
Submitted:
03 January 2024
Posted:
04 January 2024
You are already at the latest version
Abstract
SOX proteins are transcription factors which play a role in regulating the development of pro-genitor cells and tissue differentiation. Twenty members are known, clustered in eight groups named A to H, and sharing a common DNA-binding domain called the HMG (high-mobility-group) box. Eleven of the SOX genes have been associated with genetic disorders so far, covering a broad spectrum of developmental diseases.
SOX4 is a single-exon gene and belongs to the SOXC group together with SOX11 and SOX12. SOX4 variants were recently described to cause a highly penetrant but heterogeneous disorder, with a phenotypic spectrum ranging from mild developmental delay and learning difficulties to intellectual disability with congenital anomalies. Nineteen pathogenic variants were reported to date, generally de novo, heterozygous, and inactivating, either stop-gain or missense, the latter ones primarily targeting the HMG domain. Further, a bi-allelic variant was reported in a single consanguineous family. Copy number variants, leading to whole gene deletion or duplication, are rare and not clearly associated with a neurodevelopmental disorder.
Many open questions remain, regarding the definition of variants of unknown significance, a possible role of missense variants outside the HMG domain, genotype-phenotype correlation, the range of phenotypic spectrum and modifying factors, and treatment options.
Keywords:
SOX4
; SOXopathy
; intellectual disability
; developmental delay
; aortic aneurism
; semicircular canals
; hypomorphic variants
1. Introduction
Members of the SOX family of transcription factors are defined based on the presence of a DNA-binding domain with homology to the high-mobility-group (HMG) box of SRY (sex-determining region Y) [1]. It is well recognized that the SOX family plays pivotal roles in many developmental and pathological processes, including male differentiation [2,3], eye development [4], skeletogenesis [5] and neurogenesis [6,7]. Twenty members are known, clustered in eight groups (SOXA to SOXH), and half of them have been associated with human genetic disorders, termed “SOXopathies” [8].
SOX4 is a single-exon gene and belongs to the SOXC group together with SOX11 and SOX12. They are expressed in many progenitor cell types and have redundant roles to control cell survival and fate determination in response to various signalling pathways [9]. The three SOXC proteins have almost identical DNA-binding domains and show a high degree of conservation in their other known functional region, a transactivation domain located at the C terminus. Animal models first showed that SOX4 and SOX11 are essential developmental genes, since both Sox11-null and Sox4-null mice die in utero or at birth with multiple abnormalities [10,11]. Sox12-null mice do not show any apparent phenotype, thanks to functional compensation by Sox4 and Sox11 [12]. Two de novo heterozygous missense variants in the SOX11 HMG box were linked to a human disorder characterized by intellectual disability (ID), growth deficiency, facial dysmorphism, and hypoplasia of the fifth digit [13]. The disease was classified as a “Coffin-Siris syndrome-like syndrome”. More de novo heterozygous mutations were later reported in patients with similar features, including SOX11-containing 2p25 deletions, a nonsense variant, and additional HMG-domain missense variants [14], but deeper phenotyping and analysis of DNA-methylation profiles proved that this condition is distinct from Coffin-Siris syndrome (CSS) [15]. Whole Exome Sequencing led to the identification of the first heterozygous SOX4 mutations in 2019, when four de novo missense variants in the HMG box were reported in individuals with developmental delay and mild facial dysmorphism [16].
2. SOX4 structure and function
SOX4 is a single-exon gene and the open reading frame encodes a 474-amino acid protein, which includes two functionally important domains; namely, an HMG box and a C-terminal transactivation domain (TAD). The HMG box facilitates DNA binding, bending, and nuclear trafficking, whereas TAD mediates the interaction with different cofactors [17], although knowledge of critical residues within TAD is still missing. Binding to the minor groove of DNA, SOX4 alters chromatin architecture leading to changes in transcriptional activities of downstream genes. SOX4 (and SOX11) has pleiotropic functions, which are likely mediated by distinct regulatory elements and downstream target genes that are involved in multiple developmental processes, including neurogenesis [18,19], heart development [10], skeletal patterning [20], but also lymphocyte maturation [21] and more recently development of the inner ear [22]. The specific target genes of SOX4 have not yet been fully defined but, among the genes that were shown to be regulated by SOX4, some are important for neurodevelopment and disease (e.g. RELN, DCX and WDR45) [19,23]. In human brain, SOX4 expression was found to be high in several regions (dorsolateral prefrontal cortex, striatum and cerebellar cortex) during the first two trimesters of embryonic gestation, and then to decrease progressively to reach a very low level by the 4th decade of postnatal life; the expression was higher in areas of active neurogenesis [16].
The widespread involvement in developmental processes is consistent with the heterogeneous set of anomalies which can be observed in individuals carrying SOX4 pathogenic variants.
3. SOX4 single nucleotide variants
3.1. Heterozygous pathogenic variants
In line with other SOXopathies, most SOX4 causative variants reported to date are dominant, either loss of function, or missense variants which target the HMG box [16,24,25]. Globally, 13 pathogenic missense variants and six stop-gain (one frameshift, five nonsense) variants have been described (Table 1 and Figure 1). These variants were all identified through exome or genome sequencing studies and were defined as pathogenic/likely pathogenic based on a) in silico assessments (modification of highly conserved amino acids in the HMG box, in different vertebrates orthologos and in other human SOX proteins, or generation of premature stop codons, absence in gnomAD, predictions on the effects of missense variants on protein structure and function) and b) functional assessments (weak or absent DNA binding and loss of transactivation activity in reporter gene assays).
Missense variants were defined as causative mainly based on their inability to bind DNA and activate transcription. Nonetheless, differences in functional tests were reported among distinct variants. For instance, most HMG variants were severely underrepresented in the nucleus when compared with SOX4 wild-type protein and did not bind DNA, whereas p.Leu99Pro was only slightly underrepresented, and p.Leu99Pro and p.Ala112Thr weakly bound DNA. Furthermore, p.Leu99Pro and p.Ala112Thr were weak in transactivation assays whereas other HMG variants were fully inactive [24].
These differences may be a source of phenotype heterogeneity, but many more variants will need to be assessed to draw any conclusion.
Since SOX4 is a single exon gene, mutations that result in premature stop codons are expected to escape nonsense-mediated m-RNA decay (NMD) and produce a truncated shorter protein, that may exert partial functions and/or impair functioning of the wild-type SOX4 copy through a dominant-negative effect. p.Gly44Argfs*2 and p.Glu27* variants fall very early in the open reading frame and encode a peptide of unknown function, but that would lack both functional DNA-binding and transactivation domains. The p.Tyr325*, p.Ser333* and p.Ser347* variants removed 128–150 residues, including the functionally essential TAD, and p.Glu445* deprived TAD of its C-terminal segment. While the p.Gly44Argfs*2 peptide was undetectable in cytoplasm and nucleus of transfected cells with SOX expression plasmids, p.Tyr325* and p.Glu445* were over-represented, especially in the nucleus, and p.Glu445*, although weak, was not fully inactive in the transactivation assay [24].
Pathogenic missense and truncating variants were also found to interfere with the activity of wild-type SOX4, suggesting a dominant-negative effect.
Parents were available to be tested in most (18/19) cases: 14 variants were de novo (4 stop-gain mutations, 10 missense), 4 transmitted (2 stop-gain, 2 missense). SOX4 variants may thus be not fully penetrant, although it is not possible to draw definite conclusions. Only DNA samples from peripheral blood were tested; p.Gly44Argfs*2 was present with low-level mosaicism in the mother’s patient, and this woman was reported to suffer neurocognitive issues. Also, the p.Asn64Lys variant was present in patient’s father, a man with learning difficulties, mild facial dysmorphism and limited extension of 5th finger. Two parents (mother of patient with p.His119Tyr and father of patient with p.Glu445*) carried the variant in non-mosaic state in blood and did not have ID or other relevant health issues.
3.2. Variants of uncertain significance and possible bi-allelic inheritance
Five missense variants are reported in the medical literature as variants of uncertain significance (VUS) [24], but many more are present in the ClinVar database; we describe here also a single variant that may exert its pathogenic effect only in a homozygous state [26] (Table 2 and Figure 1).
Of the five heterozygous variants, two are in the HMG box domain, whereas three involve different regions of the protein. Again, both in silico and functional assessments were implemented for variant classification. These five variants are extremely rare (absent in GnomAD with the exception of p.Asp461Glu - one single allele reported). Regarding p.Lys132Arg (last position of the HMG box), it is interesting to note that Lys132 is conserved in SOXC proteins, but replaced by Arg in other SOX proteins including SRY. p.Ala316Ser, outside the HMG box, lies in an unstructured and functionally uncharacterised region, and Ala316 is poorly conserved. Two variants (p.Asp461Glu and p.Ser466Gly) targeted the SOX4 TAD. Neither of these five variants was found to be less (or more) abundant in transfected cells than wild-type SOX4, p.Ala112Gly and p.Lys132Arg (although they affect the HMG box) could bind DNA, and all five showed normal activity in the transactivation assay.
The Ala112 position deserves a specific comment: four distinct variants have been described involving this residue. p.Ala112Pro and p.Ala112Val were described in affected individuals and were shown to alter SOX4 function, whereas p.Ala112Gly (affected individual) and p.Ala112Thr (in gnomAD, thus presumably unaffected or mildly affected) showed normal activity on functional tests. This is an interesting proof of the limits of in silico assessment and the need to perform functional studies to ascertain variant pathogenicity.
Parents were available to be tested in 3/5 cases (p.Ala112Gly, p.Asp461Glu and p.Ser466Gly) and the variant was always transmitted. The mother who carries the p.Asp461Glu variant had epilepsy, whereas the mother carrying the p.Ser466Gly had learning difficulties, childhood epilepsy and a mood disorder. The mother with the p.Ala112Gly variant is also reported to be mildly affected.
On average, the clinical features associated with VUS were reported to be milder when compared with those defined as (likely) pathogenic, but the number of cases is still too limited. It is possible that these variants, although rare, are irrelevant with respect to these patients’ clinical features; otherwise, some/all of these variants may exert a pathogenic effect, possibly milder, so that they could be defined as “hypomorphic”. No functional tests are currently available to prove this hypothesis.
Further, a bi-allelic variant, an in-frame microdeletion [c.730_753del; p.(Ala244_Gly251del)], was reported in a single consanguineous family [26]. Two affected siblings had global developmental delay, moderate to severe ID, hypotonia, and mild facial dysmorphism. The parents were heterozygous for the p.(Ala244_Gly251del) variant and had normal IQ levels with no history of hypotonia or facial dysmorphism. This variant removes eight evolutionary conserved amino acids within a functionally unknown SOX4 domain, suggesting that sequences outside the DNA-binding and transactivation domains could modulate SOX4 activity and thus undergo pathogenic alterations. Again, the same authors hypothesize that the p.(Ala244_Gly251del) variant might be a hypomorphic allele of SOX4.
Functional studies were not performed, but it is tempting to think about the possibility that SOX4 activity needs to reach a threshold not to compromise normal development. Either monoallelic loss of function variants or bi-allelic hypomorphic variants may thus be disease-causing. Bi-allelic inheritance is very rare in SOXopathies. A homozygous SOX10 deletion was found to cause a severe form of four-limb arthrogryposis but, rather than representing hypomorphic variants, it was a co-dominant occurrence and parents were both affected by Waardenburg syndrome [27]. A closer scenario may be found with SOX18 variants causing Hypotrichosis-Lymphedema-Telangiectasia: this is a very rare disorder caused by heterozygous loss of function variants affecting SOX18, but the first report also described unrelated individuals with homozygous missense variants (p.Ala104Pro and p.Trp95Arg) and healthy heterozygous parents [28]; functional studies were not performed, however these may represent hypomorphic variants.
3.3. Co-occurrence of variants in other genes
The presence of pathogenic variations at two distinct loci that lead to the expression of two Mendelian conditions, which segregate independently, has been appreciated as a relatively frequent phenomenon after the introduction of large-scale sequencing studies. It is termed “dual molecular diagnosis”, and several reports demonstrated that this scenario occurs in a percentage of approximately 5% among patients who received a molecular diagnosis [29]. The two diagnoses can be “distinct”, when the two conditions have different phenotypes, or “overlapping”, when at least some of the clinical features are common to the two disorders [30].
At least three SOX4 patients have a second causative variant: a) the proband carrying the p.Trp69Gly has a distinct molecular diagnosis, a bleeding disorder caused by a stop-gain mutation (p.Gln106*) in F11; b) the patent with p.Gly44Argfs*2 has a TTN-related cardiomyopathy caused by a stop-gain variant (p.Arg18985*) and, since heart defects are frequently associated with SOX4, the presence of cardiomyopathy may be considered an overlapping phenotype; c) the proband carrying the p.Arg466Gly VUS has an associated autosomal dominant myopia caused by a frameshift variant (p.Arg179Valfs*224) in SLC39A5, and it is possible that a liability to developmental delay was worsened or unveiled by an associated disorder causing poor eye-sight.
Furthermore, two VUS of potential interest were reported, p.Pro639Arg in PHF8 in the patient carrying the p.Tyr325* variant, and p.R1406H in CHD4 in the patient carrying p.Ala112Gly. Both PHF8 and CHD4 encode chromatin remodellers, associated with human developmental disorders [31,32], that might interact with SOX4 and contribute to the clinical phenotype.
4. Copy number variants involving SOX4
SOX4-containing 6p22.3 copy number variants (CNVs) are rare [33,34,35,36]. Genome coordinates and clinical information for seven individuals that we could retrieve from the medical literature are summarized in Table 3. Six patients harbor deletions: four carry a similar deletion of approximately 2 Mb involving SOX4 and three further genes (MBOAT1, E2F3, CDKAL1), and share a specific skeletal phenotype, mesomelic dysplasia "Savarirayan type". A larger deletion (encompassing ID4) and a smaller one (encompassing SOX4 only) were NOT associated with a skeletal dysplasia. It was hypothesized that the 2 Mb deletions bring limb enhancers into close proximity of ID4, due to deletion of a topologically associated domain, resulting in the aberrant activation and misexpression of ID4 in the limb bud, and causing a skeletal dysplasia [34]. Thus, this skeletal phenotype is seemingly unrelated to SOX4 deletion. Three of these individuals are reported to suffer developmental delay, namely the ones with the largest and smallest deletion, and only one of the four patients with the 2 Mb deletion causing mesomelic dysplasia. Indeed, SOX4 haploinsufficiency may be the cause of developmental delay in these patients, considering that the individual carrying the smallest deletion (involving SOX4 only) had mild psychomotor delay (plus facial dysmorphism and cleft palate) [35] and the other genes involved in larger deletions have never been associated to abnormal brain development. On the other hand, two patients were reported to show normal psychomotor development with no speech delay (although they were small children) [34] and a third one [33] had congenital anomalies (heart defect and bilateral neurosensorial hearing loss due to malformed semicircular canals) but neurological development is not reported.
The role of somatic SOX4 variation in tumour biology is beyond the scope of this work, but many research studies correlated increased expression of SOX4 with tumorigenesis and progression in several cancer types (reviewed in [37]). It is intriguing that the only SOX4 whole gene duplication reported to date was identified in a boy with dysmorphisms and paediatric cancer (neuroblastoma), although this CNV was of paternal inheritance [36]. Furthermore, the patient carrying the smallest deletion (389 kb, SOX4 only) had nephroblastoma, with a paradoxical increased tumor/normal kidney SOX4 expression ratio. Larger case series will be needed to understand whether constitutional CNVs involving SOX4 indeed predispose carriers to develop paediatric cancer.
5. Phenotype
All affected individual had speech delay, often in the context of global developmental delay; ID can be border-line to (rarely) severe, and hypotonia and behavioural concerns are very common. Facial dysmorphisms are reported in almost every patient, but full pictures are available in the medical literature for three patients only [16], a few others are masked. A fairly specific facial phenotype was present in the first individuals reported in the literature: horizontal palpebral fissures, bulbous tip of the nose with anteverted nares, long philtrum, wide mouth with thin upper lip and cupid bow, posteriorly rotated ears [16]. A much larger dataset of facial images would be needed to define the facial phenotype as specific for this condition.
Associated congenital anomalies are also very common but heterogeneous. More than 50% of individuals with pathogenic variants have cardiovascular anomalies, especially ventricular septal defects, and it is worth mentioning that vascular anomalies are frequent and deserve a follow-up: three patients (all with a missense variant) have aortic aneurism, one (nonsense variant) has double aortic arch and another one (nonsense variant) had anomalous coronary artery that required surgery. Also, a patient with a SOX4 VUS had mild aortic dilatation (with a bicuspid aortic valve). Long term follow-up studies will be needed to assess whether aortic aneurism is more frequent in older SOX4 patients and whether they are progressive.
Otitis media and hearing loss are present in 5/19 patients, but one patient has dysplastic semicircular canals and a second one dysplastic ossicle. Hearing loss with “malformed” semicircular canals was reported also in a patient with a CNV deleting SOX4 [33]. It is interesting that hearing loss due to hypoplasia/dysplasia of the semicircular canals is very rare, but is a typical sign of another SOXopathy (Waardenburg syndrome caused by SOX10 mutations) [7], and may hint to a connection between these two conditions.
Poor eye-sight, palatal abnormalities and genitourinary findings (especially hypospadias) are further common concerns.
Overall, the phenotype of patients with SOX4 variants resembled patients with other neurodevelopmental diseases, including the SOX11-related disorder [14], but some facial features in the context of a developmental delay, together with inner ear and/or vascular anomalies, should help to address the diagnosis and facilitate interpretation of exome/genome sequencing data.
6. Discussion
Heterozygous variants that abolish SOX4 transcriptional activity in vitro cause a human neurodevelopmental syndrome with associated dysmorphic features and inconstant congenital anomalies. The clinical phenotype is consistent with the fundamental roles of SOX4 in development and is likely associated to reduced expression of SOX4 target genes at critical points in embryonic and early postnatal development.
Initially, this syndrome was classified under the umbrella of CSS, similarly to SOX11-neurodevelopmental disorder, but both conditions lacked the most specific features of CSS (e.g., fifth-finger nail hypoplasia, corpus callosum agenesis, and hypertrichosis and hirsutism).
In OMIM (#618506), SOX4-related disorder is now classified as “intellectual developmental disorder with speech delay and dysmorphic facies” (CSS10 being listed as alternative title).
Many open questions remain, some of which are common to most recently identified rare genetic conditions, some specific to the role which is played by SOX4.
First of all, how to define SOX4 VUS. Seventy such variants are reported in the ClinVar database, often in individuals reported to have ID. It is possible to perform functional studies to define the role of missense variants in the HMG box domain, but at the moment it is much harder to define missense and in-frame variants in other regions.
A help to define SOX4 VUS may come from DNA methylation profiling, which has been proved to be a useful biomarker for clinical diagnosis of many rare disorders, among them SOX11-related neurodevelopmental syndrome [15]. SOX11 pathogenic variants were found to associate with a distinct DNA methylation profile, which was also useful to separate this disorder from CSS. Given the redundant roles in development of SOX4 and SOX11, it would be extremely useful to test patients with SOX4 pathogenic variants to understand if they have a distinct methylation profile. This would then turn as a useful tool to classify VUS.
An alternative approach that can help to define a clinical diagnosis and thus interpret atypical genetic variation could be computational facial analysis: thanks to the advancements in computer vision and machine learning, so called “next-generation phenotyping” approaches have been developed, such as GestaltMatcher, which could facilitate the diagnosis of facial image analysis [38]. This approach was proposed to be integrated into exome variant prioritization pipelines. Almost every patient with a SOX4 pathogenic variant has facial dysmorphisms and we expect that more variants will be identified and described, allowing reverse phenotyping and definition of possibly typical facial features. GestaltMatcher or similar approaches may help to recognize facial similarities, determine a specific facial phenotype and eventually be used to prioritize or reclassify uncertain variants [39]. Integrated approaches may be especially useful in a disorder where incomplete penetrance, or mild clinical expression, could hamper the detection of a causative variant, if is transmitted by a seemingly healthy parent.
Other open issues are pathogenetic mechanism and genotype-phenotype correlations. Single nucleotide pathogenic variants have been shown to abolish SOX4 transcriptional activity, so are pathogenic missense and nonsense variants primarily resulting in null alleles? Or do these variants maintain some functions with respect to true haploinsufficiency? On the other hand, as functional studies seem to indicate [24], some variants may be dominant-negative, precluding normal functioning of the wild type allele. Clinical data on patients with CNVs causing SOX4 haploinsufficiency could help to clarify this point. Three out of five patients with large deletions involving SOX4 were NOT reported to have a delay in neurological development and, if this data were confirmed, they would indicate a milder phenotype caused by haploinsufficiency. An example of haploinsufficiency exerting a milder effect than recurrent truncating variants escaping NMD, and acting as dominant-negative, is Townes-Brockes syndrome [40].
The intrinsic function of the mutated SOX4 peptide (haploinsufficiency, total or partial lack of specific functions, dominant-negative effect) can of course determine clinical heterogeneity, but many others sources can be envisaged. First of all, considering that genetic analyses are usually performed on DNA from blood samples, some de novo variants may be mosaics. Mosaicism is relatively frequent in other SOXopathies (e.g. Lamb-Shaffer syndrome) [41] and can be associated to milder (or absent) phenotypes. Ideally, other tissues should be tested. At the same time, parents found to be carriers (mosaic or not) should receive a clinical assessement.
Furthermore, “expansion” of the clinical spectrum of a known disorder may often hide a dual molecular diagnosis [42], as variants in other genes can modify the clinical phenotype. At least two patients with SOX4 pathogenic variants were reported to carry a causative variant in a distinct gene (F11, TTN), others have VUS that may contribute to phenotype.
Some variants may be hypomorphic, exerting their effect when bi-allelic or on a multigenic/oligogenic background. To our knowledge, one single instance of SOX4 bi-allelic variant was ever reported [26], never of SOX11. Given the additive and redundant roles of SOX4 and SOX11 (and SOX12) in development, it is tempting to speculate that a source of clinical variability may be a synergistic heterozygosity for hypomorphic variants affecting these genes.
Treatment options and the best possible management for individuals with SOX mutation is the ultimate issue. No specific treatment is yet available, and if gene therapy could be an option for SOX4, or if SOX4 protein could be druggable, remain unanswered questions.
Yet, individuals with SOX4 variants predicted to be damaging deserve prompt intervention for behavioural problems, which are common and can compromise development and social interactions in children and adults, although they often have only a mild degree of ID. Hearing and visual impairments are common and may compromise the intrinsic developmental potential of SOX4 patients. A close follow-up should be recommended for cardiovascular problems, with a possibly high risk to develop aortic aneurism, that should alert care-givers especially at the time of adulthood transition. Finally, there are no reports of cancer in patients with single nucleotide variants, but two carriers of 6p22.3 CNVs involving SOX4 developed embryonal tumours; thus, we believe that individuals with such CNVs should be monitored during childhood.
In conclusion, these are exciting years for the definition of genetic disorders, including rare SOXopathies. Larger cohorts of patients will allow a better clinical definition, personalized follow-up, and hopefully pave the way to targeted therapies.
Author Contributions
CG conceptualized and drafted this manuscript. MG prepared the tables and figures. Both authors wrote and edited the final version. Both authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding
Institutional Review Board Statement
Not applicable
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Schepers, G.E.; Teasdale, R.D.; Koopman, P. Twenty Pairs of Sox: Extent, Homology, and Nomenclature of the Mouse and Human Sox Transcription Factor Gene Families. Developmental Cell 2002, 3, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Harley, V.R.; Clarkson, M.J.; Argentaro, A. The Molecular Action and Regulation of the Testis-Determining Factors, SRY (Sex-Determining Region on the Y Chromosome) and SOX9 [SRY-Related High-Mobility Group (HMG) Box 9]. Endocrine Reviews 2003, 24, 466–487. [Google Scholar] [CrossRef] [PubMed]
- Sutton, E.; Hughes, J.; White, S.; Sekido, R.; Tan, J.; Arboleda, V.; Rogers, N.; Knower, K.; Rowley, L.; Eyre, H.; et al. Identification of SOX3 as an XX Male Sex Reversal Gene in Mice and Humans. J Clin Invest 2011, 121, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Fantes, J.; Ragge, N.K.; Lynch, S.-A.; McGill, N.I.; Collin, J.R.O.; Howard-Peebles, P.N.; Hayward, C.; Vivian, A.J.; Williamson, K.; van Heyningen, V.; FitzPatrick, D.R. Mutations in SOX2 Cause Anophthalmia. Nat Genet 2003, 33, 462–463. [Google Scholar] [CrossRef] [PubMed]
- Wagner, T.; Wirth, J.; Meyer, J.; Zabel, B.; Held, M.; Zimmer, J.; Pasantes, J.; Bricarelli, F.D.; Keutel, J.; Hustert, E.; et al. Autosomal Sex Reversal and Campomelic Dysplasia Are Caused by Mutations in and around the SRY-Related Gene SOX9. Cell 1994, 79, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Lamb, A.N.; Rosenfeld, J.A.; Neill, N.J.; Talkowski, M.E.; Blumenthal, I.; Girirajan, S.; Keelean-Fuller, D.; Fan, Z.; Pouncey, J.; Stevens, C.; et al. Haploinsufficiency of SOX5 at 12p12.1 Is Associated with Developmental Delays with Prominent Language Delay, Behavior Problems, and Mild Dysmorphic Features. Hum Mutat 2012, 33, 728–740. [Google Scholar] [CrossRef]
- Pingault, V.; Zerad, L.; Bertani-Torres, W.; Bondurand, N. SOX10: 20 Years of Phenotypic Plurality and Current Understanding of Its Developmental Function. J Med Genet 2022, 59, 105–114. [Google Scholar] [CrossRef]
- Angelozzi, M.; Lefebvre, V. SOXopathies: Growing Family of Developmental Disorders Due to SOX Mutations. Trends Genet 2019, 35, 658–671. [Google Scholar] [CrossRef] [PubMed]
- Kavyanifar, A.; Turan, S.; Lie, D.C. SoxC Transcription Factors: Multifunctional Regulators of Neurodevelopment. Cell Tissue Res 2018, 371, 91–103. [Google Scholar] [CrossRef]
- Schilham, M.W.; Oosterwegel, M.A.; Moerer, P.; Ya, J.; de Boer, P.A.J.; van de Wetering, M.; Verbeek, S.; Lamers, W.H.; Kruisbeek, A.M.; Cumano, A.; Clevers, H. Defects in Cardiac Outflow Tract Formation and Pro-B-Lymphocyte Expansion in Mice Lacking Sox-4. Nature 1996, 380, 711–714. [Google Scholar] [CrossRef]
- Sock, E.; Rettig, S.D.; Enderich, J.; Bösl, M.R.; Tamm, E.R.; Wegner, M. Gene Targeting Reveals a Widespread Role for the High-Mobility-Group Transcription Factor Sox11 in Tissue Remodeling. Mol Cell Biol 2004, 24, 6635–6644. [Google Scholar] [CrossRef] [PubMed]
- Hoser, M.; Potzner, M.R.; Koch, J.M.C.; Bösl, M.R.; Wegner, M.; Sock, E. Sox12 Deletion in the Mouse Reveals Nonreciprocal Redundancy with the Related Sox4 and Sox11 Transcription Factors. Mol Cell Biol 2008, 28, 4675–4687. [Google Scholar] [CrossRef]
- Tsurusaki, Y.; Koshimizu, E.; Ohashi, H.; Phadke, S.; Kou, I.; Shiina, M.; Suzuki, T.; Okamoto, N.; Imamura, S.; Yamashita, M.; et al. De Novo SOX11 Mutations Cause Coffin–Siris Syndrome. Nat Commun 2014, 5, 4011. [Google Scholar] [CrossRef]
- Hempel, A.; Pagnamenta, A.T.; Blyth, M.; Mansour, S.; McConnell, V.; Kou, I.; Ikegawa, S.; Tsurusaki, Y.; Matsumoto, N.; Lo-Castro, A.; et al. Deletions and de Novo Mutations of SOX11 Are Associated with a Neurodevelopmental Disorder with Features of Coffin–Siris Syndrome. J Med Genet 2016, 53, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Al-Jawahiri, R.; Foroutan, A.; Kerkhof, J.; McConkey, H.; Levy, M.; Haghshenas, S.; Rooney, K.; Turner, J.; Shears, D.; Holder, M.; et al. SOX11 Variants Cause a Neurodevelopmental Disorder with Infrequent Ocular Malformations and Hypogonadotropic Hypogonadism and with Distinct DNA Methylation Profile. Genet Med 2022, 24, 1261–1273. [Google Scholar] [CrossRef]
- Zawerton, A.; Yao, B.; Yeager, J.P.; Pippucci, T.; Haseeb, A.; Smith, J.D.; Wischmann, L.; Kühl, S.J.; Dean, J.C.S.; Pilz, D.T.; Holder, S.E.; McNeill, A.; Graziano, C.; Lefebvre, V. De Novo SOX4 Variants Cause a Neurodevelopmental Disease Associated with Mild Dysmorphism. Am J Hum Genet 2019, 104, 246–259. [Google Scholar] [CrossRef]
- Van de Wetering, M.; Oosterwegel, M.; van Norren, K.; Clevers, H. Sox-4, an Sry-like HMG Box Protein, Is a Transcriptional Activator in Lymphocytes. EMBO J 1993, 12, 3847–3854. [Google Scholar] [CrossRef] [PubMed]
- Bhattaram, P.; Penzo-Méndez, A.; Sock, E.; Colmenares, C.; Kaneko, K.J.; Vassilev, A.; DePamphilis, M.L.; Wegner, M.; Lefebvre, V. Organogenesis Relies on SoxC Transcription Factors for the Survival of Neural and Mesenchymal Progenitors. Nat Commun 2010, 1, 1–12. [Google Scholar] [CrossRef]
- Shim, S.; Kwan, K.Y.; Li, M.; Lefebvre, V.; Šestan, N. Cis-Regulatory Control of Corticospinal System Development and Evolution. Nature 2012, 486, 74–79. [Google Scholar] [CrossRef]
- Bhattaram, P.; Penzo-Méndez, A.; Kato, K.; Bandyopadhyay, K.; Gadi, A.; Taketo, M.M.; Lefebvre, V. SOXC Proteins Amplify Canonical WNT Signaling to Secure Nonchondrocytic Fates in Skeletogenesis. J Cell Biol 2014, 207, 657–671. [Google Scholar] [CrossRef]
- Sun, B.; Mallampati, S.; Gong, Y.; Wang, D.; Lefebvre, V.; Sun, X. Sox4 Is Required for the Survival of Pro-B Cells. J Immunol 2013, 190, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Llamas, J.; Trecek, T.; Shi, T.; Tao, L.; Makmura, W.; Crump, J.G.; Segil, N.; Gnedeva, K. SoxC Transcription Factors Shape the Epigenetic Landscape to Establish Competence for Sensory Differentiation in the Mammalian Organ of Corti. Proceedings of the National Academy of Sciences 2023, 120, e2301301120. [Google Scholar] [CrossRef] [PubMed]
- Scharer, C.D.; McCabe, C.D.; Ali-Seyed, M.; Berger, M.F.; Bulyk, M.L.; Moreno, C.S. Genome-Wide Promoter Analysis of the SOX4 Transcriptional Network in Prostate Cancer Cells. Cancer Res 2009, 69, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Angelozzi, M.; Karvande, A.; Molin, A.N.; Ritter, A.L.; Leonard, J.M.M.; Savatt, J.M.; Douglass, K.; Myers, S.M.; Grippa, M.; Tolchin, D.; et al. Consolidation of the Clinical and Genetic Definition of a SOX4-Related Neurodevelopmental Syndrome. J Med Genet 2022, 59, 1058–1068. [Google Scholar] [CrossRef] [PubMed]
- Grosse, M.; Kuechler, A.; Dabir, T.; Spranger, S.; Beck-Wödl, S.; Bertrand, M.; Haack, T.B.; Grasemann, C.; Manka, E.; Depienne, C.; Kaiser, F.J. Novel Variants of SOX4 in Patients with Intellectual Disability. Int J Mol Sci 2023, 24, 3519. [Google Scholar] [CrossRef]
- Ghaffar, A.; Rasheed, F.; Rashid, M.; van Bokhoven, H.; Ahmed, Z.M.; Riazuddin, S.; Riazuddin, S. Biallelic In-Frame Deletion of SOX4 Is Associated with Developmental Delay, Hypotonia and Intellectual Disability. Eur J Hum Genet 2022, 30, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, R.E.; Vincent, V.; Spellicy, C.J.; Friez, M.J.; Chaubey, A. Biallelic Deletions of the Waardenburg II Syndrome Gene, SOX10, Cause a Recognizable Arthrogryposis Syndrome. American Journal of Medical Genetics Part A 2018, 176, 1968–1971. [Google Scholar] [CrossRef]
- Irrthum, A.; Devriendt, K.; Chitayat, D.; Matthijs, G.; Glade, C.; Steijlen, P.M.; Fryns, J.-P.; Van Steensel, M.A.M.; Vikkula, M. Mutations in the Transcription Factor Gene SOX18 Underlie Recessive and Dominant Forms of Hypotrichosis-Lymphedema-Telangiectasia. Am J Hum Genet 2003, 72, 1470–1478. [Google Scholar] [CrossRef] [PubMed]
- Posey, J.E.; Harel, T.; Liu, P.; Rosenfeld, J.A.; James, R.A.; Coban Akdemir, Z.H.; Walkiewicz, M.; Bi, W.; Xiao, R.; Ding, Y.; et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N Engl J Med 2017, 376, 21–31. [Google Scholar] [CrossRef]
- Smith, E.D.; Blanco, K.; Sajan, S.A.; Hunter, J.M.; Shinde, D.N.; Wayburn, B.; Rossi, M.; Huang, J.; Stevens, C.A.; Muss, C.; et al. A Retrospective Review of Multiple Findings in Diagnostic Exome Sequencing: Half Are Distinct and Half Are Overlapping Diagnoses. Genet Med 2019, 21, 2199–2207. [Google Scholar] [CrossRef]
- Sobering, A.K.; Bryant, L.M.; Li, D.; McGaughran, J.; Maystadt, I.; Moortgat, S.; Graham, J.M.; van Haeringen, A.; Ruivenkamp, C.; Cuperus, R.; et al. Variants in PHF8 Cause a Spectrum of X-Linked Neurodevelopmental Disorders and Facial Dysmorphology. HGG Adv 2022, 3, 100102. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.; Terhal, P.A.; Cohen, L.; Bruccoleri, M.; Irving, M.; Martinez, A. F.; Rosenfeld, J.A.; Machol, K.; Yang, Y.; Liu, P.; et al. De Novo Mutations in CHD4, an ATP-Dependent Chromatin Remodeler Gene, Cause an Intellectual Disability Syndrome with Distinctive Dysmorphisms. Am J Hum Genet 2016, 99, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Ladinsky, H.T.; Elizalde, A.; Schickler, R.; Dees, P.B.; Crenshaw, M.L.; Sleasman, J.W. Hypereosinophilic Syndrome and Hemimelia in a Patient with Chromosome 6p22.3 Deletion. Pediatr Allergy Immunol 2014, 25, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Flöttmann, R.; Wagner, J.; Kobus, K.; Curry, C.J.; Savarirayan, R.; Nishimura, G.; Yasui, N.; Spranger, J.; Esch, H.V.; Lyons, M.J.; et al. Microdeletions on 6p22.3 Are Associated with Mesomelic Dysplasia Savarirayan Type. Journal of Medical Genetics 2015, 52, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Trimouille, A.; Barouk-Simonet, E.; Charron, S.; Bouron, J.; Bernhard, J.-C.; Lacombe, D.; Fergelot, P.; Rooryck, C. Deletion of the Transcription Factor SOX4 Is Implicated in Syndromic Nephroblastoma. Clinical Genetics 2017, 92, 449–450. [Google Scholar] [CrossRef] [PubMed]
- Gambale, A.; Russo, R.; Andolfo, I.; Quaglietta, L.; De Rosa, G.; Contestabile, V.; De Martino, L.; Genesio, R.; Pignataro, P.; Giglio, S.; et al. Germline Mutations and New Copy Number Variants among 40 Pediatric Cancer Patients Suspected for Genetic Predisposition. Clinical Genetics 2019, 96, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Hanieh, H.; Ahmed, E.A.; Vishnubalaji, R.; Alajez, N.M. SOX4: Epigenetic Regulation and Role in Tumorigenesis. Seminars in Cancer Biology 2020, 67, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.-C.; Lesmann, H.; Krawitz, P.M. Facilitating the Molecular Diagnosis of Rare Genetic Disorders Through Facial Phenotypic Scores. Current Protocols 2023, 3, e906. [Google Scholar] [CrossRef] [PubMed]
- Forwood, C.; Ashton, K.; Zhu, Y.; Zhang, F.; Dias, K.-R.; Standen, K.; Evans, C.-A.; Carey, L.; Cardamone, M.; Shalhoub, C.; et al. Integration of EpiSign, Facial Phenotyping, and Likelihood Ratio Interpretation of Clinical Abnormalities in the Re-Classification of an ARID1B Missense Variant. American Journal of Medical Genetics Part C: Seminars in Medical Genetics 2023, 193, e32056. [Google Scholar] [CrossRef]
- Innoceta, A.M.; Olivucci, G.; Parmeggiani, G.; Scarano, E.; Pragliola, A.; Graziano, C. Chromosomal Microarray Analysis Identifies a Novel SALL1 Deletion, Supporting the Association of Haploinsufficiency with a Mild Phenotype of Townes–Brocks Syndrome. Genes (Basel) 2023, 14, 258. [Google Scholar] [CrossRef]
- Innella, G.; Greco, D.; Carli, D.; Magini, P.; Giorgio, E.; Galesi, O.; Ferrero, G.B.; Romano, C.; Brusco, A.; Graziano, C. Clinical Spectrum and Follow-up in Six Individuals with Lamb–Shaffer Syndrome (SOX5). American J of Med Genetics Pt A 2021, 185, 608–613. [Google Scholar] [CrossRef] [PubMed]
- Severi, G.; Bonora, E.; Perri, A.; Scarano, E.; Mazzanti, L.; Isidori, F.; Zuntini, R.; Menabò, S.; Graziano, C. HDAC8 Loss of Function and SHOX Haploinsufficiency: Two Independent Genetic Defects Responsible for a Complex Phenotype. Cytogenetic and Genome Research 2018, 157, 135–140. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Representation of SOX4 protein with the two known domains (HMG: High-mobility Group; TAD: Transactivation Domain). Known causative variants are reported above the protein, variants of uncertain significance and the bi-allelic variant below.
Figure 1.
Representation of SOX4 protein with the two known domains (HMG: High-mobility Group; TAD: Transactivation Domain). Known causative variants are reported above the protein, variants of uncertain significance and the bi-allelic variant below.


Table 1.
Heterozygous SOX4 causative variants and clinical phenotype. Dev: development; OSA: obstructive sleep apnea; NA: not assessed; ID: intellectual disability; WPW: Wolf-Parkinson-White; VSD: ventricular septal defect; Uk: unknown; ASD: atrial septal defect.
Table 1.
Heterozygous SOX4 causative variants and clinical phenotype. Dev: development; OSA: obstructive sleep apnea; NA: not assessed; ID: intellectual disability; WPW: Wolf-Parkinson-White; VSD: ventricular septal defect; Uk: unknown; ASD: atrial septal defect.
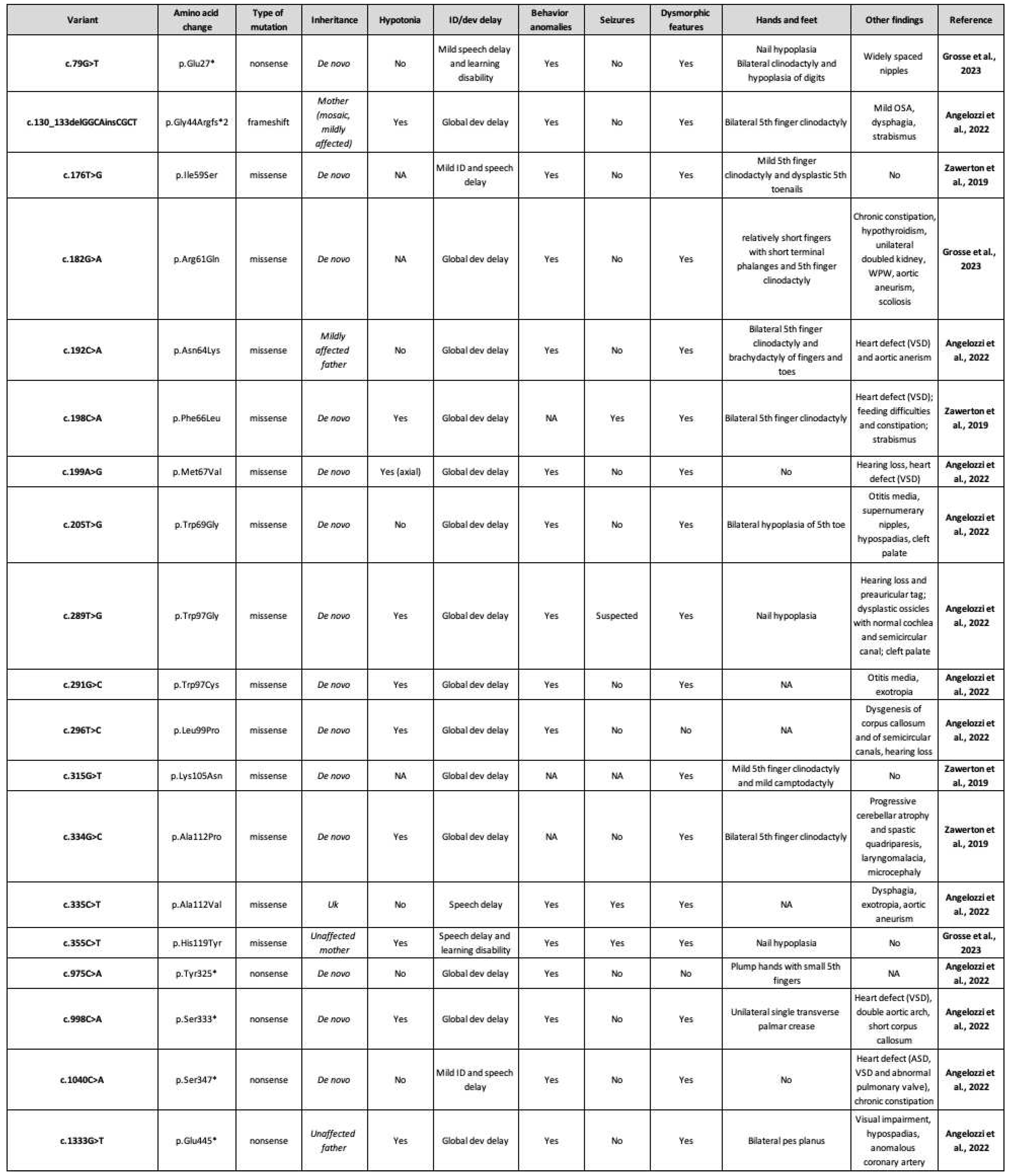 |
Table 2.
SOX4 variants of uncertain significance, biallelic variant, and clinical phenotype. NR: not reported; ID: intellectual disability; Uk: unknown; Dev: development; Het: heterozygous; OSA: obstructive sleep apnea.
Table 2.
SOX4 variants of uncertain significance, biallelic variant, and clinical phenotype. NR: not reported; ID: intellectual disability; Uk: unknown; Dev: development; Het: heterozygous; OSA: obstructive sleep apnea.
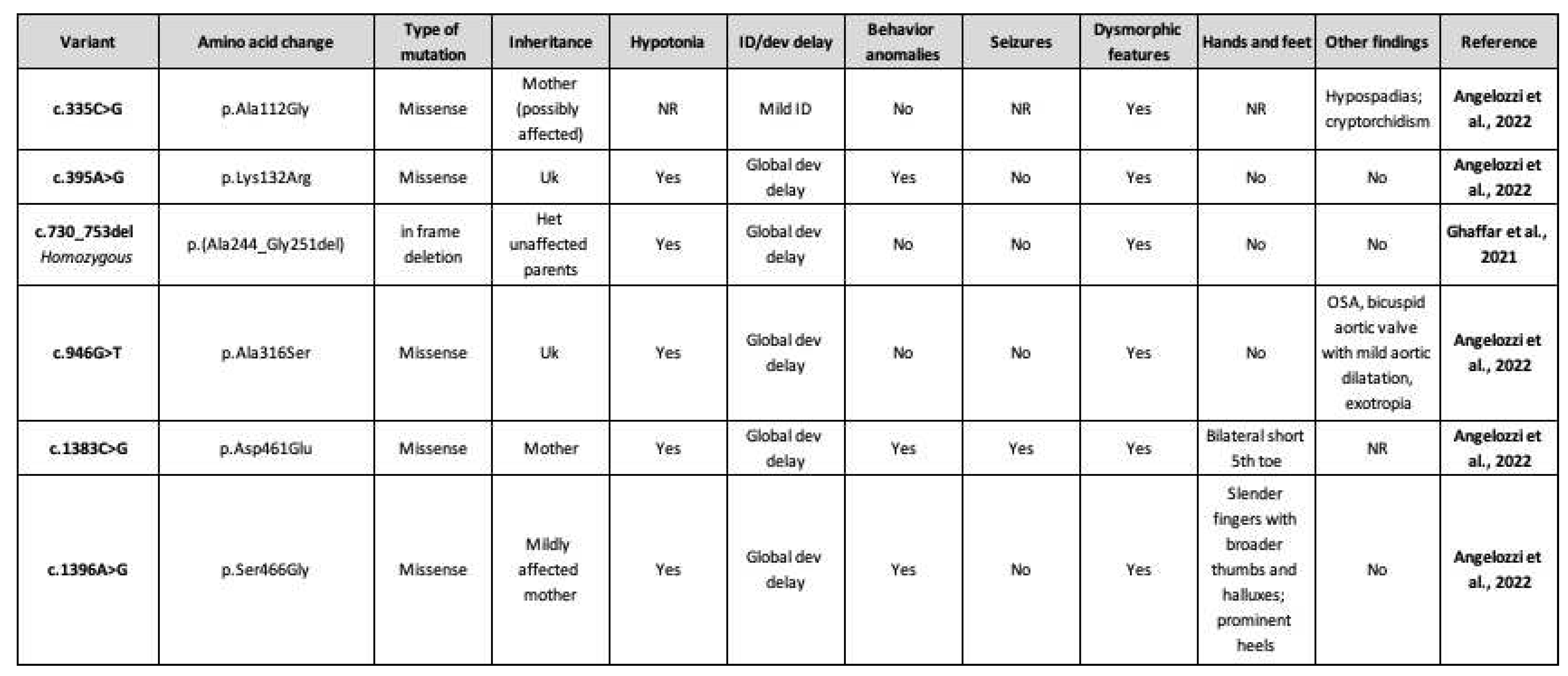 |
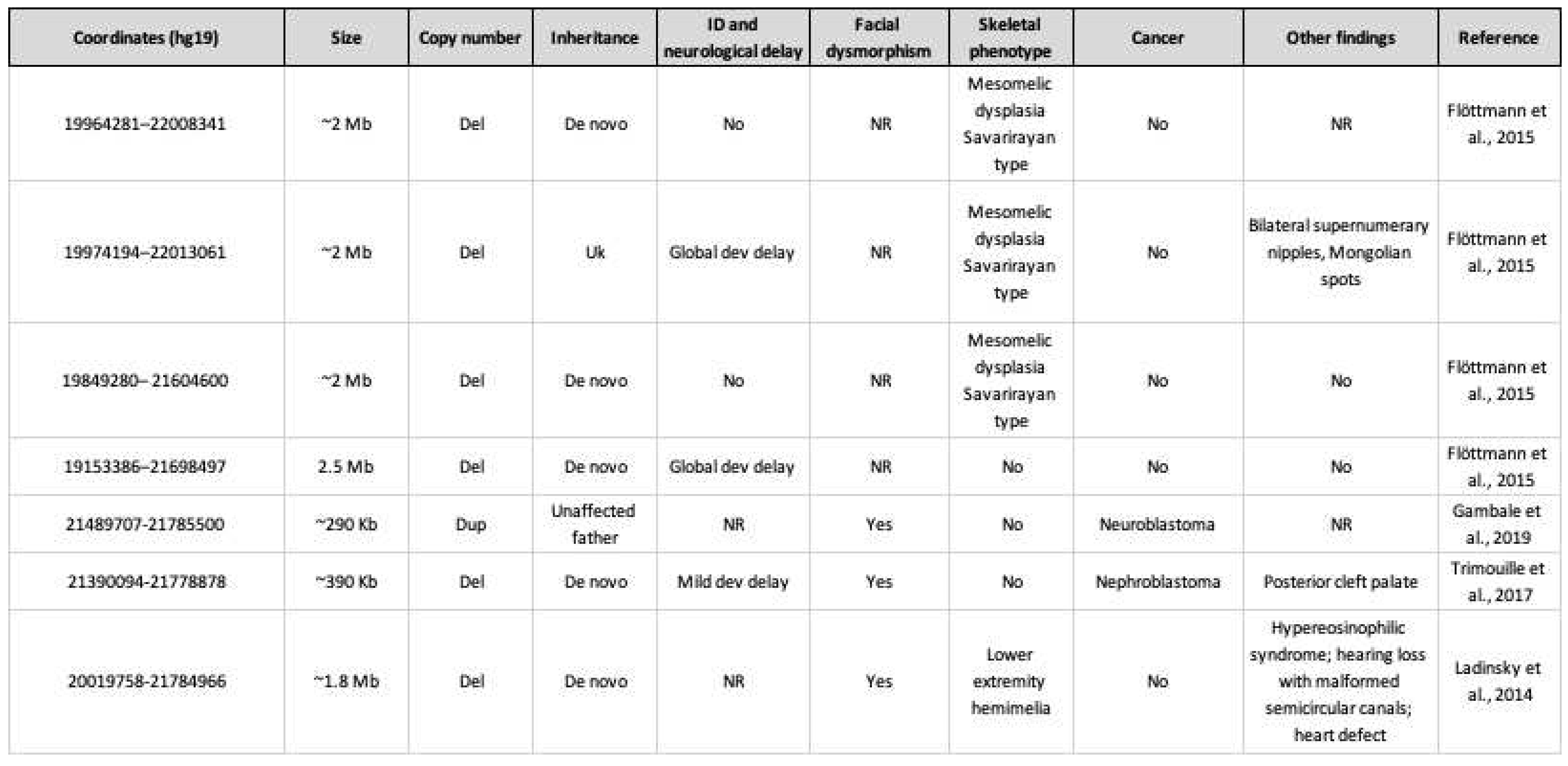
Table 3.
6p22.3 copy number variants including SOX4 and clinical phenotype. ID: intellectual disability; Del: deletion; Dev: development; NR: not reported; Uk: unknown; Dup: duplication.
Table 3.
6p22.3 copy number variants including SOX4 and clinical phenotype. ID: intellectual disability; Del: deletion; Dev: development; NR: not reported; Uk: unknown; Dup: duplication.
 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



