
Submitted:
03 January 2024
Posted:
04 January 2024
You are already at the latest version
Abstract
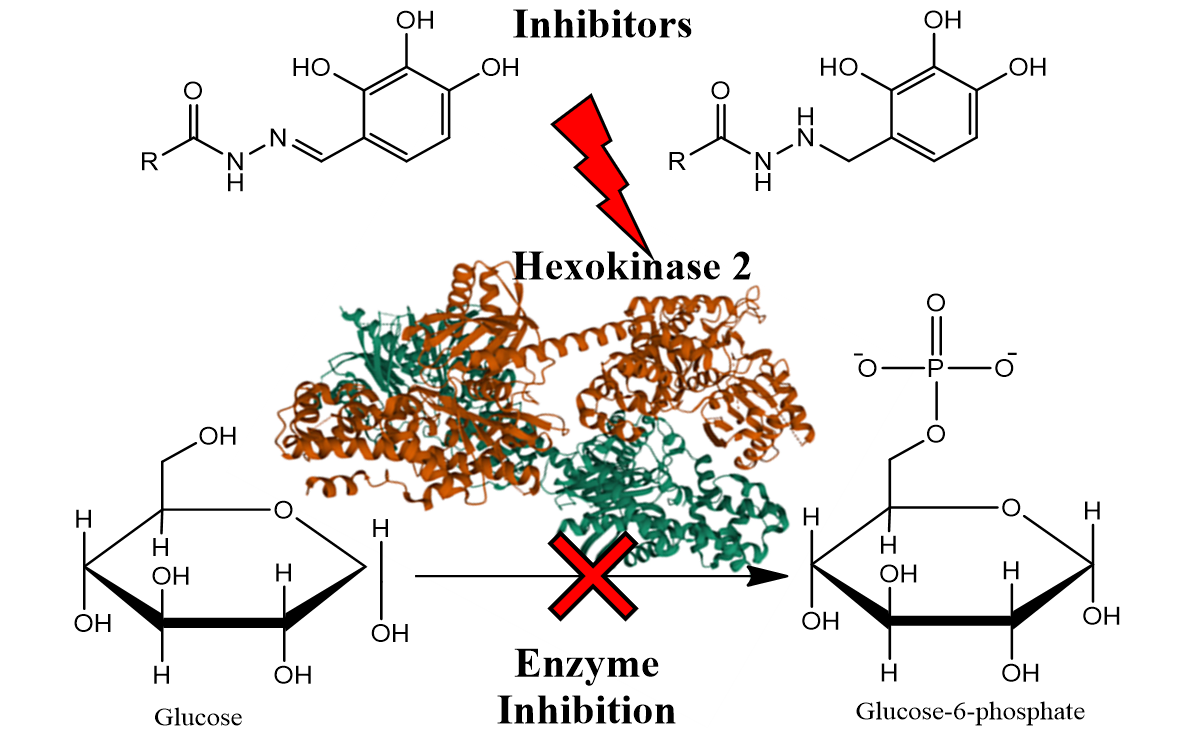
Schiff bases attract research interest due to their applications in chemical synthesis and medicinal chemistry. In recent years, benitrobenrazide and benserazide containing imine moiety, have been synthesized and characterized as promising inhibitors of hexokinase 2 (HK2) an enzyme overexpressed in most cancer cells. Both compounds possess a common structural fragment, a 2,3,4-trihydroxybenzaldehyde moiety connected through hydrazone or hydrazine linker with acylated on N’-nitrogen atom by serine or 4-nitrobenzoic acid fragment. To avoid presence of toxicophoric nitro group in benitrobenrazide molecule we introduced common pharmacophores 4-fluorophenyl or 4-aminophenyl substituents. Modification of benserazide regards an introduction of other endogenous amino acids instead of serine. Herein we report the synthesis of benitrobenrazide and benserazide analogues and preliminary results of inhibitory activity against HK2 evoked by these structural changes. The derivatives contain a fluorine atom or amino group instead of a nitro group in BNB, exhibited the most potent inhibitory effects against HK2 at a concentration of 1 µM, with HK2 inhibition rates of 60% and 54%, respectively.
Keywords:
schiff bases
; benitrobenrazide
; benserazide
; hexokinase 2
; enzyme inhibition
1. Introduction
Carbonyl compounds easily react with N-centered nucleophiles such as amines, hydrazine, and its derivatives. Schiff bases, a product of condensation of carbonyl compounds with primary amines have a wide range of applications in pharmaceuticals [1-3], and in coordination chemistry as ligands and chelating agents [4,5]. The versatile pharmacophore C=N is present in biologically active compounds exhibiting antioxidant [3,6], antimicrobial [3,6,7] and anticancer [8] properties.
N-Acylhydrazides R(CO)NHNH2 are structurally like amides, and a relocation of a proton can occur, forming an iminol form (Figure 1) [1]. Compounds containing an amide-imine bridge -C(=O)-NH-N=CH- can be considered as hybrid structures of hydrazide or hydrazones [1]. These hybrid compounds have several applications in pharmacology, exhibiting antimicrobial activity [9-11], anticancer activity [8-9] and free radical scavenging properties [12].
Benserazide (BND) and benitrobenrazide (BNB) (Figure 1) are compounds belonging to the group of hydrazine derivatives containing common structural fragments, an N-acyl rest of serine or 4-nitrobenzoic acid and a 2,3,4-trihydroxybenzaldehyde (pyrogallol-4-carboxaldehyde) fragment attached to N’ nitrogen atom by a methylene or methine carbon atom. Both compounds BNB and BND have promising inhibitory activity against hexokinase 2 (HK2) [13-15], an enzyme involved in the phosphorylation of glucose in glycolysis, a first step of glucose metabolism [16]. An increased requirement for glucose is observed in cancer cells, especially in rapidly growing and drug-resistant tumors [17-22]. Among all human hexokinase isoenzymes, HK2 shows overexpression in cancer cells, which makes it interesting in the context of molecularly targeted therapy [23-28]. Currently several HK2 potent inhibitors are recognized like metformin, 2-deoxy-D-Glucose, 3-bromopyruvate or strepantibins A C [24, 29-32].
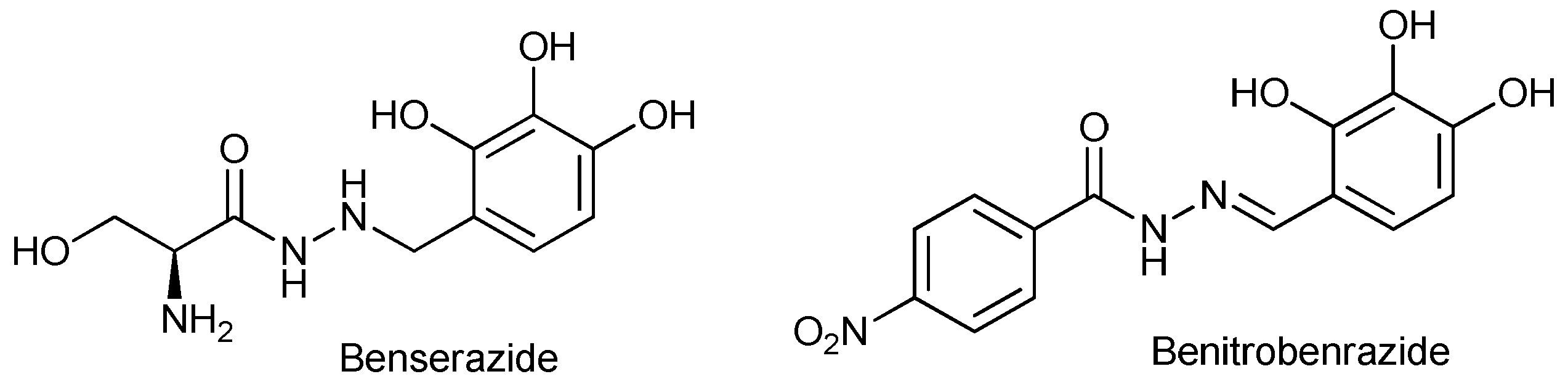
Figure 2.
The chemical structures of reported HK2 inhibitors.
Benserazide is an FDA-approved drug for the treatment of Parkinson's disease, but it was recently also recognized as a strong HK2 inhibitor [14]. Benitrobenrazide was identified as a potential HK2 inhibitor by structure-based virtual ligand screening [15]. According to our previous study the key structural feature is the presence of three hydroxy group in benzene ring, which occupies the same HK2-binding pocket as the natural substrate, glucose [29]. The influence of other structural elements, namely -NH-NH-CH2- and -NH=N-CH- on HK2 enzymatic activity has not yet been determined and requires investigation to explain their impact on enzymatic activity.
Herein, we present our primary attempts to explore an influence of structural features observed in benserazide and benitrobenrazide in their biological activity against HK2. The serine originally present in benserazide has been exchange with other amino acid fragments e.g., glycine, tyrosine, and cysteine. Application of two endogenic amino acids, threonine, and cysteine in synthesis of benserazide analogues should deliver an answer about an importance of serine moiety on benserazide inhibitory activity. In BNB molecule the 4-nitrophenyl moiety has been substitute by an alkyl chain or aromatic rings of different molecular areas (benzene, naphthalene, and anthracene), to judge their downstream influence on HK2 activity and gauge HK2 active site volume. We decide to exchange toxicophoric nitro group present in BNB moiety by two common substituents, namely fluorine atom and amino group [33, 34]. These substituents have opposite effect on electron density in benzene ring of BNB and were introduced to explain eventual influence of electronic conditions on its activity. Fluorine atom like nitro group decreasing the electron density whereas amino group is typical electron-donating group. The novel analogues of benserazide and benitrobenrazide were examined for their inhibitory activity against HK2 to get information about influence of structural variations on inhibition activity.
2. Results and Discussion
2.1. Chemistry of benitrobenrazide and benserazide derivatives
Benitrobenrazide and benserazide contain an N-acyl fragment, 4-nitrophenyl, or a serine moiety, respectively [13, 15]. We modified these regions of each molecule, incorporating instead aromatic rings of different molecular areas, to probe the volume of the active site pocket of HK2. In further modifications, a nitro group suspected as a genotoxic substituent, was replaced by substituents of different polarity, constituting either a fluorine atom or an amino group. Introduction of fluorine and amino substituent as pharmacophore groups into drugs molecules increasing their therapeutic effectiveness [35, 36]. Additionally, the imine bond in hydrazone analogue was reduced to a single carbon-nitrogen bond to identify the effect of the imine double bond (C=N) on HK2 activity.
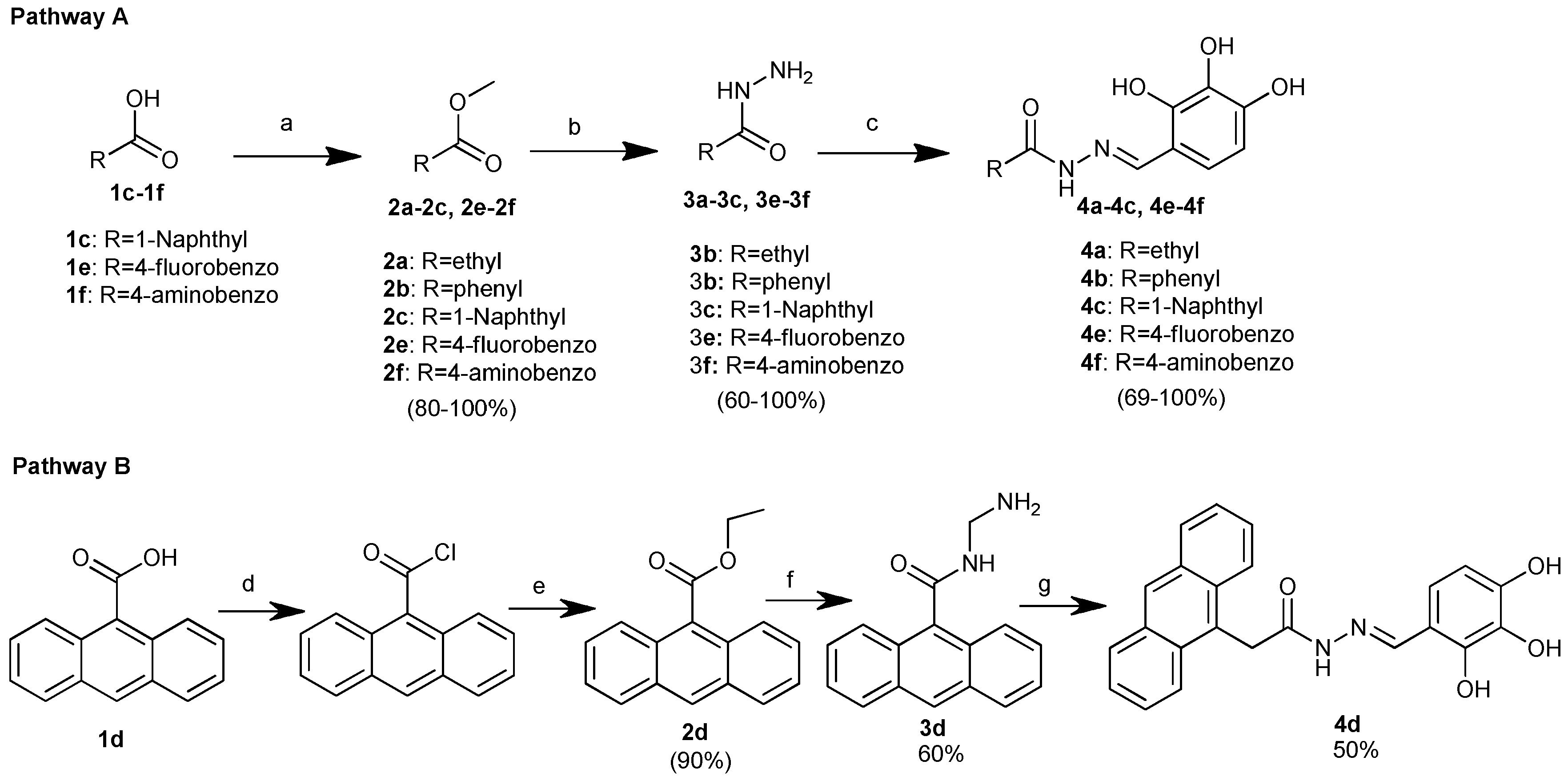
The final benitrobenrazide analogues synthesis is depicted below (Scheme 1). Commercially available methyl propionate and benzoate 2a and 2b were use. Methyl esters of other aromatic acids 1c, 1e-1f, were obtained in a one-pot synthesis involving the primary transformation of each appropriate carboxylic acid into its respective acyl chloride, by treatment with thionyl chloride in excess, followed by esterification in the presence of MeOH [37] (Scheme 1. Pathway A). The ester of anthracene-9-carboxylic acid 1d was synthesized by a two-step synthesis. The first step was conversion of anthracene-9-carboxylic acid into its acyl chloride, in the presence of an excess of thionyl chloride and a small amount of DMF, as a catalyst. The separated acyl chloride was then subjected to esterification with EtOH in the presence of triethylamine, according to the synthesis pathway B shown in Scheme 1.
Hydrazides 3a-3f were obtained with a good yield from appropriate esters in nucleophilic substitution on carbonyl carbon atom, in the presence of an excess of hydrazine monohydrate (four equivalents) in anhydrous boiling methanol [38, 39]. Conducting the hydrazide synthesis 3b-3c, 3f at room temperature led to a purer product and a good yield (60-77%). The preparation of hydrazide 3d needs harsher conditions, so the reaction was conducted in an excess of boiling hydrazine hydrate, with a prolonged reaction time (Scheme 1. B pathway). Hydrazides 3b-3f were obtained in the form of white solids, which precipitated during the reaction process and crystallized from ethanol or aqueous ethanol. Compound 3a was purified via silica gel column chromatography using MeOH/CHCl3 (1:1 v/v) as an eluent. The hydrazide structure was confirmed by 1H NMR spectra. The absence of a singlet assigned to the methyl group in the region of 3.25-3.95 ppm confirmed the conversation of the esters into hydrazides.
Compounds detailed in 3, treated with 2,3,4-trihydroxybenzaldehyde in methanol, produced the products listed in 4 at a 50-100% yield. Hydrazones 4 can adopt an E or a Z configuration at the imine double bond (-N=CH-). In the case of the E conformation, the geometrical isomer can be stabilized by formation of an intramolecular hydrogen bond between the 2-OH hydroxyl group and nitrogen atom of the imine [40], as is depicted in Figure 3. Similar behavior exhibits compounds 10.
Using quantum chemical calculations based on density function theory (DFT), we studied the possibility of the existence of equilibrium between conformers E and Z for 4a. For the conformer E, the corresponding hydrogen bond length is 2.1 Å. Hydrogen bond formation confirms our DFT calculation of stretching vibrations for the O-H group at 3224.47 cm-1. Additionally, our calculation data shows a higher stability of the E conformer over the Z conformer by 9 kcal/mol when the hydrogen bond formation is included in simulations [41]. We can assume that the E conformer is the predominant form of hydrazone 4a. 1H NMR spectroscopy verified the results of our calculation based on DFT. The chemical shifts of the protons of the 3-OH and 4-OH groups create singlet peaks at δ H 8.4–9.6 ppm, while the peak for the proton of the 2-OH group is shifted to a lower field and was observed in the region of δ H 11.3–12.4 ppm. We attribute this down-field shift of the 2-OH group to the formation of an intramolecular hydrogen bond.
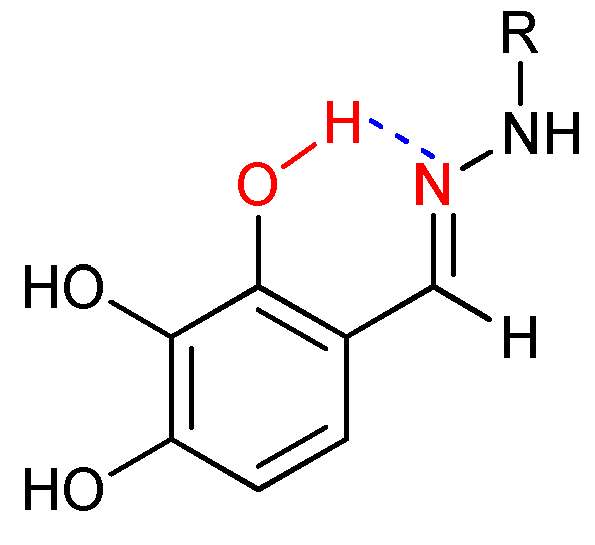
Figure 3.
Intramolecular hydrogen bonding interaction in synthesized Schiff base 4.
Based on 1H and 13C NMR spectra of 4a in DMSO, we observed separate chemical signals for the imine proton (N=CH) at 8.09 and 8.17 ppm respectively, whereas the protons of the ethyl group (CH2CH3) were present as two triplets in the region of 1.05-1.09 ppm and two quartets in the region of 2.19-2.23 and 2.51-2.54. In the 13C NMR spectrum, the carbon atom of the amide carbonyl groups (-C(O)NHN-) was detected from signals at 173.71 and 168.73 ppm. The signals for carbon from the ethyl groups (CH2CH3) were present at 8.47 and 9.41 ppm, 25.24 and 26.97. These spectra suggest the existence of another structural feature for compounds 4, namely a keto-enol tautomerism (Scheme 2).
Quantum chemical calculations using DFT were used to quantify Gibbs free energy for the constitutional isomers of 4a, which confirms the possibility of the keto and enol forms’ occurrence. According to the calculations performed on data collected without a solvent, the keto tautomer is preferred over the enol tautomer and is thermodynamically more stable than the enol form by about 11 kcal/mol. However, based on calculations performed in the presence of DMSO, the solvation process changed the Gibbs free energy of both tautomers. Our calculations clearly indicate that, in a polar aprotic solvent like DMSO, the enol form is more stable by 5 kcal/mol when compared with the keto form, due to the interaction of the OH-enol group with the oxygen atom of DMSO.
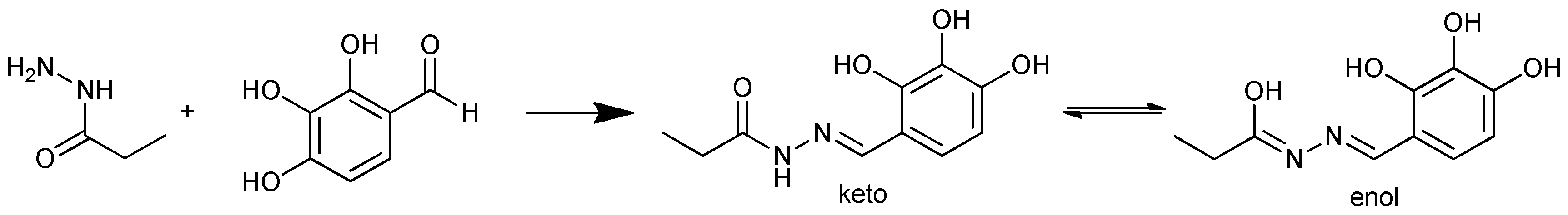
Scheme 2.
Tautomeric equilibrium in 4a benitrobenrazide.
The imine bond in derivative 4f was reduced using hydrogen in the presence of Pd(OH)2 as a catalyst, at elevated pressure (scheme 3). The reaction progress was monitored by 1HNMR spectrometry; disappearance of the signal for the 8.39 ppm imine proton region (N=CH) indicated the consumption of substrate 4f.
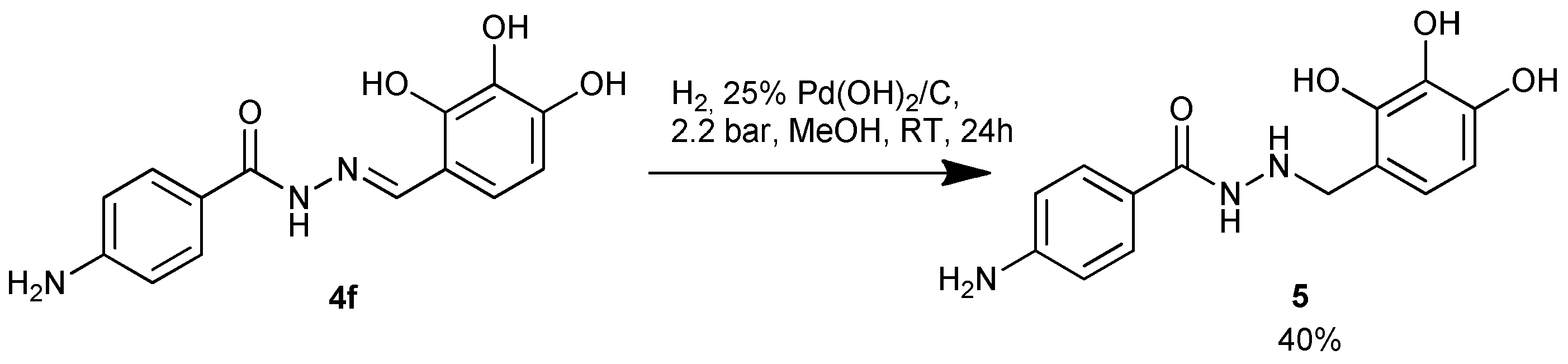
Scheme 3.
Reduction of hydrazone 4f.
Continuing our study, we synthesized benserazide analogues, in which serine was replaced with a side chain moiety of another L-amino acid glycine, tyrosine, and cysteine (Scheme 4).
Commercially available L-amino acids 6a-c were converted to methyl esters 7a-c, using thionyl chloride and methanol. A benzyloxycarbonyl group (Cbz) was used to protect the amino group of the amino acids, by reacting esters 7 with benzyl chloroformate in the presence of triethylamine (TEA) in an anhydrous methylene chloride solution [42]. This method of protection was used to facilitate the parallel deprotection of Cbz together with catalytic hydrogenation of the double bond. The reaction of N-Cbz-L-amino acid esters 8a-c with 98% hydrazine hydrate under the same conditions described in scheme 3, provided the products 9 as white crystals at a 36-97% yield. Hydrazones 10a and 10c were synthesized via the reaction of the hydrazide 9a and 9c with 2,3,4-trihydroksy- benzaldehyde in methanol at room temperature. In the case of 10b, as the above procedure failed, we repeated the condensation, in THF instead of methanol in an inert atmosphere of argon at reflux, which produced product 10b at a 55% yield. All hydrazones 10a-c were obtained with the E configuration, as confirmed by NMR spectroscopy. The best purification method for compounds 10 was crystallization with aqueous methanol (1:1 v/v). A last step in this synthesis was the elucidation of an efficient method for reduction of the imine bond of hydrazone and deprotection of the Cbz group. For the corresponding hydrazone 10b, hydrogenation was performed in a Parr’s autoclave with gaseous H2 at 2.5 bar, in the presence of palladium catalysts (mixture of 25% Pd/C and Pd(OH)2), at room temperature in methanolic solution. We observed only cleavage of the Cbz group.
Compound 10a was reduced under modified conditions; Instead of gaseous hydrogen, ammonium formate was used as a hydrogen donor and the same palladium catalysts were used, according to the reported protocol [43]. Product 11 was obtained as a hygroscopic powder, which rapidly liquefied after 5 minutes on air. For this reason, hydrazide 11 was lyophilized after the purification was complete. In the case of the cysteine analogue of benserazide 10c, hydrogenation did not occur under these conditions, or with the use of other catalysts such as Raney nickel and electrochemical reduction. The likely reason is that a compound containing sulfur in its structure causes catalyst poisoning, resulting in the loss of catalyst function [44].
2.2. Inhibitory effect of benserazide and benitrobenrazide derivatives on HK2 enzymatic activity.
We conducted in vitro studies of representative derivatives, namely N-acylhydrazones 4a-4f as benitrobenrazide derivatives, and a benitrobenrazide derivative with a single carbon-nitrogen bond 5. The benserazide derivative, hydrazones 10, 12 with imine bonds, and hydrazide 11 with single carbon-nitrogen bond, which, like benserazide, have an L-amino acid fragment (Figure 4), were selected for in vitro experiments. The selected intermediates of hydrazide 9a-c, the potential peptidomimetics, were also used to evaluate any inhibition of enzyme activity (Figure 4). The chosen compounds subjected to this enzyme activity assay were purified by preparative HPLC.
For the inhibitory activity estimation, the used a colorimetric method. The most popular HK2 enzymatic activity assays reported in the literature are tests based on spectroscopic, measuring/detecting the changes of NADPH absorbance at 320-490 nm [13-15, 31]. The alternative assays to determine the HK2 activity by reverse-phase high-performance liquid chromatography (RP-HPLC) was reported by Guan et al. [45]. According to this method concentration of released ADP during conversion of glucose into 6-glucose phosphate is measured at 254 nm. We excluded RP-HPLC as an alternative assay because tested compounds contain chromophore exhibiting absorption in UV-Vis in a region of 200-250 nm what enders the assay results unreliable. To evaluate the potential of hexokinase 2 inhibitors we used a commercially available Hexokinase II Inhibitor Screening Kit, which uses a spectrophotometric method by measurement of absorbance at 450 nm, where tested compounds no interfering with assay. This HK2 activity assay is based on HK2's ability to convert glucose into glucose-6-phosphate. Glucose-6-phosphate is oxidized by glucose-6-phosphate dehydrogenase to produce NADPH, which reduces the probe, showing strong absorbance at 450 nm.
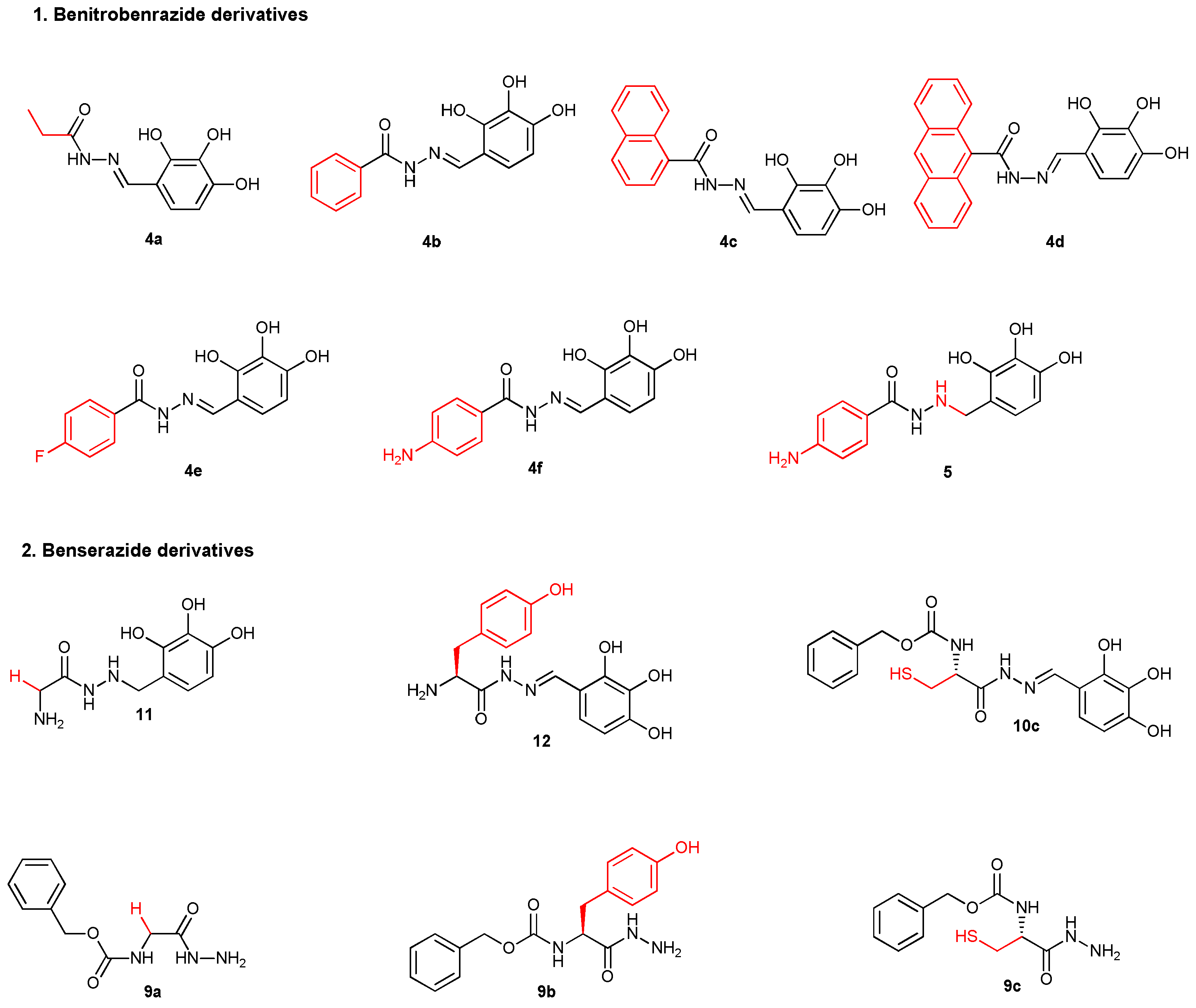
Figure 4.
Structure of the final derivatives evaluated for inhibition of HK2.
Figure 5 and Figure 6 show the results of the HK2 activity inhibition assay of synthesized compounds. Primary measurements were performed at 50 and 5 µM concentrations. Compounds 4a, 4b, 4c, 4e, and 4f, which showed the best inhibition of HK2 activity, were selected for the next assessment at a lower concentration of 1 µM. The data in Figure 5 illustrate the inhibition of HK2-mediated phosphorylation of glucose by the test compounds, through changes in absorption detection at λmax 450 nm observed in kinetic mode after 5-45 minutes.
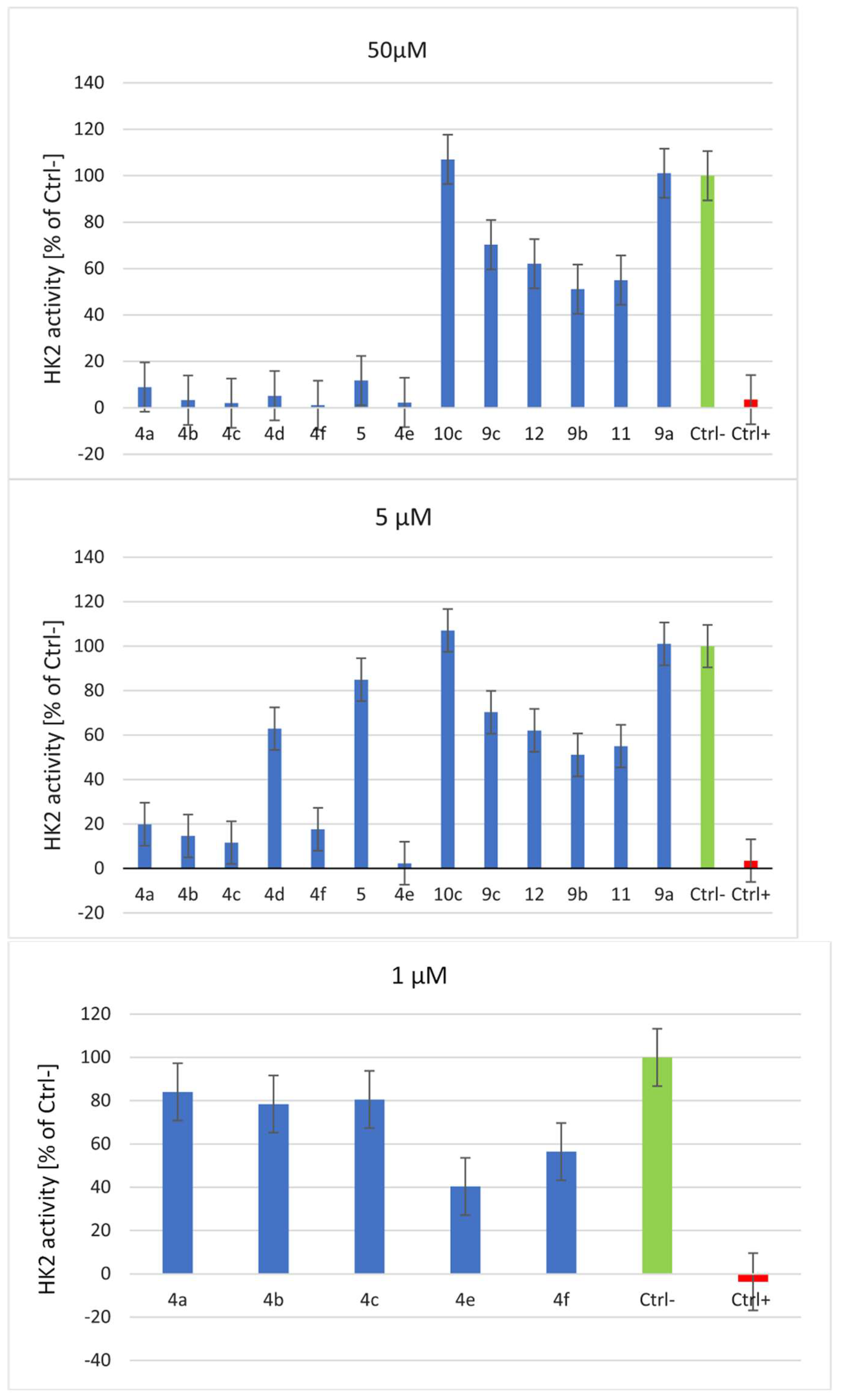
As shown in Figure 6, at a concentration of 50 µM, most of the compounds show significant HK2 inhibition activity, apart from hydrazides 9, that show moderate HK2 inhibition. According to the in vitro study, which considered the effect of HK2 inhibition by the tested compounds at a concentration of 5 µM, derivatives 4a-4f exhibited stronger HK2 inhibition activity than the benserazide derivatives 10c, 11, 12 which have a modified L-amino acid fragment in their structure. The maximum inhibition by benserazide derivatives 10c, 11 and 12 was approximately 50%. Hydrazides 9 have no significant effect on HK2 activity at a concentration of 5 µM, which confirms that the presence of three hydroxy groups is required for the inhibition of HK2 activity.
A comparison of the inhibition results for derivatives 4f and 5, for which HK2 inhibition was 92% and 15%, respectively, clearly indicates that the essential feature causing the HK2 inhibitory effect of benitrobenrazide derivatives is the presence of the imine bond (-CH=N-). Referring to studies on the pocket size of the HK2 binding site, it can be assumed that a bulky group, like the anthracenyl group in the structure of the Schiff base 4d, does not fit efficiently into the active site of HK2; Compared to other Schiff bases with smaller volume substituents 4a-c, compound 4d did not exhibit inhibitory activity against HK2 at a 5 µM concentration.
The most promising HK2 inhibitors assayed, 4a, 4b, 4c, 4e, 4f, were evaluated against human HK2 at a concentration of 1 µM. The derivatives 4e and 4f, which contain a fluorine atom or amino group instead of a nitro group, exhibited the most potent inhibitory effects against HK2 at a concentration of 1 µM, with HK2 inhibition rates of 60% and 54%, respectively. The highest efficacy among the derivatives assessed against HK2 was recorded for Schiff base 4e, in which the fluorine atom acts as an electron-withdrawing substituent in the para position of the benzene ring.
4. Materials and Methods
3.1. Chemistry
The 1H NMR and 13C NMR spectra were recorded using a Varian NMR system 600 spectrometer at 600 MHz in DMSO-d6, with tetramethylsilane (TMS) as the reference standard. NMR chemical shifts are reported in ppm (δ) and coupling constants (J) in Hz. Melting points were measured on a Boethius PHMK apparatus (VEB Analytik Dresden, Germany). The progress pf reaction was monitored by thin layer chromatography (TLC) using Merck TLC silica gel 60 F254 plates. Column chromatography was conducted using silica gel 40–60 μm 60A with methanol-chloroform mixtures as eluents. Preparative high-performance liquid chromatography was performed with the LaboACE LC-5060 system (Japan Analytical Industry Co., Ltd., Tokyo, Japan), with an ODS column (JAIGEL-ODS-AP, model SP-12-10). The compounds were eluted with a mobile phase of MeOH, at a flow rate of 9 mL/min. High resolution mass spectroscopy (HRMS) was measured on a Waters Corporation Xevo G2 QTOF apparatus using electrospray ionization (ESI).
3.1.1. General procedure for the synthesis of esters 2e-f
Methyl esters 2a and 2b were purchased from Merck. Methyl esters 2c, 2e-2f were obtained by the well-known method for esterification of acids using thionyl chloride in methanol [37]. To a stirred and cooled (0°C) solution of the required acid 1c, 1e-1f (20 mmol) in anhydrous methanol (30 mL) thionyl chloride (1.10 equiv.) was added dropwise while stirring. The mixture was warmed up to room temperature and stirred for 24 h. After that, the excess of methanol was removed under diminished pressure, and then dried under reduced pressure. In the case of 2f synthesis, after completion of the esterification reaction, the solution was neutralized by adding saturated aqueous NaHCO3 solution until no further gas evolution was observed. Solid 2f was filtered under reduced pressure.
Synthesis of ethyl anthracene-9-carboxylate 2d: To SOCl2 (7 mL) we added anthracene-9-carboxylic acid (1.80 mmol), and DMF (0.4 mL) as a catalyst. The reaction mixture was stirred at room temperature under argon atmosphere for 4h. The excess of SOCl2 was removed under reduced pressure. The residue was washed with toluene. To obtain orange solid acid chloride, EtOH (10 mL) and TEA (1.3 equiv.) were added sequentially. The reaction mixture was stirred at room temperature for 24 h. EtOH was removed under reduced pressure, then chloroform (10 mL) was added to the residue and the organic layer was washed with water (2x8 mL) and dried over anhydrous Na2SO4. After filtration and evaporation under reduced pressure, the residue was purified on a silica gel packed column using (AcOEt:n-hexane 1:1, v/v) as an eluent, obtaining solid 2d at a yield of 90%.
3.1.2. General procedure for the synthesis of hydrazides 3a-3f.
Hydrazides 3a-3f were synthesized from their corresponding esters 2a-2f, followed by reaction with hydrazine according to the method reported by Khan et al. [10].
Synthesis of hydrazides 3b-3c, 3f: To a solution of methyl ester 2b, c and f (20 mmol) in methanol (25 ml), hydrazine monohydrate (80 mmol, 4 equiv.) was added. The reaction mixture was stirred at room temperature for 72 h, and then cooled to -20°C. The formed precipitate was filtered off and dried under reduced pressure. Crystallization of crude solid from an ethanol:water (1:2, v/v) solution, producing white crystals.
Benzohydrazide (3b): Yield 77%. Mp 114-115 °C. (lit. Mp 110-113 °C [46]). 1H NMR (600 MHz, DMSO-d6): δ 4.48 (s, 2H) 7.53-7.46 (m, 2H), 7.49-7.51 (m, 1H), 7.81-7.83 (m, 2H), 9.76 (s, 1H). 13C NMR (600 MHz, DMSO-d6): δ 132.08, 133.44, 136.18, 138.46, 171.02. HRMS (ESI-TOF): m/z calcd for C7H8N2O [M+H]+ 137.0709; found: 137.0724.
1-naphthohydrazide (3c): Yield 69%. Mp 156-157 °C (lit. Mp 160-163 °C [38]). 1H NMR (600 MHz, DMSO-d6): δ 4.59 (d, J = 3.53 Hz, 2H), 7.51-7.58 (m, 4H), 7.96-7.98 (m, 1H), 8.00-8.01 (m, 1H), 8.20-8.22 (m, 1H), 9.68 (s, 2H) 13C NMR 150 MHz, DMSO-d6): 130.16, 130.51, 130.60, 131.40, 131.80, 133.35, 135.02, 135.18, 135.138.30, 138.55, 173.14. HRMS (ESI-TOF): m/z calcd for C11H10N2O [M+H]+ 187.0866; found: 187.0610.
4-aminobenzohydrazide (3f): Yield 60%. Mp 224-226 °C (lit. Mp 225-227 °C [48]) 1H NMR (600 MHz, DMSO-d6): δ 4.29 (s, 2H), 5.57 (s, 2H), 6.52 (d, J=8.72, 2H), 7.54 (d, J=8.67, 2H), 9.246 (s, 1H). 13C NMR (600 MHz, DMSO-d6): 113.04, 120.40, 128.84, 151.94, 166.88. HRMS (ESI-TOF): m/z calcd for C7H9N3O [M+H]+ = 152.0818. Found = 152.0036.
Synthesis of hydrazides 3a, 3e: To a solution of methyl propionate 2a (5.50 mmol) or methyl 4-fluorobenzoate 2e (5.50 mmol) in methanol (15 ml), we added hydrazine monohydrate (22 mmol, 4 equiv.). The reaction mixture was stirred at reflux for 24 h and evaporated under diminished pressure. The oily residue 3a was purified by silica gel column chromatography and eluted with 3% MeOH/CHCl3 (v/v). Product 3e was crystallized from an ethanol:water (1:2, v/v) solution.
Propanoic acid hydrazide (3a): Yield 100%. Mp 36-38°C (lit. Mp 38-40 °C [47]). 1H NMR (600 MHz, DMSO-d6): δ 0.99 (q, J=7.62, 3H), 2.01 (t, J=7.61, 2H), 4.13 (s, 2H), 8.90 (s, 1H), 13C NMR (600 MHz, DMSO-d6): δ 10.37, 27.07, 172.86. HRMS (ESI-TOF): m/z calcd for C3H8N2O [M+H]+ 89.0709; found: 89.0736.
4-Fluorobenzhydrazide (3e): Yield 60%. Mp 164-165 °C. (lit. Mp 160-163 °C [38]) δ 1H NMR (600 MHz, DMSO-d6): δ 4.48 (s, 2H), 7.24-7.27 (m, 2H), 7.86-7.88 (m, 2H), 9.76 (s, 1H). 13C NMR (600 MHz, DMSO-d6): 115.66 (d, J=21.75), 129.96 (d, J=8.98), 163.36, 165.01, 165.31. C7H7FN2O HRMS (ESI-TOF): m/z calcd for C7H7FN2O [M+H]+ = 155.0615. Found = 155.0623.
Synthesis of hydrazide 3d: To hydrazine monohydrate (98%, 10 mL), methyl anthracene-9-carboxylate (1.60 mmol) was added. The reaction mixture was stirred at reflux for 72 h. The residual hydrazine was removed under reduced pressure. A crude solid was purified by flash column chromatography on silica gel, using 100% CHCl3 followed by 20% MeOH/CHCl3 (v/v) as an eluent. The additional purification by the high-performance liquid chromatography (HPLC) method, using an ODS column with methanol as an eluent was performed.
Anthracene-9-carbohydrazide (3d): Yield 60%. Td (thermal decomposition temperature) 242 °C. 1H NMR (600 MHz, DMSO-d6): δ 4.81 (d, J = 3.10 Hz, 2H), 7.52-7.57 (m, 4H), 7.98-8.00 (m, 2H), 8.10-8.12 (m, 2H), 8.65 (s, 1H), 9.83 (s, 1H). 13C NMR 600 MHz, DMSO-d6): 125.92, 126.02, 126.76, 127.83, 128.33, 128.77, 131.10, 132.51, 167.96. HRMS (ESI-TOF): m/z calcd for C15H12N2O [M+H]+ 237.1028; found: 237.1031.
3.1.3. General procedure for the synthesis of hydrazones 4a-4f and hydrazide 5
Synthesis of hydrazones 4b-4c and 4e-4f: an appropriate hydrazide 3b,c,e,f (4.40 mmol) and 2,3,4-trihydroxybenzaldehyde (4.40 mmol) were dissolved in anhydrous methanol (15 mL), and the reaction mixture stirred at room temperature for 24 h. After the completion of the reaction, the formed solid was filtered and purified by recrystallisation from EtOH:H2O (1:1, v/v) [13].
(E)-N'-(2,3,4-trihydroxybenzylidene)benzohydrazide 4b: Yield 85%. Td 187-189 °C. 1H NMR (600 MHz, DMSO-d6): δ 6.41 (d, J=8.39 Hz, 1H), 6.80 (d, J=8.48 Hz, 1H), 7.53-7.56 (m, 2H), 7.59-7.62 (m, 1H) 7.93-7.94 (m, 2H)), 8.48 (s, 1H), 9.46 (s, 1H), 11.56 (s, 1H), 11.97 (s, 1H). 13C NMR (600 MHz, DMSO-d6): δ 108.10, 111.28, 121.63, 127.98, 128.96, 132.27, 133.16, 133.38, 147.97, 149.21, 150.63, 162.94. HRMS (ESI-TOF): m/z calcd for C14H12N2O4 [M+H]+ 273.0870; found: 273.0882.
(E)-N'-(2,3,4-trihydroxybenzylidene)-1-naphthohydrazide (4c): Yield 69%. Td 109-110 °C. 1H NMR (600 MHz, DMSO-d6): δ 6.38 (d, J=8.42), 6.77 (d, J=8.50), 7.58-7.61 (m, 3H), 7.76 (dd, J=1.12, J=7.03 1H), 8.00-8.02 (m, 1H), 8.09 (d, J=8.27, 1H), 8.23 (dd, J= 1.51, 8.15 1H), 8.35 (s, 1H), 8.49 (s, 1H), 9.48 (s, 1H), 11.47 (s, 1H), 12.07 (s, 1H) 13C NMR (600 MHz, DMSO-d6): δ 108.14, 111.23, 121.60, 125.42, 125.59, 126.44, 126.90, 127.56, 128.81, 130.41, 131.08, 132.77, 133.16, 133.62, 147.99,149.26, 150.51, 164.49. HRMS (ESI-TOF): m/z calcd for C18H14N2O4 [M+H]+ 323.1026; found: 323.1033.
(E)-4-fluoro-N'-(2,3,4-trihydroxybenzylidene)benzohydrazide 4e: Yield 100%. Td 217-219 °C. 1H NMR (600 MHz, DMSO-d6): 6.41(d, J=8.42, 1H), 6.80 (d, J=8.49, 1H) 7.38 (t, J= 8.84 2H), 7.99-8.03 (m, 2H), 8.47 (s, 1H) 11.98 (s, 1H) 13C NMR (600 MHz, DMSO-d6): δ 108.11, 111.24, 115.95 (d, JC-F =21.87), 121.66, 130.70 (d, JC-F =9.12), 133.13, 147.93, 149.21, 150.75, 161.99, 163.80, 165.46. HRMS (ESI-TOF): m/z calcd. for C14H11FN2O4 [M+H]+ 291.0781; found: 291,0875.
(E)-4-amino-N'-(2,3,4-trihydroxybenzylidene)benzohydrazide 4f: Yield 72%. Mp >250 °C. 1H NMR (600 MHz, DMSO-d6): δ 5.79 (s, 2H) 6.38 (d, J = 8.39 Hz, 1H), 6.60 (d, J = 8,63, 2H) 6.72 (d, J = 8.48 Hz, 1H), 7.66 (d, J = 8.41 Hz, 2H), 8.39 (s, 1H), 8.44 (s, 1H), 9.36 (s,1H), 11.55 (s, 1H). 11.78 (s, 1H) 13C NMR (600 MHz, DMSO-d6): δ 107.93, 111.47, 113.11, 119.41, 121.41, 129.68, 133.13, 147.79, 148.79, 149.01, 152.81, 162.85. HRMS (ESI-TOF): m/z calcd. for C14H13N3O4 [M+H]+ 288.0979; found: 288.0988.
Synthesis of hydrazones 4a, 4d: To a solution of anthracene-9-carbohydrazide 3d (1.14 mmol) or propanoic acid hydrazide 3a (1.14 mmol) in methanol (15 ml), 2,3,4-trihydroxybenzaldehyd (1.26 mmol, 1.1 equiv.) was added. The reaction mixture was stirred at reflux for 24 h. After consumption of the substrate, the solvent was removed under reduced pressure. The crude solid was crystallized from a solution of ethanol:water (1:1 v/v).
(E)-N'-(2,3,4-trihydroxybenzylidene)propionohydrazide 4a: Yield 89%. Td 184-186 °C. 1H NMR (600 MHz, DMSO-d6): δ 1.08 (t, J = 7.57, 3H), 2.21 (q, J = 7.55, 2H) 6.37 (d, J=8.41), 6.73 (d, J=8.46), 8.17 (s, 1H), 8.44 (s, 1H), 9.40 (s, 1H), 11.07 (s, 1H), 11.39 (s, 1H). 13C NMR (150 MHz, DMSO-d6): 9.41, 26.97, 107.40, 110.66, 120.86, 132.54, 147.20, 148.14, 148.38, 168.70 ( HRMS (ESI-TOF): m/z calcd for C10H12N2O4 [M+H]+ 225.0869; found: 225.0874.
(E)-N'-(2,3,4-trihydroxybenzylidene)anthracene-9-carbohydrazide 4d. Yield 50%. Td 167-169 °C. 1H NMR (600 MHz, DMSO-d6): δ 6.43 (d, J=8.37, 1H), 6.82 (d, J=8.48, 1H) 7.54-7.64 (m, 4H), 8.01-8.02 (m, 2H), 8.16-8.19 (m, 2H), 8.33 (s, 1H), 8.57 (s, 1H), 8.76 (s, 1H), 9.57 (s, 1H), 11.43 (s, 1H), 12.33 (s, 1H). 13C NMR 600 MHz, DMSO-d6): δ 108.26, 111.25, 121.61, 125.39, 126.22, 127.32, 127.48, 128.37, 128.99, 129.11, 131.09, 133.22, 148.05, 149.40, 150.59, 164.30. HRMS (ESI-TOF): m/z calcd for C22H16N2O4 [M+H]+ 373.1188; found: 373.1185.
Synthesis of hydrazide 5: (E)-4-amino-N'-(2,3,4-trihydroxybenzylidene)benzohydrazide 4f (3 mmol), anhydrous methanol (10 mL) and 25% of the weight of hydrazone 4f 20% Pd(OH)2/C were added to a reaction vessel. The vessel was placed in a Parr hydrogenator, and the reaction mixture was treated with hydrogen at 2.2 bar at room temperature for 6 hours. The catalyst was filtered from the solution and the reaction mixture was concentrated under reduced pressure. The crude solid was crystallized from an ethanol solution.
4-amino-N'-(2,3,4-trihydroxybenzyl)benzohydrazide 5: Yield 40%. Td 188-190 °C. 1H NMR (600 MHz, DMSO-d6): δ 3.80 (d, J=3.80, 2H), 5.18 (d, J=5.07, 1H), 5.63 (s, 2H) 6.20 (d, J=8.11, 1H), 6.42 (d, J=8.18, 1H) 6.53 (d, J=8.54, 2H), 7.54 (d, J=8.50, 2H) 8.12 (s, 1H), 8.74 (s, 1H), 9.20 (s, 1H), 9.74 (d, J=3.27, 1H) 13C NMR (600 MHz, DMSO-d6): δ 52.50, 106.56, 113.00, 115.65, 119.58, 119.85, 129.07, 133.56, 145.81, 145.98, 152.25, 166.39. HRMS (ESI-TOF): m/z calcd. for C14H15N3O4 [M+Na]+ 312.0955; found: 312.0955.
3.1.4. General procedure for the synthesis of amino acid methyl ester hydrochloride 7a-7c
Methyl esters 7a-7c were obtained by the well-known amino acid esterification method which uses thionyl chloride in methanol [25]. To a stirred and cooled (0°C) solution of required L-amino acid 6 (30 mmol) in anhydrous methanol (40 mL), SOCl2 (1.10 equiv.) was added dropwise. The reaction mixture was warmed up to room temperature and stirred for 24 h. After completion of the reaction, excess methanol was removed and dried under reduced pressure.
3.1.5. General procedure for the synthesis of N-benzyloxycarbonyl-L-amino acid methyl esters 8a-8c
An amino group of derivatives 7a-7c was protected using benzyl chloroformate [25]. To a solution of amino acid methyl ester hydrochloride salt 7a-7c (25 mmol) in dichloromethane (30 mL), triethylamine (2.5 equiv.) was added. After 10 minutes, benzyl chloroformate (1.2 equiv.) was added dropwise to the reaction mixture at 0°C. The reaction mixture was stirred at room temperature for 24 h, then, water was added to solubilize all salts. The organic layer was washed with water (2x30 mL) and dried over anhydrous Na2SO4. After filtration, the organic layer was concentrated under reduced pressure. The residue was purified on a silica gel column using a mixture of (AcOEt:n-hexane, 1:1 v/v), obtaining colorless oil 8a-8c.
3.1.6. General procedure for the synthesis of N-benzyloxycarbonyl-L-amino acid hydrazides 9a-9c
To a solution of N-Cbz-amino acid methyl esters 8a-8c (16.5 mmol) in anhydrous methanol (30mL), hydrazine hydrate (98% 4.0 equiv.) was added. The reaction mixture was stirred for 24 h at room temperature. The insoluble product formed was filtered off and was recrystallized from a solution of ethanol: H2O 1:1, v/v [25].
N-benzyloxycarbonyl-L-glycine hydrazide 9a: Yield 97%. Mp 114-115 °C (lit. Mp 112-114 °C [49]). 1H NMR (600 MHz, DMSO-d6): δ 3.55 (d, J=6.16, 2H), 4.17(s,2H), 5.00 (s, 2H), 7.28-7.39 (m, 5H), 9.02 (s, 1H), 13C NMR (600 MHz, DMSO-d6): δ 42.65, 65.91, 128.13, 128.23, 128.77, 137.44, 156.86, 168.93. HRMS (ESI-TOF): m/z calcd. for C10H13O3N3 [M+H]+ 224.1030; found: 224.1046.
N-benzyloxycarbonyl-L- tyrosine hydrazide 9b: Yield 36%. Mp 218-219 °C. (lit. Mp 219-221 °C [50]). 1H NMR (600 MHz, DMSO-d6): δ 2.62-2.66 (m,1H), 2.77-2.81 (m, 1H), 4.08-4.12 (m, 1H), 4.21 (s, 2H) 4.91-4.96 (m, 2H), 5.04 (s, 1H), 6.65 (d, J=8.36, 2H), 7.04 (d, J=8.30, 2H), 7.24-7.35 (m, 3H), 7.44 (d, J=8.71, 2H), 9.17 (s, 1H), 9.18 (s, 1H) 13C NMR (600 MHz, DMSO-d6): δ 37.48, 55.72, 65.57, 115.30, 127.86, 128.07, 128.49, 128.70, 128,76, 130.52, 137.52, 156.13, 156.19 171.29. (ESI-TOF): m/z calcd for C11H15N3O3S [M+H]+ 330.1454; found: 330.1441.
N-benzyloxycarbonyl-L-cysteine hydrazide 9c: Yield 67%. Mp 138-139 °C (lit. Mp 141-143 °C [51]). 1H NMR (600 MHz, DMSO-d6): δ 1.23 (s, SH), 2.84-2.88 (m, 1H), 3.03-3.06 (m, 1H), 4.26 (s, 2H), 5.00-5.04 (m, 2H), 7.31-7.36 (m, 4H), 7.56 (d, J=8.53, 1H) 9.30 (s, 2H) 13C NMR (600 MHz, DMSO-d6): δ 53.00, 56.47, 66.00, 128.16, 128.23, 128.75, 137.33, 156.24, 169.60. HRMS (ESI-TOF): m/z calcd. for C7H8ON2 [M-H]- 268.0761; found: 268.0763.
3.1.7. General procedure for the synthesis of hydrazones of N-benzyloxycarbonyl-amino acids 10a-10c.
Synthesis of (E)-benzyl(2-oxo-2-(2-(2,3,4-trihydroxybenzylidene)hydrazinyl)ethyl) carbamate : 2,3,4-trihydroxybenzaldehyd (11.7 mmol; 1.20 equiv.) was added to a stirring solution N-Cbz-L-glycine hydrazide 8a (9.8 mmol) in methanol (25 mL). The mixture solution was stirred at room temperature for 24 h. After completion of the reaction, the obtained solid was filtered off. The product was obtained as a solid, and purified by recrystallisation from solution of methanol: H2O 1:1 v/v.
Synthesis of (E)-benzyl(3-(4-hydroxyphenyl)-1-oxo-1-(2-(2,3,4-trihydroxybenzylidene)hydrazinyl) propan-2-yl)carbamate 10b: 2,3,4-trihydroxybenzaldehyd (1.20 mmol; 1.20 equiv.) was added to a stirring solution of N-benzyloxycarbonyl-L-tyrosine hydrazide 8b (1.00 mmol) in THF (10 mL). The resulting solution was stirred at reflux for 72 h, in an inert atmosphere of argon. The reaction mixture was concentrated under reduced pressure. The residue was crystallized from a 1:1, (v/v) solution of methanol:H2O, at a 55% yield.
Synthesis of (E)-benzyl(3-mercapto-1-oxo-1-(2-(2,3,4-trihydroxybenzylidene)hydrazinyl)propan-2-yl)carbamate 10c: 2,3,4-trihydroxybenzaldehyd (16.92 mmol; 1.20 equiv.) was added to a stirring solution of N-Cbz-L-cysteine hydrazide 8c (14.10 mmol) in methanol (30 mL). The mixture solution was stirred at room temperature for 24 and concentrated under reduced pressure. The residue was crystallized from an ethanol:H2O 1:1, v/v solution.
(E)-benzyl(2-oxo-2-(2-(2,3,4-trihydroxybenzylidene)hydrazinyl)ethyl)carbmate 10a: Yield 97%. Mp 213-215 °C (Mp 211-212 °C [52]). 1H NMR (600 MHz, DMSO-d6): δ 3.75 (d, J = 4.41, 2H), 5.06 (s, 2H), 6.38 (d, J = 7.92, 1H) 6.77 (d, J = 7.87, 1H), 7.32-7.37 (m, 4H), 7.60 (s, 1H), 8.25 (s, 1H), 8.47 (s, 1H) 9.45, (s, 1H), 9.52 (d, J=15.50, 1H) (m, 1H), 11.32 (s, 1H), 11.54 (s, 1H) 13C NMR (600 MHz, DMSO-d6): δ 43.09 66.03, 108.04, 111.16, 121.48, 128.11, 128.19, 128.26, 128.79, 133.11, 137.41, 147.80, 149.12, 149.62, 157.00, 165.65. HRMS (ESI-TOF): m/z calcd. for C17H17N3O6 [M+H]+ 360.1190; found: 360.1197.
(E)-benzyl(3-(4-hydroxyphenyl)-1-oxo-1-(2-(2,3,4-trihydroxybenzylidene)hydrazinyl)propan-2-yl)carbamate 10b: Yield 55%. Mp 106-108 °C. 1H NMR (600 MHz, DMSO-d6): δ 2.72-2.746 (m,1H), 2.88-2.91 (m, 1H), 4.20-4.21 (m, 1H), 4.94-5.00 (m, 2H), 6.66 (d, J=8.16, 2H), 6.38 (d, J=8.40, 1H), 6.77 (d, J=8.47, 1H), 7.08 (d, J=8.00, 2H), 8.24 (s, 1H) 8.46 (s, 1H), 9.21 (s, 1H) 9.46 (s, 1H), 11.27 (s, 1H) 13C NMR (600 MHz, DMSO-d6): δ 36.96, 56.11, 65.76, 108.05, 111.18, 115.37, 121.45, 127.87, 127.94, 128.15, 128.70 128.73, 130.56, 133.10, 137.40, 147.81, 149.14, 149.86, 156.29, 168.02. HRMS (ESI-TOF): m/z calcd. for C24H23N3O7 [M+H]+ 466.1609 ; found: 466.1610.
(E)-benzyl(3-mercapto-1-oxo-1-(2-(2,3,4-trihydroxybenzylidene)hydrazinyl)propan-2-yl)carbamate 10c: Yield 49%. Mp 134-135 °C. 1H NMR (600 MHz, DMSO-d6): δ 1.24 (s, 1H), 2.96-2.99 (m,1H), 3.18-3.21 (m, 1H), 4.39 (dd, J=8.54, 14.07 1H), 5.05 (s, 2H), 6.37 (d, J=8.41, 1H), 6.75 (d, J=8.41, 1H, 7.30-7.37 (m, 4H), 7.84 (d, J=8.21, 1H), 8.31 (s, 1H), 8.48 (s, 1H), 9.14 (s, 1H), 9.48 (s, 1H), 11.25 (s, 1H), 11.78 (s, 1H). 13C NMR (600 MHz, DMSO-d6): δ 53.50, 65.97, 66.18, 108.10, 111.13, 121.56, 128.21, 128.29, 128.78, 133.10, 137.21, 147.88, 149.26, 150.40, 156.40, 166.49. HRMS (ESI-TOF): m/z calcd. for C18H19N3O6S [M-H]- 404.0921; found: 404.0916.
3.1.8. General procedure for the synthesis of substituted amino acid hydrazide 11 and substituted amino acid hydrazone 12.
Synthesis of 2-amino-N’-(2,3,4-trihydroxybenzyl)acetohydrazide 11: Ammonium formate (1 mmol, 1 equiv.), 40% (w/v) hydrazone 10a and 10% PdOH2 (145 mg) were added to a stirring solution of N-(N-Cbz-L-glycine)-2,3,4-trihydroxybenzaldehyde hydrazone 10a (1 mmol) in methanol (10 mL). The reaction mixture was stirred at 50°C under an argon atmosphere for 12 h, then was cooled down to room temperature, filtered to remove the catalyst, and concentrated under reduced pressure. The residue was purified by crystallization from diethyl ether, and the pure powder was lyophilized. Hygroscopic powder obtained in 48% yield.
Synthesis of (E)-2-amino-3-(4-hydroxyphenyl)-N'-(2,3,4-trihydroxybenzylidene)propanehydrazide 12: Hydrazone 10b (0.5 mmol), anhydrous methanol (30 mL) and 25% (w/v) hydrazone 10b were added to a mixture of 10% Pd/C and Pd(OH)2. The reaction vessel was placed in a Parr shaker hydrogenator, at pressures of up to 2.5 bar at room temperature for 6 h. The solid catalyst was filtered off, and the reaction solution was concentrated under reduced pressure. The product was purified by reverse phase preparative HPLC on an ODS column using 70% MeOH:H2O (v/v).
2-amino-N'-(2,3,4-trihydroxybenzyl)acetohydrazide 11: Yield 48%. Hygroscopic powder 1H NMR (600 MHz, DMSO-d6): δ 1.99 (s, 2H), 3.06 (s, 2H), 3.49 (s, 2H), 3.60 (s, 1H), 6.12 (d, J=8.06, 1H), 6.28 (d, J= 8.09), 8.86 (s, 1H). 13C NMR (600 MHz, DMSO-d6): δ 43.17, 44.11, 106.75, 115.31, 119.81, 133.78, 145.00, 145.16, 172.52. HRMS (ESI-TOF): m/z calcd. for C9H13N3O4 [M+H]+: 228.0978; found: 228.0331.
(E)-2-amino-3-(4-hydroxyphenyl)-N'-(2,3,4-trihydroxybenzylidene)propanehydrazide 12: Yield 20%. Td 176-178 °C 1H NMR (600 MHz, DMSO-d6): δ 2.58-2.61 (m, 1H), 2.76-2.79 (m, 1H), 3.65-3.69 (m, 1H), 4.41 (d, J=7.43, 1H) 4.51 (d, J= 7.37, 1H), 6.18-6.21 (m, 1H), 6.36-6.40 (m, 1H), 6.62-6.68 (m, 2H), 6.91-7.07 (m, 2H), 8.04 (s, 1H), 8.31 (s, 1H) 13C NMR (600 MHz, DMSO-d6): δ 29.46, 55.21, 106.66, 115.39, 119.68, 128.29, 128.45, 130.57, 130.65, 133.46, 145.63, 146.01, 156.26, 172.87 HRMS (ESI-TOF): m/z calcd. for C16H17N3O5 [M+H]+: 332.1241; found: 332.1253.
3.2. Computational methods
Calculations were performed by the Orca 4.2.1 package [Neese, F. (2017) Software update: the ORCA program system, version 4.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 8, e1327.] on the DFT level (B3LYP/def2-SVP). The accuracy of the optimization process was determined using the Hessian eigenvalue analysis. All the calculated Hessian eigenvalues were positive for the compounds evaluated. In the case of calculations in a solvent environment, the CPCM continuous solvation model for DMSO was used.
3.3. Hexokinase activity assay
For studying the potential of HK2 inhibitors, a commercially available assay test (ab211114) Hexokinase II Inhibitor Screening (colorimetric) was used. The in vitro hexokinase activity assay was conducted according to the manufacturer’s instructions. Briefly, the enzyme and substrate solution were prepared, the enzyme solution was added to the wells containing sample compounds and incubated for 5 minutes at 25 °C, then the substrate solution was added to the wells and the absorbance was measured at 450 nm every 5 minutes for 45 minutes, using the Thermo Scientific™ Varioskan™ LUX multimode microplate reader. The test compounds were dissolved in DMSO at 50µM, 5 µM and 1 µM concentrations, with the final concentration of solvent not exceeding 1% by volume.
5. Conclusions
In our research, we synthesized benitrobenrazide and benserazide analogues. We identified that some of these compounds, namely compounds 4e and 4f, represent a promising class of HK2 inhibitors, inhibiting HK2 at a concentration of 5 µM by 98% and 82%, respectively. HK2 was inhibited by 4e and 4f at a concentration of 5 µM, by 98% and 82%, respectively. At the lower concentration of 1 µM, 4e and 4f inhibited HK2 by 60% and 54%, respectively. We have confirmed that the presence of a bulky anthracenyl group in 4d have no significant effect on HK2 enzymatic activity. The exchanging of serine by glycine or threonine in benserazide analogues has minor effect on their inhibition activity against HK2. Compounds 11 and 12 reduce the enzymatic activity of HK2 by approx. 40% in comparison with negative control. The presented findings suggest that the imine scaffold -CH=N-, in the structure of the potent HK2 inhibitors, helps to enhance and regulate their biological activities. The -CH=N- core is responsible for the possible binding of various groups with nucleophilic and electrophilic properties, and thus can interact with targeted enzymes and inhibit their enzymatic activity.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Figure S1: hydrogen bonding interaction in compound 4a; Figure S2-S28: 1H and 13C NMR spectrum of synthesized compounds.
Author Contributions
Conceptualization, K.J. and K.W; methodology, K.J. and K.W.; formal analysis, software, W.Sz.; investigation, K.J.; resources, K.J and KW; writing—original draft preparation, K.J.; writing—review and editing, K.W; supervision, K.W.; All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by STU internal fund, grant number 04/020/BKM22/1061.
Institutional Review Board Statement
Not Applicable
Informed Consent Statement
Not Applicable.
Data Availability Statement
Not Applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Raczuk E.; Dmochowska B.; Samaszko-Fiertek J.; Madaj J. Different Schiff Bases-Structure, Importance and Classification. Molecules, 2022, 27, 787. [CrossRef]
- Uddin N.; Faisal R.; Saqib A.; Tirmizi S. A.; Ahmad I.; Zaid S.; Zubair M.; Diaconescu P. L.; Tahir M. N.; Iqbal J.; Haider A. Synthesis, characterization, and anticancer activity of Schiff bases. J. Biomol. Struct. Dyn. 2020, 38, 3246–3259. [CrossRef]
- Kajal A.; Bala S.; Kamboj S.; Sharma N.; Saini V. Schiff Bases: A Versatile Pharmacophore. J. Catal. 2013, 2013, 1–14. [CrossRef]
- Mounika K.; Anupama B.; Pragathi J.; Gyanakumari C. Synthesis, characterization and biological activity of a Schiff base derived from 3-ethoxy salicylaldehyde and 2-amino benzoic acid and its transition metal complexes. J. Scient. Res. 2010, 2 (3), 513–524. [CrossRef]
- Kargar H.; Fallah-Mehrjardi M.; Behjatmanesh-Ardakani R.; Tahir M. N.; Ashfaqc M.; Munawar K. S. Synthesis, crystal structure determination, Hirshfeld surface analysis, spectral characterization, theoretical and computational studies of titanium(IV) Schiff base complex, J. Coord. Chem. 2021, 74 (16). 2682–2700. [CrossRef]
- Awantu A. F.; Fongang Y. S. F.; Ayimele G. A.; Nantia E. A.; Fokou P. V. T.; Boyom F. F.; Ngwang C. K.; Bruno N. Lenta B. N.; Ngouela, S. A. Novel Hydralazine Schiff Base Derivatives and Their Antimicrobial, Antioxidant and Antiplasmodial Propertie Int. J. Org. Chem. 2020, 10 (1), 1-16. [CrossRef]
- Taha M.; Ismail N. H.; Imran S.; Anouar H.; Selvaraj M.; Jamil W.; Ali M.; Kashif S. M.; Rahim F.; Khan K. M.; Adenan M. I. Synthesis and molecular modelling studies of phenyl linked oxadiazole-phenylhydrazone hybrids as potent antileishmanial agents. Eur. J. Med. Chem. 2017, 126, 1021-1033. [CrossRef]
- Li L-Y.; Peng J-D.; Zhou W.; Qiao H.; Deng X.; Li, Z-H.; Li J-D.; Fu Y-D.; Li S.; Sun K.; Liu H-M.; Zhao W. Potent hydrazone derivatives targeting esophageal cancer cells. Eur. J. Med. Chem. 2018, 148, 359-371. [CrossRef]
- Küçükgüzel S. G.; Mazi A.; Sahin F. Öztürk, S.; Stables S. Synthesis and biological activities of diflunisal hydrazide-hydrazones. Eur. J. Med. Chem. 2003, 38, 1005–1013. [CrossRef]
- Khan K. M.; Rasheed M.; et al. Synthesis and in vitro leishmanicidal activity of some hydrazides and their analogues. Bioorg. Med. Chem. 2003, 11, 1381–1387. [CrossRef]
- Durcik M.; Tammela P.; et al. Synthesis and Evaluation of N-Phenylpyrrolamides as DNA Gyrase B Inhibitors. Chem. Med. Chem. 2018, 13, 186–198. [CrossRef]
- Kareem H. S.; Ariffin A.; Nordin N.; Heidelberg T.; Azlina Abdul-Aziz A.; Kong K. W.; Yehye W.A. Correlation of antioxidant activities with theoretical studies for new hydrazone compounds bearing a 3,4,5-trimethoxy benzyl moiety. Eur. J. Med. Chem. 2015, 103, 497-505. [CrossRef]
- Liu Y.; Li M.; Zhang Y.; Wu C.; Yang K.; Gao S.; Zheng M.; , Li X.; Li H.; Chen L. Structure based discovery of novel hexokinase 2 inhibitors. Bioorg. Chem. 2020, 96, 103609. [CrossRef]
- Wei L.; Mengzhu Z.; Shuangping W.; Gao S.; Yang M.; Li Z.; Min Q.; Sun W.; Chen L.; Xiang G.; Li H. Benserazide, a dopa decarboxylase inhibitor, suppresses tumor growth by targeting hexokinase 2. J. Exp. Clin. Cancer Res. 2017, 36(58), 1–12. [CrossRef]
- Zheng M.; Wu C.; Yang K.; Yang Y.; Liu Y.; Gao S.; Wang Q.; Li Ch.; Chen L.; Li H. Novel selective hexokinase 2 inhibitor Benitrobenrazide blocks cancer cells growth by targeting glycolysis. Pharmacol. Res. 2021, 164, 105367. [CrossRef]
- Pelicano H.; DS Martin D. S.; Xu R-H.; Huang P. Glycolysis inhibition for anticancer treatment. Oncogene 2006, 25, 4633-4646. [CrossRef]
- Hay N. Aerobic glycolysis Oxidative phosphorylation Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat. Publ. Gr. 2016, 16, 635. [CrossRef]
- Heiden M. G. V.; Cantley L. C.; Thompson C. B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science, 2009, 324, no. 5930, 1029. [CrossRef]
- Semenza G.L. HIF-1: upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51-56. [CrossRef]
- Wu J.; Hu L.; Wu F.; Zou J.; He T. Poor prognosis of hexokinase 2 overexpression in solid tumors of digestive system: a meta-analysis. Oncotarget, 2017, 8, 32332. [CrossRef]
- Ciscato F.; Ferrone L.; Masgras I.; Laquatra C.; Rasola A. Hexokinase 2 in Cancer: A Prima Donna Playing Multiple Characters. Int. J. Mol. Sci. 2021, 22, 4716. [CrossRef]
- Counihan J. L.; Grossman E. A.; Nomura D. K. Cancer Metabolism: Current Understanding and Therapies. Chem. Rev. 2018, 118, 6893-6923. [CrossRef]
- Wilson J. E. Isozymes of mammalian hexokinase: structure, subcellular localization, and metabolic function. J. Exp. Biol. 2003, 206, 2049-2057. [CrossRef]
- Garcia S. N.; Guedes R. C.; Marques M. M. Unlocking the Potential of HK2 in Cancer Metabolism and Therapeutics. Curr. Med. Chem. 2019, 26, 7285-7322. [CrossRef]
- Tsai J.H.; Wilson J. E. Functional organization of mammalian hexokinases: characterization of the rat type III isozyme and its chimeric forms, constructed with the N- and C-terminal halves of the type I and type II isozymes. Arch. Biochem. Biophys. 1997, 338, 183-192. [CrossRef]
- Zhang F. Angelova A.; Garamus V. M.; Angelov B.; Tu, S.; Kong L.; Zhang X.; Li N.; Zou A. Mitochondrial Voltage-Dependent Anion Channel 1−Hexokinase-II Complex-Targeted Strategy for Melanoma Inhibition Using Designed Multiblock Peptide Amphiphiles. ACS Appl. Mater. Interfaces 2021, 13, 35281-35293. [CrossRef]
- Lin H.; Zeng J.; Xie R.; Schulz M. J.; Tedesco R.; Qu J.; Erhard K. F.; Mack J. F.; Raha K.; Rendina A. R.; Szewczuk L. M.; Kratz P. M.; Jurewicz A. J.; Cecconie T.; Martens S.; McDevitt P. J.; Martin J. D.; Chen S. B.; Jiang Y.; Nickels L.; Schwartz B. J.; Smallwood A.; Zhao B.; Campobasso N.; Qian Y.; Briand J.; Rominger C. M.; Oleykowski C.; Hardwicke M. N.; Luengo J. I. Discovery of a Novel 2,6-Disubstituted Glucosamine Series of Potent and Selective Hexokinase 2 Inhibitors. ACS Med. Chem. Lett. 2016, 7, 217-222. [CrossRef]
- Tanbin, S.; Fuad F. A. A.; Hamid A. A. A. Virtual Screening for Potential Inhibitors of Human Hexokinase II for the Development of Anti-Dengue Therapeutics. Bio. Tech. 2021, 10, 1-28. [CrossRef]
- Juszczak K.; Kubicka A.; Kitel R.; Zawadzki S.; Marczak A.; Dzido G.; Walczak K.; Łabieniec-Watała M.; Matczak K.; Tomczyk M. D. Hexokinase 2 Inhibition and Biological Effects of BNBZ and Its Derivatives: The Influence of the Number and Arrangement of Hydroxyl Groups. Int. J. Mol. Sci. 2022, 23, 2616. [CrossRef]
- Song Y-J.; Zeng H-B.; Peng A-H.; Ma J-H.; Lu D-D.; Li X.; Zhang H-Y.; Xie W-D. Strepantibins A-C: Hexokinase II Inhibitors from a Mud Dauber Wasp Associated Streptomyces sp. J. Nat. Prod. 2019, 82, 1114–1119. [CrossRef]
- Agnihotri S.; Mansouri S.; Burrell K.; et al. Ketoconazole and Posaconazole Selectively Target HK2-expressing Glioblastoma Cells. Clin. Cancer Res. 2019, 25, 844-855. [CrossRef]
- Salani B.; Del Rio A.; Marini C.; Sambuceti G.; Cordera R.; Maggi D. Metformin, cancer, and glucose metabolism. Endocrine-Related Cancer 2014, 21, 461-471. [CrossRef]
- Nepali K.; Lee H. Y.; Liou J. P. Nitro-Group-Containing Drugs. J. Med. Chem. 2019, 62, 2851-2893. [CrossRef]
- Nishiwaki N. A Walk through Recent Nitro Chemistry Advances. Molecules, 2020, 25, 3680. [CrossRef]
- Wang Q.; Jianlin Han J.; Sorochinsky A.; Landa A.; Soloshonok V. A. The Latest FDA-Approved Pharmaceuticals ContainingFragments of Tailor-Made Amino Acids and Fluorine. Pharmaceuticals 2022, 15, 999. [CrossRef]
- Al-Harthy T.; Zoghaib W.; Abdel-Jalil R. Importance of Fluorine in Benzazole Compounds. Molecules 2020, 25, 4677. [CrossRef]
- Hosangadi B. D.; Dave. R. H. An efficient general method for esterification of aromatic carboxylic acids. Tetrah. Lett. 1996, 37 (35), 6375-6378. [CrossRef]
- Li X.; Zhao Z.; Li G.; Shi P.; Design and Synthesis of Novel Molecular Tweezer Anion Receptors based on Diphenic Acid Carbonyl Thiosemicarbazide. J. Chem. Res, 2010, 410-413. [CrossRef]
- Rohane S. H.; Chauhan A. J.; Fuloria N. K.; Fuloria S. Synthesis and invitro antimycobacterial potential of novel hydrazones of eugenol. Arab. J. Chem. 2020, 13, 4495-4504. [CrossRef]
- Gloaguen E.; Brenner V.; Alauddin M.; Tardivel B.; Mons M.; Zehnacker-Rentien A.; Declerck V.; Aitkenet D. J. al. Direct spectroscopic evidence of hyperconjugation unveils the conformational landscape of hydrazides. Angew. Chem. Int. Ed. Engl. 2014, 53, 13756–13759. [CrossRef]
- Özen A. S.; De Proft F.; Aviyente V.; Geerlings P. Interpretation of hydrogen bonding in the weak and strong regions using conceptual DFT descriptors. J. Phys. Chem. A. 2006, 110, 5860-5868. [CrossRef]
- Green T. W.; Wuts P. G. M. Protective Groups in Organic Synthesis. Wiley-Interscience, NY 1999, 531-537, 736-739.
- Kozioł A.; Lendzion-Paluch A.; Manikowski A. A fast and effective hydrogenation process of protected pentasaccharide: A key step in the synthesis of fondaparinux sodium. Org. Process Res. Dev. 2013, 17, 869-875. [CrossRef]
- Bartholomew C. H. Mechanisms of catalyst deactivation. Appl. Catal. A Gen. 2001, 212(1-2), 17-60. [CrossRef]
- Guan Y.; Wang J.; Sun J. A method for determination of hexokinase activity by RP-HPLC. J. Nat. Sci. 2011, 16, 535-540. [CrossRef]
- Rodrigues D. A.; et al. Design, Synthesis, and Pharmacological Evaluation of First-in-Class Multitarget N-Acylhydrazone Derivatives as Selective HDAC6/8 and PI3Kα Inhibitors. ChemMedChem, 2020, 15, 539–551. [CrossRef]
- Saha A.; Kumar R.; Kumar R.; Devakumar C. Development and assessment of green synthesis of hydrazides. Indian J Chem. 2010, 49B, 526-531.
- Younis A.; Awad G. E. A.Utilization of Ultrasonic as an Approach of Green Chemistry for Synthesis of Hydrazones and Bishydrazones as Potential Antimicrobial Agents. Egypt.J.Chem. 2020, 63(2), 599-610. [CrossRef]
- de Fátima S. Barreto A.; dos Santos V. A.; Andrade C. K. Z. Synthesis of acylhydrazino-peptomers, a new class of peptidomimetics, by consecutive Ugi and hydrazino-Ugi reactions. Beilstein J. Org. Chem. 2016, 12, 2865-2872. [CrossRef]
- Schröder E.; Gibian H. Über Peptidsynthesen, XII. Synthese von Glukagon-Teilsequenzen. Liebigs Ann. Chem. 1962, 656 (1), 190-204. [CrossRef]
- Eugen Schnabel. E. Nebenreaktionen bei der Synthese von Peptiden nach dem Azidverfahren von Curtius. Liebigs Ann. Chem. 1962, 659 (1), 168-184. [CrossRef]
- Bartholini G.; Hegedues B. Hoffmann La Roche. Ein Hydrazid und dessen Saeureadditionssalze. CPC C07C243/34 (EP); C07C251/72 (EP); DE1941261, 1970, A1 [Chem.Abstr., vol. 81, # 91225].
Figure 1.
Tautomerism in a hydrazide molecule.
Scheme 1.
Preparation of benitrobenrazide derivatives. Reagent and conditions: (A) a: SOCl2 (1.2 equiv.), MeOH, 0°C−>RT, 24h; b: N2H4·H2O (98%, 4 equiv.), MeOH, RT/reflux 24/72h.; c: (HO)3C6H2CHO; (1.0 equiv.), MeOH, RT/reflux, 24h. (B) d: SOCl2, (53.6 equiv.), DMF, RT, Ar, 4h; e: EtOH, TEA (1.2 equiv.), RT, 24h; g: N2H4·H2O (98%, 100 equiv.), reflux, 72h; g: (HO)3C6H2CHO; (1.1 equiv.), MeOH, reflux; 24h.
Scheme 1.
Preparation of benitrobenrazide derivatives. Reagent and conditions: (A) a: SOCl2 (1.2 equiv.), MeOH, 0°C−>RT, 24h; b: N2H4·H2O (98%, 4 equiv.), MeOH, RT/reflux 24/72h.; c: (HO)3C6H2CHO; (1.0 equiv.), MeOH, RT/reflux, 24h. (B) d: SOCl2, (53.6 equiv.), DMF, RT, Ar, 4h; e: EtOH, TEA (1.2 equiv.), RT, 24h; g: N2H4·H2O (98%, 100 equiv.), reflux, 72h; g: (HO)3C6H2CHO; (1.1 equiv.), MeOH, reflux; 24h.
Scheme 4.
Preparation of Benserazide analogues. Reagent and conditions: a: MeOH, SOCl2 (1.2 equiv.), 0°C−>RT, 24h; b: CbzCl (1,2 equiv.), TEA (2,5 equiv.), DCM, RT, 24h; c: 98% N2H4.H2O (4.0 equiv.), MeOH, RT, 24h d: (HO)3C6H2CHO, (1.2 equiv.), MeOH, RT, 24h/(HO)3C6H2CHO; (1.2 equiv.), THF, reflux, 72h; e: HCOONH₄, 25% Pd(OH)2/C, MeOH, 50°C, Ar, 12h; f: H2, 25% mixture Pd(OH)2/C and Pd/C, 2.5 bar, MeOH, RT, 6h.
Scheme 4.
Preparation of Benserazide analogues. Reagent and conditions: a: MeOH, SOCl2 (1.2 equiv.), 0°C−>RT, 24h; b: CbzCl (1,2 equiv.), TEA (2,5 equiv.), DCM, RT, 24h; c: 98% N2H4.H2O (4.0 equiv.), MeOH, RT, 24h d: (HO)3C6H2CHO, (1.2 equiv.), MeOH, RT, 24h/(HO)3C6H2CHO; (1.2 equiv.), THF, reflux, 72h; e: HCOONH₄, 25% Pd(OH)2/C, MeOH, 50°C, Ar, 12h; f: H2, 25% mixture Pd(OH)2/C and Pd/C, 2.5 bar, MeOH, RT, 6h.
Figure 5.
Inhibition of HK2 activity, demonstrated as a reduction in rate or extent of generation of HK2 dependent absorbance (OD) at 450 nm by a synthesized compound. Ctrl- is an enzyme control, without inhibitor, which shows the normal rate of phosphorylation by HK2. All experiments were performed independently in triplicate.
Figure 5.
Inhibition of HK2 activity, demonstrated as a reduction in rate or extent of generation of HK2 dependent absorbance (OD) at 450 nm by a synthesized compound. Ctrl- is an enzyme control, without inhibitor, which shows the normal rate of phosphorylation by HK2. All experiments were performed independently in triplicate.
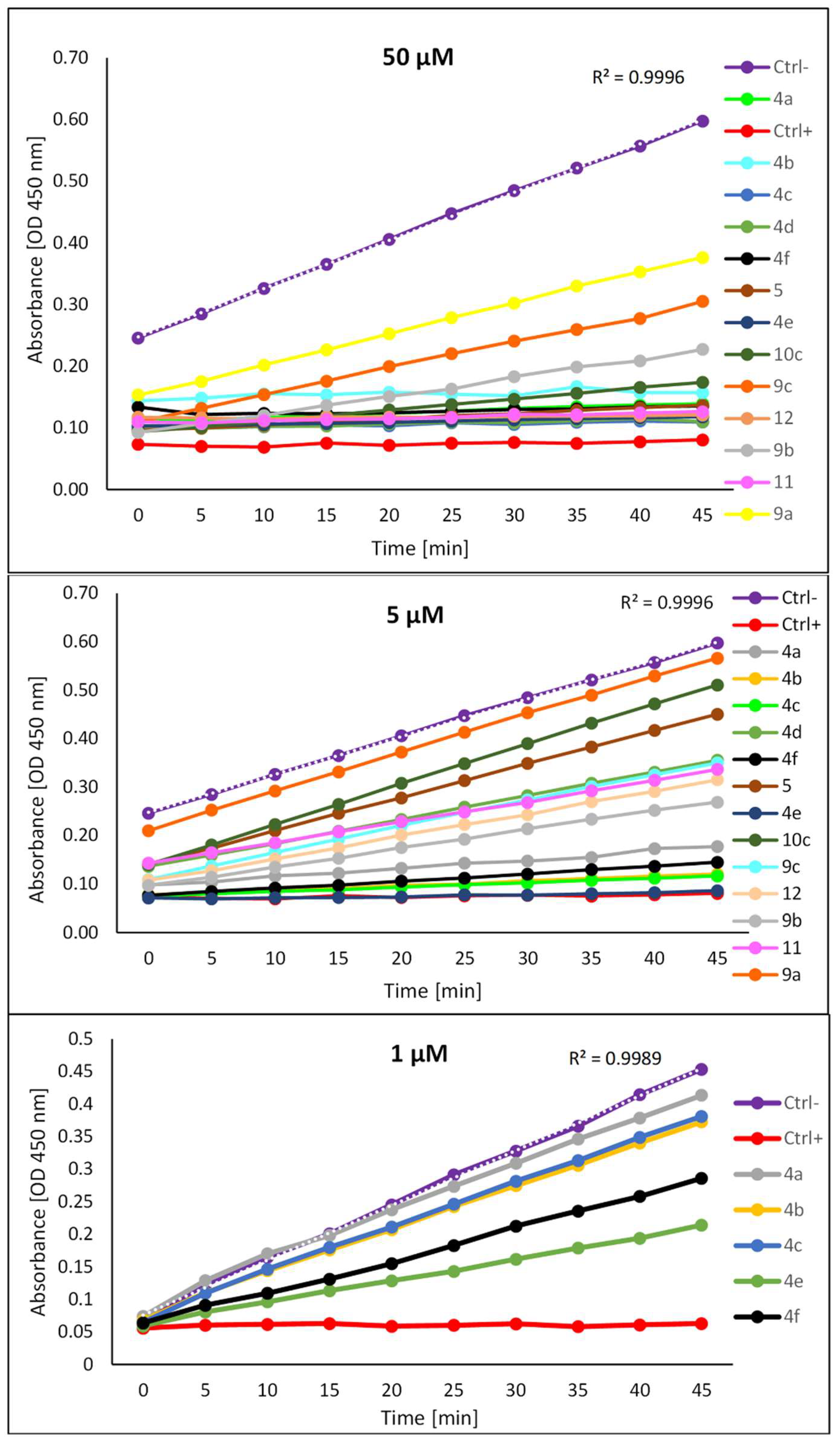
Figure 6.
Inhibitory effect of synthesized compounds on HK2 enzyme activity. Bromopyruvic acid was used as a positive HK2 inhibitor control. The relative activity of the negative enzyme control (without inhibitor) was set at 100%. Results are displayed as the mean ± SD from three independent experiments.
Figure 6.
Inhibitory effect of synthesized compounds on HK2 enzyme activity. Bromopyruvic acid was used as a positive HK2 inhibitor control. The relative activity of the negative enzyme control (without inhibitor) was set at 100%. Results are displayed as the mean ± SD from three independent experiments.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



