Submitted:
04 January 2024
Posted:
04 January 2024
You are already at the latest version
Abstract
Long read sequencers, known for their effectiveness in detecting genomic structural variations (SV), are becoming a standard in comprehensive genetic analysis. In preimplantation genetic testing (PGT) for SV carriers, information on breakpoint junctions is required for the determination of carrier status in embryo selection. These sequencers are employed for challenging cases involving SVs that are difficult to analyze with conventional cytogenetical methods or detailed junctions of chromosomal translocations, providing valuable insights. They also play a crucial role in acquiring simultaneous information on surrounding single-nucleotide polymorphisms (SNPs) around causative variants in PGT. Despite their advantages, challenges related to sequencing accuracy and testing costs exist. Thus, understanding long read sequencers’ characteristics is essential for their effective utilization. This review summarizes the advanced applications of long read sequencers in preclinical workups and their integration into PGT. It also highlights in-house clinical cases, showing the implementation of long-read sequencing and discussing prospects in the field.
Keywords:
long read sequencer
; sequel sequencer
; nanopore sequencer
; preimplantation genetic testing
1. Introduction
Long-read sequencing is a cutting-edge comprehensive genetic analysis method. Unlike short-read next-generation sequencing (NGS), which typically reads DNA sequences of approximately 50-600 base pairs (bp), long-read sequencing allows for the reading of long DNA sequences ranging from tens of kilobases (kb) to megabases (Mb) [1]. In short-read sequencing, it is difficult to analyze repeat sequences and complex structural variations because of this technique’s characteristics with regard to piecing together short fragments. However, long-read sequencing overcomes this limitation by obtaining DNA reads long enough to assemble these repeat sequences and structural variations. This not only makes the analysis easier from a technical standpoint but also leads to the discovery of novel causative pathological variants [2]. Long-read sequencing is essential for analyzing so-called "genome gaps," which are unreadable genomic regions that were previously challenging to sequence. Assembly using long-read sequencing, along with BioNano's Optical Genome Mapping method [3], played a crucial role in the Telomere-to-Telomere (T2T) project for complete human genome sequencing [4]. Although it may seem that long-read sequencing is displacing conventional comprehensive analysis methods, it has its weaknesses, such as lower sequencing accuracy compared to short-read sequencing and a higher cost per sequencing run. Therefore, like other cytological genetic testing methods such as microarray technology and short-read sequencers, it is necessary to choose the appropriate application based on an understanding of the test characteristics.
When implementing preimplantation genetic testing (PGT), how can long read sequencers be beneficial? PGT is currently primarily used in three types of tests: preimplantation genetic testing for aneuploidy (PGT-A), preimplantation genetic testing for structural rearrangement (PGT-SR), and preimplantation genetic testing for monogenic disorders (PGT-M).[5] Among these PGT types, the critical factor is the need to conduct tests on a minimal number of cells, requiring whole-genome amplification as a prerequisite [6,7,8]. It is essential to note the need for addressing allele dropouts and misdiagnosis due to contamination associated with genome amplification and the limitations of quantitative genetic testing methods such as quantitative PCR (qPCR) and multiple ligation-dependent probe amplification (MLPA) [9]. Therefore, preclinical workups for PGT often demand more detailed analysis than routine clinical genetic diagnostics. PGT-SR is the most straightforward application for understanding the comprehensive and precise analyses that long read sequencers excel at. PGT-SR is a method for avoiding embryo transfer with chromosomal imbalances resulting from chromosomal structural abnormalities, such as parental balanced translocations [10,11]. Conventional techniques like low-coverage whole-genome sequencing via next-generation sequencing (NGS) and aCGH used in PGT-SR can only confirm large genomic imbalances associated with chromosomal rearrangements [5,8]. The accuracy of these techniques is limited, especially for detecting small segments, and sometimes additional methods like FISH probes are necessary [12]. Therefore, the expertise of long read sequencers in genomic structural analysis can be envisioned to enhance precision and expand the scope of testing. Indeed, researchers have already used long-read sequencing for breakpoint-junction analysis in PGT-SR [13,14,15,16,17,18]. In the case of PGT-M, which involves embryo diagnosis for monogenic disorders, the association between pathological variants and genomic rearrangements makes long read sequencers valuable [19,20,21,22,23,24]. Reports on preclinical workups for PGT-M involve associations with SNVs and complex chromosomal structural abnormalities and the detection of deletions. PGT-A is the most widely used form of PGT. It is designed to avoid miscarriage and implantation failure associated with chromosomal aneuploidy [11,25,26,27]. Initially conducted using the FISH method and deemed ineffective, PGT-A gained recognition with the advent of the comprehensive aCGH analysis method, and current next-generation sequencers further improve testing sensitivity and expand capabilities [5]. However, caution is advised in selecting candidates for PGT-A, as indicated by some randomized controlled trials, and screening-based implementation for younger individuals may not be desirable [28,29]. In a Japanese prospective cohort study, PGT-A was particularly effective for application to those of advanced maternal age [11,30]. While it may seem there is no apparent benefit in using a costly long read sequencer for determining chromosomal aneuploidy, the emergence of the STORK method utilizing the rapid sequencing capability of Nanopore sequencers suggests potential development towards clinical use [31]. In this article, we will provide detailed information on long-read sequencing based on the content of previous reports and future prospects as well as discuss self-experiments regarding PGT-M.
2. Types and features of long read sequencers
Long read sequencers are primarily provided by two companies: Pacific Biosciences (PacBio), with their Sequel system, and Oxford Nanopore Technologies (ONT), with their Nanopore system. The Sequel system is based on Single-Molecule Real-Time (SMRT)-sequencing technology [32]. This system enables the real-time observation of DNA polymerase binding to DNA strands, allowing for continuous base sequence reading of long DNA chains, typically several kilobases in length. The latest Sequel II system achieves high accuracy long-read sequencing (HiFi reads) by repeatedly reading circularized DNA (a process known as Circular Consensus Sequencing), resulting in a sequence accuracy of Q30+ (>99.9%). While this sequencing accuracy is sufficient for confirming single-nucleotide variants (SNVs), the Sequel system has limitations in terms of the achievable read length compared to the Nanopore system. Additionally, the cost per sequence is relatively higher than nanopore sequencing. On the other hand, ONT utilizes nanopore-sequencing technology. Nanopores consist of tiny pores formed by membrane proteins, and they detect ion current changes corresponding to a base sequence as DNA molecules pass through them, enabling base calling. Theoretically, there is no limit to the length of DNA that can be measured using Nanopore sequencing [1,33]. One of the significant advantages of Nanopore sequencers is their portability. The initially released product, MinION (https://nanoporetech.com/products/sequence/minion), is a device smaller than a human palm and even a smartphone. It is a groundbreaking device that can perform long-read sequencing by simply connecting it to a PC through a USB port with an HDD cable. Due to its portability, genome sequencing using the same device has been conducted not only for outdoor use [34] but also for use in space stations [35]. When introducing NGS facilities into medical institutions, the high cost and space requirements of conventional sequencers pose significant challenges for small to medium-sized facilities. However, Nanopore sequencers are expected to significantly lower this hurdle, especially in point-of-care testing.[36,37,38,39]
Another major feature of Nanopore sequencers is the frequent updates to their software libraries and kits [33]. The base caller software (Guppy) is updated every few months, and consumables like flow cells and ligation kits are also released frequently [40]. Thus, remaining updated regarding both wet lab experimentation and bioinformatics dry analysis, including with respect to knowledge and techniques related to peripheral analysis software, becomes necessary [33]. On the other hand, many of the provided technologies have high compatibility between various kits, making the latest technology accessible to researchers with minimal potential for equipment investment. Additionally, while consumables for Nanopore sequencing may be expensive, the initial investment cost is very low. The MinION starter pack, which includes one flow cell and a ligation kit, is available for as little as USD 1,000, including the sequencer itself (https://store.nanoporetech.com/minion-basic.html). This low entry barrier may be one of the reasons why the majority of reported long-read-sequencing studies related to PGT have utilized Nanopore sequencers.
3. Application of long-read sequencing to PGT
3.1. Long-read sequencing for PGT-SR
PGT-SR using whole-genome short-read sequencing with low depth (approximately ×0.1) has shown higher scalability and signal-to-noise ratios with respect to detecting copy number variations (CNVs) compared to conventional array comparative genomic hybridization (aCGH) methods [41,42]. This technique enables the efficient detection of embryos with unbalanced translocations. PGT-SR is commonly not intended to exclude embryos with balanced translocations, which may not affect implantation, and is considered necessary and sufficient for the current forms of testing [12]. However, the handling of embryos with balanced translocations is not widely discussed due to technical difficulties in setting up efficient protocols for individual cases. In addition to short-read NGS mentioned earlier, PGT-SR analysis methods also include the use of aCGH and fluorescence in situ hybridization (FISH) for detecting translocations involving small regions [12,43]. However, both NGS and aCGH have difficulty detecting embryos with balanced translocations. Even with FISH, individual setups are required, and incidental chromosomal aneuploidy cannot be accommodated. The 2020 Good Practice Recommendation released by the ESHRE recommends the use of SNP arrays as a method for detecting embryos with balanced translocations [12]. In this regard, a question arises: what developments can be expected when applying long-read sequencing to PGT-SR? In 2019, Zhang et al. used a nanopore sequencer for PGT-SR for a couple with a balanced translocation [13]. They were able to detect not only embryos with unbalanced translocations but also distinguish between balanced inversions and normal embryos via the accurate detection of junctions of translocations and haplotyping using SNP detection in the areas surrounding these junctions. Chow et al. [14] and Liu et al. [15] in 2020 and 2021, respectively, also used a nanopore sequencer to detect breakpoints of balanced translocations, allowing PGT-SR testing based on junction-specific PCR. In 2021, analysis of breakpoints or pathological variants and surrounding SNPs was performed on 15 cases of PGT-SR and 2 cases of PGT-M related to maple syrup urine disease and amyotrophic lateral sclerosis 4 using Sequel SMRT sequencing [16]. Haplotyping for PGT-M around pathogenic variants was also conducted in this report. In 2023, Xia et al. confirmed complete concordance between nanopore sequencing and the Mapping Allele with Resolved Carrier Status (MaReCs) system [17], demonstrating the ability to estimate translocation breakpoints at an error level of approximately 200 kb using the MaReCs system, while nanopore sequencing enabled breakpoint mapping at the single-base level [18]. However, the authors highlighted the limitations of nanopore sequencing for PGT-SR, citing challenges such as the inability to detect junctions of Robertsonian translocations located in genomic gap regions around alpha-satellite DNA and the fact that the correct mapping of sequence reads is still difficult. Despite the aforementioned limitations, there is significant anticipation for the improvement in accuracy and scalability brought about by applying long-read sequencing to PGT-SR. However, due to cost considerations associated with long-read sequencing, this test is likely to remain primarily within the realm of research as it stands now.
3.2. Long-read sequencing for PGT-M
There are several related reports on PGT-M, which, like the reports on PGT-SR, mainly highlight its high analytical capacity for analyzing chromosomal structural variations and ability to simultaneously detect variants and their surrounding SNPs for haplotyping. In 2018, Miao et al. detected a 7.1 kb microdeletion using a Nanopore sequencer in the G6PC gene [19]. This microdeletion was not detected through whole-exome sequencing in short-read sequencing, and long-read sequencing enabled the preclinical workup of PGT-M for glycogen storage disease type Ia. In 2022, Watson et al. reported that they identified breakpoints in a fine-deletion region of exon 23 in the RB1 gene using amplicon sequencing with a small Flongle nanopore flow cell (https://nanoporetech.com/products/sequence/flongle), allowing the design of junction-specific PCR [20]. There are also reports on the use of the PacBio Sequel system for preclinical workup for PGT-M. In 2021, Wu et al. performed an analysis using SMRT sequencing for three couples carrying β-thalassemia variants in 17 reported pathological variants of the HBB gene in China [21]. They conducted haplotyping based on the analysis of the pathological variants and surrounding SNP information, and 68.75% of the embryos were estimated without information from the affected individuals. Tsuiko et al. performed long-read sequencing using Sequel I or Nanopore sequencers to sequence amplicons of 5-10 kb spanning the targeted region in couples where one partner had a de novo pathogenic variant [22]. They efficiently detected informative SNPs around the variants using sequencing and demonstrated the feasibility of haplotyping based on SNPs even in de novo variants. There is a similar report from 2023 wherein the authors also utilized PromethION (https://nanoporetech.com/products/sequence/promethion), a maximum throughput model of a Nanopore sequencer, to conduct de novo PKD1 variant analysis and surrounding SNP haplotyping in the proband [23].
4.3. Preclinical workup using long-read sequencing for PGT-M conducted at our institution
4.3.1. Materials and Methods
The long-read sequencing method used for our preclinical workups in the cases shown in the present article was performed as previously reported. The libraries were prepared using a ligation-sequencing kit (SQK-LSK110) in accordance with the manufacturer's protocol (ONT). A GridION sequencer using an R9.4.1 flow cell was used. To obtain sufficient data, we washed flowcells (EXP-WSH004) and applied libraries more than twice in each flow cell in accordance with the manufacturer's protocol (ONT). The sequencing library was prepared using 1μg of gDNA from peripheral blood each extracted just before a starting sequencing. One wash and two libraries (2μg, gDNA) for the PMD case (Figure 1) and two washes and three libraries (3μg, gDNA) for the OTC deficiency case and (Figure 2) DMD case (Figure 3) were used. To perform adaptive sampling [44,45], we prepared the following FASTA files of the targeted region for enrichment from the T2T CHM13 reference to improve the outcomes of the analysis [46]. Base calling and fastq conversion were conducted using MinKNOW ver. 4.2.8. Subsequently, fastq files were used for mapping against the GRCh38/hg38 or T2T CHM13 human reference genomes using minimap2 with soft clipping for supplementary alignments [47]. The breakpoints and junction of the structural rearrangements and the informative SNPs were identified manually using IGV (Integrative Genomic Viewer) software [48].
4.3.2. Targeted long-read sequencing using a Nanopore sequencer with adaptive sampling conducted at our institution
We also have data on self-experiments conducted using a nanopore sequencer in the preclinical workup of PGT-M. These cases involved chromosomal structural abnormalities for which breakpoint detection was challenging using conventional cytogenetic testing, and we attempted structural analysis using long-read sequencing. In particular, we reported a case involving complex chromosome X structural abnormalities (Figure 1) [24]. We performed targeted sequencing on chromosome X using the adaptive sampling method [49,50] implemented in the desktop model GridION of the nanopore series (https://nanoporetech.com/gridion). Adaptive sampling is a nanopore-specific technology based on Readfish software [50] that allows the real-time selection of target sequences while sequencing a DNA library. By specifying the targets in a FASTA file, it is possible to easily perform targeted sequencing without requiring specialized library adjustments and to obtain sufficient sequencing depths for genomic structural analysis. Adaptive sampling has already demonstrated utility in the analysis of the human genome, being applied to the diagnosis of Mendelian diseases with missing pathogenic variants [44,51,52] and targeting sequences for hereditary tumors [53,54]. Furthermore, its applicability extends to non-human genomes including metagenome analysis [55], showcasing a broad range of applications.
The case shown in Figure 1 is a case of Pelizaeus–Merzbach disease (PMD) caused by a duplication of the PLP1 gene via complex genomic rearrangement, and it is the first case wherein a long read sequencer was used in the setup of PGT-M at our institute. PMD is known to frequently exhibit complex genomic rearrangements, such as a DUP-TRP/INV-DUP structure, formed through flanking segmental duplications [56,57]. As the positions of junctions vary widely, the direct confirmation of the correct junction location requires the combination of cytogenetical analysis methods and is time consuming. Despite the complex chromosomal rearrangement, we were amazed that we directly detected the breakpoints and junctions in a single sequencing run using Nanopore sequencing (Figure 1A) [24]. The sequenced reads of the folded rearrangement of Junction1 were challenging to map due to the presence of segmental duplications with over 99.9% similarity. However, the genomic structure could be readily surmised from the sequence data (Figure 1B). By designing a specific PCR method based on the sequenced data from the reads spanning Junction2, we were able to design a direct method for detecting the pathogenic variants only in the carrier mother and previous child (Figure 1C). Additionally, similar to the previous report, we attempted to employ a method that involves the simultaneous detection of structural variations or pathogenic SNVs along with the detection of surrounding SNPs for haplotyping. The previously reported methods allowed for the detection of pathogenic variants, as well as the simultaneous detection of surrounding SNPs and haplotyping, using a high-read-depth sequence with PromethION, SMRT sequencing or target amplicon sequences [19,20,21,22,23]. However, due to the exorbitant cost per sequence of high-throughput long read sequencers, routine clinical use of this approach for PGT-M is challenging. It remains unclear whether adaptive sampling using the relatively cost-effective GridION sequencer can be utilized for the preclinical workup of PGT-M.
We carried out long-read sequencing using GridION with adaptive sampling to detect pathogenic variants and surrounding SNPs for haplotyping at the same time in several cases. The first case involved Ornithine Transcarbamylase Deficiency (OTC; Figure 2). The proband's wife carried a de novo pathogenic missense variant (OTC:c.643C>T) in the OTC gene. In this case, we set the target FASTA for adaptive sampling to include the OTC gene and its surrounding region within 100kbp upstream and downstream. Ornithine transcarbamylase (OTC) deficiency is an X-linked genetic disorder affecting the urea cycle, leading to the accumulation of ammonia and causing neurological deficit [58,59], and the couple wished for PGT-M. Since the proband had no affected offspring in this case, implementing preclinical workup for haplotyping was challenging. We previously performed haplotyping of pathogenic variants using STR markers for PGT-M via trio genetic analysis of couples and their embryos in de novo cases. However, when using this method, it is difficult to haplotype the post-zygotic de novo germline mosaicism of parents precisely [12]. We believed that long-read sequencing could address challenges associated with de novo cases, as mentioned earlier, even for the adaptive sampling method. Informative reads around the pathogenic variant are shown in the figure (Figure 2A). SNP candidates distinguishing pathogenic and benign alleles for use in PGT-M were identified upstream and downstream. Validation using Sanger sequencing confirmed the existence of each SNP, and primer design was carried out in preparation for embryo testing (Figure 2B). The result of the proband's SNP haplotyping is presented in Figure 2C. This adaptive-sampling-based long-read sequencing method was expected to improve the specificity and reliability of the test compared to the STR haplotyping we traditionally employed since it allows for a more precise search for SNP haplotyping markers in the vicinity. Moreover, a preclinical workup using adaptive sampling was conducted for Duchenne muscular dystrophy (DMD), with one case shown in Figure 3. In this case, we set the target FASTA for adaptive sampling to include the DMD gene and its surrounding region within 5kbp upstream and downstream. Although an exon2-44 deletion was confirmed via MLPA in this case, the detailed genomic position of the deletion region was unclear, making the direct detection of the pathogenic variant via PGT-M challenging. Multiple attempts were made using long-range PCR, but the wide intron region made it difficult to identify the junction. The identified deletion and surrounding genomic regions are shown in Figure 3A, where a decrease in read depth in the deletion region is evident in IGV view. The observation of discordant reads (Figure 3B,C) facilitated the easy identification of junctions spanning the deletion and surrounding SNPs, allowing for simultaneous haplotyping (Figure 3D) and structural analysis. We believe that a preclinical workup for PGT-M using adaptive sampling with GridION, as well as high-throughput sequencing with PromethION or a Sequel system, is also feasible.
In the implementation of PGT-M in Japan, both the direct detection of pathogenic variants and the haplotyping of pathogenic variants using informative STRs or SNPs are required for each case. This is because the number of cells obtained via biopsy is very low, typically 5-10 cells, and the process of whole-genome amplification is necessary [5,7,12]. In whole-genome amplification, it is necessary to consider allele dropout and amplification bias, and relying on a single marker can lead to misdiagnosis. Therefore, a combination of variant detection methods is employed to ensure the accuracy of PGT-M testing. However, as indicated in the guidelines issued by the ESHRE [12], many countries and facilities also permit embryo determination based solely on haplotyping with some additional conditions. In our self-experiments, we encountered a case with complex chromosomal rearrangements for which junction detection was difficult, and even haplotyping using STR markers was challenging. In this case, an intrachromosomal insertion had occurred, and the duplicated region involving MECP2 was inserted 45 Mb proximal to the original position [24]. If PGT-M was performed only via STR haplotyping at the original MECP2 site in this case, it may have led to misdiagnosis due to meiotic recombination. A preclinical workup of PGT-M using long-read sequencing may help minimize the risk of such a misdiagnosis.
4.3.3. Figures of cytogenetical analysis with long-read sequencing and SNP haplotyping
Figure 1.
Structural analysis of chromosome X complex rearrangement (Pelizaeus–Merzbacher disease) using long-read sequencing (Case 1) [24]. A. Result of mapping the sequence reads in IGV software view. Collating discordant reads revealed multiple sequence reads spanning across Junction2. (Genomic alteration is based on GRCh38.). B. Schematic representation of the inferred genomic structure around the PLP1 gene. The DUP-TRP/INV-DUP structure involving flanking segmental duplications is complex, but it is a typical genomic rearrangement observed in Pelizaeus–Merzbacher disease or MECP2 duplication syndrome. C. Results of junction-specific PCR designed based on read information spanning Junction2. PCR products were only detected in the mother and proband; these products can be used to detect a pathogenic variant directly.
Figure 1.
Structural analysis of chromosome X complex rearrangement (Pelizaeus–Merzbacher disease) using long-read sequencing (Case 1) [24]. A. Result of mapping the sequence reads in IGV software view. Collating discordant reads revealed multiple sequence reads spanning across Junction2. (Genomic alteration is based on GRCh38.). B. Schematic representation of the inferred genomic structure around the PLP1 gene. The DUP-TRP/INV-DUP structure involving flanking segmental duplications is complex, but it is a typical genomic rearrangement observed in Pelizaeus–Merzbacher disease or MECP2 duplication syndrome. C. Results of junction-specific PCR designed based on read information spanning Junction2. PCR products were only detected in the mother and proband; these products can be used to detect a pathogenic variant directly.
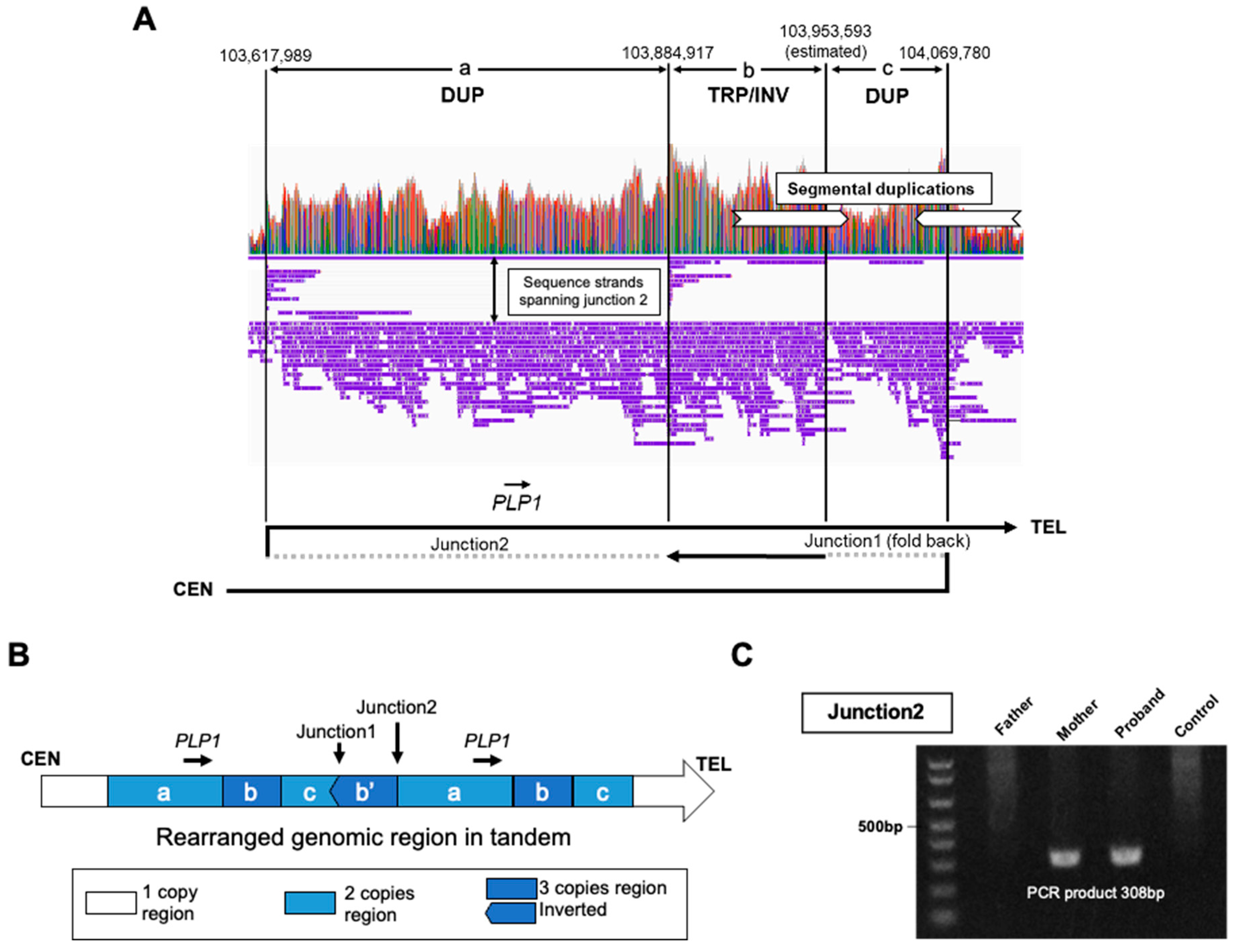
Figure 2.
SNP haplotyping of de novo pathogenic variant of OTC gene using long-read sequencing (Case 2). A. Informative SNPs around the causative pathological variant (OTC:c.643>T(p.Leu215Phe)). Haplotyping was possible without trio sequencing via confirming SNPs on the same sequence reads with a pathogenic variant. (Genomic alteration is based on CHM13.). B. Confirmation of each informative SNP through Sanger sequencing. It was determined that the read depth of adaptive sampling was insufficient for the determination of SNPs, and confirmation via Sanger sequencing is essential for preclinical workup. C. SNP haplotyping around the pathogenic variant. Three informative SNPs were identified upstream and downstream of the causative pathogenic variant in the OTC gene, respectively.
Figure 2.
SNP haplotyping of de novo pathogenic variant of OTC gene using long-read sequencing (Case 2). A. Informative SNPs around the causative pathological variant (OTC:c.643>T(p.Leu215Phe)). Haplotyping was possible without trio sequencing via confirming SNPs on the same sequence reads with a pathogenic variant. (Genomic alteration is based on CHM13.). B. Confirmation of each informative SNP through Sanger sequencing. It was determined that the read depth of adaptive sampling was insufficient for the determination of SNPs, and confirmation via Sanger sequencing is essential for preclinical workup. C. SNP haplotyping around the pathogenic variant. Three informative SNPs were identified upstream and downstream of the causative pathogenic variant in the OTC gene, respectively.
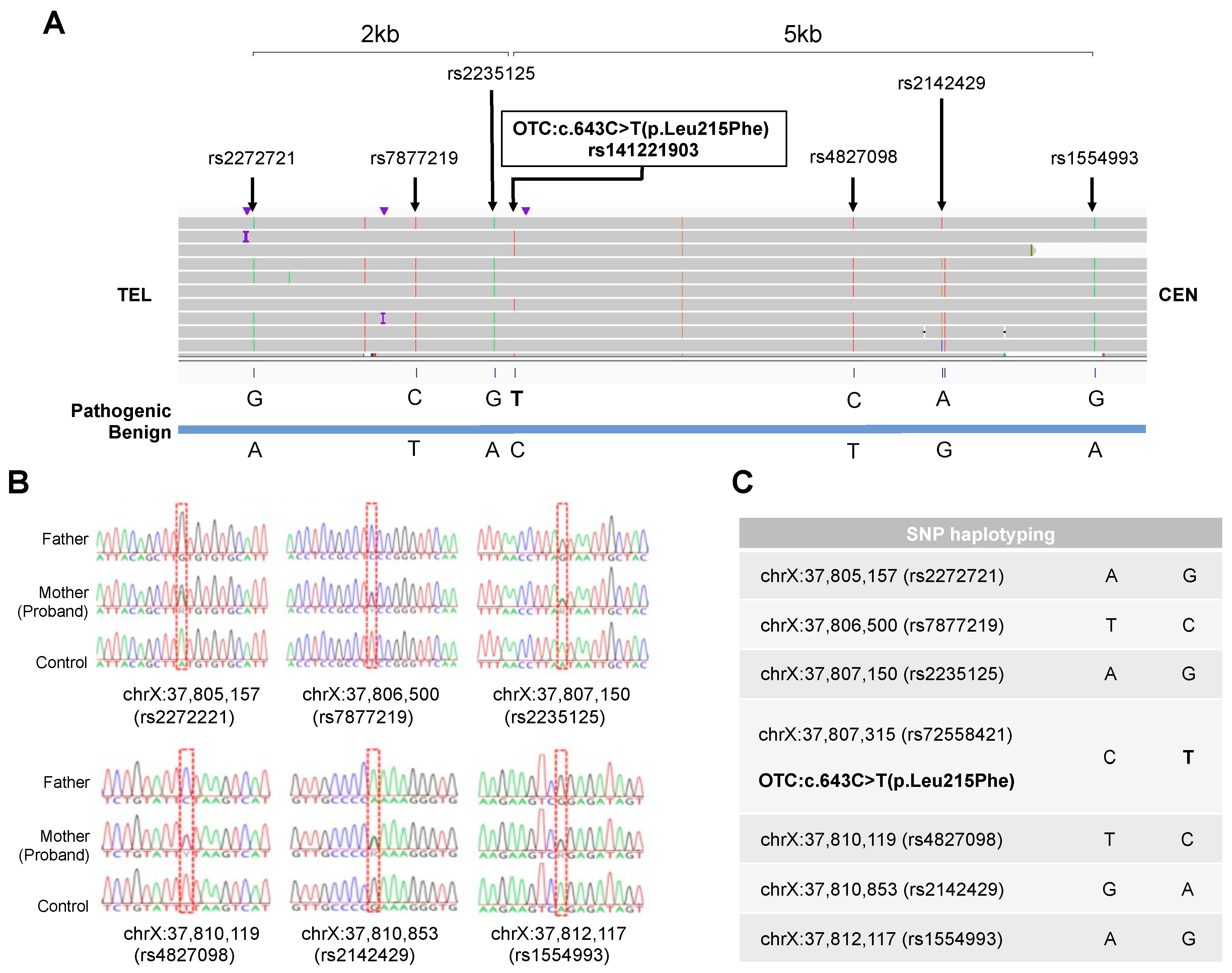
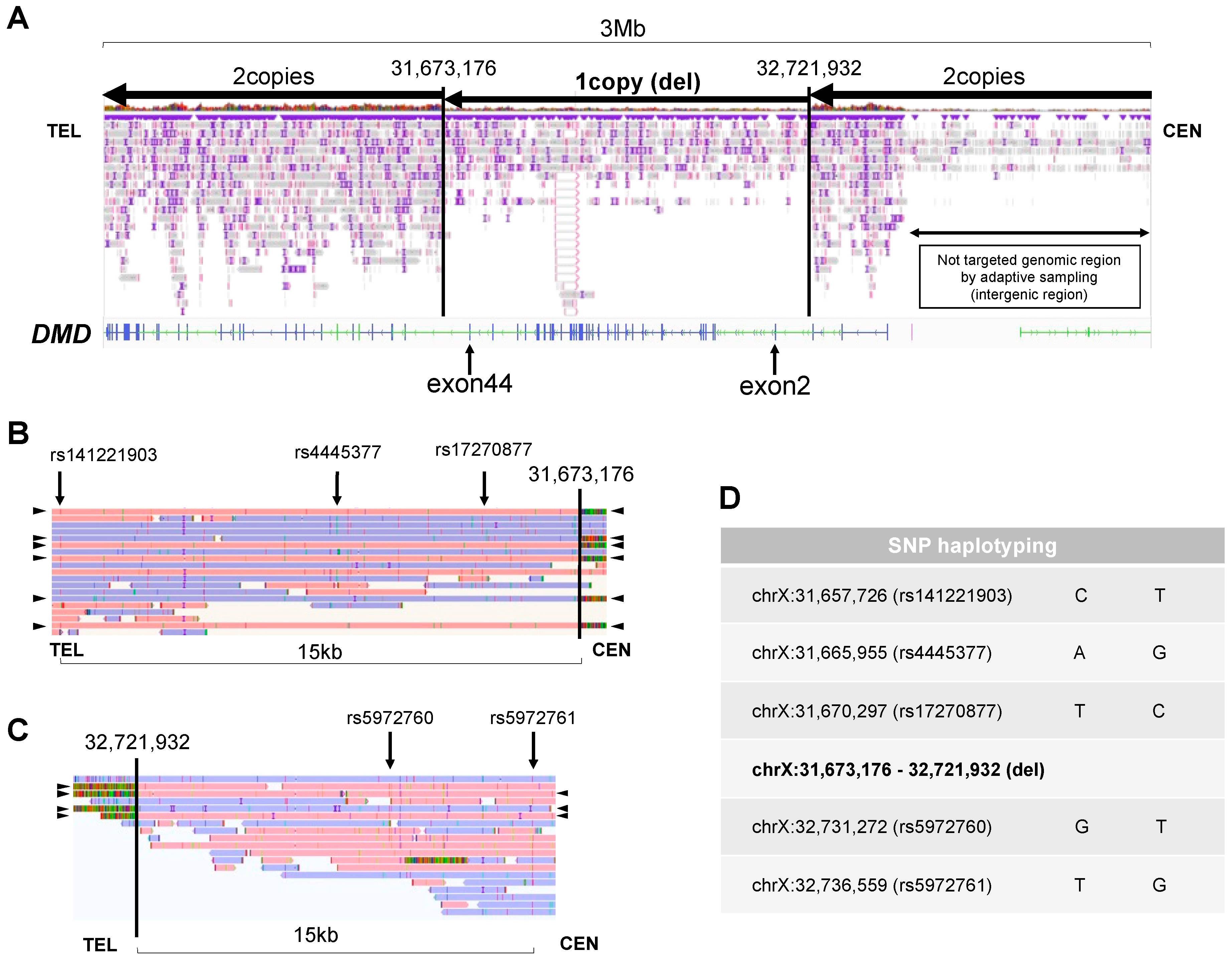
Figure 3.
Analysis of the deletion region within the DMD gene and surrounding SNPs (Case 3). A. While it was confirmed through MLPA that the region up to exon 2-44 was heterozygous deleted, long-read sequencing allowed for the precise delineation of the deleted region. Additionally, areas not targeted by intergenic adaptive sampling showed a significant decrease in read depth (the targeted region expanded by 5kbp around the regions up- and downstream of the DMD gene), indicating the effectiveness of target enrichment. (Genomic alteration is based on CHM13.). B-C. The detection status of SNPs on the telomeric side (B) and centromeric side (C) of the deletion region is presented. Informative SNPs were detected within 15 kbp in both cases, and the breakpoint is clearly indicated by discordant reads (black triangles). Structural abnormalities and haplotyping are feasible through single sequencing analysis in adaptive sampling. D. Haplotyping based on the detected informative five SNPs around the deletion region.
Figure 3.
Analysis of the deletion region within the DMD gene and surrounding SNPs (Case 3). A. While it was confirmed through MLPA that the region up to exon 2-44 was heterozygous deleted, long-read sequencing allowed for the precise delineation of the deleted region. Additionally, areas not targeted by intergenic adaptive sampling showed a significant decrease in read depth (the targeted region expanded by 5kbp around the regions up- and downstream of the DMD gene), indicating the effectiveness of target enrichment. (Genomic alteration is based on CHM13.). B-C. The detection status of SNPs on the telomeric side (B) and centromeric side (C) of the deletion region is presented. Informative SNPs were detected within 15 kbp in both cases, and the breakpoint is clearly indicated by discordant reads (black triangles). Structural abnormalities and haplotyping are feasible through single sequencing analysis in adaptive sampling. D. Haplotyping based on the detected informative five SNPs around the deletion region.
4.4. New nanopore sequencers and PGT-A with the STORK method
Nanopore sequencers theoretically have the ability to sequence long DNA libraries without length limitations, and reports have shown ultra-long sequences of up to 4.15 Mb per read [60]. However, there has been recent interest in utilizing Nanopore sequencers as high-throughput short-read sequencers. Long read sequencers are generally considered to have lower sequencing accuracy compared to short-read sequencers. Although the latest flow cell, R10.4, used in nanopore sequencers can achieve Q20+ (≥99%) accuracy, it is not sufficient for the accurate confirmation of single-nucleotide variations (SNVs). To confirm the existence of SNVs without relying on combination with methods such as Sanger sequencing, it is essential to use a high-throughput model of a Nanopore sequencer, namely, PromethION [61].
So, what does it mean to use nanopore sequencers as short-read sequencers? Wei et al. proposed rapid preimplantation genetic testing for aneuploidy (PGT-A) using nanopore sequencers in 2018 [62] and released the completed version, the short-read transpore rapid-karyotyping (STORK) method, in 2022 [31]. The setup using Nanopore sequencers, particularly MinION or Flongle, offers the advantage of low device cost, but it has limited readable sequence read numbers compared to short-read sequencers, resulting in limited accuracy for detecting copy number variations (CNVs). To address this issue, the cited authors performed library adjustments on short DNA molecules of up to 500 bp via sonication of DNA samples and size selection. In other words, they intentionally utilized the Nanopore system as a short-read sequencer to improve the utilization efficiency of each pore per read, creating a high-throughput and easily set up system. They demonstrated that chromosome aneuploidy determination in embryos could be achieved in approximately 4-5 hours, even with the addition of the whole-genome amplification process. They also demonstrated high-precision and rapid determination of chromosome aneuploidy in chorionic villus and amniotic fluid samples. Similar methods also have been employed in the field of cancer research, wherein Baslan et al. demonstrated an efficient method for calling Copy Number Alterations (CNAs) using nanopore sequencers in 2021 [63]. Using Nanopore sequencers as a powerful genomic analysis tool for point-of-care Testing applications is considered a potential breakthrough in rapid determination for preimplantation and prenatal diagnosis.
5. Conclusion
In conclusion, long read sequencers are useful tools for analyzing genomic structural variations. Furthermore, they can be used for the preclinical workup of PGT-M/SR as well as for detecting pathogenic SNVs or SVs in embryos, including haplotyping around causative variants. Moreover, the novel approach of using Nanopore sequencers as high-throughput short-read sequencers for CNV detection has a wide range of applications, and the ease with which it can be setup holds great promise for future developments in clinical settings. With further technological advancements, it is anticipated that long read sequencers will become a standard genomic analysis platform that offers higher accuracy and cost-effectiveness, bringing us closer to their routine use in clinical diagnostics.
Author Contributions
Conceptualization, H.K.; methodology, T.M.; software, H.I., Y.S.; resources, Y.M.; data curation, T.M.; writing—original draft preparation, T.M.; writing—review and editing, H.K.; visualization, T.M., Y.S.; supervision, H.K.; funding acquisition, H.K. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology (17H03616 to H.K.); from the Ministry of Health, Welfare, and Labor (H30-Nan-Ippann-012 to H.K.), Japan; and from the Japan Agency for Medical Research and Development (18ek0109301s2201 to H.K.; 23gn0110073s0101 to H.K. and T.M).
Institutional Review Board Statement
This study was reviewed and approved by the Ethics Committee of Fujita Health University.
Informed Consent Statement
DNA samples were collected from the study patients with PMD, DMD, and OTC deficiency and from their family members after obtaining written informed consent.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to the inclusion of individual genomic information.
Acknowledgments
We thank the patients and their families for agreeing to participate in our investigation.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Logsdon, G. A., M. R. Vollger, and E. E. Eichler. "Long-Read Human Genome Sequencing and Its Applications." Nat Rev Genet 21, no. 10 (2020): 597-614. [CrossRef]
- Zhao, X., R. L. Collins, W. P. Lee, A. M. Weber, Y. Jun, Q. Zhu, B. Weisburd, Y. Huang, P. A. Audano, H. Wang, M. Walker, C. Lowther, J. Fu, Consortium Human Genome Structural Variation, M. B. Gerstein, S. E. Devine, T. Marschall, J. O. Korbel, E. E. Eichler, M. J. P. Chaisson, C. Lee, R. E. Mills, H. Brand, and M. E. Talkowski. "Expectations and Blind Spots for Structural Variation Detection from Long-Read Assemblies and Short-Read Genome Sequencing Technologies." Am J Hum Genet (2021). [CrossRef]
- Dremsek, P., T. Schwarz, B. Weil, A. Malashka, F. Laccone, and J. Neesen. "Optical Genome Mapping in Routine Human Genetic Diagnostics-Its Advantages and Limitations." Genes (Basel) 12, no. 12 (2021).
- Nurk, Sergey, Sergey Koren, Arang Rhie, Mikko Rautiainen, Andrey V. Bzikadze, Alla Mikheenko, Mitchell R. Vollger, Nicolas Altemose, Lev Uralsky, Ariel Gershman, Sergey Aganezov, Savannah J. Hoyt, Mark Diekhans, Glennis A. Logsdon, Michael Alonge, Stylianos E. Antonarakis, Matthew Borchers, Gerard G. Bouffard, Shelise Y. Brooks, Gina V. Caldas, Haoyu Cheng, Chen-Shan Chin, William Chow, Leonardo G. de Lima, Philip C. Dishuck, Richard Durbin, Tatiana Dvorkina, Ian T. Fiddes, Giulio Formenti, Robert S. Fulton, Arkarachai Fungtammasan, Erik Garrison, Patrick G. S. Grady, Tina A. Graves-Lindsay, Ira M. Hall, Nancy F. Hansen, Gabrielle A. Hartley, Marina Haukness, Kerstin Howe, Michael W. Hunkapiller, Chirag Jain, Miten Jain, Erich D. Jarvis, Peter Kerpedjiev, Melanie Kirsche, Mikhail Kolmogorov, Jonas Korlach, Milinn Kremitzki, Heng Li, Valerie V. Maduro, Tobias Marschall, Ann M. McCartney, Jennifer McDaniel, Danny E. Miller, James C. Mullikin, Eugene W. Myers, Nathan D. Olson, Benedict Paten, Paul Peluso, Pavel A. Pevzner, David Porubsky, Tamara Potapova, Evgeny I. Rogaev, Jeffrey A. Rosenfeld, Steven L. Salzberg, Valerie A. Schneider, Fritz J. Sedlazeck, Kishwar Shafin, Colin J. Shew, Alaina Shumate, Yumi Sims, Arian F. A. Smit, Daniela C. Soto, Ivan Sović, Jessica M. Storer, Aaron Streets, Beth A. Sullivan, Françoise Thibaud-Nissen, James Torrance, Justin Wagner, Brian P. Walenz, Aaron Wenger, Jonathan M. D. Wood, Chunlin Xiao, Stephanie M. Yan, Alice C. Young, Samantha Zarate, Urvashi Surti, Rajiv C. McCoy, Megan Y. Dennis, Ivan A. Alexandrov, Jennifer L. Gerton, Rachel J. O’Neill, Winston Timp, Justin M. Zook, Michael C. Schatz, Evan E. Eichler, Karen H. Miga, and Adam M. Phillippy. "The Complete Sequence of a Human Genome." (2021). [CrossRef]
- Vermeesch, J. R., T. Voet, and K. Devriendt. "Prenatal and Pre-Implantation Genetic Diagnosis." Nat Rev Genet 17, no. 10 (2016): 643-56. [CrossRef]
- Zhou, X., Y. Xu, L. Zhu, Z. Su, X. Han, Z. Zhang, Y. Huang, and Q. Liu. "Comparison of Multiple Displacement Amplification (Mda) and Multiple Annealing and Looping-Based Amplification Cycles (Malbac) in Limited DNA Sequencing Based on Tube and Droplet." Micromachines (Basel) 11, no. 7 (2020).
- Li, N., L. Wang, H. Wang, M. Ma, X. Wang, Y. Li, W. Zhang, J. Zhang, D. S. Cram, and Y. Yao. "The Performance of Whole Genome Amplification Methods and Next-Generation Sequencing for Pre-Implantation Genetic Diagnosis of Chromosomal Abnormalities." J Genet Genomics 42, no. 4 (2015): 151-9. [CrossRef]
- Kurahashi, H., T. Kato, J. Miyazaki, H. Nishizawa, E. Nishio, H. Furukawa, H. Miyamura, M. Ito, T. Endo, Y. Ouchi, H. Inagaki, and T. Fujii. "Preimplantation Genetic Diagnosis/Screening by Comprehensive Molecular Testing." Reprod Med Biol 15, no. 1 (2016): 13-19. [CrossRef]
- Eijk-Van Os, P. G., and J. P. Schouten. "Multiplex Ligation-Dependent Probe Amplification (Mlpa®) for the Detection of Copy Number Variation in Genomic Sequences." Methods Mol Biol 688 (2011): 97-126.
- Nakano, Tatsuya, Michiko Ammae, Manabu Satoh, Satoshi Mizuno, Yoshiharu Nakaoka, and Yoshiharu Morimoto. "Analysis of Clinical Outcomes and Meiotic Segregation Modes Following Preimplantation Genetic Testing for Structural Rearrangements Using Acgh/Ngs in Couples with Balanced Chromosome Rearrangement." Reproductive Medicine and Biology 21, no. 1 (2022). [CrossRef]
- Iwasa, T., A. Kuwahara, T. Takeshita, Y. Taniguchi, M. Mikami, and M. Irahara. "Preimplantation Genetic Testing for Aneuploidy and Chromosomal Structural Rearrangement: A Summary of a Nationwide Study by the Japan Society of Obstetrics and Gynecology." Reprod Med Biol 22, no. 1 (2023): e12518. [CrossRef]
- Committee, Eshre Pgt Consortium Steering, F. Carvalho, E. Coonen, V. Goossens, G. Kokkali, C. Rubio, M. Meijer-Hoogeveen, C. Moutou, N. Vermeulen, and M. De Rycke. "Eshre Pgt Consortium Good Practice Recommendations for the Organisation of Pgt." Hum Reprod Open 2020, no. 3 (2020): hoaa021. [CrossRef]
- Zhang, S., F. Liang, C. Lei, J. Wu, J. Fu, Q. Yang, X. Luo, G. Yu, D. Wang, Y. Zhang, D. Lu, X. Sun, Y. Liang, and C. Xu. "Long-Read Sequencing and Haplotype Linkage Analysis Enabled Preimplantation Genetic Testing for Patients Carrying Pathogenic Inversions." J Med Genet 56, no. 11 (2019): 741-49. [CrossRef]
- Chow, J. F. C., H. H. Y. Cheng, E. Y. L. Lau, W. S. B. Yeung, and E. H. Y. Ng. "Distinguishing between Carrier and Noncarrier Embryos with the Use of Long-Read Sequencing in Preimplantation Genetic Testing for Reciprocal Translocations." Genomics 112, no. 1 (2020): 494-500. [CrossRef]
- Liu, S., H. Wang, D. Leigh, D. S. Cram, L. Wang, and Y. Yao. "Third-Generation Sequencing: Any Future Opportunities for Pgt?" J Assist Reprod Genet 38, no. 2 (2021): 357-64.
- M, M. Yc, Q. Yu, M. Ma, H. Wang, S. Tian, W. Zhang, M. Jz M, Y. Liu, Q. Yang, X. Pan, H. Liang, L. Wang, D. Leigh, D. S. Cram, and Y. Yao. "Variant Haplophasing by Long-Read Sequencing: A New Approach to Preimplantation Genetic Testing Workups." Fertil Steril 116, no. 3 (2021): 774-83. [CrossRef]
- Xu, J., Z. Zhang, W. Niu, Q. Yang, G. Yao, S. Shi, H. Jin, W. Song, L. Chen, X. Zhang, Y. Guo, Y. Su, L. Hu, J. Zhai, Y. Zhang, F. Dong, Y. Gao, W. Li, S. Bo, M. Hu, J. Ren, L. Huang, S. Lu, X. S. Xie, and Y. Sun. "Mapping Allele with Resolved Carrier Status of Robertsonian and Reciprocal Translocation in Human Preimplantation Embryos." Proc Natl Acad Sci U S A 114, no. 41 (2017): E8695-E702. [CrossRef]
- Xia, Q., S. Li, T. Ding, Z. Liu, J. Liu, Y. Li, H. Zhu, and Z. Yao. "Nanopore Sequencing for Detecting Reciprocal Translocation Carrier Status in Preimplantation Genetic Testing." BMC Genomics 24, no. 1 (2023): 1. [CrossRef]
- Miao, H., J. Zhou, Q. Yang, F. Liang, D. Wang, N. Ma, B. Gao, J. Du, G. Lin, K. Wang, and Q. Zhang. "Long-Read Sequencing Identified a Causal Structural Variant in an Exome-Negative Case and Enabled Preimplantation Genetic Diagnosis." Hereditas 155 (2018): 32. [CrossRef]
- Watson, C. M., D. L. Holliday, L. A. Crinnion, and D. T. Bonthron. "Long-Read Nanopore DNA Sequencing Can Resolve Complex Intragenic Duplication/Deletion Variants, Providing Information to Enable Preimplantation Genetic Diagnosis." Prenat Diagn 42, no. 2 (2022): 226-32.
- Wu, Haitao, Dongjia Chen, Qiang Zhao, Xiaoting Shen, Yongbin Liao, Ping Li, Philip C. N. Chiu, and Canquan Zhou. "Long-Read Sequencing on the Smrt Platform Enables Efficient Haplotype Linkage Analysis in Preimplantation Genetic Testing for Β-Thalassemia." Journal of Assisted Reproduction and Genetics (2022). [CrossRef]
- Tsuiko, O., Y. El Ayeb, T. Jatsenko, J. Allemeersch, C. Melotte, J. Ding, S. Debrock, K. Peeraer, A. Vanhie, A. De Leener, C. Pirard, C. Kluyskens, E. Denayer, E. Legius, J. R. Vermeesch, H. Brems, and E. Dimitriadou. "Preclinical Workup Using Long-Read Amplicon Sequencing Provides Families with De Novo Pathogenic Variants Access to Universal Preimplantation Genetic Testing." Hum Reprod (2023).
- Peng, C., H. Chen, J. Ren, F. Zhou, Y. Li, Y. Keqie, T. Ding, J. Ruan, H. Wang, X. Chen, and S. Liu. "A Long-Read Sequencing and Snp Haplotype-Based Novel Preimplantation Genetic Testing Method for Female Adpkd Patient with De Novo Pkd1 Mutation." BMC Genomics 24, no. 1 (2023): 521. [CrossRef]
- Mariya, T., Y. Shichiri, T. Sugimoto, R. Kawamura, S. Miyai, H. Inagaki, E. Sugihara, K. Ikeda, T. Baba, A. Ishikawa, M. Ammae, Y. Nakaoka, T. Saito, A. Sakurai, and H. Kurahashi. "Clinical Application of Long-Read Nanopore Sequencing in a Preimplantation Genetic Testing Pre-Clinical Workup to Identify the Junction for Complex Xq Chromosome Rearrangement-Related Disease." Prenat Diagn (2023). [CrossRef]
- Neal, S. A., S. J. Morin, J. M. Franasiak, L. R. Goodman, C. R. Juneau, E. J. Forman, M. D. Werner, and R. T. Scott, Jr. "Preimplantation Genetic Testing for Aneuploidy Is Cost-Effective, Shortens Treatment Time, and Reduces the Risk of Failed Embryo Transfer and Clinical Miscarriage." Fertil Steril 110, no. 5 (2018): 896-904. [CrossRef]
- Franasiak, J. M., E. J. Forman, K. H. Hong, M. D. Werner, K. M. Upham, N. R. Treff, and R. T. Scott, Jr. "The Nature of Aneuploidy with Increasing Age of the Female Partner: A Review of 15,169 Consecutive Trophectoderm Biopsies Evaluated with Comprehensive Chromosomal Screening." Fertil Steril 101, no. 3 (2014): 656-63 e1. [CrossRef]
- Reig, A., J. Franasiak, R. T. Scott, Jr., and E. Seli. "The Impact of Age Beyond Ploidy: Outcome Data from 8175 Euploid Single Embryo Transfers." J Assist Reprod Genet 37, no. 3 (2020): 595-602.
- Yan, Junhao, Yingying Qin, Han Zhao, Yun Sun, Fei Gong, Rong Li, Xiaoxi Sun, Xiufeng Ling, Hong Li, Cuifang Hao, Jichun Tan, Jing Yang, Yimin Zhu, Fenghua Liu, Dawei Chen, Daimin Wei, Juanjuan Lu, Tianxiang Ni, Wei Zhou, Keliang Wu, Yuan Gao, Yuhua Shi, Yao Lu, Ting Zhang, Wei Wu, Xiang Ma, Hailan Ma, Jing Fu, Junqiang Zhang, Qingxia Meng, Heping Zhang, Richard S. Legro, and Zi-Jiang Chen. "Live Birth with or without Preimplantation Genetic Testing for Aneuploidy." New England Journal of Medicine 385, no. 22 (2021): 2047-58. [CrossRef]
- Munné, S., B. Kaplan, J. L. Frattarelli, T. Child, G. Nakhuda, F. N. Shamma, K. Silverberg, T. Kalista, A. H. Handyside, M. Katz-Jaffe, D. Wells, T. Gordon, S. Stock-Myer, and S. Willman. "Preimplantation Genetic Testing for Aneuploidy Versus Morphology as Selection Criteria for Single Frozen-Thawed Embryo Transfer in Good-Prognosis Patients: A Multicenter Randomized Clinical Trial." Fertil Steril 112, no. 6 (2019): 1071-79.e7. [CrossRef]
- Sato, T., M. Sugiura-Ogasawara, F. Ozawa, T. Yamamoto, T. Kato, H. Kurahashi, T. Kuroda, N. Aoyama, K. Kato, R. Kobayashi, A. Fukuda, T. Utsunomiya, A. Kuwahara, H. Saito, T. Takeshita, and M. Irahara. "Preimplantation Genetic Testing for Aneuploidy: A Comparison of Live Birth Rates in Patients with Recurrent Pregnancy Loss Due to Embryonic Aneuploidy or Recurrent Implantation Failure." Hum Reprod 34, no. 12 (2019): 2340-48. [CrossRef]
- Wei, Shan, Alexandre Djandji, Miriam T. Lattin, Odelia Nahum, Nataly Hoffman, Claudia Cujar, Refik Kayali, Cengiz Cinnioglu, Ronald Wapner, Mary D’Alton, Brynn Levy, and Zev Williams. "Rapid Nanopore Sequencing–Based Screen for Aneuploidy in Reproductive Care." New England Journal of Medicine 387, no. 7 (2022): 658-60. [CrossRef]
- Chin, C. S., P. Peluso, F. J. Sedlazeck, M. Nattestad, G. T. Concepcion, A. Clum, C. Dunn, R. O'Malley, R. Figueroa-Balderas, A. Morales-Cruz, G. R. Cramer, M. Delledonne, C. Luo, J. R. Ecker, D. Cantu, D. R. Rank, and M. C. Schatz. "Phased Diploid Genome Assembly with Single-Molecule Real-Time Sequencing." Nat Methods 13, no. 12 (2016): 1050-54. [CrossRef]
- Wang, Y., Y. Zhao, A. Bollas, Y. Wang, and K. F. Au. "Nanopore Sequencing Technology, Bioinformatics and Applications." Nat Biotechnol 39, no. 11 (2021): 1348-65.
- Parker, J., A. J. Helmstetter, D. Devey, T. Wilkinson, and A. S. T. Papadopulos. "Field-Based Species Identification of Closely-Related Plants Using Real-Time Nanopore Sequencing." Sci Rep 7, no. 1 (2017): 8345. [CrossRef]
- Castro-Wallace, S. L., C. Y. Chiu, K. K. John, S. E. Stahl, K. H. Rubins, A. B. R. McIntyre, J. P. Dworkin, M. L. Lupisella, D. J. Smith, D. J. Botkin, T. A. Stephenson, S. Juul, D. J. Turner, F. Izquierdo, S. Federman, D. Stryke, S. Somasekar, N. Alexander, G. Yu, C. E. Mason, and A. S. Burton. "Nanopore DNA Sequencing and Genome Assembly on the International Space Station." Sci Rep 7, no. 1 (2017): 18022. [CrossRef]
- Greninger, A. L., S. N. Naccache, S. Federman, G. Yu, P. Mbala, V. Bres, D. Stryke, J. Bouquet, S. Somasekar, J. M. Linnen, R. Dodd, P. Mulembakani, B. S. Schneider, J. J. Muyembe-Tamfum, S. L. Stramer, and C. Y. Chiu. "Rapid Metagenomic Identification of Viral Pathogens in Clinical Samples by Real-Time Nanopore Sequencing Analysis." Genome Med 7 (2015): 99. [CrossRef]
- Sun, X., L. Song, W. Yang, L. Zhang, M. Liu, X. Li, G. Tian, and W. Wang. "Nanopore Sequencing and Its Clinical Applications." Methods Mol Biol 2204 (2020): 13-32.
- Dhesi, Z., V. I. Enne, J. O'Grady, V. Gant, and D. M. Livermore. "Rapid and Point-of-Care Testing in Respiratory Tract Infections: An Antibiotic Guardian?" ACS Pharmacol Transl Sci 3, no. 3 (2020): 401-17.
- Lastra, L. S., V. Sharma, N. Farajpour, M. Nguyen, and K. J. Freedman. "Nanodiagnostics: A Review of the Medical Capabilities of Nanopores." Nanomedicine 37 (2021): 102425. [CrossRef]
- Wick, R. R., L. M. Judd, and K. E. Holt. "Performance of Neural Network Basecalling Tools for Oxford Nanopore Sequencing." Genome Biol 20, no. 1 (2019): 129. [CrossRef]
- Lai, H. H., T. H. Chuang, L. K. Wong, M. J. Lee, C. L. Hsieh, H. L. Wang, and S. U. Chen. "Identification of Mosaic and Segmental Aneuploidies by Next-Generation Sequencing in Preimplantation Genetic Screening Can Improve Clinical Outcomes Compared to Array-Comparative Genomic Hybridization." Mol Cytogenet 10 (2017): 14. [CrossRef]
- Fiorentino, F., S. Bono, A. Biricik, A. Nuccitelli, E. Cotroneo, G. Cottone, F. Kokocinski, C. E. Michel, M. G. Minasi, and E. Greco. "Application of Next-Generation Sequencing Technology for Comprehensive Aneuploidy Screening of Blastocysts in Clinical Preimplantation Genetic Screening Cycles." Hum Reprod 29, no. 12 (2014): 2802-13. [CrossRef]
- Shetty, Sachin, Jiny Nair, Jnapti Johnson, Navya Shetty, Ajay Kumar J, Nirmala Thondehalmath, Deepanjali Ganesh, Vidyalakshmi R. Bhat, Sajana M, Anjana R, Rajsekhar Nayak, Devika Gunasheela, Jayarama S. Kadandale, and Swathi Shetty. "Preimplantation Genetic Testing for Couples with Balanced Chromosomal Rearrangements." Journal of Reproduction & Infertility (2022). [CrossRef]
- Miller, D. E., A. Sulovari, T. Wang, H. Loucks, K. Hoekzema, K. M. Munson, A. P. Lewis, E. P. A. Fuerte, C. R. Paschal, T. Walsh, J. Thies, J. T. Bennett, I. Glass, K. M. Dipple, K. Patterson, E. S. Bonkowski, Z. Nelson, A. Squire, M. Sikes, E. Beckman, R. L. Bennett, D. Earl, W. Lee, R. Allikmets, S. J. Perlman, P. Chow, A. V. Hing, T. L. Wenger, M. P. Adam, A. Sun, C. Lam, I. Chang, X. Zou, S. L. Austin, E. Huggins, A. Safi, A. K. Iyengar, T. E. Reddy, W. H. Majoros, A. S. Allen, G. E. Crawford, P. S. Kishnani, Genomics University of Washington Center for Mendelian, M. C. King, T. Cherry, J. X. Chong, M. J. Bamshad, D. A. Nickerson, H. C. Mefford, D. Doherty, and E. E. Eichler. "Targeted Long-Read Sequencing Identifies Missing Disease-Causing Variation." Am J Hum Genet (2021). [CrossRef]
- Mariya, Tasuku, Takema Kato, Takeshi Sugimoto, Syunsuke Miyai, Hidehito Inagaki, Tamae Ohye, Eiji Sugihara, Yukako Muramatsu, Seiji Mizuno, and Hiroki Kurahashi. "Target Enrichment Long-Read Sequencing with Adaptive Sampling Can Determine the Structure of the Small Supernumerary Marker Chromosomes." Journal of Human Genetics (2022). [CrossRef]
- Aganezov, S., S. M. Yan, D. C. Soto, M. Kirsche, S. Zarate, P. Avdeyev, D. J. Taylor, K. Shafin, A. Shumate, C. Xiao, J. Wagner, J. McDaniel, N. D. Olson, M. E. G. Sauria, M. R. Vollger, A. Rhie, M. Meredith, S. Martin, J. Lee, S. Koren, J. A. Rosenfeld, B. Paten, R. Layer, C. S. Chin, F. J. Sedlazeck, N. F. Hansen, D. E. Miller, A. M. Phillippy, K. H. Miga, R. C. McCoy, M. Y. Dennis, J. M. Zook, and M. C. Schatz. "A Complete Reference Genome Improves Analysis of Human Genetic Variation." Science 376, no. 6588 (2022): eabl3533. [CrossRef]
- Li, H. "Minimap2: Pairwise Alignment for Nucleotide Sequences." Bioinformatics 34, no. 18 (2018): 3094-100. [CrossRef]
- Thorvaldsdóttir, H., J. T. Robinson, and J. P. Mesirov. "Integrative Genomics Viewer (Igv): High-Performance Genomics Data Visualization and Exploration." Brief Bioinform 14, no. 2 (2013): 178-92. [CrossRef]
- Loose, M., S. Malla, and M. Stout. "Real-Time Selective Sequencing Using Nanopore Technology." Nat Methods 13, no. 9 (2016): 751-4. [CrossRef]
- Payne, A., N. Holmes, T. Clarke, R. Munro, B. J. Debebe, and M. Loose. "Readfish Enables Targeted Nanopore Sequencing of Gigabase-Sized Genomes." Nat Biotechnol 39, no. 4 (2021): 442-50. [CrossRef]
- Mastrorosa, F. K., D. E. Miller, and E. E. Eichler. "Applications of Long-Read Sequencing to Mendelian Genetics." Genome Med 15, no. 1 (2023): 42. [CrossRef]
- Miller, D. E., L. Lee, M. Galey, R. Kandhaya-Pillai, M. Tischkowitz, D. Amalnath, A. Vithlani, K. Yokote, H. Kato, Y. Maezawa, A. Takada-Watanabe, M. Takemoto, G. M. Martin, E. E. Eichler, F. M. Hisama, and J. Oshima. "Targeted Long-Read Sequencing Identifies Missing Pathogenic Variants in Unsolved Werner Syndrome Cases." J Med Genet (2022). [CrossRef]
- Yamaguchi, K., R. Kasajima, K. Takane, S. Hatakeyama, E. Shimizu, R. Yamaguchi, K. Katayama, M. Arai, C. Ishioka, T. Iwama, S. Kaneko, N. Matsubara, Y. Moriya, T. Nomizu, K. Sugano, K. Tamura, N. Tomita, T. Yoshida, K. Sugihara, Y. Nakamura, S. Miyano, S. Imoto, Y. Furukawa, and T. Ikenoue. "Application of Targeted Nanopore Sequencing for the Screening and Determination of Structural Variants in Patients with Lynch Syndrome." J Hum Genet (2021). [CrossRef]
- Filser, M., M. Schwartz, K. Merchadou, A. Hamza, M. C. Villy, A. Decees, E. Frouin, E. Girard, S. M. Caputo, V. Renault, V. Becette, L. Golmard, N. Servant, D. Stoppa-Lyonnet, O. Delattre, C. Colas, and J. Masliah-Planchon. "Adaptive Nanopore Sequencing to Determine Pathogenicity of Brca1 Exonic Duplication." J Med Genet 60, no. 12 (2023): 1206-09. [CrossRef]
- Martin, S., D. Heavens, Y. Lan, S. Horsfield, M. D. Clark, and R. M. Leggett. "Nanopore Adaptive Sampling: A Tool for Enrichment of Low Abundance Species in Metagenomic Samples." Genome Biol 23, no. 1 (2022): 11. [CrossRef]
- Beck, C. R., C. M. Carvalho, L. Banser, T. Gambin, D. Stubbolo, B. Yuan, K. Sperle, S. M. McCahan, M. Henneke, P. Seeman, J. Y. Garbern, G. M. Hobson, and J. R. Lupski. "Complex Genomic Rearrangements at the Plp1 Locus Include Triplication and Quadruplication." PLoS Genet 11, no. 3 (2015): e1005050. [CrossRef]
- Bahrambeigi, V., X. Song, K. Sperle, C. R. Beck, H. Hijazi, C. M. Grochowski, S. Gu, P. Seeman, K. J. Woodward, C. M. B. Carvalho, G. M. Hobson, and J. R. Lupski. "Distinct Patterns of Complex Rearrangements and a Mutational Signature of Microhomeology Are Frequently Observed in Plp1 Copy Number Gain Structural Variants." Genome Med 11, no. 1 (2019): 80. [CrossRef]
- Choi, J. H., B. H. Lee, J. H. Kim, G. H. Kim, Y. M. Kim, J. Cho, C. K. Cheon, J. M. Ko, J. H. Lee, and H. W. Yoo. "Clinical Outcomes and the Mutation Spectrum of the Otc Gene in Patients with Ornithine Transcarbamylase Deficiency." J Hum Genet 60, no. 9 (2015): 501-7. [CrossRef]
- Kido, J., K. Sugawara, T. Sawada, S. Matsumoto, and K. Nakamura. "Pathogenic Variants of Ornithine Transcarbamylase Deficiency: Nation-Wide Study in Japan and Literature Review." Front Genet 13 (2022): 952467. [CrossRef]
- "Oxford Nanopore Tech Update: New Duplex Method for Q30 Nanopore Single Molecule Reads, Promethion 2, and More." @nanopore, https://nanoporetech.com/about-us/news/oxford-nanopore-tech-update-new-duplex-method-q30-nanopore-single-molecule-reads-0 (.
- Marx, V. "Method of the Year: Long-Read Sequencing." Nat Methods 20, no. 1 (2023): 6-11. [CrossRef]
- Wei, S., Z. R. Weiss, P. Gaur, E. Forman, and Z. Williams. "Rapid Preimplantation Genetic Screening Using a Handheld, Nanopore-Based DNA Sequencer." Fertil Steril 110, no. 5 (2018): 910-16.e2.
- Baslan, T., S. Kovaka, F. J. Sedlazeck, Y. Zhang, R. Wappel, S. Tian, S. W. Lowe, S. Goodwin, and M. C. Schatz. "High Resolution Copy Number Inference in Cancer Using Short-Molecule Nanopore Sequencing." Nucleic Acids Res (2021). [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



