
Submitted:
03 January 2024
Posted:
05 January 2024
You are already at the latest version
Abstract
Phosphatase and tensin homolog (PTEN) is a tumor suppressor due to its ability to regulate cell survival, growth and proliferation by down-regulating PI3K/AKT signaling pathway. In addition, PTEN plays an essential role in other physiological events associated with cell growth demands, such as ischemia-reperfusion, nerve injury and immune response. Therefore, recently PTEN inhibition has emerged as a potential therapeutic intervention in these situations. Increasing evidence demonstrates that reactive oxygen species (ROS), especially hydrogen peroxide (H2O2), are produced and required for the signaling in many important cellular processes under such physiological conditions. ROS have been shown to oxidize PTEN at the cysteine residue of its active site, consequently inhibiting its function. Here we provide an overview of studies that highlight the role of oxidative inhibition of PTEN in physiological processes.
Keywords:
PTEN
; redox regulation
; oxidative inhibition
; ROS
; cell signaling
1. Introduction
Phosphatase and tensin homolog (PTEN) belongs to the protein tyrosine phosphatase (PTP) family and was initially identified as a tumor suppressor with a specific role in regulating cell growth. The structure of human PTEN consists of an N-terminal-phosphatidylinositol (4,5)-bisphosphate (PIP2)-binding/phosphatase catalytic domain followed by a C2-lipid-binding domain, which enables its membrane-associated function, a C-terminal tail domain and a PDZ-binding domain. The distinctive feature phosphatase function of PTEN, in comparison with other PTPs, is counteracting the activity of class I phosphoinositide 3-kinases (PI3Ks) through the dephosphorylation of phosphatidylinositol-3,4,5-triphosphate (PIP3) to PIP2 [1,2,3,4]. Through this mechanism, PTEN acts as a suppressor of the phosphoinositide 3-kinases /protein kinase B (PI3K/AKT) pathway. Since the PI3K/AKT signaling pathway promotes protein synthesis, cell survival, proliferation, and migration [5,6], PTEN dysfunction can contribute to the development of certain hereditary tumorigenesis disorders such as Cowden syndrome, Proteus syndrome, Bannayan–Riley–Ruvalcaba syndrome, and Lhermitte-Duclos disease [7], as well as various cancers including breast [8], thyroid [9], endometrium [10], prostate [11], brain [12], and skin cancer [13].
PTEN expression can be regulated through genetic, epigenetic, post-transcriptional, and post-translational mechanisms that influence the PTEN gene, mRNA, and protein [14]. Kinases such as Glycogen Synthase Kinase GSK3, Casein Kinase CK2, and Serine Threonine Kinase STK11 can inactivate PTEN by phosphorylating serine and threonine residues on the C-terminal tail region. Biperoxovanadium compounds are extensively used as specific PTEN inhibitors [5]. The elevated PI3K/AKT signaling pathway has been demonstrated to be beneficial in physiological processes that require cell regeneration. Therefore, inhibiting PTEN, a negative regulator of the PI3K/AKT pathway, has been considered a prospective therapy for neurodegenerative diseases, ischemia, infection, and insulin-resistant metabolic disorders [14].
Like other members of the PTP family that contain a cysteine residue in their active site, PTEN can undergo oxidative inactivation by reactive oxygen species (ROS) [15]. ROS are generated through endogenous sources such as NADPH oxidase (NOX), nitric oxide synthase (NOS), xanthine oxidase, aldehyde oxidase, cyclo-oxygenase, cytochrome P450 2E1 and electron leakage from mitochondria, as well as exogenous sources such as smoke, ultraviolet light, radiation and drugs [16,17]. Superoxide (O2-) can react with nitric oxide (NO) to form ONOO- or be transformed to hydrogen peroxide (H2O2) by superoxide dismutase (SOD), vitamin E, or vitamin C. Oxidative inactivation of PTEN, which can serve as a physiological regulatory mechanism is executed by ROS not only from oxidative stress but also from cellular signaling transductions, for example, growth factor stimulation-derived NOXs [18]. A growing body of evidence has indicated that ROS are produced and utilized in physiological circumstances to function as significant signaling messengers, facilitating the coordination of various fundamental processes, including inflammation, survival, proliferation, differentiation, apoptosis, signal transduction, and other critical events [19,20,21,22,23]. The ROS that have such cellular physiological functions are predominantly generated at the cell's plasma membrane and endomembrane through the activity of NOXs [24]. H2O2 is the major ROS responsible for initiating redox-dependent signaling within the cell's cytosol [25], and the source of this physiological H2O2 is also related to the activities of membrane-associated complex NOXs and specialized cells such as phagocytes [26] [27]. Lee et al. were the first to demonstrate the reversible inactivation of PTEN by H2O2. During this process, the Cys124 catalytic residue in the active site of PTEN is oxidized and forms a disulfide bond with Cys71 [28]. Furthermore, the oxidative inhibition of PTEN by H2O2 has been experimentally demonstrated to increase the PI3K/AKT signaling pathway [29]. Peroxynitrite (ONOO-) can also oxidize cysteine residues within the PTPs, leading to such oxidative inhibition. This process might be considerably faster and more effective in inactivating PTPs at lower concentrations than H2O2. This suggests that peroxynitrite may be the primary biological mediator responsible for PTPs’ inactivation, consequently enhancing tyrosine phosphorylation in situations related to oxidative stress [30]. However, the impact of peroxynitrite on phosphotyrosine-dependent signaling can manifest as either activation or inhibition. The upregulation of this signaling could arise from PTPs’ inactivation by a low concentration of peroxynitrite, and this feature has typical characteristics of cell signaling, being transient and reversible. Yet, how peroxynitrite affects the PI3K/AKT pathway is still controversial [31].
The oxidative inactivation of PTEN leads to an increase in PI3K/AKT downstream signaling, which subsequently induces its physiological effects [29,32,33]. Recently, bicarbonate/carbon dioxide (HCO3-/CO2) has emerged as a pivotal factor in promoting the oxidative reactivity of H2O2 by creating a higher reactive form called peroxymonocarbonate (HCO4-) [34,35,36]. Since there are several meticulous and comprehensive reviews about the regulators of PTEN and their impacts on the PI3K/AKT signaling pathway, as well as their implications in physiology and diseases, we focus on the role of oxidative inhibition of PTEN in physiological processes. In addition, we also mention the role of bicarbonate/carbon dioxide in the oxidation of PTPs by H2O2.
2. Oxidative inhibition of PTEN by ROS in physiological processes
2.1. Cardiovascular remodeling
Studies indicate the involvement of the serine/threonine kinase AKT as a mediator in the process of ischemic preconditioning, a short transient period of sustenance during ischemia reperfusion injury [37,38,39,40]. In ischemic preconditioning, AKT signaling is upregulated and prevents cardiomyocytes from undergoing apoptosis [39,40,41,42]. The PI3K/AKT/mTOR pathway plays a significant role in protecting against ischemia reperfusion injury, particularly in the context of ischemic preconditioning in cardiac tissue. Accordingly, reversible PTEN down-regulation has been suggested as a viable therapeutic approach to mitigate ischemia reperfusion-related cardiac damage [43]. A study revealed that PTEN plays a pivotal role in the post-myocardial infarction remodeling process: Partial PTEN inactivation, through regulating the AKT signaling pathway, can increase interleukin IL-10 and consequently decrease tumor necrosis factor TNFα and matrix metalloproteinase MMP2 expression in the heart. However, the authors were not able to determine the exact source of generated IL-10, apart from immune cells. It probably comes from endothelial cells and fibroblasts [44]. Several research studies demonstrate that IL-10 can eventually attenuate apoptosis and facilitate cardiac remodeling after myocardial infarction [45,46,47,48]. Hence, PTEN inhibition could be an effective approach for improving cardiac conservation after ischemia [49,50].
During acute myocardial infarction, the heart suffers from an oxidative stress with an increased ROS level [17]. In the acute and chronic cellular response to this event, NOX2 is overexpressed in human cardiomyocytes, which may not absolutely interfere with the activity of macrophages [51,52,53]. Since PTEN oxidation is likely to occur near the site of ROS formation and both PIP3 and the NOXs complex are located in the plasma membrane, H2O2 generated from NOXs is the primary candidate for inhibiting the PI3K/AKT pathway through PTEN oxidation. There is substantial supporting evidence indicating that elevated PIP3 signaling contributes to the activation of the NOXs complex in both phagocytic and non-phagocytic cells. The increase in PIP3 levels is proposed to be a key factor in initiating the activation of the NOXs complex [29,54]. This may create a circular impact, where ROS generated from NOXs can inhibit PTEN and enhance the PI3K/AKT pathway, which, in turn, promotes NOXs activity.
Cai and Semenza were the first to describe the modulation of PTEN during ischemia-reperfusion injury. During the first 15 minutes of ischemia, PTEN undergoes dephosphorylation and proteasomal degradation. However, the kinetics reveal that not all PTEN activity is impaired during this initial phase and the AKT phosphorylation increases without any significant changes. This indicates that the dephosphorylation and degradation do not greatly hinder PTEN function. However, in the subsequent initial phase of reperfusion, there is a notable increase in oxidized PTEN and consequently phosphorylated AKT. Their findings clarify that the surge in AKT phosphorylation during this short reperfusion period is caused by the oxidative inhibition of the remaining PTEN [55]. Simultaneously, elevated levels of ROS have been observed in both injured cardiomyocytes and intact hearts during ischemia-reperfusion events [55,56]. Therefore, the oxidation of PTEN during the initial reperfusion period is related to the concurrent rise in ROS levels. (Figure 1).
One vital mechanism of injured tissue in cases of blood supply shortage, due to ischemia or infarction events, is angiogenesis. Angiogenesis is defined as the formation of new blood vessels [57]. Vascular endothelial growth factor (VEGF) is associated with promoting angiogenesis. Upregulation of VEGF can be a potential treatment approach to induce axonal outgrowth and following angiogenesis after cerebral ischemia [58], as well as restore blood flow in ischemic tissues after myocardial infarction [59]. Experimental data reported by Connor et al. indicate that the overexpression of manganese superoxide dismutase (SOD2) increases the production of mitochondrial H2O2, which triggers angiogenic activity. In this process, mitochondrial H2O2 can oxidize PTEN and up-regulate the PI3K/AKT signaling axis, subsequently activating VEGF production [60]. (Figure 1).
2.2. Vascular constriction
Accumulating evidence highlights the significant role of PI3K/AKT-dependent signaling pathways in various fundamental cellular functions within the cardiovascular system. These functions include processes such as the maturation and growth, mechanotransduction, contractility, and proliferation and migration of both cardiac and vascular smooth muscle cells [61,62,63,64,65]. Dysfunction of this signaling pathway plays an essential role in cardiovascular pathophysiological conditions, such as heart failure, atherosclerosis, and hypertension [66,67,68,69]. Wu et al. observed that in the rostral ventrolateral medulla of spontaneously hypertensive rats, ROS originating from NOXs and mitochondrial oxidative stress reduce the catalytic ability of PTEN through oxidation. Consequently, the ensuing activation of PI3K/AKT signaling pathway may lead to neurogenic hypertension [69].
Maintaining a consistent cerebral blood flow distribution through myogenic tone development is vital for neurons, which lack glucose storage and rely solely on a continuous blood supply of glucose and oxygen for normal metabolic function and under conditions of increased demands [70]. The role PI3K in mediating the impact of physical forces, such as pressure, shearing and stretching on vascular smooth muscle cells and various other cell types, is well-recognized [71]. Gebremedhin et al. found that elevated intraluminal pressure in cerebral arteries leads to an increase in ROS generation, leading to oxidative inactivation of PTEN. This, in turn, results in an up-regulation of PI3K/AKT activity and a release of IP3. The activation of AKT can induce the inhibition of arterial calcium-activated potassium channels, membrane depolarization, and L-type calcium channel. In addition, the formation of inositol (3,4,5)-triphosphate (IP3) stimulates the sarcoplasmic reticulum to release of Ca2+, resulting in an increase in intracellular Ca2+ levels and the initiation of pressure-dependent myogenic constriction in cerebral arteries [70]. (Figure 1).
2.3. Neuro-regeneration and neuro-survival
PTEN activity has been shown to substantially limit cell survival in the challenging context of cerebral ischemia [50,72]. Numerous studies have demonstrated that inhibiting PTEN to activate the PI3K/AKT pathway provides protection to the brains during stroke [73,74,75,76] [77,78]. The reduction of the PI3K/AKT/GSK-3β/mTOR signaling pathway by neuronal PTEN impairs axon growth and nerve regeneration in both the peripheral and central nervous systems, post-neuronal injuries and ischemic conditions. Strong evidence consistently supports PTEN's inhibitory role in critical neurological processes in pathological contexts [79,80,81,82,83,84,85]. Enhancing the activity of the PI3K/AKT pathway has been shown to increase axon growth [86]. Therefore, it is clear that PTEN, an intrinsic inhibitor of the PI3K pathway, plays a significant role in regulating the growth of central axons. PTEN's activity also impedes nerve regeneration following neuronal injury, which is crucial for neural function recovery [83]. Hence, deliberately inhibiting PTEN activity emerges as a strategically advantageous approach with pronounced benefits for facilitating the neuronal regeneration following injury. Empirical evidence shows that deleting PTEN in the spinal cord or optic nerve significantly enhances nerve regeneration after injury [87]. Targeted application of local pharmacological agents to suppress PTEN or the precise utilization of siRNA-based techniques to specifically downregulate PTEN expression at injury site serves as a potent and effective strategy for accelerating the intricate axon outgrowth process and, expediting overall neuronal recovery [88]. Even in genetic diseases, such as spinal muscular atrophy, managing protein synthesis in motor neurons through PTEN depletion could be a therapeutic strategy [89,90]. Experimental data demonstrate that ROS signaling plays an essential role in promoting self-renewal, proliferation and differentiation of neural stem cells and neural progenitor cells through a regulatory mechanism in which the oxidation of PTEN by ROS upregulates the PI3K/AKT signaling pathway [91].
After neuronal injury, the injured axons are exposed to a highly oxidative and inflammation-driven environment. Under these conditions, growth cones, which are crucial for axon extension, initially collapse and retract. This process involves the oxidation of actin and produces ROS [92]. In a study, two experimental models were used to investigate role of ROS generation in neuronal death and the involvement of PTEN in neurodegenerative diseases. Oxygen-glucose deprivation and the neurotoxin 1-methyl-4-phenylpyridinium iodide were applied on neural cells to simulate cerebral ischemia and Parkinson's disease. However, it was found that ROS generated under these conditions did not cause oxidative inactivation to all cellular PTEN, allowing PTEN to maintain its functional activity. The suggested explanation is that the deactivation of PTEN phosphatase by ROS requires suitable intracellular co-localization with the site where these ROS are actively produced [93].
Hervera et al. have shown that non-mitochondrial sources of ROS are essential and sufficient for promoting axonal outgrowth and regenerating sensory axons. ROS signaling plays a crucial role in driving the regeneration of both peripheral and central nervous system axons in response to sciatic nerve injury. Importantly, NOXs signaling emerges as a key regulatory mechanism in response to injury, particularly in ROS-dependent neuron regeneration. Membrane-bound NOX enzymes generate O2-, is subsequently converted to H2O2 by SOD. Interestingly, NOX2 can originate from extracellular vesicles released by cytokine-recruited inflammatory macrophages. These NOX2-cointaining exosomes are then transported retrogradely in axonal endosomes post-injury and produce ROS for cellular signaling. In other words, macrophages release NOX2-containing exosomes that, subsequently enter the neuron and produce ROS, serving as a regeneration signal. These pathways involve key regulatory proteins whose activity can be modulated through oxidation of cysteine residues. PTEN, notably, emerges as the most oxidized protein in such neurons following sciatic nerve injury. The downregulation of PTEN, mediated by NOX2 activity in association with nerve injury, leads to increased activation of the PI3K/AKT pathway, promoting neuron outgrowth. The PTEN oxidative inactivation following nerve injury plays an important role in regulating nerve regeneration and is therefore a prospective mechanism in studies of neuronal pathology [94].
In Alzheimer's disease (AD), the accumulation of misfolded, hyperphosphorylated tau proteins is closely associated with the loss of neurons and cognitive dysfunction [95]. Tau normally plays a crucial role in assembling and maintaining microtubules in neuronal axons [96]. Abnormal hyperphosphorylation of tau alters its shape and impairs its ability to bind to microtubules, resulting in the destabilization of microtubules and the formation of neurofibrillary tangles, which contribute to neuronal dysfunction and cell death [97]. GSK-3β, a downstream kinase of the PI3K/AKT signaling pathway, is known for its role in phosphorylating tau in AD pathogenesis [98]. Impaired PI3K/AKT pathway leads to GSK-3β hyperactivity and excessive tau phosphorylation, which is linked to the progression of AD [99]. Treatment with insulin or curcumin can improve memory and cognitive ability in AD patients, possibly through the regulation of the PI3K/AKT pathway [100]. Stimulation with growth factors such as epidermal growth factor, platelet-derived growth factor, or insulin, leads to the formation H2O2 as a result of the activation of NOXs and the oxidation of PTEN, which increases PI3K/AKT signaling pathway [101]. These findings indicate that the oxidative inhibition of PTEN can be a possible method for improving AD patients’ condition.
Experimental data demonstrate that the presence of peroxynitrite can prevent etoposide-induced apoptotic cell death in primary cortical neurons. This effect is primarily due to the oxidation of PTEN and the subsequent up-regulation of the PI3K/AKT signaling pathway. Although the anti-apoptotic implication of peroxynitrite is subject to dispute, this data concurrently strengthens the potential of PTEN oxidation in promoting neuroprotection [102]. (Figure 2).
2.4. Immune response
Granulopoiesis is an emergency response to acute infection or inflammation, in which neutrophils are rapidly and massively produced and deployed from the bone marrow. Cytokines such as IL-6 and granulocyte colony-stimulating factor (G-CSF) are usually elevated during acute inflammation and may play a role in emergency granulopoiesis by inducing granulocyte differentiation [103,104]. In acute myocardial infarction, the myocardium also releases IL-6 and TNFα and plasma levels of these cytokines increase after a brief episode of coronary artery blockage [105,106,107]. Kwak et al. demonstrate that an increase in ROS levels in the bone marrow alone is sufficient to trigger granulopoiesis. The elevated ROS concentration is important to promote the proliferation and differentiation of myeloid progenitor cells through up-regulated AKT signal transduction, which occurs due to oxidative inhibition of PTEN’s phosphatase activity. During emergency granulopoiesis, these ROS are mainly produced by myeloid cells through phagocytic NOX2 activity, which can be induced by the cytokines G-CSF and TNFα. Therefore, the oxidative inactivation of PTEN by NADPH oxidase-dependent ROS is an essential mechanism for prompting emergency granulopoiesis [108]. The PI3K/AKT activity has also been shown to be a robust pivotal factor in the development of ROS-producing macrophages [109]. (Figure 3)
2.5. Insulin-related metabolism
Insulin resistance, which is characterized by a reduced sensitivity to insulin in regulating blood glucose levels, is the primary pathological feature of Type 2 Diabetes mellitus. The role of ROS in insulin sensitivity is complex, with a dual effect: promoting insulin sensitivity in the early stages of disease but contributing to insulin resistance as hyperglycemia progresses. The transient and controlled ROS production by NOXs in response to insulin is likely to be beneficial, while the chronic ROS generation by mitochondria during the context of prolonged nutrient overload in the later stages of the disease might be detrimental to insulin responsiveness [110,111]. Insulin stimulation can lead to this temporary increase in ROS levels by activating NOX and subsequently trigger insulin-mediated AKT activation. PIP3 and NOXs are located in the cell's plasma membrane, suggesting that upon insulin stimulation, PTEN is oxidatively inactivated in close proximity to NOXs, and recruited PI3K can elevate PIP3 levels [112]. PIP3, in turn, activates the downstream AKT signaling pathway, which subsequently phosphorylates various targets, initiating the anabolic effects of insulin stimulation [113]. In some studies, inhibiting PTEN’s PIP3-phosphatase activity has been proposed a potential therapeutic approach for Type 2 diabetes [114] . Loh et al. demonstrated that a slight increase in physiological ROS levels in muscle cells can induce PTEN oxidation and eventually enhance insulin-induced glucose uptake through the PI3K/AKT pathway [111]. As a negative regulator of PIP3, PTEN can decrease the activity of this downstream signal. Therefore, the redox regulation of PTEN holds promise as a method for managing Type 2 Diabetes mellitus. (Figure 4)
2.6. Myogenic autophagy in muscle differentiation
Autophagy is a crucial intracellular recycling process that eliminates old and dysfunctional cellular proteins and organelles. This process involves the formation of autophagosomes, which envelop parts of the cell's cytoplasm that contain unnecessary components. By doing so, autophagy functions as a dynamic mechanism for maintaining cellular health and resource efficiency [115,116]. Kim et al. demonstrate that the PI3K/AKT/mTOR signaling pathway is up-regulated by mitochondrial ROS-derived H2O2, which subsequently implicates myogenesis-specific autophagy during muscle differentiation. In this scenario, PTEN is inactivated through oxidation [117]. (Figure 4)
3. Role of bicarbonate in the oxidation of PTPs by H2O2
H2O2 serves as a signaling molecule that participates in cellular responses triggered by various factors such as growth factors, hormones, and cytokines, including platelet-derived growth factor, epidermal growth factor, VEGF, insulin, TNFα, and interleukin-1β. During signal transduction, PTPs are key targets of growth factor-mediated H2O2. These PTPs play a significant role in regulating multiple critical signaling pathways in mammalian cells by catalyzing the removal of phosphate groups from specific tyrosine residues on target proteins [25,118,119,120]. PTEN, which belongs to the PTPs family and possesses the ability to dephosphorylate PIP3, can also be inactivated by physiological H2O2 [28].
The activation of receptor-tyrosine kinase is a crucial event in the transmission of phosphorylation signals in response to growth factor stimulation, and it holds significant physiological importance [119]. When receptor tyrosine kinases are activated, they trigger a transient production of H2O2 by membrane NOXs [27]. This H2O2, in turn, leads to the reversible oxidative inhibition of PTPs [121]. However, the process by which PTPs undergo oxidation within a cellular environment has raised questions, particularly because other thiol proteins from the peroxiredoxin family are more significantly reactive and likely to interact with intracellular H2O2. In addition, both oxidized PTPs, including PTEN, and peroxiredoxin can be converted back to their active reduced forms by the Thioredoxin/Thioredoxin Reductase/NADPH system, which is abundantly expressed in cells. Under mild oxidative stress conditions, peroxiredoxin I not only protects PTEN from oxidation and but also enhances its capabilities through direct interaction [54] [122].
H2O2 can react with bicarbonate/CO2 to form peroxymonocarbonate (HCO4-), a highly reactive oxidant that has a much higher reactivity than H2O2 when reacting with low-molecular-weight thiols [123,124]. Zhou et al. demonstrated that the presence of bicarbonate augments the oxidative inactivation of PTPs, particularly PTP1B and SHP-2, caused by H2O2 probably through generating HCO4- [121]. Growth factor receptor stimulation also up-regulates activity of sodium-bicarbonate cotransporters (NBC) and carbonic anhydrase (CA) to increase the cellular concentration of bicarbonate. CA IX, a transmembrane enzyme with an extracellular active domain, can catalyzes the reaction: CO2+H2O⇌HCO3- [125]. NBCs uptake bicarbonate into the cell [126]. Through this mechanism, Dagnell et al. provide an the explanation for growth factor receptor stimulation-mediated oxidation of PTP1B: with the increase level of bicarbonate, more HCO4- are formed from H2O2, boosting the oxidative reaction rate [34]. Since PTEN’s molecular structure contains a cysteine residue in its active site as other PTPs, the H2O2-mediated oxidative inhibition of PTEN can be affected by bicarbonate. In the future, further experiments would be conducted to fortify the role of bicarbonate in redox regulation of PTEN by H2O2.
4. Concluding remarks
In conclusion, PTEN oxidative inactivation by ROS, particularly NOX-derived H2O2, has been shown to be essential in various physiological processes such as cardiovascular remodeling, vascular constriction, neuro-regeneration, immune response, insulin-related metabolism and myogenesis-specific autophagy. This PTEN inactivation increases the activity of PI3K/AKT signaling pathway, subsequently prevents apoptosis and promotes proliferation of cardiomyocytes following ischemia as well as increases the vascular angiogenesis and constriction. In the neuro-regeneration process, the ROS that oxidize PTEN could originates from extracellular NOX2 delivery vesicles of macrophages. During acute ischemia or inflammation, ROS derived from NOX2 in myeloid cells can inhibit PTEN and induce granulopoiesis. The elevated PI3K/AKT downstream signaling through redox regulation of PTEN could also mitigate insulin resistance. ROS also initiate the cellular autophagic rebuilding in the process of muscle differentiation through PTEN-mediated mTOR augmentation. Moreover, the bicarbonate can react with H2O2 to form HCO4- and therefore accelerate the oxidation of PTPs. Further studies would substantiate the importance of bicarbonate in facilitating the H2O2-mediated PTEN redox regulation and its role in physiological processes. (Figure 5)
Author Contributions
Conceptualization, S.-R.L.; methodology, V.H.T.; validation, S.-R.L.; writing—original draft preparation, V.H.T.; writing—review and editing, V.H.T., T.N.H., D.K.S., J.M.C., H.J.Y., S.C.P. and J.Y.S.; visualization, D.K.S.; supervision, S.-R.L.; funding acquisition, S.-R.L. and S.C.P. All authors have read and agreed to the published version of the manuscript.
Acknowledgments
This work was supported by the Basic Research Program (NRF-2022M3A9E4017151 to S.-RL) through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Technology and by KBRI basic research program through Korea Brain Research Institute (23-BR-03-05 to S.-RL and S.C.P.). This research was also supported by the National Research Foundation of Korea (2018R1D1A1B06051438), Republic of Korea. Thang Nguyen Huu is supported in part by Center for Global Future Biomedical Scientists at Chonnam National University. Figures were created with Biorender.com.
Conflicts of interest
The authors declare no conflict of interest.
References
- Zhang, Y.; et al., Redox regulation of tumor suppressor PTEN in cell signaling. Redox biology 2020, 34, 101553.
- Zhang, Y.; et al., Redox regulation of the tumor suppressor PTEN by hydrogen peroxide and tert-butyl hydroperoxide. International journal of molecular sciences 2017, 18, 982. [CrossRef] [PubMed]
- Han, S.-J.; et al., Redox regulation of the tumor suppressor PTEN by the thioredoxin system and cumene hydroperoxide. Free Radical Biology and Medicine 2017, 112, 277–286. [CrossRef] [PubMed]
- Han, S.-J.; et al., Assay of the redox state of the tumor suppressor PTEN by mobility shift. Methods 2015, 77, 58–62.
- Boosani, C.S., P. Gunasekar, and D.K. Agrawal, An update on PTEN modulators–a patent review. Expert opinion on therapeutic patents 2019, 29, 881–889. [CrossRef] [PubMed]
- Lee, Y.-R., M. Chen, and P.P. Pandolfi, The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nature reviews Molecular cell biology 2018, 19, 547–562. [CrossRef] [PubMed]
- Blumenthal, G.M. and P.A. Dennis, PTEN hamartoma tumor syndromes. European journal of human genetics 2008, 16, 1289–1300. [CrossRef] [PubMed]
- Baig, R.M.; et al., Genetic changes in the PTEN gene and their association with breast cancer in Pakistan. Asian Pac J Cancer Prev 2011, 12, 2773–2778.
- Liaw, D.; et al., Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nature genetics 1997, 16, 64–67. [CrossRef]
- Norimatsu, Y.; et al., Immunohistochemical expression of PTEN and β-catenin for endometrial intraepithelial neoplasia in Japanese womenomen. Annals of diagnostic pathology 2007, 11, 103–108. [CrossRef]
- Patel, R.; et al. Sprouty2, PTEN, and PP2A interact to regulate prostate cancer progression. The Journal of clinical investigation 2013, 123, 1157–1175. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; et al., Combined PTEN mutation and protein expression associate with overall and disease-free survival of glioblastoma patients. Translational oncology 2014, 7, 196–205. [CrossRef] [PubMed]
- Romano, C. and C. Schepis, PTEN gene: A model for genetic diseases in dermatology. The Scientific World Journal 2012, 2012.
- Pulido, R., PTEN inhibition in human disease therapy. Molecules 2018, 23, 285. [CrossRef] [PubMed]
- Denu, J.M. and J.E. Dixon, Protein tyrosine phosphatases: Mechanisms of catalysis and regulation. Curr Opin Chem Biol 1998, 2, 633–641. [CrossRef] [PubMed]
- Nguyen Huu, T.; et al., The Role of Oxidative Inactivation of Phosphatase PTEN and TCPTP in Fatty Liver Disease. Antioxidants (Basel) 2023, 12.
- Sun, Y., Oxidative stress and cardiac repair/remodeling following infarction. The American journal of the medical sciences 2007, 334, 197–205. [CrossRef]
- Meng, T.-C., T. Fukada, and N.K. Tonks, Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Molecular cell 2002, 9, 387–399. [CrossRef]
- Rhee, S.G.; et al., Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxinss. Curr Opin Cell Biol 2005, 17, 183–189. [CrossRef]
- Rhee, S.G.; et al., Cellular regulation by hydrogen peroxide. J Am Soc Nephrol 2003, 14 Suppl. S3, S211–S215. [CrossRef]
- Rhee, S.G., Redox signaling: Hydrogen peroxide as intracellular messenger. Experimental & Molecular Medicine 1999, 31, 53–59.
- Zhang, L.; et al., Biochemical basis and metabolic interplay of redox regulation. Redox Biol 2019, 26, 101284.
- Gupta, S.C.; et al., Upsides and downsides of reactive oxygen species for cancer: The roles of reactive oxygen species in tumorigenesis, prevention, and therapy. Antioxid Redox Signal 2012, 16, 1295–1322. [CrossRef] [PubMed]
- Veal, E.A., A.M. Day, and B.A. Morgan, Hydrogen peroxide sensing and signaling. Molecular cell 2007, 26, 1–14. [CrossRef] [PubMed]
- Rhee, S.G., H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [CrossRef] [PubMed]
- Lambeth, J.D.; et al., Novel homologs of gp91phox. Trends Biochem Sci 2000, 25, 459–461. [CrossRef]
- Lambeth, J.D., NOX enzymes and the biology of reactive oxygen. Nature Reviews Immunology 2004, 4, 181–189. [CrossRef] [PubMed]
- Lee, S.-R.; et al., Reversible inactivation of the tumor suppressor PTEN by H2O2. Journal of Biological Chemistry 2002, 277, 20336–20342. [CrossRef] [PubMed]
- Leslie, N.R.; et al., Redox regulation of PI 3-kinase signalling via inactivation of PTEN. The EMBO journal 2003, 22, 5501–5510. [CrossRef]
- Takakura, K.; et al., Rapid and irreversible inactivation of protein tyrosine phosphatases PTP1B, CD45, and LAR by peroxynitrite. Arch Biochem Biophys 1999, 369, 197–207. [CrossRef]
- Pacher, P., J.S. Beckman, and L. Liaudet, Nitric oxide and peroxynitrite in health and disease. Physiological reviews 2007, 87, 315–424. [CrossRef] [PubMed]
- Leslie, N.R., The redox regulation of PI 3-kinase-dependent signaling. Antioxid Redox Signal 2006, 8, 1765–1774. [CrossRef] [PubMed]
- Downes, C.P.; et al., Stimulation of PI 3-kinase signaling via inhibition of the tumor suppressor phosphatase, PTEN. Adv Enzyme Regul 2007, 47, 184–194. [CrossRef] [PubMed]
- Dagnell, M.; et al., Bicarbonate is essential for protein-tyrosine phosphatase 1B (PTP1B) oxidation and cellular signaling through EGF-triggered phosphorylation cascades. Journal of Biological Chemistry 2019, 294, 12330–12338. [CrossRef] [PubMed]
- Winterbourn, C.C.; et al., Carbon dioxide/bicarbonate is required for sensitive inactivation of mammalian glyceraldehyde-3-phosphate dehydrogenase by hydrogen peroxide. Proc Natl Acad Sci U S A 2023, 120, e2221047120. [CrossRef] [PubMed]
- Radi, R., Interplay of carbon dioxide and peroxide metabolism in mammalian cells. Journal of Biological Chemistry 2022, 298. [CrossRef] [PubMed]
- Murphy, E., Primary and secondary signaling pathways in early preconditioning that converge on the mitochondria to produce cardioprotection. Circulation research 2004, 94, 7–16. [CrossRef] [PubMed]
- Jonassen, A.K.; et al., Myocardial protection by insulin at reperfusion requires early administration and is mediated via Akt and p70s6 kinase cell-survival signaling. Circulation research 2001, 89, 1191–1198. [CrossRef] [PubMed]
- Matsui, T.; et al., Adenoviral gene transfer of activated phosphatidylinositol 3′-kinase and Akt inhibits apoptosis of hypoxic cardiomyocytes in vitro. Circulation 1999, 100, 2373–2379. [CrossRef]
- Uchiyama, T.; et al. Role of Akt signaling in mitochondrial survival pathway triggered by hypoxic preconditioning. Circulation 2004, 109, 3042–3049. [Google Scholar] [CrossRef]
- Tong, H.; et al. Ischemic preconditioning activates phosphatidylinositol-3-kinase upstream of protein kinase C. Circulation research 2000, 87, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Mocanu, M.M., R.M. Bell, and D.M. Yellon, PI3 kinase and not p42/p44 appears to be implicated in the protection conferred by ischemic preconditioning. Journal of molecular and cellular cardiology 2002, 34, 661–668. [CrossRef]
- Mocanu, M. and D. Yellon, PTEN, the Achilles' heel of myocardial ischaemia/reperfusion injury? British journal of pharmacology 2007, 150, 833–838. [CrossRef]
- Parajuli, N.; et al. Phosphatase PTEN is critically involved in post-myocardial infarction remodeling through the Akt/interleukin-10 signaling pathway. Basic Research in Cardiology 2012, 107, 1–15. [Google Scholar] [CrossRef]
- Burchfield, J.S.; et al. Interleukin-10 from transplanted bone marrow mononuclear cells contributes to cardiac protection after myocardial infarction. Circulation research 2008, 103, 203–211. [Google Scholar] [CrossRef]
- Krishnamurthy, P.; et al. IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR. Circulation research 2009, 104, e9–e18. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, C.; et al. Interleukin-10 improves left ventricular function in rats with heart failure subsequent to myocardial infarction. European journal of heart failure 2008, 10, 733–739. [Google Scholar] [CrossRef]
- Yang, Z., B. Zingarelli, and C. Szabó, Crucial role of endogenous interleukin-10 production in myocardial ischemia/reperfusion injury. Circulation 2000, 101, 1019–1026. [CrossRef] [PubMed]
- Keyes, K.T.; et al. Pharmacological inhibition of PTEN limits myocardial infarct size and improves left ventricular function postinfarction. American Journal of Physiology-Heart and Circulatory Physiology 2010, 298, H1198–H1208. [Google Scholar] [CrossRef]
- Ruan, H.; et al. Inducible and cardiac specific PTEN inactivation protects ischemia/reperfusion injury. Journal of molecular and cellular cardiology 2009, 46, 193–200. [Google Scholar] [CrossRef]
- Fukui, T.; et al. Expression of p22-phox and gp91-phox, essential components of NADPH oxidase, increases after myocardial infarction. Biochem Biophys Res Commun 2001, 281, 1200–1206. [Google Scholar] [CrossRef]
- Krijnen, P.; et al. Increased Nox2 expression in human cardiomyocytes after acute myocardial infarction. Journal of clinical pathology 2003, 56, 194–199. [Google Scholar] [CrossRef]
- Sirker, A.; et al. Cell-specific effects of Nox2 on the acute and chronic response to myocardial infarction. J Mol Cell Cardiol 2016, 98, 11–17. [Google Scholar] [CrossRef]
- Nguyen Huu, T.; et al. Redox Regulation of PTEN by Peroxiredoxins. Antioxidants (Basel) 2021, 10. [Google Scholar] [CrossRef]
- Cai, Z. and G.L. Semenza, PTEN activity is modulated during ischemia and reperfusion: Involvement in the induction and decay of preconditioning. Circulation research 2005, 97, 1351–1359. [Google Scholar] [CrossRef]
- Xiang, M.; et al., Role of oxidative stress in reperfusion following myocardial ischemia and its treatments. Oxidative medicine and cellular longevity 2021, 2021. [CrossRef] [PubMed]
- Lee, S.H.; et al. Early expression of angiogenesis factors in acute myocardial ischemia and infarction. New England Journal of Medicine 2000, 342, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, M.; et al. Angiogenesis in the ischemic core: A potential treatment target? Journal of Cerebral Blood Flow & Metabolism 2019, 39, 753–769. [Google Scholar]
- Zaitone, S.A. and N.M. Abo-Gresha, Rosuvastatin promotes angiogenesis and reverses isoproterenol-induced acute myocardial infarction in rats: Role of iNOS and VEGF. European journal of pharmacology 2012, 691, 134–142. [Google Scholar] [CrossRef]
- Connor, K.M.; et al. Mitochondrial H2O2 regulates the angiogenic phenotype via PTEN oxidation. Journal of Biological Chemistry 2005, 280, 16916–16924. [Google Scholar] [CrossRef]
- Latronico, M.V.; et al. Regulation of cell size and contractile function by AKT in cardiomyocytes. Annals of the New York Academy of Sciences 2004, 1015, 250–260. [Google Scholar] [CrossRef]
- Saward Peter Zahradka, L., Angiotensin II activates phosphatidylinositol 3-kinase in vascular smooth muscle cells. Circulation research 1997, 81, 249–257. [CrossRef] [PubMed]
- Sugden, P.H., Ras, Akt, and mechanotransduction in the cardiac myocyte. Circulation research 2003, 93, 1179–1192. [CrossRef] [PubMed]
- McDowell, S.A.; et al. Phosphoinositide 3-kinase regulates excitation-contraction coupling in neonatal cardiomyocytes. American Journal of Physiology-Heart and Circulatory Physiology 2004, 286, H796–H805. [Google Scholar] [CrossRef] [PubMed]
- Goncharova, E.A.; et al. PI3K is required for proliferation and migration of human pulmonary vascular smooth muscle cells. American Journal of Physiology-Lung Cellular and Molecular Physiology 2002, 283, L354–L363. [Google Scholar] [CrossRef] [PubMed]
- Perrino, C.; et al. Dynamic regulation of phosphoinositide 3-kinase-γ activity and β-adrenergic receptor trafficking in end-stage human heart failure. Circulation 2007, 116, 2571–2579. [Google Scholar] [CrossRef] [PubMed]
- Namgaladze, D. and B. Brüne, Phospholipase A2–modified low-density lipoprotein activates the phosphatidylinositol 3-kinase-akt pathway and increases cell survival in monocytic cells. Arteriosclerosis, thrombosis, and vascular biology 2006, 26, 2510–2516. [Google Scholar] [CrossRef] [PubMed]
- Northcott, C.A., J.S. Hayflick, and S.W. Watts, PI3-Kinase upregulation and involvement in spontaneous tone in arteries from DOCA-salt rats: Is p110δ the culprit? Hypertension 2004, 43, 885–890. [CrossRef] [PubMed]
- Wu, K.L.; et al. Redox-sensitive oxidation and phosphorylation of PTEN contribute to enhanced activation of PI3K/Akt signaling in rostral ventrolateral medulla and neurogenic hypertension in spontaneously hypertensive rats. Antioxidants & Redox Signaling 2013, 18, 36–50. [Google Scholar]
- Gebremedhin, D.; et al. Redox signaling via oxidative inactivation of PTEN modulates pressure-dependent myogenic tone in rat middle cerebral arteries. PLoS ONE 2013, 8, e68498. [Google Scholar] [CrossRef]
- Carnevale, D.; et al. PI3Kγ inhibition reduces blood pressure by a vasorelaxant Akt/L-type calcium channel mechanism. Cardiovascular research 2012, 93, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Ning, K.; et al. Dual neuroprotective signaling mediated by downregulating two distinct phosphatase activities of PTEN. J Neurosci 2004, 24, 4052–4060. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; et al. Delayed administration of a PTEN inhibitor BPV improves functional recovery after experimental stroke. Neuroscience 2013, 231, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; et al. PTEN inhibition prevents rat cortical neuron injury after hypoxia–ischemia. Neuroscience 2013, 238, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; et al. Rho-kinase inhibitor, fasudil, prevents neuronal apoptosis via the Akt activation and PTEN inactivation in the ischemic penumbra of rat brain. Cellular and molecular neurobiology 2012, 32, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.-Y.; et al. Dose-dependent protective effect of bisperoxovanadium against acute cerebral ischemia in a rat model of ischemia/reperfusion injury. International journal of molecular sciences 2013, 14, 12013–12022. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.-N.; et al. Down-regulation of PTEN by sodium orthovanadate inhibits ASK1 activation via PI3-K/Akt during cerebral ischemia in rat hippocampus. Neuroscience letters 2006, 404, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H., R.M. Sapolsky, and G.K. Steinberg, Phosphoinositide-3-kinase/akt survival signal pathways are implicated in neuronal survival after stroke. Molecular neurobiology 2006, 34, 249–269. [CrossRef] [PubMed]
- Christie, K.J. and D. Zochodne, Peripheral axon regrowth: New molecular approaches. Neuroscience 2013, 240, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Junco-Clemente, P. and P. Golshani, PTEN: A master regulator of neuronal structure, function, and plasticity. Commun Integr Biol 2014, 7, e28358. [Google Scholar] [CrossRef]
- Knafo, S. and J.A. Esteban, PTEN: Local and Global Modulation of Neuronal Function in Health and Disease. Trends Neurosci 2017, 40, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, Y., U. Hayat, and S. Li, PTEN inhibition and axon regeneration and neural repair. Neural Regen Res 2015, 10, 1363–1368. [PubMed]
- Park, K.K.; et al. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 2008, 322, 963–966. [Google Scholar] [CrossRef] [PubMed]
- Park, K.K.; et al. PTEN/mTOR and axon regeneration. Exp Neurol 2010, 223, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; et al. Therapeutic Targets for Cerebral Ischemia Based on the Signaling Pathways of the GluN2B C Terminus. Stroke 2015, 46, 2347–2353. [Google Scholar] [CrossRef] [PubMed]
- Soltoff, S.P.; et al. Nerve growth factor promotes the activation of phosphatidylinositol 3-kinase and its association with the trk tyrosine kinase. J Biol Chem 1992, 267, 17472–17477. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; et al. PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat Neurosci 2010, 13, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Christie, K.J.; et al. PTEN inhibition to facilitate intrinsic regenerative outgrowth of adult peripheral axons. J Neurosci 2010, 30, 9306–9315. [Google Scholar] [CrossRef]
- Little, D.; et al. PTEN depletion decreases disease severity and modestly prolongs survival in a mouse model of spinal muscular atrophy. Mol Ther 2015, 23, 270–277. [Google Scholar] [CrossRef]
- Ning, K.; et al. PTEN depletion rescues axonal growth defect and improves survival in SMN-deficient motor neurons. Hum Mol Genet 2010, 19, 3159–3168. [Google Scholar] [CrossRef]
- Le Belle, J.E.; et al. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell stem cell 2011, 8, 59–71. [Google Scholar] [CrossRef]
- Giridharan, S.S. and S. Caplan, MICAL-family proteins: Complex regulators of the actin cytoskeleton. Antioxid Redox Signal 2014, 20, 2059–2073. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; et al. Implication of PTEN in production of reactive oxygen species and neuronal death in in vitro models of stroke and Parkinson's disease. Neurochem Int 2007, 50, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Hervera, A.; et al. Reactive oxygen species regulate axonal regeneration through the release of exosomal NADPH oxidase 2 complexes into injured axons. Nat Cell Biol 2018, 20, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Isla, T.; et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol 1997, 41, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Trinczek, B.; et al. Domains of tau protein, differential phosphorylation, and dynamic instability of microtubules. Mol Biol Cell 1995, 6, 1887–1902. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, M., V.M. Lee, and J.Q. Trojanowski, Tau and axonopathy in neurodegenerative disorders. Neuromolecular Med 2002, 2, 131–150. [CrossRef] [PubMed]
- Cavallini, A.; et al. An unbiased approach to identifying tau kinases that phosphorylate tau at sites associated with Alzheimer disease. J Biol Chem 2013, 288, 23331–23347. [Google Scholar] [CrossRef]
- Hernandez, F., J.J. Lucas, and J. Avila, GSK3 and tau: Two convergence points in Alzheimer's disease. J Alzheimers Dis 2013, 33 Suppl 1, S141–S144.
- Matsuda, S.; et al. Implications of PI3K/AKT/PTEN Signaling on Superoxide Dismutases Expression and in the Pathogenesis of Alzheimer's Disease. Diseases 2018, 6. [Google Scholar] [CrossRef]
- Kwon, J.; et al. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc Natl Acad Sci U S A 2004, 101, 16419–16424. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Esteban, M.; et al. Inhibition of PTEN by peroxynitrite activates the phosphoinositide-3-kinase/Akt neuroprotective signaling pathway. J Neurochem 2007, 102, 194–205. [Google Scholar] [CrossRef]
- Manz, M.G. and S. Boettcher, Emergency granulopoiesis. Nature Reviews Immunology 2014, 14, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Walker, F.; et al. IL6/sIL6R complex contributes to emergency granulopoietic responses in G-CSF–and GM-CSF–deficient mice. Blood, The Journal of the American Society of Hematology 2008, 111, 3978–3985. [Google Scholar] [CrossRef] [PubMed]
- Gwechenberger, M.; et al. Cardiac myocytes produce interleukin-6 in culture and in viable border zone of reperfused infarctions. Circulation 1999, 99, 546–551. [Google Scholar] [CrossRef]
- Kleinbongard, P., G. Heusch, and R. Schulz, TNFα in atherosclerosis, myocardial ischemia/reperfusion and heart failure. Pharmacology & therapeutics 2010, 127, 295–314.
- Frangogiannis, N.G., The mechanistic basis of infarct healing. Antioxid Redox Signal 2006, 8, 1907–1939. [CrossRef]
- Kwak, H.-J.; et al. Myeloid cell-derived reactive oxygen species externally regulate the proliferation of myeloid progenitors in emergency granulopoiesis. Immunity 2015, 42, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; et al. Constitutively activated Akt-1 is vital for the survival of human monocyte-differentiated macrophages: Role of Mcl-1, independent of nuclear factor (NF)-κB, Bad, or caspase activation. The Journal of experimental medicine 2001, 194, 113–126. [Google Scholar] [CrossRef]
- Tiganis, T., Reactive oxygen species and insulin resistance: The good, the bad and the ugly. Trends in pharmacological sciences 2011, 32, 82–89. [CrossRef]
- Loh, K.; et al. Reactive oxygen species enhance insulin sensitivity. Cell metabolism 2009, 10, 260–272. [Google Scholar] [CrossRef]
- Seo, J.H.; et al. The major target of the endogenously generated reactive oxygen species in response to insulin stimulation is phosphatase and tensin homolog and not phosphoinositide-3 kinase (PI-3 kinase) in the PI-3 kinase/Akt pathway. Molecular biology of the cell 2005, 16, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Z., A. Di Cristofano, and M. Woo, Metabolic role of PTEN in insulin signaling and resistance. Cold Spring Harbor Perspectives in Medicine 2020, 10. [CrossRef] [PubMed]
- Rosivatz, E., Inhibiting PTEN. Biochem Soc Trans 2007, 35 Pt 2, 257–259. [CrossRef]
- Mizushima, N. and M. Komatsu, Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Harris, H. and D.C. Rubinsztein, Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol 2011, 8, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; et al. Mitochondrial ROS-derived PTEN oxidation activates PI3K pathway for mTOR-induced myogenic autophagy. Cell Death Differ 2018, 25, 1921–1937. [Google Scholar] [CrossRef]
- Lee, S.-R.; et al. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. Journal of Biological Chemistry 1998, 273, 15366–15372. [Google Scholar] [CrossRef]
- Tonks, N.K., Protein tyrosine phosphatases: From genes, to function, to disease. Nature reviews Molecular cell biology 2006, 7, 833–846. [CrossRef]
- Tanner, J.J.; et al. Redox regulation of protein tyrosine phosphatases: Structural and chemical aspects. Antioxidants & redox signaling 2011, 15, 77–97. [Google Scholar]
- Zhou, H.; et al. The biological buffer bicarbonate/CO2 potentiates H2O2-mediated inactivation of protein tyrosine phosphatases. Journal of the American Chemical Society 2011, 133, 15803–15805. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; et al. Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. Embo j 2009, 28, 1505–1517. [Google Scholar] [CrossRef] [PubMed]
- Bakhmutova-Albert, E.V.; et al. Kinetics and mechanism of peroxymonocarbonate formation. Inorganic chemistry 2010, 49, 11287–11296. [Google Scholar] [CrossRef]
- Trindade, D.F., G. Cerchiaro, and O. Augusto, A role for peroxymonocarbonate in the stimulation of biothiol peroxidation by the bicarbonate/carbon dioxide pair. Chem Res Toxicol 2006, 19, 1475–1482. [CrossRef] [PubMed]
- Dorai, T.; et al. The role of carbonic anhydrase IX overexpression in kidney cancer. European journal of cancer 2005, 41, 2935–2947. [Google Scholar] [CrossRef]
- Aalkjaer, C.; et al. Cation-coupled bicarbonate transporters. Comprehensive Physiology 2014, 4, 1605. [Google Scholar]
Figure 1.
Oxidation of PTEN in cardiovascular remodeling and myogenic constriction. Ischemia or elevated blood pressure conditions induce the production of ROS. These ROS deactivate PTEN, leading to an increase in the AKT signaling pathway. The activation of the AKT pathway enhances cell survival, proliferation, and differentiation. Furthermore, PTEN-mediated AKT activation upregulates IL-10 expression, promoting cardiac remodeling and preventing apoptosis. It also elevates VEGF expression, facilitating angiogenesis. This mechanism also involves L-type calcium channel activity and the formation of IP3, which stimulates Ca2+ secretion, thus increasing intracellular Ca2+ levels and promoting myogenic constriction.
Figure 1.
Oxidation of PTEN in cardiovascular remodeling and myogenic constriction. Ischemia or elevated blood pressure conditions induce the production of ROS. These ROS deactivate PTEN, leading to an increase in the AKT signaling pathway. The activation of the AKT pathway enhances cell survival, proliferation, and differentiation. Furthermore, PTEN-mediated AKT activation upregulates IL-10 expression, promoting cardiac remodeling and preventing apoptosis. It also elevates VEGF expression, facilitating angiogenesis. This mechanism also involves L-type calcium channel activity and the formation of IP3, which stimulates Ca2+ secretion, thus increasing intracellular Ca2+ levels and promoting myogenic constriction.
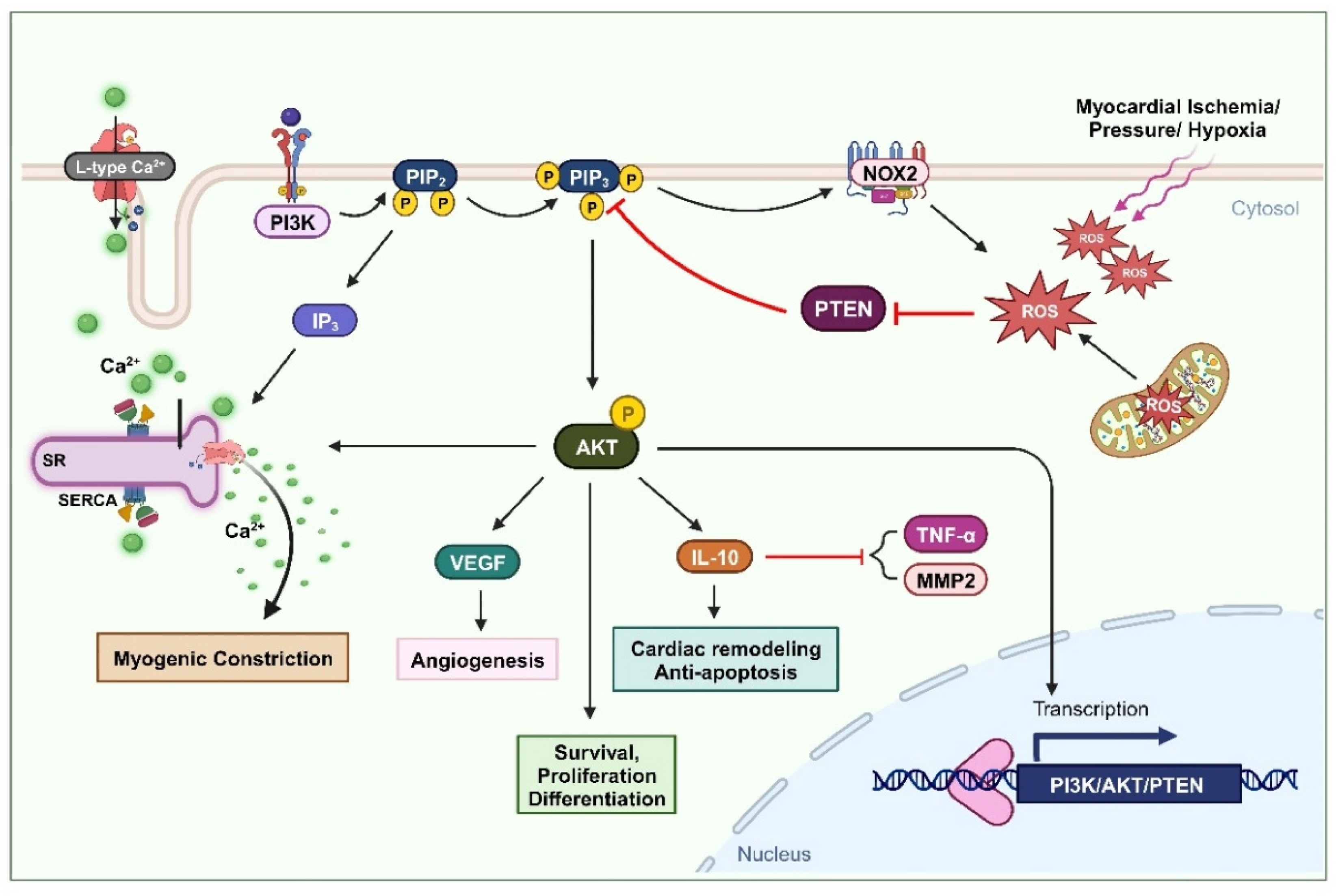
Figure 2.
Oxidative inactivation of PTEN in nerve survival and regeneration. During neuronal injury, the NOX2-derived ROS concentration increases due to receptor kinase stimulation or extracellular vesicles released by macrophages. These ROS oxidize PTEN, leading to the activation of the PIP3/AKT signaling pathway, which promotes nerve regeneration. This mechanism can also promote self-renewal, proliferation, and differentiation in neuronal stem and progenitor cells. In the context of Alzheimer's disease, the activation of the AKT pathway can downregulate GSK3β activity and subsequent phosphorylation of Tau protein, providing neuroprotection.
Figure 2.
Oxidative inactivation of PTEN in nerve survival and regeneration. During neuronal injury, the NOX2-derived ROS concentration increases due to receptor kinase stimulation or extracellular vesicles released by macrophages. These ROS oxidize PTEN, leading to the activation of the PIP3/AKT signaling pathway, which promotes nerve regeneration. This mechanism can also promote self-renewal, proliferation, and differentiation in neuronal stem and progenitor cells. In the context of Alzheimer's disease, the activation of the AKT pathway can downregulate GSK3β activity and subsequent phosphorylation of Tau protein, providing neuroprotection.
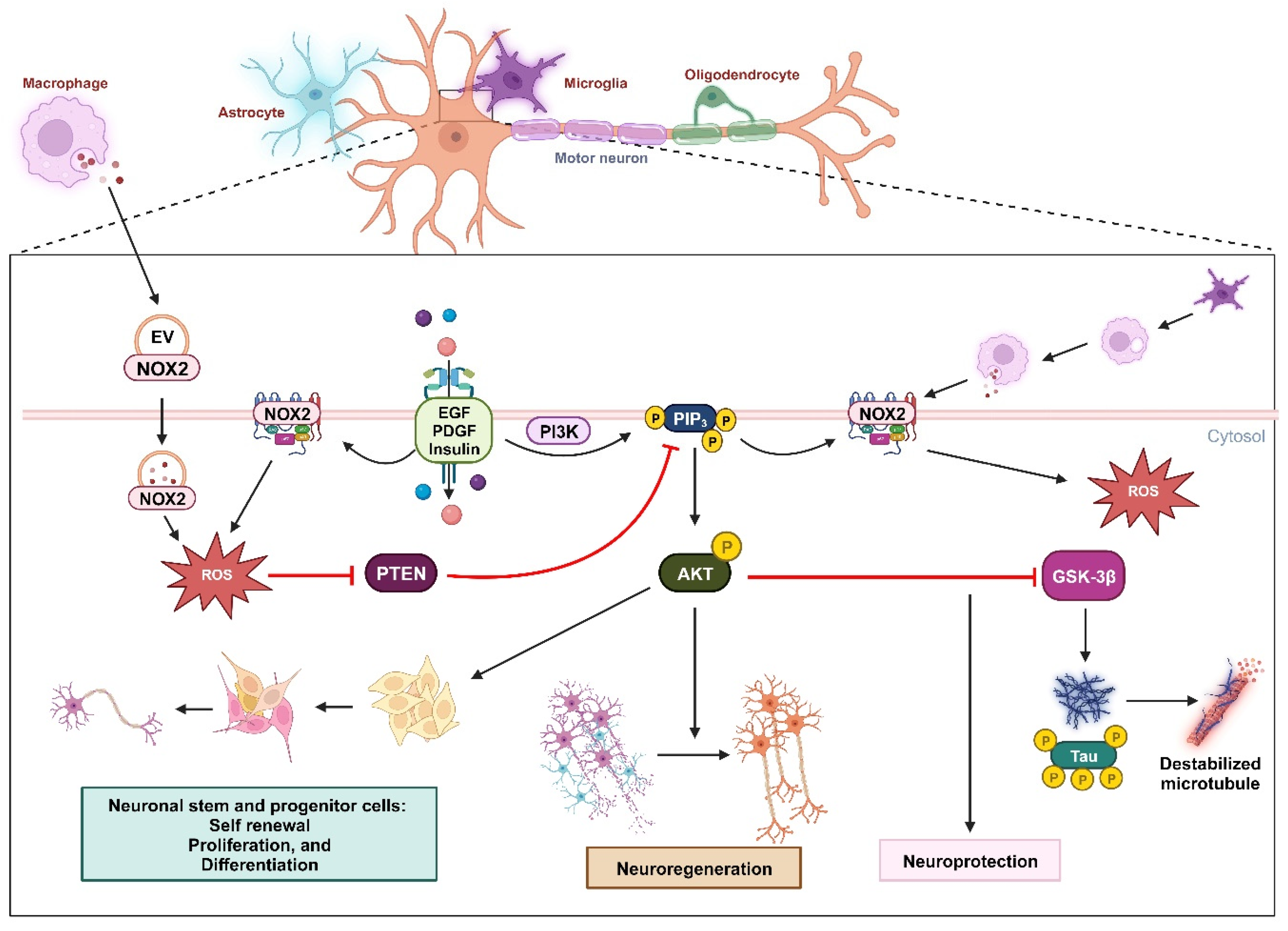
Figure 3.
Oxidative inactivation of PTEN in the immune response: Ischemia or inflammation can lead to elevated plasma cytokines, which stimulate myeloid cells to produce NOX2-derived ROS. These ROS mediate the AKT signaling pathway by inhibiting PTEN and trigger granulopoiesis, promoting the proliferation and differentiation of immune cells. These cells engage in immune reactions while also contributing to anti-apoptosis and remodeling processes.
Figure 3.
Oxidative inactivation of PTEN in the immune response: Ischemia or inflammation can lead to elevated plasma cytokines, which stimulate myeloid cells to produce NOX2-derived ROS. These ROS mediate the AKT signaling pathway by inhibiting PTEN and trigger granulopoiesis, promoting the proliferation and differentiation of immune cells. These cells engage in immune reactions while also contributing to anti-apoptosis and remodeling processes.
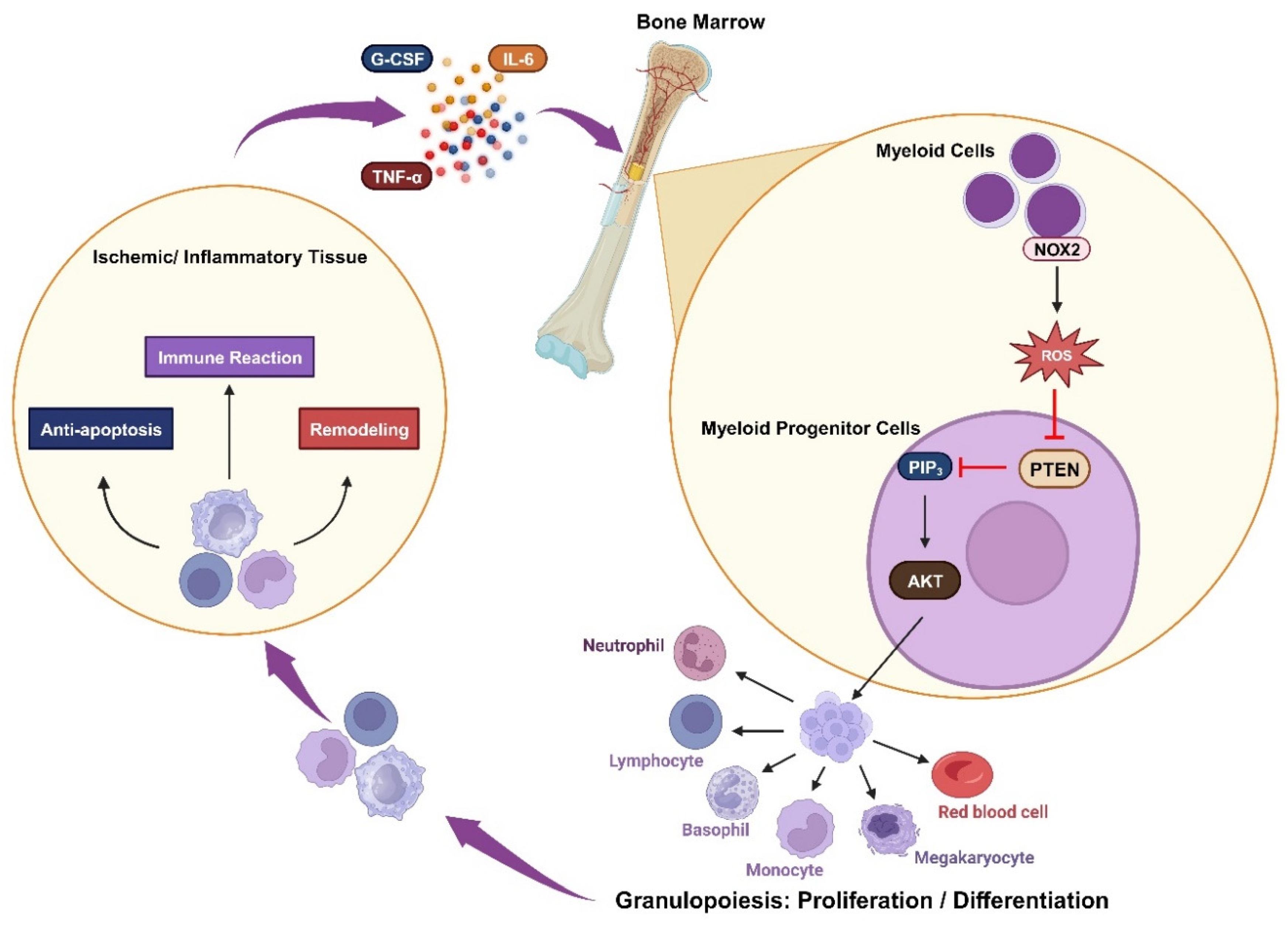
Figure 4.
Oxidative inactivation of PTEN in insulin-related metabolism and muscle differentiation. Stimulation of growth factor receptors induces NOX2 activity and the production of ROS, which can oxidize PTEN and upregulate the PI3K/AKT signaling pathway. As a result, glucose uptake and insulin sensitivity are increased. During muscle differentiation, mitochondria-derived ROS can also oxidize PTEN and promote mTOR-induced myogenic autophagy.
Figure 4.
Oxidative inactivation of PTEN in insulin-related metabolism and muscle differentiation. Stimulation of growth factor receptors induces NOX2 activity and the production of ROS, which can oxidize PTEN and upregulate the PI3K/AKT signaling pathway. As a result, glucose uptake and insulin sensitivity are increased. During muscle differentiation, mitochondria-derived ROS can also oxidize PTEN and promote mTOR-induced myogenic autophagy.
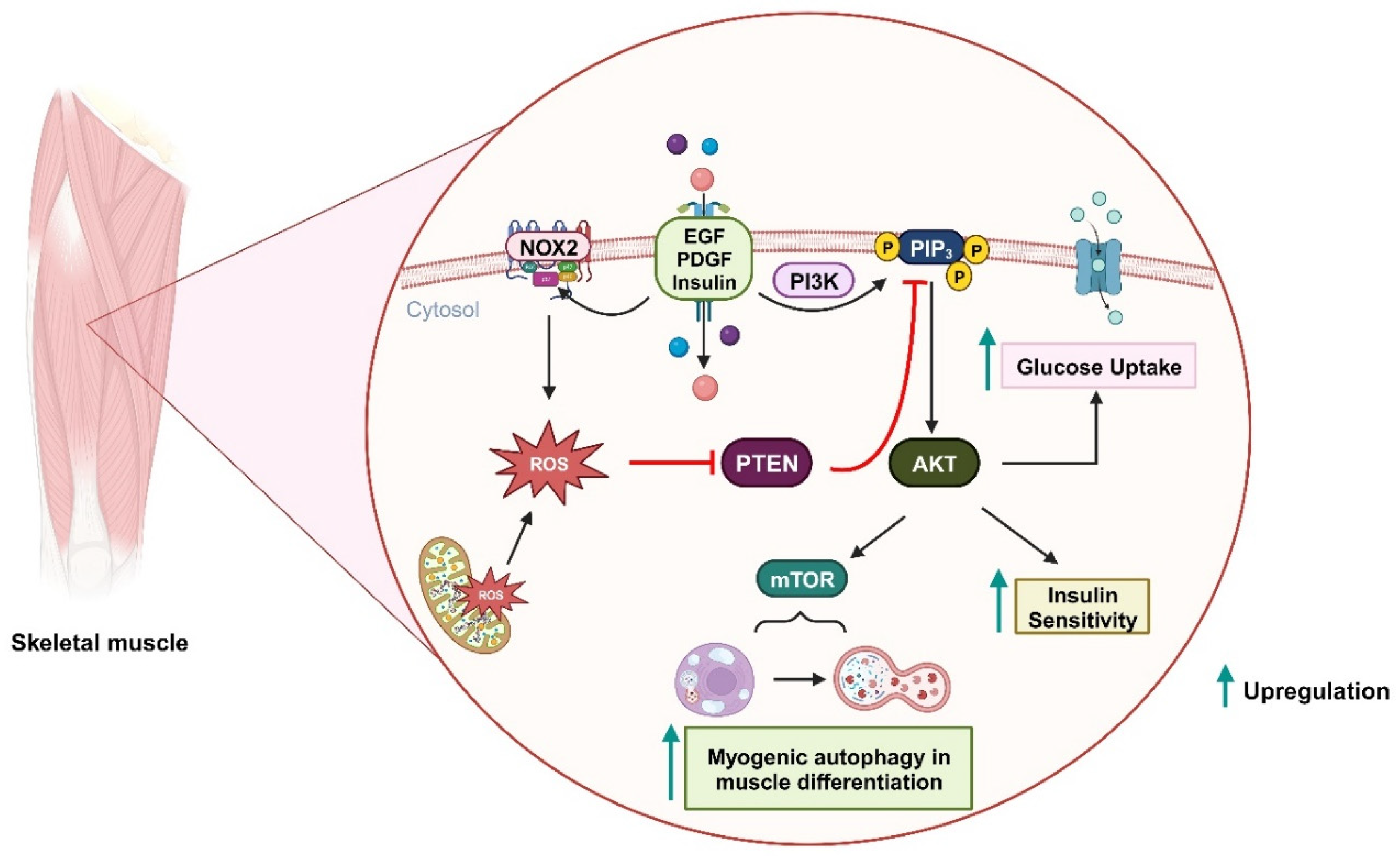
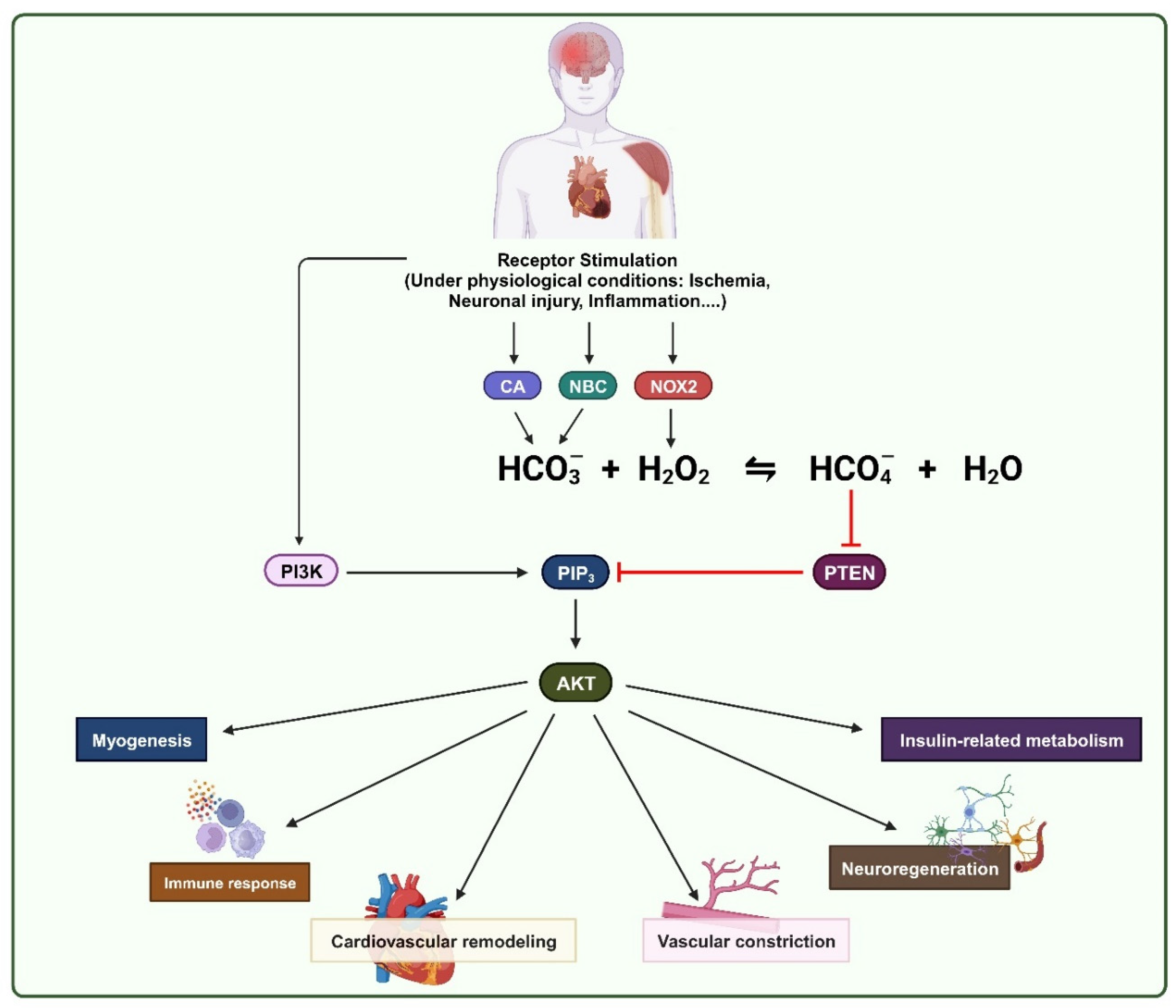
Figure 5.
Graphical abstract. Stimulation of receptor-tyrosine kinases can trigger PI3K/AKT signaling pathway and promote H2O2 production through NOX2 activity. In addition, CAs and NBCs are also activated and subsequently the concentration of HCO3- increases. H2O2 can react with HCO3- to form HCO4- and inhibit PTEN, the negative regulator of PI3K/AKT pathway. This mechanism plays crucial role in physiological processes such as the cardiovascular remodeling, vascular constriction, neuronal regeneration, immune response, insulin-related metabolism and myogenesis.
Figure 5.
Graphical abstract. Stimulation of receptor-tyrosine kinases can trigger PI3K/AKT signaling pathway and promote H2O2 production through NOX2 activity. In addition, CAs and NBCs are also activated and subsequently the concentration of HCO3- increases. H2O2 can react with HCO3- to form HCO4- and inhibit PTEN, the negative regulator of PI3K/AKT pathway. This mechanism plays crucial role in physiological processes such as the cardiovascular remodeling, vascular constriction, neuronal regeneration, immune response, insulin-related metabolism and myogenesis.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



