
Submitted:
03 January 2024
Posted:
04 January 2024
You are already at the latest version
Abstract
Autosomal dominant polycystic kidney disease (ADPKD) is the most common hereditary kidney disorder but kidneys are not the only organs involved in this systemic disorder. Individuals with the condition may display additional manifestations beyond the renal system, involving liver, pancreas and brain in the con-text of cystic manifestations, while vascular system, gastrointestinal tract, bones, and cardiac valves in the context of non-cystic manifestations. Despite kidney involvement remain the main feature of the disease, thanks to new generation therapies, early diagnosis and better management of kidney related problems, a new wave of complications must be faced from clinicians who treated polycystic patients. Involvement of the liver represents the most prevalent extrarenal manifestation and grows importance in symptom burden and quality of life. Vascular abnormalities are a key factor for patients’ life expectancy and there’s still de-bate whether to screen or not to screen all patients. Arterial hypertension is often the earliest onset among polycystic patients leading to frequent cardiovascular complications. Although cardiac valvular abnormali-ties is a frequent complication, it rarely leads to relevant problems in the clinical history of polycystic pa-tients. One of the new relevant aspects concern bone disorders, that can exert a considerable influence on the clinical course of these patients. This review aims to provide the “state of art” among extrarenal mani-festation of ADPKD.
Keywords:
ADPKD
; Ciliopathies
; Cystic kidney disease
; Genetic
; Extrarenal cystic involvement
; PLD
; Intracranial aneurysms
; Bone disorders
1. Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is the most prevalent hereditary kidney disorder, estimated to affect approximately one in every 1000 to 2500 individuals [1]. The majority of ADPKD cases, around 78%, result from mutations in the PKD1 gene, while 15% are attributed to mutations in the PKD2 gene. However, a small portion of families exhibit mutations in genes such as GANAB, ALG9, or DNAJB11 [2,3].
PKD1 is situated on chromosome 16 (16p13.3) and codes for polycystin-1 (PC1), a large glycoprotein featuring multiple domains that undergo cleavage at a G protein-coupled receptor proteolytic site. Conversely, PKD2, located on chromosome 4 (4q21), encodes polycystin-2 (PC2), a member of the transient receptor potential family of calcium-regulated cation channels. Both PC1 and PC2 are present on primary cilia, antenna-like organelles crucial for mechano transduction. It is hypothesized that PC1 and PC2 convey information from the external environment to the cell. Cystogenesis occurs when the concentration of PC1 or PC2 falls below a certain threshold, while higher levels of PC1 and PC2 inhibit cyst formation in a dose-dependent manner [4,5]. The ADPKD mutation database has cataloged over 1500 distinct mutations in PKD1 and PKD2. The pace of disease advancement in individual patients is primarily determined by the specific causative gene mutation. Patients with PKD2 mutations tend to develop fewer cysts and experience slower progression as compared to individuals with PKD1 mutations [6]. Furthermore, truncating mutations in PKD1 typically result in a more severe form of the disease than missense mutations [7].
Clinically, ADPKD is characterized by both cystic and non-cystic manifestations. The main clinical characteristic involves the formation and gradual enlargement of multiple fluid-filled cysts scattered throughout the renal parenchyma. This leads to a gradual decline in renal function over several decades, often resulting in end-stage renal disease (ESRD) around or after the sixth decade of life. Hypertension is a frequently observed early manifestation in ADPKD, occurring in 50% to 70% of cases before any substantial decline in glomerular filtration rate (GFR). The average age of onset for hypertension is around 30 years and appears to be more prevalent in patients with enlarged kidneys or reduced GFR [8,9].
Additional complications associated with renal cyst growth and expansion include urinary tract infections, concentrating defects, hematuria, nephrolithiasis, and acute or chronic flank and abdominal pain. Proteinuria is not a typical feature of the disease, but its presence as a manifestation of chronic kidney disease (CKD) could worsen the prognosis [10]. Although less common, renal cell carcinoma (RCC) can also be a complication of ADPKD [11].
The kidney, however, is not the only organ damaged in ADPKD: affected individuals might exhibit extrarenal manifestations, involving liver and pancreas in the context of cystic manifestations, and in the gastrointestinal tract, vascular system, bones, and cardiac valves in the context of non-cystic manifestations.
Further, by causing a reduction in glomerular filtration, ADPKD is also characterized by the usual complications of CKD, such as anaemia, secondary hyperparathyroidism, metabolic bone disease, inadequate nutrition and increased cardiovascular risk.
2. Pathogenesis
ADPKD is classified as a ciliopathy and primarily arises from mutations in two genes: PKD1 (polycystic kidney disease-1) and PKD2 (polycystic kidney disease-2). These genes encode polycystin-1 (PC1) and polycystin-2 (PC2), respectively [12]. Cilia are conserved structures found in various organisms, and mutations affecting primary cilia, known as immotile cilia, can lead to ADPKD. Immotile cilia possess an axoneme, which is a cytoskeletal scaffold composed of nine microtubule doublets. Intra-flagellar transport (IFT) allows the movement of components into and out of cilia [13]. The axoneme is tethered to the cell through a basal body originating from the centriole [14].
Primary cilia, typically non-motile monocilia, are situated on the surface of differentiated cells that are not actively dividing. They often exhibit a "9+0" microtubule configuration, featuring nine pairs of microtubules located in the outer part of the cell without a central apparatus or dynein arms. Primary cilia act as sensory organelles, detecting extracellular signals and functioning as surface mechano- or chemoreceptors. They sense changes in osmolality, light, temperature, gravity, and play critical roles in development and tissue differentiation. Various essential receptors, such as sonic hedgehog (SHH), epidermal growth factor receptor (EGFR), and platelet-derived growth factor receptor (PDGFR), are expressed on the surface of primary cilia. Signaling pathways associated with primary cilia include calcium, SHH, Wnt, mTOR, JAK/STAT, and MAPK, which play crucial roles in growth, distinctiveness, control of the cell cycle, programmed cell death, tissue homeostasis and alignment of cell orientation [15,16,17,18,19,20,21].
ADPKD belongs to a category of conditions referred to as primary ciliopathies, which encompass a range of syndromes and diseases involving multiple systems. Other examples of primary ciliopathies include Jeune syndrome, nephronophthisis, and Bardet-Biedl syndrome [13].
Primary cilia function as cellular antennae, responding to external stimuli and converting them into intracellular signals to regulate cellular functions. These stimuli can include physical stresses such as flow and pressure, as well as chemical substances like ligands, growth factors, and morphogens. The primary cilium possesses mechanosensing abilities, allowing it to detect fluid flow. Polycystin-1 and polycystin-2 are among the ciliary proteins responsible for mechanosensing [22,23].
PC1 is a receptor-like protein featuring an extensive extracellular N terminus, 11 membrane-spanning domains, and a brief cytoplasmic C terminus. While PC1 exhibits high expression in fetal renal tissue, its expression is subdued in adult tissue. It is found in the cilium, plasma membrane, and adhesion complex in polarized epithelial cells, suggesting its involvement in protein-protein interactions, cell-cell adhesion, and cell-matrix interactions [24,25,26]. PC2 is an integral six-transmembrane protein with intracellular N and C termini. It functions as a nonselective calcium-permeable transient receptor potential channel [27,28]. While PC2 is co-localized with PC1 both in cilium and plasma membrane, a significant part of PC2 is found inside the cell, where it plays a role in releasing calcium from intracellular stores [29,30,31]. PC1 and PC2 establish a complex by means of their C-terminal tails, contributing to intracellular calcium regulation. Activation of the PC1-PC2 channel complex occurs in response to ciliary bending, leading to signal transduction triggered by chemical or mechanical stimuli [23]. PC2, together with inositol 1, 4, 5-triphosphate receptor (IP3R) and ryanodine receptor, indirectly regulates cytoplasmic calcium levels [31,36,37].
The complete pathological mechanisms of ADPKD are not yet fully understood, but the loss of function of PC1 and/or PC2 proteins contributes to its pathogenesis through various signaling pathways [35]. The PC1-PC2 complex acts as a transient receptor potential channel involved in maintaining intracellular calcium homeostasis and calcium release. Interruption of the interaction of PC1/PC2 leads to decreased intracellular calcium levels, resulting in upregulated cyclic adenosine monophosphate (cAMP) signaling and increased cell proliferation [38,39,40,41]. PC1 and PC2 engage with numerous pathways, including the mTOR and JAK-STAT pathways, to inhibit cell growth. PC2 also reduces cell proliferation through its binding to eukaryotic translation elongation initiation factor 2a (eIF2a) and pancreatic ER-resident eIF2a kinase [42,43,44]. PC1 and PC2 also play roles in the Wnt signaling pathway, which regulates cell profliferation, specialization and orientation. Defects in planar cell polarity can trigger renal tubule expansion and cyst formation [45].
PKD1 gene’s mutation more often result in a more severe form of ADPKD and appear first compared to mutations in PKD2 [46,47]. The ADPKD mutation database has indexed over 1500 different mutations in PKD1 and PKD2, with the position of each mutation determining the gravity. Truncating mutations often lead to more severe phenotypes than non-truncating mutations [49,50]. ADPKD exhibits genetic dominance at the organismal level but operates with a recessive mechanism at the cellular level. Cysts specifically develop in certain kidney tubules and hepatic bile ducts. Nevertheless, within adult tissues, both alleles of the mutated polycystic gene experience a recessive loss of function, leading to the development of cysts in a subset of tubular epithelial cells. This phenomenon is illustrated by the "second hit" hypothesis, where the presence of an inherited PKD1 or PKD2 mutation leads to cysts only if the remaining normal copy of the gene acquires a somatic mutation [52,53,54].
3. Extrarenal Cystic Manifestations
3.1. Liver
The liver is the most common organ involved in ADPKD, with liver cysts present in more than 90% of ADPKD patients. Current literature generally considers Polycystic liver disease (PLD) as the existance of over 20 liver cysts [55]. PLD development is linked to structural alterations in the biliary tree, resulting in cyst formation, that normally occour early in the disease process [56,57,58]. Symptoms are due to cysts growth and tipically occours in adulthood. [59]. Cysts distribution could be focal or diffuse to the whole organ. [60] The prevalence of PLD has increased due to factors such as longer life expectancy and improved kidney survival. PLD associated with ADPKD is genetically distinct from isolated PLD but follows a similar clinical course, featuring hepatomegaly caused by multiple cysts while preserving liver function [61,62,63]. Several risk factors plays a role in the formation of more severe PLD. These include the severity of the renal lesion, feminine gender, exogenous estrogen exposure and repeated pregnancies. Although both males and females with ADPKD have a similar overall prevalence of PLD, cysts in females tend to be bigger and to appear earlier. The growth of hepatic cysts in females may be accelerated due to steroid hormones, as evidenced by the association between postmenopausal estrogen and selective enlargement of hepatic cysts and parenchyma [63,64,65,66]. Furthermore, studies have shown that 58% of females older than 48 years with severe PLD, experience a regression in liver volume, while the liver continues to enlarge in males. Therefore, women with severe polycystic liver disease should refrain from hormone replacement therapies and contraceptives containing estrogen [67,68,69]. The risk of developing severe PLD is is not influenced by the ADPKD genotype but is associated with the gravity of renal disease [64]. Symptoms in PLD depends on the number, size, location, and distribution of cysts, which contribute to hepatomegaly. Liver cysts often remain asymptomatic and rarely lead to hepatic function impairment. However, approximately 20% of these patients experience symptomatic PLD. Symptoms are typically associated with progressive liver enlargement and may include pain, dispepsia, gastroesophageal reflux, and, when the size compress the portal vein can lead to portal hypertension and ascites. Extensive enlargement of the liver can exert pressure on the nearby gastrointestinal tract, blood vessels, and diaphragm, resulting in diverse symptoms. Moreover, complications like cyst hemorrhage, rupture, or infection may give rise to additional symptoms. Highly symptomatic PLD has become more common due to the increased life expectancy of ADPKD patients[70,71,72,73,74,75,76,77].
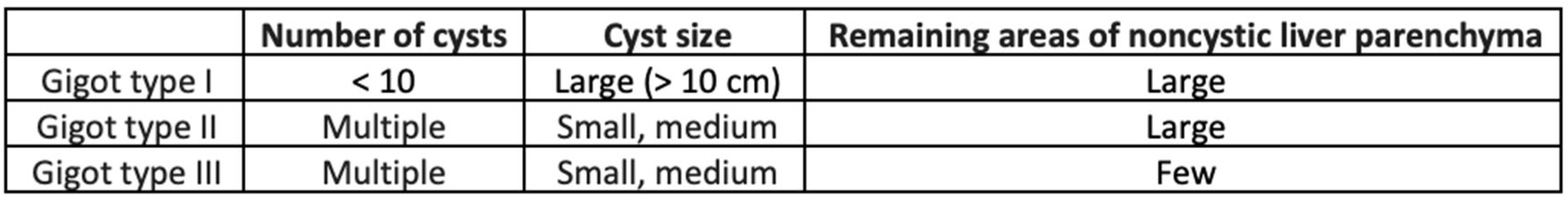
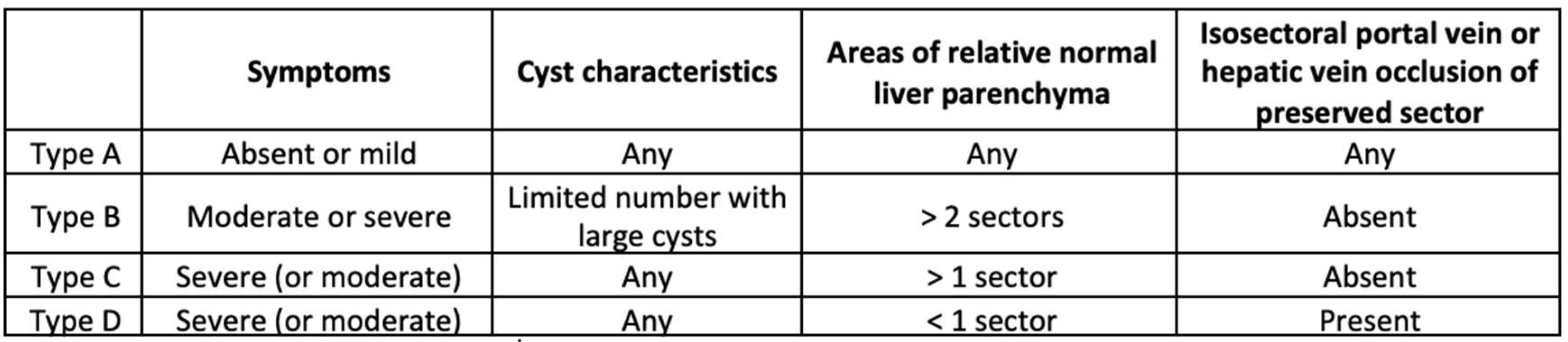
The most used clinical classifications on PLD were written by Gigot and Schnelldorfer [78,79] (respectively Table 1 and Table 2)
The Gigot classification serves to differentiate between phenotypes by considering the number, the size and the amount of liver involved [78]. Gigot classification relies on imaging studies, but has the limits not to include symptoms and not to evaluate advancement of the condition.

Table 1.
Gigot Classification.
 |
The Schnelldorfer classification incorporates factors such as the quantity and size of cysts, the volume of remaining liver parenchyma, the input and output of previously preserved liver segments, and the presence of symptoms [74].
Table 2.
Schnelldorfer Classification.
 |
For the assessment of PLD symptoms, two specific questionnaires have been created and validated: POLCA (PLD complaint-specific assessment) [80] and PLD-Q (PLD questionnaire) [81]. Following completion, the total score is determined by summing individual symptom scores, with a higher total score indicating a greater disease burden. Patients actively participated in the development of PLD-Q, unlike the development of POLCA, resulting in distinct sets of items. Nevertheless, both questionnaires are applicable for gauging disease burden and evaluating changes in symptom burden post-treatment. Since treatment is recommended exclusively for symptomatic patients with hepatomegaly, both instruments can serve as novel clinical endpoints [82]. Liver volume, measured through CT or MRI volumetry using (semi-)automatic software, acts as a prognostic marker impacting both symptom burden and quality of life [82]. Two classifications are available to differentiate between mild, moderate, and severe phenotypes based on htTLV [62,83]. In the classification introduced by Hogan MC et al., mild PLD is arbitrarily defined as height-adjusted total liver volume (htLV) <1000 mL/m, while htLV between 1000 - 1800 mL/m is considered moderate PLD. They designate severe PLD as htLV >1800 mL/m, aligned with the plateauing of height-adjusted liver parenchymal volume (htLPV) at this cutoff point. This point approximately corresponds to height-adjusted liver cyst volume (htLCV) >700 mL/m [62]. In the classification put forth by Kim H et al., they categorize patients with PLD into three groups based on their htLV: no or mild PLD (htTLV < 1.600 mL/m), moderate PLD (1.600 ≤ htLV < 3.200 mL/m), and severe PLD (htLV ≥ 3.200 mL/m) [83].
In the management of PLD, therapeutic approaches are tailored to the individual patient based on their specific needs. One common complaint among patients is related to symptoms caused by progressive hepatic enlargement. The only medical therapy that has been shown to reduce liver dimension and improve quality of life in these symptomatic patients is the use of somatostatin analogues (SAs). Somatostatin hinders the production of cyclic adenosine monophosphate (cAMP) in cystic cholangiocytes. This compound is excessively produced in PLD and contributes to cellular growth and cystic fluid production. By decreasing fluid secretion and cell proliferation, SAs can reduce liver volume. Studies have demonstrated that six months of lanreotide injections can lead to a decrease in liver volume, and extending the treatment for an additional six months may lead to the stabilization of hepatic volume. Conversely, the use of vasopressin receptor 2 (V2) antagonists does not seem to reduce liver volume [84,85,86].
When symptoms are caused by a dominant cyst, patients may be eligible for aspiration sclerotherapy (AS). AS is a less invasive procedure that entails puncturing the cyst with radiological guidance, aspirating the cyst fluid, and then injecting a sclerosing agent. This process aims to reduce the cyst volume [87].
In cases where symptoms are caused by multiple larger cysts and if these cysts are accessible, fenestration can be considered. Fenestration involves both aspirating and surgically deroofing liver cysts. However, this approach is suitable only when the distribution of cysts permits access [82].
If the distribution of cysts does not allow for AS or fenestration, segmental hepatic resection may be considered. This procedure is reserved for cases of symptomatic and severe hepatomegaly, where a few liver segments are significantly affected by multiple cysts while other segments are less affected. It is important to note that segmental hepatic resection carries a higher risk of perioperative complications and may complicate future liver transplants due to the formation of adhesions [82].
Liver transplantation is the only curative treatment for severe and advanced cases of PLD. However, only a minority of patients will qualify for this intervention. Patients with massive hepatomegaly who suffer from severe malnutrition, low serum albumin levels, sarcopenia, or severe and recurrent complications such as cyst infections or portal hypertension may be considered for liver transplantation. The Model for End-Stage Liver Disease (MELD) score, which assesses the three-month prognosis in patients with liver failure, is an important tool for selecting patients for liver transplantation. In cases of ADPKD with severe renal impairment, combined liver-kidney transplantation should be considered [82].
It's worth noting that the choice of therapy should be made in consultation with healthcare professionals who specialize in the management of PLD, taking into consideration the individual patient's condition, symptoms, and other factors.
3.2. Pancreas and Spleen
In ADPKD, the pancreas can be affected by the development of cysts in around 7% to 36% of affected patients. Loss of the proteins PC1 or PC2 are responsible for cysts formation in mouse model. PKD2 mutations are more prone (5.9 times) to develop those cysts. Cilia, which are cellular structures important for various functions, including tissue organization, are found exclusively in islet and ductal cells in pancreatic tissues. Their absence or disorganization can lead to abnormalities in the pancreatic ducts, loss of acinar cells, polarity defects, and dysregulated insulin secretion [88,89,90,91].
In terms of splenic involvement in ADPKD, the incidence of splenic lesions appears to be similar between individuals with ADPKD and those without ADPKD (7% vs. 5%). The prevalence of splenic lesions does not appear to be influenced by the type of mutated gene. However, the median spleen volume (SV) is notably larger in patients with ADPKD compared to the general population (236 ml vs. 176 ml). Height-adjusted SV (htSV) is closely linked to height-adjusted total kidney volume (htTKV), indicating a relationship between kidney and spleen size in ADPKD patients. It didn’t seem to be associated with PLD or cysts complications. [92].
Additional investigation is required to comprehensively grasp the mechanisms and clinical implications of pancreatic and splenic involvement in ADPKD.
3.3. Other Involvements
In addition to the more commonly known manifestations of ADPKD, such as liver and kidney cysts, there are several other extrarenal manifestations that can occur in ADPKD patients.
Arachnoid membrane cysts, although rare, can occur in approximately 8% of ADPKD patients. These cysts are typically asymptomatic and found incidentally, but they may increase the risk for subdural hematomas, which are collections of blood between the brain and its outermost covering.
Spinal meningeal diverticula, which are outpouchings of the spinal meninges, can occur more frequently in ADPKD patients. However, they rarely present with intracranial hypotension, which is a condition characterized by low cerebrospinal fluid pressure due to a leak [93,94].
Cysts in the seminal vesicles are observed in approximately 40% of male ADPKD patients. While they are rarely responsible directly of infertility, they can contribute to defective sperm motility, leading to the same result. Prostate median cysts near the ejaculatory ducts have also been associated with ADPKD [95].
There is evidence suggesting a higher prevalence of bronchiectasis, a condition characterized by damage and widening of the airways in the lungs, in ADPKD patients [96].
Colonic diverticula, which are small pouches that develop in the colon, as well as abdominal wall and inguinal hernias, have been documented to occur more frequently in patients with ADPKD. However, it's important to note that the studies supporting this association are dated, and there is a lack of recent data available on this topic [97,98,99,100].
Contrary to some reports, ovarian cysts are not associated with ADPKD [101].
It's worth noting that additional research is necessary to gain a comprehensive understanding of the prevalence, clinical consequences and underlying mechanisms of these extrarenal manifestations in ADPKD.
4. Extra-Renal Non Cystic Manifestations
4.1. Vascular
Patients with ADPKD are known to have vascular abnormalities, including dissections and aneurysms in various large arteries throughout the body. Among the vascular complications, intracranial aneurysms (IAs) are the most prevalent in individuals with ADPKD. The existence of these vascular irregularities has given rise to the hypothesis that polycystins play a role in maintaining vascular integrity [102,103]. It is hypothesized that polycystins play a role in the development and maintenance of the vascular system. Impairment of Pkd1 and Pkd2 function influences endothelial cell responses to fluid shear stress, resulting in diminished release of nitric oxide. This has been proposed as a potential contributor to hypertension in these patients. [23,104,105,106,107,108,109]. Interestingly, while the impairment of Pkd1 and Pkd2 yields similar phenotypes in endothelial cells, they appear to have antagonistic effects in vascular smooth muscle cells (VSMc). Deleting Pkd1 in VSMc reduces arterial myogenic tone, and knocking down Pkd2 in Pkd1-deficient arteries restores the myogenic response. The precise mechanisms through which specific PKD mutations make individuals prone to a vascular phenotype are still not fully understood [108,109,110].
The incidence of IAs in ADPKD is reported to be 4-5 times greater than in the general population, with estimates ranging from 9% to 12% compared to 2-3%. The IAs in these patients are usually more numerous and rupture 10 years earlier than in the general population [103,111]. There are modifiable and non modifiable risk factors associated with IAs rupture. Nonmodifiable factors include female gender, older age, personal or family history of aneurysm or subarachnoid hemorrhage (SAH), and certain ethnicities. Factors that can be modified include smoking habit, hypertension, and excessive alcohol consumption. Having a positive family history of IAs or SAH is a significant risk factor, and certain ethnic populations, such as Chinese, Japanese, or European, may be at elevated risk [112,113].
It's important for ADPKD patients to undergo appropriate screening and monitoring for the presence of IAs to prevent potential complications such as rupture and SAH [114]. In patients with ADPKD, individuals with a positive family history have a higher prevalence of intracranial aneurysms (21.6%). Nevertheless, even among those with a negative family history, the prevalence remains elevated at 11%. Additionally, the prevalence of IAs tends to increase with age in ADPKD patients [114,115]. ADPKD patients also exhibit a higher frequency of anatomical variants in the arterial system, such as fenestrations, duplications, or azygos variants, compared to the general population. These variants may contribute to an increased susceptibility to developing aneurysms due to alterations in arterial wall structure and turbulent blood flow. Fenestrations, in particular, can create turbulent flow and defects in the arterial segment, increasing the risk of aneurysm formation. Other arterial variants like duplications and azygos configurations may also promote aneurysm development through turbulent flow [111,116,117]. The rupture of intracranial aneurysms in ADPKD can lead to severe neurological complications and high morbidity and mortality rates, exceeding 50% of cases. Aneurysm size, location, and previous SAH are strong predictors of rupture.[118]. The PHASES scoring system has been proposed to rate the risk of aneurysm rupture based on population, hypertension, age, aneurysm size and location, and previous SAH. This scoring system helps in prognostication and decision-making for appropriate management [119].
Time-of-flight magnetic resonance angiography (MRA) is the recommended screening approach for IAs as it does not require contrast agents. However, current guidelines do not universally recommend brain MRA for screening in ADPKD patients, suggesting its use primarily for those with a positive family history. Nevertheless, recent studies suggest that screening for IAs should be considered even in ADPKD patients without a known familial history to optimize patient management [111].
Management decisions for unruptured IAs in ADPKD are complex and involve several factors, including the patient's age, overall health, aneurysm characteristics, and the feasibility of intervention. ADPKD patients have shown a greater incidence of iatrogenic complications such as hemorrhage, infarction, and dissection during aneurysm treatment compared to non ADPKD. Conservative management is often appropriate for small asymptomatic aneurysms (<7 mm) in the anterior circulation, especially in patients with no history of SAH. Regular imaging follow-up is recommended initially, with longer intervals once aneurysm stability has been established. Lifestyle modifications, including smoking cessation, reducing alcohol consumption, strict control of blood pressure and dyslipidemia, are important in minimizing the risk of aneurysm growth and rupture [112,120].
4.2. Brain
Arachnoid cysts (ACs) are indeed another extrarenal manifestation that can occur in patients with ADPKD. Most ACs are considered congenital and asymptomatic. However, their prevalence is greater in ADPKD patients compared to non ADPKD [111,121]. Reported prevalence of ACs in ADPKD ranges from 4.8% to 12.8%, whereas in the broader population is around 0.5% to 1.1%. The main sites of these cystic lesions in ADPKD are the middle cranial fossa (61%) and the posterior cranial fossa (39%), with no differences in ADPKD patients [122].
PKD1 mutations seems to have a higher risk and earlier age of diagnosis of ACs compared to those with PKD2 mutations. However, most ACs in ADPKD patients are neurologically asymptomatic and rarely associated with subdural hematoma.
The dimensions of ACs can differ, ranging from tiny to large cysts that encompass a significant portion of the cranial cavity. However, unlike kidney cysts, the alterations in volume for ACs are minor and do not correspond to the consistent and more pronounced expansion observed in kidneys. Furthermore, it appears that arachnoid cysts may be more prevalent in advanced stages of ADPKD.
4.3. Heart
Arterial hypertension (AH) is indeed a common extrarenal manifestation of ADPKD, with an early onset typically occurring before the decline in renal function. AH in ADPKD is complex. Enlarged kidney cysts can exert pressure on kidney vessels, leading to regional ischemia and activation of the intrarenal renin-angiotensin-aldosterone system (RAAS) and the renal ortho-sympathetic nerve endings. Additionally, PC are structural part of the vessel, so their alterations can lead to endothelial dysfunction and impaired contractility of VSMc [123,124]. However, persistent pressure burden and the loss of daily blood pressure decrease can contribute to heightened occurrence of cardiovascular events in ADPKD [124]. The HALT PKD study demonstrated that strict control of blood pressure is linked to a slower progression of total kidney volume, no significant alteration in eGFR, a more pronounced decrease in left ventricular mass index, and a more substantial reduction in urinary albumin excretion. Monotherapy with an ACE inhibitor has been shown to be effective in achieving AH management in most ADPKD patients with CKD, making it a recommended first-choice therapy for young patients [125].
Valvular abnormalities are also observed in a notable portion (25-30%) of individuals with ADPKD. The most frequent irregularities are represented by mild mitral valve prolapse and aortic regurgitation, while mitral and/or tricuspid regurgitation are less frequent. These valve diseases in ADPKD may be attributed to generalized abnormalities in collagen or extracellular matrix. The size of the aortic root and the pressure gradient across the aortic valve have been found to correlate with kidney volume adjusted for height, and left ventricular septal wall thickness correlates with eGFR. The increased prevalence of mitral valve regurgitation may be influenced by hypertension, as sustained elevated blood pressure can increase the risk of mitral valve regurgitation. However, it is also suggested that mitral valve prolapse could be considered a characteristic manifestation of ADPKD, as it can occur in children and young adults independent of blood pressure [126,127,128,129,130]. Although mitral valve prolapse is a frequent complication in ADPKD, it rarely leads to severe cardiac dysfunction or significant clinical problems in polycystic patients. Therefore, routine screening echocardiography for mitral valve prolapse is not typically performed [131].
In addition to valvular abnormalities, other cardiovascular abnormalities have been associated with PKD1 mutation in ADPKD patients, including left ventricular hypertrophy (LVH), arrhythmias, and dilated cardiomyopathy.
LVH is present in roughly 65% of ADPKD vs 55% in non ADPKD. It is worth noting that cardiovascular abnormalities in ADPKD are primarily limited to the left heart district, with minimal changes observed in the right part. Mild tricuspid regurgitation has approximately the same prevalence in ADPKD as in non ADPKD, with over 80% of individuals in both groups exhibiting this condition[129,132,133].
Notably, significant differences have been observed in the aortic root diameter among ADPKD patients based on the severity of cysts. Patients with a limited renal involvement (Mayo classes 1A/B) tend to have smaller aortic root diameters, while those with graver cysts (Mayo classes 1C-E) have larger aortic root diameters. This suggests a relationship between cyst severity and aortic root diameter in ADPKD [134].
Cardiovascular abnormalities related to ADPKD can vary among individuals and may not be present in all patients. Regular cardiovascular evaluations and monitoring are recommended to assess and manage these potential complications in ADPKD patients [135].
4.4. Bone
Bone disorders have emerged as a new area of study in the context of ADPKD, as they can have a significant impact on the clinical history of patients. The presence of primary cilia and polycystins in osteoblasts and osteocytes indicates their involvement in bone metabolism. PC1, in particular, acts as a mechanosensor and regulates osteoblastic gene transcription and bone cell differentiation. Dysregulation of PKD1 expression within the skeletal framework may result in atypical bone growth and morphology, diminished bone mineral density, decreased cortical thickness, and the onset of osteopenia [136,137,138,139]. PC1 also contributes to chondrocyte and osteoblast differentiation and development, and adipogenesis. Its inactivation due to a missense mutation can result in delayed bone formation. Animal models have shown that PC1 deficiency is associated with reduced mineralized bone content in both the calvaria and long bones, indicating a role in intramembranous and endochondral bone formation. This leads to decreased bone mineral density due to a decline in bone formation as opposed to an augmentation in bone resorption [136,137,140]. However, bone manifestations in humans with ADPKD are generally subtle or absent compared to animal models. It is important to consider that other syndromes with skeletal malformations can mimic ADPKD (examples: oral-facial-digital syndrome type 1, serpentine fibula-polycystic kidney syndrome). In children with ADPKD, bone developmental defects have not been identified, and their growth and stature are typically normal [141].
Different types of PKD mutations result in distinct responsiveness of cilia. Nontruncating PKD2 mutations, associated with milder kidney disease, are linked to less responsive cilia compared to truncating PKD2 or PKD1 mutations. Truncating PKD1 mutations result in osteoblasts exhibiting heightened cilia responsiveness and an expedited rate of mineralized matrix deposition [141].
Additionally, ADPKD patients with CKD stage 1-2 often have elevated levels of fibroblast growth factor 23 (FGF23), a hormone secreted by osteocytes. This drives to hypophosphatemia, renal phosphate wasting and low circulating bone-specific alkaline phosphatase (BAP) levels. Interestingly, there is evidence of peripheral resistance to FGF23 in ADPKD patients, likely due to the disease and its klotho deficiency, considering that the tubular maximum phosphorus reabsorption per glomerular filtration rate surpasses the anticipated levels based on FGF23 exposure [137,141,142,143].
In summary, the observed bone defect in patients with ADPKD is discernibly different from the presentation observed in individuals with other etiologies of CKD. It is marked by adynamic bone disorder, which refers to reduced bone turnover and mineralization. This bone defect manifests during the initial phases of CKD in individuals with ADPKD, even before significant deterioration of renal function occurs. Nevertheless, even with the existence of the bone defect, individuals with ADPKD undergoing hemodialysis do not seem to exhibit a heightened risk of fractures when compared to non ADPKD patients [137].
Since the study of bone abnormalities in ADPKD is relatively new, there is limited available data. Additional studies are required to investigate the changes occurring in the bones of ADPKD patients and to investigate the underlying aberrant regulatory pathways involved. Subsequent investigations will contribute to an enhanced comprehension of the specific bone lesions observed in ADPKD and their implications for patient care.
5. Conclusions
- ADPKD is a systemic disease that can involve several districts with cystic and non cystic involvement
- Despite kidney remains the main clinical feature, liver, vascular, heart and bone involvement could impact on patient’s quality of life
- Liver volume is a prognostic marker and it impacts both on symptom burden and quality of life
- IA rupture is the most serious complication that can occour in ADPKD patients. Early detection and appropriate treatment is highly desirable
- Although valvular abnormalities are common in ADPKD patients, rarely leads to clinical problems therefore screening echocardiography is not compulsory
- Bone defect in individuals with ADPKD aligns with adynamic bone disorder and it appears since earliest stages of CKD.
Author Contributions
RM concept and designed the work. MR and BM performed the literature search, choosing the studies that have to be considered in the review. RM and MR wrote the manuscript. BA edited and critically reviewed the manuscript. All authors read and approved the final manuscript.
Funding
The authors received no funding for the research reported.
Conflicts of Interests
The authors declare that they have no conflict of interests.
References
- Cornec-Le Gall, E.; Alam, A.; Perrone, R.D. Autosomal dominant polycystic kidney disease. Lancet 2019, 393, 919–935. [Google Scholar] [CrossRef] [PubMed]
- Cornec-Le Gall, E.; Torres, V.E.; Harris, P.C. Genetic Complexity of Autosomal Dominant Polycystic Kidney and Liver Diseases. J Am Soc Nephrol 2018, 29, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Besse, W.; et al. Mutation Carriers Develop Kidney and Liver Cysts. J Am Soc Nephrol 2019, 30, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Hopp, K.; et al. Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J Clin Invest 2012, 122, 4257–73. [Google Scholar] [CrossRef] [PubMed]
- Lantinga-van Leeuwen, I.S.; et al. Lowering of Pkd1 expression is sufficient to cause polycystic kidney disease. Hum Mol Genet 2004, 13, 3069–77. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, S.; et al. The position of the polycystic kidney disease 1 (PKD1) gene mutation correlates with the severity of renal disease. J Am Soc Nephrol 2002, 13, 1230–7. [Google Scholar] [CrossRef]
- Rossetti, S.; Harris, P.C. Genotype-phenotype correlations in autosomal dominant and autosomal recessive polycystic kidney disease. J Am Soc Nephrol 2007, 18, 1374–80. [Google Scholar] [CrossRef]
- Bergmann, C.; et al. Polycystic kidney disease. Nat Rev Dis Primers 2018, 4, 50. [Google Scholar] [CrossRef]
- Schrier, R.W. Renal volume, renin-angiotensin-aldosterone system, hypertension, and left ventricular hypertrophy in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2009, 20, 1888–93. [Google Scholar] [CrossRef]
- Rizk, D.; Chapman, A.B. Cystic and inherited kidney diseases. Am J Kidney Dis 2003, 42, 1305–17. [Google Scholar] [CrossRef]
- Grantham, J.J. Clinical practice. Autosomal dominant polycystic kidney disease. N Engl J Med 2008, 359, 1477–85. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Park, J.H. Genetic Mechanisms of ADPKD. Adv Exp Med Biol 2016, 933, 13–22. [Google Scholar] [PubMed]
- Horani, A.; Ferkol, T.W. Understanding Primary Ciliary Dyskinesia and Other Ciliopathies. J Pediatr 2021, 230, 15–22.e1. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.R. The evolution of eukaryotic cilia and flagella as motile and sensory organelles. Adv Exp Med Biol 2007, 607, 130–40. [Google Scholar]
- Nauli, S.M.; Jin, X.; Hierck, B.P. Jin, and B.P. Hierck, The mechanosensory role of primary cilia in vascular hypertension. Int J Vasc Med 2011, 2011, 376281. [Google Scholar] [PubMed]
- Abdul-Majeed, S. and S.M. Nauli, Calcium-mediated mechanisms of cystic expansion. Biochim Biophys Acta 2011, 1812, 1281–90. [Google Scholar] [CrossRef] [PubMed]
- Nauli, S.M. and J. Zhou, Polycystins and mechanosensation in renal and nodal cilia. Bioessays 2004, 26, 844–56. [Google Scholar] [CrossRef] [PubMed]
- Caspary, T., C. E. Larkins, and K.V. Anderson, The graded response to Sonic Hedgehog depends on cilia architecture. Dev Cell 2007, 12, 767–78. [Google Scholar] [CrossRef]
- Christensen, S.T.; et al. The primary cilium coordinates signaling pathways in cell cycle control and migration during development and tissue repair. Curr Top Dev Biol 2008, 85, 261–301. [Google Scholar]
- Gerdes, J.M., E. E. Davis, and N. Katsanis, The vertebrate primary cilium in development, homeostasis, and disease. Cell 2009, 137, 32–45. [Google Scholar] [CrossRef]
- Satir, P., L. B. Pedersen, and S.T. Christensen, The primary cilium at a glance. J Cell Sci 2010, 123, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Praetorius, H.A. and K.R. Spring, Removal of the MDCK cell primary cilium abolishes flow sensing. J Membr Biol 2003, 191, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Nauli, S.M.; et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 2003, 33, 129–37. [Google Scholar] [CrossRef] [PubMed]
- Chauvet, V.; et al. Expression of PKD1 and PKD2 transcripts and proteins in human embryo and during normal kidney development. Am J Pathol 2002, 160, 973–83. [Google Scholar] [CrossRef] [PubMed]
- Ibraghimov-Beskrovnaya, O.; et al. Polycystin: in vitro synthesis, in vivo tissue expression, and subcellular localization identifies a large membrane-associated protein. Proc Natl Acad Sci U S A 1997, 94, 6397–402. [Google Scholar] [CrossRef] [PubMed]
- Huan, Y. and J. van Adelsberg, Polycystin-1, the PKD1 gene product, is in a complex containing E-cadherin and the catenins. J Clin Invest 1999, 104, 1459–68. [Google Scholar] [CrossRef]
- Mochizuki, T.; et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 1996, 272, 1339–42. [Google Scholar] [CrossRef] [PubMed]
- González-Perrett, S.; et al. Polycystin-2, the protein mutated in autosomal dominant polycystic kidney disease (ADPKD), is a Ca2+-permeable nonselective cation channel. Proc Natl Acad Sci U S A 2001, 98, 1182–7. [Google Scholar] [CrossRef] [PubMed]
- Yoder, B.K., X. Hou, and L.M. Guay-Woodford, The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol 2002, 13, 2508–16. [Google Scholar] [CrossRef]
- Yu, Y.; et al. Structural and molecular basis of the assembly of the TRPP2/PKD1 complex. Proc Natl Acad Sci U S A 2009, 106, 11558–63. [Google Scholar] [CrossRef]
- Vassilev, P.M.; et al. Polycystin-2 is a novel cation channel implicated in defective intracellular Ca(2+) homeostasis in polycystic kidney disease. Biochem Biophys Res Commun 2001, 282, 341–50. [Google Scholar] [CrossRef] [PubMed]
- Nims, N., D. Vassmer, and R.L. Maser, Transmembrane domain analysis of polycystin-1, the product of the polycystic kidney disease-1 (PKD1) gene: evidence for 11 membrane-spanning domains. Biochemistry 2003, 42, 13035–48. [Google Scholar] [CrossRef] [PubMed]
- Tsiokas, L.; et al. Homo- and heterodimeric interactions between the gene products of PKD1 and PKD2. Proc Natl Acad Sci U S A 1997, 94, 6965–70. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; et al. PKD1 interacts with PKD2 through a probable coiled-coil domain. Nat Genet 1997, 16, 179–83. [Google Scholar] [CrossRef]
- Gallagher, A.R., G. G. Germino, and S. Somlo, Molecular advances in autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis 2010, 17, 118–30. [Google Scholar] [CrossRef] [PubMed]
- Koulen, P.; et al. Polycystin-2 is an intracellular calcium release channel. Nat Cell Biol 2002, 4, 191–7. [Google Scholar] [CrossRef] [PubMed]
- Anyatonwu, G.I. and B.E. Ehrlich, Organic cation permeation through the channel formed by polycystin-2. J Biol Chem 2005, 280, 29488–93. [Google Scholar] [CrossRef]
- Masyuk, A.I.; et al. Cholangiocyte cilia detect changes in luminal fluid flow and transmit them into intracellular Ca2+ and cAMP signaling. Gastroenterology 2006, 131, 911–20. [Google Scholar] [CrossRef] [PubMed]
- Masyuk, T.V.; et al. Octreotide inhibits hepatic cystogenesis in a rodent model of polycystic liver disease by reducing cholangiocyte adenosine 3',5'-cyclic monophosphate. Gastroenterology 2007, 132, 1104–16. [Google Scholar] [CrossRef]
- Kip, S.N.; et al. [Ca2+]i reduction increases cellular proliferation and apoptosis in vascular smooth muscle cells: relevance to the ADPKD phenotype. Circ Res 2005, 96, 873–80. [Google Scholar] [CrossRef]
- Banizs, B.; et al. Altered pH(i) regulation and Na(+)/HCO3(-) transporter activity in choroid plexus of cilia-defective Tg737(orpk) mutant mouse. Am J Physiol Cell Physiol 2007, 292, C1409–16. [Google Scholar] [CrossRef] [PubMed]
- Shillingford, J.M.; et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A 2006, 103, 5466–71. [Google Scholar] [CrossRef]
- Bhunia, A.K.; et al. PKD1 induces p21(waf1) and regulation of the cell cycle via direct activation of the JAK-STAT signaling pathway in a process requiring PKD2. Cell 2002, 109, 157–68. [Google Scholar] [CrossRef]
- Liang, G.; et al. Polycystin-2 down-regulates cell proliferation via promoting PERK-dependent phosphorylation of eIF2alpha. Hum Mol Genet 2008, 17, 3254–62. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E.; et al. Defective planar cell polarity in polycystic kidney disease. Nat Genet 2006, 38, 21–3. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, S.; et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2007, 18, 2143–60. [Google Scholar] [CrossRef]
- Hateboer, N.; et al. Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 Study Group. Lancet 1999, 353, 103–7. [Google Scholar] [CrossRef]
- Rossetti, S. and P.C. Harris, The genetics of vascular complications in autosomal dominant polycystic kidney disease (ADPKD). Curr Hypertens Rev 2013, 9, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; et al. A missense mutation in PKD1 attenuates the severity of renal disease. Kidney Int 2012, 81, 412–7. [Google Scholar] [CrossRef]
- Cornec-Le Gall, E.; et al. Type of PKD1 mutation influences renal outcome in ADPKD. J Am Soc Nephrol 2013, 24, 1006–13. [Google Scholar] [CrossRef]
- Chapin, H.C. and M.J. Caplan, The cell biology of polycystic kidney disease. J Cell Biol 2010, 191, 701–10. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; et al. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell 1996, 87, 979–87. [Google Scholar] [CrossRef] [PubMed]
- Watnick, T.J.; et al. Somatic mutation in individual liver cysts supports a two-hit model of cystogenesis in autosomal dominant polycystic kidney disease. Mol Cell 1998, 2, 247–51. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; et al. Somatic PKD2 mutations in individual kidney and liver cysts support a "two-hit" model of cystogenesis in type 2 autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1999, 10, 1524–9. [Google Scholar] [CrossRef]
- Gevers, T.J. and J.P. Drenth, Diagnosis and management of polycystic liver disease. Nat Rev Gastroenterol Hepatol 2013, 10, 101–8. [Google Scholar] [CrossRef] [PubMed]
- Roskams, T. and V. Desmet, Embryology of extra- and intrahepatic bile ducts, the ductal plate. Anat Rec (Hoboken) 2008, 291, 628–35. [Google Scholar] [CrossRef] [PubMed]
- Raynaud, P.; et al. Biliary differentiation and bile duct morphogenesis in development and disease. Int J Biochem Cell Biol 2011, 43, 245–56. [Google Scholar] [CrossRef] [PubMed]
- Strazzabosco, M. and L. Fabris, Development of the bile ducts: essentials for the clinical hepatologist. J Hepatol 2012, 56, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Drenth, J.P., M. Chrispijn, and C. Bergmann, Congenital fibrocystic liver diseases. Best Pract Res Clin Gastroenterol 2010, 24, 573–84. [Google Scholar] [CrossRef]
- Desmet, V.J. , Ludwig symposium on biliary disorders--part I. Pathogenesis of ductal plate abnormalities. Mayo Clin Proc 1998, 73, 80–9. [Google Scholar] [CrossRef]
- Roediger, R.; et al. Polycystic Kidney/Liver Disease. Clin Liver Dis 2022, 26, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Hogan, M.C.; et al. Liver involvement in early autosomal-dominant polycystic kidney disease. Clin Gastroenterol Hepatol 2015, 13, 155–64. [Google Scholar] [CrossRef] [PubMed]
- Bae, K.T.; et al. Magnetic resonance imaging evaluation of hepatic cysts in early autosomal-dominant polycystic kidney disease: the Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease cohort. Clin J Am Soc Nephrol 2006, 1, 64–9. [Google Scholar] [CrossRef] [PubMed]
- Gabow, P.A.; et al. Risk factors for the development of hepatic cysts in autosomal dominant polycystic kidney disease. Hepatology 1990, 11, 1033–7. [Google Scholar] [CrossRef] [PubMed]
- van Aerts, R.M.M.; et al. Severity in polycystic liver disease is associated with aetiology and female gender: Results of the International PLD Registry. Liver Int 2019, 39, 575–582. [Google Scholar] [CrossRef]
- Sherstha, R.; et al. Postmenopausal estrogen therapy selectively stimulates hepatic enlargement in women with autosomal dominant polycystic kidney disease. Hepatology 1997, 26, 1282–6. [Google Scholar]
- Chebib, F.T.; et al. Effect of genotype on the severity and volume progression of polycystic liver disease in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2016, 31, 952–60. [Google Scholar] [CrossRef] [PubMed]
- Aapkes, S.E.; et al. Estrogens in polycystic liver disease: A target for future therapies? Liver Int 2021, 41, 2009–2019. [Google Scholar] [CrossRef]
- van Aerts, R.M.M.; et al. Estrogen-Containing Oral Contraceptives Are Associated With Polycystic Liver Disease Severity in Premenopausal Patients. Clin Pharmacol Ther 2019, 106, 1338–1345. [Google Scholar] [CrossRef]
- Alvaro, D.; et al. The intrahepatic biliary epithelium is a target of the growth hormone/insulin-like growth factor 1 axis. J Hepatol 2005, 43, 875–83. [Google Scholar] [CrossRef]
- Alvaro, D.; et al. Estrogens stimulate proliferation of intrahepatic biliary epithelium in rats. Gastroenterology 2000, 119, 1681–91. [Google Scholar] [CrossRef] [PubMed]
- Koduri, S., A. S. Goldhar, and B.K. Vonderhaar, Activation of vascular endothelial growth factor (VEGF) by the ER-alpha variant, ERDelta3. Breast Cancer Res Treat 2006, 95, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Chauveau, D., F. Fakhouri, and J.P. Grünfeld, Liver involvement in autosomal-dominant polycystic kidney disease: therapeutic dilemma. J Am Soc Nephrol 2000, 11, 1767–1775. [Google Scholar] [CrossRef] [PubMed]
- Patch, C.; et al. Use of antihypertensive medications and mortality of patients with autosomal dominant polycystic kidney disease: a population-based study. Am J Kidney Dis 2011, 57, 856–62. [Google Scholar] [CrossRef]
- Orskov, B.; et al. Improved prognosis in patients with autosomal dominant polycystic kidney disease in Denmark. Clin J Am Soc Nephrol 2010, 5, 2034–9. [Google Scholar] [CrossRef]
- Orskov, B.; et al. Changes in causes of death and risk of cancer in Danish patients with autosomal dominant polycystic kidney disease and end-stage renal disease. Nephrol Dial Transplant 2012, 27, 1607–13. [Google Scholar] [CrossRef] [PubMed]
- Ecder, T.; et al. Reversal of left ventricular hypertrophy with angiotensin converting enzyme inhibition in hypertensive patients with autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 1999, 14, 1113–6. [Google Scholar] [CrossRef]
- Gigot, J.F.; et al. Adult polycystic liver disease: is fenestration the most adequate operation for long-term management? Ann Surg 1997, 225, 286–94. [Google Scholar] [CrossRef] [PubMed]
- Schnelldorfer, T.; et al. Polycystic liver disease: a critical appraisal of hepatic resection, cyst fenestration, and liver transplantation. Ann Surg 2009, 250, 112–8. [Google Scholar] [CrossRef]
- Temmerman, F.; et al. Development and validation of a polycystic liver disease complaint-specific assessment (POLCA). J Hepatol 2014, 61, 1143–50. [Google Scholar] [CrossRef]
- Neijenhuis, M.K.; et al. Development and Validation of a Disease-Specific Questionnaire to Assess Patient-Reported Symptoms in Polycystic Liver Disease. Hepatology 2016, 64, 151–60. [Google Scholar] [CrossRef] [PubMed]
- van Aerts, R.M.M.; et al. Clinical management of polycystic liver disease. J Hepatol 2018, 68, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; et al. Clinical Correlates of Mass Effect in Autosomal Dominant Polycystic Kidney Disease. PLoS One 2015, 10, e0144526. [Google Scholar] [CrossRef]
- van Keimpema, L.; et al. Lanreotide reduces the volume of polycystic liver: a randomized, double-blind, placebo-controlled trial. Gastroenterology 2009, 137, 1661–8. [Google Scholar] [CrossRef] [PubMed]
- Chrispijn, M.; et al. The long-term outcome of patients with polycystic liver disease treated with lanreotide. Aliment Pharmacol Ther 2012, 35, 266–74. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, H.; et al. Potential effect of tolvaptan on polycystic liver disease for patients with ADPKD meeting the Japanese criteria of tolvaptan use. PLoS One 2022, 17, e0264065. [Google Scholar] [CrossRef] [PubMed]
- Wijnands, T.F.; et al. Efficacy and Safety of Aspiration Sclerotherapy of Simple Hepatic Cysts: A Systematic Review. AJR Am J Roentgenol 2017, 208, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; et al. Perinatal lethality with kidney and pancreas defects in mice with a targetted Pkd1 mutation. Nat Genet 1997, 17, 179–81. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; et al. Conditional mutation of Pkd2 causes cystogenesis and upregulates beta-catenin. J Am Soc Nephrol 2009, 20, 2556–69. [Google Scholar] [CrossRef]
- Kim, J.A.; et al. Pancreatic Cysts in Autosomal Dominant Polycystic Kidney Disease: Prevalence and Association with PKD2 Gene Mutations. Radiology 2016, 280, 762–70. [Google Scholar] [CrossRef]
- diIorio, P.; et al. Role of cilia in normal pancreas function and in diseased states. Birth Defects Res C Embryo Today 2014, 102, 126–38. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; et al. Spleen phenotype in autosomal dominant polycystic kidney disease. Clin Radiol 2019, 74, 975–e17. [Google Scholar] [CrossRef] [PubMed]
- Wijdicks, E.F., V. E. Torres, and W.I. Schievink, Chronic subdural hematoma in autosomal dominant polycystic kidney disease. Am J Kidney Dis 2000, 35, 40–3. [Google Scholar] [CrossRef] [PubMed]
- Schievink, W.I. and V.E. Torres, Spinal meningeal diverticula in autosomal dominant polycystic kidney disease. Lancet 1997, 349, 1223–4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; et al. Relationship of Seminal Megavesicles, Prostate Median Cysts, and Genotype in Autosomal Dominant Polycystic Kidney Disease. J Magn Reson Imaging 2019, 49, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; et al. PKD1 deficiency induces Bronchiectasis in a porcine ADPKD model. Respir Res 2022, 23, 292. [Google Scholar] [CrossRef] [PubMed]
- Fick, G.M. and P.A. Gabow, Hereditary and acquired cystic disease of the kidney. Kidney Int 1994, 46, 951–64. [Google Scholar] [CrossRef] [PubMed]
- Scheff, R.T.; et al. Diverticular disease in patients with chronic renal failure due to polycystic kidney disease. Ann Intern Med 1980, 92, 202–4. [Google Scholar] [CrossRef] [PubMed]
- Sharp, C.K.; et al. Evaluation of colonic diverticular disease in autosomal dominant polycystic kidney disease without end-stage renal disease. Am J Kidney Dis 1999, 34, 863–8. [Google Scholar] [CrossRef]
- Morris-Stiff, G.; et al. Abdominal wall hernia in autosomal dominant polycystic kidney disease. Br J Surg 1997, 84, 615–7. [Google Scholar]
- Heinonen, P.K.; et al. Ovarian manifestations in women with autosomal dominant polycystic kidney disease. Am J Kidney Dis 2002, 40, 504–7. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; et al. Polycystin 1 is required for the structural integrity of blood vessels. Proc Natl Acad Sci U S A 2000, 97, 1731–6. [Google Scholar] [CrossRef]
- Perrone, R.D., A. M. Malek, and T. Watnick, Vascular complications in autosomal dominant polycystic kidney disease. Nat Rev Nephrol 2015, 11, 589–98. [Google Scholar] [CrossRef]
- Boulter, C.; et al. Cardiovascular, skeletal, and renal defects in mice with a targeted disruption of the Pkd1 gene. Proc Natl Acad Sci U S A 2001, 98, 12174–9. [Google Scholar] [CrossRef] [PubMed]
- Qian, Q.; et al. Analysis of the polycystins in aortic vascular smooth muscle cells. J Am Soc Nephrol 2003, 14, 2280–7. [Google Scholar] [CrossRef]
- Torres, V.E.; et al. Vascular expression of polycystin-2. J Am Soc Nephrol 2001, 12, 1–9. [Google Scholar] [CrossRef]
- Garcia-Gonzalez, M.A.; et al. Pkd1 and Pkd2 are required for normal placental development. PLoS One 2010, 5. [Google Scholar] [CrossRef]
- Nauli, S.M.; et al. Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation 2008, 117, 1161–71. [Google Scholar] [CrossRef]
- AbouAlaiwi, W.A.; et al. Ciliary polycystin-2 is a mechanosensitive calcium channel involved in nitric oxide signaling cascades. Circ Res 2009, 104, 860–9. [Google Scholar] [CrossRef]
- Sharif-Naeini, R.; et al. Polycystin-1 and -2 dosage regulates pressure sensing. Cell 2009, 139, 587–96. [Google Scholar] [CrossRef]
- Capelli, I.; et al. MR Brain Screening in ADPKD Patients : To Screen or not to Screen? Clin Neuroradiol 2022, 32, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Sanchis, I.M.; et al. Presymptomatic Screening for Intracranial Aneurysms in Patients with Autosomal Dominant Polycystic Kidney Disease. Clin J Am Soc Nephrol 2019, 14, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; et al. Is Regular Screening for Intracranial Aneurysm Necessary in Patients with Autosomal Dominant Polycystic Kidney Disease? A Systematic Review and Meta-analysis. Cerebrovasc Dis 2017, 44, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.W.; et al. Screening for intracranial aneurysm in 355 patients with autosomal-dominant polycystic kidney disease. Stroke 2011, 42, 204–6. [Google Scholar] [CrossRef] [PubMed]
- Niemczyk, M.; et al. Intracranial aneurysms in autosomal dominant polycystic kidney disease. AJNR Am J Neuroradiol 2013, 34, 1556–9. [Google Scholar] [CrossRef]
- Guo, X.; et al. Intracranial Arterial Fenestration and Risk of Aneurysm: A Systematic Review and Meta-Analysis. World Neurosurg 2018, 115, e592–e598. [Google Scholar] [CrossRef] [PubMed]
- Pascalau, R.; et al. The Geometry of the Circle of Willis Anatomical Variants as a Potential Cerebrovascular Risk Factor. Turk Neurosurg 2019, 29, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Wiebers, D.O.; et al. Unruptured intracranial aneurysms: natural history, clinical outcome, and risks of surgical and endovascular treatment. Lancet 2003, 362, 103–10. [Google Scholar] [CrossRef]
- Schievink, W.I.; et al. Saccular intracranial aneurysms in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1992, 3, 88–95. [Google Scholar] [CrossRef]
- Rozenfeld, M.N.; et al. Autosomal Dominant Polycystic Kidney Disease and Intracranial Aneurysms: Is There an Increased Risk of Treatment? AJNR Am J Neuroradiol 2016, 37, 290–3. [Google Scholar] [CrossRef]
- Shigemori, K.; et al. PKD1-Associated Arachnoid Cysts in Autosomal Dominant Polycystic Kidney Disease. J Stroke Cerebrovasc Dis 2021, 30, 105943. [Google Scholar] [CrossRef] [PubMed]
- Krauer, F.; et al. Growth of arachnoid cysts in patients with autosomal dominant polycystic kidney disease: serial imaging and clinical relevance. Clin Kidney J 2012, 5, 405–11. [Google Scholar] [CrossRef] [PubMed]
- Kuo, I.Y. and A.B. Chapman, Polycystins, ADPKD, and Cardiovascular Disease. Kidney Int Rep 2020, 5, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Turgut, F.; et al. Ambulatory blood pressure and endothelial dysfunction in patients with autosomal dominant polycystic kidney disease. Ren Fail 2007, 29, 979–84. [Google Scholar] [CrossRef] [PubMed]
- Schrier, R.W. , Blood pressure in early autosomal dominant polycystic kidney disease. N Engl J Med 2015, 372, 976–7. [Google Scholar] [CrossRef] [PubMed]
- Timio, M.; et al. The spectrum of cardiovascular abnormalities in autosomal dominant polycystic kidney disease: a 10-year follow-up in a five-generation kindred. Clin Nephrol 1992, 37, 245–51. [Google Scholar] [PubMed]
- Hossack, K.F.; et al. Echocardiographic findings in autosomal dominant polycystic kidney disease. N Engl J Med 1988, 319, 907–12. [Google Scholar] [CrossRef]
- Pfeferman, M.B.; et al. Echocardiographic Abnormalities in Autosomal Dominant Polycystic Kidney Disease (ADPKD) Patients. J Clin Med 2022, 11. [Google Scholar] [CrossRef]
- Arjune, S.; et al. Cardiac Manifestations in Patients with Autosomal Dominant Polycystic Kidney Disease (ADPKD): A Single-Center Study. Kidney360 2023, 4, 150–161. [Google Scholar] [CrossRef]
- Rahimi, K.; et al. Elevated blood pressure and risk of mitral regurgitation: A longitudinal cohort study of 5.5 million United Kingdom adults. PLoS Med 2017, 14, e1002404. [Google Scholar] [CrossRef]
- Lumiaho, A.; et al. Mitral valve prolapse and mitral regurgitation are common in patients with polycystic kidney disease type 1. Am J Kidney Dis 2001, 38, 1208–16. [Google Scholar] [CrossRef] [PubMed]
- Ecder, T. and R.W. Schrier, Cardiovascular abnormalities in autosomal-dominant polycystic kidney disease. Nat Rev Nephrol 2009, 5, 221–8. [Google Scholar] [CrossRef] [PubMed]
- Chebib, F.T.; et al. Autosomal Dominant Polycystic Kidney Patients May Be Predisposed to Various Cardiomyopathies. Kidney Int Rep 2017, 2, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Kuo, I.Y. , Defining Cardiac Dysfunction in ADPKD. Kidney360 2023, 4, 126–127. [Google Scholar] [CrossRef]
- Gigante A, Perrotta AM, Tinti F, Assanto E, Muscaritoli M, Lai S, Cianci R. Assessment of cardiovascular disease in ADPKD. Applied Sciences 2023, 13, 7175. [Google Scholar]
- Xiao, Z.S. and L.D. Quarles, Role of the polycytin-primary cilia complex in bone development and mechanosensing. Ann N Y Acad Sci 2010, 1192, 410–21. [Google Scholar] [CrossRef] [PubMed]
- Gitomer, B.; et al. Mineral bone disease in autosomal dominant polycystic kidney disease. Kidney Int 2021, 99, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; et al. Polycystin-1 regulates skeletogenesis through stimulation of the osteoblast-specific transcription factor RUNX2-II. J Biol Chem 2008, 283, 12624–34. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; et al. Cilia-like structures and polycystin-1 in osteoblasts/osteocytes and associated abnormalities in skeletogenesis and Runx2 expression. J Biol Chem 2006, 281, 30884–95. [Google Scholar] [CrossRef]
- Mekahli, D. and J. Bacchetta, From bone abnormalities to mineral metabolism dysregulation in autosomal dominant polycystic kidney disease. Pediatr Nephrol 2013, 28, 2089–96. [Google Scholar] [CrossRef]
- Pereira, R.C.; et al. Characterization of Primary Cilia in Osteoblasts Isolated From Patients With ADPKD and CKD. JBMR Plus 2021, 5, e10464. [Google Scholar] [CrossRef] [PubMed]
- Pavik, I.; et al. Patients with autosomal dominant polycystic kidney disease have elevated fibroblast growth factor 23 levels and a renal leak of phosphate. Kidney Int 2011, 79, 234–40. [Google Scholar] [CrossRef] [PubMed]
- Pavik, I.; et al. Soluble klotho and autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 2012, 7, 248–57. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



