
Submitted:
03 January 2024
Posted:
05 January 2024
You are already at the latest version
Abstract
The accumulation of waste plastics has a severe impact on the environment, and therefore, the development of efficient chemical recycling methods has become an extremely important task. In this regard, a new strategy of degradation product-promoted depolymerization process was proposed. Using N,N'-dimethyl-ethylenediamine (DMEDA) as depolymerization reagent, an efficient chemical recycling of poly(bisphenol A carbonate) (BPA-PC) material was achieved under mild conditions. The degradation product 1,3-dimethyl-2-imidazolidinone (DMI) was proven to be the critical factor in facilitating the depolymerization process. This strategy does not require catalysts or auxiliary solvents, which is truly green process. This method improves the recycling efficiency of BPA-PC and promotes the development of plastic reutilization.
Keywords:
BPA-PC
; bisphenol A
; plastic wastes
; depolymerization
; chemical recycling
1. Introduction
Plastics are essential materials in our daily life and have been used for various purposes [1]. However, the accumulation of waste plastics, caused by improper use and indiscriminate disposal, is having a serious impact on the environment. Hence, it is necessary to develop an environmentally sustainable plastics economy that recycles these materials at the end of their life-cycle [2,3,4,5,6]. Currently, the main methods of plastics recycling are categorized into physical and chemical recycling. Physical recycling is downcycling mode due to the thermo-mechanical degradation caused by the harsh melt conditions, thus ultimately prevents the material from being reused in the manufacture of high-end products [7,8,9]. In contrast, chemical recycling can convert waste plastics into initial monomers or high-value chemicals for upcycling, while combining multiple advantages such as resource utilization, environmental friendliness, quality control and economic benefits [10,11,12,13,14]. Therefore, the development of efficient chemical recycling strategies has become an extremely important task.
In this regard, researchers have made great efforts in recent years. For example, Niu et al. proposed a biomimetic catalyst strategy to promote depolymerization. They designed a dual-core zinc catalyst with biomimetic Zn-Zn sites that activate the plastic, stabilize key intermediates and enable intramolecular hydrolysis. This catalyst was stable over a wide range of operating temperatures (30-340 °C) and pH values (8-14), allowing for sustainable recycling of poly (ethylene terephthalate) (PET) [15]. Besides, Odelius et al. reported a solvent-facilitated depolymerization strategy. The reduced Tc was observed by selecting a suitable solvent, such as dimethylformamide, that strongly interacts with the monomer, which allowed the high molecular weight poly(L-lactide) to be directly chemically recycled to L-lactide within 1-4 hours at 140 °C, with a reaction selectivity of up to 98-99% [16]. Recently, Wang's team established a new method to accelerate mass transfer depolymerization by mixing solvents. Complete hydrolysis of unsaturated polyester resin (UPR) was achieved by selective cleavage of ester bonds. Under the synergistic effect of tetrahydrofuran (THF) and H2O, the depolymerization could be carried out at a mild condition of 100 °C [17]. In addition, strategies that drive polymerization-depolymerization to proceed rapidly at low energies by designing systems in thermodynamic near-equilibrium have also received attention [18,19,20,21]. For example, Lu et al. designed a thermodynamically neutral system based on β-thiolactones by introducing gem-dimethyl group on a four-membered ring. The obtained polythioesters (PTEs) could be rapidly recycled into pristine enantiomerically pure β-thiolactones at room temperature with low energy input [22]. Despite these advances, there is still an urgent need to develop more type of depolymerization strategies, given the diversity of plastic and the complexity of practical realities of degradation.
Our research group has been committed to the study of chemical depolymerization strategies. For example, a "DE-RE polymerization" strategy was proposed to achieve the recycling process from waste poly(lactide) (PLA) plastic to new virgin-quality PLA materials in a "polymer-to-polymer" mode [23,24]. The selective depolymerization of various mixed plastics was achieved by sequential or "one-pot" depolymerization strategies to obtain the initial monomers or value-added chemicals [25]. To further expand the research field of plastic depolymerization, in this study, we proposed a new degradation product-promoted depolymerization strategy. N,N'-dimethyl-ethylenediamine (DMEDA) was used as a depolymerizing agent to efficiently convert discarded BPA-PC material into the original monomer, bisphenol A (BPA), and a value-added chemical, 1,3-dimethyl-2-imidazolidinone (DMI), under the promotion of 1 equivalent of DMI. The promoting effect of product DMI on depolymerization was systematically studied. It is noteworthy that various commercial BPA-PC plastics could be applied to this strategy, as well as to the selective depolymerization of BPA-PC/ABS and BPA-PC/PET mixed plastics. This strategy provides a new idea and method for solving the recycling of engineering plastic polycarbonate.
2. Results and Discussion
2.1. Depolymerization of BPA-PC under Solvent-Free Conditions
In our previous work, amino-alcoholysis of BPA-PC materials under solvent-free and catalyst-free conditions was achieved with 1 equiv. of amino alcohol utilizing the nucleophilicity of depolymerizing reagent [26]. This success motivated us to further investigate the ammonolysis of BPA-PC materials, in which higher nucleophilicity of amine may lead to milder reaction conditions. Therefore, in this paper, N,N'-dimethyl-ethylenediamine (DMEDA) as a common diamine compound was chosen as the depolymerizing agent. To minimize energy consumption, the solvent-free ammonolysis experiments of BPA-PC were conducted at a mild temperature of 80 °C.
As shown in Figure 1, it was found that the depolymerization proceeded rapidly at the beginning, providing bisphenol A (BPA) and 1,3-dimethyl-2-imidazolidinone (DMI) in 73% and 74% yields, respectively, after 4 h. However, with the subsequent extension of the reaction time, the yields of degradation products hardly increased. The limit of yields could only be maintained at about 70-80%, which seemed to have reached an equilibrium state. By analyzing the 1H NMR spectrum of the reaction at 24 h (Figure S1: 24 h, BPA 80%, DMI 82%), it was found that the remaining DMEDA is still present in the system, but it no longer undergoes depolymerization with BPA-PC.
The unexpected phenomenon of BPA-PC being difficult to fully convert drove us to further investigate the underlying reasons. Compared to ethanolamine (amino alcohol), the depolymerization reagent DMEDA (diamine) showed a stronger basicity. From this feature, we deduced that the amino groups of the feedstock DMEDA may form strong hydrogen bonding with the phenolic hydroxyl groups of the product BPA, especially when a large amount of BPA was generated in the late stage of the reaction, thus hindering the smooth progress of the depolymerization. To verify this hypothesis, the binding constant (K) between BPA and DMEDA through the O-H···N interaction was determined using concentration variation experiments with 1H NMR titration [27,28]. As shown in Figure 2, a high value of K was calculated as 54.9±2.2, which was significantly greater than the O-H···O hydrogen bonding constants (K = 0.9-10.9) reported for a series of hydroxylated biomass compounds [29,30]. This result indicated that there was a strong interaction between BPA and DMEDA. When they were tightly bound together, the amine group of DMEDA could not be exposed to attack the carbonyl group of the polymer chains, resulting in the BPA-PC material cannot be completely depolymerized.
2.2. Effect of Solvent Type on the Reaction
Next, to achieve complete conversion of BPA-PC, the influence of adding auxiliary solvent on the reaction was investigated (Table 1). A series of solvents with good solubility for BPA-PC, such as dichloromethane (DCM) and tetrahydrofuran (THF), were prioritized. However, even with an extended reaction time of 24 h and solvent assistance, the yield of BPA was only 77% and 68%, respectively (entries 1, 2). In the poor solvent case, similar degradation results were observed when ethyl acetate (EtOAc), toluene (Tol.), and acetone (Ace.) were employed, the BPA yield of 70-75% indicating incomplete conversion as well (entries 3, 4, 5). It is worth noting that the product DMI could be used as a solvent in various organic synthesis and transformations, thus it was also taken into account. Surprisingly, when using DMI as the solvent, a high yield of 87% for BPA was reached, showing that the previous reaction limit was broken (entry 6). Surprisingly, by monitoring the reaction process, a significant amount of unclosed products was identified at the early stage of the reaction in the case of other solvents, while these products were not obvious in DMI (entries 1-5, Figure S3). This phenomenon indicated that the ring-closing rate was also accelerated in DMI (Figure S4). Among all the results mentioned above, it was found that DMI performed obviously better than other solvents and had a favorable promoting effect on the depolymerization.
2.3. Probe into the Role of DMI
After finding that DMI had outstanding effect, a series of 1H NMR experiments were conducted to investigate the role of DMI during the depolymerization. Deuterated benzene was used as a weak hydrogen-bond acceptor solvent to minimize the effects of deuterated solvents. From Figure 3 (1), it could be observed that when BPA was mixed in a 1:1 molar ratio with DMEDA, the chemical shift of -OH proton in BPA was downfield. For example, the chemical shift of the phenol hydroxyl proton of free BPA was 3.75 ppm, while it shifted to 4.65 ppm when interacting with DMEDA (Figure 3 (1a)). It is well known that hydrogen bonding causes the depletion of electron density around the proton and the de-shielding of the nucleus, thus protons participating in hydrogen bonding display a low field shift in resonance frequency [31]. The change of chemical shift further proved the existence of hydrogen bond interaction between BPA and DMEDA as mentioned before. When 1 equiv. DMI was added to BPA and BPA-DMEDA system, the chemical shifts of the phenol hydroxyl proton of BPA were shifted to 6.81 ppm and 4.83 ppm, respectively (Figure 3 (1b, c)). It could be explained that the carbonyl oxygen of the amide portion of DMI, which acts as a good proton acceptor for BPA and DMEDA, thus leading to the chemical shift change of hydroxyl proton. Besides, when DMI was added, the chemical shift of the proton on the DMEDA amino group was observed from 0.94 ppm to 1.02 ppm as well, which proved that DMI readily forms hydrogen bonds with DMEDA (Figure 3 (1d, e)). The above results indicated that the addition of DMI changed the original hydrogen bonding network in the system.
In addition to the significant changes in the chemical shifts of the active hydrogen protons of BPA and DMEDA, Figure 3 (2) also showed that the chemical shifts of the -CH3 proton of BPA and the -CH2 and -CH3 protons of DMEDA underwent considerable changes. When 1 equiv. of DMI was added to the BPA-DMEDA system with a molar ratio of 1:1, the Δ δ value of the -CH3 proton of BPA was 0.13 (Figure 3(2a)). As the amount of DMI gradually increased to 30 equiv., the Δ δ value decreased to 0.02 (Figure 3(2i)). Similarly, with increasing amounts of DMI, the chemical shift changes of DMEDA also weakened. The Δ δ value of the -CH3 proton on DMEDA decreased from 0.19 to 0.01. When the amount of DMI was 30 equiv., the chemical shifts of BPA and DMEDA almost returned to their initial positions, similar to their free states. These results indicated that DMEDA and BPA were released, when DMI disrupted the hydrogen bonding interaction between DMEDA and BPA.
From the above results, a possible reaction mechanism of product-promoted depolymerization was proposed. The amino group of DMEDA nucleophilically attacked the carbon atom of the carbonyl group, yielding the intermediate monoaminocarbamate. Subsequently, due to the strong polarity of the added product DMI, the intermediate underwent an intramolecular cyclization pathway to form DMI and BPA. This step was very rapid, with little occurrence of oligomeric intermediates. On the other hand, DMI had a high donor number (DN) and acted as a strong electron donor (Table S1). The carbonyl oxygen in DMI was an excellent hydrogen bond acceptor, which disrupted the hydrogen bonding network between BPA and DMEDA in the system, causing DMEDA to exist in its free state. This allowed the ammonolysis reaction of BPA-PC to proceed completely (Figure 4).
2.4. Optimization of DMI Equivalent
After clarifying the mechanism of DMI, DMI additions were further reduced to improve the economics of the reaction process. The addition of DMI was adjusted to 1 equiv., and the depolymerization process was monitored as well. As shown in Figure 5 (1), with the addition of only 1 equiv. DMI, the reaction process was significantly promoted compared with that without the addition (Figure 1), and the final product yield increased from 70% to 90%. This result indicated that the addition of 1 equiv. DMI was sufficient to break the original upper limit of the reaction and promote the reaction to some extent. In addition, as shown in Figure 5 (2), the yields of BPA and DMI were 70% and 72%, respectively, for 3 h of reaction under solvent-free conditions, and after continuing the reaction for 2 h, the yields of the two products remained almost unchanged (72% and 73%, respectively). However, when 1 equiv. of DMI was added to the solvent-free system after 3 h of reaction and allowed to continue for 2 h, complete degradation of BPA-PC was achieved, and the yields of BPA and DMI increased to 96% and 98%, respectively. This phenomenon further proved that the addition of DMI does indeed have promoting effect on the depolymerization of BPA-PC. Compared with previous work, although the ammonolysis of BPA-PC has been achieved by using DMEDA as a nucleophilic reagent and DMI as a solvent, in this work, the crucial role of the depolymerization product DMI in promoting the reaction was revealed for the first time [32].
2.5. Degradation of Common BPA-PC Waste Plastics
In order to demonstrate the efficiency and practicality of the product-promoted depolymerization strategy for BPA-PC materials, degradation experiments were conducted on common PC products and PC/ABS or PC/PET mixed plastics (Figure 6), such as PC buckets, face shields, goggles, and CDs etc., under optimized conditions (80 °C, 1 equiv. DMI). The results showed that these products could be rapidly depolymerized, and the corresponding BPA was obtained in 92-99% yields and DMI was in 94-99% yields, respectively. It is noteworthy that the molecular weights of these BPA-PC commodities (Mn = 12.7 kg mol-1-19.2 kg mol-1) and the dispersities (Đ = 2.31-2.80) (Figure S5) were different, but all were suitable for this strategy. Furthermore, selective depolymerization of PC/ABS and PC/PET mixed plastics could also be achieved, providing BPA and DMI with >99% yields (Figure 6). These results offer a promising solution to address the challenges posed by plastic accumulation. Notably, chemical degradation of gram-scale BPA-PC tube material from post-consumer could also be successful in recovering high yields of BPA (91%) and DMI (83%) (Figure 7). These experimental results indicated that even with different additives present in commercial BPA-PC products, they have few or no impact on this depolymerization method, making this strategy highly applicable.
3. Materials and Methods
3.1. Materials and Reagents
The BPA-PC commodities (pellet, bucket, veil, goggle, disc, tube), polyethylene terephthalate (PET) bottle materials and acrylonitrile-butadiene-styrene copolymer (ABS) granular material were purchased on Taobao.com or amazon.cn and used without further purification. N,N'-dimethyl-ethylenediamine (DMEDA) (98%, Macklin), 1,3-dimethyl-2-imidazolidinone (DMI) (> 99%, Aladdin), bisphenol A (99%, Macklin) were used without further purification. Dibromomethane (98%, Energy Chemical) and hexamethylbenzene (98%, Energy Chemical) were used as internal standard. Dichloromethane (DCM) (> 99%), tetrahydrofuran (THF) (> 99%), ethyl acetate (EtOAc) (> 99%), toluene (Tol.) (> 99%), acetone (Ace.) (> 99%) were purchased as solvents at Sinopharm Chemical Reagent Co., Chloroform-d (> 99%) was purchased at Cambridge Isotope laboratories, Inc. Dimethyl sulfoxide-d6 (> 99%) and benzene-d6 (> 99%) were purchased at Energy Chemical. Chromatographic THF was purchased from Honeywell LTD for the analysis of GPC measurements.
3.2. Depolymerization of BPA-PC
The depolymerization of BPA-PC under solvent-free condition was carried out in a Schlenk flask at 80 °C. BPA-PC pellets (508 mg, 2 mmol based on BPA unit) were added first, followed by DMEDA (229 μL, 2.12 mmol). After the reaction to a certain time, dibromomethane (140 μL, 2 mmol) was added as the internal standard and acetic acid as the quencher. The reaction was monitored by 1H NMR spectrum. After 4 h, the yield of BPA was 73% and the yield of DMI was 74%. The depolymerization experiments of BPA-PC under the condition of DCM as a solvent were carried out at 50 °C, which was almost the same as the reaction conditions of the solvent-free depolymerization of BPA-PC mentioned above, and only an additional 2 mL of solvent was added.
Similarly, for the depolymerization of BPA-PC material under the condition of 1 equiv. DMI, an additional 216 µL DMI was required on the basis of solvent-free reaction condition. Furthermore, when degradation experiments were carried out with 1 gram of BPA-PC commercial tube material under this condition, it was observed that the fragments of BPA-PC tube disappeared after 1 h. The product was separated by silica gel column chromatography, the developing agent was n-hexane and ethyl acetate system, the volume ratio = 50-1:1, and then the products were vacuum dried and weighed, after that the yield was calculated.
3.3. The Determination of the Binding Constant
At room temperature, dry solid powder of BPA was dissolved in deuterobenzene (purified by vacuum distillation) to form a dilute solution of a certain concentration (0.0026 M), and 0.5 mL of it was loaded into a dry NMR tube to test its 1H NMR spectrum. Different equivalents of DMI (purified by vacuum distillation) were gradually dropped into the NMR tube using a micro-sampler, and their 1H NMR spectra were tested after shaking well. The ratio between the two substances was determined by integration of the 1H NMR spectra. The changes in the chemical shifts of the 1H NMR spectra after the interaction between the two substances were observed and recorded. The binding constant (K) was finally obtained by curve fitting in Origin [27,28].
3.4. Product Characterization
Nuclear magnetic resonance measurements were performed at room temperature on Bruker Advance instrument at 400 MHz (1H NMR), 100 MHz (13C NMR), CDCl3 and (CD3)2SO were used as an internal reference. Molecular weight (Mn) and dispersity (Đ) of the polymers were determined by gel permeation chromatography (GPC, Agilent 1260 LC, USA) using THF as the eluent (flow rate: 1 mL/min, at 40 °C). The chemical structures of the recovered products BPA and DMI were also confirmed by infrared spectroscopy and high-resolution mass spectrometry [33,34]. The FT-IR spectra were obtained on a Bruker Tensor 27 spectrophotometer. The high resolution mass spectrometry tests were carried out on the Bruke Maxis UHR TOF instrument.
4. Conclusions
In summary, a product-promoted depolymerization strategy for chemical recycling of BPA-PC was demonstrated. Using DMEDA as the depolymerization reagent, the reaction could be efficiently carried out under mild conditions with the promotion of the degradation product, DMI. The incorporation of 1 equiv. DMI destroyed the hydrogen bonding interactions between DMEDA and BPA, thus breaking the upper limit of the reaction and achieving a conversion of >95% for depolymerization. Different types of commercial BPA-PC plastics and PC/ABS or PC/PET mixed plastics, despite having different molecular weights and dispersities, were suitable for this strategy. This work is of great significance in promoting the chemical recycling of engineering plastic BPA-PC, and provides new idea and method for the sustainable use of plastic resources in the future.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org: 1H NMR spectrum of BPA-PC degradation reaction under solvent-free conditions for 24 h. (400 MHz, DMSO-d6, 298 K); Figure S2: Dissolution of 254 mg (1 mmol based on BPA unit) BPA-PC in 4 mL of different solvents at 50 °C; Figure S3: 1H NMR spectra of BPA-PC degradation in different solvents for 2 h. (400 MHz, DMSO-d6, 298 K); Figure S4: Proposed mechanism; Figure S5: GPC analysis for BPA-PC disc, goggle, veil, bucket; Figure S6: 1H NMR spectrum of BPA (400 MHz, DMSO-d6, 298 K); Figure S7: 13C NMR spectrum of BPA. (400 MHz, DMSO-d6, 298 K); Figure S8: 1H NMR spectrum of DMI. (400 MHz, DMSO-d6, 298 K); Figure S9: 13C NMR spectrum of DMI. (400 MHz, DMSO-d6, 298 K); Figure S10: IR spectrum of BPA; Figure S11: IR spectrum of DMI; Figure S12: MS spectrum of BPA; Figure S13: MS spectrum of DMI; Table S1. Donor Number values of common compounds.
Author Contributions
Conceptualization, G.X. and Q.W.; methodology, H.S.; validation, M.C. and R.Y.; formal analysis, G.X. and Q.W.; investigation, M.C.; resources, Q.W.; data curation, M.C.; writing—original draft preparation, M.C.; writing—review and editing, G.X., M.C. and R.Y.; supervision, Q.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China, grant number 21901249; Taishan Scholars Program of Shandong Province, grant number tsqn201812112; the Scientific Research and Innovation Fund Project of Shandong Energy Research Institute, grant number SEI I202004, and the Major Science and Technology Innovation Program of Shandong Province, grant number 2022CXGC020604.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Potter, H. V. Plastics and some recent applications. Nature 1939, 143, 785-787. [CrossRef]
- Bergmann, M.; Collard, F.; Fabres, J.; Gabrielsen, G. W.; Provencher, J. F.; Rochman, C. M.; Sebille, E.; Tekman, M. B. Plastic pollution in the arctic. Nat. Rev. Earth Environ. 2022, 3, 323-337. [CrossRef]
- Fan, Y. V.; Jiang, P.; Tan, R. R.; Aviso, K.B.; You, F.; Zhao, X.; Lee, C. T.; Klemeš, J. J. Forecasting plastic waste generation and interventions for environmental hazard mitigation. J. Hazard. Mater. 2022, 424, 127330. [CrossRef]
- Zhao, X.; You, F. Life cycle assessment of microplastics reveals their greater environmental hazards than mismanaged polymer waste losses. Environ. Sci. Technol. 2022, 56, 11780-11797. [CrossRef]
- Zhao, X.; Korey, M.; Li, K.; Copenhaver, K.; Tekinalp, H.; Celik, S.; Kalaitzidou, K.; Ruan, R.; Ragauskas, A. J.; Ozcan, S. Plastic waste upcycling toward a circular economy. Chem. Eng. J. 2022, 428, 131928. [CrossRef]
- Garcia, J. M.; Robertson, M. L. The future of plastics recycling. Science 2017, 358, 870-872. [CrossRef]
- Ignatyev, I. A.; Thielemans, W.; Vander Beke, B. Recycling of polymers: a review. ChemSusChem 2014, 7, 1579-1593. [CrossRef]
- Vollmer, I.; Jenks, M. J. F.; Roelands, M. C. P.; White, R. J.; Harmelen, T.; Wild, P.; Laan, G. P.; Meirer, F.; Keurentjes, J. T. F.; Weckhuysen, B. M. Beyond mechanical recycling: giving new life to plastic waste. Angew. Chem. Int. Ed. 2020, 59, 15402-15423. [CrossRef]
- Badia, J. D.; Ribes-Greus, A. Mechanical recycling of polylactide, upgrading trends and combination of valorization techniques. Eur. Polym. J. 2016, 84, 22-39. [CrossRef]
- Petrus, R.; Bykowski, D.; Sobota, P. Solvothermal alcoholysis routes for recycling polylactide waste as lactic acid esters. ACS Catal. 2016, 6, 5222-5235. [CrossRef]
- Xu, G.; Wang, Q. Chemically recyclable polymer materials: polymerization and depolymerization cycles. Green Chem. 2022, 24, 2321-2346. [CrossRef]
- McKeown, P.; Kamran, M.; Davidson, M. G.; Jones, M. D.; Román-Ramírez, L. A.; Wood, J. Organocatalysis for versatile polymer degradation. Green Chem. 2020, 22, 3721-3726. [CrossRef]
- Chen, X.; Wang, Y.; Zhang, L. Recent progress in the chemical upcycling of plastic wastes. ChemSusChem 2021, 14, 4137-4151. [CrossRef]
- Chen, H.; Wan, K.; Zhang, Y.; Wang, Y. Waste to wealth: chemical recycling and chemical upcycling of waste plastics for a great future. ChemSusChem 2021, 14, 4123-4136. [CrossRef]
- Zhang, S.; Hu, Q.; Zhang, Y.-X.; Guo, H.; Wu, Y.; Sun, M.; Zhu, X.; Zhang, J.; Gong, S.; Liu, P.; et al. Depolymerization of polyesters by a binuclear catalyst for plastic recycling. Nat. Sustain. 2023, 6, 965-973. [CrossRef]
- Cederholm, L.; Wohlert, J.; Olsén, P.; Hakkarainen, M.; Odelius, K. "Like recycles like”: selective ring-closing depolymerization of poly(L-lactic acid) to L-lactide. Angew Chem. Int. Ed. 2022, 61, e202204531. [CrossRef]
- An, W.; Liu, X.; Li, J.; Zhao, X.; Long, Y.; Xu, S.; Wang, Y.-Z. Water-solvent regulation on complete hydrolysis of thermosetting polyester and complete separation of degradation products. J. Hazard. Mater. 2023, 453, 131423. [CrossRef]
- Höcker, H.; Keul, H. Ring-opening polymerization and ring-closing depolymerization. Adv. Mater. 1994, 6, 21-36. [CrossRef]
- Endo, T.; Nagai, D. A novel construction of ring-opening polymerization and chemical recycling system. Macromol. Symp. 2005, 226, 79-86. [CrossRef]
- Olsén, P.; Odelius, K.; Albertsson, A.-C. Thermodynamic presynthetic considerations for ring-opening polymerization. Biomacromolecules 2016, 17, 699-709. [CrossRef]
- Olsén, P.; Undin, J.; Odelius, K.; Keul, H.; Albertsson, A.-C. Switching from controlled ring-opening polymerization (cROP) to controlled ring-closing depolymerization (cRCDP) by adjusting the reaction parameters that determine the ceiling temperature. Biomacromolecules 2016, 17, 3995-4002. [CrossRef]
- Xiong, W.; Chang, W.; Shi, D.; Yang, L.; Tian, Z.; Wang, H.; Zhang, Z.; Zhou, X.; Chen, E.-Q; Lu, H. Geminal dimethyl substitution enables controlled polymerization of penicillamine-derived β-thiolactones and reversed depolymerization. Chem 2020, 6, 1831-1843. [CrossRef]
- Yang, R.; Xu, G.; Dong, B.; Hou, H.; Wang, Q. A “polymer to polymer” chemical recycling of PLA plastics by the “DE–RE polymerization” strategy. Macromolecules 2022, 55, 1726-1735. [CrossRef]
- Zhou, X.; Liu, Q.; Xu, G.; Yang, R.; Sun, H.; Wang, Q. Chemical upcycling of poly(lactide) plastic waste to lactate ester, lactide and new poly(lactide) under Mg-catalysis condition. Chin. Chem. Lett. 2023, 34, 108158. [CrossRef]
- Yang, R.; Xu, G.; Dong, B.; Guo, X.; Wang, Q. Selective, sequential, and “one-pot” depolymerization strategies for chemical recycling of commercial plastics and mixed plastics. ACS Sustainable Chem. Eng. 2022, 10, 9860-9871. [CrossRef]
- Zhou, X.; Chai, M.; Xu, G.; Yang, R.; Sun, H.; Wang, Q. Catalyst-free amino-alcoholysis depolymerization strategy: a facile and powerful tool for chemical recycling of poly(bisphenol A carbonate). Green Chem. 2023, 5, 952-959. [CrossRef]
- Monaco, M. R.; Prévost, S.; List, B. Catalytic asymmetric synthesis of thiols. J. Am. Chem. Soc. 2014, 136, 16982-16985. [CrossRef]
- Monaco, M. R.; Poladura, B.; Diaz de Los Bernardos, M.; Leutzsch, M.; Goddard, R.; List, B. Activation of carboxylic acids in asymmetric organocatalysis. Angew. Chem. Int Ed. 2014, 53, 7063-7067. [CrossRef]
- Luo, Y.; Ma, H.; Sun, Y.; Che, P.; Nie, X.; Wang, T.; Xu, J. Understanding and measurement for the binding energy of hydrogen bonds of biomass-derived hydroxyl compounds. J. Phys. Chem. A 2018, 122, 843-848. [CrossRef]
- Luo, Y.; Ma, H.; Zhang, S.; Zheng, D.; Che, P.; Liu, X.; Zhang, M.; Gao, J.; Xu, J. Binding energy as driving force for controllable reconstruction of hydrogen bonds with molecular scissors. J. Am. Chem. Soc. 2020, 142, 6085-6092. [CrossRef]
- Scheiner, S. Assessment of the presence and strength of H-Bonds by means of corrected NMR. Molecules 2016, 21, 1426. [CrossRef]
- Hata, S.; Goto, H.; Yamada, E.; Oku, A. Chemical conversion of poly(carbonate) to 1,3-dimethyl-2-imidazolidinone (DMI) and bisphenol A: a practical approach to the chemical recycling of plastic wastes, Polymer 2002, 43, 2109-2116. [CrossRef]
- Yang, J.; Li, Y.; Wang, J.; Sun, X.; Shah, S. M.; Cao, R.; Chen, J. Novel sponge-like molecularly imprinted mesoporous silica material for selective isolation of bisphenol A and its analogues from sediment extracts. Anal. Chim. Acta 2015, 853, 311-319. [CrossRef]
- Mizuno, T.; Nakai, T.; Mihara, M. Efficient solvent-free synthesis of urea derivatives using selenium-catalyzed carbonylation of amines with carbon monoxide and oxygen. Synthesis 2010, 2010, 4251-4255. [CrossRef]
Figure 1.
The degradation of BPA-PC under solvent-free conditions at 80 °C: BPA-PC pellets (sizes: 3-4 mm, Mn = 23.8 kg/mol, Đ = 2.78, 508 mg, 2 mmol based on BPA unit), N,N'-Dimethyl-1,2-ethanediamine (DMEDA) (229 μL, 2.12 mmol), acetic acid as quencher, dibromomethane (140 μL, 2 mmol) as an internal standard to calculate yields.
Figure 1.
The degradation of BPA-PC under solvent-free conditions at 80 °C: BPA-PC pellets (sizes: 3-4 mm, Mn = 23.8 kg/mol, Đ = 2.78, 508 mg, 2 mmol based on BPA unit), N,N'-Dimethyl-1,2-ethanediamine (DMEDA) (229 μL, 2.12 mmol), acetic acid as quencher, dibromomethane (140 μL, 2 mmol) as an internal standard to calculate yields.
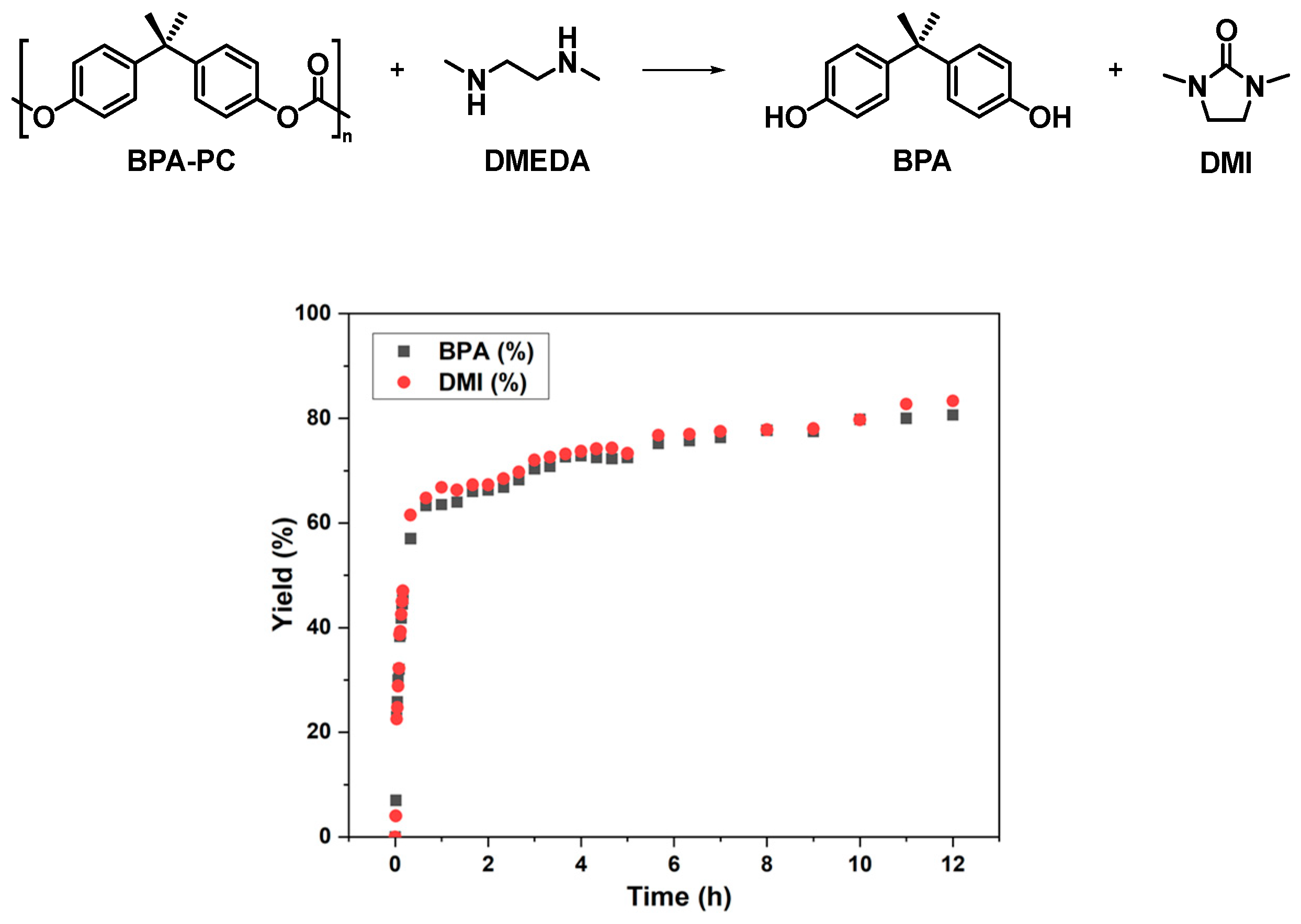
Figure 2.
Determination of binding constant: BPA and N,N'-dimethyl-1,2-ethanediamine (DMEDA).
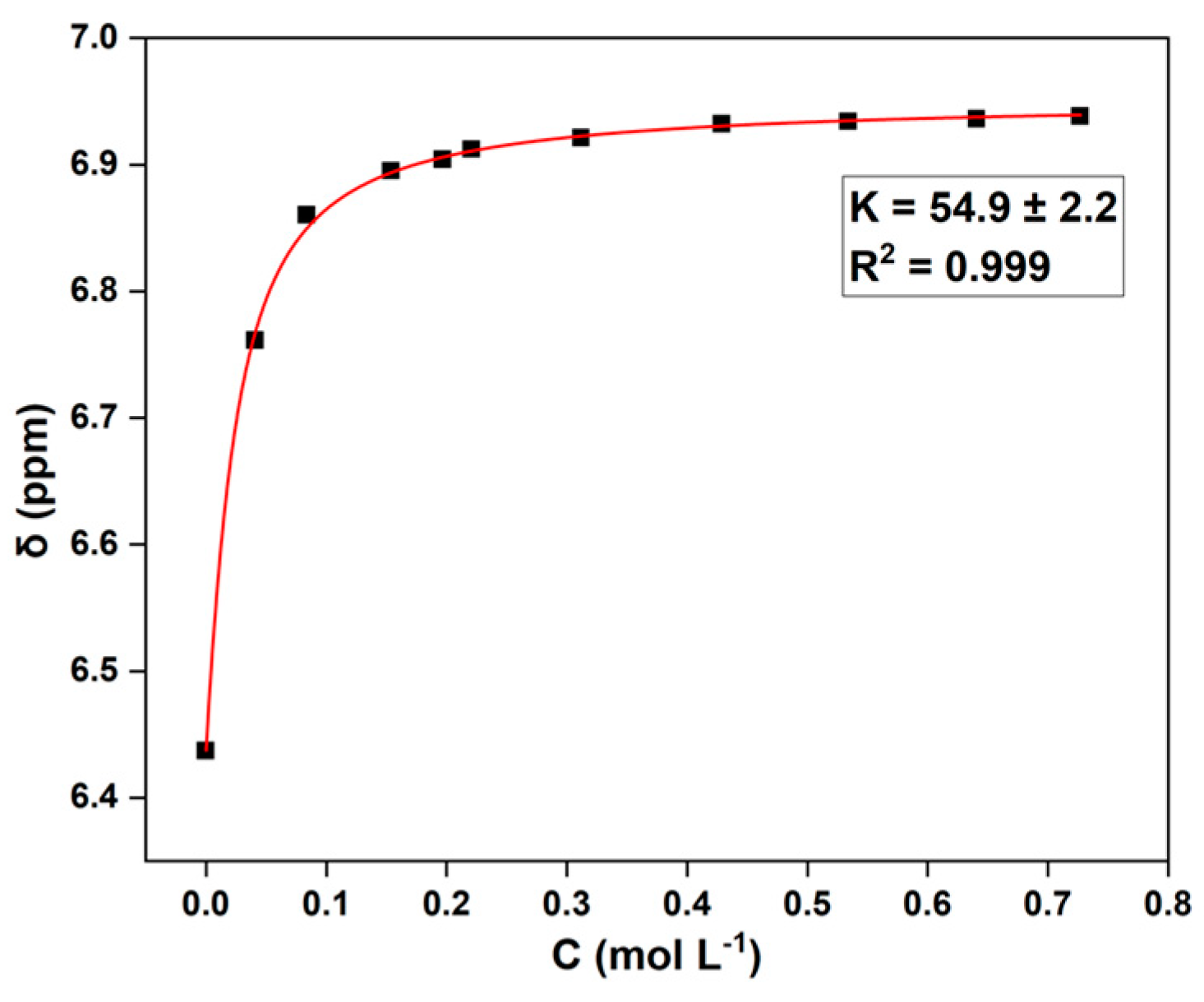
Figure 3.
The 1H NMR overlapped spectra of chemical shifts of BPA, DMEDA and DMI at different DMI concentrations in benzene-d6.
Figure 3.
The 1H NMR overlapped spectra of chemical shifts of BPA, DMEDA and DMI at different DMI concentrations in benzene-d6.
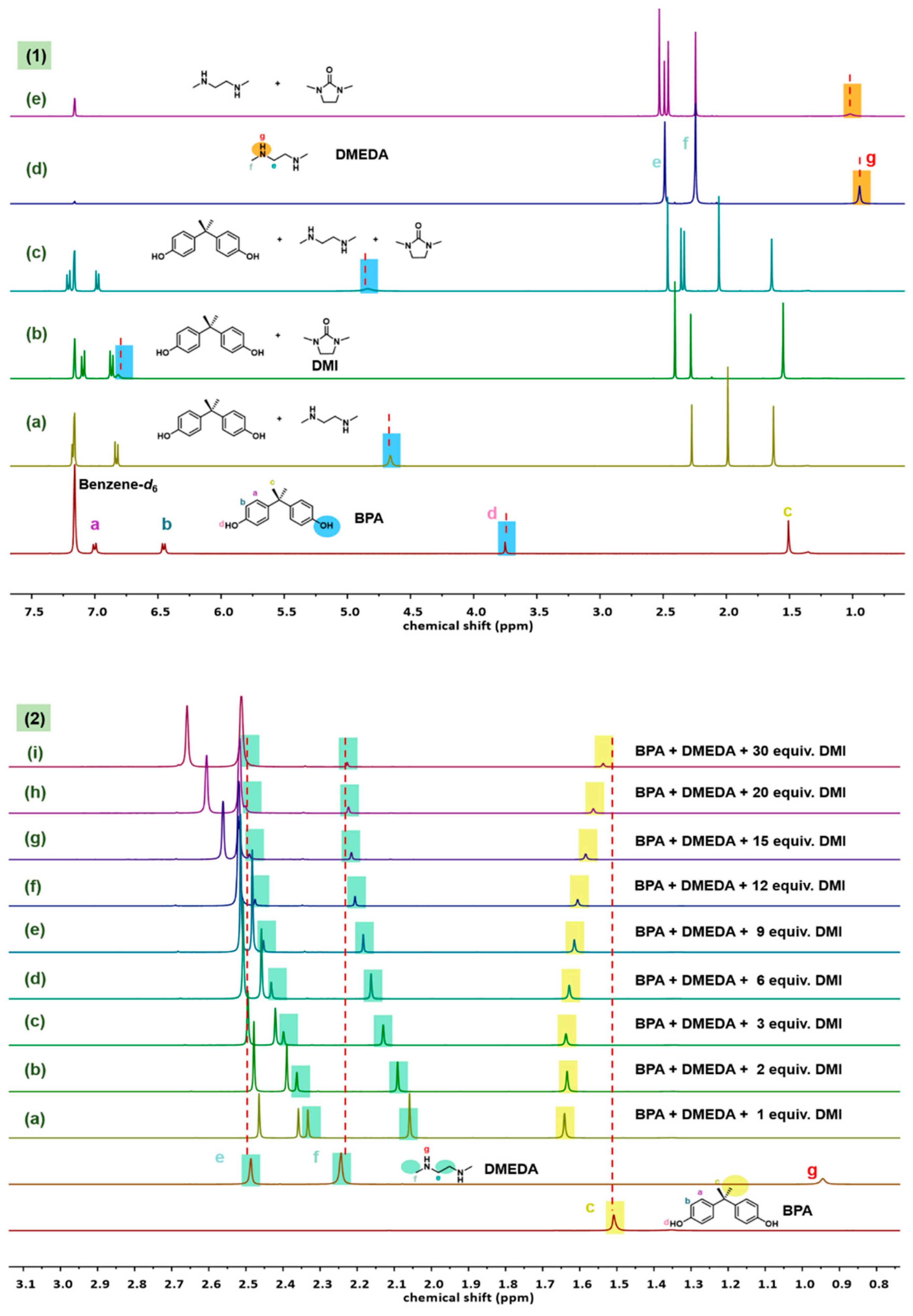
Figure 4.
Schematic diagram of H-bond breaking after adding DMI molecules to BPA and DMEDA system.
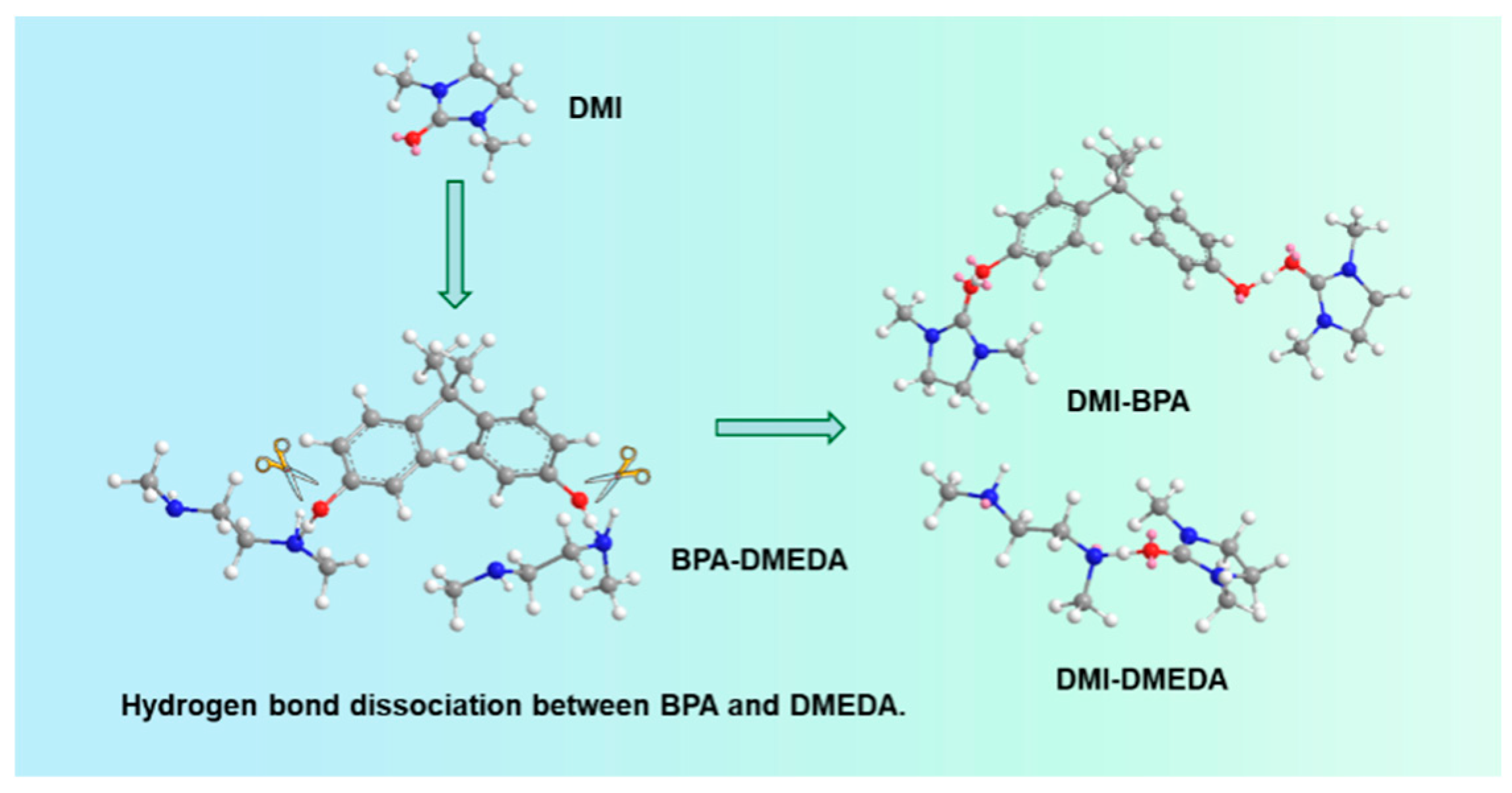
Figure 5.
(1) The degradation of BPA-PC under 1 equiv. DMI conditions at 80 °C: BPA-PC pellets (508 mg, 2 mmol based on BPA unit), N,N'-dimethyl-1,2-ethanediamine (DMEDA) (229 μL, 2.12 mmol), dibromomethane (140 μL, 2 mmol) as an internal standard, acetic acid as quencher. (2) The depolymerization of BPA-PC by DMEDA under solvent-free and after adding 1 equiv. DMI conditions.
Figure 5.
(1) The degradation of BPA-PC under 1 equiv. DMI conditions at 80 °C: BPA-PC pellets (508 mg, 2 mmol based on BPA unit), N,N'-dimethyl-1,2-ethanediamine (DMEDA) (229 μL, 2.12 mmol), dibromomethane (140 μL, 2 mmol) as an internal standard, acetic acid as quencher. (2) The depolymerization of BPA-PC by DMEDA under solvent-free and after adding 1 equiv. DMI conditions.
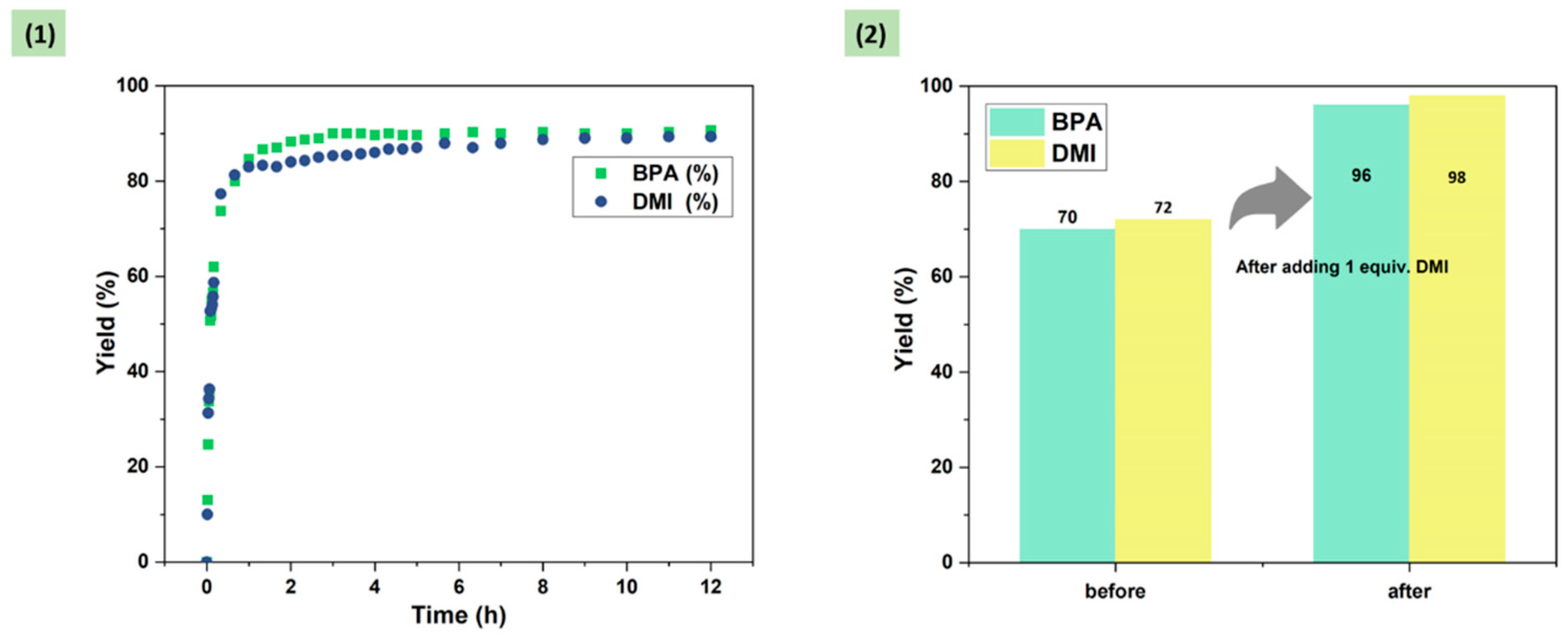
Figure 6.
The degradation of BPA-PC commodities. Depolymerization conditions: BPA-PC commodities (254 mg, 1 mmol based on BPA unit), N,N'-dimethyl-1,2-ethanediamine
Figure 6.
The degradation of BPA-PC commodities. Depolymerization conditions: BPA-PC commodities (254 mg, 1 mmol based on BPA unit), N,N'-dimethyl-1,2-ethanediamine
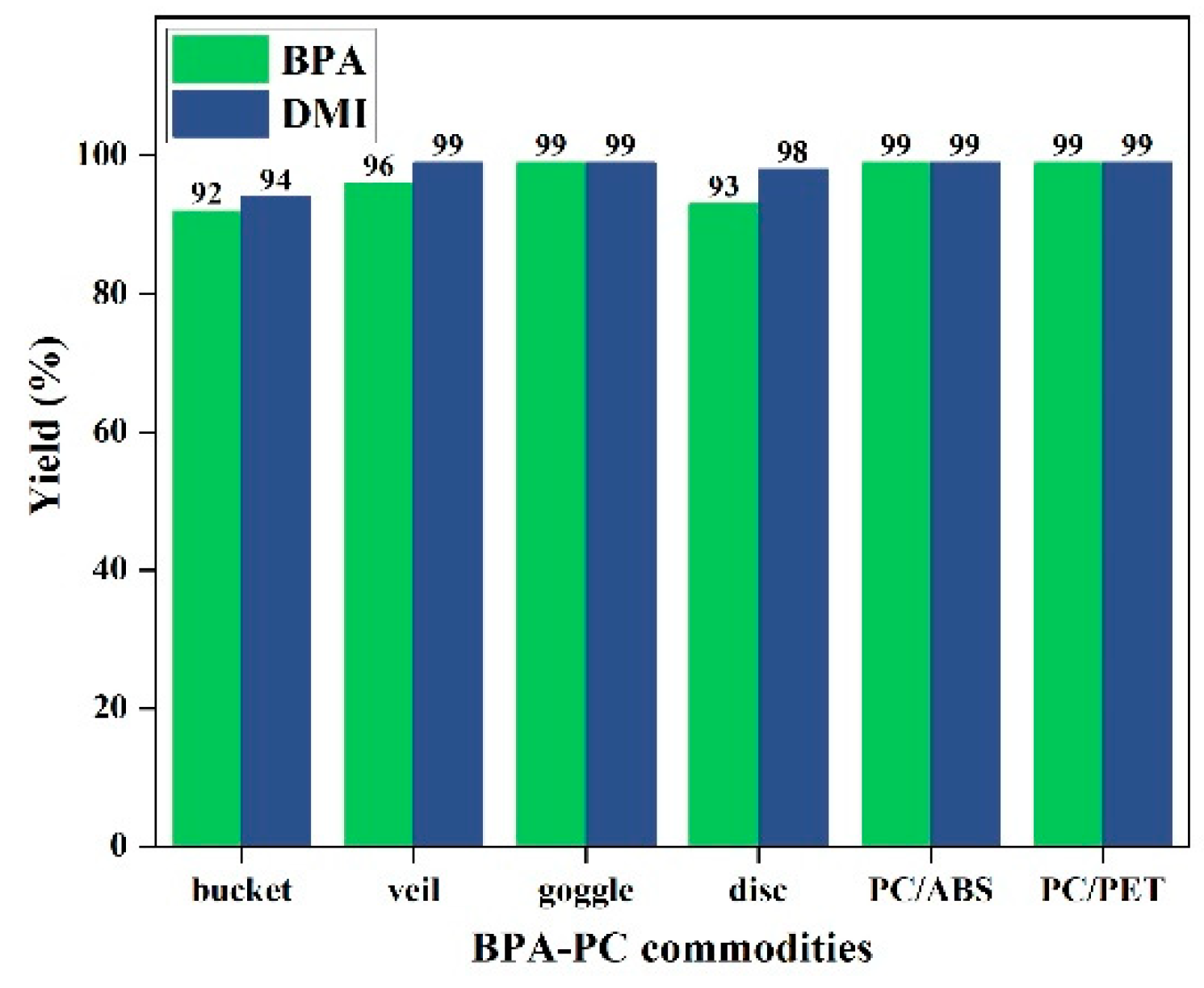
Figure 7.
The depolymerization of after-used BPA-PC tube.
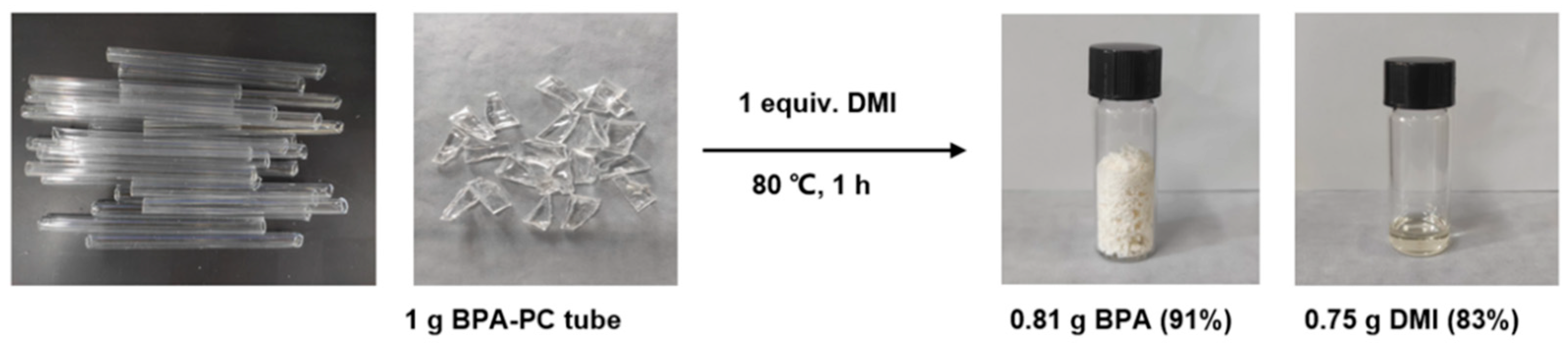

Table 1.
The degradation of BPA-PC in different solvents.
| Entry | Solvent | Yield of BPA (%) | Yield of DMI (%) |
|---|---|---|---|
| 1 | DCM | 77 | 79 |
| 2 | THF | 68 | 66 |
| 3 | EtOAc | 75 | 75 |
| 4 | Tol. | 72 | 71 |
| 5 | Ace. | 70 | 68 |
| 6 | DMI | 87 | 85 |
Depolymerization conditions: BPA-PC pellets (508 mg, 2 mmol based on BPA unit), N,N'-dimethyl-1,2-ethanediamine (DMEDA) (229 μL, 2.12 mmol), hexamethylbenzene (64 mg, 0.4 mmol) as an internal standard to calculate yields, acetic acid as quencher, Vsolvent = 2 mL. Reaction at 50 °C for 24 h.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



