
Submitted:
28 December 2023
Posted:
04 January 2024
You are already at the latest version
Abstract
Abstract The relationship between cancer and neurodegeneration has spurred extensive scientific discourse, challenging their historical classification as distinct disorders. While cancer signifies uncontrolled cellular proliferation and neurodegeneration marks progressive neuronal loss, recent insights highlight intriguing interconnections between these seemingly disparate conditions. Cancer entails perpetual signaling for growth, evasion of growth inhibitors, resistance to cell death, acquisition of replicative immortality, stimulation of blood vessel growth (angiogenesis), and initiation of invasion and metastasis. Conversely, investigations emphasize disrupted cellular energy regulation and evasion of immune surveillance as key traits arising from genome instability, mutations, and inflammation driving tumorigenesis. Neurodegeneration involves neuronal malfunction and depletion, synaptic impairment, and protein abnormalities aggregations contributing to muscle atrophy and cognitive deficits. Varied clinical studies underscore contrasting correlations between cancer and neurodegeneration, hinting at mirrored molecular pathways that foster cell resilience or vulnerability. Research explores cellular signaling in tumorigenesis, shared with neurodegenerative disorders. Aberrant expression or mutations in crucial genes implicated in neurodegeneration also surface in cancer contexts. Debates persist on the nature of the relationship between cancer and neurodegeneration, contemplating inverse associations or shared pathways. Understanding their complex interplay, encompassing genetics, cellular mechanisms, and environmental influences, remains pivotal in unraveling their connections. This review explores the intriguing perspective that neurodegeneration and cancer might share fundamental genetic links, delving into potential implications for their onset and progression.
Keywords:
Neurodegeneration
; inflammation
; cancer
; Parkinson's Disease
; neuron
; cell biology
; interleukins
; disease progression
; genetic and somatic mutations
; double hit hypothesis
1. Introduction
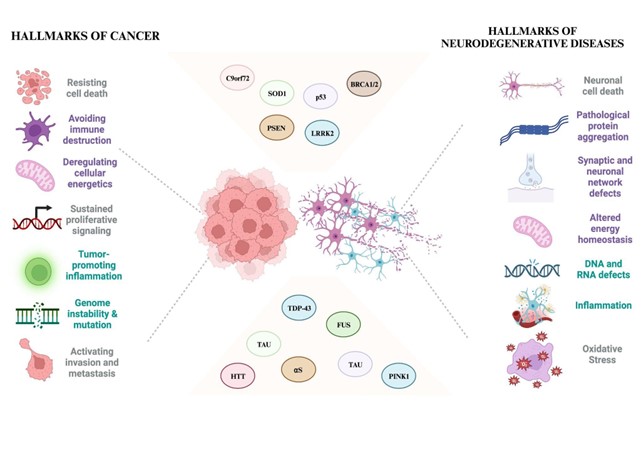
The intricate relationship between cancer and neurodegeneration has sparked extensive debates within the scientific community. While traditionally viewed as separate entities, cancer is characterized by uncontrolled cell growth and neurodegeneration marked by the progressive loss of neurons; recent research has unveiled intriguing connections between these seemingly disparate conditions [1,2]. Cancer is linked to broad features encompassing continual signals for cell proliferation, circumvention of growth inhibitors, resilience against cell demise, attainment of perpetual replication, stimulation of blood vessel growth (angiogenesis), and instigation of invasion and spread to distant sites (metastasis) [3]. Intriguingly, ongoing investigations have pinpointed attributes like disrupted cellular energy regulation and avoidance of immune system attacks as significant and unique traits [4]. These characteristics stem from genome instability, mutations, and inflammation facilitating tumor development [4].
Neurodegeneration involves the malfunction and depletion of neurons, impaired synaptic adaptability, protein irregularities like amyloid-β (Aβ) and misfolded tau in Alzheimer's disease (AD), α-synuclein (αS) in Parkinson's disease (PD), and their aggregations, alongside gradual muscle decline or wasting, contributing to memory deficiencies, cognitive lapses, and movement impairments [5,6]. Many clinical and epidemiological studies have underscored contrasting correlations between cancer and neurodegenerative conditions [1,2,7,8,9,10]. Current studies propose that the replication of genetic material, minus the subsequent cell division, leads to intrinsic molecular defects that manifest as changes triggering apoptosis [5]. The outcomes of irregular cell cycle activation followed by apoptosis are showcased through heightened levels of molecular stress response and markers associated with apoptosis [11]. These ailments exhibit mirrored molecular pathways, where one fosters resilience against cell demise. At the same time, the other accentuates vulnerability to cellular breakdown potentially serving as diagnostic and prognostic indicators at a physiological level. Research into tumorigenesis extensively investigates various cellular signaling routes regulating cell survival and death, encompassing DNA damage, irregularities in cell cycle regulation, inflammation, immune responses, and oxidative stress [12,13,14,15]. Notably, these pathways are now evident in connection with neurodegenerative disorders. Additionally, irregular expression or mutations in critical genes like αS, phosphatase and tensin homolog (PTEN), PTEN-induced kinase 1 (PINK1 or PARK6), DJ-1 (PARK7), leucine-rich repeat kinase 2 (LRRK2 or PARK8), microtubule-associated protein tau (MAPT), amyloid precursor protein (APP), presenilin 1/2 (PSEN1/2), and cyclin-dependent kinase 5 (CDK5) crucial in neurodegeneration have also been identified in cancer contexts [2,8]. However, the exact nature of the relationship between cancer and neurodegeneration remains a subject of ongoing debate and investigation. Some studies propose contrasting perspectives, suggesting a potential inverse relationship, where a lower incidence of certain cancers might coincide with a higher prevalence of neurodegenerative disorders in specific populations [1,16,17,18]. Others explore the possibility of shared molecular pathways or the impact of environmental factors influencing these conditions. Understanding the complex interplay between genetics, cellular mechanisms, and environmental influences in cancer and neurodegeneration is crucial for unraveling their connections [12,13,19]. Research in this field continues to evolve, aiming to elucidate the intricate network of factors contributing to these conditions, ultimately paving the way for potential therapeutic interventions or preventive strategies targeting shared pathways or genetic predispositions. This review proposes a view that neurodegeneration and cancer may be two sides of the same coin. It also delves into the possible link genes between the two mechanisms and discusses the potential implications of their dysregulation in the onset and progression of neuropathological events.
1.1. Converging Pathways: Unveiling Shared Risk Factors in Cancer and Neurodegeneration
Neurodegenerative diseases and cancer are multifactorial disorders where numerous genes contribute to conditions like AD, PD, and Huntington's disease (HD) [20,21,22,23,24]. Alongside genetic factors, a spectrum of elements, including mutations, single nucleotide polymorphisms, epigenetic variations (non-coding RNA, histone modifications, DNA methylation), oxidative stress, protein degradation, mitochondrial function, cellular trafficking, abnormal proteins, and oligomeric protein propagation, shape the pathophysiological mechanisms involved in the risk of specific cancer types within PD, HD, and AD [25,26,27,28,29,30,31,32].
Disruptions in genetic mutations and transcriptional, protein, and mitochondrial regulations hold substantial sway in these conditions [28,32,33]. For instance, the tumor suppressor gene p53 exhibits contrasting activity in tumors and PD, AD, and HD [7], suggesting a plausible link between cancer and neurodegeneration [2]. This connection stems from shared genes and biological pathways, marked by misregulated cell cycle activation leading to opposing outcomes [1,12].
Proposed mechanisms vary, involving molecular shifts in genes like PARKIN, PINK1, and P53 or the translation of short-interfering RNA (siRNA) in non-coding areas that could be toxic to tumor cells. Additionally, replacement therapies like levodopa (L-DOPA) in PD demonstrate effects on reducing tumor angiogenesis [34,35,36,37,38]. While the direct association between neurodegeneration and cancer remains unconfirmed, specific genes manage critical roles in both realms, spanning cell cycle control, DNA repair, and kinase signaling [39]. Conversely, specific reported tumors escalate their incidence between these diseases, such as breast and skin cancers [40].
Shared risk factors between cancer and neurodegeneration uncover common biological mechanisms predisposing individuals to both conditions [12,41]. Chronic inflammation, known to influence various cancers, also appears to play a role in AD and PD [42,43,44]. Oxidative stress, arising from an imbalance between free radicals and antioxidants, impacts cell survival in cancer development and neurodegenerative processes [9,14,30]. Disruptions in cellular mechanisms, such as impaired DNA repair or protein misfolding, constitute commonalities between cancer and neurodegeneration [15,45]. Additionally, lifestyle factors like diet, exercise, and exposure to environmental toxins influence the risk of both conditions, highlighting the multifaceted nature of shared risk factors [46]. Understanding these commonalities offers prospects for targeted strategies and potentially intersecting therapies for these seemingly distinct diseases [1].
1.2. Parallel Pathogenesis, Divergent Destinies: Double Hit
Despite their divergent outcomes, the intriguing parallel pathogenesis between cancer and neurodegeneration delves into intricate molecular pathways that influence cellular behavior [1,2]. Shared factors like chronic inflammation, oxidative stress, and genetic mutations contribute to both cancer development and neurodegenerative processes [47,48,49]. Inflammation, for instance, triggers immune responses that can either promote tumor growth or contribute to neuronal damage and degeneration in the brain [10,50]. Similarly, oxidative stress, stemming from an imbalance between reactive oxygen species and antioxidants, impacts cellular health in both cancerous and neurodegenerative conditions, albeit through different mechanisms [49].
Moreover, disruptions in critical cellular processes such as DNA repair and protein quality control mechanisms are common threads in cancer and neurodegeneration. Dysfunctional DNA repair pathways can lead to genetic mutations promoting cancer or neuronal dysfunction in neurodegenerative disorders [51,52,53]. Meanwhile, protein misfolding and aggregation, seen in diseases like AD or PD, mirror aberrant protein signaling in specific cancers [54,55,56].
The divergence in clinical outcomes emerges from the unique tissue contexts affected by these shared pathogenic processes. Cancer manifests as uncontrolled cell growth that forms tumors, impacting various organs and tissues [57]. On the contrary, neurodegeneration manifests as the progressive loss of specific neuronal populations, resulting in cognitive decline or motor impairments particular to the affected brain regions [7].
Understanding the intricate interplay between shared pathogenic mechanisms and the divergent fates of cancer and neurodegeneration offers a challenging yet promising opportunity for tailored therapeutic strategies. By dissecting these dual facets of pathogenesis, researchers aim to develop treatments that account for the distinct trajectories of these conditions, ultimately aiming for more effective interventions that address the complexities of both diseases.
2. Exploring Genetic Intersections: Parkinson's Associated Genes and Potential Links to Cancer Pathways
PD is a neurodegenerative condition marked by resting tremors, bradykinesia, and rigidity [58]. In PD, non-motor symptoms such as sleep disturbances, cognitive changes, and autonomic dysfunction often significantly impact patients' quality of life alongside the more recognized motor symptoms [59].
The impact of PD extends beyond patients to their caregivers and society, contributing significantly to disability-adjusted life years and ranking among the leading causes of years lived with disability [58]. Interestingly, research has unearthed a potential correlation between PD diagnosis and the development of cancer. Some studies have unveiled a positive association linking PD with subsequent occurrences of melanoma [60,61], although specific investigations have reported a lack of correlation [62,63]. Additionally, there are observations of melanoma onset after the use of levodopa, the primary pharmacological therapy for PD [64].
The connection between PD and cancer, particularly involving the LRRK2 gene, is an area of growing interest in medical research [65]. LRRK2 regulates various cellular processes, including cell signaling, vesicle trafficking, and autophagy [65].
The LRRK2 gene is associated with familial and sporadic PD forms [65]. Mutations in this gene have been identified as significant genetic contributors to PD development, affecting cellular processes such as protein degradation and mitochondrial function in neurons [66,67,68].
Interestingly, studies have revealed that specific mutations in the LRRK2 gene elevate the risk of PD and confer increased susceptibility to certain cancers [69,70,71,72]. Notably, these mutations have been linked to an elevated risk of lung cancer and various digestive tract cancers, such as colorectal cancer [73]. The exact mechanisms underlying how LRRK2 mutations contribute to cancer development are not fully understood but may involve alterations in cell proliferation, survival pathways, or immune responses [66,67,68]. For instance, the impact of LRRK2 mutations on cellular processes like autophagy, inflammation, or DNA repair mechanisms might contribute to the pathogenesis of PD and cancer [66,73]. The PINK1 (PTEN-induced putative kinase 1) gene is another significant genetic factor associated with PD [74,75].
Mutations in the PINK1 gene have been identified in cases of early-onset PD. PINK1 is critical in maintaining mitochondrial function, regulating mitochondrial quality control, and initiating the clearance of damaged mitochondria through mitophagy[74,75,76]. The PINK1 protein plays a crucial role in mitochondrial quality control and the regulation of cell death pathways [77,78,79,80,81]. Mutations in the PINK1 gene are associated with some instances of PD, leading to mitochondrial dysfunction and impaired removal of damaged mitochondria, contributing to neuronal degeneration [74,75]. A study involving mice lacking the PINK1 gene unveiled intriguing insights into the dual role of caspase-3 (CASP3), a pivotal protein governing programmed cell death. The absence of PINK1 led to adjustments in CASP3 activity. While excessive activation of CASP3 triggers cell demise, moderate activation seems to regulate vital physiological processes, including the modulation of corticostriatal synaptic plasticity [82]. Furthermore, in these PINK1-deficient mice, the suppression of PTEN protein intensified apoptosis rates and raised the levels of Bax and cleaved CASP3. These alterations were associated with heightened cancer-related characteristics like increased cell proliferation, colony formation, and invasiveness [83].
Interestingly, while the PINK1 gene is primarily linked to PD, some research suggests potential cancer implications [84,85]. Studies exploring the broader roles of PINK1 have indicated its involvement in cellular processes beyond PD, including aspects related to cancer biology [86]. Initially linked to cancer biology through its modulation by the tumor suppressor PTEN in a cancer cell model, PINK1 gained prominence as it exhibited robust expression in highly metastatic melanoma and colon carcinoma mouse cancer cell lines [87,88]. Subsequent studies further elucidated PINK1's multifaceted involvement in various aspects of cancer biology and metabolism, echoing pathways relevant to neurodegeneration and oncogenic transformation [89]. Intriguingly, epidemiological investigations uncovered a decreased risk of specific cancers in individuals with PD, aligning with this concept [89]. Notably, PINK1 emerged as a critical factor in tumor cell survival and resistance to chemotherapy in independent RNA interference screens, presenting itself as a promising target for cancer therapy [90,91]. Moreover, analysis of human ovarian carcinoma unveiled a negative correlation between elevated PINK1 mRNA expression and favorable patient outcomes [87,90,91]. These findings consolidate multiple studies highlighting PINK1's pivotal role in sustaining cell proliferation and thwarting cell death mechanisms. Its impact spans fundamental processes such as controlling the cell cycle, regulating apoptosis, managing protein degradation systems, maintaining mitochondrial homeostasis, and modulating cell metabolism [84].
Another gene that has drawn attention due to its potential association with PD and certain cancers is the SNCA gene, which encodes αS. This protein aggregates in the brains of individuals with PD [92].
Mutations or duplications in the SNCA gene have been linked to familial forms of PD. In addition to its role in PD, αS has been implicated in the pathology of various cancers [93,94]. It's been observed that elevated levels of αS could influence tumor growth and metastasis in particular cancer types, including breast cancer and colorectal cancer [95].
However, the relationship between SNCA mutations and cancer risk remains a subject of ongoing research and debate. Several other genes have been implicated in PD and have shown potential associations or interactions with certain aspects of cancer (Table 1). Parkin, a protein encoded by the PARK2 gene, is vital in removing damaged or unnecessary proteins within cells, a ubiquitination mechanism [96,97,98]. Mutations in the PARK2 gene are linked to familial forms of PD, impairing Parkin's function and accumulating toxic proteins contributing to neuronal degeneration [99].
While primarily linked to PD, studies have suggested a potential role for Parkin in specific cancers. Some research has explored its involvement in cellular processes related to tumor suppression and apoptosis, indicating a possible connection to cancer biology [96,100,101]. Parkin-7 (PARK7), also known as DJ-1, protects cells from oxidative stress, maintains mitochondrial function, and regulates cellular pathways associated with cell survival [102,103]. Mutations in the PARK7 gene are implicated in familial forms of PD, compromising cellular defenses against oxidative damage, and contributing to neuronal degeneration. However, the relationship between PARK7 and cancer remains unclear and warrants further investigation [104]. Some studies propose a potential role of PARK7 in certain cancer types, indicating its involvement in modulating cell proliferation, apoptosis, and tumor progression [104]. While these genes primarily feature in the context of Parkinson's disease, their broader implications or potential involvement in aspects of cancer biology are areas of ongoing investigation.
3. Exploring Genetic Intersections: Alzheimer's Associated Genes and Potential Links to Cancer Pathways
AD stands as a progressive neurodegenerative disorder marked by cognitive decline, memory impairment, and behavioral changes [105]. The correlation between AD and cancer presents a complex interplay, with emerging research shedding light on potential genetic intersections [106,107]. One of the genes that have garnered attention in both AD and certain cancers is the Apolipoprotein E (APOE) gene. [108]. The APOE gene encodes a protein involved in lipid metabolism and transportation, playing a crucial role in regulating cholesterol levels in the body [109]. In AD, specific variants of the APOE gene, notably the APOE ε4 allele, are known to significantly elevate the risk and influence the age of onset of the disease[108]. Intriguingly, studies exploring the role of APOE in cancer have shown diverse outcomes [108]. While some research suggests that the APOE ε4 allele might confer a decreased risk of certain cancers, particularly in breast and prostate cancers, other studies propose associations between APOE ε4 and an increased risk or poorer prognosis in certain malignancies, including ovarian and liver cancers [110,111,112,113]. The precise mechanisms underlying the dual roles of APOE in AD and cancer remain unclear.
Beyond APOE, several other genes and molecular pathways have been implicated in AD and certain cancers, contributing to the intriguing correlation between these seemingly disparate conditions (Table 2). TP53 (tumor protein p53), a critical tumor suppressor gene, is commonly mutated in various cancers [110]. Interestingly, studies suggest that alterations in the TP53 gene might also influence neurodegenerative processes, including AD [114,115]
[114,115]. Mutations or dysregulation of TP53 have been observed in the brains of individuals with AD, indicating a potential link between TP53 and the neuropathology of AD [114,116]. Mutations in the Presenilin 1 (PSEN1) and Presenilin 2 (PSEN2) genes are strongly linked to early-onset familial AD, promoting the accumulation of amyloid-β peptides, a hallmark of AD pathology [117,118]. PSENs play pivotal roles in various cellular functions, including processing certain proteins like amyloid precursor (APP) [119,120,121]. However, the direct involvement of PSEN in cancer is less understood. Studies suggest potential roles of PSENs in regulating cell proliferation, apoptosis, and cellular signaling pathways relevant to tumorigenesis [122,123]. Still, their exact contributions to cancer development remain a subject of ongoing research and debate.
Interestingly, abnormal APP and amyloid-β accumulation processing have also been implicated in certain cancers, suggesting a potential intersection in both molecular mechanisms [124].
Beta-secretase 1 (BACE1) is a critical enzyme that produces Aβ peptides, which aggregate to form plaques, a hallmark of AD pathology [125]. BACE1 cleaves APP to generate these Aβ fragments, contributing to the neurotoxicity seen in AD [126,127]. However, the link between BACE1 and cancer remains a subject of investigation. Some studies suggest potential implications of BACE1 in cancer biology, highlighting its involvement in regulating cell proliferation, migration, and tumor growth [128,129].
4. Exploring Genetic Intersections: amyotrophic lateral sclerosis (ALS)'s-Associated Genes and Potential Links to Cancer Pathways
The correlation between amyotrophic lateral sclerosis (ALS) and cancer remains an area of interest, with potential genetic intersections providing insight into this complex relationship [130,131]. One gene has sparked attention in both ALS and cancer is the C9orf72 gene.
The C9orf72 gene plays a crucial role in cellular functions, including vesicle trafficking, autophagy, and RNA metabolism [132,133]. Expansions of the hexanucleotide repeat in the C9orf72 gene are ALS's most common genetic cause [134,135,136]. These expansions lead to toxic RNA and protein aggregates contributing to neuronal degeneration in ALS [134,135,137]. Intriguingly, alterations in C9orf72 have also been implicated in specific cancers [137,138,139]. Studies have identified associations between C9orf72 mutations and an increased risk of developing various malignancies, including brain tumors and certain types of lymphoma [140,141]. The precise mechanisms by which C9orf72 mutations contribute to both ALS and cancer are not fully understood. Alongside the C9orf72 gene, several other genes and molecular pathways have been proposed to have potential links between ALS and cancer (Table 3). Fused in Sarcoma (FUS) gene is involved in various cellular functions, including RNA processing, transport, and DNA repair [142,143,144]. Mutations in the FUS gene are linked to familial and sporadic cases of ALS, where aberrant FUS proteins form toxic aggregates contributing to neuronal damage [145,146,147]. Aberrant FUS aggregation and pathology have also been associated with certain cancers, albeit in a relatively limited context compared to its involvement in ALS [139,148,149,150]. Superoxide dismutase 1 (SOD1) is an enzyme that is crucial in neutralizing harmful free radicals in cells by converting superoxide radicals into less toxic molecules [151,152]. Mutations in the SOD1 gene are associated with familial cases of ALS, where altered SOD1 proteins contribute to motor neuron degeneration [153,154]. While primarily studied in the context of ALS, some research has hinted at a potential role of SOD1 in modulating oxidative stress, inflammation, and cell survival pathways that might have implications in specific cancers [155,156].
5. Exploring Genetic Intersections: Huntington's Disease’s Associated Genes and Potential Links to Cancer Pathways
HD is a hereditary neurodegenerative disease caused by a polyglutamine (polyQ) expansion in the huntingtin (HTT) gene that profoundly impacts both movement control and cognitive function [157,158,159]. HD leads to the brain's gradual breakdown of nerve cells, particularly affecting the basal ganglia and cortex [160]. Studies investigating the relationship between HD and cancer have indicated intriguing links beyond the HTT gene mutations characteristic of HD [18,161,162,163]. Research has suggested a potential association between the HTT gene and specific cancer-related pathways [164,165]. The HTT gene produces the huntingtin protein, and while its primary role is linked to neuronal damage in HD, it might also have implications in cancer [166]. Some studies propose that alterations in the HTT gene could impact cell survival, DNA repair mechanisms, or cellular processes relevant to tumorigenesis [167,168]. Additionally, alterations in other genes associated with HD, such as genes involved in DNA repair pathways or cell cycle regulation, might contribute to the observed correlation between HD and certain cancers [162,165,166,167]. However, the precise mechanisms and the extent of this genetic overlap between HD and cancer remain areas of active investigation in scientific research. While the primary gene associated with HD is HTT, additional genetic factors have been suggested to potentially influence the correlation between HD and cancer [166] (Table 4). For instance, genes involved in DNA repair mechanisms, such as breast cancer type 1 (BRCA1) and type 2 (BRCA2), have been implicated in cancer susceptibility and neurodegenerative disorders like HD [169,170]. The BRCA1 protein exhibits various roles across diverse cellular functions, including DNA repair, transcriptional activation, cell cycle control, and chromatin modification. Similarly, BRCA2 contributes to transcriptional and cell cycle regulation, DNA repair, mitophagy, and fortifying the stability of replication forks within cells [169,170]. BRCA1 and BRCA2 are extensively studied due to their pivotal roles in maintaining DNA integrity, particularly in repairing damaged DNA [170,171]. While their involvement in HD remains less understood, both genes are strongly associated with cancer susceptibility [170,171]. Mutations in BRCA1 and BRCA2 significantly elevate the risk of developing breast, ovarian, and other cancers. In the context of HD, while direct links between BRCA1/2 mutations and the disease are not well-established, exploring their functions in DNA repair mechanisms may shed light on potential intersections with HD pathology [170,171]. Their crucial role in DNA repair pathways suggests a possibility of shared pathways or processes implicated in both cancer and neurodegeneration, necessitating further investigation to unravel their precise roles in Huntington's disease and cancer development [170,171].
Additionally, gene alterations related to cell cycle regulation, such as TDP-43, have been observed in both HD pathogenesis and various cancer types [172,173]. In HD, abnormal aggregates of TDP-43 have been detected in the brain tissues of individuals with the disease [174]. However, the exact role of TDP-43 in the development or progression of HD is still being researched, and its specific contributions to the pathology of HD remain to be fully elucidated [174]. In cancer, alterations in TDP-43 expression or function have been observed in various cancer types, affecting cellular processes like RNA metabolism, splicing, and stability [174]. Dysregulation of TDP-43 has been linked to tumor growth, invasion, and metastasis in some cancers [175]. While its exact role in tumorigenesis is not fully understood, aberrant TDP-43 expression may contribute to the molecular mechanisms underlying certain cancers [175,176]. Other genes involved in oxidative stress response, apoptosis, or DNA damage repair pathways, such as ATM, may also play roles in HD and cancer development [172,177,178]. The intricate interplay of these genetic factors underscores the complex relationship between HD and specific cancer pathways, warranting further investigation into their shared molecular mechanisms and potential implications for disease development and progression [179].

Table 4.
Outlines the functions and prospective roles of key genes associated with HD and their possible implications in cancer development.
Table 4.
Outlines the functions and prospective roles of key genes associated with HD and their possible implications in cancer development.
| GENE | FUNCTION | ROLE IN NEURODEGENERATION | ROLE IN CANCER |
|---|---|---|---|
|
HTT |
Signaling, transporting materials,binding proteins and other structures, and protecting against apoptosis. |
Primary role is linked to neuronal damage in HD. | Alterations in the HTT gene could impact cell survival, DNA repair mechanisms, or cellular processes relevant to tumorigenesis |
|
BRCA1 BRCA2 |
The BRCA1 protein has multiple functions in different cellular processes, including DNA repair, transcriptional activation, cell cycle regulation and chromatin remodeling. BRCA2 plays a role in transcriptional and cell cycle regulation, DNA repair, mitophagy and replication fork stabilization. |
Direct links between BRCA1/2 mutations and the disease are not well-established; exploring their functions in DNA repair mechanisms may shed light on potential intersections with HD pathology. |
Mutations in BRCA1 and BRCA2 significantly elevate the risk of developing breast, ovarian, and other cancers. |
| TDP-43 | Cell cycle regulation | Abnormal aggregates of TDP-43 have been detected in the brain tissues of individuals with the disease. However, the exact role of TDP-43 in the development or progression of HD is still being researched | Alterations in TDP-53 expression or function have been observed in various cancer types, affecting cellular processes like RNA metabolism, splicing, and stability. Dysregulation of TDP-53 has been linked to tumor growth, invasion, and metastasis in some cancers. |
6. Conclusion and Future Prospective
The convergence of genes implicated in cancer and neurodegeneration represents a captivating area of exploration, offering glimpses into shared molecular pathways that influence these seemingly disparate conditions [2,7,13]. Genes showcase the intricate genetic intersections between cancer and neurodegeneration. Understanding these shared genetic factors presents an avenue for uncovering underlying mechanisms that drive both disease processes. However, these genes' dual roles in different cellular contexts underscore the complexity of their contributions to cancer and neurodegeneration. Future research endeavors are poised to delve deeper into these genetic intersections, deciphering how gene alterations influence diverse cellular processes leading to cancerous growth or neurodegenerative pathology. Further exploration of shared genetic signatures, coupled with advancements in technology and analytical approaches, holds promise for identifying novel therapeutic targets with broader applicability across both conditions. Leveraging this knowledge could pave the way for precision medicine strategies tailored to individual genetic profiles, aiming for targeted interventions that address the intricacies of cancer and neurodegeneration, potentially transforming treatment paradigms and improving patient outcomes.
Abbreviations
- ❖ Aβ: amyloid-β
- ❖ AD: Alzheimer's disease
- ❖ αS: α-synuclein
- ❖ PD: Parkinson's disease
- ❖ PTEN: phosphatase and tensin homolog
- ❖ PINK1 or PARK6: PTEN-induced kinase 1
- ❖ LRRK2 or PARK8: leucine-rich repeat kinase 2
- ❖ MAPT: microtubule-associated protein tau
- ❖ APP: amyloid precursor protein
- ❖ PSEN1/2: presenilin 1/2
- ❖ CDK5: cyclin-dependent kinase 5
- ❖ HD: Huntington's disease
- ❖ siRNA: short-interfering RNA
- ❖ L-DOPA: levodopa
- ❖ PolyQ: polyglutamine
- ❖ Huntingtin (HTT)
- ❖ BRCA1: breast cancer type 1
- ❖ BRCA2: breast cancer type 2
- ❖ APOE: Apolipoprotein E
- ❖ TP53: tumor protein p53
- ❖ Presenilin 1: PSEN
- ❖ Presenilin 2: PSEN2
- ❖ Amyloid precursor protein: APP
- ❖ Beta-secretase 1: BACE1
- ❖ PTEN-induced putative kinase 1: PINK1
- ❖ Caspase-3: CASP3
- ❖ Amyotrophic lateral sclerosis: ALS
- ❖ Fused in Sarcoma: FUS
- ❖ Superoxide dismutase 1: SOD1
Author Contributions
Conceptualization, MA.MO., MA. ME; and G.M.; Resources, G.M.; Literature revision, MA.MO., MA. ME, A.R.; and I.E. Writing – Original Draft Preparation, MA.MO; Writing – Review & Editing: MA.MO., MA. ME; and G.M.; Visualization, MA.MO., MA. ME; A.R.; I.E. and G.M.; Supervision, ME; and G.M.; Project Administration, MA.MO; and G.M.; Funding Acquisition, M.G.
Acknowledgments
We would to acknowledge all members of Laboratories that have supported this work. Also we acknowledge all the FSL animal facility staff.
References
- Driver, J.A. Understanding the link between cancer and neurodegeneration. Journal of Geriatric Oncology 2012, 3, 58–67. [Google Scholar] [CrossRef]
- Plun-Favreau, H.; Lewis, P.A.; Hardy, J.; Martins, L.M.; Wood, N.W. Cancer and neurodegeneration: between the devil and the deep blue sea. PLoS Genet. 2010, 6, e1001257. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.C. Multiple-mutation theory of carcinogenesis. Nature 1958, 181, 651–652. [Google Scholar] [CrossRef] [PubMed]
- Crusz, S.M.; Balkwill, F.R. Inflammation and cancer: advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef]
- Gao, H.-M.; Hong, J.-S. Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol. 2008, 29, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.C.; LeVine, H. Corruption and spread of pathogenic proteins in neurodegenerative diseases. J. Biol. Chem. 2012, 287, 33109–33115. [Google Scholar] [CrossRef] [PubMed]
- Houck, A.L.; Seddighi, S.; Driver, J.A. At the crossroads between neurodegeneration and cancer: A review of overlapping biology and its implications. Curr. Aging Sci. 2018, 11, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Park, M. Molecular crosstalk between cancer and neurodegenerative diseases. Cell. Mol. Life Sci. 2020, 77, 2659–2680. [Google Scholar] [CrossRef]
- Ganguly, G.; Chakrabarti, S.; Chatterjee, U.; Saso, L. Proteinopathy, oxidative stress and mitochondrial dysfunction: cross talk in Alzheimer’s disease and Parkinson’s disease. Drug Des. Devel. Ther. 2017, 11, 797–810. [Google Scholar] [CrossRef]
- Sankowski, R.; Mader, S.; Valdés-Ferrer, S.I. Systemic inflammation and the brain: novel roles of genetic, molecular, and environmental cues as drivers of neurodegeneration. Front. Cell. Neurosci. 2015, 9, 28. [Google Scholar] [CrossRef]
- Joseph, C.; Mangani, A.S.; Gupta, V.; Chitranshi, N.; Shen, T.; Dheer, Y.; Kb, D.; Mirzaei, M.; You, Y.; Graham, S.L.; Gupta, V. Cell cycle deficits in neurodegenerative disorders: uncovering molecular mechanisms to drive innovative therapeutic development. Aging Dis. 2020, 11, 946–966. [Google Scholar] [CrossRef]
- Rojas, N.G.; Cesarini, M.; Etcheverry, J.L.; Prat, G.A.D.; Arciuch, V.A.; Gatto, E.M. Neurodegenerative diseases and cancer: sharing common mechanisms in complex interactions. J. Integr. Neurosci. 2020, 19, 187–199. [Google Scholar] [CrossRef]
- Klus, P.; Cirillo, D.; Botta Orfila, T.; Gaetano Tartaglia, G. Neurodegeneration and cancer: where the disorder prevails. Sci. Rep. 2015, 5, 15390. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative stress in neurodegenerative diseases: from molecular mechanisms to clinical applications. Oxid. Med. Cell. Longev. 2017, 2017, 2525967. [Google Scholar] [CrossRef]
- Madabhushi, R.; Pan, L.; Tsai, L.-H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Mogavero, M.P.; Silvani, A.; DelRosso, L.M.; Salemi, M.; Ferri, R. Focus on the Complex Interconnection between Cancer, Narcolepsy and Other Neurodegenerative Diseases: A Possible Case of Orexin-Dependent Inverse Comorbidity. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Engel, P.A. Is age-related failure of metabolic reprogramming a principal mediator in idiopathic Parkinson’s disease? Implications for treatment and inverse cancer risk. Med. Hypotheses 2016, 93, 154–160. [Google Scholar] [CrossRef]
- Coarelli, G.; Diallo, A.; Thion, M.S.; Rinaldi, D.; Calvas, F.; Boukbiza, O.L.; Tataru, A.; Charles, P.; Tranchant, C.; Marelli, C.; Ewenczyk, C.; Tchikviladzé, M.; Monin, M.-L.; Carlander, B.; Anheim, M.; Brice, A.; Mochel, F.; Tezenas du Montcel, S.; Humbert, S.; Durr, A. Low cancer prevalence in polyglutamine expansion diseases. Neurology 2017, 88, 1114–1119. [Google Scholar] [CrossRef]
- Ibáñez, K.; Boullosa, C.; Tabarés-Seisdedos, R.; Baudot, A.; Valencia, A. Molecular evidence for the inverse comorbidity between central nervous system disorders and cancers detected by transcriptomic meta-analyses. PLoS Genet. 2014, 10, e1004173. [Google Scholar] [CrossRef]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: a clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Nutt, J.G.; Wooten, G.F. Clinical practice. Diagnosis and initial management of Parkinson’s disease. N. Engl. J. Med. 2005, 353, 1021–1027. [Google Scholar] [CrossRef]
- Bano, D.; Zanetti, F.; Mende, Y.; Nicotera, P. Neurodegenerative processes in Huntington’s disease. Cell Death Dis. 2011, 2, e228. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, S.; Safia, *!!! REPLACE !!!*; Haque, E.; Mir, S.S. Neurodegenerative diseases: multifactorial conformational diseases and their therapeutic interventions. J. Neurodegener. Dis. 2013, 2013, 563481. [Google Scholar] [CrossRef] [PubMed]
- Prasher, D.; Greenway, S.C.; Singh, R.B. The impact of epigenetics on cardiovascular disease. Biochem. Cell Biol. 2020, 98, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Preeti, K.; Tryphena, K.P.; Srivastava, S.; Singh, S.B.; Khatri, D.K. Proteostasis in Parkinson’s disease: Recent development and possible implication in diagnosis and therapeutics. Ageing Res. Rev. 2023, 84, 101816. [Google Scholar] [CrossRef] [PubMed]
- Hernaiz, A.; Toivonen, J.M.; Bolea, R.; Martín-Burriel, I. Epigenetic Changes in Prion and Prion-like Neurodegenerative Diseases: Recent Advances, Potential as Biomarkers, and Future Perspectives. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef]
- Chesnokova, E.; Beletskiy, A.; Kolosov, P. The role of transposable elements of the human genome in neuronal function and pathology. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef]
- Griess, B.; Tom, E.; Domann, F.; Teoh-Fitzgerald, M. Extracellular superoxide dismutase and its role in cancer. Free Radic. Biol. Med. 2017, 112, 464–479. [Google Scholar] [CrossRef]
- Maldonado, E.; Morales-Pison, S.; Urbina, F.; Solari, A. Aging hallmarks and the role of oxidative stress. Antioxidants (Basel) 2023, 12. [Google Scholar] [CrossRef]
- Missiroli, S.; Genovese, I.; Perrone, M.; Vezzani, B.; Vitto, V.A.M.; Giorgi, C. The role of mitochondria in inflammation: from cancer to neurodegenerative disorders. J. Clin. Med. 2020, 9. [Google Scholar] [CrossRef]
- Corrado, M.; Scorrano, L.; Campello, S. Mitochondrial dynamics in cancer and neurodegenerative and neuroinflammatory diseases. Int. J. Cell Biol. 2012, 2012, 729290. [Google Scholar] [CrossRef] [PubMed]
- Jagtap, Y.A.; Kumar, P.; Kinger, S.; Dubey, A.R.; Choudhary, A.; Gutti, R.K.; Singh, S.; Jha, H.C.; Poluri, K.M.; Mishra, A. Disturb mitochondrial associated proteostasis: Neurodegeneration and imperfect ageing. Front. Cell Dev. Biol. 2023, 11, 1146564. [Google Scholar] [CrossRef] [PubMed]
- Wahabi, K.; Perwez, A.; Rizvi, M.A. Parkin in Parkinson’s Disease and Cancer: a Double-Edged Sword. Mol. Neurobiol. 2018, 55, 6788–6800. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Sun, W.; Wang, Y.-F.; Li, J.; Li, D.-W. Association of p53 with Neurodegeneration in Parkinson’s Disease. Parkinsons Dis 2022, 2022, 6600944. [Google Scholar] [CrossRef] [PubMed]
- Drapalo, K.; Jozwiak, J. Parkin, PINK1 and DJ1 as possible modulators of mTOR pathway in ganglioglioma. Int. J. Neurosci. 2018, 128, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Barodia, S.K.; Creed, R.B.; Goldberg, M.S. Parkin and PINK1 functions in oxidative stress and neurodegeneration. Brain Res. Bull. 2017, 133, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Checler, F.; Alves da Costa, C. p53 in neurodegenerative diseases and brain cancers. Pharmacol. Ther. 2014, 142, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Ramazi, S.; Daddzadi, M.; Sahafnejad, Z.; Allahverdi, A. Epigenetic regulation in lung cancer. MedComm 2023, 4, e401. [Google Scholar] [CrossRef]
- Cives, M.; Mannavola, F.; Lospalluti, L.; Sergi, M.C.; Cazzato, G.; Filoni, E.; Cavallo, F.; Giudice, G.; Stucci, L.S.; Porta, C.; Tucci, M. Non-Melanoma Skin Cancers: Biological and Clinical Features. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Salemi, M.; Mogavero, M.P.; Lanza, G.; Mongioì, L.M.; Calogero, A.E.; Ferri, R. Examples of Inverse Comorbidity between Cancer and Neurodegenerative Diseases: A Possible Role for Noncoding RNA. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Jo, M.; Kim, Y.R.; Lee, C.-K.; Hong, J.T. Roles of peroxiredoxins in cancer, neurodegenerative diseases and inflammatory diseases. Pharmacol. Ther. 2016, 163, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Santoro, A.; Monti, D.; Crupi, R.; Di Paola, R.; Latteri, S.; Cuzzocrea, S.; Zappia, M.; Giordano, J.; Calabrese, E.J.; Franceschi, C. Aging and Parkinson’s Disease: Inflammaging, neuroinflammation and biological remodeling as key factors in pathogenesis. Free Radic. Biol. Med. 2018, 115, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Vendramini-Costa, D.B.; Carvalho, J.E. Molecular link mechanisms between inflammation and cancer. Curr. Pharm. Des. 2012, 18, 3831–3852. [Google Scholar] [CrossRef] [PubMed]
- Morris, L.G.T.; Veeriah, S.; Chan, T.A. Genetic determinants at the interface of cancer and neurodegenerative disease. Oncogene 2010, 29, 3453–3464. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Ma, Y.; Luo, Y.; Song, Y.; Xiong, G.; Ma, Y.; Sun, X.; Kan, C. Metabolic diseases and healthy aging: identifying environmental and behavioral risk factors and promoting public health. Front. Public Health 2023, 11, 1253506. [Google Scholar] [CrossRef] [PubMed]
- Migliore, L.; Coppedè, F. Genetic and environmental factors in cancer and neurodegenerative diseases. Mutation Research/Reviews in Mutation Research 2002, 512, 135–153. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial dysfunction, oxidative stress, and neuroinflammation: intertwined roads to neurodegeneration. Antioxidants (Basel) 2020, 9. [Google Scholar] [CrossRef]
- Thanan, R.; Oikawa, S.; Hiraku, Y.; Ohnishi, S.; Ma, N.; Pinlaor, S.; Yongvanit, P.; Kawanishi, S.; Murata, M. Oxidative stress and its significant roles in neurodegenerative diseases and cancer. Int. J. Mol. Sci. 2014, 16, 193–217. [Google Scholar] [CrossRef]
- Inflammatory Mechanisms and Oxidative Stress as Key Factors Respo...: Ingenta Connect. Available online: https://www.ingentaconnect.com/content/ben/cnsnddt/2016/00000015/00000003/art00008 (accessed on 16 December 2023).
- Jeppesen, D.K.; Bohr, V.A.; Stevnsner, T. DNA repair deficiency in neurodegeneration. Prog. Neurobiol. 2011, 94, 166–200. [Google Scholar] [CrossRef]
- Martin, L.J. DNA damage and repair: relevance to mechanisms of neurodegeneration. J. Neuropathol. Exp. Neurol. 2008, 67, 377–387. [Google Scholar] [CrossRef]
- Maynard, S.; Fang, E.F.; Scheibye-Knudsen, M.; Croteau, D.L.; Bohr, V.A. DNA damage, DNA repair, aging, and neurodegeneration. Cold Spring Harb. Perspect. Med. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Surguchov, A.; Surguchev, A. Synucleins: new data on misfolding, aggregation and role in diseases. Biomedicines 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Guerra, F.; Calvani, R.; Romano, R.; Coelho-Júnior, H.J.; Bucci, C.; Marzetti, E. Mitochondrial dysfunction, protein misfolding and neuroinflammation in parkinson’s disease: roads to biomarker discovery. Biomolecules 2021, 11. [Google Scholar] [CrossRef]
- Bai, Y.; Zhang, S.; Dong, H.; Liu, Y.; Liu, C.; Zhang, X. Advanced techniques for detecting protein misfolding and aggregation in cellular environments. Chem. Rev. 2023, 123, 12254–12311. [Google Scholar] [CrossRef] [PubMed]
- Reddy, G.P.; Reddy, L.V.; Kim, S. Cancer biology and pathology. In Cancer: prevention, early detection, treatment and recovery; Stein, G.S., Luebbers, K.P., Eds.; Wiley, 2019; pp. 13–52. ISBN 9781118962886. [Google Scholar]
- Jankovic, J.; Tan, E.K. Parkinson’s disease: etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatr. 2020, 91, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Montanari, M.; Imbriani, P.; Bonsi, P.; Martella, G.; Peppe, A. Beyond the Microbiota: Understanding the Role of the Enteric Nervous System in Parkinson’s Disease from Mice to Human. Biomedicines 2023, 11. [Google Scholar] [CrossRef] [PubMed]
- Rugbjerg, K.; Friis, S.; Lassen, C.F.; Ritz, B.; Olsen, J.H. Malignant melanoma, breast cancer and other cancers in patients with Parkinson’s disease. Int. J. Cancer 2012, 131, 1904–1911. [Google Scholar] [CrossRef]
- Constantinescu, R.; Romer, M.; Kieburtz, K. ; DATATOP Investigators of the Parkinson Study Group Malignant melanoma in early Parkinson’s disease: the DATATOP trial. Mov. Disord. 2007, 22, 720–722. [Google Scholar] [CrossRef] [PubMed]
- Fiala, K.H.; Whetteckey, J.; Manyam, B.V. Malignant melanoma and levodopa in Parkinson’s disease: causality or coincidence? Parkinsonism Relat. Disord. 2003, 9, 321–327. [Google Scholar] [CrossRef]
- Sun, L.-M.; Liang, J.-A.; Chang, S.-N.; Sung, F.-C.; Muo, C.-H.; Kao, C.-H. Analysis of Parkinson’s disease and subsequent cancer risk in Taiwan: a nationwide population-based cohort study. Neuroepidemiology 2011, 37, 114–119. [Google Scholar] [CrossRef]
- Sandyk, R. Accelerated growth of malignant melanoma by levodopa in parkinson’s disease and role of the pineal gland. International Journal of Neuroscience 1992, 63, 137–140. [Google Scholar] [CrossRef]
- Thakur, G.; Kumar, V.; Lee, K.W.; Won, C. Structural insights and development of LRRK2 inhibitors for parkinson’s disease in the last decade. Genes (Basel) 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Vetchinova, A.S.; Kapkaeva, M.R.; Ivanov, M.V.; Kutukova, K.A.; Mudzhiri, N.M.; Frumkina, L.E.; Brydun, A.V.; Sukhorukov, V.S.; Illarioshkin, S.N. Mitochondrial Dysfunction in Dopaminergic Neurons Derived from Patients with LRRK2- and SNCA-Associated Genetic Forms of Parkinson’s Disease. Curr. Issues Mol. Biol. 2023, 45, 8395–8411. [Google Scholar] [CrossRef] [PubMed]
- Wallings, R.; Manzoni, C.; Bandopadhyay, R. Cellular processes associated with LRRK2 function and dysfunction. FEBS J. 2015, 282, 2806–2826. [Google Scholar] [CrossRef] [PubMed]
- Hur, E.-M.; Lee, B.D. LRRK2 at the crossroad of aging and parkinson’s disease. Genes (Basel) 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Mata, I.F.; Farrer, M.J. LRRK2: a common pathway for parkinsonism, pathogenesis and prevention? Trends Mol. Med. 2006, 12, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Ortega, R.A.; Wang, C.; Raymond, D.; Bryant, N.; Scherzer, C.R.; Thaler, A.; Alcalay, R.N.; West, A.B.; Mirelman, A.; Kuras, Y.; Marder, K.S.; Giladi, N.; Ozelius, L.J.; Bressman, S.B.; Saunders-Pullman, R. Association of dual LRRK2 G2019S and GBA variations with parkinson disease progression. JAMA Netw. Open 2021, 4, e215845. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Li, X.; Jankovic, J. The association between Parkinson’s disease and melanoma. Int. J. Cancer 2011, 128, 2251–2260. [Google Scholar] [CrossRef]
- Chittoor-Vinod, V.G.; Nichols, R.J.; Schüle, B. Genetic and environmental factors influence the pleomorphy of LRRK2 parkinsonism. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef]
- Ejma, M.; Madetko, N.; Brzecka, A.; Guranski, K.; Alster, P.; Misiuk-Hojło, M.; Somasundaram, S.G.; Kirkland, C.E.; Aliev, G. The Links between Parkinson’s Disease and Cancer. Biomedicines 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Kelm-Nelson, C.A.; Brauer, A.F.L.; Barth, K.J.; Lake, J.M.; Sinnen, M.L.K.; Stehula, F.J.; Muslu, C.; Marongiu, R.; Kaplitt, M.G.; Ciucci, M.R. Characterization of early-onset motor deficits in the Pink1-/- mouse model of Parkinson disease. Brain Res. 2018, 1680, 1–12. [Google Scholar] [CrossRef] [PubMed]
- van der Merwe, C.; Jalali Sefid Dashti, Z.; Christoffels, A.; Loos, B.; Bardien, S. Evidence for a common biological pathway linking three Parkinson’s disease-causing genes: parkin, PINK1 and DJ-1. Eur. J. Neurosci. 2015, 41, 1113–1125. [Google Scholar] [CrossRef]
- Tassone, A.; Meringolo, M.; Ponterio, G.; Bonsi, P.; Schirinzi, T.; Martella, G. Mitochondrial bioenergy in neurodegenerative disease: huntington and parkinson. Int. J. Mol. Sci. 2023, 24. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed]
- Gispert, S.; Ricciardi, F.; Kurz, A.; Azizov, M.; Hoepken, H.-H.; Becker, D.; Voos, W.; Leuner, K.; Müller, W.E.; Kudin, A.P.; Kunz, W.S.; Zimmermann, A.; Roeper, J.; Wenzel, D.; Jendrach, M.; García-Arencíbia, M.; Fernández-Ruiz, J.; Huber, L.; Rohrer, H.; Barrera, M.; Auburger, G. Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS ONE 2009, 4, e5777. [Google Scholar] [CrossRef] [PubMed]
- Brunelli, F.; Valente, E.M.; Arena, G. Mechanisms of neurodegeneration in Parkinson’s disease: keep neurons in the PINK1. Mech. Ageing Dev. 2020, 189, 111277. [Google Scholar] [CrossRef]
- Thomas, K.J.; McCoy, M.K.; Blackinton, J.; Beilina, A.; van der Brug, M.; Sandebring, A.; Miller, D.; Maric, D.; Cedazo-Minguez, A.; Cookson, M.R. DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum. Mol. Genet. 2011, 20, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; Cai, H.; Borsche, M.; Klein, C.; Youle, R.J. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Imbriani, P.; Tassone, A.; Meringolo, M.; Ponterio, G.; Madeo, G.; Pisani, A.; Bonsi, P.; Martella, G. Loss of Non-Apoptotic Role of Caspase-3 in the PINK1 Mouse Model of Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Wang, M.; Luan, S.; Fan, X.; Wang, J.; Huang, J.; Gao, X.; Han, D. The emerging multifaceted role of PINK1 in cancer biology. Cancer Sci. 2022, 113, 4037–4047. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-L.; Wang, Z.-X.; Ying, C.-Z.; Zhang, B.-R.; Pu, J.-L. Decoding the Role of Familial Parkinson’s Disease-Related Genes in DNA Damage and Repair. Aging Dis. 2022, 13, 1405–1412. [Google Scholar] [CrossRef]
- Koros, C.; Simitsi, A.-M.; Bougea, A.; Papagiannakis, N.; Antonelou, R.; Pachi, I.; Angelopoulou, E.; Prentakis, A.; Zachou, A.; Chrysovitsanou, C.; Beratis, I.; Fragkiadaki, S.; Kontaxopoulou, D.; Eftymiopoulou, E.; Stanitsa, E.; Potagas, C.; Papageorgiou, S.G.; Karavasilis, E.; Velonakis, G.; Prassopoulos, V.; Stefanis, L. Double Trouble: Association of Malignant Melanoma with Sporadic and Genetic Forms of Parkinson’s Disease and Asymptomatic Carriers of Related Genes: A Brief Report. Medicina (Kaunas) 2023, 59. [Google Scholar] [CrossRef] [PubMed]
- O’Flanagan, C.H.; O’Neill, C. PINK1 signalling in cancer biology. Biochim. Biophys. Acta 2014, 1846, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, A.; Kataoka, K.; Hong, M.; Sakaguchi, M.; Huh, N. BRPK, a novel protein kinase showing increased expression in mouse cancer cell lines with higher metastatic potential. Cancer Lett. 2003, 201, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Arena, G.; Valente, E.M. PINK1 in the limelight: multiple functions of an eclectic protein in human health and disease. J. Pathol. 2017, 241, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Inzelberg, R.; Jankovic, J. Are Parkinson disease patients protected from some but not all cancers? Neurology 2007, 69, 1542–1550. [Google Scholar] [CrossRef]
- MacKeigan, J.P.; Murphy, L.O.; Blenis, J. Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat. Cell Biol. 2005, 7, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.A.; Hewish, M.; Sims, D.; Lord, C.J.; Ashworth, A. Parallel high-throughput RNA interference screens identify PINK1 as a potential therapeutic target for the treatment of DNA mismatch repair-deficient cancers. Cancer Res. 2011, 71, 1836–1848. [Google Scholar] [CrossRef]
- Ibáñez, P.; Bonnet, A.M.; Débarges, B.; Lohmann, E.; Tison, F.; Pollak, P.; Agid, Y.; Dürr, A.; Brice, A. Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 2004, 364, 1169–1171. [Google Scholar] [CrossRef]
- Zhou, L.-X.; Zheng, H.; Tian, Y.; Luo, K.-F.; Ma, S.-J.; Wu, Z.-W.; Tang, P.; Jiang, J.; Wang, M.-H. SNCA inhibits epithelial-mesenchymal transition and correlates to favorable prognosis of breast cancer. Carcinogenesis 2022, 43, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Xu, Z.; Hu, X.; Qian, L.; Li, Z.; Zhou, Y.; Dai, S.; Zeng, S.; Gong, Z. SNCA Is a Functionally Low-Expressed Gene in Lung Adenocarcinoma. Genes (Basel) 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Zanotti, L.C.; Malizia, F.; Cesatti Laluce, N.; Avila, A.; Mamberto, M.; Anselmino, L.E.; Menacho-Márquez, M. Synuclein proteins in cancer development and progression. Biomolecules 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, J.P.; Lazarou, M.; Dewson, G. Parkin and mitophagy in cancer. Oncogene 2017, 36, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Suzuki, T.; Chiba, T.; Shimura, H.; Hattori, N.; Mizuno, Y. Parkin is linked to the ubiquitin pathway. J. Mol. Med. 2001, 79, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Suzuki, T.; Hattori, N.; Mizuno, Y. Ubiquitin, proteasome and parkin. Biochim. Biophys. Acta 2004, 1695, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-L.; Feng, S.-T.; Wang, Z.-Z.; Yuan, Y.-H.; Chen, N.-H.; Zhang, Y. Parkin, an E3 ubiquitin ligase, plays an essential role in mitochondrial quality control in parkinson’s disease. Cell. Mol. Neurobiol. 2021, 41, 1395–1411. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Denison, S.; Lai, J.-P.; Philips, L.A.; Montoya, D.; Kock, N.; Schüle, B.; Klein, C.; Shridhar, V.; Roberts, L.R.; Smith, D.I. Parkin gene alterations in hepatocellular carcinoma. Genes Chromosomes Cancer 2004, 40, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Denison, S.R.; Wang, F.; Becker, N.A.; Schüle, B.; Kock, N.; Phillips, L.A.; Klein, C.; Smith, D.I. Alterations in the common fragile site gene Parkin in ovarian and other cancers. Oncogene 2003, 22, 8370–8378. [Google Scholar] [CrossRef]
- Naren, P.; Cholkar, A.; Kamble, S.; Khan, S.S.; Srivastava, S.; Madan, J.; Mehra, N.; Tiwari, V.; Singh, S.B.; Khatri, D.K. Pathological and therapeutic advances in parkinson’s disease: mitochondria in the interplay. J Alzheimers Dis 2023, 94, S399–S428. [Google Scholar] [CrossRef]
- Olanow, C.W.; McNaught, K.S.P. Ubiquitin-proteasome system and Parkinson’s disease. Mov. Disord. 2006, 21, 1806–1823. [Google Scholar] [CrossRef] [PubMed]
- Jin, W. Novel Insights into PARK7 (DJ-1), a Potential Anti-Cancer Therapeutic Target, and Implications for Cancer Progression. J. Clin. Med. 2020, 9. [Google Scholar] [CrossRef]
- Perl, D.P. Neuropathology of Alzheimer’s disease. Mt Sinai J Med 2010, 77, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Nudelman, K.N.H.; McDonald, B.C.; Lahiri, D.K.; Saykin, A.J. Biological hallmarks of cancer in alzheimer’s disease. Mol. Neurobiol. 2019, 56, 7173–7187. [Google Scholar] [CrossRef] [PubMed]
- Nudelman, K.N.H.; Risacher, S.L.; West, J.D.; McDonald, B.C.; Gao, S.; Saykin, A.J. ; Alzheimer’s Disease Neuroimaging Initiative Association of cancer history with Alzheimer’s disease onset and structural brain changes. Front. Physiol. 2014, 5, 423. [Google Scholar] [CrossRef]
- Fernandez, H.R.; Varma, A.; Flowers, S.A.; Rebeck, G.W. Cancer chemotherapy related cognitive impairment and the impact of the alzheimer’s disease risk factor APOE. Cancers (Basel) 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W. Central nervous system lipoproteins: apoe and regulation of cholesterol metabolism. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Darwish, N.M.; Al-Hail, M.K.; Mohamed, Y.; Al Saady, R.; Mohsen, S.; Zar, A.; Al-Mansoori, L.; Pedersen, S. The role of apolipoproteins in the commonest cancers: A review. Cancers (Basel) 2023, 15. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Chen, J.; Ma, Y.; Chen, H. Apolipoproteins: New players in cancers. Front. Pharmacol. 2022, 13, 1051280. [Google Scholar] [CrossRef]
- Miao, G.; Zhuo, D.; Han, X.; Yao, W.; Liu, C.; Liu, H.; Cao, H.; Sun, Y.; Chen, Z.; Feng, T. From degenerative disease to malignant tumors: Insight to the function of ApoE. Biomed. Pharmacother. 2023, 158, 114127. [Google Scholar] [CrossRef]
- Grant, W.B. A multicountry ecological study of risk-modifying factors for prostate cancer: apolipoprotein E epsilon4 as a risk factor and cereals as a risk reduction factor. Anticancer Res. 2010, 30, 189–199. [Google Scholar] [PubMed]
- Paniri, A.; Hosseini, M.M.; Akhavan-Niaki, H. Alzheimer’s Disease-Related Epigenetic Changes: Novel Therapeutic Targets. Mol. Neurobiol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.S.; Kayed, R.; Abate, G.; Uberti, D.; Kinnon, P.; Piccirella, S. Post-translational Modifications of the p53 Protein and the Impact in Alzheimer’s Disease: A Review of the Literature. Front. Aging Neurosci. 2022, 14, 835288. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.D.; Ehrlich, B.E. Cellular mechanisms and treatments for chemobrain: insight from aging and neurodegenerative diseases. EMBO Mol. Med. 2020, 12, e12075. [Google Scholar] [CrossRef] [PubMed]
- Lardelli, M. An alternative view of familial alzheimer’s disease genetics. 2023. [Google Scholar] [CrossRef]
- Yang, Y.; Bagyinszky, E.; An, S.S.A. Presenilin-1 (PSEN1) Mutations: Clinical Phenotypes beyond Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24. [Google Scholar] [CrossRef]
- Vetrivel, K.S.; Zhang, Y.; Xu, H.; Thinakaran, G. Pathological and physiological functions of presenilins. Mol. Neurodegener. 2006, 1, 4. [Google Scholar] [CrossRef]
- Hunter, S.; Brayne, C. Understanding the roles of mutations in the amyloid precursor protein in Alzheimer disease. Mol. Psychiatry 2018, 23, 81–93. [Google Scholar] [CrossRef]
- Palihati, N.; Tang, Y.; Yin, Y.; Yu, D.; Liu, G.; Quan, Z.; Ni, J.; Yan, Y.; Qing, H. Clusterin is a potential therapeutic target in alzheimer’s disease. Mol. Neurobiol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Cheng, L.; Dai, H.; Zhang, R.; Wang, M.; Shi, T.; Sun, M.; Cheng, X.; Wei, Q. Variants in Notch signalling pathway genes, PSEN1 and MAML2, predict overall survival in Chinese patients with epithelial ovarian cancer. J. Cell. Mol. Med. 2018, 22, 4975–4984. [Google Scholar] [CrossRef]
- Ham, S.; Kim, T.K.; Ryu, J.; Kim, Y.S.; Tang, Y.-P.; Im, H.-I. Comprehensive MicroRNAome Analysis of the Relationship Between Alzheimer Disease and Cancer in PSEN Double-Knockout Mice. Int. Neurourol. J. 2018, 22, 237–245. [Google Scholar] [CrossRef]
- Holohan, K.N.; Lahiri, D.K.; Schneider, B.P.; Foroud, T.; Saykin, A.J. Functional microRNAs in Alzheimer’s disease and cancer: differential regulation of common mechanisms and pathways. Front. Genet. 2012, 3, 323. [Google Scholar] [CrossRef] [PubMed]
- Laird, F.M.; Cai, H.; Savonenko, A.V.; Farah, M.H.; He, K.; Melnikova, T.; Wen, H.; Chiang, H.-C.; Xu, G.; Koliatsos, V.E.; Borchelt, D.R.; Price, D.L.; Lee, H.-K.; Wong, P.C. BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J. Neurosci. 2005, 25, 11693–11709. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, H.; Li, R.; Sterling, K.; Song, W. Amyloid β-based therapy for Alzheimer’s disease: challenges, successes and future. Signal Transduct. Target. Ther. 2023, 8, 248. [Google Scholar] [CrossRef] [PubMed]
- Liebsch, F.; Kulic, L.; Teunissen, C.; Shobo, A.; Ulku, I.; Engelschalt, V.; Hancock, M.A.; van der Flier, W.M.; Kunach, P.; Rosa-Neto, P.; Scheltens, P.; Poirier, J.; Saftig, P.; Bateman, R.J.; Breitner, J.; Hock, C.; Multhaup, G. Aβ34 is a BACE1-derived degradation intermediate associated with amyloid clearance and Alzheimer’s disease progression. Nat. Commun. 2019, 10, 2240. [Google Scholar] [CrossRef]
- Zhai, K.; Huang, Z.; Huang, Q.; Tao, W.; Fang, X.; Zhang, A.; Li, X.; Stark, G.R.; Hamilton, T.A.; Bao, S. Pharmacological inhibition of BACE1 suppresses glioblastoma growth by stimulating macrophage phagocytosis of tumor cells. Nat. Cancer 2021, 2, 1136–1151. [Google Scholar] [CrossRef] [PubMed]
- Farris, F.; Matafora, V.; Bachi, A. The emerging role of β-secretases in cancer. J. Exp. Clin. Cancer Res. 2021, 40, 147. [Google Scholar] [CrossRef] [PubMed]
- Dash, B.P.; Naumann, M.; Sterneckert, J.; Hermann, A. Genome Wide Analysis Points towards Subtype-Specific Diseases in Different Genetic Forms of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, R.L.; Schijven, D.; van Rheenen, W.; van Eijk, K.R.; O’Brien, M.; Kahn, R.S.; Ophoff, R.A.; Goris, A.; Bradley, D.G.; Al-Chalabi, A.; van den Berg, L.H.; Luykx, J.J.; Hardiman, O.; Veldink, J.H. Project MinE GWAS Consortium; Schizophrenia Working Group of the Psychiatric Genomics Consortium Genetic correlation between amyotrophic lateral sclerosis and schizophrenia. Nat. Commun. 2017, 8, 14774. [Google Scholar] [CrossRef]
- Pang, W.; Hu, F. Cellular and physiological functions of C9ORF72 and implications for ALS/FTD. J. Neurochem. 2021, 157, 334–350. [Google Scholar] [CrossRef]
- Nassif, M.; Woehlbier, U.; Manque, P.A. The enigmatic role of C9ORF72 in autophagy. Front. Neurosci. 2017, 11, 442. [Google Scholar] [CrossRef]
- Brown, C.A.; Lally, C.; Kupelian, V.; Flanders, W.D. Estimated prevalence and incidence of amyotrophic lateral sclerosis and SOD1 and c9orf72 genetic variants. Neuroepidemiology 2021, 55, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Westergard, T.; Pasinelli, P.; Trotti, D. Pathogenic determinants and mechanisms of ALS/FTD linked to hexanucleotide repeat expansions in the C9orf72 gene. Neurosci. Lett. 2017, 636, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.A.; Frick, P.; Neumann, M. The neuropathology associated with repeat expansions in the C9ORF72 gene. Acta Neuropathol. 2014, 127, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Lafita-Navarro, M.C.; Conacci-Sorrell, M. Nucleolar stress: From development to cancer. Semin. Cell Dev. Biol. 2023, 136, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Hautbergue, G.M. RNA nuclear export: from neurological disorders to cancer. Adv. Exp. Med. Biol. 2017, 1007, 89–109. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, I.; Azuma, Y.; Yamaguchi, M. Cancer-related genes and ALS. Front Biosci (Landmark Ed) 2019, 24, 1241–1258. [Google Scholar] [CrossRef] [PubMed]
- Ingre, C.; Roos, P.M.; Piehl, F.; Kamel, F.; Fang, F. Risk factors for amyotrophic lateral sclerosis. Clin. Epidemiol. 2015, 7, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Perera, R.M. Lysosomes as coordinators of cellular catabolism, metabolic signalling and organ physiology. Nat. Rev. Mol. Cell Biol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hegde, M.L. New Mechanisms of DNA Repair Defects in Fused in Sarcoma-Associated Neurodegeneration: Stage Set for DNA Repair-Based Therapeutics? J. Exp. Neurosci. 2019, 13, 1179069519856358. [Google Scholar] [CrossRef]
- Dormann, D.; Haass, C. Fused in sarcoma (FUS): an oncogene goes awry in neurodegeneration. Mol. Cell. Neurosci. 2013, 56, 475–486. [Google Scholar] [CrossRef]
- Sukhanova, M.V.; Singatulina, A.S.; Pastré, D.; Lavrik, O.I. Fused in Sarcoma (FUS) in DNA Repair: Tango with Poly(ADP-ribose) Polymerase 1 and Compartmentalisation of Damaged DNA. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Sun, Z.; Diaz, Z.; Fang, X.; Hart, M.P.; Chesi, A.; Shorter, J.; Gitler, A.D. Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol. 2011, 9, e1000614. [Google Scholar] [CrossRef]
- Deng, H.; Gao, K.; Jankovic, J. The role of FUS gene variants in neurodegenerative diseases. Nat. Rev. Neurol. 2014, 10, 337–348. [Google Scholar] [CrossRef]
- Shang, Y.; Huang, E.J. Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis. Brain Res. 2016, 1647, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, Y.H.; Wang, H. Genetic Association between Amyotrophic Lateral Sclerosis and Cancer. Genes (Basel) 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Haile, S.; Lal, A.; Myung, J.-K.; Sadar, M.D. FUS/TLS is a co-activator of androgen receptor in prostate cancer cells. PLoS ONE 2011, 6, e24197. [Google Scholar] [CrossRef]
- Ghanbarpanah, E.; Kohanpour, M.A.; Hosseini-Beheshti, F.; Yari, L.; Keshvari, M. Structure and function of FUS gene in prostate cancer. Bratisl. Lek. Listy 2018, 119, 660–663. [Google Scholar] [CrossRef]
- Bukowska, B.; Michalowicz, J.; Pieniazek, D.; Sicinska, P.; Duda, W. Superoxide Dismutases and Their Inhibitors-the Role in Some Diseases. cei 2006, 2, 379–397. [Google Scholar] [CrossRef]
- Rosa, A.C.; Corsi, D.; Cavi, N.; Bruni, N.; Dosio, F. Superoxide dismutase administration: A review of proposed human uses. Molecules 2021, 26. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.J.; McKeown, S.R.; Rashid, S. Mutant SOD1 mediated pathogenesis of Amyotrophic Lateral Sclerosis. Gene 2016, 577, 109–118. [Google Scholar] [CrossRef]
- Kirby, J.; Halligan, E.; Baptista, M.J.; Allen, S.; Heath, P.R.; Holden, H.; Barber, S.C.; Loynes, C.A.; Wood-Allum, C.A.; Lunec, J.; Shaw, P.J. Mutant SOD1 alters the motor neuronal transcriptome: implications for familial ALS. Brain 2005, 128, 1686–1706. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Su, X.; Burley, S.K.; Zheng, X.F.S. Nuclear SOD1 in growth control, oxidative stress response, amyotrophic lateral sclerosis, and cancer. Antioxidants (Basel) 2022, 11. [Google Scholar] [CrossRef]
- Gomez, M.; Germain, D. Cross talk between SOD1 and the mitochondrial UPR in cancer and neurodegeneration. Mol. Cell. Neurosci. 2019, 98, 12–18. [Google Scholar] [CrossRef]
- Roos, R.A.C. Huntington’s disease: a clinical review. Orphanet J. Rare Dis. 2010, 5, 40. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Trojanowski, J.Q.; Lee, V.M.-Y. Protein transmission in neurodegenerative disease. Nat. Rev. Neurol. 2020, 16, 199–212. [Google Scholar] [CrossRef]
- Waldvogel, H.J.; Kim, E.H.; Tippett, L.J.; Vonsattel, J.-P.G.; Faull, R.L.M. The neuropathology of huntington’s disease. Curr. Top. Behav. Neurosci. 2015, 22, 33–80. [Google Scholar] [CrossRef] [PubMed]
- Eje, O. 29 Journal of Chemical Reviews Article info: Journal of Chemical Reviews.
- Turner, M.R.; Goldacre, R.; Goldacre, M.J. Reduced cancer incidence in Huntington’s disease: record linkage study clue to an evolutionary trade-off? Clinical genetics 2013, 83, 588–590. [Google Scholar] [CrossRef]
- Thion, M.S.; Tézenas du Montcel, S.; Golmard, J.-L.; Vacher, S.; Barjhoux, L.; Sornin, V.; Cazeneuve, C.; Bièche, I.; Sinilnikova, O.; Stoppa-Lyonnet, D.; Durr, A.; Humbert, S. CAG repeat size in Huntingtin alleles is associated with cancer prognosis. Eur. J. Hum. Genet. 2016, 24, 1310–1315. [Google Scholar] [CrossRef]
- Malla, B.; Guo, X.; Senger, G.; Chasapopoulou, Z.; Yildirim, F. A systematic review of transcriptional dysregulation in huntington’s disease studied by RNA sequencing. Front. Genet. 2021, 12, 751033. [Google Scholar] [CrossRef]
- Yalçin, M.; El-Athman, R.; Ouk, K.; Priller, J.; Relógio, A. Analysis of the Circadian Regulation of Cancer Hallmarks by a Cross-Platform Study of Colorectal Cancer Time-Series Data Reveals an Association with Genes Involved in Huntington’s Disease. Cancers (Basel) 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Leavitt, B.R.; Kordasiewicz, H.B.; Schobel, S.A. Huntingtin-Lowering Therapies for Huntington Disease: A Review of the Evidence of Potential Benefits and Risks. JAMA Neurol. 2020, 77, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Kryston, T.B.; Georgiev, A.B.; Pissis, P.; Georgakilas, A.G. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat. Res. 2011, 711, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Suwaki, N.; Klare, K.; Tarsounas, M. RAD51 paralogs: roles in DNA damage signalling, recombinational repair and tumorigenesis. Semin. Cell Dev. Biol. 2011, 22, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Impaired DNA Damage Repair as a Common Feature of Neurodegenerati...: Ingenta Connect. Available online: https://www.ingentaconnect.com/content/ben/cmm/2015/00000015/00000002/art00003 (accessed on 14 December 2023).
- Gupta, S.; You, P.; SenGupta, T.; Nilsen, H.; Sharma, K. Crosstalk between Different DNA Repair Pathways Contributes to Neurodegenerative Diseases. Biology (Basel) 2021, 10. [Google Scholar] [CrossRef]
- van der Meer, L.B.; van Duijn, E.; Wolterbeek, R.; Tibben, A. Adverse childhood experiences of persons at risk for Huntington’s disease or BRCA1/2 hereditary breast/ovarian cancer. Clin. Genet. 2012, 81, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Montalto, F.I.; De Amicis, F. Cyclin D1 in cancer: A molecular connection for cell cycle control, adhesion and invasion in tumor and stroma. Cells 2020, 9. [Google Scholar] [CrossRef]
- Monti, S.; Chapuy, B.; Takeyama, K.; Rodig, S.J.; Hao, Y.; Yeda, K.T.; Inguilizian, H.; Mermel, C.; Currie, T.; Dogan, A.; Kutok, J.L.; Beroukhim, R.; Neuberg, D.; Habermann, T.M.; Getz, G.; Kung, A.L.; Golub, T.R.; Shipp, M.A. Integrative analysis reveals an outcome-associated and targetable pattern of p53 and cell cycle deregulation in diffuse large B cell lymphoma. Cancer Cell 2012, 22, 359–372. [Google Scholar] [CrossRef]
- Cohen, T.J.; Lee, V.M.Y.; Trojanowski, J.Q. TDP-43 functions and pathogenic mechanisms implicated in TDP-43 proteinopathies. Trends Mol. Med. 2011, 17, 659–667. [Google Scholar] [CrossRef]
- Alqurashi, Y.E.; Al-Hetty, H.R.A.K.; Ramaiah, P.; Fazaa, A.H.; Jalil, A.T.; Alsaikhan, F.; Gupta, J.; Ramírez-Coronel, A.A.; Tayyib, N.A.; Peng, H. Harnessing function of EMT in hepatocellular carcinoma: From biological view to nanotechnological standpoint. Environ. Res. 2023, 227, 115683. [Google Scholar] [CrossRef]
- Deng, K.; Yao, J.; Huang, J.; Ding, Y.; Zuo, J. Abnormal alternative splicing promotes tumor resistance in targeted therapy and immunotherapy. Transl. Oncol. 2021, 14, 101077. [Google Scholar] [CrossRef] [PubMed]
- Barzilai, A.; Rotman, G.; Shiloh, Y. ATM deficiency and oxidative stress: a new dimension of defective response to DNA damage. DNA Repair (Amst) 2002, 1, 3–25. [Google Scholar] [CrossRef]
- Yan, S.; Sorrell, M.; Berman, Z. Functional interplay between ATM/ATR-mediated DNA damage response and DNA repair pathways in oxidative stress. Cell. Mol. Life Sci. 2014, 71, 3951–3967. [Google Scholar] [CrossRef] [PubMed]
- Kazanets, A.; Shorstova, T.; Hilmi, K.; Marques, M.; Witcher, M. Epigenetic silencing of tumor suppressor genes: Paradigms, puzzles, and potential. Biochim. Biophys. Acta 2016, 1865, 275–288. [Google Scholar] [CrossRef] [PubMed]
Table 1.
Outlines the functions and prospective roles of key genes associated with PD and their possible implications in cancer development.
Table 1.
Outlines the functions and prospective roles of key genes associated with PD and their possible implications in cancer development.
| GENE | FUNCTION | ROLE IN NEURODEGENERATION | ROLE IN CANCER |
|---|---|---|---|
|
LRRK2 |
LRRK2 regulates various cellular processes, including cell signaling, vesicle trafficking, and autophagy | The LRRK2 gene is associated with familial and sporadic PD forms. Mutations in this gene have been identified as significant genetic contributor to PD and development, affecting cellular processes such as protein degradation and mitochondrial function in neurons. |
Involve alterations in cell proliferation, survival pathways, or immune responses for instance, the impact of LRRK2 mutations on cellular processes like autophagy, inflammation, or DNA repair mechanisms might contribute to the pathogenesis of PD and cancer |
|
PINK1 |
Critical role in maintaining mitochondrial function, regulating mitochondrial quality control, and initiating the clearance of damaged mitochondria through a process called mitophagy. Plays a crucial role in mitochondrial quality control and the regulation of cell death pathways. |
Mutations in the PINK1 gene are associated with certain cases of PD, leading to mitochondrial dysfunction and impaired removal of damaged mitochondria, contributing to neuronal degeneration | Studies exploring the broader roles of PINK1 have indicated its involvement in cellular processes beyond PD, including aspects related to cancer biology Initially linked to cancer biology through its modulation by the tumor suppressor PTEN in a cancer cell model, PINK1 gained prominence as it exhibited robust expression in highly metastatic melanoma and colon carcinoma mouse cancer cell lines |
|
SNCA |
Encodes αS |
Mutations or duplications in the SNCA gene have been linked to familial forms of PD | It's been observed that elevated levels of αS could influence tumor growth and metastasis in particular cancer types, including breast cancer and colorectal cancer.. |
| PARK2 |
Parkin, a protein encoded by the PARK2 gene, is vital in removing damaged or unnecessary proteins within cells, a mechanism called ubiquitination | Mutations in the PARK2 gene are linked to familial forms of PD, impairing Parkin's function and accumulating toxic proteins contributing to neuronal degeneration | Some research has explored its involvement in cellular processes related to tumor suppression and apoptosis, indicating a possible connection to cancer biology |
|
PARK 7 |
Parkin-7 (PARK7), also known as DJ-1, protects cells from oxidative stress, maintains mitochondrial function, and regulates cellular pathways associated with cell survival | Mutations in the PARK7 gene are implicated in familial forms of PD, compromising cellular defenses against oxidative damage and contributing to neuronal degeneration. | Involvement in modulating cell proliferation, apoptosis, and tumor progression. While these genes primarily feature in the context of Parkinson's disease, their broader implications or potential involvement in aspects of cancer biology are areas of ongoing investigation. |
Table 2.
Outlines the functions and prospective roles of key genes associated with AD and their possible implications in cancer development.
Table 2.
Outlines the functions and prospective roles of key genes associated with AD and their possible implications in cancer development.
| GENE | FUNCTION | ROLE IN NEURODEGENERATION | ROLE IN CANCER |
|---|---|---|---|
|
APOE |
Encodes a protein involved in lipid metabolism and transportation, playing a crucial role in regulating cholesterol levels in the body. |
Specific variants of the APOE gene, notably the APOE ε4 allele, are known to significantly elevate the risk and influence the age of onset of the disease. |
Alterations in the HTT gene could impact cell survival, DNA repair mechanisms, or cellular processes relevant to tumorigenesis. |
| TP53 | A critical tumor suppressor gene, is commonly mutated in various cancers. | Mutations of TP53 have been observed in the brains of individuals with AD, | Tumor suppressor genes |
| PSEN | Play pivotal roles in various cellular functions, including processing certain proteins like APP. | Mutations in the PSEN1 and PSEN2 genes are strongly linked to early-onset familial AD, promoting the accumulation of amyloid-β peptides, | Studies suggest potential roles of PSENs in regulating cell proliferation, apoptosis, and cellular signaling pathways relevant to tumorigenesis |
| BACE1 | Critical enzyme that produces Aβ peptides | BACE1 cleaves APP to generate these Aβ fragments, contributing to the neurotoxicity seen in AD | Potential implications of BACE1 in cancer biology, highlighting its involvement in regulating cell proliferation, migration, and tumor growth |
Table 3.
Outlines the functions and prospective roles of key genes associated with ALS and their possible implications in cancer development.
Table 3.
Outlines the functions and prospective roles of key genes associated with ALS and their possible implications in cancer development.
| GENE | FUNCTION | ROLE IN NEURODEGENERATION | ROLE IN CANCER |
|---|---|---|---|
|
C9orf72 |
Crucial role in cellular functions, including vesicle trafficking, autophagy, and RNA metabolism |
Expansions of the hexanucleotide repeat in the C9orf72 gene are ALS's most common genetic cause. These expansions lead to toxic RNA and protein aggregates contributing to neuronal degeneration in ALS | Studies have identified associations between C9orf72 mutations and an increased risk of developing various malignancies, including brain tumors and certain types of lymphoma |
|
FUS |
Is involved in various cellular functions, including RNA processing, transport, and DNA repair. |
Mutations in the FUS gene are linked to familial and sporadic cases of ALS, where aberrant FUS proteins form toxic aggregates contributing to neuronal damage |
Aberrant FUS aggregation and pathology have also been associated with certain cancers, albeit in a relatively limited context compared to its involvement in ALS |
|
SOD1 |
Enzyme that is crucial in neutralizing harmful free radicals in cells by converting superoxide radicals into less toxic molecules | Mutations in the SOD1 gene are associated with familial cases of ALS, where altered SOD1 proteins contribute to motor neuron degeneration | Potential role of SOD1 in modulating oxidative stress, inflammation, and cell survival pathways that might have implications in specific cancers |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



