
Submitted:
25 December 2023
Posted:
08 January 2024
You are already at the latest version
Abstract
Background A diet rich in red meat, and poor in fruits and vegetables, can hinder the body's ability to detoxify, therefore, an alternative vegetarian diet maintained over a certain period, determines the elimination of excess toxins and accumulated residues, helping the body to regenerate and recharge energy. Following this simple rule, there are a multitude of benefits for our health. Significance of the autophagy process An even better regeneration of the body takes place during the black fast, in which senescent cells, (dead cells that no longer exercise any function, but instead produce inflammation in the body), are eliminated by autophagy, and in their place, the cells are stimulated stem. Aging can be associated with various pathological conditions if autophagy is not promoted by a healthy lifestyle. In elderly people, cells can show DNA mutations and accumulations of protein aggregates that impair the body's physiological processes. Several studies have shown that the activation of autophagy slows down the ageing process. Conclusions Aging, and its associated conditions of illness, are due to genomic instability caused by DNA damage, telomere shortening and epigenome changes that modulate gene expression.
Keywords:
autophagy
; adenosine triphosphate (ATP)
; Atg gene
; p53 protein
Introduction
A restrictive diet represents a period of self-discipline designed to purify the body from a physical and spiritual point of view. The nutritional advantages of fasting on health are explained by the more effective detoxification of tissues and the regeneration of the body. Whether you choose to fast for religious reasons or health reasons (there are some "fasting" diets, with longer or shorter periods of not eating), self-initiated fasting can be a great way to revitalize the body, mind and soul. In general, is recommended that certain people do not fast, such as children, pregnant women, and the elderly with health problems. A person without health problems can create a diversified menu and in principle can follow the fast without worries.
Relevant Sections
The process of autophagy refers to the ability of cells to destroy their contents, which they then transport in sac-shaped vesicles to a recycling compartment, the lysosomes, where the actual destruction of the cellular material occurs. Through autophagic processes, the body also cleans toxins and recycles the components of damaged cells. Cells create membranes that hunt down the debris of dead, diseased, or worn-out cells and use the resulting molecules for energy or to regenerate other cells, [1].
Disturbances in the autophagy process can lead to metabolic chronic diseases, such as Parkinson's disease, type 2 diabetes, cancer and other diseases that occur with ageing. Reduction of Beclin 1 protein, a product of the ATG-6 gene, autophagy promoter molecule, results in decreased levels of phagocytic receptor recycling and phagocytosis, an example being Alzheimer's disease (AD), with the stored peptide β-amyloid (Aβ) inter-neuronal [2].
The release of ATP stimulates the microglial cells and the release of auto-page-lysosomes in the extracellular space. In cancer cells from solid undernourished tumors, response to radiotherapy or DNA-damaging chemotherapy triggers ATP production by autophagy may have an essential role in DNA repair. The addition of pyruvate rescued ATP levels and prevented mitotic catastrophe, suggesting that autophagy, sustained ATP generation, could be employed by mechanisms that promote genomic integrity, such as DNA repair [3,4].
The efficacy of autophagy in favoring cell death is demonstrated in many other cancer models, such as breast cancer, leukemia, prostate cancer, and myeloma. Currently, protocols targeting autophagy induction instead of autophagy blockade are under intense investigation in oncology.
In preclinical studies, dietary restriction (DR) shown to extend the lifespan and reduce the development of age-related diseases such as diabetes, cancer, and neurodegenerative and cardiovascular diseases. DR promotes metabolic and cellular changes in organisms from prokaryotes to humans that allow adaptation to periods of limited nutrient availability, [5], [Scheme 1].
The main changes include decreased blood glucose, levels of growth factor signaling and the activation of stress resistance pathways affecting cell growth, energy metabolism, and protection against oxidative stress, inflammation and cell death.
Nutrient starvation also activates autophagy in most cultured cells and organs, such as the liver and muscle, as an adaptive mechanism to stressful conditions. Studies demonstrate that dietary interventions can reduce tumor incidence and potentiate the effectiveness of chemo- and radiotherapy in different tumor models, highlighting dietary manipulation as a possible adjunct to standard cancer therapies. Among the many diet regimens that are assessed, caloric restriction (CR) and fasting are the methods under intense investigation in oncology.
CR is defined as a chronic reduction in the daily caloric intake by 20-40% without the incurrence of malnutrition and with the maintenance of meal frequency. In contrast, fasting is characterized by the complete deprivation of food but not water, with intervening periods of normal food intake. Based on the duration, fasting can be classified as (i) intermittent fasting as alternate day fasting (≥16 hours) or 48 hours of fasting/week) or (ii) periodic fasting minimum of 3 days of fasting every 2 or more weeks.
The specific diet called the Bredesen diet, is a method that combines intermittent fasting, eating healthy foods and avoiding sugar. In a weight loss treatment, the recommendation for a restrictive diet can include 2 meals/day, in intermittent time, according to a well-established plan, with a fixed schedule after 16 hours of the last meal, from the previous day, more precisely breakfast at 10 a.m. and the next meal at 6 AM, on the same day. In the weight loss diet, we must avoid losing muscle mass, but we must have the main objective of a vegetarian diet, with white meat consumption, especially for people over 65 years old. A total fast of 24-48 hours, once a year, with daily hydration, 30 ml/Kg body, approximately 2.5 L/day, triggers the beneficial mechanisms of autophagy to prevent carcinogenesis, [6].
Discussions
Autophagy is required for the fulfilment of cellular metabolic demands, preservation of genomic integrity and adaptive immune processes, and regulation of pro-inflammatory mediators for cell survival. Autophagy is evolutionarily conserved and serves as a self-degradative catabolic process that removes long-lived, damaged molecules and organelles, aggregated proteins, and intracellular pathogens.
The types of autophagy include macro-autophagy, micro-autophagy and chaperone-mediated autophagy. Autophagy is used for the removal of damaged organelles including mitochondria (mitophagy), endoplasmic reticulum (ER), (ER autophagy) and lysosome (mitophagy).
Macro-autophagy applies to subcellular organelles within cytosolic double-membrane-bound autophagosomes fused with lysosomes. Micro-autophagy and micro-(ER autophagy) can occur with high precision by allowing specific cargoes to be captured before they enter the invaginations of endo-lysosome membranes (self-eating). By such actions, autophagy is an essential mechanism for the prevention of ageing, disease and cancer.
Protein aggregates are removed by macro-autophagy and lipophagy to maintain energy balance by the breakdown of lipids. The core proteins that regulate the biogenesis of autophagosomes have been not identified yet and it is not yet clear how these structures are assembled, [7].
The lysosomal compartment is a key element of the autophagic machinery, accounting for the exhaustive degradation of the engulfed substrates [8]. First characterized as the main degrading organelles, lysosomes are now granted complex regulatory functions in both normal and pathological conditions, including cancer development and progression [9].
Lysosomes are known to account for the lower susceptibility of cancer cells to chemotherapeutics which are weak bases by favoring their intra-lysosomal sequestration and ensuing removal from the cell by exocytosis [10,11]. In this lysosome can be a potential target for cancer therapy. This was mainly linked to evidence that the lysosome plays a significant role in cell death, as well as its ability to fuel cancer cells' energy needs.
Previous studies have shown that activated B cells release antibodies and label tumor cells to be recognized and attacked by other cells in the immune system which may suggest a more anti-tumor relationship between lysosomes and the presence of immune cells. Furthermore, the study found that higher levels of type interferon-2 (IFN), response, HLA (human leukocyte antigen), as a major histocompatibility complex (MHC) product in humans modulate the immune response to cancer by presenting antigens. It has been shown that type-2 IFN can directly trigger apoptosis and cell cycle arrest by impairing autophagosome-lysosomal fusion in cancer cells having an important role in tumor development, which may be related to immune cells and cytokines, [12].
However, intra-lysosomal accumulation of drugs to high concentrations, when not followed by their extracellular release, has also been reported to trigger lysosomal membrane permeabilization and activation of lysosomal death pathways. Thus, in this view, lysosomal destabilization may represent both a putative intrinsic mechanism of action of these drugs and an effective strategy to increase the susceptibility of cancer cells to anticancer therapy, [13].
Beclin-1 protein, the product of the Atg-6 gene, which allows the activation of autophagy, is a positive regulator of autophagy, due to its ability to create a link between cytoskeletal motor proteins and the cytoplasmic complex phosphatidyl-inositol triphosphate kinase, (PI3K), class III that triggers the autophagy process. Defective autophagy is implicated in tumorigenesis and an essential autophagy regulator, the Beclin-1 gene, is mono-allelically deleted in human breast, ovarian, and prostate cancers.
Beclin-1 refers to a centromeric region of BRCA1 on chromosome 17q21 that is generally deleted in 75, 50 and 40% of ovarian, breast and prostate cancers. Depending on the nutritional conditions, the activities of the ULK-kinase enzyme complex, the product of the ATG-6 gene, can be regulated by the mTOR enzyme complex. Beclin-1 the mammalian orthologue of Atg6 is cloned as a Bcl-2 interacting protein. Bcl-2 blocked the Beclin-1. In starved cells, Beclin-1 delivered from the Bcl-2 protein induces signals for the autophagic process, [14].
The molecular interactions between the p53 protein and autophagy, in cells with damaged DNA, lead to cell apoptosis. In addition to mitigating DNA damage by controlling ROS production, autophagy can also influence the dynamics of DNA repair by recycling key proteins involved in the processing of lesions. Autophagy may also provide metabolic precursors for the generation of ATP which is employed in several steps of DNA repair
The tumor suppressor protein p53, the product of the P53 gene, is a critical control protein in mammalian cells that is activated under conditions of genotoxic stress, including DNA damage, hypoxia, and oncogene activation and responds by initiating tumor suppressor mechanisms such as cell cycle arrest, senescence and apoptosis.
The tumor suppressor gene, p53, which is uniformly activated in response to DNA damage by radiation, can induce autophagy through the activation of autophagy-inducing genes, particularly ULK1 and ULK2.
Activation of gene complexes, ULK1 and ULK2, as a result of DNA damage, causes increased levels of autophagy and contributes to the triggering of apoptosis. Under the appropriate circumstances, mammalian rapamycin target protein complex 1 (mTOR1) can phosphorylate ULK1 and Atg13, preventing ULK1 from binding to Atg13, FIP200, and Atg101, and therefore suppressing autophagy. Activation of the ULK1 gene inhibits the mTOR gene, promoting the autophagy process.
Conversely, deletion of the PTEN gene is followed by an increase in mTOR activity and leads to an increase in clonogenic survival. Studies suggest that inhibition of mTOR signaling could be an effective strategy for increasing the radiosensitivity of malignant tumors. By another mechanism, the p53 protein, the product of the P53 gene, in the isoform state, accelerates the forms of cancer from the mild form to the severe form., [Scheme 2].
The native protein p53 is a control protein in the normal functioning of the cell and is activated by cellular stress, including DNA damage, hypoxia, and restrictive diet, thus contributing to the inhibition of malignant, by stopping the cell cycle in the Go-1 phase, as a rest before cell division. The p53 protein has been shown to contribute and to trigger the autophagy process by inhibiting the mTOR protein, and AMP gene-activated protein kinase (AMPK), [15], [Scheme 3].
At the same time, genetic or pharmacological inactivation of cytoplasmic p53 protein triggers autophagy indicating that the non-nuclear p53 protein pool is a potent autophagic repressor. Thus, autophagy is activated as a stress mitigation mechanism, both by stress-mediated p53 protein induction and by stress-exacerbated p53 loss. In this context, the nuclear protein p53 has been shown to protect the cell from a malignant process and only the cytoplasmic protein p53, through its isoforms, phosphorylated in multiple sites, in a modified cytoplasmic environment, through a high concentration of anaerobic ATP, leads to cancer.
In Chronic Lymphocytic Leukemia, (CLL), blocked apoptosis from malignant diseases may be due to a high ATP concentration originating from an anaerobic metabolism. The difference of energy between anaerobic ATP in B lymphocytes from CLL and aerobic ATP in activated T lymphocytes from normal status and non-malignant diseases was 2.68 μM ATP, as an energetic transfer between T and B cells initiates carcinogenesis by suppression of anti-oncogene proteins, especially p53 protein, [16].
Through genetic research for longevity, the family genes, SIRT1 to SIRT7 were discovered for the development of drugs, to extend the life span of humans. The SIR2 gene was discovered in animals, a gene that was discovered in nutritional yeast. The Sirtuins in mitochondria, such as SIRT3, SIRT4, and SIRT5 are among the seven mammalian Sirtuins. SIRT3 is a deacetylase that regulates mitochondrial activity and is one of the most significant deacetylases in mitochondria. SIRT3 deacetylates the PDC (pyruvate dehydrogenase complex) in glycolysis, allowing pyruvate to take part in the Krebs cycle and increasing glucose absorption by triggering protein kinase B [17].
Through a process of visualizing DNA and grouping it into tight bundles of protein, marking genes with chemical tags called methyl and acetyls made up of carbon, oxygen, and hydrogen atoms, the epigenome can use the genome to create the music of our lives. Sirtuins are not the only longevity genes. Two other sets of genes currently under study perform similar roles, which have also been shown to be manipulable in how one could live up to 113 years with a healthier life. Currently, life expectancy in the developed world is just over 80 years, [18].
To anticipate a cancer diagnosis, with the genes still "silent", the Whole Genome Sequencing Genetic Test (WGS) can be performed, which identifies in the patient's DNA the genetic mutations of multiple genes that are responsible for the carcinogenesis process followed by, Next Generation Sequencing (NSG) and Single Nuclear Polypeptide, (SNG) techniques, for the analysis of epigenetic variations, [19]
The other pathway involved in autophagy is a metabolic control enzyme known as AMPK, which is activated at low energy levels. The AMPK enzyme contributes to preventing cell damage and sustaining apoptosis, under conditions of a low-calorie diet, combined with certain types of physical exercise and exposure to intermittent hot and cold temperatures, called hormesis, [19,20].
The pro-apoptotic proteins Bad, Bid and Bax can activate on the cell membrane triggering apoptosis with the release of cytochrome C, promoting autophagy by dissociation of Beclin-1 from molecular complex, [21], [Scheme 4].
In established solid tumors, autophagy has been shown to favor tumor development by enhancing tumor growth, cell survival, resistance to platinum-based chemotherapy and metastasis formation. Autophagy may also interfere with immunotherapy, since some studies showed a link between autophagy and immune checkpoint activity and/or expression, including CTLA-4, IDO and PD1/PD-L1. Autophagy also has a critical function in tumor immune cells and tumor immune response, promoting the immunogenic cell death of tumor cells and favoring immune cell activation and proliferation. Meanwhile, autophagy in cancer-associated fibroblasts (CAFs) promotes tumorigenesis by providing nutrients to the cancerous cells and by favoring epithelial-to-mesenchymal transition, angiogenesis and stemness [23].
The PTEN gene demonstrates a tumor-suppressive role for autophagy when considering its genetic inactivation, either indirectly through constitutive activation of the PI3K/AKT pathway through activating PI3K m mutations, AKT amplifications, or PTEN gene mutation. Multiple mutations of the PTEN gene, or loss of function, affect lipid phosphatase activity, leading to the development of a variety of cancers. Of the three residues in the PTEN component, the R-335 mutation is the most important for interaction with the cell membrane, in common with several other germline mutations, and has been associated with inherited cancer, [24].
One of the greatest technological advances of this century has been the discovery of precise and programmable 'genome editing', CRISPR technology, (short regularly spaced palindromic repeats) with the DNA strand scissor enzyme Cas9, which can introduce healthy genes or edit mutated genes, [25].
Researchers have identified a gene, called Arc, that plays a role in modulating cell activity. This gene packs functional genetic material with the power to change the fate of other cells by transferring genetic information, including turning them into cancer, [26]. [Figure 1].
Many studies of hematological malignant disease demonstrated that autophagy allows T and B lymphocytes to survive under nutrient-deprivation conditions or during stress stimuli. Since cytoplasmic calcium levels are essential for the activation of TCR signaling pathways, autophagy-dependent regulation of calcium flux could influence lymphocyte activation. Inhibition of autophagy causes defects in T-cell activation.
Deletion of the Atg7 gene results in decreased levels of IL-2 mRNA, interleukin-2, and ATP generation, suggesting that autophagy is required to provide an adequate level of energy for cell activation T. Similar data were obtained in B lymphocytes, demonstrating that autophagy is essential for the maturation process and the subsequent maintenance of the. lymphocyte repertoire in the periphery, [27].
Molecules that alter these pathways, such as metformin, resveratrol and boosting the enzyme nicotinate deaminase (NAD), responsible for cellular respiration. For example, resveratrol activates autophagic cell death in prostate cancer cells via the downregulation of STIM1 and the mTOR pathway. Metformin is a well-known mitochondrial inhibitor that has shown beneficial effects on animal models of neurodegeneration by inducing autophagy through AMPK activation. It has been shown that modulating cyclic AMP and Inositol 3 –phosphate (cAMP/IP3) positively regulates the autophagy process.
Resveratrol is a polyphenol found in grapes, nuts, berries, and other plants that has pleiotropic effects such as antioxidant activity, neuroprotective properties, anti-inflammatory activity, and cytoprotective effect. Resveratrol may influence cellular functions by triggering critical metabolic sensors/effectors such as AMPK, and SIRT1and protects against chronic diseases, by promoting autophagy and suppressing inflammatory cytokines like interleukin 1 beta (IL-1), tumor necrosis factor-alpha (TNF-α), and nuclear factor kappa-β (NF-κβ), [28].
The autophagy process can effectively remove the misfolded and damaged proteins which can drive, starvation, tumor development and suppression. Autophagy is a kind of intracellular degradative process, and it can be initiated in the presence of poor nutritional and hypoxia conditions or upon exposure of cancer cells to chemotherapy. When autophagy-related proteins such as ATG7 and ATG5 were knocked down Overall, both autophagy and apoptosis have been reported to play an important role in protection against cellular damage, [29].
The signal transducer and activator of transcription 5 (STAT5) have been known to play a significant role in regulating cellular survival and homeostasis. However, its aberrant activation has been reported to mediate tumorigenesis. The different STAT family members including STAT5, can promote the development of various cancers such as solid tumors and hematological malignancies. Activation of the STAT5 signaling pathway is initiated upon its phosphorylation by intracellular kinases like JAK1, JAK2, and Src gene.
Additionally, previous studies have suggested that pharmacological inhibition of STAT5 gene phosphorylation could induce programmed cell death mechanism(s) like autophagy and apoptosis in human lung and mesangial cancer cells. Autophagy is a type of cellular autolysis initiated by various stimuli, one of which is cytoplasmic deprivation, which allows cells to eliminate cytoplasmic components. Autophagy’s understanding as a mechanism of resistance to metabolic stress-inducing therapies or as a means of cell death is fast developing, indicating that a new therapeutic starvation paradigm is emerging, [30].
Physical activity may positively influence lysosomal disintegration and mitochondrial quality control, reducing age-related cognitive decline. Exercise is a well-known physiological therapy that can preserve tissue integrity, reduce inflammation, and activate the direct signaling route for the cellular response. It has also been shown to significantly enhance diverse mitochondrial gene transcripts and autophagy-associated genes, promote mitophagy in mitochondria and increase autophagy and mitophagy flux.
A large body of research supports that obesity, diabetes, and cardiovascular diseases are comorbidities of neurodegenerative disorders, and they are becoming a big concern in today’s society, with a significant impact on socioeconomic status. Nevertheless, the fact that autophagy can be cytotoxic in function indicates the importance of determining the mode of autophagy that is being induced by a particular treatment modality before autophagy inhibition can be considered as a potential clinical strategy for enhancing the response of malignancies to therapy, [31].
Autophagy inhibition in status of malignant cells, concurrently with chemotherapy or radiotherapy, has emerged as a novel approach in cancer treatment because the tumors cells survival under drug and radiation induced stress. Based on the fact that approximately 70% of clinical trials focus on the role of autophagy in cancer, it indicates that the potential for autophagy modulation in cancer treatment is promising. Clinical studies involving autophagy modulation in cancers have been designed to evaluate the effect of autophagy inhibition in combination with other conventional therapy. The use of autophagy inhibitors in combination with chemotherapy can suppress tumor growth and trigger cell death in a higher percentage than alone chemotherapy.
According to multiple clinical trials, depending on the type of tumor and its stage of development, activation or inactivation of autophagy may contribute differently to tumorigenesis. In this regard, if increased autophagy confers tumor resistance to death-inducing agents, its inhibition will allow an improved response to treatment.
There are two types of autophagy inhibitors: the early stage which blocks the formation of auto phagosomes (3-methyladenine-3 MA, wortmannin and LY294002) and late stage inhibitors present in the auto phagosome lysosome fusion and degradation phases (hydroxychloroquine- HCQ). Pharmacologic manipulation of autophagy for cancer prevention and treatment will depend on the ability of doctors to successfully recognize the functional status of autophagy in tumors and on the availability of specific autophagy modulators, [32].
Conclusions
Autophagy levels decrease with age but the activity of Atg gene complexes, through autophagic proteins (Atg), may contribute to improving lifespan by delaying ageing. In principle, the four longevity genes, mTOR, AMPK, PTEN and SIRTUINS, protect the body during periods of cellular stress by activating survival mechanisms.
Prospective Strategy for Cancer Therapy
When these genes are activated, either through diets with fewer calories and a low content of fatty amino acids and carbohydrates, combined with physical exercise, the body becomes healthier and more resistant to disease. The study of preclinical models for the promotion of autophagy, through genetic or pharmacological means, revealed the possibility of destroying malignant tumors cells and triggering apoptosis with the eventual regeneration of cell vitality.
Abbreviations
Apoptosis - the programmed death of cells
Atg-Autophagy-related genes
AMPK; 5'-AMP-activated protein kinase
MAPK- mitogen-activated protein kinases;
mTOR-mammalian target of rapamycin
PI3K-phosphatidyl inositol-4,5-bisphosphate-3-kinase
Protein p-53 - protein of the suppressor gene P-53
ULK1-UNC-51-like kinase 1
References
- Baehrecke, E.H. Autophagy: Dual roles in life and death? Nat. Rev. Mol. Cell. Biol. 2005, 6, 505-10. [Google Scholar] [CrossRef] [PubMed]
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008, 132, 27. [CrossRef]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069. [CrossRef] [PubMed]
- Carew, J.S., Kelly, K.R. and Nawrocki S.T. Autophagy as a target for cancer therapy: new developments. Cancer Manag. Res. 2012, 40(90), 357-65.
- Rabinowitz JD, White E. Autophagy and metabolism. Science 2010, 330, 34. [CrossRef]
- Antunes F, Erustes AG, Costa AJ, Nascimento AC, Bincoletto C, Ureshino RP, Pereira GJS, Smaili SS. Autophagy and intermittent fasting: the connection for cancer therapy?.2018; 10;73(suppl 1): e814s. [CrossRef]
- Tabibzadeh, S. Role of autophagy in ageing: The good, the bad, and the ugly. Aging Cell 2023, 22, e13753. [Google Scholar] [CrossRef] [PubMed]
- Brunelleschi S, Franconi F. On the role of autophagy in human diseases: a gender perspective. J. Cell. Mol. Med 2011, 7(15), 1443-57. [CrossRef]
- Feng, Z.; Zhang, H.; Levine, J.A.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2015, 102, 8204-09. [Google Scholar] [CrossRef]
- Udristioiu A, Nica Badea D. Signification of protein p-53 isoforms and immune therapeutic success in chronic lymphocytic leukemia. Biomedicine & Pharmacotherapy 2018, 106, 50–53. [CrossRef]
- Hosokawa N, Hara T, Kaizuk T, Kish, C, Takamura A, Miura Y, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13- FIP200 complex required for autophagy. Mol. Biol. Cell. 2009, 20(7), 1981-91. [CrossRef]
- Song, D., Zhao, L., Zhao, G. et al. Identification and validation of eight lysosome-related gene signatures and correlation with immune cell infiltration in lung adenocarcinoma. Cancer Cell 2023, 23, 322. [CrossRef]
- Actis C, Muzio G, Autelli R. Autophagy Triggers Tamoxifen Resistance in Human Breast Cancer Cells by Preventing Drug-Induced Lysosomal Damage. Cancers 2021, 13(6), 1252. [CrossRef]
- Feng, Z., Hu, W., Stanchina, E., Teresky, A.K., Jin, S. and Lowe, S., et al. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer. Res. 2007, 67, 3043-53. [CrossRef]
- Chen, N.; Karantza-Wadsworth, V. Role and regulation of autophagy in cancer. Biophys. Acta. 2009, 1793(9), 1516-23. [Google Scholar] [CrossRef]
- Udristioiu A, Florescu C, Radu-Popescu MA, Cojocaru M. High Concentrations of Anaerobic Adenosine-Triphosphate May Impair Apoptosis in Malignant B Cells From Patients With Chronic Lymphocytic Leukemia, Laboratory Medicine. 2010, (41)4: 203–08. [CrossRef]
- Selvaraj, S., Sun, Y., Sukumaran, P. and Singh, B.B. Resveratrol activates autophagic cell death in prostate cancer cells via downregulation of STIM1 and the mTOR pathway. Mol. Carcinog. 2016, 55, 818-30. [CrossRef]
- Sinclair DA, LaPlante MD. Life span, why we age and why don’t have to. Atria Books. New York, 2023.
- Arico S, Petiot., Bauvy C, Dubbelhuis FP, Meijer JA, Codogno P. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 2013, 276, 5243-46. [CrossRef]
- B Miller, B.C., Zhao, Z., Stephenson, L.M., Cadwell, K., Pua, H.H. and Lee, H.K., et al. The autophagy gene Atg5 plays an essential role in B lymphocyte development. Autophagy 2008, 4(3), 309-14. [CrossRef]
- Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy 2008, 4(5), 600-06. [CrossRef]
- Gimm O1, Attié-Bitach T, Lees JA, Vekemans M, Eng C. Expression of the PTEN tumor suppressor protein during human development. Hum Mol Genet. 2000, 9(11), 1633-9. [CrossRef]
- Leonardi L, Siberil S, Alifano M, Cremer I, Joubert PE. Autophagy-Related Gene Signature Highlights Metabolic and Immunogenic Status of Malignant Cells in Non-Small Cell: 2023; 16(14): 610. [CrossRef]
- Mondal SK, Sen MK. Loss of phosphatase activity in PTEN (phosphatase and tensin homolog deleted on chromosome ten) results in endometrial carcinoma in humans: An in-silico study. Heliyon. 2020, 6(1), e03106. [CrossRef]
- Worthington AK, Forsberg EK. A CRISPR view of hematopoietic stem cells: Moving innovative bioengineering into the clinic. American Journal of Hematology 2022, 97(9), 1226-35. [CrossRef]
- Barncard, C. All Science & Technology stories. University of Wisconsin–Madison. 2024 Board of Regents of the University of Wisconsin System, Accessed in December, 2023.
- Jimenez AG, Lalwani S, Cipolli W. Effects of metformin, rapamycin, and resveratrol on cellular metabolism of canine primary fibroblast cells isolated from large and small breeds as they age. Geroscience 2021, 43(4), 1669-82. [CrossRef] [PubMed]
- Wang Y, Zhou K, Li T, Xu Y, Xie C, Sun Y, Rodriguez J, Zhang S, Song J, Wang X, Blomgren K, Zhu C. Selective Neural Deletion of the Atg7 Gene Reduces Irradiation-Induced Cerebellar White Matter Injury in the Juvenile Mouse Brain by Ameliorating Oligodendrocyte Progenitor Cell Loss. Front Cell Neurosci. 2019, 13, 241. [CrossRef]
- Liu, S., Yao, S., Yang, H. et al. Autophagy: Regulator of cell death. Cell Death 2023. Dis 14, 648. [CrossRef]
- Jung YY, Kim C, Ha IJ, Lee SG, Lee J, Um JY, Ahn KS. Pyrimethamine Modulates Interplay between Apoptosis and Autophagy in Chronic Myelogenous Leukemia Cells. Int J Mol Sci. 2021, 22(15), 8147. [CrossRef] [PubMed]
- Mishra J, Bhatti GK, Sehrawat A, et al. Modulating autophagy and mitophagy as a promising therapeutic approach in neurodegenerative disorders. Life Sciences 2022, 311 Pt A, 121153. [CrossRef]
- Marinković M, Šprung M, Buljubašić M, Novak I. Autophagy Modulation in Cancer: Current Knowledge on Action and Therapy. Oxid Med Cell Longev. 2018; 31:802382. [CrossRef]
Scheme 1.
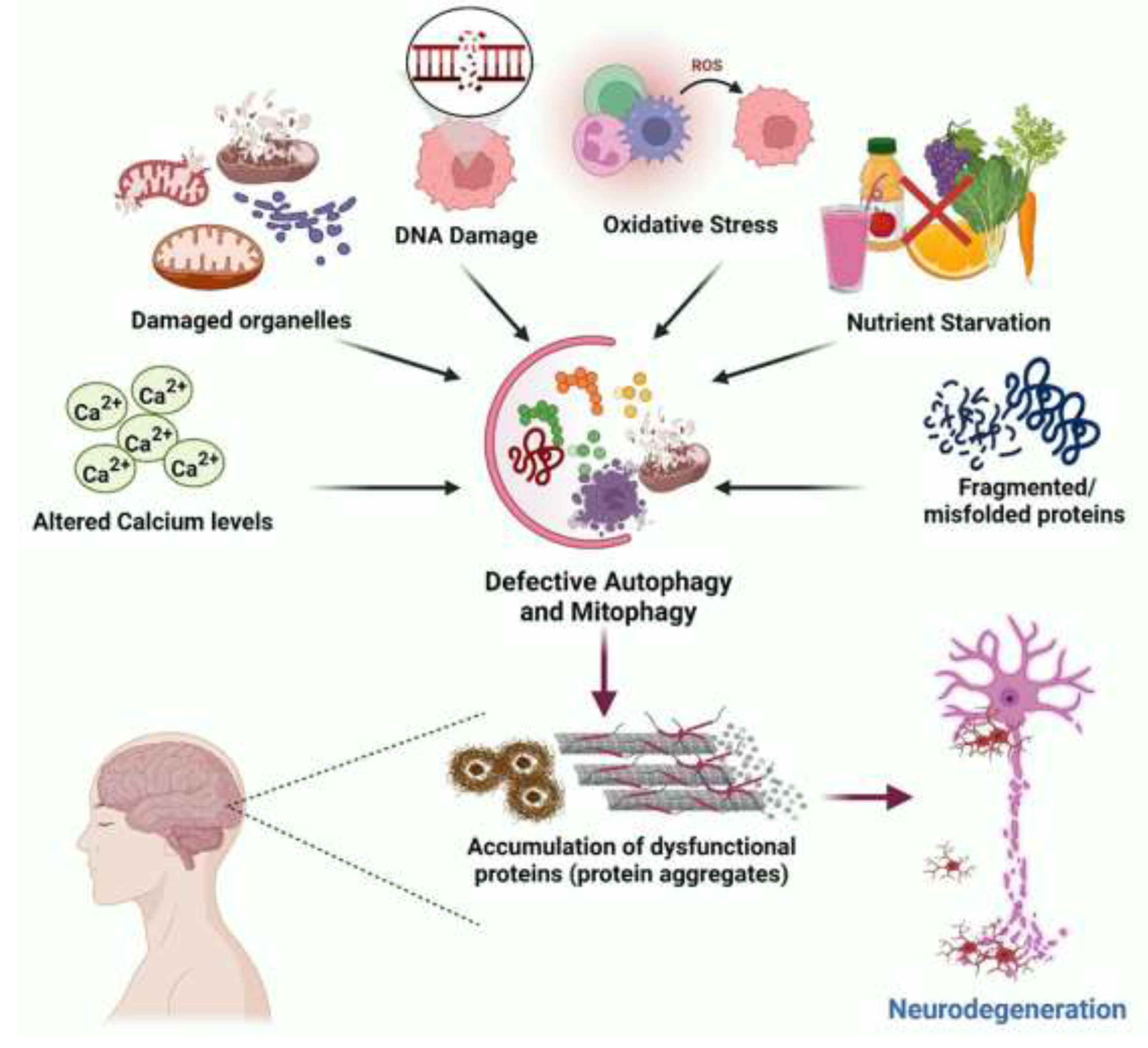
Multiple risk factors include increased ROS production, nutrient deprivation, oxidative stress, DNA damage, accumulation of misfolded proteins, damaged organelles etc., representing the complex nature of chronic diseases.
Scheme 1.
Multiple risk factors include increased ROS production, nutrient deprivation, oxidative stress, DNA damage, accumulation of misfolded proteins, damaged organelles etc., representing the complex nature of chronic diseases.
Scheme 2.
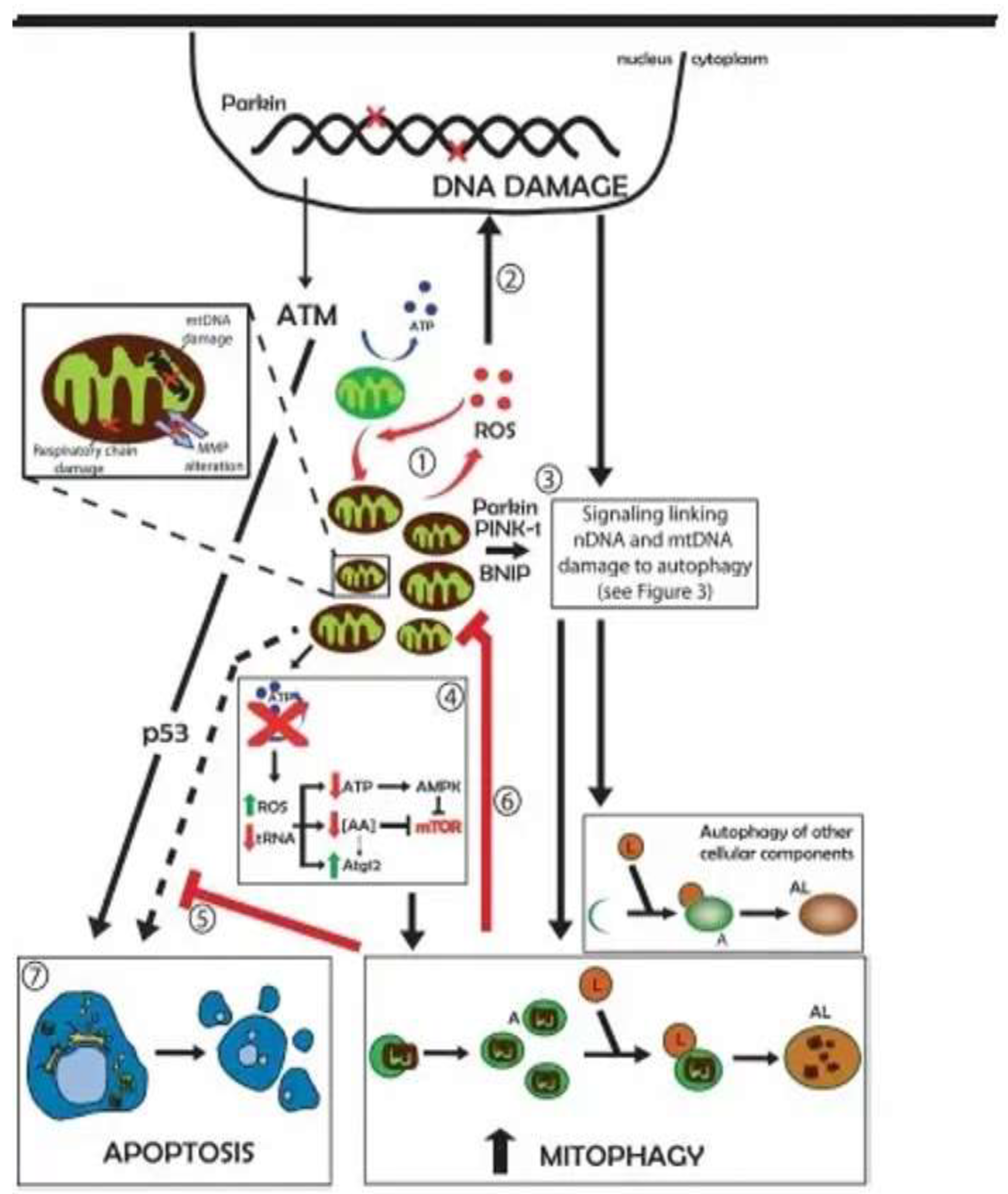
P53 protein in the isoform state amplifies DNA damage and even accelerates forms of cancer.
Scheme 2.
P53 protein in the isoform state amplifies DNA damage and even accelerates forms of cancer.

Scheme 3.
Molecular interaction between p53 proteins and autophagy in cancer cell.
Scheme 4.
Autophagy, apoptosis and Beclin-1 protein in cancer Autophagy confers stress tolerance, limits damage, and confers a survival advantage on cancer cells. In addition, autophagy may allow a quiescent state in residual cancer cells after treatment, which may contribute to cancer recurrence and metastasis. Autophagy is one of the most important survival mechanisms under stress conditions and is involved in cellular homeostasis and proliferation. Several studies have demonstrated links between autophagy and carcinogenesis, highlighting a dual role for autophagy in cancer. Depending on the tumor model and/or tumor state, autophagy may have pro- or anti-tumor effects. In the initial stage of cancer, autophagy protects normal cells from tumorigenesis by preventing DNA damage and mutations [22].
Scheme 4.
Autophagy, apoptosis and Beclin-1 protein in cancer Autophagy confers stress tolerance, limits damage, and confers a survival advantage on cancer cells. In addition, autophagy may allow a quiescent state in residual cancer cells after treatment, which may contribute to cancer recurrence and metastasis. Autophagy is one of the most important survival mechanisms under stress conditions and is involved in cellular homeostasis and proliferation. Several studies have demonstrated links between autophagy and carcinogenesis, highlighting a dual role for autophagy in cancer. Depending on the tumor model and/or tumor state, autophagy may have pro- or anti-tumor effects. In the initial stage of cancer, autophagy protects normal cells from tumorigenesis by preventing DNA damage and mutations [22].
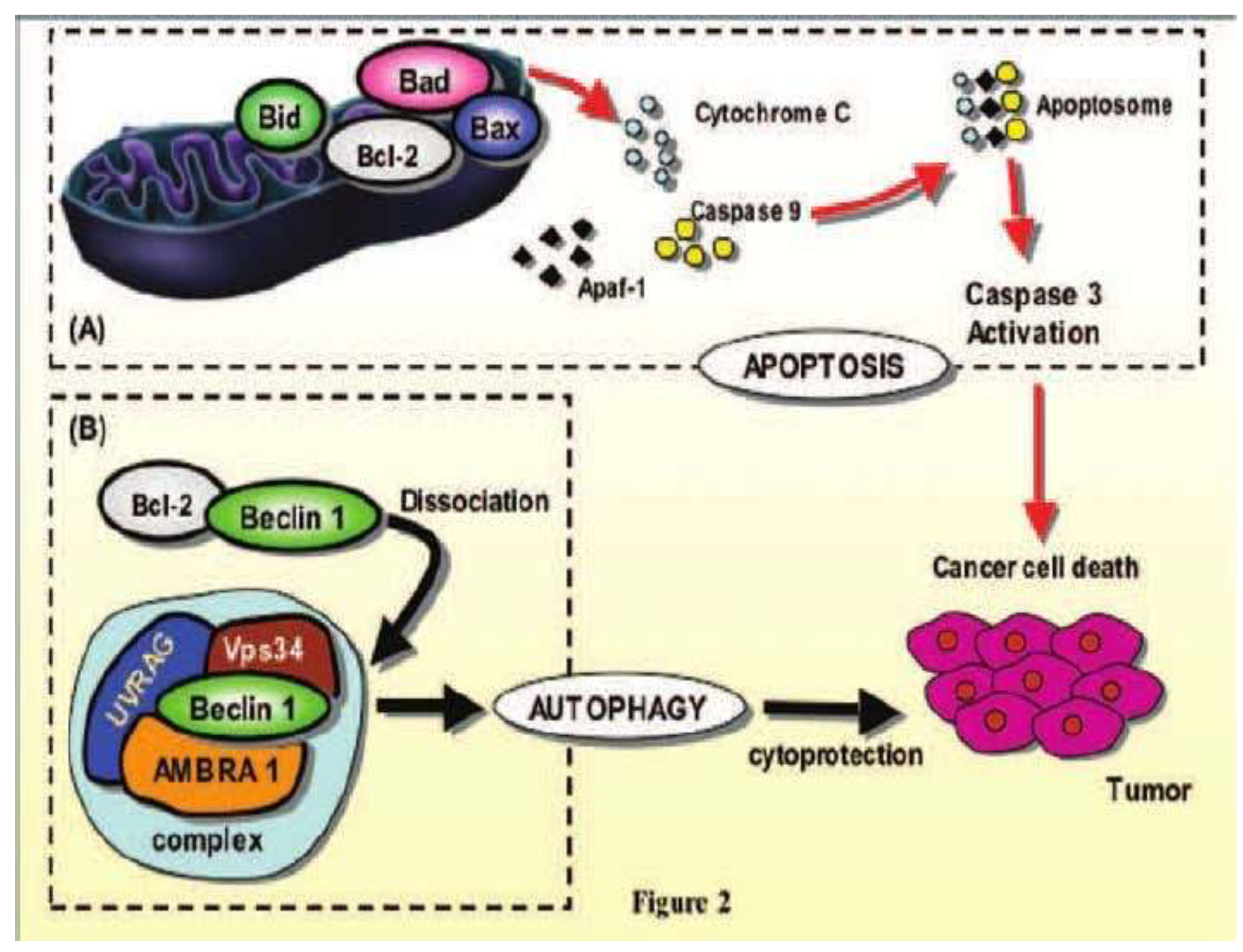
Figure 1.
The memory gene is important for all cells to communicate RNA information.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



