
Submitted:
05 January 2024
Posted:
05 January 2024
You are already at the latest version
Abstract
The extended cleavage specificities of two hematopoietic serine proteases originating from the ray-finned fish spotted gar (Lepisosteus oculatus) have been characterized using substrate phage display. The preference for particular amino acids at, and surrounding the cleavage site was further validated using a panel of recombinant substrates. For one of the enzymes, the gar granzyme G, a strict preference for the aromatic amino acid Tyr was observed at the cleavable P1 position. Using a set of recombinant substrates showed that the gar granzyme G had a high selectivity for Tyr, but also with lower activity cleaving after Phe but not after Trp. The second enzyme, gar DDN1, instead showed a high preference for Leu in the P1 position of substrates. This latter enzyme also showed high preference for Pro in the P2 position and Arg in both P4 and P5 positions. The selectivity for the two Arg residues in positions P4 and P5, suggest a highly specific substrate selectivity of this enzyme. Screening of the gar proteome with the consensus sequences obtained by substrate phage display for these two proteases resulted in a very diverse set of potential targets. Due to this diversity, a clear candidate for a specific immune function of these two enzymes cannot yet be identified.
Antisera developed against the recombinant gar enzymes were used to study their tissue distribution. Tissue sections from juvenile fish showed expression of both proteases in cells in Peyer´s patch-like structures in the intestinal region, indicating they may be expressed in T or NK cells. However, due to the lack of antibodies to specific surface markers in the gar, it has not been possible to specify the exact cellular origin. A marked difference in abundance was observed for the two proteases where gar DDN1 was expressed at higher levels than gar granzyme G. However, both appear to be expressed in the same or similar cells, having a lymphocyte-like appearance.
Keywords:
fish
; serine protease
; cleavage specificity
; tryptase
; macrophage
; evolution.
1. Introduction
Serine proteases constitute the major granule content of several hematopoietic cell types. Large amounts of active chymotryptic and tryptic enzymes are stored in cytoplasmic granules of mammalian mast cells for rapid released upon activation. Mammalian neutrophils instead store primarily elastolytic enzymes, as exemplified by neutrophil elastase and proteinase 3, but also a tryptic enzyme, NSP-4 and a chymotryptic enzyme, cathepsin G [1,2,3,4]. Human cytotoxic T cells and natural killer cells (NK cells) express and store several granzymes with tryptase, asp-ase and met-ase specificities, as exemplified by granzymes A, B and M respectively [1]. A relatively detailed picture has been established of how these enzymes have appeared and diversified during mammalian evolution [2,3,5,6,7,8,9,10,11,12,13,14,15]. However, the information concerning the presence and specificity of such enzymes in reptiles, amphibians, and fishes is still only fragmentary [16,17,18,19,20,21,22,23]. There has been an increase in the use of non-rodent animal models with much focus on non-mammalian vertebrates including fish species, notably the zebrafish. However, there are few studies on the details regarding the characterisation of hematopoietic serine proteases in other fishes. Here, the (spotted) gar is of particular interest as it represents an basally diverging branch of the ray-finned fish evolutionary tree and has been found to have a genome that has undergone relatively limited number of rearrangements and amplifications compared to many other fish lineages including the zebrafish, which is often used as a model organism [24]. The gar genome thereby represents a less derived configuration than most other fish genomes and has interestingly been found show major similarities to mammalian genomes [24]. In order to look closer into early events in the expansion and diversification of these hematopoietic serine protease, this analysis focuses on two hematopoietic serine proteases from the spotted gar.
In mammals, serine proteases play important and diverse roles in a number of physiological processes including blood coagulation, food digestion, fertilisation, immunity and tissue repair [25]. This large chymotrypsin/trypsin family all share a common mechanism for cleaving peptide bonds, based on their catalytic triad, with three vital residues: His57, Asp102 and Ser195 (chymotrypsinogen numbering) [26] (Figure 1A). These key amino acids are located near a substrate binding pocket (termed S1), typically made up of residues 189, 216 and 226 (also chymotrypsinogen numbering) [26] (Figure 1B). Together they form the specificity conferring triplet and provide clues as to the primary specificity of the serine proteases (Figure 1B). For mammalian enzymes and also other tetrapods, these three residues provide a relatively good indication for the primary specificity of an enzyme. However, for the majority of fish proteases, excluding the granzyme A/K members, these three residues give little guidance to their primary specificities. This is primarily due to large sequence divergences making the positioning of the relevant residues difficult to determine based only on the primary sequence. To determine the specificity of the fish proteases we therefore have to rely on direct experimental analysis.
The primary specificity of a protease is the amino acid after which the cleavage occurs and this residue is named the P1 residue (Figure 1A). The residues N-terminally of the cleavage site are numbered P2, P3, P4 etc and the residues C-terminally of the cleavage site P1´, P2´, P3´ etc (Figure 1A). The primary specificity determines the main specificity of the enzyme. If the enzyme cleaves after a basic amino acid, such as Arg or Lys, it has tryptic activity, when it cleaves after large aromatic amino acids, such as Phe, Tyr or Trp it has a chymotryptic activity (Figure 1B). The extended specificity is determined by the selectivity in residues surrounding the cleavage site, most often the P2, P3, P4, P1´, P2´and P3´residues (Figure 1A).
To determine the cleavage specificity of an enzyme different techniques can be used, including chromogenic substrates, peptide libraries and phage display. Substrate phage display is a method that can provide detailed information concerning both the primary and extended specificity of an enzyme. This method has been used to study the extended specificities of two serine proteases from the spotted gar. These two proteases were extracted from the genomic database depending on the distance of the relationship with other known hematopoietic serine proteases. Both enzymes showed very stringent both primary and extended specificities. One being a Tyr-specific chymase and the other being a Leu-specific Leu-ase. Antisera developed against these two proteases detected cells in Peyer´s patch-like structures of the intestinal region of the gar, with a pattern of expression indicating that the proteases are expressed in cytotoxic T cells or NK cells. However, it is yet not possible to more specifically identify the cell origin as very few reagents are available for studies of immune cells in the gar.
2. Results
2.1. Phylogenetic Analyses
Human and mouse hematopoietic serine protease sequences were used as query sequences to identify similar sequences in a large panel of vertebrate genomes in the NCBI database using the TBLASTN algorithm. The alignment using MAFFT and the MrBayes program, generating a Bayesian phylogenetic tree that is depicted in Figure 2A. An enlarged version of the fish proteases clustering in a separate branch of the tree is shown in Figure 2B. The phylogenetic analyses were performed essentially as described in a previous publication using the same strategy and sequences [2].
2.2. Production, Purification and Activation of Gar Granzyme G and Gar Duodenase 1
DNA constructs containing the coding regions for the gar granzyme G (Gzm-G) and the gar duodenase 1 (DDN-1), an N-terminal His6-tag followed by an enterokinase (EK) site were designed and ordered from Genscript. These fragments were subsequently cloned into the mammalian expression vector pCEP-Pu2 for expression in HEK 293-EBNA cells [27]. Here, the His6-tag facilitates purification on Ni2+ chelating immobilized metal ion affinity chromatography columns and cleavage with EK activates the enzyme, whilst simultaneously removing the His6-tag and the EK-site (Figure 3).
2.3. Substrate Phage Display
To determine the extended cleavage specificity of the two gar enzymes a phage T7 based system was used where individual peptide sequences are displayed on the surface of the phage. This system enables the characterization of a region covering both 4-5 amino acids N-terminally and C-terminally of the cleavage site. The library used had a complexity of approximately 50 million different peptide sequences. After 7 selection rounds, the gar enzymes selected phages which showed at least a three order of magnitude increase compared to the PBS control. One hundred and twenty phage plaques from the last selection round were picked from both of these proteases, the region encoding the peptide sequence was amplified using PCR, and the 96 clones with best quality PCR bands from both of them were sent for sequencing. Following decoding of the DNA sequence the amino acid sequence of the variable linker region was aligned. Each row represents an individually sequenced random region and multiple similar sequences are shown to the right where necessary (Figure 4).
The alignment showed a highly specific selection, with an apparent preference for Tyr in the P1 position for gar granzyme G, indicating a highly selective chymase activity (Figure 4). There were also fairly strict preferences in the extended specificity: aromatic amino acids or Pro in the P2 position, Val in the P3 position, Ala or Gly in P4, Ser or Met in P1´, no negatively charged residues in P2´, P3´anf P4´positions, Leu or Gly in the P2’ position and a slight preference for Val in the P3’ position (Figure 4).
The gar DDN1 was found to be a strict Leu-ase with a high preference for Leu in the P1 position, Pro in the P2 position, Leu in the P3 and Arg in both the P4 and P5 positions (Figure 4). A preference for Leu, Ser or Met in the P1´position and a lower selectivity in both the P3´ and P4´positions (Figure 4).
2.4. Phage Display Sequence Verification using Recombinant Substrates
In order to validate the phage display sequence data and to address small variations of amino acids in the aligned phages a system has been developed in our lab where by a number of sequences were analysed by the cleavage of recombinant substrates in a two-trx system (Figure 5). This is based on the expression of selected cleavable amino acid sequences derived from the phage display data placed in a linker region between two E. coli trx molecules. Various amino acids within the sequenced were changed to pinpoint the efficiency and selectivity of the two gar enzymes (Figure 5A,B). The analysis of the selectivity in the P1 position for gar granzyme G showed a 3-5 fold higher preference for Tyr over Phe in the P1 position and that the enzyme did not tolerate another aromatic amino acid, Trp, in this position (Figure 5C). In the P1´position the negatively charged amino acid Glu could replace the Ser of the consensus sequence without a drop in cleavage activity (Figure 5C). Phe could also replace the most preferred Pro in the P2 position without any change in cleavage activity and also an Arg could replace the Val in the P3´position (Figure 5D). However, introducing a larger amino acid, in this case a Trp in the P3 position instead of the Val of the consensus, resulted in an approximately 10-fold reduction in cleavage (Figure 5D).
When analysing the gar DDN1, exchanging the Leu for a Ser in the P1 position completely block cleavage, which was expected based on the phage display result (Figure 6A). However, only a minor reduction in cleavage activity was seen when changing the Pro in the P2 position with an Ala (Figure 6A). A more pronounced effect was seen when changing the Pro with an Arg in the P2 position, resulting in an approximately 3-5 fold reduction in cleavage efficiency (Figure 6A). Changing the P2 Pro with a Leu also resulted in a marked reduction in cleavage (Figure 6C second sequence). Interestingly changing the P1´ Leu for a Met enhanced cleavage quite substantially, almost tenfold (Figure 6B). This phenomenon is rarely seen as the phage display usually gives the most preferred cleavage site. However, Leu is encoded by six different codons and Met with only one, and Leu is therefore much more frequently occurring in a random sequence, which may be the reason for this relatively high number of Leu in the P1´position in the phage display, although we also found a significant number of Met in this position in the phage display sequences (Figure 4). The Arg residues in positions P4 and P5 appear to be very important for cleavage as a very marked reduction occurred by changing P4 Arg with a Gly (Figure 6B). Exchanging both Arg P4 and Arg P5 with Gly resulted in an almost total block in cleavage (Figure 6C). The positions of these two Arg residues in P4 and P5 also seemed to be very important as seen from the first sequence in Figure 6C.
2.5. Screening for Potential In Vivo Substrates
The initial screening was confined to the gar proteome with the consensus sequences (Ala-Val-Phe-Tyr-Ser-Leu) and (Arg-Arg-Leu-Pro-Leu-Leu) for gar granzyme G and DDN1, respectively. Both of these two sequences gave a number of hits in the gar proteome. However, no clear indication to what the potential function could be deduced with these potential target proteins. The AVFYSL consensus sequence obtained for gar granzyme G resulted in a number of extracellular proteins and cell surface receptors including protocadherin Fat 1 isoforms, neogenin isoforms, SID-1 transmembrane family members, D3 dopamine receptor isoforms, platelet activating receptor isoforms and the sodium dependent noradrenalin and the glycin transporter isoforms. The RRLPLL consensus sequence obtained for gar DDN1 resulted in another diverse set of potential targets protein including diphamide biosynthesis protein 2 isoforms, tyrosine-protein kinase BAZ1B, adenylate cyclase type 10, RNA-binding protein 10-like, tubulin polyglutamylase TTLL5, caspase recruitment domain-containing protein 11, E3 ubiquitin-protein ligase RNF216, sodium bicarbonate transporter-like protein 11 isoforms, zinc finger homeobox 4 isoforms, kelch-like protein-1 isoforms as well as additional targets.
2.6. Analysing Gar Tissue for Sites of Expression for the Two Gar Enzymes
To look at the tissue distribution of these two gar enzymes gar tissue was required. The upper part of the body of five juvenile gars were therefore obtained. Tissue sections originating from the region of the fish marked by a red arrow in the tissue stained with haematoxylin-eosin (Figure 7B). Typical villi of the intestinal wall were seen and a white arrowhead marks a Peyer’s patch-like structure in the intestinal wall (Figure 7C). An enlargement of this region is shown in Figure 7D. In Figure 7E,F a section of the head kidney, a hematologic organ in fishes, was analysed. In this organ a marked division in separate regions can be observed. There were regions with almost 100% red blood cells and other regions where an heterogenous mix of different cells, most likely immune cells were observed. In these regions there were only a few red blood cells, which seem to come in small clusters, possible in blood vessels (Figure 7E,F).
Tissue sections from the same region were used for immunohistochemical analyses with two rat polyclonal antibodies directed against the two gar enzymes. As seen in Figure 8A,B, a number of positive cells in the Peyer’s patch-like region was observed for gar DDN1. Only much lower intensity signals and fewer cells stained positive for the gar granzyme G antisera in the same Peyer’s patch region (Figure 8C). White arrow heads mark positive cells in Figure 8A–C. No staining was seen with a pre-immune serum (Figure 8D).
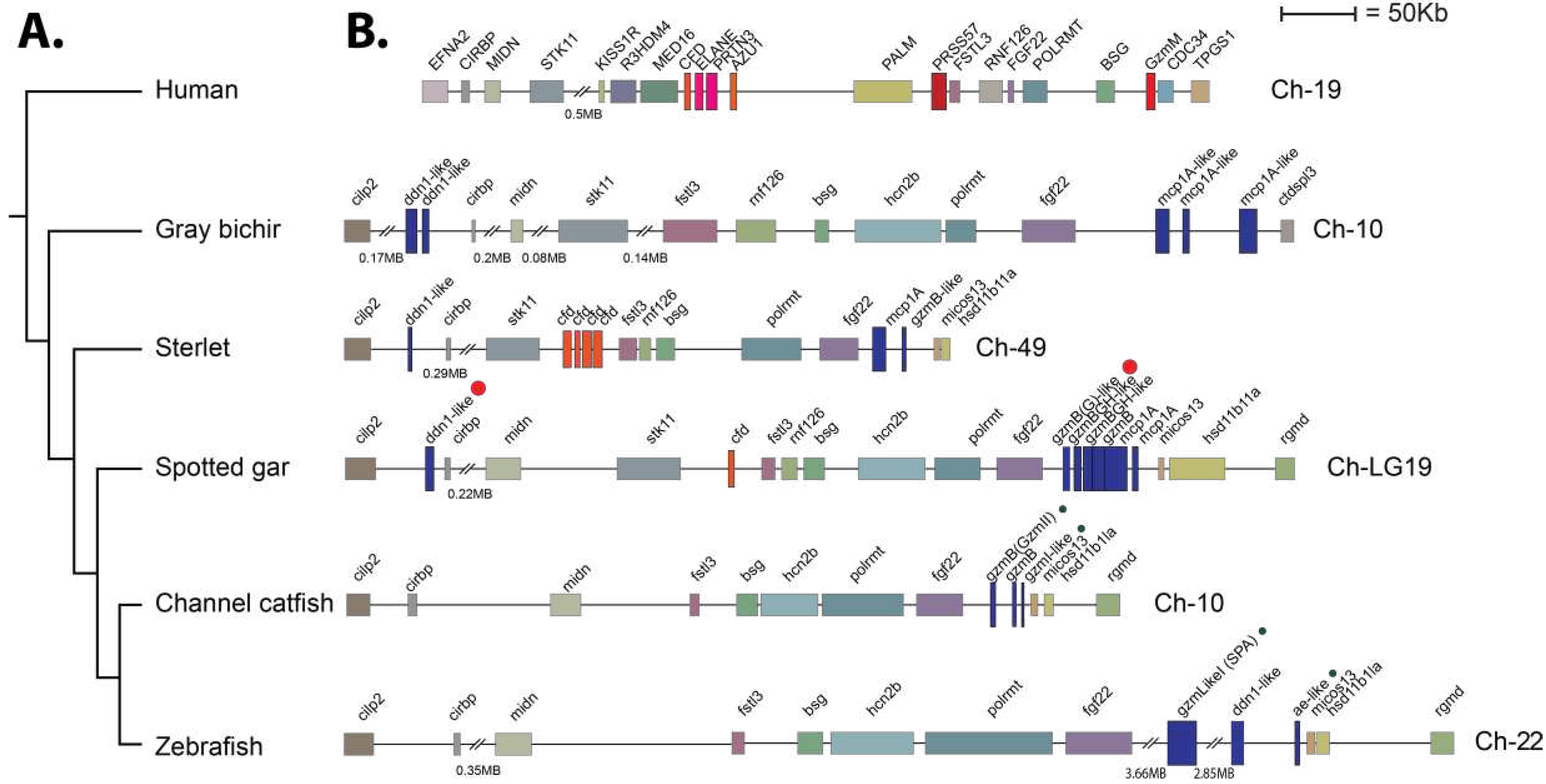
2.8. Gene Loci for Gar Hematopoietic Serine Proteases
Both of the analysed gar enzymes were located in the met-ase locus, the locus which in humans encode the majority of the neutrophil proteases, including N-elastase (ELANE), proteinase 3 (PRTN3), neutrophil serine protease 4 (NSP-4 or PRSS57) and the inactive azurocidin (AZU1), but also complement factor D (CFD) and granzyme M (GZMM) (Figure 9). Interestingly only one enzyme, the gar DDN1 was found in the region orthologous to the human locus where four serine proteases are found including CFD, ELANE, PRTN3 and AZU1 whereas five protease genes are found in a region of the locus where only one gene is found in the human locus the PRSS57 (NSP4) (Figure 9). This indicated that separate gene duplication events have resulted in an increase in serine protease numbers in this locus in fish and tetrapods. An inversion also seems to have occurred most likely in the human locus as all three early branches of fishes have the same order in this locus. We did not find any counterpart of granzyme M in the fish genome.
3. Discussion
Hematopoietic serine proteases perform a number of important functions in mammalian immunity. Granzyme B, which is an enzyme found in cytotoxic T cells and NK cells, induces apoptosis in target cells via cleavage of caspases [1,28,29]. Mast cells use the chymases to regulate blood pressure via angiotensin cleavage, to regulate types of immunity by cleaving selective sets of cytokines, to combat blood-feeding parasites such as mosquitos and ticks via the cleavage of anti-coagulant proteins and to inactivate toxins from snakes and scorpions [4,30,31,32,33,34,35,36,37,38,39]. Neutrophil proteases help these cells to migrate through tissues to reach the site of infection and cleave the pathogen-associated molecules of the infectious organisms [1]. However, which functions the corresponding proteases have among fishes and what their targets are, are almost completely unknown. Relatively solid evidence for granzyme A/K equivalents in fish are present. However, the function of mammalian granzyme A and K are have not been well elucidated [40]. However, they are the most well conserved of all hematopoietic serine proteases and are found in essentially all species from cartilaginous fish to humans but major question still remain concerning their primary targets [40]. Major similarities are also found among different tetrapods including amphibians, reptiles and birds, and mammals concerning cleavage specificities and cell origin of other hematopoietic serine proteases. However, in ray-finned fishes we know very little concerning this large family of proteases. Almost all of the non-granzyme A/K members of the hematopoietic serine proteases form their own branch in the phylogenetic tree, indicating that they have experienced an independent expansion and diversification compared to tetrapods (Figure 2). The structure of the ray-finned fish enzymes are also sufficiently different from the mammalian enzymes, so it’s not possible to use the positions of the three residues that form the S1 pocket of the tetrapod enzymes to obtain information concerning the primary specificity of the fish proteases [1,2].
What is known about hematopoietic serine proteases in fishes? Granzyme A from the catfish has previously been shown to have cytolytic activity [23]. However, caution around this interpretation maybe needed as the human enzyme, despite apoptotic activity being reported, when more physiological amounts of the enzyme were used this activity was lost [40,41,42]. A more detailed analysis of this phenomenon maybe needed before definitive conclusions can be drawn. A second catfish enzyme, catfish granzyme like-I, has recently been shown to cleave a sequence in catfish caspase 6 that corresponds to the region in human caspase 3 that is cleaved by human granzyme B [17]. However, also here no direct proof is yet available for that this cleavage occurs in vivo. The analysis has here been hampered by difficulties in producing recombinant catfish caspase 6, which is needed to enable cleavage analysis with catfish granzyme-like I. However, this enzyme is expressed by catfish NK-like cells indicating that it may be involved in apoptosis induction of cells infected with intracellular parasites. Detailed information concerning the cleavage specificities of three additional fish hematopoietic serine proteases are now available, catfish granzyme-like II, which is also expressed by the same NK-like cells and the two enzymes from the spotted gar presented here [43]. The catfish granzyme-like II is a highly specific elastase cleaving after Ala with a strong preference for several basic amino acids, primarily Arg, just N-terminally of the cleavage site [43]. To our knowledge no elastase is expressed by human NK cells, which is why the function of this enzyme may turn out to be a fish-specific immune function. The two gar enzymes are, based on their tissue location and cell shape, expressed in what appears to be NK-like cells or cytotoxic T cells. However, it is not yet clear in what cell these two proteases are expressed due to the lack of suitable reagents to determine the actual cellular origin in gar. Their specificities currently provide little clues to their function. Both enzymes are highly specific. One is a Tyr-specific chymase and the second enzyme a Leu- specific Leu-ase, similar to the mast cell chymases of rabbit and guinea pig, but both appears to be expressed by a different cell type than these latter proteases (Figure 4). Neither chymases nor Leu-ases seem to be expressed by NK or cytotoxic T cells in mammals. Screening of the total proteome with the consensus sequences for these two gar proteases did not result in any clear candidates for their potential immune functions. A relatively broad panel of different potential targets were obtained for both enzymes with no clear identified immune candidate. A more in-depth analysis involving direct cleavage of a cell extract and possibly 2-D gel analysis of cleavage products could be informative. An alternative explanation could be that these enzymes are not aimed at cleaving host proteins but instead pathogen derived proteins such as bacterial toxins.
The analysis of the intestinal region and the head kidney resulted in an interesting finding concerning the immune organs in this branch of fish evolution. Clear indications for Peyer’s patch-like structures in the intestinal region were observed. Here, the expression of the two gar enzymes was seen, which did not occur in other regions of the fish, including the head kidney, indicating that the proteases are expressed in cells that appear at these sites of immune activity in the intestinal region and not during early expansion of cells in the head kidney.
A clear separation of regions where red blood cells is formed was seen from the gar head kidney sections and other regions within the same organ where most likely the majority of other hematopoietic cells are residing was also an interesting finding that differs from the organization of human bone marrow. The clusters of red blood cells in the regions of other hematopoietic cells may be due to a rich blood supply to support rapidly expanding cell populations in need for oxygen and nutrient supply.
The analyses of the genomic organization of the two gar enzymes also gave insight into the evolution of these proteases. Both of the gar enzymes were located in the met-ase locus (Figure 9). This locus is known in mammals to encode the majority of neutrophil proteases including N-elastase, proteinase 3, azurocidin and neutrophil protease 4, but also the NK-cell expressed granzyme M and complement factor D. We also observed that independent amplifications had occurred in mammals compared to gar and sterlet sturgeon (Figure 9). In mammals it is most likely complement factor D that is the origin of the majority of the neutrophil proteases, including N-elastase, proteinase 3 and azurocidin, whereas in the gar it is most likely the PRSS57, encoding NSP4, which has been duplicated forming a small subfamily of six different proteases in the gar, two in sturgeon, and three in the bichir (Figure 9). These three fish species are representatives of non-teleost branches of the fish evolutionary tree, indicating that the gene duplications of PRSS57 are early expansions in the met-ase locus in fish [44]. In this regard, the relation and differences between the teleost catfish and zebrafish, and the non-teleost gar, sterlet, and bichir is also interesting. The one or two DDN-like enzymes in one end of the locus found in all three non-teleost fish branches including gar, sterlet and bichir have been lost in both catfish and zebrafish (Figure 9). The three genes that most likely originate from gene duplications of PRSS57 are still present in the teleosts catfish and zebrafish (Figure 9). Both the catfish granzyme-like II and gar granzyme G are located as the first gene of these PRSS57 duplicated genes, however they have very different cleavage specificities, where catfish granzyme-like II is an Ala-specific elastase and the gar enzyme is a Tyr- specific chymase. Currently there is no information concerning the other catfish and gar enzymes in this cluster except for catfish granzyme-like I, which is a met-ase. There are striking similarities between catfish and zebrafish as both catfish granzyme-like I and zebrafish AE-like, as well as also catfish granzyme-like II and zebrafish SPA have very similar specificities [43]. Future studies will hopefully be able to distinguish the similarities and differences in specificities between these enzymes in non-teleost compared to teleost fishes. Such information may guide us into key primary immune-related targets for these enzymes during fish evolution and how this relates to their mammalian counterparts.
4. Materials and Methods
4.1. Phylogenetic Analyses
The phylogenetic analyses aimed at determining the relationship between the two gar enzymes and other hematopoietic serine proteases from fish were performed essentially as described in a previous publication using the same strategy and sequences [2]. Sequences relating to the two gar enzymes were systematically uncovered by BLASTp searching of all animal NCBI databases. The mature two gar enzymes were used as the query sequence and all novel derived sequences were analysed using the multiple alignment programme MAFFT with G-INS-i strategy and default parameters to determine whether they belonged to the serine protease family. To visualise the relationship between the two gar enzymes with those from other species, a phylogenetic tree using the Bayesian interference of phylogeny algorithm with posterior probabilities in the MRBAYES program was constructed and viewed in FigTree (v1.4). The amino acid sequences for mature proteins of serine proteases branching with the two gar enzymes were aligned using MAFFT.
4.2. Production and Purification of Recombinant Gar Gzm-G and Gar DDN1
The gar granzyme sequences (GenBank accession numbers: Gar Gzm-G-like (XP_015220993) and Gar DDN1-like (XP_006640095)) were designed and ordered from GenScript (Piscataway, NJ, USA). The synthesized construct was cloned in the pU57 cloning vector, containing EcoRI and XhoI sites. The gar granzyme sequences were subsequently transferred to a pCEP-Pu2 vector, used for expression in mammalian cells [27]. The enzymes were produced as inactive recombinant proteins, with an N-terminal His6-tag followed by an enterokinase (EK) site. HEK 293 cells were grown to 70% confluency in a 25 cm3 tissue culture flask (BD VWR) with Dulbecco’s Modified Eagles Medium (DMEM) (GlutaMAX, Invitrogen, Carlsbad, CA, USA) supplemented with 5% fetal bovine serum (FBS) and 50 µg/ml gentamicin. Following transfection with lipofectamine (Invitrogen, Carlsbad, CA, USA), using approximately 25 µg of the gar constructs in pCEP-Pu2, puromycin was added to the DMEM (0.5 µg/mL) to select for cells which had taken up the DNA. Heparin (5 µg/ml) was added to the culture medium to enhance recovery of secreted protein. Cells were expanded and conditioned media collected.
After collecting sufficient amount of media, in general around 750 mL, the conditioned media was filtered (Munktell 00H 150 mm, Falun, Sweden) and 500 µL nickel nitrilotriacetic acid (Ni-NTA) beads were added (Qiagen, Hilden, Germany) to purify the recombinant enzymes. The media with Ni-NTA beads were rotated for 45 mins at 4 °C. Subsequently, the Ni-NTA beads were collected by centrifugation and transferred to a column containing a glass filter (Sartorius, Goettingen, Germany). To remove unbound protein the columns were washed with PBS tween 0.05 % + 10 mM imidazole + 1 M NaCl. Following this wash the recombinant protein was eluted in PBS tween 0.05 % + 100 mM imidazole fractions. The first fraction volume was half the Ni-NTA bead width (200 µL) and further fractions eluted with a full bead width (400 µL). Individual fractions were run on SDS-PAGE gel, their concentrations estimated from a bovine serum albumin standard (BSA) and the most concentrated were pooled and kept at 4 °C.
4.3. Activation of Recombinant Gar Granzymes
To activate the enzymes the initial concentrations of the gar enzymes were first determined by SDS-PAGE and the level of EK (Roche, Mannheim, Germany) adjusted for activation of the enzymes. To 70 µL of the eluted recombinant enzyme we added 1 µL EK and incubated for 3 hrs at 37 °C to activate the enzyme. The activated fractions were stored at 4 °C until use.
4.4. Substrate Phage Display
A T7 phage library containing 5 x 107 variants was used for the phage display analysis to determine the extended cleavage specificity of the two gar enzymes. In this library each phage displays a unique nine amino acid sequence. The nine amino acid region had been inserted into the C-terminal of the capsid 10 protein, followed by a His6-tag. 125 µL Ni-NTA agarose beads via their His6-tags for 1 hr at 4°C with gentle rotation (Qiagen, Hilden, Germany). To these 125 ul Ni-NTA we estimate that approximately 109 plaque forming units (pfu) were bound. Unbound phages were removed by washing ten times with PBS tween 0.05 % + 1 M NaCl, followed by two washes with PBS. The beads were re-suspended in 375 µL PBS and approximately 250 ng of the recombinant gar enzyme was added. This reaction was incubated overnight or for approximately 16 hours at 37 °C with gentle rotation. The enzyme cleavage results in that susceptible phages detach from the Ni-NTA beads. From the supernatant released phages was recovered after centrifugation. Thirty µL was used in a plaque assay to determine the number of released phages. Briefly, ten-fold serial dilutions were made, mixed with (E.coli) BLT5615 (for propagation and visualization of plaques on a bacterial lawn) and plated on LA-Amp (50 µg/mL) plates, incubated for 2.5 hrs at 37 °C and then counted. The remaining supernatant was added to 10 ml BLT5615 bacteria (OD600 0.5) and the culture was incubated at 37 °C for approximately 75 mins until the culture had cleared by phage lysis for phage expansion. From this, 1.5 mL was centrifuged to remove bacterial debris and 800 µL transferred to a microcentrifuge tube containing 100 µL PBS and 100 µL 5M NaCl. This solution was bound to 125 µL Ni-NTA beads directly after centrifugation to start the next selection cycle. The complete process was repeated a further 6 times, constituting 7 selection rounds. Individual plaques were isolated from the final selection round in 100 µL phage buffer before vortexing for 30 mins and stored at 4 °C. The random nine amino acid regions contained in these phages were amplified by PCR (T7Select primers, Novagen, Sacramento, CA, USA) and sequenced by Eurofins (Sequencing centre, Cologne, Germany). The resulting sequences were translated using CLC viewer and aligned using Adobe Illustrator. A parallel control reaction without enzyme (only PBS) was also run under the same conditions and plaque numbers were compared to the enzyme sample.
4.5. Phage Display Sequence Verification using a Two-Thioredoxin (trx) Approach
In order to verify the phage display data, a recombinant trx system was used. This system has been developed in our laboratory and used multiple times to study other proteases with great success. Here, the random nine amino acid cleaved region determined from the phage display was introduced between two adjacent E. coli trx proteins. Originally, a pET21 vector containing a single trx protein was modified to contain a second trx with BamHI and SalI sites in the intervening region. Here the random region was synthesised as oligonucleotides (Sigma, St Louis, USA). These oligos generated overhand suitable for ligation between BamHI and SalI restriction sites of the vector. This resulted in a vector containing a first trx followed by the random cleavable region then a second trx with His6-tag (facilitating purification).
To produce the recombinant protein the construct was expressed in E.coli Rosetta gami (Novagen, Sacramento, CA, USA). Ten ml of an overnight culture was added to 90 mL LB+Amp (50 µg/mL) and 500 µL 20 % glucose. After approximately 1 hr (reaching OD600 0.5), 100 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) was added and the culture placed on a shaker (moderate shaking) at 37 °C for 3 hrs. The bacterial cells of the culture were pelleted by centrifugation at 10,000 rpm for 3 mins and supernatant discarded. The pellet containing the bacteria with the expressed recombinant protein was washed in 10 mL PBS tween 0.05 %, centrifuged and pelleted again followed by resuspension in 1/100th starting volume (i.e., 1 mL) PBS. To obtain the intracellularly expressed protein, the resuspended pellet was sonicated for 6 x 30 secs on ice. The supernatant was transferred to a new microcentrifuge tube after centrifugation at 10,000 rpm for 10 mins at 4 °C. To purify the protein, 125 µL Ni-NTA beads were added and incubated for 45 mins at 4 °C with gentle agitation (Qiagen, Hilden Germany). The solution with Ni-NTA beads was then transferred to a 2 mL column (Terumo, Leuven, Belgium) containing a glass filter (Sartorius, Goettingen, Germany). The column was then first washed with 3 x 2 mL and 2 x 1 mL PBS tween 0.05 % + 10 mM imidazole. To elute the protein, fractions were collected after passing through PBS tween 0.05 % + 100 mM imidazole. The first fraction volume was only half the Ni-NTA bead volume (75 µL) and further fractions eluted with a full bead volume (150 µL). Individual fractions were run on SDS-PAGE gel and their concentrations estimated from a BSA standard (and Bradford assay (Bio-rad, CA, USA)). The most concentrated fractions were then pooled and kept at 4 °C. until use in the assay
For cleavage analysis, approximately 250 ng of recombinant gar enzyme was added to 20 µg of the pooled 2-trx protein (containing different sequences based on the phage display data) and aliquots of 5 µg removed after 0, 15, 45 and 150 mins after enzyme addition. The reactions were run at room temperature and the time point aliquots analysed on SDS-PAGE gel under denaturing conditions using pre-cast 4-12 % Bis-Tris gels (Invitrogen, Carlsbad, CA, USA) and 1x MES buffer (Invitrogen, Carlsbad, CA, USA). Gels were stained with Coomassie colloidal solution to visualize the protein bands [45].
4.6. Screening for Potential In Vivo Substrates
The derived consensus sequences for the two gar proteases (Ala-Val-Phe-Tyr-Ser-Leu) and (Arg-Arg-Leu-Pro-Leu-Leu), respectively were used in a screening for potential in vivo substrates using a standard protein Basic Local Alignment Search Tool (BLASTp).
4.7. Spotted Gar Husbandry and Tissue Sampling
Spotted gars were raised to juvenile stages from fertilized eggs obtained from hormone-induce spawns of wild-caught broodstock from bayous near Thibodaux, Louisiana. Five unsexed individuals were euthanized at the age of 4 months (average total length: 15.9cm) using MS-222 (250–500 mg/liter water) and fixed overnight in 4% Paraformaldehype/PBS. All animal work was approved by the Michigan State University Institutional Animal Care and Use Committee (PROTO202200298).
4.8. Immunohistochemistry
Approximately 100 µg of the recombinant enzymes were mixed with Freund’s complete adjuvant and injected in one male 20-week Sprague Dawley rat for each enzyme. Three weeks after the first injection the rats were boosted with 20 µg of the recombinant protein in Freunds incomplete adjuvant. After 6 weeks another booster dose was given with 20 µg of recombinant protein in Freund’s incomplete adjuvant. Two weeks after the last booster injection the rats were tail blead to obtain approximately 1 mL of blood. The blood was left overnight to coagulate and thereafter spun at 3000 rpm in an Eppendorf centrifuge and the serum was transferred to a new tube and centrifuged a second time to remove remaining red blood cells. The serum was then aliquoted and kept at -80oC until use in immunohistochemical staining.
The fish were preserved in a formalin solution for several days. Following preservation, the tissue underwent decalcification using 0.5 M ethylenediaminetetraacetic acid at pH 8.0 (Sigma Aldrich, Saint Louis, Missouri, USA) for a duration of one month, during which the solution was replaced on a weekly basis. Subsequently, the tissue was immersed in a 30% sucrose solution in 1x PBS for 72 hours before sectioning. A transverse incision was then performed at the tissue’s midpoint, extracting a section of approximately 1 cm² around the intestinal cavity with the use of a scalpel. The dissected tissue was then embedded in optical cutting compound (OCT, VWR, Radnor, Pennsylvania, USA). Sections of 15 µm thickness were obtained using a cryostat and mounted onto SuperFrost slides (VWR, Radnor, Pennsylvania, USA).
For immunostaining, the tissue sections were initially blocked for a period of 1 hour at room temperature with 1x protein-free blocking buffer (Pierce solution, Thermo Fisher, Waltham, Massachusetts, USA) containing 0.3 % Triton X-100 in 0.1 M PBS. Staining with the rat anti gar-DDN1 and anti-GzmG, (1:100) primary antibody was performed overnight at 4oC. After the primary antibody incubation, the tissue sections underwent triple washing with a PBS solution. Subsequently, the sections were incubated with a donkey anti-rat secondary antibody conjugated to Alexa 488 (1:500, Thermo Fisher, Waltham, Massachusetts, USA) for 2 hours at room temperature. Nuclei were visualized using DAPI (4′,6-diamidino-2-phenylindole)(Thermo Fisher, Waltham, Massachusetts, USA) and mounted using Fluoromount G (Thermo Fisher, Waltham, Massachusetts, USA). Pictures were taken in an epifluorescence Nikon microscope (Nikon Eclipse 90i Upright).
Acknowledgments
This study was supported by grants from the Knut and Alice Wallenberg Foundation (Grant number KAW2017.0022). We thank Allyse Ferrara (Nicholls State University) for providing gar spawns and Jamily Lorena (Michigan State University) for help with gar sampling. Gar work in the Braasch Lab is supported by the National Science Foundation (award # 2029216).
References
- Hellman L, Thorpe M. Granule proteases of hematopoietic cells, a family of versatile inflammatory mediators - an update on their cleavage specificity, in vivo substrates, and evolution. Biol Chem. 2014;395(1):15-49. [CrossRef]
- Akula S, Thorpe M, Boinapally V, Hellman L. Granule Associated Serine Proteases of Hematopoietic Cells - An Analysis of Their Appearance and Diversification during Vertebrate Evolution. PLoS One. 2015;10(11):e0143091. [CrossRef]
- Fu Z, Thorpe M, Akula S, Chahal G, Hellman L. Extended cleavage specificity of human neutrophil elastase, human proteinase 3 and their distant orthologue clawed frog PR3 - three elastases with similar primary but different extended specificities and stability. Frontiers in immunology. 2018;9(Article 2387):1-19. [CrossRef]
- Fu Z, Thorpe M, Alemayehu R, Roy A, Kervinen J, de Garavilla L, et al. Highly Selective Cleavage of Cytokines and Chemokines by the Human Mast Cell Chymase and Neutrophil Cathepsin G. J Immunol. 2017;198(4):1474-83. [CrossRef]
- Akula S, Fu Z, Wernersson S, Hellman L. The Evolutionary History of the Chymase Locus -a Locus Encoding Several of the Major Hematopoietic Serine Proteases. International journal of molecular sciences. 2021;22(20). [CrossRef]
- Reimer JM, Samollow PB, Hellman L. High degree of conservation of the multigene tryptase locus over the past 150-200 million years of mammalian evolution. Immunogenetics. 2010;62(6):369-82. [CrossRef]
- Andersson MK, Enoksson M, Gallwitz M, Hellman L. The extended substrate specificity of the human mast cell chymase reveals a serine protease with well-defined substrate recognition profile. Int Immunol. 2009;21(1):95-104. [CrossRef]
- Andersson MK, Karlson U, Hellman L. The extended cleavage specificity of the rodent beta-chymases rMCP-1 and mMCP-4 reveal major functional similarities to the human mast cell chymase. Mol Immunol. 2008;45(3):766-75. [CrossRef]
- Andersson MK, Pemberton AD, Miller HR, Hellman L. Extended cleavage specificity of mMCP-1, the major mucosal mast cell protease in mouse-High specificity indicates high substrate selectivity. Mol Immunol. 2008;45(9):2548-58. [CrossRef]
- Reimer JM, Enoksson M, Samollow PB, Hellman L. Extended substrate specificity of opossum chymase-Implications for the origin of mast cell chymases. Mol Immunol. 2008;45(7):2116-25. [CrossRef]
- Fu Z, Akula S, Qiao C, Ryu J, Chahal G, de Garavilla L, et al. Duodenases are a small subfamily of ruminant intestinal serine proteases that have undergone a remarkable diversification in cleavage specificity. PLoS One. 2021;16(5):e0252624. [CrossRef]
- Fu Z, Akula S, Thorpe M, Chahal G, de Garavilla L, Kervinen J, et al. Extended cleavage specificity of sheep mast cell protease-2: A classical chymase with preference to aromatic P1 substrate residues. Developmental and comparative immunology. 2019;92:160-9. [CrossRef]
- Fu Z, Akula S, Thorpe M, Hellman L. Extended cleavage specificities of two mast cell chymase-related proteases and one granzyme B-like protease from the platypus, a monotreme. International journal of molecular sciences. 2020;21(1). [CrossRef]
- Fu Z, Thorpe M, Akula S, Hellman L. Asp-ase Activity of the Opossum Granzyme B Supports the Role of Granzyme B as Part of Anti-Viral Immunity Already during Early Mammalian Evolution. PLoS One. 2016;11(5):e0154886. [CrossRef]
- Fu Z, Thorpe M, Hellman L. rMCP-2, the Major Rat Mucosal Mast Cell Protease, an Analysis of Its Extended Cleavage Specificity and Its Potential Role in Regulating Intestinal Permeability by the Cleavage of Cell Adhesion and Junction Proteins. PLoS One. 2015;10(6):e0131720. [CrossRef]
- Wernersson S, Reimer JM, Poorafshar M, Karlson U, Wermenstam N, Bengten E, et al. Granzyme-like sequences in bony fish shed light on the emergence of hematopoietic serine proteases during vertebrate evolution. Developmental and comparative immunology. 2006;30(10):901-18. [CrossRef]
- Thorpe M, Akula S, Hellman L. Channel catfish granzyme-like I is a highly specific serine protease with metase activity that is expressed by fish NK-like cells. Developmental and comparative immunology. 2016;63:84-95. [CrossRef]
- Fu Z, Akula S, Olsson AK, Hellman L. Chicken cathepsin G-like - A highly specific serine protease with a peculiar tryptase specificity expressed by chicken thrombocytes. Developmental and comparative immunology. 2021;129:104337. [CrossRef]
- Ryu J, Fu Z, Akula S, Olsson AK, Hellman L. Extended cleavage specificity of a Chinese alligator granzyme B homologue, a strict Glu-ase in contrast to the mammalian Asp-ases. Developmental and comparative immunology. 2021;128:104324. [CrossRef]
- Praveen K, Evans DL, Jaso-Friedmann L. Evidence for the existence of granzyme-like serine proteases in teleost cytotoxic cells. J Mol Evol. 2004;58(4):449-59. [CrossRef]
- Praveen K, Leary JH, 3rd, Evans DL, Jaso-Friedmann L. Molecular characterization and expression of a granzyme of an ectothermic vertebrate with chymase-like activity expressed in the cytotoxic cells of Nile tilapia (Oreochromis niloticus). Immunogenetics. 2006;58(1):41-55. [CrossRef]
- Praveen K, Leary JH, 3rd, Evans DL, Jaso-Friedmann L. Nonspecific cytotoxic cells of teleosts are armed with multiple granzymes and other components of the granule exocytosis pathway. Mol Immunol. 2006;43(8):1152-62. [CrossRef]
- Chaves-Pozo E, Valero Y, Lozano MT, Rodriguez-Cerezo P, Miao L, Campo V, et al. Fish Granzyme A Shows a Greater Role Than Granzyme B in Fish Innate Cell-Mediated Cytotoxicity. Frontiers in immunology. 2019;10:2579. [CrossRef]
- Braasch I, Gehrke AR, Smith JJ, Kawasaki K, Manousaki T, Pasquier J, et al. The spotted gar genome illuminates vertebrate evolution and facilitates human-teleost comparisons. Nat Genet. 2016;48(4):427-37. [CrossRef]
- Neurath H. The versatility of proteolytic enzymes. Journal of cellular biochemistry. 1986;32(1):35-49. [CrossRef]
- Perona JJ, Craik CS. Structural basis of substrate specificity in the serine proteases. Protein science: a publication of the Protein Society. 1995;4(3):337-60. [CrossRef]
- Vernersson M, Ledin A, Johansson J, Hellman L. Generation of therapeutic antibody responses against IgE through vaccination. Faseb J. 2002;16(8):875-7. [CrossRef]
- Rousalova I, Krepela E. Granzyme B-induced apoptosis in cancer cells and its regulation (review). Int J Oncol. 2010;37(6):1361-78. [CrossRef]
- Ewen CL, Kane KP, Bleackley RC. A quarter century of granzymes. Cell Death Differ. 2012;19(1):28-35. [CrossRef]
- Caughey GH, Raymond WW, Wolters PJ. Angiotensin II generation by mast cell alpha- and beta-chymases. Biochimica et biophysica acta. 2000;1480(1-2):245-57. [CrossRef]
- Berglund P, Akula S, Fu Z, Thorpe M, Hellman L. Extended Cleavage Specificity of the Rat Vascular Chymase, a Potential Blood Pressure Regulating Enzyme Expressed by Rat Vascular Smooth Muscle Cells. International journal of molecular sciences. 2020;21(22). [CrossRef]
- Takai S, Sakaguchi M, Jin D, Yamada M, Kirimura K, Miyazaki M. Different angiotensin II-forming pathways in human and rat vascular tissues. Clin Chim Acta. 2001;305(1-2):191-5. [CrossRef]
- Fu Z, Akula S, Thorpe M, Hellman L. Highly Selective Cleavage of TH2-Promoting Cytokines by the Human and the Mouse Mast Cell Tryptases, Indicating a Potent Negative Feedback Loop on TH2 Immunity. International journal of molecular sciences. 2019;20(20). [CrossRef]
- Fu Z, Akula S, Olsson AK, Kervinen J, Hellman L. Mast Cells and Basophils in the Defense against Ectoparasites: Efficient Degradation of Parasite Anticoagulants by the Connective Tissue Mast Cell Chymases. International journal of molecular sciences. 2021;22(23). [CrossRef]
- Metz M, Piliponsky AM, Chen CC, Lammel V, Abrink M, Pejler G, et al. Mast cells can enhance resistance to snake and honeybee venoms. Science. 2006;313(5786):526-30. [CrossRef]
- Akahoshi M, Song CH, Piliponsky AM, Metz M, Guzzetta A, Abrink M, et al. Mast cell chymase reduces the toxicity of Gila monster venom, scorpion venom, and vasoactive intestinal polypeptide in mice. J Clin Invest. 2011;121(10):4180-91. [CrossRef]
- Galli SJ, Metz M, Starkl P, Marichal T, Tsai M. Mast cells and IgE in defense against lethality of venoms: Possible "benefit" of allergy[]. Allergo J Int. 2020;29(2):46-62. [CrossRef]
- Starkl P, Gaudenzio N, Marichal T, Reber LL, Sibilano R, Watzenboeck ML, et al. IgE antibodies increase honeybee venom responsiveness and detoxification efficiency of mast cells. Allergy. 2022;77(2):499-512. [CrossRef]
- Anderson E, Stavenhagen K, Kolarich D, Sommerhoff CP, Maurer M, Metz M. Human Mast Cell Tryptase Is a Potential Treatment for Snakebite Envenoming Across Multiple Snake Species. Frontiers in immunology. 2018;9:1532. [CrossRef]
- Aybay E, Ryu J, Fu Z, Akula S, Enriquez EM, Hallgren J, et al. Extended cleavage specificities of human granzymes A and K, two closely related enzymes with conserved but still poorly defined functions in T and NK cell-mediated immunity. Frontiers in immunology. 2023;14:1211295. [CrossRef]
- Lieberman, J. Granzyme A activates another way to die. Immunol Rev. 2010;235(1):93-104. [CrossRef]
- Metkar SS, Menaa C, Pardo J, Wang B, Wallich R, Freudenberg M, et al. Human and mouse granzyme A induce a proinflammatory cytokine response. Immunity. 2008;29(5):720-33. [CrossRef]
- Thorpe, M., Akula S., Fu Z., Hellman L. The extended cleavage specificity of channel catfish granzyme-like II, a highly specific elastase, expressed by natural killer-like cells. IJMS. 2024;25. [CrossRef]
- Bi X, Wang K, Yang L, Pan H, Jiang H, Wei Q, et al. Tracing the genetic footprints of vertebrate landing in non-teleost ray-finned fishes. Cell. 2021;184(5):1377-91 e14. [CrossRef]
- Neuhoff V, Arold N, Taube D, Ehrhardt W. Improved staining of proteins in polyacrylamide gels including isoelectric focusing gels with clear background at nanogram sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis. 1988;9(6):255-62. [CrossRef]
- Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun. 1967;27(2):157-62. [CrossRef]
- Aybay E, Elkhalifa M, Akula S, Wernersson S, Hellman L. Two granzyme A/K homologs in Zebra mbuna have different specificities, one classical tryptase and one with chymase activity. Developmental and comparative immunology. 2023;148:104920. [CrossRef]
- Zhongwei Y, Akula S, Fu Z, de Garavilla L, Kervinen J, Thorpe M, et al. Extended Cleavage Specificities of Rabbit and Guinea Pig Mast Cell Chymases: Two Highly Specific Leu-Ases. International journal of molecular sciences. 2019;20(24). [CrossRef]
Figure 1.
Nomenclature of the amino acids surrounding the cleavage site and the amino acids forming the active site pocket. In panel (A), the nomenclature of the amino acids surrounding the cleaved peptide bond is shown. The amino acids N-terminal from the cleaved bond are termed P1 (where cleavage occurs, depicted by scissors), P2, P3 etc. Amino acids C-terminal of the cleaved bond are termed P1’ (adjacent to P1), P2’, P3’ etc. The corresponding interacting sub-sites in the enzyme are denoted with S. In panel (B), the three amino acids forming the active site pocket (S1 pocket) are shown. These three residues correspond to positions 189, 216 and 226 in bovine pancreatic chymotrypsinogen have been found to determining the primary specificity of the enzyme as either chymotrypsin-, trypsin- or elastase-like specificity [46]. The preferred amino acids in the P1 position of the corresponding substrates are illustrated in green, yellow and blue, respectively.
Figure 1.
Nomenclature of the amino acids surrounding the cleavage site and the amino acids forming the active site pocket. In panel (A), the nomenclature of the amino acids surrounding the cleaved peptide bond is shown. The amino acids N-terminal from the cleaved bond are termed P1 (where cleavage occurs, depicted by scissors), P2, P3 etc. Amino acids C-terminal of the cleaved bond are termed P1’ (adjacent to P1), P2’, P3’ etc. The corresponding interacting sub-sites in the enzyme are denoted with S. In panel (B), the three amino acids forming the active site pocket (S1 pocket) are shown. These three residues correspond to positions 189, 216 and 226 in bovine pancreatic chymotrypsinogen have been found to determining the primary specificity of the enzyme as either chymotrypsin-, trypsin- or elastase-like specificity [46]. The preferred amino acids in the P1 position of the corresponding substrates are illustrated in green, yellow and blue, respectively.
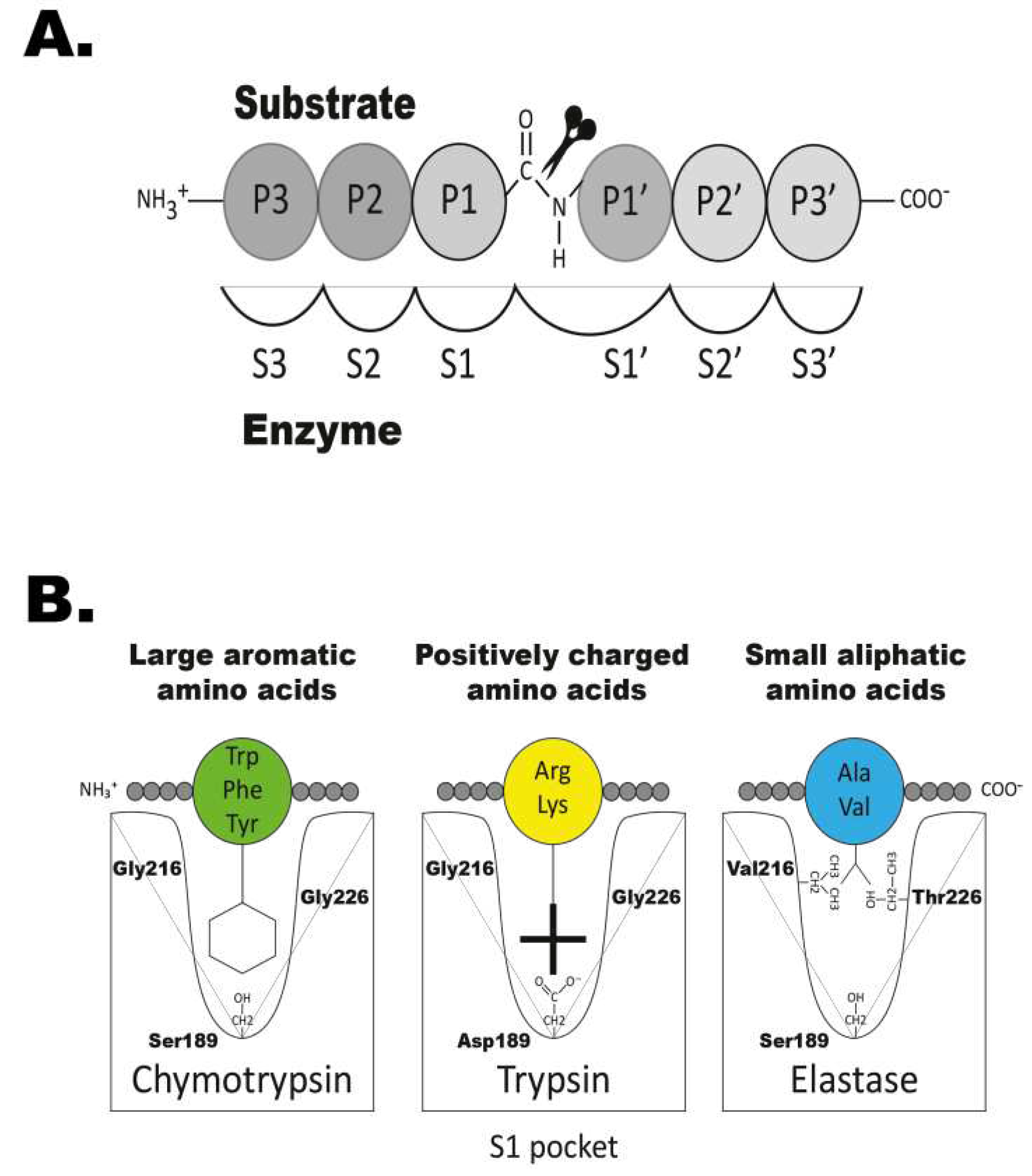
Figure 2.
Phylogenetic relationship between gar granzyme G, gar DDN1 and other hematopoietic serine proteases. All sequences were run in the multiple alignment programme MAFFT to verify they belonged to the serine protease family. The tree was constructed using MRBAYES with a Bayesian interference of phylogeny algorithm (with posterior probabilities), opened with FigTree (v1.4) and annotated in Adobe illustrator (CS5). Panel (A) shows the entire analysis involving a total of 368 vertebrate serine protease sequences. Panel (B) shows an enlargement of the branch of the major tree where the majority of the fish proteases are found, except the granzyme A/K related fish proteases. The proteases of particular interest for this study are marked with red arrows. All genes for proteins that were produced as recombinant proteins are marked with red stars.
Figure 2.
Phylogenetic relationship between gar granzyme G, gar DDN1 and other hematopoietic serine proteases. All sequences were run in the multiple alignment programme MAFFT to verify they belonged to the serine protease family. The tree was constructed using MRBAYES with a Bayesian interference of phylogeny algorithm (with posterior probabilities), opened with FigTree (v1.4) and annotated in Adobe illustrator (CS5). Panel (A) shows the entire analysis involving a total of 368 vertebrate serine protease sequences. Panel (B) shows an enlargement of the branch of the major tree where the majority of the fish proteases are found, except the granzyme A/K related fish proteases. The proteases of particular interest for this study are marked with red arrows. All genes for proteins that were produced as recombinant proteins are marked with red stars.

Figure 3.
Recombinant gar granzyme G and gar DDN1. The enzymes were produced as an inactive protein (left lane) in HEK293 cells with a N-terminal His6-tag and enterokinase (EK) site, facilitating purification and activation, respectively. Addition of EK cleaves the N-terminal sites resulting in an active enzyme and a subsequent drop in size (right lane). The enzymes were run on a 4-12 % pre-cast SDS-PAGE gel (Invitrogen, Carlsbad, USA) and stained with colloidal Coomassie brilliant blue.
Figure 3.
Recombinant gar granzyme G and gar DDN1. The enzymes were produced as an inactive protein (left lane) in HEK293 cells with a N-terminal His6-tag and enterokinase (EK) site, facilitating purification and activation, respectively. Addition of EK cleaves the N-terminal sites resulting in an active enzyme and a subsequent drop in size (right lane). The enzymes were run on a 4-12 % pre-cast SDS-PAGE gel (Invitrogen, Carlsbad, USA) and stained with colloidal Coomassie brilliant blue.
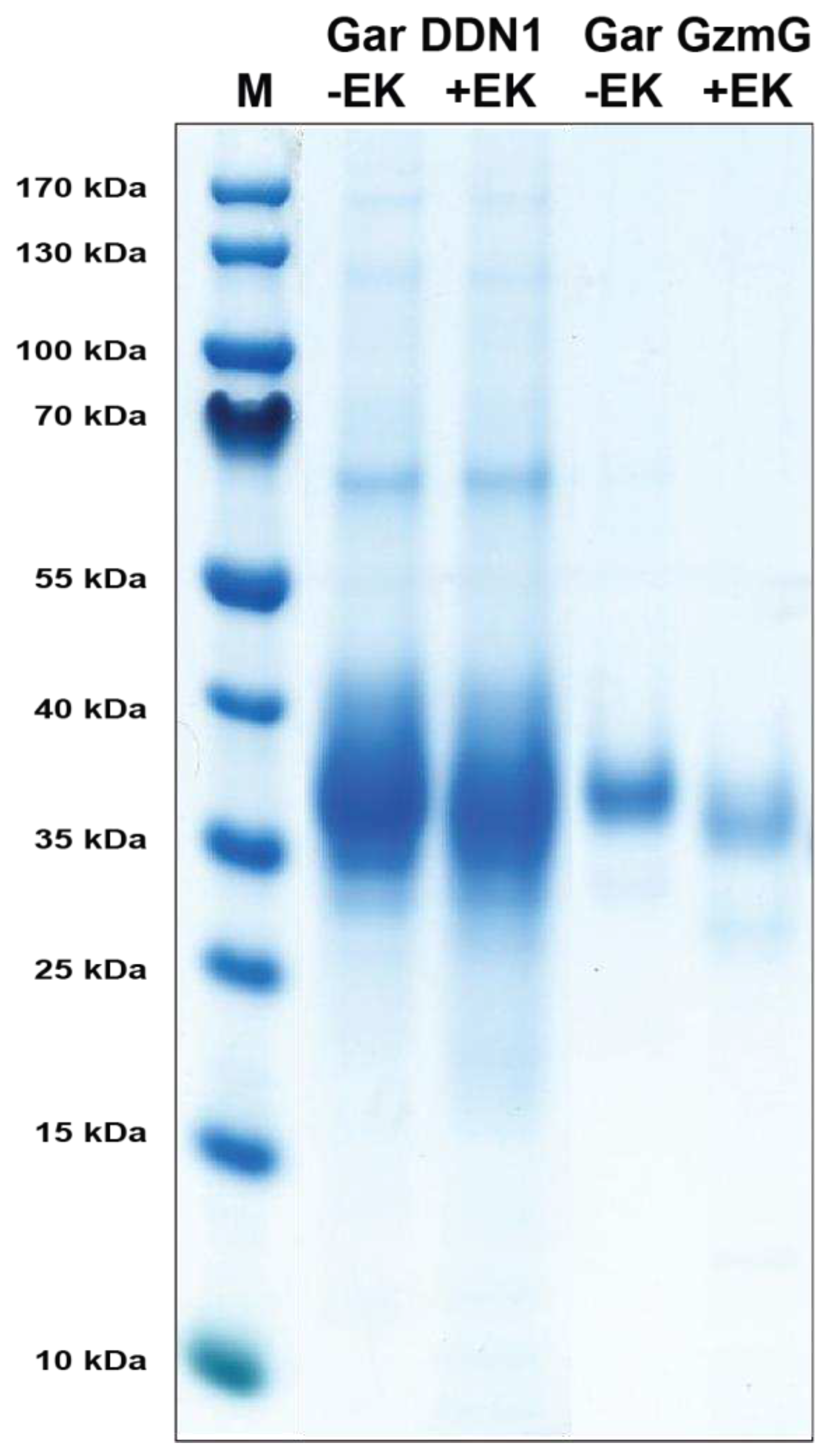
Figure 4.
Substrate phage display sequences. A T7 phage library was subject to seven selection rounds with the gar granzyme G and seven rounds with gar DDN1. After the final round, phage plaques were isolated and their random region amplified by PCR and sequenced. The sequences were decoded and aligned. For comparison two previous phage display analyses with specificities similar to what was observed for gar granzyme G and gar DDN1 are included. Gar granzyme G, which has a P1 preference for Tyr shows similarities to a recently analysed Zebra mbuna enzyme named granzyme A2 [47]. The gar DDN1 shows some similarities to rabbit and guinea pig leu-ases, as they share the same P1 preference [48].
Figure 4.
Substrate phage display sequences. A T7 phage library was subject to seven selection rounds with the gar granzyme G and seven rounds with gar DDN1. After the final round, phage plaques were isolated and their random region amplified by PCR and sequenced. The sequences were decoded and aligned. For comparison two previous phage display analyses with specificities similar to what was observed for gar granzyme G and gar DDN1 are included. Gar granzyme G, which has a P1 preference for Tyr shows similarities to a recently analysed Zebra mbuna enzyme named granzyme A2 [47]. The gar DDN1 shows some similarities to rabbit and guinea pig leu-ases, as they share the same P1 preference [48].

Figure 5.
Verification of phage display sequences using the 2xTrx system. A number of phage display-derived sequences and variants of these sequences were added in between two adjacent trx proteins (panel A), expressed in E.coli and subjected to gar granzyme G (panels C and D). The results were run on pre-cast 4-12 % SDS-PAGE gels (Invitrogen, Carlsbad, USA). Hypothetical cleavage is shown (panel B) to highlight possible cleavage patterns. The individual lanes represent various time points after addition of the enzyme, in minutes.
Figure 5.
Verification of phage display sequences using the 2xTrx system. A number of phage display-derived sequences and variants of these sequences were added in between two adjacent trx proteins (panel A), expressed in E.coli and subjected to gar granzyme G (panels C and D). The results were run on pre-cast 4-12 % SDS-PAGE gels (Invitrogen, Carlsbad, USA). Hypothetical cleavage is shown (panel B) to highlight possible cleavage patterns. The individual lanes represent various time points after addition of the enzyme, in minutes.
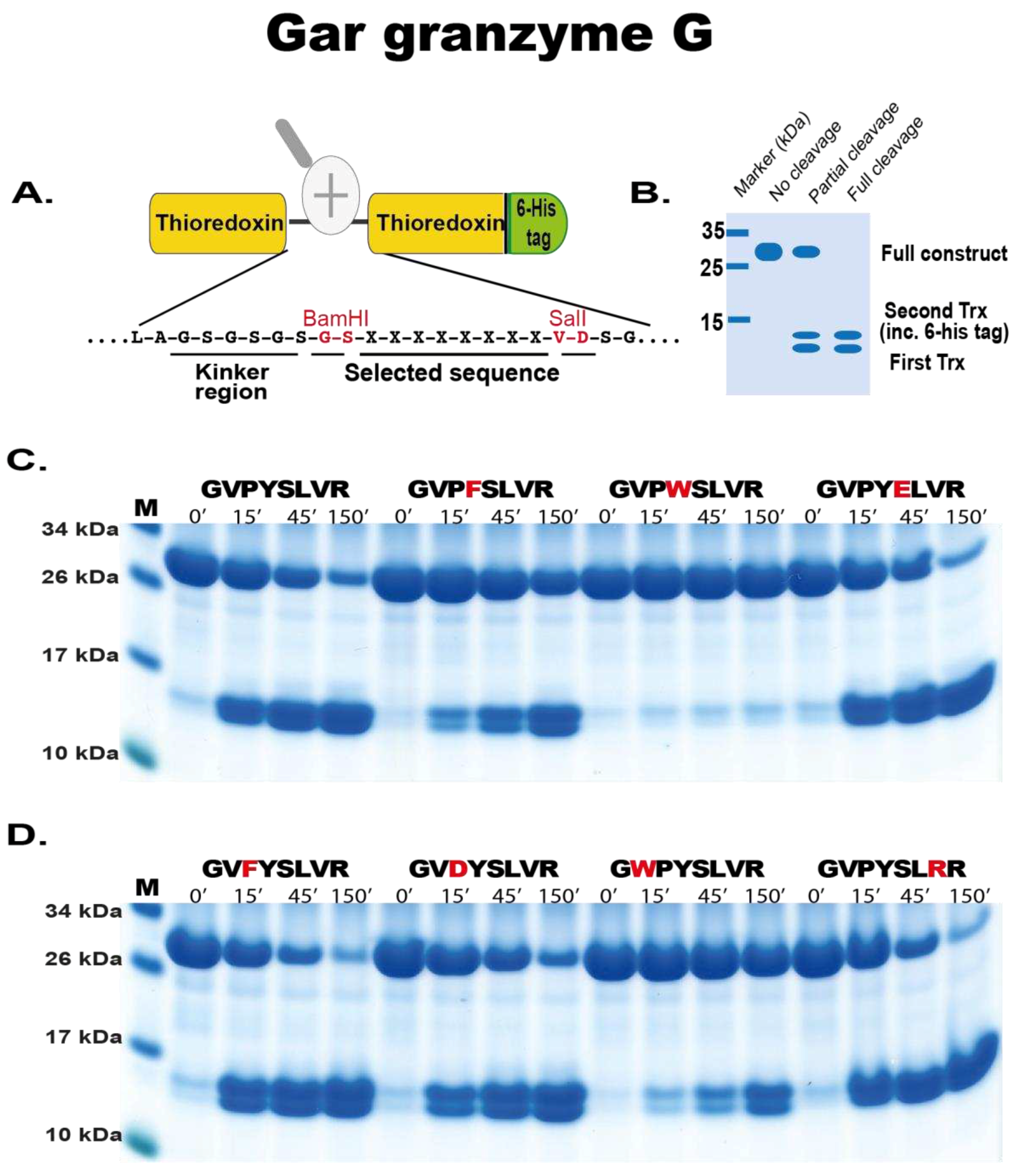
Figure 6.
Verification of phage display sequences of gar DDN1 using the 2xTrx system. A number of phage display-derived sequences and variants of these sequences were added in between two adjacent trx proteins, expressed in E.coli and subjected to gar DDN1 (panels A–C). The results were run on pre-cast 4-12 % SDS-PAGE gels (Invitrogen, Carlsbad, USA). The individual lanes represent various time points after addition of the enzyme, in minutes.
Figure 6.
Verification of phage display sequences of gar DDN1 using the 2xTrx system. A number of phage display-derived sequences and variants of these sequences were added in between two adjacent trx proteins, expressed in E.coli and subjected to gar DDN1 (panels A–C). The results were run on pre-cast 4-12 % SDS-PAGE gels (Invitrogen, Carlsbad, USA). The individual lanes represent various time points after addition of the enzyme, in minutes.
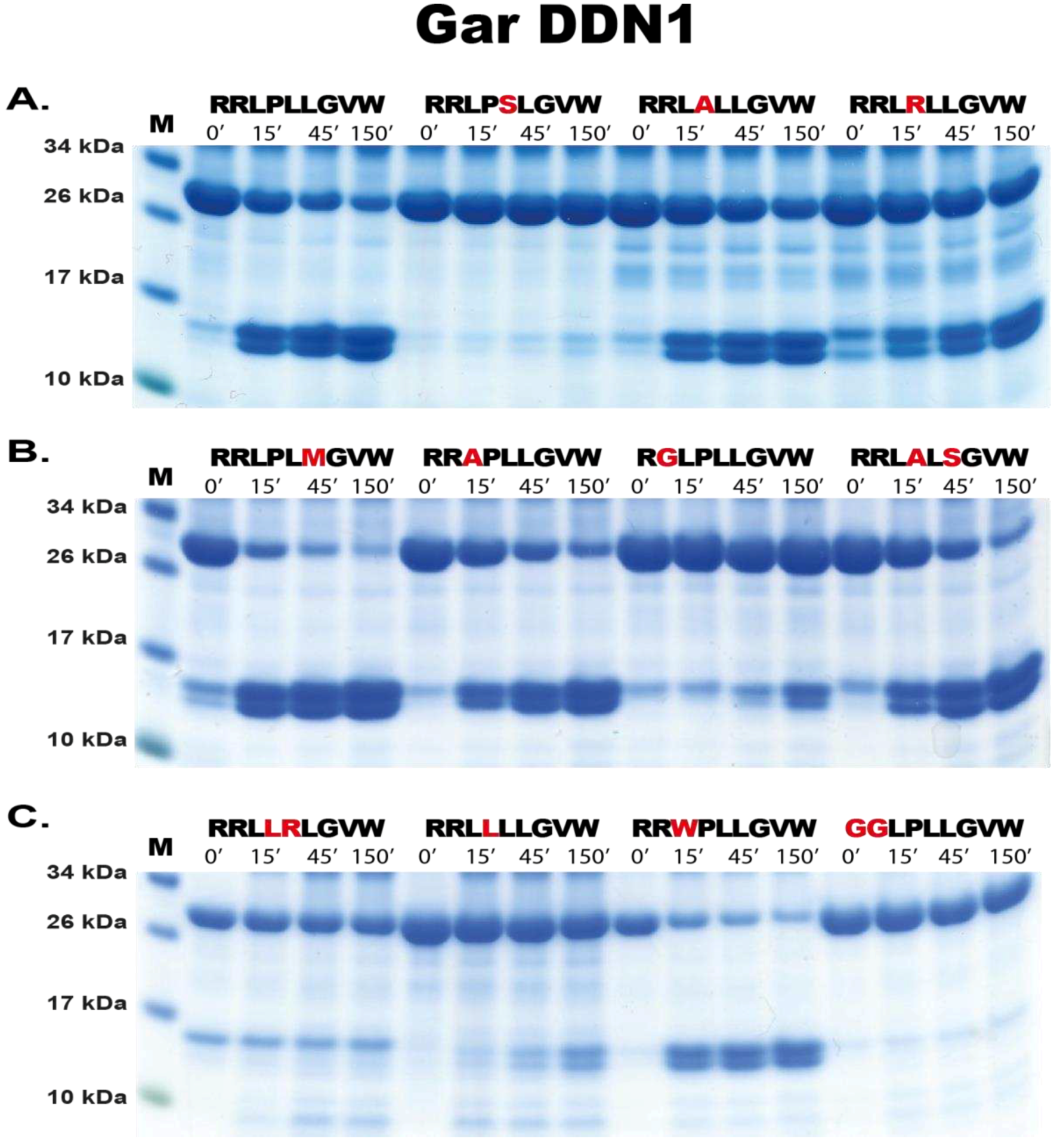
Figure 7.
Histological analysis of the gar tissues. Panels (A) shows a picture of a gar (source: Wikipedia). Panel (B) shows a picture of the juvenile gar used for histological analysis of head kidney and intestinal regions for immune cells. The section of the animal that was used for analysis is marked with a white line and a red arrow head. Panels (C–F) shows haematoxylin-eosin stained tissue sections. Panel C shows primarily the intestinal region with a white arrow head pointing at a Peyer´s patch-like structure in the intestinal wall. Panel (D) shows an enlargement of this Peyer´s patch region. Panels (E) and (F) shows the head kidney region with arrow heads marking the pure red blood cell rich regions of the head kidney. In panel (E) and arrow head number 4 shows an enlargement of this red blood region and arrow head 5 instead the immune cell rich region of the head kidney.
Figure 7.
Histological analysis of the gar tissues. Panels (A) shows a picture of a gar (source: Wikipedia). Panel (B) shows a picture of the juvenile gar used for histological analysis of head kidney and intestinal regions for immune cells. The section of the animal that was used for analysis is marked with a white line and a red arrow head. Panels (C–F) shows haematoxylin-eosin stained tissue sections. Panel C shows primarily the intestinal region with a white arrow head pointing at a Peyer´s patch-like structure in the intestinal wall. Panel (D) shows an enlargement of this Peyer´s patch region. Panels (E) and (F) shows the head kidney region with arrow heads marking the pure red blood cell rich regions of the head kidney. In panel (E) and arrow head number 4 shows an enlargement of this red blood region and arrow head 5 instead the immune cell rich region of the head kidney.
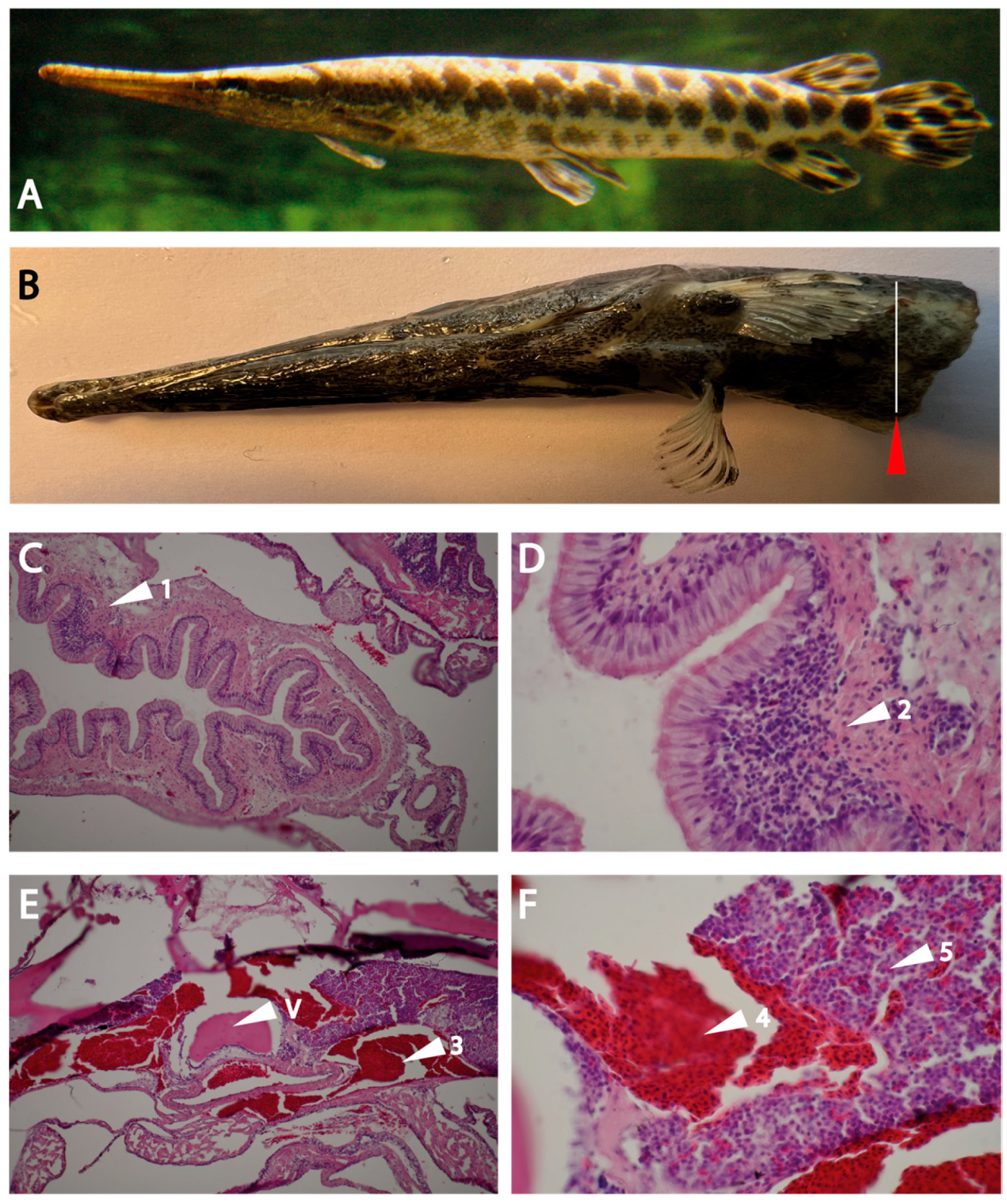
Figure 8.
Staining using DDN1 and GZM-G antibodies. In panel (A) a representative section showing DDN1 staining and in (B) a magnification for clarity is shown. In panel (C) a representative section showing the staining of gzm-G antibody and in panel (D) no primary antibody control in fish tissue are shown. Nuclei was stained using DAPI in blue.
Figure 8.
Staining using DDN1 and GZM-G antibodies. In panel (A) a representative section showing DDN1 staining and in (B) a magnification for clarity is shown. In panel (C) a representative section showing the staining of gzm-G antibody and in panel (D) no primary antibody control in fish tissue are shown. Nuclei was stained using DAPI in blue.
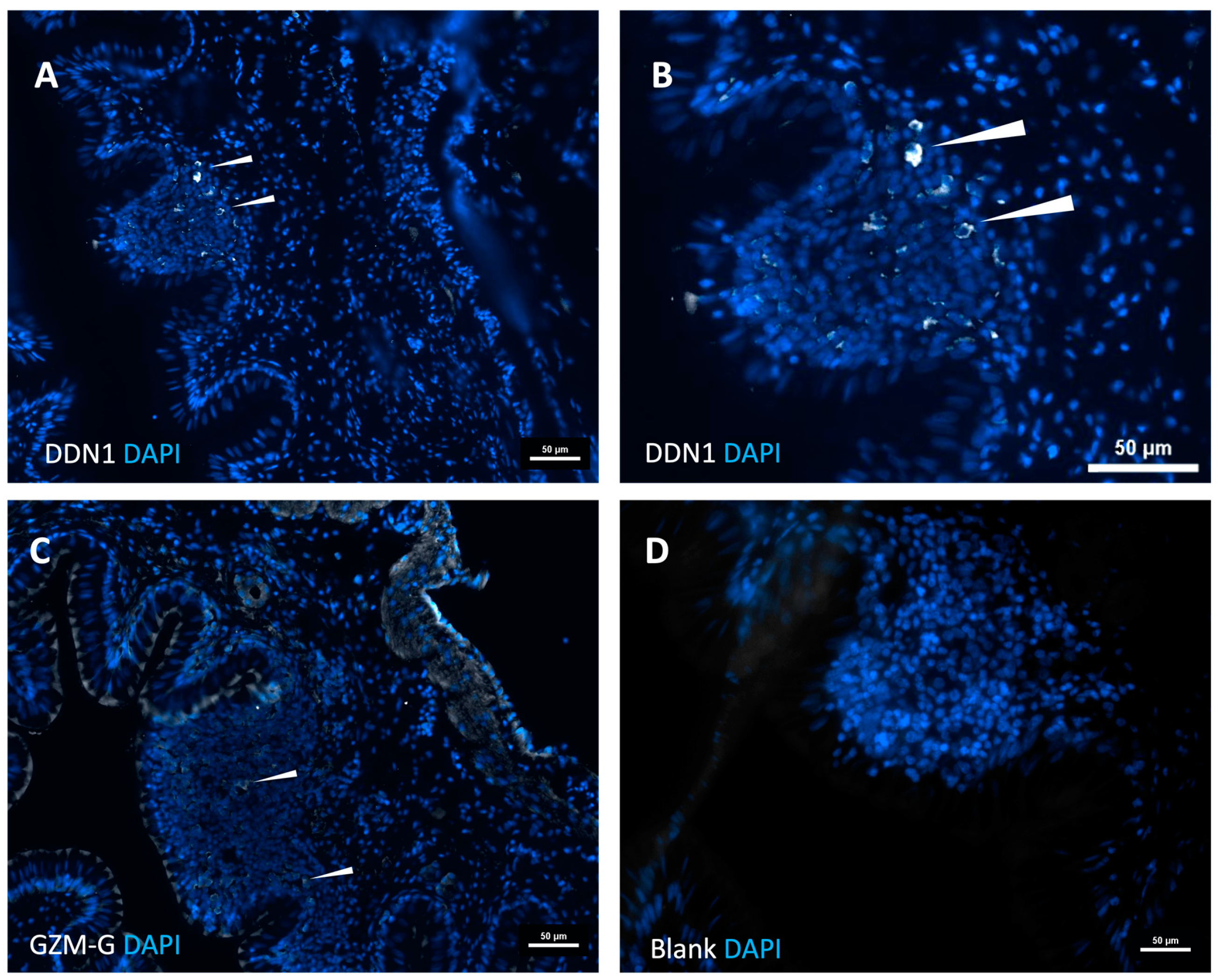
Figure 9.
Genomic loci encoding fish hematopoietic serine proteases. The locus encoding both of the enzymes are encoded in the ray-finned fish met-ase locus. This locus also exists in mammals, including humans, with a slightly different organisation. The human locus has most likely experienced an inversion and also an internal expansion, marked with a yellow oval just below the human locus. Serine protease genes are in double height for visual identification. Red dots mark the two enzymes analysed in this communication and blue dots marks enzymes previously analysed. Panel (A) shows a tree of fish evolution based on both genetic and morphological information where bichir is the first to branch out from the common fish branch followed by starlet and later of the gar and finally the teleosts [24,44]. Panel (B) shows the loci of the different fish species specified in panel (A).
Figure 9.
Genomic loci encoding fish hematopoietic serine proteases. The locus encoding both of the enzymes are encoded in the ray-finned fish met-ase locus. This locus also exists in mammals, including humans, with a slightly different organisation. The human locus has most likely experienced an inversion and also an internal expansion, marked with a yellow oval just below the human locus. Serine protease genes are in double height for visual identification. Red dots mark the two enzymes analysed in this communication and blue dots marks enzymes previously analysed. Panel (A) shows a tree of fish evolution based on both genetic and morphological information where bichir is the first to branch out from the common fish branch followed by starlet and later of the gar and finally the teleosts [24,44]. Panel (B) shows the loci of the different fish species specified in panel (A).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



