
Submitted:
05 January 2024
Posted:
08 January 2024
You are already at the latest version
Abstract
During the Renaissance, Leonardo Da Vinci was the first person to successfully detail the anatomy of the aortic root and its adjacent structures. Ever since, novel insights into morphology, function and their interplay have accumulated resulting in advanced knowledge on complex functional characteristics of the aortic valve (AV) and root. This shifted the vision from the AV as being a static structure towards that of a dynamic interconnected apparatus within the aortic root as a functional unit, exhibiting a complex interplay with adjacent structures via both humoral and mechanical stimuli. This paradigm shift has stimulated surgical treatment strategies of valvular disease that seek to recapitulate healthy AV function, whereby AV disease can no longer be seen as an isolated morphological pathology which needs to be replaced. As prostheses still cannot reproduce the complexity of human nature, treatment of diseased AVs, whether stenotic or insufficient, has tremendously evolved with a similar shift towards treatments options that are more hemodynamically centered such as the Ross procedure and valve-conserving surgery. Native AV and root components allow for an efficient Venturi effect over the valve to allow for optimal opening during the cardiac cycle, while also alleviating the left ventricle. Next to that, several receptors are present on native AV leaflets, enabling messenger pathways based on their interaction with blood and other shear-stress related stimuli. Many of these physiological and hemodynamical processes are under acknowledged but may hold important clues for innovative treatment strategies, or as potential novel targets for therapeutic agents that halt or reverse the process of valve degeneration. A structured overview of these pathways and their implications for cardiothoracic surgeons and cardiologists is lacking. As such, we provide an overview on embryology, hemodynamics, and messenger pathways of the healthy and diseased AV and its implications for clinical practice, by relating this knowledge to current treatment alternatives and clinical decision-making.
Keywords:
Aortic valve
; Embryology
; Hemodynamics
; Second messenger pathways
; Clinical decision-making
1. Introduction
Heart valve disease stands for a major worldwide burden and is associated with substantial mortality and morbidity(1). Absolute numbers of deaths attributed to aortic valve disease (AVD) have increased over the past 30 years(2), making AVD responsible for the largest proportion of deaths within the spectrum of valvular heart disorders. When left untreated, the natural course of AVD, especially aortic valve stenosis (AS), is progressive(3, 4) and associated with high mortality rates across the entire spectrum(5).
Taking a deeper look into the process of valve degeneration, this appears a multifactorial process which includes embryological, genetic, hemodynamic, structural, and cellular factors(6). Importantly, healthy aortic valve (AV) leaflets consist of multiple layers which communicate with surrounding tissues through humoral(7, 8) and mechanical stimuli(9). Neurofilaments, (myo)fibroblasts, endothelial cells, extracellular matrix (ECM) components and even smooth muscle cells (SMCs) play crucial roles in these regulatory processes(7), which are adaptive to shear stress (mechanotransduction) and blood compositions (humoral). Chronic or acute disturbance of an AV’s homeostatic state may induce AS and/or aortic valve regurgitation (AR) through various mechanisms(10). More than just the visible, clinically documentable structural defects to the AV which are measured using current routine clinical imaging techniques, are associated with AV dysfunction and it is acknowledged that pathophysiology induces morphologic changes. Messenger pathways that are disturbed by altered flow dynamics or humoral stimuli may in itself induce an effect on a functional level(7, 11). Importantly, the AV is known to generate contractile, secretory and proliferative responses to these stimuli and the interplay between physics and biology as well as the interplay between form and function play essential roles.
Treatment of AVD is commonly limited to prosthetic AV replacement (AVR). However, insights into the benefits of preserving one’s native valve and functional abilities have led to an expansion of the surgical armamentarium by development of innovative techniques, including the Ross procedure(12) and AV repair techniques (13). A perfect solution does not exist and the preferred treatment for an individual with AVD should be patient-tailored. Understanding the healthy and diseased AV on a cellular and biomechanical level is essential to appreciate the benefits of particular treatment options. The aim of this review is to provide an overview of the embryology, genetics, structure, pathophysiology, messenger pathways and hemodynamics of the AV, while linking this to its implications its treatment.
2. Embryology of the Aortic Valve
Normal outflow tract and valve formation
Twenty to thirty percent of congenital cardiovascular malformations contain some form of defective heart valves. Its incidence has been estimated as high as 5% of live births(14). Several seminal papers have been published on the different steps of cardiac development(15-17) and within the scope of this paper, we will solely focus on the embryogenesis of the heart valves.
The formation of endocardial cushions in the atrioventricular canal (AVC) and outflow tract (OFT) marks the start of valvulogenesis in the primitive looped heart at approximately 31-35 days after conception(18, 19). Contrary to the endocardial cushion and valvular leaflet relationship in the AVC, less is known about how the semilunar valves arise from the complex arrangement of the endocardial cushions in the OFT(14). Opposing dextro-superior and sinistro-inferior endocardial cushions grow at the cephalad portion of the truncus arteriosus(20). Simultaneously to the creation of these conotruncal cushions, two intercalated cushions form in between the aforementioned cushions. Upon fusion of the conotruncal cushions, the truncal septum is formed. At the early developmental stages, those conotruncal cushions appear bulky and cellularized, because the endothelial cells overlying the primitive endocardial cushions invade the conotruncal cushion matrix(21, 22). This highly proliferative state of endocardial cushions is lost in later remodeling and mature valves(14, 21). The growth of these valvular primordia continues, leading to the formation of thin fibrous cusps for the semilunar valves until they have become highly organized structures containing a rich collagen, proteoglycan and elastin ECM by the end of gestation(21). Valve maturation and microarchitectural remodeling will continue into juvenile stages in all mammalian species(14, 21, 23) with similar stratification patterns between species(21). The evolutionary basis for this similar semilunar valvulogenesis throughout mammalian species resides on highly preserved molecular pathways and physiological processes that were present in species with tubular hearts driving unidirectional flow and have been maintained throughout the formation of a four-chambered heart(16).
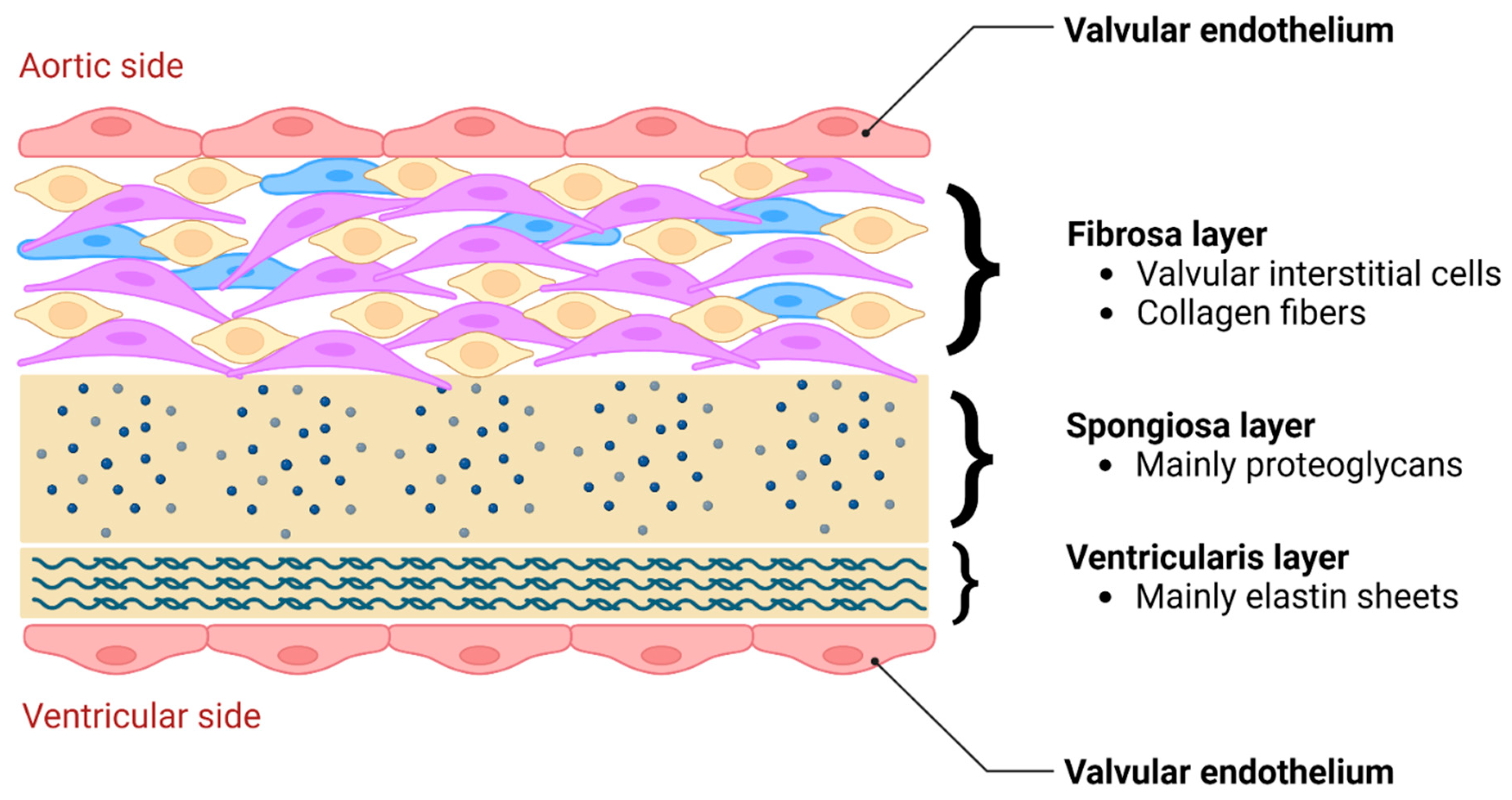
During truncal septal differentiation with conotruncal rotation and caudal shift, mesenchymal derivation from the endocardium takes place(24) where dedifferentiation from a myosin-heavy chain to an alpha-smooth muscle actin phenotype takes place. This allows for the formation of (mature) semilunar valves from the conotruncal and intercalated cushions of the OFT (Figure 1). The conotruncal cushions give rise to the semilunar right and left leaflets; that is, the right and left coronary cusp leaflets for the aorta. The opposing right and left intercalated cushions develop into the posterior aortic (non-coronary cusp) and anterior pulmonic leaflet, respectively. During endocardial cushion fusion, cavities are formed through apoptosis leading to the formation of a central lumen of each of the cushions separating the three valves and creating the wall of the supporting sinuses through peripheral arterialization(22, 25). During this process of cavitation the muscular portion of the proximal OFT contributes to the formation of valves and sinuses in a paracrine, rather than a direct cellular fashion(22). Eventually, rudimentary valves will elongate and the endocardial cushions thin out, thus remodeling the valves with a compartmentalized microarchitecture consisting of 5 layers: endothelium, fibrosa (comprising mainly valvular interstitial cells (VICs) and collagen fibers), spongiosa (comprising mainly proteoglycans), ventricularis (comprising mainly elastin sheets) and endothelium(21) (Figure 2). The microarchitectural composition of the valve leaflets together with the biomechanical properties and vasoactivity are critical for normal valve function allowing for optimal interplay between form and function. A striking example of this interplay is the occurrence of hypoplastic left heart syndrome during heart development, where the obstruction or atresia of left-sided valves produces a lack of blood flow, in turn affecting the functional stimuli – i.e. flow – to form a compact left ventricle (LV)(26, 27). Such findings emphasize the role of blood flow perturbations as a causal factor in, not only early organogenesis, but also pathogenesis of valvular (structural) disease.
Figure 1.
Leaflet development of the semilunar valves (aortic and pulmonary). The left and right valve leaflets of both the pulmonary and aortic valve arise from the conotruncal cushions, whereas the non-coronary aortic leaflet and anterior pulmonic leaflet arise from the intercalated cushions. Adapted from Martin et al. 2015(22), distributed under the terms and conditions of the Creative Commons Attribution license (CC BY 4.0 DEED, Attribution 4.0 International), 2015, the authors..
Figure 1.
Leaflet development of the semilunar valves (aortic and pulmonary). The left and right valve leaflets of both the pulmonary and aortic valve arise from the conotruncal cushions, whereas the non-coronary aortic leaflet and anterior pulmonic leaflet arise from the intercalated cushions. Adapted from Martin et al. 2015(22), distributed under the terms and conditions of the Creative Commons Attribution license (CC BY 4.0 DEED, Attribution 4.0 International), 2015, the authors..
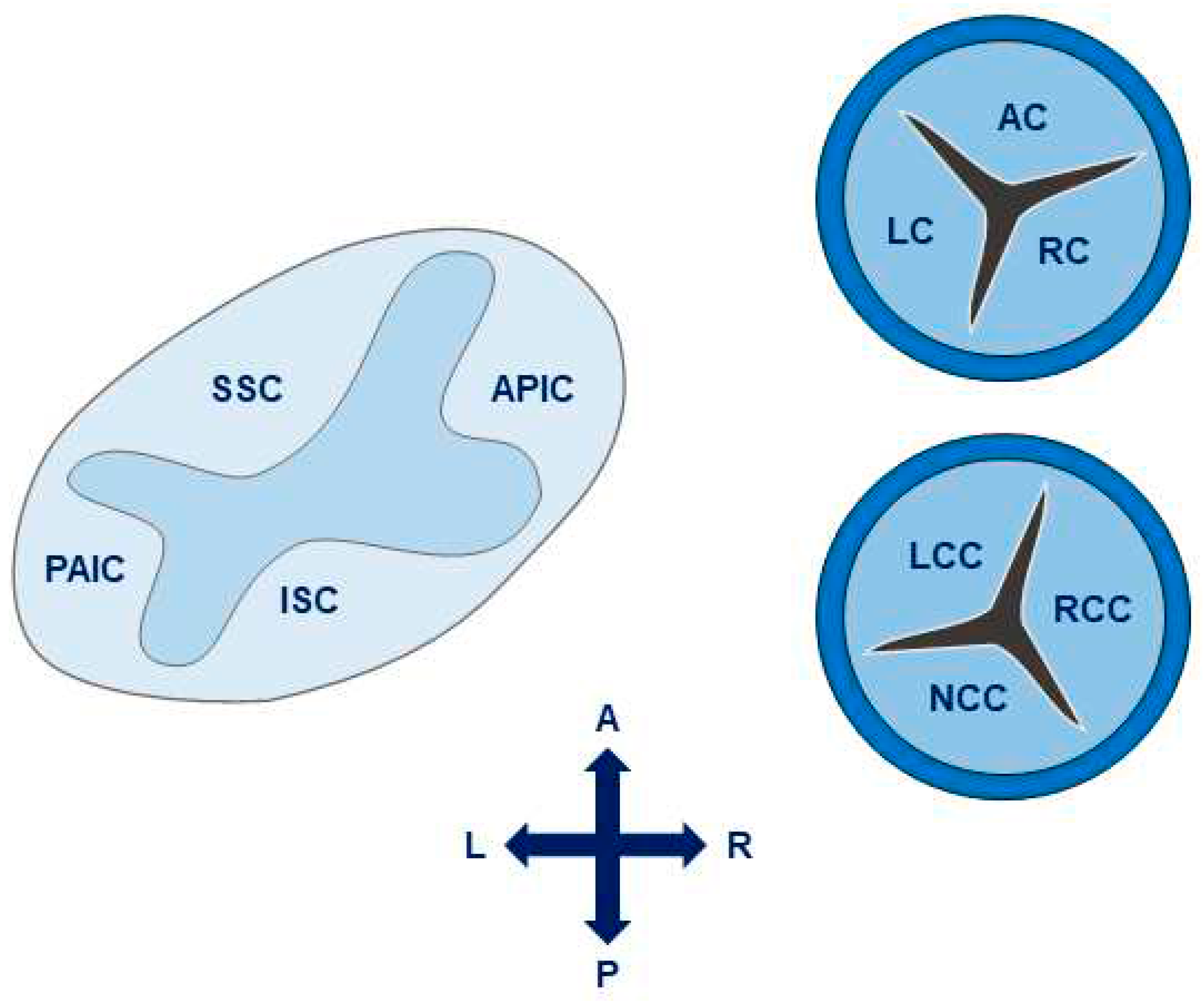
Figure 2.
Microarchitectural composition of aortic valve leaflets, consisting of a bilayer of endothelial cells (aortic and ventricular side), fibrosa layer, spongiosa layer and ventricularis layer.
Figure 2.
Microarchitectural composition of aortic valve leaflets, consisting of a bilayer of endothelial cells (aortic and ventricular side), fibrosa layer, spongiosa layer and ventricularis layer.
3. Transcriptional Regulation of Valvulogenesis
Normal development and function require tightly regulated interactions between molecules and gene transcription, and any genetic defect or signaling alteration may disturb this process, hence leading to structural malformations. There is a complex genetic regulatory network that generates valve progenitor cells through endothelial-to-mesenchymal transformation (EMT), effectuates ECM remodeling and is involved in leaflet stratification (Figure 3). Importantly, calcific aortic valve disease (CAVD), a progressive condition often stimulated by inflammatory processes, in which a tricuspid AV or a congenital bicuspid AV (BAV) becomes thickened, fibrosed, and, consequently, calcified(28), is characterized by the expression of transcription factors also involved in valve development and comparable to osteogenesis(20). Moreover, mechanosensitive systems, e.g. RhoA/ROCK and YAP/TAZ, detect substrate changes and initiate mineralization pathways leading to CAVD(29). Figure 4 plays a central role in this synopsis, as it possesses important clues on several aspects of CAVD pathophysiology and progression. During EMT, cells on the endocardial side of the primitive heart tube undergo a phenotypic switch from a endothelial-like to a mesenchymal-like phenotype(30). These cells migrate into the cardiac jelly portion of the tube, where they form the cardiac cushions which are essential for, among other things, OFT formation(31). Many transcription factors and signaling pathways have been linked to EMT, as well as neural crest (NC) cell migration to the distal cushions(31). In absence of NC-cells, OFT septation will not occur(32, 33). After fulfilling their role in septation, most NC-cells go into apoptosis(34). Their further role beyond this developmental stage remains largely unknown. Some signaling pathways that play a crucial role during heart valve development include Notch, transforming growth factor beta (TGF-β), vascular endothelial growth factor (VEGF) and Wnt/beta-catenin pathways(14). Besides signaling pathways, transcription factors expressed in the endocardial cushions represent progenitors of AV leaflets, such a Tbx20, Msx1, Msx2 and Twist1, as well as ECM protein regulators, such as Sox9 and NFATc1(35). There has been recent interest for EMT, as it has been shown that adult cardiovascular patients also experience similar transitions when presenting with degenerative diseases of their cardiovascular system(30, 36).
Figure 3.
Aortic valve formation, starting with cushion formation (1), proceeding with matrix remodeling (2) and ending with leaflet stratification (3). Adapted from Martin et al. 2015(22), distributed under the terms and conditions of the Creative Commons Attribution license (CC BY 4.0 DEED, Attribution 4.0 International), 2015, the authors.
Figure 3.
Aortic valve formation, starting with cushion formation (1), proceeding with matrix remodeling (2) and ending with leaflet stratification (3). Adapted from Martin et al. 2015(22), distributed under the terms and conditions of the Creative Commons Attribution license (CC BY 4.0 DEED, Attribution 4.0 International), 2015, the authors.
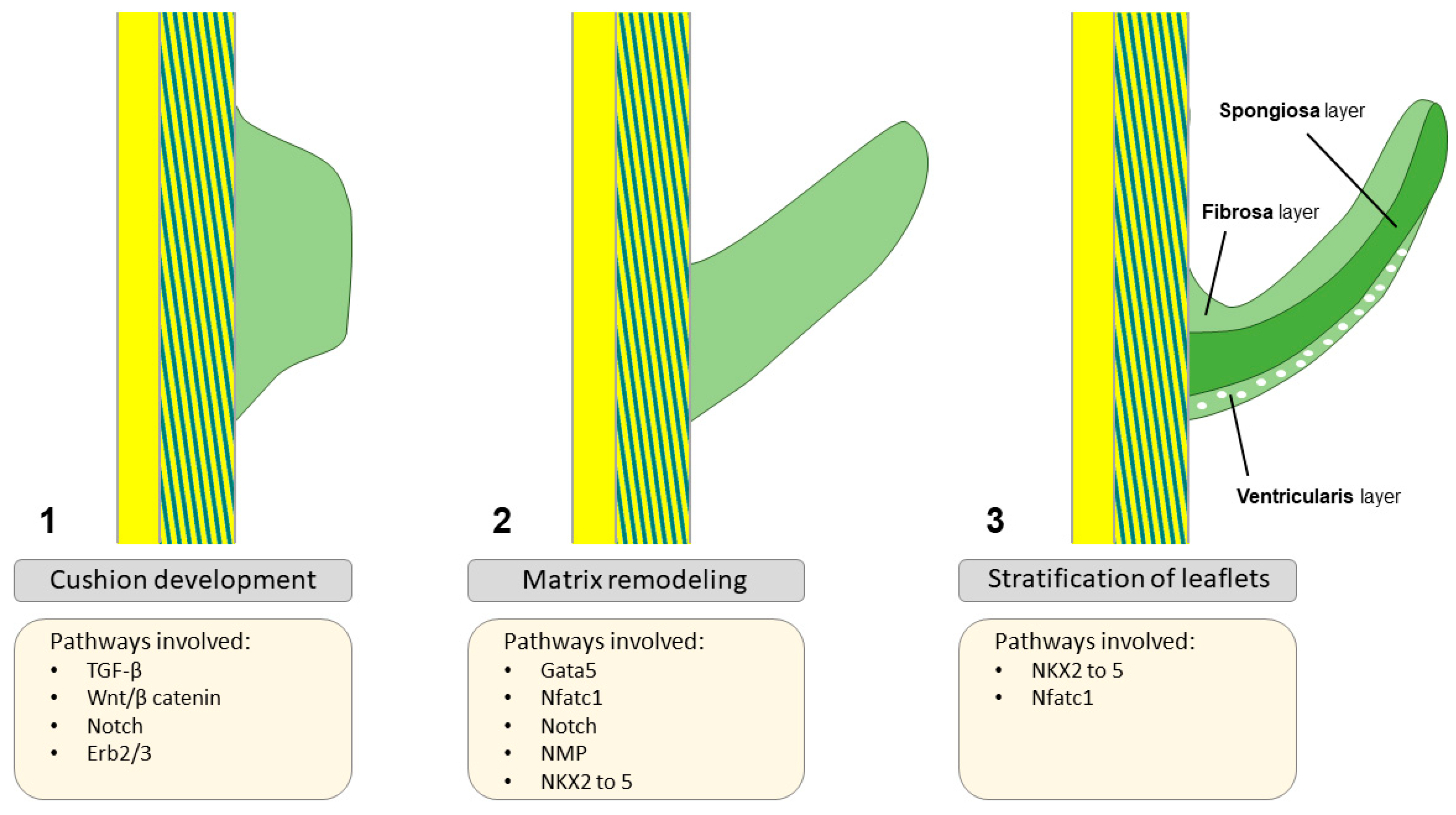
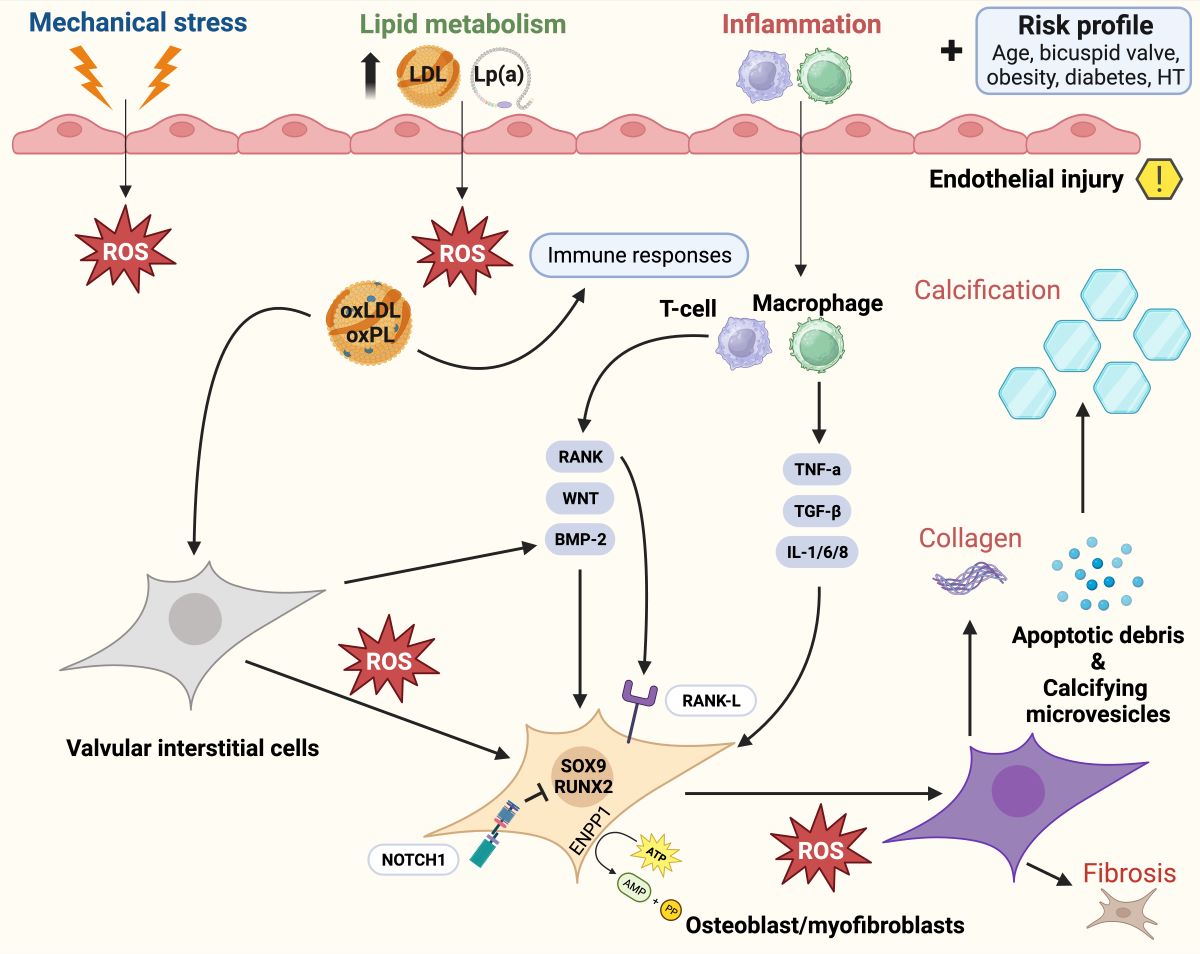
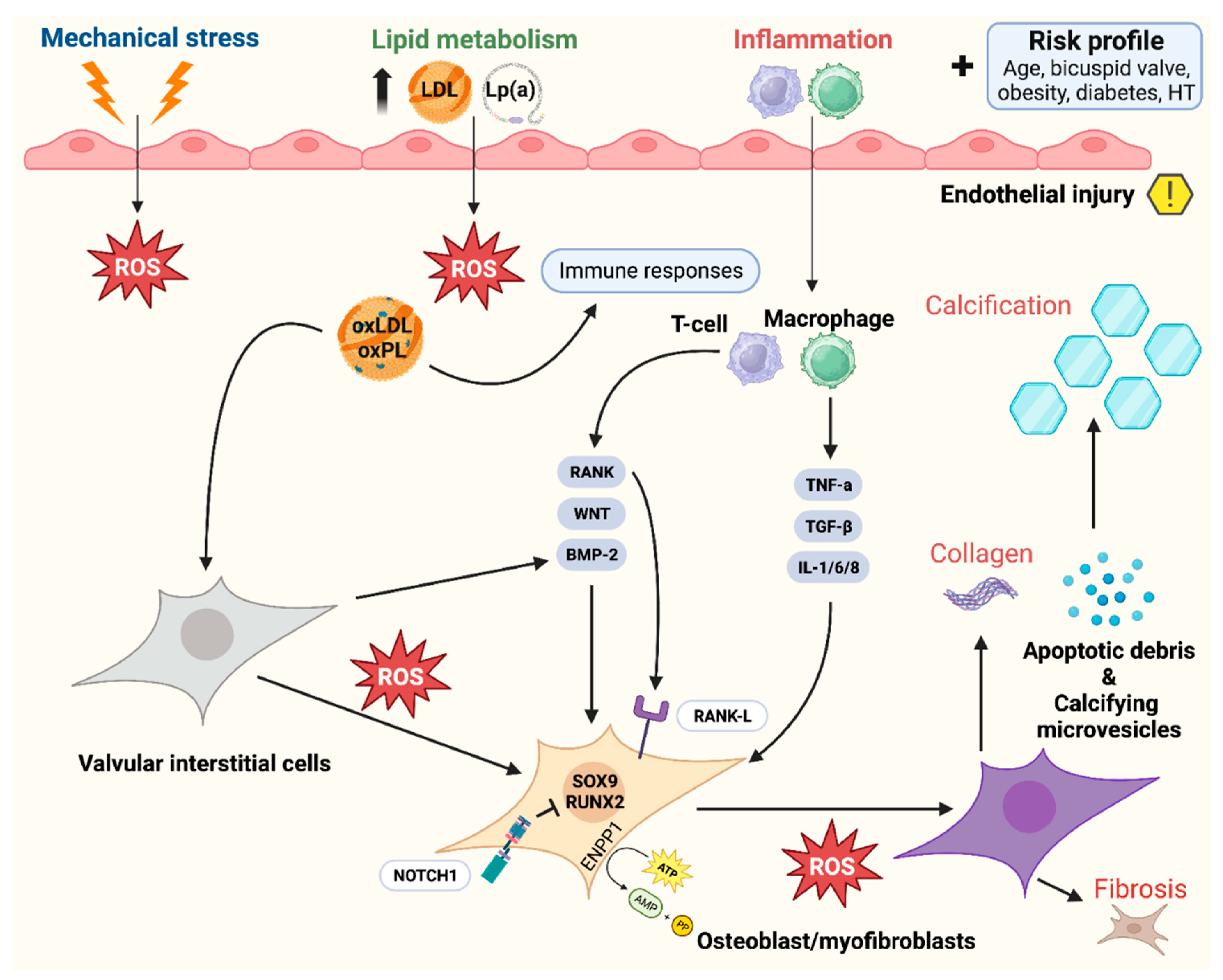
Figure 4.
Pathophysiology of calcific aortic valve disease: activation of bone-formation pathways induces aortic valve calcification through various ligands, ranging from inflammation to metabolism. Adapted from Greenberg et al.(28), distributed under the terms of the Creative Commons CC BY license, 2021, the authors. Legend: Calcific aortic valve disease is initiated by endothelial injury (right upper quadrant), and propagated by a complex cascade of signaling involving osteoblasts and myofibroblasts (bone formation pathways). Abbreviations (alphabetical): ATP = Adenosine triphosphate; AMP = Adenosine monophosphate; BAV = Bicuspid aortic valve; BMP2 = Bone morphogenic protein 2; ENPP1 = Ectonucleotide pyro phosphatase / phosphodiesterase family member 1; IL = Interleukin; LDL = Low-density lipoprotein; Lp(a) = Lipoprotein a; ROS = Reactive oxygen species; NOTCH1 = Notch homolog 1; PP = Inorganic pyrophosphate; RUNX2 = Runt-related transcription factor 2; SOX9 = SRY-box 9; TGF-β = Transforming growth factor beta; TNF = Tumor necrosis factor alpha.
Figure 4.
Pathophysiology of calcific aortic valve disease: activation of bone-formation pathways induces aortic valve calcification through various ligands, ranging from inflammation to metabolism. Adapted from Greenberg et al.(28), distributed under the terms of the Creative Commons CC BY license, 2021, the authors. Legend: Calcific aortic valve disease is initiated by endothelial injury (right upper quadrant), and propagated by a complex cascade of signaling involving osteoblasts and myofibroblasts (bone formation pathways). Abbreviations (alphabetical): ATP = Adenosine triphosphate; AMP = Adenosine monophosphate; BAV = Bicuspid aortic valve; BMP2 = Bone morphogenic protein 2; ENPP1 = Ectonucleotide pyro phosphatase / phosphodiesterase family member 1; IL = Interleukin; LDL = Low-density lipoprotein; Lp(a) = Lipoprotein a; ROS = Reactive oxygen species; NOTCH1 = Notch homolog 1; PP = Inorganic pyrophosphate; RUNX2 = Runt-related transcription factor 2; SOX9 = SRY-box 9; TGF-β = Transforming growth factor beta; TNF = Tumor necrosis factor alpha.
Timing/expression of transcription factors and deficient aortic valve formation
Valvulogenisis regulatory systems are similar in AVC and OFT development and AVC endocardial cushion formation precedes OFT endocardial cushion formation by one day(14, 37). Given the presumed similarities, the larger AVC endocardial cushions in mouse or chick embryos, and the fact that examination of the OFT cushions is complicated due to the presence of NC derived progenitors (responsible for the formation of the aorto-pulmonary septum), many research extrapolates results from the AVC to the OFT regulatory system(38). This assumption holds true and has been discussed elsewhere(37, 39, 40); here we highlight some of the pathways that have a more OFT specificity.
Cardiac progenitor cells of the second heart field give rise to several cell lines that play a role during formation of the aortic outflow tract, mediated by Nkx2.5, vascular smooth muscle cells (SMC), endocardial cushion cells and OFT myocardium(41, 42). Secondary heart field deficiencies preferentially compromise semilunar valve (but not AV Valve) defects that are related to defects in the formation of the OFT(14, 40). Next to that, vascular SMCs of the aortic root originate from these progenitor cells deriving from the second heart field as well as NCC, whereas in the ascending aorta and aortic arch, these cells form solely out of neural crest cells(43). Multiple cell lines and signalling systems are involved in AV and ascending aortic formation and defects in these pathways may induce malformations of the AV, such as BAV. Despite the vast importance of Nkx2.5 in secondary heart field development and the fact that this homeodomain factor is the most commonly mutated single gene in congenital heart disease (CHD), many of its actions remain to be elucidated(44).
The TGF-β superfamily consists of BMPs (BMP2-7) and TGF-β in the embryonic heart where BMPs are responsible for the promotion of endocardial cushion growth. Next to that, BMP, Notch and TGF-β promote EMT, cell invasion into the cardiac cushions and remodeling of the valves(22, 37, 45). Furthermore, TGF-β has been linked to activation of VICs and their transformation into myofibroblasts in the adult valve(46). Importantly, Notch signalling disruption markedly decreased the Snail transcription factor responsible for the initial steps in EMT and cushion development(47) placing the Snail pathway at the center of valve development. Notch 1, 2 and 4 receptors and their ligands (Jag 1/2 and Dll4) are specifically expressed in the OFT and its cushions(48-50) where inactivation of this pathway leads to a multitude of CHD including BAV(51-53). From a hemodynamical point of view, Notch and downstream pathways have also been linked to a process called valve polarity distinguishing the lamina fibrosa from the flow side of the valve(14, 51) allowing for valve leaflet maturation(54) (Figure 3). However not yet fully established, reports emphasize the role of Notch signaling in altered shear stress possibly promoting valve stratification and even calcification(50, 55) (Figure 4). A recently published study pointed out the role of the MIB1 gene, an essential regulator for Notch ligands signaling, in the pathophysiology of non-syndromic BAV(56). Future research should further investigate its potential as a treatment target, along with other genes linked to BAV formation such as Jag1(56, 57).
Genome-wide association studies furthermore suggest a role for transcription factors with important embryological functions in the development of AS(58) and bicuspid AS(59), which may help prioritize gene and pathway targets for medical CAVD therapy.
4. Anatomy and Hemodynamics
The heart valves open and close around 100.000 times a day adding up to around 3 billion cycles in a 75-year lifespan and are subject to a variety of stresses. For many years, the AV has been seen as a “static” structure responsible for unidirectional flow of blood coming from the LV and aiding in coronary perfusion. However, evolving insights shifted our perception of the AV and is now seen as functionally complex regulatory system necessary for optimal mechano-biological coupling of the heart.
4.1. The aortic valve and root
Located in the middle of the heart, the AV is commonly referred to as the center of the heart. This is likely no coincidence, as it has several advantages for this particular valve that is subject to the highest levels of pressure and shear stress of all heart valves in a physiological situation. Supported by the fibrous skeleton of the heart and atrioventricular valves, the AV is able to transduce mechanical stress like no other valve evidenced by its continuity with the mitral valve (MV). The pulmonary valve has no fibrous support to it whatsoever, as it is pushed upwards by the underlying infundibular muscle separating the two right-sided valves during OFT formation(15). Infundibular muscle, in its turn, is not seen on the left side where the aortomitral curtain connects the two left-sided valves(60, 61). The AV apparatus was previously described as “a tale of dynamism and crosstalk” by Yacoub(62), which is also underpinned by several treatment modalities respecting the morphology and function of native roots, i.e. the Ross(12), remodeling(63) and reimplantation operations(64).
Throughout evolution, functionality has clearly dictated its morphological counterparts and as such, the AV can be divided into several functional units (annulus, cusps, sinuses of Valsalva and sinotubular junction) accumulating into one biomechanical unit. The geometrical, crown-shaped structure of the semilunar valves allows for optimal responsiveness and efficacy during the cardiac cycle as different forces are exerted on the valves(65, 66). Next to that, the precise shape and building plan of the leaflets (including the nodules of Arantius) allow for competent seal and force distribution so that the AV remains competent throughout life. Microarchitectural and geometrical changes may therefore result in biomechanical dysfunction and lead to valvular disease(66), through pathways illustrated in Figure 4.
4.2. Valvular fluid dynamics
A complex interplay between cardiac cycle, AV biomechanics, transvalvular hemodynamics and compliant properties of the aorta all represent different mechanical stresses that act individually or combined to exert a physiological response through the cellular components of the AV (Figure 5).
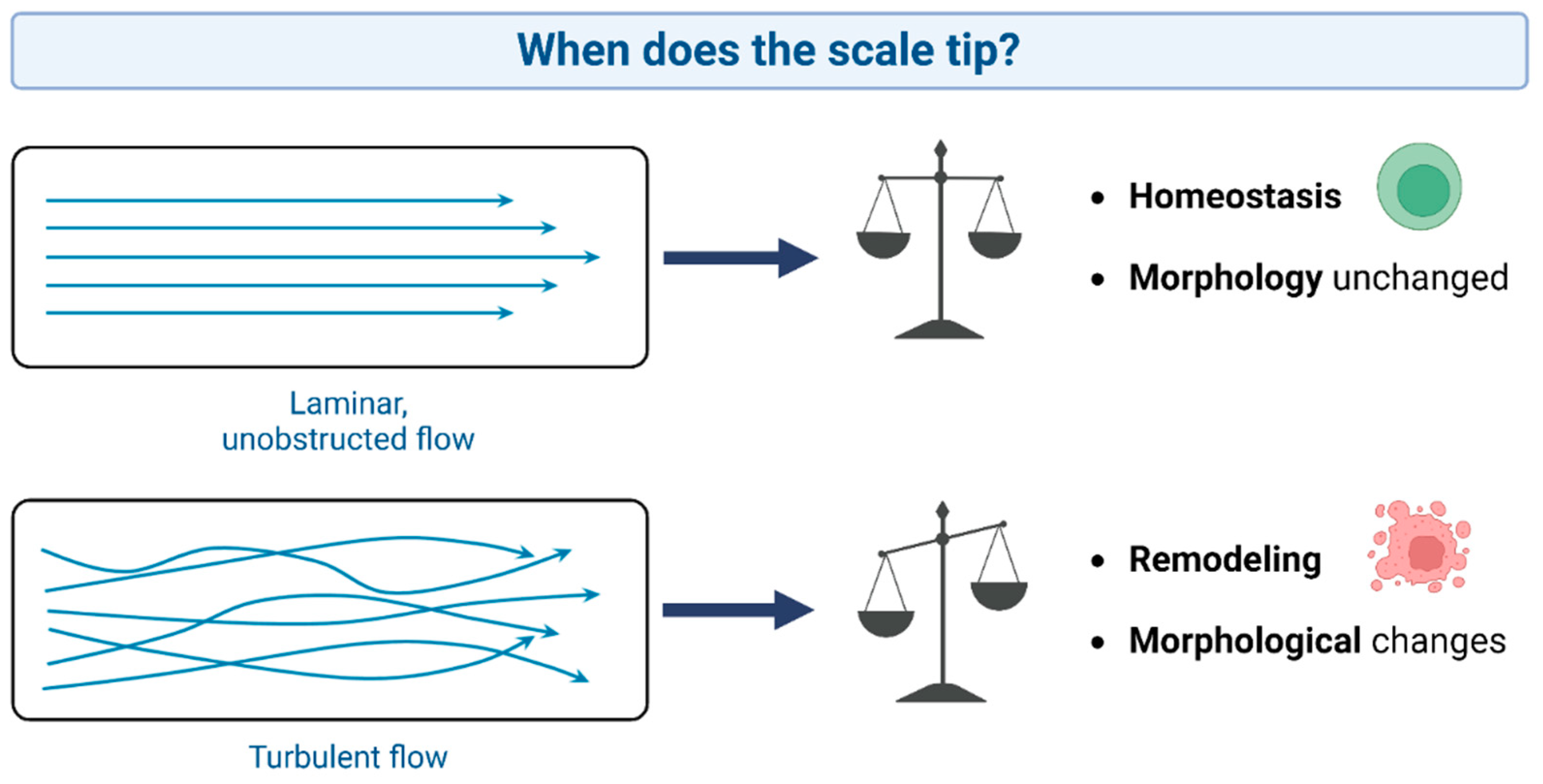
Figure 5.
The concept of laminar and turbulent flow and their implications for aortic valve remodeling.
Figure 5.
The concept of laminar and turbulent flow and their implications for aortic valve remodeling.
The AV is embedded in a crown-shaped annulus (60, 61), providing optimal support to the AV under high pressures and dynamic flow patterns. The opening of the AV is a harmonized process and this is crucial to guarantee unimpeded blood flow(67). Coordinated and competent opening of the valve is essential to decrease afterload and, therefore, ventricular systolic workload. Nonetheless, alterations in mechanical stress or blood flow near the AV may activate several pathways that in turn may lead to situations in which function deteriorates, as a result of which the LV is exposed to pressure (e.g., in case of AS) or volume (e.g., in case of AR) overload. The opening of the AV lasts for about 330ms at a heart rate of 70 bpm where blood rapidly accelerates through the valve reaching a peak velocity of 1.2m/s(68, 69). The ensuing deceleration causes a pressure gradient of only several millimeters of mercury with preferential flow at the center of the aorta and low momentum fluid near the aortic wall, thereby causing flow reversal at the sinus regions(68, 70, 71). As such, vortices of blood are created by the end of systole, aiding efficient and swift AV closure with an estimated volume (closing volume) to be less than 1%(68, 72).
In order to aid fluid dynamics and dictated by transvalvular pressure, the aortic annulus changes shape during cardiac cycle, albeit in an asymmetric fashion, with the greatest expansion during isovolumetric contraction at the left coronary cusp annular region in respect to the non-coronary cusp annular region. Due to its morphological anchoring and continuity with the MV at the site of the non-coronary cusp, annular stability is provided and allows for energetic transfer from the MV. Next to that, physiological consequence of this anatomical feature translates into a circumferential increase in diameter at the commissural level which is proportional to the end-diastolic volume. This biomechanical behavior, where the annulus reaches its minimum size at the end of systole and maximizes in size at the end of diastole, is a teleonomic hallmark of evolutionary hemodynamics in all mammals. Systolic workload of the ventricle should be as low as reasonably possible and, by anticipating the accommodation of each stroke volume, transvalvular hemodynamics are optimized, thereby minimizing the possibility of turbulent damage to the valvular cusps. Furthermore, compliant properties of the surrounding aorta are of vital importance to facilitate ejection as, from a physiological perspective, energetics from cyclic LV contraction need to be addressed in order to provide a continuous flow and pressure downstream in the arterioles(73). This allows for optimal ventriculo-arterial coupling, and the vertical motion of the aortic root during the heart cycle is important for absorbing stress. In such, not only the annulus but also the ascending aorta expands to dampen the pressure and flow during systole. This so-called Windkessel effect allows for better energetics where a portion of stroke volume is temporarily maintained by the expanding aorta and later propelled into the circulation by the recoil of the elastic aortic wall(74). Next to that, another compliancy mechanism takes place regarding the topographical anatomy of the aortic root as it sits at an angle of around 16 degrees to posterior and the left (angle between basal and commissural planes) during diastole. During systole, an alignment of the LV outflow tract (LVOT) and the aorta takes place reducing this angle to around 7 degrees, thereby straightening the tube and thus aiding ejection(65, 71, 73).
It might be clear that the underlying mechanobiology of the AV is very complex and no sole intervention can preserve all its aspects. Prosthetic valves in the aortic position fix the annulus, are intrinsically obstructive and therefore associated with suboptimal fluid dynamics through the OFT(75). Even a mild gradient over the valve may have major implications in the long run(5, 76), although not directly life-threatening. Image a large closed system, e.g., a bowl, filled with water; should you have a large opening at the bottom, blood will flow out seamlessly, but if it has a small opening, pressure must be increased to maintain equal flow over the defect. The same holds true for AS; the smaller the opening, the more difficult blood flows out under the same workload. To increase flow, one should increase pressure (workload) before the stenosis or increase the diameter of the opening. Consequently, the velocity of the fluid through the opening has to increase to achieve equal flow, which is in accordance with Bernoulli’s law(77).
In this light, a lifetime of suboptimal gradients and loss of root dynamics, as seen after mechanical or bioprosthetic valve replacement(75), will undoubtedly translate to a higher ventricular workload(78). In patients undergoing AVR, small reductions in mean transvalvular gradients are associated with significant reductions in heart failure(78). In such, the Ross procedure allows for natural gradients and hemodynamics, coupled with optimal coronary perfusion and ventricular mass regression(79, 80). Physiologic flow patterns and low wall shear stresses after the Ross procedure (full root technique) and valve-sparing root replacement, whether or not combined with reconstruction of neosinuses, are major benefits of these reconstructive approaches(81). With the current discussions on the lifetime approach to patients with AVD, this holds significant importance.
5. Biomechanics and Cellular Responses
The opening of the AV should be atraumatic as well as symmetric to ensure retained morphology of the apparatus in the long run. Any failure to comply to these terms, i.e., in situations with non-physiological pressures, resistance or volumes, leads to activation of second messenger pathways based on mechanical stimuli such as stretch, shear and transvalvular pressure (Figure 6). Mechanotransduction is the translation of mechanical processes to biological signals, which also affects the aortic valve and root. In a normally functioning AV, the ultimate goal is to efficiently transfer these mechanical stimuli into a well-orchestrated cascade of complex signals. This is reportedly regulated through the cooperative action of valve endothelial cells and VICs(8). Intrinsic nerve networks are likely responsible for part of these adaptations by regulating synthesis, contraction, repair and homeostasis within the valve, although evidence is not in abundance.
Figure 6.
Mechanical stimuli experienced by valvular endothelial cells as well as valvular interstitial cells (fibrosa layer). Adapted/modified from Balachandran et al. 2011 (68). , distributed under the CC BY 3.0 DEED Attribution 3.0 Unported licence.
Figure 6.
Mechanical stimuli experienced by valvular endothelial cells as well as valvular interstitial cells (fibrosa layer). Adapted/modified from Balachandran et al. 2011 (68). , distributed under the CC BY 3.0 DEED Attribution 3.0 Unported licence.
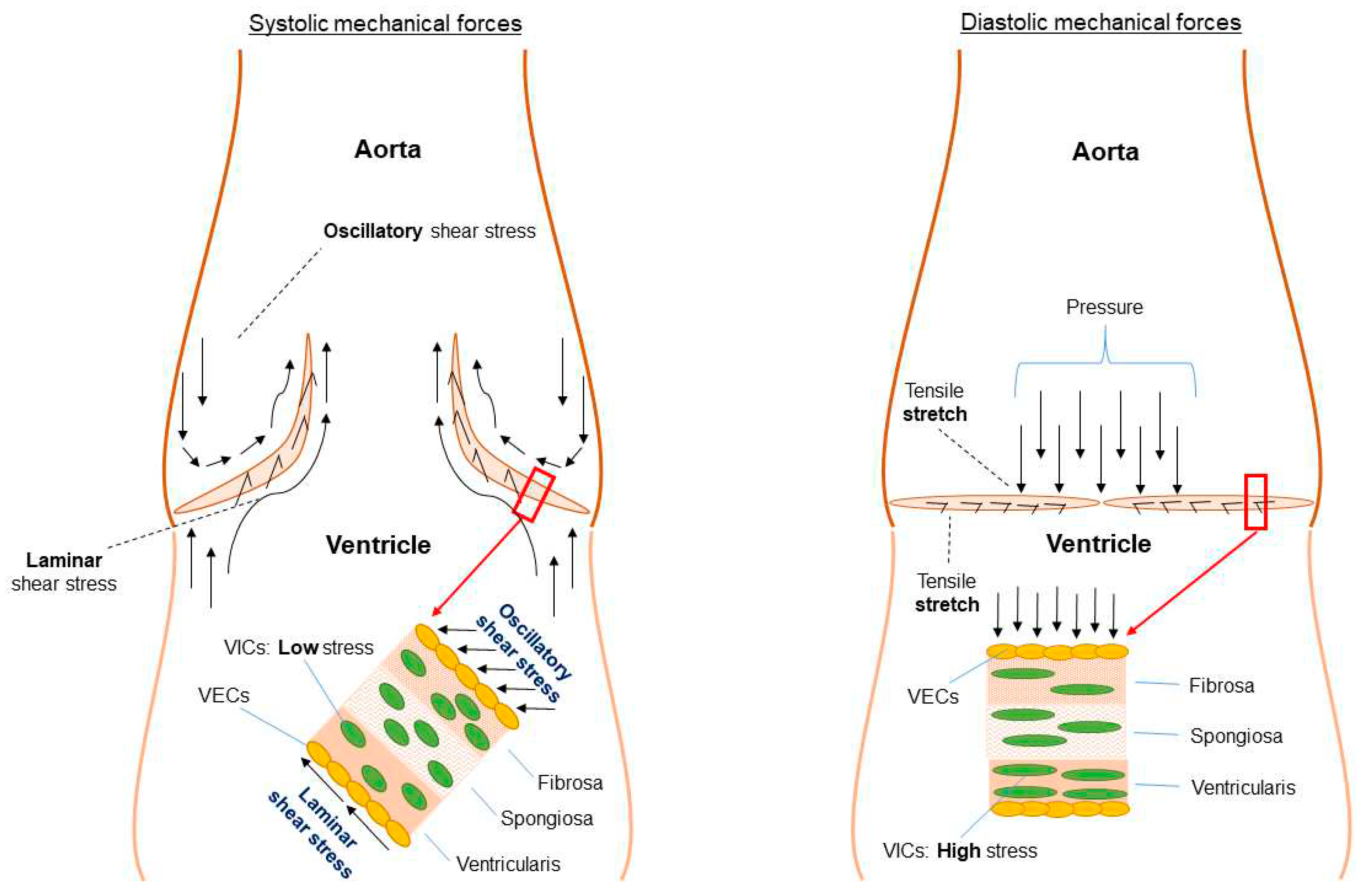
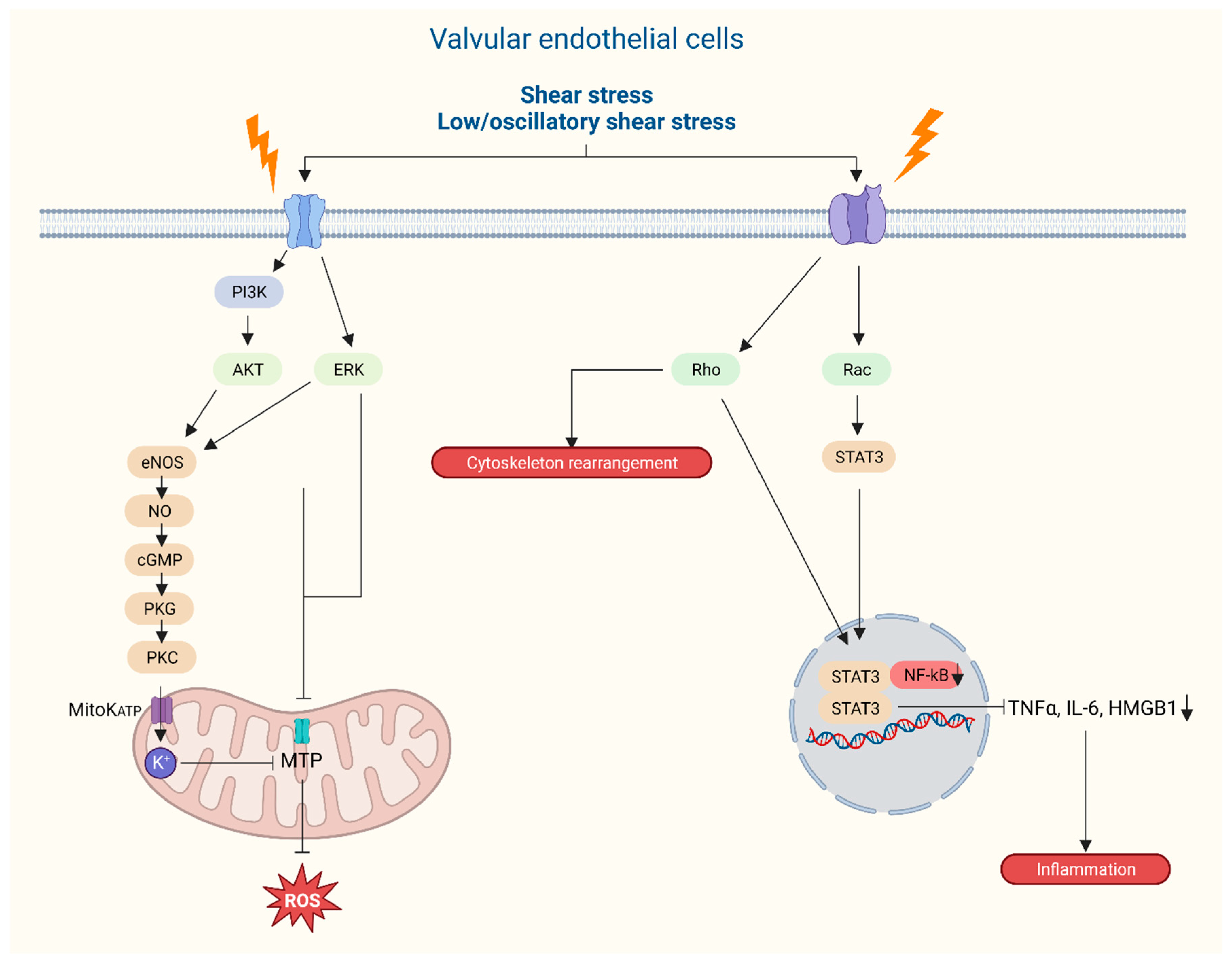
Figure 7.
Graphical representation of intracellular responses of aortic valvular endothelial cells following external mechanical stress on the aortic valve leaflets.
Figure 7.
Graphical representation of intracellular responses of aortic valvular endothelial cells following external mechanical stress on the aortic valve leaflets.
5.1. Functional morphology and mechanical stimuli
Several studies have already highlighted the intricate relationship between hemodynamics and the mechanical microenvironment(71, 73, 82, 83), corroborating the fact that AV degradation is not a passive process. The active interplay between hemodynamics and valvular biomechanical function is attributed to a dense and highly organized network of ECM and is defined by the building plan of the leaflets. Different layers are specifically built to cope with different mechanical stresses such as leaflet strain, laminar shear stress, oscillatory shear stress and pressure as evidenced by the ECM composition of the leaflets that are packed with interstitial cells for structural stability, collagen, elastic fibers, proteoglycans and glycosaminoglycans, which are lined on both sides with endothelial cells.
Endothelium forms a monolayer of cells providing a barrier function for the blood and the underlying cells and are the first to be exposed to shear stress (Figure 4 and Figure 6). Based on a bulk of endothelial function research throughout the body, it is rather peculiar that it took quite some time to place endothelial dysfunction at the basis of valvular degeneration(84). Studies on functional properties of AV endothelium show unique properties compared to other vascular endothelium where the alignment of endothelial cells with regards to the orientation of flow form the most striking difference(85, 86). AV endothelial cells show a perpendicular alignment to flow, a process that is also present in studies using valvular endothelial cells without the presence of an aligned substrate(65, 85). This particular alignment was shown to be dependent on cytoskeletal reorientation, however, stimulated by specific endothelial derived mechanotransduction pathways and differential gene expression. Next to that, compared to vascular endothelial cells, valvular endothelium shows a higher proliferative rate(87) and location of the valvular endothelial (ventricular or aortic side) also seems to play a role in pathway activation. Specific biomechanical profiles and types of shear on the aortic side lead to higher levels of calcification-associated gene and BMP-4 expression whilst on the ventricular side, inflammation associated gene expression together with BMP-4 expression are upregulated(86, 88, 89). Importantly, different shear stresses are exerted onto the two sides of the AV leaflets where the aortic side is exposed to interrupted low-shear stress as compared to the high-shear stress ventricular side with a peak of 70 dynes/m2(90). Those differences in flow patterns on either sides of the valve leaflets are sensed by the glycocalyx activating signal pathways thereby releasing endothelium-derived vasoactive substances, such as nitric oxide, that play a role in valvular stiffness(9). The endothelial dysfunction, initiated by lipid deposition, inflammation, mechanical stimuli and other risk factors – e.g. smoking – produces reactive molecules called reactive oxygen species (ROS)(28), and stimulates a cascade of signaling molecules through valvular interstitial cells (VICs), including TGF-β, interleukin-6, TNF and BMP-2(58, 91). The accumulation of these ROS induces several ROS-mediated mechanisms that, in turn, stimulate calcification, mineralization, apoptosis and osteogenesis, clinically encountered as CAVD (Figure 4). This figure captures key aspects of risk factors, origin and actionable targets of AVD.
VICs are highly plastic cells that can alter phenotype, form the dominant AV cell type and play a crucial role in architectural maintenance and biomechanical functionality of the valve(92). Optimal biomechanics can be attributed to the ECM as it functions as an integrator between form and function and provides several signaling molecules.
In healthy adults, extracellular homeostasis is regulated by these interstitial cells and mediate valvular remodeling through a balanced secretion of matrix degradation enzymes, including matrix metalloproteinases (MMPs) and their inhibitors (TIMPs), and deposition of structural ECM components within the layers(84), which also play a role in thoracic aortic aneurysm formation(93). VICs and inflammatory cells stimulate expression of MMP1,2,9 and cathepsins resulting in abnormal ECM remodeling which lies at the basis of valvular deterioration(86, 92, 94). Deterioration of the valve is based on activated MMP’s and cathepsins degrading collagen and elastin with ensuing pro-inflammatory response leading to calcification(95). Furthermore, VICs secrete ECM components such as hyaluronan and collagen which will deposit in a disorganized fashion thereby altering valvular stiffness and biomechanical profile which is aggravated by a VIC transition to osteoblast-like cells(96). It may be clear that not only altered shear stresses but the whole complex interaction between endothelial (dys)function and the ECM will have their effect on valvular phenotype resulting in inflammation, degeneration and calcification.
5.2. Adaptive remodeling
Prosthetic valve replacement may not sufficiently restore natural valve function, whereas the Ross procedure allows for a living substitute, even showing growth abilities in children(97, 98). It has been correctly postulated that the pulmonary autograft possesses the ability to adapt to the systemic environment by a phenotypic switch to an aortic phenotype after the Ross procedure, as the pulmonary and aortic roots share a common embryological genesis – the conotruncus(99). Explanted autografts are populated by viable valvular interstitial and endothelial cells after several years(100). This process is called adaptive remodeling and is facilitated by gradually exposing the autograft to the systemic environment, for example by systolic blood pressure management below 110mmHg during the first 6-12 months postoperatively(101). These adaptive capabilities all result from the concepts of mechanotransduction and activation of adaptive messenger pathways, as described here. This is also suggested by expression of the gene EphrinB2 in autograft endothelium after the Ross procedure, which is a biomarker of left-sided, but not right-sided, heart valve endothelium. This induced expression of EphrinB2 stimulates ECM remodeling, leading to increased production of smooth muscle actin(100, 102). Surgical modifications – i.e., autograft reinforcement(101, 103, 104) – and adjuncts to conventional postoperative management strategies – i.e., meticulous beta-blocker-driven blood pressure regulation – have been proposed, some to promote adaptive remodeling and all to prevent autograft dilatation. Given the concepts put forward by this review, it appears salient to realize that no technique is perfect and all choices will affect outcomes, and there should be a balance between the support provided and dynamism preserved. The growing data favoring the Ross procedure support a reevaluation of current guidelines for the treatment of AVD, although the adoption of the Ross procedure should be carefully balanced with its increased technical complexity in Ross centers of excellence(105, 106).
From a biomechanical standpoint, suboptimal alignment of the components of the AV will increase shear (oscillatory) stress on the AV leaflets and proximal aorta, additional to systolic loss of energy in the LV(107). As an example, chronic oscillatory stress of the different component parts is thought to cause premature deterioration after the subcoronary Ross operation(107). Adjuncts performed to the total root technique of the Ross procedure should thoughtfully balance support with maintenance of valve dynamism.
5.3. Calcific aortic valve disease
Portrayed in Figure 4, CAVD is governed by important and unalterable risk factors, such as BAV morphology and patient age. An additional risk is posed by elevated blood pressure, elevated plasma lipoprotein(a) levels and the presence of diabetes mellitus or obesity – i.e. truly modifiable factors. Specifically, key studies support a principal, and perhaps causal, role for lipoprotein(a) in CAVD(108, 109) and its progression(110). There have been seminal genetic and molecular studies that have claimed the WNT–β-catenin, Notch and MYOCD pathways to be involved in the control and commitment of heart valve cells to a fibrocalcific lineage(111). The activation of these pathways, also involved in the varying embryological steps of AV formation, may contribute to CAVD. Endothelial activation of second messenger pathways related to inflammation, metabolism and bone formation, instigating this fibrocalcific lineage of the valvular cells, have been shown to lie at the basis of CAVD (Figure 4). Furthermore, circulating osteoprogenitors, likely arising from the bone marrow, that are recruited and are capable of creating a bone-like microenvironment, contribute to valve ossification(112-114). Indeed, the effects of such pathways of valvar degeneration have been described more than a decade ago. The next step is the identification of actionable targets based on these hypotheses, which is the subject of extensive ongoing studies(111).
Several examples of potential therapeutic targets contributing to CAVD initiation include 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, lipoprotein(a), angiotensin II, angiotensin-converting enzyme, and matrix metalloproteinases(6, 108, 115). Toll-like receptors (TLRs) function at the interface between tissue repair and innate immunity pathways(116) and it was very recently shown that the TLR3 pathway is an evolutionarily conserved pathway that regulates CAVD later in life(116).
With increasing understanding of regulatory cellular and genetic pathways, therapeutic targets may be identified and further investigated in a (pre)clinical setting. The yet unmet potential of these targets is to be elucidated by research focusing on cellular mechanisms of these drugs.
6.(. Surgical) Treatment
6.1. Evidence-based medicine and clinical decision-making
Evidence-based medicine has previously been elegantly illustrated as a three-legged stool, which exemplifies that the best available evidence is just one leg of this stool(117). The other two legs, physician’s skills and expertise and patient values and expectations, cannot be left out of the equation during clinical decisions. In other words, evidence-based medicine is not “cookbook” medicine(117, 118). Prosthetic valve selection still carries several challenges pertaining to a lack of robust evidence and widely varying patient values and expectations between individuals. Current options for AVR include bioprosthetic AVR (surgically or transcatheter), mechanical AVR, the Ross procedure and homograft AVR(119, 120). Bioprostheses are commercially available and do not require lifelong anticoagulation, but exhibit limited durability(119). Mechanical prostheses are designed to last a lifetime, but produce a ticking sound and have a thrombogenic surface, therefore requiring lifelong anticoagulation, translating to increased bleeding and thrombo-embolism hazards(121). Homografts come from human donor tissue and do not require anticoagulation, but they show premature calcifications and early failure(122, 123). The Ross procedure is the only living aortic valve substitute available(12), translating to optimal hemodynamics, requires no anticoagulation, and has excellent long-term outcomes in experienced hands(106, 122). However, it transforms single-valve disease to double-valve disease and is technically demanding(124). Unique benefits and drawbacks of all substitutes become immediately clear, but it remains a challenge to implement this into the decision-making process. Prostheses still cannot reproduce the complexity of human nature, and Donald Ross correctly postulated that a living valve substitute was necessary to ensure longevity of a valve substitute(12). The Ross procedure has known times of little acceptance and adoption in clinical practice, with a trough around the year 2010(125). Previous data on the long-term outcomes of this operation have been discouraging in some instances, but novel, contemporary data show excellent long-term results with survival that is comparable to the matched general population(106, 122, 126). Insights into the technical success factors and improvements in patient selection have led to a standardized operation that is reproducible. It is essential to thoughtfully balance the recent enthusiasm for the Ross procedure with its technical complexity by concentrating them in Ross centers of excellence(106, 127).
Decision-making in AVD is complex and entails much more than available evidence and the clinical state of the patient. There is an ongoing shift toward tailoring treatment to the individual patient’s needs and circumstances, taking patient values and goals into account, as well as the short and long-term advantages and disadvantages of different treatment options (survival and complications, quality of life). The lifetime aspect of AVD adds another dimension to the decision-making process, as decisions made now will undoubtedly influence later decisions and outcome. This makes one appreciate the potential harm of avoiding risk in the short term, since this may produce higher risks in the long term(128). So, individuals do not only benefit from tailored treatment options at the present day, but also in the future, all while taking into account patient values and expectations. In this regard, one should aim for strategic planning of interventions over a lifetime, bearing in mind also the options for a second and perhaps third intervention during index procedure planning, in an informed shared decision-making process together with the patient.
We can acknowledge that no valve substitute or treatment solution is perfect. Circling back to the basis of this review, on the other hand, we can now appreciate that the Ross procedure comes closest to a perfect solution in terms of its biomechanics, embryological origin, anatomy/geometry, gene transcription and cellular responses. Novel treatment options are direly needed to meet the needs of patients with AVD. For the future, tissue engineering of heart valves (TEHV), to be regarded a byproduct of the Ross procedure, meets great interest. TEHV can produce a living valve able to emulate the sophisticated functions of a native valve(129, 130). The concept of in-situ regeneration, which uses the microenvironment as a natural bioreactor, has produced encouraging results in recent years(131-133). Cellular repopulation of an acellular scaffold has recently been successfully shown in a sheep model(133), with endothelial cells and nerves connecting with contractile cells and blood vessels, just like in a native valve(133).
A method able to simulate individual patient lives and generate age- and sex-specific estimates of patient outcome, microsimulation models may fulfill a role in objectifying these different lifetime treatment pathways and their outcomes(97, 119, 121, 134, 135). For the future, these models should become more patient-tailored and its potential for modeling sequential treatments over a lifetime should be explored.
7. Conclusions and Directions
The interplay seen in a healthy, native aortic root and valve is a perfectly synchronized, almost magical, dynamic process and the undeniable clinical benefits of a living valve have been repetitively addressed. All the more the conclusions of this review support this paradigm by providing an integrated approach to embryological, genetic, and biomechanical background to valve morphology, function and benefits of hemodynamically-centered treatment strategies. Clinical decision-making in AVD remains complex and is based on much more than scientific evidence, but the Ross procedure comes closest to offering patients a durable solution while allowing multiple options for future therapies. This review furthermore identified gaps that invite research on cellular, (epi)genetic and mechanical factors in AVD formation and progression.
Author Contributions
Supervision: Takkenberg, Rega, Taverne, Concept and design: Taverne, Rega, Van Hoof, Takkenberg, Schuermans, Notenboom, Administrative, technical, or material support: Takkenberg, Rega, Taverne Data acquisition: analysis & interpretation: -Drafting of the manuscript: Taverne, Notenboom, Van Hoof, Rega, Schuermans, Takkenberg. Statistical analysis: -Critical revision of the manuscript for intellectual content: Taverne, Notenboom, Van Hoof, Rega, Schuermans, Takkenberg.
Funding
The authors (MLN, LvH, AS, JJMT, FRR, YJHJT) received no funding for this research.
Disclosures
The authors (MLN, LvH, AS, JJMT, FRR, YJHJT) have nothing to disclose.
References
- d'Arcy JL, Prendergast BD, Chambers JB, Ray SG, Bridgewater B. Valvular heart disease: the next cardiac epidemic. Heart. 2011;97(2):91-3. [CrossRef]
- Coffey S, Cox B, Williams MJ. Lack of progress in valvular heart disease in the pre-transcatheter aortic valve replacement era: increasing deaths and minimal change in mortality rate over the past three decades. Am Heart J. 2014;167(4):562-7 e2. [CrossRef]
- Rosenhek R, Zilberszac R, Schemper M, Czerny M, Mundigler G, Graf S, et al. Natural history of very severe aortic stenosis. Circulation. 2010;121(1):151-6. [CrossRef]
- Ross J, Jr., Braunwald E. Aortic stenosis. Circulation. 1968;38(1 Suppl):61-7.
- Philippe G, Rahul PS, Robert JC, Lucy A, Omar MA, Konstantinos PK, et al. The Mortality Burden of Untreated Aortic Stenosis. Journal of the American College of Cardiology. 2023;82(22):2101-9.
- Goldbarg SH, Elmariah S, Miller MA, Fuster V. Insights Into Degenerative Aortic Valve Disease. Journal of the American College of Cardiology. 2007;50(13):1205-13. [CrossRef]
- El-Hamamsy I, Chester AH, Yacoub MH. Cellular regulation of the structure and function of aortic valves. Journal of Advanced Research. 2010;1(1):5-12. [CrossRef]
- El-Hamamsy I, Yacoub MH, Chester AH. Neuronal regulation of aortic valve cusps. Curr Vasc Pharmacol. 2009;7(1):40-6. [CrossRef]
- El-Hamamsy I, Balachandran K, Yacoub MH, Stevens LM, Sarathchandra P, Taylor PM, et al. Endothelium-dependent regulation of the mechanical properties of aortic valve cusps. J Am Coll Cardiol. 2009;53(16):1448-55. [CrossRef]
- Boodhwani M, de Kerchove L, Glineur D, Poncelet A, Rubay J, Astarci P, et al. Repair-oriented classification of aortic insufficiency: impact on surgical techniques and clinical outcomes. J Thorac Cardiovasc Surg. 2009;137(2):286-94. [CrossRef]
- Misfeld M, Chester AH, Sievers HH, Yacoub MH. Biological mechanisms influencing the function of the aortic root. J Card Surg. 2002;17(4):363-8. [CrossRef]
- Ross, DN. Replacement of aortic and mitral valves with a pulmonary autograft. Lancet. 1967;2(7523):956-8. [CrossRef]
- Lansac E, de Kerchove L. Aortic valve repair techniques: state of the art. Eur J Cardiothorac Surg. 2018;53(6):1101-7.
- Combs MD, Yutzey KE. Heart valve development: regulatory networks in development and disease. Circ Res. 2009;105(5):408-21.
- Gittenberger-de Groot AC, Bartelings MM, Deruiter MC, Poelmann RE. Basics of cardiac development for the understanding of congenital heart malformations. Pediatr Res. 2005;57(2):169-76.
- Moorman AF, Christoffels VM. Cardiac chamber formation: development, genes, and evolution. Physiol Rev. 2003;83(4):1223-67. [CrossRef]
- Lamers WH, Moorman AF. Cardiac septation: a late contribution of the embryonic primary myocardium to heart morphogenesis. Circ Res. 2002;91(2):93-103.
- Soufan AT, van den Hoff MJ, Ruijter JM, de Boer PA, Hagoort J, Webb S, et al. Reconstruction of the patterns of gene expression in the developing mouse heart reveals an architectural arrangement that facilitates the understanding of atrial malformations and arrhythmias. Circ Res. 2004;95(12):1207-15. [CrossRef]
- Schroeder JA, Jackson LF, Lee DC, Camenisch TD. Form and function of developing heart valves: coordination by extracellular matrix and growth factor signaling. J Mol Med (Berl). 2003;81(7):392-403. [CrossRef]
- Wirrig EE, Yutzey KE. Conserved transcriptional regulatory mechanisms in aortic valve development and disease. Arterioscler Thromb Vasc Biol. 2014;34(4):737-41.
- Hinton RB, Jr., Lincoln J, Deutsch GH, Osinska H, Manning PB, Benson DW, et al. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ Res. 2006;98(11):1431-8.
- Martin PS, Kloesel B, Norris RA, Lindsay M, Milan D, Body SC. Embryonic Development of the Bicuspid Aortic Valve. Journal of Cardiovascular Development and Disease. 2015;2(4):248-72. [CrossRef]
- Rabkin-Aikawa E, Farber M, Aikawa M, Schoen FJ. Dynamic and reversible changes of interstitial cell phenotype during remodeling of cardiac valves. J Heart Valve Dis. 2004;13(5):841-7.
- de Lange FJ, Moorman AF, Anderson RH, Männer J, Soufan AT, de Gier-de Vries C, et al. Lineage and morphogenetic analysis of the cardiac valves. Circ Res. 2004;95(6):645-54. [CrossRef]
- Butcher JT, Markwald RR. Valvulogenesis: the moving target. Philos Trans R Soc Lond B Biol Sci. 2007;362(1484):1489-503.
- Gobergs R, Salputra E, Lubaua I. Hypoplastic left heart syndrome: a review. Acta Med Litu. 2016;23(2):86-98. [CrossRef]
- Rahman A, DeYoung T, Cahill LS, Yee Y, Debebe SK, Botelho O, et al. A mouse model of hypoplastic left heart syndrome demonstrating left heart hypoplasia and retrograde aortic arch flow. Dis Model Mech. 2021;14(11). [CrossRef]
- Greenberg HZE, Zhao G, Shah AM, Zhang M. Role of oxidative stress in calcific aortic valve disease and its therapeutic implications. Cardiovascular Research. 2021;118(6):1433-51. [CrossRef]
- Dayawansa NH, Baratchi S, Peter K. Uncoupling the Vicious Cycle of Mechanical Stress and Inflammation in Calcific Aortic Valve Disease. Front Cardiovasc Med. 2022;9:783543. [CrossRef]
- Peng Q, Shan D, Cui K, Li K, Zhu B, Wu H, et al. The Role of Endothelial-to-Mesenchymal Transition in Cardiovascular Disease. Cells. 2022;11(11). [CrossRef]
- Henderson DJ, Eley L, Turner JE, Chaudhry B. Development of the Human Arterial Valves: Understanding Bicuspid Aortic Valve. Front Cardiovasc Med. 2021;8:802930. [CrossRef]
- Hutson MR, Kirby ML. Model systems for the study of heart development and disease: Cardiac neural crest and conotruncal malformations. Seminars in Cell & Developmental Biology. 2007;18(1):101-10. [CrossRef]
- George RM, Maldonado-Velez G, Firulli AB. The heart of the neural crest: cardiac neural crest cells in development and regeneration. Development. 2020;147(20). [CrossRef]
- Poelmann RE, Mikawa T, Gittenberger-de Groot AC. Neural crest cells in outflow tract septation of the embryonic chicken heart: differentiation and apoptosis. Dev Dyn. 1998;212(3):373-84.
- Chakraborty S, Combs MD, Yutzey KE. Transcriptional regulation of heart valve progenitor cells. Pediatr Cardiol. 2010;31(3):414-21. [CrossRef]
- Maleki S, Poujade FA, Bergman O, Gådin JR, Simon N, Lång K, et al. Endothelial/Epithelial Mesenchymal Transition in Ascending Aortas of Patients With Bicuspid Aortic Valve. Front Cardiovasc Med. 2019;6:182. [CrossRef]
- Armstrong EJ, Bischoff J. Heart valve development: endothelial cell signaling and differentiation. Circ Res. 2004;95(5):459-70.
- Camenisch TD, Molin DG, Person A, Runyan RB, Gittenberger-de Groot AC, McDonald JA, et al. Temporal and distinct TGFbeta ligand requirements during mouse and avian endocardial cushion morphogenesis. Dev Biol. 2002;248(1):170-81.
- Ayoub S, Ferrari G, Gorman RC, Gorman JH, Schoen FJ, Sacks MS. Heart Valve Biomechanics and Underlying Mechanobiology. Compr Physiol. 2016;6(4):1743-80.
- Rochais F, Mesbah K, Kelly RG. Signaling pathways controlling second heart field development. Circ Res. 2009;104(8):933-42. [CrossRef]
- Grewal N, Gittenberger-de Groot AC, Lindeman JH, Klautz A, Driessen A, Klautz RJM, et al. Normal and abnormal development of the aortic valve and ascending aortic wall: a comprehensive overview of the embryology and pathology of the bicuspid aortic valve. Ann Cardiothorac Surg. 2022;11(4):380-8. [CrossRef]
- Harmon AW, Nakano A. Nkx2-5 lineage tracing visualizes the distribution of second heart field-derived aortic smooth muscle. Genesis. 2013;51(12):862-9. [CrossRef]
- Sawada H, Rateri DL, Moorleghen JJ, Majesky MW, Daugherty A. Smooth Muscle Cells Derived From Second Heart Field and Cardiac Neural Crest Reside in Spatially Distinct Domains in the Media of the Ascending Aorta-Brief Report. Arterioscler Thromb Vasc Biol. 2017;37(9):1722-6. [CrossRef]
- George V, Colombo S, Targoff KL. An early requirement for nkx2.5 ensures the first and second heart field ventricular identity and cardiac function into adulthood. Dev Biol. 2015;400(1):10-22. [CrossRef]
- Garside VC, Chang AC, Karsan A, Hoodless PA. Co-ordinating Notch, BMP, and TGF-β signaling during heart valve development. Cell Mol Life Sci. 2013;70(16):2899-917.
- Walker GA, Masters KS, Shah DN, Anseth KS, Leinwand LA. Valvular myofibroblast activation by transforming growth factor-beta: implications for pathological extracellular matrix remodeling in heart valve disease. Circ Res. 2004;95(3):253-60.
- Niessen K, Fu Y, Chang L, Hoodless PA, McFadden D, Karsan A. Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J Cell Biol. 2008;182(2):315-25. [CrossRef]
- Loomes KM, Taichman DB, Glover CL, Williams PT, Markowitz JE, Piccoli DA, et al. Characterization of Notch receptor expression in the developing mammalian heart and liver. Am J Med Genet. 2002;112(2):181-9. [CrossRef]
- Varadkar P, Kraman M, Despres D, Ma G, Lozier J, McCright B. Notch2 is required for the proliferation of cardiac neural crest-derived smooth muscle cells. Dev Dyn. 2008;237(4):1144-52. [CrossRef]
- Timmerman LA, Grego-Bessa J, Raya A, Bertrán E, Pérez-Pomares JM, Díez J, et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004;18(1):99-115. [CrossRef]
- Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437(7056):270-4. [CrossRef]
- Kamath BM, Bauer RC, Loomes KM, Chao G, Gerfen J, Hutchinson A, et al. NOTCH2 mutations in Alagille syndrome. J Med Genet. 2012;49(2):138-44.
- Fu Y, Chang A, Chang L, Niessen K, Eapen S, Setiadi A, et al. Differential regulation of transforming growth factor beta signaling pathways by Notch in human endothelial cells. J Biol Chem. 2009;284(29):19452-62.
- Kokubo H, Miyagawa-Tomita S, Tomimatsu H, Nakashima Y, Nakazawa M, Saga Y, et al. Targeted disruption of hesr2 results in atrioventricular valve anomalies that lead to heart dysfunction. Circ Res. 2004;95(5):540-7.
- Cripe L, Andelfinger G, Martin LJ, Shooner K, Benson DW. Bicuspid aortic valve is heritable. J Am Coll Cardiol. 2004;44(1):138-43. [CrossRef]
- Tessler I, Albuisson J, Piñeiro-Sabarís R, Verstraeten A, Kamber Kaya HE, Siguero-Álvarez M, et al. Novel Association of the NOTCH Pathway Regulator MIB1 Gene With the Development of Bicuspid Aortic Valve. JAMA Cardiology. 2023;8(8):721-31. [CrossRef]
- MacGrogan D, D'Amato G, Travisano S, Martinez-Poveda B, Luxán G, Del Monte-Nieto G, et al. Sequential Ligand-Dependent Notch Signaling Activation Regulates Valve Primordium Formation and Morphogenesis. Circ Res. 2016;118(10):1480-97. [CrossRef]
- Small AM, Peloso GM, Linefsky J, Aragam J, Galloway A, Tanukonda V, et al. Multiancestry Genome-Wide Association Study of Aortic Stenosis Identifies Multiple Novel Loci in the Million Veteran Program. Circulation. 2023;147(12):942-55. [CrossRef]
- Gehlen J, Stundl A, Debiec R, Fontana F, Krane M, Sharipova D, et al. Elucidation of the genetic causes of bicuspid aortic valve disease. Cardiovascular Research. 2022;119(3):857-66. [CrossRef]
- Anderson, RH. Clinical anatomy of the aortic root. Heart. 2000;84(6):670-3.
- Anderson, RH. The surgical anatomy of the aortic root. Multimed Man Cardiothorac Surg. 2007;2007(102):mmcts 2006 002527. [CrossRef]
- Yacoub MH, Kilner PJ, Birks EJ, Misfeld M. The aortic outflow and root: a tale of dynamism and crosstalk. Ann Thorac Surg. 1999;68(3 Suppl):S37-43. [CrossRef]
- Sarsam MA, Yacoub M. Remodeling of the aortic valve anulus. J Thorac Cardiovasc Surg. 1993;105(3):435-8. [CrossRef]
- David TE, Feindel CM. An aortic valve-sparing operation for patients with aortic incompetence and aneurysm of the ascending aorta. J Thorac Cardiovasc Surg. 1992;103(4):617-21; discussion 22.
- Chester AH, El-Hamamsy I, Butcher JT, Latif N, Bertazzo S, Yacoub MH. The living aortic valve: From molecules to function. Glob Cardiol Sci Pract. 2014;2014(1):52-77. [CrossRef]
- Rajamannan, NM. Bicuspid aortic valve disease: the role of oxidative stress in Lrp5 bone formation. Cardiovasc Pathol. 2011;20(3):168-76. [CrossRef]
- Fries R, Graeter T, Aicher D, Reul H, Schmitz C, Böhm M, et al. In vitro comparison of aortic valve movement after valve-preserving aortic replacement. The Journal of Thoracic and Cardiovascular Surgery. 2006;132(1):32-7. [CrossRef]
- Arjunon S, Rathan S, Jo H, Yoganathan AP. Aortic valve: mechanical environment and mechanobiology. Ann Biomed Eng. 2013;41(7):1331-46. [CrossRef]
- Bellhouse BJ, Talbot L. The fluid mechanics of the aortic valve. Journal of Fluid Mechanics. 1969;35:721 - 35. [CrossRef]
- Campinho P, Vilfan A, Vermot J. Blood Flow Forces in Shaping the Vascular System: A Focus on Endothelial Cell Behavior. Front Physiol. 2020;11:552. [CrossRef]
- Yacoub MH, Aguib H, Gamrah MA, Shehata N, Nagy M, Donia M, et al. Aortic root dynamism, geometry, and function after the remodeling operation: Clinical relevance. J Thorac Cardiovasc Surg. 2018;156(3):951-62 e2. [CrossRef]
- Bellhouse B, Bellhouse F. Fluid mechanics of model normal and stenosed aortic valves. Circ Res. 1969;25(6):693-704.
- Cheng A, Dagum P, Miller DC. Aortic root dynamics and surgery: from craft to science. Philos Trans R Soc Lond B Biol Sci. 2007;362(1484):1407-19. [CrossRef]
- Bruno RM, Climie R, Gallo A. Aortic pulsatility drives microvascular organ damage in essential hypertension: New evidence from choroidal thickness assessment. J Clin Hypertens (Greenwich). 2021;23(5):1039-40. [CrossRef]
- Um KJ, McClure GR, Belley-Cote EP, Gupta S, Bouhout I, Lortie H, et al. Hemodynamic outcomes of the Ross procedure versus other aortic valve replacement: a systematic review and meta-analysis. J Cardiovasc Surg (Torino). 2018;59(3):462-70. [CrossRef]
- Stewart S, Afoakwah C, Chan Y-K, Strom JB, Playford D, Strange GA. Counting the cost of premature mortality with progressively worse aortic stenosis in Australia: a clinical cohort study. The Lancet Healthy Longevity. 2022;3(9):e599-e606. [CrossRef]
- Harris P, Kuppurao L. Quantitative Doppler echocardiography. BJA Education. 2015;16(2):46-52. [CrossRef]
- Chan V, Rubens F, Boodhwani M, Mesana T, Ruel M. Determinants of persistent or recurrent congestive heart failure after contemporary surgical aortic valve replacement. J Heart Valve Dis. 2014;23(6):665-70.
- Duebener LF, Stierle U, Erasmi A, Bechtel MF, Zurakowski D, Böhm JO, et al. Ross procedure and left ventricular mass regression. Circulation. 2005;112(9 Suppl):I415-22.
- Hauser M, Bengel FM, Kühn A, Sauer U, Zylla S, Braun SL, et al. Myocardial blood flow and flow reserve after coronary reimplantation in patients after arterial switch and ross operation. Circulation. 2001;103(14):1875-80. [CrossRef]
- Gaudino M, Piatti F, Lau C, Sturla F, Weinsaft JW, Weltert L, et al. Aortic flow after valve sparing root replacement with or without neosinuses reconstruction. J Thorac Cardiovasc Surg. 2019;157(2):455-65. [CrossRef]
- Balachandran K, Sucosky P, Yoganathan AP. Hemodynamics and mechanobiology of aortic valve inflammation and calcification. Int J Inflam. 2011;2011:263870. [CrossRef]
- Rego BV, Sacks MS. A functionally graded material model for the transmural stress distribution of the aortic valve leaflet. J Biomech. 2017;54:88-95. [CrossRef]
- Kodigepalli KM, Thatcher K, West T, Howsmon DP, Schoen FJ, Sacks MS, et al. Biology and Biomechanics of the Heart Valve Extracellular Matrix. J Cardiovasc Dev Dis. 2020;7(4). [CrossRef]
- Butcher JT, Penrod AM, García AJ, Nerem RM. Unique morphology and focal adhesion development of valvular endothelial cells in static and fluid flow environments. Arterioscler Thromb Vasc Biol. 2004;24(8):1429-34. [CrossRef]
- Bäck M, Gasser TC, Michel J-B, Caligiuri G. Biomechanical factors in the biology of aortic wall and aortic valve diseases. Cardiovascular Research. 2013;99(2):232-41. [CrossRef]
- Farivar RS, Cohn LH, Soltesz EG, Mihaljevic T, Rawn JD, Byrne JG. Transcriptional profiling and growth kinetics of endothelium reveals differences between cells derived from porcine aorta versus aortic valve. Eur J Cardiothorac Surg. 2003;24(4):527-34. [CrossRef]
- Simmons CA, Grant GR, Manduchi E, Davies PF. Spatial heterogeneity of endothelial phenotypes correlates with side-specific vulnerability to calcification in normal porcine aortic valves. Circ Res. 2005;96(7):792-9. [CrossRef]
- Butcher JT, Tressel S, Johnson T, Turner D, Sorescu G, Jo H, et al. Transcriptional profiles of valvular and vascular endothelial cells reveal phenotypic differences: influence of shear stress. Arterioscler Thromb Vasc Biol. 2006;26(1):69-77.
- Yap CH, Saikrishnan N, Yoganathan AP. Experimental measurement of dynamic fluid shear stress on the ventricular surface of the aortic valve leaflet. Biomech Model Mechanobiol. 2012;11(1-2):231-44. [CrossRef]
- Yu Chen H, Dina C, Small AM, Shaffer CM, Levinson RT, Helgadóttir A, et al. Dyslipidemia, inflammation, calcification, and adiposity in aortic stenosis: a genome-wide study. European Heart Journal. 2023;44(21):1927-39. [CrossRef]
- Scott AJ, Simon LR, Hutson HN, Porras AM, Masters KS. Engineering the aortic valve extracellular matrix through stages of development, aging, and disease. J Mol Cell Cardiol. 2021;161:1-8. [CrossRef]
- El-Hamamsy I, Yacoub MH. Cellular and molecular mechanisms of thoracic aortic aneurysms. Nat Rev Cardiol. 2009;6(12):771-86.
- Leopold, JA. Cellular mechanisms of aortic valve calcification. Circ Cardiovasc Interv. 2012;5(4):605-14. [CrossRef]
- Chen JH, Simmons CA. Cell-matrix interactions in the pathobiology of calcific aortic valve disease: critical roles for matricellular, matricrine, and matrix mechanics cues. Circ Res. 2011;108(12):1510-24.
- Yip CY, Chen JH, Zhao R, Simmons CA. Calcification by valve interstitial cells is regulated by the stiffness of the extracellular matrix. Arterioscler Thromb Vasc Biol. 2009;29(6):936-42. [CrossRef]
- Notenboom ML, Schuermans A, Etnel JRG, Veen KM, van de Woestijne PC, Rega FR, et al. Paediatric aortic valve replacement: a meta-analysis and microsimulation study. Eur Heart J. 2023. [CrossRef]
- Schoof PH, Cromme-Dijkhuis AH, Bogers JJ, Thijssen EJ, Witsenburg M, Hess J, et al. Aortic root replacement with pulmonary autograft in children. J Thorac Cardiovasc Surg. 1994;107(2):367-73. [CrossRef]
- Van Hoof L, Verbrugghe P, Jones EAV, Humphrey JD, Janssens S, Famaey N, et al. Understanding Pulmonary Autograft Remodeling After the Ross Procedure: Stick to the Facts. Front Cardiovasc Med. 2022;9:829120. [CrossRef]
- Rabkin-Aikawa E, Aikawa M, Farber M, Kratz JR, Garcia-Cardena G, Kouchoukos NT, et al. Clinical pulmonary autograft valves: pathologic evidence of adaptive remodeling in the aortic site. J Thorac Cardiovasc Surg. 2004;128(4):552-61. [CrossRef]
- Mazine A, El-Hamamsy I, Verma S, Peterson MD, Bonow RO, Yacoub MH, et al. Ross Procedure in Adults for Cardiologists and Cardiac Surgeons: JACC State-of-the-Art Review. J Am Coll Cardiol. 2018;72(22):2761-77.
- Gorczynski A, Trenkner M, Anisimowicz L, Gutkowski R, Drapella A, Kwiatkowska E, et al. Biomechanics of the pulmonary autograft valve in the aortic position. Thorax. 1982;37(7):535-9. [CrossRef]
- Mazine A, El-Hamamsy I. The Ross procedure is an excellent operation in non-repairable aortic regurgitation: insights and techniques. Ann Cardiothorac Surg. 2021;10(4):463-75. [CrossRef]
- Tanaka D, Mazine A, Ouzounian M, El-Hamamsy I. Supporting the Ross procedure: preserving root physiology while mitigating autograft dilatation. Curr Opin Cardiol. 2022;37(2):180-90. [CrossRef]
- Ismail E-H, Patrick TOG, David HA. The Ross Procedure. Journal of the American College of Cardiology. 2022;79(10):1006-9.
- Notenboom ML, Melina G, Veen K, De Robertis F, Coppola G, De Siena P, et al. Long-Term Clinical and Echocardiographic Outcomes Following the Ross Procedure: A Post Hoc Analysis of a Randomized Controlled Trial. JAMA Cardiol. 2023;In press.
- Latif N, Mahgoub A, Nagy M, Sarathchandra P, Yacoub MH. Severe degeneration of a sub-coronary pulmonary autograft in a young adult. Glob Cardiol Sci Pract. 2021;2021(2):e202114. [CrossRef]
- Thanassoulis G, Campbell CY, Owens DS, Smith JG, Smith AV, Peloso GM, et al. Genetic Associations with Valvular Calcification and Aortic Stenosis. New England Journal of Medicine. 2013;368(6):503-12. [CrossRef]
- Torzewski M, Ravandi A, Yeang C, Edel A, Bhindi R, Kath S, et al. Lipoprotein(a) Associated Molecules are Prominent Components in Plasma and Valve Leaflets in Calcific Aortic Valve Stenosis. JACC Basic Transl Sci. 2017;2(3):229-40. [CrossRef]
- Capoulade R, Chan KL, Yeang C, Mathieu P, Bossé Y, Dumesnil JG, et al. Oxidized Phospholipids, Lipoprotein(a), and Progression of Calcific Aortic Valve Stenosis. Journal of the American College of Cardiology. 2015;66(11):1236-46. [CrossRef]
- Moncla LM, Briend M, Bossé Y, Mathieu P. Calcific aortic valve disease: mechanisms, prevention and treatment. Nat Rev Cardiol. 2023;20(8):546-59. [CrossRef]
- Khosla S, Eghbali-Fatourechi GZ. Circulating cells with osteogenic potential. Ann N Y Acad Sci. 2006;1068:489-97. [CrossRef]
- Egan KP, Kim JH, Mohler ER, 3rd, Pignolo RJ. Role for circulating osteogenic precursor cells in aortic valvular disease. Arterioscler Thromb Vasc Biol. 2011;31(12):2965-71.
- Eghbali-Fatourechi GZ, Lamsam J, Fraser D, Nagel D, Riggs BL, Khosla S. Circulating osteoblast-lineage cells in humans. N Engl J Med. 2005;352(19):1959-66. [CrossRef]
- Goel SS, Kleiman NS, Zoghbi WA, Reardon MJ, Kapadia SR. Renin-Angiotensin System Blockade in Aortic Stenosis: Implications Before and After Aortic Valve Replacement. J Am Heart Assoc. 2020;9(18):e016911. [CrossRef]
- Gollmann-Tepeköylü C, Graber M, Hirsch J, Mair S, Naschberger A, Pölzl L, et al. Toll-Like Receptor 3 Mediates Aortic Stenosis Through a Conserved Mechanism of Calcification. Circulation. 2023. [CrossRef]
- Mokhles S, Takkenberg JJM, Treasure T. Evidence-Based and Personalized Medicine. It’s [AND] not [OR]. The Annals of Thoracic Surgery. 2017;103(1):351-60.
- David LS, William MCR, Gray JAM, Haynes RB, Richardson WS. Evidence based medicine: what it is and what it isn't. Bmj. 1996;312(7023):71-2. [CrossRef]
- Etnel JRG, Huygens SA, Grashuis P, Pekbay B, Papageorgiou G, Roos Hesselink JW, et al. Bioprosthetic Aortic Valve Replacement in Nonelderly Adults: A Systematic Review, Meta-Analysis, Microsimulation. Circ Cardiovasc Qual Outcomes. 2019;12(2):e005481.
- Goldstone AB, Chiu P, Baiocchi M, Lingala B, Patrick WL, Fischbein MP, et al. Mechanical or Biologic Prostheses for Aortic-Valve and Mitral-Valve Replacement. N Engl J Med. 2017;377(19):1847-57. [CrossRef]
- Korteland NM, Etnel JRG, Arabkhani B, Mokhles MM, Mohamad A, Roos-Hesselink JW, et al. Mechanical aortic valve replacement in non-elderly adults: meta-analysis and microsimulation. Eur Heart J. 2017;38(45):3370-7. [CrossRef]
- El-Hamamsy I, Eryigit Z, Stevens LM, Sarang Z, George R, Clark L, et al. Long-term outcomes after autograft versus homograft aortic root replacement in adults with aortic valve disease: a randomised controlled trial. Lancet. 2010;376(9740):524-31. [CrossRef]
- Yacoub M, Rasmi NR, Sundt TM, Lund O, Boyland E, Radley-Smith R, et al. Fourteen-year experience with homovital homografts for aortic valve replacement. J Thorac Cardiovasc Surg. 1995;110(1):186-93; discussion 93-4. [CrossRef]
- Klieverik LM, Takkenberg JJ, Bekkers JA, Roos-Hesselink JW, Witsenburg M, Bogers AJ. The Ross operation: a Trojan horse? Eur Heart J. 2007;28(16):1993-2000.
- Reece TB, Welke KF, O'Brien S, Grau-Sepulveda MV, Grover FL, Gammie JS. Rethinking the ross procedure in adults. Ann Thorac Surg. 2014;97(1):175-81. [CrossRef]
- El-Hamamsy I, Toyoda N, Itagaki S, Stelzer P, Varghese R, Williams EE, et al. Propensity-Matched Comparison of the Ross Procedure and Prosthetic Aortic Valve Replacement in Adults. J Am Coll Cardiol. 2022;79(8):805-15. [CrossRef]
- Yacoub MH, Notenboom ML, Melina G, Takkenberg JJM. Surgical Heritage: You Had to be There, Ross: The Comeback Kid. Seminars in Thoracic and Cardiovascular Surgery: Pediatric Cardiac Surgery Annual. 2023.
- Tom T, Asif H, Magdi Y. Is there a risk in avoiding risk for younger patients with aortic valve disease? Bmj. 2011;342:d2466.
- Fioretta ES, Motta SE, Lintas V, Loerakker S, Parker KK, Baaijens FPT, et al. Next-generation tissue-engineered heart valves with repair, remodelling and regeneration capacity. Nat Rev Cardiol. 2021;18(2):92-116. [CrossRef]
- Huygens SA, Rutten-van Mölken M, Noruzi A, Etnel JRG, Corro Ramos I, Bouten CVC, et al. What Is the Potential of Tissue-Engineered Pulmonary Valves in Children? Ann Thorac Surg. 2019;107(6):1845-53.
- van Haaften EE, Bouten CVC, Kurniawan NA. Vascular Mechanobiology: Towards Control of In Situ Regeneration. Cells. 2017;6(3).
- Wissing TB, Bonito V, Bouten CVC, Smits A. Biomaterial-driven in situ cardiovascular tissue engineering-a multi-disciplinary perspective. NPJ Regen Med. 2017;2:18. [CrossRef]
- Yacoub MH, Tseng YT, Kluin J, Vis A, Stock U, Smail H, et al. Valvulogenesis of a living, innervated pulmonary root induced by an acellular scaffold. Commun Biol. 2023;6(1):1017. [CrossRef]
- Meccanici F, Notenboom ML, Meijssen J, Smit V, van de Woestijne PC, van den Bosch AE, et al. Long-term surgical outcomes of congenital supravalvular aortic stenosis: a systematic review, meta-analysis and microsimulation study. Eur J Cardiothorac Surg. 2023. [CrossRef]
- Notenboom ML, Rhellab R, Etnel JRG, van den Bogerd N, Veen KM, Taverne Y, et al. Aortic Valve Repair in Neonates, Infants and Children: A Systematic Review, Meta-Analysis and Microsimulation Study. Eur J Cardiothorac Surg. 2023. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



