
Submitted:
04 January 2024
Posted:
05 January 2024
You are already at the latest version
Abstract
Quambalaria eucalypti is a fungal pathogen that causes leaf spot, shoot blight, and stem canker disease in Eucalyptus spp. Although the advanced stages of the disease show clear symptoms, diagnosis of the early stages can be challenging. To enable fast and sensitive screening of asymptomatic or latent infected plant material for Q. eucalypti, a specific SYBR green-based real-time PCR assay targeting the partial histone-H3 region was developed. The assay was demonstrated to be specific for Q. eucalypti and did not give cross-reactivity with any of the other Quambalaria species examined or other eucalyptus pathogens. The primers developed in this study ensured high analytical sensitivity, allowing the detection of Q. eucalypti DNA concentrations as low as 10 fg DNA from asymptomatic plants. Comparable results were obtained in interlaboratory testing, demonstrating the assay's robustness and effectiveness in other laboratories. This newly developed quantitative PCR assay can be used for more comprehensive epidemiological investigations, testing plant material in known Q. eucalypti distribution areas for early management strategies, or collecting data for resistance breeding programs.
Keywords:
Quambalaria eucalypti
; Quambalaria shoot blight
; qPCR
; diagnostics
; molecular detection
; Eucalyptus.
1. Introduction
The genus Eucalyptus L’Hér., which includes about 900 species in the family Myrtaceae, is native to Australia and its neighbouring islands [1]. Most eucalypts plants are evergreen woody perennial shrubs and tall trees that grow rapidly, reaching heights of up to about 90 metres. Todays, eucalyptus plantations are the most widely planted broadleaf forests in the world, with the majority located in tropical and temperate regions. They are also widely used in afforestation programmes [2]. It is estimated that the total area of eucalyptus plantations worldwide exceeds 22.57 million hectares based on a survey conducted in 65 countries with extensive plantations [3]. Eucalyptus plants have been well known since ancient times for the numerous uses of their components (wood, fibres, cellulose, dyes, pulp, rubber, resin, essential oils) which are used in various sectors such as construction, pharmaceutical and plant protection [4].
Among the fungal diseases affecting eucalyptus forestry in various regions of the world are those caused by species of the genus Quambalaria (Quambalariaceae, Basidiomycota). Of particular relevance is Quambalaria eucalypti (M.J. Wingfield, Crous, & W.J. Swart) J.A. Simpson [5], which commonly infects eucalyptus plants in nurseries but also in plantations [6,7]. In particularly, the pathogen is known to cause significant productivity losses clonal cuttings of mini hedge plants used for vegetative propagation in nurseries [8].
This fungal species was originally described as Sporothrix eucalypti causing leaf spots and shoot dieback on a clone of Eucalyptus grandis in South Africa [5]. The same fungus was later associated with stem cankers in plantations of Eucalyptus globulus in Uruguay [9], while in Brazil it was reported to cause stem girdling on seedlings of E. globulus and leaf and shoot blight on ministumps of a hybrid of Eucalyptus saligna x E. maidenii [10]. It has since been recognized as a basidiomycete and placed in the genus Quambalaria [11]. Disease caused by Q. eucalypti has subsequently been reported from Australia [12], Portugal [13], China [14] and, most recently, Indonesia [7].
Q. eucalypti is currently identified through the isolation of the fungus and species recognition via morphological examination and phylogenetic analysis by using both the ITS and LSU regions of ribosomal DNA. However, these techniques are laborious, costly and time-consuming. Quantitative PCR (qPCR) is one of the most widely used molecular methods, as it does not require post-amplification processing steps (e.g. gel electrophoresis) which are required in conventional endpoint PCR (cPCR), and allows a specific quantification of the target DNA in the sample [15]. As knowledge of the life cycle and epidemiology of Q. eucalypti in the host is still limited, early detection and quantification of this pathogen is crucial in order to plan an appropriate disease management strategy.
To date, no studies have been conducted to rapidly detect and quantify Q. eucalypti in eucalyptus plant tissues by using a molecular approach. Therefore, the aim of this study was to develop and validate a specific and sensitive real-time PCR assay for the early detection and quantification of Q. eucalypti in eucalyptus plants.
2. Materials and Methods
2.1. Fungal Isolates and Plant Materials
To validate the real-time PCR assay, 30 isolates of Q. eucalypti and 19 isolates of nontarget species from Eucalyptus spp. and various geographical regions were used (Table 1). The Q. eucalypti isolates from Brazil were obtained from leaves of E. globulus seedlings in two nurseries located in the states of Espírito Santo and Rio Grande do Sul in 2021 and 2022, respectively. Uruguayan isolates were obtained in 2004 from twigs with cankers of E. globolus in a plantation located in the west-littoral region. The non-target isolates included 4 other Quambalaria species, and 9 isolates from different species of genera associated with foliar diseases of Eucalyptus spp. The identifications of the Quambalaria isolates were confirmed by sequencing the ITS and LSU regions using primer pairs ITS1F-ITS4 [16,17] and LR0R-LR5 [18,19], respectively. Isolates from species of other genera were identified by ITS sequencing using the ITS5-ITS4 primer pair [17] and, for some of them, the beta-tubulin and elongation factor genes were also sequenced using the primers Bt2a and Bt2b [20], and EF1-728F [21] and EF2 [22], respectively. The isolates were maintained as monosporic cultures into tubes containing malt extract agar (MEA) and stored at 4±1°C.
To confirm the sensitivity of the qPCR assay using infected plant material, three randomly selected Q. eucalypti isolates (i.e. CBS 118844, EGES-5, and UY198) were inoculated on 3-month-old potted E. globulus plants. For each isolate, five plants were inoculated by manually spraying a suspension of 1 × 106 ml-1 spores onto the surface that was previously pricked with a sterile 0.5 mm diameter syringe needle. The spores were obtained from 7-day-old fungal colonies grown on potato dextrose agar (PDA). Five control plants were sprayed with sterilised distilled water instead. Each plant was covered for 48 hours with a clear plastic bag to maintain high relative humidity. Each seedling was covered for 48 hours with a clear plastic bag and kept in the growth chamber at 30°C for 14 days under a 12-hour photoperiod and watered as needed. The material from the inoculated plants was then collected by sorting the samples into 4 different batches based on disease severity. An empirical symptom scale from 0 to 3 was assessed visually since the appearance of first symptoms as leaf spot or stem canker (0 = no symptoms, 1 = 1-25% leaf spot or stem canker, 2 = 26-75% leaf spot or stem canker, 3 = 76-100% shoot and leaf blight or clear signs of the pathogen. Symptomatic and non-symptomatic plant material was kept refrigerated until DNA extraction and then processed either separately or pooled and combined with healthy stem tissues sampled from the control plant in a 1:10 weight ratio.
2.2. Genomic DNA Extraction and Quantification
DNA extraction of fungal isolates was carried out from mycelium grown on PDA for 9 days at 24±1°C in Petri dishes, and that of eucalyptus plants from leaf and stem samples. Both fungal and plant samples were frozen in liquid nitrogen and ground in a a mortar with pestle. DNA was extracted from 50 mg and 100 mg of each mycelium and plant tissue sample respectively, by using the Plant/Fungi DNA Isolation Kit (Norgen Biotek Corp, Canada) according to the manufacturer’s instructions. Extract DNA was quantified using a NanoDrop 2000 spectrophotometer (Thermo Fischer Scientific, Waltham, MA) and adjusted to a concentration of 10 ng µl–1. Genomic DNA with an A260/A280 ratio between 1.8 and 2.0 was used for PCR analyses.
2.3. Primer Design and cPCR Amplification
The Primer-Blast program (https://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi) [23] was used to design the specific primers for a histone-H3 gene selected as the target locus. The sequences of the primers were searched against the whole draft genome of Q. eucalypti isolate CBS 118844 (Genbank accession no. RRYC01000000) [24]. The expected PCR product size parameter was set at 150-400 bp, and the optimal melting temperature at 60±3°C. In silico specificity was verified using Primer-Blast, while the potential formation of dimers and hairpin structures was checked using the tool OligoAnalyzer (https://eu.idtdna.com/calc/analyzer). PCR primers were synthesised and supplied by Invitrogen.
The designed primer pair was tested for their ability to amplify the expected product and to confirm specificity by gradient endpoint cPCR using DNA samples from all fungal isolates listed in Table 1. The amplification reactions were performed on an Arktik thermocycler (Thermo Fischer Scientific, Waltham, MA) in a final volume of 25 µl, containing 12.5 µl of AccuPower PCR Premix (Bioneer, Alameda, CA), 1 µl of each primer (10 µM), 1 µl of DNA (10 ng µl-1), and DNase-free water to make up the final volume. Cycling conditions were as follows: an initial preheat at 95°C for 2 min, followed by 35 cycles of denaturation at 94°C for 1 min, annealing at 58, 59, 60, 61, or 62°C for 30 s, and extension at 72°C for 3 min, followed by a final extension at 72°C for 7 min. PCR products were detected on 2% agarose gel containing SYBR safe DNA gel stain (Life Technologies) in a Tris-borate-EDTA buffer with lane markers and a 100-bp small fragment ladder (Fermentas). Amplicons were purified with ExoSAP-IT (Affymatrix Inc., Santa Clara, CA) and sequenced in both directions by an external service (Macrogen, Amsterdam, The Netherlands). All the chromatograms generated were checked for correct base calling using FinchTV v.1.4.0. The sequences were aligned to the target histone-H3 sequence of Q. eucalypti isolate CBS 118844 using Clustal 2.1 software to confirm the match and to detect for possible polymorphic bases.
A duplex PCR assay was also performed to reveal any false negatives that might have occurred due to PCR inhibition. The primers from this study and those of the cyclin-dependent kinase (CDK8) gene [25] were used to co-amplify both fungal and plant DNA in a single reaction, with the CDK8 (196 bp) serving as a plant internal control. The PCR reaction followed the same procedure as the cPCR, but with 1.5 μl of a DNA mixture from Q. eucalypti isolate CBS 118844 and eucalyptus leaf and stem used as the template. The DNA mixture was composed of 0.5 μl fungal DNA, 0.5 μl leaf DNA (100 ng μl-1) and 0.5 μl stem DNA (100 ng μl-1). Five 10-fold dilutions (10-2 to 102) of fungal DNA (10 ng μl-1) were tested.
2.4. SYBR Green Real-Time PCR Optimization
To optimise the assay, the qPCR reactions were performed in a gradient using an iCycler iQ Real-Time PCR DetectionSystem (Bio-Rad) in a final volume of 25 μl, which contained 12.5 μl of Platinum SYBR Green qPCR SuperMix-UDG (Invitrogen), 1 µl of each primer (8, 10 or 12 µM), 1 µl of DNA (10 ng µl-1) from CBS 118844, and DNase-free water to make up the final volume. Thermal cycling parameters consisted of an initial preheating step for 5 min at 95°C followed by 40 cycles at 95°C for 30 s, annealing at 59, 60, 60.5, 61, 61.5, 62, or 62.5°C for 30 s, and 72°C for 30 s for data collection and real-time analysis; then, the set point temperature was increased after cycle 2 from 72 to 95°C for 30 s in 0.5°C increments to construct a melting curve and perform data collection and analysis.
2.5. Real-Time PCR Assay Specificity and Sensitivity
The qPCR assay’s specificity was tested using DNA samples from Q. eucalypti isolates listed in Table 1. The real-time PCR reaction conditions and thermocycling settings were optimized as follows: initial denaturation at 95°C for 3 min, followed by 40 cycles of denaturation at 95°C for 30 s, annealing at 60°C, and extension at 72°C for 30 s. The sensitivity of the qPCR assay was determined by testing six 10-fold dilutions (100 to 10-5) of DNA (100 pg μl-1) from Q. eucalypti isolate CBS 118844 in sterile water or in DNA extracted from eucalyptus leaf (10 ng μl-1) or stem (10 ng μl-1). The sensitivity test was conducted in triplicate for each dilution and three real-time PCR runs were carried out for each sample using the previously mentioned thermocycle conditions and DNA templates. Standard curves were generated by graphing the mean cycle threshold (Ct) values on the y-axis and the concentration of fungal DNA on the x-axis. The efficiency of the PCR assay was determined using the following equation: E = (101/-S) -1 [26].
2.6. Interlaboratory Tests
The mean Ct values and standard deviation of the intra- and inter-assay results were used to calculate the coefficient of variation (CV). The CV for intra-assay repeatability was assessed by conducting ten technical replicates in the same reaction using four dilutions of CBS 118844 isolate DNA in water (10, 1, 0.1, and 0.01 pg μl-1). The CV between assays was determined from the results of three independent blind panel experiments (three technical replicates in each assay) performed on different real-time PCR machines. The reproducibility test was performed by three distinct operators using the same series of dilutions of the DNA as described previously. Additionally, to validate the applicability of the qPCR assay, the test was also carried out in an external laboratory on a different real-time PCR instrument (i.e. StepOnePlus qPCR system, Applied Biosystems) using different set of reagents.
3. Results
3.1. Primer Design and cPCR Amplification
A primer pair was successfully developed for a histone-H3, which is used as a target locus in the genome of Q. eucalypti isolate CBS 118844. The designed specific primers (QEH3-F: 5'-CTTAGGACTTCTCGCCTCGG-3' and QEH3-R: 5'-CTGAGCTCCTCATCCGCAAG-3') amplify a DNA segment of 237 bp (Figure 1). The comparative sequence analysis, conducted on nucloeotide database of NCBI GenBank, showed that the designed primers are specific to Q. eucalypti and do not share any identical or similar sequences with those of other fungi.
The QEH3-F/QEH3-R primer pair successfully generated an amplicon of the expected lenght for all Q. eucalypti isolates tested in this study using gradient end-point PCR. In contrast, the non-target isolates of other Quambalaria species and those of different genera were not amplified. Direct sequencing of the PCR products from these isolates showed 100% identity with the targeted histone-H3 region and no ambiguous nucleotide sites, demonstrating the specificity of the primers and the absence of polymorphisms.
The duplex PCR assay amplified both the Q. eucalypti histone-H3 and the eucalyptus CDK8 targets from mixed DNA samples of the fungus and plant tissues with no evidence of inhibition.
3.2. SYBR Green Real-Time PCR Optimization
The target was successfully amplified using a gradient real-time PCR with newly designed primers. At different annealing temperatures (59 to 62.5°C) slight variations in Ct values were observed. For further assay development, an annealing temperature of 60°C was selected. At this temperature, the lowest Ct values and detection curves with high relative fluorescence were obtained. By assaying different primer concentrations (8, 10 and 12 µM), it was observed that the 8 µM concentration resulted in lower Ct values. Therefore, a concentration of 8 µM was selected for subsequent assay validation.
3.3. Real-Time PCR Assay Specificity and Sensitivity
Likewise the cPCR, the new real-time PCR assay detected all 30 Q. eucalypti isolates without cross-reacting with any of the 19 non-target fungal isolates or plant DNA. The primers QEH3-F and QEH3-R generated a single absorbance intensity peak for all Q. eucalypti isolates. Melting curve analysis of the target DNA samples showed melting points ranging from 86.5 to 87.3. No rise in absorbance was observed for the non-target fungal isolates or the negative control (Table 1).
The assay’s analytical sensitivity using a 10-fold serial dilution of Q. eucalypti DNA in water or in eucalyptus leaf or stem DNA revealed that the assay was capable of detecting pathogen DNA levels as low as 10 fg. The sensitivity test showed a linear curve response from 100 pg to 10 fg DNA (Figure 2) with PCR efficiencies of 96 and 97% for pathogen DNA diluted in water and plant tissue, respectively.
The pathogen was successfully detected using DNA obtained from Q. eucalypti-infected leaves and stems artificially inoculated in eucalyptus seedlings. The Ct values obtained from the tissues of symptomatic plants were lower than those of asymptomatic plants, decreasing from 27.70 to 24.81 with increasing disease severity. However, amplification was also observed in asymptomatic samples with an average Ct value of 33, equivalent to about 18 fg of pathogen DNA. No significant differences in Ct values were observed between the Q. eucalypti isolates assayed (Table 2). The qPCR assay detected Q. eucalypti even in infected tissues mixed with healthy plant material in a ratio of 1:10. The fungus was not detected in DNA samples from non-inoculated eucalyptus plants.
3.4. Interlaboratory Tests
The test repeatability of the qPCR assay resulted in none or minimal Ct value variation. Furthermore, the intra-assay CV for the four tested dilutions of Q. eucalypti DNA remained consistently low. The assay’s reproducibility testing showed only a slight difference in Ct values, with a low inter-assay CV (Table 3). Independent laboratory testing of the assay further confirmed its reproducibility.
4. Discussion
Among the molecular diagnostic techniques, qPCR is more specific, higher sensitive and less time-consuming than conventional endpoint PCR [27,28]. This technique enables quantification of pathogens and has been used for various fungal plant pathogens [29,30,31], including Q. eucalypti [32]. Duong et al. (2022) developed a qPCR assay for Q. eucalypti using the multi-copy ITS region, but it has not been tested on infected host tissue. In this study, a SYBR green real-time PCR assay for the detection and quantification of Q. eucalypti in eucalyptus plants was successfully developed. The use of a qPCR detection method based on single-copy genes allows for higher accuracy in quantitative analyses [15], therefore the single-copy histone-H3 gene was selected for the preliminary endpoint PCR assay. The primers (QEH3-F and QEH3-R) were designed against a portion of the histone-H3 ORF identified in the draft genome of Q. eucalypti isolate CBS 118844. The newly developed assay was shown to be specific for Q. eucalypti by testing a panel of 30 Q. eucalypti isolates and 19 non-target species associated with Eucalyptus spp.
High-sensitivity detection methods depend on the pathogen’s abundance in the plant, particularly in cases of asymptomatic latent infections. This assay was able to detect the pathogen in both symptomatic and asymptomatic infected eucalyptus plants. The use of the histone H3 region ensured high analytical sensitivity, allowing the detection of Q. eucalypti DNA concentrations in symptomatic plants ranging from approximately 0.3 to 1.6 pg, depending on the disease severity. While, in asymptomatic plants the pathogen can be detected at levels as low as 10 fg DNA, which is the limit of detection for the assay. Additionally, the pathogen was also detected when asymptomatic infected eucalyptus leaf and stem tissues were mixed with healthy plant material in a ratio of 1:10, demonstrating the high sensitivity of the assay. To enable the simultaneous detection of pathogen and plant internal control, the cPCR assay was duplexed with an assay targeting the plant CDK8 gene [25]. Although duplexing or multiplexing can compromise the sensitivity of pathogen-specific assays [33], the duplexed cPCR assay was able to detect both Q. eucalypti and the plant internal control in a single reaction without any interference or reduction in sensitivity. Furthermore, the robustness and effectiveness of the test in other laboratories was demonstrated by comparable results obtained in interlaboratory tests.
5. Conclusions
In conclusion, this newly developed real-time PCR assay can be used for conducting more detailed epidemiological studies, testing plant material in known Q. eucalypti distribution areas for early management practices, or gathering information for resistance breeding programs. In addition, this SYBR Green-based qPCR assay also offers cost advantages, being much less expensive than other diagnostic tools such as qPCR using fluorescent probe chemistry or real-time lamp.
Author Contributions
Conceptualization, R.F.; methodology, R.F. and G.B.S.; software, R.F. and G.B.S.; validation, R.F. and G.B.S.; formal analysis, R.F. and G.B.S.; investigation, R.F. and G.B.S.; resources, R.F. and G.B.S.; data curation, R.F. and G.B.S.; writing—original draft preparation, R.F.; writing—review and editing, R.F.; supervision, R.F.; project administration, G.B.S.; funding acquisition, G.B.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors thank Hui Yang for kindly providing several Q. eucalypti isolates and Barbara Gonçalves, Júlio de Castilhos, and Ricardo Gomes completed blind panel testing.
Conflicts of Interest
All the authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Naithani, H.B. Botany of genus Eucalyptus. In: Eucalyptus in India, Bhojvaid, P.P., Kaushik, S., Singh, Y.P., Kumar, D., Thapliyal, M., Barthwal, S., Eds.; ICFRE Dehra Dun, 2014; pp. 1–20.
- Trabado, G.I., Wilstermann, D. Eucalyptus global map 2008: Cultivated forests worldwide—EUCALYPTOLOGICS: GIT Forestry consulting information resources on Eucalyptus cultivation worldwide. Available online: http://git-forestry-blog.blogspot.com/2008/09/eucalyptus-global-map-2008-cultivated.html (accesssed on).
- Wen, Y.; Zhou, X.; Yu, S.; Zhu, H. The predicament and countermeasures of development of global eucalyptus plantations. Guangxi Sci. 2018, 25, 107–116. [Google Scholar]
- Salehi, B.; Sharifi-Rad, J.; Quispe, C.; Llaique, H.; Villalobos, M.; Smeriglio, A.; Trombetta, D.; Ezzat, S.M.; Salem, M.A.; Zayed, A.; Salgado Castillo, C.M.; Yazdi, S.E.; Sen, S.; Acharya, K., Sharopov, F.; Martins, N. Insights into Eucalyptus genus chemical constituents, biological activities and health-promoting effects. Trends Food Sci. Technol. 2019, 91, 609–624. [CrossRef]
- Wingfield, M.J.; Crous, P.W.; Swart, W.J. Sporothrix eucalypti (sp. nov.), a shoot and leaf pathogen of Eucalyptus in South Africa. Mycopathologia 1993, 123, 159–164. [Google Scholar] [CrossRef]
- Santos, G.S.; Mafia, R.G.; Zarpelon, T.G.; Soares, T.P.F., Ferreira, M.A. First report of Quambalaria eucalypti in young eucalyptus plants in field conditions in Brazil. For. Pathol. 2020, 50, e12633. [CrossRef]
- Tarigan, M.; Wingfield, M.J.; Marpaung, Y.M.A.N.; Durán, A.; Pham, N.Q. Quambalaria eucalypti found on Eucalyptus in Indonesia. Forest Pathol. 2023, 53, e12829. [Google Scholar] [CrossRef]
- Alfenas, A.C.; Zauza, E.A.V.; Mafia, R.G.; Assis, T.F. Clonagem e Doenças do Eucalipto, 2nd ed.; Universidade Federal de Viçosa: Viçosa, Brazil, 2009; p. 500. [Google Scholar]
- Bettucci, L.; Alonso, R.; Tiscornia, S. Endophytic mycobiota of healthy twigs and the assemblage of species associated with twig lesions of Eucalyptus globulus and E. grandis in Uruguay. Mycol. Res. 1999, 103, 468–472. [Google Scholar] [CrossRef]
- Alfenas, A.C.; Zauza, E.A.; Rosa, O.P.; Assis, T.F. Sporothrix eucalypti, a new pathogen of eucalyptus in Brazil. Fitopatol. Bras. 2001, 26, 221. [Google Scholar] [CrossRef]
- Simpson, J.A. Quambalaria, a new genus of eucalypt pathogens. Australas. Mycol. 2000, 19, 57–62. [Google Scholar]
- Pegg, G.S.; O’Dwyer, C.; Carnegie, A.J.; Burgess, T.I.; Wingfield, M.J.; Drenth, A. Quambalaria species associate with plantation and native eucalypts in Australia. Plant Pathol. 2008, 57, 702–714. [Google Scholar] [CrossRef]
- Bragança, H.; Diogo, E.L.F.; Neves, L.; Valente, C.; Araujo, C.; Bonifácio, L.; Phillips, A.J.L. Quambalaria eucalypti a pathogen of Eucalyptus globulus newly reported in Portugal and in Europe. Forest Pathol. 2016, 46, 67–75. [Google Scholar] [CrossRef]
- Chen, S.F.; Liu, Q.; Li, G.; Wingfield, M.J. Quambalaria species associated with eucalypt diseases in southern China. Front. Agric. Sci. Eng. 2017, 4, 433–447. [Google Scholar] [CrossRef]
- Schena, L.; Li Destri Nicosia, M.G.; Sanzani, S.M.; Faedda, R.; Ippolito, A.; Cacciola, S.O. Development of quantitative PCR detection methods for phytopathogenic fungi and oomycetes. J. Plant Pathol. 2013, 95, 7–24. [Google Scholar] [CrossRef]
- Gardes, M.; Bruns, T.D. ITS primers with enhanced specificity for basidiomycetes-application to the identification of mycorrhizae and rusts. Mol. Ecol. 1993, 2, 113–118. [Google Scholar] [CrossRef] [PubMed]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for Phylogenetics. In PCR protocols: A sequencing guide to methods and applications, Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press, 1990; pp. 315–322.
- Rehner, S.A.; Samuels, G.J. Taxonomy and phylogeny of Gliocladium analysed from nuclear large subunit ribosomal DNA sequences. Mycol. Res. 1994, 98, 625–634. [Google Scholar] [CrossRef]
- Vilgalys, R.; Hester, M. Rapid genetic identification and mapping of enzymatically amplified ribosomal DNA from several Cryptococcus species. J. Bact. 1990, 172, 4238–4246. [Google Scholar] [CrossRef] [PubMed]
- Glass, N.L.; Donaldson, G.C. Development of Primer Sets Designed for Use with the PCR To Amplify Con-served Genes from Filamentous Ascomycetes. Appl. Environ. Microbiol. 1995, 61, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Carbone, I.; Kohn, L.M. A method for designing primer sets for speciation studies in filamentous ascomycetes. Mycologia 1999, 91, 553–556. [Google Scholar] [CrossRef]
- O’Donnell, K.; Kistler, H.C.; Cigelnik, E.; Ploetz, R.C. Multiple evolutionary origins of the fungus causing Panama disease of banana: Concordant evidence from nuclear and mitochondrial gene genealogies. Proc. Natl. Acad. Sci. USA 1998, 95, 2044–2049. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef]
- Wingfield, B.D.; Liu, M.; Nguyen, H.D.T.; Lane, F.A.; Morgan, S.W.; De Vos, L.; Wilken, P.M.; Duong, T.A.; Aylward, J.; Coetzee, M.P.A.; et al. Nine draft genome sequences of Claviceps purpurea s.lat., including C. arundinis, C. humidiphila, and C. cf. spartinae, pseudomolecules for the pitch canker pathogen Fusarium circinatum, draft genome of Davidsoniella eucalypti, Grosmannia galeiformis, Quambalaria eucalypti, and Teratosphaeria destructans. IMA Fungus 2018, 9, 401–418. [Google Scholar] [CrossRef]
- de Oliveira, L.A.; Breton, M.C.; Bastolla, F.M.; Camargo, S.d.S.; Margis, R.; Frazzon, J.; Pasquali, G. Reference genes for the normalization of gene expression in Eucalyptus species. Plant Cell Physiol. 2012, 53, 405–422. [Google Scholar] [CrossRef] [PubMed]
- Ginzinger, D.G. Gene amplification using real-time quantitative PCR: An emerging technology hits the mainstream. Exp. Hematol. 2002, 30, 503–512. [Google Scholar] [CrossRef] [PubMed]
- McCartney, H.A.; Poster, S. J.; Fraaije, B.A.; Ward, E. Molecular diagnostics for fungal plant palhogens. Pest Manage. Sci. 2003, 59, 129–142. [Google Scholar] [CrossRef]
- Zijlstra, C.; Lund, I.; Justesen, A.F.; Nicolaisen, M.; Jensen, P.K.; Bianciotto, V.; Posta, K.; Balestrini, R.; Przetakiewicz, A.; Czembor, E.; van de Zande, J. Combining novel monitoring tools and precision application technologies for integrated high-tech crop protection in the future (a discussion document). Pest Manage. Sci. 2011, 67, 616–625. [Google Scholar] [CrossRef]
- Schena, L.; Nigro, F.; Ippolito, A.; Gallitelli, D. Real-time quantitative PCR: A new technology to detect and study phytopathogenic and antagonistic fungi. Eur. J. Plant Pathol. 2004, 110, 893–908. [Google Scholar] [CrossRef]
- Sanzani, S.M.; Li Destri Nicosia, M.G.; Faedda, R.; Cacciola, S.O.; Schena, L. Use of quantitative PCR detection methods to study biocontrol agents and phytopathogenic fungi and oomycetes in environmental samples. J. Phytopathol. 2014, 162, 1–13. [Google Scholar] [CrossRef]
- Hariharnn, G.; Prasannath, K. Recent advances in molecular diagnostics of fungal plant pathogens: A mini review. Front. Cell. Lnfect. Microbiol. 2021, 10, 600234. [Google Scholar] [CrossRef]
- Duong, H.T.; Williams, B.; White, D.; Burgess, T.I.; Hardy, G.E.S.J. qPCR assays for sensitive and rapid detection of Quambalaria species from plant tissues. Plant Dis. 2022, 106, 107–113. [Google Scholar] [CrossRef]
- Baskarathevan, J.; Taylor, R.K.; Ho, W.; McDougal, R.L.; Shivas, R.G.; Alexander, B.J.R. Real-time PCR assays for the detection of Puccinia psidii. Plant Dis. 2016, 100, 617–624. [Google Scholar] [CrossRef]
Figure 1.
The nucleotide sequence of histone-H3 of Quambalaria eucalypti and the flanking regions of the designed primers shown in the red box.
Figure 1.
The nucleotide sequence of histone-H3 of Quambalaria eucalypti and the flanking regions of the designed primers shown in the red box.

Figure 2.
The Real-Time PCR assay standard curve of serial dilutions of Quambalaria eucalypti DNA in water (a) and in combination with eucalyptus DNA (b). A linear trend was observed for both dilution series from 10 to 105 fg DNA. The standard deviation is indicated by the vertical bars, while Ct corresponds to the quantification cycle.
Figure 2.
The Real-Time PCR assay standard curve of serial dilutions of Quambalaria eucalypti DNA in water (a) and in combination with eucalyptus DNA (b). A linear trend was observed for both dilution series from 10 to 105 fg DNA. The standard deviation is indicated by the vertical bars, while Ct corresponds to the quantification cycle.
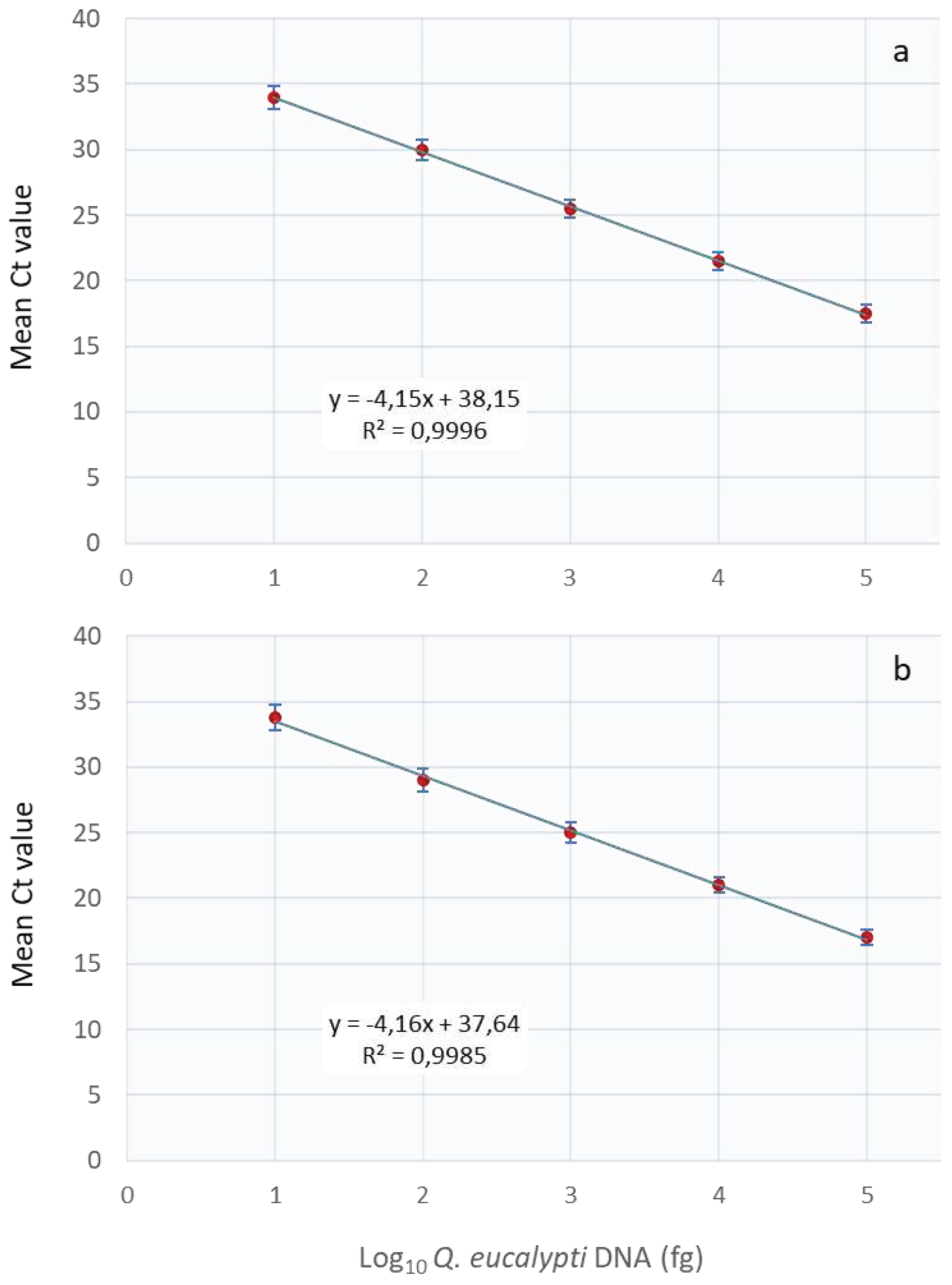

Table 1.
Fungal isolates examined in this study and results of specificity testing of the qPCR assay.
Table 1.
Fungal isolates examined in this study and results of specificity testing of the qPCR assay.
| Species | Isolatea | Host | Origin | Ctb value ± SDc | Tm (°C)d |
|---|---|---|---|---|---|
| Quambalaria eucalypti | CBS 118844e | Eucalyptus grandis | South Africa | 21.54±0.71 | 87.2 |
| CBS 119680f | 21.30±0.52 | 87.0 | |||
| ITEF/1937 | 20.48±0.35 | 86.1 | |||
| ITEF/1999 | 20.43±0.14 | 86.8 | |||
| EGES-1 | Eucalyptus globulus | Brazil | 21.14±0.35 | 86.1 | |
| EGES-2 | 20.87±0.62 | 87.1 | |||
| EGES-5 | 20.91±0.53 | 87.0 | |||
| EGES-10 | 22.07±0.26 | 86.0 | |||
| EGES-11 | 22.09±0.88 | 86.2 | |||
| EGES-13 | 21.30±0.13 | 87.0 | |||
| EGES-17 | 21.58±0.44 | 86.9 | |||
| EGES-19 | 22.43±0.46 | 86.1 | |||
| EGES-20 | 22.46±0.35 | 86.2 | |||
| EGES-22 | 22.45±0.21 | 87.2 | |||
| RGS-1 | 21.41±0.57 | 86.3 | |||
| RGS-2 | 21.35±0.22 | 87.0 | |||
| RGS-3 | 21.47±0.31 | 87.2 | |||
| RGS-4 | 21.29±0.81 | 86.2 | |||
| RGS-5 | 20.93±0.73 | 87.0 | |||
| RGS-6 | 21.02±0,54 | 86.6 | |||
| SUL/01 | Uruguay | 21.27±0.52 | 86.7 | ||
| SUL/03 | 22.05±0.54 | 86.8 | |||
| UY198 | 21.24±0.31 | 86.3 | |||
| UY199 | 21.63±0.61 | 87.0 | |||
| MATY 4665 | 19.98±1.07 | 86.5 | |||
| MATY 4751 | 20.15±0.86 | 86.5 | |||
| MATY 4752 | 21.73±0.24 | 86.7 | |||
| MATY 5001 | 21.65±0.35 | 86.6 | |||
| 3421-S | 19.82±1.13 | 87.0 | |||
| 3422-T | 21.36±0.52 | 87.1 | |||
| Quambalaria cyanescens | CBS 876.73 | Eucalyptus pauciflora | Australia | n/a | n/a |
| IMI 178848 | n/a | n/a | |||
| EU-PA | n/a | n/a | |||
| Quambalaria pitereka | CERC/03/08 | Eucalyptus sp. | China | n/a | n/a |
| CERC/04/08 | n/a | n/a | |||
| CERC/05/05 | n/a | n/a | |||
| Quambalaria simpsonii | CBS 124772f | Eucalyptus tintinnans | Australia | n/a | n/a |
| EU-TI3 | n/a | n/a | |||
| Quambalaria tasmaniae | CBS 145602a | Eucalyptus sp. | Australia | n/a | n/a |
| 7608 | n/a | n/a | |||
| Alternaria alternata | CYC-60 | Eucalyptus grandis | Uruguay | n/a | n/a |
| Calonectria spathulata | CS11340 | Eucalyptus grandis | Brazil | n/a | n/a |
| Colletotrichum boninense | MWJ-43 | Eucalyptus grandis | South Africa | n/a | n/a |
| Cryphonectria havanensis | GSEG_95 | Eucalyptus grandis | Brazil | n/a | n/a |
| Cylindrocladium candelabrum | 11356 | Eucalyptus sp. | Brazil | n/a | n/a |
| Cylindrocladium pteridis | IMI 354530 | Eucalyptus grandis | Brazil | n/a | n/a |
| Neopestalotiopsis eucalyptorum | 912/88 | Eucalyptus globulus | Portugal | n/a | n/a |
| Phomopsis arnoldiae | CR 345-96 | Eucalyptus grandis | Uruguay | n/a | n/a |
| Pseudocercospora eucalyptorum | BBR 5689 | Eucalyptus globulus | Spain | n/a | n/a |
a CBS = cultures from Westerdijk Fungal Biodiversity Institute, Netherlands; b Ct = cycle treshold; c SD = standard deviation; d melting temperature ± standard deviation; e ex-holotype; f ex-epitype; n/a = no amplification.
Table 2.
Quantitative PCR detection of Quambalaria eucalypti artificially inoculated in eucalyptus plants.
Table 2.
Quantitative PCR detection of Quambalaria eucalypti artificially inoculated in eucalyptus plants.
| Isolate | Disease severity | Tm (°C) | Cta value ± SDb | DNA conc. (fg) ± SDb |
|---|---|---|---|---|
| CBS 118844 | 0 | 87.2 | 33.10±0.02 | 16.48±2.40 |
| 1 | 86.9 | 27.70±0.05 | 329.66±5.24 | |
| 2 | 87.3 | 26.75±0.03 | 558.45±11.07 | |
| 3 | 87.0 | 24.89±0.02 | 1,567.40±14.85 | |
| EGES-5 | 0 | 87.3 | 32.87±0.18 | 18.72±0.65 |
| 1 | 86.8 | 27.43±0.27 | 382.94±5.18 | |
| 2 | 87.1 | 26.73±0.02 | 564.69±11.67 | |
| 3 | 86.9 | 24.95±0.03 | 1,516.08±12.36 | |
| UY198 | 0 | 87.0 | 32.84±0.26 | 19.03±0.15 |
| 1 | 87.0 | 27.53±0.02 | 362.27±4.73 | |
| 2 | 86.9 | 26.71±0.04 | 570.99±10.45 | |
| 3 | 87.1 | 24.81±0.06 | 1,638.54±21.74 |
a Ct = cycle threshold; b SD = standard deviation.
Table 3.
The test results for repeatability (intra-assay) and reproducibility (inter-assay) using four dilutions of DNA in water from Quambalaria eucalypti isolate CBS 118844.
Table 3.
The test results for repeatability (intra-assay) and reproducibility (inter-assay) using four dilutions of DNA in water from Quambalaria eucalypti isolate CBS 118844.
| DNA conc. (pg) |
Cta value ± SDb (intra-assay) |
Cta value ± SDb (inter-assay) | Coefficient of variance (%) | |||
|---|---|---|---|---|---|---|
| Operator 1 | Operator 2 | Operator 3 | Intra-assay | Inter-assay | ||
| 10 | 21.49±0.15 | 21.40±0.54 | 20.98±0.66 | 20.78±0.15 | 0.3 | 0.9 |
| 1 | 25.50±0.11 | 25.18±0.36 | 25.34±0.12 | 26.03±0.24 | 0.4 | 1.5 |
| 0.1 | 30.01±0.12 | 29.87±0.21 | 29.91±0.15 | 30.05±0.45 | 0.6 | 1.3 |
| 0.01 | 34.01±0.13 | 33.85±0.18 | 34.13±0.11 | 33.97±0.21 | 0.8 | 2.4 |
a Ct = cycle threshold; b SD = standard deviation.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



