
Submitted:
08 January 2024
Posted:
08 January 2024
You are already at the latest version
Abstract
This article provides a thorough overview of biomarkers, pathophysiology, and molecular pathways involved in the transition from acute kidney injury (AKI), acute kidney disease (AKD) to chronic kidney disease (CKD). It categorizes the biomarkers of AKI into stress, damage, and functional markers, highlighting their importance in early detection, prognosis, and clinical application. The pathophysiological mechanisms underlying AKI, AKD, including renal hypoperfusion, sepsis, nephrotoxicity, and immune responses, with the impact on renal injury. In addition, various molecules play pivotal roles in inflammation and hypoxia, triggering maladaptive repair, mitochondrial dysfunction, immune system reactions, and cellular senescence of renal cell. Key signaling pathways, such as Wnt/β-Catenin, TGF-β/SMAD, and Hippo/YAP/TAZ, promote fibrosis and impact renal function. The Renin-Angiotensin-Aldosterone System (RAAS) triggers a cascade leading to renal fibrosis, with aldosterone exacerbating oxidative stress and cellular changes that promote fibrosis. Clinical evidence suggests RAS inhibitors may protect against CKD progression, especially post-AKI, though more extensive trials are needed to confirm their full impact.
Keywords:
Acute kidney injury
; Acute kidney disease
; biomarker
; chronic kidney disease
1. Introduction
Impaired renal function is a disease with spectrum according to the duration and progression of renal deterioration. Broadly, this can be divided into three stages: acute kidney injury (AKI), acute kidney disease (AKD) and chronic kidney disease (CKD) [1]. According to the guideline of 2012 KDIGO, the diagnosis of AKI could be defined as 1.5 times increased serum creatinine from baseline or less urine output (<0.5ml/kg/h) within 7 days [2]. AKD is indicated as acute or subacute damage and/or loss of kidney function between 7 and 90 days [3]. CKD is defined as abnormalities of kidney structure or function, present for more than 3 months, with implications for health [4]. Nowadays, kidney disease may classify as the continuum of AKI, AKD, and CKD [5].
Those with AKI had higher risk of incidence of AKD, CKD or even progression to end stage kidney disease (ESKD) [6]. It is believed that there’s a causal relationship between AKI and CKD [7,8]. According to a cohort study revealed that 24.6% patients with AKI develop CKD with 3-year follow-up period [9]. A meta-analysis displayed that the pooled rate of CKD development among patients with AKI is 25.8 per 100 person-years [8]. Besides, it is also believed that patients with AKD had higher risk to progress to CKD [10]. The meta-analysis, encompassing a large cohort of 1,114,012 patients with AKD, demonstrated that 37.2% of these patients are likely to progress to CKD. This finding underscores that the risk of developing CKD is notably higher in patients with AKD compared to those with non-AKD [11].
With the rising incidence, the economic and healthcare burden that cause by AKI or the extent complication is increasing [12]. To prevent the progression of renal function and the further induced complication from AKI, early detection of AKI become quite an important issue in clinical practice. Nevertheless, the routine diagnostic tool for AKI remained serum creatinine, which is a delayed and unreliable biomarker for AKI due to various reasons [13]. It remains a clinical dilemma for direct evaluating the etiologies of AKI apart from renal biopsy despite that numerous biomarkers were proposed within recent decades as indicator for specific damage site of nephron, including Neutrophil Gelatinase-associated Lipocalin (NGAL), Cystatin C, Liver-type Fatty Acid-binding Protein(L-FABP), Kidney Injury Molecule-1(KIM-1), and Interleukin 18(IL-18) and so on [14,15]. Besides, the process of AKI to CKD transition involving numerous complicated molecule mechanisms, which is basically the consequence of cellular injury and maladaptive repair, is still not well elucidated [16,17]. Hence, the current review would focus on the issue of the biomarker including NGAL, Tissue inhibitor of metalloproteinases 2 X Insulin-like growth factor binding protein 7 ([TIMP-2]X[IGFBP7]), KIM-1, L-FABP for early detection of AKI, the pathophysiology of AKI, and the molecular pathway of AKI to CKD transition.
2. Biomarkers of AKI
Extensive research is currently underway to explore novel biomarkers for the early detection and prognosis of AKI. As outlined by the Acute Disease Quality Initiative Consensus Conference on AKI biomarkers, these biomarkers are categorized into three main categories: stress markers, damage markers, and functional markers [18]. Stress markers serve as early indicators of cellular stress, offering insights into the prediction of AKI. Conversely, damage markers, indicate structural damage, which may or may not result in a reduction of renal function. Lastly, functional markers are linked to changes in glomerular filtration, thereby offering a measure of renal function alterations.
2.1. Stress markers
Urinary Dickkopf-3 (DKK3), a glycoprotein originating from kidney tubular epithelial cells (TECs), is utilized in the risk assessment and prediction of AKI. Preoperative levels of urinary DKK3 have been identified as an independent predictor for the occurrence of postoperative AKI [19]. The urinary levels of TIMP-2 and IGFBP-7 serve as markers indicating G1 cell cycle arrest. These markers may show a rapid increase after cellular stress, typically within 4 to 12 hours, even before the occurrence of injury [20,21].
2.2. Damage markers
Alanine aminopeptidase, alkaline phosphatase, and γ-glutamyl transpeptidase are enzymes located on the brush border villi of the proximal tubular cell [21]. When these cells sustain damage, these enzymes are released into the urine and can be detected, indicating tubular injury in patients [22].
Calprotectin is a cytosolic calcium-binding complex derived from neutrophils and monocytes. In cases of intrinsic AKI, the levels of calprotectin in urine are significantly elevated [23,24]. C–C motif chemokine ligand 14 (CCL14) is a pro-inflammatory chemokine released into urine in response to stress or damage to tubular cells. According to findings from the RUBY study elevated levels of CCL14 serve as a predictive marker for persistent AKI in critically ill patients, particularly those with severe AKI [25]. NGAL exists in three different types: a monomeric glycoprotein form derived from neutrophils and TECs, a homodimeric protein originating from neutrophils, and a heterodimeric protein produced by tubular cells. These forms of NGAL can be detected in both serum and urine during the development of AKI, particularly after ischemic or toxicity-induced damage to the kidney [22,23,26] and NGAL had the best predictive accuracy for the occurrence of AKI [27].
Urine KIM-1, a transmembrane glycoprotein produced by proximal tubular cells, is released into the urine following tubular damage. It has been established as a proven marker of AKI in adults [15,26]. Indeed, additional biomarkers are released in response to tubular damage, including L-FABP, a protein located in the cytoplasm of renal proximal tubules. IL-18, a pro-inflammatory cytokine, is also among the biomarkers associated with tubular damage. Monitoring the levels of these biomarkers can contribute to the assessment and diagnosis of AKI [26,28].
2.3. Functional markers
Cystatin C, a cysteine protease inhibitor produced by nucleated human cells, exhibits an increased level within 12–24 hours following renal injury [29]. It is considered to have better accuracy than serum creatinine in identifying individuals with reduced glomerular filtration rate (GFR) [30]. Firstly, serum creatinine is not capable of promptly reflecting changes in the glomerular filtration rate, especially in situations where the GFR is not in a steady state [31]. Additionally, the clearance of serum creatinine from the body is not solely through glomerular filtration; it also involves partial secretion by the renal tubules. This widely recognized process can lead to a significant overestimation of the GFR. Therefore, Cystatin C is particularly useful for detecting even mild declines in GFR [32].
Proenkephalin A is an endogenous polypeptide hormone found in various tissues such as the adrenal medulla, nervous system, immune system, and renal tissue [33]. It has been reported that Proenkephalin A serves as a useful biomarker for early detection of AKI and predicting a shorter duration and successful liberation from renal replacement therapy (RRT) [34,35]. Both Cystatin C and Proenkephalin A are considered as damage biomarkers, along with functional biomarkers. When used in combination, they contribute to a more comprehensive assessment and accurate diagnosis of AKI [21] (Table 1).
3. Pathophysiology of acute kidney injury
AKI arises from various insults, such as renal hypoperfusion, sepsis, major surgery, immunological diseases affecting kidney parenchyma, administration of radiocontrast or nephrotoxic agents, and post-renal causes [46]. The pathophysiology of AKI varies depending on numerous conditions [47]. Here, we provide a concise overview of the aforementioned pathophysiology.
- In cases of renal hypoperfusion induced by hypovolemia, autoregulation and neurohumoral mechanisms are triggered to maintain GFR. Nevertheless, persistent renal hypoperfusion can lead to sustained inadequate oxygen delivery and depletion of adenosine triphosphate (ATP), causing cellular injury to the epithelium [48]. This can subsequently activate inflammatory responses, induce endothelial injury, and ultimately result in renal damage [49,50].
- In sepsis, inflammatory cytokines can induce leukocyte activation, recruit neutrophils, and trigger endothelial injury and coagulation. Additionally, these inflammatory mediators may bind to specific receptors expressed by renal endothelial and tubule epithelial cells, causing direct injury [51]. The release of damage-associated molecular patterns (DAMPs) by damaged cells further contributes to vasodilation, increased vascular permeability, and a pro-thrombotic environment [52]. Furthermore, filtered DAMPs and Pathogen-Associated Molecular Patterns (PAMPs) may activate Toll-like receptors 2 (TLR2) and Toll-like receptors 4 (TLR4) on proximal tubules, subsequently triggering interstitial inflammation. Vascular dysfunction, endothelial injury, immunological dysregulation, and abnormal cellular responses to injury collectively contribute to the development of AKI in sepsis [53].
- AKI resulting from major surgery can be attributed to fluid depletion, including blood loss and the extravasation of fluid into the third space [54]. Additionally, anesthetic agents may induce peripheral vasodilation and myocardial depression, thereby influencing renal perfusion. In case of AKI associated with cardiac surgery, ischemia–reperfusion injury (IRI) may occur due to extracorporeal circulation, leading to cell injury and death by increasing mitochondrial permeability [54,55]. Renal IRI stands as the primary cause of AKI, contributing to tubular epithelial apoptosis, necrosis, and inflammation during the peri-operative period [56].
- In individuals genetically predisposed to autoimmune activation, the renal consequences may involve glomerular inflammation and injury, such as rapidly progressive glomerulonephritis [58].
- Extrarenal or intrarenal obstruction has the potential to elevate intratubular pressure, compromise renal blood flow, and trigger inflammatory processes, ultimately leading to AKI [59].
4. Molecular mechanism involved in AKI to CKD transition
4.1. Inflammation
While AKI is linked to various mechanisms outlined earlier, it is primarily considered a complex clinical syndrome driven by inflammatory diseases with systemic effects [60]. Following an acute insult, stressed cells and injured tissues may release DAMPs, which interact with pattern recognition receptors (PRRs) such as TLRs would activate the innate immune pathway results in the production of proinflammatory cytokines, chemokines, and reactive oxygen species (ROS), which eventually leading to further cell necrosis and tissue damage [61,62]. Intracellular molecules like high mobility group box 1 (HMGB1), histones, heat kinin, and fibronectin released from necrotic renal tubular cells enter the extracellular space, exacerbating inflammatory cascades [63]. Moreover, the sustained release of inflammation-associated fibrotic cytokines such as transforming growth factor-β (TGF-β) and Interleukin-13 (IL-13) can trigger epithelial-mesenchymal transition (EMT), potentially leading to renal fibrosis and chronic renal insufficiency [64].
The most prominent signaling pathways involved at expression of inflammation-associated genes include nuclear factor kappa-B (NF-κB), mitogen-activated protein kinase (MAPK), and STAT pathways. NF-κB, a crucial nuclear transcription factor, plays a key role in regulating genes associated with the inflammatory response and influencing the release of inflammatory cytokines, chemokines, and adhesion factors. The NF-κB family comprises five related protein members: p50, p52, RelA (p65), RelB, and c-Rel. The inactivation of NF-κB is regulated by and IκB kinase (IΚΚ) [65]. IκB undergoes phosphorylation and rapid degradation in response to stimulation by Reactive oxygen species (ROS) and cytokines. This process results in the liberation of the free NF-κB dimer, which then undergoes phosphorylation and translocation to the nucleus. Subsequently, this translocation promotes the transcription of inflammation-related genes [66]. However, research has revealed that Silent Information Regulator Transcript 1 (SIRT1), also known as Sirtuin1, has the potential to mitigate kidney injury [67]. SIRT1 is a histone deacetylase and overexpressing SIRT1 in renal TECs would inhibits NF-κB activation. This inhibition occurs through the deacetylation of the Lys310 residue on the RelA/p65 subunit or by reducing the activity of the acetyltransferase P300/CBP [68]. Hence, the SIRT1 pathway presents a potential therapeutic strategy for mitigating inflammatory damage in AKI.
4.2. Hypoxia
Renal tubular epithelium cells rely on ATP from the mitochondrial respiratory chain for maintaining the renal function through the Na-K-ATPase, and this process is highly oxygen-dependent. Insufficient oxygen during AKI can lead to mitochondrial dysfunction, increased ROS production, and endothelial inflammation, which would further induce peritubular capillary rarefaction, worsening tissue hypoxia and perpetuating this cycle [69]. Hypoxia was shown to significantly increase miR-493, leading to the suppression of Stathmin-1 (STMN-1), a cell cycle regulator. This induction resulted in G2/M cell cycle arrest and the release of profibrotic cytokines in vitro [70]. Besides, Hypoxia-induced factors (HIFs) are also involved in the process of post-AKI renal fibrosis. Normally, there are various subunits that comprise the HIF, including varies α subunits (HIF1/2/3α) and a shared HIF1β. In normal physiology condition, HIF-1α undergoes hydroxylation by prolyl hydroxylase domain (PHD) proteins, followed by binding to Von Hippel-Lindau (VHL) E3 ubiquitin ligase and subsequent degradation in proteasomes [71]. In hypoxia condition, inhibition of PHDs results in the translocation of HIF-1/2α into the cell nucleus and binds with HIF-1β, which further initiates gene transcription, including vascular endothelial growth factor (VEGF), erythropoietin (EPO), glucose transporter 1 (GLUT1) [72]. Whether HIF is a protective factor for renal fibrosis is still under debate. Some studies demonstrated that HIF are participant in in regulating pro-fibrotic genes and promoting renal fibrosis via TGF-β , NF-κB, phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) pathway or via G2/M cell cycle arrest pathway through p53 upregulation [73,74]. On the contrary, some authors displayed that under hypoxia condition, HIF-1α would bind to FoxO3, which is thought to be a renoprotection factor after AKI, and subsequently inhibit hydroxylation and degradation of FoxO3 [75]. SerpinA3K is another novel biomarker for AKI to CKD transition in animal models, and knockout of SerpinA3K demonstrate higher FoxO3 expression with improved cellular responses to hypoxic injury, suggesting SerpinA3K involvement in renal oxidant response, HIF1α pathway, and cell apoptosis [76,77]. Studies explores the effectiveness of PHD inhibitors in renal injury and highlights the positive impact of HIF on renal fibrosis [78].
4.3. Signal pathways involved in the process of renal fibrosis
Maladaptive repair after renal tubule injury inducing progressive renal fibrosis and destruction of normal architecture of kidney is thought to be the key pathological mechanism contributing to CKD [79,80]. Various local stromal cells in kidney transform to myofibroblast plays important roles in the progressive kidney fibrosis, including resident fibroblasts, pericytes/perivascular fibroblasts, EMT, endothelial to mesenchymal transition (EndMT), macrophage (bone-marrow-derived) to myofibroblast transition (MMT) [81]. Myofibroblast attribute to excessive extracellular matrix production and deposition in renal parenchyma, which eventually lead to chronic kidney fibrosis and loss of renal function [16]. There are severe molecules involved in the complicated process of renal fibrosis, mainly including Wnt/β-Catenin signaling pathway, TGF-β1/SMAD signaling pathway, and hippo signaling pathway (Figure 1).
4.3.1. Wnt/β-Catenin signal pathway
Wnt/β-Catenin signaling pathway is heavily involved in the initiation and signal transmit of renal fibrosis [82,83]. Normally, Wnt/β-Catenin activation is in charge for cellular repairment regeneration during acute insult of renal tissue, however, constant expression of this pathway would contribute to renal fibrosis and eventually induce CKD [84]. In normal physiology process, a protein complex composed of five proteins would inactivate the Wnt/β-Catenin by phosphorylation and prevent overexpression of this pathway and renal fibrosis. However, in the pathologic status, the Wnt ligants would bind to frizzled protein (FZD), LDL receptor-associated protein 5/6 (LRP5/6) and LRP, which were mainly derived from the cytoplasm of tubular cell, leading β-catenin unphosphorylated and make β-catenin become an active form. The downstream signal including TGF-β1/SMAD signals, RAS, Snail1, Twist1, matrix metalloproteinase 7 (MMP-7), transient receptor potential canonical 6 (TRPC6), plasminogen activator inhibitor-1 (PAI-1), fibroblasts activation would transmit subsequently after the β-catenin translocated into the nucleus and binds to T cell factor/lymphoid enhancer factor (TCF/LEF) transcription factors [85,86]. The sustained expression of Wnt ligands eventually induced myofibroblast transformation and resulted in fibrosis [16].(Figure 2). Additionally, WNT-β-catenin signaling is involved in CKD-associated vascular calcification and mineral bone disease. The WNT-β-catenin pathway is tightly regulated, for example, by proteins of the DKK family. In particular, DKK3 is released by 'stressed' TECs; DKK3 drives kidney fibrosis and is associated with short-term risk of CKD progression and acute kidney injury [84].
4.3.2. TGF-β1/SMAD signal pathway
TGF-β1/SMAD pathway is another important mechanism contributing to the transition to myofibroblast and renal fibrosis [87]. TGF-β1 is produced in an inactive state, where it is bound to latency-associated peptide (LAP) and latent TGF-β1 binding protein (LTBP). Various triggers, such as ROS, have the capability to liberate TGF-β1 from LAP and LTBP, leading to the activation of TGF-β1. The active form TGF-β1 would bind to type II TGF-β1 receptor (TβRII) and further bind to type I TGF-β1 receptor (TβRI) and induced phosphorylation of SMAD2/SMAD3 complex, which would further activate the SMAD4. SMAD4 serves for the translocation of SMAD2/SMAD3 from cytoplasm to nucleus and the complex would eventually activate miRNA-21, miRNA-192 and make a consequence of extracellular matrix production and renal fibrosis [88]. In contrast, SMAD7, an inhibitory SMAD which is induced by Smad3 transcriptionally, regulate the function of SMAD3 and serves as a negative feedback mechanism of the TGF-β1/SMAD pathway [89,90]. In normal physiology, the amount of Smad7 is tremendous and would inhibit the TGF-β1/SMAD pathway by degradation of TβRI via an ubiquitin proteasome degradation mechanism [91]. In pathologic state, overexpression of SMAD3 would induce Smad ubiquitination regulatory factor 1 (Smurf1), Smad ubiquitination regulatory factor2 (Smurf2), and arkadia and degrade SMAD7 protein [92,93]. This process would further contribute to the profibrogenic expression, induce the transition to myeloblast and progress the renal fibrosis [94,95,96] (Figure 2).
4.3.3. Hippo/ Yes-associated protein (YAP)/ Tafazzin (TAZ) Signaling
The Hippo pathway was first identified 20 years ago and is thought to be involved in cell growth, proliferation, and apoptosis. It plays a key role in regulating organ size, tissue regeneration, and tumor development [97]. Various physiological and pathological signals can induce the Hippo signaling pathway, including extracellular matrix (ECM) stiffness, cell polarity, and energy stress [98]. The upstream membrane receptor serves as a receptor for the extracellular growth inhibition signal. When the inhibitory signal binds to the receptor, the TAO kinase would activate the Hippo pathway via phosphorylation of STE20-like serine/threonine kinase 1/2(MST1/2) which would further form a complex with adaptor protein Salvador 1 (SAV1). Subsequently, this complex phosphorylates large tumor suppressor (LATS1/2) and LATS1/2-interacting protein MOB kinase activator 1 (MOB1), and then the phosphorylated LATS1/2–MOB1 complex phosphorylates YAP and TAZ, which promotes cytoplasmic polyubiquitination and consequent degradation of YAP/TAZ by proteasomes [99]. On the contrary, inactivation of the Hippo pathway results in the dephosphorylation of upstream kinases, causing active YAP/TAZ form migration into the nucleus. There, they interact with various transcription factors, including TEA domain DNA-binding family members (TEAD1–4), to regulate cell proliferation [100]. YAP/TAZ would also bind to other transcription factors, including TCF/LEF transcription factors, SMAD1, SMAD2/3, and p37 [101] (Figure 3).
Studies had demonstrated that Hippo pathway is associated with AKI [102]. YAP protein levels rise in both the cytoplasm and nucleus of renal TECs during the AKI repair stage, and they also indicate a positive correlation between the changes in YAP expression and TEAD expression [103]. Moreover, the Hippo pathway is linked to EMT in renal TECs, a pivotal process in the transition from AKI to CKD. EMT occurs through the activation of the TGF-β/Smad pathway and lose of tubular epithelial cell polarity. This polarity is primarily maintained by the Crumbs (CRB)/PALS1 complex, which also regulates the Hippo signaling pathway [104,105].
4.4. Innate and Adaptive Immunity
Except the myofibroblast transition, immune system also plays a crucial role in the pathophysiology of kidney injury and repair. In the acute phase of injury, components of the innate immune system, such as macrophages and neutrophils, are recruited to the injured site. They release pro-inflammatory cytokines like Interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α), thereby amplifying the inflammatory response. At the latter stage of the AKI, the immune system also involves in the repair process. For example, macrophage, which presents as M1-phenotype during the acute insult exacerbate the inflammation process by releasing Interleukin-1 (IL-1), IL-6 and TNF-α, would switch to M2-phenotype for mediating the inflammation cascade and propagating the repair process [106,107]. However, M2-type macrophages are implicated in renal fibrosis via MMT or numerous growth factors [95]. TECs release Wnt ligands, prompting a transition in macrophage phenotype from M1 to M2, thereby promoting fibrosis [108]. Macrophages would also secrete Wnt proteins, TGF-β1, and TIMPs, exerting a pivotal influence on kidney fibrosis through their involvement in the synthesis and deposition of extracellular matrix [109].
Increasing complement level in kidney cells also implicate in the pathogenesis of kidney fibrosis. For example, elevated C1q levels in PDGFRβ-positive pericytes lead to heightened inflammation and renal scarring. This is attributed to increased production of cytokines like IL-6, monocyte chemoattractant protein-1 (MCP-1; CCL2), and macrophage Inflammatory Protein-1 Alpha (MIP1-α; CCL3) [110]. Both mRNA expression and protein of C1r and C1s are also increased during fibrosis, and this contribute to the increasing level of C3 fragments which eventually propagate the downstream reaction of complement system and lead to activation of myofibroblast transition and renal fibrosis [111]. Animal studies had demonstrated that C5 knockout and C5R antagonist usage is associated with reducing tissue fibrosis [112]. Additional studies have demonstrated that inhibition of C1r serine protease or C3a/C3aR, effectively alleviated interstitial fibrosis in diverse murine models [113,114].
The adaptive immune system is also engaged in the AKI process. During the initial insult, renal dendritic cells present antigens to T cells. Activated T cells subsequently release pro-inflammatory cytokines, including interferon-gamma (IFN-γ), contributing to the overall inflammatory cascade [115]. Much like the observed shift in phenotype seen in macrophages, regulatory T cells, which are powerful mediators for immune system, become discernible in the kidney. These regulatory T cells suppress the activation of diverse immune cells through both contact-dependent mechanisms and the release of soluble mediators such as Interleukin-10 (IL-10) which would induce phosphorylation of STAT1, 3 and 5, thereby promoting the repair and anti-fibrotic process following injury [116,117,118].
4.5. Mitochondria dysfunction
AKI, especially ischemic injury, would lead to mitochondria dysfunction because hypoxia disrupt the electron transport chain in mitochondria and produce excessive free radicles containing substance. These harmful molecules would further damage the tubular cell [119]. Mitochondria dysfunction is also recognized increasingly as a crucial contributor in the process of AKI to CKD transition in recent studies [16,120]. Basically, Mitochondrial homeostasis is completed through three processes: mitochondrial dynamics, mitochondrial mitophagy and mitochondrial biogenesis [121]. Mitochondrial dynamics including two different processes: fission (regulated by DRP1) and fusion (regulated by MFN1, MFN2, and OPA1). Mitophagy is the process that damaged mitochondria are selectively degraded by autophagy, which is regulated by PINK1-PARK2 Pathway, BNIP3 and NIX pathway and FUNDC1 pathway. Mitochondrial biogenesis which is mainly regulated by peroxisome proliferator- activated receptor γ coactivator -1α (PGC-1α) address the heightened cellular energy demands and replenishing mitochondrial content in newly formed cells during the process of cell proliferation [119,122]. Animal studies demonstrated that adjust these genes involved in the process of mitochondria homeostasis would influence the process of kidney inflammation, kidney fibrosis and tubular epithelial cell apoptosis [56,123,124,125]. For instance, elevated PGC-1α in tubular cells boosts mitochondrial mass, providing kidney protection post-ischemic and inflammatory injury without cell death [126]. Conversely, global PGC-1α deficiency leads to severe renal dysfunction in septic AKI [125].
4.6. G2/M Arrest Pathway and cellular senescence
The initiation of DNA damage response (DDR) signaling is essential for the reparative mechanisms in renal proximal epithelial cells following AKI. When the repairment is not completely accomplished, the injured cell would arrest at the G2/M phase for stabilizing genetic factors [127,128]. These cells would subsequently marked upregulate messenger RNA expression of fibrogenic growth factors encoding TGF-β1 and connective tissue growth factor (CTGF) and eventually lead to interstitial fibrosis [80,129]. Target of rapamycin (TOR)-autophagy special coupling compartments (TASCCs), a novel complex that is present in the G2/M arrest tubular cell, is associated with cellular senescence [130]. Cyclin G1 (CG1) plays a pivotal role in the formation of TASCC, and studies have indicated that the deletion of CG1 or the specific deletion of Raptor (a key component of the TASCC complex) in proximal tubules markedly mitigated renal fibrosis. Epithelial toll-like and interleukin 1 receptor (TLR/IL-1R) are other factors that mediate the cellular senescence and G2/M arrest pathway [17]. After AKI, activation of TLRs by IL-1 and DAMPs can trigger an excessive inflammatory response and facilitate interstitial fibrosis, in contrast, deletion of Myd88, an TLR/IL-1 downstream protein and an NF-κB upstream protein, revealed ameliorated renal fibrosis in kidney damage [17,131]. Hence, targeting tubules experiencing G2/M cell-cycle arrest represents a promising therapeutic approach, with several documented interventions. For example, pifithrin-α, an inhibitor for crucial cell cycle regulator-p53, is a promising drug candidate that could be used after acute kidney injury for reducing post AKI renal fibrosis [132]. PTBA analog methyl-4-(phenylthio) butanoate (M4PTB), a histone deacetylase inhibitors, have also been demonstrated to decrease G2/M arrest in injured tubular cells, thereby reducing interstitial fibrosis[133]. Other studies have revealed the effectiveness of specific inhibitors targeting cyclin-dependent kinases 4/6, pivotal mediators of cell-cycle progression from G1 to S phase, in optimizing the cell-cycle stages of injured tubules [134,135].
4.7. Renin-Angiotensin-Aldosterone System
The Renin-Angiotensin-Aldosterone System (RAAS) initiated from the release of renin from juxtaglomerular cells in the kidneys. It converts the liver formed angiotensinogen into angiotensin I, which subsequently transformed to angiotensin II by angiotensin converting enzyme. Prolonged and overactivation of RAAS after AKI would lead to CKD progression through various mechanisms [136]. Angiotensin II binding to angiotensin II receptor type 1 (AT1R) activate the cascade of RhoGEF/RhoA/ROCK and make the NF-κB, PAI-1, MAPK/ Extracellular signal-Regulated Kinases (ERK) 1/2 and NADPH oxidases overexpression. NADPH oxidases, a ROS generating enzymes, increased the oxidative stress inside the cell and contribute to overexpression of TGF-β/SMAD and MAPK/ERK1/2 [137,138]. All these molecules mentioned above would result in organ remodeling tissue fibrosis. Besides, AT1R activation mediate the balance of intracellular level of nitric oxide (NO) and calcium through PI3K/Akt pathway. The suppression of endothelial nitric oxide synthase (eNOS) and increasing cytosol calcium through inositol 1,4,5-trisphosphate (IP3) would elevate the resistance of efferent arteriole and disrupt the autoregulation of afferent arteriole, which would eventually leading to glomerular hyperfiltration and sclerosis [137,139,140] (Figure 3). Aldosterone is a mineralocorticoid hormone is thought to be involve in the process of renal fibrosis apart from its actions to increase blood pressure by mediating salt retention. Aldosterone exerts its influence on kidney via inducing the production of ROS, upregulates the expression of EGFR and AT1R, and activates NF-κB, activator protein-1 (AP-1). These molecular events further contribute to cell proliferation, apoptosis, and phenotypic transformation of epithelial cell which further triggers the expression of TGF-β, CTGF and PAI-1, ultimately culminating in the development of renal fibrosis [141,142].
Numerous clinical trials support the renoprotective effects of RAS inhibitors like ACE inhibitors and AT1a receptor blockers in diabetic or proteinuric non-diabetic CKD patients [143,144]. A meta-analysis enrolled 70,801 patients revealed that exposure to ACEi/ARB after AKI is associated with lower risks of recurrent AKI, and progression to incident CKD [145]. However, there’s only certain observational studies continue to explore the impact of RAS inhibitors on the AKI to CKD transition. Besides, there hasn't been a large-scale randomized controlled trial to assess the impact of RAS blockade on AKI and the subsequent development of CKD. The role of RAS activity in the acute phase and severity of AKI is still uncertain.
5. Conclusion
The classification of kidney disease into AKI, AKD, and CKD, highlighting the progression risks and the causal relationship between AKI/AKD to CKD. It addresses the need for better biomarkers for early AKI detection, beyond traditional serum creatinine, and the complex molecular pathways from AKI to CKD transition. The biomarkers for early detection of AKI, categorizing them into stress, damage, and functional markers. AKI emerges from diverse insults, leading to complex pathophysiological events such as cellular hypoxia, inflammation, and nephrotoxicity, with subsequent renal damage. The inflammatory response plays a central role in AKI, where cellular stress leads to the release of DAMPs and activation of innate immunity, causing further injury and fibrosis. Molecular pathways including NF-κB, MAPK, and STAT are pivotal in the inflammation and fibrosis associated with AKI. Maladaptive repair mechanisms after AKI, involving the transition of various renal cells to myofibroblasts, contribute significantly to the progression of CKD. The Wnt/β-Catenin, TGF-β/SMAD, and Hippo signaling pathways are critical in this transition, promoting fibrosis and affecting renal function. Mitochondrial dysfunction, cellular senescence, hypoxia, and the RAAS are also key contributors to AKI progression and CKD transition, with the RAAS in particular playing a role in renal remodeling and fibrosis. Despite the recognized importance of the RAAS, more research is needed to understand its role in AKI and subsequent CKD development fully.
Author Contributions
Conceptualization, JYC.; methodology, JYC, software, JYC.; validation, JYC, KCT.; formal analysis, JHY.; investigation, HYW.; resources, HYW.; data curation, KCT.; writing—original draft preparation, JYC, KCT.; writing—review and editing, JHY.; visualization, JYC, JHY; supervision, JYC.; project administration, JYC.; funding acquisition, JYC. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
This study did not require ethical approval.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data is available.
Acknowledgments
We would like to express my heartfelt gratitude to my colleagues at the Chi Mei Nephrology Department for their invaluable assistance and support throughout the course of this research. Their expertise, insights, and dedication have been instrumental in the successful completion of this paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kellum, J.A.; et al. Acute kidney injury. Nat Rev Dis Primers, 2021. 7(1): p. 52.
- Khwaja, A. KDIGO Clinical Practice Guidelines for Acute Kidney Injury. Nephron Clin. Pr. 2012, 120, c179–c184. [Google Scholar] [CrossRef]
- Chawla, L.S.; Bellomo, R.; Bihorac, A.; Goldstein, S.L.; Siew, E.D.; Bagshaw, S.M.; Bittleman, D.; Cruz, D.; Endre, Z.; Fitzgerald, R.L.; et al. Acute kidney disease and renal recovery: consensus report of the Acute Disease Quality Initiative (ADQI) 16 Workgroup. Nat. Rev. Nephrol. 2017, 13, 241–257. [Google Scholar] [CrossRef]
- Levey, A.S.; Eckardt, K.-U.; Tsukamoto, Y.; Levin, A.; Coresh, J.; Rossert, J.; Zeeuw, D.D.; Hostetter, T.H.; Lameire, N.; Eknoyan, G. Definition and classification of chronic kidney disease: A position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2005, 67, 2089–2100. [Google Scholar] [CrossRef]
- Lameire, N.H.; Levin, A.; Kellum, J.A.; Cheung, M.; Jadoul, M.; Winkelmayer, W.C.; Stevens, P.E.; Caskey, F.J.; Farmer, C.K.; Fuentes, A.F.; et al. Harmonizing acute and chronic kidney disease definition and classification: report of a Kidney Disease: Improving Global Outcomes (KDIGO) Consensus Conference. Kidney Int. 2021, 100, 516–526. [Google Scholar] [CrossRef]
- Levey, A.S. Defining AKD: The Spectrum of AKI, AKD, and CKD. Nephron 2021, 146, 302–305. [Google Scholar] [CrossRef]
- Rifkin, D.E., S. G. Coca, and K. Kalantar-Zadeh, Does AKI truly lead to CKD? J Am Soc Nephrol, 2012. 23(6): p. 979-84.
- Coca, S.G.; Singanamala, S.; Parikh, C.R. Chronic kidney disease after acute kidney injury: A systematic review and meta-analysis. Kidney Int. 2012, 81, 442–448. [Google Scholar] [CrossRef]
- Horne, K.L.; Packington, R.; Monaghan, J.; Reilly, T.; Selby, N.M. Three-year outcomes after acute kidney injury: results of a prospective parallel group cohort study. BMJ Open 2017, 7, e015316. [Google Scholar] [CrossRef]
- James, M.T.; Levey, A.S.; Tonelli, M.; Tan, Z.; Barry, R.; Pannu, N.; Ravani, P.; Klarenbach, S.W.; Manns, B.J.; Hemmelgarn, B.R. Incidence and Prognosis of Acute Kidney Diseases and Disorders Using an Integrated Approach to Laboratory Measurements in a Universal Health Care System. JAMA Netw. Open 2019, 2, e191795–e191795. [Google Scholar] [CrossRef]
- Su, C.-C.; Chen, J.-Y.; Chen, S.-Y.; Shiao, C.-C.; Neyra, J.A.; Matsuura, R.; Noiri, E.; See, E.; Chen, Y.-T.; Hsu, C.-K.; et al. Outcomes associated with acute kidney disease: a systematic review and meta-analysis. EClinicalMedicine 2022, 55, 101760. [Google Scholar] [CrossRef]
- Silver, S.A.; Chertow, G.M. The Economic Consequences of Acute Kidney Injury. Nephron 2017, 137, 297–301. [Google Scholar] [CrossRef]
- Zhang, W.R.; Parikh, C.R. Biomarkers of Acute and Chronic Kidney Disease. Annu. Rev. Physiol. 2019, 81, 309–333. [Google Scholar] [CrossRef]
- Bhosale, S.J. and A.P. Kulkarni, Biomarkers in Acute Kidney Injury. Indian J Crit Care Med, 2020. 24(Suppl 3): p. S90-s93.
- Kane-Gill, S.L.; Meersch, M.; Bell, M. Biomarker-guided management of acute kidney injury. Curr. Opin. Crit. Care 2020, 26, 556–562. [Google Scholar] [CrossRef]
- Sato, Y.; Takahashi, M.; Yanagita, M. Pathophysiology of AKI to CKD progression. Semin. Nephrol. 2020, 40, 206–215. [Google Scholar] [CrossRef]
- Yu, S.M.-W.; Bonventre, J.V. Acute kidney injury and maladaptive tubular repair leading to renal fibrosis. Curr. Opin. Nephrol. Hypertens. 2020, 29, 310–318. [Google Scholar] [CrossRef]
- Ostermann, M.; et al. Recommendations on Acute Kidney Injury Biomarkers From the Acute Disease Quality Initiative Consensus Conference: A Consensus Statement. JAMA Netw Open, 2020. 3(10): p. e2019209.
- Schunk, S.J.; Zarbock, A.; Meersch, M.; Küllmar, M.; A Kellum, J.; Schmit, D.; Wagner, M.; Triem, S.; Wagenpfeil, S.; Gröne, H.-J.; et al. Association between urinary dickkopf-3, acute kidney injury, and subsequent loss of kidney function in patients undergoing cardiac surgery: an observational cohort study. Lancet 2019, 394, 488–496. [Google Scholar] [CrossRef]
- Kashani, K.; Al-Khafaji, A.; Ardiles, T.; Artigas, A.; Bagshaw, S.M.; Bell, M.; Bihorac, A.; Birkhahn, R.; Cely, C.M.; Chawla, L.S.; et al. Discovery and validation of cell cycle arrest biomarkers in human acute kidney injury. Crit. Care 2013, 17, R25. [Google Scholar] [CrossRef]
- Yoon, S.-Y.; Kim, J.-S.; Jeong, K.-H.; Kim, S.-K. Acute Kidney Injury: Biomarker-Guided Diagnosis and Management. Medicina 2022, 58, 340. [Google Scholar] [CrossRef]
- Coca, S.; Yalavarthy, R.; Concato, J.; Parikh, C. Biomarkers for the diagnosis and risk stratification of acute kidney injury: A systematic review. Kidney Int. 2008, 73, 1008–1016. [Google Scholar] [CrossRef]
- Charlton, J.R.; Portilla, D.; Okusa, M.D. A basic science view of acute kidney injury biomarkers. Nephrol. Dial. Transplant. 2014, 29, 1301–1311. [Google Scholar] [CrossRef]
- Heller, F.; Frischmann, S.; Grünbaum, M.; Zidek, W.; Westhoff, T.H. Urinary Calprotectin and the Distinction between Prerenal and Intrinsic Acute Kidney Injury. Clin. J. Am. Soc. Nephrol. 2011, 6, 2347–2355. [Google Scholar] [CrossRef]
- Hoste, E., et al., Identification and validation of biomarkers of persistent acute kidney injury: the RUBY study. Intensiv. Care Med. 2020, 46, 943–953. [CrossRef]
- Ho, J.; Tangri, N.; Komenda, P.; Kaushal, A.; Sood, M.; Brar, R.; Gill, K.; Walker, S.; MacDonald, K.; Hiebert, B.M.; et al. Urinary, Plasma, and Serum Biomarkers’ Utility for Predicting Acute Kidney Injury Associated With Cardiac Surgery in Adults: A Meta-analysis. Am. J. Kidney Dis. 2015, 66, 993–1005. [Google Scholar] [CrossRef]
- Pan, H.-C.; Yang, S.-Y.; Chiou, T.T.-Y.; Shiao, C.-C.; Wu, C.-H.; Huang, C.-T.; Wang, T.-J.; Chen, J.-Y.; Liao, H.-W.; Chen, S.-Y.; et al. Comparative accuracy of biomarkers for the prediction of hospital-acquired acute kidney injury: a systematic review and meta-analysis. Crit. Care 2022, 26, 1–30. [Google Scholar] [CrossRef]
- Kamijo-Ikemori, A.; Ichikawa, D.; Matsui, K.; Yokoyama, T.; Sugaya, T.; Kimura, K. [Urinary L-type fatty acid binding protein (L-FABP) as a new urinary biomarker promulgated by the Ministry of Health, Labour and Welfare in Japan]. . 2013, 61, 635–40. [Google Scholar]
- Risch, L.; Blumberg, A.; Huber, A. Rapid and accurate assessment of glomerular filtration rate in patients with renal transplants using serum cystatin C. Nephrol. Dial. Transplant. 1999, 14, 1991–1996. [Google Scholar] [CrossRef]
- Delanaye, P.; et al. Detection of decreased glomerular filtration rate in intensive care units: serum cystatin C versus serum creatinine. BMC Nephrol, 2014. 15: p. 9.
- Chen, S. Retooling the Creatinine Clearance Equation to Estimate Kinetic GFR when the Plasma Creatinine Is Changing Acutely. J. Am. Soc. Nephrol. 2013, 24, 877–888. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, H.; Du, L.; Wan, J.; Li, X. Serum Cystatin C Predicts AKI and the Prognosis of Patients in Coronary Care Unit: a Prospective, Observational Study. Kidney Blood Press. Res. 2017, 42, 961–973. [Google Scholar] [CrossRef]
- Legrand, M.; Hollinger, A.; Vieillard-Baron, A.; Dépret, F.; Cariou, A.; Deye, N.; Fournier, M.-C.; Jaber, S.; Damoisel, C.; Lu, Q.; et al. One-Year Prognosis of Kidney Injury at Discharge From the ICU: A Multicenter Observational Study. Crit. Care Med. 2019, 47, e953–e961. [Google Scholar] [CrossRef]
- von Groote, T.; et al. Proenkephalin A 119-159 predicts early and successful liberation from renal replacement therapy in critically ill patients with acute kidney injury: a post hoc analysis of the ELAIN trial. Crit Care, 2022. 26(1): p. 333.
- Lin, L.-C.; Chuan, M.-H.; Liu, J.-H.; Liao, H.-W.; Ng, L.L.; Magnusson, M.; Jujic, A.; Pan, H.-C.; Wu, V.-C.; Forni, L.G. Proenkephalin as a biomarker correlates with acute kidney injury: a systematic review with meta-analysis and trial sequential analysis. Crit. Care 2023, 27, 1–12. [Google Scholar] [CrossRef]
- Marchewka, Z.; Długosz, A.; Kuźniar, J. Diagnostic application of AAP isoenzyme separation. Int. Urol. Nephrol. 1999, 31, 409–416. [Google Scholar] [CrossRef]
- Marchewka, Z., J. Kuźniar, and A. Długosz, Enzymuria and beta2-mikroglobulinuria in the assessment of the influence of proteinuria on the progression of glomerulopathies. Int Urol Nephrol, 2001. 33(4): p. 673-6.
- Westenfelder, C. Earlier diagnosis of acute kidney injury awaits effective therapy. Kidney Int. 2011, 79, 1159–1161. [Google Scholar] [CrossRef]
- Oh, D.-J. A long journey for acute kidney injury biomarkers. Ren. Fail. 2020, 42, 154–165. [Google Scholar] [CrossRef]
- Seibert, F.S.; Rosenberger, C.; Mathia, S.; Arndt, R.; Arns, W.; Andrea, H.; Pagonas, N.; Bauer, F.; Zidek, W.; Westhoff, T.H. Urinary Calprotectin Differentiates Between Prerenal and Intrinsic Acute Renal Allograft Failure. Transplantation 2017, 101, 387–394. [Google Scholar] [CrossRef]
- McMahon, B.A.; Galligan, M.; Redahan, L.; Martin, T.; Meaney, E.; Cotter, E.J.; Murphy, N.; Hannon, C.; Doran, P.; Marsh, B.; et al. Biomarker Predictors of Adverse Acute Kidney Injury Outcomes in Critically Ill Patients: The Dublin Acute Biomarker Group Evaluation Study. Am. J. Nephrol. 2019, 50, 19–28. [Google Scholar] [CrossRef]
- Geng, J.; Qiu, Y.; Qin, Z.; Su, B. The value of kidney injury molecule 1 in predicting acute kidney injury in adult patients: a systematic review and Bayesian meta-analysis. J. Transl. Med. 2021, 19, 1–13. [Google Scholar] [CrossRef]
- Medić, B.; Rovčanin, B.; Jovanović, G.B.; Radojević-Škodrić, S.; Prostran, M. Kidney Injury Molecule-1 and Cardiovascular Diseases: From Basic Science to Clinical Practice. BioMed Res. Int. 2015, 2015, 1–10. [Google Scholar] [CrossRef]
- Manabe, K.; Kamihata, H.; Motohiro, M.; Senoo, T.; Yoshida, S.; Iwasaka, T. Urinary liver-type fatty acid-binding protein level as a predictive biomarker of contrast-induced acute kidney injury. Eur. J. Clin. Investig. 2011, 42, 557–563. [Google Scholar] [CrossRef]
- Araki, S.; Haneda, M.; Koya, D.; Sugimoto, T.; Isshiki, K.; Chin-Kanasaki, M.; Uzu, T.; Kashiwagi, A. Predictive impact of elevated serum level of IL-18 for early renal dysfunction in type 2 diabetes: an observational follow-up study. Diabetologia 2007, 50, 867–873. [Google Scholar] [CrossRef]
- Turgut, F.; Awad, A.S.; Abdel-Rahman, E.M. Acute Kidney Injury: Medical Causes and Pathogenesis. J. Clin. Med. 2023, 12, 375. [Google Scholar] [CrossRef]
- Ronco, C., R. Bellomo, and J.A. Kellum, Acute kidney injury. Lancet, 2019. 394(10212): p. 1949-1964.
- Xu, K.; Rosenstiel, P.; Paragas, N.; Hinze, C.; Gao, X.; Shen, T.H.; Werth, M.; Forster, C.; Deng, R.; Bruck, E.; et al. Unique Transcriptional Programs Identify Subtypes of AKI. J. Am. Soc. Nephrol. 2016, 28, 1729–1740. [Google Scholar] [CrossRef]
- Makris, K.; Spanou, L. Acute Kidney Injury: Definition, Pathophysiology and Clinical Phenotypes. Clin. Biochem. Rev. 2016, 37, 85–98. [Google Scholar]
- Meola, M.; et al. Clinical Scenarios in Acute Kidney Injury: Post-Renal Acute Kidney Injury. Contrib Nephrol, 2016. 188: p. 64-8.
- Cantaluppi, V.; Quercia, A.D.; Dellepiane, S.; Ferrario, S.; Camussi, G.; Biancone, L. Interaction between systemic inflammation and renal tubular epithelial cells. Nephrol. Dial. Transplant. 2014, 29, 2004–2011. [Google Scholar] [CrossRef]
- Parthasarathy, U.; Martinelli, R.; Vollmann, E.H.; Best, K.; Therien, A.G. The impact of DAMP-mediated inflammation in severe COVID-19 and related disorders. Biochem. Pharmacol. 2021, 195, 114847–114847. [Google Scholar] [CrossRef]
- Gómez, H. and J.A. Kellum, Sepsis-induced acute kidney injury. Curr Opin Crit Care, 2016. 22(6): p. 546-553.
- Wang, Y.; Bellomo, R. Cardiac surgery-associated acute kidney injury: risk factors, pathophysiology and treatment. Nat. Rev. Nephrol. 2017, 13, 697–711. [Google Scholar] [CrossRef]
- Gameiro, J.; Fonseca, J.A.; Neves, M.; Jorge, S.; Lopes, J.A. Acute kidney injury in major abdominal surgery: incidence, risk factors, pathogenesis and outcomes. Ann. Intensive Care 2018, 8, 22. [Google Scholar] [CrossRef]
- Tang, C.; Han, H.; Liu, Z.; Liu, Y.; Yin, L.; Cai, J.; He, L.; Liu, Y.; Chen, G.; Zhang, Z.; et al. Activation of BNIP3-mediated mitophagy protects against renal ischemia–reperfusion injury. Cell Death Dis. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Virzì, G.M.; Breglia, A.; Brocca, A.; de Cal, M.; Bolin, C.; Vescovo, G.; Ronco, C. Levels of Proinflammatory Cytokines, Oxidative Stress, and Tissue Damage Markers in Patients with Acute Heart Failure with and without Cardiorenal Syndrome Type 1. Cardiorenal Med. 2018, 8, 321–331. [Google Scholar] [CrossRef]
- Dalfino, L.; et al. Intra-abdominal hypertension and acute renal failure in critically ill patients. Intensive Care Med, 2008. 34(4): p. 707-13.
- Praga, M. and E. González, Acute interstitial nephritis. Kidney Int, 2010. 77(11): p. 956-61.
- He, Z., H. Wang, and L. Yue, Endothelial progenitor cells-secreted extracellular vesicles containing microRNA-93-5p confer protection against sepsis-induced acute kidney injury via the KDM6B/H3K27me3/TNF-α axis. Exp Cell Res, 2020. 395(2): p. 112173.
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, Y.; Li, L.; Liu, S.; Wang, C.; Yuan, Y.; Yang, G.; Chen, Y.; Cheng, J.; Lu, Y.; et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics 2021, 11, 1845–1863. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, H.; Chen, Q.; Jiao, F.; Shi, C.; Pei, M.; Lv, J.; Zhang, H.; Wang, L.; Gong, Z. TNF-α/HMGB1 inflammation signalling pathway regulates pyroptosis during liver failure and acute kidney injury. Cell Prolif. 2020, 53, e12829. [Google Scholar] [CrossRef]
- Sato, Y.; Yanagita, M. Immune cells and inflammation in AKI to CKD progression. Am. J. Physiol. Physiol. 2018, 315, F1501–F1512. [Google Scholar] [CrossRef]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol, 2009. 1(6): p. a001651.
- Oeckinghaus, A. M. S. Hayden, and S. Ghosh, Crosstalk in NF-κB signaling pathways. Nat Immunol, 2011. 12(8): p. 695-708.
- Peasley, K.; Chiba, T.; Goetzman, E.; Sims-Lucas, S. Sirtuins play critical and diverse roles in acute kidney injury. Pediatr. Nephrol. 2021, 36, 3539–3546. [Google Scholar] [CrossRef]
- Yeung, F.; et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. Embo j, 2004. 23(12): p. 2369-80.
- Honda, T.; Hirakawa, Y.; Nangaku, M. The role of oxidative stress and hypoxia in renal disease. Kidney Res. Clin. Pr. 2019, 38, 414–426. [Google Scholar] [CrossRef]
- Liu, T.; et al. MicroRNA-493 targets STMN-1 and promotes hypoxia-induced epithelial cell cycle arrest in G(2)/M and renal fibrosis. Faseb j, 2019. 33(2): p. 1565-1577.
- Gunaratnam, L.; Bonventre, J.V. HIF in Kidney Disease and Development. J. Am. Soc. Nephrol. 2009, 20, 1877–1887. [Google Scholar] [CrossRef]
- Haase, V.H. Hypoxia-inducible factor–prolyl hydroxylase inhibitors in the treatment of anemia of chronic kidney disease. Kidney Int. Suppl. 2021, 11, 8–25. [Google Scholar] [CrossRef]
- Liu, J.; Wei, Q.; Guo, C.; Dong, G.; Liu, Y.; Tang, C.; Dong, Z. Hypoxia, HIF, and Associated Signaling Networks in Chronic Kidney Disease. Int. J. Mol. Sci. 2017, 18, 950. [Google Scholar] [CrossRef]
- Liu, L.; et al. p53 upregulated by HIF-1α promotes hypoxia-induced G2/M arrest and renal fibrosis in vitro and in vivo. J Mol Cell Biol, 2019. 11(5): p. 371-382.
- Li, L.; Kang, H.; Zhang, Q.; D’agati, V.D.; Al-Awqati, Q.; Lin, F. FoxO3 activation in hypoxic tubules prevents chronic kidney disease. J. Clin. Investig. 2019, 129, 2374–2389. [Google Scholar] [CrossRef]
- Sánchez-Navarro, A.; Mejía-Vilet, J.M.; Pérez-Villalva, R.; Carrillo-Pérez, D.L.; Marquina-Castillo, B.; Gamba, G.; Bobadilla, N.A. SerpinA3 in the Early Recognition of Acute Kidney Injury to Chronic Kidney Disease (CKD) transition in the rat and its Potentiality in the Recognition of Patients with CKD. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- González-Soria, I.; Soto-Valadez, A.D.; Martínez-Rojas, M.A.; Ortega-Trejo, J.A.; Pérez-Villalva, R.; Gamba, G.; Sánchez-Navarro, A.; Bobadilla, N.A. SerpinA3K Deficiency Reduces Oxidative Stress in Acute Kidney Injury. Int. J. Mol. Sci. 2023, 24, 7815. [Google Scholar] [CrossRef]
- Sakashita, M., T. Tanaka, and M. Nangaku, Hypoxia-Inducible Factor-Prolyl Hydroxylase Domain Inhibitors to Treat Anemia in Chronic Kidney Disease. Contrib Nephrol, 2019. 198: p. 112-123.
- Matovinović, M.S. 1. Pathophysiology and Classification of Kidney Diseases. 2009, 20, 2–11. [Google Scholar]
- Ferenbach, D.A.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef]
- Grgic, I.; Duffield, J.S.; Humphreys, B.D. The origin of interstitial myofibroblasts in chronic kidney disease. Pediatr. Nephrol. 2011, 27, 183–193. [Google Scholar] [CrossRef]
- Huang, P.; Yan, R.; Zhang, X.; Wang, L.; Ke, X.; Qu, Y. Activating Wnt/β-catenin signaling pathway for disease therapy: Challenges and opportunities. Pharmacol. Ther. 2018, 196, 79–90. [Google Scholar] [CrossRef]
- Xie, H.; Miao, N.; Xu, D.; Zhou, Z.; Ni, J.; Yin, F.; Wang, Y.; Cheng, Q.; Chen, P.; Li, J.; et al. FoxM1 promotes Wnt/β-catenin pathway activation and renal fibrosis via transcriptionally regulating multi-Wnts expressions. J. Cell. Mol. Med. 2021, 25, 1958–1971. [Google Scholar] [CrossRef]
- Schunk, S.J.; et al. WNT-β-catenin signalling - a versatile player in kidney injury and repair. Nat Rev Nephrol, 2021. 17(3): p. 172-184.
- Zhou, D.; Li, Y.; Lin, L.; Zhou, L.; Igarashi, P.; Liu, Y. Tubule-specific ablation of endogenous β-catenin aggravates acute kidney injury in mice. Kidney Int. 2012, 82, 537–547. [Google Scholar] [CrossRef]
- Li, S.-S.; Sun, Q.; Hua, M.-R.; Suo, P.; Chen, J.-R.; Yu, X.-Y.; Zhao, Y.-Y. Targeting the Wnt/β-Catenin Signaling Pathway as a Potential Therapeutic Strategy in Renal Tubulointerstitial Fibrosis. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef]
- Meng, X.M.; et al. TGF-β/Smad signaling in renal fibrosis. Front Physiol, 2015. 6: p. 82.
- Wu, W.; Wang, X.; Yu, X.; Lan, H.-Y. Smad3 Signatures in Renal Inflammation and Fibrosis. Int. J. Biol. Sci. 2022, 18, 2795–2806. [Google Scholar] [CrossRef]
- von Gersdorff, G.; et al. Smad3 and Smad4 mediate transcriptional activation of the human Smad7 promoter by transforming growth factor beta. J Biol Chem, 2000. 275(15): p. 11320-6.
- Nagarajan, R.P.; Zhang, J.; Li, W.; Chen, Y. Regulation of Smad7 Promoter by Direct Association with Smad3 and Smad4. J. Biol. Chem. 1999, 274, 33412–33418. [Google Scholar] [CrossRef]
- Lan, H.Y.; Chung, A.C.-K. TGF-β/Smad Signaling in Kidney Disease. Semin. Nephrol. 2012, 32, 236–243. [Google Scholar] [CrossRef]
- Inoue, Y. and T. Imamura, Regulation of TGF-beta family signaling by E3 ubiquitin ligases. Cancer Sci, 2008. 99(11): p. 2107-12.
- Lan, H.Y. Smad7 as a therapeutic agent for chronic kidney diseases. Front. Biosci. 2008, ume, 4984–92. [Google Scholar] [CrossRef]
- Tang, P.M.-K.; Nikolic-Paterson, D.J.; Lan, H.-Y. Macrophages: versatile players in renal inflammation and fibrosis. Nat. Rev. Nephrol. 2019, 15, 144–158. [Google Scholar] [CrossRef]
- Wang, Y.Y.; et al. Macrophage-to-Myofibroblast Transition Contributes to Interstitial Fibrosis in Chronic Renal Allograft Injury. J Am Soc Nephrol, 2017. 28(7): p. 2053-2067.
- Chen, J.; Xia, Y.; Lin, X.; Feng, X.-H.; Wang, Y. Smad3 signaling activates bone marrow-derived fibroblasts in renal fibrosis. Mod. Pathol. 2014, 94, 545–556. [Google Scholar] [CrossRef]
- Edgar, B.A. From Cell Structure to Transcription: Hippo Forges a New Path. Cell 2006, 124, 267–273. [Google Scholar] [CrossRef]
- Meng, Z.; Moroishi, T.; Guan, K.-L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef]
- Fang, C.-Y.; Lai, T.-C.; Hsiao, M.; Chang, Y.-C. The Diverse Roles of TAO Kinases in Health and Diseases. Int. J. Mol. Sci. 2020, 21, 7463. [Google Scholar] [CrossRef]
- Lei, D.; Chengcheng, L.; Xuan, Q.; Yibing, C.; Lei, W.; Hao, Y.; Xizhi, L.; Yuan, L.; Xiaoxing, Y.; Qian, L. Quercetin inhibited mesangial cell proliferation of early diabetic nephropathy through the Hippo pathway. Pharmacol. Res. 2019, 146, 104320. [Google Scholar] [CrossRef]
- Sun, Y.; Jin, D.; Zhang, Z.; Jin, D.; Xue, J.; Duan, L.; Zhang, Y.; Kang, X.; Lian, F. The critical role of the Hippo signaling pathway in kidney diseases. Front. Pharmacol. 2022, 13, 988175. [Google Scholar] [CrossRef]
- Habshi, T.; Shelke, V.; Kale, A.; Lech, M.; Gaikwad, A.B. Hippo signaling in acute kidney injury to chronic kidney disease transition: Current understandings and future targets. Drug Discov. Today 2023, 28, 103649. [Google Scholar] [CrossRef]
- Xu, J.; Li, P.-X.; Wu, J.; Gao, Y.-J.; Yin, M.-X.; Lin, Y.; Yang, M.; Chen, D.-P.; Sun, H.-P.; Liu, Z.-B.; et al. Involvement of the Hippo pathway in regeneration and fibrogenesis after ischaemic acute kidney injury: YAP is the key effector. Clin. Sci. 2016, 130, 349–363. [Google Scholar] [CrossRef]
- Yang, J.; Liu, Y. Dissection of Key Events in Tubular Epithelial to Myofibroblast Transition and Its Implications in Renal Interstitial Fibrosis. Am. J. Pathol. 2001, 159, 1465–1475. [Google Scholar] [CrossRef]
- Iwakura, T.; Fujigaki, Y.; Fujikura, T.; Tsuji, T.; Ohashi, N.; Kato, A.; Yasuda, H. Cytoresistance after acute kidney injury is limited to the recovery period of proximal tubule integrity and possibly involves Hippo-YAP signaling. Physiol. Rep. 2017, 5, e13310. [Google Scholar] [CrossRef]
- Lee, S.; Huen, S.; Nishio, H.; Nishio, S.; Lee, H.K.; Choi, B.-S.; Ruhrberg, C.; Cantley, L.G. Distinct Macrophage Phenotypes Contribute to Kidney Injury and Repair. J. Am. Soc. Nephrol. 2011, 22, 317–326. [Google Scholar] [CrossRef]
- Yang, L. How Acute Kidney Injury Contributes to Renal Fibrosis. Adv Exp Med Biol, 2019. 1165: p. 117-142.
- Feng, Y.; Ren, J.; Gui, Y.; Wei, W.; Shu, B.; Lu, Q.; Xue, X.; Sun, X.; He, W.; Yang, J.; et al. Wnt/β-Catenin–Promoted Macrophage Alternative Activation Contributes to Kidney Fibrosis. J. Am. Soc. Nephrol. 2017, 29, 182–193. [Google Scholar] [CrossRef]
- Yang, Y.; Feng, X.; Liu, X.; Wang, Y.; Hu, M.; Cao, Q.; Zhang, Z.; Zhao, L.; Zhang, J.; Guo, R.; et al. Fate alteration of bone marrow-derived macrophages ameliorates kidney fibrosis in murine model of unilateral ureteral obstruction. Nephrol. Dial. Transplant. 2018, 34, 1657–1668. [Google Scholar] [CrossRef]
- Xavier, S.; Sahu, R.K.; Landes, S.G.; Yu, J.; Taylor, R.P.; Ayyadevara, S.; Megyesi, J.; Stallcup, W.B.; Duffield, J.S.; Reis, E.S.; et al. Pericytes and immune cells contribute to complement activation in tubulointerstitial fibrosis. Am. J. Physiol. Physiol. 2017, 312, F516–F532. [Google Scholar] [CrossRef]
- Portilla, D.; Xavier, S. Role of intracellular complement activation in kidney fibrosis. Br. J. Pharmacol. 2021, 178, 2880–2891. [Google Scholar] [CrossRef]
- Boor, P.; Konieczny, A.; Villa, L.; Schult, A.-L.; Diaeresis]Cher, E.B.; Rong, S.; Kunter, U.; van Roeyen, C.R.; Polakowski, T.; Hawlisch, H.; et al. Complement C5 Mediates Experimental Tubulointerstitial Fibrosis. J. Am. Soc. Nephrol. 2007, 18, 1508–1515. [Google Scholar] [CrossRef]
- Xavier, S.; Sahu, R.K.; Bontha, S.V.; Mas, V.; Taylor, R.P.; Megyesi, J.; Thielens, N.M.; Portilla, D. Complement C1r serine protease contributes to kidney fibrosis. Am. J. Physiol. Physiol. 2019, 317, F1293–F1304. [Google Scholar] [CrossRef]
- Ye, J.; Qian, Z.; Xue, M.; Liu, Y.; Zhu, S.; Li, Y.; Liu, X.; Cai, D.; Rui, J.; Zhang, L. Aristolochic acid I aggravates renal injury by activating the C3a/C3aR complement system. Toxicol. Lett. 2019, 312, 118–124. [Google Scholar] [CrossRef]
- Dong, X.; Swaminathan, S.; Bachman, L.A.; Croatt, A.J.; Nath, K.A.; Griffin, M.D. Antigen presentation by dendritic cells in renal lymph nodes is linked to systemic and local injury to the kidney. Kidney Int. 2005, 68, 1096–1108. [Google Scholar] [CrossRef]
- Kinsey, G.R.; Sharma, R.; Okusa, M.D. Regulatory T Cells in AKI. J. Am. Soc. Nephrol. 2013, 24, 1720–1726. [Google Scholar] [CrossRef]
- Sharma, R.; Kinsey, G.R. Regulatory T cells in acute and chronic kidney diseases. Am. J. Physiol. Physiol. 2018, 314, F679–F698. [Google Scholar] [CrossRef]
- Wei, W.; Zhao, Y.; Zhang, Y.; Jin, H.; Shou, S. The role of IL-10 in kidney disease. Int. Immunopharmacol. 2022, 108, 108917. [Google Scholar] [CrossRef]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Galvan, D.L.; Green, N.H.; Danesh, F.R. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. 2017, 92, 1051–1057. [Google Scholar] [CrossRef]
- Wang, Y.; Cai, J.; Tang, C.; Dong, Z. Mitophagy in Acute Kidney Injury and Kidney Repair. Cells 2020, 9, 338. [Google Scholar] [CrossRef]
- Yang, M.; Linn, B.S.; Zhang, Y.; Ren, J. Mitophagy and mitochondrial integrity in cardiac ischemia-reperfusion injury. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2019, 1865, 2293–2302. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, L.; Zhang, Y.; Yu, X.; Sun, X.; Zhu, T.; Li, X.; Liang, W.; Han, Y.; Qin, C. PINK1 Deficiency Ameliorates Cisplatin-Induced Acute Kidney Injury in Rats. Front. Physiol. 2019, 10, 1225. [Google Scholar] [CrossRef]
- Li, N.; Wang, H.; Jiang, C.; Zhang, M. Renal ischemia/reperfusion-induced mitophagy protects against renal dysfunction via Drp1-dependent-pathway. Exp. Cell Res. 2018, 369, 27–33. [Google Scholar] [CrossRef]
- Tran, M.; Tam, D.; Bardia, A.; Bhasin, M.; Rowe, G.C.; Kher, A.; Zsengeller, Z.K.; Akhavan-Sharif, M.R.; Khankin, E.V.; Saintgeniez, M.; et al. PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Investig. 2011, 121, 4003–4014. [Google Scholar] [CrossRef]
- Tran, M.T.; Zsengeller, Z.K.; Berg, A.H.; Khankin, E.V.; Bhasin, M.K.; Kim, W.; Clish, C.B.; Stillman, I.E.; Karumanchi, S.A.; Rhee, E.P.; et al. PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 2016, 531, 528–532. [Google Scholar] [CrossRef]
- Koyano, T.; Namba, M.; Kobayashi, T.; Nakakuni, K.; Nakano, D.; Fukushima, M.; Nishiyama, A.; Matsuyama, M. The p21 dependent G2 arrest of the cell cycle in epithelial tubular cells links to the early stage of renal fibrosis. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Matos, D.A.; Zhang, J.-M.; Ouyang, J.; Nguyen, H.D.; Genois, M.-M.; Zou, L. ATR Protects the Genome against R Loops through a MUS81-Triggered Feedback Loop. Mol. Cell 2019, 77, 514–527. [Google Scholar] [CrossRef]
- Yang, L.; et al. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med, 2010. 16(5): p. 535-43, 1p following 143.
- Canaud, G.; Brooks, C.R.; Kishi, S.; Taguchi, K.; Nishimura, K.; Magassa, S.; Scott, A.; Hsiao, L.-L.; Ichimura, T.; Terzi, F.; et al. Cyclin G1 and TASCC regulate kidney epithelial cell G 2 -M arrest and fibrotic maladaptive repair. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Lemos, D.R.; McMurdo, M.; Karaca, G.; Wilflingseder, J.; Leaf, I.A.; Gupta, N.; Miyoshi, T.; Susa, K.; Johnson, B.G.; Soliman, K.; et al. Interleukin-1β Activates a MYC-Dependent Metabolic Switch in Kidney Stromal Cells Necessary for Progressive Tubulointerstitial Fibrosis. J. Am. Soc. Nephrol. 2018, 29, 1690–1705. [Google Scholar] [CrossRef]
- Yang, L., B. D. Humphreys, and J.V. Bonventre, Pathophysiology of acute kidney injury to chronic kidney disease: maladaptive repair. Contrib Nephrol, 2011. 174: p. 149-155.
- Novitskaya, T.; McDermott, L.; Zhang, K.X.; Chiba, T.; Paueksakon, P.; Hukriede, N.A.; de Caestecker, M.P.; Skrypnyk, N.I.; Voziyan, P.; Yang, H.; et al. A PTBA small molecule enhances recovery and reduces postinjury fibrosis after aristolochic acid-induced kidney injury. Am. J. Physiol. Physiol. 2014, 306, F496–504. [Google Scholar] [CrossRef]
- Fang, Y.; Gong, A.Y.; Haller, S.T.; Dworkin, L.D.; Liu, Z.; Gong, R. The ageing kidney: Molecular mechanisms and clinical implications. Ageing Res. Rev. 2020, 63, 101151–101151. [Google Scholar] [CrossRef]
- Pabla, N.; et al. Mitigation of acute kidney injury by cell-cycle inhibitors that suppress both CDK4/6 and OCT2 functions. Proc Natl Acad Sci U S A, 2015. 112(16): p. 5231-6.
- Chou, Y.-H.; Huang, T.-M.; Chu, T.-S. Novel insights into acute kidney injury–chronic kidney disease continuum and the role of renin–angiotensin system. J. Formos. Med Assoc. 2017, 116, 652–659. [Google Scholar] [CrossRef]
- Seccia, T.M.; Rigato, M.; Ravarotto, V.; Calò, L.A. ROCK (RhoA/Rho Kinase) in Cardiovascular–Renal Pathophysiology: A Review of New Advancements. J. Clin. Med. 2020, 9, 1328. [Google Scholar] [CrossRef]
- AlQudah, M.; Hale, T.M.; Czubryt, M.P. Targeting the renin-angiotensin-aldosterone system in fibrosis. Matrix Biol. 2020, 91-92, 92–108. [Google Scholar] [CrossRef]
- Rüster, C.; Wolf, G. Renin-Angiotensin-Aldosterone System and Progression of Renal Disease. J. Am. Soc. Nephrol. 2006, 17, 2985–2991. [Google Scholar] [CrossRef]
- Ravarotto, V.; Pagnin, E.; Fragasso, A.; Maiolino, G.; Calò, L.A. Angiotensin II and Cardiovascular-Renal Remodelling in Hypertension: Insights from a Human Model Opposite to Hypertension. High Blood Press. Cardiovasc. Prev. 2015, 22, 215–223. [Google Scholar] [CrossRef]
- Shrestha, A., R. C. Che, and A.H. Zhang, Role of Aldosterone in Renal Fibrosis. Adv Exp Med Biol, 2019. 1165: p. 325-346.
- Brown, N.J. Aldosterone and end-organ damage. Curr. Opin. Nephrol. Hypertens. 2005, 14, 235–241. [Google Scholar] [CrossRef]
- Lewis, E.J.; Hunsicker, L.G.; Clarke, W.R.; Berl, T.; Pohl, M.A.; Lewis, J.B.; Ritz, E.; Atkins, R.C.; Rohde, R.; Raz, I.; et al. Renoprotective Effect of the Angiotensin-Receptor Antagonist Irbesartan in Patients with Nephropathy Due to Type 2 Diabetes. N. Engl. J. Med. 2001, 345, 851–860. [Google Scholar] [CrossRef]
- Molnar, M.Z.; et al. Angiotensin-converting enzyme inhibitor, angiotensin receptor blocker use, and mortality in patients with chronic kidney disease. J Am Coll Cardiol, 2014. 63(7): p. 650-658.
- Chen, J.-Y.; Tsai, I.-J.; Pan, H.-C.; Liao, H.-W.; Neyra, J.A.; Wu, V.-C.; Chueh, J.S. The Impact of Angiotensin-Converting Enzyme Inhibitors or Angiotensin II Receptor Blockers on Clinical Outcomes of Acute Kidney Disease Patients: A Systematic Review and Meta-Analysis. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef]
Figure 1.
Mechanism involved in the transition from acute kidney injury to chronic kidney disease.
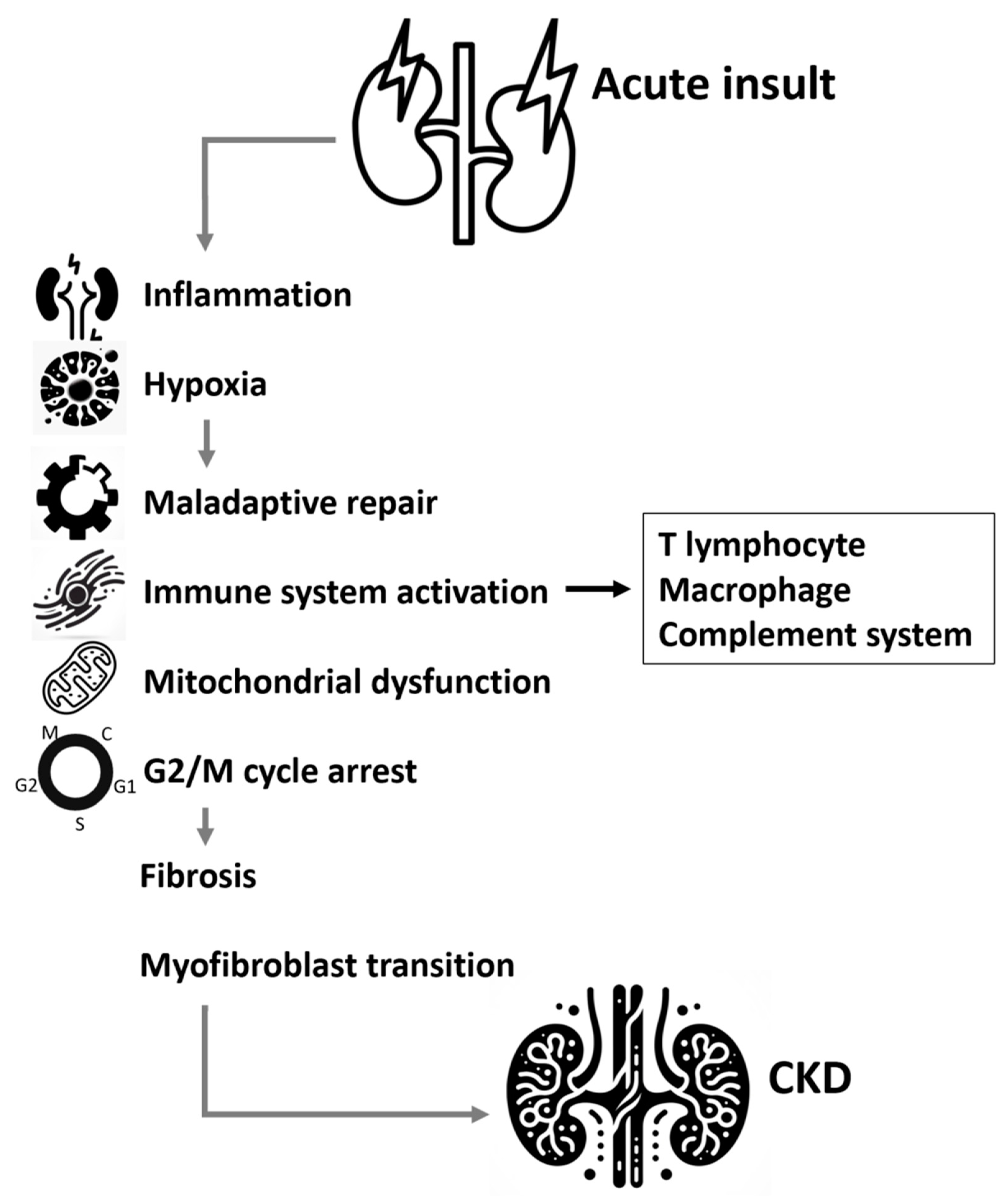
Figure 2.
Under normal conditions, in the absence of Wnt ligand interaction, LRP contributes to the phosphorylation of β-catenin, resulting in its retention in the cytoplasm. However, during pathological processes, Wnt ligands bind to the FZD/LRP complex, leading to the prevention of β-catenin phosphorylation. This allows β-catenin to migrate into the nucleus and initiate downstream pathways, thereby promoting renal fibrosis (2) After an acute insult, TGF-β1 becomes activated and binds to its receptor, which in turn phosphorylates SMAD2/3. The phosphorylated SMAD2/3, along with SMAD4, then translocates into the nucleus to activate miRNA-21 and miRNA-192. This activation ultimately leads to renal fibrosis. Concurrently, Smurf1/2 is activated by the SMAD2/3/4 complex, which diminishes the inhibitory capability of SMAD7 (3) The activation of the Hippo pathway leads to the phosphorylation of MST1/2, SAV1, LAST1/2, and MOB1, which contributes to the degradation of YAP/TAZ. Conversely, the inactivation of the Hippo pathway results in the activation of YAP/TAZ, allowing them to migrate into the nucleus. This migration initiates cellular proliferation and contributes to the development of renal fibrosis. Abbreviations: LRP: LDL receptor-associated protein; FZD: frizzled protein; TCF/LEF: T cell factor/lymphoid enhancer factor transcription factors; MMP-7: matrix metalloproteinase 7; TRPC6: Transient receptor potential canonical 6; TEAD1–4: TEA domain DNA-binding family members; MST1/2: STE20-like serine/threonine kinase 1/2; SAV1: Salvador 1; LATS1/2: large tumor suppressor; MOB1: MOB kinase activator 1; YAP: Yes-associated protein; TAZ: Tafazzin; LAP: latency-associated peptide; LTBP: latent TGF-β1 binding protein; TGF-β1:Transforming growth factor-β; Smurf: Smad ubiquitination regulatory factor.
Figure 2.
Under normal conditions, in the absence of Wnt ligand interaction, LRP contributes to the phosphorylation of β-catenin, resulting in its retention in the cytoplasm. However, during pathological processes, Wnt ligands bind to the FZD/LRP complex, leading to the prevention of β-catenin phosphorylation. This allows β-catenin to migrate into the nucleus and initiate downstream pathways, thereby promoting renal fibrosis (2) After an acute insult, TGF-β1 becomes activated and binds to its receptor, which in turn phosphorylates SMAD2/3. The phosphorylated SMAD2/3, along with SMAD4, then translocates into the nucleus to activate miRNA-21 and miRNA-192. This activation ultimately leads to renal fibrosis. Concurrently, Smurf1/2 is activated by the SMAD2/3/4 complex, which diminishes the inhibitory capability of SMAD7 (3) The activation of the Hippo pathway leads to the phosphorylation of MST1/2, SAV1, LAST1/2, and MOB1, which contributes to the degradation of YAP/TAZ. Conversely, the inactivation of the Hippo pathway results in the activation of YAP/TAZ, allowing them to migrate into the nucleus. This migration initiates cellular proliferation and contributes to the development of renal fibrosis. Abbreviations: LRP: LDL receptor-associated protein; FZD: frizzled protein; TCF/LEF: T cell factor/lymphoid enhancer factor transcription factors; MMP-7: matrix metalloproteinase 7; TRPC6: Transient receptor potential canonical 6; TEAD1–4: TEA domain DNA-binding family members; MST1/2: STE20-like serine/threonine kinase 1/2; SAV1: Salvador 1; LATS1/2: large tumor suppressor; MOB1: MOB kinase activator 1; YAP: Yes-associated protein; TAZ: Tafazzin; LAP: latency-associated peptide; LTBP: latent TGF-β1 binding protein; TGF-β1:Transforming growth factor-β; Smurf: Smad ubiquitination regulatory factor.
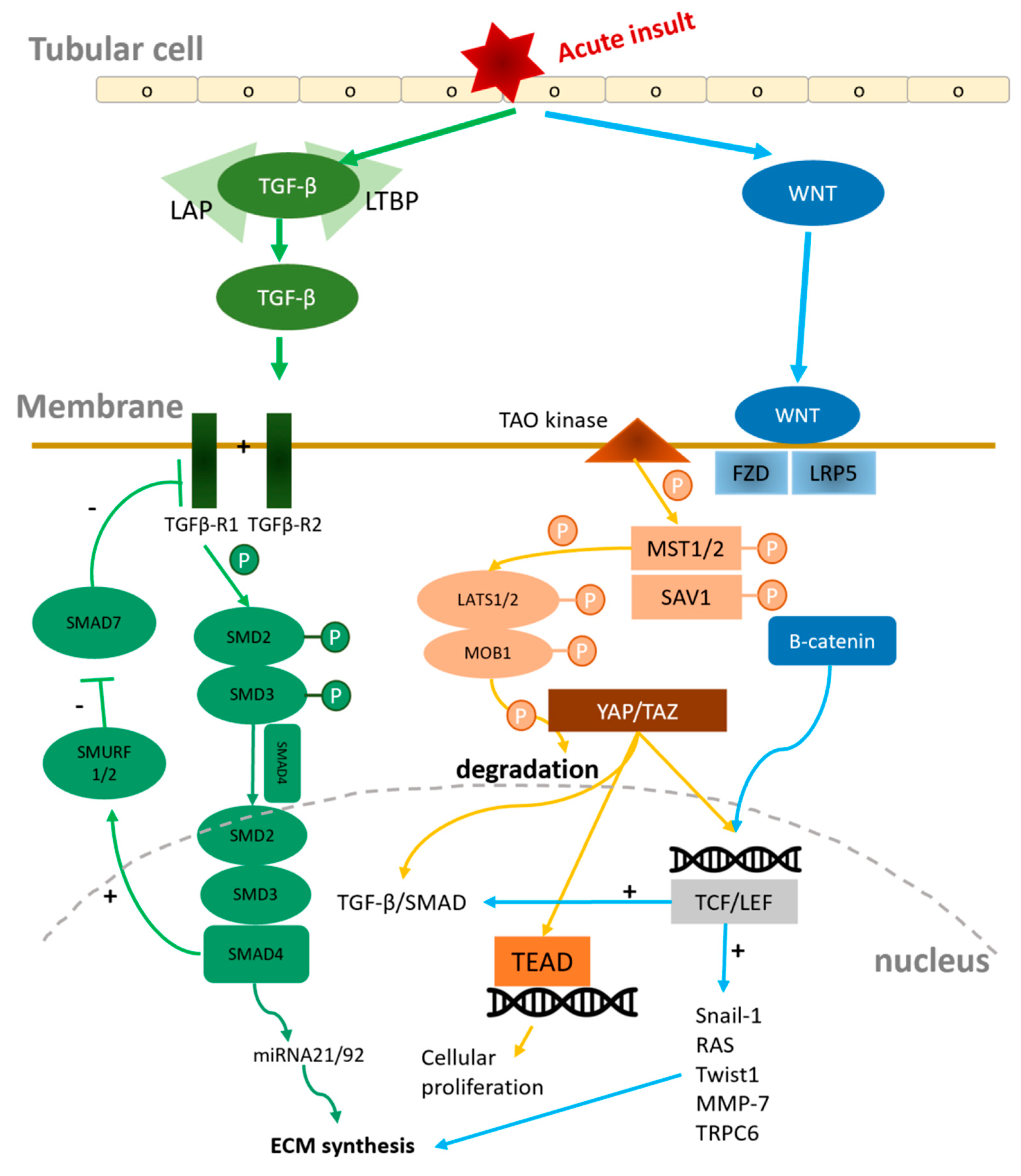
Figure 3.
Binding of Angiotensin II to the AT1 receptor activates the RhoGEF/RhoA/ROCK cascade, leading to overexpression of NF-κB, PAI-1, MAPK/ERK 1/2, and NADPH oxidases. These oxidases, as ROS-generating enzymes, increase oxidative stress, thereby upregulating TGF-β/SMAD and MAPK/ERK pathways. This cascade results in organ remodeling and tissue fibrosis. Additionally, AT1R activation alters intracellular NO and calcium levels via the PI3K/Akt pathway and disrupt the autoregulation of kidney by arteriole vasocontraction. Abbreviations: MAPK: mitogen-activated protein kinase; AT1R: angiotensin II receptor type 1; TGF-β: transforming growth factor-β; ERK: Extracellular signal-Regulated Kinases; NF-κB: nuclear factor kappa-B; PAI-1: Plasminogen Activator Inhibitor-1; eNOS : endothelial nitric oxide synthase; PI3K/Akt: phosphatidylinositol 3-kinase/protein kinase B; NO: nitric oxide.
Figure 3.
Binding of Angiotensin II to the AT1 receptor activates the RhoGEF/RhoA/ROCK cascade, leading to overexpression of NF-κB, PAI-1, MAPK/ERK 1/2, and NADPH oxidases. These oxidases, as ROS-generating enzymes, increase oxidative stress, thereby upregulating TGF-β/SMAD and MAPK/ERK pathways. This cascade results in organ remodeling and tissue fibrosis. Additionally, AT1R activation alters intracellular NO and calcium levels via the PI3K/Akt pathway and disrupt the autoregulation of kidney by arteriole vasocontraction. Abbreviations: MAPK: mitogen-activated protein kinase; AT1R: angiotensin II receptor type 1; TGF-β: transforming growth factor-β; ERK: Extracellular signal-Regulated Kinases; NF-κB: nuclear factor kappa-B; PAI-1: Plasminogen Activator Inhibitor-1; eNOS : endothelial nitric oxide synthase; PI3K/Akt: phosphatidylinositol 3-kinase/protein kinase B; NO: nitric oxide.
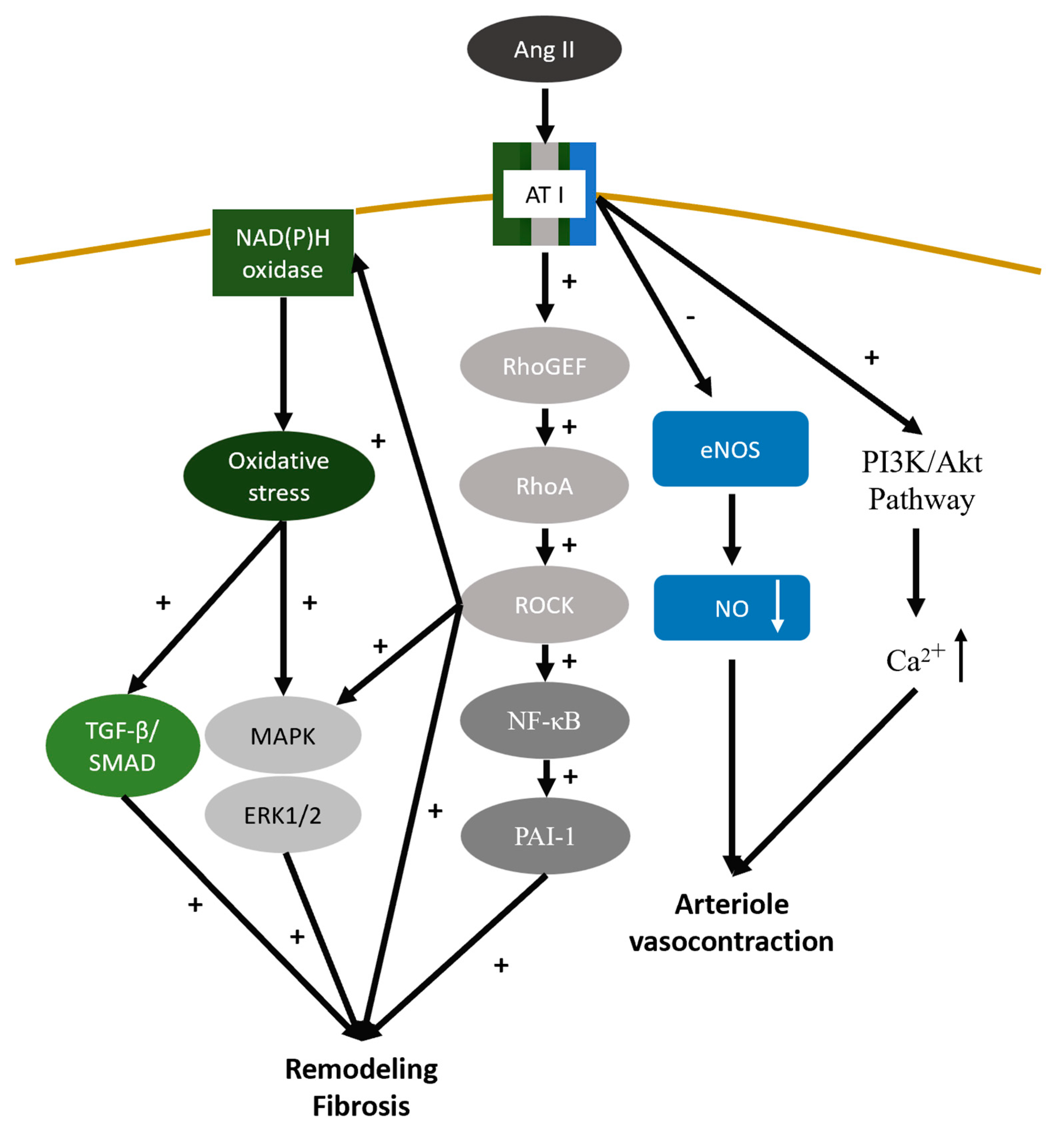

Table 1.
Biomarkers for acute kidney injury.
| Types of Markers | Markers | Clinical application |
|---|---|---|
| Stress marker | Urine | |
| DKK3 | Preoperative levels of urinary DKK3 have been identified as an independent predictor for the occurrence of postoperative AKI [19]. | |
| TIMP-2 IGFBP-7 |
These markers may show a rapid increase after cellular stress, typically within 4 to 12 hours, even before the occurrence of injury [20,21]. | |
| Damage marker | Urine | |
| Alanine aminopeptidase | Diagnostic relevance in nephrolithiasis [36]. Positive correlation between urinary Alanine aminopeptidase concentrations and glomerulonephritis [37]. |
|
| Alkaline phosphatase | Endre et al. took Alkaline phosphatase as biomarker of acute kidney biomarker in the EARLYARF trial [38]. | |
| γ-glutamyl transpeptidase | The Translational Research Investigating Biomarker Endpoints in AKI (TRIBE-AKI) study evaluated γ-glutamyl transpeptidase in AKI diagnosis [39]. | |
| Calprotectin | Calprotectin indicates primary intrinsic AKI causes [40]. | |
| CCL14 | predictive marker for persistent AKI in critically ill patients in the RUBY study [25]. | |
| NGAL | Elevated levels of urinary NGAL are useful for predicting AKI, differentiating intrinsic AKI from pre-renal AKI, predicting renal non-recovery, in-hospital mortality, long-term CKD progression [41]. | |
| KIM-1 | Indicator of renal tubular damage [42]. Elevated levels of KIM-1 in patients with AKI may manifest prior to histological changes [43]. |
|
| L-FABP | Indicator of ischemic or toxic insults that result in tubulointerstitial damage [44]. | |
| IL-18 | Indicators of severity of albuminuria, and deterioration of kidney function and associated with diabetic nephropathy [45]. | |
| Serum | ||
| NGAL | NGAL can be detected in ischemic or toxicity-induced damage to the kidney [22,23,26] and had the best predictive accuracy for the occurrence of AKI [27]. | |
| Functional marker | Serum | |
| Cystatin C | Better accuracy than serum creatinine in identifying individuals with reduced GFR [30] and increased level within 12–24 hours following renal injury [29]. | |
| Proenkephalin A | Proenkephalin A serves as a useful biomarker for early detection of AKI and predicting a shorter duration and successful liberation from renal replacement therapy [34,35]. | |
Abbreviations: DKK-3, Dickkopf-3; TIMP2, Tissue inhibitor of metalloproteinase-2; IGFBP-7, Insulin-like growth factor binding protein 7; CCL14, C-C motif chemokine ligand 14; NGAL, Neutrophil gelatinase-associated lipocalin; KIM-1, Kidney Injury Molecule-1; L-FABP, Liver-type fatty acid binding protein; IL-18: Interleukin 18.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



