by dietary administration of this substance. The process of CoQ10 absorption and bioavailability is complex and strongly depends on the formulation of the preparation [155]. CoQ10 and its analogues, idebenone and MitoQ, are used in the treatment of mitochondrial disorders and in the supportive treatment of neurodegenerative diseases associated with mitochondrial dysfunction, such as AD [108]. CoQ10 is well tolerated and safe, but not approved for treatment of AD [156]. The therapeutic application of CoQ10 is limited by its insolubility in water. Various preparations of CoQ10 have been developed to improve solubility and bioavailability.
The synthetic analogue of CoQ10 idebenone (hydroxydyecylubiquinone) acts as an antioxidant and an electron carrier in ETS. Idebenone has protective effects against many toxins [157], but inhibits complex I [158]. In some studies it showed therapeutic effects on AD progression [159], but in other studies no effect of idenbenone on cognitive decline in AD [160] or on biomarkers related to Aβ and tau pathology in AD was demonstrated [91].
The administration of mitoquinone (MitoQ), which passes through membranes more easily and concentrates in the mitochondria thanks to the attached triphenylphosphonium to ubiquinone, seems promising. MitoQ has protective effects against mitochondrial damage [161] and appears promising in suppressing AD symptoms. In experiments with cell cultures and a mouse model of AD, MitoQ was found to increase synaptic connectivity and neurite outgrowth, prevented Aβ induced oxidative stress, and improved memory retention [162,163,164].
In summary, To evaluate the effect of CoQ10 on the activity of the OXPHOS system, or on mitochondrial dysfunction associated with impaired electron transport in the ETS in AD, data from in vivo measurements are not yet available. However, in vitro measurements using isolated mitochondria suggest that exogenously supplied CoQ10 can increase mitochondrial respiratory rate. It can be hypothesized that the antioxidant activity of CoQ10, which protects mitochondrial membranes from oxidative damage, and the increase in electron transfer efficiency due to the incorporation of CoQ10 into the inner mitochondrial membrane contribute to this increase in ETS efficiency. From this point of view, it appears as a perspective synthesis and testing of (i) analogues of CoQ10 with good bioavailability in brain mitochondria, (ii) substances aimed at regulating CoQ10 biosynthesis, and (iii) substances aimed at increasing the activity of CoQ10 in the Q cycle, including those that affect the formation of supercomplexes containing complex III.
5. Discussion and Conclusions
Based on the findings on which the mitochondrial hypothesis of AD and the oxidative stress hypothesis are based, mitochondrial ETS, especially CoQ10 and the processes mediated by it, appear to be a promising target for new AD drugs. The uptake of dietary CoQ10 into tissues is limited, as CoQ10 is localized in the membranes of the central hydrophobic part of the lipid bilayer; thus, the space available for CoQ10 and similar lipophilic compounds is limited. CoQ10 diffusion in lipid bilayer may represent the rate-limiting step of electron transfer [165].
CoQ10 concentration decreases with aging, with the availability and redox status of CoQ10 playing a role in the oxidative stress associated with aging [166,167,168,169]. Increased availability of CoQ10 may have neuroprotective effects through antioxidant and bioenergetic effects. However, a systematic review did not confirm a decrease in plasma CoQ10 in AD patients [133]. Also, the decrease in mitochondrial respiratory rate in platelets with age was not different in AD patients compared to age-matched healthy controls [61]. These results indicate that mitochondrial dysfunction (potentially associated with reduced availability and activity of CoQ10) in AD is associated with the onset of the disease rather than its progression. CoQ10 supplementation may then slow age-related neurodegeneration, but does not act as a causal cure for AD.
When we tested in vitro the effect of CoQ10 and other antioxidants on mitochondrial respiration using a model of isolated brain mitochondria, only the addition of CoQ10 caused an increase in respiratory rate [170]. It can be hypothesized that the increase in ETS activity may be achieved by direct incorporation of CoQ10 into the IMM, rather than the antioxidant action of CoQ10. It can be assumed that the improvement of mitochondrial function in AD is possible by increased CoQ10 biosynthesis and increased mitochondrial biogenesis, rather than dietary CoQ10 supplementation, which does not ensure an increase in CoQ10 in IMM brain mitochondria. To increase the availability of CoQ10 in the brain and in brain mitochondria, its exogenous administration and biosynthesis stimulation can be combined, as exogenous supplementation of CoQ10 is safe and does not affect its endogenous biosynthesis [171].
Age-associated mitochondrial dysfunction (measurable as a decrease in ETS capacity or a decrease in respiratory reserve) together with the effects of ApoE4 may be at the start of Aβ and tau pathology, oxidative stress, and neuroinflammation in the late-onset sporadic form of AD. Targeting new AD drugs on the activity and efficiency of ETS, specifically on the availability and activity of CoQ10 in the inner mitochondrial membrane and on the regulation of redox processes in the ETS associated with CoQ10, can therefore be considered a promising research approach. The direct in vitro effects of CoQ10 on increasing mitochondrial respiration [170] suggest that the regulation of CoQ10 biosynthesis could be a promising direction in the development of new AD drugs.
In conclusion, the specific pathophysiology of AD is primarily associated with Aβ and tau pathology. According to the mitochondrial hypothesis and the oxidative stress hypothesis, mitochondrial dysfunction and oxidative stress are involved in the pathogenesis of AD, which can be both initiating and accompanying processes in the pathophysiology of AD and the development of neurodegenerative processes in AD. The approach of targeting new AD drugs to the availability and activity of CoQ10 is underpinned by the role of CoQ10 in the cellular antioxidant system and in mitochondrial bioenergetics. Increasing the availability and activity of CoQ10 is possible by its exogenous administration; using biological models of isolated brain mitochondria, cell cultures, and animal models of AD, both antioxidant and bioenergetic effects of CoQ10 have been demonstrated. However, the effects of antioxidants are shown to be insufficiently effective in AD therapy in humans, and exogenous administration of CoQ10 does not yet allow its reliable increased utilization by brain mitochondria. From this point of view, targeting new AD drugs to increase mitochondrial bioenergetics by regulating mitochondrial biogenesis or CoQ10 biosynthesis in the brain appears to be a more appropriate pharmacological strategy. Regulation of the activity of the OXPHOS system through increasing the efficiency of electron transfer in the ETS using CoQ10 and cyt c appears promising. However, targeted pharmacological intervention in this electron transfer requires a deeper understanding of normal and pathological processes in the OXPHOS system, including those associated with the assembly of respiratory complexes into respirasomes and function of supercomplexes in electron transfer efficiency by CoQ10.
Considering the role of CoQ10 in bioenergetics and lipid peroxidation, it is advisable to continue studying the possibilities of AD therapy by regulating the activity of CoQ10 in the OXPHOS system. In the context of the mitochondrial hypothesis and the oxidative stress hypothesis, stimulation of mitochondrial biogenesis and CoQ10 biosynthesis appears to be a promising target for new AD drugs. In the early stages of AD development, the stimulation of mitochondrial bioenergetics and the antioxidant action of CoQ10 could prevent the development of Aβ and tau neurotoxicity. However, even in the later stages of the disease, the effects of CoQ10 on mitochondrial bioenergetics could slow the progression of the disease.
Author Contributions
Conceptualization, Z.F. and J.H.; writing—original draft preparation, Z.F.; writing—review and editing, Z.F. and J.H.. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Supported by Charles University, Prague, Czech Republic (project Cooperatio, research area Neurosciences) and by the projects of Ministry of Health, Czech Republic (grant number MH CZ-DRO VFN64165 and grant number AZV CR NU23-04-00032).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Mattson, M.P.; Arumugam, T.V. Hallmarks of Brain Aging: Adaptive and Pathological Modification by Metabolic States. Cell Metab 2018, 27, 1176–1199. [Google Scholar] [CrossRef] [PubMed]
- Zia, A.; Pourbagher-Shahri, A.M.; Farkhondeh, T.; Samarghandian, S. Molecular and cellular pathways contributing to brain aging. Behav Brain Funct 2021, 17, 6. [Google Scholar] [CrossRef]
- Tanaka, M.; Vecsei, L. Editorial of Special Issue 'Dissecting Neurological and Neuropsychiatric Diseases: Neurodegeneration and Neuroprotection'. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction, Oxidative Stress, and Neuroinflammation: Intertwined Roads to Neurodegeneration. Antioxidants (Basel) 2020, 9. [Google Scholar] [CrossRef]
- Hroudová, J.; Singh, N.; Fišar, Z. Mitochondrial dysfunctions in neurodegenerative diseases: relevance to Alzheimer's disease. Biomed Res Int 2014, 2014, 175062. [Google Scholar] [CrossRef]
- Grimm, A.; Eckert, A. Brain aging and neurodegeneration: from a mitochondrial point of view. J Neurochem 2017, 143, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Hroudová, J.; Fišar, Z. Control mechanisms in mitochondrial oxidative phosphorylation. Neural Regen Res 2013, 8, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Rauchova, H. Coenzyme Q10 effects in neurological diseases. Physiol Res 2021, 70, S683–S714. [Google Scholar] [CrossRef]
- 2023 Alzheimer's disease facts and figures. Alzheimers Dement 2023 Alzheimer's disease facts and figures, 19, 1598-1695. [CrossRef]
- Liu, P.P.; Xie, Y.; Meng, X.Y.; Kang, J.S. History and progress of hypotheses and clinical trials for Alzheimer's disease. Signal Transduct Target Ther 2019, 4, 29. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer's disease. Alzheimers Dement 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011, 7, 263–269. [Google Scholar] [CrossRef]
- Wareham, L.K.; Liddelow, S.A.; Temple, S.; Benowitz, L.I.; Di Polo, A.; Wellington, C.; Goldberg, J.L.; He, Z.; Duan, X.; Bu, G.; et al. Solving neurodegeneration: common mechanisms and strategies for new treatments. Mol Neurodegener 2022, 17, 23. [Google Scholar] [CrossRef] [PubMed]
- Rysz, J.; Franczyk, B.; Rysz-Gorzynska, M.; Gluba-Brzozka, A. Ageing, Age-Related Cardiovascular Risk and the Beneficial Role of Natural Components Intake. Int J Mol Sci 2021, 23. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef] [PubMed]
- Fišar, Z. Linking the Amyloid, Tau, and Mitochondrial Hypotheses of Alzheimer’s Disease and Identifying Promising Drug Targets. Biomolecules 2022, 12, 1–43. [Google Scholar] [CrossRef] [PubMed]
- Troutwine, B.R.; Hamid, L.; Lysaker, C.R.; Strope, T.A.; Wilkins, H.M. Apolipoprotein E and Alzheimer's disease. Acta Pharm Sin B 2022, 12, 496–510. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef]
- Gao, X.; Chen, Q.; Yao, H.; Tan, J.; Liu, Z.; Zhou, Y.; Zou, Z. Epigenetics in Alzheimer's Disease. Front Aging Neurosci 2022, 14, 911635. [Google Scholar] [CrossRef]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med 2012, 367, 795–804. [Google Scholar] [CrossRef] [PubMed]
- van der Flier, W.M.; Scheltens, P. Epidemiology and risk factors of dementia. J Neurol Neurosurg Psychiatry 2005, 76 Suppl 5, v2-7. [CrossRef]
- Demetrius, L.A.; Eckert, A.; Grimm, A. Sex differences in Alzheimer's disease: metabolic reprogramming and therapeutic intervention. Trends Endocrinol Metab 2021, 32, 963–979. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.J.; Fulton-Howard, B.; Goate, A. Interpretation of risk loci from genome-wide association studies of Alzheimer's disease. Lancet Neurol 2020, 19, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Cejudo, J.; Wisniewski, T.; Marmar, C.; Zetterberg, H.; Blennow, K.; de Leon, M.J.; Fossati, S. Traumatic Brain Injury and Alzheimer's Disease: The Cerebrovascular Link. EBioMedicine 2018, 28, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Gaugler, J.; James, B.; Johnson, T.; Reimer, J.; Solis, M.; Weuve, J.; Buckley, R.F.; Hohman, T.J. 2022 Alzheimer's disease facts and figures. Alzheimers & Dementia 2022, 18, 700–789. [Google Scholar] [CrossRef]
- Bellou, V.; Belbasis, L.; Tzoulaki, I.; Middleton, L.T.; Ioannidis, J.P.A.; Evangelou, E. Systematic evaluation of the associations between environmental risk factors and dementia: An umbrella review of systematic reviews and meta-analyses. Alzheimers Dement 2017, 13, 406–418. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.A.; Hebert, L.E.; Beckett, L.A.; Scherr, P.A.; Albert, M.S.; Chown, M.J.; Pilgrim, D.M.; Taylor, J.O. Education and other measures of socioeconomic status and risk of incident Alzheimer disease in a defined population of older persons. Arch Neurol 1997, 54, 1399–1405. [Google Scholar] [CrossRef] [PubMed]
- Flicker, L. Modifiable lifestyle risk factors for Alzheimer's disease. J Alzheimers Dis 2010, 20, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Douros, A.; Santella, C.; Dell'Aniello, S.; Azoulay, L.; Renoux, C.; Suissa, S.; Brassard, P. Infectious Disease Burden and the Risk of Alzheimer's Disease: A Population-Based Study. J Alzheimers Dis 2021, 81, 329–338. [Google Scholar] [CrossRef]
- Luchsinger, J.A.; Reitz, C.; Honig, L.S.; Tang, M.X.; Shea, S.; Mayeux, R. Aggregation of vascular risk factors and risk of incident Alzheimer disease. Neurology 2005, 65, 545–551. [Google Scholar] [CrossRef]
- Yan, X.; Hu, Y.; Wang, B.; Wang, S.; Zhang, X. Metabolic Dysregulation Contributes to the Progression of Alzheimer's Disease. Front Neurosci 2020, 14, 530219. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.N.; Chorawala, M.R.; Shah, M.B.; Shah, K.C.; Dave, B.P.; Shah, M.P.; Patel, T.M. Emerging Pathophysiological Mechanisms Linking Diabetes Mellitus and Alzheimer's Disease: An Old Wine in a New Bottle. J Alzheimers Dis Rep 2022, 6, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Baldeiras, I.; Santana, I.; Leitao, M.J.; Vieira, D.; Duro, D.; Mroczko, B.; Kornhuber, J.; Lewczuk, P. Erlangen Score as a tool to predict progression from mild cognitive impairment to dementia in Alzheimer's disease. Alzheimers Res Ther 2019, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- Marquez, F.; Yassa, M.A. Neuroimaging Biomarkers for Alzheimer's Disease. Mol Neurodegener 2019, 14, 21. [Google Scholar] [CrossRef] [PubMed]
- Leuzy, A.; Chiotis, K.; Lemoine, L.; Gillberg, P.G.; Almkvist, O.; Rodriguez-Vieitez, E.; Nordberg, A. Tau PET imaging in neurodegenerative tauopathies-still a challenge. Mol Psychiatry 2019, 24, 1112–1134. [Google Scholar] [CrossRef] [PubMed]
- Vlassenko, A.G.; Benzinger, T.L.; Morris, J.C. PET amyloid-beta imaging in preclinical Alzheimer's disease. Biochim Biophys Acta 2012, 1822, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Gordon, B.A.; Blazey, T.M.; Su, Y.; Hari-Raj, A.; Dincer, A.; Flores, S.; Christensen, J.; McDade, E.; Wang, G.; Xiong, C.; et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer's disease: a longitudinal study. Lancet Neurol 2018, 17, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Nistico, R.; Seyfried, N.T.; Levey, A.I.; Modeste, E.; Lemercier, P.; Baldacci, F.; Toschi, N.; Garaci, F.; Perry, G.; et al. Omics sciences for systems biology in Alzheimer's disease: State-of-the-art of the evidence. Ageing Res Rev 2021, 69, 101346. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Reed, T.; Newman, S.F.; Sultana, R. Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer's disease and mild cognitive impairment. Free Radic Biol Med 2007, 43, 658–677. [Google Scholar] [CrossRef]
- Fišar, Z.; Hroudová, J.; Hansiková, H.; Spáčilová, J.; Lelková, P.; Wenchich, L.; Jirák, R.; M., Z.; Zeman, J.; Martásek, P.; et al. Mitochondrial respiration in the platelets of patients with Alzheimer's disease. Curr Alzheimer Res 2016, 13, 930-941.
- Fišar, Z.; Hansíková, H.; Křížová, J.; Jirák, R.; Kitzlerová, E.; Zvěřová, M.; Hroudová, J.; Wenchich, L.; Zeman, J.; Raboch, J. Activities of mitochondrial respiratory chain complexes in platelets of patients with Alzheimer's disease and depressive disorder. Mitochondrion 2019, 48, 67–77. [Google Scholar] [CrossRef]
- Zheng, C.; Zhou, X.W.; Wang, J.Z. The dual roles of cytokines in Alzheimer's disease: update on interleukins, TNF-alpha, TGF-beta and IFN-gamma. Transl Neurodegener 2016, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R., Jr.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011, 7, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Khan, S.M. A "mitochondrial cascade hypothesis" for sporadic Alzheimer's disease. Med Hypotheses 2004, 63, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer's disease mitochondrial cascade hypothesis. J Alzheimers Dis 2010, 20 Suppl 2, S265–279. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer's Disease. J Alzheimers Dis 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed]
- Pires, M.; Rego, A.C. Apoe4 and Alzheimer's Disease Pathogenesis-Mitochondrial Deregulation and Targeted Therapeutic Strategies. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Yang, K.; Chen, Z.; Gao, J.; Shi, W.; Li, L.; Jiang, S.; Hu, H.; Liu, Z.; Xu, D.; Wu, L. The Key Roles of GSK-3beta in Regulating Mitochondrial Activity. Cell Physiol Biochem 2017, 44, 1445–1459. [Google Scholar] [CrossRef]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther 2015, 148, 114–131. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, D.; Sehgal, A.; Singh, S.; Sharma, N.; Zengin, G.; Andronie-Cioara, F.L.; Toma, M.M.; Bungau, S.; Bumbu, A.G. Role of Monoamine Oxidase Activity in Alzheimer's Disease: An Insight into the Therapeutic Potential of Inhibitors. Molecules 2021, 26. [Google Scholar] [CrossRef]
- Vance, J.E. MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim Biophys Acta 2014, 1841, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Giacomello, M.; Pellegrini, L. The coming of age of the mitochondria-ER contact: a matter of thickness. Cell Death Differ 2016, 23, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, D.; Suski, J.M.; Oules, B.; Debayle, D.; Gay, A.S.; Lacas-Gervais, S.; Bussiere, R.; Bauer, C.; Pinton, P.; Paterlini-Brechot, P.; et al. Localization and Processing of the Amyloid-beta Protein Precursor in Mitochondria-Associated Membranes. J Alzheimers Dis 2017, 55, 1549–1570. [Google Scholar] [CrossRef] [PubMed]
- Eysert, F.; Kinoshita, P.F.; Mary, A.; Vaillant-Beuchot, L.; Checler, F.; Chami, M. Molecular Dysfunctions of Mitochondria-Associated Membranes (MAMs) in Alzheimer's Disease. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Schon, E.A.; Area-Gomez, E. Mitochondria-associated ER membranes in Alzheimer disease. Mol Cell Neurosci 2013, 55, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Vera, J.; Wolkenhauer, O. The systems biology of mitochondrial fission and fusion and implications for disease and aging. Biogerontology 2014, 15, 1–12. [Google Scholar] [CrossRef]
- Santos, R.X.; Correia, S.C.; Zhu, X.; Smith, M.A.; Moreira, P.I.; Castellani, R.J.; Nunomura, A.; Perry, G. Mitochondrial DNA oxidative damage and repair in aging and Alzheimer's disease. Antioxid Redox Signal 2013, 18, 2444–2457. [Google Scholar] [CrossRef] [PubMed]
- Fišar, Z.; Hroudová, J.; Zvěřová, M.; Jirák, R.; Raboch, J.; Kitzlerová, E. Age-Dependent Alterations in Platelet Mitochondrial Respiration. Biomedicines 2023, 11. [Google Scholar] [CrossRef]
- Brown, M.R.; Geddes, J.W.; Sullivan, P.G. Brain region-specific, age-related, alterations in mitochondrial responses to elevated calcium. J Bioenerg Biomembr 2004, 36, 401–406. [Google Scholar] [CrossRef]
- Braidy, N.; Poljak, A.; Grant, R.; Jayasena, T.; Mansour, H.; Chan-Ling, T.; Guillemin, G.J.; Smythe, G.; Sachdev, P. Mapping NAD(+) metabolism in the brain of ageing Wistar rats: potential targets for influencing brain senescence. Biogerontology 2014, 15, 177–198. [Google Scholar] [CrossRef]
- Lakatos, A.; Derbeneva, O.; Younes, D.; Keator, D.; Bakken, T.; Lvova, M.; Brandon, M.; Guffanti, G.; Reglodi, D.; Saykin, A.; et al. Association between mitochondrial DNA variations and Alzheimer's disease in the ADNI cohort. Neurobiol Aging 2010, 31, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, R.K.; Flint Beal, M. Mitochondrial diseases of the brain. Free Radic Biol Med 2013, 63, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R.; Barnes, K.; Hastings, C.; Mortiboys, H. Mitochondrial abnormalities in Parkinson's disease and Alzheimer's disease: can mitochondria be targeted therapeutically? Biochemical Society transactions 2018, 46, 891–909. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Sun, P.; Zhang, H.; Yang, H. Mitochondrial quality control in the brain: The physiological and pathological roles. Front Neurosci 2022, 16, 1075141. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Jo, M.G.; Kim, S.Y.; Chung, C.G.; Lee, S.B. Dietary Antioxidants and the Mitochondrial Quality Control: Their Potential Roles in Parkinson's Disease Treatment. Antioxidants (Basel) 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Guo, L.; Yan, S.; Sosunov, A.A.; McKhann, G.M.; Yan, S.S. Early deficits in synaptic mitochondria in an Alzheimer's disease mouse model. Proc Natl Acad Sci U S A 2010, 107, 18670–18675. [Google Scholar] [CrossRef] [PubMed]
- Calkins, M.J.; Reddy, P.H. Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer's disease neurons. Biochim Biophys Acta 2011, 1812, 507–513. [Google Scholar] [CrossRef]
- Pavlov, P.F.; Hansson Petersen, C.; Glaser, E.; Ankarcrona, M. Mitochondrial accumulation of APP and Abeta: significance for Alzheimer disease pathogenesis. J Cell Mol Med 2009, 13, 4137–4145. [Google Scholar] [CrossRef]
- Pagani, L.; Eckert, A. Amyloid-Beta interaction with mitochondria. Int J Alzheimers Dis 2011, 2011, 925050. [Google Scholar] [CrossRef]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A 2009, 106, 14670–14675. [Google Scholar] [CrossRef]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet 2006, 15, 1437–1449. [Google Scholar] [CrossRef] [PubMed]
- Caspersen, C.; Wang, N.; Yao, J.; Sosunov, A.; Chen, X.; Lustbader, J.W.; Xu, H.W.; Stern, D.; McKhann, G.; Yan, S.D. Mitochondrial A beta: a potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. Faseb Journal 2005, 19, 2040. [Google Scholar] [CrossRef]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Hum Mol Genet 2011, 20, 2495–2509. [Google Scholar] [CrossRef] [PubMed]
- Rhein, V.; Song, X.; Wiesner, A.; Ittner, L.M.; Baysang, G.; Meier, F.; Ozmen, L.; Bluethmann, H.; Drose, S.; Brandt, U.; et al. Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer's disease mice. Proc Natl Acad Sci U S A 2009, 106, 20057–20062. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: a theory based on free radical and radiation chemistry. J Gerontol 1956, 11, 298–300. [Google Scholar] [CrossRef]
- Harman, D. The aging process. Proc Natl Acad Sci U S A 1981, 78, 7124–7128. [Google Scholar] [CrossRef]
- Harman, D. Origin and evolution of the free radical theory of aging: a brief personal history, 1954-2009. Biogerontology 2009, 10, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24. [Google Scholar] [CrossRef]
- Ristow, M.; Schmeisser, S. Extending life span by increasing oxidative stress. Free Radic Biol Med 2011, 51, 327–336. [Google Scholar] [CrossRef]
- Jang, J.Y.; Blum, A.; Liu, J.; Finkel, T. The role of mitochondria in aging. J Clin Invest 2018, 128, 3662–3670. [Google Scholar] [CrossRef]
- Son, J.M.; Lee, C. Mitochondria: multifaceted regulators of aging. BMB Rep 2019, 52, 13–23. [Google Scholar] [CrossRef]
- Son, J.M.; Lee, C. Aging: All roads lead to mitochondria. Semin Cell Dev Biol 2021, 116, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Martini, H.; Passos, J.F. Cellular senescence: all roads lead to mitochondria. FEBS J 2023, 290, 1186–1202. [Google Scholar] [CrossRef] [PubMed]
- Padurariu, M.; Ciobica, A.; Lefter, R.; Serban, I.L.; Stefanescu, C.; Chirita, R. The oxidative stress hypothesis in Alzheimer's disease. Psychiatr Danub 2013, 25, 401–409. [Google Scholar] [PubMed]
- Miles, E.A.; Calder, P.C. Effects of Citrus Fruit Juices and Their Bioactive Components on Inflammation and Immunity: A Narrative Review. Front Immunol 2021, 12, 712608. [Google Scholar] [CrossRef]
- Plascencia-Villa, G.; Perry, G. Roles of Oxidative Stress in Synaptic Dysfunction and Neuronal Cell Death in Alzheimer's Disease. Antioxidants (Basel) 2023, 12. [Google Scholar] [CrossRef]
- Mattson, M.P. Roles of the lipid peroxidation product 4-hydroxynonenal in obesity, the metabolic syndrome, and associated vascular and neurodegenerative disorders. Exp Gerontol 2009, 44, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Galasko, D.R.; Peskind, E.; Clark, C.M.; Quinn, J.F.; Ringman, J.M.; Jicha, G.A.; Cotman, C.; Cottrell, B.; Montine, T.J.; Thomas, R.G.; et al. Antioxidants for Alzheimer disease: a randomized clinical trial with cerebrospinal fluid biomarker measures. Arch Neurol 2012, 69, 836–841. [Google Scholar] [CrossRef] [PubMed]
- Testai, L.; Martelli, A.; Flori, L.; Cicero, A.F.G.; Colletti, A. Coenzyme Q(10): Clinical Applications beyond Cardiovascular Diseases. Nutrients 2021, 13. [Google Scholar] [CrossRef]
- Aberg, F.; Appelkvist, E.L.; Dallner, G.; Ernster, L. Distribution and redox state of ubiquinones in rat and human tissues. Arch Biochem Biophys 1992, 295, 230–234. [Google Scholar] [CrossRef]
- Barcelos, I.P.; Haas, R.H. CoQ10 and Aging. Biology (Basel) 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Bentinger, M.; Tekle, M.; Dallner, G. Coenzyme Q--biosynthesis and functions. Biochem Biophys Res Commun 2010, 396, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Manzar, H.; Abdulhussein, D.; Yap, T.E.; Cordeiro, M.F. Cellular Consequences of Coenzyme Q10 Deficiency in Neurodegeneration of the Retina and Brain. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Ernster, L.; Dallner, G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim Biophys Acta 1995, 1271, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Trumpower, B.L. New concepts on the role of ubiquinone in the mitochondrial respiratory chain. J Bioenerg Biomembr 1981, 13, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Genova, M.L.; Lenaz, G. New developments on the functions of coenzyme Q in mitochondria. Biofactors 2011, 37, 330–354. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo-Gutierrez, A.; Gonzalez-Garcia, P.; Diaz-Casado, M.E.; Barriocanal-Casado, E.; Lopez-Herrador, S.; Quinzii, C.M.; Lopez, L.C. Metabolic Targets of Coenzyme Q10 in Mitochondria. Antioxidants (Basel) 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Bentinger, M.; Brismar, K.; Dallner, G. The antioxidant role of coenzyme Q. Mitochondrion 2007, 7 Suppl, S41–50. [Google Scholar] [CrossRef]
- Echtay, K.S.; Winkler, E.; Klingenberg, M. Coenzyme Q is an obligatory cofactor for uncoupling protein function. Nature 2000, 408, 609–613. [Google Scholar] [CrossRef]
- Walter, L.; Miyoshi, H.; Leverve, X.; Bernard, P.; Fontaine, E. Regulation of the mitochondrial permeability transition pore by ubiquinone analogs. A progress report. Free Radic Res 2002, 36, 405–412. [Google Scholar] [CrossRef]
- Agmo Hernandez, V.; Eriksson, E.K.; Edwards, K. Ubiquinone-10 alters mechanical properties and increases stability of phospholipid membranes. Biochim Biophys Acta 2015, 1848, 2233–2243. [Google Scholar] [CrossRef]
- Li, X.; Zhan, J.; Hou, Y.; Chen, S.; Hou, Y.; Xiao, Z.; Luo, D.; Lin, D. Coenzyme Q10 suppresses oxidative stress and apoptosis via activating the Nrf-2/NQO-1 and NF-kappaB signaling pathway after spinal cord injury in rats. Am J Transl Res 2019, 11, 6544–6552. [Google Scholar] [PubMed]
- Linnane, A.W.; Kios, M.; Vitetta, L. Coenzyme Q(10)--its role as a prooxidant in the formation of superoxide anion/hydrogen peroxide and the regulation of the metabolome. Mitochondrion 2007, 7 Suppl, S51–61. [Google Scholar] [CrossRef]
- Dallner, G.; Sindelar, P.J. Regulation of ubiquinone metabolism. Free Radic Biol Med 2000, 29, 285–294. [Google Scholar] [CrossRef]
- Pradhan, N.; Singh, C.; Singh, A. Coenzyme Q10 a mitochondrial restorer for various brain disorders. Naunyn Schmiedebergs Arch Pharmacol 2021, 394, 2197–2222. [Google Scholar] [CrossRef]
- Staiano, C.; Garcia-Corzo, L.; Mantle, D.; Turton, N.; Millichap, L.E.; Brea-Calvo, G.; Hargreaves, I. Biosynthesis, Deficiency, and Supplementation of Coenzyme Q. Antioxidants (Basel) 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Del-Rio, L.; Clarke, C.F. Coenzyme Q Biosynthesis: An Update on the Origins of the Benzenoid Ring and Discovery of New Ring Precursors. Metabolites 2021, 11. [Google Scholar] [CrossRef]
- Stefely, J.A.; Pagliarini, D.J. Biochemistry of Mitochondrial Coenzyme Q Biosynthesis. Trends Biochem Sci 2017, 42, 824–843. [Google Scholar] [CrossRef] [PubMed]
- Hunte, C.; Palsdottir, H.; Trumpower, B.L. Protonmotive pathways and mechanisms in the cytochrome bc1 complex. FEBS Lett 2003, 545, 39–46. [Google Scholar] [CrossRef]
- Crofts, A.R. The cytochrome bc1 complex: function in the context of structure. Annu Rev Physiol 2004, 66, 689–733. [Google Scholar] [CrossRef]
- Barragan, A.M.; Crofts, A.R.; Schulten, K.; Solov'yov, I.A. Identification of ubiquinol binding motifs at the Qo-site of the cytochrome bc1 complex. J Phys Chem B 2015, 119, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Fišar, Z.; Hroudová, J. Measurement of Mitochondrial Respiration in Platelets. Methods Mol Biol 2021, 2277, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Hroudová, J.; Fišar, Z. Assessment of the Effects of Drugs on Mitochondrial Respiration. Methods Mol Biol 2021, 2277, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Enriquez, J.A. Supramolecular Organization of Respiratory Complexes. Annu Rev Physiol 2016, 78, 533–561. [Google Scholar] [CrossRef]
- Genova, M.L.; Lenaz, G. Functional role of mitochondrial respiratory supercomplexes. Biochim Biophys Acta 2014, 1837, 427–443. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Mileykovskaya, E.; Dowhan, W. Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J Biol Chem 2002, 277, 43553–43556. [Google Scholar] [CrossRef] [PubMed]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acin-Perez, R.; Latorre-Pellicer, A.; Colas, C.; Balsa, E.; Perales-Clemente, E.; Quiros, P.M.; Calvo, E.; Rodriguez-Hernandez, M.A.; et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 2013, 340, 1567–1570. [Google Scholar] [CrossRef]
- Lenaz, G.; Genova, M.L. Supramolecular organisation of the mitochondrial respiratory chain: a new challenge for the mechanism and control of oxidative phosphorylation. Adv Exp Med Biol 2012, 748, 107–144. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Loshuertos, R.; Enriquez, J.A. Respiratory supercomplexes and the functional segmentation of the CoQ pool. Free Radic Biol Med 2016, 100, 5–13. [Google Scholar] [CrossRef]
- Hernansanz-Agustin, P.; Enriquez, J.A. Functional segmentation of CoQ and cyt c pools by respiratory complex superassembly. Free Radic Biol Med 2021, 167, 232–242. [Google Scholar] [CrossRef]
- Hackenbrock, C.R.; Chazotte, B.; Gupte, S.S. The random collision model and a critical assessment of diffusion and collision in mitochondrial electron transport. J Bioenerg Biomembr 1986, 18, 331–368. [Google Scholar] [CrossRef] [PubMed]
- Schagger, H.; Pfeiffer, K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J 2000, 19, 1777–1783. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, S.; Cabrera-Alarcon, J.L.; Enriquez, J.A. Regulation and functional role of the electron transport chain supercomplexes. Biochemical Society transactions 2021, 49, 2655–2668. [Google Scholar] [CrossRef] [PubMed]
- Enriquez, J.A.; Lenaz, G. Coenzyme q and the respiratory chain: coenzyme q pool and mitochondrial supercomplexes. Mol Syndromol 2014, 5, 119–140. [Google Scholar] [CrossRef] [PubMed]
- Acin-Perez, R.; Enriquez, J.A. The function of the respiratory supercomplexes: the plasticity model. Biochim Biophys Acta 2014, 1837, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G.; Genova, M.L. Kinetics of integrated electron transfer in the mitochondrial respiratory chain: random collisions vs. solid state electron channeling. Am J Physiol Cell Physiol 2007, 292, C1221–1239. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Quinzii, C.M.; Hirano, M. Mutations in coenzyme Q10 biosynthetic genes. J Clin Invest 2007, 117, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Park, H.H.; Koh, S.H.; Choi, N.Y.; Yu, H.J.; Park, J.; Lee, Y.J.; Lee, K.Y. Coenzyme Q10 protects against amyloid beta-induced neuronal cell death by inhibiting oxidative stress and activating the P13K pathway. Neurotoxicology 2012, 33, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, K.; Ikeda, A.; Moriyama, Y.; Chei, C.L.; Noda, H.; Umesawa, M.; Cui, R.; Nagao, M.; Kitamura, A.; Yamamoto, Y.; et al. Serum coenzyme Q10 and risk of disabling dementia: the Circulatory Risk in Communities Study (CIRCS). Atherosclerosis 2014, 237, 400–403. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Jimenez, F.J.; Alonso-Navarro, H.; Garcia-Martin, E.; Agundez, J.A.G. Coenzyme Q10 and Dementia: A Systematic Review. Antioxidants (Basel) 2023, 12. [Google Scholar] [CrossRef]
- Fišar, Z.; Jirák, R.; Zvěřová, M.; Setnička, V.; Habartová, L.; Hroudová, J.; Vaníčková, Z.; Raboch, J. Plasma amyloid beta levels and platelet mitochondrial respiration in patients with Alzheimer's disease. Clin Biochem 2019, 72, 71–80. [Google Scholar] [CrossRef]
- Fišar, Z.; Hroudová, J.; Hansíková, H.; Spáčilová, J.; Lelková, P.; Wenchich, L.; Jirák, R.; Zvěřová, M.; Zeman, J.; Martásek, P.; et al. Mitochondrial Respiration in the Platelets of Patients with Alzheimer's Disease. Current Alzheimer Research 2016, 13, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.; Carvalho, C.; Correia, S.C.; Seica, R.M.; Moreira, P.I. Alzheimer's Disease: From Mitochondrial Perturbations to Mitochondrial Medicine. Brain Pathol 2016, 26, 632–647. [Google Scholar] [CrossRef]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-beta Pathway in Alzheimer's Disease. Mol Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef]
- Cummings, J.; Zhou, Y.; Lee, G.; Zhong, K.; Fonseca, J.; Cheng, F. Alzheimer's disease drug development pipeline: 2023. Alzheimers Dement (N Y) 2023, 9, e12385. [Google Scholar] [CrossRef] [PubMed]
- Frontinan-Rubio, J.; Rabanal-Ruiz, Y.; Duran-Prado, M.; Alcain, F.J. The Protective Effect of Ubiquinone against the Amyloid Peptide in Endothelial Cells Is Isoprenoid Chain Length-Dependent. Antioxidants (Basel) 2021, 10. [Google Scholar] [CrossRef]
- Cummings, J.; Aisen, P.S.; DuBois, B.; Frolich, L.; Jack, C.R., Jr.; Jones, R.W.; Morris, J.C.; Raskin, J.; Dowsett, S.A.; Scheltens, P. Drug development in Alzheimer's disease: the path to 2025. Alzheimers Res Ther 2016, 8, 39. [Google Scholar] [CrossRef]
- Llanos-Gonzalez, E.; Sancho-Bielsa, F.J.; Frontinan-Rubio, J.; Rabanal-Ruiz, Y.; Garcia-Carpintero, S.; Chicano, E.; Ubeda-Banon, I.; Flores-Cuadrado, A.; Gimenez-Llort, L.; Alcain, F.J.; et al. Spatial and Temporal Protein Modules Signatures Associated with Alzheimer Disease in 3xTg-AD Mice Are Restored by Early Ubiquinol Supplementation. Antioxidants (Basel) 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Imagawa, M.; Naruse, S.; Tsuji, S.; Fujioka, A.; Yamaguchi, H. Coenzyme Q10, iron, and vitamin B6 in genetically-confirmed Alzheimer's disease. Lancet 1992, 340, 671. [Google Scholar] [CrossRef]
- Santa-Mara, I.; Santpere, G.; MacDonald, M.J.; Gomez de Barreda, E.; Hernandez, F.; Moreno, F.J.; Ferrer, I.; Avila, J. Coenzyme q induces tau aggregation, tau filaments, and Hirano bodies. J Neuropathol Exp Neurol 2008, 67, 428–434. [Google Scholar] [CrossRef]
- Duran-Prado, M.; Frontinan, J.; Santiago-Mora, R.; Peinado, J.R.; Parrado-Fernandez, C.; Gomez-Almagro, M.V.; Moreno, M.; Lopez-Dominguez, J.A.; Villalba, J.M.; Alcain, F.J. Coenzyme Q10 protects human endothelial cells from beta-amyloid uptake and oxidative stress-induced injury. PLoS One 2014, 9, e109223. [Google Scholar] [CrossRef]
- Kleinová, L.; Cerman, J.; Hlávka, J.; Hort, J. New pharmacological options in the treatment of Alzheimer‘s disease (Nové farmakologické možnosti v léčbě Alzheimerovy nemoci). Cesk Slov Neurol N 2022, 85, 462–469. [Google Scholar] [CrossRef]
- Butler, M.; Nelson, V.A.; Davila, H.; Ratner, E.; Fink, H.A.; Hemmy, L.S.; McCarten, J.R.; Barclay, T.R.; Brasure, M.; Kane, R.L. Over-the-Counter Supplement Interventions to Prevent Cognitive Decline, Mild Cognitive Impairment, and Clinical Alzheimer-Type Dementia: A Systematic Review. Ann Intern Med 2018, 168, 52–62. [Google Scholar] [CrossRef]
- Cenini, G.; Voos, W. Mitochondria as Potential Targets in Alzheimer Disease Therapy: An Update. Front Pharmacol 2019, 10, 902. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Mitochondrial dysfunction and oxidative damage in Alzheimer's and Parkinson's diseases and coenzyme Q10 as a potential treatment. J Bioenerg Biomembr 2004, 36, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, A.; Kamyab, A.; Hosseini, S.; Ebrahimi, S.; Ashkani-Esfahani, S. Involvement of Coenzyme Q10 in Various Neurodegenerative and Psychiatric Diseases. Biochem Res Int 2023, 2023, 5510874. [Google Scholar] [CrossRef]
- Dumont, M.; Kipiani, K.; Yu, F.; Wille, E.; Katz, M.; Calingasan, N.Y.; Gouras, G.K.; Lin, M.T.; Beal, M.F. Coenzyme Q10 decreases amyloid pathology and improves behavior in a transgenic mouse model of Alzheimer's disease. J Alzheimers Dis 2011, 27, 211–223. [Google Scholar] [CrossRef]
- Takahashi, M.; Takahashi, K. Water-soluble CoQ10 as A Promising Anti-aging Agent for Neurological Dysfunction in Brain Mitochondria. Antioxidants (Basel) 2019, 8. [Google Scholar] [CrossRef]
- Komaki, H.; Faraji, N.; Komaki, A.; Shahidi, S.; Etaee, F.; Raoufi, S.; Mirzaei, F. Investigation of protective effects of coenzyme Q10 on impaired synaptic plasticity in a male rat model of Alzheimer's disease. Brain Res Bull 2019, 147, 14–21. [Google Scholar] [CrossRef]
- Mantle, D.; Heaton, R.A.; Hargreaves, I.P. Coenzyme Q10, Ageing and the Nervous System: An Overview. Antioxidants (Basel) 2021, 11. [Google Scholar] [CrossRef]
- Hooff, G.P.; Wood, W.G.; Muller, W.E.; Eckert, G.P. Isoprenoids, small GTPases and Alzheimer's disease. Biochim Biophys Acta 2010, 1801, 896–905. [Google Scholar] [CrossRef]
- Mantle, D.; Dybring, A. Bioavailability of Coenzyme Q(10): An Overview of the Absorption Process and Subsequent Metabolism. Antioxidants (Basel) 2020, 9. [Google Scholar] [CrossRef]
- Raizner, A.E. Coenzyme Q(10). Methodist Debakey Cardiovasc J 2019, 15, 185–191. [Google Scholar] [CrossRef]
- Gueven, N.; Ravishankar, P.; Eri, R.; Rybalka, E. Idebenone: When an antioxidant is not an antioxidant. Redox Biol 2021, 38, 101812. [Google Scholar] [CrossRef]
- Rauchova, H.; Drahota, Z.; Bergamini, C.; Fato, R.; Lenaz, G. Modification of respiratory-chain enzyme activities in brown adipose tissue mitochondria by idebenone (hydroxydecyl-ubiquinone). J Bioenerg Biomembr 2008, 40, 85–93. [Google Scholar] [CrossRef]
- Gutzmann, H.; Hadler, D. Sustained efficacy and safety of idebenone in the treatment of Alzheimer's disease: update on a 2-year double-blind multicentre study. J Neural Transm Suppl 1998, 54, 301–310. [Google Scholar] [CrossRef]
- Thal, L.J.; Grundman, M.; Berg, J.; Ernstrom, K.; Margolin, R.; Pfeiffer, E.; Weiner, M.F.; Zamrini, E.; Thomas, R.G. Idebenone treatment fails to slow cognitive decline in Alzheimer's disease. Neurology 2003, 61, 1498–1502. [Google Scholar] [CrossRef]
- Murphy, M.P.; Smith, R.A. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol 2007, 47, 629–656. [Google Scholar] [CrossRef]
- Manczak, M.; Mao, P.; Calkins, M.J.; Cornea, A.; Reddy, A.P.; Murphy, M.P.; Szeto, H.H.; Park, B.; Reddy, P.H. Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer's disease neurons. J Alzheimers Dis 2010, 20 Suppl 2, S609–631. [Google Scholar] [CrossRef]
- McManus, M.J.; Murphy, M.P.; Franklin, J.L. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer's disease. J Neurosci 2011, 31, 15703–15715. [Google Scholar] [CrossRef]
- Young, M.L.; Franklin, J.L. The mitochondria-targeted antioxidant MitoQ inhibits memory loss, neuropathology, and extends lifespan in aged 3xTg-AD mice. Mol Cell Neurosci 2019, 101, 103409. [Google Scholar] [CrossRef]
- Lenaz, G.; Fato, R.; Di Bernardo, S.; Jarreta, D.; Costa, A.; Genova, M.L.; Parenti Castelli, G. Localization and mobility of coenzyme Q in lipid bilayers and membranes. Biofactors 1999, 9, 87–93. [Google Scholar] [CrossRef]
- Wada, H.; Goto, H.; Hagiwara, S.; Yamamoto, Y. Redox status of coenzyme Q10 is associated with chronological age. J Am Geriatr Soc 2007, 55, 1141–1142. [Google Scholar] [CrossRef]
- Onur, S.; Niklowitz, P.; Fischer, A.; Metges, C.C.; Grune, T.; Menke, T.; Rimbach, G.; Doring, F. A comparative study into alterations of coenzyme Q redox status in ageing pigs, mice, and worms. Biofactors 2014, 40, 346–354. [Google Scholar] [CrossRef]
- Niklowitz, P.; Onur, S.; Fischer, A.; Laudes, M.; Palussen, M.; Menke, T.; Doring, F. Coenzyme Q10 serum concentration and redox status in European adults: influence of age, sex, and lipoprotein concentration. J Clin Biochem Nutr 2016, 58, 240–245. [Google Scholar] [CrossRef]
- Nagase, M.; Yamamoto, Y.; Mitsui, J.; Tsuji, S. Simultaneous detection of reduced and oxidized forms of coenzyme Q10 in human cerebral spinal fluid as a potential marker of oxidative stress. J Clin Biochem Nutr 2018, 63, 205–210. [Google Scholar] [CrossRef]
- Fišar, Z.; Hroudová, J.; Singh, N.; Kopřivová, A.; Macečková, D. Effect of Simvastatin, Coenzyme Q10, Resveratrol, Acetylcysteine and Acetylcarnitine on Mitochondrial Respiration. Folia Biol (Praha) 2016, 62, 53–66. [Google Scholar]
- Hidaka, T.; Fujii, K.; Funahashi, I.; Fukutomi, N.; Hosoe, K. Safety assessment of coenzyme Q10 (CoQ10). Biofactors 2008, 32, 199–208. [Google Scholar] [CrossRef]
Figure 1.
Biological hypotheses of Alzheimer's disease.
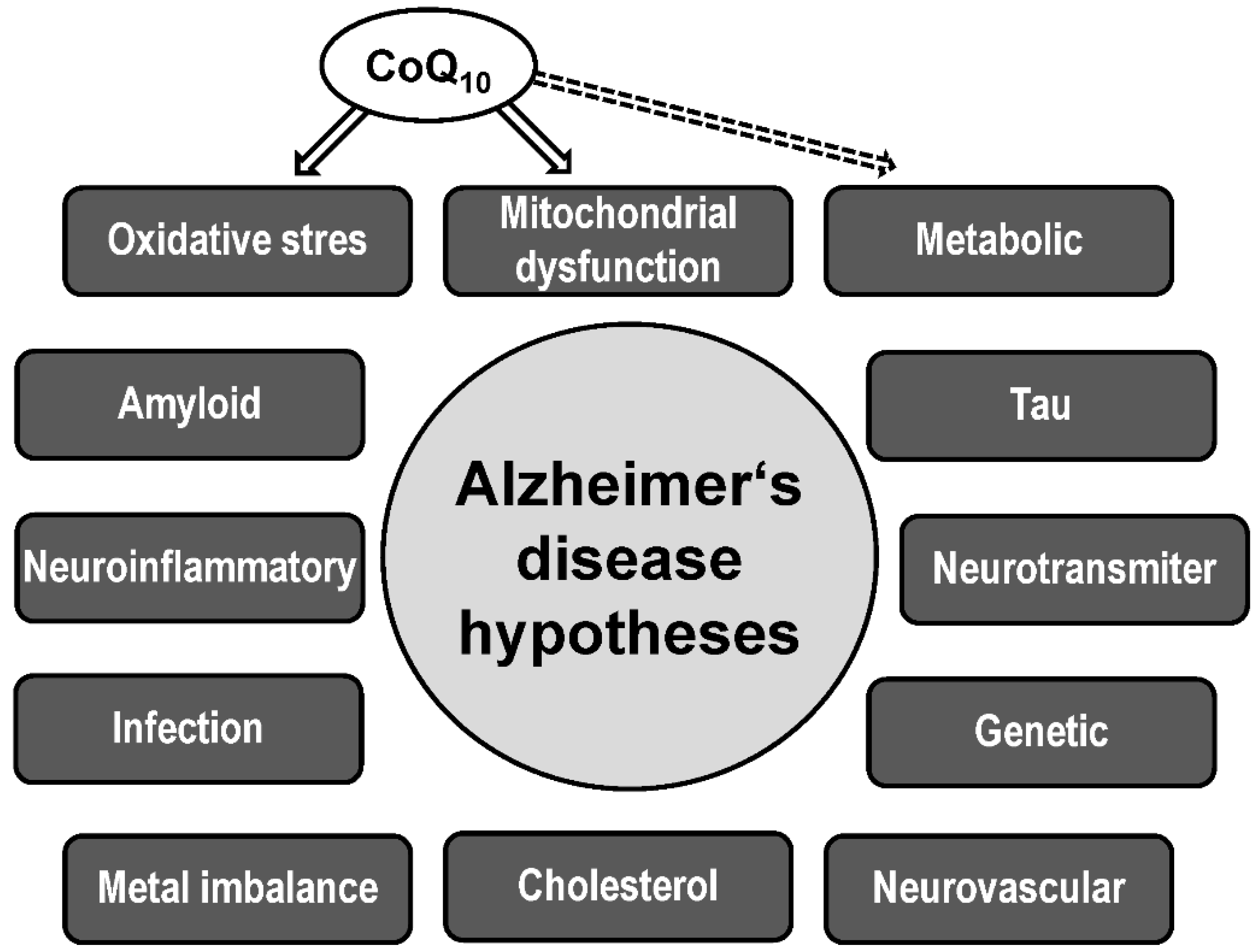
Figure 2.
Basic steps in coenzyme Q10 biosynthesis and three redox isoforms. HMG-CoA – β-Hydroxy β-methylglutaryl-coenzyme A.
Figure 2.
Basic steps in coenzyme Q10 biosynthesis and three redox isoforms. HMG-CoA – β-Hydroxy β-methylglutaryl-coenzyme A.
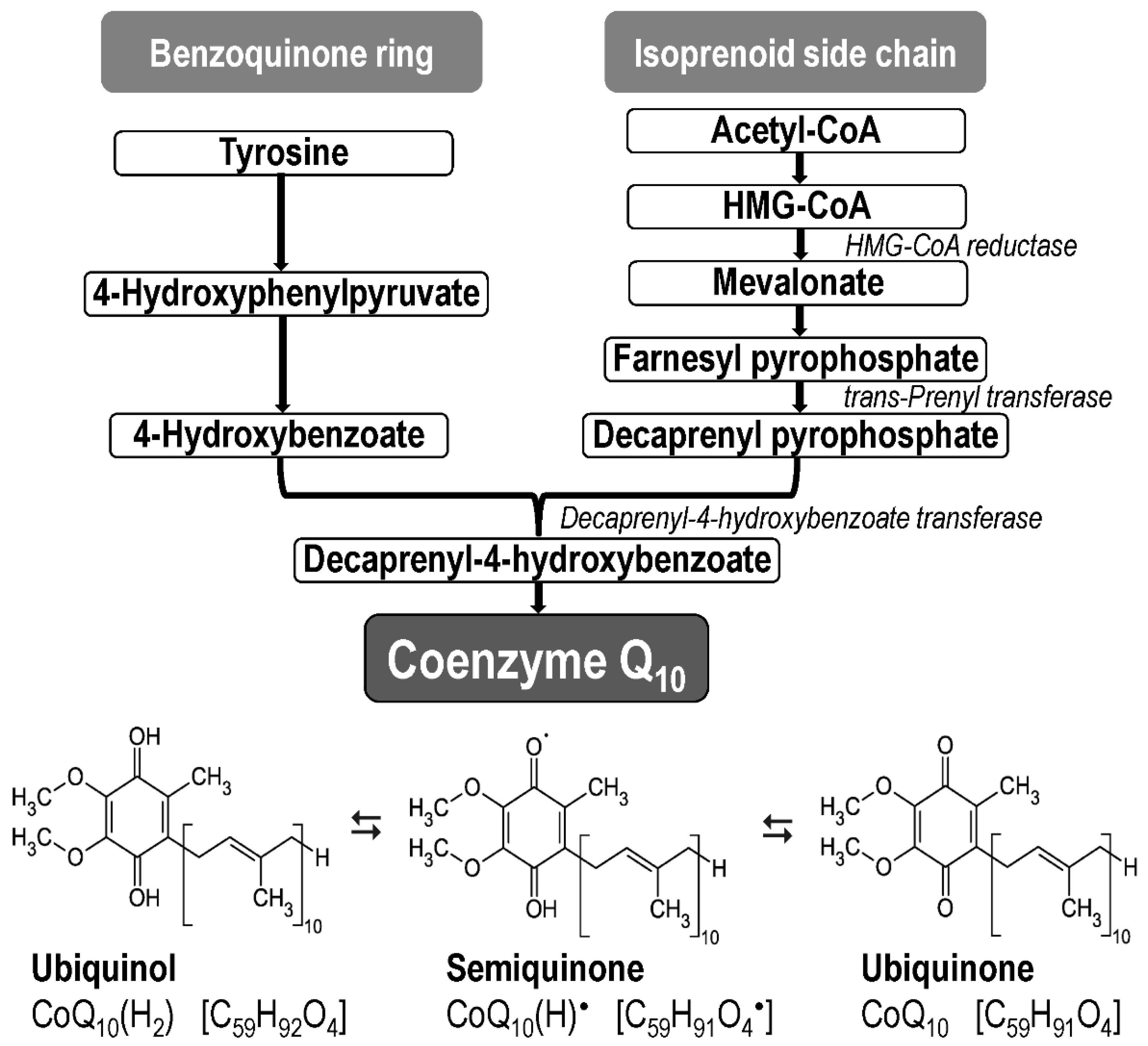
Figure 3.
A simplified diagram of the mitochondrial electron transport system (ETS) with electron transfer in the Q-cycle of complex III. Electrons enter the ETS via complex I or via complex II and are transferred by coenzyme Q10 (CoQ10) to complex III. CoQ10 can also transfer electrons from dehydrogenases (DH) located on the outer or inner surface of the inner mitochondrial membrane (IMM). In the Qo-site complex III near the outer side of IMM, two electrons from one ubiquinol (CoQ10(H2)) and subsequently two more electrons from the second ubiquinol pass into two bifurcated transfer chains: (1) acceptor of two electrons is iron –sulfur cluster (Fe2S2 center) of the Rieske protein, which passes electrons via cytochrome c1 (cyt c1) to cytochrome c (cyt c) on the outer surface of the IMM. Cyt c transfers electrons to complex IV (cytochrome c oxidase), where oxygen is finally reduced to water; (2) the acceptor of the second two electrons in the Q-cycle is cytochrome b containing low (cyt bL) and a higher (cyt bH) potential hemes; this chain supplies electrons to the Qi-site at the matrix side of the IMM, where they reduce one ubiquinone (CoQ10) to semiquinone and then to ubiquinol [113]. Complexes I and III (Qo-site) are sources of superoxide (O2•–).
Figure 3.
A simplified diagram of the mitochondrial electron transport system (ETS) with electron transfer in the Q-cycle of complex III. Electrons enter the ETS via complex I or via complex II and are transferred by coenzyme Q10 (CoQ10) to complex III. CoQ10 can also transfer electrons from dehydrogenases (DH) located on the outer or inner surface of the inner mitochondrial membrane (IMM). In the Qo-site complex III near the outer side of IMM, two electrons from one ubiquinol (CoQ10(H2)) and subsequently two more electrons from the second ubiquinol pass into two bifurcated transfer chains: (1) acceptor of two electrons is iron –sulfur cluster (Fe2S2 center) of the Rieske protein, which passes electrons via cytochrome c1 (cyt c1) to cytochrome c (cyt c) on the outer surface of the IMM. Cyt c transfers electrons to complex IV (cytochrome c oxidase), where oxygen is finally reduced to water; (2) the acceptor of the second two electrons in the Q-cycle is cytochrome b containing low (cyt bL) and a higher (cyt bH) potential hemes; this chain supplies electrons to the Qi-site at the matrix side of the IMM, where they reduce one ubiquinone (CoQ10) to semiquinone and then to ubiquinol [113]. Complexes I and III (Qo-site) are sources of superoxide (O2•–).
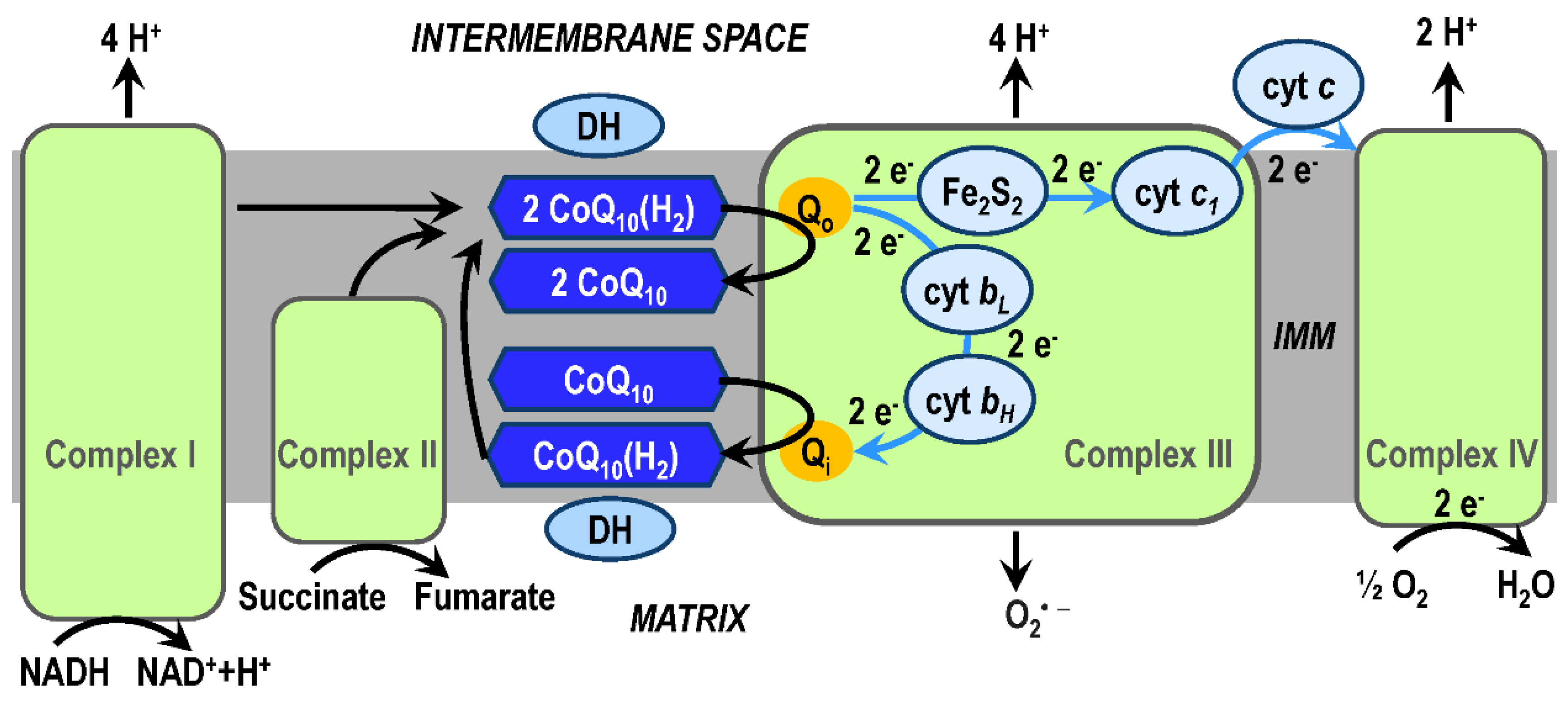
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



